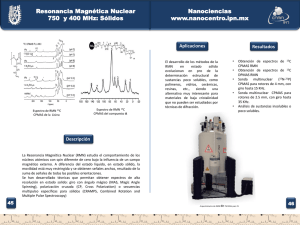
síntesis y caracterización de poli(amida)sy po
Anuncio
sy po")
DIRECCIÓN DE INVESTIGACIÓN Y POSTGRADO DEPARTAMENTO DE QUÍMICA ORGÁNICA “SÍNTESIS Y CARACTERIZACIÓN DE POLI(AMIDA)S Y POLI(IMIDA)S FLEXIBLES BASADAS EN UNIDADES DIFENILSILANO Y/O OXIARENOS. CORRELACIÓN ESTRUCTURA-PROPIEDAD” ALAIN TUNDIDOR CAMBA Tesis para optar al Grado Académico de Doctor en Química. Profesor Tutor Profesor Co-Tutor : : Santiago de Chile, Junio de 2013 Dr. Claudio Terraza I. Dr. Luis Hernán Tagle D. DEDICATORIA A mi familia: A mi esposa Deysma, por todo el amor y la paciencia que ha sabido darme durante estos años. ¿Sabes que te amo? A mi hijo Marcelo, porque tu sonrisa es como caminar por las calles de Varadero, nunca te cansas. A mi hijo Marcos, por recordarme nuevamente lo que es ser papá. A los dos, porque la alegría que desprenden me ayuda a continuar adelante. A mi mamá Zuleika y mi papá Eugenio, por ser mis padres y quererme tanto. A los dos por apoyarme incondicionalmente en todas mis decisiones. A mi hermano Raydel, el hermano que cualquier persona quisiera tener, a ti, porque te quiero. A mis sobrinos, a Marila y a Mirian, porque han sabido formar parte de mi familia. A Deysi, por ser la suegra que todos quisieran. A Edenia, por comportarte como una madre en Chile. Gracias por todo. A mis amigos A Julio, porque nos criamos juntos en Cuba y seguimos juntos en Chile, por aconsejarme estudiar química. A Alex, porque me enseñaste que la amistad perdura en el tiempo. A Ibel y Maryorie, porque no puedo mencionar a una sin dejar de decir la otra, esta tesis también es de ustedes. A Yamilé, Ileana y Ana Paula, porque saben que son parte de la familia. A Ignacio, porque tú sabes que estamos destinados a seguir cerca, de eso no me queda duda. A Danahe, Julito, Johana, Ana, Luis, Pedro, Gielen, Carlos, Ana (Che), a mis amigos del laboratorio (especialmente a Pablo) y algunos que ya no están, porque son una pequeña parte de mi vida. A mis profesores Al Profe Terraza, como suelo llamarle, esta tesis es suya aunque aún no sepa pronunciar bien mi nombre. Al Profesor Tagle, porque desde que nos conocimos en su oficina supimos que lo lograríamos. Muchas gracias. 4 AGRADECIMIENTOS Este trabajo de tesis fue desarrollado gracias a muchas personas y entidades y quisiera dejar plasmado con unas simples palabras lo agradecido que estoy a: La Facultad de Química de la Pontificia Universidad Católica de Chile por haber hecho posible que pudiera desarrollar mis estudios de Doctorado en esta prestigiosa Universidad. La Comisión Nacional de Ciencia y Tecnología (CONICYT) por las becas que permitieron, tanto mi manutención en Chile como el financiamiento para el desarrollo de este trabajo de investigación, a través de: 1. Beca Nacional de Doctorado 2009. 2. Beca a la Participación a Eventos y Cursos Cortos en el Extranjero 2010. 3. Beca Apoyo a la Realización de Tesis Doctoral 2011. 4. Beca de Pasantía en el Extranjero 2012. 5. Beca de Término de Tesis 2013. A la VRAID por la beca de Inscripción a Congresos Internacionales 2011 Al Dr. Javier de Abajo por aceptarme en su grupo de investigación en el Instituto de Ciencia y Tecnología de Polímeros del CSIC, España. Al Dr. Toro-Labbe por indicarme el camino del doctorado en el año 2006, cuando recién llegué a Chile. A mis profesores, Dr. Claudio Terraza y Dr. Luis Hernán Tagle por ser mis directores y tutores en este trabajo y por todo su apoyo y todo lo que me enseñaron. A Chile, por acogerme y finalmente concederme la Nacionalidad. 6 LISTA DE ABREVIACIONES NMP = N-metil-2-pirrolidona DMSO = dimetilsulfóxido DMAc = N,N-dimetilacetamida DMF = N,N-dimetilformamida Py = piridina CDCl3 = cloroformo deuterado THF = tetrahidrofurano DMSO-d6 = dimetilsulfóxido deuterado Acetona-d6 = acetona deuterada Ac2O = anhídrido acético Ph = fenil Me = metil TDT = temperatura de descomposición térmica Tg = temperatura de transición vítrea Tf = temperatura de fusión Te = temperatura de ebullición DSC = calorimetría diferencial de barrido Rc = residuo carbónico T400 = transmitancia a 400 nm ηinh = viscosidad inherente Mn = masa molecular promedio en número Mw = masa molecular promedio en peso P = presión GPC = cromatografía de permeación de gel PAs = poli(amida)s PIs = poli(imida)s 8 ÍNDICE GENERAL Página Dedicatoria 3 Agradecimientos 5 Lista de Abreviaciones 7 Índice de Tablas 12 Índice de Figuras 14 Resumen 25 Abstract 27 INTRODUCCIÓN 29 I. Breve reseña histórica 30 II. Tipos de polímeros según su mecanismo de polimerización 32 III. Polímeros de alto rendimiento 33 IV. Poli(amida)s (PAs) 34 V. Poli(imida)s (PIs) 39 VI. Poli(amida)s y poli(imida)s con estructuras modificadas 42 VII. Polímeros que contienen silicio en su estructura 45 VIII. Hipótesis 50 IX. Objetivos generales 51 X. Objetivos específicos 51 PARTE EXPERIMENTAL 52 I. Información general 53 II. Síntesis de precursores 55 III. Síntesis de monómeros 65 IV. Síntesis de poli(amida)s 85 V. Síntesis de poli(imida)s 92 RESULTADOS Y DISCUSIÓN 98 I. Síntesis de precursores indirectos 99 II. Síntesis de monómeros 108 III. Síntesis de poli(amida)s 135 IV. Síntesis de poli(imida)s 146 10 V. Viscosidad inherente 158 VI. Cromatografía de exclusión por tamaños (SEC) 161 VII. Solubilidad 165 VIII. Propiedades térmicas 173 IX. Transparencia óptica 184 CONCLUSIONES 190 BIBLIOGRAFÍA 193 ANEXOS 202 PUBLICACIONES 303 11 ÍNDICE DE TABLAS Página Tabla 1. Valores de viscosidad inherente de las PAs y PIs 160 sintetizadas. Tabla 2. Datos obtenidos del análisis de cromatografía de 162 exclusión por tamaño para PI-I. Tabla 3. Peso molecular viscosimétrico para las poli(imida)s de la 164 Serie A. Tabla 4. Valores de parámetro de solubilidad de algunos 166 disolventes. Tabla 5. Solubilidad de PAs en algunos disolventes orgánicos 167 comunes. Tabla 6. Solubilidad de PIs en algunos disolventes orgánicos 170 comunes. Tabla 7. Propiedades térmicas de las PAs. 175 Tabla 8. Propiedades térmicas de las PIs. 179 Tabla 9. Propiedades ópticas de las PAs y PIs sintetizadas. 186 13 ÍNDICE DE FIGURAS SECCIÓN INTRODUCCIÓN Figura 1. Estructura cíclica de un dímero de isopreno en agregados Página 31 de n unidades. Figura 2. Interacciones por puente de hidrógeno entre los grupos 35 amida de las cadenas poliméricas. Figura 3. Esquema de síntesis del nylon-6,6 y el nylon-6. 36 Figura 4. Síntesis de poli(amida)s a baja temperatura empleando 37 N,N-dimetilacetamida (DMAc) como solvente (A) y mediante policondensación directa en solución de N-metil-2-pirrolidona (NMP) (B). Figura 5. Formación de complejos de transferencia de carga en 40 poli(imida)s. Figura 6. Esquema general de síntesis de poli(imida)s, donde se 40 resalta su precursor inmediato; el ácido poliámico (a). Figura 7. Ejemplos de monómeros difuncionales con fragmentos 44 flexibles en su estructura. Figura 8. Poli(amida)s y poli(imida)s aromáticas comerciales que 45 presentan modificaciones estructurales. Figura 9. Poli(amida-imidas) que contienen silicio. 47 Figura 10. Ejemplos de poli(amida)s sililadas solubles en disolventes 47 orgánicos comunes. Figura 11. Polímeros semiconductores organosililados. 48 Figura 12. Material sililado transparente en la región UV-vis. 48 Figura 13. Ejemplo de poli(imida) que contiene silicio con aplicación 49 en el campo de membranas separadoras de gases. SECCIÓN RESULTADOS Y DISCUSIÓN Figura 1. Ruta I para la síntesis de los compuestos 1 y 2. 99 Figura 2. Ruta II para la síntesis de los difenoles 1 y 2. 101 Figura 3. Rutas III y IV alternativas para la preparación del difenol 2. 102 15 Figura 4. Ruta de síntesis para la obtención del compuesto 6. 103 Figura 5. Ruta de síntesis para la obtención del compuesto A. 104 Figura 6. Ruta de síntesis para la obtención del compuesto B. 105 Figura 7. Ruta de síntesis para la obtención del compuesto C. 106 Figura 8. Ruta de síntesis para la obtención de los bis(4-(4- 108 aminofenoxi)fenil)diorganosilano. Figura 9. Ruta de síntesis para la obtención de los ácidos bis(4-(4- 111 carboxifenoxi)fenil)diorganosilano. Figura 10. Mecanismo aceptado para la hidrólisis básica del grupo 113 nitrilo. Figura 11. Ruta I propuesta para la obtención de los anhídridos bis(4- 115 (3,4-dicarboxifenoxi)fenil)diorganosilano. Figura 12. Ruta II para la obtención de los anhídridos bis(4-(3,4- 116 dicarboxifenoxi)fenil)diorganosilano. Figura 13. Reacción colateral debido a la presencia del ion nitrito. 117 Figura 14. Mecanismo de deshidratación química usando anhídrido 118 acético como agente deshidratante. Figura 15. Ruta de síntesis para la obtención de los monómeros 21 y 120 22. Figura 16. Formación del agente nitrante en la reacción de nitración 120 de dibenzofurano. Figura 17. Reacción entre el ácido nítrico y el anhídrido acético a 121 temperaturas superiores a 50 °C. Figura 18. Curvas de energía potencial calculadas en función de la 122 distancia entre el átomo de carbono del dibenzofurano y el átomo de nitrógeno del ion nitronio. Figura 19. Mecanismos propuestos para la nitración de 122 dibenzofurano. Figura 20. Ruta I para la obtención del monómero 24. 124 Figura 21. Efecto resonante del átomo de oxígeno en el sustrato 6 y 125 16 en 4-bromofenol. Figura 22. Ruta II propuesta para la obtención del monómero 24. 126 Figura 23. Mecanismo propuesto en la reacción de acoplamiento C-C 127 para la obtención del compuesto 23. Figura 24. Ruta de síntesis para la obtención del monómero 26. 129 Figura 25. Ruta de síntesis para la obtención del monómero 29. 130 Figura 26. Monómeros a obtener a partir de los precursores 132 difenólicos 1 y 2. Figura 27. Curso del ataque nucleofílico promovido por el difenolato 133 derivado de 1 sobre los sustratos A y B propuestos. Figura 28. Dianhídridos de ácido disililados propuestos en el proyecto 134 de tesis. Figura 29. Ruta de síntesis para la obtención de las poli(amida)s PA- 135 I, PA-II, PA-III y PA-IV pertenecientes a la Serie A. Figura 30. Mecanismo propuesto por Higashi en la reacción de 136 poliamidación directa. Figura 31. Mecanismo propuesto por Aharoni en presencia de 136 piridina. Figura 32. Mecanismo propuesto por Aharoni en ausencia de piridina. 137 Figura 33. Diaminas propuestas en el proyecto de tesis que contienen 140 cuatro grupos flexibles de oxiarenos. Figura 34. Ruta de síntesis para la obtención de la poli(amida) PA-V 140 (Serie B). Figura 35. Poli(amida)s propuestas en el proyecto de tesis. 141 Figura 36. Ruta de síntesis para la obtención de las poli(amida)s PA- 143 VI, PA-VII, PA-VIII y PA-IX. Figura 37. Ruta de síntesis para la obtención de los ácidos 144 dicarboxílicos MM, EM, MP y PP. Figura 38. Ruta de síntesis para la obtención de PI-I, PI-II, PI-III y PIIV. 17 146 Figura 39. Mecanismo de formación del ácido poliámico. 146 Figura 40. Mecanismo propuesto para la imidación química usando 149 anhídrido acético y piridina. Figura 41. Ruta de síntesis para la obtención de PI-V y PI-VI. 152 Figura 42. Ruta de síntesis para la obtención de PI-VII. 153 Figura 43. Ruta de síntesis para la obtención de PI-VIII y PI-IX. 155 Figura 44. Diaminas y dianídridos de ácido propuestos en el proyecto 157 de tesis que contienen cuatro grupos flexibles de oxiarenos cada uno (R= Me o Ph). Figura 45. Esquema que representa la relación existente entre la 158 viscosidad reducida e inherente con la concentración polimérica. Figura 46. A. Curvas TGA de las PAs sintetizadas (20 °C/min en 174 atmósfera de N2); B. Curvas DTGA para las distintas Series de PAs. Figura 47. A. Curvas TGA de las PIs (20 °C/min en atmósfera de 178 nitrógeno); B. Curvas DTGA para las distintas Series de PIs. Figura 48. Curvas DSC de las PAs sintetizadas (segundo barrido a 180 10 °C/min en atmósfera de nitrógeno). Figura 49. Curvas DSC de las PIs sintetizadas (segundo barrido a 10 183 °C/min en atmósfera de nitrógeno). Figura 50. Curvas de transmitancia para las PAs. 187 Figura 51. Curvas de transmitancia de las PIs. 189 SECCIÓN ANEXO Figura 1. Espectro FTIR del compuesto 1. 203 Figura 2. Espectro RMN 1H del compuesto 1. 203 13 C del compuesto 1. 204 Figura 4. Espectro RMN 29Si del compuesto 1. 204 Figura 5. Espectro FTIR del compuesto 2. 205 Figura 6. Espectro RMN 1H del compuesto 2. 205 Figura 7. Espectro RMN 13C del compuesto 2. 206 Figura 8. Espectro RMN 29Si del compuesto 2. 206 Figura 3. Espectro RMN 18 Figura 9. Espectro FTIR del compuesto 3. 207 1 Figura 10. Espectro RMN H del compuesto 3. 207 Figura 11. Espectro RMN 13C del compuesto 3. 208 Figura 12. Espectro FTIR del compuesto 4. 209 Figura 13. Espectro RMN 1H del compuesto 4. 209 Figura 14. Espectro RMN 13C del compuesto 4. 210 Figura 15. Espectro RMN 29Si del compuesto 4. 210 Figura 16. Espectro FTIR del compuesto 5. 211 Figura 17. Espectro RMN 1H del compuesto 5. 211 Figura 18. Espectro RMN 13C del compuesto 5. 212 Figura 19. Espectro RMN 29Si del compuesto 5. 212 Figura 20. Espectro FTIR del compuesto 6. 213 Figura 21. Espectro RMN 1H del compuesto 6. 213 Figura 22. Espectro RMN 13 C del compuesto 6. 214 Figura 23. Espectro FTIR del compuesto A. 215 Figura 24. Espectro RMN 1H del compuesto A. 215 Figura 25. Espectro RMN 13C del compuesto A. 216 Figura 26. Espectro FTIR del compuesto ácido 4-(4- 217 bromofenoxi)ftálico. Figura 27. Espectro RMN 1 RMN 13 H del compuesto ácido 4-(4- 217 del compuesto ácido 4-(4- 218 bromofenoxi)ftálico. Figura 28. Espectro C bromofenoxi)ftálico. Figura 29. Espectro FTIR del compuesto B. 1 219 Figura 30. Espectro RMN H del compuesto B. 219 Figura 31. Espectro RMN 13C del compuesto B. 220 Figura 32. Espectro FTIR del compuesto C. 221 Figura 33. Espectro RMN 1H del compuesto C. 221 Figura 34. Espectro RMN 13C del compuesto C. 222 Figura 35. Espectro RMN 29Si del compuesto C. 222 19 Figura 36. Espectro FTIR del compuesto 7. 1 223 Figura 37. Espectro RMN H del compuesto 7. 223 Figura 38. Espectro RMN 13C del compuesto 7. 224 Figura 39. Espectro RMN 29Si del compuesto 7. 224 Figura 40. Espectro FTIR del compuesto 8. 225 Figura 41. Espectro RMN 1H del compuesto 8. 225 Figura 42. Espectro RMN 13C del compuesto 8. 226 Figura 43. Espectro RMN 29 Si del compuesto 8. 226 Figura 44. Espectro FTIR del compuesto 9. 227 Figura 45. Espectro RMN 1H del compuesto 9. 227 Figura 46. Espectro RMN 13C del compuesto 9. 228 Figura 47. Espectro RMN 29Si del compuesto 9. 228 Figura 48. Espectro FTIR del compuesto 10. 229 1 Figura 49. Espectro RMN H del compuesto 10. 229 Figura 50. Espectro RMN 13C del compuesto 10. 230 Figura 51. Espectro RMN 29Si del compuesto 10. 230 Figura 52. Espectro FTIR del compuesto 11. 231 Figura 53. Espectro RMN 1H del compuesto 11. 231 Figura 54. Espectro RMN 13 C del compuesto 11. 232 Figura 55. Espectro RMN 29 Si del compuesto 11. 232 Figura 56. Espectro FTIR del compuesto 12. 233 Figura 57. Espectro RMN 1H del compuesto 12. 233 Figura 58. Espectro RMN 13C del compuesto 12. 234 Figura 59. Espectro RMN 29Si del compuesto 12. 234 Figura 60. Espectro FTIR del compuesto 13. 235 Figura 61. Espectro RMN 1H del compuesto 13. 235 Figura 62. Espectro RMN 13C del compuesto 13. 236 Figura 63. Espectro RMN 29Si del compuesto 13. 236 Figura 64. Espectro FTIR del compuesto 14. 237 Figura 65. Espectro RMN 1H del compuesto 14. 237 20 Figura 66. Espectro RMN 13C del compuesto 14. Figura 67. Espectro RMN 29 238 Si del compuesto 14. 238 Figura 68. Espectro FTIR del compuesto 15. 239 Figura 69. Espectro RMN 1H del compuesto 15. 239 Figura 70. Espectro RMN 13C del compuesto 15. 240 Figura 71. Espectro RMN 29Si del compuesto 15. 240 Figura 72. Espectro FTIR del compuesto 16. 241 1 Figura 73. Espectro RMN H del compuesto 16. 241 Figura 74. Espectro RMN 13C del compuesto 16. 242 Figura 75. Espectro RMN 29Si del compuesto 16. 242 Figura 76. Espectro FTIR del compuesto 17. 243 Figura 77. Espectro RMN 1H del compuesto 17. 243 Figura 78. Espectro RMN 13C del compuesto 17. 244 Figura 79. Espectro RMN 29 Si del compuesto 17. 244 Figura 80. Espectro FTIR del compuesto 18. 245 Figura 81. Espectro RMN 1H del compuesto 18. 245 Figura 82. Espectro RMN 13C del compuesto 18. 246 Figura 83. Espectro RMN 29Si del compuesto 18. 246 Figura 84. Espectro FTIR del compuesto 19. 247 1 Figura 85. Espectro RMN H del compuesto 19. 247 Figura 86. Espectro RMN 13C del compuesto 19. 248 Figura 87. Espectro RMN 29Si del compuesto 19. 248 Figura 88. Espectro FTIR del compuesto 20. 249 Figura 89. Espectro RMN 1H del compuesto 20. 249 13 C del compuesto 20. 250 Figura 91. Espectro RMN 29Si del compuesto 20. 250 Figura 90. Espectro RMN Figura 92. Espectro FTIR de la mezcla de 2,7 y 2,8- 251 dinitrodibenzofurano. Figura 93. Espectro FTIR del compuesto 21. 252 Figura 94. Espectro RMN 1H del compuesto 21. 252 21 Figura 95. Espectro RMN 13C del compuesto 21. 253 Figura 96. Espectro FTIR del compuesto 22. 254 Figura 97. Espectro RMN 1H del compuesto 22. 254 Figura 98. Espectro RMN 13C del compuesto 22. 255 Figura 99. Espectro FTIR del compuesto 23. 256 Figura 100. Espectro FTIR del compuesto 24. 257 Figura 101. Espectro RMN 1H del compuesto 24. 257 Figura 102. Espectro RMN 13 C del compuesto 24. 258 Figura 103. Espectro FTIR del compuesto 25. 259 Figura 104. Espectro FTIR del compuesto 26. 260 Figura 105. Espectro RMN 1H del compuesto 26. 260 Figura 106. Espectro RMN 13C del compuesto 26. 261 Figura 107. Espectro FTIR del compuesto 27. 262 1 Figura 108. Espectro RMN H del compuesto 27. 262 Figura 109. Espectro RMN 13C del compuesto 27. 263 Figura 110. Espectro FTIR del compuesto 28. 264 Figura 111. Espectro RMN 1H del compuesto 28. 264 Figura 112. Espectro RMN 13C del compuesto 28. 265 Figura 113. Espectro FTIR del compuesto 29. 266 1 Figura 114. Espectro RMN H del compuesto 29. 266 Figura 115. Espectro RMN 13C del compuesto 29. 267 Figura 116. Espectro FTIR de la PA-I. 268 Figura 117. Espectro RMN 1H de la PA-I. 268 Figura 118. Espectro RMN 13C de la PA-I. 269 Figura 119. Espectro RMN 29 Si de la PA-I. 269 Figura 120. Espectro FTIR de la PA-II. 270 Figura 121. Espectro RMN 1H de la PA-II. 270 Figura 122. Espectro RMN 13C de la PA-II. 271 Figura 123. Espectro RMN 29Si de la PA-II. 271 Figura 124. Espectro FTIR de la PA-III. 272 22 Figura 125. Espectro RMN 1H de la PA-III. 13 272 C de la PA-III. 273 Figura 127. Espectro RMN 29Si de la PA-III. 273 Figura 128. Espectro FTIR de la PA-IV. 274 Figura 129. Espectro RMN 1H de la PA-IV. 274 Figura 130. Espectro RMN 13C de la PA-IV. 275 Figura 131. Espectro RMN 29Si de la PA-IV. 275 Figura 132. Espectro FTIR de la PA-V. 276 Figura 133. Espectro RMN 1H de la PA-V. 276 Figura 134. Espectro RMN 13C de la PA-V. 277 Figura 135. Espectro RMN 29Si de la PA-V. 277 Figura 136. Espectro FTIR de la PA-VI. 278 Figura 137. Espectro RMN 1H de la PA-VI. 278 Figura 126. Espectro RMN Figura 138. Espectro RMN 13 C de la PA-VI. 279 Figura 139. Espectro RMN 29Si de la PA-VI. 279 Figura 140. Espectro FTIR de la PA-VII. 280 Figura 141. Espectro RMN 1H de la PA-VII. 280 Figura 142. Espectro RMN 13C de la PA-VII. 281 Figura 143. Espectro RMN 29 Si de la PA-VII. 281 Figura 144. Espectro FTIR de la PA-VIII. 282 Figura 145. Espectro RMN 1H de la PA-VIII. 282 Figura 146. Espectro RMN 13C de la PA-VIII. 283 Figura 147. Espectro RMN 29Si de la PA-VIII. 283 Figura 148. Espectro FTIR de la PA-IX. 284 1 Figura 149. Espectro RMN H de la PA-IX. 284 Figura 150. Espectro RMN 13C de la PA-IX. 285 Figura 151. Espectro RMN 29Si de la PA-IX. 285 Figura 152. Espectro FTIR de la PI-I. 286 Figura 153. Espectro RMN 1H de la PI-I. 286 Figura 154. Espectro RMN 13C de la PI-I. 287 23 Figura 155. Espectro RMN 29Si de la PI-I. 287 Figura 156. Espectro FTIR de la PI-II. 288 Figura 157. Espectro RMN 1H de la PI-II. 288 Figura 158. Espectro RMN 13C de la PI-II. 289 Figura 159. Espectro RMN 29Si de la PI-II. 289 Figura 160. Espectro FTIR de la PI-III. 290 Figura 161. Espectro RMN 1H de la PI-III. 290 Figura 162. Espectro RMN 13 C de la PI-III. 291 Figura 163. Espectro RMN 29Si de la PI-III. 291 Figura 164. Espectro FTIR de la PI-IV. 292 Figura 165. Espectro RMN 1H de la PI-IV. 292 Figura 166. Espectro RMN 13C de la PI-IV. 293 Figura 167. Espectro RMN 29Si de la PI-IV. 293 Figura 168. Espectro FTIR de la PI-V. 294 Figura 169. Espectro FTIR de la PI-VI. 295 Figura 170. Espectro RMN 1H de la PI-VI. 295 Figura 171. Espectro RMN 13C de la PI-VI. 296 Figura 172. Espectro RMN 29Si de la PI-VI. 296 Figura 173. Espectro FTIR de la PI-VII. 297 1 Figura 174. Espectro RMN H de la PI-VII. 297 Figura 175. Espectro RMN 13C de la PI-VII. 298 Figura 176. Espectro RMN 29Si de la PI-VII. 298 Figura 177. Espectro FTIR de la PI-VIII. 299 Figura 178. Espectro RMN 1H de la PI-VIII. 299 Figura 179. Espectro RMN 13 C de la PI-VIII. 300 Figura 180. Espectro FTIR de la PI-IX. 301 Figura 181. Espectro RMN 1H de la PI-IX. 301 Figura 182. Espectro RMN 13C de la PI-IX. 302 24 RESUMEN Los polímeros de alto rendimiento o altas prestaciones son materiales que presentan propiedades bien definidas, como elevada resistencia térmica, mecánica y química. Sin embargo, la baja solubilidad en disolventes orgánicos comunes, los elevados valores de temperatura de transición vítrea y de fusión, o bien, la infusibilidad de dichos materiales ha limitado sus aplicaciones. La incorporación de unidades flexibles, heteroátomos, sustituyentes voluminosos o estructuras asimétricas, son algunas de las modificaciones estructurales que se pueden realizar en la cadena principal del polímero para mejorar su procesabilidad industrial. En este sentido, las poli(amida)s (PAs) y las poli(imida)s (PIs) aromáticas que incorporan unidades difenilsilano en su estructura, así como unidades flexibles de oxiareno, presentan valores bajos de Tg y al mismo tiempo, un alto grado de transparencia en la región UV-visible. Estas modificaciones en la estructura polimérica permiten mantener aún elevadas temperaturas de descomposición térmica a la vez que modifican positivamente los parámetros de solubilidad de estos materiales, características deseables en cualquier aplicación industrial. En este trabajo se realiza la síntesis y caracterización de PAs y PIs aromáticas que contienen en su estructura unidades difenilsilano y/o oxiarenos. Los monómeros se preparan a partir de tres estructuras básicas: 9,9-bis(4-hidroxi-3-metilfenil)fluoreno, bis(4-hidroxifenil)diorganosilano y dibenzofurano. Las propiedades de estos materiales se correlacionan con la estructura de la unidad repetitiva de los polímeros. 26 ABSTRACT High performance polymers are materials that exhibit well defined properties, such as high thermal, mechanical and chemical resistance. However, the low solubility in common organic solvents, the high glass transition temperature and melting point, or the infusibility of these materials has limited their applications. The incorporation of flexible units, heteroatoms, bulky substituents or asymmetric structures are some of the structural modifications that can be made in the polymer backbone to improve its industrial processability. In this sence, aromatic poly(amide)s (PAs) and poly(imide)s (PIs) having diphenylsilane units in their structure, as well as, diphenylether units exhibits low Tg values, and the same time, a high degree of transparency in the UVvisible region. These estructural modifications maintain high thermal decomposition temperatures and modify positively the solubility parameters of the materials, desirable characteristics in any industrial application. This work report the synthesis and characterization of aromatic PAs and PIs containing diphenylsilane and/or diphenylether units in their structure. The monomers are prepared from three basic structures: 9,9-bis(4-hydroxy-3methylphenyl)fluorene, bis(4-hydroxyphenyl)diorganosilano and dibenzofuran. The properties of these materials are correlated with the structure of the repeating unit of the polymers. 28 INTRODUCCIÓN I. BREVE RESEÑA HISTÓRICA Los polímeros se definen como estructuras macromoleculares formadas por un gran número de unidades repetitivas ligadas entre sí por enlaces covalentes, concepto que no fue aceptado hasta la década de 1920 -1930. Los primeros estudios de esta ciencia se remontan a la necesidad del ser humano por imitar las propiedades de los polímeros naturales, sustancias que han estado presentes en la biósfera desde su origen. Casi toda sustancia animal o vegetal está formada por una estructura biológica polimérica compleja. El avance en el desarrollo de la química de los polímeros estuvo detenido durante muchos años fundamentalmente por dos factores objetivos. El primero de ellos fue por parte de los químicos orgánicos al no aceptar obtener sustancias resinosas, no destilables y poco solubles, en vez de compuestos puros, preferiblemente cristalinos a los cuales se les podía registrar con plena confianza su composición química, propiedades físicas, etc. El segundo fue una hipótesis química-física que se iba definiendo por esa época acerca de un cuarto estado de la materia: el estado coloidal. Los estudios de Thomas Gram (1) y Wolfgang Ostwald (2) mostraban que el estado coloidal era una dispersión de partículas, cada una de ellas formadas por un gran número de moléculas agregadas debido a interacciones físicas, y como los polímeros en disolución presentaban un comportamiento muy parecido al de estas partículas, se creyó que polímeros como la celulosa o el almidón debían sus propiedades gracias a que sus moléculas se encontraban asociadas formando agregados muy difíciles de separar. Una vez desarrollada las leyes de las disoluciones y los métodos coligativos por Raoult (3) y Van’t Hoff (4), se determinó la masa molecular de varios polímeros, arrojando valores elevadísimos, por lo que prevaleció la opinión 30 de que esos resultados estaban equivocados, y que lo que se estaba midiendo era la masa de la partícula coloidal que a su vez estaba formada por cientos de miles de moléculas, en vez de la masa molecular de una sola molécula. Al aplicar las técnicas de análisis elemental a los polímeros, se obtenían valores iguales a los de los monómeros. Asimismo, pensar en cadenas lineales no era correcto, debido a que los grupos terminales no aparecían en el análisis elemental. Debido a esto, se proponían estructuras cíclicas en agregados de n unidades (5). Figura 1. Estructura cíclica de un dímero de isopreno en agregados de n unidades. No fue hasta 1926, donde el químico alemán Hermann Staudinger (6) implantó una interpretación correcta y aceptada acerca de la estructura de las sustancias poliméricas, como macromoléculas físicamente indivisibles. Sus trabajos experimentales y argumentaciones teóricas por sus descubrimientos en el campo de la química de los polímeros, lo llevaron a ganar el Premio Nobel de Química en 1953. El avance en esta materia se refleja en el desarrollo vertiginoso de la industria de los materiales plásticos, lo que ha provocado que la ciencia de los polímeros sea de imprescindible dominio para un número cada vez mayor de científicos. Anualmente se producen en el mundo millones de toneladas 31 de estos materiales que se utilizan tanto en aplicaciones corrientes como especiales (electrónica, tecnología espacial, medicina, entre otros). II. TIPOS DE POLÍMEROS SEGÚN SU MECANISMO DE POLIMERIZACIÓN Los polímeros se pueden clasificar de acuerdo a varios criterios, como por ejemplo, según su origen en naturales y sintéticos; según su estructura en lineales, ramificados, estrellados o bien entrecruzados; según el tipo de monómero que lo forma se encuentran los homopolímeros y los copolímeros. En particular, los co-polímeros se pueden clasificar según su microestructura como al azar, en bloques o alternados. Finalmente, según el mecanismo de formación, los polímeros pueden ser agrupados en materiales poliadicionados o policondensados. Este último criterio propone que un polímero es un poliadicionado si en la reacción de polimerización que le dio origen se obtiene un polímero cuya unidad repetitiva presenta la misma composición química que la del monómero o monómeros de partida. Por otro lado, un polímero se considera un policondensado si la composición química de la unidad repetitiva carece de algunos átomos presentes en la estructura del monómero o monómeros de partida. Esta clasificación fue introducida por Carothers (7) en 1929. Así, las reacciones de polimerización donde se obtienen poliadicionados se denominan reacciones de poliadición, mientras que las reacciones donde se obtienen policondensados se conocen como reacciones de policondensación. La mayoría de los monómeros que polimerizan por un mecanismo de poliadición son los llamados monómeros vinílicos. Este mecanismo transcurre a través de una reacción en cadena, donde de forma general, como primer paso ocurre una reacción de iniciación, activando una pequeña parte de los monómeros (centros activos), luego ocurre la propagación que 32 no es más que la adición a estos centros activos de otras moléculas de monómeros y como última etapa se encuentra la reacción de terminación, donde se desactivan los centros originando el polímero. Los polímeros de condensación son obtenidos a través de un mecanismo de polimerización por pasos o por etapas. El monómero o los monómeros presentan en su estructura las funciones químicas necesarias para que la reacción se lleve a cabo. Por ejemplo, el ácido 3-hidroxipropanoico contiene las dos funciones necesarias para una reacción de esterificación, por lo que podría reaccionar intermolecularmente para formar un poli(éster), siempre y cuando se den las condiciones necesarias. Sin embargo, dicha reacción se puede llevar a cabo usando cantidades estequiométricas de un diol y un ácido dicarboxílico. Por lo tanto, para realizar la síntesis de un polímero de condensación es necesario utilizar monómeros con dos o más funciones complementarias, capaces de unirse para formar un enlace químico y eliminar una pequeña molécula producto de la condensación, por lo general, agua, cumpliéndose así la clasificación propuesta por Carothers. III. POLÍMEROS DE ALTO RENDIMIENTO Los polímeros sintéticos han experimentado un extraordinario desarrollo durante los últimos sesenta años, y se vienen aplicando en innumerables especialidades tecnológicas, de tal manera que se puede afirmar que estos materiales están presentes en cualquier tipo de actividad productiva. Analizando las aplicaciones tecnológicas de los nuevos materiales poliméricos, se hace imprescindible obtener una adecuada combinación de las propiedades de estos. Algunas de estas propiedades son intrínsecas a la familia a la que pertenece el polímero y otras se manifiestan debido a las modificaciones llevadas a cabo sobre la estructura de la unidad repetitiva. De 33 esta manera, se ha desarrollado una gran variedad de materiales para la ingeniería que se orientan a satisfacer las demandas específicas de la industria moderna denominados polímeros de alto rendimiento (High performance polymers). (8,9) Los polímeros de alto rendimiento, también conocidos como polímeros de alto desempeño o altas prestaciones, son materiales que se caracterizan por presentar propiedades específicas, tales como, elevada resistencia térmica, mecánica y química, baja densidad específica, elevado aislamiento eléctrico y sonoro, buena resistencia a la llama, entre otras, y son fabricados en función del uso que se les desea dar (10,11). Estos materiales actualmente presentan aplicaciones en el área aeroespacial, automotriz, electrónica, optoelectrónica, construcción, textil, pinturas, alimentaria, separación de gases, óptica, medicina y muchas más. Existen numerosas familias de polímeros que son utilizadas para fabricar materiales de alto desempeño, entre ellas se pueden encontrar poli(cetona)s, poli(uretano)s, poli(amida)s, poli(imida)s, poli(carbonato)s, poli(éstere)s, poli(benzoxazole)s, y combinaciones de ellas, como los poli(amida-imida)s, poli(éter-cetona)s, poli(imida-éstere)s, poli(éter-imida)s, etc. Sin embargo, dos de estas familias han alcanzado especial importancia en el mundo de los polímeros de alto desempeño; las poli(amida)s y las poli(imida)s. IV. POLI(AMIDA)S (PAs) Las poli(amida)s son productos de policondensación y fueron los primeros polímeros semicristalinos, las primeras fibras sintéticas y los primeros materiales considerados como termoplásticos para ingeniería. Esto último debido a las excelentes propiedades mecánicas a baja y alta temperatura y también a la nula reactividad frente a diferentes sustancias químicas, específicamente disolventes. 34 Las excelentes propiedades de las poli(amida)s se pueden explicar al analizar las interacciones existentes entre las cadenas poliméricas, tal como se muestra en la figura 2. Como se puede ver, debido a la presencia de grupos amida a lo largo de la cadena, se origina la formación de enlaces por puente de hidrógeno con los otros grupos cercanos de otras cadenas, dando lugar a una fuerte atracción entre ellas. Esto conlleva a la obtención de una elevada resistencia mecánica, una elevada cristalinidad y una alta temperatura de fusión en estos materiales. R R C H O H N O C N C R H C O O H O O H N O C N C N H C O R H R N N H C O R R N O R R C N H R H C C N N R H O R R O R C H N R R Figura 2. Interacciones por puente de hidrógeno entre los grupos amida de las cadenas poliméricas. Las poli(amida)s pueden ser alifáticas o aromáticas. Las primeras son los materiales que se conocen como nylons y debido a su estructura alifática pueden fundir, por lo que a nivel industrial se pueden preparar en fundido con el consiguiente ahorro de disolventes. Los nylons de mayor interés comercial son el nylon-6,6 y el nylon-6, los cuales abarcan cerca del 80 % de la producción total de este tipo de materiales (12). El primero de ellos se obtiene haciendo reaccionar un diácido de seis átomos de carbono con una diamina de igual largo de 35 cadena, mientras que el segundo se prepara a partir de una lactama de seis átomos de carbono (Figura 3). O O C H2N (CH2)4 C HO (CH2)6 NH2 O O C (CH2)4 C OH N (CH2)6 N H H n Nylon-6,6 O H O N (CH2)5 C N H n Nylon-6 Figura 3. Esquema de síntesis del nylon-6,6 y el nylon-6. Existe otro método para obtener nylons a partir de ω-aminoácidos. El nylon11 y nylon-12, que se obtienen a partir del ácido 11-aminoundecanoico y el ácido 12-aminododecanoico, respectivamente, son los terceros con interés industrial. Debido a la gran polaridad del grupo amida, los nylons presentan gran capacidad para absorber agua, lo que provoca distorsiones dimensionales importantes, aumentando la movilidad de las cadenas y disminuyendo la rigidez y la resistencia a la tracción. Sin embargo, como los nylons 11 y 12 presentan una proporción mucho menor de enlaces amida en sus cadenas respecto a los homólogos 6 y 6,6, la absorción de agua también es mucho menor. Las aplicaciones de las poli(amida)s alifáticas se han dividido en dos grandes campos, la fabricación en forma de fibras y en forma de piezas extruidas o inyectadas. La industria automotriz es una de las que más ha utilizado los nylons como materiales termoplásticos y han encontrado su uso en aislamiento de cables, conectores eléctricos, tubos para combustible, rejillas protectoras para radiadores, depósitos para líquido de frenos, parachoques, 36 etc. Otro mercado que demanda la fabricación de nylons es la industria alimenticia, principalmente para el envasado de cierto tipo de alimentos. Las poli(amida)s aromáticas o también llamadas aramidas presentan propiedades muy diferentes a los nylons, a pesar de pertenecer a la misma familia de polímeros, y esto se debe a la combinación de los enlaces por puente de hidrógeno entre los grupos amida y a la elevada rigidez molecular que poseen los anillos aromáticos de la cadena polimérica. A diferencia de los nylons, las aramidas no se pueden fabricar en estado fundido, debido a las elevadísimas temperaturas de fusión que poseen y en muchos casos descomponen antes de fundir. Las poli(amida)s aromáticas se obtienen por lo general a partir de dos métodos: una reacción de policondensación entre un dicloruro de ácido aromático y una diamina aromática a baja temperatura en disolución (13), o bien, una reacción de policondensación directa entre un diácido aromático y una diamina aromática usando agentes condensantes, como por ejemplo trifenilfosfito (TPP) y piridina (Py), también en disolución (14) (Figura 4). Figura 4. Síntesis de poli(amida)s a baja temperatura empleando N,Ndimetilacetamida (DMAc) como solvente (A) y mediante policondensación directa en solución de N-metil-2-pirrolidona (NMP) (B). Existen otros métodos alternativos, menos utilizados, para sintetizar poli(amida)s aromáticas, por ejemplo, a partir de la reacción entre un diácido 37 con un diisocianato, la policondensación directa de diácidos con diaminas utilizando cloruro de tionilo como agente condensante o activante, la condensación de diácidos con sales de formamidinium de diaminas aromáticas y la policondensación-carbonilación catalizada por Pd entre compuestos dihaloarilos y diaminas aromáticas (15-20). Las poli(amida)s aromáticas son consideradas polímeros de alto rendimiento, pues debido a su estructura molecular, poseen una excelente combinación de propiedades, tales como, alta resistencia a la tracción y bajo peso, baja elongación a rotura y alto módulo (rigidez estructural), baja conductividad eléctrica, alta resistencia química, alta tenacidad, excelente estabilidad dimensional, alta resistencia al corte y a la llama y excelente estabilidad térmica. Las primeras poli(amida)s aromáticas y las más conocidas comercialmente fueron la poli(p-fenilentereftalamida) (PPTA) y la poli(m-fenilenisoftalamida) (MPIA). Ambas fueron desarrolladas por la empresa DuPont® bajo los nombres Kevlar y Nomex, respectivamente. La MPIA, debido a la sustitución de tipo meta en sus anillos aromáticos, puede plegarse fácilmente y romper la linealidad, dificultando el empaquetamiento de cadenas, por lo que es más soluble que la PPTA. De hecho, las fibras de MPIA se pueden hilar a partir de una disolución de DMAc, mientras que la PPTA solo se disuelve en ácido sulfúrico. Estas poli(amida)s aromáticas tuvieron y tienen aplicaciones en tejidos avanzados, revestimientos, aislamiento eléctrico, fabricación de chalecos antibalas, filtros industriales, refuerzos de neumáticos, velas de navíos, como productos antifricción remplazando al amianto, etc. Las características físicas de las fibras de aramidas comerciales y las propiedades de las películas de 38 estas fueron bien resumidas por Gallini (21), Ozawa y Matsuda (22), Tanner et al. (23) y Yarn (24). V. POLI(IMIDA)S (PIs) Las poli(imida)s también son policondensados y fueron los primeros materiales poliméricos en los cuales se registró una alta estabilidad térmica. En la actualidad, las poli(imida)s son prácticamente los únicos polímeros útiles para aplicación donde el material debe resistir elevadas temperaturas (10), y esto se debe a los complejos de transferencia de carga que se forman entre sus cadenas. Un complejo de transferencia de carga no es más que una asociación química entre moléculas o partes de una macromolécula, donde existen grupos donores y aceptores de electrones muy cercanos, pudiendo existir una atracción electrostática, lo que favorece en el caso de los polímeros, la atracción entre las cadenas aumentando su resistencia térmica, mecánica y química (25). En la figura 5 se pueden evidenciar estas interacciones electrónicas en sus versiones intra e intercadenas. Las regiones azules aceptoras sustraen densidad electrónica de las regiones rojas donoras, lo que origina las interacciones intramoleculares. Mientras que las regiones aceptoras de una cadena interaccionan con las regiones donoras de otra originando las atracciones intermoleculares. 39 Figura 5. Formación de complejos de transferencia de carga en poli(imida)s. Las poli(imida)s se obtienen generalmente mediante la reacción entre un dianhidrido de ácido y una diamina, lo que lleva a la obtención de los ácidos poliámicos. Una posterior ciclodeshidratación térmica o química da lugar a la formación de la función imida. (Figura 6). El grupo imida puede ser cíclico o lineal, aunque en general las poli(imida)s están compuestas de anillos de imida de cinco miembros unidos a ciclos aromáticos. Sin embargo, pueden existir fragmentos alifáticos a lo largo de la cadena polimérica. Figura 6. Esquema general de síntesis de poli(imida)s, donde se resalta su precursor inmediato; el ácido poliámico (a). 40 Las poli(imida)s que contienen fragmentos alifáticos se preparan a partir de dianhídridos de ácido aromáticos y diaminas alifáticas, ya sea en disolución o en estado fundido (26-28). Sin embargo, el interés por este tipo de poli(imida)s no duró muchos años debido a que la incorporación de grupos alifáticos disminuye marcadamente la resistencia térmica del material, pero por otra parte se gana en mejorar su solubilidad y procesabilidad. Por otro lado, las poli(imida)s que contienen fragmentos alifáticos muestran elevada transparencia y baja constante dieléctrica, lo que las hace útiles en la industria electrónica y fotoelectrónica (29). Pese a esto, la baja resistencia térmica antes mencionada es un problema grave en este tipo de aplicaciones (30). Las poli(imida)s completamente aromáticas, obtenidas a partir de dianhídridos de ácido aromáticos y diaminas aromáticas, han encontrado una amplia gama de aplicaciones en tecnologías avanzadas. Al igual que las poli(amida)s aromáticas, la fabricación de poli(imida)s aromáticas no es posible en el estado fundido, debido a que presentan temperaturas de fusión extremadamente altas y descomponen antes de fundir. Las poli(imida)s aromáticas convencionales no muestran propiedades de fluidez adecuadas para moldeo por inyección o extrusión. Además, su rigidez estructural extrema hace que sean insolubles en cualquier disolvente orgánico. La poli(imida)s aromáticas no pueden procesarse en su forma cíclica, por lo general, su aplicación se debe hacer en forma de ácido poliámico en disolución, para luego eliminar el disolvente y el agua producida en el proceso de ciclación, por ello su uso se reduce a películas delgadas, barnices y recubrimientos, donde la eliminación de los productos volátiles puede llevarse a cabo sin oclusión de burbujas en el sistema (31). 41 Las poli(imida)s aromáticas han sido demandadas durante los últimos años por las industrias eléctrica y electrónica debido a las magníficas propiedades aislantes. La principal aplicación ha sido como capas dieléctricas en la producción de semiconductores, o bien, en aplicaciones de microelectrónica donde el uso de poli(imida)s aromáticas fotosensibles se ha ido desarrollando (12). VI. POLI(AMIDA)S Y POLI(IMIDA)S CON ESTRUCTURAS MODIFICADAS Las dificultades en el procesamiento de ambas familias de polímeros aromáticos se deben a las características moleculares inherentes de cada una de ellas, tales como, la elevada rigidez molecular, alta polaridad y altas fuerzas de asociación intermoleculares, lo que hace que sean polímeros prácticamente insolubles en cualquier medio orgánico, temperaturas de fusión muy por encima de las temperaturas de descomposición, o bien elevadas temperaturas de transición vítrea, entre otras. Debido a esto, en los últimos años se han desarrollado nuevos tipos de polímeros con estructuras modificadas químicamente, principalmente mediante la preparación de nuevos monómeros, que proporcionan menor orden molecular, aumentando la movilidad de torsión y disminuyendo las uniones intermoleculares. Algunas de estas modificaciones se han adoptado universalmente como: Introducción de fragmentos alifáticos u otro tipo de segmento flexible, con el objetivo de reducir la rigidez molecular. Introducción de sustituyentes laterales voluminosos, que ayuden a la separación de las cadenas poliméricas y dificulten el empaquetamiento molecular y la cristalización. El uso de monómeros que contengan enlaces angulares, lo cual suprime o bien disminuye la coplanaridad estructural. 42 El uso de monómeros 1,3-disustituidos en lugar de 1,4-disustituidos, y/o monómeros asimétricos, los cuales reducen la regularidad y el orden molecular. La preparación de co-polímeros con el objetivo de romper la simetría y regularidad de las cadenas. El grupo CF3 es un ejemplo clásico de un sustituyente que mejora la solubilidad y la estabilidad térmica de estos materiales (32,33), debido a la elevada polaridad de los enlaces C-F y al volumen que ocupa. En cuanto al uso de anillos heterocíclicos, es posible encontrar poli(amida)s, poli(imida)s y poli(amida-imida)s basadas en una estructura del tipo “spiro” (34). En todos los casos, la inclusión de fragmentos flexibles de oxiarenos en la cadena principal (35,36) en combinación con grupos oxiarenos-CF3 (37-40) o grupos heterocíclicos-oxiarenos-CF3 (41), permite obtener polímeros con buena solubilidad en disolventes orgánicos comunes, buenas propiedades mecánicas, alta estabilidad térmica, elevada transparencia en la región UVvisible y moderados valores de temperatura de transición vítrea (Tg), características convenientes para los procesos industriales. La figura 7 muestra monómeros difuncionales que presentan algunas de estas modificaciones. 43 Figura 7. Ejemplos de monómeros difuncionales con fragmentos flexibles en su estructura. Todas estas modificaciones y otras más puntuales, hacen que disminuyan ciertas propiedades del material, pero la ganancia en procesabilidad es tal que es perfectamente asumible en muchas aplicaciones, por lo tanto debe existir un compromiso entre la procesabilidad y las propiedades finales del material. En la figura 8, se presentan poli(amida)s y poli(imida)s aromáticas con algunas de estas modificaciones lo que las ha llevado a alcanzar interés industrial. Estos polímeros son solubles en disolventes orgánicos comunes y procesables por técnicas convencionales (inyección, moldeo por compresión, etc). 44 Figura 8. Poli(amida)s y poli(imida)s aromáticas comerciales que presentan modificaciones estructurales. VII. POLÍMEROS QUE CONTIENEN SILICIO EN SU ESTRUCTURA Las primeras investigaciones sobre compuestos orgánicos que contienen silicio fueron realizadas exclusivamente por curiosidad, estableciendo así las vías de síntesis y el estudio sobre su comportamiento. En el campo de los polímeros, la inclusión de silicio en la cadena principal tuvo como intención mejorar algunas cualidades y también conferir nuevas propiedades al material. Los polímeros orgánicos que contienen silicio se pueden subdividir en cuatro categorías principales, dependiendo del elemento enlazado al átomo de silicio: poli(silanos) (uniones Si-Si), poli(siloxanos) (uniones Si-O), poli(silazanos) (uniones Si-N) y poli(sililalquilenos) o poli(sililarilenos) (uniones Si-C) (42). De estas familias, los poli(siloxanos) o siliconas han sido los más estudiados y son los que presentan mayor interés comercial (43-48). 45 Las otras familias han encontrado aplicaciones en campos más especializados. Sin embargo, las nuevas investigaciones están siendo dirigidas a los polímeros que presentan uniones Si-C. Desde hace unos años, existe un gran interés por parte de los científicos acerca de los polímeros aromáticos que contienen uniones Si-C debido a sus aplicaciones potenciales para la producción de materiales optoelectrónicos, ya que los electrones pueden moverse a lo largo de toda la cadena macromolecular conjugada utilizando como puente la interacción entre los orbitales p de los anillos aromáticos y el orbital d del átomo de silicio, cuando este se encuentra entre dos anillos aromáticos. Este hecho ha sido investigado utilizando diferentes técnicas espectroscópicas (49). Vignollet y Marie (50) informaron que cuando se introduce un grupo electrodonante fuerte en posición para del anillo aromático con respecto al átomo de silicio, este utiliza sus orbitales 3d para formar un enlace pπ – dπ con el anillo fenílico. Jadhav et al. (51), en 1985, informaron la síntesis de cuatro nuevas poli(amida-imidas) a partir de una reacción de policondensación entre dianhídridos de ácido aromáticos y una dihidrazida que contiene silicio como átomo central (Figura 9). Estos polímeros resultaron ser solubles en dimetilformamida (DMF), DMAc, dimetilsulfóxido (DMSO) y NMP, mientras que los datos obtenidos de la curva de termogravimétrica (TGA) mostraron que las poli(amida-imida)s poseían una excelente estabilidad térmica. 46 Figura 9. Poli(amida-imida)s que contienen silicio. Bruma et al. han sintetizado y caracterizado poli(amida)s que contienen anillos de difenilquinoxalina (52–54) y oxadiazol (55), donde han incorporado grupos difenilsililo (Figura 10). Además, estos autores informaron la síntesis de poli(imida)s que contienen unidades difenilsilano y fenilquinoxalina (56). Los resultados muestran que los polímeros son solubles en disolventes orgánicos comunes (NMP, DMAc, piridina y cloroformo) y presentan temperaturas de descomposición (TDT) por encima de 450 ºC. Además, se han preparado poli(imida)s con estructuras similares (57,58) y se han propuesto avanzadas aplicaciones en el campo de la microelectrónica y optoelectrónica. Figura 10. Ejemplos de poli(amida)s sililadas solubles en disolventes orgánicos comunes. Yi et al. (59) informaron la síntesis de polímeros semiconductores de tiofeno que contenían unidades organosililadas. La conductividad eléctrica de estos 47 materiales fue de 10-4 – 10-6 S/cm, demostrando así la capacidad que posee el átomo de silicio para facilitar el paso de los electrones de un anillo de tiofeno a otro. Figura 11. Polímeros semiconductores organosililados. Algunas poli(amida-imida)s que contienen silicio se han sintetizado mostrando buena solubilidad en disolventes polares apróticos. Estos polímeros se pueden obtener como películas delgadas flexibles o recubrimientos a partir de una disolución, los cuales presentan TDT por encima de 400 °C y valores de Tg entre 220-280 °C (60,61). Otros ejemplos de polímeros que contienen silicio son los poli(ésteresimidas), donde se utilizan varias unidades flexibles con el fin de aumentar la solubilidad y disminuir la Tg del material (62). En general, las poli(imida)s que contienen silicio basadas en cadenas flexibles de oxidianilinas (3,4- y 4,4-disustituidas) o estructuras similares (Figura 12) presentan buenas propiedades mecánicas y alta transparencia óptica (63). Figura 12. Material sililado transparente en la región UV-vis. 48 Stern et al. (64) estudiaron la permeabilidad y selectividad que presentaron membranas de poli(imida)s que contenían silicio, en la separación de gases como hidrógeno, oxígeno, nitrógeno, dióxido de carbono y metano (Figura 13). La permeabilidad a estos gases fue sustancialmente mayor en estos polímeros que en las membranas análogas basadas en carbono, debido probablemente al mayor tamaño de los átomos de silicio presentes en la cadena principal, lo que favorece el aumento del volumen libre del polímero. Figura 13. Ejemplo de poli(imida) que contiene silicio con aplicación en el campo de membranas separadoras de gases. El trabajo de tesis propuesto implica la síntesis de nuevas series de poli(amida)s y poli(imida)s aromáticas que presenten algunas de las modificaciones estructurales descritas, con el objeto de aumentar la solubilidad de estos materiales en disolventes orgánicos comunes y modular los valores de Tg, manteniendo o incrementando la resistencia térmica del polímero. El logro de este objetivo entregará materiales con solubilidad adecuada y una amplia ventana térmica para su procesamiento. Una vez sintetizadas y caracterizadas las series de polímeros, se podrán proponer eventuales aplicaciones dependiendo de las propiedades específicas registradas. 49 VIII. HIPÓTESIS Los nuevos monómeros difuncionales; diaminas, ácidos dicarboxílicos y dianhídridos de ácidos propuestos en este proyecto contienen unidades difenilsilano y/o oxiareno en diversas proporciones. Estas características estructurales permitirán la síntesis de poli(amida)s y poli(imida)s con alta resistencia térmica y al mismo tiempo permitirán una modulación adecuada de los valores de Tg y de los parámetros de solubilidad, mejorando las características de procesamiento del material final. Además, la flexibilidad de las cadenas también conllevará a un aumento de la transparencia en la región UV-visible. El registro sistemático de estas propiedades permitirá establecer relaciones con las modificaciones estructurales específicas realizadas en la cadena principal. La suma de estas propiedades, hará de estos polímeros materiales prometedores de gran uso industrial. 50 IX. OBJETIVOS GENERALES 1. Sintetizar nuevas poli(amida)s y poli(imida)s cuya cadena principal contenga unidades difenilsilano y/o oxiarenos. 2. Caracterizar los polímeros usando técnicas espectroscópicas, térmicas y de transparencia óptica en la región UV-visible. 3. Establecer relaciones estructura-propiedad con el fin de proponer eventuales aplicaciones a los materiales preparados. X. OBJETIVOS ESPECÍFICOS 1. Sintetizar y caracterizar los precursores que darán origen a los monómeros diaminas, ácidos dicarboxílicos y dianhídridos de ácidos. 2. Sintetizar y caracterizar diaminas monoméricas bis(4-(4-aminofenoxi)fenil) dimetilsilano (9), bis(4-(4-aminofenoxi)fenil)difenilsilano (10), 2,7-diamino dibenzofurano (21), 2,8-diaminodibenzofurano (22), 2,8-di(4-aminofenil) dibenzofurano (24), 9,9-bis(4-(4-aminofenoxi)-3-metilfenil)fluoreno (26). 3. Sintetizar y caracterizar ácidos dicarboxílicos monoméricos bis(4-(4carboxifenoxi)fenil)dimetilsilano (13) y bis(4-(4-carboxifenoxi)fenil)dimetil silano (14). 4. Sintetizar y caracterizar los dianhídridos de ácidos monoméricos anhídrido bis(4-(3,4-dicarboxifenoxi)fenil)dimetilsilano (19), anhídrido bis(4-(3,4-dicarboxifenoxi)fenil)difenilsilano (20) y anhídrido 9,9-bis(4-(3,4dicarboxifenoxi)-3-metilfenil)fluoreno (29). 5. Sintetizar poli(amida)s (Serie A, B y C) y poli(imida)s (Serie A, B, C y D) aromáticas a partir de los monómeros difuncionales obtenidos. 6. Caracterizar los polímeros mediante técnicas espectroscópicas (FT-IR, RMN), análisis térmico (DSC, TGA), solubilidad, microanálisis elemental, viscosidad inherente y transparencia en la región UV-visible. 7. Establecer relaciones estructura-propiedad de acuerdo con los detalles estructurales de las unidades repetitivas correspondientes. 51 PARTE EXPERIMENTAL I. INFORMACIÓN GENERAL Los reactivos y disolventes empleados en las síntesis fueron de calidad p.a. o p.s. de procedencia Aldrich, Merck, J. T. Baker y Across. Los espectros infrarrojos fueron registrados empleando pastillas de KBr en un espectrómetro Perkin-Elmer (Fremont CA) 1310 en un intervalo entre 4000-250 cm-1. Los espectros de RMN 1H, 13 C, 29 Si y DEPT-135° fueron realizados en un equipo Bruker AC-200 a 400 MHz, usando DMSO-d6, CDCl3 o acetona-d6 como disolvente y tetrametilsilano (TMS) como referencia interna. Las medidas de viscosidad se llevaron a cabo en un viscosímetro de dilución Desreux-Bischof a 25 °C y una concentración de 0,3 g/dL. Los valores de temperatura de transición vítrea (Tg) fueron obtenidos en un calorímetro DSC 821 Mettler-Toledo (Greifensee, Switzerland), a partir del segundo ciclo de calentamiento y a una velocidad de 10 °C/min. Los análisis termogravimétricos fueron llevados a cabo en un calorímetro TA3000 Mettler (Switzerland) equipado con un procesador TC-10A y una termobalanza TG-50 con una microbalanza Mettler MT5. Los análisis fueron realizados bajo flujo de nitrógeno a una velocidad de calentamiento de 20 °C/min. Los registros de análisis elemental se realizaron en un equipo Fisons EA 1108-CHNS-O. La transmitancia óptica en la región UV-visible se midió en un espectrofotómetro UV-3101PC UV-Vis-NIR (Shimadzu, Japan). Para esto se emplearon soluciones a una concentración de 0,5 g/L. 53 El registro de las dimensiones moleculares se llevó a cabo en un cromatógrafo conectado en línea con una bomba isocrática Perkin-Elmer 250 y con un refractómetro viscotek modelo VE3580, y en paralelo con un detector dual viscotek modelo 270. Este detector consta de un viscosímetro diferencial y un detector de dispersión de luz a 90 º (RALS). La muestra se eluyó a través de tres columnas de poliestireno-divinilbenceno PL-gelTM de 500, 103 y 104 Å de tamaño de poro (Polymer Laboratories) a 70 ºC conectadas en serie. La fase móvil fue DMF con 0,1 % (p/v) de LiBr y el flujo de elución fue de 1 mL/min. Para la calibración del equipo se utilizó poliestireno estándar de peso molecular 60 000 Da. La concentración de las muestras fue de 5 mg/mL. 54 II. SÍNTESIS DE PRECURSORES 1. Síntesis de bis(4-hidroxifenil)diorganosilano (1, 2) Estos precursores fueron obtenidos por dos rutas de síntesis: Ruta I En un balón de 500 mL de capacidad y tres bocas equipado con termómetro, agitación magnética, embudo isobárico y atmósfera de nitrógeno se añaden 0,091 mol de n-butillitio (n-BuLi) 1,6 M en hexano. En el embudo isobárico se disuelven 0,045 mol de 4-bromofenol en 108 mL de THF anhidro. La disolución de n-BuLi se enfría a -70 °C en un baño de etanol-nitrógeno líquido y se deja gotear la disolución de 4-bromofenol sobre el n-BuLi cuidando que la temperatura no supere los -60 °C. Una vez concluida la adición, se deja que la temperatura ascienda lentamente. Sobre -30 ºC se observa la formación de un gel que continúa hasta 6 ºC, momento en el cual la mezcla de reacción se agita por 30 minutos adicionales. La mezcla nuevamente se enfría a -70 ºC y se añaden 0,014 mol (65 % de la cantidad estequeométrica) de diclorodiorganosilano disueltos en 16 mL de THF anhidro y se deja agitando durante toda la noche. La mezcla de reacción se hidroliza con 50 mL de HCl (ac) al 40 % y se realiza una extracción en éter etílico. La fase orgánica se seca con sulfato de magnesio anhidro, se filtra y se concentra en un evaporador rotatorio a 40 ºC. El aceite resultante se vuelca sobre n-hexano bajo agitación precipitando un sólido de color blanco, el cual se filtra, se lava con abundante n-hexano y se seca en una estufa a vacío a 50 °C durante 12 h. 55 Bis(4-hidroxifenil)dimetilsilano (1): 5 CH3 HO Si 3 2 1 4 OH 6 CH3 R.: 70 %. T.f.: 170-173 °C (Lit (65) 173-174 °C). IR (KBr, cm-1): 3288 (O-H); 3028 (C-H arom.); 2953 (C-H alif.); 1601, 1582, 1423 (C=C arom.); 1503, 1178 (Si-C arom.); 1365 (Si-C alif.); 835 (arom. p-sust.). RMN 1H (acetonad6, δ, ppm): 0,45 (s, 6H(5)); 6,84 (d, J= 8.4 Hz, 4H(2)); 7,35 (d, J= 8.4 Hz, 4H(3)); 8,44 (s, 2H(6)). NMR 13 C (acetona-d6, δ, ppm): -1,6 (C5); 115,9 (C2); 128,9 (C4); 136,4 (C3); 159,2 (C1). NMR 29 Si (acetona-d6, δ, ppm): -9,3. Análisis elemental. Calculado: C14H16O2Si; (244,36): C, 68,81 %; H, 6,60 %. Encontrado: C, 68,25 %; H, 6,45 %. Bis(4-hidroxifenil)difenilsilano (2): 8 7 6 3 5 HO Si 4 2 1 OH 9 R.: 50 %. T.f.: 219-222 °C (Lit (65) 221-222 °C). IR (KBr, cm-1): 3382 (O-H); 3021 (C-H arom.); 1597, 1580, 1427 (C=C arom.); 1501, 1177 (Si-C arom.); 826 (arom. p-sust.); 742 (arom. mono-sust.). RMN 1H (acetona-d6, δ, ppm): 6,92 (d, J= 8.5 Hz, 4H(2)); 7,44 (m, 14H(3,6,7,8)); 8,59 (s, 2H(9)). NMR 13 C (acetona-d6, δ, ppm): 116,1 (C2); 124,6 (C4); 128,7 (C7); 130,3 (C8); 136,3 (C3); 137,0 (C5); 138,7 (C6); 159,8 (C1). NMR 29 Si (acetona-d6, δ, ppm): -14,5. Análisis elemental. Calculado: C24H20O2Si; (368,50): C, 78,22 %; H, 5,47 %. Encontrado: C, 78,15 %; H, 5,33 %. 56 Ruta II a) Síntesis de 4-bromofenilbencil éter (3) En un balón de 250 mL de capacidad se añaden 57,8 mmol de 4bromofenol, 57,6 mmol de K2CO3 anhidro, 63,4 mmol de cloruro de bencilo y 40 mL de DMF. La mezcla se calienta a reflujo durante 6 h. Al enfriar, se vierte sobre 1 L de agua con agitación vigorosa. El sólido de color blanco se filtra, se lava con abundante agua y se seca en una estufa a vacío a 50 °C durante 12 h. 4-bromofenilbencil éter (3): R: 100 %. T.f.: 62-64 °C. (Lit (66) 64 °C). IR (KBr, cm-1): 3060, 3031 (C-H arom.); 2915, 2887 (C-H alif.); 1587, 1575, 1497 (C=C arom.); 1247 (C-O-C); 825 (arom. p-sust.); 734 (arom. mono-sust.); 694 (C-Br). RMN 1H (CDCl3, δ, ppm): 5,02 (s, 2H(5)); 6,84 (d, J= 9,0 Hz, 2H(3)); 7,31-7,40 (m, 7H(2,7,8,9)). RMN 13 C (CDCl3, δ, ppm): 70,6 (C5); 113,5 (C1); 117,1 (C3); 127,8 (C7); 128,5 (C9); 129,0 (C8); 132,7 (C2); 136,9 (C6); 158,3 (C4). Análisis elemental. Calculado: C13H11BrO: (263,13): C, 59,34 %; H, 4,21 %. Encontrado: C, 59,01 %; H, 4,12 %. b) Síntesis de bis(4-(benciloxi)fenil)diorganosilano (4, 5) Una mezcla de 38 mmol de 4-bromofenil bencil éter (3) y 200 mL de THF enfriada a -70 °C se trata con 38 mmol de n-BuLi (1,6 M en hexano) y se deja reaccionar con agitación durante 1 h. A continuación, se añaden 19 mmol del diclorodiorganosilano correspondiente, disueltos en 20 mL de THF. 57 La reacción se mantiene a -70 ºC durante 2 h y luego se retira el baño de enfriamiento. Cuando la reacción está a temperatura ambiente se vierte sobre 200 mL de éter etílico. La fase orgánica se lava con agua (3 x 200 mL) y después se seca sobre Na2SO4 anhidro. Luego de filtrar, se elimina el disolvente a presión reducida para obtener un aceite, el cual se vuelca sobre n-hexano obteniéndose un sólido de color blanco. Este sólido se filtra, se lava con n-hexano y se seca a 50 °C a vacío durante 12 h. Bis(4-(benciloxi)fenil)dimetilsilano (4): R: 75 %. T.f.: 106-108 °C. IR (KBr, cm-1): 3061, 3031 (C-H arom.); 2954 (C-H alif.); 1592, 1561, 1501 (C=C arom.); 1454, 1182 (Si-C arom.); 1313 (Si-C alif.); 1241 (C-O-C); 861 (arom. p-sust.); 776 (arom. mono-sust.). RMN 1H (CDCl3, δ, ppm): 0,49 (s, 6H(10)); 5,04 (s, 4H(5)); 6.96 (d, J= 8,4 Hz, 4H(3)); 7,37 (m, 14H(2,7,8,9)). RMN 13 C (CDCl3, δ, ppm): -2,0 (C10); 69,7 (C5); 114,4 (C3); 127,4 (C7); 127,9 (C9); 128,5 (C8); 129,7 (C1); 135,6 (C2); 136,9 (C6); 159,6 (C4). RMN 29 Si (CDCl3, δ, ppm): -8,8. Análisis elemental. Calculado: C28H28O2Si; (424,60): C, 79,20 %; H, 6,65 %. Encontrado: C, 79,03 %; H, 6,43 %. Bis(4-(benciloxi)fenil)difenilsilano (5): 13 12 7 11 2 10 O Si 58 1 3 5 4 O 6 8 9 R: 60 %. T.f.: 124-128 °C. IR (KBr, cm-1): 3063, 3024 (C-H arom.); 2907 (C-H alif.); 1591, 1563, 1500 (C=C arom.); 1452, 1181 (Si-C arom.); 1241 (C-OC); 829 (arom. p-sust.); 753 (arom. mono-sust.). RMN 1H (CDCl3, δ, ppm): 5,05 (s, 4H(5)); 6.98 (d, J= 8,5 Hz, 4H(3)); 7,41 (m, 24H(2,7,8,9,11,12,13)). RMN 13 C (CDCl3, δ, ppm): 69,7 (C5); 114,4 (C3); 125,5 (C1); 127,5 (C7); 127,7 (C9); 127,9 (C8); 128,5 (C12); 129,4 (C13), 134,9 (C10); 136,2 (C2); 136,8 (C6); 137,8 (C11); 160,0 (C4). RMN 29 Si (CDCl3, δ, ppm): -14,7. Análisis elemental. Calculado: C38H32O2Si; (548,74): C, 83,17 %; H, 5,88 %. Encontrado: C, 82,98 %; H, 5,56 %. c) Síntesis de bis(4-hidroxifenil)dimetilsilano (1): Una mezcla de 10,3 mmol de bis(4-(benciloxi)fenil)dimetilsilano (4), 0,5 g de Pd/C (10 % en peso) y 100 mL de THF se coloca bajo atmósfera de hidrógeno (40 psi) y agitación durante 24 horas. Pasado este tiempo, se filtra sobre celite y el disolvente se elimina a presión reducida. El aceite resultante se vierte sobre n-hexano precipitando un sólido de color blanco. Este sólido se filtra, se lava con n-hexano y se seca a 50 ° C bajo vacío durante 12 h. d) Síntesis de bis(4-hidroxifenil)difenilsilano (2): La metodología de síntesis de este compuesto sigue igual procedimiento al descrito para la obtención de (1), empleando como precursor inmediato el derivado (5). La hidrogenación catalítica se realiza a una presión de hidrógeno de 65 psi y 40 °C durante 5 días. Los resultados de los análisis espectrocópicos de ambos difenoles sililados han sido entregados en el desarrollo de la Ruta I de síntesis. 59 2. Síntesis de 2,8-diyododibenzofurano (6) En un balón de 500 mL de capacidad equipado con refrigerante y agitación magnética se añaden 0,05 mol de dibenzofurano, 0,04 mol de yodo y 0,022 mol de ácido yódico. A esta mezcla de sólidos se le agregan 100 mL de ácido acético glacial, 8 mL de agua, 1 mL de ácido sulfúrico y 5 mL de tetracloruro de carbono. La mezcla se calienta a 65 °C y se agita durante 48 h. El producto precipita durante la reacción y el color de la disolución cambia de púrpura a café. Una vez dejado enfriar a temperatura ambiente, el sólido se filtra y se lava con ácido acético (15 mL) y luego con abundante agua. El producto se seca a 50 °C a vacío durante 12 h. 2,8-diyododibenzofurano (6): R.: 65 %. T.f.: 176-178 °C (Lit (67) 178 °C). IR (KBr, cm-1): 3063 (C-H arom.); 1450, 1435, 1403 (C=C arom.); 1243 (C-O-C); 623 (C-I.). RMN 1H (CDCl3, ppm): 7,30 (d, J= 8,7 Hz, 2H(2)); 7,72 (dd, J= 8,6 y 1,7 Hz, 2H(3)); 8,16 (d, J= 1,4 Hz, 2H(5)). RMN 13C (CDCl3, ppm): 86,1 (C4); 113,8 (C2); 125,4 (C6); 129,7 (C5); 136,3 (C3); 155,6 (C1). Análisis elemental. Calculado: C12H6I2O; (419,98): C, 34,32 %; H, 1,44 %. Encontrado: C, 34,27 %; H, 1,56 %. Nota aclaratoria: los siguientes tres compuestos se nombraron con letras debido a que son precursores de monómeros propuestos en el proyecto de tesis. Sin embargo, la síntesis de dichos monómeros no fue exitosa, pero estos precursores sí quedaron preparados y caracterizados adecuadamente. 60 3. Síntesis de 4-(4-bromofenoxi)nitrobenceno (A) En un balón de 250 mL de capacidad se agregan 63 mmol de 1-fluoro-4nitrobenceno, 63 mmol de 4-bromofenol, 72 mmol de K2CO3 y 60 mL de DMF. La mezcla de reacción se calienta a temperatura de reflujo y se agita por 24 horas. Luego, se enfría y se vuelva sobre 1 L de una mezcla de aguaetanol (1:1 vol/vol) con agitación. El sólido de color amarillo se filtra, se lava con abundante agua y se seca a 40 °C y a vacío durante 12 horas. El producto se recristaliza desde etanol. 4-(4-bromofenoxi)nitrobenceno (A): R.: 90 %. T.f.: 71-73 °C. IR (KBr, cm-1): 3107, 3074 (C-H arom.); 1609, 1595, 1480 (C=C arom.); 1510, 1341 (NO2), 1245 (C-O-C); 845 (arom. p-sust.), 506 (C-Br). RMN 1H (CDCl3, ppm): 8,21 (d, J= 9,2 Hz, 2H(2)); 7,55 (d, J= 8,9 Hz, 2H(7)); 7,03 (d, J= 9,2 Hz, 2H(3)), 7,00 (d, J = 8,9 Hz, 2H(6)). RMN 13 C (CDCl3, ppm): 162,8 (C4); 154,0 (C5); 143,1 (C1); 133,4 (C7); 126,1 (C2); 122,3 (C6), 118,3 (C8), 117,4 (C3). Análisis elemental. Calculado: C12H8BrNO3; (294,10): C, 49,01 %; N, 4,76 %, H, 1,44 %. Encontrado: C, 48,85 %; N, 4,35, H, 1,52 %. 4. Síntesis de anhídrido 4-(4-bromofenoxi)ftálico (B) La síntesis de este precursor consta de dos etapas: Etapa I: una mezcla de 5,7 mmol de 4-bromofenol, 5,7 mmol de anhídrido 4fluoroftálico, 11,4 mmol de K2CO3 anhidro y 10 mL de sulfolano se calienta a temperatura de reflujo (285 °C) bajo atmósfera de nitrógeno. La suspensión de color amarillo se mantiene en reflujo por 14 horas y luego se deja enfriar 61 a temperatura ambiente. El sólido obtenido se filtra y se seca a 160 ºC en una estufa a vacío. Luego, el sólido de color blanco es disuelto en una mezcla de HCl-agua (1:1 vol/vol) y se refluja por 3,5 horas. La aparición de un sólido color beige indica la formación del diácido. Este sólido se separa por filtración y se lava con abundante agua. Luego, se seca en una estufa a 100 ºC al vacío. Ácido 4-(4-bromofenoxi)ftálico: 11 9 Br 10 8 3 7 O 2 COOH 1 4 COOH 12 5 6 R.: 80 %. T.f.: 290-293 °C. IR (KBr, cm-1): 3280 (O-H), 3091 (C-H arom.); 1748, 1686 (C=O), 1602, 1575 (C=C arom.); 1240 (C-O-C); 833 (arom. psust.), 738 (arom. tri-sust.), 561 (C-Br). RMN 1H (acetona-d6, ppm): 7,88 (d, J= 8,6 Hz, 1H(6)); 7,62 (d, J= 9,0 Hz, 2H(9)); 7,25 (d, J= 2,5 Hz, 1H(3)), 7,18 (dd, J = 8,5; 2,6 Hz, 1H(5)), 7,12 (d, J= 9,0 Hz, 2H(8)). RMN 13C (acetona-d6, ppm): 168,6 (C12), 167,8 (C11), 160,5 (C4); 156,0 (C7); 137,3 (C2); 134,1 (C9); 132,7 (C6); 127,1 (C1), 122.9 (C5), 120,2 (C8), 118,5 (C3), 117,8 (C10). Análisis elemental. Calculado: C14H9BrO5; (337,12): C, 49,88 %; H, 2,69 %. Encontrado: C, 49,35 %; H, 2,83 %. Etapa II: una mezcla de 4,36 mmol de ácido 4-(4-bromofenoxi)ftálico y 5 mL de anhídrido acético se calentaron a reflujo durante 2 horas. Pasado ese tiempo, se retira el disolvente mediante destilación a presión reducida y el residuo aceitoso se disuelve en 1 mL de acetato de etilo y se vuelca lentamente sobre 150 mL de n-hexano. El sólido obtenido se filtra, se lava con abundante n-hexano y se seca a 100 °C durante 12 horas. 62 Ácido 4-(4-bromofenoxi)ftálico (B): O 11 9 Br 10 8 3 7 O O 2 1 4 12 O 5 6 R.: 45 %. T.f.: 150-152 °C. IR (KBr, cm-1): 3090 (C-H arom.); 1825, 1750 (C=O), 1601, 1591, 1570 (C=C arom.); 1260 (C-O-C); 830 (arom. p-sust.), 740 (arom. tri-sust.), 560 (C-Br). RMN 1H (CDCl3, ppm): 7,95 (d, J= 8,4 Hz, 1H(6)); 7,57 (d, J= 8,9 Hz, 2H(9)); 7,44 (dd, J= 8,4; 2,2 Hz, 1H(5)), 7,37 (d, J = 2,1 Hz, 1H(3)), 7,01 (d, J= 8,9 Hz, 2H(8)). RMN 13 C (CDCl3, ppm): 164,6 (C4), 162,3 (C12), 160,0 (C11); 153,2 (C7); 134,0 (C2); 133,7 (C9); 127,7 (C6); 124,9 (C5), 124.7 (C1), 122,4 (C8), 119,0 (C10), 112,7 (C3). Análisis elemental. Calculado: C14H7BrO4; (319,11): C, 52,69 %; H, 2,21 %. Encontrado: C, 52,25 %; H, 2,43 %. 5. Síntesis de cloro (3,4-dimetilfenil)dimetilsilano (C) En un balón equipado con agitación magnética se añaden 88 mmol de litio y 30 mL de éter dietílico anhidro bajo atmósfera de nitrógeno. A dicha mezcla se le agrega 44 mmol de 4-bromo-o-xileno disuelto en 30 mL de éter dietílico anhidro. Una vez terminada la adición, se calienta a 40 °C durante 2 ó 3 h y luego se enfría a 15 °C (Solución I). En otro balón se adicionan 44 mmol de diclorodimetilsilano disueltos en 30 mL de éter dietílico anhidro bajo atmósfera de nitrógeno y se gotea la Solución I lentamente a través de una jeringa. La mezcla de reacción se deja agitando durante toda la noche. Luego, se filtran las sales y la disolución se destila a presión reducida. El compuesto destila a 80 °C a una presión de 0,1 mmHg. 63 Cloro (3,4-dimetilfenil)dimetilsilano (C): R.: 63 %. T.e.: 80 °C (0,1 mmHg). IR (KBr, cm-1): 3006 (C-H arom.); 2965, 2922 (C-H alif.), 1601, 1557, 1498 (C=C arom.); 1380, 1108 (Si-C arom.), 787 (C-Si alif.), 743 (arom. tri-sust.). RMN 1H (CDCl3, ppm): 7,37 (s, 1H(3)); 7,33 (d, J= 6,9 Hz, 1H(6)); 7,12 (d, J= 6,9 Hz, 1H(5)), 2,24 (s, 3H(8)), 2,22 (s, 3H(9)), 0,63 (s, 6H(7)). RMN 13 C (CDCl3, ppm): 139,0 (C1), 136,2 (C2), 134,2 (C3); 133,1 (C4); 130,7 (C5); 129,4 (C6); 19,7 (C8); 19,6 (C9), 2,1 (C7). RMN 29 Si (CDCl3, ppm): -20,1. Análisis elemental. Calculado: C10H15ClSi; (198,76): C, 60,43 %; H, 7,61 %. Encontrado: C, 59,95 %; H, 7,85 %. 64 III. SÍNTESIS DE MONÓMEROS 1. Síntesis de las diaminas bis(4-(4-aminofenoxi)fenil)diorganosilano (9, 10) La síntesis de estas diaminas monoméricas consta de dos etapas: Etapa I: Una mezcla de 4,09 mmol de bis(4-hidroxifenil)diorganosilano (1 ó 2), 8,18 mmol de K2CO3 anhidro, 8,18 mmol de 1-fluoro-4-nitrobenceno y 5 mL de DMF se calienta a 60 °C durante 24 horas. Al enfriar, la mezcla se vierte sobre 600 mL de una disolución de etanol-agua (1:1 vol/vol). El sólido de color amarillo se filtra, se lava con abundante agua y se seca a 50 °C a vacío durante 12 h. Bis(4-(4-nitrofenoxi)fenil)dimetilsilano (7): R: 90 %. T.f.: 110-113 °C. IR (KBr) (cm-1): 3107, 3079 (C-H arom.), 2950 (CH alif.), 1597, 1579, 1487 (C=C arom); 1509, 1342 (NO2); 1390, 1110 (Si-C arom.), 775 (Si-C alif.), 1236 (C-O-C); 874 (arom. p-sust.). RMN 1H (DMSOd6, δ, ppm), 8,26 (d, J= 9,2 Hz, 4H(2)), 7,66 (d, J= 8,5 Hz, 4H(7)), 7,21 (d, J= 8,5 Hz, 4H(6)), 7,16 (d, J= 9,2 Hz, 4H(3)), 0.59 (s, 6H(9)). RMN 13 C (DMSO- d6, δ, ppm), -2.59 (C9), 117,6 (C3); 119,7 (C6); 126,1 (C2); 134,4 (C8); 136,1 (C7); 142,3 (C1); 155,4 (C5); 162,3 (C4). RMN 29Si (DMSO-d6, δ, ppm), -8,90. Análisis elemental. Calculado: C26H22N2O6Si; (486,55): C, 64,18 %; H, 4,56 %; N, 5,76 %. Encontrado: C, 63,96 %; H, 4,08 %; N, 5,58 %. 65 Bis(4-(4-nitrofenoxi)fenil)difenilsilano (8): R: 71 %. T.f.: 174-175 °C. IR (KBr) (cm-1): 3100, 3050 (C-H arom.), 1590, 1571 (C=C arom); 1522, 1344 (NO2); 1395, 1100 (Si-C arom.), 1245 (C-O-C); 878 (arom. p-sust.); 750 (arom. mono-sust). RMN 1H (DMSO-d6, δ, ppm), 8,26 (d, J= 9,2 Hz, 4H(2)), 7,59 (d, J= 8,4 Hz, 4H(7)), 7,49 (m, 10H(10,11,12)); 7,27 (d, J= 8,4 Hz, 4H(6)), 7,21 (d, J= 9,2 Hz, 4H(3)). RMN 13 C (DMSO-d6, δ, ppm), 117,8 (C3); 119,6 (C6); 125,9 (C2); 128,0 (C8); 129,7 (C11); 129,8 (C12); 132,9 (C7); 135,5 (C9); 137,9 (C10); 142,4 (C1); 155,9 (C5); 161,8 (C4). RMN 29 Si (DMSO-d6, δ, ppm), -14,8. Análisis elemental. Calculado: C36H26N2O6Si; (610,69): C, 70,80 %; H, 4,29 %; N, 4,59 %. Encontrado: C, 70,76 %; H, 4,18 %; N, 4,47 %. Etapa II: Una mezcla de 3,08 mmol de bis(4-(4-nitrofenoxi)fenil)diorgano silano (7 ó 8), 0,37 g de Pd/C (10 % en peso) y 20 mL de etanol se calienta a reflujo. Luego, se añaden 10 mL de hidracina monohidratada al 80 % gota a gota durante un período de 1 hora. Después de la adición, se continúa el reflujo durante 24 horas. La mezcla de reacción se filtra en caliente y se vuelca sobre 800 mL de agua bajo agitación. El sólido de color blanco obtenido se filtra y se seca a 50 °C a vacío durante 12 h. Bis(4-(4-aminofenoxi)fenil)dimetilsilano (9): 66 R: 67 %. T.f.: 86-89 °C. IR (KBr) (cm-1): 3402, 3333 (N-H), 3015 (C-H arom.), 2959 (C-H alif.), 1619 (N-H flexión), 1587, 1508, 1493 (C=C arom.); 1394, 1108 (Si-C arom.), 1311 (Si-C alif.), 1233 (C-O-C); 871 (arom. p-sust.). RMN 1 H (CDCl3, δ, ppm), 7,41 (d, J= 8,5 Hz, 4H(7)), 6,89 (d, J= 8,5 Hz, 4H(6)), 6,86 (d, J= 8,7 Hz, 4H(3)); 6,66 (d, J= 8,7 Hz, 4H(2)), 3,49 (s, 4H(10)), 0,49 (s, 6H(9)). RMN 13 C (CDCl3, δ, ppm), -2.05 (C9), 116,2 (C3); 116,4 (C6); 121,4 (C2); 130,9 (C8); 135,6 (C7); 142,8 (C1); 148,0 (C4); 160,0 (C5). RMN 29 Si (CDCl3, δ, ppm), -7.68. Análisis elemental. Calculado: C26H26N2O2Si; (426.58): C, 73.20 %; H, 6.14 %; N, 6.57 %. Encontrado: C, 73,66 %; H, 5,80 %; N, 6,18 %. Bis(4-(4-aminofenoxi)fenil)difenilsilano (10): R: 71 %. T.f.: 83-85 °C. IR (KBr) (cm-1): 3320, 3380 (NH), 3090, 3010 (C-H arom.), 1625 (NH flexión), 1590, 1500, 1498 (C=C arom.); 1390, 1090 (Si-C arom.), 1235 (C-O-C); 870 (arom. p-sust.); 743 (arom. mono-sust.). RMN 1H (CDCl3, ppm), 7,62 (d, J= 6,5 Hz, 4H(7)), 7,46 (m, 10H(10,11,12)), 6,98 (d, J= 8,6 Hz, 4H(3)); 6,94 (d, J= 6,0 Hz, 4H(6)), 6,71 (d, J= 8,7 Hz, 4H(2)); 3,62 (s, 4H(13)). RMN 13 C (CDCl3, ppm), 116,2 (C3); 116,4 (C6); 121,6 (C2); 126,7 (C8); 127,9 (C11); 129,5 (C12); 134,7 (C7); 136,3 (C9); 137,9 (C10); 143,0 (C1); 147,8 (C4); 160,5 (C5). 29 Si RMN (CDCl3, ppm), -14,4. Análisis elemental. Calculado: C36H30N2O2Si; (550,72): C, 78,51 %; H, 5,49 %; N, 5,09 %. Encontrado: C, 78,21 %; H, 5,28 %; N, 4,99 %. 67 2. Síntesis de los diácidos bis(4-(4-carboxifenoxi)fenil)diorganosilano (13, 14) La síntesis de estos monómeros ácidos dicarboxílicos consta de dos etapas: Etapa I: Una mezcla de 4,09 mmol de bis(4-hidroxifenil)diorganosilano (1 ó 2), 8,18 mmol de K2CO3 anhidro, 8,18 mmol de 4-fluorobenzonitrilo y 10 mL de DMF se calienta a reflujo durante 24 h. Al enfriar, se vierte sobre 600 mL de una mezcla etanol-agua (1:1 vol/vol). El sólido de color blanco se filtra, se lava con abundante agua y se seca a 50 °C a vacío durante 12 h. Bis(4-(4-cianofenoxi)fenil)dimetilsilano (11): R: 85 %. T.f.: 125-127 °C. IR (KBr) (cm-1): 3025 (C-H arom.), 2953 (C-H alif.), 2225 (CN), 1606, 1583, 1493 (C=C arom.); 1390, 1109 (Si-C arom.), 1293 (Si-C alif.), 1242 (C-O-C); 872 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 7,85 (d, J= 8,8 Hz, 4H(2)), 7,62 (d, J= 8,3 Hz, 4H(7)), 7,16 (d, J= 8,4 Hz, 4H(6)), 7,13 (d, J= 8,7 Hz, 4H(3)), 0,57 (s, 6H(9)). RMN 13 C (DMSO-d6, δ, ppm), -2,03 (C9), 105,8 (C1); 118,8 (C6); 119,1 (C10); 119,9 (C3); 134,5 (C8); 135,1 (C7); 136,6 (C2); 156.1 (C5); 161,1 (C4). RMN 29 Si (DMSO-d6, δ, ppm), -8,05. Análisis elemental. Calculado: C28H22N2O2Si; (446.57): C, 75.31 %; H, 4.97 %; N, 6.27 %. Encontrado: C, 74.68 %; H, 4.68 %; N, 6.08 %. 68 Bis(4-(4-cianofenoxi)fenil)difenilsilano (12): R: 57 %. T.f.: 133-135 °C. IR (KBr) (cm-1): 3069, 3019 (C-H arom.), 2226 (CN), 1606, 1584, 1492 (C=C arom.); 1390, 1116 (Si-C arom.), 1246 (C-O-C); 872 (arom. p-sust.); 699 (arom. mono-sust). RMN 1H (DMSO-d6, δ, ppm), 7,48 - 6,91 (m, 26H(2,3,6,7,10,11,12)). RMN 13 C (DMSO-d6, δ, ppm), 106,1 (C1); 117,8 (C13); 118,5 (C6); 119,4 (C3); 127,9 (C8); 129,8 (C11); 130,2 (C12); 133,4 (C7); 134,0 (C2); 136,0 (C9); 138,2 (C10); 156.4 (C5); 160,7 (C4). RMN 29 Si (DMSO-d6, δ, ppm), -14,5. Análisis elemental. Calculado: C38H26N2O2Si; (570,71): C, 79,97 %; H, 4,59 %; N, 4,91 %. Encontrado: C, 78,25 %; H, 4,38 %; N, 4,56 %. Etapa II: Una mezcla de 3,35 mmol de bis(4-(4-cianofenoxi)fenil) diorganosilano (11 ó 12), 67 mmol de KOH, 50 mL de etanol y 50 mL de agua se calienta a reflujo durante aproximadamente 2 días, hasta que la evolución de amoniaco cese. La mezcla se filtra y el filtrado se diluye con 50 mL de agua. Luego, se acidifica con una mezcla de HCl/Agua (1:1 vol/vol) hasta pH 2-3. El precipitado de color blanco formado se filtra, se lava con abundante agua y se seca 50 °C a vacío durante 12 h. El sólido se disuelve en una disolución de hidróxido de sodio al 5 %, se filtra y se acidifica nuevamente con una mezcla de HCl/Agua (1:1 vol/vol) hasta pH 2-3. El sólido obtenido se filtra, se lava con abundante agua y se seca a 50 °C a vacío durante 24 horas. 69 Bis(4-(4-carboxifenoxi)fenil)dimetilsilano (13): 9 CH3 HOOC O Si 8 7 6 3 5 O 4 2 1 10 COOH CH3 R: 60 %. T.f.: 230-233 °C. IR (KBr) (cm-1): 3400 (OH), 3075, 3019 (C-H arom.), 2956 (C-H alif.), 1688 (C=O), 1607, 1585, 1494 (C=C arom.); 1108 (Si-C arom.), 1310 (Si-C alif.), 1242 (C-O-C); 874 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 7,96 (d, J= 8,5 Hz, 4H(2)), 7,60 (d, J= 8,1 Hz, 4H(6)), 7,12 (d, J= 8,1 Hz, 4H(7)), 7,06 (d, J= 8,5 Hz, 4H(3)), 0,56 (s, 6H(9)). RMN 13 C (DMSO-d6, δ, ppm), -1,98 (C9), 118,1 (C3); 119,5 (C6); 126,0 (C1); 132,1 (C2); 133,9 (C8); 136,4 (C7); 156,9 (C5); 160,9 (C4), 167,1 (C10). RMN 29 Si (DMSO-d6, δ, ppm), -7,7. Análisis elemental. Calculado: C28H24O6Si; (484,57): C, 69,40 %; H, 4,99 %. Encontrado: C, 68,89 %; H, 4,77 %. Bis(4-(4-carboxifenoxi)fenil)dimetilsilano (14): R: 70 %. T.f.: 265-267 °C. IR (KBr) (cm-1): 3424 (OH), 3068, 3023 (C-H arom.); 1688 (C=O), 1606, 1585, 1494 (C=C arom.); 1390; 1108 (Si-C arom.), 1243 (C-O-C); 874 (arom. p-sust.); 700 (arom. mono-sust). RMN 1H (DMSOd6, δ, ppm), 12,9 (s, 2H(13)); 7,1 – 8,0 (m, 26H(2,3,6,7,10,1,12)). RMN 13 C (DMSO-d6, δ, ppm), 118,3 (C3); 119,4 (C6); 126,1 (C1); 128,5 (C8); 129,2 (C11); 130,3 (C12); 132,0 (C2); 133,8 (C7); 136,1 (C9); 138,3 (C10); 157,4 (C5); 160,4 (C4), 167,0 (C13). RMN 29 Si (DMSO-d6, δ, ppm), -14,8. Análisis 70 elemental. Calculado: C38H28O6Si; (608,71): C, 74,98 %; H, 4,64 %. Encontrado: C, 74,01 %; H, 4,56 %. 3. Síntesis de los anhídridos bis(4-(3,4-carboxifenoxi)fenil) diorganosilano (17, 18) La síntesis de estos dianhídridos de ácidos monoméricos consta de tres etapas: Etapa I: En un balón de 50 mL se añaden 4,09 mmol de bis(4hidroxifenil)diorganosilano (1 ó 2), 8,18 mmol de K2CO3 anhidro, 8,18 mmol de 4-nitroftalonitrilo y 5 mL de DMF. La mezcla se calienta a 60 °C en un baño de aceite por 48 horas y con agitación. Luego, la mezcla se enfría y se deja caer sobre 800 mL de agua con agitación. El sólido de color café obtenido se filtra, se lava con abundante agua y se seca a vacío a 50 °C por 12 horas. Bis(4-(3,4-dicianofenoxi)fenil)dimetilsilano (15): R: 87 %. T.f.: 89-91 °C. IR (KBr) (cm-1): 3079 (C-H arom.), 2956 (C-H alif.), 2232 (CN), 1602, 1583, 1562 (C=C arom.); 1419, 1107 (Si-C arom.), 1310 (Si-C alif.), 1250 (C-O-C); 833 (arom. p-sust.). RMN 1 H (DMSO-d6, δ, ppm), 7,74 (d, J= 8,6 Hz, 2H(6)), 7,46 (s, 2H(3)), 7,30 (d, J= 7,5 Hz, 4H(9)), 7,05 (m, 2H(5)), 6,85 (d, J= 7,4 Hz, 4H(8)), 0,23 (s, 6H(11)). RMN 13 C (DMSO-d6, δ, ppm), -3,35 (C11), 107,6 (C1), 114,5 (C12), 115,0 (C13), 115,9 (C2), 118,7 (C8), 121,5 (C3), 122,1 (C5), 134,0 (C10), 134,4 (C6), 135,5 (C9), 154,1 (C7), 159,7 (C4). RMN 29 Si (DMSO-d6, δ, ppm), -8,67. Análisis elemental. 71 Calculado: C30H20N4O2Si; (496,59): C, 72,56 %; H, 4,06 %, N, 11,28 %. Encontrado: C, 72,21 %; H, 4,16 %, N, 11,08 %. Bis(4-(3,4-dicianofenoxi)fenil)difenilsilano (16): R: 89 %. T.f.: 91-93 °C. IR (KBr) (cm-1): 3067, 3024 (C-H arom.), 2231 (CN), 1602, 1582, 1562, 1486 (C=C); 1427, 1109 (Si-C arom.), 1250 (C-O); 829 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 8,13 (d, J= 8,7 Hz, 2H(6)), 7,91 (d, J= 2,5 Hz, 2H(3)), 7,61 (d, J= 8,4, 4H(9)), 7,51 (m, 12H(5,12,13,14)), 7,28 (d, J= 8,4 Hz, 4H(8)). RMN 13 C (DMSO-d6, δ, ppm), 109,0 (C1), 115,7 (C16), 116,2 (C15), 117,1 (C2), 120,0 (C8), 123,1 (C3), 123,6 (C5), 128,6 (C10), 130,6 (C13), 133,4 (C14), 136,2 (C9), 136,6 (C11), 136,8 (C6), 138,5 (C12), 156,0 (C7), 160,6 (C4). RMN 29 Si (DMSO-d6, δ, ppm), -14,83. Análisis elemental. Calculado: C40H24N4O2Si; (620,73): C, 77,40 %; H, 3,90 %, N, 9,03 %. Encontrado: C, 77,22 %; H, 3,75 %, N, 8,98 %. Etapa II: En un balón de 100 mL se añaden 2,01 mmol de bis(4-(3,4dicianofenoxi)fenil)diorganosilano (15 ó 16), 80 mmol de KOH, 15 mL de agua, 15 mL de etanol y se calienta a reflujo. La suspensión de color rojo se disuelve a medida que pasa el tiempo y la reacción se detiene cuando cesa la emisión de amoniaco, lo cual toma, por lo general, entre 48 y 72 horas. La disolución se filtra y el filtrado se acidifica con HCl-agua (1:1 vol/vol) hasta pH 2-3. El sólido obtenido se filtra, se lava con agua y se seca a vacío a 50 °C durante 12 horas. El sólido se disuelve en una disolución de hidróxido de sodio al 5 %, se filtra y se acidifica nuevamente con una mezcla de HCl/Agua 72 (1:1 vol/vol) hasta pH 2-3. El sólido obtenido se filtra, se lava con abundante agua y se seca a 50 °C a vacío durante 24 horas. Ácido bis(4-(3,4-dicarboxifenoxi)fenil)dimetilsilano (17): R: 85 %. T.f.: > 300 °C. IR (KBr) (cm-1): 3425 (OH), 3023 (C-H arom.), 2955 (C-H alif.), 1709 (C=O), 1605, 1577, 1495 (C=C); 1421, 1108 (Si-C arom.), 1389 (Si-C alif.), 1231 (C-O-C); 833 (arom. p-sust.). RMN 1 H (DMSO-d6, δ, ppm), 7,84 (d, J= 8,6 Hz, 2H(6)), 7,37 (d, J= 8,0 Hz, 4H(9)), 7,29 (d, J= 2,2 Hz, 2H(3)), 6,94 (dd, J= 2,3, 8,6 Hz, 2H(5)), 6,88 (d, J= 8,1 Hz, 4H(8)), 0,33 (s, 6H(11)). RMN 13 C (DMSO-d6, δ, ppm), -2,49 (C11), 118,9 (C8), 119,4 (C6), 119,5 (C3), 128,1 (C5), 133,3 (C1), 133,8 (C10), 136,0 (C9), 137,1 (C2), 156,4 (C7), 158,3 (C4), 167,4 (C13), 167,8 (C12). RMN 29 Si (DMSO-d6, δ, ppm), -8,08. Análisis elemental. Calculado: C30H24O10Si; (572,59): C, 62,93 %; H, 4,22 %. Encontrado: C, 62,85 %; H, 4,15 %. Ácido bis(4-(3,4-dicarboxifenoxi)fenil)difenilsilano (18): R: 77 %. T.f.: > 300 °C. IR (KBr) (cm-1): 3431 (OH), 3068, 3019 (C-H arom.), 1708 (C=O), 1604, 1578, 1494 (C=C); 1427, 1108 (Si-C arom.), 1231 (C-OC); 829 (arom. p-sust.). RMN 1 H (DMSO-d6, δ, ppm), 14,3 (s, 4H(15,16)), 73 7,93 (d, J= 8,4 Hz, 2H(6)), 7,52 (m, 20H(3,8,9,12,13,14)), 7,20 (d, J= 8,4 Hz, 2H(5)). RMN 13 C (DMSO-d6, δ, ppm), 119,3 (C6), 120,0 (C3), 128,6 (C8), 129,2 (C5), 130,3 (C1), 130,7 (10), 133,0 (13), 133,7 (C14), 134,8 (C9), 136,1 (C2), 137,2 (C11), 138,3 (C12), 157,4 (C7), 158,5 (C4), 167,8 (C16), 168,4 (C15). RMN 29 Si (DMSO-d6, δ, ppm), -14,9. Análisis elemental. Calculado: C40H28O10Si; (696,73): C, 68,95 %; H, 4,05 %. Encontrado: C, 68,78 %; H, 4,25 %. Etapa III: En un balón de 100 mL equipado con agitación magnética se agregan 1,74 mmol de ácido bis(4-(3,4-carboxifenoxi)fenil)diorganosilano (17 ó 18) y 7 mL de anhídrido acético. La mezcla se calienta a reflujo suavemente durante 24 horas y se deja enfriar. El anhídrido acético y el ácido acético formado se eliminan por destilación a presión reducida. Al finalizar la destilación se obtiene un aceite de color rojo oscuro el cual se disuelve en 2 mL de acetato de etilo y se deja caer sobre 300 mL de nhexano. El precipitado obtenido de color amarillo se filtra, se lava con abundante n-hexano y finalmente se seca en una estufa a 140 °C durante 3 días. Ánhídrido bis(4-(3,4-dicarboxifenoxi)fenil)dimetilsilano (19): O O CH3 O O 13 11 O Si 10 CH3 9 8 3 7 O O 2 4 1 12 O 5 6 R: 42 %. T.f.: 149-152 °C. IR (KBr) (cm-1): 3021 (C-H arom.), 2954 (C-H alif.), 1848, 1780 (C=O), 1618, 1585, 1480 (C=C); 1418, 1019 (Si-C arom.), 1385 (Si-C alif.), 1283 (C-O-C); 836 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 8,08 (d, J= 8,4 Hz, 2H(6)), 7,69 (d, J= 8,4 Hz, 4H(9)), 7,53 (dd, J= 8,4, 2,2 Hz, 2H(5)), 7,49 (d, J= 2,0 Hz, 2H(3)), 7,23 (d, J= 8,4 Hz, 4H(8)), 0,61 (s, 6H(11)). RMN 13 C (DMSO-d6, δ, ppm), -3,00 (C11), 112,6 (C6), 119,3 (C8), 124,3 74 (C3), 124,5 (C5), 127,5 (C1), 133,6 (C2), 134,4 (C10), 135,9 (C9), 154,9 (C7), 162,0 (C4), 162,1 (C13), 163,0 (C12). RMN 29 Si (DMSO-d6, δ, ppm), -7,98. Análisis elemental. Calculado: C30H20O8Si; (536,56): C, 67,15 %; H, 3,76 %. Encontrado: C, 67,08 %; H, 3,68 %. Ánhídrido bis(4-(3,4-dicarboxifenoxi)fenil)difenilsilano (20): R: 62 %. T.f.: 150-155 °C. IR (KBr) (cm-1): 3070 (C-H arom.), 1851, 1777 (C=O), 1618, 1585, 1480 (C=C); 1428, 1016 (Si-C arom.), 1281 (C-O-C); 892 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 8,09 (d, J= 9,1 Hz, 2H(6)), 7,56 (d, J= 8,5 Hz, 4H(9)); 7,46 (m, 14H(3,5,12,13,14)), 7,29 (d, J= 8,4 Hz, 4H(8)). RMN 13 C (DMSO-d6, δ, ppm), 113,4 (C6), 119,7 (C8), 125,1 (C3), 127,8 (C5), 128,2 (C1), 130,0 (C10), 130,2 (C2), 133,0 (C13), 134,0 (C9), 134,4 (C11), 135,7 (C12), 138,2 (C14), 156,0 (C7), 162,4 (C4), 162,5 (C16), 163,0 (C15). RMN 29 Si (DMSO-d6, δ, ppm), -14,8. Análisis elemental. Calculado: C40H24O8Si; (660,70): C, 72,72 %; H, 3,66 %. Encontrado: C, 72,68 %; H, 3,56 %. 4. Síntesis de diaminas derivadas de dibenzofurano (21, 22) La síntesis de estas dos diaminas monoméricas consta de dos etapas: Etapa I: En un balón de 500 mL se disuelven 0,09 mol de dibenzofurano en 250 mL de anhídrido acético. La disolución se enfría en un baño de hielo y se agrega lentamente una mezcla de 50 mL de ácido nítrico fumante y 10 mL de 75 ácido sulfúrico concentrado, controlando que la temperatura de la disolución no sobrepase los 10 °C. Una vez concluida la adición, la mezcla se agita a dicha temperatura durante 15 minutos y luego se vierte sobre 1 L de agua fría con agitación. El sólido de color amarillo se filtra, se lava con abundante agua y se seca a vacío a 50 °C durante 12 horas. 2,8-Dinitrobenzofurano y 2,7-dinitrodibenzofurano (en mezcla): R: 65 %. IR (KBr) (cm-1): 3063 (C-H arom.), 1508, 1490 (C=C); 1519, 1344 (NO2), 1200 (C-O-C). La mezcla de isómeros no se caracterizó en disolución (RMN) debido a que el análisis de FT-IR ratificó la nitración deseada. La separación de los isómeros se realizó posterior a la reducción tal como se señala en el punto siguiente. Etapa II: En un balón de 250 mL se agregan 0,04 mol de la mezcla del derivado dinitrado obtenida en la etapa anterior, 0,2 g de Pd/C activado (10 % en peso), 120 mL de etanol y 15 mL de hidracina monohidratada al 80 %. La disolución se agita vigorosamente por media hora a temperatura ambiente y luego se calienta a reflujo durante dos horas. Terminado ese tiempo, la mezcla se enfría, el catalizador remanente se elimina por filtración sobre celite y la disolución se vierte sobre 500 mL de agua. El precipitado de color café se filtra y se seca a vacío a 50 °C durante 12 horas. El sólido, consistente en dos isómeros, se somete a tratamiento empleando una columna cromatográfica (silica gel 60) y acetato de etilo como eluyente. El primer isómero separado, con Rf = 0,76, consistió en 2,7- diaminodibenzofurano (65 % de la masa total), mientras que el segundo 76 isómero con Rf = 0,58 consistió en 2,8-diaminodibenzofurano (35 %). Ambos isómeros se purifican mediante la técnica de sublimación. 2,7-diaminodibenzofurano (21): R: 91 %. T.f: 148-150 °C (Lit (68). 150 °C). IR (KBr) (cm-1): 3397, 3296, 3199 (NH2), 1627 (NH flexión), 3063 (C-H arom.), 1597, 1489, 1462 (C=C); 1188 (C-O-C). RMN 1H (DMSO-d6, δ, ppm), 7,55 (d, J= 8,3 Hz, 1H(1)), 7,20 (d, J= 8,6 Hz, 1H(9)), 6,98 (d, J= 2,1 Hz, 1H(12)), 6,70 (d, J= 1,5 Hz, 1H(4)); 6,58 (m, 2H(2,10)); 5,45 (s, 2H(14)); 4,90 (s, 4H(13)). RMN 13 C (DMSO-d6, δ, ppm), 95,7 (C4); 103,5 (C12), 110,6 (C2); 111,1 (C9), 112,1 (C10); 113,2 (C6); 121,2 (C1), 125,5 (C7), 144,7 (C11), 148,0 (C8); 149,5 (C3); 158,3 (C5). Análisis elemental. Calculado: C12H10N2O; (198,22): C, 72,71 %; H, 5,08 %, N, 14,13 %. Encontrado: C, 72,66 %; H, 4,96 %, N, 14,08 %. 2,8-diaminodibenzofurano (22): O 5 6 4 3 2 H2N 1 7 NH2 R: 91 %. T.f: 210-212 °C (Lit. (68) 212 °C). IR (KBr) (cm-1): 3397, 3296, 3199 (NH2), 1627 (NH flexión), 3063 (C-H arom.), 1597, 1489, 1462 (C=C); 1188 (C-O-C). RMN 1H (DMSO-d6, δ, ppm), 7,27 (d, J= 8,7 Hz, 2H(4)), 7,06 (d, J= 2,0 Hz, 2H(1)), 6,74 (dd, J= 8,7, 2,2 Hz, 2H(3)), 4,97 (s, 4H(7)). RMN 13 C (DMSO-d6, δ, ppm), 104,3 (C1), 111,7 (C3), 115,0 (C4), 124,7 (C6), 144,5 (C5), 149,1 (C2). Análisis elemental. Calculado: C12H10N2O; (198,22): C, 77 72,71 %; H, 5,08 %, N, 14,13 %. Encontrado: C, 72,68 %; H, 5,01 %, N, 14,10 %. 5. Síntesis de 2,8-di(4-aminofenil)dibenzofurano (24) La síntesis de esta diamina monomérica consta de dos etapas: Etapa I: En un balón de 50 mL se añaden 1,19 mmol de 2,8diyododibenzofurano, 2,85 mmol de ácido 4-nitrofenilborónico, 0,18 mmol de cloruro de bis(trifenilfosfina)paladio (II) manteniendo el sistema en atmósfera de nitrógeno. A esta mezcla sólida se adicionan 10 mL de dioxano y se agita a temperatura ambiente durante 30 minutos. Luego, se añaden 4 mL de una disolución acuosa de carbonato de potasio (1 M) y la suspensión de color amarillo se calienta a reflujo. Después de media hora de calentamiento, la suspensión cambia su apariencia a color café. El reflujo se mantiene por 24 horas adicionales. Pasado este tiempo, se enfría el sistema, se añade agua y el sólido de color café obtenido se filtra y se seca en una estufa a vacío a 50 °C durante 12 horas. 2,8-di(4-nitrofenil)dibenzofurano (23): R: 95 %. T.f.: 253-255 °C. IR (KBr) (cm-1): 3080 (C-H arom.), 1594, 1474, 1453 (C=C); 1514, 1344 (NO2), 1202 (C-O-C). No se realiza la caracterización en disolución (RMN) dada la insolubilidad del compuesto en los disolventes orgánicos y disolventes deuterados ensayados a temperatura 78 ambiente y a 40 °C. Análisis elemental. Calculado: C24H14N2O5; (410,38): C, 70,24 %; H, 3,44 %, N, 6,83 %. Encontrado: C, 70,18 %; H, 3,35 %, N, 6,78 %. Etapa II: En un balón de 100 mL se agrega 1,0 mmol de 2,8di(nitrofenil)dibenzofurano (23), 0,1 g de Pd/C activado (10 % en peso) y 10 mL de etanol. El sistema se calienta a reflujo y se añaden 7 mL de hidracina monohidratada al 80 % gota a gota por un período de 30 minutos. La mezcla se calienta a reflujo durante 24 horas, se filtra sobre celite en caliente y se vuelca sobre 300 mL de agua con agitación. El precipitado de color blanco se filtra y se seca a vacío a 50 °C por 12 horas. 2,8-di(4-aminofenil)dibenzofurano (24): R: 70 %. T.f.: 210-212 °C. IR (KBr) (cm-1): 3438, 3396, 3316, (NH2), 3029 (CH arom.) 1619 (NH flexión), 1520, 1488, 1426 (C=C); 1199 (C-O-C). RMN 1H (DMSO-d6, δ, ppm), 8,42 (d, J= 0,7 Hz, 2H(10)), 7,68 (m, 4H(6,7)), 7,51 (d, J= 8,5 Hz, 4H(3)), 6,73 (d, J= 8,4 Hz, 4H(2)), 5,28 (s, 4H(11)). RMN 13 C (DMSO-d6, δ, ppm), 111,5 (C7), 114,2 (C9), 117,7 (C10), 124,3 (C2), 125,1 (C6), 127,3 (C3), 127,5 (C4), 136,1 (C5), 148,0 (C1), 154,5 (C8). Análisis elemental. Calculado: C24H18N2O; (350,41): C, 82,26 %; H, 5,18 %, N, 7,99 %. Encontrado: C, 82,16 %; H, 5,06 %, N, 7,88 %. 6. Síntesis de 9,9-bis(4-(4-aminofenoxi)-3-metilfenil)fluoreno (26) La síntesis de esta diamina monomérica consta de dos etapas: 79 Etapa I: Una mezcla de 2,6 mmol de 9,9-bis(4-hidroxi-3-metilfenil)fluoreno, 7,80 mmol de K2CO3 anhidro, 5,2 mmol de 1-fluoro-4-nitrobenceno y 4 mL de DMF se calienta a reflujo por 24 horas. La suspensión de color verde obtenida se enfría y se deja caer sobre una mezcla de 200 mL de etanolagua (1:1 vol/vol). El sólido de color amarillo se filtra, se lava con abundante agua y se seca en una estufa a 120 °C. 9,9-bis(4-(4-nitrofenoxi)-3-metilfenil)fluoreno (25): R: 88 %. T.f.: 231-233 °C. IR (KBr) (cm-1): 3104, 3073 (C-H arom.); 2924, 2852, 1446 (C-H alif.); 1610, 1487 (C=C arom.); 1589, 1339 (NO2); 1253 (CO-C); 851 (arom. p-sust.). No se realiza la caracterización en disolución (RMN) dada la insolubilidad del compuesto en los disolventes orgánicos y disolventes deuterados ensayados a temperatura ambiente y a 40 °C. Análisis elemental. Calculado: C39H28N2O6; (620,65): C, 75,47 %; H, 4,55 %; N, 4,51 %. Encontrado: C, 76,00 %; H, 4,29 %; N, 4,72 %. Etapa II: Una mezcla de 1,33 mmol de 9,9-bis(4-(4-nitrofenoxi)-3metilfenil)fluoreno (25), 0,11 g de Pd/C (10 % en peso) y 8 mL de etanol se calienta a reflujo y se añade gota a gota 8 mL de hidracina monohidratada al 80 % por un período de 1 hora. Una vez concluida la adición, se deja reflujar por 24 horas. La mezcla se filtra en caliente sobre celite y la disolución se vuelca sobre 300 mL de agua con agitación vigorosa. El sólido de color blanco obtenido se filtra, se lava con abundante agua y se seca a vació a 50 °C durante 12 h. 80 9,9-bis(4-(4-aminofenoxi)-3-metilfenil)fluoreno (26): 19 2 H2N 18 CH3 CH3 6 O 5 7 13 3 11 8 O 1 NH2 4 10 9 12 14 15 17 16 R: 67 %. T.f.: 124-126 °C. IR (KBr) (cm-1): 3441, 3364 (NH2); 3037, 3015 (CH arom.); 2919 (C-H alif.); 1619 (NH2 flexión); 1508, 1490 (C=C arom.); 1242 (C-O-C); 822 (arom. p-sust.) RMN 1H (CDCl3, ppm): 7,73 (d, J= 7,5 Hz, 2H(16)); 7,46 (d, J= 7,6 Hz, 2H(13)); 7,31 (m, 2H(15)); 7,21 (m, 2H(14)); 7,08 (d, J= 1,5 Hz, 2H(7)); 6,91 (dd, J= 8,6; 1,8 Hz, 2H(9)); 6,74 (d, J= 8,6 Hz, 4H(3)); 6,57 (m, 6H(2,10)); 3,45 (s, 4H(19)); 2,14 (s, 6H(18)). RMN 13 C (CDCl3, ppm): 155,1 (C5); 151,6 (C4); 149,2 (C17); 142,0 (C12); 139,9 (C8); 139,8 (C1); 130,6 (C7); 127,8 (C6); 127,6 (C13); 127,3 (C14); 126,6 (C15); 126,1 (C16); 120,07 (C9); 120,0 (C3); 116,4 (C10); 116,1 (C2); 64,3 (C11); 16,4 (C18). Análisis elemental. Calculado: C39H32N2O2; (560,68): C, 83,54 %; H, 5,75 %; N, 5,00 %. Encontrado: C, 82,88 %; H, 6,11 %; N, 5,34 %. 7. Síntesis de anhídrido 9,9-bis(4-(3,4-dicarboxifenoxi)-3- metilfenil)fluoreno (29) La síntesis de este dianhídrido de ácido monomérico consta de tres etapas: Etapa I: En un balón de 50 mL se añaden 1,32 mmol de 9,9-bis(4-hidroxi-3metilfenil)fluoreno, 2,64 mmol de K2CO3 anhidro, 2,64 mmol de 4nitroftalonitrilo y 5 mL de DMF. La mezcla se calienta a 60 °C en un baño de aceite por 48 horas bajo agitación constante. Luego, la mezcla se enfría y se deja caer sobre 800 mL de agua con agitación. El sólido obtenido se filtra, se lava con abundante agua y se seca a vacío a 50 °C por 12 horas. 81 9,9-bis(4-(3,4-dicianofenoxi)-3-metilfenil)fluoreno (27): 22 CN CN NC 20 CH3 8 O 7 9 10 15 14 3 CH3 O 2 4 21 CN 1 6 5 12 11 13 16 17 19 18 R: 95 %. T.f.: 231-233 °C. IR (KBr) (cm-1): 3071 (C-H arom.), 2922 (C-H alif.), 2231 (CN), 1594, 1564, 1485 (C=C arom.); 1250 (C-O); 832 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 8,05 (d, J= 8,8 Hz, 2H(6)); 7,94 (d, J= 7,3 Hz, 2H(18)); 7,70 (d, J= 2,5 Hz, 2H(3)); 7,55 (d, J= 7,4 Hz, 2H(15)); 7,40 (m, 4H(16,17)); 7,24 (dd, J= 8,8 y 2,5 Hz, 2H(5)); 7,11 (m, 4H(9,11)); 7,02 (d, J= 8,4 Hz, 2H(12)); 2,01 (s, 6H(20)). RMN 13 C (DMSO-d6, δ, ppm): 160,8 (C4); 150,3 (C7); 150,1 (C14); 143,2 (C19); 139,4 (C10); 136,3 (C6); 130,8 (C9); 129,5 (C15); 128,0 (C8); 127,9 (C16); 127,4 (C17); 126,1 (C18); 121,8 (C11); 120,9 (C5); 120,6 (C3); 116,7 (C12); 115,9 (C2); 115,4 (C21), 115,3 (C22); 107,8 (C1); 64,0 (C13); 15,7 (C20). Análisis elemental. Calculado: C43H26N4O2; (630,69): C, 81,89 %; H, 4,16 %; N, 8,88 %. Encontrado: C, 81,56 %; H, 4,08 %; N, 8,68 %. Etapa II: En un balón de 100 mL se añaden 1,11 mmol de 9,9-bis(4-(3,4dicianofenoxi)-3-metilfenil)fluoreno (27), 22 mmol de KOH, 15 mL de agua, 15 mL de etanol y se calienta a reflujo. La suspensión se disuelve a medida que pasa el tiempo y la reacción se detiene cuando cesa la emisión de amoniaco, por lo general esto ocurre entre las 48 y 72 horas. La disolución se filtra y el filtrado se acidifica con HCl-agua (1:1 vol/vol) hasta pH 2-3. El sólido obtenido se filtra, se lava con agua y se seca a vacío a 50 °C durante 12 horas. El sólido se disuelve en una disolución de hidróxido de sodio al 5 %, 82 se filtra y se acidifica nuevamente con una mezcla de HCl/Agua (1:1 vol/vol) hasta pH 2-3. El sólido obtenido se filtra, se lava con abundante agua y se seca a 50 °C a vacío durante 24 horas. Ácido 9,9-bis(4-(3,4-dicarboxifenoxi)-3-metilfenil)fluoreno (28): R: 92 %. T.f.:> 280 °C. IR (KBr) (cm-1): 3408 (OH), 3061 (C-H arom.), 2923 (C-H alif.), 1707 (C=O), 1598, 1574, 1492 (C=C); 1243 (C-O-C); 828 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 7,95 (d, J= 7,4 Hz, 2H(18)); 7,75 (d, J= 8,5 Hz, 2H(6)); 7,56 (d, J= 7,4 Hz, 2H(15)); 7,40 (m, 4H(16,17)); 7,04 (m, 10H(3,5,9,11,12)); 2,05 (s, 6H(20)). RMN 13 C (DMSO-d6, δ, ppm): 168,5 (C21); 167,3 (C22); 159,2 (C4); 151,3 (C7); 150,3 (C14); 142,4 (C19); 139,4 (C10); 136,6 (C2); 131,4 (C9); 130,6 (C15); 129,3 (C8); 128,0 (C16); 127,8 (C17); 127,1 (C18); 126,1 (C1); 125,1 (C11); 120,6 (C5); 120,2 (C12); 117,4 (C3); 115,5 (C6); 64,0 (C13); 15,8 (C20). Análisis elemental. Calculado: C43H30O10; (706,69): C, 73,08 %; H, 4,28 %. Encontrado: C, 72,96 %; H, 4,18 %. Etapa III: En un balón de 100 mL equipado con agitación magnética se agregan 2,17 mmol de ácido 9,9-bis(4-(3,4-dicarboxifenoxi)-3- metilfenil)fluoreno (28) y 7 mL de anhídrido acético. La mezcla se somete a reflujo suave durante 24 horas y se deja enfriar. El anhídrido acético y ácido acético formado se eliminan por destilación a presión reducida. Al finalizar la 83 destilación se obtiene un aceite, el cual se disuelve en 2 mL de acetato de etilo y se deja caer sobre 300 mL de n-hexano. El precipitado obtenido de color amarillo se filtra, se lava con abundante n-hexano y finalmente se seca en una estufa a 100 °C a vacío durante 12 horas. Anhídrido 9,9-bis(4-(3,4-dicarboxifenoxi)-3-metilfenil)fluoreno (29): O O O O 20 CH3 CH3 8 O 7 9 10 15 14 3 13 O 22 2 4 O 21 O 1 6 5 12 11 16 17 19 18 R: 83 %. T.f.:250-252 °C. IR (KBr) (cm-1): 3066 (C-H arom.), 2921 (C-H alif.), 1852, 1777 (C=O), 1618, 1480, 1448 (C=C); 1283 (C-O); 891 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 8,04 (d, J= 8,4 Hz, 2H(6)); 7,75 (d, J= 7,3 Hz, 2H(18)); 7,59 (d, J= 7,5 Hz, 2H(15)); 7,41 (m, 6H(12,16,17)); 7,32 (s, 2H(3)); 7,17 (s, 2H(9)); 7,15 (d, J= 9,3 Hz, 2H(11)); 7,06 (d, J= 8,3 Hz; 2H(5)); 2,03 (s, 6H(20)). RMN 13 C (DMSO-d6, δ, ppm): 163,7 (C21); 162,6 (C22); 162,4 (C4); 150,7 (C7); 150,1 (C14); 143,3 (C19); 139,4 (C10); 134,1 (C9); 130,9 (C2); 129,6 (C15); 128,1 (C8); 127,9 (C16); 127,4 (C17); 126,1 (C18); 124,4 (C11); 123,5 (C1); 120,8 (C5); 120,6 (C12); 118,2 (C3); 111,5 (C6); 64,0 (C13); 15,8 (C20). Análisis elemental. Calculado: C43H26O8; (670,66): C, 77,01 %; H, 3,91 %. Encontrado: C, 76,89 %; H, 3,85 %. 84 IV. SÍNTESIS DE POLI(AMIDA)S Una mezcla de 0,47 mmol de bis(4-(4-aminofenoxi)fenil)diorganosilano (9 ó 10), 0,47 mmol de ácido bis(4-(4-carboxifenoxi)fenil)diorganosilano (13 ó 14), 0,24 g of CaCl2, 0,30 mL de TPP, 1,20 mL de piridina y 1,80 mL de NMP se calienta con agitación a una temperatura de 120 °C durante 3 horas. Pasado este tiempo, la mezcla se enfría y se vierte sobre 300 mL de metanol bajo agitación constante. El precipitado de color blanco se filtra, se lava con abundante metanol y se seca. El producto (PA-I – PA-IV) se disuelve en DMSO y se reprecipita en metanol. El sólido se filtra y se seca a vacío a 100 °C durante 12 horas. Las poli(amida)s PA-V – PA-IX se prepararon siguiendo la misma metodología, variando solo la naturaleza de la diamina y del ácido dicarboxílico empleado. PA-I: R: 89 %. IR (KBr, cm-1), 3421 (N-H), 3067 (C-H arom.), 2926 (C-H alif.), 1647 (C=O), 1606, 1587, 1492 (C=C), 1407, 1014 (Si-C arom.), 1309 (Si-C alif.), 1228 (C-O-C), 874 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 10,28 (s, 2H(20)), 8,03 (d, J= 7,7 Hz, 4H(2)); 7,83 (d, J= 7,8 Hz, 4H(11)); 7,51 (d, J= 7,3 Hz, 4H(16)); 7,45 (d, J= 7,1 Hz, 4H(3)); 7,19 (m, 8H(6,7)); 7,06 (d, J= 8,1 Hz, 4H(12)); 6,99 (d, J= 7,3 Hz, 4H(15)), 0,52 (s, 12H(9,18)). RMN 13 C (DMSO-d6, δ, ppm), 164,5 (C19); 159,6 (C4); 158,4 (C14); 155,5 (C5); 151,3 (C13); 135,6 (C7); 135,3 (C17); 131,4 (C8); 130,1 (C16); 129,8 (C2); 129,4 (C1); 124,2 (C10); 121,9 (C11); 119,5 (C3); 119,4 (C6); 117,3 (C15); 117,0 (C12); -2,42 (C9 y C18), RMN 29 Si (DMSO-d6, δ, ppm), -8,98. Análisis elemental. Calculado: [C54H46N2O6Si2]n (875,12)n: C, 74,11 %; H, 5,30 %; N, 3,20 %. Encontrado: C, 73,98 %; H, 5,18 %; N, 3,13%. 85 * H N CH3 O Si 17 12 16 15 14 O 13 CH3 20 11 10 O H N C 19 O Si CH3 7 8 6 3 5 O 4 2 1 O * C CH3 18 n 9 PA-II: R: 93 %. IR (KBr, cm-1), 3423 (N-H), 3068 (C-H arom.), 2954 (C-H alif.), 1655 (C=O), 1606, 1585, 1492 (C=C), 1406, 1014 (Si-C arom.), 1308 (Si-C alif.), 1227 (C-O-C), 874 (arom. p-sust.), 700 (arom. mono-sust). RMN 1 H (DMSO-d6, δ, ppm), 10,25 (s, 2H(23)), 7,98 (d, J = 6,9 Hz, 4H(2)); 7,76 (d, J= 8,0 Hz, 4H(14)); 7,46 (m, 18H(3,10,11,12,19)); 7,12 (s, 8H(6,7); 6,99 (s, 4H(15)); 6,91 (s, 4H(18)); 0,45 (s, 6H(21)). RMN 13 C (DMSO-d6, δ, ppm), 165,0 (C22); 159,2 (C4); 159,0 (C17); 158,0 (C5); 151,9 (C16); 138,3 (C10); 136,2 (C7); 136,1 (C19); 135,7 (C9); 133,9 (C20); 132,0 (C13); 130,4 (C12); 129,8 (C1); 128,9 (C8); 128,7 (C11); 122,5 (C14); 120,1 (C2); 119,1 (C3); 118,7 (C6); 117,6 (C18); 115,6 (C15); -1,9 (C21), RMN 29 Si (DMSO-d6, δ, ppm), -8,1 (-Ph-Si(CH3)2-Ph-) y -15,0 (-Ph-Si(Ph)2-Ph-). Análisis elemental. Calculado: [C64H50N2O6Si2]n (999,26)n: C, 76,93 %; H, 5,04 %; N, 2,80 %. Encontrado: C, 76,83 %; H, 4,98 %; N, 2,75%. PA-III: R: 83 %. IR (KBr, cm-1), 3422 (N-H), 3067 (C-H arom.), 2954 (C-H alif.), 1655 (C=O), 1606, 1586, 1492 (C=C), 1405, 1014 (Si-C arom.), 1312 (Si-C alif.); 1228 (C-O-C), 873 (arom. p-sust.); 703 (arom. mono-sust). RMN 1 H (DMSO-d6, δ, ppm), 10,30 (s, 2H(23)), 8,03 (d, J= 6,9 Hz, 4H(2)); 7,84 (d, J= 7,7 Hz, 4H(11)); 7,59 (d, J= 6,3 Hz, 4H(16)); 7,47 (m, 18H(3,12,19,20,21)); 7,11 (s, 8H(6,7)); 7,04 (d, J= 6,3 Hz, 4H(15)); 0,56 (s, 6H(9)). RMN 13 C (DMSO-d6, δ, ppm), 164,5 (C22); 159,2 (C4); 159,1 (C14); 156,8 (C5); 151,0 86 (C13); 137,6 (C19); 135,9 (C7); 135,7 (C16); 135,5 (C18); 133,8 (C17); 133,0 (C10); 130,2 (C1); 129,9 (C21); 129,7 (C20); 128,1 (C11); 122,0 (C2); 119,9 (C3); 119,4 (C8); 118,6 (C6); 117,9 (C15); 117,1 (C12); -2,4 (C9), RMN 29 Si (DMSO-d6, δ, ppm), -7,3 (-Ph-Si(CH3)2-Ph-) y -14,2 (-Ph-Si(Ph)2-Ph-). Análisis elemental. Calculado: [C64H50N2O6Si2]n (999,26)n: C, 76,93 %; H, 5,04 %; N, 2,80 %. Encontrado: C, 76,79 %; H, 4,86 %; N, 2,68%. PA-IV: R: 93 %. IR (KBr, cm-1), 3422 (N-H), 3068 (C-H arom.), 2954 (C-H alif.), 1655 (C=O), 1606, 1585, 1492 (C=C), 1407, 1015 (Si-C arom.), 1229 (C-O-C), 873 (arom. p-sust.); 702 (arom. mono-sust). RMN 1H (DMSO-d6, δ, ppm), 10,32 (s, 2H(26)), 8,02 (s, 4H(2)); 7,82 (s, 4H(14)); 7,49 (m, 28H(3,10,11,12,19,22,23,24)); 7,10 (m, 16H(6,7,15,16)). RMN 13 C (DMSO-d6, δ, ppm), 164,9 (C25); 159,5 (C4); 159,1 (C17); 157,9 (C5); 151,4 (C16); 138,3 (C10); 138,2 (C22); 138,0 (C7); 136,1 (C19); 134,1 (C9); 134,0 (C21); 133,8 (C20); 130,3 (C12); 130,2 (C24); 129,7 (C13); 128,6 (C11); 128,5 (C23); 128,4 (C14); 127,3 (C1); 122,4 (C2); 120,3 (C3); 119,0 (C6); 118,6 (C18); 117,4 (C15); 115.5 (C8). RMN 29 Si (DMSO-d6, δ, ppm), -14,9 y -15,0. Análisis elemental. Calculado: [C74H54N2O6Si2]n (1123,40)n: C, 79,12 %; H, 4,84 %; N, 2,49 %. Encontrado: C, 79,00 %; H, 4,78 %; N, 2,35%. 87 PA-V: R: 92 %. IR (KBr, cm-1), 3422 (N-H), 3026 (C-H arom.), 2956 (C-H alif.), 1650 (C=O), 1608, 1582, 1519 (C=C), 1403, 1020 (Si-C arom.), 1309 (Si-C alif.); 1225 (C-O-C), 805 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 10,35 (s, 2H(21)), 8,59 (s, 2H(19)); 8,07 (d, J= 7,9 Hz, 4H(2)); 7,96 (d, J= 8,1 Hz, 4H(12)); 7,79 (m, 8H(11,15,16)); 7,60 (d, J= 7,5 Hz, 4H(7)); 7,12 (m, 8H(3,6)); 0,56 (s, 6H(9)). RMN 13 C (DMSO-d6, δ, ppm), 164,1 (C20); 158,6 (C17); 156,3 (C4); 154,8 (C5); 138,0 (C10); 135,4 (C7); 134,6 (C8); 132,5 (C13); 129,7 (C1); 129,4 (C15); 129,3 (C14); 126,4 (C2); 123,8 (C12); 120,1 (C18); 119,0 (C19); 118,5 (C11); 118,1 (C6); 117,4 (C3); 111,3 (C16); 115.5 (C8); -2,9 (C9). RMN 29 Si (DMSO-d6, δ, ppm), -7,6. Análisis elemental. Calculado: [C52H38N2O5Si]n (798,95)n: C, 78,17 %; H, 4,79 %; N, 3,51 %. Encontrado: C, 77,95 %; H, 4,56 %; N, 3,37%. O 18 17 16 15 12 19 14 13 11 * N H 9 O CH3 HN C 8 10 21 O 20 Si CH3 7 6 3 5 O 4 2 1 O C * n PA-VI: R: 95 %. IR (KBr, cm-1), 3417 (N-H), 3062 (C-H arom.), 2953, 2920 (C-H alif.), 1659 (C=O), 1602, 1506, 1491 (C=C), 1407, 1017 (Si-C arom.), 1311 (Si-C alif.); 1248 (C-O-C), 814 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 10,23 (s, 2H(24)), 7,88 (m, 6H(2,20)); 7,66 (m, 8H(3,17,19)); 7,36 (m, 6H(6,18)); 6,93 (m, 8H(7,11,13)); 6,68 (m, 2H(14)); 2,05 (s, 6H(22)); 0,57 (s, 6H(25)). RMN 13 C (DMSO-d6, δ, ppm), 164,2 (C23); 152,4 (C9); 151,5 (C8); 149,6 (C16); 140,4 (C21); 139,5 (C12); 138,4 (C5); 138,3 (C4); 134,6 (C1); 133,2 (C10); 132,7 (C3); 129,2 (C19); 127,0 (C11); 126,7 (C18); 126,6 (C13); 126,5 (C17); 125,7 (C2); 124,9 (C20); 120,9 (C6); 119,3 (C7); 116,8 (C14); 62,8 (C15); 14,9 (C22); -4,1 (C25). RMN 88 29 Si (DMSO-d6, δ, ppm), -7,2. Análisis elemental. Calculado: [C55H44N2O4Si]n (824.66)n: C, 80,10 %; H, 5,33 %; N, 3,40 %. Encontrado: C, 80,05 %; H, 5,17 %; N, 3,23 %. 24 H * N 6 7 22 CH3 CH3 10 O 11 17 12 15 16 9 O 5 H N 25 O CH3 C 4 23 Si 3 2 1 O C * CH3 8 n 14 13 18 19 21 20 PA-VII: R: 86 %. IR (KBr, cm-1), 3417 (N-H), 3062 (C-H arom.), 2953, 2920 (C-H alif.), 1654 (C=O), 1601, 1506, 1491 (C=C), 1408, 1011 (Si-C arom.), 1313 (Si-C alif.); 1248 (C-O-C), 828 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 10,25 (s, 2H(24)), 7,88 (m, 6H(2,20)); 7,67 (m, 8H(3,17,19)); 7,37 (m, 6H(6,18)); 6,94 (m, 8H(7,11,13)); 6,69 (m, 2H(14)); 2,05 (s, 6H(22)); 1,00 (m, 5H(26,27)); 0,56 (s, 3H(25)). RMN 13 C (DMSO-d6, δ, ppm), 164,2 (C23); 152,4 (C9); 151,5 (C8); 149,6 (C16); 139,5 (C21); 139,4 (C12); 138,3 (C5); 138,0 (C4); 134,7 (C1); 133,3 (C10); 133,0 (C3); 129,2 (C19); 126,9 (C11); 126,7 (C18); 126,6 (C13); 126,1 (C17); 125,7 (C2); 124,9 (C20); 120,9 (C6); 119,3 (C7); 116,9 (C14); 62,8 (C15); 14,9 (C22); 6,0 (C27); 3,7 (C26); -6,7 (C25). RMN 29 Si (DMSO-d6, δ, ppm), -5,0. Análisis elemental. Calculado: [C56H46N2O4Si]n (838,67)n, C, 80,19 %; H, 5,48 %; N, 3,35 %. Encontrado: C, 79,92 %; H, 5,05 %; N, 3,19 %. 89 PA-VIII: R: 95 %. IR (KBr, cm-1), 3423 (N-H), 3065 (C-H arom.), 2955, 2919 (C-H alif.), 1670 (C=O), 1602, 1506, 1491 (C=C), 1407, 1017 (Si-C arom.), 1309 (Si-C alif.); 1248 (C-O-C), 828 (arom. p-sust.); 742 (arom. mono-sust.). RMN 1H (DMSO-d6, δ, ppm), 10,23 (s, 2H(24)), 7,90 (m, 6H(2,20)); 7,65 (m, 8H(3,17,19)); 7,38 (m, 11H(6,18,27,28,29)); 6,94 (m, 8H(7,11,13)); 6,70 (m, 2H(14)); 2,05 (s, 6H(22)); 0,88 (s, 3H(25)). RMN 13 C (DMSO-d6, δ, ppm), 165,3 (C23); 153,5 (C9); 152,6 (C8); 150,7 (C16); 140,6 (C21); 139,6 (C12); 139,4 (C5); 139,3 (C4); 136,6 (C27); 136,2 (C29); 135,5 (C1); 134,8 (C10); 134,4 (C3); 133,6 (C26); 130,5 (C28); 130,3 (C19); 128,1 (C11); 127,0 (C18); 126,8 (C13); 126,0 (C2); 125,8 (C17); 125,4 (C20); 122,0 (C6); 120,5 (C7); 118,1 (C14); 63,9 (C15); 16,1 (C22); -4,0 (C25). RMN 29 Si (DMSO-d6, δ, ppm), -10,8. Análisis elemental. Calculado: [C60H46N2O4Si]n (886,71)n, C, 81,27 %; H, 5,19 %; N, 3,16 %. Encontrado: C, 80,98 %; H, 4,99 %; N, 3,00 %. 90 PA-IX: R: 86 %. IR (KBr, cm-1), 3423 (N-H), 3067 (C-H arom.), 2919 (C-H alif.), 1663 (C=O), 1603, 1506, 1491 (C=C), 1407, 1018 (Si-C arom.), 1310 (Si-C alif.); 1247 (C-O-C), 828 (arom. p-sust.); 744 (arom. mono-sust.). RMN 1 H (DMSO-d6, δ, ppm), 10,38 (s, 2H(24)), 7,92 (m, 6H(2,20)); 7,67 (m, 8H(3,17,19)); 7,41 (m, 16H(6,18,26,27,28)); 6,95 (m, 8H(7,11,13)); 6,70 (m, 2H(14)); 2,07 (s, 6H(22)). RMN 13 C (DMSO-d6, δ, ppm), 165,3 (C23); 153,5 (C9); 152,7 (C8); 150,7 (C16); 140,6 (C21); 139,4 (C12); 137,1 (C5); 136,5 (C4); 135,8 (C26); 134,5 (C28); 134,4 (C1); 132,5 (C3); 130,3 (C10); 130,2 (C25); 129,3 (C27); 128,3 (C19); 128,1 (C11); 127,9 (C18); 127,6 (C13); 127,1 (C2); 126,7 (C17); 126,0 (C20); 122,0 (C6); 120,5 (C7); 118,0 (C14); 63,9 (C15); 16,0 (C22). RMN 29 Si (DMSO-d6, δ, ppm), -14,8. Análisis elemental. Calculado: [C65H48N2O4Si]n (948,76)n, C, 82,28 %; H, 5,06 %; N, 2,95 %. Encontrado: C, 81,03 %; H, 4,89 %; N, 2,67 %. 91 V. SÍNTESIS DE POLI(IMIDA)S A una mezcla de 0,37 mmol de bis(4-(4-aminofenoxi)fenil)diorganosilano (9 ó 10) y 0,8 mL de DMAc se añaden 0,37 mmol de anhídrido bis(4-(3,4carboxifenoxi)fenil)diorganosilano (19 ó 20) y se agita a temperatura ambiente durante 6 horas. Pasado este tiempo, se añaden 0,20 mL de anhídrido acético y 0,15 mL de piridina. La mezcla se agita durante 2 h a temperatura ambiente y durante 1 h a 60 °C. Una vez concluido el calentamiento, la mezcla se vierte sobre 300 mL de agua bajo agitación constante. El precipitado obtenido (PI-I – PI-IV) se filtra, se lava con abundante agua y se seca a vacío a 100 °C durante 12 horas. Las poli(imida)s PI-V – PI-IX se prepararon siguiendo la misma metodología, variando solo la naturaleza de la diamina y del dianhídrido empleado. PI-I: R: 87 %. IR (KBr, cm-1), 3060, 3021 (C-H arom.), 2956 (C-H alif.), 1777, 1720 (C=O), 1585, 1506, 1493, 1477 (C=C), 1444, 1106 (Si-C arom.), 1307 (Si-C alif), 1235 (C-O-C), 808 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 7,94 (m, 2H(6)); 7,54 (m, 18H(3,9,12,16,17)); 7,12 (m, 10H(5,8,13)); 0,58 (s, 6H(20)); 0,52 (s, 6H(19)). RMN 13 C (DMSO-d6, δ, ppm), 165,8 (C21); 165,6 (C22); 161,7 (C4); 156,8 (C15); 155,4 (C7); 155,3 (C14); 135,7 (C9); 135,3 (C17); 133,7 (C10); 133,6 (C18); 132,0 (C2); 128,4 (C5); 126,5 (C1); 125,3 (C11); 125,0 (C6); 122,7 (C12); 118,9 (C13); 118,3 (C8), 117,7 (C16); 111,4 (C3); -2,9 (C20); -3,0 (C19). RMN 29 Si (DMSO-d6, δ, ppm), -8,63. Análisis elemental. Calculado: [C56H42N2O8Si2]n (927,11)n, C, 72,55 %; H, 4,57 %; N, 3,02 %. Encontrado: C, 71,56 %; H, 4,32 %; N, 2,95 %. 92 PI-II: R: 89 %. IR (KBr, cm-1), 3020 (C-H arom.), 2957 (C-H alif.), 1778, 1720 (C=O), 1585, 1506, 1493, 1477 (C=C), 1429, 1106 (Si-C arom.), 1307 (Si-C alif), 1236 (C-O-C), 810 (arom. p-sust.); 700 (arom. mono-sust.). RMN 1H (DMSO-d6, δ, ppm), 7,92 (m, 2H(6)); 7,50 (m, 28H(3,9,12,16,17,21,22,23)); 7,13 (m, 10H(5,8,13)); 0,56 (s, 6H(19)). RMN 13 C (DMSO-d6, δ, ppm), 166,2 (C24); 166,1 (C25); 162,1 (C4); 157,9 (C15); 155,8 (C7); 155,3 (C14); 137,7 (C9); 136,1 (C17); 135,6 (C21); 134,4 (C10); 134,2 (C20); 134,1 (C2); 133,5 (C1); 129,7 (C23); 128,9 (C22); 128,0 (C5); 127,9 (C11); 127,2 (C18); 125,7 (C6); 123,1 (C12); 119,3 (C13); 119,2 (C16), 118,1 (C8); 111,9 (C3); -2,5 (C19). RMN 29 Si (DMSO-d6, δ, ppm), -7,8 y -14,7. Análisis elemental. Calculado: [C66H46N2O8Si2]n (1050,28)n, C, 75,41 %; H, 4,41 %; N, 2,66 %. Encontrado: C, 74,87 %; H, 4,12 %; N, 2,53 %. PI-III: R: 74 %. IR (KBr, cm-1), 3068 (C-H arom.), 2969 (C-H alif.), 1777, 1720 (C=O), 1585, 1506, 1493, 1477 (C=C), 1429, 1106 (Si-C arom.), 1307 (Si-C alif), 1236 (C-O-C), 810 (arom. p-sust.); 699 (arom. mono-sust.). RMN 1H (DMSO-d6, δ, ppm), 7,97 (m, 2H(6)); 7,52 (m, 24H(3,9,12,16,17,21,23)); 7,15 (m, 14H(5,8,13,22)); 0,52 (s, 6H(19)). RMN 13 C (DMSO-d6, δ, ppm), 165,8 (C24); 165,6 (C25); 161,2 (C4); 156,8 (C7); 156,1 (C15); 155,3 (C14); 137,6 (C17); 135,3 (C9); 135,2 (C18); 132,7 (C20); 129,5 (C21); 129,1 (C23); 128,5 (C2); 127,7 (C1); 126,5 (C11); 125,3 (C22); 123,0 (C10); 120,0 (C5); 119,3 (C6); 118,9 (C12); 118,3 (C13); 117,7 (C8), 116,4 (C16); 112,0 (C3); -2,9 (C19). RMN 29 Si (DMSO-d6, δ, ppm), -8,1 y -14,9. Análisis elemental. Calculado: [C66H46N2O8Si2]n (1050,28)n, C, 75,41 %; H, 4,41 %; N, 2,66 %. Encontrado: C, 74,92 %; H, 4,25 %; N, 2,60 %. 93 PI-IV: R: 86 %. IR (KBr, cm-1), 3068, 3067 (C-H arom.), 1778, 1721 (C=O), 1585, 1506, 1493, 1476 (C=C), 1428, 1106 (Si-C arom.), 1234 (C-O-C), 825 (arom. p-sust.); 698 (arom. mono-sust.). RMN 1H (DMSO-d6, δ, ppm), 7,96 (m, 2H(6)); 7,53 14H(5,8,13,25)). RMN (m, 13 34H(3,9,12,16,17,20,21,22,24,26)); 7,16 (m, C (DMSO-d6, δ, ppm), 166,6 (C27); 166,5 (C28); 162,1 (C4); 158,4 (C15); 157,0 (C7); 155,8 (C14); 138,5 (C9); 138,2 (C17); 136,15 (C24); 136,12 (C20); 135,1 (C26); 134,6 (C22); 133,9 (C23); 133,6 (C19); 130,2 (C2); 129,4 (C1); 129,3 (C11); 128,6 (C25); 128,5 (C21); 127,7 (C10); 126,2 (C5); 123,9 (C18); 120,9 (C12); 120,5 (C6), 119,8 (C13); 119,6 (C8); 118,6 (C16); 112,9 (C3). RMN 29 Si (DMSO-d6, δ, ppm), -14,7 y -14,8. Análisis elemental. Calculado: [C76H50N2O8Si2]n (1174,31)n, C, 77,66 %; H, 4,29 %; N, 2,38 %. Encontrado: C, 77,23 %; H, 4,18 %; N, 2,28 %. PI-V: R: 94 %. IR (KBr, cm-1), 3046 (C-H arom.), 2958 (C-H alif.); 1775, 1717 (C=O), 1586, 1520, 1495, 1476 (C=C), 1412, 1107 (Si-C arom.), 1307 (Si-C alif.); 1237 (C-O-C), 811 (arom. p-sust.). No se realiza la caracterización en disolución (RMN) dada la insolubilidad del compuesto en los disolventes orgánicos y disolventes deuterados ensayados a temperatura ambiente y a 94 40 °C. Análisis elemental. Calculado: [C54H34N2O7Si]n (850,94)n, C, 76,22 %; H, 4,03 %; N, 3,29 %. Encontrado: C, 75,86 %; H, 3,89 %; N, 3,08 %. PI-VI: R: 93 %. IR (KBr, cm-1), 3049 (C-H arom.), 1777, 1721 (C=O), 1587, 1520, 1477 (C=C), 1429, 1114 (Si-C arom.), 1238 (C-O-C), 812 (arom. psust.); 698 (arom. mono-sust.). RMN 1 H (DMSO-d6, δ, ppm), 8,58 (m, 2H(20)); 7,82 (m, 12H(3,6,16,17,22)); 7,50 (m, 20H(5,9,12,13,23,24)); 7,26 (m, 4H(8)). RMN 13 C (DMSO-d6, δ, ppm), 166,6 (C25); 166,5 (C26); 162,2 (C4); 157,0 (C7); 156,0 (C18); 140,1 (C14); 139,0 (C21); 138,5 (C9); 136,1 (C11); 134,8 (C22); 134,0 (C24); 133,6 (C2); 131,3 (C1); 130,7 (C16); 130,4 (C15); 130,1 (C23); 128,6 (C19); 128,4 (C13); 128,2 (C5); 127,9 (C10); 127,6 (C12); 126,2 (C20); 124,6 (C6); 119,8 (C8), 119,7 (C17); 112,4 (C3). RMN 29 Si (DMSO-d6, δ, ppm), -13,6. Análisis elemental. Calculado: [C64H38N2O7Si]n (975,08)n, C, 78,83 %; H, 3,93 %; N, 2,87 %. Encontrado: C, 78,06 %; H, 3,56 %; N, 2,75 %. PI-VII: R: 86 %. IR (KBr, cm-1), 3068, 3023 (C-H arom.), 2954 (C-H alif.), 1776, 1722 (C=O), 1587, 1544, 1479 (C=C), 1413, 1106 (Si-C arom.), 1375 95 (Si-C alif), 1238 (C-O-C), 811 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 8,17 (m, 2H(16)); 7,95 (m, 2H(6)); 7,82 (m, 2H(13)); 7,62 (m, 6H(9,12)); 7,41 (m, 4H(3,5)); 7,18 (m, 4H(8)); 0,57 (s, 6H(17)). RMN 13 C (DMSO-d6, δ, ppm), 166,1 (C18); 166,0 (C19); 162,0 (C4); 155,8 (C7); 154,9 (C14); 135,9 (C9); 134,1 (C10); 133,9 (C11); 127,0 (C5); 125,5 (C12); 123,9 (C2); 123,3 (C1); 123,1 (C6); 120,5 (C15); 120,0 (C16); 119,8 (C3); 119,1 (C8); 111,9 (C13); -2,7 (C17). RMN 29 Si (DMSO-d6, δ, ppm), -8,1. Análisis elemental. Calculado: [C42H26N2O7Si]n (698,75)n, C, 72,19 %; H, 3,75 %; N, 4,01 %. Encontrado: C, 71,86 %; H, 3,46 %; N, 3,85 %. O 14 12 15 * 13 O 11 16 O 17 CH3 N O O Si 10 19 9 8 3 7 O CH3 1 4 * N 2 18 O n 5 6 PI-VIII: R: 85 %. IR (KBr, cm-1), 3064 (C-H arom.), 2922 (C-H alif.), 1777, 1724 (C=O), 1619, 1479, 1444 (C=C), 1240 (C-O-C), 808 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 8,13 (m, 2H(26)); 7,92 (m, 6H(3,6,18)); 7,58 (m, 4H(15,23)); 7,21 (m, 14H(5,9,11,12,16,17,22)); 2,04 (s, 6H(20)). RMN 13 C (DMSO-d6, δ, ppm), 166,8 (C27); 166,8 (C28); 163,0 (C4); 155,4 (C7); 151,5 (C24); 150,6 (C14); 143,3 (C19); 139,8 (C10); 135,6 (C21); 134,6 (C1); 131,2 (C2); 129,9 (C8); 128,2 (C25); 127,7 (C17); 127,6 (C9); 126,4 (C16); 125,3 (C5); 124,4 (C11); 123,8 (C22); 123,3 (C15); 122,1 (C6); 121,0 (C18); 120,7 (C26); 112,2 (C12); 111,3 (C3); 110,9 (C23); 64,4 (C13); 16,6 (C20). Análisis elemental. Calculado: [C55H32N2O7]n (832,85)n, C, 79,32 %; H, 3,87 %; N, 3,36 %. Encontrado: C, 78,84 %; H, 3,65 %; N, 3,23 %. 96 PI-IX: R: 90 %. IR (KBr, cm-1), 3065 (C-H arom.), 2923 (C-H alif.), 1779, 1721 (C=O), 1621, 1498, 1475, 1444 (C=C), 1240 (C-O-C), 802 (arom. p-sust.). RMN 1H (DMSO-d6, δ, ppm), 8,21 (m, 2H (22,31)); 7,89 (m, 6H(3,6,18)); 7,31 (m, 18H(5,9,11,12,15,16,17,25,26,28,29)); 2,07 (s, 6H(20)). RMN 13 C (DMSO-d6, δ, ppm), 166,4 (C33); 166,1 (C34); 162,6 (C4); 155,4 (C23); 155,1 (C7); 151,1 (C27); 150,2 (C14); 142,9 (C19); 139,4 (C10); 134,2 (C21); 131,2 (C30); 130,7 (C1); 129,5 (C2); 127,9 (C8); 127,8 (C32); 127,3 (C17); 127,2 (C9); 126,0 (C16); 125,9 (C5); 124,9 (C11); 123,3 (C29); 122,6 (C15); 122,5 (C6); 121,6 (C24); 120,6 (C25); 119,6 (C18); 117,9 (C31); 114,7 (C26); 112,0 (C12); 111,6 (C3); 110,9 (C28); 110,5 (C22); 64,0 (C13); 15,8 (C20). Análisis elemental. Calculado: [C55H32N2O7]n (832,85)n, C, 79,32 %; H, 3,87 %; N, 3,36 %. Encontrado: C, 78,75 %; H, 3,59 %; N, 3,18 %. 97 RESULTADOS Y DISCUSIÓN I. SÍNTESIS DE PRECURSORES INDIRECTOS 1. Síntesis de los bis(4-hidroxifenil)diorganosilano (1, 2) Figura 1. Ruta I para la síntesis de los compuestos 1 y 2. Esta metodología fue propuesta por Davidsohn y colaboradores (65), donde 4-bromofenol (4-BF) reacciona con dos equivalentes de n-BuLi en THF anhídro a -70 °C. La adición de la disolución de 4-BF sobre n-BuLi debe ser lo más lenta posible, de forma tal que la temperatura no supere -60 °C y que no se forme turbidez en la mezcla de la reacción. Una vez concluida la adición, se deja subir la temperatura lentamente hasta 6 °C, procedimiento que se realiza sin retirar el baño de etanol-nitrógeno líquído que refrigera el sistema. Sobre los -30 °C se comienza a observar la formación de un gel muy fino que poco a poco afecta la agitación magnética, por lo que se debe controlar muy bien dicha agitación en este punto. Davidsohn plantea que a -70 °C ocurre la reacción ácido base entre el n-BuLi y 4-BF, consumiéndose 1 equivalente de n-BuLi y formándose 4bromofenóxido de litio, mientras que a -30 °C ocurre el intercambio metalhalógeno dando lugar a la formación de la sal 4-litiofenóxido de litio, la cual es poco soluble en el medio de reacción y a la temperatura de trabajo. Una vez alcanzada la temperatura de 6 °C se debe mantener una agitación enérgica durante 30 minutos y luego descender la temperatura nuevamente 99 hasta -50 °C y añadir la disolución de diclorodiorganosilano correspondiente en THF anhídro. Concluida la adición del derivado sililado se deja que ascienda la temperatura hasta el ambiente y se puede dejar agitando toda la noche. Al día siguiente, se añade HCl acuoso al 20 % y se observa un cambio de coloración de anaranjado a amarillo. Una vez que se extrae en éter dietílico la fase orgánica y se procesa, se obtiene un aceite, el cual se deja caer sobre n-hexano con agitación vigorosa. En este punto debe precipitar el producto. Por lo general, es necesario cambiar varias veces el nhexano por decantación con el fin de que precipite el compuesto. El compuesto bis(4-hidroxifenil)dimetilsilano precipita como un sólido de color blanco (en algunas ocasiones presenta tonalidades rosadas), mientras que la obtención de bis(4-hidroxifenil)difenilsilano es más dificultosa y es necesario realizar muchos cambios de n-hexano y esperar varios días su precipitación, apareciendo finalmente un sólido de color blanco. En la mayoría de los casos, este compuesto no se llega a obtener como un sólido, sino que se mantiene como un aceite denso y pegajoso. Davidsohn informa que la purificación de ambos difenoles se puede hacer mediante recristalización en benceno o tolueno, sin embargo, las veces en esto se intentó se terminó perdiendo por completo el producto, ya que una vez disuelto no vuelve a precipitar, contaminándose aún más el aceite. Esta reacción es altamente sensible a los cambios de temperatura, lo que provoca que sea de baja reproducibilidad, además, tal como lo muestra la ecuación correspondiente (Figura 1), consume dos equivalentes de n-BuLi, encareciendo la síntesis. A esto se debe sumar el hecho de que la importación de n-BuLi está controlada-restringida para Chile a partir del año 2012. Por las razones expuestas, se exploró una nueva ruta sintética para la obtención de estos difenoles, la que se presenta en la Figura 2. 100 Figura 2. Ruta II para la síntesis de los difenoles 1 y 2 (Bn = bencilo). Esta vía consume solo un equivalente de n-BuLi y los cambios de temperatura no afectan marcadamente la reproducibilidad como en la Ruta I. Como se puede ver, 4-BF se protege con cloruro de bencilo, luego se trata con n-BuLi a -70 °C y luego con el compuesto diclorodiorganosilano correspondiente, para obtener el difenol protegido. Hasta este punto, los rendimientos son elevados y la síntesis no presenta ninguna dificultad. El último paso de síntesis consiste en una hidrogenólisis catalizada por Pd/C activado en THF bajo enérgica agitación. El tiempo de reacción, la presión de hidrógeno y la temperatura a emplear son dependientes de la naturaleza del difenol a obtener. En el caso del bis(4-hidroxifenil)dimetilsilano se trabaja a 30 psi, temperatura ambiente y 24 horas de reacción. Después de procesar la mezcla de reacción como se indica en la Parte Experimental, el aceite obtenido se vuelca sobre n-hexano con agitación y el producto precipita casi instantáneamente en forma de sólido de color blanco. Por otro lado, las condiciones de hidrogenólisis para la obtención de bis(4hidroxifenil)difenilsilano son diferentes. Se debe operar a 65 psi, 45 °C y 5 días de reacción. Sin embargo, el aceite obtenido luego de remover el solvente precipita como sólido blanco una vez que se deja caer sobre nhexano con agitación. 101 Dada las condiciones de alta presión de hidrógeno, temperatura y el largo tiempo de reacción, son atendibles dos variantes de síntesis que podrían ser empleadas en la obtención de este derivado (69, 70). La Ruta III propone la síntesis de bis(4-bromofenil)difenilsilano a partir de la reacción entre 1,4dibromobenceno con n-BuLi y luego con diclorodifenilsilano (Figura 3). Finalmente, el difenol 2 se obtiene a partir de una secuencia de reacciones, las cuales se basan en sustituir el átomo de bromo por un éster borónico y luego oxidarlo al alcohol correspondiente. La Ruta IV comienza con la protección del 4-BF con cloruro de triisopropilsilano. La reacción de este con n-BuLi, luego con diclorodifenilsilano y finalmente con fluoruro de tetrabutilamonio origina el difenol 2. Figura 3. Rutas III y IV como alternativas para la preparación del difenol 2. Respecto a la caracterización de los difenoles obtenidos, los datos espectroscópicos de FTIR, RMN de 1 H, 13 C y 29 Si comprobaron las estructuras propuestas, las que se resumen en la Parte Experimental. Brevemente, se puede decir que en los espectros de FTIR de ambos compuestos (1 y 2) se observa la banda de estiramiento O-H entre los 3280 y 3380 cm-1, mientras que en el espectro de RMN 1H del compuesto 1, se observa a 6,84 y 7,35 ppm, el clásico patrón de sustitución para de los hidrógenos aromáticos H2 y H3, como dobletes con J= 8,4 Hz. La señal de 102 los hidrógenos del grupo metilo aparece a 0,45 ppm evidenciando el desapantallamiento generado por el átomo de silicio vecino y la del hidrógeno del grupo OH a 8,44 ppm, ambos como singletes. El espectro de RMN 1H del compuesto 2 es más complejo debido a que las señales aromáticas aumentan por el reemplazo de los grupos metilo por fenilos, sin embargo se pudo asignar la totalidad de las señales, apareciendo el hidrógeno del grupo OH en 8,59 ppm. En el RMN 29 Si, para el compuesto 1 se observa una señal a -9,3 ppm, mientras que para el compuesto 2 dicha señal aparece a campo más alto (-14,5 ppm). Dicho corrimiento se debe al aumento de la densidad electrónica sobre el átomo de silicio cuando está unido a cuatro anillos aromáticos (71). 2. Síntesis de 2,8-diyododibenzofurano (6) Figura 4. Ruta de síntesis para la obtención del compuesto 6. La reacción de yodación en compuestos orgánicos aromáticos directamente con yodo molecular no se ve favorecida. Esto se debe, por una parte, a la baja electrofilia que presenta el reactivo (72), y por otra, a que el HI formado es un agente reductor muy fuerte y puede reaccionar con el producto yodado que se desea obtener (73). Para evitar esto último, la técnica actualmente en uso adiciona un agente oxidante fuerte, tal como ácido nítrico, ácido sulfúrico, ácido yódico, trióxido de azufre o peróxido de hidrógeno, entre otros (74 – 76), a fin de oxidar el HI formado. Para la obtención de 2,8-diyododibenzofurano se eligió seguir una metodología descrita por Hilal y colaboradores (67) donde se emplea ácido 103 yódico como agente oxidante fuerte y ácido acético como medio de reacción. Según Butler (77), el electrófilo responsable de la reacción de yodación en presencia de agentes oxidantes fuertes es incierto. Sin embargo, en trabajos realizados por Ogata y colaboradores (76) se plantea la posibilidad que el agente electrofílico sea el acetato de yodo, el cual se forma según las siguientes ecuaciones químicas: La posición de sustitución sobre el dibenzofurano depende marcadamente de la naturaleza del sustituyente y de las condiciones de reacción (78). Así, las halogenaciones y sulfonaciones ocurren fundamentalmente en la posición 2, las nitraciones en la posición 3 y las metalaciones en la posición 4. En el espectro de FTIR del compuesto 6 se observa una banda a 623 cm-1 correspondiente al estiramiento C-I, mientras que en el espectro de RMN 1H se puede observar el doblete de doblete que presenta el hidrógeno H3, con acoplamientos a corta distancia con H2 (J= 8,6 Hz) y a larga distancia, del tipo W con el hidrógeno H5 (J= 1,7 Hz). Por su parte, en el espectro de RMN 13 C se observa el desplazamiento del carbono unido directamente al átomo de yodo, el cual aparece a 86,1 ppm. 3. Síntesis de 4-(4-bromofenoxi)nitrobenceno (A) Figura 5. Ruta de síntesis para la obtención del compuesto A. 104 La síntesis del precursor A se lleva a cabo mediante una reacción de sustitución nucleofílica aromática entre 4-BF y 1-fluoro-4-nitrobenceno en presencia de K2CO3 como base y DMF como disolvente. Este compuesto estaba propuesto en el proyecto de tesis como precursor de monómeros que finalmente no se lograron obtener, como se explicará más adelante. Debido a esto, se prefirió discutir los detalles mecanísticos de esta reacción en las páginas 108 y 109 donde ocurre una reacción similar. Vale destacar que el rendimiento de esta reacción es elevado (90 %) y el producto se aísla fácilmente por filtración después que precipita en una mezcla de agua-etanol (1:1 vol/vol). El espectro de FTIR confirma la formación del producto A con las señales más características; a 1510 y 1341 cm-1 y la señal a 1245 cm-1 correspondientes al estiramiento simétrico y asimétrico del grupo nitro y a la flexión del enlace C-O-C, respectivamente. En el espectro de RMN 1H se observan los patrones característicos de sustitución para de ambos anillos aromáticos. 4. Síntesis de anhídrido 4-(4-bromofenoxi)ftálico (B) O O O Br OH F 1) K2CO3; Sulfolano O 2) HCl 3) Ac2O O Br O O (B) Figura 6. Ruta de síntesis para la obtención del compuesto B. Esta reacción se realizó empleando distintas variables, tales como disolvente (DMF, DMSO), base (K2CO3, Cs2CO3, NaH) y distintos catalizadores de cobre (Cu, CuI), debido a que la formación de diaril éteres se conoce como condensación de Ullmann (79) y se emplea este tipo de catalizadores metálicos. Sin embargo, en todos los casos se obtuvo el anhídrido de partida en su forma ácida. 105 En una patente (80) se informa el empleo de sulfolano como disolvente en este tipo de reacciones, debido a su elevada temperatura de ebullición (285 °C). Por lo tanto, se probó dicha reacción con K2CO3 y a la temperatura de reflujo del sulfolano y se obtuvo un 80 % de rendimiento de la forma ácida del precursor B. Vale destacar que el sólido que se obtiene primeramente es la disal potásica, la que luego genera el ácido 4-(4-bromofenoxi)ftálico por tratamiento con HCl al 20 %. Por último, la reacción de ciclación química empleando anhídrido acético origina el precursor B con un 45 % de rendimiento. El espectro de FTIR presenta las señales características del compuesto B a 1825 y 1750 cm-1 correspondientes a los estiramientos de los carbonilos del grupo anhídrido y la flexión del enlace C-O-C a 1260 cm-1. Por su parte, el espectro de RMN 1H presenta el patrón de sustitución para de los hidrógenos H8 y H9. En este también se puede observar el acoplamiento doble que presenta el hidrógeno H5 a corta distancia con H6 y a larga distancia con H3. 5. Síntesis de cloro (3,4-dimetilfenil)dimetilsilano (C) Figura 7. Ruta de síntesis para la obtención del compuesto C. En esta reacción se debe tener un estricto control estequiométrico y se tiene que emplear una adición inversa. 4-Bromo-o-xileno reacciona con litio metálico generando una especie intermedia litiada, la cual se añade de forma 106 inversa sobre un exceso de diclorodimetilsilano. Con la posterior destilación a presión reducida se logra obtener el precursor C con un 63 % de rendimiento. En el espectro del FTIR se observan las señales correspondientes a los estiramientos Si-C aromático a 1380 y 1108 cm-1 y Si-C alifático a 787 cm-1. Por otro lado, en el espectro RMN 1H se observan los dobletes a 7,33 y 7,12 ppm acoplados entre sí (J= 6,9 Hz), correspondientes a los hidrógenos H5 y H6. Además, el singlete a 7,37 ppm se asigna al hidrógeno H3. Un aspecto importante en el RMN 29 Si es el desplazamiento químico del átomo de silicio, el cual aparece a -20,1 ppm. Los desplazamientos químicos del átomo de silicio han sido estudiados por el grupo de trabajo y se ha establecido una clara relación estructural con la densidad electrónica que recibe el heteroátomo por parte de los grupos sustituyentes enlazados a él (71) y este resultado se suma a los obtenidos. 107 II. SÍNTESIS DE MONÓMEROS 1. Síntesis de los bis(4-(4-aminofenoxi)fenil)diorganosilano (9, 10) Figura 8. Ruta de síntesis para la obtención de los bis(4-(4- aminofenoxi)fenil)diorganosilano. La síntesis de las diaminas monoméricas 9 y 10 comienza con la reacción de sustitución nucleofílica aromática entre el difenol (1 ó 2) y 1-fluoro-4nitrobenceno en presencia de carbonato de potasio como base. Este tipo de reacción, es decir, la formación de diaril éteres, se conoce como condensación de Ullmann (79) y por lo general se lleva a cabo utilizando un catalizador de cobre. Sin embargo, debido a la presencia del grupo nitro en el anillo aromático y al flúor en la posición 1 de este, dicha molécula se encuentra activada frente a una reacción de sustitución nucleofílica aromática directa, sin la presencia de un catalizador. De todas formas, a modo de prueba, dicha reacción se realizó en presencia de CuI y los rendimientos fueron similares que en ausencia de este. Para explicar por qué se usa un halogenuro de arilo fluorado en vez de uno clorado, bromado o yodado se debe recordar lo siguiente. En las reacciones de sustitución nucleofílica de los halogenuros de alquilo, el orden de reactividad en cuanto al desplazamiento del halógeno es I > Br > Cl > F (81), donde se propone un mecanismo en el cual la reacción ocurre en una sola 108 etapa y, de forma concertada se forma el enlace Nu-C y se rompe el enlace C-X (Nu = nucleófilo; X = halógeno). Como en el estado de transición está presente la ruptura del enlace C-X, la velocidad de la reacción será mayor cuanto más fácil se rompa dicho enlace y si se comparan las energías de enlace C-X de los diferentes halogenuros de alquilo (C-F = 552 KJ/mol; C-Cl = 397 KJ/mol; C-Br = 280 KJ/mol; C-I = 209 KJ/mol (82)) se puede entender el orden dado anteriormente. Sin embargo, la evidencia experimental observada en las reacciones de sustitución nucleofílica de halogenuros de arilo demuestra que el orden de reactividad en cuanto al desplazamiento del halógeno es F >> Cl ~ Br ~ I. Bunnett y colaboradores (83, 84) determinaron que la velocidad de la reacción no depende de la ruptura del enlace C-X y por lo tanto, el mecanismo debe ser en dos etapas, donde la primera etapa consiste en la formación del enlace Nu-C (paso lento de la reacción) y la segunda etapa contempla la ruptura del enlace C-X. Vale decir, la formación y ruptura de los enlaces involucrados no es un proceso concertado. La sustitución de flúor es mil veces más rápida que la de los otros halógenos en los halogenuros de arilo, lo que lleva a pensar que la presencia del átomo de flúor sobre el carbono insaturado del anillo aromático, acelera el ataque del agente nucleofílico. Como último detalle, se debe mencionar que por lo general estas reacciones se llevan a cabo a altas temperaturas (> 150 °C). Sin embargo, se ha informado también que a temperaturas mayores a 150 °C los bis(4hidroxifenil)diorganosilano sufren un reordenamiento donde se forman derivados del tipo fenoxisilanos (Si-O) (85). Por esta razón, la temperatura de reacción, que promueve el ataque nucleofílico, debe ser inferior a 60 °C. En ambas reacciones, las diaminas (9 y 10) se obtuvieron por reducción del compuesto dinitrado (obtenido en la primera etapa de reacción) mediante 109 hidrogenación catalítica con paladio soportado sobre carbono activado como catalizador e hidracina monohidratada como fuente de hidrógeno. El catalizador metálico, finamente dividido y en suspensión en el medio de reacción, promueve la descomposición de la hidracina monohidratada en nitrógeno, hidrógeno y amoníaco (86). El hidrógeno obtenido in situ es el encargado de reducir el grupo nitro a amino. El factor limitante de la velocidad en esta reacción es la transferencia de hidrógeno al catalizador (87). Es por esto que se debe emplear una cantidad mayor de catalizador de la que establece la estequiometría, conduciendo a una rápida reducción del grupo nitro. Las estructuras de los compuestos nitrados (7 y 8) fueron confirmadas mediante análisis elemental y espectroscopía FTIR y de RMN. Las bandas más características en el espectro de FTIR para ambos compuestos son los estiramientos asimétricos y simétricos del grupo nitro, los cuales aparecen a 1509 y 1342 cm-1 para el compuesto 7 y a 1522 y 1344 cm-1 para el compuesto 8. En el espectro de RMN 1H del compuesto 7 se observa el patrón de sustitución para característico que presentan los hidrógenos de ambos anillos aromáticos, los cuales aparecen como dobletes muy bien definidos. La señal en 0,59 ppm corresponde a los hidrógenos del grupo metilo unido al átomo de silicio. Por su parte, el compuesto 8 presenta una mayor complejidad en la zona correspondiente a los hidrógenos aromáticos, debido a la incorporación de anillos bencénicos sobre el átomo de silicio, sin embargo es posible detectar también el patrón de sustitución para. En el espectro FTIR de ambas diaminas (9 y 10) se observa la desaparición de las bandas correspondientes al grupo nitro y aparecen las bandas de los estiramientos N-H entre los 3300 y 3400 cm-1. Por otro lado, en el espectro de RMN 1H para ambos compuestos se observa un desplazamiento a campo 110 alto del hidrógeno H2 en relación a lo observado en los derivados dinitrados. El cambio de un grupo que retira densidad electrónica por medio de un efecto inductivo y también resonante (nitro) por uno que entrega densidad electrónica a través de resonancia (amino), hace que exista un mayor apantallamiento en dicha posición. Los hidrógenos correspondientes al grupo amino se observan como un singlete a 4,98 ppm para el compuesto 9 y a 3,62 ppm para el compuesto 10. 2. Síntesis de los ácidos bis(4-(4-carboxifenoxi)fenil)diorganosilano (13, 14) Figura 9. Ruta de síntesis para la obtención de los ácidos bis(4-(4carboxifenoxi)fenil)diorganosilano. La síntesis de los ácidos dicarboxílicos 13 y 14 comienza con la misma metodología empleada para obtener los compuestos nitrados anteriores, solo que ahora se hace reaccionar el difenol (1 ó 2) con 4-fluorobenzonitrilo. Básicamente el mecanismo de esta reacción transcurre de la misma forma que para los compuestos nitrados, donde la base (carbonato de potasio) extrae el hidrógeno ácido del difenol formando difenolato de potasio, nucleófilo capaz de atacar y sustituir al átomo de flúor en una reacción de sustitución nucleofílica aromática. 111 Sin embargo, esta reacción no ocurre a 60 °C y es necesario emplear temperaturas más altas, lo que hace que el difenol puede sufrir la transformación estructural por acción del reordenamiento mencionado anteriormente (85). Debido a esto, la reproducibilidad de esta reacción resulta ser baja y, por lo general, cuando se emplea el difenol 1 es mucho menor que cuando se emplea el difenol 2. El proceso de reordenamiento, promovido por la temperatura de reacción, la cual es equivalente a la de reflujo de la DMF, consume aproximadamente el 60 % del sustrato en ambas reacciones. Sin embargo, en una oportunidad se logró obtener un 85 % de rendimiento en la obtención del compuesto 11. La diferencia de reactividad entre 1-fluoro-4-nitrobenceno y 4- fluorobenzonitrilo radica en que, a diferencia del grupo nitro, el grupo ciano no posee la misma extensión en su efecto desactivante sobre el anillo aromático y por ende, la reacción de sustitución nucleofílica aromática buscada no se ve favorecida a 60 °C. Los compuestos dinitrilo 11 y 12 se transforman en ácidos dicarboxílicos por hidrólisis alcalina, usando hidróxido de potasio en una mezcla de etanol-agua (1:1 vol/vol). Por lo general, esta reacción se puede monitorear detectando el amoníaco gaseoso que se desprende como subproducto de la reacción, al colocar un papel para registro de acidez humedecido en agua a la salida del refrigerante. Una vez que el papel indicador no detecta la emanación de amoniaco, se detiene la reacción y se lleva a pH 3 añadiendo una solución de HCl acuoso al 20 %. El mecanismo que se propone para esta reacción es bien conocido (88), donde en una primera etapa el ión hidroxilo se adiciona nucleofílicamente al carbono del grupo nitrilo, y luego la hidroxi-imina formada isomeriza, por acción del agua, a una amida. Como las condiciones de hidrólisis se mantienen, el ion hidroxilo se adiciona al carbono carbonílico de la amida, 112 seguido por una desprotonación del grupo OH y una eliminación del ion amiduro (Figura 10). OH N C N C O Si O N C O O C R H2N Si O Si NH2 R R O Si C O O R R O NH2 R Si OH NH2 NH2 - NH3 O H O Si O O C O R O C R O H2N H2N O R O H C O O OH O O C O H O C N R H OH O OH C N H2O H OH O R C N O R H Si OH R OH R O O H OH H O O H+ NH4 R O O HO Si O R O OH (13): R = Me; (14): R = Ph) Figura 10. Mecanismo aceptado para la hidrólisis básica del grupo nitrilo. El ion amiduro reacciona rápidamente con el agua del medio dando lugar a la formación de hidróxido de amonio el cual, por acción del calor, evoluciona a amoniaco, gas que es detectado por su reacción con el papel indicador. Por último, la disal formada se transforma en ácido dicarboxílico mediante la acidificación del medio con ácido clorhídrico diluido. Las bandas más características observadas en el espectro FTIR de ambos nitrilo derivados (11 y 12) son el estiramiento del grupo ciano alrededor de los 2225 y 2226 cm-1 y la flexión del grupo C-O-C sobre los 1242 y 1246 cm-1, respectivamente. En el espectro de RMN 1H del compuesto 11 se puede observar claramente el patrón de sustitución para de los hidrógenos aromáticos de ambos anillos y los hidrógenos del grupo metilo a 0,57 ppm. 113 Esta misma definición no se logró en el espectro RMN 1H del compuesto 12 debido al aumento de las señales aromáticas producto de la incorporación de los grupos fenilos sobre el átomo de silicio. Sin embargo, en los espectros RMN 13 C de ambos productos se observa que el carbono unido al grupo ciano (C1) aparece alrededor de los 106 ppm. Este valor era esperado y se debe al fuerte efecto apantallante del grupo CN sobre su carbono . En el espectro FTIR de los ácidos dicarboxílicos 13 y 14 se observa la desaparición de la banda del grupo ciano y la aparición de las bandas anchas correspondientes al estiramiento O-H del grupo ácido entre los 3400 y los 3450 cm-1. De igual forma se observa la banda de estiramiento del grupo carbonilo sobre los 1688 cm-1. En el espectro de RMN 1H del compuesto 13, el singlete a 0,56 ppm, claramente corresponde a los hidrógenos de los grupos metilo y entre 7,06 y 7,96 ppm se observa el clásico patrón de sustitución para de los hidrógenos de ambos anillos aromáticos. Como es común para muchos análisis de ácidos carboxílicos, los hidrógenos ácidos no se observaron, probablemente debido al intercambio isotópico con el deuterio proveniente del solvente de análisis. En el espectro de RMN 13 C del mismo compuesto se observan los 8 picos correspondientes a los carbonos de los anillos aromáticos y la señal a 167 ppm asignada al grupo carbonilo. La señal a -1,98 ppm corresponde al carbono metílico unido al átomo de silicio. En el espectro de RMN 1H del compuesto 14, no se observa el patrón de sustitución para debido a las señales adicionales de los hidrógenos de los anillos aromáticos unidos al átomo de silicio. Sin embargo, la señal correspondiente a los hidrógenos ácidos se observa a 12,9 ppm. En el espectro de RMN 13C del mismo compuesto se observa la señal a 167,0 ppm asignada al carbono carbonílico. 114 3. Síntesis de los anhídridos bis(4-(3,4-dicarboxifenoxi)fenil)diorgano silano (19, 20) La Ruta I, propuesta originalmente en el proyecto de tesis para la obtención de estos anhídridos de ácidos, consistía en hacer reaccionar el difenol 1 ó 2, con el anhídrido 4-fluoroftálico en presencia de una base y DMF como disolvente (Figura 11). O O R HO Si R (1): R = Me; (2): R = Ph F OH O K2CO3, DMF HOOC HOOC COOH R O (17): R = Me; (18): R = Ph Si O COOH R Ac2O O O R O O O (19): R = Me; (20): R = Ph Si O O O R Figura 11. Ruta I propuesta para la obtención de los anhídridos bis(4-(3,4dicarboxifenoxi)fenil)diorganosilano. Esta ruta está basada en experiencias informadas previamente donde difenolatos o ditiofenolatos en base a carbono, sustituyen eficientemente el halógeno del anhídrido de ácido (89, 90). Sin embargo, esta síntesis no entregó resultados positivos, pues una vez formado el difenolato, este no realiza la sustitución deseada. Informes de literatura indicarían que el uso de bajas temperaturas (60 °C) no favorece esta reacción (91). La opción de elevar la temperatura está restringida por la descomposición del difenol en medio básico (85). Esta reacción se probó modificando ciertas variables como temperatura (60 °C y temperatura de reflujo), base (K2CO3, Cs2CO3, NaH), disolvente (DMF, sulfolano) y catalizador (CuI, Cu, CuSO4) en todos los intentos realizados el producto mayoritario obtenido fue ácido 4- 115 fluoroftálico, vale decir, las condiciones empleadas promueven la apertura del anillo del anhídrido. Debido a los problemas que presenta la Ruta I, se realizó una búsqueda bibliográfica que llevó a probar una síntesis alternativa donde se sustituye el anhídrido 4-bromoftálico por el sustrato 4-nitroftalonitrilo de mayor reactividad frente a la reacción de sustitución (92) (Figura 12). Figura 12. Ruta II para la obtención de los anhídridos bis(4-(3,4dicarboxifenoxi)fenil)diorganosilano. La obtención de los anhídridos de ácidos 19 y 20 siguiendo la Ruta II consta de tres etapas de reacción, donde en las dos primeras se ocupan metodologías similares a las discutidas hasta el momento. La primera etapa consiste en una reacción de sustitución nucleofílica aromática entre el difenol (1 ó 2) y 4-nitroftalonitrilo en medio básico. La velocidad de la reacción de sustitución del grupo nitro depende, al igual que la del flúor ya discutida en las páginas 108 y 109, de la formación del enlace C-nucleófilo y no de la ruptura del enlace C-NO2 (83, 84). De hecho, el grupo nitro es desplazado más rápidamente que el flúor y a su vez más rápido que los demás halógenos, lo que se puede explicar por la gran electronegatividad que posee. De esta forma, disminuye la densidad electrónica sobre el átomo de carbono al cual está enlazado, lo que hace que el nucleófilo ataque más rápido. 116 Es muy importante que la temperatura de reacción no supere 60 °C, no solo por el reordenamiento del difenol mencionado anteriormente, sino también, porque la reacción pasa de un color rojo a un color negro intenso, indicando la presencia de reacciones colaterales (93). Estas reacciones colaterales han sido explicadas y estudiadas por Parker y colaboradores (94, 95) donde se plantea que la presencia del ion nitrito generado en el medio de reacción puede atacar a 4-nitroftalonitrilo desplazando el grupo nitro y formando un nitritoderivado, el cual reacciona nuevamente con el ion nitrito para formar un fenóxido (Figura 13). Este fenóxido compite con el difenol y puede atacar al 4-nitroftalonitrilo para formar un derivado diariléter. Figura 13. Reacción colateral debido a la presencia del ion nitrito. El grupo nitro no es desplazado fácilmente por el ion nitrito (95), pero a temperaturas altas se favorece dicha reacción, de ahí la importancia de trabajar por debajo de los 60 °C. La segunda etapa para la obtención de los monómeros 19 y 20, consiste en la reacción de hidrólisis básica de los tetraciano derivados 15 y 16 usando la misma metodología de síntesis que se empleó para la obtención de los ácidos dicarboxílicos 13 y 14, la cual fue explicada ampliamente en dicha sección. Nuevamente, el tratamiento final con HCl acuoso al 20 % origina los ácidos tetracarboxílicos 17 y 18. 117 Como último paso de síntesis se emplea la ciclación química de estos tetraácidos empleando anhídrido acético como agente deshidratante. Esta reacción ocurre debido a que los grupos ácidos se encuentran en posición orto uno respecto al otro, y el mecanismo que se propone para este tipo de reacción es el que se muestra en la figura 14. Figura 14. Mecanismo de deshidratación química usando anhídrido acético como agente deshidratante. La extracción de anhídrido acético en exceso y del ácido acético formado durante la reacción se realiza mediante destilación a presión reducida, hasta casi sequedad. La masa obtenida se disuelve en acetato de etilo y se deja caer sobre n-hexano, precipitando así el producto. Sin embargo, el producto obtenido después de filtrar y lavar varias veces con n-hexano aún puede presentar ocluido restos de anhídrido acético (estos restos se observaron en varias ocasiones en el espectro de RMN 1H), por lo que el proceso de secado debe ser a temperaturas elevadas (140 °C) y durante un tiempo prolongado. En caso de que estas trazas de impureza no sean removidas desde el monómero final influirán negativa y considerablemente en el proceso de polimerización, en términos de su rendimiento y peso molecular alcanzado. Otra forma de obtener los dianhídridos de ácidos 19 y 20, es calentando los ácidos tetracarboxílicos 17 y 18 a 300 °C en presencia de pentóxido de fósforo como agente desecante en un equipo de sublimación a vacío. 118 Mediante esta técnica ocurre la deshidratación térmica obteniéndose los dianhídridos correspondientes. Sin embargo, la temperatura de trabajo es cercana a la temperatura de descomposición de los ácidos (> 300 °C) y se puede perder gran parte de la masa por degradación térmica. Las estructuras de los compuestos 15, 16, 17, 18, 19 y 20 fueron confirmadas mediante análisis elemental y espectroscopía FTIR y RMN. Brevemente, los espectros FTIR de los tetracianos derivados (15 y 16) muestran la señal intensa del estiramiento C-N del grupo ciano a 2231 y 2232 cm-1, respectivamente. Dicha señal desaparece en los espectros de los ácidos (17 y 18) observándose el estiramiento O-H a 3425 y 3431 cm-1 y el estiramiento del grupo carbonilo a 1709 y 1708 cm-1, respectivamente. Por otro lado, en el espectro de los dianhídridos (19 y 20) desaparece la señal del grupo O-H y aparece el patrón de estiramiento simétrico y asimétrico de los carbonilos a 1848, 1780 cm-1 y a 1851, 1777 cm-1, respectivamente. En el espectro de RMN 1H del compuesto 19 se puede asignar cada una de las señales con el tipo de hidrógeno correspondiente. Así, el hidrógeno H5 presenta un desdoblamiento de la señal debido al acomplamiento a corta distancia con H6 y a larga distancia con H3. También es posible observar el patrón de sustitución para de los hidrógenos H8 y H9 con constante de acoplamiento de 8,4 Hz. La señal a 0,61 ppm corresponde a los hidrógenos del grupo metilo unido al átomo de silicio. La señales recogidas del espectro de RMN 1H del compuesto 20 muestran también el patrón de sustitución para de los hidrógenos H8 y H9. Sin embargo, las señales de los otros hidrógenos aromáticos se solapan unas con otras debido al aumento de núcleos de este tipo por la presencia de los grupos fenilos sobre el átomo de silicio. En los espectros de RMN 13 C de ambos compuestos se observan dos señales entre 162 y 163 ppm correspondientes a los carbonos carbonílicos del dianhídrido de ácido. La 119 aparición de estas dos señales implica necesariamente que estos dos carbonos presentan entornos magnéticos ligeramente diferentes. 4. Síntesis de los isómeros 2,7- y 2,8-diaminodibenzofurano (21, 22) Figura 15. Ruta de síntesis para la obtención de los monómeros 21 y 22. La reacción de nitración de dibenzofurano se intentó en varias ocasiones utilizando una mezcla de ácido nítrico y ácido sulfúrico a tres temperaturas distintas; 0 °C, 25 °C y 60 °C. En todos los casos, al realizar cromatografía de capa fina, se obtuvo como resultado diversas manchas indicando la formación de numerosos compuestos, tal vez, mononitraciones, dinitraciones o polinitraciones; mezcla de alta complejidad y difícil separación. Después de una extensa revisión bibliográfica se encontró que si esta reacción se hacía en anhídrido acético como disolvente se obtenían mejores resultados (96). La mezcla de anhídrido acético y ácido nítrico genera un agente nitrante mucho más suave, el nitrato de acetilo o acetato de nitronio, además de producir ácido acético, como se indica en la figura 16. Figura 16. Formación del agente nitrante en la reacción de nitración de dibenzofurano. 120 Esta reacción hay que realizarla a bajas temperaturas debido a que por encima de los 50 °C el ácido nítrico ataca al anhídrido acético, formando tetranitrometano el cual puede ser explosivo (97). Figura 17. Reacción entre el ácido nítrico y el anhídrido acético a temperaturas superiores a 50 °C. Como se dijo en la página 104, las reacciones de sustitución electrofílica sobre los anillos del dibenzofurano dependen de la naturaleza del grupo que se va a introducir y de las condiciones de reacción (78). En el caso de las nitraciones, los resultados experimentales muestran que la posición 3 es la más favorecida, ya que dicho isómero es el que se obtiene en mayor proporción. Con el objetivo de comprender este comportamiento, Takashi y colaboradores (98) realizaron un estudio y determinaron que la nitración en la posición 3 del dibenzofurano no podía ocurrir mediante un mecanismo de formación del complejo σ, ya que dicho mecanismo favorecía la nitración en la posición 2, al poseer una energía de estabilización menor (Figura 18). Por ello, es que estos autores proponen que la nitración del dibenzofurano ocurre a través de un mecanismo de transferencia de carga, y para demostrarlo realizaron la reacción de nitración usando tetranitrometano en ácido trifluoroacético en presencia de radiación usando una lámpara de mercurio. Bajo estas condiciones la nitración solo puede ocurrir mediante un mecanismo de transferencia de carga. 121 Figura 18. Curvas de energía potencial calculadas en función de la distancia entre el átomo de carbono del dibenzofurano y el átomo de nitrógeno del ion nitronio (imagen tomada de la referencia 96). Los resultados fueron los mismos que cuando se realiza la nitración usando ácido nítrico, es decir, el isómero 3 fue el producto mayoritario. Además, cuando se hizo la reacción en ausencia de luz, no se obtuvo producto alguno. Estos resultados llevaron a plantear los siguientes mecanismos: Figura 19. Mecanismos propuestos para la nitración de dibenzofurano. 122 A pesar de la elevada regioselectividad que presenta esta reacción por la posición 3 del dibenzofurano, también está presente en menor medida la formación del isómero 2, lo que explica la mezcla resultante. Esta mezcla solo se caracterizó mediante FTIR para determinar las bandas características del grupo nitro, las cuales se encontraron a 1519 y 1344 cm-1. La separación de estos isómeros se prefirió hacer en su forma reducida. La posterior reducción mediante reacción con hidracina y paladio soportado en carbono como catalizador origina la mezcla isomérica de diaminas, la cual se separa mediante columna cromatográfica usando acetato de etilo como eluyente. El primer isómero en eluir fue 2,7-diaminodibenzofurano (21) con un Rf de 0,76, mientras que 2,8-diaminodibenzofurano (22) tuvo un Rf de 0,58. Como era de esperar, en la mezcla de isómeros hubo un 65 % de 2,7diaminodibenzofurano y 35 % de 2,8-diaminodibenzofurano. El análisis elemental de ambas diaminas entrega valores concordantes con los esperados, mientras que los espectros de FTIR son similares mostrando tres bandas de estiramiento N-H a 3397, 3296, 3199 cm-1 y la banda de flexión del grupo N-H a 1627 cm-1. Diferencias apreciables pueden observarse en los espectros de RMN de ambos compuestos debido a la pérdida de simetría de la diamina 21. Los singletes a 5,45 y 4,90 ppm en el espectro de RMN 1H de la diamina 21 corresponden a los hidrógenos de lo grupos aminos, los cuales presentan entornos magnéticos diferentes. Los hidrógenos aromáticos aparecen como dobletes exceptuando los hidrógenos H2 y H10, donde sus desplazamientos químicos son muy parecidos y se solapan las señales. Por su parte, en el espectro de RMN 13 C del mismo monómero se pueden observar las señales para los 12 carbonos aromáticos, siendo los carbonos C3, C5, C8 y C11 los más desplazados a campo bajo debido a que se encuentran enlazados directamente a átomos más electronegativos que carbono (O y N). 123 El patrón de señales evidenciado en el espectro de RMN 1H del compuesto 22 es más sencillo debido a la simetría de la molécula. La señal del hidrógeno H3 a 6,74 ppm aparece como un doblete de doblete con constantes de acoplamiento de 8,7 y 2,2 Hz, correspondientes al acoplamiento a corta distancia con H4 y a larga distancia con H1, respectivamente. A diferencia del compuesto 21, el monómero 22 presenta una sola señal a 4,97 ppm correspondiente a los cuatro hidrógenos de los grupos amino. Con respecto al análisis de RMN 13 C, este permite observar las seis señales de los carbonos aromáticos y nuevamente los carbonos C2 y C5 son los más desplazados a campo bajo debido al efecto desapantallante de los átomos de nitrógeno y oxígeno. 5. Síntesis de 2,8-di(4-aminofenil)dibenzofurano (24) La ruta de síntesis propuesta en el proyecto de tesis para la obtención de este monómero se muestra en la figura 20. Figura 20. Ruta I para la obtención del monómero 24. Las experiencias desarrolladas permiten asegurar que la mezcla del sustrato 6 con litio metálico en éter etílico anhidro no produce la sustitución buscada, pese a las modificaciones de tiempo y temperatura empleadas. La metalación de un anillo aromático por sustitución de un átomo de halógeno, ocurre más fácilmente cuando este está activado con algún grupo que presente efecto inductivo del tipo +I, como los grupos alquilos por ejemplo. Al parecer, la presencia del átomo de oxígeno, de alta 124 electronegatividad en 2,8-diyododibenzofurano, no ayuda a la activación de los anillos pese a que manifestaría un efecto resonante +M hacia el carbono sustituido (Figura 21) en forma similar a lo que ocurre en 4-BF. Figura 21. Efecto resonante del átomo de oxígeno en el sustrato 6 y en 4-BF. En todo caso, aunque la reacción anterior se hubiese producido, la posterior reacción de acoplamiento con el 4-cloronitrobenceno tampoco hubiese sido exitosa, ya que informes en la literatura (99 y 100) revelan que este tipo de reacción de acoplamientos C-C entre un arillitio (o arilmagnesio) y un haluro de arilo no ocurren fácilmente, a pesar que el 4-cloronitrobenceno está activado para recibir un ataque nucleofílico por parte del carbono enlazado al metal. En este tipo de reacciones es obligatorio el uso de catalizadores de niquel, paladio y sus combinaciones con ligandos de difenilfosfina, en una reacción conocida como acoplamiento de Kumada-Tamao-Corriu. Debido a lo expuesto, se decidió explorar una metodología más limpia y recientemente desarrollada por Susuki-Miyaura (101), la cual es específica para la formación de acoplamientos C-C en compuestos aromáticos y se muestra en la figura 22. 125 Figura 22. Ruta II propuesta para la obtención del monómero 24. La obtención de la diamina monomérica 24 mediante la Ruta II consiste en una reacción de acoplamiento C-C empleando un derivado halogenado, un ácido borónico y un catalizador de un complejo de paladio (0) en presencia de una base. En el caso estudiado, el compuesto halogenado es el precursor 6 y el derivado borónico es el ácido 4-nitrofenilborónico, el cual aporta el grupo nitro para su posterior reducción a amino. Existen numerosos catalizadores de paladio que se utilizan en este tipo de reacción, sin embargo, el cloruro de bis(trifenilfosfina)paladio (II) ((PPh3)2PdCl2) y una disolución de carbonato de potasio en dioxano funciona correctamente en la mayoría de los casos. El mecanismo propuesto para esta reacción (102) comienza con la adición oxidativa de 2,8-diyododibenzofurano (6) al complejo de paladio (0), para formar el intermediario (I) (Figura 23). Luego, ocurre un desplazamiento del ion yoduro por la base, formando el intermediario (II). Posteriormente, una transmetalación promovida por el derivado activado del ácido borónico origina el complejo diarilpaladio (III), que mediante una eliminación reductiva forma el nuevo enlace C-C regenerando el catalizador. Se debe destacar, que el reemplazo de los átomos de yodo a fin de generar el complejo (II), aunque esté representado en forma simultánea en la figura, lo más probable 126 es que sea un proceso por etapas, reemplazándose uno primero y luego el segundo. O Cl Ph3P Pd PPh3 Cl NO2 O2N O I Ph3P Pd PPh3 Eliminación reductiva I Adición oxidativa O Ph3P Pd Pd PPh3 Ph3P PPh3 PPh3 O Pd I Ph3P (I) (III) NO2 O2N Ph3P Pd PPh3 I 2 B(OH)4K KHCO3 + O2N K OH B OH OH O Ph3P HO Pd base Ph3P PPh3 PPh3 Pd OH (II) O2N B OH OH base: K2CO3 + H2O KOH + KHCO3 2 KI K2CO3 + H2O Figura 23. Mecanismo propuesto en la reacción de acoplamiento C-C para la obtención del compuesto 23. El papel de la base dentro del mecanismo es fundamental, debido a que además de desplazar el ion yoduro en la formación del complejo (II), también activa el ácido 4-nitrofenilborónico, ya que el anión RB(OH)-3 formado es 106 veces más reactivo que la especie neutra (103). La especie de Pd(0) inicial se crea in situ a partir de cloruro de bis(trifenilfosfina)paladio (II). Aunque este proceso no está del todo claro, existen evidencias de especies Pd(0) aniónicas, tales como, Pd(0)L2Cl- (104). El compuesto dinitrado 23 se obtiene con un rendimiento elevado (95 %) y es insoluble en diversos disolventes orgánicos incluyendo 1,4-dioxano. Por esta razón, la diamina precipita a medida que se forma, lo que hace que su aislamiento y purificación no sea mediante técnicas cromatográficas, como 127 es usual en este tipo de reacciones. La caracterización en disolución tampoco es posible debido a su insolubilidad en disolventes deuterados, sin embargo el espectro de FTIR muestra claramente las señales correspondientes al grupo nitro a 1514 y 1344 cm-1. La reducción del compuesto dinitrado 23 con hidracina monohidratada en presencia de paladio soportado sobre carbono activado produce finalmente el monómero deseado 24. El espectro FTIR de la diamina 24 muestra los estiramientos N-H a 3438, 3396 y 3316 cm-1 y a 1619 cm-1 la flexión de dicho grupo. En el espectro de RMN 1H se observa el patrón de sustitución para de los hidrógenos H2 y H3 en forma de dobletes con constantes de acoplamientos de 8,4 y 8,5 Hz, respectivamente. La señal a 8,42 ppm presenta un débil doblete con J= 0,7 Hz correspondiente al acoplamiento a larga distancia del hidrógeno H10 con H6. El singlete a 5,28 ppm corresponde a los hidrógenos del grupo amino los cuales son magnéticamente equivalentes dada la simetría de la estructura. Por su parte, el espectro de RMN 13 C presenta las diez señales aromáticas esperadas. Las desplazadas a campo bajo corresponden a los carbonos C1 y C8, debido a la influencia de los átomos de N y O enlazados directamente a estos. 6. Síntesis de 9,9-bis(4-(4-aminofenoxi)-3-metilfenil)fluoreno (26) La síntesis y uso de esta diamina no estaba considerada en la formulación del proyecto de tesis original. Sin embargo, y dado que la nueva diamina sintetizada (26) aporta al concepto de introducir unidades de flexibilización conjuntamente con alto contenido aromático y una porción estructural de gran volumen, es que se incorporó al desarrollo de la tesis. Esta diamina se hizo reaccionar con diácidos sililados previamente informados por el grupo de investigación. 128 CH3 O2N CH3 HO NO2 CH3 OH CH3 O F O NO2 K2CO3, DMF NH2-NH2 Pd/C H2N (25) NH2 CH3 CH3 O O (26) Figura 24. Ruta de síntesis para la obtención del monómero 26. La metodología empleada para la obtención de esta diamina monomérica es la misma que la utilizada en la síntesis de los compuestos 9 y 10 (página 108). Brevemente, el compuesto dinitro 25 se obtiene por la reacción de fluoro-desplazamiento metilfenil)fluoreno y entre dos el difenol equivalentes comercial de 9,9-bis(4-hidroxi-3- 1-fluoro-4-nitrobenceno en presencia de carbonato de potasio como base y DMF como disolvente. Este compuesto no se puede caracterizar en disolución debido a la insolubilidad que posee en los disolventes deuterados empleados, sin embargo, el espectro FTIR evidencia claramente las bandas características del grupo nitro a 1589 y 1339 cm-1. La diamina 26 se obtiene mediante reacción del compuesto dinitrado con hidracina monohidratada en presencia de paladio soportado sobre carbono activado a reflujo en etanol. El espectro FTIR muestra las bandas de estiramiento N-H a 3441 y 3364 cm-1, y la banda de flexión de dicho grupo a 1619 cm-1. En el espectro de RMN 1H se observan los hidrógenos del grupo metilo y amino como singletes a 2,14 y 3,45 ppm, respectivamente. Los hidrógenos H16 y H13 aparecen como dobletes a 7,73 y 7,46 ppm debido al acoplamiento con los hidrógenos H15 y H14, respectivamente. A 7,08 ppm 129 se observa un doblete con una baja constante de acoplamiento de 1,5 Hz correspondiente al hidrógeno H7. El hidrógeno H9 presenta una señal en forma de doblete de doblete debido al acoplamiento a corta distancia con H10 y a larga distancia con H7. El patrón de sustitución para entre los hidrógenos H2 y H3 aparece a 6,57 y 6,74 ppm, respectivamente, sin embargo la señal a 6,65 ppm integra para seis hidrógenos, indicando que existe un solapamiento de las señales de los cuatro hidrógenos H2 y los dos hidrógenos H10. En el espectro de RMN 13 C, los carbonos C8 y C1 aparecen muy cercanos (139,9 y 139,8 ppm, respectivamente), al igual que los carbonos C9 y C3 (120,07 y 120,00 ppm, respectivamente). El resto de los carbonos aromáticos muestran buena resolución entre los picos. La señal a 64,5 ppm corresponde al carbono C11, mientras que el carbono de los grupos metilo aparece a 16,6 ppm. 7. Síntesis de anhídrido 9,9-bis(4-(3,4-dicarboxifenoxi)-3-metilfenil) fluoreno (29) Figura 25. Ruta de síntesis para la obtención del monómero 29. 130 Este dianhídrido de ácido, al igual que la diamina 26 derivada de 9,9-bis(4hidroxi-3-metilfenil)fluoreno tampoco estaba considerado originalmente en el proyecto de tesis, sin embargo, fue incorporado por las mismas razones esgrimidas para la diamina: aporte de unidades flexibles, alto contenido aromático y porción estructural de gran volumen. La metodología empleada para la obtención del compuesto 29 es la misma que la utilizada en la síntesis de los compuestos 19 y 20 (página 116). Brevemente, la reacción entre el difenol comercial 9,9-bis(4-hidroxi-3metilfenil)fluoreno y 4-nitroftalonitrilo en presencia de carbonato de potasio como base, DMF como disolvente y a 60 °C, origina el compuesto tetraciano 27. El cual se transforma en el tetraácido correspondiente a través de una hidrólisis básica empleando hidróxido de potasio en una mezcla de etanol:agua (1:1) y posterior acidificación. Finalmente, se obtiene el dianhídrido de ácido 29 mediante reacción de deshidratación empleando anhídrido acético como agente deshidratante. El resultado del análisis elemental practicado a los compuestos 27 y 28, está de acuerdo con los valores porcentuales esperados. Desde el punto de vista espectroscópico, las señales recogidas desde el espectro FTIR del compuesto 27 muestra los estiramientos de los nitrilos a 2231 cm-1, mientras que en el del compuesto 28 se observa la desaparición de esta señal y la aparición de los estiramientos O-H a 3408 cm-1 y carbonilo a 1707 cm-1. Estas dos observaciones evidencian claramente la formación de ambos compuestos. Las señales observadas en los espectros de RMN (1H y 13 C) de ambos compuestos se correlacionan con las estructuras propuestas. El espectro de FTIR del dianhídrido de ácido 29 muestra la desaparición de la banda de estiramiento O-H y la aparición de los estiramientos de los carbonilos del grupo anhídrido a 1852 y 1777 cm-1. En el espectro de RMN 1 H se observa a 8,05 ppm un doblete (J= 8,4 Hz) correspondiente al 131 hidrógeno H6 el cual acopla directamente con H5, que se encuentra a 7,06 ppm. Este último aparece en forma de doblete (J= 8,3 Hz), sin embargo, es de esperar que se vea un acoplamiento débil a larga distancia con H3. Por su parte, H3 aparece como un singlete a 7,32 ppm, por lo que evidentemente el acoplamiento tipo W no se ve bajo la resolución del equipo de RMN empleado. El análisis de RMN 13 C muestra los picos de los carbonos carbonílicos del grupo anhídrido a 163,7 y 162,6 ppm. Los carbonos C4 y C7 son los más desplazados a campo bajo debido a la influencia del átomo de oxígeno enlazado directamente a estos. El carbono cuaternario C13 aparece a 64,0 ppm mientras que el carbono de los grupos metilo aparece a 15,8 ppm. 8. Trabajos sintéticos que no condujeron a la preparación de los monómeros respectivos En el proyecto de tesis se proponía la síntesis de cuatro nuevos monómeros: dos diaminas y dos dianhídridos de ácidos, los cuales incorporaban cuatro enlaces oxiéter cada uno como unidades flexibilizantes y serían empleados en la preparación de poli(amida)s y/o poli(imida)s. La estructura de estos y sus relaciones sintéticas se resumen en la figura 26. Figura 26. Monómeros a obtener a partir de los precursores difenólicos 1 y 2. 132 En la reacción entre el difenol 1 con el sustrato A o B, se esperaba que se sustituyera el átomo de bromo a través de una reacción de sustitución nucleofílica aromática en forma similar a lo realizado en la síntesis de derivados dinitrados por ejemplo (Figura 8). Sin embargo, en ambas reacciones, el ataque nucleofílico ocurrió en la posición 4 señalada en la figura 27, obteniéndose los compuesto 7 y 15, respectivamente. Vale decir, el grupo 4-bromofenoxi actuó como grupo saliente en ambos casos. Figura 27. Curso del ataque nucleofílico promovido por el difenolato derivado de 1 sobre los sustratos A y B propuestos. Este resultado confirma que la obtención de monómeros con más de un grupo flexible de oxiareno no ocurre por esta vía, lo que debe llevar a investigar y diseñar una nueva ruta sintética. En la figura 28 se muestran los dos últimos monómeros ofrecidos en el proyecto de tesis, cuya obtención, pese a las experiencias realizadas, no fue posible. En todos los casos, al tratar de obtener el compuesto D, el producto final consistía mayoritariamente al material de partida 6. 133 O O I (6) 1) n-BuLi I CH3 R 2) Cl Si R R R H3C D Si H3C CH3 R R CH3 CH3 Si C O O O O R R Si Si R R O O O Figura 28. Dianhídridos de ácido disililados propuestos en el proyecto de tesis. Se ha informado que la reacción entre n-BuLi y dibenzofurano produce la extracción de los hidrógenos ácidos adyacentes al átomo de oxígeno (105). Esto explicaría la falta de reactividad frente al intercambio metal-halógeno que muestra el sustrato 6. Probablemente, la reacción ácido-base es la predominante frente al intercambio metal-halógeno. Sin embargo, la adición de la sal dianiónica (ya sea por el intercambio metal-halógeno o intercambio metal-hidrógeno) sobre el sustrato C no produjo reacción alguna. Prats et al. (106) dieron evidencia de la dificultad que posee una reacción de este tipo con el mismo sustrato C, donde 1,4-dibromobenceno reacciona con n-BuLi y luego con el sustrato C, obteniéndose solamente el monoacoplamiento y no la disustitución como se esperaba. Estos monómeros eran de gran interés desde el punto de vista sintético y su aporte a los estudios de estructura-propiedad propuestos. Asimismo, está el hecho que este tipo de monómero no se ha informado previamente, vale decir, dianhídridos de ácido que contengan dos átomos de silicio unidos a cuatro átomo de carbono cada uno en su estructura. 134 III. SÍNTESIS DE POLI(AMIDA)S 1. Poli(amida)s derivadas de las diaminas 9 y 10 y los ácidos dicarboxílicos 13 y 14 (Serie A). Figura 29. Ruta de síntesis para la obtención de las poli(amida)s PA-I, PA-II, PA-III y PA-IV pertenecientes a la Serie A. Las cuatro PAs fueron preparadas usando la metodología de fosforilación desarrollada por Yamasaki-Higashi (14). Esta técnica involucra la poliamidación directa entre una diamina y un ácido dicarboxílico usando trifenilfosfito (TPP) y piridina como agentes condensantes. El disolvente utilizado es NMP y se agregan pequeñas cantidades de sales metálicas, tales como, cloruro de calcio o cloruro de litio con el objeto de aumentar la solubilidad de las cadenas poliméricas que se van formando, ya que estas sales evitan la formación de los enlaces por puente de hidrógeno de las especies en crecimiento. La temperatura de la reacción no debe superar los 120 °C y el tiempo de reacción oscila entre 3 a 6 horas. El mecanismo por el cual ocurre la poliamidación directa no está del todo dilucidado, sin embargo, Higashi propone que la piridina juega un papel fundamental en dicha reacción (107). Como se puede ver en la figura 30, la piridina es constituyente de un intermediario formado entre el TPP y el ácido dicarboxílico monómero, el cual es atacado por la diamina para generar la 135 poli(amida) correspondiente. Es decir, el paso limitante en este mecanismo de reacción es la formación de dicho intermediario. Figura 30. Mecanismo propuesto por Higashi en la reacción de poliamidación directa. Sin embargo, Aharoni y colaboradores (108), en el año 1984, proponen un nuevo mecanismo basado en evidencias derivadas de estudios de RMN, en el cual la piridina no juega un papel importante en la velocidad de la reacción y el paso limitante es la formación de una aminofenoxifosfina. Como se ve en la figura 31, primero ocurre la reacción entre el TPP y la diamina formando el intermediario aminofenoxifosfina, el cual reacciona posteriormente con el ácido dicarboxílico obteniéndose la poli(amida). Figura 31. Mecanismo propuesto por Aharoni en presencia de piridina. 136 La piridina presente en el medio de reacción actúa como extractor de hidrógeno. Sin embargo, esta reacción se puede llevar a cabo en ausencia de piridina y el mecanismo propuesto es el mismo, excepto que no hay abstracción del hidrógeno (Figura 32). Figura 32. Mecanismo propuesto por Aharoni (108) en ausencia de piridina. Actualmente se sigue usando piridina en este tipo de reacciones, pues si bien no influye en el paso determinante de la reacción, favorece la formación y evolución del intermediario fósforo-amina-ácido para la posterior transformación a amida y difenilfosfito. Como se dijo en la sección introductoria, las PAs también se pueden obtener a baja temperatura y en disolución, mediante reacción entre una diamina y un dicloruro de ácido, sin embargo, actualmente se prefiere utilizar el método desarrollado por Yamazaki-Higashi, debido a varios factores, tales como; evitar un paso extra de reacción (la preparación del cloruro de ácido), se disminuye la toxicidad ambiental al no trabajar con agentes halogenantes como el cloruro de tionilo, los monómeros son más estables (el ácido carboxílico es mucho más estable que el cloruro de ácido frente a la 137 humedad) y por lo general los rendimientos de las reacciones siempre son elevados. Las poli(amida)s PA-I, PA-II, PA-III y PA-IV se obtuvieron con rendimientos entre 83 y 93 % y el análisis elemental de cada una de ellas entrega valores concordantes con los esperados. Por otro lado, el espectro de FTIR de estos polímeros presenta el estiramiento del grupo N-H de la amida entre los 3421 - 3423 cm-1, y el estiramiento del grupo carbonilo se observa entre los 1647 1655 cm-1. Los espectros de RMN 1H de las cuatro poli(amida)s están de acuerdo a las estructuras planteadas en cuanto al número de señales, multiplicidad e integración. En particular, la evidencia de la formación del grupo amida se puede observar a través de la presencia de los hidrógenos amídicos, los cuales aparecen como singlete entre 10,25 y 10,32 ppm. En el caso de la PA-I se observa un singlete relativamente ancho a 0,52 ppm, que integra para 12 hidrógenos. Esto demuestra que el entorno magnético de los dos tipos de hidrógenos unidos a silicio; H9 provenientes de la porción diácido y H18 aportados por la diamina, son similares. Para las muestras PAII y PA-III, la señal correspondiente a los hidrógenos del grupo metilo aparecen centradas en 0,45 y 0,56 ppm, respectivamente. PA-IV no presenta hidrógenos alifáticos, concentrando sus señales en la zona aromática. En el espectro de RMN 13 C de las cuatro PAs se observa el carbono carbonílico del grupo amida alrededor de los 165,0 ppm. PA-I presenta una señal a -2,42 ppm correspondiente a los carbonos de los grupos metilos. Es claro que estos dos carbonos (C9 y C18) no son magnéticamente equivalentes pero sí muy similares, por lo que se asume que presentan desplazamientos muy próximos, sin poder diferenciarse en el experimento desarrollado. Esta evidencia se repite al analizar el espectro de RMN 29 Si de PA-I, donde también deberían aparecer dos señales muy cercanas y sin embargo solo se observa una a -8,98 ppm. 138 En el espectro de RMN 13 C de PA-II y PA-III aparece una señal a -1,9 y -2,4 ppm, respectivamente, las que se asocian a los carbonos del grupo metilo aportado por la diamina en PA-II y por el ácido dicarboxílico en PA-III. En los espectros de RMN 29 Si para PA-II y PA-III se observan dos señales, una entre -7,3 y -8,1 ppm y otra entre -14,2 y -15,0 ppm. La primera de ella corresponde al silicio rodeado de dos anillos aromáticos y dos grupos metilos (Ph-Si(Me)2-Ph), mientras que la segunda corresponde al silicio cuando está rodeado de cuatro anillos bencénicos (Ph-Si(Ph)2-Ph). Estos desplazamientos químicos están acorde con las evidencias experimentales observadas por el grupo de trabajo para variados tipo de monómeros y polímeros sililados, las que guardan relación con la densidad electrónica que recibe el heteroátomo por parte de los grupos sustituyentes enlazados a él (71). PA-IV no presenta fragmentos alifáticos por lo que solo existen las señales correspondientes a los carbonos aromáticos y carbonilo. Sin embargo, los dos anillos aromáticos enlazados al átomo de silicio proveniente del fragmento diamina no son equivalentes a los enlazados al silicio proveniente del monómero ácido dicarboxílico, por lo que se debería esperar 24 picos aromáticos. Al igual que en PA-I, probablemente la resolución del equipo de RMN empleado en el análisis no es capaz de distinguir estas señales, por lo que solo se observan 20 correspondientes a los carbonos aromáticos. En cambio, en el espectro de RMN 29 Si es posible observar dos señales a -14,9 y -15,0 ppm correspondientes a los dos átomos de silicio. La aparición de estas señales corrobora el hecho de que estos dos heteroátomos, y por ende sus entornos directos, no son magnéticamente equivalentes, y que su detección dependería de la resolución del equipo de RMN utilizado. En la Serie A de poli(amida)s estaban propuestas otros cuatro polímeros que serían sintetizados a partir de la reacción entre los ácidos dicarboxílicos 13 y 139 14 y las diaminas que contendrían cuatro fragmentos flexibles de oxiareno (Figura 33). Figura 33. Diaminas propuestas en el proyecto de tesis que contienen cuatro grupos flexibles de oxiarenos. Como estas diaminas no se sintetizaron según lo expuesto en el punto 8 (página 132) de esta sección, no se pudo llevar a cabo la preparación de estas últimas PAs. 2. Poli(amida) derivada de la diamina 24 y el ácido dicarboxílico 13 (Serie B). Figura 34. Ruta de síntesis para la obtención de la poli(amida) PA-V (Serie B). 140 La PA-V se obtuvo con un 92 % de rendimiento, empleando para ello la policondensación directa, metodología explicada anteriormente. La estructura propuesta como unidad repetitiva fue confirmada mediante análisis elemental y empleando técnicas espectroscópicas. Así, el espectro de FTIR da evidencias de la formación de dicho polímero por medio de la visualización del estiramiento N-H del grupo amida a 3422 cm-1 y el estiramiento del grupo carbonilo a 1650 cm-1. Por su parte, en el espectro de RMN 1H se observa el singlete del hidrógeno amídico a 10,35 ppm y los hidrógenos de los grupos metilos a 0,56 ppm. Los demás picos correspondientes a los hidrógenos aromáticos se correlacionan con la estructura planteada en términos de su desplazamiento e integración. En el espectro de RMN 13 C se encuentra el carbono carbonílico a 164,1 ppm y los carbonos del grupo metilo a -2,9 ppm. Las 18 señales restantes corresponden a los 18 carbonos aromáticos, siendo los más desplazados a campo bajo los carbonos C4, C5 y C10 debido al efecto del átomo de oxígeno enlazado directamente a estos. En el espectro de RMN 29 Si se observa una señal a -7,6 ppm que corresponde al único átomo de silicio presente. Este desplazamiento es concordante con el entorno inmediato del heteroátomo y la densidad electrónica que recibe de él; dos anillos aromáticos y dos grupos metilos. Al igual que lo mencionado para la Serie A de PAs, en esta Serie estaban propuestos otros tres polímeros que se prepararían a partir de la reacción entre los ácidos dicarboxílicos 13 y 14 y la diamina 22, así como, el ácido dicarboxílico 14 y la diamina 24 (Figura 35). Figura 35. Poli(amida)s propuestas en el proyecto de tesis. 141 Estas tres PAs no se lograron sintetizar a pesar de que a lo largo del desarrollo de este trabajo se obtuvieron los cuatro monómeros correspondientes para su preparación. Sin embargo, los monómeros ácidos dicarboxílicos 13 y 14 tuvieron serios problemas de reproducibilidad, en especial este último. El díacido 14 se prepara a partir del difenol 2, el cual y tal como se mencionó anteriormente, se obtiene mediante una reacción de hidrogenólisis asistida por calentamiento. El sistema de calefacción del equipo de hidrogenación estuvo sin funcionar por problemas técnicos durante gran parte del último año de tesis, por lo que la obtención de dicho difenol se vio limitada. Actualmente, el grupo de investigación continúa con serios problemas para su obtención. Con respecto al diácido 13, ya se mencionó en el punto 2 (página 112) de esta sección, que su preparación involucra una reacción a alta temperatura a fin de obtener el diciano derivado que le da origen. Si bien la síntesis funciona, la condición térmica impuesta lleva a la obtención de bajos rendimientos, probablemente debido a la descomposición del precursor difenol ya comentada. Este hecho llevó a que la cantidad de diácido 13 que se obtuvo y se purificó adecuadamente durante el desarrollo del trabajo de tesis, alcanzara para la obtención de las PAs de la Serie A y la PA de la Serie B. A fin de compensar esta situación, se decidió incorporar en el trabajo de tesis otras cuatro PAs derivadas de la diamina 26 y cuatro ácidos dicarboxílicos preparados con anterioridad por el grupo de investigación, todos ellos monómeros que aportan en el concepto de flexibilización y contenido aromático. Todo esto lleva a que los resultados derivados de los análisis practicados a las PAs obtenidas, los cuales resaltan los distintos efectos estructurales incorporados en sus unidades repetitivas, permitan dar cumplimiento al objetivo de establecer eficientes correlaciones estructurapropiedad y verificar así, la hipótesis propuesta. 142 3. Poli(amida)s derivadas de la diamina 26 y los ácidos dicarboxílicos MM, EM, MP y PP (Serie C). Figura 36. Ruta de síntesis para la obtención de las poli(amida)s PA-VI, PAVII, PA-VIII y PA-IX. Estas cuatro PAs no estaban consideradas en la formulación del proyecto original, pero, y tal como se mencionó en el punto precedente, problemas técnicos y experimentales hicieron que no se pudieran sintetizar algunas de las PAs propuestas en las Series. La nueva diamina sintetizada (26) introduce unidades flexibles conjuntamente con alto contenido aromático y una unidad voluminosa basada en la porción 9,9-difenilfluoreno. Por esta razón, se decidió sintetizar cuatro nuevas PAs al hacer reaccionar dicha diamina con diácidos sililados previamente informados por el Grupo de Investigación. La metodología empleada en la síntesis de estas poli(amida)s es la policondensación directa y los ácidos dicarboxílicos utilizados, que tal como se mencionó anteriormente, fueron sintetizados en años anteriores por el 143 grupo de trabajo (109, 110). Brevemente, el p-bromotolueno reacciona con litio metálico en éter dietílico anhidro y luego con el correspondiente dicloroorganosilano para obtener el di(p-tolil)derivado, el cual es oxidado eficientemente al correspondiente ácido dicarboxílico (Figura 37). Figura 37. Ruta de síntesis para la obtención de los ácidos dicarboxílicos MM, EM, MP y PP. Estas PAs se obtuvieron con rendimientos entre 86 y 95 % y las estructuras fueron verificadas mediante análisis elemental. Como era de esperar, en el espectro FTIR de las cuatro poli(amida)s se observan las bandas específicas asociadas al grupo amida. El estiramiento para el enlace N-H aparece en 3417 y 3423 cm-1, mientras que el del grupo carbonilo se encuentra entre 1654 y 1670 cm-1. En el espectro de RMN 1H se observan señales con desplazamientos químicos similares para cada una de las poli(amida)s. Así, los hidrógenos amídicos aparecen entre 10,23 y 10,37 ppm, mientras que el singlete a 2,05 ppm corresponde a los hidrógenos de los grupos metilos unidos a la unidad 9,9´-difenilfluoreno. Las señales correspondientes a los hidrógenos aromáticos evidencian un patrón más complejo a medida que aumenta el carácter aromático sobre el átomo de silicio del monómero ácido dicarboxílico, haciendo dificultoso la asignación de señales acopladas y su magnitud (J). Respecto a los hidrógenos de los grupos metilos enlazados directamente a silicio, estos se desplazan a campo bajo en relación a los metilos enlazados a la porción 9,9´-difenilfluoreno. De esta forma, PA-VI presenta una señal a 144 0,57 ppm para estos hidrógenos, mientras que PA-VIII los muestra, por medio de un singlete a 0,88 ppm. Por otro lado, PA-VII presenta dos señales en la zona alifática: Una señal a 0,56 ppm que se asigna a los hidrógenos del grupo metilo directamente enlazado a silicio y una señal ancha a 1,00 ppm que integra para cinco hidrógenos correspondiente al grupo etilo. En el espectro de RMN 13 C de las cuatro PAs se observa el carbono carbonílico del grupo amida alrededor de 164 ppm, mientras que los grupos metilos unidos directamente al átomo de silicio aparecen con desplazamientos negativos. Los diversos carbonos aromáticos aparecen agrupados entre 116 y 155 ppm aproximadamente. Por otro lado, en el espectro de RMN 29 Si se observa una señal para cada polímero y el desplazamiento químico de cada señal se corresponde con la naturaleza, en particular con el aporte electrónico, de los sustituyentes enlazados al heteroátomo que la origina. 145 IV. SÍNTESIS DE POLI(IMIDA)S 1. Poli(imida)s derivadas de las diaminas 9 y 10 y los dianhídridos de ácidos 19 y 20 (Serie A). Figura 38. Ruta de síntesis para la obtención de PI-I, PI-II, PI-III y PI-IV. El método usual para la preparación de PIs aromáticas consta de dos etapas. La primera de ellas es la formación del ácido poliámico a partir de la reacción entre la diamina y el dianhídrido de ácido a temperatura ambiente y en disolución, empleando para ello un disolvente polar aprótico. El segundo paso de la síntesis es la ciclodeshidratación intramolecular para generar la PI final. Como se puede ver en la figura 39, el ataque nucleofílico del par de electrones desapareados del nitrógeno amínico sobre el carbono carbonílico del dianhídrido de ácido ocasiona la apertura de este, originando el ácido poliámico intermedio. Figura 39. Mecanismo de formación del ácido poliámico. 146 Esta reacción es simple, sin embargo existen diversos parámetros que afectan la obtención de altos pesos moleculares (111). Así, la pureza de los monómeros debe ser mayor que 99,9 %, la estequiometría de los reaccionantes debe ser exactamente uno es a uno, los grupos reactivos deben estar lo más accesibles posibles, el tiempo de reacción debe ser suficientemente largo para lograr una mayor conversión y no deben existir reacciones secundarias o bien producto de impurezas del disolvente. Como dicha reacción es reversible (112), la reacción inversa se considera secundaria y comienza transfiriendo el protón del grupo carboxílico al nitrógeno del grupo amida. Sin embargo, esto se puede disminuir empleando disolventes polares apróticos (113), ya que estos pueden formar enlaces por puente de hidrógeno con dicho protón desplazando el equilibrio hacia la derecha. Otra reacción secundaria es la que se origina al introducir pequeñas cantidades de agua a través del disolvente y esto conlleva a la hidrólisis de los grupos anhídridos terminales o bien la hidrólisis del ácido poliámico originando los monómeros de partida. Esto afectaría el crecimiento de la cadena polimérica y el rendimiento. Por lo tanto, es necesario el uso de disolventes con alto grado de pureza. Los ácidos poliámicos pueden ser aislados como productos de estabilidad intermedia. Muchas veces, cuando la poli(imida) final es altamente insoluble, se emplean como criterio de determinación estructural en solución (RMN) de las mismas, ya que su solubilidad suele ser mayor. El segundo paso de síntesis es la ciclodeshidratación del ácido poliámico. Esta puede ser mediante tres metodologías diferentes (112). La primera de ellas es la imidación térmica con evaporación simultánea de disolvente y agua. La disolución de ácido poliámico se coloca sobre una placa y se 147 calienta gradualmente hasta los 350 °C. Este método es el más usado en la industria para preparar películas de polímero o bien, cuando la poli(imida) resulta insoluble en disolventes orgánicos, la cual impedirá su procesamiento en forma de polvo. Otra forma de realizar la imidación es mediante una ciclación térmica en disolución utilizando un disolvente de elevada temperatura de ebullición (150 a 220 °C). El tercer método consiste en una imidación química en disolución usando un agente deshidratante y una base terciaria como piridina o trietilamina. Este método se usa mucho a nivel de laboratorio para poder obtener las poli(imida)s en forma de polvo y procesarlas posteriormente. El requisito para este método es que la poli(imida) tiene que ser soluble en el medio de reacción empleado. Como en el grupo de investigación se postula que las poli(imida)s modificadas estructuralmente, en el sentido de incorporar unidades de flexibilización, serían solubles en disolventes orgánicos comunes, se optó por utilizar este último método para realizar la ciclación del ácido poliámico. El mecanismo propuesto para esta reacción (114) se muestra en la figura 40. El primer paso es la N-acetilacion de la piridina, lo cual cataliza la reacción de imidación. Para esto, la piridina reacciona con el anhídrido acético formando el catión acilpiridinium y el anión acetato (reacción 1). Como el ácido poliámico (pKa = 8,8 en DMF) (115) es un ácido más fuerte que el ácido acético (pKa = 13,5 en DMF) (115), el ion acetato puede ser protonado rápidamente desplazando el equilibrio de la reacción 1 hacia la derecha (reacción 2). Por lo tanto, la alta concentración del ion acilpiridinium promueve la reacción con el ion conjugado del ácido poliámico para generar el intermediario anhídrido-amida y reconstituir la piridina base (reacción 3). Como es esperado, el equilibrio de esta reacción se ve favorecido con una alta concentración de ácido poliámico. 148 Posteriormente, el intermediario anhídrido-amida es desprotonado por la piridina para formar dos estructuras aniónicas resonantes diferentes (reacción 4). El ataque nucleofílico intramolecular del nitrógeno cargado negativamente produce la imida correspondiente, mientras que el ataque del oxígeno cargado negativamente produce el isómero isoimida. Sin embargo, como el nitrógeno cargado negativamente es mejor nucleófilo que el oxígeno cargado negativamente, la formación de la imida se ve favorecida. O O N H3C O O O CH3 N O H O O O N O CH3 O H k2 N O O N O O O CH3 N N k3 O 3 CH3 O H 2 OH N k-2 O H3C O H N O O k-1 O 1 O N k1 CH3 H3C H O O O CH3 k-a H N O ka O k-3 O O O CH3 O N O H k4 N k-5 k5 O CH3 4 O N O N O O O O CH3 O CH3 O N H O CH3 O O kb N k-b H3C OH 5 Figura 40. Mecanismo propuesto para la imidación química usando anhídrido acético y piridina. El ion acetato que se genera de la reacción 4 es protonado rápidamente por el ácido poliámico presente en el medio de la reacción, lo que desplaza el equilibrio hacia la derecha e impide que exista la reacción inversa. Una vez 149 que se consume el ácido poliámico, el ion acetato se genera nuevamente al reaccionar el ácido acético formado con la piridina (k-b, reacción 5). Este ion acetato puede reaccionar con la isoimida formada para generar nuevamente el intermediario anhídrido-amida (k-5, reacción 4), el cual se cicla a la forma imida más estable. Como se pudo ver, el agua no está presente en este mecanismo lo que disminuye las posibles reacciones secundarias de hidrólisis. Las poli(imida)s PI-I, PI-II, PI-III y PI-IV se obtuvieron con rendimientos entre 74 y 89 % y los resultados de análisis elemental de cada una de ellas, concuerdan con las estructuras propuestas para las unidades repetitivas. En el espectro FTIR de estos polímeros no se observan bandas anchas en la zona entre los 3200 y 3500 cm-1, lo que corrobora el éxito de la reacción de cilclación de las cadenas del ácido poliámico. La presencia de dos bandas intensas alrededor de 1780 y 1720 cm-1 confirma la formación del grupo imida, debido a que estas frecuencias se asocian a los estiramientos simétricos y antisimétricos de dicho grupo. Los espectros de RMN 1H de las cuatro PIs están de acuerdo con las estructuras planteadas. PI-I, PI-II y PI-III presentan señales correspondientes a los hidrógenos del grupo metilo cercano a los 0,5 ppm. PI-I, en particular, presenta dos señales a 0,58 ppm y 0,52 ppm debido a que los grupos metilos unidos a los átomos de silicio no son magnéticamente equivalentes. Uno proviene del monómero diamina y el otro proviene del monómero dianhídrido de ácido. PI-IV no presenta señal alguna en la zona alifática debido a que no existen hidrógenos unidos a carbonos sp3, sin embargo los anillos aromáticos presentes sobre los átomos de silicio no son equivalentes, lo cual hace complejo cualquier intento de asignación particular de señales. Esta noequivalencia se evidencia mejor en el espectro de RMN 150 13 C, donde se logra identificar y separar las señales correspondientes a los carbonos C19, C20, C21 y C22 de los carbonos C23, C24, C25 y C26. En los cuatro espectros de RMN 13 C se observan los carbonos carbonílicos del anillo imida alrededor de 166,0 ppm. PA-I presenta dos señales a -2,9 ppm y -3,0 ppm correspondientes a los carbonos de los grupos metilos. Sin embargo, en el espectro de RMN 29 Si de PI-I solo se observa una señal a -8,63 ppm, cuando se esperaban dos. Es probable que aparezcan solapados ya que dicha señal aparece más ancho que lo normal (ver Anexo). En los espectros de RMN 29 Si de PI-II, PI-III y PI-IV se observa lo esperado, es decir, dos señales para cada una: -7,8 y -14,7 ppm para PI-II, -8,1 y -14,9 ppm para PI-III y 14,7 y -14,8 ppm para PI-IV. En la Serie A de poli(imida)s estaban propuestos otros cuatro polímeros que iban a ser sintetizados a partir de la reacción entre los dianhídridos de ácidos 19 y 20 y las diaminas que contendrían cuatro fragmentos flexibles de oxiareno (Figura 33). Estas diaminas no se sintetizaron según lo explicado en el punto 8 (página 132) razón por la cual no se pudo llevar a cabo la preparación de las poli(imida)s propuestas. 151 2. Poli(imida)s derivadas de la diamina 24 y los dianhídridos de ácidos 19 y 20 (Serie B). Figura 41. Ruta de síntesis para la obtención de PI-V y PI-VI. La síntesis de las poli(imida)s PI-V y PI-VI se realiza siguiendo la misma metodología explicada anteriormente, vale decir, formando inicialmente el ácido poli(ámico) y luego sometiéndolo a una imidación química en disolución. Estas poli(imida)s se obtuvieron con un 94 y 93 % de rendimiento, respectivamente y el resultado del análisis elemental practicado a cada una de ellas, está de acuerdo con la unidad repetitiva propuesta. En cuanto a lo experimental, se debe destacar que la PI-V comenzó a precipitar durante el proceso de ciclación química, lo que hace que su caracterización en disolución no sea posible, ya que resultó ser insoluble en todos los disolventes deuterados utilizados. En este sentido, los resultados de análisis elemental antes mencionados, avalan fuertemente la naturaleza estructural de la unidad repetitiva propuesta para esta poli(imida). El espectro FTIR de ambas PIs muestra las bandas intensas de estiramiento simétrico y antisimétrico del grupo carbonilo del anillo imida cercano a los 152 1780 y 1720 cm-1. En el espectro de RMN 1H de PI-VI, se logran distinguir tres grupos de señales. La señal desplazada a campo bajo corresponde a los hidrógenos H20 del fragmento diamina. Por otro lado, la señal que se encuentra a campo alto corresponde a los hidrógenos H8 del fragmento dianhídrido de ácido, mientras que las señales ubicadas entre las dos antes señaladas corresponden a los hidrógenos aromáticos restantes. En el espectro de RMN 13 C de PI-VI se observa los picos a 166,6 y 166,5 ppm correspondientes a los carbonos carbonílicos del anillo imida, los cuales no son equivalentes. Los carbonos C4, C7 y C18 aparecen a campo bajo debido al efecto desapantallante que ejerce el átomo de oxígeno. Los desplazamientos observados para los carbonos aromáticos restantes están de acuerdo con la estructura planteada. En el espectro de RMN 29 Si se observa una sola señal a -13,6 ppm, desplazamiento químico esperado para el átomo de silicio cuando se encuentra rodeado de cuatro anillos aromáticos. 3. Poli(imida) derivada de la diamina 22 y el dianhídrido de ácido 19 (Serie C). Figura 42. Ruta de síntesis para la obtención de PI-VII. 153 Esta poli(imida) se obtuvo con un rendimiento de 86 % empleando la misma metodología de síntesis planteada anteriormente y los resultados de análisis elemental corroboran la estructura propuesta. En el espectro de FTIR se aprecian las bandas de estiramiento de los carbonilos del grupo imida a 1726 y 1722 cm-1. En el espectro de RMN 1H se observa el singlete a 0,57 ppm correspondiente a los hidrógenos de los grupos metilo unidos al átomo de silicio. Los hidrógenos aromáticos restantes aparecen en forma de grupos de señales, siendo el hidrógeno H16 el más desplazado a campo bajo y el hidrógeno H8 el más desplazado a campo alto. El espectro de RMN 13 C muestra las señales de los carbonos carbonílicos a 166,1 y 166,0 ppm. Los carbonos C4, C7 y C14 aparecen a campo bajo debido al efecto del átomo de oxígeno, mientras que el desplazamiento de los otros carbonos aromáticos se correlaciona con la estructura planteada. La señal a -2,7 ppm corresponde a los carbonos del grupo metilo, los cuales son magnéticamente equivalentes. Como era de esperar, en el espectro de RMN 29 Si aparece una sola señal a -8,1 ppm, desplazamiento propio al entorno electrónico del núcleo estudiado. A fin de completar esta Serie debería haberse preparado la poli(imida) derivada de la reacción entre la diamina 22 y el dianhídrido de ácido 20. Sin embargo, el compuesto 20 proviene del difenol 2, cuya obtención como ya se mencionó anteriormente, se ha visto impedida por problemas técnicos con el equipo de hidrogenación con calentamiento. 154 4. Poli(imida)s derivadas de la diaminas 21 y 22 y el dianhídrido de ácido 29 (Serie D). O O NH2 O (21) H2N CH3 O CH3 O O o H2N O O O NH2 1) DMAc, Tamb 2) Ac2O, Py, 60 °C O (22) O O O N * O PI-VIII: 22 + 29 (29) CH3 O * N CH3 O O n PI-IX: 21 + 29 Figura 43. Ruta de síntesis para la obtención de PI-VIII y PI-IX. Estas dos PIs no estaban consideradas originalmente en el proyecto de tesis, sin embargo, debido a los problemas técnicos y experimentales que provocaron que no se pudiera sintetizar las poli(imida)s propuestas en el proyecto, así como también el hecho de que el monómero 29 aporte unidades flexibles y un alto contenido aromático y además el monómero 21 aporta asimetría a la cadena polimérica, se decidió incorporar estas dos PIs al proyecto y así evaluar el efecto de la ruptura de la simetría provocado por el monómero 21. La metodología de síntesis seguida fue equivalente a la descrita anteriormente alcanzando un rendimiento de 85 % para PI-VIII y 90 % para PI-IX. Ambas poli(imida)s fueron caracterizadas según análisis elemental y los resultados concuerdan satisfactoriamente con los porcentajes de H, C y N esperados. Asimismo, en los espectros de FTIR se observa el estiramiento 155 simétrico y asimétrico de los carbonilos del anillo imida cercano a 1780 y 1720 cm-1, lo que es otra prueba de la consistencia estructural. El espectro de RMN 1 H de PI-VIII es más sencillo que los patrones observados en el espectro de PI-IX, debido a la falta de simetría que presenta la diamina 21 en esta última poli(imida). La señal desplazada a campo bajo corresponde a los hidrógenos H26, mientras que el desplazamiento del resto de los hidrógenos aromáticos se correlaciona con la estructura planteada. Por otro lado, el singlete centrado en 2,04 ppm corresponde a los hidrógenos de los grupos metilos del fragmento 9,9´difenilfluoreno los cuales, dada la simetría de esta 13 magnéticamente equivalentes. En el espectro de RMN porción, son C de PI-VIII se observan los dos carbonos carbonílicos del grupo imida solapados a 166,8 ppm, mientras que los carbonos C4, C7 y C24 son los más desplazados a campo bajo debido a la electronegatividad con la cual afecta el átomo de oxígeno. El carbono C13 a pesar de ser alifático, aparece a 64,4 ppm, debido al efecto desapantallante de los cuatro anillos aromáticos que lo rodean. Por último, el pico a 16,6 ppm corresponde a los carbonos del grupo metilo del fragmento 9,9´-difenilfluoreno. Como se dijo anteriormente, el espectro de RMN 1H y de 13 C de PI-IX es de mayor complejidad dada la asimetría estructural de uno de los monómeros constituyentes de la unidad repetitiva. En este caso, los hidrógenos más desplazados a campo bajo en el espectro de RMN 1H son H22 y H31, los demás hidrógenos aromáticos aparecen entre 7,89 y 7,31 ppm en forma de multipletes complejos. El singlete a 2,07 ppm que integra para 6 hidrógenos es asignado inequívocamente a los hidrógenos de los grupos metilo del fragmento 9,9´-difenilfluoreno. En el espectro de RMN 13 C se aprecia una mayor cantidad de señales respecto a lo observado para PI-VIII. Esto se debe a que ahora existen seis carbonos adicionales debido a la asimetría del fragmento diamina. A 166,4 y 166,1 ppm aparecen los carbonos carbonílicos 156 del grupo imida, mientras que a campo más bajo aparecen los cuatro carbonos unidos directamente al átomo de oxígeno; C4, C7, C23 y C27. Al igual que en PI-VIII, el carbono C13 aparece a 64,0 ppm y los carbonos de los grupos metilo se encuentran a 15,8 ppm. Por último, cabe destacar que en el proyecto de tesis se proponían otras dos series de PIs donde estaban involucrados los monómeros diamina y dianhídridos de ácidos con cuatro grupos flexibles de oxiarenos cada uno (Figura 44), los cuales no se sintetizaron por las razones expuestas anteriormente. Figura 44. Diaminas y dianídridos de ácido propuestos en el proyecto de tesis que contienen cuatro grupos flexibles de oxiarenos cada uno (R= Me o Ph). Es necesario enfatizar el hecho de que las preparaciones de PIs no logradas, no influencian las conclusiones a las cuales se llegaron gracias a la síntesis de los polímeros preparados y que estaban inicialmente propuestos, como también de los que se incorporaron por razones dadas oportunamente. Los trabajos realizados permiten dar cumplimiento al objetivo de establecer eficientes correlaciones estructura-propiedad y verificar así, la hipótesis propuesta. 157 V. VISCOSIDAD INHERENTE Las medidas de viscosidad de una disolución polimérica es una herramienta altamente útil para determinar el peso molecular de este. Desde que Staudinger, en 1930, demostró la utilidad de esta técnica, la comunidad científica la ha seguido ocupando hasta la actualidad. Para los polímeros lineales, la viscosidad de una disolución se relaciona con el tamaño de las cadenas poliméricas y por lo tanto con el peso molecular. En este sentido, se han definido distintos tipos de viscosidad: viscosidad relativa (ηr), específica (ηsp), reducida (ηred), inherente (ηinh) e intrínseca ([η]). El la figura 45 se esquematizan las curvas de viscosidad reducida e inherente frente a la concentración de un polímero en disolución. Como se puede ver, la viscosidad intrínseca no depende de la concentración, debido a que se determina extrapolando los datos obtenidos de viscosidad inherente o reducida, a concentración cero (dilución infinita). Si bien este parámetro se independiza de la concentración, es función de la estructura del polímero, de la temperatura y del disolvente empleado. Figura 45. Esquema que representa la relación existente entre la viscosidad reducida e inherente con la concentración polimérica. 158 La viscosidad intrínseca se relaciona con la masa molecular del polímero a través de la ecuación Mark-Houwink-Sakurada ([η] = KMa), donde K y a son constantes que dependen de la interacción polímero-disolvente. Por lo general, para polímeros flexibles el valor de a (adimensional) oscila entre 0,5 y 0,8, mientras que K varía de 0,5 a 5x10 -4 (dL/g) (116). En muchos casos, la viscosidad inherente para una concentración determinada, por lo general entre 0,3 a 0,5 g/dL, se utiliza como una aproximación de la viscosidad intrínseca (116). Esto se debe a que si se trabaja a concentraciones muy diluidas, más se aproxima a la concentración cero. Sin embargo, la viscosidad intrínseca siempre es mayor que la viscosidad inherente. Estas medidas de viscosidad inherente en un solo punto a concentraciones muy diluidas han sido aceptadas para estimar y comparar las masas moleculares de polímeros lineales que presenten estructuras similares, sin tener que medir dicha viscosidad a diferentes concentraciones. Vale destacar que si el valor de ηinh de un polímero Y es 0,3 dL/g y el de otro polímero W, de estructura similar medido en el mismo disolvente a la misma temperatura, es 0,6 dL/g, no se puede concluir que el polímero W tiene el doble de masa molecular que el polímero Y, solo se concluye que el polímero W debe tener una masa molecular mayor que el polímero Y. En este trabajo se mide la viscosidad inherente en un solo punto, a una concentración de 0,5 g/dL en disolución de DMSO a 25 ± 0,1 °C. Los valores obtenidos, tanto para las poli(amida)s como para las poli(imida)s se presentan en la tabla 1. 159 Tabla 1. Valores de viscosidad inherente de las PAs y PIs sintetizadas. Poli(amida)s ηinh (dL/g) Serie A Poli(imida)s ηinh (dL/g) Serie A PA-I 0,10 PI-I 0,27 PA-II 0,20 PI-II 0,20 PA-III 0,18 PI-III 0,10 PA-IV 0,16 PI-IV 0,15 Serie B PA-V Serie B 0,18 Serie C PI-V n.d. PI-VI 0,14 PA-VI 0,21 Serie C PA-VII 0,14 PI-VII PA-VIII 0,20 Serie D PA-IX 0,23 PI-VIII 0,15 PI-IX 0,18 0,20 n.d.: No determinada dada la insolubilidad de la muestra. Como se puede ver, los valores de viscosidad inherente fueron similares y bajos (entre 0,10 y 0,27 dL/g), por lo que se puede estimar que los polímeros formados presentan un bajo a moderado peso molecular, vale decir, cadenas poliméricas de corta longitud. En el caso de PI-I, que presenta el mayor valor de viscosidad inherente, se pudo determinar la masa molecular mediante técnicas de cromatografía de exclusión por tamaño (SEC). Dichos resultados se comentan a continuación. 160 VI. CROMATOGRAFÍA DE EXCLUSIÓN POR TAMAÑOS (SEC) Uno de los métodos más empleados en la actualidad para determinar la distribución de masas moleculares de un polímero es la cromatografía de exclusión por tamaño (SEC, Size Exclusion Cromatography), la cual separa las cadenas poliméricas en función de su tamaño. Esta técnica también se conoce como cromatografía de permeación de gel (GPC, Gel Permeation Cromatography), debido a que las columnas de separación están rellenas con un gel poroso rígido. Para el análisis PI-I se utilizaron tres columnas conectadas en series donde el gel poroso fue una mezcla de poliestirenodivinilbenceno. Cuando se inyecta una disolución diluida del polímero (5 mg/mL) las cadenas de este comienzan a interactuar con los poros según el tamaño de cada una de ellas. Las cadenas más pequeñas pueden penetrar en los poros mientras que las más grandes avanzarán más rápido, dando como resultado que los tamaños moleculares mayores eluyen primero que los más pequeños. Así se obtiene una distribución basada en el tamaño y no en la masa molecular del polímero (117). Como el tamaño del polímero no depende solamente de la masa molecular de este, es necesario realizar una calibración previa con un polímero que presente una distribución estrecha de masas moleculares. En este caso, esta operación se realizó con un patrón de poliestireno de masa molecular 60 000 Da. Esto hace que la técnica sea clasificada como relativa y no absoluta. Si dos moléculas presentan igual volumen de retención o tiempo de retención se dice que estas presentan igual volumen hidrodinámico. Este parámetro es directamente proporcional al producto de la viscosidad intrínseca por la masa molecular ([η]M) (117). Si se grafica el logaritmo de este producto en función 161 del volumen de retención para distintos polímeros se puede obtener lo que se conoce como curva de calibración universal. La viscosidad intrínseca se puede determinar experimentalmente usando un viscosímetro de flujo y registrando el tiempo de elución de la solución polimérica y del solvente empleado. Por otro lado, a partir del volumen de retención y la curva de calibración universal se puede determinar la masa molecular. Si al equipo de SEC se le coloca un detector de dispersión de luz se pueden determinar las masas moleculares absolutas. Vale destacar que el equipo de SEC utilizado para medir la masa molecular promedio en número y en peso de la muestra PI-I tenía acoplado un detector de índice de refracción, un viscosímetro diferencial y un detector de dispersión de luz. Los datos obtenidos se muestran en la tabla 2. Por razones ajenas a la voluntad del grupo de investigación, solo se pudo medir una muestra, por lo que se escogió la de mayor viscosidad inherente. Tabla 2. Datos obtenidos del análisis de cromatografía de exclusión por tamaño para PI-I. Poli(imida) Mn Mw Mw/Mn (g/mol) (g/mol) PI-I 6870 11 990 [η] a (dL/g) 1,74 0,28 K (dL/g) 0,585 1,27 10-3 Como se puede ver, los valores de masa molecular promedio no son altos, sino más bien representan un peso molecular moderado. Teniendo en cuenta que la masa de la unidad repetitiva de la PI-I es de 927,11 g/mol y que la masa molecular promedio en número es de 6870 (g/mol), el largo promedio de las cadenas es de siete unidades repetitivas (heptámero), aunque como se observa en el valor de la masa molecular promedio en peso 162 existen cadenas más largas (aproximadamente 13 unidades repetitivas) que consecuentemente aportan más al peso total del polímero. El valor de la viscosidad intrínseca entregado por el equipo fue similar al de la viscosidad inherente calculado mediante técnicas viscosimétricas (0,28 dL/g y 0,27 dL/g, respetivamente). Sin embargo, se debe destacar que esta similitud probablemente es accidental debido a que, si bien dichas viscosidades fueron medidas a igual temperatura, los disolventes empleados en los respectivos análisis fueron diferentes. Otro hecho importante derivado del análisis SEC es la obtención de los parámetros de Mark-Houwink-Sakurada a y K. Por lo general, estas constantes se encuentran tabuladas en handbooks de polímeros y son diferentes para cada tipo de polímero y dependen de la temperatura, de la interacción polímero-disolvente y también de la distribución del peso molecular. Sin embargo, de forma aproximada se puede decir que para familias de polímeros que guardan una estructura base común, como lo son las PIs de la Serie A, se pueden usar estas constantes para determinar los valores de masa molecular conociendo la viscosidad intrínseca de estos polímeros. Si se toma la viscosidad inherente en un solo punto para un sistema diluido como una aproximación de la viscosidad intrínseca, tal como lo sugiere Billmeyer (116) y se sustituyen en la ecuación de Mark-Houwink-Sakurada los valores de a y K, es posible obtener los valores de masa molecular promedio viscosimétrico para todas las poli(imida)s de la Serie A (Tabla 3). 163 Tabla 3. Peso molecular viscosimétrico para las poli(imida)s de la Serie A. Poli(imida) PI-I PI-II PI-III PI-IV Mv* (g/mol) 9520 5700 1740 3490 *Valor calculado empleando la ecuación de Mark-Houwink-Sakurada con K = 1,27 10-3 y a = 0,585. A partir de estos valores se reafirma la idea de que las cadenas poliméricas alcanzaron longitudes bajas. Así, PI-I aparece como un decámero, PI-II como un pentámero, mientras que PI-III y PI-IV se representarían como un dímero y un trímero, respectivamente. Debe dejarse claro que en este cálculo, se han despreciado los factores hidrodinámicos que podrían ser diferentes entre las PIs debido a las sustituciones practicadas sobre el átomo de silicio. 164 VII. SOLUBILIDAD La solubilidad de los polímeros en un disolvente determinado ocurre de forma lenta. A simple vista se observa un hinchamiento del material, debido a que las moléculas del disolvente difunden entre las cadenas poliméricas y comienzan a romper las interacciones que existen entre ellas, y luego con agitación vigorosa comienza la disolución total del polímero. Mientras mayor es la masa molecular de este, más fuertes son las interacciones polímeropolímero y el proceso de disolución se dificulta más, pudiendo durar varias horas o días, dependiendo de la cantidad a disolver. La disolución de un polímero depende de varios factores tales como temperatura, naturaleza del disolvente, topología del polímero, tipo de polímero, masa molecular, entre otros. Sin embargo, la semejanza entre la naturaleza del disolvente y el polímero en cuanto a naturaleza química y polaridad, permite escoger a priori un disolvente en el cual el polímero pudiera resultar soluble. Existe un parámetro que relaciona la capacidad que posee un disolvente para disolver una sustancia, denominado parámetro de solubilidad (δ) (118). Este parámetro fue introducido por Charles M. Hansen y también se conoce como parámetro de solubilidad de Hansen. Matemáticamente se representa por la suma de los cuadrados de tres contribuciones: contribución de las fuerzas de dispersión (δD), contribución de las fuerzas dipolo-dipolo (δP) y contribución de las interacciones por enlace por puente de hidrógeno (δH). Físicamente se puede interpretar de la siguiente forma: un disolvente pudiera disolver un polímero cuanto más próximos estén los parámetros de 165 solubilidad de ambos. En la siguiente tabla se muestran los valores del parámetro de solubilidad de algunos disolventes polares apróticos. Tabla 4. Valores de parámetro de solubilidad* de algunos disolventes. Disolvente D P H (MPA)1/2 (MPA)1/2 (MPA)1/2 (MPA)1/2 DMSO 18,4 16,4 10,2 26,7 DMAc 16,8 11,5 7,2 21,6 DMF 17,4 13,7 11,3 24,8 NMP 18,0 12,3 7,2 23,0 m-cresol 18,0 5,1 12,9 22,7 * Valores tomados de la referencia 117. Estos disolventes son los más utilizados para disolver polímeros aromáticos que presentan grupos polares, como poli(éter-imida)s, poli(amida-imida)s, poli(éter-sulfona)s, etc, debido a que los parámetros de solubilidad de estas familias de polímeros son muy cercanos en valor a los de estos disolventes (118). Por lo tanto, es de esperar que las poli(amida)s y poli(imida)s aromáticas sintetizadas en este trabajo sean solubles en estos disolventes. Sin embargo, la similitud en el valor de parámetro de solubilidad polímerodisolvente no es un criterio absoluto, ya que la estructura molecular del polímero es un factor importante. Por ejemplo, el Kevlar o poli(pfenilentereftalamida) presenta un δ= 23 MPA1/2 (119), y no se disuelve en ninguno de los disolventes anteriores, debido a su alta rigidez estructural. En la tabla 5 se muestran los resultados obtenidos al probar la solubilidad de las poli(amida)s sintetizadas en distintos disolventes orgánicos. 166 Tabla 5. Solubilidad de PAs en algunos disolventes orgánicos comunes. Poli(amida)s Disolvente DMSO DMAc DMF NMP m-cresol THF PA-I + + + + + + PA-II + + + + + + PA-III + + + + + + PA-IV + + + + + + + + + + + + PA-VI + + + + + +/- PA-VII + + + + + +/- PA-VIII + + + + + +/- PA-IX + + + + + +/- - +* - +* - - + + + + n.i. n.i. Serie A Serie B PA-V Serie C Kevlara Nomex a b Poli(p-fenilentereftalamida) y b Poli(m-fenilenisoftalamida) empleados como referencia (119). +: Soluble a temperatura ambiente. +/-: Parcialmente soluble a temperatura ambiente y con calor. +*: Soluble con un 5 % en peso de LiCl. -: Insoluble. n.i.: No informada. a b Como se observa, la mayoría de las PAs resultaron solubles en los disolventes orgánicos aromáticas comerciales empleados que no en comparación presentan con grandes poli(amida)s modificaciones estructurales como son Kevlar y Nomex. Sin embargo, lo novedoso de estos 167 resultados es que la mayoría de las PAs sintetizadas se disuelven en THF, un disolvente que presenta temperatura de ebullición baja. Desde el punto de vista de procesabilidad esto último resulta ser altamente positivo, pues da pie a la fácil formación de películas delgadas. La Serie A de PAs presenta elevada solubilidad en todos los disolventes empleados a temperatura ambiente. Esto puede estar relacionando con la incorporación de unidades flexibles tipo éter, lo que provoca el llamado “efecto bisagra” en la cadena polimérica, aumentando la flexibilidad y con esto la solubilidad. En la Serie A ambos monómeros aportan flexibilidad debido a las uniones tipo éter. Las PAs de la Serie C también presentan buena solubilidad, pero resultaron ser solo parcialmente solubles en THF. Si se compara la estructura molecular de las PAs de la Serie A con aquellas de la Serie C, se puede ver que en esta última, solo uno de los monómeros presenta las uniones tipo éter (fragmento diamina), a diferencia de las PAs de la Serie A, donde cada monómero incorporó uniones flexibilizantes. De esta forma, en la Serie C, el factor de empaquetamiento de cadenas debe ser mayor y por lo tanto disminuyen las interacciones con THF. Este hecho lleva finalmente a una reducida solubilidad incluso con la aplicación de calor. En el caso de la Serie B (PA-V), también se tiene que solo uno de los monómeros aporta las uniones flexibles tipo éter. Sin embargo, la flexibilidad aportada por el fragmento ácido dicarboxílico en esta cadena sería mayor que el aporte hecho por el fragmento diamina en las PAs de la Serie C. Esta afirmación provendría del hecho de que la longitud del enlace Si-C es mayor que la del enlace C-C, por lo que la rotación sobre dicho enlace está más favorecida que la rotación de la cadena sobre la unión C12-C15 del fragmento diamina de la Serie C. De igual modo, la porción fluoreno de esta 168 última diamina provoca un alto impedimento estérico a las rotaciones de cadena De estos resultados se puede concluir que la incorporación de unidades flexibles tipo éter y unidades difenilsilano en la cadena principal de todas las poli(amida)s sintetizadas, ejerce un efecto positivo en el proceso de solubilización de los polímeros en disolventes orgánicos comunes al compararlas con PAs que presentan elevada rigidez estructural. Es conocido que el bajo peso molecular contribuye al aumento de la solubilidad de una muestra polimérica. Sin embargo, en el presente estudio, todas las PAs presentan un bajo peso molecular y no todas se comportan igual frente a los disolventes empleados. Por ejemplo, las PAs de la Serie C presentan valores de viscosidad inherente muy parecidos a las de la Serie A y B, sin embargo no se disuelven completamente en THF, incluso con la aplicación de calor. Esto lleva a concluir que lo que se está observando y por ende, está siendo usado para establecer correlaciones, proviene de las modificaciones estructurales practicadas al interior de las distintas unidades repetitivas. Por su parte, la solubilidad de las poli(imida)s aromáticas sintetizadas también fue muy alta en los mismos disolventes anteriores. En la siguiente tabla se muestran los resultados obtenidos. 169 Tabla 6. Solubilidad de PIs en algunos disolventes orgánicos comunes. Poli(imida)s Disolvente DMSO DMAc DMF NMP m-cresol THF PI-I + + + + + + PI-II + + + + + + PI-III + + + + + + PI-IV + + + + + + PI-V - - - - - - PI-VI +* +* +* +* +* +* + + + + + + + + + + + +* + + + + + + - - - - - - Serie A Serie B Serie C PI-VII Serie D PI-VIII PI-IX Kapton a a Poli(piromelitimida-1,4-difenil éter) incluido como referencia (119). +: Soluble a temperatura ambiente. +*: Soluble con calor. -: Insoluble. a O N O O N O n O Como se puede ver en la tabla 6, la mayoría de las PIs resultaron solubles en los disolventes ensayados, excepto PI-V. Vale destacar que la solubilidad de las PIs es mucho menor que la de las PAs de estructura similar debido a la formación de complejos de transferencia de carga, hecho discutido en la sección introductoria. Sin embargo, las PIs de la Serie A resultaron solubles a temperatura ambiente, incluso frente en THF. Probablemente, las uniones 170 tipo éter y las unidades difenilsilano presentes a lo largo de las cadenas polimérica ayudaron a disminuir la formación de estos complejos debido al aumento de la flexibilidad molecular. Al analizar la estructura de las PIs de la Serie B se ve que estas están formadas por dos tipos de monómeros: uno que aporta flexibilidad a través de las uniones tipo éter y difenilsilano (fragmento dianhídrido de ácido) y otro que aporta rigidez debido a la planaridad del heterociclo dibenzofurano y a las uniones bifenilos que posee (fragmento diamina). Al parecer, esta última situación tiene una mayor contribución que los efectos flexibilizantes, logrando en suma, una rigidez estructural que se manifiesta al momento de realizar las pruebas de solubilidad. De esta forma, PI-V es insoluble incluso con aplicación de calor, mientras que PI-VI se disuelve solo al alcanzar temperaturas cercanas a los 40 °C. La diferencia estructural entre PI-V y PI-VI radica en los sustituyentes presentes sobre el átomo de silicio de la unidad difenilsilano. Los grupos fenilos de la PI-VI, los cuales son más voluminosos que los grupos metilos presentes sobre el átomo de silicio en la PI-V, deben estar actuando como separadores de cadena, permitiendo que en presencia de calor, el disolvente pueda penetrar entre esos espacios y promover la disolución del polímero. Por otro lado, la PI-VII se puede comparar tanto con PI-VIII de la Serie D como con PI-V de la Serie B de acuerdo a la fracción de monómero presente en la unidad repetitiva. La diferencia en cuanto a solubilidad entre las PI-VII y PI-VIII radica en que esta última se disuelve en THF solo con la aplicación de calor. Esto puede deberse al mismo factor explicado anteriormente para las PAs, donde el fragmento dianhídrido de ácido en la PI-VII es de mayor flexibilidad que el fragmento dianhídrido de ácido de la PI-VIII el cual se basa en la unidad 9,9´-difenilfluoreno Al comparar PI-VII con PI-V, estas se diferencian solo en el fragmento diamina. Al parecer, la planaridad y las 171 uniones bifenilos que se suman estructuralmente al sistema dibenzofurano en la diamina constituyente de la PI-V aumentan la rigidez de la cadena haciéndola menos soluble que la PI-VII, la cual se basa en una diamina con estructura base en dibenzofurano. Al analizar la solubilidad de las PIs de la Serie D se observa que PI-IX es más soluble que PI-VIII debido, probablemente, a la asimetría que presenta el fragmento diamina en la primera. De esta forma, se reduciría la regularidad y el orden molecular, y por ende se minimizan las interacciones intercadena lo cual aumenta su solubilidad. Por último, se puede concluir que en general las modificaciones estructurales relacionadas con el aumento de la flexibilidad y/o la reducción de la regularidad de las cadenas, aumentan la solubilidad de las PIs sintetizadas al compararlas con la solubilidad de una poli(imida) de estructura más rígida como el Kapton. 172 VIII. PROPIEDADES TÉRMICAS La caracterización de polímeros por sus propiedades térmicas implica generalmente la determinación de la resistencia a la descomposición a alta temperatura y las transiciones térmicas. La primera se investiga mediante análisis termogravimétrico (TGA) y las transiciones térmicas con métodos de calorimetría diferencial de barrido (DSC). Análisis termogravimétrico (TGA) En un análisis termogravimétrico se registra la pérdida de masa con respecto al aumento de temperatura. Por lo general, la velocidad de calentamiento es de forma lineal con el tiempo y se realiza en atmósfera controlada. Los resultados se registran en un termograma, en el que se representa gráficamente el porcentaje de masa respecto a la masa inicial en función de la temperatura de análisis. Existen varios criterios para definir la temperatura de descomposición de una muestra y todas son válidas siempre y cuando se comparen de igual manera: TDTonset: es la temperatura de descomposición térmica a la que comienza el proceso de degradación. TDT(5 %): es la temperatura a la cual se ha perdido el 5 % de la masa inicial. TDT(10 %): es la temperatura a la cual se ha perdido el 10 % de la masa inicial. TDT(50 %): es la temperatura a la cual se ha perdido la mitad de la masa inicial. En este estudio se informa la temperatura a la cual se pierde el 10 % de la masa inicial. En la figura 46 se representan las curvas TGA de las PAs con la 173 derivada de dicha curva (DTGA) y en la tabla 7 se resumen los valores obtenidos. A B Figura 46. A. Curvas TGA de las PAs sintetizadas (20 °C/min en atmósfera de nitrógeno); B. Curvas DTGA para las distintas Series de PAs. Tal como se observa en la tabla 7, todas las PAs sintetizadas se clasifican como termoestables, teniendo en cuenta que este criterio se define para polímeros que pierden el 10 % de su masa a una temperatura no inferior a los 400 °C. Estos resultados confirman que la incorporación de unidades flexibles tipo éter y difenilsilano, contribuye a mejorar la solubilidad sin afectar considerablemente la estabilidad térmica del material, como se propuso en la hipótesis de trabajo. Esta elevada estabilidad térmica se puede deber al gran contenido aromático del polímero, ya que en todos los casos las unidades flexibles incorporan 174 anillos bencénicos adicionales (Ph-O, Ph-Si(R1,R2)-Ph). Por otra parte, las unidades difenilsilano presentes a lo largo de las cadenas poliméricas manifiestan un segundo e importante aporte a esta propiedad; aumentar el carácter iónico de las muestras. Esto se debe a la diferencia de electronegatividad que existe entre carbono y el átomo de silicio. Este fenómeno también puede producir un aumento en la estabilidad térmica del material. Tabla 7. Propiedades térmicas de las PAs. TDT10% Tg Rc (°C) (°C) (%) PA-I 419 43 16 PA-II 439 79 33 PA-III 412 120 34 PA-IV 428 146 41 433 189 47 PA-VI 461 198 47 PA-VII 419 205 33 PA-VIII 465 188 51 PA-IX 424 211 45 Kevlara 500 425 n.i. Nomexb 458 280 n.i. Poli(amida)s Serie A Serie B PA-V Serie C a Poli(p-fenilentereftalamida) y b Poli(m-fenilenisoftalamida) incorporadas como referencias (119). n.i.: No informado. 175 La derivada de la curva de TGA representa la velocidad con que varía la masa de la muestra con respecto a la temperatura de análisis, por lo que al no haber pérdida de masa dicha velocidad es nula. Sin embargo, cuando se comienza a perder masa se registra una velocidad, siendo máxima en algunos puntos críticos donde ocurren transformaciones químicas en función de la temperatura. Dichas transformaciones químicas tienen que ver con la ruptura de enlaces en las cadenas del polímero debido a la temperatura aplicada, siendo esto un proceso irreversible. Mardorsky y Straus (120) proponen dos tipos de degradación térmica en los polímeros: despolimerización de las cadenas y degradación aleatoria. La primera de ellas consiste en la liberación sucesiva de unidades de monómero de un extremo de la cadena o de un eslabón débil. Mientras que la degradación aleatoria se produce por la rotura de la cadena en puntos aleatorios a lo largo de esta, dando una mezcla dispersa de fragmentos que son generalmente de gran tamaño en comparación con la unidad de monómero. La despolimerización de las cadenas es a menudo el proceso de degradación dominante en los polímeros vinílicos, mientras que la degradación en los polímeros de condensación es fundamentalmente debido a la ruptura de cadenas al azar. Las derivadas de las curvas de TGA de las PAs sintetizadas (DTGA) muestran patrones similares, donde se observan dos pérdidas fundamentales de masa, las que deben estar gobernadas por las entalpías de enlace de los fragmentos que se pierden en forma de gas. En el caso de las PAs aromáticas, la primera pérdida consiste fundamentalmente en agua, óxidos de carbono, amoníaco y metano (siempre y cuando la cadena tenga grupos metilos). A temperaturas más altas, se siguen produciendo los óxidos de carbono y agua, pero las cadenas se destruyen completamente y se pierden unidades de benceno, benzonitrilo, cianuro de hidrógeno, entre otros (121). 176 Los valores de residuo carbónico (Rc) de las PAs sintetizadas son elevados y siempre diferentes de cero. Esto se debe a varios factores. El primero de ellos es que el experimento se lleva a cabo en atmósfera inerte de nitrógeno por lo que la combustión completa del material nunca se alcanza. El segundo factor es que como las cadenas poliméricas presentan átomo de silicio, deben existir óxidos de silicio como productos de degradación, los cuales no son volátiles a 900 °C. El resto de la masa debe ser carbono. En la tabla 7 también se muestra la temperatura de descomposición térmica del Kevlar y el Nomex, poli(amida)s comerciales. Ninguna de las PAs sintetizadas alcanzan valores tan altos como el del Kevlar, sin embargo, PAVI y PA-VIII superan la estabilidad térmica declarada para Nomex. Las curvas de TGA y DTGA de las PIs preparadas se encuentran en la figura 47 y los datos obtenidos se resumen en la tabla 8. Las PIs aromáticas, por lo general, son más resistentes térmicamente que las PAs aromáticas. Este comportamiento también se manifiesta en las PIs sintetizadas. Como se puede observar, todas estas presentan elevados valores para la TDT10%. Estos registros fluctúan entre 473 y 562 °C, a excepción de PI-IX que presenta un valor cercano a 390 °C. Nuevamente, la incorporación de fragmentos flexibles de oxiareno no afecta negativamente la estabilidad térmica de los polímeros. En el caso de la PI-IX se utiliza una diamina asimétrica (2,7diaminodibenzofurano), por lo que la baja estabilidad térmica que posee debe estar asociada a la disminución de la regularidad y el orden molecular, que si bien contribuye a aumentar la solubilidad no logra mantener una alta estabilidad térmica, ya que las interacciones entre cadenas deben estar disminuidas. 177 A B Figura 47. A. Curvas TGA de las PIs (20 °C/min en atmósfera de nitrógeno); B. Curvas DTGA para las distintas Series de PIs. PI-I presenta una estabilidad similar a la de Kapton, poli(imida) comercializada por la empresa DuPont®, mientras que PI-VII la supera largamente (525 °C y 562 °C, respectivamente). En cuanto a las derivadas de las curvas de TGA de las PIs se observa mayormente una sola pérdida de masa crítica. Esto debe estar asociado a que como las cadenas resisten una mayor temperatura, al momento de descomponerse el material lo hace de una sola vez. En otras palabras, la pérdida de los primeros fragmentos volátiles como agua, monóxido de carbono, dióxido de carbono, amoníaco y metano ocurre junto con la degradación completa del material (122). En estos polímeros también existe 178 un elevado residuo carbónico (Rc) debido a los mimos factores que se explicaron para las poli(amida)s. Tabla 8. Propiedades térmicas de las PIs. TDT10% Tg Rc (°C) (°C) (%) PI-I 526 169 52 PI-II 498 164 41 PI-III 473 169 50 PI-IV 488 184 54 PI-V 491 218 55 PI-VI 490 205 41 562 213 57 PI-VIII 479 165 55 PI-IX 389 95 53 Kaptona 525 363 - 410 n.i. Poli(imida)s Serie A Serie B Serie C PI-VII Serie D a Poli(piromelitimida-1,4-difenil éter) incluido como referencia (119). n.i.: No informado. Calorimetría diferencial de barrido (DSC) El método de calorimetría diferencial de barrido detecta transiciones de fase como la fusión, la cristalización y/o la temperatura de transición vítrea (Tg). Este último parámetro, es la temperatura por encima de la cual las cadenas poliméricas comienzan a manifestar movimientos segmentales aislados 179 producto del aumento de la energía cinética consecuente con el aumento de la temperatura. El comienzo de este movimiento va a depender, por una parte, de la rigidez estructural de la cadena polimérica y por otra, de la intensidad de las interacciones entre las cadenas o segmentos de ellas (123). Otro factor importante que afecta la Tg es la masa molecular del polímero, por lo general a medida que aumenta la masa molecular, la Tg aumenta, pero, por encima de una masa molecular crítica, la Tg no varía. En las tablas 7 y 8 se presentaron los valores de Tg para las PAs y PIs, respectivamente y en la figura 48 y 49 se muestran las curvas obtenidas para ambas familias de polímeros. Figura 48. Curvas DSC de las PAs sintetizadas (segundo barrido a 10 °C/min en atmósfera de nitrógeno). 180 Los valores de Tg obtenidos para las PAs se ajustan a lo esperado. Como se dijo anteriormente, los dos monómeros que forman las PAs de la Serie A aportan flexibilidad a la cadena debido a las uniones tipo éter (PI-I - PI-IV). Su mayor flexibilidad estructural hace que estas PAs presenten los valores de Tg más bajos respecto al resto de las muestras analizadas. Además, dentro de la propia Serie A, PA-I tiene el menor valor de Tg y PA-IV el valor mayor. Esto puede deberse a que PA-IV presenta un mayor contenido aromático, lo que implica una mayor rigidez, debido a los dos grupos fenilos unidos a cada átomo de silicio de la unidad repetitiva. A diferencia de esto, PA-I presenta grupos metilos como sustituyentes sobre los heteroátomos. PA-V tiene una Tg de 189 °C. Este valor es más elevado que el de las PAs de la Serie A, debido a la rigidez que presenta el fragmento proveniente de la diamina. Por su parte, las PAs de la Serie C también presentan valores de Tg más altos que los observados al interior de la Serie A. Probablemente, el aumento de rigidez estructural de la porción diamina con base en la unidad 9,9´-difenilfluoreno, es la explicación a lo observado experimentalmente. En este mismo sentido se aprecia que PA-IX presenta el mayor valor de Tg dentro de la Serie C, lo cual, probablemente, es resultado del mayor contenido aromático. Sin embargo, PA-VIII tiene el menor valor lo cual debe estar asociado a la ruptura de la simetría debido a que el fragmento ácido dicarboxílico presenta una sustitución asimétrica sobre el átomo de silicio (Me y Ph). Por otro lado, PA-VII también presenta una asimetría estructural con grupos metilo y etilo enlazados al átomo de silicio de la porción diácido carboxílico de la unidad repetitiva. Sin embargo, esta asimetría tiene una menor contribución a la disminución de la Tg, probablemente porque la diferencia estructural que posee un grupo metilo y un grupo fenilo es mucho mayor que la que se deduce al comparar un grupo metilo y un grupo etilo. 181 Aunque algunos valores de Tg sean más altos que otros vale destacar que todas las PAs sintetizadas presentan valores de Tg muy inferiores a los de las poli(amida)s comerciales, Kevlar y Nomex. Por su parte, en el análisis de PIs se espera que estas presenten valores de Tg mayores que las PAs de estructura similar, debido a la mayor rigidez de las cadenas poliméricas. Las PIs de la Serie A presentan valores de Tg entre 164 y 184 °C. El valor más alto lo evidencia la PI-IV probablemente debido, al aumento de la rigidez estructural que aportan los anillos aromáticos unidos a cada átomo de silicio, como ya se mencionó anteriormente. Las PIs de la Serie B presentan valores de Tg por encima de 200 °C. Las uniones bifenilos y la cuasi planaridad del heterociclo dibenzofurano deben aumentar, tal como se comentó con anterioridad, la rigidez de las cadenas y las interacciones entre ellas, lo que justificaría el aumento en los valores de la Tg. De hecho, PI-V presenta el mayor valor de Tg entre todas las PIs sintetizadas y es la única que no resultó soluble en los disolventes empleados, como ya se informó en la sección de análisis de solubilidad de las muestras. Si se compara PI-VII y PI-VIII se observa que el fragmento diamina de la unidad repetitiva es el mismo, mientras que la flexibilidad del fragmento dianhídrido de ácido de la PI-VII es mayor que el de la PI-VIII. Esta diferencia de flexibilidad hace que esta última presente un valor de Tg más alto (213 °C) que el de PI-VII (165 °C). Por otra parte, si se compara la estructura de PI-VIII y PI-IX se ve que existe una asimetría estructural en esta última lo que implica, como ya se ha dicho antes, un rompimiento de la regularidad de la cadena y esto hace que aumente la flexibilidad y por lo tanto los valores de Tg sean más bajos. Este fenómeno se evidencia en la PI-IX la cual presenta solo 95 °C de Tg. 182 Todas las PIs sintetizadas presentaron valores de Tg marcadamente menores que la poli(imida) comercial Kapton. Por último, se debe destacar que debido a las modificaciones estructurales tanto en las PAs como en las PIs, se logra ampliar la ventana térmica de trabajo. Esto se traduce en que existe un intervalo de temperaturas amplio entre la Tg y la TDT10% del material, lo cual es altamente positivo desde el punto de vista de la necesaria procesabilidad en aplicaciones tecnológicas. Figura 49. Curvas DSC de las PIs sintetizadas (segundo barrido a 10 °C/min en atmósfera de nitrógeno). 183 IX. TRANSPARENCIA ÓPTICA Esta propiedad se puede cuantificar a partir de la medida de transmitancia utilizando un espectrofotómetro. Básicamente, la transmitancia de una muestra se mide haciendo incidir un haz de luz de intensidad conocida (I0) sobre el material y luego se mide la intensidad de la luz que la atraviesa (I). La fracción I/I0 es lo que se conoce como transmitancia (T). Por lo general, este valor es menor o igual a 1 por lo que si se multiplica por 100, se puede expresar la transmitancia en unidades porcentuales (124). Así, mientras más alto sea este valor, mayor será la transparencia óptica. La transparencia óptica depende de la longitud de onda a la cual se mide. Por ejemplo, un material puede presentar 90 % de transmitancia a 700 nm y 20 % a 400 nm. Debido a esto, esta propiedad se mide haciendo un barrido desde longitudes de onda grandes (1000 nm) hasta longitudes de onda pequeñas (190 nm) y así se detecta a partir de qué longitud de onda comienza a disminuir su transmitancia. Debido a la importancia desde el punto de vista de aplicabilidad, en la mayoría de los trabajos con polímeros se busca que estos presenten una elevada transparencia en la región UV-visible. Más específicamente, un material se considera transparente en esta región, si a 400 nm mantiene más del 80 % de transmitancia (T400). En un polímero aromático, las cadenas se encuentran muy cercanas unas de otras por lo que existe una interacción entre la nube de electrones π conjugados de los anillos, los cuales absorben energía de la región visible y UV-visible. Esta absorción produce color en el material, disminuyendo de esta forma la transparencia óptica. En el caso de las PIs, además de este fenómeno, la formación de los complejos de transferencia de carga también contribuye negativamente a la transparencia óptica (125). Por lo tanto, 184 cualquier modificación estructural que disminuya estas interacciones aumentaría la transparencia del material analizado. La medición de esta propiedad se puede realizar de dos formas. La primera de ellas es haciendo una película delgada a partir del polímero y midiendo directamente la transmitancia. Para poder comparar distintas películas de polímeros, la superficie de estas no debe presentar irregularidades y deben poseer el mismo espesor. De no cumplirse esto último, las medidas deben ser normalizadas, lo cual muchas veces es una operación que realiza directamente el software con el que opera el instrumento. La segunda forma es en disolución, siempre y cuando se preparen concentraciones iguales de los polímeros a medir, se utilice el mismo disolvente y se haga a la misma temperatura. En este caso, el efecto adicional que pueda aportar el disolvente se elimina midiendo con un patrón del disolvente puro (126). Así, la transmitancia medida la aporta solo el soluto. A fin de analizar la transparencia de las muestras obtenidas, tanto las PAs como las PIs que mostraron solubilidad en los análisis respectivos, se trataron con THF y se intentó obtener películas delgadas por evaporación lenta del disolvente. Sin embargo, las películas formadas resultaron frágiles y quebradizas, haciendo dificultosa su manipulación. Este comportamiento es comúnmente observado en polímeros con baja viscosidad inherente, debido al carácter oligomérico de las cadenas. Debido a esto, las mediciones se realizaron en disolución de NMP a 25 °C y a una concentración fija de 0,5 g/L. Los datos obtenidos para las PAs y PIs se resumen en la tabla 9 y los espectros de transmitancia se muestran en la figura 50 y figura 51, respectivamente. 185 Tabla 9. Propiedades ópticas de las PAs y PIs sintetizadas. Poli(amida)s T400 (%) λT= 80% λcutoff (nm) (nm) Serie A Poli(imida)s T400 λT= 80% λcutoff (%) (nm) (nm) Serie A PA-I 92 373 323 PI-I 84 396 318 PA-II 96 359 319 PI-II 84 386 311 PA-III 73 405 317 PI-III 89 386 314 PA-IV 95 369 316 PI-IV 85 370 269 PI-V n.d. n.d. n.d. PI-VI 60 413 334 80 400 335 Serie B PA-V Serie B 78 410 348 Serie C PA-VI 90 384 330 Serie C PA-VII 93 375 329 PI-VII PA-VIII 89 385 328 Serie D PA-IX 91 378 329 PI-VIII 80 400 332 PI-IX 60 466 334 n.d.: No determinado por insolubilidad de la muestra. Como se puede ver, la mayoría de las PAs sintetizadas presentan una elevada transparencia óptica en la región ultravioleta visible, exceptuando PA-III y PA-V, las cuales presentan una moderada transparencia a 400 nm. Las modificaciones estructurales realizadas en las PAs de la Serie A aumentan la flexibilidad de las cadenas lo que hace que las interacciones entre los anillos aromáticos disminuyan, por lo que es de esperar elevados valores de transmitancia. PA-III solo alcanza un 73 % a 400 nm, pero a 410 nm su tranparencia aumenta a 85 %. Es probable que en esta poli(amida) las cadenas presenten un leve aumento en sus interacciones, sin llegar a ser significativo. 186 La PA-V por otro lado, también presenta un valor moderado de la transmitancia a 400 nm y a 410 nm solo alcanza 80 %. Es probable que la planaridad del fragmento diamina aumente las interacciones entre los anillos aromáticos disminuyendo así, la transparencia óptica. Por último, todas las PAs de la Serie C muestran una elevada transmitancia a 400 nm a pesar de la mayor rigidez que presentan según los valores de Tg registrados. Sin embargo, el gran volumen que tiene el fragmento diamina puede estar actuando como separador de cadenas dejando volumen libre por donde puede atravesar el haz de luz. Figura 50. Curvas de transmitancia para las poli(amida)s (c = 0,5 g/L en NMP a 25 °C). La longitud de onda de corte (λcutoff) es un valor que se informa para cada polímero y significa que a partir de ese valor se comienza a observar cierta transparencia, mientras que por debajo de este el material es completamente opaco. En el caso de las PAs, estos valores son similares dentro de una misma Serie. El intervalo en el cual se mueve la longitud de corte registrada es relativamente amplio (316 nm para PA-IV y 348 nm para PA-V). Esto implica que las modificaciones estructurales afectan también el punto en el cual el 187 material deja de transmitir la luz. En esta ocasión coincide que la muestra con menor λcutoff presenta a su vez uno de los mayores valores de transparencia a 400 nm, mientras que la PA con mayor λcutoff evidencia la menor transparencia. Las PIs por lo general, presentan menor transparencia que las poli(amida)s debido a los complejos de transferencia de carga presentes, interacciones que se suman a las evidenciadas por los anillos aromáticos. Como se puede observar en la tabla 9, ninguna PI supera el 90 % de transmitancia al ser analizada a 400 nm. Sin embargo, la presencia de unidades flexibles en las PIs de la Serie A permite que estas alcancen valores entre 84 y 89 %, lo que las clasifica como transparentes. De las PIs de la Serie B, solo se pudo medir la transmitancia a la PI-VI debido a la insolubilidad que presenta la PI-V. Al igual que en la PA-V, la elevada planaridad y rigidez del fragmento diamina contribuye negativamente a la transparencia, por lo que PI-VI solo alcanza un 60 % de transmitancia a 400 nm. Si se registra igual propiedad a 413 nm, dicho valor aumenta a 80 %. PI-VII y PI-VIII presentan igual valor de transmitancia a 400 nm (80 %) quedando así en el límite para ser clasificadas de transparentes. Esto puede deberse a la planaridad que aporta el fragmento diamina en ambos polímeros, lo que podría estar aumentando, en cierto nivel, la formación de complejos de transferencia de carga. La PI-IX de la Serie D presenta un comportamiento anómalo, ya que, tal como se mencionó anteriormente, la falta de simetría que presenta el fragmento diamina contribuye a disminuir la rigidez de la cadena (Tg= 95 °C). Por otra parte, el gran volumen que presenta el fragmento anhídrido de ácido en base a la unidad 9,9´-difenilfluoreno, ayudaría a aumentar la transparencia 188 y sin embargo, esta PI solo presenta un 60 % de transmitancia a 400 nm. Este resultado indicaría que en disolución existe un mayor empaquetamiento de las cadenas, contradictorio al valor tan bajo de Tg registrado en estado sólido. Figura 51. Curvas de transmitancia de las PIs (c = 0,5 g/L NMP a 25 °C). La longitud de onda de corte de las PIs, al igual que lo observado para las PAs, presentan valores cercanos, siendo los más bajos los de la Serie A, familia de polímeros a los que se le asigna la mayor flexibilidad de cadena. Finalmente, en esta tesis fue imposible realizar pruebas mecánicas a los polímeros obtenidos debido a que no forman películas delgadas, ya que su masa molecular es muy baja y estas se quiebran. La ausencia de este análisis no permite proponer aplicaciones específicas a los materiales. Sin embargo, con los resultados obtenidos de las demás propiedades se pudo conocer las relaciones estructura-propiedad de los polímeros, lo que permitirá establecer futuras relaciones en la preparación de nuevos polímeros dentro del Grupo de Investigación. 189 CONCLUSIONES En este trabajo de tesis se sintetizaron y caracterizaron un total de 32 compuestos y 18 polímeros. Los polímeros fueron caracterizados usando técnicas térmicas y ópticas, así como también se midió la viscosidad inherente, se ensayó la solubilidad en diferentes disolventes orgánicos comunes y se determinó la masa molecular de uno de los polímeros usando técnicas de GPC. En general, las modificaciones estructurales realizadas no afectaron la alta resistencia térmica de las PAs y PIs sintetizadas, probablemente el contenido aromático de las unidades difenilsilano y oxiarenos ayudó a mantener dicha resistencia térmica, mientras que sí se logró disminuir los valores de temperatura de transición vítrea, los cuales suelen ser elevados para este tipo de polímeros. La mayoría de las PAs y PIs resultaron ser transparentes en la región UVvisible a 400 nm, demostrando así que la flexibilidad de las cadenas poliméricas, causada por la incorporación de fragmentos flexibles, originó un menor empaquetamiento de los polímeros en disolución. Además, dichas modificaciones también ayudaron a que la mayoría de los polímeros fueran solubles en disolventes orgánicos comunes, incluso en THF, un disolvente que posee una baja temperatura de ebullición, lo que desde el punto de vista industrial es muy favorable. Los valores de viscosidad inherente medidos sirvieron para comparar y estimar el largo de las cadenas poliméricas, dando como resultado que se está frente a cadenas pequeñas, del tipo oligoméricas. Esto se corroboró con un análisis de GPC al polímero que presentó el mayor valor de viscosidad inherente, cuyo resultado fue una masa molecular promedio en número correspondiente a siete unidades repetitivas y una masa molecular promedio en peso correspondiente a cadenas con trece unidades repetitivas. 191 Todos los resultados obtenidos dieron cumplimiento a la hipótesis planteada en este trabajo. Solo hay que destacar que debido a las bajas masas moleculares no se pudo fabricar las películas de polímeros para realizar pruebas mecánicas, por lo que no es posible proponer aplicaciones específicas por la falta de este tipo de análisis, el cual es decisivo para conocer si un material puede ser utilizado y manejado sin que se quiebre. 192 BIBLIOGRAFÍA 1. T. Graham, Trans Roy Soc (London), 151, 183, 1861. 2. W. Otswald, Kolloid Z. 1, 331, 1907. 3. F. M. Raoult, Compt Rend 95, 1030, 1882. 4. J. H. van’t Hoff, Z. Physik Chem 1, 481, 1887. 5. C. Harries, Ber, 37, 2708, 1904. 6. H. Staudinger, Ber, 59 3019, 1926. 7. W. H. Carothers, J Am Chem Soc, 51, 2548, 1929. 8. J. M. Margolis, Engineering Thermoplastics, Ed, Dekker, New York, 1985. 9. H. G. Elias, F. Vohwinkel, New Comercial Polymers. Gordon and Breach, New York, 1986. 10. Polyimides: Synthesis, Characterization and Application, Vols 1 & 2; K. L. Mittal., Ed, Plenum, New York, 1984. 11. T. L. ST. Clair, Structure-Property Relationships in Linear Aromatic Polyimides and Polyimides, D. Wilson, H. D. Stenzenberger, P. M. Hergenrother, Eds., Blackie & Son, Glasgow, 1990. 12. J. G. de la Campa, J. de Abajo, Ciencia y Tecnología de Materiales Poliméricos., Vol II., Cap. 3., Ed. Instituto de Ciencia y Tecnología de Polímeros (CSIC), Madrid, 2004. 13. J. M. García, F. C. García, F. Serna, J. L. de la Peña, Progress Polym Sci, 35, 623, 2010. 14. N. Yamazaki, M. Matsumot, F. Higashi, J Polym Sci Polym Chem Ed, 13, 1373, 1975. 15. Y. Kubota, S. Nakada, Y. Sugi, Mater Trans, 43, 326, 2002. 16. G. Rabani, A. Kraft, Macromol Rapid Commun, 23, 375, 2002. 17. R. J. Gaymans. Polyamides, Ed. Rogers ME and Long TE. Synthetic Methods in Step-growth Polymers. Hoboken: John Wiley & Sons, Inc. New Jersey, pp.135–196, 2003. 18. J. K. Fink, High Performance Polymers, William Andrew Inc., New York, 2008. 194 19. H. Sekiguchi, B. Coutin, Polyamides. Ed Kricheldorf HR. Handbook of Polymer Synthesis. Part A, Marcel-Dekker Inc., 807–939, cap. 14, New York, 1992. 20. L. Vollbracht, Aromatic Polyamides. Ed Allen G, Bevington B, Eastmond GV, Ledwith A, Russo S, Sigwald P. Comprehensive Polymer Science, vol. 5, Pergamon Press, pp. 373–83 cap. 22, Oxford, 1989. 21. J. Gallini. Aromatic Polyamides. Encyclopedia of Polymer Science and Technology, vol. 3, John Wiley & Sons, pp. 558–584, New York, 2005 22. S. Ozawa, K. Matsuda. Aramid Copolymer Fibers. Ed, Lewin M, Preston J, Handbook of Fiber Science and Technology. Vol III. High Technology Fibers. Part B, Marcel-Dekker Inc. pp. 1–34, cap. 1, New York, 1989. 23. D. Tanner, J. A. Fitzgerald, P. G. Riewald, W. F. Knoff. Aramid Structure/ Property Relationships and their role in Applications Development. Ed, Lewin M, Preston J. Handbook of Fiber Science and Technology. Vol III. High Technology Fibers. Part B, Marcel- Dekker Inc., pp. 35–80, cap. 2, New York, 1989. 24. H. Yarn, Nomex Aramid Fiber. Ed, Lewin M, Preston J. Handbook of Fiber Science and Technology. Vol III. High Technology Fibers. Part C, MarcelDekker Inc., pp. 77–178, cap. 2, New York, 1993. 25. S. Shah, Solution processible aromatic polyimides via diels alder precursor., A Master Thesis in Industrial Chemistry in the Department of Chemistry in the College of Sciences at the University of Central Florida Orlando, Florida, 2008. 26. H. Lee, Stoffey D, Neville K, New Linear Polymers. McGraw-Hill, New York, 1967. 27. W. Edwards, I. Robinson, US Pat 2,710,853, 1955. 28. C. Sroog, J Polym Sci C, 16, 1191, 1967. 29. Y. Watanabe, Y. Sakai, Y. Shibasaki, S. Ando, M. Ueda, Y. Oish., K. Mori, Macromolecules, 35, 2277, 2002. 30. A. Mathews, L. Kim, C. Ha, Macromol Res, 15(2), 114, 2007 195 31. C. Sroog, Polyimides. Ed, Mark F, Gaylord NG, Bikales NM, Encyclopedia of Polymer Science and Technology, vol 11. Interscience, New York, pp. 247, 1969. 32. Z. Ge, L. Fan, S. Yang, Eur Polym J, 44, 1252, 2008. 33. S. Hsiao, W. Chen, J Polym Sci Part A Polym Chem, 41, 420, 2003. 34. S. Hsiao, C. Yang, J Polym Sci Part A Polym Chem, 35, 1487, 1997. 35. S. Hsiao, C. Yang, K. Chu, J Polym Sci Part A Polym Chem, 35, 1469, 1997. 36. S. Hsiao, Y. Chang, J Polym Sci Part A Polym Chem, 42, 4056, 2004. 37. C. Yang, Y. Su, Polymer, 46, 5797, 2005. 38. M. Madhra, M. Sharma, S. Banerjee, J Appl Polym Sci, 93, 235, 2004. 39. D. Liaw, J Polym Sci Part A Polym Chem, 43, 4559, 2005. 40. Y. Chen, C. Yang, S. Hsiao, Macromol Chem Phys, 207, 1888, 2006. 41. S. Banerjee, M. Madhra, V. Kute, J Appl Polym Sci, 93, 821, 2004. 42. J. E, Mark, in Silicon-Based Polymer Science, Advances in Chemistry Series 224, Ed, J.M. Zeigler and F. W. G. Fearon. American Chemical Society, Washington. DC, 1990. 43. M. Zeldin. K.J. Wynne and H. R. Alleock, Inorganic and Organometallic Polymers. ACS Symposium Series 360, American Chemical Society, Washington. DC, 1988. 44. W Noll. Chemistry and Technology of the Silicones. Academic Press, New York, 1968. 45. W. J. Bobear, in Rubber Technology, Ed, M. Morton. Van Nostrand Reinhold, New York, 1973. 46. A. L. Smith. Analysis of Silicones. Krieger, Malabar. FL. 1983. 47. E. L. Warrick, O. R. Pierce. K. E. Polmanteer. and J. C. Saam, Rubber Chem Technol, 52, 437, 1979. 48. E. G. Rochow, Silicon and Silicones, Springer-Verlag, Berlin, 1987. 49. S. Thames, K. Panjnani, J Inorg Organometall Polym, 6(2), 59, 1996. 50. Y. Vignollet and J. C. Marie. J Organomet Chem, 17, 43, 1969. 196 51. J. Y. Jadhav. Makromol Chem Rapid Commun, 6(7), 457, 1985. 52. M. Bruma, B. Schulz, T. Hopnick, B. Stiller, N. Belomoina, F. Mercer, Eur Polym J, 35, 1253, 1996. 53. I. Sava, M. Bruma, B. Schulz, F. Mercer, V. Reddy, N. Belomoina, J Appl Polym Sci, 65, 1533, 1997. 54. I. Sava, M. Szesztay, M. Bruma, F. Mercer, B. Schulz, Angew Makromol Chem, 253, 169, 1997. 55. M. Iosip, M. Bruma, J. Robinson, Y. Kaminorz, B. Schulz, High Perform Polym, 13, 133, 2001. 56. E. Hamciuc, B. Schulz, T. Kopnick, M. Bruma, High Perform Polym, 14(1), 63, 2002. 57. M. Bruma, E. Hamciuc, I. Sava, N. Belomoina, Russ Chem Bull Intern Ed, 53(9), 1813, 2004. 58. M. Bruma, Revue Roum Chim, 52(4), 309, 2007. 59. S. H. Yi. J. Nagase, H. Sato. Synth Met, 58(3) 353, 1993. 60. I. Sava, E. Hamciuc, Revue Roum Chim, 52(1-2), 127, 2007. 61. I. Sava, M. Bruma, B. Schulz, T. Kopnick, High Perform Polym, 17, 438, 2005. 62. M. Bruma, B. Schulz, T. Kopnick, J. Robinson, High Perform Polym, 12, 429, 2000. 63. B. Lin, Y. Pan, Y. Qian, C. Yuan, J Appl Polym Sci, 94, 2363, 2004. 64. S. A. Stern and R. Vaidyanathan. J Membr Sci, 49, 1, 1990. 65. W. Davidsohn, B. Laliberte, C. Goddard, C. Henry, J Organomet Chem, 36, 283, 1972. 66. B. Venkataramanan, Cristal Growth & Design, 4(3), 553, 2004. 67. A. Hilal, R. Steven, L. LaBrenz, L. Woo, C. Serpell, W. Jeffery, J Am Chem Soc, 122, 5262, 2000. 68. T. Matsumoto, K. Nishimura, T. Kurosaki, European Polymer Journal, 35, 1529, 1999. 197 69. W. Zhonggang, Z. Bufeng, Aromatic cyanate ester monomer containing silicon and preparation method thereof; Patente, CN101585850 (A), 2009. 70. H. Arishima, H. Hotaka, Polymerizable compound, photocurable composition, optical element and optical head device. Patente. WO2009139476 (A1), 2009. 71. C. A. Terraza; L. H. Tagle; F. Concha; L. Poblete, Desig Monom Polym 10(3), 253, 2007. 72. J. March, Advanced Organic Chemistry, 3rd edition, Wiley-Interscience, New York, pp. 478, 1985. 73. P. Arvind, Z. Sainath, V. Arcana, S. Kalyankar, Inter Multidisciplinary Res J, 2(6), 07, 2012. 74. W. Kimmler, J Amer Chem Soc, 60, 855, 1938. 75. L. Jurd, J Sci Res 2A, 111, 1949; 3A, 587, 1950. 76. Y. Ogata, K. Nakajima, Tetrahedron, 20, 2751, 1960. 77. A. R. Butler, J Chem Educ, 36, 508, 1971. 78. H. Gilman, G. E. Brown, W. G. Bywater, W. H. Kirkpatrick; J Am Chem Soc, 56(11), 2473, 1934. 79. V. Steven, W. Andrew, Angew Chem Int Ed, 42, 5400, 2003. 80. W. Schwartz, United State Patent: 4827000, 1989. 81. J. McMurry, Organic Chemistry, Fifth Edition, Jennifer Huber, Chapter 11, Pacific Grobe, CA, pp. 398, 2000. 82. The Periodic Table on the web http://www.webelements.com (accessed Dic, 2012). 83. J. Bunnett, R. Zahler, Chem Rev, 49, 273, 1951. 84. J. Bunnett, W. Edgar, Jr. Garbisch, M. Kenneth, J Amer Chem Soc, 77, 340, 1955. 85. J. Speier, J Org Chem, 74, 1003, 1952. 86. B. Han, D. Shin, S. Cho, Tetrahedron Lett, 26(50), 6233, 1985. 87. P. N. Rylander, Hydrogenation of Nitro Compounds. Hydrogenation Methods, Academic Press, New York, 1985. 198 88. J. McMurry, Organic Chemistry, Fifth Edition, Jennifer Huber, Chapter. 21, Pacific Grobe, CA, pp. 872, 876, 2000. 89. C. A. Terraza, J-G. Liu, Y. Nakamura, Y. Shibasaki, S. Ando, M. Ueda, J Polym Sci Part A Polym Chem, 46, 1510, 2008. 90. Z. Gomurashvili, S. Allan, J Macromol Sci Part A-Pure and Appl Chem, 42,127, 2005. 91. F. Williams, H. Relles, P. Donahue, J. Manello, J Org Chem, 42(21), 3425, 1977. 92. A. Nas, H. Kantekin, Polyhedron 30, 1085, 2011. 93. D. Liaw, B. Yang, P. Hsu, C. Hwang, Chem Mater, 13, 1811, 2001. 94. T. J. Broxton, D. M. Muir, A. J. Parker, J Org Chem, 40(22), 3230, 1975. 95. T. J. Broxton, D. M. Muir, A. J. Parker, J Org Chem, 40(14), 2037, 1975. 96. P. García, S. Vázquez, C. Escolano, Fundamentos de Síntesis de Fármacos, Universidad de Barcelona, 1 Ed, Cap 4, 175, 2005. 97. H. Beyer, W. Walter; Manual de Química Orgánica, Reverté S. A. 19a Ed, Cap. 1, pp. 168, Barcelona, 1987. 98. T. Keumi, K. Hamanaka, H. Hasegawua, N. Minamide, Y. Inoue, H. Kitajima, Chem Lett, 17(8), 1285, 1988. 99. R. J. P. Corriu, J. P. Masse, J Chem Soc Chem Commun, 144, 1972. 100. K. Tamao, K. Sumitani, M. Kumada, J Am Chem Soc, 94, 4374, 1972. 101. M. Norio; S, Akira, Chem Rev 95(7), 2457, 1995. 102. S, Akira, J Organomet Chem, 576, 147, 1999. 103. G. Smith, G. Dezeny, D. Hughes, A. King, T. Verhoeven, J Org Chem, 59, 8151, 1994. 104. A. Christian, J. Anny, Acc Chem Res, 33, 314, 2000. 105. H. Gilman, R. Young, J Am Chem Soc, 56(6), 1415, 1934. 106. J. R. Pratt, S. Thames, Journal of Organic Chemistry, 38(26), 4271, 1973. 107. F. Higashi, M. Goto, H. Kakiniki, J Polym Sci Chem Ed, 18, 1711, 1980. 199 108. S. M. Aharoni, W. B. Hammond, J. S. Szobota, D. Masilamani, J Polym Sci Chem Ed, 22, 2579, 1984. 109. L. H. Tagle, C. A. Terraza, L Lopez, A. Leiva, J Chil Chem Soc, 51, 1041, 2006. 110. L. H. Tagle, F. Diaz, J. Vega, P. Valenzuela, Eur Polym J, 39, 407, 2003. 111. J. E. McGrath, Polymers Synthesis. Encyclopedia of Physical Science and Technology, Vol. 13; Academic Press, New York, 1992. 112. F. W. Harris, Polyimides. Synthesis of Aromatic Polyimides from Dianhydrides and Diamines. Ed. D. Wilson, H. D. Stenzenberger, P. M. Hergenrother, Blackie, Glasgow, 1990. 113. M. Koton, V. Kudryavtsev, N. Adrova, K. Kalnin, A. Dubnova, V. Svetlichnyi, Polym Sci USSR, 16(9), 2411, 1974. 114. M. Kailani, C. Sung, Macromolecules, 31, 5771, 1998. 115. J. Dean, Langes Handbook of Chemistry, 13th Ed.; McGraw-Hill, Tables 5-8, Section 5, New York, 1985. 116. F. W. Billmeyer, Ciencia de los Polímeros. Ed. Reverté S. A. España. pp 86. Barcelona, 2004. 117. S. Mori, H. G. Barth, Size Exclusion Chromatography; Springer-Verlag, Berlín-Heidelberg, 1999. 118. C. M. Hansen, Hansen Solubility Parameters: A User’s Handbook; CRC Press, New York, 2000. 119. J. E. Mark, Physical Properties of Polymers Handbook, Woodbury, New York, 1996. 120. S. Mardosky, S. Straus, J Res Natl Bur Standards, 53, 361, 1954. 121. C. L. Beyler, Handbook of Fire Protection Engineering, Chapter 7, 3rd Ed, M. Marcelo, Philadelphia, 2001. 122. W. Xie, W. Pan, K. C. Chuang, Journal of Thermal Analysis and Calorimetry, 64, 477, 2001. 123. B. Vollmert, Polymer Chemistry; Springer-Verlag, Berlín, 1973. 200 124. D. Skoog, M. West, J. Holler, Fundamentos de Química Analítica, Editorial Reverte S. A. 4ta Ed. Barcelona, 2003. 125. H. Sheng-Hsiang, H. Hsin-Ling, L. Jihperng, Seminar papers, http://www.mse.nctu.edu.tw/people/writing_seminar.php?Sn=50. 126. J. W. Verhoeven, Pure and Appl Chem, 68(12), 2223, 1996. 201 ANEXOS Bis(4-hidroxifenil)dimetilsilano (1): 5 CH3 HO Si 3 4 CH3 Figura 1. Espectro FTIR del compuesto 1. Figura 2. Espectro RMN 1H del compuesto 1. 203 2 1 OH 6 Figura 3. Espectro RMN 13C del compuesto 1. Figura 4. Espectro RMN 29Si del compuesto 1. 204 Bis(4-hidroxifenil)difenilsilano (2): Figura 5. Espectro FTIR del compuesto 2. Figura 6. Espectro RMN 1H del compuesto 2. 205 Figura 7. Espectro RMN 13C del compuesto 2. Figura 8. Espectro RMN 29Si del compuesto 2. 206 4-bromofenilbencil éter (3): Figura 9. Espectro FTIR del compuesto 3. Figura 10. Espectro RMN 1H del compuesto 3. 207 Figura 11. Espectro RMN 13C del compuesto 3. 208 Bis(4-(benciloxi)fenil)dimetilsilano (4): Figura 12. Espectro FTIR del compuesto 4. Figura 13. Espectro RMN 1H del compuesto 4. 209 Figura 14. Espectro RMN 13C del compuesto 4. Figura 15. Espectro RMN 29Si del compuesto 4. 210 Bis(4-(benciloxi)fenil)difenilsilano (5): Figura 16. Espectro FTIR del compuesto 5. Figura 17. Espectro RMN 1H del compuesto 5. 211 Figura 18. Espectro RMN 13C del compuesto 5. Figura 19. Espectro RMN 29Si del compuesto 5. 212 2,8-diyododibenzofurano (6): Figura 20. Espectro FTIR del compuesto 6. Figura 21. Espectro RMN 1H del compuesto 6. 213 Figura 22. Espectro RMN 13C del compuesto 6. 214 4-(4-bromofenoxi)nitrobenceno (A): Figura 23. Espectro FTIR del compuesto A. Figura 24. Espectro RMN 1H del compuesto A. 215 Figura 25. Espectro RMN 13C del compuesto A. 216 Ácido 4-(4-bromofenoxi)ftálico: Figura 26. Espectro FTIR del compuesto ácido 4-(4-bromofenoxi)ftálico. Figura 27. Espectro RMN 1H del compuesto ácido 4-(4-bromofenoxi)ftálico. 217 Figura 28. Espectro RMN 13C del compuesto ácido 4-(4-bromofenoxi)ftálico. 218 Ácido 4-(4-bromofenoxi)ftálico (B): Figura 29. Espectro FTIR del compuesto B. Figura 30. Espectro RMN 1H del compuesto B. 219 Figura 31. Espectro RMN 13C del compuesto B. 220 Cloro (3,4-dimetilfenil)dimetilsilano (C): Figura 32. Espectro FTIR del compuesto C. Figura 33. Espectro RMN 1H del compuesto C. 221 Figura 34. Espectro RMN 13C del compuesto C. Figura 35. Espectro RMN 29Si del compuesto C. 222 Bis(4-(4-nitrofenoxi)fenil)dimetilsilano (7): Figura 36. Espectro FTIR del compuesto 7. Figura 37. Espectro RMN 1H del compuesto 7. 223 Figura 38. Espectro RMN 13C del compuesto 7. Figura 39. Espectro RMN 29Si del compuesto 7. 224 Bis(4-(4-nitrofenoxi)fenil)difenilsilano (8): Figura 40. Espectro FTIR del compuesto 8. Figura 41. Espectro RMN 1H del compuesto 8. 225 Figura 42. Espectro RMN 13C del compuesto 8. Figura 43. Espectro RMN 29Si del compuesto 8. 226 Bis(4-(4-aminofenoxi)fenil)dimetilsilano (9): Figura 44. Espectro FTIR del compuesto 9. Figura 45. Espectro RMN 1H del compuesto 9. 227 Figura 46. Espectro RMN 13C del compuesto 9. Figura 47. Espectro RMN 29Si del compuesto 9. 228 Bis(4-(4-aminofenoxi)fenil)difenilsilano (10): Figura 48. Espectro FTIR del compuesto 10. Figura 49. Espectro RMN 1H del compuesto 10. 229 Figura 50. Espectro RMN 13C del compuesto 10. Figura 51. Espectro RMN 29Si del compuesto 10. 230 Bis(4-(4-cianofenoxi)fenil)dimetilsilano (11): Figura 52. Espectro FTIR del compuesto 11. Figura 53. Espectro RMN 1H del compuesto 11. 231 Figura 54. Espectro RMN 13C del compuesto 11. Figura 55. Espectro RMN 29Si del compuesto 11. 232 Bis(4-(4-cianofenoxi)fenil)difenilsilano (12): Figura 56. Espectro FTIR del compuesto 12. Figura 57. Espectro RMN 1H del compuesto 12. 233 Figura 58. Espectro RMN 13C del compuesto 12. Figura 59. Espectro RMN 29Si del compuesto 12. 234 Bis(4-(4-carboxifenoxi)fenil)dimetilsilano (13): Figura 60. Espectro FTIR del compuesto 13. Figura 61. Espectro RMN 1H del compuesto 13. 235 Figura 62. Espectro RMN 13C del compuesto 13. Figura 63. Espectro RMN 29Si del compuesto 13. 236 Bis(4-(4-carboxifenoxi)fenil)dimetilsilano (14): Figura 64. Espectro FTIR del compuesto 14. Figura 65. Espectro RMN 1H del compuesto 14. 237 Figura 66. Espectro RMN 13C del compuesto 14. Figura 67. Espectro RMN 29Si del compuesto 14. 238 Bis(4-(3,4-dicianofenoxi)fenil)dimetilsilano (15): Figura 68. Espectro FTIR del compuesto 15. Figura 69. Espectro RMN 1H del compuesto 15. 239 Figura 70. Espectro RMN 13C del compuesto 15. Figura 71. Espectro RMN 29Si del compuesto 15. 240 Bis(4-(3,4-dicianofenoxi)fenil)difenilsilano (16): Figura 72. Espectro FTIR del compuesto 16. Figura 73. Espectro RMN 1H del compuesto 16. 241 Figura 74. Espectro RMN 13C del compuesto 16. Figura 75. Espectro RMN 29Si del compuesto 16. 242 Ácido bis(4-(3,4-dicarboxifenoxi)fenil)dimetilsilano (17): Figura 76. Espectro FTIR del compuesto 17. Figura 77. Espectro RMN 1H del compuesto 17. 243 Figura 78. Espectro RMN 13C del compuesto 17. Figura 79. Espectro RMN 29Si del compuesto 17. 244 Ácido bis(4-(3,4-dicarboxifenoxi)fenil)difenilsilano (18): Figura 80. Espectro FTIR del compuesto 18. Figura 81. Espectro RMN 1H del compuesto 18. 245 Figura 82. Espectro RMN 13C del compuesto 18. Figura 83. Espectro RMN 29Si del compuesto 18. 246 Ánhídrido bis(4-(3,4-dicarboxifenoxi)fenil)dimetilsilano (19): Figura 84. Espectro FTIR del compuesto 19. Figura 85. Espectro RMN 1H del compuesto 19. 247 Figura 86. Espectro RMN 13C del compuesto 19. Figura 87. Espectro RMN 29Si del compuesto 19. 248 Ánhídrido bis(4-(3,4-dicarboxifenoxi)fenil)difenilsilano (20): Figura 88. Espectro FTIR del compuesto 20. Figura 89. Espectro RMN 1H del compuesto 20. 249 Figura 90. Espectro RMN 13C del compuesto 20. Figura 91. Espectro RMN 29Si del compuesto 20. 250 Mezcla de 2,7-dinitrodibenzofurano y 2,8-dinitrodibenzofurano: Figura 92. Espectro FTIR de la mezcla de 2,7 y 2,8-dinitrodibenzofurano. 251 2,7-diaminodibenzofurano (21): Figura 93. Espectro FTIR del compuesto 21. Figura 94. Espectro RMN 1H del compuesto 21. 252 Figura 95. Espectro RMN 13C del compuesto 21. 253 2,8-diaminodibenzofurano (22): O 5 4 6 3 2 H2N 1 Figura 96. Espectro FTIR del compuesto 22. Figura 97. Espectro RMN 1H del compuesto 22. 254 7 NH2 Figura 98. Espectro RMN 13C del compuesto 22. 255 2,8-di(4-nitrofenil)dibenzofurano (23): Figura 99. Espectro FTIR del compuesto 23. 256 2,8-di(4-aminofenil)dibenzofurano (24): Figura 100. Espectro FTIR del compuesto 24. Figura 101. Espectro RMN 1H del compuesto 24. 257 Figura 102. Espectro RMN 13C del compuesto 24. 258 9,9-bis(4-(4-nitrofenoxi)-3-metilfenil)fluoreno (25): Figura 103. Espectro FTIR del compuesto 25. 259 9,9-bis(4-(4-aminofenoxi)-3-metilfenil)fluoreno (26): 19 2 H2N 3 18 CH3 CH3 6 O 5 7 11 13 8 4 O 10 9 12 14 15 17 16 Figura 104. Espectro FTIR del compuesto 26. Figura 105. Espectro RMN 1H del compuesto 26. 260 1 NH2 Figura 106. Espectro RMN 13C del compuesto 26. 261 9,9-bis(4-(3,4-dicianofenoxi)-3-metilfenil)fluoreno (27): Figura 107. Espectro FTIR del compuesto 27. Figura 108. Espectro RMN 1H del compuesto 27. 262 Figura 109. Espectro RMN 13C del compuesto 27. 263 Ácido 9,9-bis(4-(3,4-dicarboxifenoxi)-3-metilfenil)fluoreno (28): Figura 110. Espectro FTIR del compuesto 28. Figura 111. Espectro RMN 1H del compuesto 28. 264 Figura 112. Espectro RMN 13C del compuesto 28. 265 Anhídrido 9,9-bis(4-(3,4-dicarboxifenoxi)-3-metilfenil)fluoreno (29): Figura 113. Espectro FTIR del compuesto 29. Figura 114. Espectro RMN 1H del compuesto 29. 266 Figura 115. Espectro RMN 13C del compuesto 29. 267 PA-I: Figura 116. Espectro FTIR de la PA-I. Figura 117. Espectro RMN 1H de la PA-I. 268 Figura 118. Espectro RMN 13C de la PA-I. Figura 119. Espectro RMN 29Si de la PA-I. 269 PA-II: Figura 120. Espectro FTIR de la PA-II. Figura 121. Espectro RMN 1H de la PA-II. 270 Figura 122. Espectro RMN 13C de la PA-II. Figura 123. Espectro RMN 29Si de la PA-II. 271 PA-III: Figura 124. Espectro FTIR de la PA-III. Figura 125. Espectro RMN 1H de la PA-III. 272 Figura 126. Espectro RMN 13C de la PA-III. Figura 127. Espectro RMN 29Si de la PA-III. 273 PA-IV: Figura 128. Espectro FTIR de la PA-IV. Figura 129. Espectro RMN 1H de la PA-IV. 274 Figura 130. Espectro RMN 13C de la PA-IV. Figura 131. Espectro RMN 29Si de la PA-IV. 275 PA-V: Figura 132. Espectro FTIR de la PA-V. Figura 133. Espectro RMN 1H de la PA-V. 276 Figura 134. Espectro RMN 13C de la PA-V. Figura 135. Espectro RMN 29Si de la PA-V. 277 PA-VI: Figura 136. Espectro FTIR de la PA-VI. Figura 137. Espectro RMN 1H de la PA-VI. 278 Figura 138. Espectro RMN 13C de la PA-VI. Figura 139. Espectro RMN 29Si de la PA-VI. 279 PA-VII: Figura 140. Espectro FTIR de la PA-VII. Figura 141. Espectro RMN 1H de la PA-VII. 280 Figura 142. Espectro RMN 13C de la PA-VII. Figura 143. Espectro RMN 29Si de la PA-VII. 281 PA-VIII: Figura 144. Espectro FTIR de la PA-VIII. Figura 145. Espectro RMN 1H de la PA-VIII. 282 Figura 146. Espectro RMN 13C de la PA-VIII. Figura 147. Espectro RMN 29Si de la PA-VIII. 283 PA-IX: Figura 148. Espectro FTIR de la PA-IX. Figura 149. Espectro RMN 1H de la PA-IX. 284 Figura 150. Espectro RMN 13C de la PA-IX. Figura 151. Espectro RMN 29Si de la PA-IX. 285 PI-I: Figura 152. Espectro FTIR de la PI-I. Figura 153. Espectro RMN 1H de la PI-I. 286 Figura 154. Espectro RMN 13C de la PI-I. Figura 155. Espectro RMN 29Si de la PI-I. 287 PI-II: Figura 156. Espectro FTIR de la PI-II. Figura 157. Espectro RMN 1H de la PI-II. 288 Figura 158. Espectro RMN 13C de la PI-II. Figura 159. Espectro RMN 29Si de la PI-II. 289 PI-III: Figura 160. Espectro FTIR de la PI-III. Figura 161. Espectro RMN 1H de la PI-III. 290 Figura 162. Espectro RMN 13C de la PI-III. Figura 163. Espectro RMN 29Si de la PI-III. 291 PI-IV: 22 21 26 20 17 16 19 * O Si 18 13 15 O 14 12 25 O 11 O O 28 24 N 9 23 O Si 10 8 3 7 O 1 4 Figura 165. Espectro RMN 1H de la PI-IV. 292 27 O n 5 Figura 164. Espectro FTIR de la PI-IV. * N 2 6 Figura 166. Espectro RMN 13C de la PI-IV. Figura 167. Espectro RMN 29Si de la PI-IV. 293 PI-V: Figura 168. Espectro FTIR de la PI-V. 294 PI-VI: Figura 169. Espectro FTIR de la PI-VI. Figura 170. Espectro RMN 1H de la PI-VI. 295 Figura 171. Espectro RMN 13C de la PI-VI. Figura 172. Espectro RMN 29Si de la PI-VI. 296 PI-VII: Figura 173. Espectro FTIR de la PI-VII. Figura 174. Espectro RMN 1H de la PI-VII. 297 Figura 175. Espectro RMN 13C de la PI-VII. Figura 176. Espectro RMN 29Si de la PI-VII. 298 PI-VIII: Figura 177. Espectro FTIR de la PI-VIII. Figura 178. Espectro RMN 1H de la PI-VIII. 299 Figura 179. Espectro RMN 13C de la PI-VIII. 300 PI-IX: Figura 180. Espectro FTIR de la PI-IX. Figura 181. Espectro RMN 1H de la PI-IX. 301 Figura 182. Espectro RMN 13C de la PI-IX. 302 PUBLICACIONES El presente trabajo de Tesis generó, hasta el momento, un total de 3 publicaciones en revistas indexadas. Estas publicaciones se relacionan con algunos de los monómeros sintetizados en dicho trabajo. Actualmente, se encuentra en proceso de escritura 4 publicaciones adicionales con los resultados finales de la Tesis. “Poly(amide)s obtained from 4-(4-((4-(4-aminophenoxy)phenyl)diphenylsilyl) phenoxy)benzenamine and dicarboxylic acids containing diphenylsilarylene units. Synthesis and characterization” Terraza, Claudio A.; Tagle, Luis H.; Tundidor-Camba, Alain; Gonzalez-Henriquez, Carmen; Ortiz, Pablo; Coll, Deysma. European Polymer Journal, 48 (3), 649, 2012 “Synthesis of poly(urethane)s based on diphenyl-sylane/germane and oxyphenyl units. Structure-properties relationship” A. Tundidor-Camba, C. A. Terraza, L. H. Tagle, D. Coll, P. Ortiz. Journal of Applied Polymer Science. 124 (2), 1036 -1041. 2012 “Poly(amide)s obtained by direct polycondesation of 4,4'-(4,4'-(9h-fluorene9,9-diyl)bis(2-methyl-4,1-phenylene))bis(oxy)dibenzenamine with dicarboxylic acids based on a diphenyl-silane moiety” A. Tundidor-Camba, C. A. Terraza, L. H. Tagle, D. Coll. Journal of Applied Polymer Science. 120 (4); 2381 – 2389, 2011 304