Clinical Slides (Diapositivas Clinicas) PDF version
Anuncio
 PDF version")
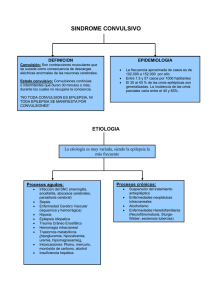
Una crisis epiléptica
p p
es la manifestación de la excitación anormal y
sincronizada de una población de neuronas corticales. Esta
descarga puede producir síntomas subjetivos o signos objetivos,
en cuyo caso se trata de una crisis epiléptica clínica, y es aparente
sólo en un electroencefalograma (EEG), en cuyo caso se trata de
una crisis electroencefalográfica (o subclínica).
Una crisis epiléptica
p p
es la manifestación de una descarga
g
anormal, hipersincrónica de una población de neuronas corticales.
Esta descarga puede producir síntomas subjetivos o signos
objetivos, en cuyo caso se trata de una crisis con manifestación
clínica, o puede ser aparente sólo en un electroencefalograma
(EEG), en cuyo caso se trata de una descarga subclínica
(electroencefalográfica).
La incidencia de crisis de nueva aparición en la población general es de aproximadamente
80 por cada 100.000 habitantes al año, aproximadamente el 60% de estas, serán
pacientes con epilepsia, o sea aquellos con una tendencia hacia la recurrencia de crisis
no provocadas.
Al menos se requieren de dos crisis no provocadas para hacer el diagnóstico de epilepsia.
En el pasado, los médicos se mostraban renuentes a hacer el diagnóstico, incluso
después de crisis repetidas, debido a las consecuencias adversas, incluida la
estigmatización social y las limitaciones en la conducción de vehículos y la consecución
d l empleo.
del
l
A pesar d
de llos avances en lla comprensión
ió social
i ld
de lla enfermedad,
f
d d estos
t
estigmas siguen siendo vigentes. El eufemismo de trastorno convulsivo ha sido empleado
con frecuencia para evitar el de epilepsia y también puede ser usado para referirse a
situaciones que se caracterizan por crisis epileptiformes recurrentes que son provocadas
por un estímulo identificable, por ejemplo, las convulsiones febriles. La definición actual de
la epilepsia es la manifestación de crisis repetitivos (al menos dos) como consecuencia de
un trastorno cerebral, es decir, eventos no provocados por una afección sistémica o
cerebral aguda
aguda. Esta definición hace hincapié en que el problema es en las funciones
cerebrales, y que el paciente tiene el potencial para tener más crisis. Esta definición
excluye los crisis debidas a factores exógenos, como el etanol o la suspensión de un
fármaco sedante o trastornos metabólicos, como la hiperglucemia no cetósica.
Las estimaciones de la incidencia anual de epilepsia la ponen en el rango de la población
general de 30 a 57 por 100.000 habitantes. Estas tasas varían con la edad, siendo más
elevada en los lactantes y niños pequeños, con una disminución en la edad adulta hasta
aproximadamente los 60 años,
años cuando nuevamente comienza a aumentar
aumentar. La prevalencia
global de la epilepsia es de aproximadamente 6 por 1000.
Las crisis epilépticas pueden ser clasificadas en función de sus características clínicas y
electroencefalográficas. El diagnóstico de epilepsia en el paciente se basa en
su historia clínica y el tipo (s) de crisis que padece.
Las crisis parciales tienen inicio en una parte del cerebro; sinónimos que se utilizan con
frecuencia incluyen ataques relacionados con la localización o focales. Las convulsiones
parciales se dividen en dos tipos principales, dependiendo de si la conciencia está
completamente preservada. Durante las crisis parciales simples, la conciencia se
conserva, la persona está alerta, puede responder a preguntas u órdenes, y puede
recordar lo que ocurrió durante el ataque. Durante las crisis parciales complejas, la
conciencia se altera o se pierde, la capacidad de prestar atención o responder a preguntas
u órdenes se deteriora o se pierde. A menudo, no hay memoria de lo sucedido durante
t d o parte
toda
t de
d la
l crisis
i i parcial
i l compleja.
l j L
La di
distinción
ti ió entre
t llas crisis
i i parciales
i l simples
i l y
complejas es fundamental ya que las actividades tales como conducir y operar maquinaria
peligrosa deben ser restringidas en pacientes con crisis parciales complejas que no estén
controladas (las restricciones para las personas con crisis parciales simples sólo dependen
de las manifestaciones específicas de la crisis y de las leyes y normas para conducir de
acuerdo al estado o país en particular). Crisis de inicio parcial pueden progresar a
convulsiones generalizadas secundariamente. Estas implican actividad motora de ambos
lados del cuerpo y puede ser difícil de distinguir de las convulsiones generalizadas
primarias.
La amplia gama de crisis parciales simples da lugar a problemas de diagnóstico. Por
ejemplo, parestesias (sensación de hormigueo) en el quinto dedo que se extienden hacia
el antebrazo puede ser resultado de una convulsión, la migraña, ataque isquémico
transitorio, o trastorno del nervio cubital. Malestar abdominal repentino puede ser
producido por un trastorno gastrointestinal, así como por una crisis derivada de las
estructuras del cerebro que inervan la función autónoma o visceral. Cuando se producen
de forma aislada, estos síntomas pueden no ser reconocidos como ataques por parte del
paciente o el médico.
Las convulsiones motoras alteran la actividad muscular. La postura localizada tónica
(endurecimiento) o movimientos clónicos (espasmos y sacudidas) pueden ocurrir. Los
movimientos anormales pueden limitarse a una parte del cuerpo o se refieren a la
extensión gradual a zonas adyacentes en el mismo lado del cuerpo (convulsion
jacksoniana) o ambos lados del cuerpo con pérdida de la conciencia (convulsión
generalizada secundariamente).
Las crisis epilépticas que ocurren en la corteza sensorial puede producir convulsiones
sensoriales que se manifiestan como alucinaciones o ilusiones, por ejemplo, una
sensación de algo que no está allí o falsear el juego de una verdadera sensación. Las
alucinaciones pueden quedar restringidas a un área (por ejemplo, parestesias en un dedo)
o extenderse a otras áreas (por ejemplo, toda la extremidad superior o lateral todo en una
marcha jacksoniana sensorial). Alucinaciones e ilusiones pueden afectar a cualquier
modalidad sensorial,
sensorial incluyendo el tacto
Continued on next page
Slide 7 continued
(por ejemplo, hormigueo, sensaciones eléctricas), el olfato o el gusto (por ejemplo, productos
químicos o sensaciones metálicas, a menudo desagradables), la visión (por ejemplo, luces
intermitentes, escenas complejas), y la audición (por ejemplo, zumbido, la voz de la persona).
Las convulsiones autonómica son comunes. Estas evocan cambios en la actividad autónoma
(por ejemplo, el corazón alterado o sudoración frecuencia respiratoria) o sensaciones viscerales
(por ejemplo, en el abdomen o el pecho).
Los ataques psíquicos afectan al modo en que sentimos, pensamos, y las cosas
experimentadas. Los pacientes pueden informar de un "estado de ensueño", de transición entre
la vigilia y la inconsciencia. Los ataques psíquicos pueden alterar la función del lenguaje, la
percepción o la memoria. También puede evocar emociones espontáneas (por ejemplo, el
miedo, la ansiedad o la depresión), la percepción alterada del tiempo o la familiaridad (déjà vualgo nuevo parece familiar, jamais vu-las cosas familiares parecen extrañas), despersonalización
( un sentimiento de que no es uno mismo), desrealización (el mundo parece irreal, de ensueño),
o autoscopia (cuerpo de una visión desde el exterior).
Las crisis parciales complejas son crisis que se asocian con alteración de la
conciencia. Un error común es el pensar que se requiere la extensión de la crisis
a ambos lados del cerebro. La mayoría de las crisis parciales complejas se
originan en el lóbulo temporal y pueden afectar a la conciencia sin dejar de ser
focales. Durante las crisis parciales complejas el paciente tiende la mirada perdida.
Esto va acompañado de la incapacidad de respuesta normal, alteración de la
función cognoscitiva, imposibilidad para recordar, a pesar de cierto grado de
capacidad de respuesta que se puede conservar (por ejemplo, la orientación hacia
un estímulo).
tí l ) Los
L
movimientos
i i t
automáticos
t áti
((automatismos)
t
ti
) son comunes y muchos
h
son en la boca (por ejemplo, chupeteo de labios, masticación, deglución), las
extremidades superiores (por ejemplo, buscando a tientas, agarrando), vocalización
/verbalización (por ejemplo, gruñidos, repetir frases), o actos complejos (por ejemplo,
, caminar arrastrando los pies). Automatismos más dramáticos se pueden presentar
ocasionalmente (por ejemplo, gritar, correr, desnudarse, empujar la pelvis). Las crisis
parciales complejas suelen durar entre 15 segundos a 3 minutos. Después de la
crisis se presenta confusión postictal la que es común y que generalmente dura
crisis,
menos de 15 minutos, aunque otros síntomas, como la fatiga, pueden persistir
durante horas.
Las crisis parciales pueden progresar a crisis generalizadas con la actividad tónico-clónica.
Una vez que una crisis parcial secundariamente se generaliza es imposible de diferenciarla
de una crisis generalizada primaria. La historia clínica, el elelectroencefalograma (EEG), el
examen neurológico (especialmente post-ictal), y los estudios de neuroimagen (TC o
resonancia magnética) a menudo ayudan a distinguir estos tipos de crisis. En las crisis
secundariamente generalizadas, los pacientes pueden recordar un aura antes de la
actividad convulsiva y los testigos pueden observar una crisis parcial simple o parcial
compleja antes de la generalización. Además, a partir de una crisis secundariamente
generalizada,
li d ell paciente
i t puede
d ttener d
debilidad
bilid d ffocall en una o en ambas
b extremidades
t
id d
(parálisis de Todd) en el lado contralateral al inicio de la crisis.
El EEG en las crisis parciales es variable. Durante las crisis parciales simples, la actividad
sobre cuero cabelludo-EEG registrado puede ser normal, o mostrar anormalidades muy
localizadas o lateralizadas sobre una actividad rítmica anormal. Durante las crisis parciales
complejas, la actividad rítmica, que a menudo se ve es bilateral. En segundo lugar las
crisis generalizadas, la actividad rítmica es por lo general de gran amplitud, bilateral y
difusa, aunque por lo general es ocultada por artefactos de la actividad muscular que es
abundante y que caracteriza a estas crisis.
El EEG en las crisis parciales es variable. Durante las crisis parciales simples, la actividad
sobre cuero cabelludo-EEG registrado puede ser normal, o mostrar anormalidades muy
localizadas o lateralizadas sobre una actividad rítmica anormal. Durante las crisis
parciales complejas, la actividad rítmica, que a menudo se ve es bilateral. En segundo
lugar las crisis generalizadas, la actividad rítmica es por lo general de gran amplitud,
bilateral y difusa, aunque por lo general es ocultada por artefactos de la actividad
muscular que es abundante y que caracteriza a estas crisis.
El EEG en las crisis parciales es variable. Durante las crisis parciales simples, la actividad
sobre cuero cabelludo-EEG registrada puede ser normal, o mostrar anormalidades muy
localizada o lateralizada sobre una actividad rítmica anormal. Durante las crisis parciales
complejas, la actividad rítmica, que a menudo se ve es bilateral. En segundo lugar las
crisis generalizadas, la actividad rítmica es por lo general de gran amplitud, bilateral y
difusa, aunque por lo general es ocultada por artefactos de la actividad muscular que es
abundante y que caracteriza a estas crisis.
El EEG en las crisis parciales es variable. Durante las crisis parciales simples, la actividad
sobre cuero cabelludo-EEG registrado puede ser normal, o mostrar anormalidades muy
localizadas o lateralizadas sobre una actividad rítmica anormal. Durante las crisis
parciales complejas, la actividad rítmica, que a menudo se ve es bilateral. En segundo
lugar las crisis generalizadas, la actividad rítmica es por lo general de gran amplitud,
bilateral y difusa, aunque por lo general es ocultada por artefactos de la actividad
muscular que es abundante y que caracteriza a estas crisis.
Las crisis generalizadas afectan a ambos hemisferios cerebrales desde el inicio de la
descarga. Se asocian a la pérdida de la conciencia, ya sea brevemente o durante un
período de tiempo más largo, y se sub-clasifican en varios tipos principales: las
ausencias, las mioclónicas, las atónicas, y las tónico-clónicas,
Las ausencias (petit mal o pequeño mal) son crisis de episodios breves, por lo
general duran 3-20 segundos, de mirada fija y perdida, con alteración de la conciencia y
pérdida de capacidad de respuesta. No hay ninguna advertencia antes de la crisis, e
inmediatamente después la persona vuelve a estar alerta y atenta. La
carencia de un período postictal es una característica clave que permite distinguir entre la
ausencia de las crisis parciales complejas. Si la duración es> 10 segundos, hay a
menudo fenómenos motores (por ejemplo, parpadeo, la boca se abre y cierra
en automático o movimientos de la mano, así como cambios en el tono muscular). Estos
casos normalmente
l
t comienzan
i
entre
t las
l edades
d d d
de 4 a 14 años,
ñ
y suelen
l d
desaparecer a
los 18 años. Las crisis de ausencia son a menudo provocadas por
hiperventilación, un medio eficaz para la reproducción de crisis en el consultorio o durante
el registro del EEG. La característica del EEG de epilepsia de crisis de ausencia en el
patrón de descarga 3 Hz de punta-onda. Los niños con crisis de ausencia típicas
suelen tener un desarrollo e inteligencia normales.
Las crisis de ausencia atípicas también se presentan predominantemente en niños, por lo
general comienzan antes de 6 años de edad. A diferencia de las ausencias típicas, las
ausencias atípicas pueden empezar y acabar de forma gradual (en segundos), por lo
general duran entre 5-30 segundos, y generalmente no son provocadas por la
hiperventilación. La mirada fija del niño y la reducción en la capacidad de respuesta suelen
ser incompletas. El parpadeo y leves movimientos de los labios pueden apreciarse. Las
crisis de ausencia atípicas ocurren a menudo en niños con deterioro cognitivo global por lo
que la crisis puede ser difícil de distinguinda de la conducta habitual del niño. El EEG suele
mostrar
t actividad
ti id d paroxística
í ti generalizada
li d de
d complejos
l j punta-onda
t
d lenta
l t (es
(
decir, <2,5 Hz). Las crisis de ausencia atípicas suelen surgir durante
la infancia, pero pueden persistir hasta la edad adulta. Las crisis atónicas y tónicas ocurren
a menudo en pacientes con crisis de ausencia atípica.
Las crisis mioclónicas consisten en una sacudida breve, similar a la contracción de un
músculo o grupo de músculos
músculos. Las mioclonías benignas se producen en personas sanas
(por ejemplo, mientras se queda dormido). Esto no es una crisis mioclónica epiléptica. Las
mioclonías patológicas pueden deberse a causas epilépticas y no epilépticas. Las
mioclonías epilépticas por lo general son sacudidas bilaterales, sincrónicas y más a
menudo afecta al cuello, hombros, brazos, tronco y muslos. La conciencia no parece verse
afectada, aunque esto es difícil de comprobar, dada la corta duración de menos de 1
segundo, si ocurren varios en una sucesión rítmica, se puede llamar un estado mioclónico,
y puede estar asociado con la alteración de la conciencia
conciencia. El EEG durante una crisis
mioclónicas generalmente muestra consiste en una descarga de polipuntas-y de ondas
lentas. Las crisis mioclónicas ocurren en una variedad de síndromes epilépticos. En raras
ocasiones pueden ser vistos como parte de una enfermedad progresiva y degenerativa (es
decir, la llamada epilepsia mioclónica progresiva).
Las crisis atónicas y las tónicas, al igual que las ausencias atípicos, son mas frecuentes
en personas que tienen otras alteraciones neurológicas que han provocado una epilepsia
secundaria.
En contraste a las crisis parciales motoras, las crisis tónicas son generalizadas e
involucran musculatura bilateral en forma simétrica o casi simétrica. Las crisis tónicas se
caracterizan por flexión de la cintura y el cuello, abducción y flexión o extensión de las
extremidades superiores y flexión o extensión de las inferiores. Tipicamente ocurren
durante el sueño y duran dos a veinte segundos. El EEG suele mostrar actividad
generalizada de polipuntas rápidas de bajo voltaje.
Atonic seizures consist of a sudden loss of postural tone, often resulting in falls, or, when
milder, head nods or jaw drops. Consciousness is usually impaired and significant injury
may occur. Duration is usually several seconds, rarely more than 1 minute. EEG often
shows an electrodecremental response.
L crisis
Las
i i atónicas
tó i
consisten
i t en una pérdida
é did súbita
úbit d
dell ttono muscular,
l ffrecuentemente
t
t
producen caídas o cuando es mas leve, caída de la mándíbula y la cabeza. Generalmente
la consciencia está afectada y pueden presentarse lesiones significativas. Dura algunos
segundos, raramente mas de un minuto. El EEG muestra una respuesta de
electrodecremento.
Las crisis de caída pueden ocurrir no solo con las crisis atónicas, sino también con las
crisis mioclónicas o tónicas, si las piernas están afectadas.
Las crisis tónico-clónicas generalizadas primarias (también llamadas crisis de gran mal o
convulsivas) causan la pérdida de conciencia asociada con una fase tónica inicial de
rigidez, caída, y muchas veces un grito evocado por aire forzado a través de la contracción
de las cuerdas vocales. Las piernas se muestran generalmente extendidas, y los brazos
puede estar extendidos, flexionados, o cada uno en sucesión. La fase clónica
posteriorconsiste consiste en sacudidas de las extremidades que se reducen gradualmente
conforme la crisis cede. Las convulsiones tónico-clónicas suelen durar entre 30 a 120
segundos. Puede haber sialorrea debido a la falta de la deglución y salivación excesiva,
mordiéndose
dié d
lla llengua, mejillas
jill o llabios,
bi
con h
hemorragia
i e iincontinencia
ti
i d
de lla vejiga
ji o d
dell
intestino. Suele haber Letargia postictal y confusión a menudo y puede ser seguido por
agitación transitoria. El EEG muestra polipuntas generalizada, pero estas suelen ser
ocultadas por el artefacto de la actividad muscular. En el postictal, hay supresión de la
actividad de fondo y a continuación enlentecimiento difuso.
La epilepsia es un término general, en las que se incluyen muchos tipos de enfermedades
y síndromes. Algunos autores distinguen entre las epilepsias y síndromes epilépticos,
dependiendo de si las crisis son el único trastorno neurológico (epilepsia) o son parte de un
grupo de síntomas (un síndrome epiléptico). Algunas de las epilepsias (por ejemplo,
epilepsia mioclónica juvenil) tiene una genética bien definida, el curso clínico, y la
respuesta a la medicación. Otros (por ejemplo, epilepsia del lóbulo temporal) tiene una
historia natural que es muy variables, y que reflejan las diferencias en la patología, así
como en la respuesta del huésped de este procesopatológico a los tratamientos
administrados.
d i i t d
La actual clasificación de las epilepsias y síndromes epilépticos intenta separar estos
trastornos de acuerdo a su origen topográfico cerebral, es decir, si se producen en una
parte circunscrita del cerebro (parcial), o al parecer comienzan de forma difusa en la
corteza y sus conexiones más profundas (generalizadas). El síndrome es idiopático
cuando el trastorno no se asocia con otra anormalidad neurológica ; sintomática indica que
hay una anomalía presente y la causa por tanto es reconocida. Criptogénica se refiere a
los síndromes que se supone que son sintomáticos, pero la causa en un paciente
específico es desconocida. Muchas epilepsias idiopáticas se presentan en niños y
adolescentes,
d l
t
y muchas
h veces continuan
ti
en la
l adolescencia
d l
i o edad
d d adulta.
d lt H
Hay pruebas
b
de que la mayoría de estos síndromes tienen una base genética, y que cuando esta base
se conoce, pasará de la causa idiopática a la categoría sintomática.
continued
Continuation of slide 25
CLASIFICACIÓN INTERNACIONAL DE LAS EPILEPSIAS Y SINDROMES EPILÉPTICOS
1. Relacionados a su localización (local, focal, parcial) Epilepsias y Síndromes
1.1 idiopática (con inicio relacionado con la edad)
La epilepsia infantil benigna con puntas centrotemporales (('epilepsia
epilepsia rolándica)
Epilepsia infantil con paroxismos occipitales
1.2 sintomática
La epilepsia Partialis crónica progresiva de la infancia (por ejemplo, 'la encefalitis de
Rasmussen)
Epilepsias del lóbulo frontal
Epilepsias del lóbulo occipital
Epilepsias del lóbulo parietal
Epilepsias del lóbulo temporal
1.3 criptogénica
2. Generalizadas Epilepsias y Síndromes
2.1 idiopática (con inicio relacionado con la edad)
Crisis neonatales benignas familiares
Crisis neonatales benignas
Epilepsia mioclónica benigna de la infancia
Infancia epilepsia de ausencia (piknolepsia)
Epilepsia de ausencia
Epilepsia mioclónica juvenil
2.2 criptogénica o sintomática
Síndrome de West
Síndrome de Lennox-Gastaut
3. Epilepsias y Síndromes en donde se ignora si fueron focales o generalizadas
4. Síndromes especiales
La evaluación inicial después de una sola crisis debe incluir: 1) el determinar si una crisis
ocurrió realmente, o si el paciente experimentó un acontecimiento transitorio de otro tipo, 2)
la búsqueda de evidencia de un inicio parcial , 3) búsqueda de evidencia de la disfunción
subyacente del sistema nervioso central , 4 ) búsqueda de evidencia de enfermedades
sistémicas o metabólicas que podrían haber precipitado la crisis; 5) intento de clasificar la
crisis y su tipo; 6) determinar cuáles son los estudios de diagnóstico apropiados, y 7)
determinar si la terapia con medicamentos debe ser instituida, y si es así, con qué agente
terapéutico debe iniciarse.
A menudo, el paciente no recuerda los acontecimientos que rodearon al evento crítico y la
descripción debe ser obtenido de los familiares
familiares, amigos u otras personas
personas. Los
observadores podrán reportar alteraciones compatibles con una crisis parcial compleja
inmediatamente antes de una convulsión generalizada. En otros casos, el paciente no
puede recuperar la actividad motora en una parte localizada, lo que sugiere una crisis
parcial simple motora antes de la perdida de la conciencia. A veces, la única evidencia de
inicio parcial puede ser un breve acto subjetivo consistente con un aura, y en este caso es
importante determinar si el aura es idéntica a algún evento previo o nunca antes se había
presentado.
presentado
El examen del paciente que ha sufrido una crisis a menudo es más reveladora cuando se
realiza tan pronto después del evento o lo mas pronto posible, y debe ser repetida con
frecuencia para determinar si los déficit observados son transitorios. Tales como la
debilidad post-ictal, la afasia, o la disfunción sensorial que pueden además proporcionar
información de gran realce para la lateralización y localización de la crisis. Las neuronas
motoras que están afectadas transitoriamente (por ejemplo, el encontrar en forma
j de Babinski)) también proporcionan
p p
datos importantes.
p
Los
transitoria y unilateral un reflejo
signos que no son transitorios pueden indicar una lesión estructural preexistente (por
ejemplo, tumor) o una nueva condición (por ejemplo, enfermedad cerebrovascular), y
puede llevar al diagnóstico de una crisis aguda sintomática, es decir, una crisis como
resultado de un nuevo insulto cerebral, que no implica necesariamente la existencia de
epilepsia (aunque más adelante se puede desarrollar esta).
Continued from slide 30
No hay signos patognomónicos físicos que demuestren que un evento fue una crisis epiléptica,
pero hay muchas asociaciones de utilidad. Las mordeduras en la lengua o en la mejilla, el
vaciamiento urinario y / o la incontinencia fecal, son más comunes después de las crisis que
después de la pérdida de la conciencia por otras causas. El examen físico general es otra cosa
útil cuando se descubre evidencia de un factor precipitante de una crisis aguda sintomática (por
ejemplo meningitis),
ejemplo,
meningitis) o una predisposición genética a sufrir una crisis epiléptica
epiléptica, como sería un
síndrome neurocutáneo (por ejemplo, la esclerosis tuberosa).
La evaluación de laboratorio de un paciente después de una crisis epiléptica solo depende de las
circunstancias que rodean el caso. Los análisis de sangre debe ser considerados de acuerdo a
la edad del paciente y las circunstancias clínicas exámenes de sangre rutinarios pueden indicar
problemas tales como hipo o hiperglucemia, alteraciones en el sodio y el calcio, o la deficiencia
de magnesio; disfunción hepática cardiorrespiratoria, o alteraciones en la función renal, o
infección Ante cualquier sospecha de meningitis o encefalitis debe hacerse una punción lumbar
infección.
(después de evaluar el potencial de herniación cerebral), pero por lo demás este procedimiento
generalmente no es necesario. Debido a que muchas drogas ilícitas pueden causar
convulsiones, evaluar en sangre y orina tóxicos , sobre todo en adolescentes y adultos jóvenes.
Los pacientes que han tenido una crisis de reciente aparición deben someterse a un
electroencefalograma (EEG) y, con pocas excepciones definibles, a imágenes por resonancia
magnética (RM). Una tomografía computarizada es útil si un proceso agudo si se sospecha (por
ejemplo, de una hemorragia intracerebral), pero no es suficiente para excluir a los pequeños
tumores o malformaciones vasculares, a la atrofia del hipocampo, y a la displasia
cortical. Algunas excepciones comunes a la necesidad de neuroimagen son los niños con crisis
febriles no complicadas si con los hallazgos clínicos y el EEG se determina un síndrome
idiopático bien definido, como la epilepsia infantil de ausencia o la epilepsia benigna con puntas
centrotemporales.
Uno o más factores precipitantes pueden contribuir a la presencia de crisis en un paciente.
El descubrir un factor precipitante no obvia la necesidad de búsqueda de patología
intracraneal o una predisposición genética hacia la crisis, pero puede conducir a un
diagnóstico de no epilepsia (por ejemplo, la crisis por abstinencia de alcohol), y es muy útil
en la orientación del paciente. Desencadenantes comunes incluyen al desequilibrio
metabólico y de electrolitos (como la glucosa baja en la sangre, baja en el sodio, calcio o
magnesio ), la reducción de la medicación antiepiléptica o un tratamiento con un fármaco
antiepiléptico no adecuado, las variaciones hormonales, el estrés, la infección, la privación
d l sueño,
del
ñ lla abstinencia
b ti
i d
dell alcohol
l h l u otros
t
sedantes
d t , y lla administración
d i i t ió d
de fá
fármacos con
propiedades pro-convulsivantes, tales como los estimulantes del sistema nervioso,
incluyendo la cocaína, los anticolinérgicos (incluyendo antihistamínicos de venta libre), casi
todos los agentes bloqueadores de la dopamina, los nuevos antipsicóticos (en particular,
clozapina), antidepresivos (especialmente bupropión), inmunosupresores como la
ciclosporina y los antibióticos como las quinolonas o imipenem-y las estatinas.
El EEG es muyy útil p
para clasificar el tipo
p de crisis yy, en muchos casos, el
síndrome de epilepsia. Un EEG normal no excluye el diagnóstico de la
epilepsia. El EEG es sólo una muestra de tiempo muy breve de la actividad
eléctrica del cerebro del paciente y perderá anomalías intermitentes o
transitorias. En la evaluación de un paciente con sospecha de haber tenido
una convulsión, un EEG muestra actividad epileptiforme interictal (entre
ataques), proporciona evidencia que lo corrobora, pero no se prueba, a
menos que el paciente tenga una ataque durante el EEG (en cuyo caso la
actividad epileptiforme ictal es más que interictales).
Actividad epileptiforme incluye puntas, ondas agudas, crisis
electroencefalográficas, y algunos otros fenómenos estereotipados que
están fuertemente asociados con las convulsiones. Espigas y ondas agudas
son eventos epileptiformes interictales. Anomalías de fondo indican
disfunción cerebral localizada o difusa, y pueden reflejar una alteración
transitoria postictal o el proceso subyacente responsable de la convulsión.
Si el tratamiento con fármacos antiepilépticos (FAE) se debe iniciar después de una
primera crisis es polémico. Dentro de los primeros 5 años después de una sola crisis no
provocada, 16-62% de los pacientes pueden volver a tener otra crisis epileptiforme. La
recurrencia es más probable si se tiene una lesión neurológica previa suficiente para
causar crisis, una anormalidad estructural en la neuroimagen, un patrón anormal
epileptiforme en particular en el EEG, o el tener antecedentes familiares de epilepsia, son
elementos importantes para tener mayor riesgo. La mayoría de estudios también sugieren
que las crisis parciales (incluyendo generalización secundaria) y las crisis generalizadas
son más
á propensas a recurrir
i principalmente
i i l
t llas generalizadas
li d tó
tónico-clónicas.
i
ló i
El
tratamiento puede reducir (quizá en un 50%), pero no eliminar el riesgo de un segundo
evento. La decisión de tratamiento debe hacerse de forma individual negociada con el
paciente, teniendo en cuenta las posibles consecuencias físicas, psicológicas y
profesionales de las crisis epilépticas y de la terapia con fármacos antiepilépticos (FAE).
Antes de que el tratamiento se inicie, el clínico debe decidir si las crisis del
paciente son parciales o generalizadas de inicio. El fármaco de elección debe tener
la mejor eficacia (capacidad de detener las crisis) y la menor probabilidad de
efectos adversos. Varios estudios han demostrado al compararlos mínimas diferencias
que son de relevancia en la eficacia de los FAE estándar. Por lo tanto, las
diferencias en el perfil esperado, efectos adversos y el perfil farmacocinético, así como de
costos, deben guiar la elección del FAE. La mayoría de los pacientes pueden
ser evaluados de forma óptima y seleccionar un solo FAE. Uno debe estar seguro de
que un determinado
d t
i d fá
fármaco ha
h fallado
f ll d antes
t de
d pasar a un medicamento
di
t alternativo
lt
ti o
dar una combinación de dos medicamentos. Si el
paciente tiene crisis persistentes pero sin efectos adversos, la dosis puede
aumentarse según la tolerancia y hasta que el control de las crisis se obtiene. El
"rango terapéutico" asociado a las concentraciones séricas es sólo para el clínico una
guía para determinar en el paciente la dosis adecuada.
Los medicamentos antiepilépticos pueden ser más o menos clasificados como de
amplio espectro o agentes para crisis parciales. Fármacos de amplio espectro son útiles
en el tratamiento tanto de las crisis generalizadas primarias como de las crisis de inicio
parcial. Fármacos de espectro parcial se utilizan para el tratamiento de las crisis de inicio
parcial, pero generalmente se evitan en la epilepsia generalizada primaria ya que
pueden agravar algunos tipos de crisis (por ejemplo, crisis de ausencia atípicas ). La
fenitoína y la carbamazepina, se utilizan a veces para las crisis tónico-clónicas
generalizadas primarias.
En las crisis de inicio parcial con generalización secundaria, carbamazepina, fenitoína,
valproato, fenobarbital y primidona suelen ser eficaces. En las crisis parciales sin
generalización secundaria, la fenitoína y la carbamazepina pueden ser un poco más
eficaces. Estas conclusiones se basan en estudios de comparación directa al azar de estos
medicamentos.
Felbamato, gabapentina, lamotrigina, levetiracetam, oxcarbazepina, tiagabina, topiramato,
zonisamida y rufinamida son los nuevos fármacos antiepilépticos aprobados por la FDA
desde 1993. Estos medicamentos marcan el inicio de nuevas opciones de tratamiento para
l epilepsia,
la
il
i varios
i nuevos medicamentos
di
t antiepilépticos
ti ilé ti
es probable
b bl que se aprueben
b
dentro de los próximos años. Después de los ensayos clínicos aleatorios clínicos
controlados, los nueve medicamentos cuentan con la aprobación de la FDA para el
tratamiento adyuvante en pacientes con crisis de inicio parcial. De los nuevos fármacos
antiepilépticos, sólo oxcarbazepina cuenta con la aprobación de la FDA para uso en
monoterapia en crisis parciales de reciente aparición. Lamotrigina, el topiramato y la
gabapentina no han sido aprobados por la FDA para la monoterapia de primera línea, sin
embargo han demostrado una eficacia similar a la de carbamazepina de liberación
embargo,
inmediata en los ensayos comparativos y se recomiendan para esta indicación por la AAN.
La FDA ha aprobado la lamotrigina en monoterapia en pacientes adultos con crisis
parciales tras el fracaso de la fenitoína o carbamazepina.
Continued from slide 42
Levetiracetam no ha sido aprobado por la FDA o en las guías de manejo ecomendadas por AAN
en las crisis parciales de novo,
no o pero ha demostrado tener una
na eficacia similar a la carbamazepina
carbama epina
en un ensayo comparativo, después de la publicación de las directrices de la AAN. Lacosamida
fue recientemente (octubre de 2008) aprobada por la FDA como tratamiento adyuvante en crisis
de inicio parcial basada en dos ensayos clínicos fase III en que demostró su eficacia frente a
placebo como terapia de adición.En la práctica, gran parte de la elección de los fármacos
antiepilépticos para las crisis parciales depende del perfil de efectos secundarios de la droga y
las preocupaciones individuales de cada paciente.
Azar and Abou
Abou-Khalil,
Khalil Seminars in Neurology,
Neurology 2008 28:305
28:305-316
316
En los pacientes con crisis generalizadas de inicio, la elección depende del síndrome
epiléptico específico, y en particular de los diferentes tipos de crisis generalizadas
asociadas. Las epilepsias generalizadas que se caracterizan por convulsiones tónicoclónicas, crisis mioclónicas, y / o crisis de ausencia, o en la epilepsia fotosensible, el
valproato se considera generalmente como el fármaco de elección. El clonazepam y el
fenobarbital o primidona puede ser útiles en las crisis generalizadas, pero a menudo tienen
más efectos sedantes y de afección del comportamiento que otros fármacos
antiepilépticos. El clonazepam, benzodiazepinas, pueden perder parte de su eficacia al
cabo
b d
de seis
i meses o menos, d
debido
bid all d
desarrollo
ll d
de lla ttolerancia.
l
i L
La lamotrigina,
l
ti i
ell
topiramato y la zonisamida puede ser eficaces contra algunos tipos de crisis generalizadas,
sobre todo, las convulsiones tónico-clónicas, las crisis de ausencia y las tónicas. El
topiramato ha recibido recientemente una indicación por la FDA como tratamiento de
primera línea de crisis generalizadas tónico-clónicas. La fenitoína y la carbamazepina, son
eficaces para tónico-clónicas, pero no para otros tipos de crisis generalizadas. La
carbamazepina puede exacerbar algunas crisis generalizadas de inicio como las ausencias
y crisis mioclónicas.
mioclónicas La lamotrigina también pueden empeorar las crisis mioclónicas.
mioclónicas Esto
pone de relieve la necesidad de clasificación para la selección del FAE apropiado.
En los niños con crisis de ausencia (sin convulsiones tónico-clónicas), la etosuximida y el
valproato son igualmente eficaces. El valproato tiene la ventaja de proteger contra las
crisis tónico-clónicas que pueden aparecer más tarde, debido al riesgo infrecuente pero
potencial fatal del valproato de provocar hepatotoxicidad inducida, por lo que,
etosuximida se considera más segura. Este riesgo valproato es máyor en los niños
menores de 2 años, especialmente los menores de 6 meses de edad y más si
cursa con trastornos metabólicos congénitos o que son tratados con medicamentos
múltiples antiepilépticos. El valproato también debe utilizarse con precaución en las
niñas,
iñ
d d su asociación
dada
i ió con ell síndrome
í d
d ovario
de
i poliquístico
li í ti y de
d teratogenicidad.
t t
i id d
La mayoría de los pacientes con epilepsia se manejan mejor con un solo fármaco. La
monoterapia puede simplificar los regímenes de tratamiento, reducir los efectos adversos y
a menudo mejorar el control de las crisis. Para la conversión a la monoterapia, ya que
incluso las personas con crisis no controladas pueden tener un mejor control, así como
menos efectos adversos con el uso de altas dosis de un único FAE en lugar de las
combinaciones de medicamentos. El clínico debe determinar primero si el paciente ha
tenido un tratamiento adecuado con un fármaco de primera línea, es decir, si las crisis
persisten incluso cuando el FAE se incrementó gradualmente hasta la presencia de efectos
adversos
d
molestos.
l t
Al pasar a llos pacientes
i t a lla monoterapia,
t
i se debe
d b ttratar
t de
d eliminar
li i
primero los medicamentos sedantes (benzodiazepinas y barbitúricos). Estos deben ser
retirados poco a poco, por lo general durante varios meses. A pesar de que la monoterapia
se prefiere, algunos pacientes con epilepsia requieren politerapia.
Los fármacos antiepilépticos (FAE) que se unen a las proteínas séricas (por ejemplo, la
fenitoína, el valproato y la tiagabina) pueden ser desplazados de los sitios de unión por
otros fármacos altamente ligables a proteínas (por ejemplo, la aspirina, la warfarina, las
fenotiazinas). En estos casos, la concentración sérica puede no reflejar exactamente la
proporción de fármaco no unido. La fracción no uniida (libre) puede medirse mediante las
concentraciones séricas y pueden ser útiles en pacientes que
toman estos medicamentos con otros fármacos con alta afinidad a proteínas o
en pacientes con enfermedad renal o hipoalbuminemia.
L mayoría
La
í de
d FAE son metabolizados
t b li d por las
l enzimas
i
h
hepáticas
áti
y pueden
d
inducir o inhibir el metabolismo hepático de otros fármacos. Las excepciones son la
gabapentina y levetiracetam que no tienen metabolismo hepático . La inducción de las
enzimas hepáticas por fármacos antiepilépticos como la carbamazepina, la fenitoína y
el fenobarbital puede causar un aumento en el metabolismo y disminución de las
concentraciones séricas de muchas otras drogas, como las hormonas esteroides (por
ejemplo, anticonceptivos orales) o la warfarina. El Felbamato y el
valproato son inhibidores del metabolismo y pueden aumentar las concentraciones séricas
de otras drogas metabolizadas en el hígado. Por el contrario, otros fármacos (por
ejemplo, la eritromicina o la fluoxetina son inhibidores potentes) pueden inhibir
el metabolismo de los antiepilépticos. A veces
es difícil de predecir qué tipo de interacción se produce cuando dos medicamentos
antiepilépticos o un FAE y otro fármaco se utilizan juntos.
El "rango terapéutico" de las concentraciones séricas de FAE se asocian a un control de
las crisis sin una toxicidad significativa, y se han determinado de los estudios de población.
Esta gama es una guía útil, pero no puede sustituir a la evaluación de la respuesta clínica
de cada paciente a un FAE. Muchos pacientes pueden experimentar un excelente control
de las crisis y sin efectos adversos con las concentraciones séricas por encima o por
debajo del rango terapéutico. Además, algunos pacientes experimentan efectos
secundarios molestos con los niveles dentro o incluso por debajo de este rango. Los
médicos no deben adherirse estrictamente a las concentraciones séricas que son la ayuda
para buscar
b
ell equilibrio
ilib i d
de lla eficacia
fi
i y lla ttoxicidad
i id d FAE
FAE.
Factores farmacocinéticos también debe tenerse en cuenta al interpretar las
concentraciones de suero de los FAE. La mayoría de las drogas necesitan cinco vidas
medias para alcanzar el estado estable. Los fármacos con vida media larga, como la
fenitoína, el fenobarbital y la zonisamida puede requerir dos semanas o más para llegar en
estado estable. Por lo tanto, las concentraciones séricas tomadas demasiado pronto
después del inició del fármaco o del cambio la dosis puede no reflejar con exactitud el
estado de equilibrio. Por el contrario, la concentración sérica de fármacos con vida media
corta puede verse significativamente afectado por el intervalo de tiempo entre la última
dosis y la toma de muestra de suero.
Antes de iniciar un FAE, el paciente debe ser informado acerca de los efectos adversos y
la probabilidad realista de la eficacia. Por ejemplo, menos del 50% de los
adultos con crisis de inicio parcial estarán libres de crisis durante más de 12 meses
después de comenzar la monoterapia de primera línea. Los pacientes deben registrar la
frecuencia de crisis y el tipo así como los efectos adversos en un calendario, con lo que la
eficacia puede ser cuantificada y comparada entre los medicamentos antiepilépticos. Los
posibles factores de recaída, como la menstruación, también puede ser registrados. La
mayoría de los FAE deben introducirse lentamente para minimizar los efectos adversos.
Ad á antes
Además,
t de
d iiniciar
i i ell FAE,
FAE debe
d b tit
titularse
l
a intervalos
i t
l durante
d
t los
l primeros
i
meses de uso, es razonable para comprobar el buen control, determinar valores de
electrolitos séricos, pruebas de función hepática y la concentración sérica del fármaco.
Los medicamentos antiepilépticos pueden eventualmente ser retirados con éxito en más
del 60% de los pacientes que permanecen libres de crisis. La mayoría de los neurólogos
requieren que los pacientes estén libres de crisis durante 2 a 4 años antes de la
interrupción del FAE, y los medicamentos son generalmente suspendidos durante un
período de 2 a 6 meses. El síndrome epiléptico subyacente también puede influir en el
éxito de la suspensión del fármaco antiepiléptico. Por ejemplo, la tasa de éxito para la
interrupción del tratamiento de la epilepsia mioclónica juvenil es sólo un 20%, mientras que
en la epilepsia benigna con puntas centrotemporales, es casi del 100%. El mejor
pronóstico
ó ti para ell eventual
t l retiro
ti d
de llos antiepilépticos
ti ilé ti
en pacientes
i t con epilepsia
il
i
idiopática generalizada (pero no la epilepsia mioclónica juvenil), un examen neurológico
normal y comprobar que no hay lesión cerebral estructural, son factores favorables, sin
embargo, nunca hay una garantía de permanecer libre de crisis y hay que comentarlo
también con el paciente, antes de la suspensión del fármaco.
Cuando las crisis repiten, las principales cuestiones a considerar son: 1) si se trata de una
manifestación de una patología progresiva, como un tumor o una enfermedad
neurodegenerativa, 2) si existe un factor precipitante que podría evitarse en el futuro;3) si
el paciente estaba recibiendo un FAE, a) si el cumplimiento o algún otro factor
farmacocinética (absorción, es decir el metabolismo) está interviniendo, b) si la dosis o la
medicación debe ser modificada, y 4) si el paciente no estaba tomando la medicación, si
esta crisis recurrente es ya una indicación para instituir el tratamiento.
En general, los pacientes con crisis parciales (con o sin generalización secundaria) la
experiencia
i
i que un cambio
bi en ell patrón
tó d
de crisis,
i i sobre
b ttodo
d un cambio
bi en lla manifestación
if t ió
inicial, deben ser evaluados por la posibilidad una lesión progresiva con un examen
neurológico aun normal, y posiblemente se requiera de la repetición de la RM y el EEG.
Para los pacientes que reciben fármacos antiepilépticos, un ataque recurrente puede ser
una indicación para solicitar niveles de concentración sérica de la droga. Esto es
especialmente útil en un paciente cuyas crisis han estado bajo control durante un período
de tiempo. Si la concentración sérica de un FAE ha caído, se debe determinar la causa de
la caída e intentar restablecer el nivel previamente eficaz (es decir
decir, incumplimiento
incumplimiento,
interacciones farmacocinéticas). Por el contrario, si el paciente tiene crisis frecuentes con
las concentraciones séricas en “ nivel terapéutico”,más mediciones no son útiles y un
cambio en la estrategia terapéutica puede estar indicada.
Aunque los FAE son la base del tratamiento, algunas modalidades alternativas de
tratamiento no farmacológico tienen diferentes grados de apoyo para el clínico y pueden
experimentarse. Modificaciones del estilo de vida, sobre todo evitar el alcohol y la
deprivación del sueño, pueden ser muy importantes en los síndromes de ciertos individuos.
La relajación, el biofeedback (retroalimentación) y otras técnicas conductuales pueden
ayudar a un subgrupo de pacientes, especialmente aquellos con un aura determinado que
predice el inicio de las crisis parciales complejas o secundariamente generalizadas. Los
suplementos dietéticos son de valor indeterminado, a excepción de la piridoxina (vitamina
B6) que es crucial
B6),
i l para ell ttratamiento
t i t d
de lla d
dependencia
d
i d
de piridoxina
i id i rara en los
l recién
ié
nacidos y lactantes y obligada en forma concomitante a la terapia antituberculosa con
isoniacida. Los remedios herbolarios están también bajo investigación.
La dieta cetogénica se ha utilizado durante más de 80 años en niños con trastornos
convulsivos graves. Se basa en la observación de que la cetosis y acidosis tener efectos
anti-crisis, aunque recientemente la estabilización de la glucosa, la restricción calórica y
dirigir los efectos anticonvulsivos de los ácidos grasos poliinsaturados han sido
reportados en modelos animales. Debido a los riesgos de graves alteraciones metabólicas
durante y después del período de ayuno inicial, esta dieta se inicia en el hospital.
Recientemente, la exigencia de un período de ayuno ha demostrado que no es ncesario
hospitalizar, sin embargo, la mayoría de centros individualizan en la actualidad. La
i
ingesta
t d
de proteínas
t í
con control
t l estricto,
t i t asíí como las
l calorías,
l í
y sobre
b todo
t d la
l restricción
t i ió
de hidratos de carbono en el contexto de una dieta alta en grasa es necesaria para la
cetosis, y puede por tanto ser difícil de mantener. En el 10% de los pacientes con
epilepsia intratable, permaneciendo en esta dieta durante meses o años puede resultar en
un control sostenido de las crisis y permitir incluso el retiro de los antiepilépticos. Los
efectos secundarios incluyen pérdida de peso, acidosis, cálculos renales (5%), detención
del desarrollo del crecimiento y dislipidemia.
La reciente aparición de las dietas cetogénicas menos restrictivas, como la dieta
modificada de Atkins, la del Johns Hopkins y el tratamiento de bajo índice glucémico del
General Hospital de Massachusetts, han añadido nuevas
opciones para niños y adultos con epilepsia intactable. Estas dietas tienen tasas de
respuesta similar (aunque en series cortas) a la dieta cetogénica tradicional, con menos
efectos secundarios. Se precisan ensayos clínicos mayores, algunos ya en curso.
La mayoría de los casos de epilepsia se controlan bien con medicamentos antiepilépticos,
sin embargo, entre el 20 - 30% de los casos, no lo son. El tratamiento quirúrgico debe ser
considerado en pacientes en los que las crisis y / o los efectos secundarios de la
medicación deterioran significativamente la calidad de vida. El tratamiento quirúrgico está
indicado en estos pacientes si los ataques se derivan de un área que se puede quitar sin
causar deficiencias neurológicas inapcetables. El candidato para la cirugía está
determinado por una constelación de pruebas, que incluyen la monitorización EEG / vídeo,
la neuroimagen y estudios neuropsicológicos. En algunos casos, se realizan
procedimientos
di i t paliativos
li ti
quirúrgicos
iú i
para reducir
d i lla ffrecuencia
i d
de llas crisis
i i o lla gravedad
d d
las mismas, a pesar de que existe una baja expectativa de curación. Estos procedimientos
implican típicamente desconexiones, como la reducción del cuerpo calloso, en lugar de
eliminar el tejido cerebral.
En general, el determinante más importante de un resultado quirúrgico exitoso es la
selección adecuada de pacientes. Esto requiere una evaluación detallada antes de la
cirugía, para caracterizar el tipo de crisis, la frecuencia y el lugar de inicio, funcionamiento
neuropsicológico, psiquiátrico, psicológico y grado de discapacidad.
La historia clínica y la exploración física se realizan en detalle para determinar, si es
posible, la etiología, curso y el impacto funcional de la epilepsia del paciente. Los detalles
de los eventos ictales pueden proporcionar información importante para localizar, por
ejemplo un aura autonómica o una psíquica-cognitiva que puede sugerir origen mesial en
ell lób
lóbulo
l ttemporal.
l Adecuación
Ad
ió de
d los
l ensayos anteriores,
t i
FAE adecuados
d
d debe
d b estar
t
garantizado.
Las técnicas de Neuoimagen son muy importantes para la planificación prequirúrgica. La
IRM es necesaria para identificar posibles lesiones sintomáticas (tumores, displasia) y en
especial la esclerosis mesial temporal. Se deben hacer protocolos específicos para mejorar
la sensibilidad de la resonancia magnética (es decir, cortes de 1,5 mm coronales para la
esclerosis mesial temporal). La tomografía por emisión de positrones (PET) y tomografía
de emisión de fotón único (SPECT) también pueden ayudar a identificar las alteraciones
funcionales que pueden apuntar a la zona epileptogénica. La magnetoencefalografía
(MEG) es una nueva técnica que utiliza el campo magnético de la actividad interictal para
localizar el foco epiléptico. También tiene aplicaciones en el mapeo cerebral funcional.
Las pruebas formales neuropsicológicas pueden revelar déficits cognitivos específicos
focales o multifocales que a veces pueden ser correlacionadas con los datos de
neuroimagen y EEG. Esta prueba puede ayudar a localizar un área del cerebro anormal
funcionamiento y también sirven como punto de referencia para la evaluación postquirúrgica.
Continued
Continued from slide 74
La evaluación psiquiátrica y la psicosocial son de vital importancia para evaluar el nivel actual de
funcionamiento y garantizar que el paciente y su familia tienen metas realistas y actitud
adecuada. Esta evaluación también establece una relación que puede ser útil en el tratamiento
de las cuestiones de ajuste hasta de fármacos que puede producirse incluso después de la
cirugía de la epilepsia con éxito.
La inyección de Amobarbital sódico durante la angiografía carotídea acompañada de pruebas de
l
lenguaje
j y llas pruebas
b d
de memoria
i (t
(testt de
d Wada)
W d ) revelan
l información
i f
ió crítica
íti con respecto
t a la
l
lateralización del lenguaje y la memoria, que es necesario evaluar para determinar si el paciente
puede tolerar la cirugía de la epilepsia sin que se afecte significativamente estas áreas de su
funcionalidad. Actualmente la resonancia magnética funcional se utiliza en algunos centros en
lugar de la tradicional prueba de Wada.
La lobectomía temporal es el procedimiento quirúrgico más común para la epilepsia y se
puede realizar tanto en el hemisferio dominante o como en el no dominante, sin alteración
del lenguaje en forma significativa. Entre los pacientes altamente seleccionables, más del
80% (en algunos estudios) estarán libres de crisis parciales complejas o secundariamente
generalizadas después de la cirugía, aunque muchos permanecen con los medicamentos.
Resecciones extra-temporales, con mayor frecuencia en el lóbulo frontal y con menor
frecuencia en la región parietal y occipital. Las resecciones extra-temporales se realizan
principalmente en pacientes con lesiones estructurales o anormalidades en el desarrollo y
ocasionalmente
i
l
t en llas epilepsias
il
i ffocales
l criptogénicas.
i t é i
C
Casos
raros asociados
i d con llas
crisis derivadas de grandes partes de un hemisferio cerebral, asociados a déficits
hemisféricos, se pueden tratar con una hemisferectomía anatómica o funcional,
especialmente niños. Estas operaciones pueden ser muy exitosa en los casos de
hemimegaloencefalia, el síndrome de Rasmussen, el síndrome de Sturge-Weber, y las
alteraciones corticales hemisfericas de gran tamaño. Los procedimientos paliativos como el
cortar el cuerpo calloso se puede realizar en pacientes con crisis atónicas (crisis de caida),
así como crisis generalizadas tónico
tónico-clónicas
clónicas refractarias y otras con generalización
secundaria persistente.
Aproximadamente dos tercios de los pacientes con epilepsia temporal mesial estarán libres
de crisis, con excepción del aura, después de la lobectomía temporal anterior. Sólo hay un
estudio clase I aleatoriozado controlado, de los resultados de la cirugía de la epilepsia
(Wiebe et al. NEJM 2001). En este estudio de 80 pacientes con epilepsia del lóbulo
temporal mesial, el 64% de los que recibieron cirugía quedaron libres de crisis, contra el
8% de los que recibieron tratamiento médico. Un meta-análisis de 24 estudios de la clase
IV demostró resultados similares (véase la diapositiva). En un meta-análisis clase IV , se
encontraron pocas variaciones cuando los datos se examinan en relación con la región
geográfica,
áfi
un seguimiento
i i t más
á prolongado,
l
d y lla cirugía
i í d
después
é d
de lla aparición
i ió d
de lla
resonancia magnética (Engel, J et al. Neurología 2003). Otros estudios sugieren que la
presencia de esclerosis mesial temporal en la RM y un historial de crisis febriles son
factores pronósticos para un mejor resultado. Si la RM y EEG son concordantes, la
posibilidad de ausencia de crisis puede ser tan alta como 77% (Gilliam et al. Epilepsia
1997).
No hay estudios clase I o II disponibles en las resecciones del lóbulo extra
extra-temporal
temporal o no
notemporal mesial. Los datos que aquí se presenta se basan en un meta-análisis de 14
series de la Clase IV (Engel, J et al. Neurología 2003). En este estudio, aproximadamente
la mitad de los pacientes sometidos a resección neocortical localizada quedó libre de crisis
discapacitantes, y el 21% mejoró. La resección neocortical abarca un grupo mucho más
heterogéneo de las cirugías y de los tipos de epilepsia. Los resultados probablemente
varían mucho según la región de la resección y la etiología. En un subgrupo de la serie que
se veía a los pacientes con lesiones discretas por separado
separado, el 63% de 131 pacientes con
lesiones quedaron libres de crisis.
Continued
Continued from slide 76
Sobre esta base de los datos la Academia Americana de Neurología, la Sociedad Americana de
Epilepsia, y la Academia Americana de Cirugía Neurológica llegó a la conclusión de que la
resección mesial anterior debe ser considerada en pacientes con crisis parciales complejas que
han fracasado a tratamiento apropiados con la primera línea de fármacos antiepilépticos y que
cumplen con los criterios establecidos para la resección del lóbulo temporal anteromesial. En lo
que respecta a resecciones localizadas neocorticales, concluyeron que no hay pruebas
suficientes en este momento para hacer una recomendación definitiva acerca de si los pacientes
se beneficiarán o no de la resección quirúrgica. (Engel 2003)
El estimulador del nervio vago (VNS) es un dispositivo que proporciona la estimulación
eléctrica intermitente del nervio vago. Se demostró en varios estudios que es eficaz en la
reducción de la frecuencia de las crisis parciales complejas, y recibió la aprobación de la
FDA para su uso en 1997. El estimulador es similar a un marcapasos cardíaco y se
implanta quirúrgicamente por vía subcutánea. La estimulación intermitente se descarga
cada 0,3-10 minutos por 7-30 segundos, los pacientes que experimentan un aura de crisis
pueden activar el dispositivo en forma manual, con un éxito en forma de anecdota para
algunos. El mecanismo por el cual la estimulación reduce las crisis no está bien
establecido.
t bl id L
Los efectos
f t adversos
d
iincluyen
l
ronquera, d
dolor
l d
de garganta
t o una sensación
ió
de disnea durante la estimulación, los cuales generalmente son leves. Los efectos
centrales de los antiepilépticos no están presentes. El estimulador se ha estudiado sólo en
combinación con el tratamiento con FAE, pero en este contexto, la eficacia frente a las
crisis parciales resistentes a los medicamentos-es comparable a la de algunos de los
nuevos fármacos antiepilépticos. El costo del dispositivo y su implantación pueden ser
factores limitantes. Los ensayos clínicos demuestran que menos del 5% de los pacientes
van a estar libres de crisis con la colocación del estimulador del nervio vago
vago, pero
aproximadamente 1 / 3 de los pacientes experimentarán una disminución clínica
significativa en la frecuencia de las crisis.
La FDA ha aprobado el dispositivo para las crisis de inicio parcial, pero que pueden tener
valor para las epilepsias generalizadas, especialmente el síndrome de Lennox-Gastaut y
crisis atónicas específicamente. Muchos centros intentan una estimulación del nervio vago
antes de la callostomía para crisis intratables atónicas. La estimulación del nervio vago
tiene una tasa de respuesta del 40% (es decir
decir,40%
40% de los pacientes tienen una
disminución del 50% o más en sus ataques). La estimulación del nervio vago se aprobó a
finales de 2005 para la depresión resistente al tratamiento médico por la FDA.
El estado epiléptico se define como: 1) un episodio de más de 10 minutos de actividad
crítica continua, o 2) dos o más crisis consecutivas que abarcan este período de tiempo
sin recuperación completa entre las crisis. Clínicamente, sin embargo, la mayoría de las
crisis duran menos de 5 minutos, y las persistentes es poco probable que cedan de forma
espontánea. Por lo tanto, se debe iniciar en estos casos el tratamiento inmediato en todas
las crisis epilépticas que duran más de 5 minutos.
La incidencia de estado epiléptico es por lo menos 60.000 casos / año en los EUA con
tasas más altas entre los niños y los adultos mayores. El estado epiléptico es una
emergencia
i y causa d
de alta
lt morbilidad
bilid d y mortalidad,
t lid d cualquier
l i titipo d
de crisis
i i se puede
d
manifestar como un estado epiléptico. El resultado de estado epiléptico convulsivo en gran
medida depende de la etiología, pero el tratamiento oportuno puede mejorar el resultado.
Desde un punto de vista práctico, el estado epiléptico puede ser dividido en convulsivo y
no convulsivo. Las formas pueden ser convulsivo generalizado o parcial. Las formas no
convulsivas son difíciles de clasificar por motivos clínicos,pero a menudo se divide
electroencefalográficamente en estado de ausencia (en la que el electroencefalograma
muestra generalizada actividad punta
punta-onda)
onda) y el estado parcial complejo (en el que el EEG
puede mostrar una variedad de descargas rítmicas localizadas) .
Un problema importante para médicos de atención primaria y también para los
neurólogos es el reconocimiento de eventos transitorios y paroxísticos que se asemejan
a las crisis epilépticas. Los ataques isquémicos transitorios, la migraña, los trastornos
del sueño, las alteraciones del movimiento, y los problemas
metabólicos pueden producir episodios de la actividad mental perturbada o alteraciones
en el movimiento. La adecuada Historia Clínica, el Examen Físico durante o después
de los ataques y hacer un abordaje de laboratorio adecuado e imagen, nos ayudan
a distinguir estos eventos de las crisis epilépticas.
El síncope vasovagal o de origen cardiogénico puede imitar los ataques epilépticos,
especialmente cuando se presenta con la extensión tónica del tronco y las extremidades o
varias sacudidas clónicas, por lo que es obligado conducir de forma correcta al
diagnóstico diferencial de una convulsión. La breve postura tónica y la presencia de
movimientos clónicos son comunes con síncope. En raras ocasiones, cuando la persona
es especialmente susceptible o la isquemia prolongada, una crisis puede causar un
síncope convulsivo, que es un trastorno cardiovascular primario no cerebral, y no debe ser
tratado como un ataque epiléptico. La pérdida de la conciencia exclusivamente en el pie o
sentado,
t d los
l estímulos
tí l dolorosos
d l
o un ambiente
bi t muy caliente
li t son ffactores
t
d
de provocación,
ió
y un pródromo de calor, náuseas, diaforesis, y la desaparición gradual de la visión
binocular sugieren al síncope en lugar de crisis epilépticas como primer diagnóstico. Un
rápido regreso a la conciencia normal también es más característico del síncope que de
crisis epilépticas.
La historia clínica puede ayudar a distinguir entre el síncope y las crisis epilépticas. Las
características más sugerentes de una crisis epiléptica generalizada son la
letargia postictal y la confusión (más útil), la mordedura de lengua, espuma en la
boca, cianosis, y mialgias difusas postictales.
.
La migraña y la epilepsia son los trastornos que comparten características similares desde
su fisiopatogenia. Aproximadamente el 6% de los migrañosos tienen epilepsia (frente al
0,5% en la población general) y 24% de los pacientes con epilepsia tienen migraña
(Ottman y Lipman, Neurología, 1996). Puede ser difícil distinguir la migraña de una crisis
parcial, sobre todo si coexisten en el mismo paciente. Las auras migrañosas tienen más
probabilidades de ser confundidas con crisis occipitales en que ambas pueden implicar un
fenómeno visual. Las auras de migraña son más largas en duración, por lo general son de
color negro y blanco, y no son seguidas por una generalización secundaria. Ambos
t t
trastornos
pueden
d o no incluir
i l i d
dolor
l d
de cabeza,
b
náuseas
á
y vómitos.
ó it
Las crisis psicógenas se observan en el 10 al 45% de los pacientes evaluados en los
centros de epilepsia. Pueden ocurrir a cualquier edad después de la infancia temprana y
son más comunes en las mujeres. El reconocimiento de ellas permite evitar las dosis
intoxicantes de FAE que se utilizan a menudo debido a que las convulsiones son
refractarias. Hay que mantener un alto grado de sospecha cuando las convulsiones son
refractarias a la terapia o cuando algunas características atípicas están presentes. Porque
los médicos generalmente se basan en el relato de los pacientes y testigos en de las crisis,
las posibilidades de un diagnóstico erróneoson altos. El diagnóstico se establece mejor
mediante
di t ell registro
i t d
de llos eventos
t tí
típicos
i
con vídeo-EEG.
íd EEG Las
L limitaciones
li it i
de
d esta
t técnica
té i
debe tenerse en cuenta, en particular la susceptibilidad a la circulación vascular, EKG y
otros artefactos, y el potencial de falsos negativos en las crisis parciales simples y algunas
crisis del lóbulo frontal (pseudo-pseudocrisis).
Una evaluación detallada de estrés psicosocial y la psicopatología subyacente es esencial.
En muchos casos, el estrés subyacente puede ser identificado (por ejemplo, la historia de
abuso físico o sexual). Una parte significativa (10-40%) de los pacientes con crisis
psicógenas también tienen epilepsia
epilepsia, una situación extremadamente difícil para el
diagnóstico y tratamiento.
Características clínicas
Advertencias/comentarios
Inicio gradual de crisis
Las crisis epilépticas inician subitamente, aunque
pueden ser precedidas
p
p
por un aura
p
Duración prolonmgada
Las crisis epilépticas duran menos de 4 min, aunque algunas crisis pueden ser mas largas; Distinguir entre el estado pre y postictal
Empujar, luchar, gritar, movimientos Pelvicos, voltear
lt
l
la cabeza
b
l d a lado, Enrrolarse, lado
l d E
l
hacer movimientos salvajes
Automatismos bizarros complejos pueden
originarse
i i
en las
l crisis parciales
ii
i l complejas
l j del d l
lóbulo frontal Actividad intermitente arrítmica Que termina
súbitamente
Los espasmos en las crisis GTC son rítmicos y corresponden a la fase clónica
Actividad clónica Que termina abruptamente
Al terminar las crisis los espasmos
Al terminar
crisis los espasmos se presentan
se presentan a a
intervalos largos
Actividad motora que para y reinicia
Extremedamente raro tener crisis aisladas; se distingue de una serie de crisis
Puede hablar mientras “debería estar
d
desconectado”
d ”
Puede haber automatismos de lenguaje en las
crisis parciales
ii
i l
La conciencia se mantiene mientras hay actividad TC
Crisis del area suplementaria motora puede
asociarse a crisis bilaterales tónicas o clónicas con lenguaje conservado
Actividad convulsiva en las extremidades Sin afección
f ió facial
f i l
La afección facial puede ser sutil
Terminación gradual de las crisis Las crisis epilépticas terminan subitamente, pero
pueden tener un periodo postictal gradual Características fluctuantes de una crisis a otra
Las crisis epilépticas son estereotipadas
No hay confusión postictal
Con frecuencia las crisis frontales y menos
frecuentemente las temporales tienen un postictal
de grito, con conducta agresiva verbal Sugestiuonables (abilidad para inducir una
nueva crisis epilépticas) crisis epilépticas)
No se lastiman después de muchas
recurrencias, aunque pueden Con frecuencia
de golpea al caer o se muerde lengua
El estrés y la sugestión pueden rara vez inducir la presencia de crisis de crisis
Presentarse en pacientes con historia de autodestrucción generalmente del mismo lado
Las crisis psicógenas son un síntoma común de la conversión o trastorno de somatización
y debe ser reconocida como una enfermedad discapacitante que requiere tratamiento
psiquiátrico .
En muchos casos, el mecanismo subyacente crisis psicógenas no se identifica, ya que los
pacientes pueden ser resistentes a la intervención psicológica o psiquiátrica. Aunque el
diagnóstico es difícil para el médico y para el paciente hacer frente a su problema, el
aprendizaje del diagnóstico y, por lo general, el llevar a cabo un buen tratamiento se logra
un buen control en aproximadamente el 50% de los pacientes
Psicógena NES debe distinguirse de la simulación, o conscientemente fingiendo ataques
epilépticos.
Para la distinción entre las pseudocrisis que imitan a la epilepsia, a menudo requiere de
monitorización mediante vídeo EEG en una unidad de vigilancia hospitalaria de Epilepsia
(UHE). El diagnóstico definitivo de los eventos no epilépticos es esencial para evitar
tratamientos innecesarios con fármacos antiepilépticos. También es importante para el
cuidado de las personas con epilepsia conocidas. Estas unidades ofrecen un entorno
seguro para la suspensión de los medicamentos antiepilépticos y la observación de las
crisis para la evaluación pre-quirúrgica. También permiten el reconocimiento de las crisis
no detectadas que es parte importante de la evaluación del paciente, sobre todo si la
conducción
d
ió d
de su ttratamiento
t i t no h
ha sido
id exitosa.
it
La epilepsia es única entre la variedad de problemas legales que crea. Entre estos, genera
tanto debate y controversia en la conducción de vehículos automotores
automotores. Todos los estados
de la Unión Americana regulan la conducción de las personas con epilepsia. El intervalo
adecuado libre de crisis antes de conducir es permitida o puede ser prescrita o sugerida
por cada Consejo Estatal, pero a menudo el privilegio de conducción se basa en un
comunicado médico sobre el paciente en particular. La mayoría de los estados dependen
de los solicitantes a divulgar su estado de salud pertinentes a la conducción. Algunos
estados (actualmente seis) requieren de presentación de informes médicos en forma
mandatoria.
mandatoria
Las declaraciones de los médicos son examinados por una junta de revisión médica, que
suele incluir neurólogos. La junta o permisos de conducir o especifica el periodo de
restricción antes de conducir y puede ser legalmente se reanuda. El tiempo requerido de
estar libre de crisis para la conducción varían de tres meses a dos años.
La conducción de vehículos por personas con epilepsia representa un riesgo pequeño pero
identificable a la seguridad pública, mientras que los riesgos individuales de los pacientes
puede ser mayor.
p
y Es una tarea difícil idear un método efectivo de control de la seguridad
g
de conducción entre las personas con epilepsia que no sea discriminatorio y al mismo
tiempo que proteja tanto al paciente como a su entorno.
Una gran variedad de defectos al nacimiento se asocian con el uso de prácticamente todos
los medicamentos antiepilépticos. Entre los defectos congénitos más graves están los
principales defectos del tubo neural (espina bífida y anencefalia). Dado que existe
evidencia de que los suplementos de ácido fólico pueden reducir el riesgo de estos
defectos, muchos neurólogos proporcionan folato suplementario a todas las mujeres en
edad fértil cuando están en tratamiento para la epilepsia.
Una de las principales preocupaciones de las mujeres en edad fértil es el potencial
teratogénico de los antiepilépticos. Considerando que la incidencia de defectos de
nacimiento graves (que requieren una intervención médica o quirúrgica) en la población
normal es de aproximadamente un 2-3%, es aproximadamente dos veces más o del 7.4%
en la descendencia de las mujeres con monoterapia Los FAE han sido reconocidos como
facilitadores de los principales defectos de nacimiento, con otro 5-10% de las anomalías
cosméticas menores tales como dedos acortados en su porción distal. Los FAE se
perciben como la razón principal del aumento del riesgo de malformaciones fetales,
aunque algunos
l
pueden
d estar
t relacionados
l i
d con lesiones
l i
provocadas
d por las
l
crisis durante el embarazo o anomalías genéticas realizadas a la madre. Mientras que los
médicos no pueden hacer mucho acerca de este problema, hay una evidente necesidad de
un control óptimo de las crisis maternas frente a la teratogenicidad de los
antiepilépticos (especialmente al principio del embarazo).
Los datos disponibles sobre las principales malformaciones congénitas y los FAE
provienen de diferentes estudios basados en la población, los registros de embarazo en los
hospitales y las compañías farmacéuticas. Estos registros utilizan diferentes métodos de
comprobación de datos lo que hace difícil comparar los datos entre los estudios y datos
sobre varios de los fármacos antiepilépticos más nuevos que es aún limitada. El Valproato,
sin embargo, ha sido consistentemente asociado con tasas más altas de malformaciones
fetales.
Las crisis neonatales pueden ser un signo neurológico. La importancia de las crisis
neonatales se encuentra en su asociación con una variedad de enfermedades
subyacentes graves, incluida la encefalopatía hipóxico-isquémica, la infección, el
traumatismo, la enfermedad cerebrovascular, los trastornos metabólicos, y las
malformaciones del desarrollo cortical. Aunque depende de la etiología subyacente, hay
una alta tasa de mortalidad y morbilidad neurológica. Las manifestaciones clínicas y
electroencefalográficas de las crisis neonatales difieren de las de más edad y de los
individuos neurológicamente mas maduros. Esto refleja las diferencias funcionales, tales
como un bajo
b j grado
d d
de mielinización
i li i
ió en ell cerebro
b en d
desarrollo.
ll
La incidencia de convulsiones neonatales varía enormemente, desde 3% a 25%. Parte de
esta variación probablemente refleja las dificultades en el diagnóstico. Las convulsiones se
asocian con mayores tasas de mortalidad y morbilidad neurológica crónica, con secuelas
hasta en el 50% al 70%. Las crisis pueden ayudar a identificar una enfermedad tratable
que puede causar daño cerebral permanente. Por ejemplo, la hipoglucemia y la meningitis
bacteriana pueden causar convulsiones neonatales. En tales circunstancias, el tratamiento
rápido y adecuado puede detener el progreso de la enfermedad y prevenir el daño
adicional al cerebro.
El diagnóstico de las crisis neonatales se ha basado históricamente en la inspección
visual, aunque los estudios con EEG vídeo, han demostrado que el diagnóstico clínico de
las crisis neonatales no es fiable. Un sistema de clasificación de crisis neonatales, basado
en signos clínicos, ha ido evolucionando, e incluye crisis clónicas, mioclónicas, tónicas, y
"sutiles“ . Es un problema relativamente frecuente el reconocimiento de crisis clónicas
focales, convulsiones tónicas focales, y estos eventos clínicos tienen una correlación fiable
con la actividad en el EEG ictal. Las crisis mioclónicas, las crisis generalizadas tónico y las
crisis sutiles son más difíciles de diagnosticar clínicamente. Aunque las conductas clínicas
para eventos
t como mioclonías,
i l í
postura
t
tónica,
tó i
apnea, empuje
j d
de lla llengua, movimientos
i i t
de succión, y nistagmo ocular pueden ser parte de la evaluación clínica, no tienen una alta
correlación con la actividad en el EEG ictal. Más bien estos eventos reciben el nombre de
"automatismos" o "signos de liberación del tallo cerebral."
La evaluación del niño con crisis neonatales no debe esperar la confirmación por EEG. La
sospechas de las crisis neonatales sigue un curso triple simultáneos: 1) confirmar
el diagnóstico con EEG, 2) evaluar en el bebé la causa(s) de las crisis, haciendo hincapié
en las etiologías tratables, y 3) poner en marcha los Fármacos antiepilépticos (FAE) para la
terapia. Muchos niños con crisis neonatales han tenido enfermedades médicas
o neurológicas subyacentes que provocan las crisis, es decir, la
manifestación aguda, sintomática, no un "umbral convulsivo disminuido“ que
intrínsecamente constituyen el sustrato de la epilepsia.
El fenobarbital sigue siendo el fármaco más comúnmente utilizado para las convulsiones
neonatales. Las alternativas incluyen la fenitoína y las benzodiacepinas. Muchos de los
fármacos antiepilépticos más nuevos tienen la experiencia anecdótica en esta población y
pueden ser utilizados clínicamente si se juzga conveniente.
Algunos recién nacidos posteriormente desarrollan enfermedades crónicas, incluyendo las
condiciones de refractarios como West o síndrome de Lennox-Gastaut. La frecuencia con
la que se informa la epilepsia crónica que se desarrolla después de las crisis neonatales
varía del 4% al 56% de aquellos que sobreviven el período neonatal.
U d
Una
de llas ttareas más
á difí
difíciles
il en ell manejo
j iintegral
t
ld
dell recién
ié nacido
id con convulsiones
l i
es
un diálogo franco, la discusión precisa y realista de pronóstico con los padres del bebé.
Las convulsiones son un signo de peligro y los riesgos de muerte posterior, parálisis
cerebral, retraso mental, epilepsia, la atención / hiperactividad, trastornos de conducta y
otros trastornos relacionados, sistema nervioso central con sede deben ser
cuidadosamente evaluados y comunicados.
Síndromes Epilépticos Selectos en Pediátrica
Los síndromes epilépticos se pueden clasificar en función de que las crisis asociadas si
son parciales o generalizadas, y si la etiología es idiopática o sintomática /
criptogénica. Varios síndromes pediátricos importantes se pueden clasificar de acuerdo a
la edad de aparición y el pronóstico. Estos pueden dividirse en las encefalopatías
epilépticas de la infancia y la niñez temprana, las crisis febriles y los síndromes benignos
parciales y generalizados de la niñez y la adolescencia.
Síndromes Catastróficos Epilépticos de los prescolares y escolares
E t encefalopatías
Estas
f l
tí epilépticas
ilé ti
característicamente
t í ti
t están
tá presentes
t en la
l vida
id
temprana, y puede resultar de una variedad de trastornos subyacentes. Si bien la edad
de inicio varía entre los diversos síndromes, su base etiológica es común y tiene
coincidencias en las características clínicas y EEG que sugieren que forman un espectro
común. 1. Síndrome de West se inicia generalmente en el primer año de vida, por lo
general entre 3 y 8 meses, y presenta una tríada distintiva electroclínica consistente en
los espasmos infantiles, la hipsarrítmia en el EEG (actividad caótica, de alto voltaje con
puntas multifocales) y el retraso psicomotor
psicomotor. Los espasmos flexores son típicos
típicos, pero la
postura extensora y funciones de coordinación motora inadecuada son comunes. Estos
suelen durar varios segundos cada uno, pero ocurren en grupos (salvas) de varios
minutos o más. Puede haber lesión cerebral prenatal y perinatal, enfermedades
metabólicas, degenerativas y trastornos neurocutáneos (eclerosis tuberosa) y
malformaciones cerebrales, las que son frecuentemente identificadas.
Continued from slide 125
Los factores asociados con un mal pronóstico incluyen la aparición antes de 3 meses de edad, la
etiología de los síntomas, y varios tipos de crisis. El desarrollo neurológico es normal en sólo 1015% de los pacientes afectados. Los agentes farmacológicos utilizados para tratar el Síndrome
d W
de
Westt son las
l benzodiazepinas,
b
di
i
ell valproato
l
t y los
l corticosteroides
ti
t id o corticotropina
ti t i
(ACTH). Más recientemente, la vigabatrina (un inhibidor de la enzima GABA-catabólico GABA
transaminasa) se ha introducido con éxito, especialmente para pacientes con esclerosis
tuberosa. Otras terapias que se informan con éxito anecdótico incluyen a la zonisamida, al
topiramato, y la dieta cetogénica. El Síndrome de West asociado con displasia cortical focal ha
sido tratada por cirugía resectiva.
2. El síndrome de Lennox-Gastaut se caracteriza por múltiples tipos de convulsiones, retraso
mental y descargas en el EEG de punta
punta-onda
onda lenta
lenta. Las crisis comienzan en las edades 1
1-7
7
años; hasta un 25% de los pacientes se presentan inicialmente con Síndrome de West. Las crisis
pueden ser tónicas, atónicas, de ausencia atípica y convulsiones tónico-clónicas son frecuentes,
mientras que las crisis mioclónicas son menos comunes. La aparición del síndrome de LennoxGastaut puede ser gradual o abrupto, pero se asocia típicamente con algún retroceso en el
desarrollo.
El pronóstico para los pacientes con el síndrome de Lennox-Gastaut es pobre. Múltiples tipos de
crisis gradualmente dan paso a un solo patrón predominante en la segunda década de la vida
vida,
mientras que la discapacidad mental y las limitaciones sociales son permanentes. episodios
recurrentes de estado epiléptico son comunes, y los FAE estándar para el control de las crisis a
menudo dan un resultado insatisfactorio. La aparición del síndrome de Lennox-Gastaut antes de
2 años de edad tiene un resultado particularmente desfavorable. La terapia con corticosteroides
puede proporcionar control a corto plazo de las crisis y la dieta cetogénica ha sido exitosa en
pacientes seleccionados. El valproato de sodio y la lamotrigina son favorecidos por muchos
estudios y trabajos preliminares con topiramato sugieren un papel beneficioso
beneficioso. El Felbamato
también es beneficioso, pero pueden tener un mayor potencial de efectos adversos graves. La
dieta cetogénica y la estimulación del nervio vago también se han utilizado con éxito.
3. epilepsias mioclónicas del prescolar y del escolar son un grupo heterogéneo de trastornos
caracterizados por diferentes manifestaciones clínicas de la epilepsia mioclónica, a menudo
asociada con crisis generalizadas o parciales, retraso del desarrollo neurológico y las
anormalidades generalizadas de EEG. La variedad de presentaciones electroclínicas ha dado
g a un sistema de clasificación complejo
p j y confuso. Todos los p
pacientes con epilepsia
p p
lugar
mioclónica, especialmente aquellos con retraso en el desarrollo o regresión, deben someterse a
una evaluación cuidadosa de las causas subyacentes incluyendo displasia cerebral, esclerosis
tuberosa, lipofuscinosis ceroide neuronal, y el síndrome de Angelman.
Dado que los datos específicos de la eficacia en la infancia son limitados, la selección de
los FAE para las crisis en la infancia a menudo se basan en la extrapolación de los
estudios de eficacia en los adultos. Afortunadamente, la eficacia de la mayoría de los
agentes en los niños parece ser paralelo a la experiencia adulta. Sin embargo, los efectos
adversos y toxicidad a menudo son el principal determinante de la selección de
medicamentos en cualquier grupo de edad. No es raro, por ejemplo, para los padres
solicitar el retirar un fármaco debido a efectos adversos, incluso a costa de la exacerbación
de las crisis.
E los
En
l llactantes
t t y niños
iñ pequeños,
ñ
llas iinteracciones
t
i
d
de llos antiepilépticos
ti ilé ti
con fó
fórmulas
l d
de
leche y el niño son un problema potencial. La administración de medicamentos
antiepilépticos, mientras que la alimentación se debe evitar en esta población. Factores de
maduración y la variabilidad en la absorción y el metabolismo puede indicar a la atención
de monitorización de drogas en su concentración sérica. Los problemas específicos con
efectos adversos también pueden requerir del control metabólico. La mayoría de los niños
no se puede esperar que trague en forma fiable la tableta o la cápsula por lo que se
requiere de formulaciones espéciales de medicamentos hasta los 8
8-10
10 años de edad
edad. Esto
hace que los preparados líquidos, tabletas masticables y el espolvorear formulaciones de
cápsulas alternativas sean atractivas en el niño más pequeño e infantil. Por último, hay
que tener en cuenta que los niños tienen en constante cambio de masa corporal y el
metabolismo que afectan a la absorción, distribución y eliminación de la medicación. Un
regular ajuste de dosis con el tiempo se necesitan y los niños más pequeños suelen
requerir una dosis relativamente mayor de la medicación en miligramos por kilogramo por
día que los niños mayores y adultos.
adultos
Manejo Psicosocial
El deterioro social y neurológico están fuertemente asociados con la aparición de la epilepsia
en la infancia. El pronóstico a largo plazo de la epilepsia refractaria es pobre y muchas veces
impide el funcionamiento normal del adulto. Si la educación se ve comprometida, menos del
5% de los pacientes seguidos en función de la edad adulta se manejan con normalidad. Por
esta razón, es esencial que aliados los profesionales de la salud tales como neuropsicólogos,
educadores especiales, y los trabajadores sociales ayudan a los padres a manejar
adecuadamente al niño con epilepsia.
Di t cetogénica
Dieta
t é i
Los niños con crisis intratables que demuestran a la medicación o que experimentan efectos
adversos intolerables de la medicación pueden ser candidatos para la terapia no
farmacológica con la dieta cetogénica. La dieta también puede permitir una simplificación de
un régimen de tratamiento de drogas y, en ocasiones, la interrupción de todos los
medicamentos antiepilépticos.
Cirugía de Epilepsia
Los niños que sufren de epilepsia refractaria son cada vez más fácil de referirse a la cirugía
cirugía.
Esta tendencia ha dado como resultado de los avances en la apreciación de las perspectivas
pronósticas a largo plazo de estos niños, la identificación de sustratos neuropatológicos de la
enfermedad, la selección de pacientes adecuados, y la localización del foco epiléptico. Una
pequeña minoría de los niños con crisis refractarias presentan remisión espontánea, pero la
ausencia de crisis no se puede predecir por muchos años, y saber cuando se producirá la
remisión es prácticamente imposible. Aunque no hay datos comparativos a largo plazo, cabría
esperar que el alivio de crisis antes que contribuir a una mayor reducción de la morbilidad
psicosocial, una mejor oportunidad para una educación significativa y una mejor calidad de
vida. Los niños con epilepsia intratable que no son candidatos a cirugía de epilepsia, pueden
beneficiarse de la estimulación del nervio vago.
Estas diapositivas
p
tienen un nivel más complicado
p
que el resto de la charla
q
¿Indicaciones del levetiracetam?