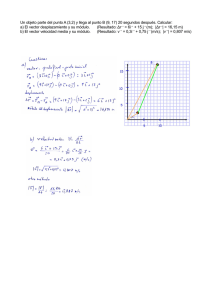
composicion antitumoral a base de polipetido inmunogeno
Anuncio

19 OFICINA ESPAÑOLA DE PATENTES Y MARCAS 11 Número de publicación: 2 229 528 51 Int. Cl. : C07K 14/025 7 C12N 15/86 A61K 39/12 ESPAÑA 12 TRADUCCIÓN DE PATENTE EUROPEA T3 86 Número de solicitud europea: 98939708 .8 86 Fecha de presentación: 17.07.1998 87 Número de publicación de la solicitud: 0989999 87 Fecha de publicación de la solicitud: 05.04.2000 54 Título: Composición antitumoral a base de polipéptido inmunógeno de localización celular modificada. 30 Prioridad: 18.07.1997 FR 97 09152 73 Titular/es: TRANSGENE S.A. 11, rue de Molsheim 67000 Strasbourg, FR 45 Fecha de publicación de la mención BOPI: 16.04.2005 72 Inventor/es: Kieny, Marie-Paule; Balloul, Jean-Marc y Bizouarne, Nadine 45 Fecha de la publicación del folleto de la patente: 74 Agente: Curell Suñol, Marcelino ES 2 229 528 T3 16.04.2005 Aviso: En el plazo de nueve meses a contar desde la fecha de publicación en el Boletín europeo de patentes, de la mención de concesión de la patente europea, cualquier persona podrá oponerse ante la Oficina Europea de Patentes a la patente concedida. La oposición deberá formularse por escrito y estar motivada; sólo se considerará como formulada una vez que se haya realizado el pago de la tasa de oposición (art. 99.1 del Convenio sobre concesión de Patentes Europeas). Venta de fascículos: Oficina Española de Patentes y Marcas. C/Panamá, 1 – 28036 Madrid ES 2 229 528 T3 DESCRIPCIÓN Composición antitumoral a base de polipéptido inmunógeno de localización celular modificada. 10 La presente invención tiene por objeto una composición antitumoral que comprende, como agente terapéutico o profiláctico, al menos un vector recombinante que contiene las secuencias que codifican un polipéptido inmunógeno modificado a fin de presentar una localización membránica en la superficie de las células en las que se expresa, diferente de su localización nativa. La presente invención se refiere asimismo a una composición antitumoral que comprende dicho polipéptido inmunógeno. Tal composición está destinada más particularmente al tratamiento o a la prevención de lesiones asociadas a los papilomavirus. 15 Se admite generalmente que el cáncer es una enfermedad que resulta de una pérdida de control de la multiplicación celular. Sus causas pueden ser múltiples y debidas especialmente a una disfunción de los genes celulares (por ejemplo, activación mediante mutación somática de genes potencialmente oncogénicos; desregulación de la expresión; inhibición de la expresión de genes supresores de tumores), o a la expresión no deseada de los genes víricos. 5 20 25 30 35 40 45 50 55 En el ser humano, los papilomavirus (HPV) se asocian a patologías tales como una infección benigna de la piel, verrugas, hasta tumores malignos. Entre los 75 tipos de HPV identificados hasta ahora, 20 aislados distintos son altamente específicos de los conductos genitales, y 5 de ellos (HPV-16 y 18, y, en menor grado, los HPV-31, 33 y 45) están claramente asociados con el cáncer de cuello uterino y de las vías bajas. Toda una serie de estudios demuestra el papel transformador de estos virus, su integración específica en el genoma de células neoplásicas, su actividad génica en las células cancerígenas y la importancia de la expresión de algunos genes víricos en el mantenimiento del fenotipo maligno de las células neoplásicas HPV positivas (Monsenego, J. Impact Medecin, 11 de marzo de 1994). De manera general, los papilomavirus son virus con ADN que posee un genoma circular de aproximadamente 7900 pares de bases rodeado por una cápsida proteica. Se ha identificado un cierto número de tipos de papilomavirus, especialmente bovinos (BPV) y humanos (HPV) (Pfister, 1987, en The papoviridae: The Papillomaviruses, edición Salzman y Howley, Plenum Press, Nueva York, p. 1-38). Sus genomas comprenden una región precoz que contiene los cuadros de lectura E1, E2, E4, E5, E6 y E7, y una región tardía que codifica las proteínas de cápsida L1 y L2. Las proteínas precoces tienen la capacidad de unir el ADN, y se encuentran de manera predominante en el núcleo. Los productos de expresión E1 y E2 regulan la replicación vírica y la expresión de los genes víricos, mientras que los de las regiones E5, E6 y E7 están implicados en los procesos de transformación oncogénica de las células infectadas. Para ello, se ha demostrado experimentalmente que la proteína E5 de BPV-1 puede transformar células in vitro (Schlegel et al., 1986, Science, 233, 464-467). Se ha demostrado el poder transformador del E7 para HPV-16 y HPV-18 (Kanda et al., 1988, J. Virol. 62, 610-613; Vousden et al., 1988, Oncogene Res., 3, 1-9; Bedell et al., 1987, J. Virol. 61, 36353640), y se ha correlacionado con su capacidad en unir el producto del gen del retinoblastoma (Rb) (Munger et al., 1989, EMBO J. 8, 4099-4105; Heck et al., 1992, Proc. Natl. Acad. Sci. USA 89, 4442-4446). Por otra parte, Crook et al. (1991, Cell 67, 547-556) han demostrado que el antígeno E6 del HPV-16 y 18 puede complejar el producto del gen p53, lo que explica su papel predominante en la transformación celular. Las patologías asociadas a los virus HPV plantean un problema terapéutico por el hecho de su naturaleza persistente y recurrente. Aunque los enfoques clásicos son la cirugía y la quimioterapia, ahora se considera la inmunoterapia para el tratamiento de estas enfermedades. El candidato ideal como vacuna debe, a título preventivo (inmunoprofilaxis), impedir que la infección se establezca de forma duradera y se propague a los tejidos vecinos, y, a título curativo (inmunoterapia), debe reducir la evolución tumoral en los pacientes infectados. Se ha propuesto hasta ahora utilizar los antígenos de cápsida para inducir la producción de anticuerpos contra los epítopos localizados en la superficie de las partículas víricas, y las proteínas precoces para establecer la inmunidad celular frente a las células infectadas después de la integración del ADN vírico. A este respecto, la patente europea EP 0.462.187 describe un enfoque terapéutico mediante administración de poxvirus que expresan los genes precoces de los papilomavirus. El enfoque de vacunación descrito en el documento WO 93/02184 se basa en la utilización de los antígenos de cápsida como agentes inmunógenos y especialmente como partículas víricas vacías de ADN (VLP del inglés partículas similares a virus) reconstituidas in vitro. La solicitud francesa 96/09584 divulga una composición que asocia el efecto preventivo proporcionado por los polipéptidos precoces y el efecto curativo conferido por los polipéptidos tardíos de los papilomavirus. Sin embargo, hasta ahora se han utilizado las proteínas víricas, eventualmente mutadas a fin de eliminar su eficacia transformadora, pero no obstante nativas desde el punto de vista de su localización celular. 60 65 Se puede igualmente citar la solicitud internacional de patente publicada con el número WO 94/21680 que describe vectores que comprenden secuencias que codifican un polipéptido inmunógeno fusionado con una secuencia señal del retículo endoplásmico, y sus utilizaciones en composiciones para el tratamiento de cánceres y de infecciones víricas, interviniendo la secuencia señal para permitir el anclaje de los polipéptidos quiméricos en la membrana del retículo endoplásmico. La presente invención propone la utilización de las proteínas inmunógenas, cuya localización ha sido modificada con el objeto de mejorar su accesibilidad al sistema inmunitario del hospedante a fin de mejorar o estimular una 2 ES 2 229 528 T3 respuesta inmunitaria atendiendo al tumor o al cáncer a tratar, que sea específica o no y de tipo humoral (producción de anticuerpos) o celular (respuesta citotóxica CTL). 5 10 15 20 25 30 35 40 45 Se han modificado ahora los antígenos nucleares E6 y E7 del HPV-6 mediante introducción de secuencias de anclaje y de secreción apropiadas a fin de conferirles una presentación transmembránica. El cambio de localización tiene un efecto beneficioso sobre la respuesta inmunitaria, que se traduce por una actividad antitumoral superior en los animales tratados con un virus de la viruela vacuna que coexpresa los antígenos E6 y E7 membránicos y la IL-2 humana, comparado con los animales que han recibido un virus equivalente que produce los antígenos nucleares. El objeto de la presente invención es poner a disposición del público composiciones antitumorales más eficaces que las composiciones de la técnica anterior, para inhibir al menos parcialmente el establecimiento o la progresión de un tumor o un cáncer. Una aplicación particularmente útil es el tratamiento de las infecciones por HPV y más particularmente de patologías graves tales como el cáncer de cuello uterino. Es por ello que la presente invención tiene por objeto una composición antitumoral que comprende, como agente terapéutico o profiláctico, al menos un vector recombinante que contiene las secuencias que codifican uno o varios polipéptidos inmunógenos, presentando naturalmente al menos uno de dichos polipéptidos una localización no membránica y estando modificado por una inserción de una secuencia de anclaje membránica y, en el caso de que el polipéptido inmunógeno nativo no la tenga, de una secuencia de secreción, a fin de presentar una localización membránica en la superficie de las células en las que se expresa, presentando dicho polipéptido inmunógeno un grado de similitud superior a 70% con todo o parte de un polipéptido codificado por una región precoz y/o tardía de un papilomavirus. En el sentido de la presente invención, la expresión “polipéptido inmunógeno” designa un polipéptido que no encuentra su equivalente en las células normales. Un ejemplo preferido está constituido por un antígeno específico de tumores. Como ilustración, se pueden citar los antígenos celulares cuya expresión aparece durante el periodo fetoembrionario y se reduce desde el parto hasta su desaparición; los antígenos que se expresan normalmente en un nivel muy bajo y que, expresados en un nivel fuerte, se vuelven característicos de un tumor; los antígenos celulares cuya estructura o conformación se modifica; o también los antígenos no celulares, particularmente víricos que derivan de un virus oncogénico. Por ejemplo, se puede tratar de productos de expresión de los genes BRCA-1 (Miki et al., 1994, Science 226, 66-71), BRCA-2 (Wooster et al., 1995, Nature 378, 789-792), MUC-1 (Hareuveni et al., 1990, Proc. Natl. Acad. Sci. USA 87, 9498-9502), CEA, etc., en los que están implicadas algunas mutaciones o la sobrexpresión en el desarrollo cancerígeno. Por lo que respecta a los antígenos víricos, se pueden citar más particularmente los productos de expresión de los genes precoces o tardíos de los papilomavirus, EBNA-1 del virus Epstein Barr, los antígenos de los virus HTLV (virus de linfocitos T humanos) I y II, o los virus de las hepatitis B y C. Los antígenos específicos de tumores se describen ampliamente en la bibliografía accesible para el experto en la técnica. La característica esencial del polipéptido para uso en la presente invención es presentar una localización diferente de su localización nativa. Los mecanismos de transporte y las señales implicadas se describen en los documentos de biología celular (véase, por ejemplo, Molecular Biology of the Cell, tercera ed., Garland Publishing Inc., NY y Londres). Brevemente, la gran mayoría de los polipéptidos se sintetiza en ribosomas libres en el citosol, en el que ejercen su actividad. Sin embargo, algunos polipéptidos tienen un destino celular diferente, generalmente determinado por la presencia de señales peptídicas apropiadas, y deben ser transportados hasta ellas. Así, los polipéptidos destinados a ser exportados hacia la membrana plasmática, o segregados en el exterior de la célula, se sintetizan mediante ribosomas asociados al retículo endoplásmico (RE), generalmente en forma de precursores que tienen en su extremo aminoterminal una secuencia de secreción (o péptido señal) que inicia su paso al RE. Después, se elimina mediante una endopeptidasa específica para dar el polipéptido maduro. Una secuencia de secreción contiene habitualmente 15 a 35 aminoácidos esencialmente hidrofobos. No existe secuencia consensual, y parece que la estructura secundaria es la que determina el reconocimiento mediante la endopeptidasa. Sin embargo, ruptura proteólica tiene lugar lo más frecuentemente después un residuo glicina, serina o alanina. 50 55 60 65 Las proteínas membránicas contienen generalmente una secuencia de anclaje de naturaleza fuertemente hidrófoba que queda insertada en la membrana plásmica. El polipéptido puede ser transmembránico con uno de sus extremos expuesto en el exterior de la célula, atravesando la secuencia de anclaje la membrana y el otro extremo del lado citosólico. En la mayoría de los casos, la cadena polipeptídica imbricada en la doble capa lipídica de la membrana tiene una conformación en hélice α (véase, por ejemplo, Branden y Tooze, 1991, en Introduction to Protein Structure, p. 202-214, NY Garland). En cuanto a la localización nuclear, se puede conferida por la presencia de una secuencia corta llamada de localización nuclear (NLS) compuesta principalmente de restos cargados positivamente, tales como lisina y arginina. A título de ejemplos, se pueden citar las señales de translocación nuclear KRKKRK y RKRRKR presentes en los polipéptidos L1 y L2 de HPV (Zhou et al., 1991, Virology 185, 625-632). Sin embargo, algunos polipéptidos que ejercen su función en el seno del núcleo no poseen secuencia NLS típica. Es el caso de los antígenos E6 y E7 del papilomavirus. El polipéptido inmunógeno comprendido en la composición según la invención puede resultar de la introducción y/o la eliminación de señales de localización apropiadas en el seno de un polipéptido nativo, de un fragmento de éste, de una quimera que contiene secuencias de orígenes diferentes o de una variante (eliminación, inserción y/o sustitución de uno o varios aminoácidos). De manera más particular, su secuencia de aminoácidos presenta un grado de similitud, con todo o parte de la secuencia del polipéptido nativo del que deriva, superior a 70%, ventajosamente 3 ES 2 229 528 T3 superior a 80% y, preferentemente, superior a 90%. El grado de similitud se puede calcular fácilmente con la ayuda de un programa informático apropiado, o alineando las secuencias a fin de obtener el grado máximo de homología y contando el número de posiciones en las que los aminoácidos de las dos secuencias son idénticos con relación al numero total de posiciones. 5 10 15 20 25 30 35 40 El experto en la técnica conoce las señales que permiten el cambio de presentación celular de un polipéptido. En el caso en el que se desee que se segregue el polipéptido inmunógeno, la adición de una secuencia de secreción en su extremo aminoterminal permitirá su transporte vía el RE hacia el exterior de la célula hospedante. La inserción tiene lugar con preferencia inmediatamente en dirección 3’ del codón iniciador de la traducción. En el ámbito de la presente invención, puede ser ventajoso mutar/eliminar todo o una parte de los restos que determinan la localización nativa, para evitar las interferencias. Por ejemplo, si el polipéptido nativo tiene un destino membránico, ya tiene una secuencia de secreción y se procederá eventualmente a la mutación o eliminación de la secuencia de anclaje hidrófoba. Además, la inactivación (por mutación/eliminación) de las señales nativas puede permitir una localización citoplásmica. La presentación citoplásmica de un péptido inmunógeno que presenta normalmente una localización celular diferente (por ejemplo, nuclear, membránica, segregada, etc.) puede favorecer la presentación peptídica mediada por los antígenos de clase I del complejo mayor de histocompatibilidad y la respuesta CTL in vivo (Tobery y Siliciano, 1997, J. Exp. Med. 5, 909-920). Otro modo de realización factible consiste en fusionar el polipéptido inmunógeno con la ubiquitina igualmente con el objetivo de estimular una respuesta CTL potente. La composición según la invención comprende al menos un vector recombinante que contiene las secuencias que codifican un polipéptido inmunógeno modificado a fin de presentar una localización membránica en la superficie de las células en las que se expresa, mediante inserción de una secuencia de anclaje membránica y, en el caso en el que el polipéptido nativo no la tenga, de una secuencia de secreción. El sitio de inserción preferido de la secuencia de secreción es el extremo N-terminal, según se indica anteriormente, y el de la secuencia de anclaje membránica es el extremo C-terminal, por ejemplo inmediatamente en dirección 5’ del codón de finalización. En este contexto, puede ser igualmente ventajoso proceder a la mutación/eliminación de todo o parte de las señales de localización nativas (por ejemplo, secuencia NLS) para no interferir con la nueva localización. La elección de la señal de localización susceptible de ser utilizada en el ámbito de la presente invención es amplia. Puede derivar de toda proteína que tenga un origen eucariota o no (virus, parásito, hongo, etc.) desde el momento en que sea reconocida por la célula a tratar. Puede ser natural o sintética, heteróloga u homóloga frente a esta última. Puede igualmente tener una o varias modificaciones con relación a la señal de la que deriva, con tal de que no afecte(n) a su función. A título indicativo, se prefiere recurrir a las secuencias de secreción y/o de anclaje membránica de la glicoproteína de la rabia, de la glicoproteína env del virus del VIH, o de la proteína F del virus de la rubéola. En el caso en el que la modificación del polipéptido inmunógeno haga intervenir varias señales de localización (por ejemplo, secuencia de secreción y de anclaje membránica), estas pueden tener un origen común o diferente. Es igualmente posible utilizar señales de localización que se dirijan a un compartimento celular particular. Se puede citar especialmente una secuencia consenso de endocitosis (por ejemplo, la presente en la región C-terminal de las cadenas pesadas de inmunoglobulina IgG1 de secuencia IPNYRNM; Kaisho et al., 1997, Science 276, 412-414), o una secuencia que permita dirigirse a la membrana del aparato de Golgi (Mochamer y Rose, 1987, J. Cell Biol. 105, 1205-1214; Mochamer, 1993, Curr. Opin. Cell Biol. 5, 606-612). Se puede contemplar particularmente el uso de secuencias derivadas de la glicoproteína E1 de Coronavirus divulgadas en la base de datos Swiss-Prot con el numero de accesión P11222. Este tipo de secuencia se puede incorporar en el extremo C-terminal del polipéptido inmunógeno. 45 Por otra parte, la modificación de la localización celular se puede realizar mediante toda técnica convencional, especialmente mediante mutagénesis dirigida, ligación de señales exógenas o PCR. 50 Según un modo de realización preferido, una composición antitumoral según la invención se destina para tratar o prevenir las infecciones con papilomavirus y los trastornos resultantes de las mismas, en particular las displasias del cuello de bajo grado y el cáncer de cuello uterino. La composición antitumoral según la invención comprende al menos un polipéptido inmunógeno originario de una región precoz y/o tardía de un papilomavirus, especialmente de un virus de riesgo, tal como HPV-16, 18, 31, 33 o también 45. 55 Conforme a los objetivos perseguidos por la presente invención, se puede utilizar uno cualquiera o varios polipéptidos inmunógenos de papilomavirus. Como se recuerda anteriormente, su genoma codifica 8 polipéptidos, dos polipéptidos tardíos L1 y L2 que componen la cápsida vírica, y 6 polipéptidos precoces (E1, E2, E4, E5, E6 y E7) implicados en la regulación, el mantenimiento del genoma vírico y la transformación de las células infectadas. 60 Tratándose de un polipéptido inmunógeno de tipo precoz, se elige ventajosamente utilizar un polipéptido E6 o E7, modificado especialmente a fin de presentar una localización membránica. Habiéndose hecho las observaciones anteriores sobre el poder transformador, se recurre preferentemente a una variante no oncogénica, mutada en el nivel de la región implicada en el proceso de transformación celular. Tales variantes se describen en la bibliografía (Munger et al., 1992, EMBO J. 8, 4099-4105; Crook et al., 1991, Cell 67, 547-556; Heck et al., 1992, Proc. Natl. Acad. Sci. USA 89, 4442-4446; Phelps et al., 1992, J. Virol. 66, 2418-2427). Un polipéptido inmunógeno que conviene particularmente a los fines de la presente invención es el antígeno E6 del HPV-16 eliminado de los restos 111 a 115 (+1, que representa el primer aminoácido del antígeno vírico nativo) y fusionado a las señales de secreción y de anclaje de la proteína F del virus de la rubéola (SEC ID nº: 1). Se puede igualmente utilizar el antígeno E7 de HPV-16 eliminado de los restos 65 4 ES 2 229 528 T3 22 a 25 y fusionado a las secuencias de anclaje y de secreción de la glicoproteína de la rabia (SEC ID nº: 2). La composición antitumoral según la invención puede igualmente comprender un polipéptido inmunógeno originario de la región tardía de un papilomavirus, que deriva del L1 o L2. 5 10 15 Bien entendido, la composición antitumoral según la invención puede comprender varios polipéptidos inmunógenos, de los cuales al menos uno presenta una localización celular diferente de su localización nativa. Se puede citar, como ilustración, una composición que asocia varios polipéptidos derivados del papilomavirus, que pueden tener un origen común o diferente (por ejemplo HPV-16 y 18 con el objetivo de ampliar el espectro de acción). Una composición que combina varios polipéptidos de origen precoz permitirá mejorar el efecto terapéutico. La combinación de los polipéptidos derivados de L1 y L2 podrían tener un efecto beneficioso sobre las propiedades preventivas de la composición. Finalmente, una composición que conviene muy particularmente a los objetivos perseguidos por la presente invención comprende al menos un polipéptido precoz y al menos un polipéptido tardío de papilomavirus, con el fin de combinar el efecto preventivo y curativo. Según un modo preferido, al menos uno de los polipéptidos inmunógenos de origen precoz se modifica a fin de presentar una localización membránica, mediante adición de secuencias de secreción y de anclaje tales como las citadas anteriormente. A este respecto, una composición preferida según la invención comprende: (1) un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 20 1, (2) un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 2, 25 (3) un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 1 y un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 2, 30 (4) un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 1, siendo un polipéptido inmunógeno la proteína L1 de un papilomavirus y/o siendo un polipéptido inmunógeno la proteína L2 de un papilomavirus, 35 (5) un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 2, siendo un polipéptido inmunógeno la proteína L1 de un papilomavirus y/o siendo un polipéptido inmunógeno la proteína L2 de un papilomavirus, o 40 (6) un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 1, un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 2, siendo un polipéptido inmunógeno la proteína L1 de un papilomavirus y/o siendo un polipéptido inmunógeno la proteína L2 de un papilomavirus. El término “homóloga” hace referencia a un grado de identidad con dicha secuencia superior a 70%, ventajosamente superior a 80%, preferentemente superior a 90% y, de manera muy preferida, superior a 95%. 45 50 55 60 65 Ventajosamente, una composición antitumoral según la invención puede comprender además al menos un compuesto que mejora su efecto antitumoral, con el objeto de aumentar la intensidad de la respuesta inmunitaria específicamente o no. Independientemente de los adyuvantes, los inmunoestimulantes representan compuestos particularmente preferidos. Por “inmunoestimulantes” se entiende un compuesto que tiene la capacidad de reforzar una respuesta inmunitaria humoral a fin de ampliar la producción de anticuerpos dirigidos contra el polipéptido inmunógeno o, por mediación celular, a fin de iniciar una respuesta citotóxica significativa contra las células tumorales o infectadas. A título indicativo, la inmunoestimulación se puede evaluar mediante un modelo animal cancerígeno comparando el porcentaje de rechazo en un animal tratado con el polipéptido inmunógeno, en presencia y en ausencia del inmunoestimulante. De manera más general, los medios para poner en evidencia una inmunoestimulación se indican en Roitt (Immunology, 4ª edición, Moby Ltd). Una de las ventajas de tal composición es que combina la inmunidad específica inducida por el polipéptido inmunógeno y la inmunidad no específica inducida por la molécula inmunoestimulante. En el ámbito de la presente invención, se puede utilizar un inmunoestimulante nativo, especialmente de origen humano, una parte de éste, una quimera que proviene de la fusión de secuencias de orígenes diversos, o también un mutante, con la condición sin embargo de conservar la función inmunoestimulante. Entre todas las moléculas factibles, se prefiere utilizar un inmunoestimulante elegido entre la interleuquina-2, la interleuquina-7, la inteleuquina-12 y las moléculas de la coadhesión B7.1 y B7.2. Se indica que la interleuquina-2 y la molécula B7.1 son particularmente preferidos. De manera general, los polipéptidos inmunógenos e inmunoestimulantes se pueden producir mediante los métodos convencionales de síntesis química, o bien mediante técnicas del ADN recombinante (véase, por ejemplo, Maniatis et al., 1989, Laboratory Manual, Cold Spring Harbor, Laboratory Press, Cold Spring Harbor, NY). Más particularmente, un procedimiento de preparación comprende cultivar una célula transformada mediante un fragmento de ADN que codifica el polipéptido en cuestión para generar una célula productora, y recolectar dicho polipéptido a partir del 5 ES 2 229 528 T3 5 10 15 20 cultivo. La célula productora puede ser, de cualquier origen y sin limitación, una bacteria, una levadura o bien una célula de mamífero, en la medida en la que el fragmento de ADN considerado se integre en su genoma o bien se integre en un vector de expresión apropiado. Bien entendido, el fragmento de ADN se pone bajo control de señales de transcripción y de traducción que permiten su expresión en la célula productora. Se conocen los vectores de expresión y las señales de control por el experto en la técnica. La presente invención se dirige así a una composición antitumoral que comprende a título de agente terapéutico o profiláctico al menos un vector recombinante que comprende las secuencias que codifican uno o varios polipéptidos inmunógenos según la presente invención, y, eventualmente, un compuesto que mejora el efecto antitumoral. Este tipo de composición presenta la ventaja de una producción poco costosa y de una gran estabilidad en diversas condiciones medioambientales. En particular, las condiciones de conservación son menos restrictivas. Los polipéptidos tienen las características tales como se definen aquí anteriormente. Las secuencias que codifican el polipéptido inmunógeno o que mejoran el efecto antitumoral se pueden obtener mediante clonación, PCR (reacción en cadena de polimerasa) o mediante síntesis química según las técnicas convencionales comúnmente en uso y a partir de datos de la bibliografía. Refiriéndose al modo de realización preferido, las secuencias que codifican los polipéptidos de papilomavirus se pueden aislar a partir de células de papilomavirus positivas obtenidas de pacientes o de colecciones. La inserción de señales de localización apropiadas se puede realizar mediante las técnicas de biología molecular. Las secuencias que codifican el inmunoestimulante se pueden clonar a partir del ADN celular o de los ARN mensajeros de una célula en la que se expresa. El experto en la técnica es capaz de generar las sondas o cebadores apropiados a partir de los datos publicados. Se indica que la secuencia nucleotídica de los genomas HPV-16 y 18 se divulga en Genbank con los números de accesión K02718 y X05015 respectivamente. La secuencia del gen humano IL-2 se describe en la patente francesa 85 09480 y en Taniguchi et al. (1983, Nature 302, 305-311), y la que codifica el antígeno B7.1 en Freeman et al. (1989, J. of Immunology 143, 2714-2722). 25 Un vector preferido en el ámbito de la invención puede ser un poxvirus. Se puede utilizar un vector plásmico o vírico, especialmente un adenovirus, un retrovirus, un virus del herpes, o un virus asociado a adenovirus. Ventajosamente, se tratará de un vector no integrante y de una virulencia atenuada. Tales vectores así como sus técnicas de preparación son conocidos por el experto en la técnica. 30 35 40 45 50 55 60 65 En el caso en el que se utilice un vector adenovírico, se recurre preferentemente a un vector no replicativo por eliminación de regiones esenciales para la replicación y, especialmente, de la mayoría de la región E1, a fin de evitar su propagación en el seno del organismo hospedante o del medioambiente. Es evidente que se puede modificar o eliminar otras regiones del genoma adenovírico, especialmente en el seno de las regiones E2, E4 y/o L1-L5, en la medida en la que las funciones esenciales defectuosas se complementen en trans. Para ilustrar estos modos de realización, se puede citar la mutación termosensible que afecta al gen DBP (del inglés Proteína de Unión a ADN) de la región E2A (Ensinger et al., 1972, J. Virol. 10, 328-339). Igualmente es factible una eliminación parcial de la región E4, con la excepción de las secuencias que codifican los cuadros de lectura abiertos (ORF) 6 y 7 (Ketner et al., 1989, Nucleic Acids Res. 17, 3037-3048). Otra posibilidad es la eliminación total de la unidad transcripcional E4. Por otra parte, el vector adenovírico según la invención puede ser desprovisto de todo o de una parte de la región no esencial E3. Según esta alternativa, puede ser interesante conservar sin embargo las secuencias E3 que codifican los polipéptidos que permiten el escape al sistema inmunitario del hospedante, especialmente la glicoproteína gp19k (Gooding et al. 1990, Critical Review of Immunology 10, 53-71). Un vector adenovírico preferido según la invención retendrá al mínimo las secuencias esenciales al encapsidamiento, a saber, las ITR (repeticiones terminales invertidas) 5’ y 3’ y la región de encapsidamiento. Se indica que puede derivar de un adenovirus humano o animal, y de un serotipo cualquiera. Los adenovirus humanos del subgrupo C y especialmente los adenovirus 2 (Ad2) y 5 (Ad5) convienen muy particularmente a la realización de la invención. Los diferentes vectores adenovíricos así como sus técnicas de preparación son convencionales y se describen en Graham y Prevect (1991, en Methods in Molecular Biology, vol. 7, p. 109-128; Ed: E.J. Murey, The Human Press Inc.) y en la solicitud internacional WO 94/28152. Por ejemplo, se puede generar in vitro en Escherichia coli (E. coli) mediante ligación o recombinación homóloga (véase, por ejemplo, la Solicitud Internacional WO 96/17070), o también mediante recombinación en una línea de complementación. Si se trata de un retrovirus, se conservan los LTR (repeticiones terminales largas) y las secuencias de encapsidamiento (véase, por ejemplo, Naviaux y Verma, 1992, Current Opinion in Biotechnology 3, 540-547). La secuencia que codifica el o los polipéptidos inmunógenos se puede poner bajo control del LTR retrovírico o de un promotor interno tales como los descritos a continuación. Puede derivar de un retrovirus de cualquier origen (murino, primate, felino, humano, etc.), y en particular del MoMuLV (virus de la leucemia murina de Moloney), MVS (virus del sarcoma murino) o retrovirus murino de Friend (Fb29). Igualmente puede tener modificaciones, especialmente a nivel de los LTR (sustitución de la región promotora por un promotor eucariota) o de la región de encapsidamiento (sustitución por una región de encapsidamiento heteróloga, por ejemplo de tipo VL30) (véanse las solicitudes francesa 94 08300 y 97 05203). Según un modo de realización ventajoso, un vector recombinante según la invención es un poxvirus y, especialmente, un poxvirus aviar, tal como el poxvirus del canario, una viruela aviar o un virus de la viruela vacuna, siendo este último el preferido. Entre todos los virus de la viruela vacuna factibles en el ámbito de la presente invención, se elige preferentemente las cepas Copenhague, Wyeth y Ankara modificada (MVA por virus Ankara modificado de la viruela vacuna). 6 ES 2 229 528 T3 5 Generalmente, el sitio de inserción se elige en una región no esencial para la replicación, de manera que las capacidades de replicación y de propagación del virus recombinante no se alteren. A título indicativo, cuando se utiliza un virus de la cepa Copenhague, el sitio de inserción preferido es el locus TK, lo que tiene por efecto inactivar éste y así facilitar la selección de los recombinantes. Se puede igualmente utilizar el locus K1L. En cuanto a un virus MVA, la inserción de las secuencias recombinantes (inmunógenas y inmunoestimulantes) se puede realizar en el seno de al menos una de las escisiones I a VI, y especialmente II ó III (Meyer et al., 1991, J. Gen. Virol. 72, 1031-1038; Sutter et al., 1994, Vaccine 12, 1032-1040). La inserción puede tener lugar igualmente en una región vírica esencial, como la región D4R, pudiendo la función defectuosa ser suministrada en trans, por ejemplo mediante una línea de complementación. 10 15 20 25 30 35 40 45 50 55 60 65 Bien entendido, en el ámbito de la presente invención las secuencias que codifican el polipéptido inmunógeno o que mejoran el efecto antitumoral son puestas bajo control de los elementos necesarios para su expresión en una célula o un organismo hospedante. Éstos incluyen los elementos de regulación de la transcripción así como señales de iniciación y de terminación de la traducción. Entre éstos, el promotor reviste una importancia particular. De manera general, se recurre a un promotor funcional en el organismo o en la célula hospedante que se quiere tratar y adaptar al vector usado. Además, se puede modificar a fin de contener las secuencias reguladoras, por ejemplo un elemento activador de la transcripción o de las secuencias que responden a algunas señales celulares. Con este fin, puede ser ventajoso utilizar un promotor específico de tejido, porque las lesiones asociadas a los papilomavirus se localizan al nivel de las vías genitales, o un promotor que responde a señales específicamente tumorales (por ejemplo, activado en presencia de factores de crecimiento generalmente sobreexpresados por células tumorales), a fin de limitar la expresión solamente a las células tumorales. Entre los promotores factibles en el ámbito de la invención, se pueden citar los promotores SV40 (virus del simio 40), HMG (hidroximetil-glutaril-coenzima A), TK (timidina quinasa), CMV (citomegalovirus), RSV (virus del sarcoma de Rous), MLP (promotor tardío principal) adaptado al vector adenovírico, y el LTR del Mo-MLV (virus de la leucemia murina de Moloney), más específico de los vectores retrovíricos. Se prefiere más particularmente el promotor precoz del citomegalovirus (CMV). Se puede igualmente tratar de un promotor que estimula la expresión en una célula tumoral o cancerígena. Se pueden citar especialmente los promotores de los genes MUC-1 sobreexpresado en los cánceres de mama y de próstata (Chen et al., 1995, J. Clin. Invest. 96, 2775-2782), de tirosinosis sobreexpresado en los melanomas (Vile et al., 1993, Cancer Res. 53, 3860-3864) y ERB-2 sobreexpresado en los cánceres de mama y de páncreas (Harris et al., 1994, Gene Therapy 1, 170-175). Los promotores en cuestión se describen en la bibliografía y se pueden clonar a partir del genoma celular o vírico mediante las técnicas clásicas. Tratándose de un vector poxvírico, se recurre a un promotor pox, por ejemplo 7,5K, H5R, TK, p.28, p.11 o también K1L del virus de la viruela vacuna. Un promotor sintético conviene igualmente a la realización de la presente invención (véase, por ejemplo, Chakrabarti et al., 1997, Biotechniques 23, 1094-1097; Hammond et al., 1997, J. Virological Methods 66, 135-138, y Kumar y Boyle, 1990, Virology 179, 151-158). Con este fin, se trata ventajosamente de un promotor quimera entre un promotor tardío y un promotor precoz. Por otra parte, los elementos necesarios para la expresión pueden igualmente contener secuencias que mejoran la expresión o el mantenimiento en la célula hospedante (intrón, secuencia terminadora de la transcripción, sitio de iniciación de la traducción, etc.). Sin embargo, en el caso de un vector poxvírico, se evita el uso de intrones. Una composición según la invención puede comprender uno o varios vectores recombinantes que expresan las secuencias correspondientes a los polipéptidos elegidos puestos bajo el control de elementos independientes o comunes. Según esta última opción, se puede recurrir a secuencias que permiten iniciar la traducción de manera interna (IRES) o a fusiones de fase de los diferentes genes. Las condiciones generales para la obtención de un vector recombinante de uso en la presente invención se describen ampliamente en la técnica anterior. Tratándose de un vector poxvírico, se puede citar la patente europea EP 83.286 cuyo contenido se incorpora aquí como referencia. Estas condiciones se pueden aplicar a otros virus aceptables como vector, que poseen una región genómica en la que se pueden incorporar los bloques de expresión. Bien entendido, se pueden insertar en el mismo locus o en un locus diferente. Conforme a los objetivos perseguidos por la presente invención, un vector recombinante puede además comprender un bloque de expresión de un gen marcador de selección a fin de facilitar las etapas de aislamiento y de purificación del virus recombinante. Se puede citar especialmente el gen neo que confiere resistencia al antibiótico G418, el gen pac de resistencia a la puromicina, el gen TK del virus del herpes simple de tipo 1 (HSV-1) que confiere sensibilidad a algunos análogos de nucleósidos tales como el ganciclovir o el aciclovir, el gen gpt (xantina guanina fosforibosil transferasa), los genes bacteriocidas LacZ que codifican la β-galactosidasa y gus A que codifica la β-glucuronidasa. Estos dos últimos marcadores enzimáticos permiten señalar los virus recombinantes mediante coloración en presencia de los substratos X-Gal (5-bromo-4-cloro-3-indolil-β-D-galactopiranósido) y XglcA (5-bromo-6-cloro-3-indolil-β-Dglucurónido), respectivamente. Una composición antitumoral preferida está destinada al tratamiento o a la prevención de una infección o tumor con papilomavirus, y comprende al menos un virus de la viruela vacuna de la cepa Copenhague o MVA en el que se insertan: 7 ES 2 229 528 T3 (1) las secuencias que codifican un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 1, 5 (2) las secuencias que codifican un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 2, (3) las secuencias que codifican un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 1, y un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 2, 10 (4) las secuencias que codifican un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 1, siendo un polipéptido inmunógeno la proteína L1 de un papilomavirus y/o siendo un polipéptido inmunógeno la proteína L2 de un papilomavirus, 15 20 25 30 (5) las secuencias que codifican un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 2, siendo un polipéptido inmunógeno la proteína L1 de un papilomavirus y/o siendo un polipéptido inmunógeno la proteína L2 de un papilomavirus, o (6) las secuencias que codifican un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 1, un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la que se muestra en la SEC ID nº: 2, siendo un polipéptido inmunógeno la proteína L1 de un papilomavirus y/o siendo un polipéptido inmunógeno la proteína L2 de un papilomavirus. Dicha composición puede igualmente comprender las secuencias que codifican un inmunoestimulante elegido preferentemente entre IL-2 ó B7.1. El inmunoestimulante puede ser llevado por uno de los vectores recombinantes que permiten la expresión del o de los genes inmunógenos, o por un vector independiente. Una composición según la invención se puede preparar según los procedimientos conocidos en el campo de las vacunas; y las dosis aplicables pueden variar en un amplio intervalo. Las dosis están en función especialmente de los polipéptidos y del vector usados, de la patología a tratar, del estado del paciente y de otros parámetros que se pueden evaluar por el médico. Sin embargo, en general, la dosis de virus será de 104 a 1013 , ventajosamente de 105 a 1012 , y preferentemente de 105 a 109 unidades formadoras de colonias (ufc), cuando el agente terapéutico es un vector vírico, y de 0,05 a 500 mg, ventajosamente de 0,5 a 200 mg, y preferentemente de 1 a 100 mg, cuando el agente terapéutico es de origen polipeptídico. 35 40 45 50 55 60 65 Una composición según la invención se puede administrar según cualquier vía convencional de administración, preferentemente sistémica y en particular por vía intramuscular, intravenosa, intrapulmonar, subcutánea o subepitelial, o bien por escarificación. En el caso de un tumor accesible, es igualmente posible recurrir a una inyección directa in situ o en la proximidad del tumor, o a una aplicación tópica. Como vacuna, una composición según la invención se puede administrar según las prácticas corrientes en este campo, por ejemplo en dosis única o repetida, una o varias veces después de un cierto intervalo. Por el contrario, en el ámbito de un tratamiento curativo, se puede administrar frecuentemente durante un periodo suficiente para que el tratamiento sea eficaz. Cuando el agente terapéutico es un vector vírico, el virus está preferentemente en forma viva. Tratándose de un vector vírico, se prefiere usar una cepa atenuada, tal como la cepa MVA o la cepa Copenhague timidina quinasa negativa. Finalmente, un vector vírico recombinante se puede atenuar mediante un tratamiento químico apropiado conocido por el experto en la técnica. Sin embargo, se puede también considerar la inyección de un vector recombinante exterminado. Según un modo de realización preferido, una composición antitumoral según la invención comprende una cantidad terapéuticamente eficaz del agente terapéutico, en asociación con un soporte aceptable desde un punto de vista farmacéutico. El soporte se elige a fin de permitir su administración mediante inyección al ser humano o al animal. Igualmente puede comprender un vehículo, un diluyente y/o un adyuvante, y se puede presentar en forma líquida o liofilizada. Con este fin, se puede considerar la asociación de una o varias sustancias susceptibles de mejorar la eficacia transfeccional y/o la estabilidad del vector. Estas sustancias están ampliamente documentadas en la bibliografía accesible al experto en la técnica (véase, por ejemplo, Felgner et al., 1989, Proc. West. Pharmacol. Soc. 32, 115121; Hodgson y Solaiman, 1996, Nature Biotechnology 14, 339-342; Remy et al., 1994, Bioconjugate Chemistry 5, 647-654). A título ilustrativo pero no limitativo, se puede tratar de polímeros, de lípidos especialmente catiónicos, de liposomas, de proteínas nucleares y de lípidos neutros. Una posible combinación es un vector plásmico asociado a lípidos catiónicos (DC-Cho1, DOGS, etc.) y lípidos neutros (DOPE). La composición se puede igualmente asociar a otras sustancias, especialmente anti-cancerígenas, pudiendo éstas ser administradas separadamente o de manera concomitante. El ligando Flt3 es un ejemplo, entre otros (Lynch et al., 1997, Nature Medicine 3, 625; Brasel et al., 1996, Blood 88, 2004-2012; Marakovsky et al., 1996, J. Exp. Med. 184, 1953-1962). Se indica que la composición puede estar en forma de seudopartículas cuando comprende los polipéptidos L1 y/o L2. La presente invención tiene igualmente por objeto un vector recombinante que comprende al menos las secuencias que codifican un polipéptido inmunógeno originario de una región precoz y/o tardía de un papilomavirus, teniendo dicho polipéptido las características definidas anteriormente. Además, puede contener otras secuencias de interés, por ejemplo que codifican uno o varios otros péptidos inmunógenos y/o inmunoestimulantes, tales como se describen anteriormente. 8 ES 2 229 528 T3 La elección de un vector según la invención es amplia. Se puede tratar de un vector plasmídico o vírico, tal como los citados anteriormente. Un modo de realización preferido consiste en un vector poxvírico y muy particularmente un virus de la viruela vacuna de la cepa Copenhague o MVA. Los sitios de inserción y los elementos necesarios para la expresión de las secuencias de interés a expresar se pueden elegir entre los mencionados anteriormente. 5 Un vector preferido es un virus de la viruela vacuna de la cepa Copenhague o MVA en el que se insertan: (1) las secuencias que codifican un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la mostrada en la SEC ID nº: 1, 10 (2) las secuencias que codifican un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la mostrada en la SEC ID nº: 2, 15 20 (3) las secuencias que codifican un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la mostrada en la SEC ID nº: 1, y un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la mostrada en la SEC ID nº: 2, (4) las secuencias que codifican un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la mostrada en la SEC ID nº: 1, un polipéptido inmunógeno siendo la proteína L1 de un papilomavirus y/o siendo un polipéptido inmunógeno la proteína L2 de un papilomavirus, (5) las secuencias que codifican un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la mostrada en la SEC ID nº: 2, un polipéptido inmunógeno siendo la proteína L1 de un papilomavirus y/o siendo un polipéptido inmunógeno la proteína L2 de un papilomavirus, 25 (6) las secuencias que codifican un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la mostrada en la SEC ID nº: 1, un polipéptido inmunógeno que tiene una secuencia homóloga o idéntica a la mostrada en la SEC ID nº: 2, siendo un polipéptido inmunógeno la proteína L1 de un papilomavirus y/o siendo un polipéptido inmunógeno la proteína L2 de un papilomavirus. 30 De manera opcional, igualmente se puede incluir las secuencias que codifican la IL2 o el polipéptido B7.1. 35 40 45 50 55 60 La presente invención se refiere igualmente a una partícula vírica preparada a partir de un vector vírico recombinante según la invención. Tratándose de un vector adenovírico, las partículas víricas se pueden generar mediante transfección del vector en una célula de complementación adaptada a sus deficiencias. Se recurre por ejemplo a la estirpe 293 establecida a partir de células de riñón embrionario humano, que complementa eficazmente la función E1 (Graham et al., 1977, J. Gen. Virol. 36, 59-72), la estirpe A549-E1 (Imler et al., 1996, Gene Therapy 3, 75-84) o una estirpe que permite una doble complementación (Yeh et al., 1996, J. Virol. 70, 559-565; Krougliak y Graham, 1995, Human Gene Therapy 6, 1575-1586; Wang et al., 1995, Gene Therapy 2, 775-783; solicitud internacional WO 97/04119). Por célula de complementación, se entiende una célula capaz de complementar en trans un vector defectuoso para generar una partícula vírica. Dicha célula puede no complementar ella sola todas las funciones defectuosas del vector, y en este caso se puede recurrir a un virus auxiliar para una complementación parcial. La partícula vírica se puede recuperar del sobrenadante del cultivo, pero igualmente de las células. Uno de los métodos frecuentemente utilizados consiste en lisar las células mediante ciclos consecutivos de congelación/descongelación para recolectar los viriones en el sobrenadante del lisado. Éstos se pueden amplificar y purificar según las técnicas anteriores (procedimiento cromatográfico, ultracentrifugación, especialmente a través de un gradiente de cloruro de cesio, etc.). Una partícula retrovírica se puede obtener mediante transfección del vector retrovírico en una estirpe apropiada, por ejemplo una estirpe capaz de proporcionar en trans los polipéptidos víricos gag, pol y/o env cuyas secuencias están eliminadas/son no funcionales en el vector. Tales estirpes se describen en la bibliografía (PA317, Psi CRIP GP + Am-12, etc.). Tratándose de un vector poxvírico, las condiciones generales de obtención de un virus de la viruela vacuna, capaz de expresar un gen heterólogo, se exponen en la patente europea EP 83.286 y la solicitud de patente EP 206.920. En cuanto al virus MVA, se describe más particularmente en Mayr et al. (1975, Infection 3, 6-14) y Sutter y Moss (1992, Proc. Natl. Acad. Sci. USA 89, 10847-10851). Brevemente, los genes a transferir, puestos bajo el control de los elementos apropiados para su expresión in vivo, se insertan en un vector de transferencia que incluye secuencias víricas a un lado y a otro del sitio de inserción. Se introduce en células infectadas por un virus de la viruela vacuna infeccioso. El gen recombinante se integra en el genoma vírico mediante recombinación homóloga entre las secuencias homólogas del virus infeccioso y del vector de transferencia. El vector recombinante o la partícula vírica se puede eventualmente asociar a una o varias sustancias que mejoran la eficacia transfeccional y/o la estabilidad del vector ya citadas. 65 En el ámbito de la presente invención, dicho polipéptido inmunógeno se puede anclar en la estructura proteica que rodea a la partícula vírica (cápsida, envoltorio, etc.). Los poxvirus son estructuras complejas rodeadas por múltiples membranas. Se producen dos tipos de partículas 9 ES 2 229 528 T3 5 10 víricas: los viriones maduros intracelulares (IMV) y los viriones envueltos extracelulares (EEV). La superficie de los IMV está compuesta de 2 membranas superpuestas, y el envoltorio de los EEV consiste en 4 membranas que incluyen la membrana plásmica. Conforme a los objetivos de la presente invención, las secuencias que codifican el polipéptido inmunógeno se pueden igualmente modificar a la vista de una inserción en una u otra de las membranas poxvíricas (IMV o EEV), con el objetivo de mejorar la respuesta inmunitaria. Según un modo de realización ventajoso, el péptido inmunógeno se ancla en la membrana externa del envoltorio de una partícula EEV (Katz et al. 1997, AIDS Res. Hum. Retrov. 13, 1497-1500). Con este fin, las secuencias que codifican la parte C-terminal de la proteína B5R, constituyente proteico de dicha membrana externa, se insertan en 3’ de la región codificante del polipéptido inmunógeno justo en dirección 5’ del codón STOP. Un ejemplo muy preferido es una fusión entre el polipéptido E7 o uno de sus mutantes no oncógenos y los 42 restos C-terminales de la proteína B5R. El efecto beneficioso sobre la respuesta inmunitaria se puede evaluar mediante estudios de inmunoprofilaxis y de inmunoterapia según el protocolo descrito en los ejemplos a continuación que comparan las diferentes formulaciones (localización nativa, anclaje en la membrana celular, anclaje en la membrana vírica). 15 La presente invención se refiere igualmente a una composición antitumoral caracterizada porque contiene uno o varios polipéptidos inmunógenos, presentando al menos uno de dichos polipéptidos las características definidas aquí anteriormente para las composiciones antitumorales según la presente invención que comprenden al menos un vector recombinante. 20 La presente invención se refiere igualmente a la utilización de una composición antitumoral, de un vector recombinante o de una partícula vírica según la invención, para la preparación de un medicamento para el tratamiento o la prevención del cáncer o de tumores y, particularmente del cáncer de cuello uterino, de una displasia del cuello de grado bajo o de una infección con papilomavirus. La utilización preferida es para la preparación de un medicamento inyectable por vía intramuscular. 25 Finalmente, se puede citar un método de tratamiento o de prevención de las patologías citadas aquí arriba, según el cual se administra a un individuo que necesita de tal tratamiento una cantidad eficaz desde un punto de vista farmacéutico de una composición antitumoral, de un vector recombinante o de una partícula vírica según la invención. 30 35 40 45 50 La presente invención se ilustra con referencia a las figuras siguientes. La Figura 1 es una representación esquemática de pTG8042. BRG3 y BRD3 representan los brazos de recombinación izquierdo y derecho que permiten la inserción al nivel de la zona de escisión III del virus MVA. El pTG8042 comprende un casete de expresión del gen de E6 de HPV16 fusionado a la secuencia señal (SF) y la región transmembránica (TMF) de la proteína F del virus de la rubéola, dirigido por el promotor 7.5K; un casete de expresión del gen de IL-2 puesto bajo control del promotor pH5R; un casete de expresión del gen de E7 de HPV16 fusionado a la secuencia señal (SR) y la región transmembránica (TMR) de la glicoproteína rábica, puesto bajo control del promotor p7.5; y un casete de expresión del gen gusA (uidA) puesto bajo control del promotor p7.5. La Figura 2 es una representación esquemática del vector pTG9936. BRG3 y BRD3 representan los brazos de recombinación izquierdo y derecho que permiten la inserción al nivel de la zona de escisión III del genoma de MVA. El pTG9936 contiene el promotor p4BK1L que dirige la expresión del gen marcador gpt, el promotor p4BK1L que dirige la expresión del gen L2 de HPV16, el promotor pH5R que dirige la expresión de la IL-2, el promotor p11k7.5 que dirige la expresión del gen L1 de HPV16, el promotor p7.5 que dirige la expresión del gen E7 de HPV16 fusionado a la secuencia señal (SR) y la región transmembránica (TMR) de la glicoproteína del virus de la rabia, seguido de una secuencia IRES, y del gen E6 de HPV16 fusionado a la secuencia señal (SF) y la región transmembránica (TMF) de la proteína F del virus de la rubéola. La Figura 3 ilustra el ensayo N121 que presenta el porcentaje de supervivientes en función del tiempo (en días) de los animales vacunados antes de la prueba tumoral con el virus MVATG8042, MVATG6090, MVAN33 o con PBS. La Figura 4 representa el porcentaje de animales sin tumores en función del tiempo (en días) después de la administración de 107 , 106 ó 105 pfu (pfu = unidades formadoras de colonias) de los virus MVAN33 o MVATG8042 (ensayo N127). 55 La Figura 5 ilustra el ensayo N134 que representa el porcentaje de supervivientes en función del tiempo (en días) de los animales tratados mediante el virus MVATG8042 o mediante el control MVAN33, por escarificación, vía subcutánea (SC), intraperitoneal (IP) e intramuscular (IM). 60 Ejemplos La presente invención se describe de forma más completa, con la ayuda de los ejemplos siguientes, sin estar limitada a los mismos. 65 Las construcciones descritas a continuación se realizan según las técnicas generales de ingeniería genética y de clonación molecular, detalladas en Maniatis et al., (1989, supra), o según las recomendaciones del fabricante cuando se utilice un kit comercial. La mutagénesis dirigida in vitro mediante oligonucleótidos sintéticos se efectúa con la ayuda del kit distribuido por Amersham. Las técnicas de amplificación por PCR son conocidas por el experto en la técnica 10 ES 2 229 528 T3 5 (véase, por ejemplo, PCR Protocols-A guide to methods and applications, 1990, publicado por Innis, Gelfand, Sninsky y White, Academic Press Inc). Haciendo referencia a la reparación de los sitios de restricción, la técnica utilizada consiste en un llenado de los extremos 5’ sobresalientes con la ayuda del gran fragmento del ADN polimerasa I de E. coli (Klenow). En lo que se refiere a las etapas de clonación, los bacteriófagos M13 recombinantes se multiplican sobre la cepa de E. coli NM522 (Stratagθne) en un medio mínimo gelificado (agar 7,5%), o en un medio rico LBM líquido. Los plásmidos recombinantes que contienen el gen de resistencia a la ampicilina se replican en las cepas de E.coli C600 (Stratagθne), BJ 5183 (Hanahan, 1983, J. Mol. Biol. 166, 557-580) y NM522 en medio gelificado o líquido suplementado con 100 µg/ml de antibiótico. Se utiliza preferentemente la cepa BJ5183 cuando la clonación se efectúa mediante recombinación homóloga (Bubeck et al., 1993, Nucleic Acid Res. 21, 3601-3602). 10 La construcción de los virus de la viruela vacuna recombinantes se efectúa según la tecnología clásica en el campo, divulgada en los documentos ya citados y en Mackett et al., (1982, Proc. Natl. Acad. Sci. USA 79, 7415-7419) y Mackett et al. (1984, J. Virol. 49, 857-864). 15 20 Los ensayos in vivo (inmunoprofilaxis o inmunoterapia) se realizan sobre ratones hembra C57B16 (C. Rivers Rouen, Francia), o ratones atímicos (Janvier, Le Genest St. Isle, Francia), que tienen de 6 a 8 semanas. Los animales se reparten generalmente en grupos de 20 (salvo cuando se indique) según los virus, la dosis o la vía de administración a ensayar. Las células tumorales TC1 (Wu, John Hopkins University, Baltimore, USA) se obtienen de células de pulmón tomadas de ratones C57 B16 transducidas con dos retrovirus, uno que expresa los genes E6 y E7 nativos de HPV-16, y el otro que expresa el oncogén ras. Las células se cultivan en medio DMEM (medio Eagles modificado de Dubelcco) en presencia de G418 (0,5 mg/ml). Las células se utilizan para el ensayo animal, después del tratamiento con tripsina y 3 lavados en medio isotónico. Ejemplo 1 25 Construcción de los vectores que contienen las secuencias que codifican los mutantes no oncogénicos de E6 y E7 de HPV-16 que tienen señales de localización transmembránica 30 35 40 45 50 55 60 65 Los genes E6 y E7 se aíslan a partir de la estirpe celular Caski como se describe en los ejemplos 2 y 3 de la patente europea EP 0.462.187. Se han derivado dos construcciones del clon M13E7/E6 que contiene los genes E6 y E7 del HPV-16 a fin de facilitar las etapas ulteriores de clonación. La primera, denominada M13TG8188, resulta de la introducción mediante mutagénesis dirigida de sitios PstI y BamHI respectivamente en dirección 5’ y en dirección 3’ del gen E7; y la segunda, M13TG8189, contiene un sitio PstI en dirección 5’ del gen E6. La introducción de mutaciones puntuales en dirección 5’ de un ATG iniciador y en dirección 3’ de un codón stop son conocidas por el experto en la técnica. La asociación de la proteína E7 de HPV-16 con el producto del gen de retinoblastoma ha sido demostrada por diversos autores (véase, por ejemplo, Munger et al., 1989, EMBO J. 8, 4099-4105), y se ha correlacionado con su poder transformador. Por razones evidentes de seguridad, se genera un mutante no oncogénico eliminado de los codones 21 a 26 de la proteína E7 nativa implicados en la función de transformación, mediante mutagenesis dirigida del vector M13TG8188 con la ayuda del oligonucleótido oTG5118 (SEC ID nº: 3). Se obtiene M13TG9104 que contiene el gen E7 mutado, denominado a continuación E7*. Las señales de secreción y de anclaje de la glicoproteína rábica se aíslan del gen de la glicoproteína rábica insertado en forma de un fragmento Bg/II en el pBR327 (pTG150 descrito en la patente francesa nº 83 15716) y subclonado en el vector M13 (M13TG177). Se introduce entre estas señales un sitio BamHI, en fase con estos últimas, mediante mutagénesis dirigida con la ayuda del oligonucleótido oTG5745 (SEC ID nº: 4). La construcción resultante se denomina M13TG9128. Después, las secuencias de secreción y de anclaje se aíslan del M13TG9128 mediante digestión PstI y se insertan en 3’ del promotor natural del virus de la viruela vacuna p7.5 en el vector pTG5003 linealizado mediante PstI, para dar pTG5016. A título indicativo, pTG5003 deriva de pTG186poli (descrito en la patente francesa nº 2.583.429) mediante digestión SalI, tratamiento con el fragmento Klenow, y después digestión mediante SmaI y religan, de manera que no contiene más que los sitios PstI y EcoRI en su poliligador. El vector M13TG9104 se modifica mediante mutagénesis dirigida con la ayuda de los oligonucleótidos oTG6390 y oTG6880 (SEC ID nº: 5 y 6) a fin de poner en fase las secuencias E7* y las señales de secreción y de anclaje de la glicoproteína rábica (denominadas a continuación E7*TMR). La construcción resultante se denomina M13TG9150. Asimismo, se ha demostrado que la proteína E6 de HPV-16 podía interaccionar con el producto de expresión del gen supresor de tumor p53 (Crook et al., 1991, Cell 67, 547-556). El campo implicado en esta interacción se ha definido claramente, y se sitúa entre los restos 111 a 115 de la proteína nativa. El vector M13TG9125 se genera mediante mutagénesis de M13TG8189 con la ayuda del oligonucleótido oTG5377 (SEC ID nº: 7). El gen E6 λ111115 se denomina en lo sucesivo E6*. Las señales de secreción y de anclaje de la proteína F del virus de la rubéola se aíslan mediante PCR a partir de la construcción plasmídica pTG2148 (descrita en la patente europea EP 0.305.229) que contiene el ADN que codifica la proteína F del virus de la rubéola. La fusión entre estas secuencias y las que codifican E6* se realiza mediante PCR directa: la secuencia de secreción se amplifica con la ayuda de los oligonucleótidos oTG10829 que contiene en 5’ un sitio XbaI (SEC ID nº: 8) y oTG10830 que cubre el extremo 5’ de E6 (SEC ID nº: 9); las secuencias que codifican el mutante E6* se amplifican a partir del M13TG9125 con la ayuda de los oligonucleótidos oTG10835, que permite 11 ES 2 229 528 T3 5 10 la fusión con el extremo 3’ de la señal de secreción de la proteína F (SEC ID nº: 10), y oTG10836, que permite la fusión con el extremo 5’ de la secuencia de anclaje de la proteína F (SEC ID nº: 11). Para la secuencia de anclaje, se utilizan los cebadores oTG10833, que permite la fusión entre el 3’ de E6* y el 5’ de la secuencia de anclaje (SEC ID nº: 12), y oTG10834, que crea en 3’ los sitios KpnI y SphI (SEC ID nº: 13). El fragmento amplificado que contiene las secuencias E6*, fusionadas en N y C-terminal, respectivamente, a las secuencias de secreción y de anclaje membránica de la proteína F (denominada en lo sucesivo E6*TMF), se digiere mediante XbaI y SphI y después se inserta en los mismos sitios en el vector M13TG131 (Kieny et al., 1983, Gene 26, 91-99). La construcción así obtenida se denomina M13TG9199. Ejemplo 2 Construcción del virus recombinante MVATG8042 que expresa los antígenos E6 y E7 de localización membránica y la IL-2 humana 15 20 25 El virus MVA deriva de la cepa del virus de la viruela vacuna Ankara. No es capaz de generar partículas infecciosas en las células de los mamíferos, pero se desarrolla correctamente en fibroblastos embrionarios de pollo. Su adaptación a estas células ha provocado la escisión de 6 regiones no esenciales para su desarrollo y su sistema infeccioso en este tipo de células (desaparición de aproximadamente 15% del genoma vírico; Meyer et al., 1991, J. Gen. Virol. 72, 1031-1038). La integración de material genético exógeno se puede realizar a nivel de una cualquiera de estas zonas de escisión. En el ámbito de la presente invención, se utilizan las escisiones II y III localizadas a nivel de los fragmentos de restricción HindIII N y A, respectivamente (Altenburger et al., 1989, Arch. Virol. 105, 15-27). En primer lugar, se construye el vector pTG6019 que permite la inserción en la zona de escisión III del virus MVA. Los brazos de recombinación homóloga a cada lado de la zona de escisión III se aíslan mediante PCR a partir del genoma vírico (véase la patente americana US 5.185.146) y los cebadores oTG7637 y oTG7638 (SEC ID nº: 14 y 15) por el brazo izquierdo, y oTG7635 y oTG7636 (SEC ID nº: 16 y 17) por el brazo derecho. Los fragmentos amplificados se clonan al sitio EcoRI del vector pTG1E, para dar pTG6019. El material genético a transferir se inserta entre los dos brazos de recombinación. El vector pTG1E es similar al pTG1H (patente francesa 2.583.429), salvo por la presencia de un adaptador EcoRI en lugar de sitios múltiples de clonación. 30 35 40 45 50 55 60 65 En primer lugar, se inserta un casete de expresión del gen marcador gus A. Se clona primero el promotor 7,5K en el sitio BamHI de pTG6019. Se obtiene pTG6021 en el sitio BamHI, del cual se inserta el gen gus A generado en forma de un fragmento Bg/II-BamHI. Éste se puede obtener a partir de la secuencia divulgada en la bibliografía. La construcción resultante se denomina pTG6022. La presencia del marcador permitirá discriminar los virus salvajes de los virus recombinantes, por detección de la actividad enzimática GUS mediante el sustrato XglcA. Una coloración roja revela actividad de β-glucuronidasa. Sin embargo, en la óptica de una aplicación clínica, puede ser útil poder eliminar este marcador bacteriano del producto final después de la selección de los virus recombinantes. Para ello, se utiliza la capacidad de la viruela vacuna para eliminar las secuencias comprendidas entre dos sitios homólogos. Es por ello que se inserta un segundo promotor p7,5K en dirección 3’ del gen gus A en una orientación sentido con relación al promotor que dirige la expresión de este último. El vector pTG6025 resulta de la inserción entre los sitios BamHI y SacI de pTG6022 de un fragmento p7,5K que tiene extremos cohesivos. Por otra parte, el ADNc que codifica la interleukina-2 humana se aísla del plásmido pTG36 (patente francesa 2.583.770) mediante digestión PstI y se inserta en el sitio PstI del plásmido pTG186 (patente fancesa 2.583.429), dando lugar a pTG188. El virus obtenido mediante recombinación homóloga se denomina VVTG188. Después de la digestión Bg/II/EcoRI, las secuencias de IL-2 se insertan en 3’ del promotor natural de la viruela vacuna pH5R en los sitios BamHI/EcoRI del M13TG9132, para dar M13TG9185. A título indicativo, el vector M13TG9132 proviene de la inserción del promotor del gen H5R aislado mediante PCR del genoma vírico en el fago M13TG6131, el cual deriva de M13TG131 (Kieny et al., 1983, supra) mediante mutación del sitio Bg/II interno situado fuera de los sitios múltiples de clonación. Después, los genes HPV-16 e IL-2 se clonan en la zona de escisión III del genoma del MVA. Las secuencias E7*TMR se aíslan mediante digestión Bg/II/HindIII y se insertan entre los sitios BamHI y HindIII del pTG6025 en 3’ del promotor de la viruela vacuna p7.5. La construcción resultante se denomina pTG6050. Se crea un sitio de inserción del casete de expresión del gen de la IL-2 humana mediante recombinación homóloga entre los sitios HindIII y KpnI del pTG6050, por inserción de los oligonucleótidos oTG10502 y oTG10503 (SEC ID nº: 18 y 19). Después, el vector generado pTG6074 se linealiza mediante digestión HindIII/KpnI, y se realiza la recombinanción homóloga con el casete de expresión pH5R-IL-2 aislado del M13TG9185 mediante digestión Bg/II/EcoRI. Finalmente, la construcción resultante pTG6088 se linealiza mediante KpnI y se liga con el gen E6*TMF, aislado del M13TG9199 mediante digestión KpnI/XbaI, y el promotor p7.5, aislado del M13TG9136 mediante digestión XbaI/KpnI. La construcción resultante se denomina pTG8042 (Figura 1). El vector M13TG9136 proviene de M13TG5107 modificado mediante mutagénesis dirigida con la ayuda de los oTG5925 (creación de un sitio PstI en 3’ del promotor p7.5; SEC ID nº: 20) y oTG5924 (creación de un sitio BamHI y KpnI en 5’ del promotor p7.5; SEC ID nº: 21). El virus MVATG8042 se genera mediante recombinación homóloga con el genoma de MVA según las técnicas anteriores. El aislamiento de los recombinantes se facilita mediante la presencia del gen marcador gusA. Se verifica que la modificación de la localización celular de los antígenos precoces de HPV no perjudica su ex12 ES 2 229 528 T3 5 presión. El análisis mediante transferencia Western de extractos celulares infectados por MVATG8042 con la ayuda de un anticuerpo anti-E7 permite la detección de 3 bandas con peso molécular comprendido entre 20 y 35 kDa. Esta heterogeneidad se puede explicar por la presencia de un sitio potencial de O-glicolización en la zona de anclaje membránica de la glicoproteína rábica. Cuando el análisis de transferencia Western se repite en presencia de fenil-GalNac (Fenil-N-acetil-α-D-galactopiranósido, Sigma, P4023), se detecta sólo una forma de 20 kDa lo que confirma la O-glicosilación del producto de expresión del gen E7*TMR. 10 La detección mediante transferencia Western con la ayuda de un anticuerpo E6 del producto de expresión del gen E6*TMF pone de manifiesto una sola banda que migra al peso molecular esperado de 20 kDa, lo que confirma la ausencia de modificaciones postraduccionales. A título indicativo, los anticuerpos anti-E6 y E7 antes citados son antisueros de conejo obtenidos mediante administración del antígeno purificado. Pero igualmente pueden ser convenientes otros anticuerpos específicos, ya sean monoclonales o policlonales. 15 La expresión del gen hIL-2 se evalúa mediante ELISA (Quantikine R&D Systems) y el ensayo de proliferación celular dependiente de IL-2. Según las condiciones de cultivo, la cantidad de IL-2 producida varía de 200 a 800 ng/ml/24 h por 106 células infectadas con 0,1 pfu/célula. Ejemplo 3 20 25 Eficacia in vivo del virus MVATG8042 (inmunoterapia) Se inoculan ratones C57BL6 con 103 células BMK-16 myc implantadas por vía subcutánea. A título indicativo, la estirpe celular deriva de células de riñón de ratones neonatos transfectadas mediante el genoma HPV-16 y el gen c myc murino. Después, se administran 107 pfu (unidades formadoras de colonias) de virus MVATG8042 igualmente de forma subcutánea en el D3, D6 y D9, y se sigue regularmente la evolución de los tumores. Los ratones tratados presentan un retraso del crecimiento tumoral de hasta D15 (15 días) con relación a los controles formados por animales que han recibidos un virus MVA no recombinante. Además, el tratamiento se acompaña de una regresión parcial de los tumores en D30-35 que no se observa cuando se utiliza un MVA equivalente que expresa los mutantes E6* y E7* de localización nuclear nativa. 30 Ejemplo 4 Construcción del virus recombinante VVTG5095 que expresa el antígeno E7* de localización transmembránica 35 Las secuencias E7*TMR se aíslan del M13TG9150 mediante digestión Bg/II/BamHI, y se insertan al sitio BamHI del pTG5016. La construcción resultante, denominada pTG5095, contiene las secuencias E7* fusionadas a las señales de secreción y de anclaje de la glicoproteína rábica, bajo control del promotor p7.5. El virus VVTG5095 se genera mediante recombinación homóloga con el genoma de la viruela vacuna. 40 Ejemplo 5 Construcción del virus recombinante VVTG6002 que expresa el antígeno E7* de localización transmembránica y el inmunoestimulante B7.1 45 50 55 El gen B7.1 humano se aísla a partir de una estirpe celular humana Daudi mediante PCR inversa (RT-PCR) con la ayuda de los cebadores oTG6353 y oTG6352 (SEC ID nº: 22 y 23), y se inserta entre los sitios Bg/II y EcoRI de M13TG6131. Se obtiene M13TG9149 del cual se aísla el fragmento Bg/II-EcoRI el cual se subclona entre los mismos sitios de M13TG5107, en 3’ del promotor p7.5 (M13TG5107 contiene las secuencias promotoras p7.5 clonadas en el sitio BamHI del M13TG130). El casete p7.5-B7.1 se aísla de la construcción así obtenida, denominada M13TG9152, mediante digestión EcoRI/PstI, y se introduce en el sitio EcoRI de pTG5095 con la ayuda del oTG1086 (5’AATTTG CA3’). La construcción resultante se denomina pTG6002 y los virus recombinantes VVTG6002 se producen mediante recombinación homóloga con el genoma de la viruela vacuna. Se realiza una construcción equivalente mediante inserción de los casetes de expresión de los genes E7* y B7.1 en la zona de escisión III del genoma de MVAN33, según la misma tecnología que la desarrollada en el ejemplo 2. Ejemplo 6 Eficacia in vivo de los virus VVTG5095 y VVTG6002 (inmunoprofilaxis) 60 65 Se han vacunado ratones C57BL6 en tres ocasiones por vía subcutánea con 107 pfu de VVTG5095 o VVTG6002. Tres días después de la última inmunización, estos animales se exponen a 103 células E7W1 implantadas por vía subcutánea. A título indicativo, las células E7W1 provienen de una estirpe de linfoma murino transfectada mediante un vector que expresa el gen E7 oncogénico de HPV-16. El porcentaje de supervivientes de los animales, en función del tiempo, se compara con el obtenido con ratones testigo tratados con 107 pfu de un virus de la viruela vacuna no recombinante VVTG186 (derivado del vector TG186 descrito aquí arriba). El seguimiento de la mortalidad muestra una diferencia entre los tres grupos. Mientras que en el grupo del testigo el 100% de los animales están muertos el D36, se observa la supervivencia de aproximadamente un cuarto de los animales vacunados por VVTG5095. La protección 13 ES 2 229 528 T3 se mejora sensiblemente con la construcción VVTG6002, que incluye las secuencias que codifican el antígeno B7.1. Ejemplo 7 5 10 15 20 Construcción de MVATG9936 que expresa los genes precoces modificados y los genes tardíos de HPV16 Se aíslan los fragmentos que codifican las proteínas L1 y L2 de HPV16, mediante PCR a partir de ADN genómico de células Caski (ATCC 1550), según las técnicas generales de la técnica anterior. El fragmento de amplificación que contiene las secuencias L1 se subclona en M13TG130 (Kieny et al., 1983, Gene 26, 91-99), para dar la construcción M13TG8171. La secuencia del gen L1 clonado revela varias mutaciones con relación a la secuencia contenida en Genebank (Accesión K02718): C en lugar de A en posición 248, C en lugar de A en posición 253, G en lugar de A en posición 537, G en lugar de C en posición 682, G en lugar de A en posición 874, inserción de un triplete ACT en posición 1393, eliminación de un triplete GAT en posición 1390. La secuencia se corrige mediante mutagénesis puntual a fin de adecuarla a la publicada por Zhou et al. (1991, Virology 185, 251-257), e introducir mutaciones silenciosas a nivel de las secuencias TTTTTNT que constituyen sitios potenciales de terminación de la transcripción precoz susceptibles de interferir con la fase precoz de desarrollo de la viruela vacuna. La técnica de mutagénesis dirigida es conocida por el experto en la técnica. El vector que contiene la secuencia corregida se denomina M13TG4041. La inserción del fragmento de PCR que contiene las secuencias L2 en el vector M13TG6131 conduce a M13TG9126. Se cuentan 5 mutaciones puntuales con relación a la secuencia divulgada en Genebank: C en lugar de T en posición 378, A en lugar de G en posición 691, A en lugar de G en posición 702, G en lugar de A en posición 990 y C en lugar de A en posición 1092. A título indicativo, el vector M13TG6131 deriva de M13TG131 (Kieny et al., 1983, Gene 26, 91-99) mediante mutación del sitio Bg/II interno situado fuera de los sitios múltiples de clonación. 25 30 35 40 45 El vector M13TG4041 se digiere mediante XbaI-SacI, y el fragmento que contiene las secuencias L1 se inserta entre los sitios XbaI-SacI de M13TG9126 en dirección 3’ del gen L2 y en dirección opuesta. La construcción así generada se denomina M13TG4042. Después, las secuencias L1 y L2 se aíslan mediante digestión Bg/II y se subclonan entre los sitios Bg/II y BamHI del vector M13TG4052 que contiene el promotor sintético p11K7.5, que resulta de la fusión de los promotores tardíos p11K y precoz p7.5. De las dos orientaciones, se selecciona la que pone el gen L1 en dirección 3’ del promotor, que se denomina M13TG4055. Después, el casete de expresión, que contiene el promotor pH5R y el gen hIL-2, se aísla de pTG8042 mediante digestión HindIII antes de ser introducido en el sitio HindIII de M13TG4055 situado entre los genes L1 y L2. Finalmente, la construcción obtenida, M13TG4057, se digiere mediante Bg/II, y el fragmento que contiene las secuencias tardías de HPV16 se inserta en el sitio BamHI de M13TG4060, el cual resulta de la clonación del promotor sintético p4BK1L. Este último es un promotor híbrido entre el promotor precoz p4B (Davidson y Moss, 1989, J. Mol. Biol. 210, 749-769) y tardío (Davidson y Moss, 1989, J. Mol. Biol. 210, 771-784). Se obtiene M13TG4062 que comprende el gen L1 bajo control de p11K7.5, el casete pH5R-IL2 y el gen L2 bajo control de p4BK1L en orientación inversa con relación a las secuencias L1. Se construye un casete policistrónico que contiene las secuencias que codifican E7*TMR y E6*TMF. En primer lugar, las secuencias IRES del virus EMC (encefalomiocardiovirus; Genbank accesión M22458) se aíslan, mediante las técnicas convencionales, en forma de fragmento EcoRI-NcoI introducido en dirección 3’ del gen E7*TMR entre los sitios EcoRI y NcoI de pTG6002 (ejemplo 5), para dar pTG8084. Después, el gen E6*TMF se amplifica mediante PCR con la ayuda de cebadores apropiados creando un sitio NcoI en su extremo 5’. El fragmento Bg/II-NcoI, que contiene el gen E7*TMR y las secuencias IRES, obtenido de pTG8084, y el fragmento PCR, que contiene el gen E6*TMF digerido mediante NcoI y SacI, se reconstituyen en el vector M13TG6131 (ejemplo 2) anteriormente digerido mediante Bg/II y SacI. Se obtiene M13TG4059. Después, el bloque E7*TMR-IRES-E6*TMF se aísla de este último en forma de un fragmento Bg/II, y se inserta en el sitio BamHI de pTG8093 en dirección 3’ del promotor p7.5K. (pTG8093 resulta de la clonación del promotor p7.5K en el vector pTG6025). La construcción así obtenida se denomina pTG9901. 50 55 60 El fragmento SacI de M13TG4062 se clona en el sitio SacI de pTG9901, para dar pTG9902. El gen de selección gpt (Falkner y Moss, 1988, J. Virol. 62, 1849-1854) se empalma con los genes tardíos e IL2 en el vector p poli II (Lathe et al., 1987, Gene 57, 193-201) de la siguiente manera. El primero se aísla de M13TG4076 (este vector contiene el gen gpt flanqueado en sus dos extremos por las secuencias promotoras p4BK1L) mediante digestión Bg/II-NcoI, y los segundos se aíslan de pTG9902 mediante digestión NcoI-SacI. Los fragmentos purificados se clonan entre los sitios Bg/II-SacI de p poli II. Se obtiene pTG9933, del cual se aísla el fragmento SacI que se introduce en el vector pTG9901 escindido mediante SacI. La construcción así obtenida, pTG9936, se ilustra en la figura 2. Los virus MVATG9936 se generan mediante recombinación homóloga con el genoma de MVAN33. El aislamiento de clones se puede realizar según las técnicas anteriores. Ejemplo 8 Eficacia de los virus en inmunoprofilaxis 65 A. Importancia de la formulación de los virus (ensayo N121) El objetivo de este estudio preclínico es comparar la cepa vírica (virus de la viruela vacuna Copenhague frente a 14 ES 2 229 528 T3 MVA) y la formulación (presentación transmembránica frente a localización nuclear nativa) en términos de protección antitumoral. 5 Se vacunan ratones C57B16 en tres ocasiones con 107 pfu de virus. Las inyecciones se llevan a cabo cada diez días (D1, D11 y D21), por vía intraperitoneal. Los animales se exponen 7 días después de la última inmunización, por administración subcutánea en su lado derecho, a 5 x 104 células TC1. Se constituyen ocho grupos de 20 animales en función del virus o de la disolución administrada, respectivamente: 1. MVATG8042 (ejemplo 2, E6*TMF, E7*TMR, IL-2), 10 2. MVATG6037 (ejemplo 5, E7*TMR, B7.1), 3. VVTG5095 (ejemplo 4, E7*TMR), 15 4. MVATG6090 (E6*, E7*, IL-2), 5. VVTG5061x188 (E6*, E7*, IL-2), 6. VVTG186 (virus de la viruela vacuna no recombinante), 20 7. MVAN33 (virus MVA no recombinante), y 8. una disolución salina (PBS). 25 30 Los grupos 1 a 3 se vacunan con los virus de la presente invención que expresan al menos un antígeno HPV transmembránico, mientras que los grupos 4 y 5 han recibido virus que expresan los antígenos precoces de HPV16 de localización nuclear nativa; y los grupos 6 a 8, virus de control no recombinantes o una disolución salina. Se indica que los virus MVATG6090 y VVTG5061x188 se describen en la Solicitud Internacional WO98/04705. El porcentaje de supervivientes de los animales de los diferentes grupos se sigue durante las 12 semanas que siguientes a la exposición tumoral. Los resultados se pueden resumir de la siguiente manera. De manera general, la mortalidad es importante en los tres grupos testigo, llegando al 95% (con los virus MVAN33 y VV186) y 81% (con el PBS). 35 40 Se observa un crecimiento significativo de la supervivencia de los animales inmunizados con los virus que expresan los antígenos nucleares de HPV. Así, el 55% de los ratones que han recibidos el virus MVATG6090 están libres de tumores, mientras que la administración de VVTG5061x188 induce un porcentaje de rechazo tumoral de 75%. Sin embargo, la gran mayoría de los animales vacunados con los virus que expresan los antígenos transmembránicos de HPV16 han rechazados su tumor, o presentan un importante retraso del crecimiento tumoral. Más precisamente, se observa 100% de rechazo con MVATG8042, 95% con MVATG6037 y 90% con VVTG5095. La Figura 3 presenta las curvas de supervivencia obtenida con los animales vacunados con MVATG8042 y MVATG6090 con relación a los controles (MVAN33 y PBS). 45 En su conjunto, estos datos ponen en evidencia la ausencia de diferencia significativa entre los virus que provienen de un virus de la viruela vacuna Copenhague y de un MVA, y la mejor inmunogenecidad conferida por la presentación membránica. De todos los virus ensayados, el virus MVATG8042 es el más eficaz porque da lugar a 100% de rechazo tumoral en este modelo animal de inmunoprofilaxis. 50 B. Estudio de la respuesta memoria (ensayo N122) 55 60 65 Se inmunizan los ratones C57B16 por vía intraperitoneal con 107 pfu de virus en D1, D11 y D21 antes de ser expuestos en el D82, mediante administración subcutánea en su lado derecho, a 5 x 104 células TC1. Se constituyen ocho grupos idénticos al ensayo anterior (N121). Se sigue la evolución de los tumores en función del tiempo. Los datos obtenidos 50 días después de la exposición tumoral confirman que el virus más eficaz en términos de rechazo tumoral es el virus MVATG8042. Su administración protege al 100% de los animales, indicando su capacidad para inducir una inmunidad a largo plazo. C. Efecto de dosis (ensayo N127) Se inmunizan los ratones C57B16 por vía intraperitoneal en D1, D11 y D21 con 105 , 106 o 107 pfu de virus MVATG8042 antes de ser expuestos en el D28, mediante administración subcutánea en su lado derecho, a 5 x 104 células TC1. Los animales del control reciben cantidades idénticas de MVAN33. Se sigue la evolución de los tumores dos veces por semana durante 12 semanas. Los resultados (Figura 4) muestran 100% de protección independientemente de la dosis de MVATG8042 administrada, mientras que la gran mayoría de los animales testigo desarrollan tumores. Estos datos confirman la eficacia del virus MVATG8042 también con dosis bajas. 15 ES 2 229 528 T3 D. Efecto de la vía de administración (ensayo N134) 5 Se inmunizan los ratones C57B16 por diferentes vías de administración (escarificación, subcutánea, intraperitoneal o intramuscular) en D1, D11 y D21 con 107 pfu de virus antes de ser expuestos en el D44, por administración subcutánea en su lado derecho, a 5 x 104 células TC1. Se sigue la evolución de los tumores dos veces por semana durante 12 semanas. Como se muestra en la figura 5, las vías intraperitoneal o intramuscular dan los porcentajes de protección más elevados con el virus MVATG8042. Ejemplo 9 10 Eficacia de los virus en inmunoterapia A. Eficacia de los virus en un contexto de inmunoterapia (ensayo N125) 15 20 El objetivo de este estudio es comparar las capacidades terapéuticas frente a un tumor preestablecido de los virus que presentan los antígenos HPV en una forma membránica o nuclear. Para ello, se administran 5 x 104 células TC1 por vía subcutánea en el lado derecho de los ratones C57B16 (D0). Después, se inyectan 107 pfu de virus por vía intraperitoneal en D7, D14 y D21. Se constituyen cuatro grupos de animales según el virus administrado: respectivamente MVAN33 (control negativo), una disolución salina de Tris-HCl/NaCl (control negativo), MVATG8042 (formulación transmembránica) y MVATG6090 (formulación nuclear). La evolución de los tumores se evalúa dos veces por semanas durante 12 semanas. Los resultados muestran una mejor eficacia de la presentación membránica en términos de protección antitumoral en un contexto terapéutico. El 100% de los ratones que han recibido MVATG8042 han sobrevivido 140 días después de la implantación de las células tumorales, mientras que el porcentaje es de aproximadamente 60% para los animales inyectados con MVATG6090, y muy inferior por los animales del control. 25 B. Estudios de toxicidad - efecto de la dosis 30 Es importante verificar la ausencia de virulencia de los virus antes de considerar su aplicación a los tumores humanos. Se administran a ratones atímicos (5 animales/grupo) 106 o 107 pfu de virus MVATG8042, MVAN33 o un virus de la viruela vacuna Copenhague salvaje (VVwt), por vía intracraneal. La supervivencia de los ratones se sigue durante 20 días. Los resultados se presentan en la tabla 1 siguiente: TABLA 1 35 Virus 106 pfu 107 pfu MVAN33 5/5 5/5 MVATG8042 5/5 5/5 VVwt - 0/5 40 45 50 Numero de ratones atímicos supervivientes No se detecta ningún efecto secundario después de la administración intracraneal de 107 pfu del virus MVATG8042, mientras que todos los ratones tratados con el virus de la viruela vacuna salvaje están muertos tres días después de la inyección. El virus MVA de control tampoco es tóxico para los animales, lo que confirma su atenuación con relación al virus de la viruela vacuna ya descrita en la bibliografía. 55 60 65 16 ES 2 229 528 T3 REIVINDICACIONES 5 10 1. Composición antitumoral que comprende al menos un vector recombinante que contiene las secuencias que codifican uno o varios polipéptidos inmunógenos, caracterizada porque al menos uno de dichos polipéptidos es un polipéptido que presenta naturalmente una localización no membránica y está modificado mediante la inserción de una secuencia de anclaje membránica y, en el caso en que el polipéptido inmunógeno nativo esté desprovisto de ella, de una secuencia de secreción a fin de presentar una localización membránica en la superficie de las células en las que se expresa, y caracterizada porque dicho polipéptido inmunógeno presenta un grado de similitud superior a 70% con la totalidad o parte de un polipéptido codificado por una región precoz y/o tardía de un papilomavirus. 2. Composición antitumoral según la reivindicación 1, caracterizada porque dicho polipéptido presenta naturalmente una localización nuclear y además se elimina de susecuencia natural de localización nuclear. 15 20 25 3. Composición antitumoral según una de las reivindicaciones 1 y 2, caracterizada porque dicha secuencia de anclaje membránica y/o la secuencia de secreción se selecciona de entre el grupo constituido por la de la glicoproteína rábica, la glicoproteína env del virus VIH, y la proteína F del virus de la rubéola. 4. Composición antitumoral según una de las reivindicaciones 2 a 3, caracterizada porque dicha secuencia de secreción se inserta en el extremo N-terminal, y la secuencia de anclaje membránica se inserta en el extremo C-terminal de dicho polipéptido inmunógeno. 5. Composición antitumoral según una de las reivindicaciones 1 a 4, caracterizada porque dicho polipéptido inmunógeno presenta un grado de similitud superior a 70% con un polipéptido codificado mediante la región precoz de un papilomavirus. 6. Composición antitumoral según la reivindicación 5, caracterizada porque dicho polipéptido inmunógeno es un polipéptido E6 o E7 de un papilomavirus. 30 35 7. Composición antitumoral según la reivindicación 6, caracterizada porque dicho polipéptido inmunógeno es una variante no oncogénica de dicho polipéptido E6 o E7 de un papilomavirus mutado a nivel de la región implicada en el proceso de transformación celular. 8. Composición antitumoral según una de las reivindicaciones 1 a 4, caracterizada porque dicho polipéptido inmunógeno presenta un grado de similitud superior a 70% con un polipéptido L1 o L2 de un papilomavirus. 9. Composición antitumoral según una de las reivindicaciones 1 a 8, caracterizada porque al menos un polipéptido inmunógeno presenta un grado de similitud superior a 70% con un polipéptido precoz, y porque al menos un polipéptido inmunógeno presenta un grado de similitud superior a 70% con un polipéptido tardío de un papilomavirus. 40 45 10. Composición antitumoral según una de las reivindicaciones 1 a 9, caracterizada porque al menos un polipéptido inmunógeno es tal que: (1) dicho polipéptido inmunógeno tiene una secuencia que presenta un grado de identidad superior a 70% o es idéntica a la que se muestra en la SEC ID nº: 1, (2) dicho polipéptido inmunógeno tiene una secuencia que presenta un grado de identidad superior a 70% o es idéntica a la que se muestra en la SEC ID nº: 2, 50 55 60 65 (3) dicho polipéptido inmunógeno tiene una secuencia que presenta un grado de identidad superior a 70% o es idéntica a la que se muestra en la SEC ID nº: 1, y un polipéptido inmunógeno que tiene una secuencia que presenta un grado de identidad superior a 70% o es idéntica a la que se muestra en la SEC ID nº: 2, (4) dicho polipéptido inmunógeno tiene una secuencia que presenta un grado de identidad superior a 70% o es idéntica a la que se muestra en la SEC ID nº: 1, siendo un polipéptido inmunógeno la proteína L1 de un papilomavirus y/o siendo un polipéptido inmunógeno la proteína L2 de un papilomavirus, (5) dicho polipéptido inmunógeno tiene una secuencia que presenta un grado de identidad superior a 70% o es idéntica a la que se muestra en la SEC ID nº: 2, siendo un polipéptido inmunógeno la proteína L1 de un papilomavirus y/o siendo un polipéptido inmunógeno la proteína L2 de un papilomavirus, o (6) dicho polipéptido inmunógeno tiene una secuencia que presenta un grado de identidad superior a 70% o es idéntica a la que se muestra en la SEC ID nº: 1, y un polipéptido inmunógeno que tiene una secuencia que presenta un grado de identidad superior a 70% o es idéntica a la que se muestra en la SEC ID nº: 2, siendo un polipéptido inmunógeno la proteína L1 de un papilomavirus y/o siendo un polipéptido inmunógeno la proteína L2 de un papilomavirus. 11. Composición antitumoral según una de las reivindicaciones 1 a 10, caracterizada porque dicho papilomavirus es un papilomavirus humano, tal como HPV-16, 18, 31, 33 ó 45. 17 ES 2 229 528 T3 12. Composición antitumoral según una de las reivindicaciones 1 a 11, caracterizada porque dicho vector recombinante comprende además las secuencias que codifican al menos para un compuesto que mejora el efecto antitumoral de dicha composición. 5 10 13. Composición antitumoral según la reivindicación 12, caracterizada porque dicho compuesto que mejora el efecto antitumoral es un inmunoestimulante. 14. Composición antitumoral según la reivindicación 13, caracterizada porque dicho compuesto inmunoestimulante se selecciona de entre el grupo constituido por la interleuquina-2, la interleuquina-7, la interleuquina-12 y las moléculas de co-adhesión B7.1 y B7.2. 15. Composición antitumoral según una de las reivindicaciones 1 a 14, caracterizada porque dicho vector recombinante es un poxvirus. 15 16. Composición antitumoral según la reivindicación 15, caracterizada porque dicho poxvirus es un MVA. 17. Composición antitumoral según una de las reivindicaciones 1 a 16, que contiene un soporte aceptable desde un punto de vista farmacéutico, que permite su administración mediante inyección al ser humano o al animal. 20 18. Vector recombinante que comprende las secuencias que codifican para uno o varios polipéptidos inmunógenos, caracterizado porque al menos uno de dichos polipéptidospresenta las características definidas en las reivindicaciones 1 a 17. 19. Partícula vírica que comprende un vector recombinante según la reivindicación 18. 25 20. Composición antitumoral caracterizada porque comprende uno o varios polipéptidos inmunógenos, caracterizada porque al menos uno de dichos polipéptidos presenta las características definidas en las reivindicaciones 1 a 11. 30 35 21. Utilización de una composición antitumoral según una de las reivindicaciones 1 a 17 y 20, de un vector recombinante según la reivindicación 18, o de una partícula vírica según la reivindicación 19, para la preparación de un medicamento destinado al tratamiento o a la prevención de un cáncer o de un tumor. 22. Utilización según la reivindicación 21, para la preparación de un medicamento destinado al tratamiento o a la prevención de un cáncer de cuello uterino, de una displasia del cuello de bajo grado o de una infección con papilomavirus. 40 45 50 55 60 65 18 ES 2 229 528 T3 19 ES 2 229 528 T3 20 ES 2 229 528 T3 21 ES 2 229 528 T3 22 ES 2 229 528 T3 23 ES 2 229 528 T3 LISTA DE SECUENCIAS (1) INFORMACIÓN GENERAL: 5 (i) DEPOSITARIO: (A) NOMBRE: Transgene SA (B) DIRECCIÓN: 11 rue de Molsheim 10 (C) CIUDAD: Estrasburgo (D) PAIS: Francia (E) CÓDIGO POSTAL: 67082 (F) TELÉFONO: (33) 03 88 27 91 00 15 (G) FAX: (33) 03 88 27 91 11 (ii) TÍTULO DE LA INVENCIÓN: Composición antitumoral a base de polipéptido inmunógeno de localización celular modificada. 20 (iii) NÚMERO DE SECUENCIAS: 23 (iv) FORMATO LEGIBLE POR ORDENADOR: (A) TIPO DE SOPORTE: Cinta 25 (B) ORDENADOR: IBM PC compatible (C) SISTEMA OPERATIVO: PC-DOS/MS-DOS (D) PROGRAMA: PatentIn Release #1.0, versión #1.25 (OEB) 30 (2) INFORMACIÓN PARA LA SEC ID nº: 1: (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 243 aminoácidos 35 (B) TIPO: aminoácido (C) CONFIGURACIÓN: lineal (ii) TIPO DE MOLÉCULA: proteína 40 (iii) HIPOTÉTICA: NO (iv) ORIGEN: (A) ORGANISMO: Papilomavirus humano 45 (B) CEPA: HPV-16 (C) CEPA CLÍNICA INDIVIDUAL: Proteína E6 fusionada a señales de la proteína F (v) FUENTE INMEDIATA: 50 (A) CLON: E6*TMF (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 1: 55 60 65 1 ES 2 229 528 T3 5 10 15 20 25 30 35 40 45 50 (3) INFORMACIÓN PARA LA SEC ID nº: 2: 55 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 185 aminoácidos (B) TIPO: aminoácido (C) CONFIGURACIÓN: lineal 60 (ii) TIPO DE MOLÉCULA: proteína (iii) HIPOTÉTICA: NO 65 (iv) ORIGEN: (A) ORGANISMO: Papilomavirus humano (B) CEPA: HPV-16 2 ES 2 229 528 T3 (C) CEPA CLÍNICA INDIVIDUAL: E7 fusionada a las señales de la glicoproteína rábica (v) FUENTE INMEDIATA: 5 (A) CLON: E7*TMF (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 2: 10 15 20 25 30 35 40 45 (4) INFORMACIÓN PARA LA SEC ID nº: 3: (i) CARACTERÍSTICAS DE LA SECUENCIA: 50 (A) LONGITUD: 36 pares de bases (B) TIPO: ácido nucleico (C) NÚMERO DE HEBRAS: Simple (D) CONFIGURACIÓN: lineal 55 (ii) TIPO DE MOLÉCULA: ADN (genómico) (iii) HIPOTÉTICA: NO (iv) ANTISENTIDO: SÍ 60 (v) ORIGEN: (A) ORGANISMO: Papilomavirus humano (B) CEPA: HPV-16 65 (C) CEPA CLÍNICA INDIVIDUAL: oligonucleótido de síntesis oTG5118 (E7 eliminado 21 26) (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 3: 3 ES 2 229 528 T3 TCTGAGCTGT CATTTAATTG AGTTGTCTCT GGTTGC 36 (5) INFORMACIÓN PARA LA SEC ID nº: 4: 5 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 42 pares de bases (B) TIPO: ácido nucleico 10 (C) NÚMERO DE HEBRAS: Simple (D) CONFIGURACIÓN: lineal (ii) TIPO DE MOLÉCULA: ADN (genómico) 15 (iii) HIPOTÉTICA: NO (iv) ANTI-SENTIDO: NO (v) ORIGEN: 20 (A) ORGANISMO: virus de la rabia (B) CEPA CLÍNICA INDIVIDUAL: oligonucleótido de mutagénesis oTG5745 (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 4: 25 TGCACTCAGT AATACATAGG ATCCAATAGG GAATTTCCCA AA 42 (6) INFORMACIÓN PARA LA SEC ID nº: 5: 30 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 38 pares de bases (B) TIPO: ácido nucleico 35 (C) NÚMERO DE HEBRAS: Simple (D) CONFIGURACIÓN: lineal (ii) TIPO DE MOLÉCULA: ADN (genómico) 40 (iii) HIPOTÉTICA: NO (iv) ANTI-SENTIDO: SÍ (v) ORIGEN: 45 (C) CEPA CLÍNICA INDIVIDUAL: oligonucleótido de síntesis oTG6390 (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 5: GTATCTCCAT GCATGGATCC TGCAGGGTTT CTCTACGT 50 (7) INFORMACIÓN PARA LA SEC ID nº: 6: (i) CARACTERÍSTICAS DE LA SECUENCIA: 55 (A) LONGITUD: 36 pares de bases (B) TIPO: ácido nucleico (C) NÚMERO DE HEBRAS: Simple 60 (D) CONFIGURACIÓN: lineal (ii) TIPO DE MOLÉCULA: ADN (genómico) (iii) HIPOTÉTICA: NO 65 (iv) ANTI-SENTIDO: SÍ (v) ORIGEN: 4 38 ES 2 229 528 T3 (C) CEPA CLÍNICA INDIVIDUAL: oligonucleótido de síntesis oTG6880 (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 6: 5 GGATCCGCCA TGGTAGATCT TGGTTTCTGA GAACAG 36 (8) INFORMACIÓN PARA LA SEC ID nº: 7: 10 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 32 pares de bases (B) TIPO: ácido nucleico (C) NÚMERO DE HEBRAS: Simple 15 (D) CONFIGURACIÓN: lineal (ii) TIPO DE MOLÉCULA: ADN (genómico) 20 (iii) HIPOTÉTICA: NO (iv) ANTI-SENTIDO: SÍ (v) ORIGEN: 25 (A) ORGANISMO: virus de la rabia (B) CEPA: HPV-16 (C) CEPA CLÍNICA INDIVIDUAL: oligonucleótido de síntesis oTG5377 (E6 eliminado 111 a 115) (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 7: 30 TGTCCAGATG TCTTTGCAGT GGCTTTTGAC AG 32 (9) INFORMACIÓN PARA LA SEC ID nº: 8: 35 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 34 pares de bases (B) TIPO: ácido nucleico 40 (C) NÚMERO DE HEBRAS: Simple (D) CONFIGURACIÓN: lineal (ii) TIPO DE MOLÉCULA: ADN (genómico) 45 (iii) HIPOTÉTICA: NO (iv) ANTI-SENTIDO: NO (v) ORIGEN: 50 (B) CEPA: oligonucleótido de síntesis oTG10829 (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 8: 55 GCGCGCTCTA GAATTATGGG TCTCAAGGTG AACG (10) INFORMACIÓN PARA LA SEC ID nº: 9: 60 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 35 pares de bases (B) TIPO: ácido nucleico (C) NÚMERO DE HEBRAS: Simple 65 (D) CONFIGURACIÓN: lineal (ii) TIPO DE MOLÉCULA: ADN (genómico) 5 34 ES 2 229 528 T3 (iii) HIPOTÉTICA: NO (iv) ANTI-SENTIDO: SÍ 5 (v) ORIGEN: (B) CEPA: oligonucleótido de síntesis oTG10830 (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 9: 10 CAGTTCTCTT TTGGTGCATG CCCCAATGGA TTTGA 35 (11) INFORMACIÓN PARA LA SEC ID nº: 10: 15 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 38 pares de bases (B) TIPO: ácido nucleico 20 (C) NÚMERO DE HEBRAS: Simple (D) CONFIGURACIÓN: lineal (ii) TIPO DE MOLÉCULA: ADN (genómico) 25 (iii) HIPOTÉTICA: NO (iv) ANTI-SENTIDO: NO (v) ORIGEN: 30 (B) CEPA: oligonucleótido de síntesis oTG10835 (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 10: ATGCTAGTGC TCGATAAACC CAGCTGGGTT TCTCTACG 38 35 (12) INFORMACIÓN PARA LA SEC ID nº: 11: (i) CARACTERÍSTICAS DE LA SECUENCIA: 40 (A) LONGITUD: 35 pares de bases (B) TIPO: ácido nucleico (C) NÚMERO DE HEBRAS: Simple (D) CONFIGURACIÓN: lineal 45 (ii) TIPO DE MOLÉCULA: ADN (genómico) (iii) HIPOTÉTICA: NO 50 (iv) ANTI-SENTIDO: SÍ (v) ORIGEN: (B) CEPA: oligonucleótido de síntesis oTG10836 55 (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 11: TCAAATCCAT TGGGGCATGC ACCAAAAGAG AACTG 60 (13) INFORMACIÓN PARA LA SEC ID nº: 12: (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 38 pares de bases 65 (B) TIPO: ácido nucleico (C) NUMERO DE HEBRAS: Simple (D) CONFIGURACIÓN: lineal 6 35 ES 2 229 528 T3 (ii) TIPO DE MOLÉCULA: ADN (genómico) (iii) HIPOTÉTICA: NO 5 (iv) ANTI-SENTIDO: NO (v) ORIGEN: (C) CEPA CLÍNICA INDIVIDUAL: oligonucleótido de síntesis oTG10833 10 (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 12: CGTAGAGAAA CCCAGCTGGG TTTATCGAGC ACTAGCAT 15 38 (14) INFORMACIÓN PARA LA SEC ID nº: 13: (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 36 pares de bases 20 (B) TIPO: ácido nucleico (C) NUMERO DE HEBRAS: Simple (D) CONFIGURACIÓN: lineal 25 (ii) TIPO DE MOLÉCULA: ADN (genómico) (iii) HIPOTÉTICA: NO (iv) ANTI-SENTIDO: SÍ 30 (v) ORIGEN: (A) CEPA CLÍNICA INDIVIDUAL: oligonucleótido de síntesis oTG10834 (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 13: 35 GCGGGCATGC GGTACCTCAG AGCGACCTTA CATAGG 36 (15) INFORMACIÓN PARA LA SEC ID nº: 14: 40 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 32 pares de bases (B) TIPO: ácido nucleico 45 (C) NUMERO DE HEBRAS: Simple (D) CONFIGURACIÓN: lineal (ii) TIPO DE MOLÉCULA: ADN (genómico) 50 (iii) HIPOTÉTICA: NO (iv) ANTI-SENTIDO: NO (v) ORIGEN: 55 (A) ORGANISMO: Virus de la viruela vacuna (A) CEPA: Ankara modificada (B) CEPA CLÍNICA INDIVIDUAL: Oligonucleótido de síntesis oTG7637 (PCR zona III) 60 (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 14: GGGGGGGAAT TCAGTAAACT TGACTAAATC TT (16) INFORMACIÓN PARA LA SEC ID nº: 15: 65 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 39 pares de bases 7 32 ES 2 229 528 T3 (B) TIPO: ácido nucleico (C) NUMERO DE HEBRAS: Simple (D) CONFIGURACIÓN: lineal 5 (ii) TIPO DE MOLÉCULA: ADN (genómico) (iii) HIPOTÉTICA: NO 10 (iv) ANTI-SENTIDO: SÍ (v) ORIGEN: (A) ORGANISMO: Virus de la viruela vacuna (B) CEPA: Ankara modificada 15 (C) CEPA CLÍNICA INDIVIDUAL: Oligonucleótido de síntesis oTG7638 (PCR zona III) (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 15: 20 GGGGGGGGAT CCGAGCTCAC CAGCCACCGA AAGAGCAAT 39 (17) INFORMACIÓN PARA LA SEC ID nº: 16: 25 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 32 pares de bases (B) TIPO: ácido nucleico (C) NÚMERO DE HEBRAS: Simple 30 (D) CONFIGURACIÓN: lineal (ii) TIPO DE MOLÉCULA: ADN (genómico) (iii) HIPOTÉTICA: NO 35 (iv) ANTI-SENTIDO: NO (v) ORIGEN: (A) ORGANISMO: Virus de la viruela vacuna 40 (B) CEPA: Ankara modificada (C) CEPA CLÍNICA INDIVIDUAL: Oligonucleótido de síntesis oTG7635 (PCR zona III) (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 16: 45 GGGGGGGGAT CCGGAAAGTT TTATAGGTAG TT (18) INFORMACIÓN PARA LA SEC ID nº: 17: 50 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 30 pares de bases (B) TIPO: ácido nucleico 55 (C) NÚMERO DE HEBRAS: Simple (D) CONFIGURACIÓN: lineal (ii) TIPO DE MOLÉCULA: ADN (genómico) 60 (iii) HIPOTÉTICA: NO (iv) ANTI-SENTIDO: SÍ (v) ORIGEN: 65 (A) ORGANISMO: Virus de la viruela vacuna (B) CEPA: Ankara modificada 8 32 ES 2 229 528 T3 (C) CEPA CLÍNICA INDIVIDUAL: Oligonucleótido de síntesis oTG7636 (PCR zona III) (vi) DESCRIPCIÓN DE LA SECUENCIA:SEC ID nº: 17: 5 GGGGGGGAAT TCTTTGTATT TACGTGAACG (19) INFORMACIÓN PARA LA SEC ID nº: 18: 10 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 77 pares de bases (B) TIPO: ácido nucleico (C) NÚMERO DE HEBRAS: Simple 15 (D) CONFIGURACIÓN: lineal (ii) TIPO DE MOLÉCULA: ADN (genómico) 20 (iii) HIPOTÉTICA: NO (iv) ANTI-SENTIDO: NO (v) ORIGEN: 25 (B) CEPA: oligonucleótido de síntesis oTG10502 (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 18: 30 AGCTTTTTAT TCTATACTTA AAAAATGAAA ATAAACTCGA GTTGTCAAAG CATCATCTCA ACACTGACTT GAGGTAC 60 77 (20) INFORMACIÓN PARA LA SEC ID nº: 19: 35 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 69 pares de bases (B) TIPO: ácido nucleico (C) NÚMERO DE HEBRAS: Simple 40 (D) CONFIGURACIÓN: lineal (ii) TIPO DE MOLÉCULA: ADN (genómico) (iii) HIPOTÉTICA: NO 45 (iv) ANTI-SENTIDO: SÍ (v) ORIGEN: (A) CEPA: oligonucleótido de síntesis oTG10503 50 (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 19: 55 CTCAAGTCAG TGTTGAGATG ATGCTTTGAC AACTCGAGTT TATTTTCATT TTTTAAGTAT AGAATAAAA (21) INFORMACIÓN PARA LA SEC ID nº: 20: (i) CARACTERÍSTICAS DE LA SECUENCIA: 60 (E) LONGITUD: 39 pares de bases (F) TIPO: ácido nucleico (G) NÚMERO DE HEBRAS: Simple 65 (H) CONFIGURACIÓN: lineal (ii) TIPO DE MOLÉCULA: ADN (genómico) 9 60 69 ES 2 229 528 T3 (iii) HIPOTÉTICA: NO (iv) ANTI-SENTIDO: SÍ 5 (v) ORIGEN: (C) CEPA CLÍNICA INDIVIDUAL: oligonucleótido de síntesis oTG5925 (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 20: 10 TCAGATCTGT CGAGGGATCT GCAGCTTCTT CTAGAGGTA 39 (22) INFORMACIÓN PARA LA SEC ID nº: 21: 15 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 44 pares de bases (B) TIPO: ácido nucleico 20 (C) NÚMERO DE HEBRAS: Simple (D) CONFIGURACIÓN: lineal (ii) TIPO DE MOLÉCULA: ADN (genómico) 25 (iii) HIPOTÉTICA: NO (iv) ANTI-SENTIDO: NO (v) ORIGEN: 30 (C) CEPA CLÍNICA INDIVIDUAL: oligonucleótido de síntesis oTG5924 (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 21: AGTGAATTGC TGCAGGTACC CGGATCCGCA TCGACTATCG ACAT 44 35 (23) INFORMACIÓN PARA LA SEC ID nº: 22: (i) CARACTERÍSTICAS DE LA SECUENCIA: 40 (A) LONGITUD: 35 pares de bases (B) TIPO: ácido nucleico (C) NÚMERO DE HEBRAS: Simple (D) CONFIGURACIÓN: lineal 45 (ii) TIPO DE MOLÉCULA: ADN (genómico) (iii) HIPOTÉTICA: NO 50 (iv) ANTI-SENTIDO: NO (v) ORIGEN: (A) ORGANISMO: Homo sapiens 55 (B) CEPA: Estirpe celular Daudi (C) CEPA CLÍNICA INDIVIDUAL: cebador PCR oTG6353 (clonación B7.1) (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 22: 60 TCAGCCCCTG AATTCTGCGG ACACTGTTAT ACAGG (24) INFORMACIÓN PARA LA SEC ID nº: 23: 65 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 33 pares de bases (B) TIPO: ácido nucleico 10 35 ES 2 229 528 T3 (C) NÚMERO DE HEBRAS: Simple (D) CONFIGURACIÓN: lineal 5 (ii) TIPO DE MOLÉCULA: ADN (genómico) (iii) HIPOTÉTICA: NO (iv) ANTI-SENTIDO: SÍ 10 (v) ORIGEN: (A) ORGANISMO: Homo sapiens (B) CEPA: Estirpe celular Daudi (C) CEPA CLÍNICA INDIVIDUAL: cebador PCR oTG6352 (clonación B7.1) 15 (vi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID nº: 23 TTGACCCTAA AGATCTGAAG CCATGGGCCA CAC 20 25 30 35 40 45 50 55 60 65 11 33