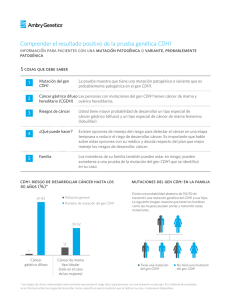
Ver/Abrir - Scriptorium Principal
Anuncio

Universidad de La Habana Facultad de Biología Selección de un modelo celular para evaluar la influencia de la sobre-expresión del gen supresor de tumores Cdh1 en células tumorales Autor: Dayana Pérez Martínez Tutores: Dr. Joel de León Delgado / Lic. Anett Rubio Torres Centro de Inmunología Molecular La Habana, junio 2015 “Lo importante en ciencia no es tanto obtener nuevos hechos como descubrir nuevas formas de pensar sobre ellos.” William Lawrence Bragg A las personas que confían y apuestan por mi Índice Índice 1. Introducción ...........................................................................................................................................1 2. Revisión bibliográfica ............................................................................................................................3 2.1 La molécula Cdh1 .................................................................................................................................3 2.1.1 Cdh1 como co-activador del APC/C ..................................................................................................3 2.1.2 Cdh1 como gen supresor de tumores ...............................................................................................5 2.2 Regulación de la actividad del APC/CCdh1 .............................................................................................6 2.3 Blancos del APC/CCdh1 .........................................................................................................................10 2.3.1 Regulación de la expresión de Plk1 .................................................................................................10 2.3.2 Regulación de la expresión de Aurora quinasa A ............................................................................12 2.3.3 Relación indirecta del APC/CCdh1 con p27 ........................................................................................13 3. Materiales y métodos..........................................................................................................................15 3.1 Líneas celulares ..................................................................................................................................15 3.2 Condiciones de cultivo celular............................................................................................................15 3.3 Sincronización en la fase G2/M del ciclo celular ................................................................................15 3.4 Ensayos de citometría de flujo ...........................................................................................................16 3.5 Ensayos de Western Blot ....................................................................................................................17 3.6 Procesamiento de los datos ...............................................................................................................18 4. Resultados............................................................................................................................................19 4.1 Comparación de los niveles de expresión de la proteína Cdh1 en líneas tumorales humanas de diferentes localizaciones. .........................................................................................................................19 4.2 Determinación de los niveles de expresión de la proteína Cdh1 en líneas tumorales humanas de origen mamario. .......................................................................................................................................19 4.3 Determinación de la expresión del inhibidor de las quinasas dependientes de ciclinas p27 y de los blancos moleculares de Cdh1, AURKA y Plk1, en líneas tumorales humanas de origen mamario. .........21 4.4 Determinación de la expresión proteica de Cdh1 en líneas tumorales murinas de origen mamario. ..................................................................................................................................................................24 5. Discusión ..............................................................................................................................................26 6. Conclusiones ........................................................................................................................................30 7. Recomendaciones ...............................................................................................................................31 8. Referencias bibliográficas ...................................................................................................................32 Introducción 1. Introducción El ciclo celular requiere de proteínas reguladoras para la correcta multiplicación de las células. Dentro de estas proteínas se pueden citar las ciclinas, las quinasas dependientes de ciclinas, las proteínas inhibidoras de estas quinasas, así como las proteínas de control que a su vez determinan el funcionamiento de las anteriores (Vermeulen y cols., 2003). Fallas en la actividad de estas moléculas conllevan al desarrollo y acumulación de aberraciones genéticas que, junto a otros factores intrínsecos y extrínsecos, favorecen el desarrollo de neoplasias (Hartwell y Kastan, 1994). La relación directa entre las afectaciones en el funcionamiento y expresión de estas moléculas reguladoras y la aparición de diferentes tipos de cáncer se ha demostrado (Sherr, 1996; Park y Lee, 2003). Por ello, la búsqueda de blancos terapéuticos relacionados directamente con la dinámica del ciclo celular representa una estrategia para combatir esta enfermedad (McDonald y el Deiry, 2000). En este marco emerge el gen supresor de tumores Cdh1 (del inglés, Cdc20 homologue 1) como blanco atractivo en la terapia antitumoral. Cdh1 es una proteína de 55 kDa que actúa como co-activador del Complejo Promotor de la Anafase/Ciclosoma (APC/C; del inglés, Anaphase Promoting Complex/Cyclosome). Al asociarse a este complejo, juega un rol fundamental en el control de la mitosis mediando la ubicuitinación y degradación de importantes reguladores mitóticos, tales como las ciclinas A y B1, securina, geminina, Polo-like quinasa 1 (Plk1, del inglés Polo-like kinase 1) y Aurora quinasa A (AURKA, del inglés Aurora A kinase) (Peters, 2006; Li y Zhang, 2009). El APC/C humano es una ubicuitín-ligasa E3 compuesto por la asociación de más de una docena de subunidades distintas, alcanzando un peso molecular aproximado de 1.5 MDa (Peters, 2006). Los dos coactivadores más estudiados que determinan la especificidad de sustratos de esta ubicuitínligasa son las proteínas adaptadoras Cdc20 (del inglés, cell division cycle 20) y Cdh1. En el ciclo celular Cdc20 se une al APC/C en la mitosis temprana para promover la transición de metafase a anafase, mientras que Cdh1 se une al APC/C en la mitosis tardía y extiende su actividad hasta la fase G1 donde evita la entrada prematura a la fase S (Li y cols., 2007; 2008; García-Higuera y cols., 2008). APC/CCdh1 promueve los eventos de diferenciación celular al coordinar la salida de la mitosis y la detención del ciclo en la fase G1 (Wäsch y cols., 2010). También contribuye a la integridad genética de las células, ya que participa en las rutas de señalización apoptóticas ante la presencia de daños en el ADN (Qiao y cols., 2010), así como en los puntos de control que evitan el progreso del ciclo celular ante estos mismos daños. La afectación de la expresión de Cdh1 provoca la acumulación de sus blancos moleculares, algunos de los cuales potencian la proliferación celular causando una menor fidelidad de la duplicación del material genético (García-Higuera y cols., 2008). Por tanto, la afectación de la 1 Introducción funcionalidad de la proteína Cdh1 conlleva a trastornos importantes en la dinámica del ciclo celular, lo que podría desencadenar el desarrollo de neoplasias. Por otra parte, se ha demostrado la relación entre APC/CCdh1 y el metabolismo energético. Esta ubicuitín-ligasa reconoce y destina a la degradación a dos enzimas metabólicas relevantes para la proliferación de las células tumorales, la PFKBR3 (del inglés, 6-phosphofructo-2kinase/fructose-2,6-bisphosphatase) y la GLS1 (glutaminasa 1) (Moncada y cols., 2012). Las células tumorales expresan preferentemente PFKBR3, lo que favorece la ruta glicolítica y el efecto Warburg (Chesney y cols., 1999; Yalcin y cols., 2009). GLS1 es crucial para el consumo de la glutamina en el paso de la fase S a la mitosis (Pérez y cols., 2005). Varias investigaciones relacionan la afectación de la expresión y funcionalidad de Cdh1 con el desarrollo de tumores. Wang y Kung en el 2012 expusieron que debido a la acción de la quinasa MAK (de inglés, Male germ cell Associated Kinase), Cdh1 es poco funcional en el cáncer de próstata. Además, se han encontrado diferencias significativas en el nivel de esta proteína entre tejidos normales y los respectivos tejidos tumorales, por ejemplo en el cáncer de recto, pulmón, ovario, próstata, colon y mama (Fujita y cols., 2008; 2009). En particular, se ha indagado sobre el valor pronóstico de Cdh1 en el cáncer de mama y se ha demostrado que altos niveles relativos de esta proteína se asocian a una mayor supervivencia en pacientes con dicha enfermedad, lo que puede convertir a Cdh1 un novedoso biomarcador para este tipo de neoplasia (Fujita y cols., 2008). El diseño de una estrategia para manipular la expresión de la molécula Cdh1 e incidir sobre la tumorigenicidad requiere de la selección de un modelo relevante de células neoplásicas. Considerando este hecho y los antecedentes antes expuestos, se propuso como objetivo general del trabajo: seleccionar un modelo celular para estudiar la influencia de Cdh1 sobre la tumorigenicidad, basado en la determinación de la expresión de este gen supresor de tumores y de moléculas relacionadas con su funcionalidad. En correspondencia, se trazaron los siguientes objetivos específicos: 1. Comparar la expresión de Cdh1 en líneas tumorales de diferentes localizaciones, sincronizadas o no en la fase G2/M del ciclo celular. 2. Evaluar la expresión de AURKA y Plk1, blancos moleculares de Cdh1, así como del inhibidor de las quinasas dependientes de ciclinas p27 en células tumorales. El presente estudio permitirá comparar la expresión de Cdh1 entre diferentes líneas tumorales y brindará información sobre la expresión de sus blancos, lo cual servirá como herramienta para la correcta selección de un posible modelo celular sobre el cual se evaluará la relevancia de la sobre-expresión de Cdh1 en la tumorigenicidad de las células. 2 Revisión bibliográfica 2. Revisión bibliográfica 2.1 La molécula Cdh1 2.1.1 Cdh1 como co-activador del APC/C El APC/C es una ubicuitín ligasa E3 que regula progresión del ciclo celular. Está compuesta por alrededor de 14 subunidades distintas que se ensamblan en un complejo llegando a alcanzar más de 1.2 MDa (Zhang y cols., 2013). Este complejo es imprescindible en las células eucariotas, puesto que de él depende la separación de las cromátidas en la anafase y por tanto la realización completa de la mitosis, así como los eventos necesarios para la replicación del ADN en la fase S (Peters, 2006). Además se ha demostrado que la inactivación genética de APC/C causa letalidad en todas las especies evaluadas (Wirth y cols., 2004). Las funciones de este complejo de proteínas dependen de su capacidad de ensamblar cadenas de poliubicuitinas sobre las proteínas diana, las que son destruidas por el complejo de proteasoma 26S. Los sustratos del APC/C pueden ser proteínas catalíticas, como es el caso de AURKA y Plk1, o activadores de estas, como por ejemplo las ciclinas, las cuales constituyen subunidades activadoras de las quinasas dependientes de ciclinas (Cdk, del inglés cyclin dependent kinase). El APC/C también puede estimular la actividad enzimática a partir de favorecer la proteólisis de sus inhibidores, como el caso de la securina, cuya degradación estimula la actividad de la enzima separasa importante en la disociación de las cromátidas hermanas durante la anafase (Peters, 2006). La actividad del APC/C es dependiente de varias proteínas co-activadoras que se vinculan al complejo durante períodos específicos del ciclo celular. Las mejores estudiadas son las proteínas Cdc20 y Cdh1, las que se codifican en todas las células eucariotas. APC/CCdc20 está activo en los momentos iniciales de la mitosis, mientras que APC/CCdh1 se activa en la etapa final de la mitosis y se mantiene durante la fase G1, para luego inactivarse nuevamente en el tránsito a la fase S (Peters, 2006). Las proteínas co-activadoras, cuya asociación con APC/C está estrictamente regulada, le confieren especificidad a la acción del APC/C al contar con motivos de reconocimiento específicos para las llamadas cajas de destrucción D (Glotzer y cols., 1991) y cajas KEN encontradas en los sustratos, las cuales determinan el tiempo de vida media de los mismos (Pfleger y Kirschner, 2000). Los sustratos con caja D son reconocidos por ambos co-activadores, mientras que los que cuentan con caja KEN solo se reconocen por Cdh1 (Pfleger y cols., 2001). Las regiones comprometidas con dicho reconocimiento se encuentran en el N-terminal de estos co-activadores y son funcionalmente activas independientemente de que estos estén unidos al APC/C (Pfleger y cols., 2001). Estudios previos demuestran que la 3 Revisión bibliográfica afectación mínima de esta región puede ser fatal para la identificación de las moléculas blancos tales como las ciclinas (Sorensen y cols., 2001). Además, Passmore y Barford evidenciaron en el 2005 que la interacción óptima de los sustratos con el APC/C es específicamente dependiente de la asociación simultánea con el co-activador Cdh1, lo cual explica la existencia de una relación estequiométrica entre ellos, donde, según estos resultados, Cdh1 representa una molécula limitante al comprarse con las cantidades del APC/C. Adicionalmente, la actividad ubicuitín-ligasa de los APC/CCdc20 y APC/CCdh1 se puede ver limitada por la fosforilación de sus sustratos en sitios específicos que impidan una adecuada interacción y reconocimiento entre estas moléculas (Mailand y Diffley, 2005). La inactivación del APC/CCdh1, al terminar la fase G1, es determinante para la acumulación de proteínas requeridas para la replicación del ADN y la mitosis. Los mecanismos descritos como inhibidores de la actividad de este complejo incluyen la degradación en la fase G1, por el propio APC/CCdh1, del componente funcional del complejo UBCH10 (del inglés, ubiquitin-conjugating catalytic human 10), lo que favorece la acumulación de sustratos como la ciclina A. Esta última es un activador de la Cdk2, la que fosforila Cdh1 y favorece su disociación del APC/C. Una vez fosforilado, Cdh1 es ubicuitinado por otra familia de ubicuitín-ligasa y degradado por el proteasoma 26S (Peters, 2006). Adicionalmente, la proteína Emi1 (del inglés, early mitotic inhibitor 1), cuya expresión se estimula en la transición G1-S, inhibe a APC/CCdh1 y APC/CCdc20, lo que favorece la acumulación de los sustratos de estos complejos en este momento del ciclo (Reimann y cols., 2001; Hsu y cols., 2002). Los mecanismos de control que operan durante la fase G0-G1 del ciclo celular influyen en la capacidad de la célula de: replicar su ADN y dividirse, o abandonar el ciclo celular y comenzar el proceso de diferenciación. Precisamente, APC/CCdh1 coordina ambos fenómenos a partir de determinar los niveles de expresión de proteínas que conducen a las células por un camino u otro. La desregulación de este complejo provoca la progresión del ciclo limitando los eventos de diferenciación y potenciando la inestabilidad genética (Qiao y cols., 2010). Por tanto, la reducción en la expresión de Cdh1 puede favorecer la tumorigénesis, a partir de estimular la proliferación de células pobremente diferenciadas y genéticamente inestables (Wäsch y cols., 2010). Lo anterior se explica por la acumulación de sustratos del APC/CCdh1 cuya expresión aberrada contribuye a la inestabilidad genética, tales como AURKA y Plk1, los que se encuentran incrementados en múltiples tipos de tumores (Wäsch y Engelbert, 2005; Lehman y cols., 2007). 4 Revisión bibliográfica 2.1.2 Cdh1 como gen supresor de tumores En la actualidad, la relevancia funcional de Cdh1 ha determinado que esta molécula sea considerada un gen supresor de tumores. La importancia de la misma ha sido evidenciada dado que se ha demostrado que las células deficientes de Cdh1 manifiestan alteraciones en la proliferación y acumulan aberraciones cromosómicas, a la par de que animales heterocigóticos de este gen desarrollan tumores espontáneos (García-Higuera y cols., 2008). La regulación negativa en la expresión de Cdh1 ha sido detectada tanto en cánceres hematológicos como en tumores sólidos (Engelbert y cols., 2008). Tal disminución, respecto a la contraparte del tejido normal, se ha demostrado además en cortes de tejidos tumorales de diversas localizaciones (Fujita y cols., 2008). Además, se ha descrito en una diversidad de tumores humanos el incremento en la expresión de la molécula Emi1, que como se comentó anteriormente ejerce un control negativo sobre la expresión de Cdh1 (Lehman y cols., 2007). En pacientes con tumores de mama no se han demostrado diferencias significativas entre la expresión de Cdh1 y el estado de la enfermedad, el tamaño del tumor o la presencia de metástasis. Sin embargo, en estos mismos pacientes, sí se ha demostrado una correlación inversa entre el intervalo libre de enfermedad y los niveles de expresión de Cdh1 (Fujita y cols., 2009). En modelos tumorales de carcinoma mamario se ha demostrado que la manipulación in vitro de Cdh1 conduce a incrementar o reducir la tumorigenicidad de líneas celulares, en dependencia de la modulación negativa o la sobre-expresión de esta molécula, respectivamente (Fujita y cols., 2009). Los mecanismos que pueden contribuir al carácter de gen supresor de tumores conferido a Cdh1 se relacionan con el efecto de este co-activador sobre eventos que rebasan el control directo del ciclo celular. Esto es debido a su relación con otras proteínas importantes en la biología tumoral, ya sean oncogenes u otros genes supresores de tumores. En este sentido, se ha demostrado la capacidad de Cdh1 de ubicuitinar la molécula Skp2 (del ingés, S phase kinase-associated protein 2), lo que determina la estabilización del gen supresor de tumores p27 quien limita la transición de G1-S y posibilita la realización de este punto de control del ciclo celular (Fujita y cols., 2008; 2009). En tumores donde el oncogén Akt (proteína quinasa B) se encuentra activado, la fosforilación de Skp2 la hace insensible a la degradación por APC/CCdh1, lo que trae como consecuencia la reducción en la expresión de p27 y la pérdida de este punto de control (Gao, 2009). A esto se suma el gen supresor de tumores retinoblastoma quien ejerce su actividad por un mecanismo dependiente de APC/CCdh1, también asociado al control de p27 (Santamaría y Pagano, 2007). Otro gen supresor de tumores, PTEN (del inglés, phosphatase and tensin homolog), cuya actividad determina el control de Akt, contribuye a estabilizar la actividad de Cdh1 por un mecanismo independiente de su actividad fosfatasa (Song y cols., 5 Revisión bibliográfica 2011). Por otra parte uno de los eventos más sorprendentes es que se ha demostrado la participación de Cdh1, tanto en células normales como tumorales, en la conexión entre la proliferación celular y los ajustes en el metabolismo energético asociados a cada etapa del ciclo celular. Estudios han evidenciado la correlación negativa entre la expresión de Cdh1 y las enzimas PFKFBR3 y GLS1, claves en las rutas de glicólisis y glutaminólisis, respectivamente (Moncada y cols., 2012). Otros eventos importantes para la biología tumoral, como la estabilización de Factor inducible por hipoxia‐1α (HIF1α, del inglés, Hypoxia Inducible Factor‐ 1α) por el gen supresor de tumores VHL (von Hippel-Lindau), también se han asociado con la actividad de APC/CCdh1 (Liu y cols., 2011). A pesar de lo atractivo que resulta el gen supresor de tumores Cdh1 como blanco en la terapia antitumoral, no hay reportes en la literatura que demuestren su manipulación in vivo para sobreexpresarlo en células tumorales e incidir sobre la progresión tumoral. En este mismo sentido, la manipulación de su modulador negativo Emi1 tampoco ha sido abordada. De hecho, las evidencias que demuestran que la alteración de la expresión de Cdh1, incremento o reducción, incide en la progresión tumoral, se han obtenido por manipulación in vitro de células tumorales humanas y la posterior implantación de estas en animales inmunodeficientes (Fujita y cols., 2008; 2009). No obstante, blancos de APC/CCdh1 cuya expresión está incrementada en varios tipos de tumores y de los que se ha descrito su valor pronóstico, como AURKA y Plk1, sí han sido dianas de estrategias terapéuticas, por ejemplo las basadas en inhibidores de pequeña talla (Harrington y cols., 2004; Strebhardt y Ulrich, 2006). 2.2 Regulación de la actividad del APC/CCdh1 La capacidad del APC/CCdh1 de reconocer selectivamente sus sustratos en el momento correcto del ciclo celular es esencial para el desarrollo de las diferentes fases de este y la preparación para la ronda de replicación del ADN (Peters, 2006). En correspondencia con su importancia, la actividad de este complejo está estrictamente controlada por mecanismos redundantes que se describen a continuación. En primer lugar, el APC/CCdh1 está básicamente regulado por reacciones sucesivas de fosforilación-desfosforilación, así como por la influencia de determinadas moléculas que actúan como pseudo-sustratos e impiden la correcta actividad del complejo. En las células normales estos mecanismos se complementan para garantizar la actividad del sistema en función del reloj bilógico del ciclo, sin embargo, en las células tumorales, muchos de estos mecanismos están alterados, por lo que rompen la dinámica necesaria para la óptima división celular. 6 Revisión bibliográfica En el tránsito de la fase G1 a S, APC/CCdh1 se inactiva debido a la acción de las ciclinas y quinasas que a este término se acumulan, principalmente la ciclina A y la quinasa Cdk 2 (Lukas y cols., 1999). Estas ciclinas/Cdks fosforilan ambos componentes del complejo, facilitando la disociación del mismo. Una vez que Cdh1 es fosforilado y disociado, se convierte en blanco de degradación del complejo de ubicuitín-ligasas E3 SCF (del inglés, Skp Cullin F-box containing complex), las cuales reconocen preferencialmente los sustratos fosforilados e hidroxilados (Petroski y Deshaies, 2005). Existen evidencias de un mecanismo de retroalimentación negativa en el cual el propio complejo ubicuitina una de sus subunidades, la UBCH-10, la cual es crucial para la degradación de la propia ciclina A (Rape y Kirschner, 2004). La inactivación del complejo en G1-S también es llevada a cabo por el pseudosustrato Emi1, el cual se activa tras la acción del factor de transcripción E2F e impide la actividad de APC/CCdh1 al bloquear la unión de Cdh1 con los verdaderos sustratos (Hsu y cols., 2002). Este mecanismo de regulación es crucial en los procesos de diferenciación de las células madres embrionarias humanas, donde se ha demostrado que la desdiferenciación de estas células es función de los niveles elevados de Emi1 (Bar-On y cols., 2010). En tumores de próstata se describió un mecanismo similar al de Emi1, basado en la actividad de un inhibidor del complejo, el represor de la transcripción Daxx, el cual se une a los co-activadores (tanto Cdh1 como Cdc20) por medio de la región consenso de reconocimiento D-box e impide la degradación de los respectivos sustratos verdaderos durante la mitosis (Kwan y cols., 2013). Otra molécula identificada como pseudosustrato del APC/CCdh1 es la proteína Acm1 (APCCdh1 modulator 1), la cual, aunque es prescindible para la inhibición de la actividad catalítica del complejo, evita las interacciones prematuras de los sustratos con el complejo durante la fase tardía de la mitosis (Martínez y cols., 2012). Otro mecanismo de regulación negativa del complejo por fosforilación es el encontrado por Wang y Kung en el 2012, en el cual Cdh1 es fosforilado e inactivado por la acción de las quinasas MAK. Estas enzimas fosforilan a Cdh1 en los mismos sitios donde actúan las Cdk y determinan la inactividad del complejo durante la fase S y el inicio de la mitosis. Esta inactivación desencadena la acumulación de sustratos como Plk1, AURKA y las ciclinas mitóticas en el cáncer de próstata, donde esta quinasa está sobre-expresada (Wang y Kung, 2012). Según lo antes expuesto, APC/CCdh1 se mantiene inactivo desde el trance de G1 a S, durante toda la fase S del ciclo (donde se requiere de la acumulación de los sustratos del mismo para la nueva ronda de replicación) y hasta la fase tardía de la mitosis, donde, por acción previa del APC/CCdc20 los niveles de las ciclinas (en este caso las tipo B) y quinasas (Cdk1 principalmente) 7 Revisión bibliográfica han disminuido. Debido a esto, los niveles de Cdh1 fosforilados decrecen, por lo que nuevamente recupera su actividad al unirse al APC/C. Es estrictamente necesaria la coordinación entre la actividad del complejo mediada por Cdc20 y Cdh1 durante toda la mitosis. En este sentido, las moléculas pertenecientes al complejo del control mitótico MAD2L2 ( del inglés, mitotic arrest deficient 2-like 2) juegan un rol fundamental al actuar como inhibidoras del complejo (al bloquear la región de reconocimiento de los sustratos) y contribuir a la activación secuencial del APC/C por sus co-activadores (Listovsky y Sale, 2013). En prometafase, MAD2L2 secuestra las moléculas de Cdh1 disociadas del complejo y a la llegada de la anafase, es rápidamente degradada por el APC/CCdc20, elevándose de esta forma los niveles de Cdh1 que pueden interactuar nuevamente con el APC/C desfosforilado. Se ha demostrado que la pérdida de MAD2L2 conlleva a una asociación prematura de Cdh1 con el complejo, a la destrucción temprana de sus sustratos y una mitosis acelerada con frecuentes errores, por lo que MAD2L2 ayuda en la coordinación del trance de metafase a anafase y contribuye a la fidelidad de la mitosis (Listovsky y Sale, 2013). Por otra parte, se sospecha que la activación de Cdh1 en la mitosis tardía también depende de la existencia de fosfatasas homólogas a la Cdc14 encontrada en levaduras (hCdc14a, del inglés human Cdc 14 a), que desfosforilan y activan a las moléculas de Cdh1 fosforiladas remanentes (Peters, 2002). La funcionalidad del APC/CCdh1 también está determinada por la forma en que se presentan los sustratos. Estudios develan que modificaciones post-transcripcionales de determinados sustratos, cercanas a la región de reconocimiento, pueden influir directamente en la capacidad de reconocimiento del complejo de los mismos y en la actividad de ubicuitinación sobre ellos (Littlepage y Ruderman, 2002; Mailand y Diffley, 2005). También se ha descrito la regulación temporal de ubicuitinación del APC/CCdh1 en función de los sustratos. Existen sustratos con mayor susceptibilidad y rapidez de ser ubicuitinados que otros, lo cual determina la existencia de una regulación intrínseca del complejo que define los sustratos tempranos y tardíos del mismo, lo cual depende de la afinidad o estabilidad entre las interacciones intermoleculares durante la reacción de ubicuitinación (Rape y cols., 2006). La acetilación/desacetilación de Cdh1 constituye otra herramienta para controlar el funcionamiento del complejo. Resultados demuestran que cuando Cdh1 está desacetilado se optimiza la unión del mismo al APC/C, mientras que la acetilación provoca el efecto contrario. En este estudio se demostró que la desacetilación y por tanto activación del complejo, es llevado a cabo por la histona desacetilasa SIRT2 (del inglés, Sirtuin-2), la cual por esta vía, emerge como proteína que mantiene la integridad del genoma y la supresión de la tumorigénesis (Kim y cols., 2011). 8 Revisión bibliográfica Respecto a la regulación espacial del complejo durante el ciclo celular, Zhou y cols. en el 2003 demostraron que Cdh1 se encuentra preferencialmente en el núcleo durante la interfase, mientras que durante la metafase y la anafase se encuentra asociado a los centrosomas. Esta localización preferencial en el núcleo durante la interfase responde a la actividad del complejo de ubicuitinar a la ciclina A. Sin embargo, como se describió anteriormente, en el tránsito de G1S, los eventos de fosforilación de la ciclina A/Cdk2 junto a la activación de otros mecanismos reguladores negativos del complejo, hacen que decaiga la actividad del mismo. O sea, durante toda la interfase se establece una relación de competencia recíproca entre Cdh1 y sus moléculas blancos. Como resultado de este estudio se reveló que esta localización nuclear es regulada precisamente por las reacciones de fosforilación de esta ciclina, las cuales determinan la exportación de Cdh1 hacia el citosol (Zhou y cols., 2003). Contrariamente, la localización nuclear del APC/CCdh1 puede estar beneficiada por otras moléculas reguladoras como por ejemplo la fosfatasa PTEN, la cual independientemente de su actividad fosfatasa, interactúa y estabiliza la asociación del complejo mejorando la actividad de ubicuitinación del mismo (Song y cols., 2011). En cuanto a la regulación transcripcional de Cdh1, se ha evidenciado que la transcripción del ARNm de este gen (Fzr1, del inglés Fizzy-related 1) comienza en la fase G1, justo antes del punto de restricción G1-S, extendiéndose hasta las fases S y G2. Posteriormente, los niveles de ARNm decaen bruscamente en el momento de la división celular y se mantienen indetectables durante los primeros momentos de la fase G1, aspectos que son contradictorios ya que el producto proteico se mantiene estable durante todo este período. Además, es en este momento de la interfase (inicio de G1) donde más actividad muestra la proteína para potenciar los eventos de diferenciación celular (Inbal y cols., 1999). Estos resultados sugieren que Fzr1 sufre una compleja regulación a diferentes niveles de síntesis y degradación. Por otra parte, cuando las células son detenidas en G0 por ausencia de nutrientes, se evidencia un aumento de la transcripción de este gen, lo cual demuestra la importancia del mismo en la quiescencia celular (Inbal y cols., 1999). Actualmente los virus han emergido como nuevas entidades reguladoras del APC/C y sus coactivadores. La existencia de diversas familias virales con la capacidad de reprogramar el ciclo celular mediante el APC/C, abre las puertas de un ámbito desconocido lleno de interrogantes. Los mecanismos que emplean los virus responden a los requerimientos celulares que los benefician, e involucran el secuestro de las células en una determinada fase del ciclo celular lo que causa diversas afectaciones en el funcionamiento del complejo (Fehr y Yu, 2013). Algunos de estos mecanismos desencadenan irreversiblemente la transformación de las células 9 Revisión bibliográfica hospederas hacia células malignas, por lo que el entendimiento de estos procesos puede constituir una herramienta útil para la terapéutica de las enfermedades relacionadas, tanto víricas como neoplásicas (Fehr y Yu, 2013). 2.3 Blancos del APC/CCdh1 El APC/C regula el progreso del ciclo celular, debido en primera instancia, a que determina los niveles proteicos de moléculas claves para el control del mismo. La regulación de la expresión de estas moléculas por el complejo puede ser de una forma directa (si las mismas son reconocidas como sustratos y son ubicuitinadas) o de una forma indirecta (si la molécula que es blanco directo del complejo, es otra molécula que determina la expresión de la primera). Son muchas las proteínas que se han identificado como blancos del APC/CCdh1, sobresaliendo aquellas que participan en la salida de la mitosis, la entrada de la anafase, el ensamblaje de las fibras del huso mitótico, la síntesis del ADN, el mantenimiento de las fases G0/G1, así como en diversas rutas de señalización (Li y Zhang, 2009). Dentro de estas moléculas, el control de la expresión de las quinasas Plk1 y AURKA es un evento crítico para la correcta división celular, dado que participan activamente en la mitosis y determinan en cuantía la estabilidad genética de las células (Floyd y cols., 2008). Se ha demostrado que la sobre-expresión de estas quinasas es evidente en transformaciones malignas de diferentes localizaciones y se asocia al desarrollo de aneuploidías y carcinogénesis (Strebhardt y Ullrich, 2006; Ice y cols., 2013). Ambas quinasas se comportan como sustratos directos del APC/CCdh1. Por otra parte, uno de los blancos indirectos del complejo es el inhibidor de las quinasas dependientes de ciclinas p27. Esta proteína es miembro de la familia de los CDKIs (del inglés, cyclin-dependent kinase inhibitors) y en el ciclo regula la transición de G1 a S al detener la célula en G1 (Koljonen y cols., 2006). Precisamente, uno de los mecanismos que tiene el APC/CCdh1 para optimizar la diferenciación celular tras la detención de la célula en esa fase, es impidiendo la degradación proteolítica de p27 (Santamaría y Pagano, 2007). 2.3.1 Regulación de la expresión de Plk1 Plk1 se expresa en células en proliferación y regula diversos procesos que incluyen la maduración de los centrosomas, la entrada en la mitosis, el puto de control de G2/M, la migración cromosómica, la formación del complejo bipolar del huso mitótico, la activación del complejo ciclina B/Cdk1, la salida de la mitosis y la citocinesis (Zitouni y cols., 2014). Estudios demuestran que la expresión exacerbada de Plk1 es evidente en tumores de pulmón, colón, estómago, cérvix, mama, próstata, ente otros, lo cual demuestra el fallo de los diferentes 10 Revisión bibliográfica mecanismos reguladores de la expresión de esta proteína durante el ciclo celular (Winkles y Alberts, 2005; Strebhardt y Ullrich, 2006; Lehman y cols., 2007; Kim y cols., 2013;). Uno de los mecanismos reguladores de Plk1 es la ubicuitinación de la misma en la anafase por el APC/CCdh1, para optimizar la salida de la mitosis (Eckerdt y Strebhardt, 2006). Paradójicamente, esta quinasa es necesaria en la citocinesis, lo cual sugiere la existencia de otros niveles de regulación (Zitouni y cols., 2014). Por ejemplo, en el 2013, Kim y cols. demostraron la existencia de una proteína llamada CIP2A ( del inglés, Cancerous Inhibitor of PP2A), la cual obstruía el reconocimiento del APC/CCdh1 por Plk1, impidiendo de esta forma la degradación de la misma. Lindon y Pines en el 2004 evidenciaron que la activación del APC/CCdh1 no se correlaciona con la disminución inmediata de los niveles de Plk1, lo que ratifica que la regulación de esta quinasa es compleja y puede involucrar mecanismos que van desde la localización celular hasta la afinidad preferencial del APC/C por sus sustratos. Plk1 también es regulada post-traduccionalmente por fosforilación. Según los resultados expuestos por Tang y cols. en el 2008, la fosforilación de Plk1 en la serina 326 (sitio cercano a la región D-box) por acción de Mk2 (del inglés, MAP kinase-activated protein kinase 2), puede conllevar a una estabilidad de la misma al hacerla insensible a la degradación proteolítica. Además, se han obtenido resultados que sugieren que la activación de Plk1 en la transición de G2 a M in vivo, puede ser atribuible mayoritariamente, sino en la totalidad, a la fosforilación (Mundt y cols., 1997). Un mecanismo que contrarresta esta activación por fosforilación es el punto de control ante daños en el ADN en G2/M, donde para impedir el avance del ciclo en este término, se desencadenan mecanismos que impiden la fosforilación de Plk1 en la Serina 137 y la Treonina 210 (Tsvetkov y Stern, 2005). Plk1 también cuenta con un mecanismo de auto-inhibición el cual depende de interacciones entre los extremos C y N terminal de la molécula y es independiente del estado en que se encuentre el ciclo celular. Esta auto-inhibición puede ser revertida por fosforilaciones en el extremo N terminal (Serina 123 y 137; Treonina 196 y 210) o por la unión de un fosfopéptido en el C terminal (Xu y cols., 2013). Además Plk1 es estrictamente regulada a nivel transcripcional. Tanto los niveles de proteínas como los de ARNm son bajos en la Interface y altos en la mitosis (Winkles y Alberts, 2005). La expresión de los genes de Plk1 están bajo el control de varios represores transcripcionales en G1 y activadores transcripcionales en G2 (Archambault y Glover, 2009). Uno de los activadores transcripcionales descritos es el factor E2F, el cual también media la expresión positiva del ya referido inhibidor del APC/C Emi1, de esta forma se justifica la convergencia de la existencia de estas moléculas en diversos tipos de tumores (Lehman y cols., 2007). Por otra parte, los niveles 11 Revisión bibliográfica intracelulares de esta quinasa parecen estar también controlados por la chaperona Hsp90 (Winkles y Alberts, 2005). 2.3.2 Regulación de la expresión de Aurora quinasa A AURKA es una serina/treonina quinasa esencial para la entrada de la mitosis, la duplicación de los centrosomas, la formación del huso mitótico, la segregación cromosómica y la citocinesis (Marumoto y cols., 2005). Se encuentra sobre-expresada en muchos tipos de cáncer (Zhou y cols., 1998) sobresaliendo los de mama, ovario, colon y próstata. Altos niveles de esta proteína se asocian a una disminución en la supervivencia de los pacientes por lo que constituye un marcador pronóstico de esta enfermedad (Ke y cols., 2003; Nadler y cols., 2008). Aunque en los carcinomas primarios de mamas invasivos el 94% tienen una sobre-expresión de esta molécula (Tanaka y cols., 1999), solo el 13.6% muestra una amplificación en estos genes, por lo que en este tipo de transformación maligna los mecanismos post-transcripcionales que determinan la estabilidad o tiempo de vida media de esta proteína son muy importantes (Kitajima y cols., 2007; Ice y cols., 2013). Al igual que Plk1, en la mitosis tardía del ciclo celular, AURKA es poliubicuitinada por el APC/CCdh1 lo que reduce los niveles de la misma (Taguchi y cols., 2002; Floyd y cols., 2008). La ubicuitinación de esta quinasa depende del reconocimiento por el APC/C de las regiones consensos D-box y A-box (Kitajima y cols., 2007). Pese a esto, las células tumorales expresan altos niveles de AURKA independientemente del momento del ciclo, lo cual sugiere que hay mecanismos que hacen escapar a esta quinasa de la degradación proteolítica dependiente de ubicuitinación (Ice y cols., 2013). El primer ejemplo lo constituye la acción de la USP2a (del inglés, Ubiquitin-specific Cysteine Protease 2a). UPS2a es miembro de la familia de deubicuitinasas de la cual se ha evaluado su potencial como oncogén ya que su sobreexpresión se asocia con la formación y progresión de tumores humanos (Priolo y cols., 2006). En el 2011 se demostró que esta deubicuitinasa interactúa directamente con AURKA tanto in vitro como in vivo sugiriéndola como uno de sus sustratos (Shi y cols., 2011). De esta forma la actividad del APC/CCdh1 es directamente revertida, por lo que la degradación de AURKA hasta este nivel, dependerá del balance de ubicuitinación/deubicuitinación de las respectivas enzimas involucradas. Recientemente se ha descrito la existencia de proteínas involucradas en la regulación de la estabilidad de AURKA (Huang y cols., 2011). En este sentido, emerge el factor traduccional PUMP2, el cual se une físicamente a la región D-box de AURKA e impide el reconocimiento y la ubicuitinación de la misma por el APC/CCdh1. La asociación de PUMP2 con AURKA alarga el 12 Revisión bibliográfica tiempo de vida media de la misma y perfecciona su actividad quinasa. Este mismo estudio sugiere que PUMP2 juega dos roles diferentes durante la progresión del ciclo celular: en la interfase, actúa como represor traduccional a través de su dominio de unión al ARN y en la mitosis se une físicamente con AURKA para mejorar su estabilidad y funcionalidad. Otra molécula que impide la destrucción de AURKA por esta vía es el marcador de genes metastásicos NEDD9 (del inglés, Neural precursor cell-expressed, developmentally downregulated 9) identificado en adenocarcinomas de mama y melanomas. Incrementos en la expresión de NEDD9 se correlacionan con incrementos de los niveles de AURKA en cáncer de mama. Esto es debido a que NEDD9 forma un complejo con esta quinasa (por la región A-box de reconocimiento por el APC/CCdh1) y regula su estabilidad. La reducción de NEDD9 por sí sola o en combinación con inhibidores de AURKA disminuye el tamaño de tumores y la metástasis de pulmón en ratones xenotrasplantados (Ice y cols., 2013). Por otra parte, Kitajima y cols. en el 2007 observaron que eventos de fosforilación sobre A-box (Serina 51) pueden impedir la ubicuitinación y por tanto la degradación por esta vía, encontrando además que esta fosforilación ocurre de forma constitutiva en células tumorales de cabeza y cuello donde esta quinasa está sobre-expresada. Además, se demostró que la proteína de unión a los microtúbulos TPX2 (del inglés, targeting protein for Xklp2) no solo incidía en la actividad y localización de AURKA, sino que también determinaba los niveles de la misma al proteger esta quinasa de la acción ubicuitinante del APC/CCdh1 (Giubettini y cols., 2011). También se identificó a la quinasa LIMK2 (del inglés, LIM domain kinase 2), la cual es importante en la oncogénesis de las células de mama debido a que estabiliza a AURKA mediante su dominio LIM. LIMK2 regula positivamente los niveles de AURKA mediante su acción quinasa, creando un mecanismo de retroalimentación positiva mediante el cual AURKA promueve la transformación celular en este tipo de cáncer (Johnson y cols., 2012). 2.3.3 Relación indirecta del APC/CCdh1 con p27 p27 es una proteína de 27 kDa que pertenece a la familia de las KIP (del inglés, kinase inhibitor protein) y al unirse a las Cdks (principalmente Cdk2) evita que estas interactúen con las ciclinas indispensables para la entrada de la célula en la duplicación de su material genético (Slingerland y Pagano, 2000). Los daños en el ADN, las señales inhibitorias del crecimiento y los procesos de proliferación y diferenciación de tejidos en desarrollo estimulan la actividad de p27 (Koljonen y cols., 2006; Egozi y cols., 2007; Cuadrado y cols., 2009), lo que conduce a bloquear la transición errónea de G1 a S. p27 es considerado como un gen supresor de tumores y en correspondencia, las células malignas con frecuencia tienen reducidos los niveles de la misma (Oh y Park, 2000; Bloom y Pagano, 2003). 13 Revisión bibliográfica Los niveles proteicos de p27 están determinados en gran medida por la acción de la proteína Skp2 en asociación con el complejo SCF las cuales la ubicuitinan y la destinan a la degradación proteolítica (Carrano y cols., 1999; Sutterluty y cols., 1999). La asociación de p27 con SCF depende de que Skp2 reconozca el residuo fosforilado de la treonina 187 del mismo, acción que ocurre a cargo de la Cdk2b (Slingerland y Pagano, 2000). De esta forma, altos niveles de Skp2 pueden conllevar a la disminución de p27 y por tanto a la progresión errada del ciclo celular, lo cual desemboca en el desarrollo de transformaciones neoplásicas. Una de las vías claves por las que el APC/CCdh1 regula el ciclo celular es precisamente reconociendo y ubicuitinando a Skp2, por lo que Cdh1 estabiliza p27 y con esto protege la fidelidad del ciclo celular (Bashir y cols., 2004; Wei y cols., 2004). En células de cáncer de mama y colon se evidenció que las cantidades de Cdh1 y p27 disminuyen significativamente con respecto al tejido normal, mientras que Skp2 se mantiene elevada (Fujita y cols., 2008; 2009). Una de las rutas de señalización que atentan contra la efectividad de la asociación del APC/CCdh1 con Skp2 es la que involucra a la quinasa Akt1. Esta quinasa fosforila a Skp2 en la serina72, provocando así su localización citoplasmática y estabilidad al impedir la unión con Cdh1. Esta primera fosforilación facilita que ocurra una segunda en la serina 75 mediada por CKI (del inglés Casein kinase I), lo que resulta en una mayor repulsión entre APC/CCdh1 y Skp2. Esto sugiere que los tumores con exacerbada actividad de Akt1 tienen bajos niveles de p27, lo cual evidencia la existencia de mecanismos recurrentes en las células malignas para garantizar su proliferación y evadir las principales vías de control del ciclo celular (Gao y cols., 2009). Interesantemente, Shirane y cols. demostraron en 1999 que junto a la degradación de p27 dependiente de ubicuitinación, existe otro mecanismo dependiente de ATP (del inglés, Adenosine triphosphate) que disminuye las cantidades proteicas de p27 en G1/S. Este consiste en la degradación proteolítica de esta proteína independientemente de su ubicuitinación (mediada por 26S), pasando por un intermediario de 22 kDa en el cual ha sido removido el dominio de unión con las ciclinas del Nterminal. Estos resultados evidencian otra forma de regular negativamente los niveles de p27, primero asegurando su inactividad y después mediando su degradación proteolítica. 14 Materiales y métodos 3. Materiales y métodos 3.1 Líneas celulares En el presente trabajo se emplearon las líneas celulares humanas: A431 (carcinoma epidermoide de vulva), H125 (carcinoma de pulmón), PC3 (adenocarcinoma de próstata), los adenocarcinomas de mama MCF7, MCF7-HER2, MDA-MB231 y MDA-MB468, así como los carcinomas mamarios ZR75, T47D y SKBR3. Además se emplearon las líneas celulares murinas de origen mamario AT3 (carcinoma), F3II (adenocarcinoma) y 4T1 (carcinoma). 3.2 Condiciones de cultivo celular Las células se cultivaron con el medio DMEM-F12 (del inglés, Dulbecco's Modified Eagle's Medium/Nutrient F-12 Ham, Gibco, EUA) excepto las T47D y las SKBR3, las cuales se cultivaron con el medio RPMI-1640 (del ingés, Roswell Park Memorial Institute, PAA, Austria). Se emplearon como suplementos adicionales 100 U/mL de penicilina, 100 µg/mL de estreptomicina y suero fetal de ternera (SFT) al 10%, todos adquiridos de la firma Gibco, EUA. Las células se cultivaron a 37ºC en atmósfera de 5% de CO2. 3.3 Sincronización en la fase G2/M del ciclo celular Para sincronizar las células en la fase G2/M del ciclo celular, se utilizaron diferentes concentraciones del agente desestabilizante de los microtúbulos nocodazol (Sigma, EUA). Las concentraciones empleadas de nocodazol y los tiempos de tratamiento para cada tipo celular se describen en la Tabla 1. En todos los casos las células se sembraron en placas de 6 pozos (Greiner BioOne, Alemania) a razón de 2x105 células por pozo y el nocodazol se añadió después de 18 h de iniciado el cultivo. Tabla 1. Tiempo de incubación y concentración de nocodazol empleado para la sincronización de las líneas celulares Líneas celulares ng/ml Tiempo de incubación (h) A431 200 12 H125 100 15 PC3 100 24 MDA-MB231 200 15 AT3 200 24 F3II 200 24 4T1 200 24 15 Materiales y métodos 3.4 Ensayos de citometría de flujo Para la determinación de la expresión de Cdh1 en las líneas celulares, sincronizadas o no en la fase G2/M del ciclo celular, las células se colectaron por tratamiento con 300μL de tripsinaEDTA (Gibco, EUA). Luego, se pasaron a viales de 1.5mL y se lavaron dos veces por centrifugación con 1mL de solución salina tamponada con fosfato (SSTF), a 555 x g por 5 min (Eppendorf, Alemania). Las células se permeabilizaron y fijaron con las soluciones Cytofix/Cytoperm, Cytofix/Cytoperm plus y Perm/Wash, incluidas en un juego de reactivos adquirido de la firma BD Bioscience (EUA). Se siguieron las indicaciones del fabricante, con ajustes menores. Brevemente, las células se resuspendieron en 100µL de Cytofix/Cytoperm y se incubaron durante 30 min en hielo. Al cabo de este tiempo, se lavaron por centrifugación a 555 x g durante 5 min. Luego se incubaron con 50μL de Cytofix/Cytoperm plus por 10 min, se lavaron por centrifugaron en similares condiciones y se incubaron nuevamente con 50μL de Cytofix/Cytoperm, por 5 min, antes de volverlas a lavar. Todos los lavados se realizaron en 500μL de Perm/Wash, diluido en agua destilada, a partir de una solución madre concentrada 10x. Posteriormente las células se marcaron durante 30 min con el anticuerpo (Ac) específico para la molécula Cdh1 (Abcam, EUA), diluido 1/20 en 40μL de la solución Perm/Wash. Al cabo de este tiempo, y después de lavar las células, estas se incubaron 30 min con el Ac antiinmunoglubulina murina conjugado a biotina (Jackson, EUA), diluido 1/800, en un volumen de 100μL de la solución Perm/Wash. Finalmente, las células se incubaron 20 min con del conjugado de estreptavidina y Ficoeritrina-Cianina 5 (PE-Cy5, del inglés: phycoerythrin- cyanine 5; BD Bioscience) diluido 1/400 en 100μL de Perm/Wash. Las células se lavaron con 1mL de Perm/Wash y se resuspendieron en 300μL de SSTF para realizar la determinación en el citómetro de flujo. Como control, se incluyeron células de cada línea permeabilizadas y marcadas con el Ac secundario y el conjugado de estreptavidina y PE-Cy5. Las células utilizadas como control de la sincronización se marcaron con el agente intercalante de las bases del ADN, yoduro de propidio (PI, del inglés Propidium iodide; Sigma). Inicialmente, estas se lavaron con SSTF y se fijaron con 500μl de una mezcla metanol:acetona (1:4) más 500μL de SSTF, durante 2 h. Se lavaron por centrifugación a 555 x g por 5 min y posteriormente se incubaron con 50μl de RNAsa (100μg/mL; Sigma) por 20 min a 37ºC. Se lavaron por centrifugación, se añadieron 300μl de PI a 100μg/mL y se incubaron bajo estas condiciones durante 20 min. Para la detección de los niveles proteicos de AURKA se realizó un ensayo de marcaje simultáneo de esta molécula con PI. Para ello se siguió exactamente el protocolo de marcaje con PI solo que las células antes de ser incubadas con el mismo, se incubaron con 100µl del Ac 16 Materiales y métodos murino anti-AURKA (1/600; Cell Signalling) durante 1 h a temperatura ambiente, se lavaron y se incubaron con 50µl de Ac anti-inmunoglubulina murina conjugado a isotiocianato de fluoresceína (FITC, del inglés Fluorescein isothiocyanate; 1/200; Abcam), 30 min a 4oC. Posteriormente las células se lavaron y se incubaron con PI. La lectura de las muestras, marcadas con anti-Cdh1, AURKA y PI, se realizó en un citómetro de flujo Gallios (Beckman Coulter, EUA). 3.5 Ensayos de Western Blot Las líneas celulares humanas de origen mamario (provenientes de frascos de 25cm2 con un 80% aproximado de confluencia) se homogenizaron por 10 min a 4ºC en un tampón de lisis que incluye: nonidet P40 1%, deoxicolato de sodio 0,5%, SDS 0,1%, NaF 50 mM, Na 3VO4 1 mM, EDTA 5 mM y fluoruro del fenilmetilsulfonilo 1 mM, todos adquiridos de Sigma. Luego se realizó una centrifugación durante 30 min a 14 000 x g a 4°C, y se colectó el sobrenadante que contiene el extracto de proteínas totales aisladas. La concentración de proteínas se determinó mediante el método del ácido becinconínico descrito por Smith y cols, en el año 1985, utilizando un juego de reactivos comerciales (Pierce, EEUU). Se aplicaron 30μg de los lisados celulares antes descritos en dos geles de SDS-PAGE al 10% en condiciones de reducción para la separación de las proteínas. Después de la electroforesis, las proteínas se transfirieron a una membrana de difluoruro de polivinilideno (PVDF del inglés polyvinylidine difluoride) (GE Healthcare Amersham Hybond-P, Reino Unido). El bloqueo de las membranas se realizó con una solución de Tween al 0.1 % y leche descremada al 5% en TBS (Tris-Base Solution), durante 2 h a 37ºC. Posteriormente, se incubaron las membranas durante otras 18 h a 4ºC con los respectivos anticuerpos primarios: primeramente se incubó una membrana con anti-Cdh1 (1/1000; Abcam) y la otra con anti-AURKA (1/1000, Cell Signaling, EUA) ambos originados en ratón. Después de tres lavados con TBS/Tween 0,1% (TBS/T) y bloqueo durante 1 h a 37ºC, las membranas se incubaron con los Acs de conejo anti-p27 (1/1000, Cell Signaling) y el Ac antiPlk1 (1/1000, Cell Signaling), respectivamente. Todos los Acs se diluyeron en TBS/Tween al 5% de BSA excepto el anti-AURKA, el cual fue diluido en la solución de bloqueo. Después de tres lavados de 5 min con TBS/T, las membranas se incubó con los Acs secundarios anti-IgG de ratón (Cell Signaling) y anti-IgG de conejo (Cell Signalling), respectivamente, conjugados a peroxidasa de rábano picante (HRP del inglés horseradish peroxidase), diluidos 1/1000 en la solución de bloqueo. Luego de tres lavados con esta misma solución, el ensayo se reveló empleando el juego de reactivos Western Blotting Luminol Reagent (Santa Cruz Biotechnologies) y se expusiero las membranas al filme fotográfico Chemiluminescence BioMax Light Film (Kodak-Industries, Francia), para visualizar las proteínas inmunorreactivas. 17 Materiales y métodos Para determinar la expresión de la proteína de referencia β-actina, las membranas se incubaron durante 30 min a 56ºC con un tampón de remoción (Tris-HCl 62,5mM [pH 6,7], βmercaptoetanol 100 mM, SDS 2%) para la eliminación de los Acs primarios y secundarios. Posteriormente, se realizó un protocolo similar al antes descrito. Las membranas se bloquearon, se incubaron con un Ac de conejo anti-β-actina (dilución 1/1000; Cell Signaling) y con el respectivo Ac secundario para la detección de las proteínas. 3.6 Procesamiento de los datos El procesamiento de los datos de la citometría de flujo se realizó empleando los programa Summit versión 4.3 y el programa Kaluza versión 1.2. Los datos se expresaron como valores porcentuales o Intensidad Media de Fluorescencia (IMF), en dependencia del análisis requerido. En el caso de los resultados obtenidos del ensayo de Western Blot, la cuantificación de proteínas totales se realizó empleando un ajuste no lineal en el editor de gráficos GraphPad Prism versión 5.0, mientras que el análisis densitométrico se realizó empleando el programa Image J versión 1.46r. Los cálculos, en todos los casos, se auxiliaron de las hojas de Excel. 18 Resultados 4. Resultados 4.1 Comparación de los niveles de expresión de la proteína Cdh1 en líneas tumorales humanas de diferentes localizaciones. Con el objetivo de comparar los niveles de expresión proteica del gen supresor de tumores Cdh1 en líneas tumorales de diferentes localizaciones, se procedió a la medición de esta molécula en células sincronizadas o no en la fase G2/M del ciclo celular, por citometría de flujo. Para ello, las líneas tumorales provenientes de vulva, pulmón, próstata y mama se colectaron tras ser cultivadas con nocodazol, y posteriormente se marcaron con PI y el Ac anti-Cdh1. Como se observa en la Figura 1, en la evaluación del ciclo celular de las líneas tumorales tratadas o no con nocodazol, ocurrió un incremento en la proporción de células detectables en la fase G2/M del ciclo celular con respecto a las células no tratadas, lo que sugiere la eficacia de las condiciones de sincronización empleadas. En cuanto a la determinación de los niveles de expresión de Cdh1, todas las líneas tumorales sincronizadas en la fase G2/M del ciclo celular mostraron un incremento en los valores de ∆IMF con respecto a las líneas celulares no sincronizadas. Entre las células no sincronizadas la línea tumoral de vulva A431 presenta los menores valores de ∆IMF, sin embargo, al hacer una comparación entre las líneas tumorales en la condición de sincronización, se observa que la línea MDA-MB231 de origen mamario, presenta los menores valores de ∆IMF, lo cual sugiere un menor nivel de expresión de Cdh1 en esa línea tumoral (Fig. 1). 4.2 Determinación de los niveles de expresión de la proteína Cdh1 en líneas tumorales humanas de origen mamario. Al considerar el menor nivel de expresión de Cdh1 en la línea tumoral humana de origen mamario MDA-MB231, nos propusimos extender la determinación de los niveles proteicos de esta molécula hacia otras líneas tumorales de igual localización, con el objetivo de seleccionar la línea tumoral de menor expresión de Cdh1 dentro de nuestro panel de líneas celulares evaluadas. Las determinaciones se realizaron mediante ensayos de Western Blot, donde se utilizaron lisados de proteínas totales que se obtuvieron a partir de cultivos celulares con alrededor de un 80% de confluencia. En la Figura 2 se muestran los resultados de la determinación de los niveles de expresión proteica de Cdh1 obtenidos por Western Blot y se grafican los valores de las intensidades relativas de las bandas correspondientes a cada línea tumoral evaluada, datos obtenidos a partir de un análisis densitométrico. Los valores de las intensidades relativas de las bandas indican que las menores cantidades proteicas de Cdh1 se encuentran en las líneas MDA- 19 Resultados MB231, MDA-MB468, T47D y MCF7-HER2, las cuales poseen similares valores de intensidades relativas entre ellas. Yoduro de Propidio Cdh1 Figura 1. Determinación de los niveles de expresión de la proteína Cdh1 en líneas tumorales de diferentes localizaciones: A431 de vulva, H125 de pulmón, PC3 de próstata y MDA-MB231 de mama. La primera columna muestra la evaluación del ciclo celular mediante el marcaje con PI de células no tratadas (histogramas insertados) o tratadas con nocodazol (histogramas mayores). El resto de las columnas corresponde al marcaje de Cdh1 en células no sincronizadas (segunda columna) y sincronizadas (tercera columna) en la fase G2/M del ciclo celular. La región delimitada como R2 representa el fondo de marcaje con el anticuerpo secundario. Se muestran los valores de ∆IMF=IMF [marcaje Cdh1] - IMF [control Ac secundario]. 20 Resultados Por su parte, las líneas ZR75, MCF7 y SKBR3 muestran los mayores niveles relativos de Cdh1. Este resultado centra nuestro interés en las cuatro líneas celulares con menores niveles de expresión de Cdh1, dado que esta condición facilitaría la evaluación futura de los efectos que tendría el hecho de sobre-expresar Cdh1 en la biología tumoral de estas células. Figura 2. Expresión proteica de Cdh1 en líneas tumorales humanas de origen mamario. La expresión de los niveles proteicos de Cdh1 en líneas humanas se determinó mediante Western Blot (panel superior). La carga total de proteína se normalizó con la proteína de referencia β‐actina y los valores de intensidad relativa de las bandas obtenidas se muestran en un gráfico de barras (panel inferior). Para el análisis de densitometría se empleó el programa Image J. 4.3 Determinación de la expresión del inhibidor de las quinasas dependientes de ciclinas p27 y de los blancos moleculares de Cdh1, AURKA y Plk1, en líneas tumorales humanas de origen mamario. Luego de identificar entre un panel de líneas tumorales humanas de mama aquellas con menor nivel de expresión proteica de Cdh1, consideramos importante evaluar sobre estas mismas células los niveles de expresión proteica de moléculas relacionadas con la funcionalidad de Cdh1. Las moléculas que se seleccionaron fueron AURKA y Plk1, blancos moleculares directos 21 Resultados de Cdh1 (Lehman y cols., 2007) y el inhibidor de las quinasas dependientes de ciclinas p27, el cual es regulado positivamente por Cdh1 de una forma indirecta (Bashir y cols., 2004). La determinación de los niveles de expresión de las proteínas AURKA, Plk1 y p27 se realizó mediante ensayos de Western Blot y los resultados se representan en la Figura 3. Como se muestra, en el caso del inhibidor de las quinasas dependientes de ciclinas p27, los valores de intensidad relativa de las bandas indican que los niveles proteicos de esta molécula se encuentran disminuidos en la línea celular MDA-MB231 con respecto a las líneas MCF7, MCF7HER2 y T47D. En estas últimas, las cantidades relativas de p27 alcanzan valores de intensidad relativa superiores al doble de las calculadas para la línea tumoral MDA-MB231. En el caso de las líneas MDA-MB468 y SKBR3, se observan valores intermedios de intensidad relativa de las bandas. En la determinación de los niveles de expresión de Plk1, los resultados densitométricos mostraron que la expresión proteica de esta quinasa es semejante entre las líneas tumorales MCF7, MCF7-HER2, MDA-MB468 y SKBR3, mientras que en la línea celular T47D los niveles de expresión son menores. En la línea tumoral MDA-MB231 se calculó el mayor valor de intensidad relativa de la banda, el cual supera aproximadamente tres veces el calculado para la línea tumoral T47D. Lo anterior indica que la línea celular MDA-MB231 presenta los mayores niveles de expresión proteica de Plk1. Los resultados obtenidos en la evaluación de AURKA demuestran que su nivel de expresión proteica es variable en el panel de líneas celulares empleadas. Al igual que en la determinación anterior, el valor de intensidad relativa de la banda calculada para MDA-MB231 se encuentra por encima del calculado para el resto de las células. En este caso, la línea SKBR3 posee la menor expresión de AURKA, ya que presenta niveles de expresión aproximadamente 5 veces inferiores a los mostrados por la línea tumoral MDA-MB231. Con el objetivo de corroborar el resultado obtenido mediante Western Blot respecto al comportamiento de la expresión de AURKA, realizamos un ensayo de citometría de doble marcaje, con un Ac anti-AURKA y PI, para visualizar la expresión de AURKA en función de las diferentes fases del ciclo celular. Para este estudio se seleccionó la línea celular MDA-MB231, la cual muestra según los resultados obtenidos por Western Blot, los mayores niveles de expresión de AURKA, así como la línea celular MDA-MB468, que presenta niveles intermedios de expresión de dicha quinasa. Como se observa en la Figura 4, el porcentaje de células de la línea MDA-MB231 positivas al marcaje de AURKA supera al obtenido para la línea MDAMB468. Este resultado ratifica lo obsevado en el ensayo de Western Blot al confirmar que la 22 Resultados expresión proteica de AURKA en MDA-MB231 es superior a la encontrada en la línea celular MDA-MB468. Al analizar de forma global los resultados obtenidos, es la línea tumoral MDA-MB231 la que presenta las condiciones más adecuadas para considerarla como un modelo celular de interés sobre el cual evaluar la relevancia de la manipulación positiva de Cdh1 sobre la tumorigenicidad, debido al patrón de expresión mostrado entre todas las moléculas estudiadas. Figura 3. Determinación de los niveles proteicos de p27, Plk1 y AURKA en líneas tumorales humanas de mama. La determinación se realizó por el ensayo de Western Blot y la carga total de proteína se normalizó con la proteína de referencia β‐actina (panel izquierdo). Se muestra en gráficos de barras los valores relativos de la intensidad de las bandas obtenidas (panel derecho), producto del análisis densitométrico realizado en el programa Image J. 23 Resultados 4.4 Determinación de la expresión proteica de Cdh1 en líneas tumorales murinas de origen mamario. Con el fin de acercarnos a un modelo celular que permita evaluar in vivo los efectos de la sobreexpresión de Cdh1 en el microambiente tumoral, decidimos determinar los niveles de expresión de esta molécula en líneas tumorales murinas de origen mamario. Para ello, las células tumorales de mama murinas AT3, F3II y 4T1 se sincronizaron en la fase G2/M del ciclo celular, se marcaron con PI y el Ac anti-Cdh1 y se evaluaron por citometría de flujo. Figura 4. Expresión de AURKA en función del ciclo celular en las líneas MDA-MB231 y MDA-MB468. La expresión de AURKA se midió mediante una citometría de flujo de doble marcaje con PI. Para cada línea se muestra el ciclo celular (columna izquierda) y el doble marcaje PI / AURKA (columna derecha). La barrera situada entre las regiones E++ y E+delimita el marcaje del Ac anti-AURKA con respecto al fondo de marcaje del Ac secundario respectivamente. Se muestra el porcentaje de las células positivas para AURKA obtenido a partir del análisis de los datos en el programa Kaluza. En las células murinas tratadas con nocodazol se incrementó el número de células en la fase G2/M del ciclo celular, lo que confirma la efectividad del proceso de sincronización (Fig. 5). Además como se puede observar en las tres líneas tumorales, en la condición de sincronización se obtuvo un incremento en el porcentaje de células positivas al marcaje de Cdh1, así como en 24 Resultados los valores de ∆IMF, con respecto a las células no sincronizadas. Sin embargo, no se evidenciaron diferencias importantes en ambos parámetros al comparar entre sí las líneas celulares, lo que sugiere que los niveles de expresión de Cdh1 en estas líneas son similares. Figura 5. Expresión proteica de Cdh1 en líneas tumorales murinas de origen mamario. La expresión de los niveles proteicos de Cdh1 en líneas murinas fue determinada mediante citometría de flujo. Las dos primeras columnas representan la medición del ciclo celular mediante PI en células no tratadas (columna izquierda) o tratadas (columna derecha) con nocodazol. Las otras columnas representan la expresión de Cdh1 en células sin y con nocodazol. Se muestran los porcentajes de células positivas al marcaje del Ac anti-Cdh1 y los valores respectivos de ∆IMF calculados en el programa Kaluza (∆IMF=IMF [marcaje Cdh1] IMF [control Ac secundario]). La barrera situada entre las regiones F++ y F-+ delimita el marcaje del Ac anti-Cdh1 con respecto al fondo de marcaje del Ac secundario respectivamente. 25 Discusión 5. Discusión La molécula Cdh1, proteína adaptadora del APC/C, es actualmente considerada como un gen supresor de tumores debido en primera instancia, a que regula los procesos de diferenciación celular e integridad del genoma. Esto justifica que en las células donde existe una afectación en su expresión, ocurra la proliferación de las mismas con aberraciones genéticas, lo cual desencadena el desarrollo de transformaciones malignas (Wäsch y cols., 2010). Además, Cdh1 regula la fidelidad de los eventos tardíos de la mitosis, al determinar el tiempo de vida media de importantes enzimas mitóticas tales como Plk1 y AURKA, las cuales se encuentran sobreexpresadas en diversos tipos de cáncer (Qiao y cols.,2010). También se ha referenciado la relación de este cofactor con otras moléculas supresoras de tumores, oncogenes y enzimas involucradas en el metabolismo energético, lo que lo convierte en un punto de convergencia de mecanismos y rutas claves que influyen en la biología tumoral (Gao, 2009; Song y cols., 2011; Moncada y cols., 2012). Todos estos aspectos convierten a Cdh1 en un atractivo blanco terapéutico antitumoral. Con este trabajo nos propusimos, sobre la base del estudio de la expresión de Cdh1 y de otras moléculas relacionadas con su funcionalidad, seleccionar una línea celular adecuada que sirva como modelo para evaluar los efectos que sobre la tumorigenicidad tendría el hecho de sobreexpresar Cdh1. En este sentido, la línea celular más adecuada sería aquella que posea bajos niveles de Cdh1 y que los niveles de expresión de sus dianas moleculares estén en correspondencia con la reducida expresión de la misma. El hecho de que exista una línea celular que cumpla con estas características, permitiría validar a Cdh1 como blanco de la terapia antitumoral al evaluar el efecto de su manipulación sobre la biología tumoral y a la vez, focalizaría la evaluación de su impacto clínico en tumores de un determinado tipo histológico y localización. Inicialmente, se compararon los niveles de expresión proteica de Cdh1 entre líneas tumorales de diferentes localizaciones. Cdh1 aunque también tiene actividad durante la fase G0/G1 del ciclo celular, tiene una alta participación en la mitosis, de modo que en este período del ciclo sus niveles de expresión proteica se elevan (Li y cols., 2007; 2008). Teniendo lo anterior en cuenta, las células se sincronizaron en la fase G2/M del ciclo celular con el fin de detectar los máximos niveles de expresión de esta proteína. Es válido señalar que la variedad de líneas celulares incluidas en esta comparación representan localizaciones tumorales para las cuales se ha demostrado una reducida expresión de Cdh1 respecto a la contraparte del tejido normal (Fujita y cols., 2009), de ahí que hemos comparado los niveles de esta molécula entre células 26 Discusión tumorales que probablemente ya contienen una afectación en la expresión de esta proteína. Los estudios realizados por Fujita y cols. en el 2009 incluyeron células provenientes de pulmón, próstata y mama, localizaciones presentes en nuestro estudio, sin embargo, estos autores no hacen alusión a células tumorales provenientes de vulva, las cuales, según los valores de ∆IMF obtenidos en la condición de sincronización, presentan los mayores niveles de expresión de Cdh1. Los resultados obtenidos apuntan a que, entre las líneas tumorales en estudio (A431 de vulva, H125 de pulmón, PC3 de próstata y MDA-MB231 de mama), la de menor nivel de expresión de Cdh1 es la de origen mamario en la condición de sincronización. Coincidentemente, los bajos niveles de expresión de Cdh1 en líneas tumorales de origen mamario ha sido reportada por Fujita y cols. en el 2008, llegando incluso a proponer a esta molécula como un factor pronóstico para este tipo de neoplasia maligna. Dado que los primeros resultados obtenidos apuntan a que las células tumorales de mama son las que presentan menores niveles de expresión de Cdh1, decidimos centrarnos en varias líneas tumorales de esta localización para hacer una comparación más extensa de los niveles de expresión de esta molécula. Los resultados obtenidos permitieron la identificación de cuatro líneas celulares con menores cantidades de Cdh1: MDA-MB231, MDA-MB468, T47D y MCF7HER2; mientras que las líneas ZR75, SKBR3 y MCF7 poseen una mayor expresión de la misma. Lo anterior nos hizo enfocarnos en estas cuatro líneas para definir, teniendo en cuenta el resto de las determinaciones, la línea más adecuada para el modelo celular que buscamos. Además de la medición de Cdh1 en las células tumorales humanas de origen mamario, evaluamos los niveles de expresión de las quinasas AURKA y Plk1 (reguladas negativamente por Cdh1) y del inhibidor de las quinasas dependientes de ciclinas p27 (regulado positivamente por Cdh1). La importancia de estos experimentos consiste en que con la determinación de la expresión de moléculas funcionalmente conectadas con Cdh1 en las líneas celulares de interés, se puede especular acerca de la presencia en mayor o menor cuantía de múltiples mecanismos de regulación que operan sobre la función de Cdh1 (Lehman y cols., 2007; Tang y cols., 2008; Gao y cols., 2009; Huang y cols., 2011; Shi y cols., 2011), los cuales van más allá de los niveles de expresión de esta molécula. Si se analizan en conjunto los resultados mostrados en las Figuras 2 y 3, podemos considerar que dentro de las líneas que tienen menores niveles de expresión de Cdh1, sólo la línea tumoral MDA-MB231 cumple la relación esperada respecto a la expresión de AURKA, Plk1 y p27. O sea, sólo en esta línea celular las cantidades de p27 fueron comparativamente inferiores con respecto al resto de las líneas, mientras que las de AURKA y Plk1 están elevadas. Además, en 27 Discusión el caso específico de AURKA corroboramos los resultados obtenidos en el ensayo de Western Blot, mediante un marcaje intracelular de citometría de flujo. Observamos que la línea tumoral MDA-MB231 muestra niveles de expresión de esta quinasa superiores a los encontrados en la línea MDA-MB468 (MDA-MB468 fue la línea tumoral seleccionada para la comparación debido a que sus niveles de expresión de Cdh1 son similares a los de MDA-MB231). Teniendo esto en cuenta, los resultados sugieren que las cantidades elevadas de AURKA en MDA-MB231, detectadas por ambas técnicas, no solo podría deberse a la expresión reducida de Cdh1 que esta posee, sino también a la existencia en estas células de mecanismos alternativos que determinan la sobre-expresión de esta quinasa. De hecho, existen estudios que evidencian que en células neoplásicas la tumorigenicidad de las mismas se refuerza por la sobre-expresión de quinasas mitóticas como Plk1 y en especial AURKA (Tanaka y cols., 1999; Strebhardt y Ullrich, 2006; Nadler y cols., 2008; Johnson y cols., 2012), por lo que es posible que mecanismos dilucidados en tales estudios estén presentes en esta línea celular. El hecho de que MDA-MB231 sea una línea celular con bajos niveles de Cdh1 y p27, y elevados niveles de Plk1 y AURKA, la señala como adecuada para evaluar los efectos directos que podría tener el acto de sobre-expresar Cdh1 en estas células, ya que permitiría comparar y corroborar mediante la medición de estas mismas moléculas la funcionalidad del producto proteico del transgén de Cdh1 incorporado. En las otras líneas tumorales humanas de mama ensayadas donde se detectaron niveles de Cdh1 similares a los de MDA-MB231, pueden estar operando algunos de los mecanismos de regulación negativa de la funcionalidad de Cdh1 que impiden relacionar directamente la expresión de esta molécula y la de sus blancos. Dentro de estos mecanismos figuran aquellos donde diferentes moléculas limitan la unión y reconocimiento del APCCdh1 por los sustratos independientemente de las cantidades proteicas de Cdh1. Ejemplo de estas moléculas son Emi1, CIP2A y NEDD9 de las cuales se ha comprobado su participación en la oncogénesis y agresividad de diferentes tipos de tumores, incluyendo los de mama (Lehman y cols., 2007; Ice y cols., 2013; Kim y cols., 2013). Por tanto, sería importante la determinación de estas moléculas en las líneas celulares en estudio, incluyendo la propia MDA-MB231. En el caso de las líneas tumorales murinas de origen mamario, el hecho de que no se encontrarse diferencias importantes entre los niveles de expresión de Cdh1, motiva la determinación de otras moléculas relacionadas. Ello permitirá obtener un modelo celular válido para realizar los experimentos in vivo en los cuales se evalúe la incidencia de la sobreexpresión de Cdh1 sobre la tumorigenicidad de las células en el contexto del microambiente 28 Discusión tumoral. El hallazgo de este modelo celular tendría gran importancia ya que no hay evidencias en la literatura que demuestren la manipulación de Cdh1 in vivo en aras de incidir negativamente sobre la progresión tumoral. De hecho, los estudios que demuestran que la alteración de la expresión de Cdh1, por incremento o reducción, inciden en la progresión tumoral, se han obtenido por manipulación in vitro de células tumorales humanas y la posterior implantación de estas en animales inmunodeficientes (Fujita y cols., 2008; 2009). Lo anterior limita el estudio e impide entender la posible relación entre la actividad de Cdh1 y la conocida capacidad de las células tumorales de evadir la vigilancia inmunológica (Schreiber y cols., 2011), por lo que con el empleo futuro de un modelo murino derivado de nuestros estudios estos obstáculos podrían superarse. Los resultados obtenidos a lo largo del presente estudio permitieron seleccionar una línea tumoral humana como potencial modelo para estudiar la influencia que la manipulación de Cdh1 puede tener en la tumorigenicidad, y validar la relevancia de esta molécula para la terapia del cáncer. La estrategia de selección evidenció que no solo la determinación de los niveles de expresión de Cdh1, sino además los de sus blancos moleculares, son requisitos claves para valorar la actividad del complejo APC/CCdh1. Esto último debe extenderse a la selección de una variante tumoral murina por la relevancia que tendría para las evaluaciones in vivo. Ambos modelos, tanto humano como murino, podrían convertirse en herramientas eficaces para futuras terapias contra el cáncer que tengan como centro de atención al gen supresor de tumores Cdh1. 29 Conclusiones 6. Conclusiones 1. La línea tumoral humana de mama MDA-MB231, sincronizada en la fase G2/M del ciclo celular, tiene menor nivel de expresión de Cdh1 que las líneas tumorales de vulva, pulmón y próstata evaluadas. 2. La línea tumoral MDA-MB231 tiene menor nivel de expresión de p27 y mayor de AURKA y PlK1 que el resto de las líneas tumorales de mama evaluadas, lo que sugiere una menor actividad de Cdh1 en esta línea y la convierte en un modelo adecuado para estudiar el efecto de la sobre-expresión de esta molécula en la tumorigenicidad de estas células. 30 Recomendaciones 7. Recomendaciones 1. Determinar la influencia de la modificación de la expresión de Cdh1 sobre la tumorigenicidad de la línea celular humana MDA-MB231. 2. Medir en la línea celular humana MDA-MB231 la expresión de moléculas relacionadas con la regulación del complejo APC/CCdh1. 3. Extender el estudio a las células murinas de mama para seleccionar un modelo celular adecuado para experimentos in vivo. 31 Referencias bibliográficas 8. Referencias bibliográficas Archambault V. & Glover D. M. (2009). Polo-like kinases: conservation and divergence in their functions and regulation. Nature Rev. Mol. Cell Biol. 10, 265-275. Bar-On O., Shapira M., Skorecki K., Hershko A., & Hershko D. D. (2010). Regulation of APC/C Cdh1 ubiquitin ligase in differentiation of human embryonic stem cells. Cell Cycle. 9, 1986-1989. Bashir T., Dorrello N.V., Amador V., Guardavaccaro D., Pagano M. (2004). Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C (Cdh1) ubiquitin ligase. Nature. 428,190-193. Bloom J. & Pagano M. (2003). Deregulated degradation of the cdk inhibitor p27 and malignant transformation. Semin. Cancer Biol. 13, 41-47. Carrano A.C., Eytan E., Hershko A., Pagano M. (1999). SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1,193 -199. Chesney J., Mitchell R., Benigni F., Bacher M., Spiegel L., Al-Abed Y., Han J. H., Metz C. &Bucala, R. (1999). An inducible gene product for 6-phosphofructo-2-kinase with an AU-rich instability element: role in tumor cell glycolysis and the Warburg effect. Proc. Natl. Acad. Sci. U.S.A. 96, 3047-3052. Cuadrado M., Gutierrez P., Swat A., Nebreda A. R., & Fernandez O. (2009). p27 Kip1 stabilization is essential for the maintenance of cell cycle arrest in response to DNA damage. Cancer Research. 69, 8726-8732. Eckerdt, F., & Strebhardt, K. (2006). Polo-Like Kinase 1: target and regulator of anaphasepromoting complex / cyclosome – dependent proteolysis. Cancer Research. 66, 6895-6898. Egozi D., Shapira M., Paor G., Ben-izhak O., Skorecki K., & Hershko D. D. (2007). Regulation of the cell cycle inhibitor p27 and its ubiquitin ligase Skp2 in differentiation of human embryonic stem cells. The FASEB Journal. 21, 2807-2817. Engelbert D., Schnerch D., Baumgarten A. & Wäsch R. (2008). The ubiquitin ligase APC (Cdh1) is required to maintain genome integrity in primary human cells. Oncogene. 27, 907-917. Fehr A. R.,& Yu D. (2013). Control the host cell cycle: viral regulation of the anaphase-promoting complex. Journal of Virology. 87, 8818-8825. Floyd S., Pines J., & Lindon C. (2008). Targets Aurora kinase APC / C to control reorganization of the mitotic spindle at anaphase. Current Biology. 18, 1649-1658. 32 Referencias bibliográficas Fujita T., Liu W. &Doihara H. (2008). Dissection of the APC Cdh1 -Skp2 cascade in breast cancer. Clinical Cancer Research. 14, 1966-1975. Fujita T., Liu W., & Doihara H. (2008). Regulation of Skp2-p27 axis by the Cdh1 / AnaphasePromoting Complex pathway in colorectal tumorigenesis. The American Journal of Pathology. 173, 217-228. Fujita T., Liu W., Doihara H., & Wan Y. (2009). An in vivo study of Cdh1 / APC in breast cancer formation. Int. J. Cancer. 125, 826-836. Gao D., Inuzuka H., Tseng A., & Wei W. (2009). Akt finds its new path to regulate cell cycle through modulating Skp2 activity and its destruction by APC / Cdh1. Cell Division. 4,11. García-Higuera I., Manchado E., Dubus P., Cañamero M., Méndez J., Moreno S., Malumbres M. (2008). Genomic stability and tumour suppression by the APC/C cofactor Cdh1. Nature Cell Biology, 10, 802-811. Giubettini M., Asteriti I. A., Scrofani J., Luca M. D., Lindon C., Lavia P., y cols. (2011). Control of Aurora-A stability through interaction with TPX2. Journal of Cell Science. 124, 113-122. Glotzer M., Murray A. W. & Kirschner M. W. (1991). Cyclin is degraded by the ubiquitin pathway. Nature, 349, 132-138. Harrington E.A., Bebbington D., Moore J., Rasmussen R.K., Ajose-Adeogun A.O, Nakayama T., Graham J.A, Demur C., Hercend T., Diu-Hercend A., Su M., Golec J.M.C & Miller K.M. (2004). VX-680, a potent and selective small-molecule inhibitor of the Aurora kinases, suppresses tumor growth in vivo. Nature Medicine. 10, 262-267. Hartwell L. H.,& Kastan M. B. (1994). Cell cycle control and cancer. Science. 266, 1821- 1828. Hsu J. Y., Reimann J. D., Sorensen C. S., Lukas J. &Jackson P. K.(2002). E2F-dependent accumulation of hEmi1 regulates S phase entry by inhibiting APCCdh1. Nature Cell Biol. 4, 358366. Huang Y., Wu C., Chou C. & Huang, C.F. (2011). A translational regulator, PUM2, promotes both protein stability and kinase activity of Aurora-A. PLoS ONE. 6, e19718. Ice R. J., Mclaughlin S. L., Livengood R. H., Ice R. J., Mclaughlin S. L., Livengood R. H., y cols. (2013). NEDD9 depletion destabilizes Aurora A kinase and heightens the efficacy of Aurora A inhibitors: implications for treatment of metastatic solid tumors. Cancer Res. 73, 3168-3180. 33 Referencias bibliográficas Inbal N., Listovsky T., & Brandeis M. (1999). The mammalian Fizzy and Fizzy-related genes are regulated at the transcriptional and post-transcriptional levels. FEBS Letters. 463, 350-354. Johnson E. O., Chang K.-hua, Ghosh S., Venkatesh C., Giger K., & Low P. S. (2012). LIMK2 is a crucial regulator and effector of Aurora-A- kinase-mediated malignancy. Journal of Cell Science. 125, 1204-1216. Ke Y., Dou Z., Zhang, J., & Yao, X. (2003). Function and regulation of Aurora / Ipl1p kinase family in cell division. Cell Research. 13, 69-81. Kim H.S., Vassilopoulos A., Wang R.H., Lahusen T., Xu X., Li C., y cols. (2011). SIRT2 maintains genome integrity and suppresses tumorigenesis through regulating APC/C activity. Cancer Cell. 20, 487-499. Kim J.-sung, Kim E. J., & Oh J. S. (2013). CIP2A modulates cell cycle progression in human cancer cells by regulating the stability and activity of PLK1. Cancer Research. 73, 6667-6678. Kitajima S., Kudo Y., Ogawa I., Tatsuka M., Kawai H., y cols. (2007). Constitutive phosphorylation of Aurora-A on Ser51 induces its stabilization and consequent overexpression in cancer. PLoSONE. 2, e944. Koljonen V., Tukiainen E., Haglund C. A. J., & Böhling T. O. M. (2006). Cell cycle control by p21, p27 and p53 in merkel cell carcinoma. Anticancer Research. 26, 2209-2212. Kwan P. S., Lau C. C., Chiu Y. T., Man C., Liu J., & Tang K. D. (2013). Daxx regulates mitotic progression and prostate cancer predisposition. Carcinogenesis. 34, 750-759. Lehman N. L., Tibshirani R., Hsu J. Y., Natkunam Y., Harris B. T., West R. B., y cols. (2007). Oncogenic Regulators and Substrates of the Anaphase Promoting Complex / Cyclosome Are Frequently Overexpressed in Malignant Tumors. The American Journal of Pathology. 170, 17931805. Li M., Shin Y-H.,, Hou L., Huang X., Wei Z.,Klann E., Zhang P. (2008).The adaptor protein of the anaphase promoting complex Cdh1 isessential in maintaining replicative lifespan and in learning and memory. Nature Cell Biology, 10, 1083-1089. Li M., York JP., Zhang P. (2007). Loss of Cdc20 causes a securin-dependent metaphase arrest in two-cell mouse embryos. Molecular and Cellular Biology. 27, 3481-3488. 34 Referencias bibliográficas Li M.,& Zhang P. (2009). The function of APC / C Cdh1 in cell cycle and beyond. Cell Division. 4,2. Lindon C., & Pines J. (2004). Ordered proteolysis in anaphase inactivates Plk1 to contribute to proper mitotic exit in human cells. The Journal of Cell Biology. 164, 233-241. ListovskyT.,& Sale J. E. (2013). Sequestration of CDH1 by MAD2L2 prevents premature APC/C activation prior to anaphase onset. J. Cell Biol. 203, 87-100. Littlepage L. E. &Ruderman J. V. (2002). Identification of anew APC/C recognition domain, the A box, which isrequired for the Cdh1-dependent destruction of the kinase Aurora-A during mitotic exit. Genes Dev. 16, 2274-2285. Liu W., Xin H., Eckert D.T., Brown J.A. & Gnarra J.R. (2011). Hypoxia and cell cycle regulation of the von Hippel-Lindau tumor suppressor. Oncogene. 30, 21-31. Lukas C. y cols. (1999). Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature. 401, 815- 818. Mailand N. & DiffleyJ.F.(2005). CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell. 122, 915-926. Martinez, J. S., Hall, H., Bartolowits, M. D., Hall, M. C., Martinez, J. S., Hall, H., y cols. (2012). Acm1 contributes to nuclear positioning by inhibiting Cdh1-substrate interactions Do not distribute. Cell Cycle. 11, 384-394. Marumoto T., Zhang D., Saya H. (2005). Aurora-A. A Guardian of poles. Nat. Rev. Cancer. 5, 42-50. McDonald E.R III., & el Deiry W.S. (2000) Cell cycle control as a basis for cancer drug development. Int. J. Oncol.16, 871-886. Moncada S., Higgs E.A. & Colombo S.L. (2012). Fulfilling the metabolic requirements for cell proliferation. Biochemical Journal. 446, 1-7. Mundt K. E., Golsteyn R. M., Lane H. A., & Nigg E. A. (1997). On the Regulation and Function of Human Polo-like Kinase 1 (PLK1): Effects of Overexpression on Cell Cycle Progression. Biochemical and Biophysical Research Communications. 239, 377-385. 35 Referencias bibliográficas Nadler Y., Camp RL., Schwartz C., Rimm D.L., Kluger H.M., Kluger Y. (2008). Expression of Aurora A (but not Aurora B) is predictive of survival in breast cancer. Clin. Cancer Res. 14, 4455-4462. Oh Y.H. & Park C.K. (2000). Expression of cyclin-dependent kinase inhibitor p27kip1 in malignant lymphomas. J. Korean Med. Sci. 15, 399-406. Park M.T., & Lee S.J. (2003). Cell cycle and cancer. Journal of Biochemistry and Molecular Biology. 36, 60-65. Passmore L. A., & Barford D. (2005). Coactivator functions in a stoichiometric complex with anaphase-promoting complex/cyclosome to mediate substrate recognition. EMBO reports, 6, 873-878. Pérez C., Campos J. A., Alonso F. J., Segura J. A., Manzanares E., Ruiz P., González M. E., Márquez J. M. & Matés J. M. (2005). Co-expression of glutaminase K and L isoenzymes in human tumour cells. Biochem. J. 386, 535-542. Peters J. M. (2002). The Anaphase-Promoting Complex: proteolysis in mitosis and beyond. Molecular Cell. 9, 931-943. Peters J. M. (2006). The anaphase promoting complex / cyclosome: a machine designed to destroy. Nature, 7, 644-656. Petroski M. D. & Deshaies R. J. (2005).Function and regulation of cullin–RING ubiquitin ligases. Nature Rev. Mol. Cell Biol. 6, 9-20. Pfleger C. M. & Kirschner M. W. (2000). The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 14, 655-665. Pfleger C. M., Lee E., & Kirschner M. W. (2001). Substrate recognition by the Cdc20 and Cdh1 components of the anaphase-promoting complex. Genes & Development. 15, 2396-2407. Priolo C., Tang D., Brahamandan M., Benassi B., Sicinska E., Ogino S., Farsetti A., Porrello A., Finn S., Zimmermann J., Febbo P. & Loda M. (2006). The Isopeptidase USP2a protects human prostate cancer from apoptosis. Cancer Res. 66, 8625-8632. Qiao X., Zhang L., Gamper A. M., Fujita T., & Wan Y. (2010). From cell cycle to cellular differentiation and genomic integrity APC / C-Cdh1. Cell Cycle. 9, 3904-3912. 36 Referencias bibliográficas Rape M. & KirschnerM. W. (2004). Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature. 432, 588-595. Rape M., Reddy S. K. & Kirschner M. W. (2006). The processivity of multiubiquitination by the APC determines the order of substrate degradation. Cell. 124, 89-103. Reimann J. D. et al. (2001). Emi1 is a mitotic regulator that interacts with Cdc20 and inhibits the anaphase promoting complex. Cell. 105, 645-655. Santamaría P. G., & Pagano M. (2007). The pRb – Cdh1 – p27 autoamplifying network. Nature Cell Biology. 9, 137-138. Schreiber R.D., Old L.J., Smyth M.J., (2011). Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 331,1565-1570. Sherr C.J. (1996). Cancer cell cycles. Science. 274, 1672-1677. Shi Y., Solomon, L. R., Pereda-Lopez, A., Giranda, V. L., Luo, Y., Johnson, E. F., y cols. (2011). Ubiquitin-specific Cysteine Protease 2a (USP2a) Regulates the Stability of Aurora-A. Journal of Biological Chemistry. 286, 38960 -38968. Shirane M., Harumiya Y., Hirai A., Miyamoto C., & Chem J. B. (1999). Down-regulation of p27 Kip1 by Two Mechanisms, Ubiquitin-mediated Degradation and Proteolytic Processing. J. Biol. Chem. 274, 13886-13893. Slingerland J., & Pagano M. (2000). Regulation of the Cdk Inhibitor p27 and Its Deregulation in Cancer. Journal of Cellular Physiology. 183, 10 -17. Song M. S., Carracedo A., Salmena L., Song S. J., & Egia A. (2011). Nuclear PTEN regulates the APC-CDH1 tumor suppressive complex in a phosphatase-independent manner. Cell. 144, 187-199. Sørensen C. S., Lukas C., Kramer E. R., Peters J. M, Bartek J., & Lukas J. (2001). A conserved cyclin-binding domain determines functional interplay between Anaphase-Promoting Complex – Cdh1 and cyclin A-Cdk2 during cell cycle progression. Molecular and Cellular Biology, 21, 36923703. Strebhardt K., & Ullrich, A. (2006). Targeting polo-like kinase 1 for cancer therapy. Nature Review Cancer, 6, 321-330. 37 Referencias bibliográficas Sutterluty H., Chatelain E., Marti A., y cols. (1999). p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat. Cell Biol. 1, 207-214. Taguchi Si, Honda K, Sugiura K, Yamaguchi A, Furukawa K, Urano T. (2002). Degradation of human Aurora-A protein kinase is mediated by hCdh1. FEBS. 519, 59–65. Tanaka T., Kimura M., Matsunaga K., Fukada D., Mori H., Okano Y.(1999) Centrosomal kinase AIK1 is overexpressed in invasive ductal carcinoma of the breast. Cancer Res. 59, 2041–2044. Tang J., Yang X., & Liu X. (2008).Phosphorylation of Plk1 at Ser326 regulates its functions during mitotic progression.Oncogene. 27, 6635–6645. Tsvetkov, L., & Stern, D. F. (2005). Phosphorylation of Plk1 at S137 and T210 is inhibited in response to DNA damage. Cell Cycle. 4, 166-171. Vermeulen K., Bockstaele D. R. V., & Berneman Z. N. (2003). The cell cycle: a review of regulation, deregulation and therapeutic targets in cancer. Cell Proliferation. 36, 131-149. Wang L. & Kung H. (2012). Male germ cell-associated kinase is overexpressed in prostatecancer cells and causes mitotic defects via deregulation of APC/C-CDH1. Oncogene. 31, 2907-2918. Wäsch R. & Engelbert D. (2005). Anaphase-promoting complex-dependent proteolysis of cell cycle regulators and genomic instability of cancer cells. Oncogene. 24, 1-10. Wäsch R., Robbins J., & Cross F. (2010). The emerging role of APC/CCdh1 in controlling differentiation, genomic stability and tumor suppression. Oncogene. 29, 1-10. Wei W., Ayad N.G., Wan Y., Zhang G.J., Kirschner M.W., Kaelin W.G., Jr. (2004). Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 428, 194-198. Winkles J. A., & Alberts G. F. (2005). Differential regulation of polo-like kinase 1, 2, 3, and 4 gene expression in mammalian cells and tissues. Oncogene. 24, 260-266. Wirth, K. G. y cols. (2004).Loss of the anaphase-promoting complex in quiescent cells causes unscheduled hepatocyte proliferation. Genes Dev. 18, 88-98. Xu J., Shen C., Wang T., & Quan J. (2013). Structural basis for the inhibition of Polo-like kinase 1. Nature Structural & Molecular Biology. 20, 1047-1053. 38 Referencias bibliográficas Yalcin A., Telang S., Clem B. & Chesney J. (2009) Regulation of glucose metabolismby 6phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Exp. Mol. Pathol.86, 174-179. Zhang Z.,Yang J., Kong E., Chao W., Morris E. y cols.(2013). Recombinant expression, reconstitution and structure of human anaphase-promoting complex (APC/C). Biochem. J. 449, 365-371. Zhou H., Kuang J., Zhong L., Kuo W.L., Gray J.W., Sahin A., y cols. (1998). Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat. Genet. 20, 189-193. Zhou Y., ChingY.P.,Chun A.C.,Jin D.Y. (2003). Nuclear localization of the cell cycle regulator CDH1 and its regulation by phosphorylation. The Journal of Biological Chemistry. 278, 1253012536. Zitouni, S., Nabais, C., Jana, S. C., Guerrero, A., & Bettencourt, D. M. (2014). Polo-like kinases: structural variations lead to multiple functions. Molecular Cell Biology. 15, 433-452. 39