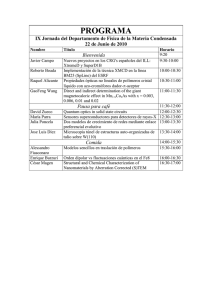
233 capítulo 6 polímeros
Anuncio

6. POLíMEROS CAPÍTULO 6 POLÍMEROS XI CONGRESO NACIONAL DE MATERIALES 233 6. POLíMEROS MODIFICACIÓN QUÍMICA DEL COPOLÍMERO DE ETILENO-ACRILATO DE BUTILO (EBA) CON GRUPOS FOTOESTABILIZANTES HALS. ESTUDIO DE FOTOESTABILIDAD L. López-Vilanova1, A. Fontecha2, M.R. Calvo2, I.A. Real2, E.Espí2, C. Peinado1, F.Catalina1. Departamento de Fotoquímica, Instituto de Ciencia y Tecnología de Polímeros (CSIC). C/ Juan de la Cierva, 3 28006 (Madrid). E-mail: [email protected] ; [email protected] 2 Centro de Investigación y Desarrollo, Repsol Dirección de Tecnología Química. Autovía A-5, Km.18, 28931 Móstoles (Madrid). 1 Resumen: Los copolímeros de etileno-acrilato de butilo (EBA), tienen en la actualidad un gran interés en el sector de los filmes agrícolas y, en concreto, para cubierta de invernadero. Para esta aplicación, a estos filmes se les exige una larga duración por lo que es necesario el empleo de fotoestabilizantes de gran eficiencia como son las aminas impedidas HALS, en concreto, las estructuras de mayor resistencia al empleo de pesticidas (HALS tipo N-OMe). En este trabajo, se ha llevado a cabo la reacción de modificación química del copolímero EBA, para anclar covalentemente estructuras HALS mediante transesterificación por su interés industrial en estado fundido. Se han obtenido modificaciones de EBA-8% con grupos fotoestabilizantes HALS tipo N-Me y N-OMe de bajo peso molecular, determinándose los contenidos de HALS (inferiores al 1%) mediante la Espectroscopía Fotoelectrónica de Rayos X (XPS). El envejecimiento acelerado en cámara climática de estos nuevos materiales modificados ha demostrado su elevada eficiencia de fotoestabilización, especialmente bajo tratamiento con los pesticidas metam-sodio y cipermetrina. Palabras clave: transesterificación, copolímeros, filme agrícola, extrusión reactiva 1. INTRODUCCION En los últimos años la aplicación de los polímeros en el sector agrícola ha experimentado un enorme crecimiento debido a las excelentes prestaciones para cumplir las necesidades específicas de cada aplicación, y en particular, la destinada al empleo de filmes para el cultivo [1]. El diseño de estos materiales se ha complicado y diversificado por su grado de especialización presentando diferencias en cuanto a su composición, formulación (polímero base y aditivos) y espesor, pero tienen en común el estar constituidos en su mayor parte por polímeros de gran consumo pertenecientes a la familia de las poliolefinas [2]. La gran durabilidad exigida a todos estos materiales salvo alguna excepción, hace necesaria su estabilización frente al ataque de determinados agentes ambientales sobre su estructura, ya que su aplicación se desarrolla a la intemperie. En concreto, los requisitos para los filmes utilizados como cubierta de invernadero son altamente exigentes ya que durante su aplicación han de estar expuestos a la luz solar (frecuentemente varias campañas) y bajo duras condiciones ambientales [3,4] tales como, el aumento de temperatura dentro del invernadero, la exposición a agentes químicos agresivos (pesticidas, insecticidas,…), humedad, lluvia, viento, y granizo. Además, el filme de invernadero debe ofrecer una combinación de propiedades entre las que caben citar, buenas propiedades mecánicas, ópticas, resistencia a la luz, calor y agentes fitosanitarios entre otras muchas, y que deben mantenerse durante todo el tiempo de servicio de la cubierta del invernadero. El objetivo del trabajo se ha centrado en desarrollar nuevos materiales para la aplicación del filme agrícola, y XI CONGRESO NACIONAL DE MATERIALES en particular de larga duración, consiguiendo nuevos sistemas fotoestabilizantes que mejoren o igualen a menor precio las prestaciones (duración, resistencia a los pesticidas y otros medios agresivos) de las formulaciones utilizadas actualmente. La reacción de transesterificación del copolímero EBA-8% con grupos HALS se muestra a continuación en la figura 1. Figura 1. Reacción de transesterificación en el estado fundido El análisis de los productos de reacción para valorar el HALS anclado se ha realizado empleando la Espectroscopía Fotoelectrónica de Rayos X, lo que supone una novedad en la detección de HALS. 2. EXPERIMENTAL Materiales Como material base se ha utilizado el copolímero al azar EBA con un contenido en acrilato de butilo del 8% suministrado por Repsol y de interés para la aplicación de filme agrícola. Recientemente este copolímero EBA-8% se emplea en la fabricación de algunos filmes agrícolas por extrusión y soplado por sus buenas propiedades y excelente procesabilidad. 234 6. POLíMEROS Para la reacción de transesterificación se han utilizado tres tipos de aminas impedidas (HALS) de bajo peso molecular con grupos hidroxilo en posición 4, (HOHALS-N-H), y (HO-HALS-N-Me) comerciales y (HOHALS-N-OMe) sintetizado previamente en este trabajo [5]. Para el estudio comparativo de estabilización de los nuevos HALS se ha empleado un HALS comercial de tipo oligomérico (HALS-NOR-371) de referencia considerado como uno de los más eficientes fotoestabilizantes empleados en la actualidad. Reacción de transesterificación en el estado fundido Las reacciones de funcionalización del EBA en estado fundido [6,7] fueron realizadas en un mezclador interno Haake Rheocord 9000. En todas las reacciones la temperatura se mantuvo constante a 190 ºC, y la velocidad de giro de los husillos fue de 64 rpm. En todos los casos, se utilizó un 1% en peso de HALS, y un 0.5% en peso de catalizador (óxido de dibutil estaño (DBTO)) [8,9] respecto a 50 g de EBA-8%. También se realizó una serie de mezclas sin catalizador a modo de control. Para la purificación de los productos se disolvieron 5 g del producto de la reacción en fundido en 200 ml de xileno a reflujo y bajo atmósfera de argón, después de 30 minutos el matraz se dejó enfriar hasta temperatura ambiente. A continuación se procedió a la purificación del polímero por precipitación en acetona y posterior filtrado y secado del sólido. Para asegurar la completa eliminación del HALS sin reaccionar y de los restos de catalizador, se repitió de nuevo este proceso. Obtención de filmes Los filmes se prepararon mediante moldeo por compresión en una prensa Collin P-200 fijando la temperatura en 170ºC y llevando a cabo dos ciclos de presión (3 minutos a 0 Bar y 1 minuto a 200 Bar), el enfriamiento de los materiales se realizó de forma controlada. Como referencia en el estudio se emplearon filmes de EBA-8% aditivados con el fotoestabilizante comercial HALS-NOR-371 al 0,5, 1 y 2% fabricados mediante un equipo de extrusión (Randcastle) con una relación L/D de 24:1. El espesor obtenido para todos los filmes fue constante de 100 micras. Espectroscopía Fotoelectrónica de Rayos X (XPS) El grado de funcionalización alcanzado en la reacción de transesterificación se ha calculado considerando el valor experimental para la relación de composición atómica entre el nitrógeno y el carbono () determinada por XPS en el copolímero EBA-8% funcionalizado con grupos HALS. La expresión para el cálculo es como sigue: = () / () donde, y son las áreas de los picos de nitrógeno y carbono obtenidos para las muestras purificadas. Los factores de sensibilidad () para el nivel energético estudiado de cada elemento descritos en la bibliografía son 0,42 para el orbital 1s del átomo de nitrógeno y 0,25 para el orbital 1s del átomo de carbono. A partir XI CONGRESO NACIONAL DE MATERIALES del tanto por ciento de HALS anclado se ha determinado el valor de () en el copolímero. Ensayo de envejecimiento acelerado en cámara climática (WOM) Los filmes se sometieron a envejecimiento acelerado en cámara climática (Weather-Ometer Atlas Ci4000 (WOM)) provisto de lámpara de xenón según el ciclo estándar ISO 4892-2:1994 fijando la intensidad de radiación (a 340 nm) en 0,47 W/m2. La temperatura de la cámara fue de 40ºC. Los pesticidas utilizados fueron metam-sodio y cipermetrina. Para el tratamiento se pulverizaron las muestras antes de su introducción en la cámara climática únicamente aplicando el metam-sodio. Posteriormente los filmes se trataron con cipermetrina (3 veces) a intervalos de tiempo de 525 horas. Determinación de especies de fotooxidación El seguimiento de la fotodegradación de los filmes se realizó a través del índice de carbonilo medido por espectroscopia infrarroja de transmisión con transformada de Fourier (FTIR). Los valores del índice de carbonilo se han obtenido empleando un calibrado previo, desarrollado por Repsol, que relaciona la absorbancia a 908 cm-1 en el EBA-8% con la absorbancia a 1710-1715 cm-1 en polietilenos de baja densidad (LDPE) cuando ambos polímeros son fotodegradados en cámara climática bajo las mismas condiciones. De esta forma se obtiene el número de carbonilos presentes en el EBA-8% por cada cien átomos de carbono, y que permite comparar la fotoestabilidad de todas las muestras sometidas a envejecimiento acelerado. 3. RESULTADOS Y DISCUSIÓN Grado de funcionalización. Estudios por XPS La baja concentración de los fotoestabilizantes HALS en los copolímeros y su estructura de carácter alifático, impiden su determinación mediante técnicas convencionales. Sin embargo, los filmes obtenidos con los materiales producto de la reacción mostraban un efecto fotoestabilizante a diferencia de los obtenidos en las reacciones llevadas a cabo sin catalizador. Este hecho fue la primera confirmación de que la estructura HALS que actuaba de esa manera tenía que ser de alto peso molecular y estar, por lo tanto, anclado covalentemente al EBA-8%. El estudio por XPS [10] realizado sobre muestras de reacción sin purificar muestra dos señales diferenciadas que aparecen como un pico asimétrico, la señal que aparece por debajo de 400 eV [11] se asocia a especies de nitrógeno protonadas, y la señal a 400eV se debe a especies no protonadas. A continuación en la figura 2 se muestran dichas señales en productos de reacción a diferentes tiempos, por su interés para el HALS-N-OMe. En el proceso de extracción del aditivo sin anclar desaparece la señal de HALS por debajo de 400 eV. Esto confirma, que solo los HALS anclados covalentemente al EBA se encuentran presentes en las muestras. 235 6. POLíMEROS Figura 3. Evolución del índice de carbonilo para filmes sin catalizador y de referencia Figura 2. Análisis XPS del orbital 1s del átomo de nitrógeno en productos de reacción del copolímero EBA 8% y el HALS reactivo OH-HALS-NOMe: A: en el producto de reacción y B: después de la extracción del aditivo. Para valorar la extensión de la reacción se procedió al estudio por XPS de las muestras (tabla 1). Tabla 1.Resultados obtenidos del contenido de HALS en los productos de reacción por XPS. Reacción en fundido t (min) N/C % HALS EBA/HALS-NH (no catalizada) 5 --- --- EBA/HALS-NMe (no catalizada) 5 --- --- EBA/HALS-NOMe (no catalizada) 5 --- --- EBA/HALS-NMe 5 --- --- EBA/HALS-NMe 10 --- --- EBA/HALS-NMe 15 0.0010 0.17 EBA/HALS-NMe 30 0.0023 0.39 EBA/HALS-NOMe 5 0.0012 0.32 EBA/HALS-NOMe 10 0.0030 0.63 EBA/HALS-NOMe 15 0.0020 0.42 EBA/HALS-NOMe 30 0.0010 0.21 La modificación del EBA se ha realizado con un contenido del 8% en acrilato de butilo por su interés aplicado en filmes agrícolas. Los grados de modificación inferiores al 1% (tabla 2) son adecuados para conseguir materiales fotoestabilizados tal y como se muestra a continuación en la siguiente sección. Estudio de fotoestabilidad por Espectroscopia Infrarroja con Transformada de Fourier (FTIR) A continuación, se muestra la evolución del índice de carbonilo para todos los filmes (figuras 3, 4 y 5) en el envejecimiento acelerado en cámara WOM y con tratamiento de pesticidas. XI CONGRESO NACIONAL DE MATERIALES Figura 4. Evolución del índice de carbonilo para filmes funcionalizados con OH-HALS-N-Me y de referencia. Figura 5. Evolución del índice de carbonilo para filmes funcionalizados con OH-HALSN-OMe y de referencia. Los filmes que contienen los fotoestabilizantes de bajo peso molecular (ver figura3) empleados como reactivos muestran una oxidación similar al EBA-8% sin aditivos HALS. Este resultado confirma el hecho, conocido desde hace mucho tiempo [12], de que los HALS de bajo peso molecular no son buenos fotoestabilizantes e incluso, en este caso, no se observan diferencias claras en el comportamiento frente a pesticidas de las diferentes funcionalidades N-H, N-Me y N-OMe. Este comportamiento contrasta con los resultados obtenidos en los filmes preparados con los materiales modificados empleando el catalizador de estaño (DBTO). Hay que señalar que el comportamiento que presentan estos 236 6. POLíMEROS materiales en el envejecimiento es idéntico en los productos de reacción y en los materiales sometidos a extracción del aditivo. Se demuestra así la nula eficiencia fotoestabilizadora de los HALS utilizados como reactivos. Por otro lado, la fotoestabilidad de los filmes de referencia aditivados con el HALS-NOR 371 comercial, es independiente de la cantidad añadida (0,5, 1 y 2%) de fotoestabilizante en el tiempo de envejecimiento (4200 horas) alcanzado en este trabajo. Todos los filmes funcionalizados con OH-HALS-NMe (ver figura 4) presentan una mayor fotoestabilidad que el EBA-8% de referencia y también mayor oxidación que las referencias con HALS-NOR 371. Se puede ver la importante influencia que tiene el tiempo de reacción, siendo la fotoestabilidad tanto mayor cuanto mayor es el tiempo de reacción. Estos resultados están de acuerdo con el grado de funcionalización determinado por XPS. A tiempos de reacción de 5 y 10 minutos no se detecta nitrógeno por XPS (Tabla 2) y la evolución del índice de carbonilo es similar a la del EBA-8% sin fotoestabilizar. Los materiales modificados para 15 y 30 minutos con contenidos de HALS de 0.17 y 0.39% respectivamente, si presentan una fotoestabilización eficiente y en clara diferencia con los anteriores. Todos los filmes funcionalizados con HO-HALS-N-OMe (ver figura 5) presentan una excelente fotoestabilidad y la evolución del índice de carbonilo se mantiene en fase estacionaria a diferencia del incremento observado en el EBA 8% sin fotoestabilizar. En esta funcionalización el tiempo de reacción no influye de forma importante en el índice de carbonilo, y el valor inicial más elevado que se observa a las 525 horas de envejecimiento, puede deberse al desarrollo de la oxidación producida durante el procesado. A partir de las 525 horas los materiales muestran una fotoestabilidad comparable a la del HALSNOR-371. Estos resultados confirman que la estructura del fotoestabilizante tipo NOMe es, como cabía esperar por los precedentes bibliográficos [13], mucho más eficiente en los foto-envejecimientos con pesticidas que las estructuras tipo NMe, este hecho, se ha correlacionado con la constante de acidez (pKa) de la amina [14]. Incluso para tiempos de reacción cortos 5 y 10 minutos se obtienen materiales excelentes desde el punto de vista de la fotoestabilidad. Estos resultados, también están de acuerdo con el grado de funcionalización determinado por XPS. Hay que señalar que cuando se habla de duración del filme para cubierta de invernadero, nos referimos al tiempo que el filme es adecuado para la aplicación en un clima como el de Almería, donde dos campañas agrícolas equivaldrían a 2800 horas en WOM. Los nuevos materiales serian adecuados para una aplicación de 3 ó 4 campañas agrícolas con tratamientos de pesticidas. 4. CONCLUSIONES necesaria la presencia del catalizador empleado en este trabajo, el óxido de dibutil estaño (DBTO). La funcionalización puede conseguirse en fundido a tiempos cortos (5, 10 minutos), hecho que tiene un gran interés industrial. La XPS permite determinar los contenidos de HALS (inferiores al 1%) anclados covalentemente al EBA. La fotoestabilidad alcanzada por introducción de funcionalidades HALS tipo N-OMe, es excelente en el intervalo de funcionalización alcanzado (0,32-0,63%) y estaría de acuerdo con las exigencias de duración (entre 3 y 4 campañas) para la aplicación agrícola como cubierta de invernadero y con tratamientos pesticidas. Los resultados obtenidos en este trabajo y la importancia científico técnica que supone el hecho de poder obtener fotoestabilizantes HALS tipo N-OMe poliméricos, de excelente eficiencia fotoestabilizante en un proceso en fundido, ha permitido presentar una solicitud de Patente [15]. 5. AGRADECIMIENTOS Este proyecto ha sido financiado por el Centro para el Desarrollo Tecnológico industrial (CDTI) dentro del proyecto CENIT MEDIODÍA. También la financiación del MICINN, Proyecto MAT2009 -09671. 6. REFERENCIAS [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] Mediante la reacción de transesterificación en estado fundido se ha conseguido modificar la estructura del copolímero EBA con un 8% de contenido en acrilato de butilo. Así se han sintetizado, por introducción de funcionalidades HALS de tipo N-Me y N-OMe, fotoestabilizantes poliméricos de gran interés para la industria. Para la reacción de transesterificación es XI CONGRESO NACIONAL DE MATERIALES [15] Catalina, F., García, Y., Salmerón, A., Espí, E., Rev, Plast. Mod 83, 547, 2002. Plastic Materials, Brydson, J.A., 6th Edition; Butterworth-Heinemann, Londres, 1995. Handbook of Material Weathering, Murphy, G., Elsevier Advanced Technology, Elsevier Science Ltd., U.K. Polymer Durability, Clough, R.L., Billingham, N.C., Gillen, K.T., Washington, DC, 1996. Galbo, J. P., Process for preparing N-methoxy derivatives of 4-hydroxy-2,2,6,6tetramethylpiperidine Ciba 1993. Moad, G., Progress in Polymer Science (Oxford) 24, 81 1999. Hu, G. H.; Lambla, M., Polymer 35, 3082, 1994. Poller, R. C., Journal of Organometallic Chemistry 239, 189, 1982. Poller, R. C.; Retout, S. P., Journal of Organometallic Chemistry 173, 1979. Watts, J.F., “X-ray photoelectron spectroscopy”, en “Surface Science Techniques”, J.M. Wallsy R. Smith, Pergamon (1994). High Resolution XPS of Organic Polymers, The Scienta ESCA 300 Database. G. Beamsonand, D. Gugumus, F., Polymer Degradation and Stability 24, 289 1989. Propisil, J., Adv.Polym. Sci, 87 1995. Lelli, A. L., Guo, M., Tan, K. H., Proceedings of the 15th Internacional Congress for Plastics in Agriculture, Hershey, Pensilvania, EE.UU., 386, 2000. Espí, E.; Fontecha, A.; Calvo, M.R.; Real, A. I.; Catalina, F.; López-Vilanova, L.; Peinado, C. Filmes para sistemas de cultivo intensivo bajo cubierta plástica que contengan copolímeros acrílicos funcionalizados con grupos fotoestabilizantes NORHALS. 08/09/2008 Solicitud nº Reg.P200802677. 237 6. POLíMEROS SÍNTESIS Y CARACTERIZACIÓN DE UN POLÍMERO MIMÉTICO A LA CUTINA VEGETAL J.A. Heredia-Guerrero1, A. Heredia2, R. García-Segura3, J.J. Benítez1 Instituto de Ciencia de Materiales de Sevilla, Centro Mixto CSIC-Universidad de Sevilla, Avda. Americo Vespuccio 49, 41092 Sevilla, España. 2 Departamento de Biología Molecular y Bioquímica, Facultad de Ciencias, Universidad de Málaga, E-29071 Málaga, España. 3 Departamento de Bioquímica, Biología Molecular y Química Orgánica, Facultad de Ciencias, Universidad de Málaga, E-29071 Málaga, España. 1 Resumen: Una estrategia interesante para la obtención de nuevos materiales consiste en mimetizar materiales biológicos manteniendo sus propiedades originales o mejorándolas. En este sentido, se ha sintetizado un polímero mimético a la cutina vegetal (un poliéster hidrófobo y biodegradable) a partir del ácido 9,10,16-trihidroxihexadecanoico (ácido aleurítico) mediante una reacción de policondensación a baja temperatura. El polímero polialeurato resultante se caracterizó por ATR-FTIR, DSC, XRD y 13C-CP/MAS NMR. También se han investigado ciertas propiedades mecánicas e hidrodinámicas. El producto obtenido es física y químicamente muy similar a la cutina. Un análisis estructural detallado revela que la arquitectura polimérica del polialeurato es más rígida y ordenada que la del polímero natural. Los enlaces de H adicionales del polialeurato se consideran los responsables de tales observaciones. Palabras clave: polímeros biomiméticos, biopolímero cutina, polialeurato. 1. INTRODUCCIÓN La obtención de materiales poliméricos completamente biocompatibles es un objetivo de muchos proyectos de investigación. En este sentido, los polihidroxialcanoatos y algunos poliésteres termoplásticos son los más estudiados [1-4]. En paralelo a los diseños sintéticos de nuevos polímeros, aproximaciones alternativas como la biomimetización de polímeros naturales se muestran muy atractivas. Éste es el caso para la cutina. El biopolímero cutina es el mayor componente de la cutícula vegetal, la membrana extracelular continua y lipídica que cubre las zonas aéreas de hojas, frutos y tallos no lignificados. Químicamente, la cutina es una red polimérica amorfa compuesta por ácidos grasos polihidroxilados C16 y C18 entrecruzados por enlaces éster. Las propiedades físicas, químicas y morfológicas de este poliéster natural han sido resumidas en una exhaustiva revisión [5]. Estudios estructurales sobre la cutina vegetal revelan un material amorfo con una textura superficial que puede ser descrita como una red interconectada de motivos similares a “gusanos” [6,7]. La biomimetización de la cutina se consiguió a través de su síntesis química in vitro a partir del ácido 9,16dihidroxihexadecanoico, el principal monómero de la cutina de fruto de tomate [8]. El proceso se basó en una reacción de policondensación a baja temperatura en un medio hidrófobo [9]. En este trabajo, un monómero muy similar, el ácido aleurítico (ácido 9,10,16-trihidroxihexadecanoico), previamente utilizado como modelo para el estudio de la formación de cutina [10,11], se emplea para obtener un poliéster análogo. La molécula de ácido aleurítico cuenta con un grupo hidroxilo extra por lo que se espera XI CONGRESO NACIONAL DE MATERIALES que aumente el rendimiento de la reacción y el grado de entrecruzamiento. En la Figura 1 se muestran las estructuras químicas del ácido aleurítico y del poliéster derivado. Ácido aleurítico OH OH HO OH O Polialeurato OR’ * O O OR’ n * O Figura 1. Estructuras químicas del ácido aleurítico y el polímero polialeurato. R’ representa un H u otro fragmento entrecruzado. 2. RESULTADOS 2.1. Síntesis de los poliésteres Una considerable cantidad de trabajo se ha realizado para la determinación de las condiciones de reacción que conducen a los mejores resultados de la polimerización del ácido aleurítico. El punto de partida se describe en la referencia [8]. Aquí, la temperatura, el disolvente, el tipo de catalizador y cebador y la cantidad relativa de cada componente se optimizaron para aumentar los resultados de la reacción. El proceso de optimización se resume en la Tabla 1. 238 6. POLíMEROS Tabla 1. Optimización de las diferentes condiciones de reacción de la policondensación de ácido aleurítico en medio orgánico. mg producto / 100 mg ácido aleurítico DBSA: 67.9 TsOH: 60.9 Amberlyst® 15: 9.9 50ºC: 0 60ºC: 0 70ºC: 60.2 80ºC: 67.9 90ºC: 51.7 Tolueno: 67.9 Cloroformo: 37.9 THF: 45.5 10 mg: 0 40 mg: 59.2 80 mg: 67.9 120 mg: 53.1 Polialeurato: 47.1 Cutina de hoja de Clivia miniata: 68.6 Pectina de manzana: 46.5 Parámetro Catalizador Temperatura Disolvente Cantidad de catalizador Cebador a b : tensión, : deformación, a: asimétrico, s: simétrico. f: fuerte, m: medio, d: débil, m: muy, an: ancho. Los espectros 13C-CP/MAS NMR, Figura 3(a), corroboran los resultados de los espectros ATR-FTIR. Además, la movilidad de ambos polímeros fue determinada por la medida del tiempo de relajación de spin, T1, de los átomos C y H del grupo metileno, Tabla 3. Tabla 3. Tiempo de relajación de spin para los átomos C y H del grupo metileno en la cutina y el polialeurato. Polialeurato T1 C T1 H (ms) (ms) 2.44 ± 1.44 ± 0.09 0.03 2.45 ± 1.49 ± 0.05 0.04 1.46 ± 1.54 ± 0.17 0.01 (ppm) 24.8 (CH)2 28.8 33.5 En consecuencia, los mejores resultados de reacción se obtienen utilizando tolueno como disolvente a 80ºC con DBSA (80 mg por 230 mg de ácido aleurítico) como catalizador y cutina de hoja de Clivia miniata como cebador. 2.2. Caracterización La Figura 2 compara los espectros ATR-FTIR de una muestra de cutina aislada de fruto de tomate y otra de polialeurato. Ambos espectros son muy similares. Los principales picos y sus asignaciones se muestran en la Tabla 2. Cutina T1 C T1 H (ms) (ms) 3.72 ± 2.16 ± 0.42 0.09 4.30 ± 2.78 ± 0.14 0.24 2.56 ± 2.31 ± 0.21 0.22 Los difractogramas del polialeurato y la cutina natural son también muy similares, Figura 3(b). La banda ancha a 0.46 nm es característica de una amplia gama de polímeros amorfos. Cutine Heredia (a) Polialeurato Cutine Peau de Tomate PLx Transmitancia Cutina Polialeurato Cutina Cutin 200 150 200 Figura 2. Espectros ATR-FTIR de una cutina aislada de fruto de tomate y el polímero polialeurato. Las principales diferencias se asocian a la presencia de hidroxilos extras en el polialeurato. Tabla 2. Principales absorciones ATR-FTIR presentes en la cutina vegetal y en el polialeurato. En ambos casos los espectros son típicos de poliésteres alifáticos. a Asignación (O-H···O) a(CH2) s(CH2) (C=O) éster (CH2) tijereteo a(C-O-C) s(C-O-C) (CH2) balanceo Número de onda (cm-1) (Intensidadb) Cutina Polialeurato 3444 (d, an) 3403 (m, an) 2925 (f) 2925 (f) 2853 (m) 2854 (m) 1729 (mf) 1730 (mf) 1462 (d) 1463 (d) 1169 (mf) 1178 (mf) 1103 (d) 1108 (d) 724 (d) 725 (d) XI CONGRESO NACIONAL DE MATERIALES 100 100 50 0 50 0 ppm (b) Intensidad Número de onda (cm-1) 150 Polialeurato Cutina 10 15 20 2 Theta (º) 25 30 0 Figura 3. (a) Espectros 13C-CP/MAS NMR para los polímeros polialeurato y cutina. El pico a 173 ppm es indicativo de grupos éster. (b) La difracción de rayos-X revela su carácter amorfo. El termograma del polialeurato, Figura 4, muestra tres eventos térmicos: una transición vítrea a -1.9ºC y dos pequeños endotérmicos a 46.0 y 72.8ºC, que pueden ser relacionados con la presencia de dominios ordenados a corto alcance. 239 Flujo de calor (mW/mg) 6. POLíMEROS 3. DISCUSIÓN 3.1. Método sintético 0.2 mW/mg -30 -10 10 30 Temperatura (ºC) 50 70 Figura 4. Termograma del polialeurato. La temperatura de transición vítrea se observa a -1.9ºC. Picos endotérmicos adicionales son detectados a 46.0 y 72.8ºC. 2.3. Propiedades hidrodinámicas y mecánicas La adsorción de agua del polialeurato desde una atmósfera saturada de agua fue de (4.0 ± 0.3) % respecto al peso de la muestra seca. La permeabilidad al agua (permeanza) calculada fue de (2.6 ± 0.4) · 10-5 m s-1. Ambos parámetros son representativos de un material hidrófobo. La Figura 5 muestra las curvas de deformación-tiempo (5a) y tensión-deformación (5b) del polialeurato a 23ºC y 40% de humedad relativa. Se observa un comportamiento bifásico definido por un régimen elástico y otro viscoelástico. A partir del régimen elástico de la curva tensión-deformación, el módulo elástico (E) del polímero polialeurato se puede calcular: E = (17 ± 3) MPa. Régimen elástico Deformación (%) Régimen viscoelástico Tiempo (s) Régimen viscoelástico Tensión (MPa) Régimen elástico Deformación (%) Figura 5. Curvas de (a) deformación vs tiempo y (b) tensión deformación de láminas de polialeurato. Los datos se obtienen a 23ºC y 40% de humedad relativa. La presencia de un comportamiento bifásico (elástico y viscoelástico) es detectada. XI CONGRESO NACIONAL DE MATERIALES La ruta de síntesis de polímeros derivados del ácido aleurítico, tomado como modelo de hidroxiácido componente de cutinas vegetales, utilizada en este trabajo de investigación ha proporcionado de un modo general resultados positivos. En la síntesis, un ácido orgánico surfactante, el ácido 4- dodecilbencenosulfónico (DBSA), ha sido el catalizador que mayor grado de polimerización y cantidad de producto ha producido al compararlo con otros catalizadores ácidos como el ácido p-toluénsulfónico o la Amberlist-15. Se ha atribuido este efecto de mejora a la capacidad de formación de micelas inversas del DBSA, en cuyo interior polar pueden alojarse moléculas de agua que podrían hidrolizar los enlaces éster formados. Se ha realizado además la optimización de condiciones concretas de reacción tales como la temperatura, la naturaleza del disolvente y la cantidad de DBSA. De los resultados puede inferirse que estas micelas inversas son críticas para la reacción de esterificación. De esta manera, a pesar de lo que podría pensarse en un principio, temperaturas elevadas son capaces de aportar la energía suficiente para romper las interacciones que mantienen la estructura de las micelas inversas, proporcionando una menor cantidad de producto. Disolventes más polares que el tolueno como el cloroformo o el tetrahidrofurano, donde el ácido aleurítico es más soluble a menores temperaturas, no mejoran los resultados como consecuencia de que en estos medios la formación de micelas inversas está desfavorecida. Por último, respecto a la cantidad de DBSA, las reacciones de deshidratación, entre ellas la esterificación, en medio acuoso utilizando DBSA han sido estudiadas y se concluyó que la cantidad de DBSA afecta a la posición del equilibrio de la reacción de esterificación [12]. En general, pequeñas cantidades de este catalizador ácido proporcionan mayores rendimientos a costa de mayores tiempos de reacción. A medida que la cantidad de DBSA se hace más grande, la hidrólisis va acentuando su participación y los rendimientos son menores, aunque el equilibrio se alcanza antes. Este fenómeno se explica porque cuanto mayor es la cantidad de catalizador, más pequeñas son las micelas que se forman, aumentando la superficie de contacto entre las fases orgánica y acuosa, favoreciendo de esta manera la reacción de hidrólisis [12]. No obstante, parece no haber concordancia entre las cantidades de producto y la cantidad del ácido sulfónico, las cuales deberían ser menores a medida que la de DBSA es más grande. El uso de cebadores de diferente naturaleza (polialeurato, cutina de Clivia minata y pectina de manzana) en las reacciones químicas de síntesis de los polímeros provocó un crecimiento brusco de los mismos originando en lugar de delgadas películas masas informes de poliéster y una disminución drástica de los tiempos de reacción. La cantidad de producto obtenido dependió del tipo de cebador. Así para la cutina vegetal se obtuvo una cantidad comparable a la obtenida sin 240 6. POLíMEROS cebador y para la pectina y el homopoliéster derivado del ácido aleurítico se obtuvieron productos en menores cantidades. La justificación de este fenómeno no parece tener fácil explicación, aunque puede estar relacionada con el bloqueo de monómeros en el interior del polímero que tan bruscamente se forma, de manera que no pueden reaccionar y que posteriormente, durante los lavados, se eliminan. 3.2. Comparación cutina-polialeurato Mecánicamente, ambos materiales tienen módulos elásticos comparables, E = (17 ± 3) MPa para el polialeurato y E = (15 ± 2) MPa para la cutina [13]. Sin embargo hay una importante diferencia mecánica, mientras que el polialeurato es un material bifásico (elástico y viscoelástico), Figura 5, la cutina natural es fundamentalmente viscoelástica [13]. La región elástica del polímero sintético está relacionada con la red de enlaces de H más amplia del mismo. La ruta sintética propuesta ha sido satisfactoria en cuanto a la creación de un poliéster derivado del ácido aleurítico muy similar a la cutina vegetal. Químicamente hay una buena correspondencia como se observa por ATR-FTIR y 13C-CP/MAS NMR. Tal similitud es consecuencia directa de la analogía entre el principal monómero de la cutina, el ácido 9,10dihidroxihexadecanoico, y la molécula de ácido aleurítico (ácido 9,10,16-trihidroxihexadecanoico). La única diferencia química destacable es la elevada presencia de grupos hidroxilos no esterificados en el polialeurato como se muestra en el espectro ATR-FTIR, Figura 2. Esto es debido obviamente a la alta ratio -OH/-COOH en la molécula de ácido aleurítico si se compara con el monómero de cutina. Después de la esterificación completa uno y dos grupos hidroxilos sin reaccionar por molécula son esperables para la cutina y el polialeurato, respectivamente. Estos grupos hidroxilos libres también explican la mayor absorción de agua del polialeurato (4% p/p) si se compara con la cutina (2.5% p/p). La ratio experimental 1.6 está en muy buena concordancia con el teórico 1.9 deducida de la fórmula química de ambas moléculas. Ambos valores de absorción de agua son bastante bajos, indicando un comportamiento muy hidrófobo de los polímeros. Esto se corrobora por valor tan pequeño de permeanza obtenido. 4. CONCLUSIONES Estructuralmente, tanto la cutina como el polialeurato son principalmente amorfos. Esto se muestra en difractogramas, Figura 3(b), donde las bandas anchas, asignables a las distancias basales entre las cadenas metilénicas, son idénticos. [4] [5] [6] Sin embargo, algunas diferencias entre ambos polímeros se han destacado por DSC y los ensayos mecánicos. La detección de dos pequeños picos endotérmicos del polialeurato, Figura 4, que no se observan para la cutina, indican la presencia de dominios ordenados a corto alcance en el poliéster sintético. Otra diferencia es su temperatura de transición vítrera, Tg, de -47.0ºC para la cutina y de -1.9ºC para el polialeurato. La elevada Tg para el poliéster sintético es asociada a la menor movilidad de las cadenas hidrocarbonadas. Esto se corrobora por los tiempos de relajación de spin, T1, para los átomos de C y H en los grupos metilénicos, Tabla 2. Los valores más bajos para el polialeurato del T1 indican que la red de polialeurato es más rígida que para la cutina. Esto es probablemente debido a la red de enlaces de H más extensa del producto sintético. [8] XI CONGRESO NACIONAL DE MATERIALES Siguiendo la ruta sintética previamente descrita por muestro grupo [8] la síntesis química de un poliéster mimético a la cutina vegetal se lleva a cabo utilizando el ácido aleurítico como monómero. Las condiciones de reacción tales como la temperatura, el disolvente, el tipo de catalizador y la presencia de cebadores se estudiaron y optimizaron. La caracterización de este nuevo polialeurato por ATR-FTIR, DSC, 13C-CP/MAS NMR y XRD revela un poliéster hidrófobo y amorfo. A pesar de su similitud química y estructural, la red de cadenas alifáticas en el polialeurato es más rígida que la de la cutina. Además, el polímero sintético muestra un comportamiento elástico extra frente al polímero natural. Ambas características se explican por la presencia de enlaces de H adicionales en el interior de la estructura del polialeurato. 5. REFERENCIAS [1] [2] [3] [7] [9] [10] [11] [12] [13] Cassagnau P, Bounor-Legare V, Fenouillot F. Int Polym Proc 2007;22(3):218-258. Verlinden RAJ, Hill DJ, Kenward MA, Williams CD, Radecka I. J Appl Microbiol 2007;102(6):1437-1449. Uyama H, Kobayashi S. Adv Polym Sci 2006;194:133-158. Okama M. Prog Polym Sci 2002;27(1):87-133. Heredia A. Biochim Biophys Acta 2003;1620:1-7. Luque P, Bruque S, Heredia A. Arch Biochem Biophys 1995;317:417-422. Benítez JJ, Matas AJ, Heredia A. J Struct Biol 2004;147:179-184. Benítez JJ, García-Segura R, Heredia A. Biochim Biophys Acta 2004;1674:1-3. Saam JC. J Polym Sci A 1998;361:341-346. Benítez JJ, Heredia-Guerrero JA, Heredia A. J Phys Chem C 2007;111(26):9465-9470. Heredia-Guerrero JA., Benítez JJ., Heredia A. Bioessays. 30:273-277. Manabe K, Iimura S, Sun XM, Kobayashi S. J Am Chem Soc 2002;124: 11971López-Casado G, Matas AJ, Domínguez E, Cuartero J, Heredia A. J Exp Bot 2007; 58:38753883. 241 6. POLíMEROS INCIDENCIA DEL ADHESIVO TERMOFUSIBLE EN BASE EVA EN EL BUEN ENCOLADO DE CANTOS DE CHAPA DE MADERA NATURAL EN EL SECTOR DEL MUEBLE Y LA MADERA M.V. Navarro Bañón1, F.J. Melero Muñoz1, R. Fernández Reyes2, J.M. Martín Martínez2 Centro Tecnológico del Mueble y la Madera (CETEM), Yecla (Murcia). [email protected] Laboratorio de Adhesión y Adhesivos, Universidad de Alicante, 03080 Alicante. [email protected] 1 2 Resumen: Las chapas de madera natural se emplean frecuentemente, en el sector del mueble y la madera, como material para el recubrimiento de cantos de tableros aglomerados de partículas o de fibras. El objetivo de estas uniones adhesivas, en las que se emplean adhesivos termofusibles en base EVA, es embellecer el canto del tablero y protegerlo frente a cambios de humedad que pueden dar lugar al desmoronamiento de la estructura interna del tablero. A nivel industrial, aparece un alto porcentaje de uniones de cantos encolados que no cumplen con los requisitos de calidad necesarios para su uso en mobiliario, produciéndose frecuentes delaminaciones (separación de los cantos del tablero). Este trabajo, muestra los resultados de investigación obtenidos durante los últimos 2 años en el marco del proyecto ENCANNTO cuyo objetivo es establecer las causas físico-químicas que producen la carencia de adhesión en cantos encolados a tableros. Palabras clave: ADHESIVO TERMOFUSIBLE, EVA, ENCOLADO, CANTOS, MADERA 1. INTRODUCCIÓN El encolado de cantos de tableros derivados de la madera se realiza para proteger y embellecer el tablero. Un canto no protegido constituye un punto débil por el que puede producirse pérdida o ganancia de humedad y provocar el desmoronamiento del tablero. Industrialmente, el proceso se realiza en aplicadoras automáticas de cantos empleando, principalmente, adhesivos termofusibles en base EVA. Los materiales empleados para cubrir el canto (cubrecantos) pueden ser de diferente naturaleza: chapas de madera natural, papel impregnado en melamina, metales y polímeros, principalmente PVC y ABS, entre otros [1]. Las uniones de cantos encolados proporcionan numerosos problemas de falta de adhesión obteniéndose productos no conformes, lo que supone un coste adicional innecesario y una imagen comercial poco adecuada para las empresas del sector del mueble. Estudios previos muestran la influencia de diferentes variables que intervienen en el proceso de encolado automatizado utilizado a nivel industrial en los resultados de adhesión [2], pero no existen estudios que muestren la influencia de los materiales empleados en las propiedades de adhesión de dichas uniones. Hoy en día, el origen de las frecuentes delaminaciones de cantos no se conoce y la solución no es siempre factible. Por este motivo, el objetivo de este trabajo es analizar la problemática de adhesión relacionada con uniones de cantos encolados, empleadas en el sector del mueble y la madera. Como primer paso se realizó un análisis exhaustivo del tipo de fallo producido para diferentes tipos de uniones empleadas en el sector del mueble para detectar los puntos más débiles de las mismas y poder establecer estrategias fundamentadas para el fortalecimiento de dichas uniones [3,4]. 2. EXPERIMENTAL 2.1. Materiales Se han empleado muestras de cantos encolados, de origen industrial, con cubrecantos de chapa natural XI CONGRESO NACIONAL DE MATERIALES (nogal, cerezo, pino). En todos los casos el interior estaba constituido por un tablero aglomerado de partículas estándar. Se han utilizado materiales de uso común en uniones de cantos encolados: Cantos de chapa de madera natural, tablero aglomerado de partículas estándar, tablero MDF y 7 formulaciones adhesivas de origen comercial. 2.2. Metodología Para comprobar la adecuación al uso de las uniones de cantos encolados se realizaron ensayos de resistencia al agua y encolado de cantos siguiendo la Norma UNE 56843 [5]. Para la determinación de la composición química y la morfología superficial en superficies se emplearon las siguientes técnicas: (i) Espectroscopía infrarroja mediante reflectancia total atenuada (IR-ATR) (Tensor 27, Bruker). Accesorio ATR de rebote simple de diamante (ii) Microscopía óptica de reflexión (MSX-500Di, Herter Instruments). Para la caracterización de las propiedades de los adhesivos se emplearon, además, las siguientes técnicas experimentales: (Q500, TA Instrument). Termogravimetría Calentamiento en atmósfera de N2 de 30 a 850 ºC a una velocidad de calentamiento de 5ºC/min. Fluorescencia de rayos X (Philips Magix Pro). Tubo de Rodio y ventana de Berilio. plato-plato (Bohlin CS 50). Reometría Enfriamiento de 200 a 30 ºC a una velocidad de enfriamiento de 5ºC/min y a una frecuencia de 1 Hz. Viscosidad Brookfield. Vástago S27, 20 rpm, 200ºC. 242 6. POLíMEROS 0 .3 0 0 .0 0 0 .2 0 0 .0 0 0 .2 5 0 .0 5 0 .0 0 0 .2 0 Absorbancia A b s o r b a n c ia 0 .2 0 0 .0 0 0 .2 0 Los resultados obtenidos muestran la problemática real existente, actualmente, con el delaminado de cantos encolados en el sector del mueble y la madera. Para poder establecer las causas de dicha delaminación se realizó un análisis exhaustivo de los materiales que intervienen en el proceso de encolado de cantos. A1 405_08/19 2916 1737 2849 A2 3500 3000 2500 2000 A3 3500 3000 2500 A4 3500 3000 A5 3500 A6 A7 0 .0 0 El tipo de fallo obtenido para cada una de las uniones que presentaron falta de adhesión, determinado a partir de análisis mediante espectroscopia IR-ATR y mediante microscopía óptica de las superficies separadas, depende de si el canto de chapa natural está unido a un soporte de forma previa o no. En el caso de cantos de chapa natural unidos a un soporte (normalmente base papel) se obtiene un fallo de adhesión al canto, mientras que para cantos de chapa de madera natural sin soporte se obtiene principalmente un fallo de cohesión en el adhesivo. Puesto que la composición química de los adhesivos A2, A4 y A5 es muy similar y también lo es la de los adhesivos A3, A6 y A7, se seleccionaron para el resto del estudio a los adhesivos A2 y A3 junto al A1 como representativos del resto de los de sus grupos. 0 .2 0 Una unión encolada de cantos es adecuada para su uso en mobiliario, según normativa vigente, si supera los ensayos de resistencia al agua y encolado de cantos. Los resultados obtenidos tras el análisis de una amplia selección de uniones encoladas procedentes de empresas del sector del mueble reflejan que un 60% de las muestras ensayadas no supera los ensayos según normativa vigente para uniones de cantos encolados de uso en mobiliario. El análisis mediante microscopía óptica en modo transmisión confirma la existencia de cargas en la formulación de todos los adhesivos termofusibles, salvo en la formulación A1. 0 .0 0 3. RESULTADOS EXPERIMENTALES 1372 1464 1238 1020 720 1500 1000 500 2000 1500 1000 500 2500 2000 1500 1000 500 3000 2500 2000 1500 1000 500 3500 3000 2500 2000 1500 1000 500 3500 3000 2500 2000 1500 1000 500 3500 3000 2500 2000 1500 1000 500 1434 Número de onda (cm-1) 874 608 748 Figura 1. Espectros IR-ATR de los adhesivos termofusibles. Propiedades de los adhesivos La Figura 1 muestra los espectros IR-ATR obtenidos para las 7 formulaciones de adhesivos termofusibles, en base EVA, seleccionados y de uso común en uniones de cantos encolados. La formulación A1 muestra las bandas de absorción características del copolímero EVA (tensión C-H de metilos y metilenos a 2916 y 2849 cm-1, tensión C=O a 1737 cm-1, flexión asimétrica y simétrica de C-H de CH3 y CH2 a 1464 cm-1y 1372 cm-1, tensión C-O de grupo éster a 1239 y 1020 cm-1 y balanceo de CH2 a 720 cm-1 [5,6]. Adicionalmente, aparecen las bandas de cera (grupos metilo y metileno) y de un éster de colofonia (grupos metilo y metileno, y grupos C=O y C-O). Las formulaciones A2, A4, A5 constituyen un segundo grupo de adhesivos puesto que sus espectros IR-ATR presentan bandas de absorción similares, bandas características del copolímero EVA, la cera y el éster de colofonia, así como otras bandas adicionales que podrían corresponder a CaCO3 como carga inorgánica adicionada en la formulación (bandas a 1434 y 874 cm-1 correspondientes a las vibraciones de tensión y flexión C-O del grupo carbonato). También se detectan otras bandas a 478 y 608 cm-1. Las formulaciones A3, A6 y A7 constituyen un tercer grupo de adhesivos ya que presentan bandas de absorción características del copolímero EVA, la cera y el éster de colofonia, y bandas de CaCO3 las cuales son mucho más intensas que las del grupo 2. XI CONGRESO NACIONAL DE MATERIALES 100 µ m 100 µ m 100 µ m A1 A2 A3 Figura 2. Imágenes de microscopio óptico de transmisión de las películas de adhesivos. Tabla 1. Resultados obtenidos del análisis termogravimétrico de los adhesivos. Adhesivo A1 A2 A3 R: Residuo T (ºC) (T1/T2/T3…) 151/334/445/R 126/334/372/446/647/R 117/337/381/445/661/R Pérdida de peso (%) 1.6/26.3/72.8/0.0 1.1/14.5/12.2/36.0/9.3/26.9 1.7/12.5/22.0/24.0/17.0/22.8 Tabla 2. Composición porcentual de cenizas de los adhesivos obtenida mediante fluorescencia de rayos X. Adhesivo A1 A2 Cenizas (%) 0.0 26.92 A3 20.26 Óxidos en cenizas (%) --14.05% BaO, 10.07% CaO, 2.28% SO3, 0.20 % SiO2, 0.17% SrO, 0.11% MgO, 0.04% Fe2O3 19.64% CaO, 0.21 % SiO2, 0.14% P2O5, 0.09% MgO, 0.07% Al2O3, 0.04% SO3, 0.03% Fe2O3, 0.01% BaO 243 6. POLíMEROS La Tabla 2 muestra los resultados obtenidos mediante calcinación de los adhesivos y el análisis de las cenizas obtenidas mediante fluorescencia de rayos X. El adhesivo A2 contiene tanto CaCO3 como BaSO4 como cargas, mientras que el adhesivo A3 sólo contiene CaCO3 como carga. Además, el adhesivo A2 contiene menos cantidad de CaCO3 que el adhesivo A3. Las propiedades reológicas de los adhesivos se relacionan con su formulación y afectan notablemente a sus propiedades de adhesión. La incorporación de cargas aumenta la viscosidad de los adhesivos, en la misma magnitud incorporando CaCO3 como BaSO4 como cargas. El adhesivo A2 presenta una viscosidad Brookfield similar a la del adhesivo A3 pero su comportamiento es menos elástico, ya que el cruce de los módulos elástico y viscoso de produce a menor temperatura y menor módulo (Tabla 3). Por otra parte, el adhesivo A3 presenta propiedades reológicas parecidas al as del adhesivo A1 y muestra mayores valores de temperatura y módulo en el cruce de los módulos elástico y viscoso. Tabla 3. Propiedades reológicas de los adhesivos. Adhesivo A1 A2 A3 ηBrookfield (mPa·s) 60.000 64.000 65.000 Tcruce (ºC) 75 55 75 G(Pa) T=Tcruce 5.34 · 104 3.07 · 104 6.55 · 104 Para evaluar las propiedades de adhesión de las tres formulaciones adhesivas seleccionadas, se realizaron ensayos de pelado en T de uniones serraje/adhesivo/serraje y ensayos de cizalla en modo de single lap shear de uniones caucho SBR lijado/adhesivo/caucho SBR lijado. Se eligieron estos ensayos para considerar el comportamiento de los adhesivos en diversos modos de solicitación mecánica. Los resultados obtenidos se incluyen en la Tabla 4. Tabla 4. Evaluación de las propiedades de adhesión de las formulaciones adhesivas. Adhesivo A1 A2 A3 Pelado en T Serraje lijado F(kN/m) Fallo 1.6 ± 0.3 A 1.0 ± 0.3 A 0.6 ± 0.1 A Resistencia a cizalla Caucho SBR lijado F (kN/m) Fallo 6.8 ± 0.2 A 6.7 ± 1.2 A 6.5± 0.5 A diferentes adhesivos. Posteriormente, las uniones sometieron a ensayos de resistencia al agua y encolado de cantos. La Tabla 5 muestra los resultados obtenidos: Tabla 5. Ensayos de adecuación al uso (resistencia al agua y encolado de cantos) de uniones entre canto de chapa mukaly con soporte y a) tablero aglomerado de partículas y b) tablero MDF, empleando los tres adhesivos representativos de cada grupo. a) Adhesivo A1 Resistencia al agua – Encolado de Cantos – Adhesivo A3 + + b) Adhesivo A1 Resistencia al agua + Encolado de Cantos – Adhesivo A3 + + Adhesivo A2 Adhesivo A2 XI CONGRESO NACIONAL DE MATERIALES – – – Únicamente las uniones de cantos encolados preparadas con el adhesivo A3 superan los requisitos de calidad específicos para su uso en mobiliario. Puesto que los resultados del análisis de las formulaciones adhesivas no se correlacionan con el comportamiento de las uniones cantos-tableros se han analizado los sustratos empleados en la formación de las uniones adhesivas. Propiedades de los sustratos que intervienen en las uniones adhesivas. a) Tableros: Aglomerado de partículas estándar y MDF Principalmente, las propiedades en superficie de los tableros son las que van a influir directamente en las propiedades de adhesión de las uniones. Como cabría esperar, el espectro IR-ATR de la superficie de ambos tableros es muy similar, y en ambos casos se observan las bandas características de la celulosa, componente principal de la composición química (figura no mostrada). En la Figura 3 se muestran las imágenes de microscopía óptica de la superficie de los tableros. El tablero MDF es un material mucho más denso con menor porosidad superficial que puede dificultar el anclaje del adhesivo. De todos modos, no se observan diferencias entre las propiedades de adhesión obtenidas para tableros aglomerados de partículas y tableros MDF. Los resultados obtenidos muestran valores de adhesión en cizalla similares para las tres formulaciones adhesivas (lo que concuerda con las propiedades reológicas de los mismos), pero bajo esfuerzos de pelado en T, la adhesión es mayor en el adhesivo sin cargas (Tabla 4). Para evaluar el comportamiento de los adhesivos en uniones de cantos encolados se prepararon, a escala semiindustrial empleando una encoladora de cantos automática manual, uniones entre tablero aglomerado de partículas y canto de mukaly con soporte empleando los + 100 µ m (a) 100 µ m (b) Figura 3. Imágenes de microscopio óptico en modo reflexión para a) Tablero aglomerado de partículas y b) Tablero MDF. 244 6. POLíMEROS 1234 1369 2854 500 603 1500 945 2000 1118 1602 2500 1000 1371 1500 1433 3345 2000 0.0 0.2 2500 2924 0.8 0.00 Absorbancia 3000 b) “Cola blanca” 0.4 0.6 3500 2919 3329 a) Canto (cara soporte) 0.04 0.08 b) Cantos: Chapas de madera natural con soporte Los cantos de chapa de madera natural, en ciertas ocasiones, suelen ir preencolados con “cola blanca” (adhesivo de acetato de polivinilo) a un material soporte (papel, tejido, etc.) con el fin de fortalecer mecánicamente el canto y evitar fracturas de éste durante el proceso automatizado de encolado de cantos. Por ello, y dado que los fallos detectados en uniones en los que intervienen cantos de chapa de madera natural preencolados a soporte son fallos de adhesión, se procedió a realizar una selección de diferentes tipos de cantos de chapa con soporte. 3500 3000 Número de onda (cm-1) 1000 500 Figura 4. Espectros IR-ATR obtenidos para a) Cara de adhesión de canto de chapa de madera natural preencolado a soporte y b) película sólida de cola blanca. El análisis IR-ATR de la cara de adhesión del canto de chapa de madera natural preencolado a soporte, muestra las bandas características del papel (soporte) y las bandas de cola blanca en la superficie del canto (Figura 4). La micrografía óptica obtenida en la superficie del canto de chapa de madera natural preencolado a soporte muestra la presencia de restos plásticos en la superficie que podrían corresponder a los restos de cola banca, detectados por espectroscopia IR-ATR (Figura 5). A nivel comercial se encuentran diferentes formulaciones de adhesivos termofusibles en base EVA que presentan contenidos importantes de cargas inorgánicas (carbonato de calcio, sulfato de bario) que dan lugar a una modificación de las propiedades reológicas de los adhesivos. Sin embargo, éstas no presentan una gran influencia en las propiedades de adhesión a cantos y tableros. Las propiedades superficiales de los tableros seleccionados como sustratos, tablero aglomerado de partículas y de fibras, no presentan una influencia clave en los resultados de adhesión obtenidos para las uniones tras ser sometidas a los ensayos de adecuación al uso, de uniones de cantos encolados. La presencia de acetato de polivinilo en la superficie que va a participar en el proceso de adhesión del canto de chapa de madera natural preencolado a soporte, aparentemente, es un factor clave y determinante de la adhesión. Su presencia puede disminuir el número de puntos de anclaje del adhesivo disminuyendo la adhesión mecánica, mecanismo principal de adhesión de los adhesivos termofusibles y, por tanto, sería de gran interés continuar el desarrollo de este estudio para determinar la influencia de la cantidad de acetato de polivinilo en la superficie de la cara del canto a encolar con las propiedades de adhesión. 5. AGRADECIMIENTOS -Al Ministerio de Ciencia en Innovación por la concesión de subvención para el desarrollo de los proyectos: DEX-560550-2008-20 y PID-560550-2009-3 que han permitido el desarrollo de este trabajo. -Al Ministerio de Ciencia e Innovación por la concesión de subvención para la contratación de Doctores (PTQ05-02). 6. REFERENCIAS [1] [2] 100 µm Figura 5. Imagen de microscopio óptico en modo reflexión de la cara de adhesión de canto de chapa de madera natural preencolado a soporte de papel. La presencia de cola blanca en la superficie del soporte preencolado a la chapa de madera natural puede dificultar el anclaje del adhesivo termofusible dando lugar a una falta de adhesión de la unión, lo que concuerda con los resultados obtenidos en los ensayos normalizados de las uniones de cantos a tableros. 4. CONCLUSIONES De los resultados experimentales obtenidos pueden extraerse las siguientes conclusiones: XI CONGRESO NACIONAL DE MATERIALES [1] [3] [4] [5] [6] W. Nutsch. Tecnología de la madera y del mueble. Reverté, Barcelona, (1992). pp. 103-114. J. Ortiz. Encolado de cantos con colas termofusibles. Proyecto de investigación desarrollado en AITIM en 1985. A. Pizzi, K.L. Mittal. Handbook of adhesive technology. 2nd ed. Marcel Dekker, New York (2003). pp. 739-747. J. M. Martín Martínez. “Conceptos básicos de adhesión y de uniones adhesivas”. Universidad de Alicante, Alicante (2000). pp. 15-31, 73-100. UNE 56843. Muebles de cocina. Ensayos físicos. Marzo 2001 B. H. Stuart. Infrared Spectroscopy: Fundamentals and applications. Wiley, New York (2004). pp. 71-93 E. Pretsch, P. Bühlmann, C. Affolter, A. Herrera, R. Martínez. Determinación structural de compuestos orgánicos. Springer, Barcelona (2001). pp. 245-312 245 6. POLíMEROS MEZCLAS POLIMÉRICAS CRISTAL LÍQUIDO/OLEFINAS: MORFOLOGÍA Y PROPIEDADES DE TRANSPORTE J. Arranz-Andrés1, M. L. Cerrada1, V. Lorenzo2, M .U. de la Orden3, J. Martínez Urreaga2,4, E. Pérez1 Instituto de Ciencia y Tecnología de Polímeros, CSIC, Juan de la Cierva 3, 28006 Madrid, España, [email protected] 2 Grupo de Investigación ‘‘POLímeros: Caracterización y Aplicaciones” (U.A. del ICTP-CSIC), E.T.S.I. Industriales, Universidad Politécnica de Madrid, José Gutiérrez Abascal 2, 28006 Madrid, España. 3 Departamento de Química Orgánica I, E. U. Óptica, Universidad Complutense de Madrid, Arcos de Jalón S/N, 28037 Madrid, España. 4 Departamento de Ingeniería Química Industrial y del Medio Ambiente, E.T.S.I. Industriales, Universidad Politécnica de Madrid, José Gutiérrez Abascal 2, 28006 Madrid, España. 1 Resumen: Se han preparado mezclas con diferentes composiciones a partir de un copolímero de etileno-1-octeno, sintetizado con un catalizador de tipo metaloceno y un polímero cristal líquido. La caracterizción estructural y morfológica de las películas obtenidas se realizó mediante difracción de rayos X, calorimetría diferencial de barrido y microscopía electrónica de barrido, mientras que el estudio de las propiedades físicas se ha llevado a cabo mediante análisis mecanodinámico, microdureza y ensayos esfuerzo-deformación, poniendo especial énfasis a la evaluación de sus propiedades de transporte de oxígeno. Palabras clave: Mezclas, cristal líquido, poliolefinas, permeabilidad. 1. INTRODUCIÓN Los polímeros termotrópicos cristal líquido (LCPs) son materiales de interés tecnológico debido a su baja viscosidad, capacidad de orientación, resistencia y propiedades barrera [1]. Sin embargo presentan algunos inconvenientes, como reducida solubilidad y temperaturas de transición muy altas, lo que dificulta su procesado. La mezcla de los LCPs con termoplásticos se está explorando como una posibilidad atractiva de extender sus aplicaciones [2, 3]. 2. MATERIALES Y MÉTODOS Se prepararon mezclas LCP-plastómeros con diferentes composiciones, denominadas CVXX, donde XX denota el porcentaje en peso de polímero cristal líquido (Tabla 1). El plastómero empleado fue un copolímero comercial de etileno-1-octeno, CEO, sintetizado con catalizador metaloceno, con un contenido de 5.2% molar de 1-octeno (suministrado por Exxon Chemical). El polímero cristal líquido fue el Vectra V400p (suministrado por Ticona). Previo al proceso de mezclado, los gránulos de Vectra se molieron empleando un molino IKA A10 yellow-line, y se secaron a vacío a 110 ºC durante 24 h. Las mezclas CV se prepararon desde el fundido por un proceso de extrusión inversa, empleando un sistema compuesto por cilindros de acero. El diámetro interno y la altura del cilindro exterior son 29,82 y 80,20 mm respectivamente. El ajuste entre las partes cilíndricas es por deslizamiento con un espaciado entre ambas de 40 µm. El sistema de cilindros está rodeado por una resistencia eléctrica cuya temperatura se controla XI CONGRESO NACIONAL DE MATERIALES mediante un termopar. Todas las mezclas se realizaron a 240 ºC. Después del mezclado se prepararon películas mediante moldeo por compresión en una prensa Collin a 170 ºC para el CEO y a 230 ºC para el resto de las muestras, utilizando una presión de 2 MPa durante 2 min, seguido de un enfriamiento rápido hasta temperatura ambiente. Las películas se prepararon entre dos placas de acero cromado. Se dispuso una película de teflón entre el acero y la muestra para evitar problemas de adhesión. Estas películas se emplearon en la completa caracterización de las muestras. Los espectros de difracción de rayos X se obtuvieron a temperatura ambiente en el modo de reflexión en un difractómetro Bruker D8 Advance. Las propiedades térmicas se analizaron en un calorímetro TA Q100 conectado a un sistema de enfriamiento. Los pesos de las muestras están comprendidos entre 5 y 7 mg. Para determinar la cristalinidad, fcDSC, se empleó un valor de 290 J/g como entalpía de fusión del cristal ortorrómbico perfecto de PE [4]. Las medidas de microindentación se realizaron con un indentador Vickers unido a un equipo Leitz de microdureza (MH). Los experimentos se realizaron a 23 ºC con una carga de 0,96 N y un tiempo de contacto de 25s. Las medidas mecanodinámicas se llevaron a cabo con un equipo Polymer DMTA II trabajando en modo de tracción. El módulo complejo y la tangente de pérdidas se determinaron a 1, 3, 10 y 30 Hz en un rango de 246 6. POLíMEROS temperatura de -140 a 130 ºC, a una velocidad de calentamiento de 1,5 ºC min-1. de los dos componentes, así como una distribución homogénea de los dominios. Los ensayos esfuerzo-deformación se realizaron con un dinamómetro Instron a una velocidad de deformación de 0,67 min-1 a 23 ºC. Los parámetros mecánicos y viscoelásticos de las muestras indican un gran aumento en módulo y microdureza con el aumento de V400p incorporado (Tabla 1). La permeación de O2 de las mezclas se midió en un dispositivo experimental fabricado en nuestro laboratorio. Las dos cámaras separadas por la película se sometieron a alto vacío (∼10−4 mbar). Posteriormente, se introdujo O2 en la cámara de alta presión, tomando tiempo cero en ese momento. La variación de la presión con el tiempo en la cámara de baja presión se registró con un sensor de presión. Antes de cada experimento se determinó la curva de pérdidas midiendo la variación de la presión con el tiempo en la cámara de baja presión. El área de permeación de la membrana es de 13,85 cm2. Los espesores de las películas se encuentran entre 400– 600 µm, dependiendo de la membrana empleada. La permeabilidad al oxígeno y el coeficiente de difusión de las mezclas son inferiores a los encontrados en el CEO, y a mayor contenido de V400p menor es la permeabilidad y el coeficiente de difusión, según lo esperado (Figura 1). 3. RESULTADOS Y DISCUSIÓN La Tabla 1 muestra el grado de cristalinidad (fcDSC) estimado por DSC del primer proceso de fusión, así como los siguientes parámetros mecánicos de las diferentes muestras analizadas a 23 ºC: módulo de Young, E; módulo de almacenamiento, E′; y microdureza, MH. Tabla 1. Propiedades de las muestras. Muestra fcDSC CEO CV10 CV20 CV30 CV50 CV90 V400p 0.32 0.32 0.33 0.30 0.30 0.24 0.00 E (MPa) 75 125 230 300 500 3010 5310 MH E′ (MPa) (MPa) 71 8 127 10 180 13 394 19 900 26 2670 88 4415 109 Los resultados de la Tabla 1 muestran que la cristalinidad del CEO permanece casi constante hasta contenidos de LCP V400p de 50 wt.%. El aumento en la composición de V400p, polímero cristal líquido de bajo orden, (muestra CV90) origina una disminución considerable en la intensidad de las difracciones, en la entalpía de fusión, así como del grado de cristalinidad de CEO (estimado por WAXS y DSC). Esta disminución puede deberse al cambio en la morfología de esta muestra, en donde dominios minoritarios de CEO se distribuyen dentro de la matriz de V400p, como se observa en las imágenes de SEM. Por medio de esta técnica también se aprecia que para todas las mezclas en las diferentes composiciones existe separación de fases XI CONGRESO NACIONAL DE MATERIALES 30 ºC 40 ºC 50 ºC 60 ºC 10 P (barrer) Los experimentos de microscopía electrónica de barrido se realizaron en un equipo XL30 ESEM PHILIPS. Las muestras se criofracturaron previamente. 1 0,1 0 10 20 30 40 50 60 wt% V400p Figura 1. Permeabilidad en función del contenido de LCP (wt.%) a diferentes temperaturas. De lo anteriormente comentado cabe concluir que la adición del LCP a la poliolefina permite aumentar su rigidez y su resistencia a la deformación y mejorar sus propiedades de transporte de gases, observando una disminución en la permeabilidad. 4. AGRADECIMIENTOS Los autores agradecen la ayuda económica al Ministerio de Educación y Ciencia (proyectos MAT2007-65519C02-01 y MAT2007-65519-C02-02). También se agradece a Ticona el suministro del Polímero Cristal Líquido V400p. 5. REFERENCIAS [1] [2] [3] [4] Collyer A. A., “Liquid Crystal Polymers: From Structures to Applications”, Ed. Elsevier Applied Science, 1992. Utracki, L. A., “Commercial Polymer Blends”, Ed. Chapman and Hall, London, 1998. Zhang, B. Y., Sun Q. J., Li Q. Y., Wang Y. J. Appl. Polym. Sci. 2006, 102(5), 4712-9. Wunderlich, B. “Macromolecular Physics”, Ed. Academic Press, New York, vol. 3. 1980, p. 42. 247 6. POLíMEROS DISEÑO Y CARACTERIZACIÓN DE MEZCLAS EVA/PLASTÓMERO PARA SU EMPLEO EN COMPUESTOS DE AISLAMIENTO ACÚSTICO M. Niubó1, A.I. Fernandez1, L. Haurie2, V. Realinho3, J.I .Velasco3 1 Departamento de Materiales e Ingeniería Metalúrgica, Universidad de Barcelona. Martí i Franquès 1, 08028 Barcelona. [email protected] 2 Departamento de Construcciones Arquitectónicas II, Universidad Politécnica de Cataluña. Avda. Dr. Marañón 50, 08028 Barcelona 3 Centro Catalán del Plástico. Universidad Politécnica de Cataluña, Colom 114, 08222 Terrassa. Resumen: En el desarrollo de materiales aislantes acústicos para sectores como la automoción, se utilizan habitualmente compuestos poliméricos con un contenido en cargas inorgánicas superior al 70% en peso. Este hecho obliga a utilizar matrices que mantengan en el compuesto la ductilidad necesaria para su termoconformado. En este trabajo se ha aplicado el diseño estadístico de mezclas al estudio del comportamiento térmico y mecánico de mezclas preparadas con dos grados de EVA de diferente contenido en acetato de vinilo y un plastómero de etileno y buteno. En particular, se ha estudiado la evolución de las entalpías y temperaturas de fusión y cristalización, así como la estabilidad térmica y el comportamiento mecánico de las mezclas en función de su composición. Palabras clave: EVA, DSC, TGA, diseño estadístico de mezclas, polímeros termoplásticos. 1. INTRODUCCIÓN En el sector de la automoción se utilizan diferentes estrategias para lograr un efectivo aislamiento acústico en los automóviles. El uso de láminas pesadas termoconformables de polímeros compuestos es una solución ampliamente utilizada por la industria automovilística para producir muchos componentes importantes del vehículo [1]. La mayoría de las fórmulas consisten en una matriz polimérica de elevada ductilidad como EPDM, EVA o LLDPE y altos porcentajes de cargas minerales como barita, carbonato de calcio, talco, etc [2,3]. Se ha comprobado que la incorporación de plastómeros a las formulaciones de EVA permite mejorar el alargamiento hasta rotura y aceptar mayores porcentajes de carga [4]. Por este motivo se han evaluado las propiedades de mezclas de un plastómero de etileno-buteno y dos grados de EVA con diferentes contenidos de acetato de vinilo (VA), 18 y 24 % pp. A fin de obtener la máxima información con el mínimo número de experimentos se ha utilizado el diseño de experimentos, en concreto el diseño de mezclas [5,6]. Esta herramienta permite asimismo identificar las posibles interacciones que existen entre los polímeros a estudio. 2. PROCEDIMENTO EXPERIMENTAL 2.1. Materiales y preparación de las mezclas Se han utilizado los siguientes polímeros: etileno-1buteno EXACT 48201 de DEX Plastomers (EB), EVA 18VA Alcudia PA-538 de Repsol YPF (EVA18) y EVA 24VA Alcudia PA-430 de Repsol YPF (EVA24). Además se ha evaluado el efecto de una carga mineral compuesta por mezclas de carbonato de calcio, sulfato de bario y polvo de acería [1]. En la Tabla 1 se muestran XI CONGRESO NACIONAL DE MATERIALES las referencias y relaciones de las distintas matrices y compuestos cargados estudiados, donde la carga mineral (CM) se expresa en % en peso de carga añadida a la matriz polimérica. Tabla 1. Referencia de las mezclas estudiadas. Referencia EVA18 EVA24 EB EVA18EVA24 EVA18-EB EVA24-EB EVA18EVA24-EB M18 MEB C18 CEB EVA 18 EVA 24 EB CM (%) 1 0 0 0 0 1 0 0 0 0 1 0 0,5 0,5 0 0 0,5 0 0 0,5 0,5 0,5 0 0 0,33 0,33 0,33 0 0,9 0,1 0,9 0,1 0 0 0 0 0,1 0,9 0,1 0,9 0 0 75 75 Los polímeros se han amasado a 150ºC en una mezcladora de palas y con una prensa de platos calientes se han obtenido placas de 3 mm de espesor. En el caso de los compuestos cargados (C18 y CEB) adicionalmente se añadió estearato de zinc (0.5%) como agente compactador y aceite mineral como aditivo de proceso. La caracterización de las propiedades térmicas de las distintas muestras se ha realizado por calorimetría diferencial de barrido (DSC) y análisis termogravimétrico (TGA). Las propiedades mecánicas 248 6. POLíMEROS de las matrices y sus mezclas se han determinado mediante ensayos de tracción a partir de probetas tipo IV troqueladas según norma ASTM D 648. 2.2. Propiedades térmicas Se han obtenido las temperaturas de fusión y cristalización, así como las entalpías de fusión por DSC, a una velocidad de calentamiento de 10ºC/min entre 0ºC y 120ºC, en atmósfera de N2 para todas las muestras preparadas. Se ha realizado un primer calentamiento para borrar la historia térmica de los materiales. El porcentaje de cristalinidad de las muestras se ha determinado a partir de la relación Hm/ Hm100, donde Hm es el calor de fusión en J/g de los segmentos de polietileno en las muestras de EVA y Hm100 es el calor de fusión en J/g de polietileno puro 100% cristalino, cuyo valor es de 281 J/g [78]. En el caso del plastómero se utilizado la misma relación considerando Hm como la entalpía del copolímero. Las entalpías de fusión se han obtenido a partir de la integración de los picos endotérmicos desde el inicio hasta el final de la fusión. Los análisis termogravimétricos se han llevado a cabo hasta 600ºC con una velocidad de calentamiento de 10ºC/min en atmósfera de N2, tanto para las mezclas de polímero como para los compuestos cargados. 2.3. Propiedades mecánicas Las propiedades mecánicas de las matrices se han determinado mediante ensayos de tracción en un dinamómetro Zwick a una velocidad de 500 mm/min, según norma ASTM D 648. Para cada muestra se ha obtenido el módulo secante al 1% de deformación (E1%), la tensión máxima a tracción (y) y la elongación a rotura (máx). Se ensayaron un mínimo de diez especímenes por muestra. 2.4. Diseño estadístico de mezclas El diseño estadístico de mezclas para tres componentes se ha desarrollado con el programa “Design Expert”. Se han obtenido las superficies de respuesta para cada una de las respuestas de las propiedades mecánicas estudiadas. 3. RESULTADOS Y DISCUSIÓN 3.1. Propiedades térmicas En la Tabla 2 se muestran los resultados obtenidos del estudio térmico por DSC. Como era de esperar, el porcentaje de cristalinidad de los polímeros EVA estudiados disminuye al aumentar el contenido en acetato de vinilo, ya que éste dificulta la cristalización de los grupos etilénicos. La fusión de los dominios cristalinos se produce a temperaturas inferiores para el EVA24 y en el caso del enfriamiento desde 120ºC se observa una cristalización más tardía, con aproximadamente 10ºC de diferencia respecto al EVA18 [9]. XI CONGRESO NACIONAL DE MATERIALES Tabla 2. Calores de fusión (Hf), Temperaturas de fusión (Tf) y cristalización (Tc), y porcentajes de cristalinidad (Xc) de las mezclas y compuestos cargados. Referencia EVA18 EVA24 EB EVA18EVA24 EVA18EB EBEVA24 EVA18EVA24EB M18 C18 MEB CEB Tf EVA (ºC) 88 79,6 -- Tf EB (ºC) --69,9 Tc EVA (ºC) 67,1 58,9 -- Tc EB (ºC) --53 82,2 -- 64,7 83,2 66,8 75,1 Hf (J/g) Xc (%) 47,9 32,4 39,8 20,7 15,2 14,2 -- 50,7 22,9 67,1 54,4 49,3 19,3 69,1 60,5 54,9 40,2 16,2 79,8 68,5 64,8 54,4 43,5 18,0 85,3 77,9 --- 63,9 65 64,3 61,2 67,2 61,7 66,4 -- --56,5 52,5 48,8 37,9 42,4 33,8 20,7 16,1 15,3 12,2 La Figura 1 muestra los resultados de la calorimetría diferencial de barrido de las matrices y de los compuestos cargados. Puede observarse que mientras los dos polímeros EVA son completamente miscibles y presentan una temperatura de cristalización intermedia, para el caso en que se emplea EB en un 50% en peso, éste no es miscible ni en EVA18 ni en EVA24. EB y EVA son polímeros termodinámicamente inmiscibles aunque mecánicamente miscibles [4]. El efecto de las cargas inorgánicas sobre la cristalización de los polímeros puede verse en la Figura 1b). Éstas retrasan la cinética de cristalización de los polímeros, que lo hacen a menor temperatura y en menor cantidad. Esto se debe a la gran cantidad de cargas inorgánicas introducidas (75%pp), que dispersadas en la matriz polimérica, lejos de facilitar la cristalización creando un efecto nucleante, la dificultan. Además las cargas tienen una superficie específica lo suficientemente alta para promover la adsorción de polímero sobre la partícula y disminuir la cristalización [1,10]. Este efecto es ligeramente más acentuado en el compuesto con mayor porcentaje de EVA en la matriz (C18). El carácter polar del EVA provoca el encapsulamiento de las partículas inorgánicas. En cambio para el compuesto CEB, en el cual predomina el carácter apolar del etileno-buteno, no se observa una disminución tan pronunciada del porcentaje de cristalinidad, lo cual podría relacionarse con una mayor dispersión de la carga mineral en la matriz [11]. Los resultados del estudio termogravimétrico de los polímeros puros y de las mezclas M18, MEB y EVA18EVA24-EB se muestran en la Figura 2a. La descomposición del etileno-buteno tiene lugar en una única etapa, entre 450-500ºC. Sin embargo, los dos grados de EVA, descomponen en dos etapas; la primera alrededor de 300-380ºC corresponde a la eliminación de los grupos de acetato de vinilo, y la segunda etapa se 249 6. POLíMEROS solapa con la descomposición del EB y se atribuye en ambos casos a la degradación de las cadenas etilénicas. conlleva una aceleración de la descomposición de los grupos de acetato de vinilo de los EVA y de la cadena etilénica de los polímeros. Flujo de Calor >> Endo 3.2. Propiedades mecánicas La Tabla 3 resume los resultados de las propiedades mecánicas de las mezclas de los polímeros. En la misma figura también se muestran las superficies de respuesta de las propiedades mecánicas en función de los polímeros a estudio. EVA18-EB EVA18-EVA24 EVA24-EB EVA18-EVA24-EB 10 Flujo de Calor >> Endo 0 20 30 40 50 60 Temperatura (ºC) 70 80 90 100 Tabla 3. Composición de las mezclas y resultados de las propiedades mecánicas. Donde max: tensión máxima, b: elongación a rotura, y Módulo: módulo secante al 1% de deformación. Referencia M18 C18 MEB CEB 0 10 20 30 40 50 60 Temperatura (ºC) 70 80 90 100 Figura 1. Resultado de los DSC de a) mezclas de polímeros y b) matrices poliméricas M18 y MEB, y sus respectivos compuestos acústicos cargados C18 y CEB. 100 % Pérdida de peso 80 60 40 20 0 230 EB EVA18 EVA24 EVA18-EVA24-EB M18 MEB 280 330 380 430 Temperatura (ºC) 480 530 580 % Pérdida de peso 100 M18 80 CEB 60 MEB 40 C18 20 0 230 280 330 380 430 Temperatura (ºC) 480 530 580 Figura 2. Análisis termogravimétrico de a) muestras poliméricas y b) polímeros y compuestos cargados. En la Figura 2b se muestran las descomposiciones de las matrices y de los correspondientes compuestos cargados. Respecto al comportamiento de los compuestos parece que la adición de cargas inorgánicas disminuye la estabilidad térmica del material [1]. El aceite mineral descompone alrededor de 300ºC, lo que XI CONGRESO NACIONAL DE MATERIALES EVA18 EVA24 EB EVA18EVA24 EVA18-EB EVA24-EB EVA18EVA24-EB 10,2 ± 0,5 10,7 ± 0,9 8,1 ± 0,9 Módulo (MPa) 904 ± 39 38,6 ± 1,8 1032 ± 98 21,6 ± 2,2 1118 ± 45 18,3 ± 1,2 12,2 ± 1,5 1066 ± 129 29,9 ± 1,6 9,0 ± 0,4 10,5 ± 0,5 974 ± 43 27,4 ± 1,4 1145 ± 48 20,6 ± 1,5 10,6 ± 1,5 1062 ± 29 25,8 ± 0,9 max (MPa) b (%) Para cada una de las tres respuestas estudiadas (tensión máxima, elongación a rotura y módulo secante al 1%), se obtienen sendos modelos matemáticos de segundo orden, que permiten por un lado predecir cuantitativamente la respuesta dentro del rango estudiado de las variables y por otro, a partir de unos requisitos el modelo propone diferentes formulaciones óptimas. Se han preparado diferentes mezclas para evaluar los modelos matemáticos propuestos. A partir de la Figura 3a se deduce que los valores de tensión máxima aumentan al introducir más porcentaje de EVA y disminuye a medida que se introduce EB en la matriz. Cabe destacar una interacción positiva significativa entre los dos polímeros EVA, esto se traduce en un aumento de la tensión máxima por parte de las mezclas EVA18-EVA24. Los dos polímeros presentan la misma naturaleza y son completamente miscibles, como puede verse en la los resultados térmicos. El modelo matemático se ilustra en la ecuación 1. Tensión máxima = +8.14090 * EB +10.21090 * EVA 18 +10.67590 * EVA24 + 6.59759 * EVA 18 * EVA24 (1) Modelo matemático para tensión máxima. En la Figura 3b se muestra la superficie de respuesta para elongación a rotura. A partir de ella se corrobora que introduciendo más cantidad de EVA24 y EB los valores de elongación aumentan, el comportamiento opuesto se obtiene al aumentar el porcentaje de EVA 18 en las formulaciones. Entre los dos EVA se repite una interacción positiva significativa que permitiría en 250 6. POLíMEROS algunos casos similares a EB. obtener resultados de elongación Elongación = + 1097.05364 * EB + 885.25364 * EVA 18 + 1008.25364 * EVA24 + 349.26897 * EB * EVA24 + 456.86897* EVA 18 * EVA24 (2) Modelo matemático para la elongación a rotura. 4. CONCLUSIONES Los dos grados de EVA dan lugar a mezclas totalmente miscibles, mientras que el etileno-buteno no es totalmente miscible en las mezclas con EVA estudiadas. En cuanto a la cristalinidad, el EB presenta un porcentaje menor que los dos grados de EVA. En ninguno de los casos se aprecia un efecto nucleante por parte de las cargas, probablemente debido al elevado porcentaje respecto a la matriz polimérica. El diseño de mezclas se ha revelado como una herramienta eficaz para detectar sinergias entre los polímeros de la mezcla. Se ha identificado una interacción positiva entre los dos EVA para la elongación y la tensión máxima. 5. AGRADECIMIENTOS Los autores agradecen al MICINN por su apoyo a través del proyecto FIS2009-13360-C03-03. 6. REFERENCIAS [1] Figura 3. Superficie de contorno de a) Tensión máxima (MPa) b) Elongación a rotura (%) y c) Módulo secante al 1% (MPa) en función de los polímeros a estudio. Donde A: EB, B: EVA 18 y C: EVA 24. La superficie de contorno para el módulo secante al 1% de deformación se resume en la Figura 3c. El resultado máximo para este parámetro se obtiene con EVA 18, los valores mínimos en cambio se obtienen introduciendo tanto EVA 24 como EB. No se aprecian interacciones significativas entre los polímeros a estudio. Módulo 1% = + 18.41253 * EB + 38.52253 * EVA 18 + 21.57253 * EVA24 (3) Modelo matemático para módulo secante al 1% de deformación. XI CONGRESO NACIONAL DE MATERIALES Niubó, M., Fernández, A.I, Chimenos, J.M., Haurie, L., J. “A Possible Recycling Method for High Grade Steels EAFD in Polymer Composites”, J. Hazardous Materials. 171 1139-1144, (2009). [2] ”Composite Material With Improved Structural, Acoustic and Thermal Properties”, Haque, E, Cheney T.L., Blinkhorn, A., U.S. Pat. Appl. (2005), US 2005115662 A1 20050602. [3] Polyon Barkai Industries, 1993 Ltd. Israel.” Isulation Monolithic Heavy Plastic Film Made of a MineralFilled Plastic”. Yanai, G., U.S. Pat. Appl. (2005), US 2005202213 A1 20050915. [4] Kontopoulou, M., Huang, L.C., “Rheology, Structure, and Properties of Ethylene-Vinyl Acetate/ Metallocene - Catalyzed Ethylene-α-Olefin Cop. Blends”, J. Applied Polymer Science, 94 881-889 (2004). [5] Bowles, M.L., Montgomery, D.C., “How to Formulate the Ultimate Margarita”, Quality Engineering, 239253 (1997). [6] Montgomery, D.C., “Design and Analysis of Experiments”,3rd ed., John Wiley & Sons, NY(1991). Shieh, Y.T., Lin, Y.G., Induced Crystallization of EVA [7] Having Various VA Contents by Compressed CO2, J. Applied Polymer Science, 87 1144-1151 (2003). [8] Brandrup, J., Immergut, E., Polymer Handbook, 3rd ed., John Wiley & Sons: New York, (1989). [9] Arsac, A., Carrot, C., Guillet, “Rheological Characterization of Ethylene Vinyl Acetate Copolymers”, J., J. Appied Polymer Science,74 2625-2630 (1999). [10] Janigová, I., Chodak, I., “Temperature Effect on Kinetics of Isothermal Crystallization of Crosslinked Filled LDPE-2. Particulate Silica With High Surface area as a Filler”, Eur. Pol. J.,(1995). [11] Premphet, K., Horanont, P., “Phase Structure of Ternary Polypropylene/Elastomers/Filler Composites: Effect of Elastomer Polarity”, Polymer, 9283-9290 (2000). 251 6. POLíMEROS PREPARACIÓN Y CARACTERIZACIÓN DE ESPUMAS MICROCELULARES NANOREFORZADAS V. Realinho, M. Antunes, O.O. Santana, M. Sánchez-Soto, J.I. Velasco Centre Català del Plàstic, Departament de Ciència dels Materials i Enginyeria Metal·lúrgica, Universitat Politècnica de Catalunya, C/ Colom 114, E-08222 Terrassa (Barcelona), España, Vera. [email protected] Resumen: Las espumas microcelulares poseen óptimas propiedades específicas que pueden ser ampliadas por incorporación de nanopartículas laminares y/o otros sustratos inorgánicos. Por ello, en el presente estudio se han preparado, por el método de espumación por disolución de gas y caída de presión, espumas microcelulares de polimetacrilato de metilo y respectivos nanocompuestos con montmorillonita e hidrotalcita. Además de la caracterización morfológica y termomecánica de las espumas se determinó la solubilidad y el coeficiente de difusión del CO2 en el PMMA, evaluando el efecto de las nanopartículas en dichas propiedades. Se ha podido constatar que la solubilidad del CO2 se incrementa con la presencia de las nanopartículas respecto a las del polímero puro y que el coeficiente de difusión se reduce marcadamente para ambos nanocompuestos. La rigidez específica de las espumas con nanopartículas es superior comparativamente con la del polímero puro debido a su estructura celular más fina y a la presencia de las nanopartículas. Palabras clave: Espumas microcelulares, nanocompuestos, montmorillonita, hidrotalcita. 1. INTRODUCCIÓN Importantes mercados como los de automoción, aeronáutica o de productos deportivos han ido introduciendo las espumas poliméricas en sus desarrollos y exigen a estos materiales trabajar bajo condiciones cada vez más exigentes. Una de las estrategias seguidas actualmente por varios investigadores es la de unir el conocimiento sobre nanocompuestos poliméricos con la del proceso de espumación [1]. La montmorillonita es una de las arcilla más empleadas como precursor de la formación de nanocompuestos en polímeros. Existen multitudes de estudios sobre los efectos de estas nanopartículas en la mejora de las propiedades en polímeros de distinta naturaleza: termoestables [2], termoplásticos [3] y de altas prestaciones [4]. Una familia de compuestos inorgánicos laminares mucho menos utilizados como precursores de nanocompuestos son los hidróxidos dobles laminares (LDHs), también llamados arcillas aniónicas por su capacidad de intercambiar aniones. Las investigaciones relacionadas con este tema se centran en evaluar la influencia de la adición de LDHs en la estabilidad térmica, en las propiedades de barrera frente a gases y en las mecánicas [5-7] La incorporación de nanopartículas en la preparación de espumas poliméricas puede promover el aumento de la densidad celular y reducir el tamaño de celda debido a su acción nucleante. Este aspecto es particularmente importante en la producción de espumas microcelulares y nanocelulares (tamaño de celda entre 1 y 100 µm y 0.1-100 nm, respectivamente) donde se requiere potenciar al máximo la nucleación heterogénea del material [8]. Adicionalmente, estas nanopartículas XI CONGRESO NACIONAL DE MATERIALES laminares con elevada rigidez y relación de aspecto pueden conllevar a una mejora significativa en las propiedades mecánicas finales de la espuma, en particular en su rigidez, resistencia y tenacidad, debido a su confinamiento en las delgadas paredes de las espumas. En los últimos años ha aumentado considerablemente el interés en utilizar procesos físicos de espumación en estado sólido o semisólido para la preparación de espumas poliméricas microcelulares [9-11]. Comparativamente a la espumación física por extrusión o inyección [12-13], aquellos se caracterizan por expandir el polímero cerca de la transición vítrea de la mezcla polímero-gas. Estas condiciones limitan la coalescencia celular debido a una elevada resistencia del material en tales condiciones de espumación, permitiendo obtener espumas con una estructura microcelular quasi-isotrópica. Se suele emplear un proceso en dos etapas, donde el gas en condiciones supercríticas es inicialmente disuelto en el polímero a elevada presión durante tiempos largos y posteriormente espumado por aumento de la temperatura a baja presión. El propósito de este trabajo ha sido preparar y caracterizar espumas microcelulares de PMMA con nanopartículas laminares de cara a poder evaluar su potencial como materiales ligeros con propiedades mejoradas. 2. MATERIALES Para preparar los precursores sólidos de las espumas nanoreforzadas se ha usado un PMMA comercial (Altuglas V 825 T), suministrado por Arkema Group Altuglas, de MFI = 2.8 dg/min determinado a 230 ºC y 3.8 kg. Como refuerzo se han empleado dos tipos de arcillas: una montmorillonita comercial orgánicamente modificada con cationes diestearildimetilamonio, 252 6. POLíMEROS [N(CH3)2(C18)2]+ (Nanofil SE 3000) suministrada por Sud-Chemie y denominada en este trabajo MMT; y una hidrotalcita comercial, suministrada por Sud-Chemie (Syntal HSA 696) posteriormente modificada por intercambio iónico con aniones dodecilsulfato sódico (C12H25O4SNa), denominada HT. 3. PROCEDIMIENTO EXPERIMENTAL Mediante el método de mezclado en estado fundido se prepararon dos nanocompuestos de PMMA, uno con un 5% en peso de MMT (PMMA-5MMT) y otro con 5% en peso de HT (PMMA-5HT). Para ello se empleó una extrusora de doble tornillo corrotatoria (Collin ZK36) con un diámetro de tornillo de 25 mm y una relación L/D = 36. Se usó un perfil de temperaturas entre 140 y 200 ºC y una velocidad de husillos de 110 rpm. El PMMA puro también fue procesado utilizando las mismas condiciones de modo a obtener una historia térmica idéntica. En la fig. 1 se presenta una micrografía típica de las nanopartículas dispersas en la matriz polimérica. Se mecanizaron probetas directamente a partir de las espumas obtenidas, con el cuidado de eliminar las pieles sólidas generadas durante el proceso de espumación, para posterior caracterización. 1ª Etapa (saturación): CO2 Temperatura y tiempo Precursor sólido Autoclave 2ª Etapa (espumación): ∆P/∆t Sólido saturado con CO2 Sólido saturado con CO2 Espuma Temperatura y tiempo Baño atemperado Figura 2. Esquema del proceso de espumación por disolución de CO2. La densidad de los nanocompuestos sólidos y espumados fue medida de acuerdo con la norma ISO 845. Para la determinación de la solubilidad y del coeficiente de difusión se realizaron medidas de la evolución de la pérdida de peso con el tiempo (curvas de desorción de CO2). Para ello, se usó una balanza analítica Mettler Toledo PB 303 con una precisión de + 1 mg. Figura 1. Micrografía obtenida por microscopía electrónica de barrido mostrando las nanopartículas dispersas en la matriz polimérica. Las probetas utilizadas en la caracterización de los sólidos fueron obtenidas a partir de discos circulares (D = 75 mm) con un espesor igual a 3.5 mm, preparadas por moldeo por compresión usando una prensa de platos calientes IQAP-LAP PL-15. Las espumas fueron preparadas por un proceso de espumación física por disolución de CO2 en estado semi-sólido por control de caída de presión. Para ello, se saturaron inicialmente en una primera etapa los precursores sólidos con CO2 en un autoclave a 200 bar y 70 ºC durante 2h, aplicando posteriormente una súbita caída de presión (∆P/∆t ). Estos sólidos saturados se acondicionaron a temperatura ambiente durante 30 min, y posteriormente, en una segunda etapa, se colocaron en un baño atemperado a 50 ºC para su espumación. En la fig. 2 se presenta un esquema simplificado de dicho proceso. En la espumación se utilizaron como precursores sólidos probetas circulares (D = 49.5 mm) mecanizadas a partir de placas cuadradas (100 x 100 mm2) con 1.6 mm de espesor, obtenidas igualmente por compresión. XI CONGRESO NACIONAL DE MATERIALES La estructura celular de las espumas fue estudiada por microscopia electrónica de barrido utilizando un microscopio JEOL JSM-5610. Las muestras fueron preparadas por fractura a baja temperatura y posterior recubrimiento con una fina capa de oro. Para estudiar el comportamiento viscoelástico de los materiales preparados se empleó el análisis térmico mecánico dinámico (DMTA). El equipo de DMTA (TA Instruments Q 800) fue calibrado de acuerdo con el procedimiento normalizado del equipo. El módulo de almacenamiento (E’), módulo de pérdidas (E’’) y factor de pérdidas (tan δ) fueron obtenidos en una geometría de single cantilever (modo empotrado simples) con una distancia entre apoyos de 17.5 mm. Los ensayos fueron realizados entre 30 y 170 ºC a 5 ºC/min y 10 Hz por control de deformación (0.02 %). Se utilizaron probetas prismáticas mecanizadas con unas dimensiones nominales de 35 x 12 x 1.5 mm. 4. RESULTADOS Y DISCUSIÓN 4.1. Curvas de desorción Para poder comparar la influencia que tienen las partículas laminares en la solubilidad del CO2 en el PMMA y posterior comportamiento de desorción, se realizaron medidas de la masa de gas presente en la muestra y su evolución con el tiempo (curvas de desorción). En la fig. 3 se presentan las curvas de desorción de los tres materiales analizados. 253 6. POLíMEROS La solubilidad del CO2 en los materiales (M0) fue determinada a partir del método de la pendiente inicial [14], extrapolando la concentración de CO2 a tiempo cero a partir de los datos iniciales de desorción (datos de pérdida de masa registrados en los primeros 7 min). Corrigiendo los valores en base a la cantidad de polímero presente en cada material, se han obtenido los siguientes valores para M0: 114.8, 173.2 y 155.1 mg de CO2/g de PMMA respectivamente para el PMMA puro, PMMA-5MMT y PMMA-5HT. El coeficiente de difusión (D) del CO2 en los tres materiales fue determinado asumiendo difusión unidimensional en placa plana [15]: (1) Donde, Mt es la concentración de CO2 en la muestra medida a tiempo t, M0 es la solubilidad (concentración de CO2 extrapolada a tiempo 0) y l es el espesor de la muestra. Se registró una reducción del valor de D del PMMA en un 40 y 43% al incorporar nanopartículas de HT y MMT respectivamente. Tabla 1. Densidad de los sólidos y espumas. Materiales PMMA PMMA-5MMT PMMA-5HT a ER=ρespuma/ρsólida ρ (g/cm3) Sólido Espuma 1.177 0.824 1.181 0.866 1.192 0.860 ERa 1.4 1.4 1.4 La presencia de una pequeña cantidad de nanopartículas laminares parece actuar como agente nucleante durante el proceso de espumación del PMMA, una vez que el tamaño de celda disminuye significativamente en este material (tamaño promedio de celda < 100 nm). En la fig. 4 se presentan micrografías típicas del PMMA puro con y sin partículas, donde se puede apreciar la diferencia de tamaño de celda (espuma microcelular frente a espuma nanocelular). Espuma microcelular Espuma nanocelular Figura 3. Curvas de desorción de los materiales analizados. Comparando las curvas de desorción se puede constatar, por una parte, que el nanocompuesto con hidrotalcita retiene mejor el CO2 en los primeros minutos del proceso de desorción de CO2 en comparación con el de MMT. Este fenómeno se puede atribuir a la bien conocida afinidad química entre los hidróxidos dobles laminares y los aniones carbonato [16]. Por otra parte, a partir de un cierto tiempo la desorción de CO2 en el nanocompuesto con MMT se iguala a la del PMMA puro, mientras que en el nanocompuesto con HT es más lenta. Estos resultados son indicativos de un mejor comportamiento de espumación del nanocompuesto de HT, ya que no solo presenta una caída más suave de pérdida de CO2 durante las primeras etapas como lo retiene a tiempos más largos. 4.2. Estructura celular En la siguiente tabla se presentan los valores de densidad de los nanocompuestos sólidos y respectivas espumas: XI CONGRESO NACIONAL DE MATERIALES Figura 4. Micrografías de muestras espumadas sin partículas (x 100) y con nanopartículas (x 5000). 4.3. Comportamiento dinámico mecánico. En la fig. 5 se presentan las curvas obtenidas por DMTA para los materiales sólidos. Se puede constatar que la incorporación de una pequeña cantidad de nanopartículas laminares disminuye la temperatura de transición vítrea (Tg) del PMMA. Esta tendencia puede ser debida a un efecto plastificante inducido por la presencia de las nanopartículas laminares orgánicamente modificadas en el seno de la matriz polimérica. Esta observación es coherente con el aumento de la solubilidad de CO2 en estos materiales durante el proceso de saturación. 254 6. POLíMEROS 4000 3500 (a) 140.7 ºC 3000 E' (MPa) 500 60 80 100 ºººº 40 T ( C) 120 140 160 (b) 3000 E' (MPa) 40 60 80 100 ºººº 3500 T ( C) 120 140 160 (c) 3000 133.2 ºC 300 1,75 E' (MPa) 2,00 300 1,75 80 100 T ( C) 120 140 160 1,50 1,25 tan δ 60 ºººº 40 0,50 350 100 500 0,75 0,00 150 1000 1,00 0,25 200 1500 1,25 0 250 2000 1,50 50 E'' (MPa) 2500 0 20 2,00 tan δ 500 4000 350 100 1000 0,50 0,00 150 1500 0,75 0 200 2000 1,00 0,25 E'' (MPa) 136.4 ºC 1,25 50 250 2500 1,50 tan δ E'' (MPa) 100 1000 0 20 1,75 150 1500 3500 300 200 2000 4000 2,00 250 2500 0 20 350 1,00 0,75 0,50 50 0,25 0 0,00 Figura 5. Evolución de E’, E’’ y tan δ con la temperatura para los materiales sólidos. (a) PMMA; (b) PMMA-5MMT; (c) PMMA-5HT. En la tabla 2 se presentan los valores del módulo de almacenamiento medido a 30 ºC, así como los respectivos valores específicos (E’esp = E’/ρespuma). Cabe destacar el aumento de los valores específicos de rigidez de las espumas al incorporar las nanopartículas. Este aumento es debido a la combinación de una estructura celular más fina junto con la presencia de las nanopartículas laminares rígidas, especialmente en el caso de la MMT. Tabla 2. Valores del módulo (E’) y respectivo valor específico (E’esp) de los sólidos y respectivas espumas. Materiales PMMA PMMA-5MMT PMMA-5HT E’ (MPa) Sólido 3390 3523 3132 Espuma 1640 2188 1720 E’esp (MPa cm3/g) Sólido 2880 2983 2628 Espuma 1991 2528 2001 5. CONCLUSIONES La incorporación de nanopartículas laminares ha tenido un efecto positivo en la nucleación de celdas durante el proceso de espumación y en la disminución del tamaño celular. XI CONGRESO NACIONAL DE MATERIALES A igualdad de condiciones de saturación, la solubilidad del CO2 en los nanocompuestos es mayor que en el PMMA. Este aumento puede ser justificado por la disminución de la temperatura de transición vítrea en los sólidos por la presencia de las nanopartículas laminares. El coeficiente de difusión del CO2 disminuye con la presencia de las nanopartículas, especialmente en el caso del nanocompuesto con hidrotalcita debido a su mayor afinidad con el CO2. Las espumas de los nanocompuestos presentan una mayor rigidez específica debido al efecto combinado de una estructura celular más fina (espumas con tamaños de celda < 100 nm - espumas nanocelulares) junto con la presencia de las nanopartículas rígidas. Por tanto, se ha demostrado que es posible preparar espumas rígidas micro y nanocelulares a base de nanocompuestos de PMMA con propiedades mejoradas para aplicaciones donde se requiera una elevada rigidez específica. 6. AGRADECIMIENTOS Los autores desean agradecer a la CICYT la ayuda económica prestada a través del proyecto MAT200762956. 7. REFERENCIAS [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] Lee, L. J., Zeng, C., Cao, X., Han, X., Shen, J., Xu, G., Comp. Sci. Tech., 65 (2005) 2344-2363. Lan, T., Kaviratna, P., Pinnavaia, T., Chem. Mater., 11 (1995) 2144-2150. Hasegawa, N., Okamoto, H., Kawasumi, M., Usuki, A., 14 (1999) 3359-3364. Tyan, H., Wu, C., Wei, K., J. Appl. Polym. Sci., 7 (2001) 1742-1747. Kuila, T., Acharya, H., Srivastava, S. K., Bhowmick, A. K., J. Appl. Polym. Sci., 3 (2007) 1845-1851. Du, L. C., Qu, B. J., Polym. Comp., 2 (2007) 131-138. Chen, W., Qu, B. J., Polym. Degrad. Stab., 1 (2005) 162-166. McClurg, R. B., Chem. Eng. Sci., 59 (2004) 5779-5786. Kumar, V., Prog. Rub. Plast. Tech. 9 (1993) 54. Goel, S. K., Beckman, E. J., Cell. Polym. 12 (1993), 251. Zeng, C., Han, ,X., Lee, L. J., Koelling, K. W., Tomasko, D. L., Adv. Mater., 15 (2003), 1743. Zheng, W., Lee, Y. H., Park, C. B., J. Cell. Plast. 42 (2006) 271. Kumar, V., Nadella, K. V., “Microcellular foams. In: Handbook of Polymer Foams”, D. Eaves Ed., Rapra Technology, UK , 2004. Nawaby, A. V., Zhang, Z., “Solubility and diffusivity in thermoplastic foam processing”, Ed. Lee, S. T., CRC Press, London, 2005. Crank, J., “The mathematics of diffusion”, Oxford University Press, London, 1956. Rives, V., “Layered Double Hydroxides: present and future”, Nova Science Publishers, New York, 2001 255 6. POLíMEROS PREPARACIÓN Y CARACTERIZACIÓN DE ESPUMAS MULTIFUNCIONALES DE POLIPROPILENO M. Antunes, V. Realinho, A.B. Martínez, M.Ll. Maspoch, J.I. Velasco Centre Català del Plàstic, Departament de Ciència dels Materials i Enginyeria Metal-lúrgica, Universitat Politècnica de Catalunya, C/Colom 114, E-08222 Terrassa (Barcelona), España, [email protected] Resumen: La combinación de la espumación junto con refuerzos funcionales permite ampliar el rango de propiedades de los sólidos, añadiendo a una elevada rigidez específica la regulación de otras características como las propiedades de transporte. Aunque las espumas poliolefínicas se encuentren extendidas, las ventajas de emplear PP en lugar de PE sólo empiezan a ser importantes para densidades > 100 kg/m3. Por ello, en este trabajo se presenta el estudio de la influencia de la espumación e incorporación de varios refuerzos en las propiedades de espumas de PP de densidad media-alta. Se muestra que la presencia de nanopartículas de montmorillonita induce estructuras celulares más isotrópicas, mejorando su comportamiento mecánico, pero sin alterar la conductividad térmica [1-2]. La incorporación de elevados porcentajes de carga mineral resulta en espumas rígidas con marcadas anisotropías celulares y térmicas, e interesantes propiedades de resistencia al fuego [3-4]. También pueden obtenerse espumas conductoras eléctricas por incorporación de nanofibras de carbono con posibilidades en aplicaciones como el pintado electrostático [5]. Palabras clave: Espumas multifuncionales, polipropileno, montmorillonita, nanofibras de carbono. 1. INTRODUCCIÓN Además de sus aplicaciones tradicionales para el envasado y embalaje o aislamiento térmico, importantes mercados como la automoción o la aeronáutica han considerado las espumas poliméricas en sus desarrollos, exigiendo a estos materiales trabajar bajo condiciones cada vez más severas. Esta razón explica la necesidad de llevar a cabo investigaciones en este campo que han de permitir optimizar los materiales y/o aprovechar mejor sus propiedades. La espumación permite ampliar el rango de valores de los materiales, creando aplicaciones que no pueden cubrirse con el uso de los respectivos sólidos continuos. Las reducidas densidades permiten el diseño de estructuras rígidas y resistentes de bajo peso, como son los paneles sándwich. Asimismo, su baja conductividad térmica permite la producción de elementos ligeros para aislamiento térmico. Las espumas flexibles son materiales ideales para aplicaciones donde resulta fundamental el confort y las rígidas pueden emplearse casi como sólidos compactos. Para la mayoría de aplicaciones estructurales, el plástico más adecuado por su elevada rigidez, resistencia térmica, facilidad de procesamiento y bajo coste, es el polipropileno (PP). Sin embargo, debido a la linealidad de sus cadenas y por ende reducida resistencia en fundido, la espumación del PP resulta sumamente difícil [6]. En los últimos años se han desarrollado nuevos grados comerciales de PP pensados para aplicaciones de espumación, designados polipropilenos de alta resistencia en fundido (PP-HMS), basados en la colocación de ramificaciones a lo largo de la cadena del PP. Estas ramificaciones incrementan su resistencia en fundido, limitando la posible coalescencia y colapso celulares, al mismo tiempo manteniendo su reprocesado y reciclabilidad [7-10]. Otra estrategia considera la incorporación de nanopartículas [11-12]. Con la XI CONGRESO NACIONAL DE MATERIALES espumación se consigue ampliar el rango de propiedades de los materiales por su extremo inferior, mientras que la incorporación de refuerzos o nanorefuerzos lo hacen por su extremo superior. Esta extensión en el rango de propiedades permite aplicaciones para ambos tipos de materiales que no pueden cubrirse con los respectivos sólidos continuos. Combinando ambas vías se pueden conseguir compuestos de PP con una adecuada procesabilidad para preparar espumas rígidas no entrecruzadas para aplicaciones estructurales (espumas multifuncionales). Las espumas multifuncionales son aquellas que, por su particular conjunto de propiedades, pueden ejercer simultáneamente una función estructural junto a otras de índole no mecánica, como puede ser la regulación de propiedades de transporte como la conductividad térmica, la conductividad eléctrica o la resistencia frente al fuego, entre otras. La funcionalidad que pueden aportar a las espumas poliméricas determinadas partículas o fibras, ya sean de dimensión micro o nanométrica, proviene de sus inherentes propiedades fisicoquímicas, de su confinamiento en las paredes celulares y de la microestructura desarrollada en la espuma. En este trabajo se investigan las propiedades que presentan nuevas espumas rígidas de densidad mediaalta (entre 0.1 y 0.6 g/cm3) constituidas por compuestos y nanocompuestos a base de mezclas de PP lineal y ramificado y diferentes refuerzos (montmorillonita, nanofibras de carbono e hidróxido de magnesio), en relación con el proceso de espumación, composición y su microestructura y estructura celular, y se comparan con los comportamientos respectivos del PP espumado sin partículas y de sus compuestos sólidos de referencia, a fin de establecer modelos útiles para su empleo en aplicaciones estructurales. 256 6. POLíMEROS 2. MATERIALES 2.1. Preparación de los nanocompuestos y precursores Los diversos compuestos y nanocompuestos fueron preparados por mezclado en fundido en una extrusora de doble husillo Collin Kneter 25X36D (L/D = 36) a 160 ºC y 160 rpm a partir de una matriz polimérica a base de una mezcla de 50 phr (partes por cien de polímero) de un PP-HMS y 50 phr de un PP lineal con 0.2 phr de ácido esteárico, 1.0 phr de talco y dos porcentajes de azodicarbonamida (ADC), un agente espumante químico: 1.5 y 3.5 phr. En el caso de los nanocompuestos con montmorillonita, ésta fue incorporada a la mezcla anterior en forma de un masterbatch comercial (Nanomer C32P) en la extrusora para obtener al final un nanocompuesto con un 5.0 phr de MMT modificada (PP-MMT). Los nanocompuestos de PP con nanofibras de carbono (PP-CNF) fueron preparados en la extrusora de doble husillo a partir de la mezcla indicada previamente añadiendo porcentajes variables de nanofibras de carbono (entre 5 y 20% en peso). Por último, los compuestos de PP con hidróxido de magnesio fueron preparados añadiendo in-situ a la extrusora el hidróxido de magnesio (Magnifin H-5 KV de Martinswerk) en dos porcentajes: 50 y 70% en peso (respectivamente PP-50Mg y PP-70Mg). Posteriormente al enfriamiento de los perfiles extruídos en una bañera de agua y granceado en línea, se prepararon los precursores sólidos de las espumas por compresión en una prensa de platos calientes IQAP LAP PL-15. Para ello, se coloca una cantidad de material considerada suficiente para rellenar la cavidad de un molde circular de 74 mm de diámetro y 3.5 mm de espesor y se procede a su moldeo a una temperatura de 170 ºC y presión de 14 bar. Al finalizar la etapa de moldeo los discos son enfriados bajo presión (14 bar). 2.2. Espumación química por compresión Preparados los precursores sólidos de todos los compuestos y nanocompuestos a base de PP, éstos son espumados por un proceso de espumación química por compresión en la misma prensa de platos calientes. Para el efecto, se colocan los precursores en un molde circular con un diámetro de 74 mm y se calienta entre 180 y 195 ºC aplicando una presión constante de 40 bar durante 15 a 25 min. Al final de la etapa de calentamiento se procede a la espumación del material por descomposición térmica del agente espumante químico y liberación brusca de la presión. 3. PROCEDIMIENTO EXPERIMENTAL La densidad de los compuestos y nanocompuestos celulares y respectivos sólidos fue determinada de acuerdo con la norma ISO 845. La estructura celular de las espumas fue analizada empleando un microscopio electrónico de barrido JEOL JSM-5610 a partir de muestras previamente fracturadas en frío y hechas conductoras por deposición de una capa de oro. A partir de las micrografías obtenidas fue posible determinar por el método de las intersecciones [13] los principales parámetros de caracterización celular: XI CONGRESO NACIONAL DE MATERIALES tamaño de celda (φ), densidad celular y grado de anisotropía (AR), este último definido como el cociente entre el tamaño de celda en la dirección de liberación de la presión (φVD) y el tamaño de celda en la dirección perpendicular (φWD). Las características cristalinas de la matriz polimérica fueron estudiadas por difracción de rayos-X empleando un difractómetro Bruker D8, con radiación CuKα (λ = 0.154 nm) operando a 45 kV y 40 mA. El barrido fue realizado de 1 a 60º con un paso de 0.033º y 0.06 s. La conductividad eléctrica de los sólidos y espumas de PP con nanofibras de carbono fue medida a temperatura ambiente entre 10-2 y 106 Hz empleando un medidor de impedancias Novocontrol HP 4192 A LF. Se emplearon muestras con un espesor de 130 µm y 1.5 mm respectivamente para los sólidos y espumas, depositando sobre su superficie un recubrimiento de oro para garantizar un contacto eléctrico óptimo con el electrodo superior del medidor. El comportamiento frente al fuego de los compuestos sólidos y espumas de PP con hidróxido de magnesio fue estudiado empleando el ensayo de epirradiador (UNE 23725-90) y midiendo el índice de oxígeno límite, LOI (ISO 4589). 4. RESULTADOS Y DISCUSIÓN 4.1. Microestructura y estructura celular La incorporación de refuerzos nanométricos, tanto montmorillonita como nanofibras de carbono, resulta en estructuras celulares más isotrópicas (valores del grado de anisotropía cercanos a uno) y de menor tamaño de celda para grados de expansión similares comparativamente con las espumas sin refuerzos, resultado directo del efecto nucleante de celdas por parte de ambos refuerzos (véase fig. 1). La presencia tanto de la montmorillonita como de las nanofibras de carbono resulta en la eliminación de la orientación preferencial del cristal α-monoclínico del PP observada por difracción de rayos-X inicialmente tanto en el precursor sólido sin partículas como en menor medida en las respectivas espumas. En el caso de las espumas con hidróxido de magnesio se obtienen estructuras celulares desde las altamente isotrópicas y de reducidos tamaños de celda de las espumas con 70% de Mg(OH)2 (fig. 1c) hasta las cada vez más orientadas en la dirección de crecimiento al incrementar la fracción de gas para las espumas con 50% de hidróxido (fig. 1d). Debido a la creciente anisotropía celular al reducir la densidad relativa, las espumas con 50% de Mg(OH)2 presentan anisotropías crecientes tanto de las partículas de hidróxido como de los cristales de polímero, con una marcada orientación perpendicular a la superficie, alcanzando valores de anisotropía de hasta un 60%. Las espumas con 70% de Mg(OH)2, a pesar de presentar una orientación preferencial de las partículas de hidróxido perpendicular a la superficie, presentan valores de la orientación de la fase cristalina prácticamente idénticos a los del respectivo sólido. 257 6. POLíMEROS (a) (b) VD (c) VD WD VD (d) VD WD WD WD Figura 1. Micrografías obtenidas por microscopía electrónica de barrido para las espumas de (a) PP-CNF, (b) PP-MMT, (c) PP-70Mg y (d) PP-50Mg. 4.2. Propiedades de transporte Aunque para porcentajes de nanofibras de carbono inferiores al 5% en peso las propiedades eléctricas de los compuestos y respectivas espumas resulten controladas por el carácter aislante eléctrico de la matriz polimérica, a porcentajes superiores se observa un comportamiento de conducción eléctrica por efecto de túnel [14-15], tal como se aprecia en la fig. 3. Las espumas presentan un valor de concentración crítica de nanofibras (φc) para conducción eléctrica inferior al de los sólidos y un comportamiento más parecido al de un sistema formado por una red conductora de nanofibras dispuestas al azar, asociada a una superior aglomeración de las nanofibras en los sólidos y superior dispersión de las mismas en las espumas. 0 Partículas esféricas Fibras dispuestas al azar Sólidos Espumas -3 -6 -1 log [σdc/(S.cm )] Como se puede apreciar por los valores de la conductividad térmica normalizada con la del respectivo sólido de referencia presentados en la fig. 2, la conductividad de las espumas de PP con y sin refuerzos viene controlada por la densidad relativa de la espuma, subiendo al incrementar ésta, siendo su valor poco dependiente de la estructura celular. estructura celular desarrollada durante la espumación afecta a la morfología de los compuestos y por ello al comportamiento térmico de los mismos. En particular, la estructura celular anisotrópica de las espumas con 50% de hidróxido tuvo consecuencias en la generación de una orientación preferencial de las partículas de hidróxido perpendicular a la superficie, induciendo una superior conductividad térmica en esa dirección. Conductividad térmica normalizada 1.00 0.80 PP PP-MMT PP-50Mg PP-70Mg -12 Comportamiento eléctrico controlado por el PP -15 0.60 0 0.40 φc (sólidos) = 12% φc (espumas) = 10% 5 10 φCNF (% en peso) 15 20 Figura 3. Conductividad eléctrica en continuo (σdc) vs. concentración de nanofibras de carbono (φCNF). 0.20 0.00 0.0 -9 0.2 0.4 0.6 Densidad relativa 0.8 1.0 Figura 2. Conductividad térmica normalizada vs. densidad relativa para las espumas de PP, PP-MMT, PP-50Mg y PP-70Mg. La incorporación de nanopartículas de MMT no afecta a la conductividad térmica del sólido y por ende al valor de la conductividad de las espumas, resultando prácticamente idéntico al de las respectivas espumas sin refuerzos. En lo que toca a las espumas con Mg(OH)2 destacan los altos valores absolutos de conductividad comparados con las conductividades de las espumas de PP de similar densidad relativa. Las espumas de 50% de Mg(OH)2 presentan valores de conductividad térmica de aproximadamente el doble que las respectivas espumas de PP y las espumas de 70% de hidróxido una conductividad de aproximadamente el doble que la conductividad del PP sólido. Pese a que las medidas globales de conductividad de las espumas sigue la tendencia típica para espumas poliméricas de media-alta densidad, la XI CONGRESO NACIONAL DE MATERIALES En las espumas con hidróxido de magnesio se observa la lógica subida de los valores de LOI al incrementar el porcentaje de hidróxido (fig. 4). La incorporación de un 50% de Mg(OH)2 resulta en un incremento marginal de este valor, tanto el sólido como las espumas presentando valores ligeramente superiores a los típicamente observados para el PP. Por el contrario, los compuestos con el 70% de hidróxido presentan valores de LOI mucho más elevados, con incrementos superiores al 50% cuando comparados con los compuestos del 50%. En particular, las espumas de los compuestos con el 70% de Mg(OH)2 presentan un LOI de 36.0%, superior al del sólido respectivo e incluso que otros sistemas similares altamente cargados [16], relacionado con la combinación de un elevado contenido en carga mineral y la estructura celular, que actúa como una capa protectora intumescente incrementando la autoextinción de la llama, como se puede apreciar por los menores valores de los tiempos de combustión, y consistencia de los residuos formados. 258 6. POLíMEROS 40 35 LOI (%) 25 20 60 15 40 10 20 5 0.2 0.4 0.6 Densidad relativa 0.8 1.0 LOI Tiempo medio de combustión 0 60 30 50 25 40 20 LOIPP = 17.8% 30 15 20 10 10 5 0 0.0 0.2 0.4 0.6 Densidad relativa 0.8 1.0 Tiempo medio de combustión (s) 80 40 35 Tiempo medio de combustión (s) 100 30 0 (b) 120 LOI Tiempo medio de combustión LOI (%) (a) 0 Figura 4. Índice de oxígeno límite (LOI) y tiempo medio de combustión para los sólidos y espumas de PP con (a) 50% y (b) 70% de Mg(OH)2. 5. CONCLUSIONES La incorporación de montmorillonita y nanofibras de carbono a una matriz polimérica de PP y posterior espumación resulta en espumas con estructuras celulares más isotrópicas y con superiores densidades celulares que las espumas sin refuerzos, demostrando el efecto nucleante de celdas de ambos nanorefuerzos. La espumación elimina la orientación preferencial del cristal α-monoclínico del PP paralela a la superficie. Por oposición, y debido a su creciente anisotropía celular al espumar, las espumas de PP con 50% de hidróxido presentan anisotropías microestructurales que resultan en espumas con conductividades térmicas más elevadas en esa dirección. Aún así, la conductividad térmica de las espumas depende sobre todo de su densidad relativa, subiendo al incrementar ésta, y es poco dependiente de la estructura celular. En el caso particular de la montmorillonita, no se observan cambios apreciables en la conductividad de las espumas comparativamente con las respectivas sin refuerzos. Las espumas con nanofibras de carbono presentan un comportamiento de conducción eléctrica por efecto de túnel a menores valores críticos de concentración de nanofibras comparativamente con los sólidos, además de un comportamiento más parecido al de un sistema formado por fibras conductoras distribuidas al azar, demostrando la eficacia de la espumación en la consecución de una red conductora a menores porcentajes de nanofibras. Las espumas de los compuestos con el 70% de Mg(OH)2 presentan un LOI superior al del sólido respectivo, indicando las posibilidades de estos materiales en aplicaciones donde se requiera la combinación de ligereza y de buenas propiedades de resistencia al fuego. 6. AGRADECIMIENTOS Agradecemos a la CICYT la ayuda económica prestada a través del Proyecto MAT2007-62956, y a los profesores M.A. Rodríguez Pérez (UVA) y M. Mudarra (UPC) su inestimable colaboración con las XI CONGRESO NACIONAL DE MATERIALES medidas de conductividad respectivamente. térmica y eléctrica 7. REFERENCIAS [1] [2] [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] Antunes, M., Velasco, J. I., Realinho, V., Solórzano, E., Polym. Eng. Sci. 49 (2009) 24002413. Antunes, M., Velasco, J. I., Realinho, V., Martínez, A. B., Rodríguez-Pérez, M. A., de Saja, J. A., Adv. Eng. Mater. 11 (2009) 811-817. Antunes, M., Haurie, L., Velasco, J. I., “Characterization of highly filled magnesium hydroxide-polypropylene composite foams”, J. Cell. Plast. (2010), accepted. Antunes, M., Realinho, V., Martínez, A. B., Solórzano, E., Rodríguez-Pérez, M. A., Velasco, J. I., Def. Diff. Forum 297-301 (2010) 990-995. Antunes, M., Velasco, J. I., Realinho, V., Arencón, D., J. Nanosci. Nanotech. 10 (2010) 1241-1250. Park, C. B., Cheung, L. K., Polym. Eng. Sci. 37 (1997) 1-10. Park, J. J., Katz, L., Gaylord, N. G. U.S. Patent 5,149,579 (1992). Park, J. J., Katz, L. Gaylord, N. G. U.S. Patent 5,116,881 (1991). Philips, E. M., McHugh, K. E., Bradley, M. B., Kunststoffe 82 (1992) 671-676. Stange, J., Münstedt, H., J. Rheo. 50 (2006) 907923. Bhattacharya, S., Gupta, R. K., Jollands, M., Bhattacharya, S. N., Polym. Eng. Sci. 49 (2009) 2070-2084. Lee, L. J., Zeng, C., Cao, X., Han, X., Shen, J., Xu, G., Comp. Sci. Tech. 65 (2005) 2344-2363. Sims, G. L. A., Khunniteekool, C., Cell. Polym. 13 (1994) 137-146. Sichel, E. K. “Carbon Black Polymer Composites”, Ed. M. Dekker, New York, 1982. Ryvkina, N., Tchmutina, I., Vilcakova, J., Peliskova, M., Saha, P., Synth. Met. 148 (2005) 141-146. Chiu, S.-H., Wang, W.-K., J. Appl. Polym. Sci. 67 (1998) 989-995. 259 6. POLíMEROS ANÁLISIS ESTRUCTURAL DE ESPUMAS AISLANTES PUR y POLIISOCIANURATO DE PORO CERRADO P. Arrué, A.C. Cárcel, A.M. Romero, B. Cárcel ITM-DIMM Universidad Politécnica de Valencia, [email protected] Resumen: En este trabajo se analizan a partir de secciones 2D e imágenes SEM, mediante análisis de imágenes, los parámetros estructurales de espumas de poliuretano rígido y poliisocianurato, con densidad relativa entre 0,03 y 0,06, empleadas como aislantes. La estructura observada es, en todos los casos, de poro cerrado, isotrópica, con tamaños de celda entre 250-400 micras, muy bajos espesores de pared (<0,50 micras) y una alta fracción de sólido en los bordes, (>80%). La aplicación de modelos topológicos indica que la estructura tipo de celda es dodecaédrica. Palabras clave: sólidos celulares, espumas, poliuretano, poliisocianurato, caracterización microestructural. 1. INTRODUCCIÓN La caracterización estructural permite determinar cómo se configura la estructura celular interna de los materiales celulares o espumas. La estructura o mesoestructura de las espumas junto con la densidad relativa, condicionan tanto el comportamiento mecánico como los fenómenos de transporte del material celular [1]. La determinación de los parámetros estructurales incluye un elevado número de aspectos: densidad, tamaño de celdas, espesores de paredes, etc. cuya determinación resulta necesaria para la modelización del comportamiento de los materiales celulares mediante modelos analíticos o mediante simulación por elementos finitos MEF [2]. También resulta necesario para la modelización MEF la determinación de la estructura celular tipo que mejor reproduce la estructura real de la espuma, y para cuya determinación se requiere del empleo de modelos topológicos adecuados capaces de llenar completamente el espacio [3]. Kraynik y de Warren (1994) revisaron los diferentes modelos regulares de espuma, de utilidad para los cálculos por simulación de las propiedades mecánicas o de transporte, y sugieren el uso de estructuras tipo dodecaédrica o tetracaidecaédrica, capaces de ocupar todo el espacio y satisfacer la mayoría de las reglas geométricas [4]. En el presente trabajo se determinan los parámetros estructurales de espumas aislantes de poliuretano rígido PUR y Poliisocianurato, que se emplean como aislantes debido a su baja conductividad térmica, a partir de imágenes 2D de secciones de las espumas. La metodología empleada puede aplicarse de manera similar a la caracterización de otros tipos de espumas o materiales celulares. 2. MATERIALES Y MÉTODO EXPERIMENTAL Se han analizado tres tipos de espumas de poliuretano (PUR) y una de poliisocianurato (CASSPIR). La tabla XI CONGRESO NACIONAL DE MATERIALES 2.1 muestra el espesor total, la conductividad térmica (10ºC), la resistencia a compresión y la densidad teórica de las muestras analizadas. Tabla 2.1. Materiales analizados. t* (mm) λ* (W/m·K) Rc (kPa) Muestra PUR-1 50 0,021 177 PUR-2 50 0,021 206 PUR-3 60 0,024 451 CASSPIR 50 0,023 245 ρ* (kg/m3) 35 40 70 40 La identificación de la estructura se realizó a partir de imágenes SEM (Jeol JSM 5410 y JSM6300) de muestras seccionadas mediante crio-fractura, según una metodología similar a la empleada por otros autores [5,6]. Las micrografías se analizan con el software Autocad para la medición de los parámetros característicos realizando una selección discrecional que evite medidas erróneas, de los elementos característicos de la muestra, especialmente en el estudio del espesor de las aristas de las celdas, en los que en multitud de casos se produce una curvatura en las paredes de las celdas. Esta selección no se puede realizar con programas de tratamiento de imágenes en los cuales todos los perfiles de las celdas los estudia como espesores de paredes. Se determina el valor promedio de las características geométricas, longitud, espesor y tamaño medio de celda, conectividad de bordes, Ze, conectividad de las caras, Zf, número medio de bordes por cara, n y el número medio de celdas por unidad de longitud, Nc. Por otro lado la densidad aparente se determina mediante medición directa. La identificación de la tipología de celdas se realiza por comparación entre la fracción del sólido en arista, φ, la relación entre la densidad relativa y las relaciones te/l y tf/l de los modelos teóricos [3] y los resultados experimentales. 260 6. POLíMEROS 3. RESULTADOS y ANÁLISIS 3.1. Relación entre espesor de pared y espesor de arista con la longitud de arista En la figura 3.3 se muestra la relación entre te/l y tf/l con la ρr*. La pendiente de la recta respecto al te es mucho mayor que respecto a tf siendo el factor determinante de su densidad puesto que todas ellas tenían el mismo espesor de pared (0,48 µm). Las micrografías obtenidas de las muestras se indican en la figura 3.1. Figura 3.3. Relación tf/l y te/l vs la densidad relativa del poliuretano y poliisocianurato. Figura 3.1. PUR-1(D-35), 100x (a), PUR-2 (D-40), 100x (b), PUR-3 (D-70), 100x (c) y CASSPIR (D-40), 200x (d). A partir de las imágenes 2D de las muestras se observa una mayor proporción de sólido en la configuración de las aristas de las celdas que en las paredes. La figura 3.2 presenta la medición del espesor de arista y pared. 3.3. Conectividad de caras, Zf, de bordes, Ze y número medio de bordes por cara, n A partir de las imágenes en 2D se obtiene el valor promedio de Zf, Ze, n y Nc. Figura 3.4. Figura 3.4. Zf, Ze, nexp y Nc, en PUR-2 (D-40). 50x. Figura 3.2. Espesor de arista equivalente en PUR-1 (D-35). 1500x. La relación te/l en las espumas, 0,1295 en PUR y 0,1681 en CASSPIR es mucho más elevada que tf/l, 0,030 en PUR y 0,0056 en CASSPIR. 3.2. Densidad relativa La densidad relativa se obtiene de la relación entre la densidad aparente y del sólido, siendo en las de celda cerrada proporcional a la relación t/l. ρr * = ρ* t = C3 · l ρs XI CONGRESO NACIONAL DE MATERIALES (3.1) El número medio de bordes por cara teórico, nteórico, está relacionada con el número de las caras f mediante la ecuación de Euler. n= Ze ·Zf 2 · 1 − Ze − 2 f (3.2) Los valores teóricos son de 5,00 en estructuras dodecaédricas y 5,14 en tetracaidecaédricas. Experimentalmente se obtiene un valor promedio de 5,4 correspondiente a caras pentagonales y hexagonales. Esto indica que se trata de estructuras de tipo preferentemente dodecaédricas y tetracaidecaédricas. 261 6. POLíMEROS El valor teórico corresponde a celdas con geometrías perfectas que configuran un empaquetamiento compacto. La identificación por este modelo no es determinante puesto que los valores de f y n dependen de cómo se ha formado la espuma y de las fuerzas que intervienen [1]. En las espumas PUR y CASSPIR, la fracción del sólido en la arista teórica para un número n teórico correspondiente a diferentes estructuras geométricas tipo se presenta en la Tabla 3.3. Tabla 3.3. Fracción del sólido en la arista teórica y experimental en PUR y CASSPIR. 3.4. Tamaño medio de celdas, L y anisotropía, R Muestra El tamaño medio de celdas se obtiene cuantificando el número de celdas, Nc por unidad de longitud. En micrografías 2D, el tamaño medio de la celda en las dos direcciones principales se obtiene con la relación: Tamaño medio de celda es L1 = 1,5 / N c (3.4) La Tabla 3.1. recoge los resultados obtenidos. Tabla 3.1. Tamaño medio de celdas, L, y ratio de anisotropía, R, en PUR y CASSPIR. Muestra PUR-1 PUR-2 PUR-3 CASSPIR Promedio R L (µm) 390,54 388,62 281,41 336,42 349,25 0,98 1,06 1,01 1,09 1,03 Los tamaños de celda se encuentran entre 250-400 micras. Los ratios de anisotropía muy próximos a la unidad indican una uniformidad aceptable y, en consecuencia, un comportamiento mecánico y térmico isotrópico. 3.5. Fracción de sólido en aristas, φ En las espumas analizadas el sólido se sitúa preferencialmente en las aristas de la celda, que son más gruesas que las caras. En esta situación se define el parámetro φ, que representa la fracción de sólido contenido en las aristas de las celdas, la fracción restante (1- φ) está en las caras. φ= t e2 Z t e2 + f ·t f ·l n (3.5) A partir de los valores experimentales se obtiene la cuantía de φexp que se indica en la tabla 3.2. Tabla 3.2. Fracción del sólido en la arista. Muestra PUR-1 PUR-2 PUR-3 CASSPIR Promedio Zf 3 3 3 3 te (µm) tf (µm) 20,10 20,20 20,79 23,53 0,48 0,48 0,48 0,79 l (µm) 199,78 183,16 117,11 140,03 XI CONGRESO NACIONAL DE MATERIALES CASSPIR (3.3) Ratio de anisotropía es R = L1 / L2 PUR-1 PUR-2 PUR-3 nexp 5,39 5,44 5,56 5,37 φexp 0,88 0,89 0,94 0,90 90,3 % Promedio φteórico Dodecaedro Tetracaidecaedro Icosaedro 0,88 0,89 0,93 0,89 89,5% 0,88 0,89 0,93 0,90 89,8% 0,88 0,89 0,93 0,90 90,2% φexp 0,88 0,89 0,94 0,90 90,3% En las espumas PUR y CASSPIR el 90% del sólido en arista es de una cuantía similar a los valores teóricos pero no resulta determinante para la identificación de la tipología de estructura. 3.6. Análisis de la tipología de celda en función de la densidad relativa A partir de los datos experimentales se ha intentado determinar cuál es el tipo de estructura probable en base a la comparación entre la densidad relativa teórica y experimental. Los resultados del modelo (3.6) se presentan en la tabla 3.4. siguiente. 2 t t ρ* = φ · C5 e + (1 − φ ) ·C6 f ρs l l 3.6) Tabla 3.4. Relación entre la densidad relativa experimental y la teórica en estructura tipo dodecaedro rómbico (a), dodecaedro pentagonal (b), tetracaidecaedro (c) e icosaedro (d). Muestra ρr (e xp) PUR-1 PUR-2 PUR-3 CASSPIR Promedio 0,0303 0,0332 0,0612 0,0426 ρr a 0,86 0,77 0,52 0,43 0,65 (exp) / ρr (teórica) b 2,37 2,12 1,42 1,18 1,77 c 3,00 2,68 1,80 1,49 2,24 d 0,40 0,36 0,24 0,20 0,30 Los resultados con los valores próximos a la unidad sugieren, en todos los casos, el tipo de celda preferentemente dodecaédrica. 3.7. Análisis de la tipología de celda en función de la densidad relativa respecto a la relación te/l La Tabla 3.5. presenta los coeficientes C8 del modelo (3.7) determinando cual es el tipo de estructura probable en base a la comparación entre los coeficientes teóricos y experimentales. 262 6. POLíMEROS 1/ 2 te ρ* = C8 · φ · l ρs (3.7) Tabla 3.5. Relación entre el coeficiente C8 experimental y teórico de estructuras tipo dodecaedro rómbico (a), dodecaedro pentagonal (b), tetracaidecaedro (c) e icosaedro (d). Muestra C8 (e xp) PUR-1 PUR-2 PUR-3 CASSPIR Promedio 0,61 0,64 0,74 0,86 0,71 C8 (exp) / C8 (teórica) a b 1,16 1,21 1,41 1,63 1,35 c 0,70 0,73 0,85 0,98 0,82 0,63 0,66 0,76 0,88 0,74 d 1,77 1,84 2,13 2,47 2,05 La tipología que mejor reproduce los resultados experimentales es de tipo dodecaédrica 3.8. Análisis de la tipología de celda en función del coeficiente C3 y C2 Algunos autores [1] sugieren el uso de una relación simple entre densidad relativa de la espuma y t/l a través del coeficiente C3 y C2. Para las celdas cerradas, la constante C3 oscila de 1,18 a 4,46 y para celdas abiertas C2 oscila entre 1,06 a 4,61. Las ecuaciones que relacionan la densidad con la relación t/l son: Espumas de celda cerrada: ρ* = ρs t 1 f C · 1 l − φ ( ) 7 (3.8) C3 ρ* t Espumas de celda abierta: = C2 · e ρs l 2 4. CONCLUSIONES Las espumas rígidas de PU y Poliisocianurato presentan una estructura de celda cerrada, en la que se aprecia una clara diferenciación entre los espesores de paredes y bordes. Debido a su carácter termoestable y al método de preparación de las muestras, las micrografías SEM presentan celdas con caras de lados rectos, sin deformación local apreciable. Los espesores de pared promedio, tf, son de 0,48µm en las PUR y de 0,79µm en la CASSPIR. Los espesores de arista promedio, te, son de 20,36 µm en PUR y de 23,53 µm en CASSPIR. Y las longitudes de arista, l, son de 166,68 µm en las espumas de PUR y 140,03 µm en la de Poliisocianurato. El parámetro determinante en las características de este tipo de espumas es la relación te/l de 0,1295 en PUR y 0,1681 en CASSPIR, mucho más elevada que tf/l, siendo más influyente el espesor de aristas que el de las paredes. Las mediciones efectuadas sobre las micrografías arrojan valores de la fracción de material en bordes de valor 0,88 a 0,94 para valores crecientes de la densidad relativa de las espumas PUR desde 0,03 a 0,06 y de 0,90 en la CASSPIR. Los tamaños de celda se encuentran entre 250-400 micras, con estructura isótropa dada por un índice de anisotropía promedio R=1,03. Los modelos que incluyen el análisis de la densidad relativa y su relación con te/l, así como los coeficientes C8 y C2, indican que la estructura celular tipo de estas espumas es preferentemente dodecaédrica. 5. REFERENCIAS [1] (3.9) [2] Tabla 3.6. Coeficiente C3 y C2 experimentales. Muestra PUR-1 PUR-2 PUR-3 CASSPIR C3 (e xp) 12,60 12,63 15,05 7,56 Promedio C2 (e xp) 13,43 7,56 2,99 2,73 1,94 1,51 Promedio 2,56 [4] 1,51 Los resultados incluidos en la Tabla 3.6. indican que para las espumas de poliuretano y poliisocianurato el coeficiente C3 se encuentra fuera de rango debido a la gran proporción de sólido en las aristas. En cambio bajo la consideración de tratarse de celdas abiertas el coeficiente C2 si se encuentra dentro del rango admisible de 1,06 a 4,61. El coeficiente teórico C2 es de 1,06 para tetracaidecaedros y 2,87 para dodecaedros rómbicos [3]. El valor experimental obtenido sugiere, como en anteriores análisis, que la estructura tipo más probable es de tipo dodecaédrica. XI CONGRESO NACIONAL DE MATERIALES [3] [5] [6] Gibson L.J. y Ashby M.F. “Cellular Solids: Structure and Properties. 2nd Ed.”, Pergamon Press, Oxford, England, (1997). James Ren X., Silberschmidt V.V. “Numerical modelling of low-density cellular materials” Computational Materials Science 43 65–74, (2008). De Hoff, R.T. and Rhines F.N. Quantitative Microscopy, Mc Graw Hill, New York. P.93, (1968). Kraynik A.M. and Warren W.E. in: N.C. Hilyard and A.C. Cunningham, Editors, Low Density Cellular Plastics, Chapman & Hall, London (1994). Das S. Dave M., Wilkes G. L. Characterization of flexible polyurethane foams based on soybeanbased polyols J. Appl. Polym. Sci. 112: 299-308, (2009). Doroudiani S. Park C.B. Kortschot M. Polymer Eng. And Science (36) 21; 2645:2662, (1996) 263 6. POLíMEROS ANÁLISIS DE LA CONTRIBUCIÓN DE LA RADIACIÓN EN LAS PROPIEDADES TÉRMICAS DE ESPUMAS DE POLIURETANO RÍGIDO P. Arrué, A.C. Cárcel, A.M. Romero, B. Cárcel ITM-DIMM Universidad Politécnica de Valencia, [email protected] Resumen: Se estudia la conductividad térmica de espumas PUR empleando un modelo aditivo de los mecanismos independientes: conducción a través del sólido, λs*; conducción a través del gas, λg*; convección dentro de las celdas, λc*; y radiación a través de las celdas, λr*, cuyas contribuciones relativas varían dependiendo de la densidad y de la estructura de las espumas. La contribución de la radiación y los valores característicos del coeficiente de extinción Cs se determinaron experimentalmente, teniendo en cuenta los valores de conductividad térmica total. Para valores de conductividad total entre 21,5-24,4 mW/mK, la conductividad por radiación λr* varió entre 5,9-7 mW/mK. La comparación de los valores experimentales con los modelos analíticos disponibles sugiere que el modelo de Rosseland es más preciso que el modelo de Gibson-Ashby para la determinación de λr*. Palabras clave: transmisión térmica, radiación, sólidos celulares, espumas de poliuretano. 1. INTRODUCCIÓN El uso de las espumas poliméricas está ampliamente extendido como aislantes térmicos en la construcción. Para controlar las propiedades térmicas es necesario conocer los mecanismos que gobiernan el comportamiento térmico de las espumas aislantes, estimar las posibilidades de optimización y mejorar la eficiencia energética con claras ventajas económicas y medioambientales. Se han determinado para las diferentes espumas analizadas las contribuciones relativas de los diferentes mecanismos que contribuyen a la conductividad térmica: conducción a través del sólido, conducción y convección a través de la fase gas y radiación y su relación con la densidad relativa. Las espumas de celda cerrada tienen la conductividad térmica más baja que cualquier aislamiento convencional no sometido al vacío. Varios factores se combinan para limitar el flujo del calor a través de la espuma: la baja fracción de volumen ocupada por la parte sólida, el pequeño tamaño de las celdas que minimizan la influencia de la convección y reducen la radiación con la absorción repetida y la reflexión en las paredes de celda; y la baja conductividad del gas del interior de las celdas. La contribución de la radiación es importante en las espumas de baja densidad, siendo uno de los campos de estudio para optimizar los sólidos celulares y sus propiedades térmicas. En este mecanismo resulta determinante la determinación adecuada de los parámetros Cs y Cs*, coeficientes de extinción del sólido y de la espuma, respectivamente, que determinan la absorción y transmisión de la radiación según la Ley de Beer. Su determinación puede llevarse a cabo experimentalmente o de modo indirecto, a partir de XI CONGRESO NACIONAL DE MATERIALES modelos analíticos apropiados, como el modelo de Rosseland propuesto por Glicksmann [1] o el modelo simplificado propuesto por Gibson y Ashby [2]. El objetivo de este trabajo es la determinación de la contribución de la radiación a la conductividad y la validación de los modelos anteriormente citados, tomando en consideración como referencia valores experimentales disponibles [3] de los coeficientes de extinción Cs en el rango IR. La validación de los modelos resulta de interés dadas las limitaciones del método experimental directo y las discrepancias entre los resultados obtenidos con diferentes modelos para otros tipos de espumas [4]. 2. MATERIALES En este trabajo se analizan materiales aislantes térmicos de base polimérica empleados en construcción seleccionados utilizando el método de Ashby [5] de la base de datos CES de Granta Design Ltd. de la Universidad de Cambridge, consistente en establecer un indicador correlacionado con una funcionalidad. La función elemento es el aislamiento térmico, el objetivo es minimizar costes y el requisito funcional es el coste total en función del espesor del material. λ *· ∆Ta + C t = Ca · e a · t · TE ea Coste de inversión (2.1) C os te energético La función objetivo se obtiene minimizando la expresión del coste en función del espesor. Ct = 1 1 · 2 · ( ∆Ta · t · TE ) 2 ( C a · λ *) 2 Parámetros var iables (2.2) Parámetros fijos 264 6. POLíMEROS Siendo: C a = Coste del material aisla n te (€/ m 3 ) e a = Espesor del material aislante (m ) λ * = Conductividad térmica del m aterial aislante ( W / m ·K ) t = T iem po de estudio ( h ) TE = Tarifa energética (2009). ∆ Ta = Salto térm ico (º C) TE = 0,11473 ( €/ kW ·h ) El coste será mínimo cuando el indicador M sea máximo. M = ( Ca · λ *) 1 − 2 (2.3) De los resultados de la selección se desprende que una opción óptima son las espumas poliméricas de celda cerrada que permiten minimizar costes asociados a la inversión y a las pérdidas energéticas. En años recientes se ha extendido el uso de espumas de poliuretano de mayor coste que presentan la ventaja de su aplicabilidad directa sobre todo tipo de superficies y también una menor conductividad térmica. En esta investigación se analizarán las espumas de celda cerrada de poliuretano (PUR). En la tabla 2.1 se indican las características de las muestras analizadas como son el espesor total, t*, la conductividad térmica de la espuma (10ºC), λ*, la densidad experimental, ρ*exp, la densidad relativa, ρr*, y la fracción del sólido en arista, φ. Tabla 2.1. Materiales analizados. Muestra PUR-1 PUR-2 PUR-3 t* (mm) 50 50 60 λ* (W/m·K) 0,0215 0,0215 0,0244 ρ*exp (kg/m3) 35,76 39,22 72,20 ρr* φ 0,0303 0,8832 0,0332 0,8935 0,0612 0,9350 Los parámetros constantes en todas las muestras son la conductividad del material sólido (0,2394 W/m·K), del gas (0,013 W/m·K), emisividad (0,95) y temperatura media absoluta (283K). sólido en las aristas, Glicksman [1] sugiere el empleo de un factor estimado para la eficacia de valor (2/3 - φ/3). En las espumas de poliuretano analizadas, dado que el tamaño de celdas oscila entre 250 y 400 µm, la contribución de la convección, λc*, a la temperatura ambiente puede despreciarse, ya que los fenómenos de convección sólo se consideran relevantes cuando el número de Grashof es mayor de 1000 [8], que en el caso de las espumas requiere tamaños de celda superiores a 10 mm. Para el análisis de la contribución de la radiación se han propuesto diferentes modelos analíticos o experimentales que permiten cuantificar su contribución a la conductividad global del sólido celular. El método experimental directo requiere la medición directa sobre muestras de espuma, de la radiación transmitida y/o absorbida por una muestra de un espesor dado en un rango de longitudes de onda, normalmente en el rango infrarrojo de 2-20 micras, mediante técnicas de espectrometría de infrarrojos FTIR. Los coeficientes de extinción espectral para cada longitud de onda Cs se obtienen a partir de la Ley de Beer. En el método experimental indirecto se determina la contribución de la radiación a partir de los valores de conductividad global, eliminando la contribución previsible de la conducción a través del sólido, gas y de la convección. Una metodología similar ha sido empleada para medir la contribución independiente de cada mecanismo en otros trabajos recientes [9]. Este método ha sido el utilizado en el desarrollo de este trabajo. Todos los mecanismos dependen de la densidad relativa, cuando es muy baja la conductividad aumenta debido a la mayor contribución de la radiación y cuando es alta también aumenta debido a la mayor contribución de la conductividad del sólido base, como se observa en la figura 3.1. correspondiente al análisis de la muestra PUR-1. 3. MODELOS 3.1. Transmisión de calor. La conductividad del material celular se ha expresado empleando el modelo aditivo de los diferentes mecanismos,conducción a través del sólido, conducción y convección a través del gas y radiación [1,5,6,7]: ρ* 2 φ ρ* λ* = λs ⋅ · − + λ g · 1 − + λ c * + λ r * (3.1) ρ 3 3 ρs Convección s Radiación Sólido Gas La conducción a través del sólido es directamente proporcional a la densidad relativa, corregida por un factor de eficacia que depende del recorrido tortuoso que debe realizar el flujo de calor a través de las paredes y bordes del sólido, y que depende de la fracción de XI CONGRESO NACIONAL DE MATERIALES Figura 3.1. Contribución de los mecanismos a la conductividad térmica respecto de la densidad relativa de la espuma de poliuretano (PUR-1). 265 6. POLíMEROS 3.2. Determinación del coeficiente de extinción. En la contribución a la conductividad de la radiación el parámetro más influyente es el coeficiente de extinción del sólido celular Cs*, el cual depende de la densidad relativa del sólido celular, del coeficiente de extinción del sólido, Cs, del tamaño de las celdas, de su geometría, del modo en que se distribuye el material entre paredes y bordes y del espesor total de la muestra. ρ* Cs * = Cs· ρs que define factores de eficacia promedio, F = 0,37 muy bajos, haciendo poco influyente la transmisión a través del sólido en las espumas de poliuretano. (3.2) El coeficiente de extinción del sólido es un parámetro característico del propio material, es decir, distintos sólidos celulares con distinta densidad relativa configurados con el mismo material deben presentar valores similares si la materia prima original o material a espumar es similar. La determinación experimental indirecta se ha realizado empleando los siguientes modelos: Figura 4.1. Contribución de los mecanismos a la conductividad térmica del sólido celular (W/m·K). Modelo Gibson y Ashby [5]: 4· ε · σ ·T 3 ·t ∗ ρ∗ ∗ Cs = ln · ·t λ∗r ρs −1 (3.3) Modelo de Rosseland: Cs = ρ* 16 · σ ·T 3 · 3· λ r * ρs −1 (3.4) Glicksman [1] sugirió este modelo para el cálculo de la transmisión de calor por radiación en espumas ópticamente gruesas en las que el medio absorbe o refleja la radiación isotrópicamente. 4. RESULTADOS 4.1.Contribución de los distintos mecanismos de transmisión de calor El mayor porcentaje de contribución corresponde a la que proporciona la conductividad del gas (56%). Al margen del control de la estructura de la espuma, el método universal para proporcionar una baja conductividad global en los sólidos celulares es utilizar en el proceso de fabricación gases de muy baja conductividad, como por ejemplo, los HFCs, que se utiliza en la fabricación de espumas poliméricas, o como el dióxido de carbono con conductividad 0,013 W/m·K inferior a la del aire de 0,024 W/m·K, como es el caso de las muestras analizadas. Esto implica que la optimización de las muestras conllevaría el reducir o controlar el siguiente mecanismo en importancia, la radiación (28%). 4.2. Coeficientes de extinción del sólido, Cs Sobre las muestras analizadas se ha obtenido el valor del coeficiente de extinción del material sólido base que constituye el sólido celular. Lógicamente, el método y los resultados obtenidos dependen del modelo empleado y de su validez para el caso considerado. La figura 4.1. siguiente muestra la contribución absoluta a la conductividad térmica global de cada uno de los mecanismos, para las muestras de espumas PUR analizadas, según el modelo (3.1). La contribución relativa de cada mecanismo presenta unos valores promedio del 15,85% para la conducción a través del sólido, 55,75% para la conducción a través del gas y 28,40% para la conducción debida a la radiación. Las proporciones obtenidas son significativamente similares a las indicadas por otros autores [1,5] para espumas de similar material y densidad. El coeficiente de extinción del sólido Cs determinado a partir del modelo de Gibson y Ashby, ecuación (3.3) tiene un valor promedio de 1.892,50 m-1. En la figura 4.2 se indican los coeficientes de extinción del material sólido, Cs, y del sólido celular, Cs*, obtenidos según el modelo Gibson y Ashby. La menor contribución la proporciona la conducción a través del sólido, debido a que se trata de sólidos celulares con densidades relativas muy bajas (0,03 a 0,06) y a la elevada distribución del sólido en las aristas de las celdas, con una fracción promedio (φ) del 90%, El coeficiente de extinción Cs según el modelo de Rosseland tiene un valor promedio de 29.058,59 m-1. En la figura 4.3 se indican los coeficientes de extinción del sólido, Cs, y del sólido celular, Cs*, obtenidos según el modelo Rosseland. XI CONGRESO NACIONAL DE MATERIALES 266 6. POLíMEROS analizados. Esta discrepancia sugiere que el modelo de Ashby no es apropiado para el análisis de la contribución de la radiación, posiblemente debido a simplificaciones que solamente serían válidas para muestras ópticamente transparentes con paredes muy delgadas. Figura 4.2. Coeficiente de extinción del poliuretano Cs* (m-1) y Cs (m-1) vs densidad relativa. Modelo Gibson y Ashby. Los valores del coeficiente de extinción del sólido obtenidos mediante el modelo de Rosseland son en cambio del mismo orden de magnitud que los valores publicados para los polímeros en el rango IR, que en valor medio presentan valores del orden de 104 m-1[2]. Para el PUR, Gibson y Ashby indican valores experimentales de 5,67·104 m-1 [5] y Shcuetz y Glicksman valores de 5,40·104 m-1 [10]. Sería factible abordar el proceso de optimización mediante el control de los valores de coeficiente de extinción del material celular, intentando llevar a cabo la optimización en el rango adecuado del espectro IR para su aplicación como aislante en edificación. 6. REFERENCIAS [1] Figura 4.3. Coeficiente de extinción del poliuretano Cs* (m-1) y Cs (m-1) vs densidad relativa. Modelo Rosseland. 5. CONCLUSIONES Respecto de la contribución de los distintos mecanismos de transmisión de calor, se observa que el mayor porcentaje de la conductividad corresponde a la que proporciona la conductividad del gas, 56% en PUR, de baja cuantía debido a que el gas utilizado (CO2) es de menor conductividad que el aire. El siguiente mecanismo de importancia es la radiación con un 28%. Y por último, el de menor cuantía es la conducción a través del sólido con un 17%, debido a que se trata de sólidos celulares con densidades relativas muy bajas y elevada fracción del sólido en arista, φ. Las proporciones obtenidas son significativamente similares a las indicadas por otros autores [1,5] para espumas de similar material y densidad. Glicksman, L. R. “Heat Transfer in foams” In Low Density Cellular Plastics: Physical Basis of Behaviour; Hilyard, N. C.; Cunningham A., Eds.; Chapman and Hall: UK, Chapter 5. (1994). [2] Gibson L.J. y Ashby M.F. “Cellular Solids: Structure and Properties. 2nd Ed.”, Pergamon Press, Oxford, England. (1997). [3] Wang Y. et al. Refractive indices and extinction coefficients of polymers for the mid-infrared region. Applied Optics 37 (30), 7091-7095. (1998). [4] De Micco C., Aldao C.M. “Radiation Contribution to the Thermal Conductivity of Plastic Foams” Journal of Polymer Science: Part B: Polymer Physics, Vol. 43, 190–192. (2005). [5] Ashby M.F. “Materials selection in Mechanical Design. 3rd Ed.” Eselvier, Oxford, England. (2007). [6] Leach, A. G. J Phys D: Appl Phys 1993, 26, 733– 742 (1993). [7] Colishaw, P. G.; Evans, J. R. G. J Mater Sci 1994, 29, 486–499. (1994). [8] Holman J.P. Heat transfer 5th Ed. McFraw Hill New York. (1981). [9] Ahern A, Verbist G. Weaire D. Phelan R and Fleurent H. “The conductivity of foams: a generalisation of the electrical to the thermal case” Colloids and Surfaces A: Physicochem. Eng. Aspects 263 275–279. (2005). [10] Schuetz M.A. Glicksman L.R. Proc. of the Society of Plastics Industry; 6th Int. Technical Conference San Diego. California. 341-347. (1983). Respecto a los coeficientes de extinción del sólido según los modelos aplicados se ha comprobado que los valores obtenidos empleando el modelo de Gibson y Ashby son un orden de magnitud inferior a los valores experimentales, para todos los tipos de espumas XI CONGRESO NACIONAL DE MATERIALES 267 6. POLíMEROS ESTUDIO COMPARATIVO DE LA PRESENCIA DE STYRENE-ETHYLENEBUTYLENE-STYRENE BLOCK COPOLIMERO (SEBS) Y POLIBUTADIENO (PB) EN EL COMPORTAMIENTO DE POLIESTIRENO DE ALTO IMPACTO. F. Parres1, J.E. Crespo1, R. Navarro, A. Nadal 1 Escuela politécnica Superior de Alcoy Plaza Ferrándiz y Carbonell, 2, [email protected] Resumen: Este trabajo se divide en tres partes. En primer lugar, el poliestireno de alto impacto (HIPS) fue sometido a diferentes ciclos de extrusión a fin de evaluar las variaciones en sus propiedades mecánicas y térmicas. Los resultados indican ligeras variaciones de las propiedades del HIPS, tanto mecánicas como térmicas. En segundo lugar, mezclas de HIPS degradado/SEBS y HIPS degradado/PB fueron preparadas para evaluar la influencia de la concentración de fase elástica en las propiedades de las mezclas. En ambos casos, la incorporación de SEBS y PB permite recuperar las propiedades iniciales que presentaba el HIPS virgen. Finalmente, mezclas de HIPS degradado/2% SEBS y HIPS degradado/2% PB fueron extruidas de forma sucesiva hasta completar cuatro ciclos. En este caso, las propiedades mecánicas permanecen constantes para las muestras de HIPS degradado con un 2% de SEBS, en cambio las mezclas de HIPS/2% PB muestran un incremento de la tensión de rotura en función de los ciclos de extrusión. Palabras clave: Poliestireno de alto impacto, reciclado, aditivo, SEBS, PB. 1. INTRODUCION La investigación sobre residuos poliméricos ha sido estudiada ampliamente desde hace muchas décadas; en estos estudios se han analizado la variación de propiedades mecánicas en función del número de ciclos de reprocesado que haya podido sufrir el material [1-3], además también existen múltiples referencias sobre la variación de propiedades térmicas e incluso estructurales [4]. Las técnicas utilizadas para realizar dichos estudios son muy amplias, abarcando desde ensayos mecánicos hasta el uso de técnicas de elevada sensibilidad como la Py – GC/MS [5-7]. Todos estos estudios han permitido conocer los fenómenos de degradación y la modificación de las propiedades por efecto de la temperatura o por los ciclos térmicos que haya podido sufrir un polímero reciclado. Por otro lado, las diversas investigaciones realizadas en el campo de la recuperación de residuos poliméricos, han revelado la existencia de impurezas tras el proceso de recuperación. Dichas impurezas ocasionan variaciones importantes en las propiedades finales del polímero recuperado [8]. Finalmente, aparecen estudios donde la incorporación de compuestos permite recuperar en cierta medida las propiedades iniciales que presentaba el polímero virgen, en algunos casos actuando directamente sobre el polímero y en otros, actuando como compatibilizador entre dos polímeros, polímero reciclado e impureza [9,10]. Pero no se encuentran estudios que analicen a largo plazo el efecto que tiene la incorporación de estos compuestos al material reciclado. El poliestireno de alto impacto está formado por dos fases, una estirénica y otra formada por butadieno. Durante el proceso de recuperación los diversos ciclos térmicos que sufre el material provocan el XI CONGRESO NACIONAL DE MATERIALES entrecruzamiento de las cadenas butadienicas y consecuentemente un incremento de la rigidez del HIPS. A nivel industrial y con el fin de recuperar la ductilidad del poliestireno de alto impacto se incorpora cierta cantidad de polibutadieno, en torno al 2%, consiguiendo buenos resultados. Teóricamente, si la pérdida de ductilidad del HIPS reciclado es consecuencia del entrecruzamiento de la fase butadiénica, la incorporación de más butadieno provocará el mismo efecto a largo plazo y por lo tanto el problema volverá a aparecer. Dada esta circunstancia, el objetivo de este trabajo es analizar el efecto a largo plazo de la incorporación de polibutadieno en el HIPS reciclado, así como analizar la posibilidad de substituir el polibutadieno por SEBS. 2. EXPERIMENTAL Preparación de muestras Para este estudio se ha utilizado PS Impact 6541 (Total Petrochemical). El proceso de degradación ha sido realizado mediante una máquina convencional de extrusión programada con las siguientes temperaturas de extrusión 183.5 – 185 – 190 – 200 ºC. La Figura 1 representa el esquema del proceso experimental en sus diferentes fases. Caracterización Las propiedades mecánicas fueron analizadas mediante el uso de una máquina universal de ensayos ELIB 30 (S.A.E. Ibertest, Madrid, España), con una célula de carga de 5 kN. Todos los ensayos fueron realizados siguiendo la norma UNE-EN ISO 527, a una velocidad de 50 mm min-1. La tensión de impacto se determine mediante un péndulo Charpy (S.A.E. Ibertest, Madrid, España) acorde con la norma ISO-179. Los valores de todos las propiedades mecánicas fueron obtenidas con la media de 5 – 7 muestras. 268 6. POLíMEROS Tabla 1. Variación de las propiedades mecánicas con el número de ciclos de extrusión. Ciclos de extrusión Figura 1. Esquema del proceso experimental en sus diferentes fases. 3. RESULTADOS Y DISCUSIÓN Efecto de los ciclos de reprocesado sobre las propiedades del HIPS Las propiedades mecánicas de cualquier material son fundamentales para su uso en una aplicación concreta. El ensayo de tracción y de impacto son de elevada importancia ya que permiten conocer propiedades tales como, la tensión de rotura, el alargamiento y la energía de impacto. La degradación térmica consecuencia de los procesos de recuperación provocan cambios en la estructura interna del polímero; estos cambios, dan lugar a una variación en las propiedades mecánicas. En el estudio llevado a cabo sobre el HIPS, la representación gráfica de los valores de la tensión de rotura con los ciclos de extrusión revela un incremento lineal de los valores obtenidos. Por otro lado, la evolución del alargamiento es diferente; en este caso, la pérdida de alargamiento es significativa entre el primer y tercer ciclo de extrusión, para mantenerse en valores constantes en ciclos posteriores de extrusión (Tabla 1). XI CONGRESO NACIONAL DE MATERIALES Tensión de rotura, MPa Alargamiento a la rotura, Energía de Impacto, % kJ m-2 Virgen 13,57 9,80 10,38 1 13,84 9,66 11,29 2 14,28 9,00 9,84 3 15,01 8,25 9,60 4 15,16 8,00 9,12 5 15,90 8,00 8,94 Inicialmente, las variaciones producidas en el HIPS no son excesivamente importantes, este comportamiento es lógico ya que las temperaturas utilizadas en el proceso de extrusión e inyección no son muy elevadas. Kalfoglou and Chaffey [11] mostró que sólo se producen grandes variaciones en las propiedades mecánicas cuando se utilizan temperaturas de procesado elevadas (290 ºC). Al igual que en el caso de la tensión de rotura y el alargamiento la energía de impacto no sufre grandes variaciones con el número de ciclos de extrusión (Tabla 1). A pesar de utilizar temperaturas de extrusión e inyección relativamente bajas, la presencia de la fase butadienica provoca cierto grado de entrecruzamiento de cadenas y consecuencia de ello un cierto incremento de la rigidez en el poliestireno de alto impacto. Otros autores realizaron estudios en este campo y observaron que no se producía variaciones significativas en las propiedades mecánicas debido a un ligero grado de entrecruzamiento a temperaturas de procesado cercanas a 190 ºC [12]. Análisis de las propiedades del HIPS reprocesado en presencia de bajos contenidos de SEBS y PB. La pérdida de ductilidad en el HIPS reciclado limita su uso en algunas aplicaciones. Dado que la variación de propiedades es fundamentalmente debido al entrecruzamiento de la fase butadiénica, es frecuente a nivel industrial la adición de polibutadieno con el fin de aumentar la ductilidad del HIPS reciclado. Como término medio se utiliza una cantidad equivalente al 2% en peso respecto al HIPS reciclado. En este caso, el estudio va mucho más allá y realiza una comparación entre el PB y el SEBS con distintos porcentajes en peso (1, 2, 4, y 8%). 269 6. POLíMEROS Para ambos compuestos, PB y SEBS, la evolución de los valores de las diferentes propiedades mecánicas (Tensión de rotura, Alargamiento a la rotura y Energía de Impacto) es muy similar, aumentando la ductilidad de las mezclas en función del contenido de PB o SEBS. En primer lugar, la tensión de rotura desciende paulatinamente hasta alcanzar valores cercanos al HIPS virgen en contenidos del 8%. En cambio, la adición de SEBS y PB provoca un efecto contrario en el alargamiento a la rotura, en este caso los valores se incrementan de forma importante en contenidos relativamente bajos; consiguiendo superar las propiedades iniciales del HIPS virgen para un 1% de PB y un 2% de SEBS (Figura 2). virgen poseía e incluso superarlas. Inicialmente, la adición de un 2% de PB y SEBS permite recuperar las propiedades que presentaba el HIPS. Los resultados obtenidos permiten concluir que los porcentajes más viables para la recuperación de propiedades en el HIPS degradado es el 2% en peso, ya que los valores de las diversas propiedades mecánicas (tensión de rotura, alargamiento a la rotura, y energía de impacto) son próximas al HIPS virgen. Estudio de las propiedades de mezclas de HIPS degradado – SEBS y HIPS degradado – PB sometido a diferentes ciclos de reprocesado. La adición de compuestos sobre un polímero reciclado provoca en la mayoría de los casos acciones beneficiosas para las propiedades del material recuperado, pero en muchas ocasiones se olvida de los efectos que tiene ese compuesto sobre las propiedades del polímero cuando este es sometido de nuevo a diversos ciclos de reprocesado. En el caso de la adición de un 2% de SEBS y PB en el HIPS extrusionado 5 veces, la evolución de los valores de tensión de rotura es distinto, así como los valores correspondientes a la adición de SEBS permanecen prácticamente constantes, en el caso de la adición de PB se observa un ligero incremento de la tensión de rotura a medida que aumentan los ciclos de extrusión. Por otro lado, en lo que se refiere al alargamiento de las muestras, los valores permanecen prácticamente constantes durante los primeros ciclos de extrusión, pero a partir del tercer ciclo los valores descienden ligeramente (Figura 3). Figura 2. Evolución de la tensión de rotura y el alargamiento a la rotura en función del contenido de PB y SEBS. Del mismo modo que en el caso anterior la energía de impacto se incrementa a medida que aumenta el porcentaje de PB y SEBS, aunque en este caso los valores de la energía de impacto no superan al HIPS virgen hasta alcanzar porcentajes del 4% (Tabla 2). Tabla 2. Variación de la energía de impacto en función del contenido de SEBS y PB. Contenido, % Energía de Impacto, kJ m-2 SEBS PB 0 08,94 08,94 1 09,60 10,16 2 09,80 10,55 4 11,10 11,86 8 11,86 14,02 La incorporación de PB y SEBS con carácter dúctil al HIPS tras su quinta extrusión permite recuperar en cierta medida las propiedades iniciales que el HIPS XI CONGRESO NACIONAL DE MATERIALES Figura 3. Evolución de las propiedades mecánicas (Tensión de rotura y alargamiento a la rotura) de las mezclas de HIPS degradado/SEBS y HIPS degradado/PB en función de los ciclos de extrusión. 270 6. POLíMEROS Finalmente, la tendencia de los valores de la energía de impacto es distinta en función de si se ha utilizado PB o SEBS; en el caso de las muestras con un 2% de PB la energía de impacto aumenta de forma lineal con el número de ciclos de extrusión. En cambio, la energía de impacto de las mezclas de HIPS con SEBS permanece constante independientemente del número de ciclos que haya sufrido (Tabla 3). Tabla 3. Variación de la energía de impacto de las mezclas con un 2% de SEBS y PB en función de los ciclos de extrusión. Contenido, % Energía de Impacto, kJ m-2 SEBS PB 0 09,80 10,55 1 09,93 10,73 2 10,16 11,29 3 10,16 12,02 4 10,20 12,50 4. CONCLUSIÓN La aplicación de sucesivos ciclos de extrusión sobre el poliestireno de alto impacto provoca ligeras variaciones en las propiedades mecánicas, térmicas y físicas debido fundamentalmente al uso de bajas temperaturas en el proceso de extrusión. A pesar de producirse pequeñas variaciones en las propiedades del HIPS, la microscopía electrónica de barrido permite observar un incremento de rugosidad en la matriz estirénica. Con respecto a las propiedades mecánicas del HIPS degradado en función del contenido en SEBS y PB, la adición de PB y SEBS permite recuperar en cierto grado las propiedades iniciales que poseían el HIPS virgen. Por otro lado, la temperatura a de reblandecimiento Vicat se mantiene constante hasta un 2% de PB y SEBS, pero decrece rápidamente cuando se excede dicho valor. Además, la microscopía electrónica de barrido permite observar cambios en la superficie de fractura de las diferentes muestras en función del contenido de SEBS y PB. En ellas puede observarse un incremento del tamaño y número de aglomerado de butadieno a medida que se incrementa el contenido de SEBS y PB. Finalmente el reprocesado de mezclas de HIPS degradado/PB (2 % en peso) y HIPS degradado/SEBS (2% en peso) provocan de nuevo variaciones en las propiedades mecánicas, aunque los valores de las mezclas con SEBS se asemejan más a los valores del HIPS virgen que las mezclas con PB. Vistos los resultados, aunque en ambos casos la adición de SEBS y PB permite recuperar las propiedades del HIPS degradado, el reprocesado de dichas mezclas indica un mejor comportamiento de las mezclas con SEBS que con PB. XI CONGRESO NACIONAL DE MATERIALES 5. AGRADECIMIENTOS Los autores agradecen al Vicerrectorado de Investigación de la Universidad Politécnica de Valencia por su apoyo y financiación del proyecto: “Investigación de sistemas ternarios aplicados a materiales poliméricos para la revalorización de residuos estirénicos.”, Ref: 20091056 englobado en el programa de primeros proyectos de investigación (PAID 06-09) donde se enmarca este trabajo. 6. REFERENCIAS [1] Christiani, C., Klason, C., Shishoo, R. "The effect of reprocessing of polypropylene on fiber spinning". J Appl Polymer Sci. 1999. [2] Grigoryeva, O. P., Fainleb, A. M., Shumskii, V. F., Vilenskii, V. A., Kozak, N. V., Babkina, N. V. "The Effect of multi-reprocessing on the structure and characteristics of thermoplastic elastomers based on recycled polymers". Polym Sci Ser A+. 2009. [3] Paukszta, D., Borysiak, S. "Influence of reprocessing on the crystallization of polypropylene in PP/PA6 blends". Polimery. 2009. [4] Santana, R. C., Manrich, S. "Studies on thermomechanical properties of post-consumer high impact polystyrene in five reprocessing steps". Prog Rubber, Plast Recyc Technol. 2002. [5] Ramirez-Vargas, E., Navarro-Rodriguez, D., Blanqueto-Menchaca, A. I., Huerta-Martinez, B. M., Palacios-Mezta, M. "Degradation effects on the rheological and mechanical properties of multiextruded blends of impact-modified polypropylene and poly(ethylene-co-vinyl acetate)". Polymer Degrad Stabil. 2004. [6] Parres, F., Balart, R., Crespo, J. E., Lopez, J. "Effects of the injection-molding temperatures and pyrolysis cycles on the butadiene phase of high-impact polystyrene". J Appl Polym Sci. 2007. [7] Yang, M., Tsukame, T., Saitoh, H., Shibasaki, Y. "Investigation of the thermal degradation mechanisms of poly(styrene-co-methacrylonitrile)s by flash pyrolysis and TG-FTIR measurements". Polymer Degrad Stabil. 2000. [8] Parres, F., Balart, R., Lopez, J., Garcia, D. "Changes in the mechanical and thermal properties of high impact polystyrene (HIPS) in the presence of low polypropylene (PP) contents". J Mater Sci. 2008. [9] Yang, W., Tang, X. G., Xie, B. H., Zhu, Y. P., Yang, M. B., Hou, M. "Heterogeneous Dispersion of the Compatibilizer in the Injection Molding of Polyamide 6/Polypropylene Blends". J Appl Polymer Sci. 2009. [10] Becker, D., Porcel, F., Hage, E., Pessan, L. A. "The Influence of the compatibilizer Characteristics on the interfacial characteristics and phase morphology of aPA/SAN blends". Polym Bull. 2008. [11] Kalfoglou, N. K., Chaffey, C. E. "Effects Of Extrusion On The Structure And Properties Of High-Impact Polystyrene". Polym. Eng. Sci. 1979. [12] Michaeli, W., Höcker, H., Berghaus, U., Heidemeyer, P., Klee, D., Günzel, R., Rathmer, P. "Extrusion von schlagzähem Polystyrol - Einfluß wiederholter Verarbeitung auf seine Eigenschaften". Plastverarbeiter.1990. 271 6. POLíMEROS CARACTERIZACIÓN MECÁNICA DE UNIONES ADHESIVAS DE SUSTRATOS DE POLIPROPILENO TRATADO CON TÉCNICAS DE FOTOPOLIMERIZACIÓN J. F. Balart1, R. Balart1, L. Sánchez-Nácher1, V. Fombuena1, J.M. España1 1 Instituto de Tecnología de Materiales (ITM), Universidad Politécnica de Valencia (UPV), Plaza Ferrándiz y Carbonell 1, 03801, Alcoy, España; [email protected] Resumen: Hoy en día numerosos sectores industriales, tanto de alto como de bajo contenido tecnológico, utilizan polipropileno. Sus propiedades adhesivas son muy débiles como consecuencia de su baja energía superficial. Muchas aplicaciones, sobre todo de alto contenido tecnológico requieren procesos de adhesión, por lo que es necesario modificar la superficie del polipropileno para que presente mayor actividad superficial. En este trabajo, se ha utilizado un proceso de fotopolimerización, mediante el cual y, con la utilización de luz ultravioleta, se insertan moléculas de monómero de metil metacrilato, sobre la superficie del polipropileno. Se ha utilizado benzofenona (BP) como agente iniciador de la reacción (fotoiniciador). Los cambios superficiales se han cuantificado mediante la medición del ángulo de contacto y el consiguiente cálculo de la energía superficial. Por último, se han caracterizado mecánicamente las uniones adhesivas de sustratos de polipropileno, previamente tratados con dicho proceso de fotopolimerización, mediante ensayos de pelado en ”T”. Palabras clave: fotopolimerización, adhesión, polipropileno, metil metacrilato. 1. INTRODUCCIÓN Dado el consumo creciente de polipropileno en sectores de alto contenido tecnológico, es necesario modificar alguna de sus propiedades para adaptarlo a aplicaciones requeridos por este sector, como por ejemplo, procesos de adhesión o procesos de pintura. Por lo tanto para poder llevar a cabo estos, es necesario modificar la actividad superficial del substrato de polipropileno, con el fin de aumentarle la energía superficial, que inicialmente es muy baja, debido a la cualidad de apolaridad de su molécula principal. Esta cualidad influye de manera negativa sobre las propiedades adhesivas. La modificación superficial se puede realizar de varias maneras. Existen los tratamientos físicos (tratamientos con plasma) y los tratamientos químicos. La utilización de unos o de otros requiere diferentes costes de implementación. Los físicos son más respetuosos con el medio ambiente, ya que no requieren el consumo de reactivos, pero por el contrario requieren una gran inversión económica. La implementación de los procesos químicos es más asequible, y según el proceso que se elija se logra controla el consumo de reactivos. El proceso que hemos elegido para modificar la actividad superficial ha sido el de fotopolimerización[13]. Posee el menor consumo de reactivos de entre los químicos. Destaca por su bajo coste de implementación y por la obtención de grandes rendimientos en tiempos relativamente cortos. El proceso en cuestión se caracteriza por la inserción de cadenas de monómero (metil metacrilato)[4, 5] de elevada polaridad, sobre la superficie de un substrato de polipropileno. El proceso se realiza en presencia de luz ultravioleta (UV) y a través de un fotoiniciador, que en nuestro caso hemos utilizado la benzofenona (BP)[6]. El estudio realizado, ha consistido en analizar la influencia de las variables que participan en el proceso. XI CONGRESO NACIONAL DE MATERIALES Los cambios en la humectabilidad superficial se han cuantificado mediante la medición del ángulo de contacto para cuatro líquidos de referencia distintos. A continuación y siguiendo el modelo de Owens-Wendt se ha calculado la energía superficial. Por último, se han caracterizado mecánicamente las uniones adhesivas de sustratos de polipropileno, previamente tratados con dicho proceso de fotopolimerización, mediante ensayos de pelado en ”T”. 2. EXPERIMENTAL El polipropileno utilizado como sustrato es ISPLEN® PB 180 G2M, proporcionado por REPSOL YPF. El monómero es de metil metacrilato extra puro y estabilizado, proporcionado por Acros Organics, con una pureza del 99%. El fotoiniciador utilizado ha sido la benzofenona con calidad para síntesis proporcionada por Scharlau Chemie S.A. Para la realización del proceso de fotopolimerización se utilizó un equipo de luz ultravioleta (UV). El modelo de la lámpara es el UVASPOT 1000RF2 de la marca HONLE UV TECHNOLOGY. La característica principal de esta lámpara es que se trata de una lámpara de alta presión de mercurio. El equipo utilizado para la medida del ángulo de contacto de las diferentes muestras de PP mediante cuatro líquidos de contacto diferentes es el EASYDROP STANDARD de la marca KRÜSS modelo FM140 110/220 V, 50/60 Hz. Para corroborar la mejora en las propiedades adhesivas de los polipropilenos tratados, se han realizado ensayos de tracción. Estos ensayos se han hecho siguiendo la normativa UNE-EN 1895 “ensayo de pelado en “T” (“T-peel test”) a 180º para unión encolada de adherente flexible sobre flexible”. El adhesivo utilizado es el Poliuretano801 Blanco, se trata de un poliuretano monocomponente de alta densidad comercializado por Kefren S.A. (Alicante, Spain) 272 6. POLíMEROS 3. RESULTADOS Y DISCUSIÓN La tabla 1, muestra la variación del ángulo de contacto en función del tiempo de exposición a la luz ultravioleta. Tiempo (s) S/tratar 30 60 90 120 150 180 210 240 θ Agua (º) 87,30 75,54 66,40 40,56 46,38 43,98 43,80 39,32 37,04 θ θ θ Glicerol Diodometano Formamida (º) (º) (º) 74,67 54,00 71,38 64,42 37,14 47,84 57,08 36,70 46,08 47,62 35,72 45,78 48,16 31,82 40,18 47,22 29,88 39,60 39,98 29,54 32,44 38,26 21,82 31,90 32,63 15,94 30,76 Tabla 1. Ángulo de contacto con respecto al tiempo de exposición UV. Tal y como se observa en la tabla, los valores de ángulo de contacto, disminuyen a medida que aumenta el tiempo de exposición. Esta tendencia de disminución se presenta de igual manera en los cuatro líquidos de referencia utilizados. A continuación, utilizando el modelo de Owens-Wendt se ha calculado la energía superficial. La figura 1, representa la energía superficial en función del tiempo de exposición a la luz ultravioleta. La figura 2, muestra la fuerza necesaria para separar dos substratos de PP unidos mediante un adhesivo de poliuretano. Figura 2. Fuerza de pelado en función del tiempo de exposición UV. Se observa que a medida que aumenta el tiempo de exposición, la fuerza necesaria para separar las muestras también aumenta. 4. CONCLUSIONES Ha quedado demostrado como el proceso de fotopolimerización consigue cambiar las propiedades superficiales del polipropileno, para dotarlo de unas características excepcionales y óptimas para un posterior proceso de adhesión. Se consigue transferir la polaridad del monómero de metil metacrilato a la superficie del PP con tiempos relativamente cortos (aproximadamente unos 210 – 240 segundos) y con costes de implementación muy bajos. Además se trata de un proceso muy en concordancia con el medio ambiente, por su reducido consumo de reactivos. Al conseguir mejorar la actividad superficial de PP, los procesos de adhesión obtienen elevados rendimientos, haciendo apto su utilización en sectores que requieren un elevado contenido tecnológico. 5. REFERENCIAS [1] Figura 1. Energía superficial en función del tiempo de exposición a luz UV. Observamos una tendencia ascendente en función del tiempo de exposición, cosa normal, ya que esta función es inversamente proporcional a la de ángulo de contacto. Destacar, que se ha utilizado el tiempo de exposición de 0 para el polipropileno sin tratar. XI CONGRESO NACIONAL DE MATERIALES [2] [3] [4] [5] [6] C. Decker and K. Zahouily. Surface modification of polyolefins by photografting of acrylic monomers. Bratislava, Slovakia: Wiley-V C H Verlag Gmbh, 1997. p. 99. J.P. Deng, Q.X. He, Z.L. Wu and W.T. Yang, J Polym Sci Pol Chem, 46, 2193 (2008) L. Tan, J.P. Deng and W.T. Yang, Polym Adv Technol, 15, 523 (2004) N. Luo, J.B. Hutchison, K.S. Anseth and C.N. Bowman, Macromolecules, 35, 2487 (2002) Y.X. Wang, W.B. Zhong, N. Jiang and W.T. Yang, Macromol Rapid Commun, 26, 87 (2005) L.B. Kong, J.P. Deng and W.T. Yang, Macromol Chem Phys, 207, 2311 (2006) 273 6. POLíMEROS RECUPERACIÓN DE LAS PROPIEDADES HIDROFÓBICAS DE UNA SUPERFICIE DE SILICONA TRAS TRATAMIENTO CON PLASMA ATMOSFÉRICO N. Encinas, B. Díaz-Benito, J.Abenojar, M.A. Martínez Departamento de Ciencia e Ingeniería de los Materiales e Ingeniería Química, IAAB Grupo de Comportamiento en Servicio de Materiales. Universidad Carlos III de Madrid. Av.Universidad, 30. 28911-Leganés, España Resumen: Las siliconas son elastómeros sintéticos con aplicaciones como lubricantes, adhesivos, impermeabilizantes o en las industrias farmacéutica y médica, gracias tanto a su resistencia a temperaturas en el rango de -60º C a 250 º C, como a propiedades aislantes, de resistencia a la tracción, flexibilidad, estabilidad química y biocompatibilidad. Sin embargo, su baja polaridad y energía superficial, hacen que no puedan ser mojadas por casi ningún líquido o adhesivo, obligando a la utilización de tratamientos superficiales para su posterior unión o pintado. En el presente estudio se evaluará el aumento de energía superficial de una superficie de silicona mediante antorcha de plasma atmosférico (APPT) y su estabilidad en el tiempo. La energía será calculada a través de medidas de ángulo de contacto, estableciéndose el tiempo de superficie activa, así como el período de recuperación de la silicona. La posible introducción de nuevos grupos funcionales en la superficie de las muestras será analizada mediante Espectroscopia Infrarroja de Reflexión Atenuada (ATR-IR), se evaluarán los cambios topográficos generados mediante Microscopía Electrónica de Barrido (SEM). Palabras clave: Silicona, Modificación superficial, Plasma atmosférico, Ángulo de Contacto, Mojado de polímeros. 1. INTRODUCCIÓN Las siliconas son polímeros elastoméricos de fórmula general – (Si(R)2-O-)n, en los que la existencia simultánea de grupos funcionales orgánicos unidos a un esqueleto de naturaleza inorgánica confiere importantes propiedades para su aplicación en diversos campos como la industria aerospacial (por su capacidad de uso en un amplio rango de temperaturas), de adhesivos, electrónica (resistencia eléctrica) o medicina (biocompatibilidad) [1-3]. Sin embargo, estos materiales presentan una mojabilidad extremadamente baja que limita procesos de pintado, recubrimiento o pegado de los mismos. El mojado de una superficie viene determinado por la ecuación de Young (Ec. 1) [4]: s = sl + l.cos El ángulo que forma dicho líquido sobre la superficie sólida determinará la mojabilidad y energía de la misma, de modo que >90º implican s< l y, por tanto, el no mojado del sólido. Entre los diversos métodos para la medida de la energía superficial de un sólido, los basados en medidas de ángulo de contacto [5-6] suponen una vía simple, rápida y sensible a las características superficiales de los sólidos, aunque poseen una baja reproducibilidad de las medidas, debido a las heterogeneidades del material y una compleja interpretación de datos. El modelo matemático de Owens-Wendt-Rable-Kaelble (OWRK) (Fig.2.) [7-8] permite el cálculo de las componentes dispersiva, polar y total de la energía superficial haciendo uso de la expresión de una recta (Fig.2): (1) En esta expresión, los términos s, sl y l, hacen referencia las tensiones o energías superficiales del sólido, de la interfase sólido-líquido (Fig.1.) y del líquido, respectivamente. Figura 1. Diagrama vectorial de tensiones superficiales de un sistema S-L-V XI CONGRESO NACIONAL DE MATERIALES Figura 2. Representación gráfica del método OWRK 274 6. POLíMEROS Por todo esto, el aumento de la energía superficial de las siliconas por encima de la tensión superficial de líquidos o adhesivos que vayan a ser usados será una condición necesaria para asegurar mecanismos de adhesión o pintura. Para ello se someten los polímeros a tratamientos superficiales físicos o químicos que, mediante la creación de grupos específicos, variación de rugosidad o por entrecruzamiento de las cadenas poliméricas, logran aumentar su energía. El tratamiento mediante plasma es un método rápido, limpio (no se genera ningún tipo de residuo ambiental) que provoca, además de una efectiva limpieza superficial, la sustitución de átomos de baja energía de la superficie polimérica por otras funcionalidades más reactivas, como podrían ser hidroxilos (-OH), aminas (-NH) o carbonilos (-CO), sin afectar a las propiedades intrínsecas del material [9-11]. Además, dentro de las distintas técnicas de plasma (térmicos, fríos, a baja presión, etc.) existentes, el uso de los equipos de APPT evita los problemas tanto de coste como seguridad que supone el uso de bajas presiones. 3. RESULTADOS Y DISCUSIÓN En primer lugar, se realiza el tratamiento con APPT sobre las piezas y se almacenan a temperatura ambiente en condiciones libres de contaminación. En la Figura 3 se presentan las imágenes de gotas de agua bisdestilada depositadas sobre superficie de silicona, tanto recibida como tratada con plasma. Como se puede observar, en los momentos iniciales tras el APPT (0,1 h), el ángulo de contacto sobre la superficie sólida desciende desde 112º hasta 86º, lo cual indica un aumento en la energía superficial de la misma debido a la introducción de grupos funcionales de carácter polar (grupos carbonilo – C=O, hidroxilo -OH, amina -NH, carboxilo -COOH, etc.) gracias a reacciones de tipo radicálico entre las especies activas del flujo de plasma y la superficie. XI CONGRESO NACIONAL DE MATERIALES 112º 2h 97º 0,1 h 1h 86º 3h 92º 6h 104º 111º Figura 3. Gotas de agua sobre silicona tratada con APPT tras diversos tiempos de envejecimiento de las piezas. Dicho valor de ángulo de contacto va aumentando con el tiempo de envejecimiento de las mismas (1, 2, 3 y 6 h), hasta recuperar prácticamente su valor inicial (111º) tras 6 h de almacenamiento. Se calculan los valores tanto de energía superficial total como de sus componentes dispersiva y polar mediante el método OWRK, llegando a los valores expuestos en la Figura 4. 2. PROCEDIMIENTO EXPERIMENTAL 20 15 2 σ (mJ / m ) Los ensayos se llevan a cabo sobre piezas silicona comercial, cuya limpieza y desengrasado se realiza con el disolvente metil-etil-cetona (MEK). Para su tratamiento superficial se utiliza un equipo de APPT (PlasmaTreat) provisto de boquilla rotatoria que expele el flujo de plasma a una presión de 2 bar. Como parámetros de trabajo se establece una velocidad de trabajo de 0,02 m/min y una distancia boquilla-muestra de 6 mm. Se realiza un estudio de la recuperación de la energía superficial y propiedades hidrofóbicas de la silicona en un período de 24 h a través de medidas de ángulo de contacto con agua desionizada, glicerol, nitrometano, diiodometano y 1,5-pentanodiol utilizando un equipo OCA 15 plus Se sigue la metodología propuesta en la prenorma prEN 828:2009, y la energía superficial es calculada mediante el método de OwensWendt-Rabel-Kaelble (OWRK). Se comprobará la introducción de nuevos grupos funcionales en la superficie de las muestras mediante ATR-IR, y los posibles cambios en la morfología de las superficies con el tratamiento con APPT se estudian mediante SEM. 0h 10 σTotal σDispersiva σPolar 5 0 0h 0,1 h 1h 2h 3h 4h 6h 24 h Tiempo de envejecimiento Figura 4. Variación de las componentes de la energía superficial (dispersiva, polar y total) de la silicona con el tiempo tras aplicar APPT. Inicialmente la silicona sin tratar muestra un carácter prácticamente dispersivo (Tabla 1). Su baja energía superficial, muy por debajo de la tensión de los líquidos de medida provoca que los líquidos apenas se extiendan por su superficie, ya que la condición de mojado de un sólido establece que s > l, no siendo muy acusada la diferencia entre ambas. El APPT provoca un aumento inmediato de la energía superficial, desde un valor de T = 15,7 hasta 20,0 mJ/m2 transcurrida 0,1 h. La hipótesis de que este aumento sea generado por la formación de funcionalidades hidrófilas en la superficie de silicona se ve corroborado por el cambio de la componente polar de la energía desde P = 0,8 a 13.0 mJ/m2 tras 0,1 h del 275 6. POLíMEROS tratamiento. Posteriormente sigue la misma tendencia que la componente total, es decir, la recuperación del valor correspondiente a la pieza sin tratar a las 6 h de envejecimiento. Tabla 1. Comparativa de las γL de los líquidos de ángulo de contacto y la energía de la silicona sin tratar. Líquido/Material Agua Glicerol 1,5-pentanodiol Diiodometano Nitrometano Silicona σT 72.1 62.7 43.3 50.0 36.8 15.7 σT 19.9 21.2 27.6 47.4 20.3 14.9 σP 52.2 41.5 15.7 2.6 16.5 0.80 La fracción dispersiva de la energía, de igual modo que las ya vistas, sufre una modificación inmediata con la aplicación del plasma y una recuperación de su valor inicial a las 6 h de almacenamiento de las piezas, pero su tendencia, como cabría esperar, es opuesta (disminuye inicialmente de σD = 14,9 hasta 7,1 mJ/m2) para más tarde volver a aumentar hasta 13,82 mJ/m2 tras 6h y 14,6 mJ/m2, prácticamente el valor inicial, a las 24 h de envejecimiento. Mediante espectroscopia ATR-IR (Figura 5) se observan los picos correspondientes a las vibraciones normales de las siliconas, es decir, los correspondientes al grupo metilo (2955, 1257, 866 y 787 cm-1) y a los enlaces Si-CH3 (1073 y 664 cm-1) y Si-O-Si (995 cm-1). Dichas bandas se mantienen en todas las condiciones, y únicamente aparece un nuevo pico a 2357 cm-1 transcurridas 3 h de envejecimiento, la cual se mantiene tras 4 h. Esta banda puede atribuirse a vibraciones (SiO)Si-H. Por último, se lleva a cabo un estudio por SEM de la topografía de las piezas. En la Figura 6 se muestran los efectos del APPT sobre la morfología de la silicona tanto sin tratar como después de 0,1, 2 y 4 h de envejecimiento. La superficie original, con morfología heterogénea (partículas y placas con tamaño aproximado de 10-30 µm) y rugosa, presenta alisamiento de la misma tras la aplicación del plasma. Como se puede observar, los cambios superficiales producidos no son muy notables, y aproximadamente se mantienen con el tiempo de envejecimiento, lo cual indica que los cambios en microrugosidad generados por el impacto del flujo de plasma no son determinantes en el aumento de energía de las piezas, al mantenerse la misma topografía pese a recuperarse las propiedades hidrofóbicas de la silicona tras 4 h de envejecimiento. a) Sin tratar b) 4 h envejecimiento c) 0,1h envejecimiento d) 2 h envejecimiento Figura 6. Análisis de SEM de las superficies de silicona, (a) sin tratar, (b) 4 h, (b) 0,1h (d) 2 h tras el tratamiento de APPT. Figura 5. Espectros de ATR-IR del envejecimiento de las piezas durante 4 h tras aplicar APPT. XI CONGRESO NACIONAL DE MATERIALES Mediante análisis sonda de Espectroscopía de Energía Dispersiva (EDX) se obtiene la composición superficial de las piezas mostrada en la Tabla 2. El polímero inicial está formado por cadenas conteniendo silicio y oxígeno mayoritariamente, con la presencia de un cierto porcentaje de carbono relativo a grupos funcionales de naturaleza orgánica y magnesio, aditivo utilizado en la síntesis del polímero en reguladores de la acidez y agentes anti-apelmazantes. Como cabría esperar, el tratamiento con plasma induce un ligero aumento en el porcentaje composicional de aquellos átomos presentes en grupos orgánicos hidrófilos, es decir, carbono y oxígeno. 276 6. POLíMEROS Tabla 2. Variación en la composición en peso (Wt%) y atómica (At%) de la superficie de silicona obtenida por EDX. Condición Sin tratar 0,1 h 24 h Elemento C O Si Mg C O Si Mg C O Si Mg Wt% 13,18 28,12 43,67 15,06 13,32 28,51 44,17 14,00 13,90 27,11 43,08 15,92 At% 21,82 34,96 30,92 12,30 22,01 35,36 31,21 11,42 22,96 33,62 30,43 12,99 4. CONCLUSIONES El estudio de la variación de energía de superficies de silicona mediante APPT lleva a la observación de un aumento de la misma, que, aunque no es muy notable en cuanto a la componente total de la misma (27%), sí modifica increíblemente su componente polar en varios órdenes de magnitud. Esto se debe a la introducción en las cadenas poliméricas superficiales de funcionalidades polares tipo carbonilo, hidroxilo, amina o carboxilo, cuya naturaleza hidrófila genera un aumento en la mojabilidad de la silicona. Dicha mejora vendrá justificada únicamente por la existencia de estos grupos funcionales hidrófilos, pues el estudio mediante SEM revela que, pese a generar una superficie algo más lisa y homogénea, el efecto del APPT en la morfología de las muestras no sigue la misma tendencia que la energía de las mismas, al mantenerse aproximadamente constante en el tiempo de estudio. La corta durabilidad de los efectos de APPT (4 h), limita la posibilidad de uso industrial del plasma para el posterior pintado o pegado de silicona a un tratamiento en serie de los productos. XI CONGRESO NACIONAL DE MATERIALES 5. AGRADECIMIENTOS Se agradece la financiación de la Fundación Universidad Carlos III de Madrid e Instituto Tecnológico de Química y Materiales “Álvaro Alonso Barba”. 6. REFERENCIAS [1] Ragborg, W. H., Badger, W. H., Wear. 1 (1958) 352. [2] De Buyl, F.,“Silicone sealants and structural adhesives”, Int. J. Adh. Adh. 21 (2001) 411-422. [3] Williams, R. L., Wilson, D. J., Rhodes, N. P.,“Stability of plasma-treated silicone rubber and its influence on the interfacial aspects of blood compatibility”. Biomaterials. 25 (2004) 46594673. [4] Young T., “An Essay on the Cohesion of Fluids”. Philos, Trans. 95, 65 (1805). [5] Mittal K. L.,“Polymer Surface Modification: Relevance to Adhesion”, Vol. 5, Ed. VSP/Brill, Leiden (2009). [6] Mittal K. L.,“Contact Angle, Wettability and Adhesion”, Vol. 6, Ed. VSP/Brill, Leiden (2009). [7] Owens D. K., Wendt R. C., “Estimation of the free energy of polymers”. J. Appl. Polym. Sci. 13, 1741-47 (1961). [8] Kaelble D. H., J. Adhesion. 2, 66 (19709. [9] Braithwaite N. S. J., Plasma Sources Sci. Technol. 9, 441-54 (2000). [10] Mittal K. L.,“Polymer Surface Modification: Relevance to Adhesion”, Vol. 4, Ed. VSP/Brill, Leiden (2007). [11] Bárdos L., Baránková H.,“Plasma processes at atmospheric and low pressures”, Vacuum. 83, 522-27 (2009). 277 6. POLíMEROS CARACTERIZACIÓN SUPERFICIAL DE CANTOS DE CHAPA DE MADERA NATURAL PREENCOLADOS. INFLUENCIA DEL PRIMER EN EL PROCESO DE ADHESIÓN R. Fernández Reyes1, M. V. Navarro Bañón2, F.J. Melero Muñoz2, J.M. Martín Martínez1. Laboratorio de Adhesión y Adhesivos, Universidad de Alicante, 03080 Alicante [email protected] 2 Centro Tecnológico del Mueble y la Madera de la Región de Murcia (CETEM), C/ Perales s/n, 30510 Yecla, Murcia [email protected] 1 Resumen: Los cantos de madera natural permiten revestir tanto las molduras como los perfiles de tableros para protegerlos contra la humedad. La unión del canto al tablero debe ser duradera y resistir los esfuerzos mecánicos que experimenta durante la vida de uso del mueble. Ante la frecuente delaminación que se produce en los cantos unidos con adhesivos termofusibles, se ha propuesto la aplicación de un primer de un material celulósico impregnado de cola poliacetato de vinilo (PVA) al canto. Hasta donde sabemos, no se ha estudiado la interacción entre el primer de PVA aplicado a la chapa de madera y el adhesivo termofusible de EVA (copolímero de etileno y acetato de vinilo) aplicado para su unión al tablero, siendo éste el objetivo principal de este trabajo. Los resultados obtenido han mostrado que los cantos de madera natural comerciales llevan un primer de papel preencolado con diferentes cantidades de PVA, siendo el PVA el que se une al adhesivo termofusible. Para analizar el efecto de los distintos contenidos de PVA en la adhesión del canto de madera al tablero con adhesivos termofusible, se han depositado películas de diferente nivel de cubrimiento, analizando su incidencia en las propiedades de los cantos de madera virgen. Palabras clave: Canto de madera, tablero, adhesivo termofusible, PVA, EVA. 1. INTRODUCCIÓN Actualmente en la industria del mueble moderno se utilizan tableros de aglomerados de partículas para abaratar costes y tiempos de producción. La utilización de chapas de madera natural como recubrimiento de los perfiles de dichos tableros así como de molduras es habitual. Este recubrimiento se realiza no sólo con fines estéticos, sino que también sirve como protección de los mismos contra la humedad, la cual puede originar el desmoronamiento de la estructura del tablero. En el proceso de adhesión de cantos encolados a tableros participan tres componentes: el sustrato o soporte cuyo perfil se desea recubrir (habitualmente una moldura o un tablero, bien de partículas de fibras, de fibras de densidad media (DM), contrachapados, etc), el canto o material de recubrimiento (canto de PVC, canto de madera natural, etc) y el adhesivo (normalmente un adhesivo termofusibles o hot melt en base EVA). Las variables experimentales que más afectan al proceso de encolado son el tipo de tablero a encolar, el tipo de canto, el tipo de adhesivo, la temperatura de aplicación de la cola, y la velocidad de trabajo, entre otras [1]. Las delaminaciones o separaciones se observan a nivel industrial en un alto porcentaje de uniones de cantos encolados, lo que impide que se cumplan los requisitos de calidad necesarios para su utilización en mobiliario [2][3]. Se ha observado que las uniones que presentan un mayor número de separaciones son las que se realizan con cantos de madera natural. XI CONGRESO NACIONAL DE MATERIALES A pesar de que los cantos comerciales se cortan en la dirección de la veta de la madera para que posean una mayor resistencia mecánica, sin embargo son muy frágiles. Para reducir la fragilidad de los cantos de madera natural se suele aplicar un recubrimiento interno de una lámina de celulosa preencolada con un adhesivo. Esta lámina de celulosa actúa además como un primer o promotor de la adhesión frente a los adhesivos termofusibles (Figura 1). Estos adhesivos son bastante viscosos y se aplican a alta temperatura, enfriando rápidamente en contacto con la superficie del tablero y del canto de madera, por lo que presentan una pobre mojabilidad; la adición del primer de papel preencolado facilita el mojado inicial por el adhesivo [4]. Superficie exterior (vista) del canto Papel Adhesivo termofusible Tablero Figura 1. Esquema de la unión canto-adhesivo-tablero. Hasta donde conocemos, no se han realizado estudios de la interacción primer-canto de madera, ni primertablero, ni tampoco se ha analizado la incidencia de la aplicación de la lámina de papel preencolado en la adhesión de cantos de madera. Por tanto, el objetivo 278 6. POLíMEROS principal de este trabajo es mostrar los resultados preliminares obtenidos en la caracterización de cantos de madera natural preencolados mediante distintas técnicas experimentales así como la influencia del contenido de primer en el proceso de adhesión canto de madera-adhesivo termofusible de EVA. Este estudio se enmarca dentro del proyecto DEX-560550-2008-20 del Programa Nacional de Proyectos de Desarrollo Experimental-2008 en el que participan el Centro Tecnológico del Mueble de Murcia (CETEM) y el Laboratorio de Adhesión y Adhesivos de la Universidad de Alicante. 2. EXPERIMENTAL (Tokio, Japón), que se encuentra en los Servicios Técnicos de Investigación de la Universidad de Alicante. Se empleó un haz incidente de electrones de energía 20 KV. Las muestras se observaron sin recubrimiento de oro y su composición química se determinó mediante análisis EDX. Aplicación de cola blanca con varilla de gramaje Se aplicaron diferentes grosores (50 a 200 micras) de cola blanca sobre canto de madera natural para analizar la incidencia del grosor en las interacciones canto de madera– adhesivo termofusible de EVA. Para ello, se utilizaron varillas de distinto gramaje. 3. RESULTADOS EXPERIMENTALES El CETEM (Yecla, Murcia) suministró todos los materiales utilizados en este estudio. Cantos de chapa de madera comerciales Se han utilizado cuatro cantos de chapa de madera natural con papel preencolado (Figura 2), cuyas referencias son 40508/24, 40508/31, 40508/35, y 40508/36; estos cantos difieren en el tipo de madera natural y en su anchura. Cantos comerciales Se han analizado químicamente los cuatro cantos comerciales realizando espectros de IR de la cara de unión al adhesivo (Figura 3). 0.04 A bs Materiales 40508-24 40508/24 0.02 3340 2844 A bs 0.03 40508-31 40508/31 3334 0.02 A bs 0.03 Figura 2. Cantos comerciales de madera natural preencolado a papel. Componentes individuales de los cantos de madera Se proporcionaron por separado los tres componentes de los cantos de madera con papel preencolado: canto de madera natural sin papel (canto virgen), lámina de papel preencolado (papel) y cola blanca de poliacetato de vinilo (cola blanca) Técnicas Experimentales Espectroscopia infrarroja con transformada de Fourier (FTIR) Para identificar la composición química de los materiales se obtuvieron espectros IR utilizando un espectrómetro Alpha P (Bruker, Madrid). Los experimentos se han llevado a cabo en modo de reflectancia total atenuada (ATR). Todos los materiales son sólidos (la cola blanca se dejó curar a temperatura ambiente durante un día hasta la formación de película), y se analizaron empleando un prisma de germanio. Se realizaron 60 barridos con una resolución de 4 cm-1. Microscopía electrónica de barrido (SEM) Para evaluar la topografía de las muestras se empleó un microscopio electrónico de barrido Hitachi S-3000N XI CONGRESO NACIONAL DE MATERIALES 1103 1163 1411 1340 40508/36 1731 0.02 4000 1058 1033 2000 873 724 848 794 1372 1162 1241 1372 1340 1446 29222840 3000 665 873 755 1243 1104 1434 29212848 40508-36 3347 725 1058 1035 2852 1732 3345 0.01 40508/36 2918 1538 40508/35 0.04 -0.00 40508/35 1247 724 703 873 792 1162 40508-35 A bs 40508/31 1725 0.02 0.01 40508/24 1103 1432 1372 1340 2925 1057 1032 1240 1733 11061058 1033 1162 665610 657 725 671 895 874788 1000 Wavenumbers (cm-1) Figura 3. Espectros IR-ATR de cantos comerciales de madera natural preencolados a papel. La Figura 3 muestra que los cuatro cantos son químicamente similares entre sí. Presentan las bandas de tensión de –CH2 y –CH3 a 2925 y 2851 cm-1 y la banda a 725 cm-1 correspondiente al balanceo del metileno; también aparecen las bandas de tensión del doble enlace C=O del grupo acetato a 1733 cm-1 y las bandas de tensión simétrica y asimétrica del enlace sencillo de CO del grupo acetato a 1240 y 1032 cm-1. La comparación de los espectros IR-ATR de los cantos comerciales (lado a unir al adhesivo) con la base de espectros IR de la biblioteca existente en el Laboratorio de Adhesión y Adhesivos de la Universidad de Alicante, muestra que corresponden a una matriz celulósica de papel y a acetato de polivinilo (PVA) que tiene el papel. La principal diferencia entre los cuatro espectros IR de los cantos de madera se debe a la diferente intensidad relativa de las bandas del grupo acetato, lo que podría indicar que existe un diferente grosor de PVA procedente posiblemente de una cola blanca aplicada al 279 6. POLíMEROS Por tanto, los cantos de madera comerciales contienen diferente cantidad de PVA en la superficie del papel preencolado. Tabla 1 . Relación de intensidades de las bandas de C=O a 1730 cm-1 y de -CH2 a 725 cm-1 en los espectros IR-ATR de los cantos. 40508-CV Canto Virgen 0.02 3365 2916 1425 1320 1732 1506 1372 1238 1614 A bs 1236 1734 0.05 0.04 2923 14351372 1022 1116 1093 946 2844 40508-Papel Cara 1 Papel cara 1 3345 1161 1410 1340 0.1 4000 3344 874 2921 2848 Cola Blanca 3000 14351373 2000 660 615 1236 1735 2923 724 12471104 40508-17 0.2 789 725 1057 1034 1717 0.02 895 2844 Papel cara 2 -0.00 40508/31 1055 0.10 40508-Papel Cara 2 3354 Para comparar la topografía de las superficies de los cantos de madera natural por el lado a encolar, se realizaron micrografías SEM a media resolución (Figura 4). 40508/24 0.04 A bs Iλ=1730/Iλ=725 1.55 0.89 1.13 1.31 Para comprobar que la cola blanca que aparece en la superficie del papel aplicada al canto corresponde a PVA y para verificar la composición química de la superficie del canto expuesta al adhesivo termofusible, se realizaron espectros de IR-ATR de los componentes individuales del mismo, canto virgen, papel y cola blanca (Figura 5). A bs Canto de madera 40508/24 40508/31 40508/35 40508/36 Componentes individuales de los cantos de madera A bs papel (Figura 3). La diferente cantidad de PVA en los cantos de madera natural se ha cuantificado mediante la relación de la intensidad de las bandas IR del acetato de vinilo a 1740 cm-1 y de la matriz celulósica del papel a 725 cm-1 (Tabla 1). El contenido de PVA ordenado de mayor a menor es 40508/24 ≅ 40508/36 40508/35>>40508/31. 1123 1022 947 848 796 605 511 1000 Wavenumbers (cm-1) Figura 5. Espectros IR-ATR de los componentes de cantos de madera natural preencolado a papel. 200µm 40508/35 200µm 40508/36 200µm 200µm Figura 4. Micrografías SEM de cantos comerciales de madera natural preencolado a papel. Las micrografías SEM de las superficies a encolar de los cuatro cantos muestran el entramado de las fibras correspondientes al papel. En la superficie del canto 40508/31 se observa que la mayor parte de las fibras están expuestas a la superficie. Sin embargo, en el resto de las superficies de los cantos a encolar se aprecia que las fibras de papel están más recubiertas de cola de PVA, particularmente el 40508/35. Esto puede ser debido a la porosidad del papel que permite una difusión de la cola hacia la superficie a encolar o bien a que se aplica un exceso de cola. Por tanto, la superficie que está en contracto con el adhesivo termofusible para su unión al tablero corresponde principalmente a la cola de PVA más que a las fibras de papel, y ello en distinta magnitud dependiendo del tipo de cantos. XI CONGRESO NACIONAL DE MATERIALES El espectro IR-ATR del canto virgen fue realizado por ambas caras comprobando que el canto no contenía ningún tipo de tratamiento, siendo su composición química similar por ambas caras y correspondiendo a celulosa (componente básico de la madera natural). En el caso del papel también se realizaron los espectros IR-ATR por las dos caras no obteniéndose espectros similares. Una de las caras (cara 1) muestra bandas más intensas del papel mientras que el espectro IR-ATR de la cara 2 muestra mayor intensidad de las bandas correspondientes al PVA (tensión del doble enlace C=O y del enlace sencillo C-O). No obstante, en ambas caras aparecen las bandas de la cola de PVA. Se realizó también el espectro IR de la cola blanca destacando la presencia de las bandas de tensión del doble enlace C=O del grupo acetato a 1735 cm-1 y de las tensiones simétrica y asimétrica del enlace sencillo C-O del grupo acetato a 1240 y 1030 cm-1, respectivamente, las cuales aparecen en los cantos comerciales. La topografía del canto virgen se analizó mediante SEM, mostrando claramente el corte transversal de los vasos de la matriz de la madera (Figura 6). Las micrografías SEM de las dos caras del papel que presentaban distinta cantidad de cola de PVA se muestran también en la Figura 6. La cara 1 del papel es más parecida a la que muestran los cantos 40508/24 y 280 6. POLíMEROS 40508/31, pero diferente a la de los cantos 40508/35 y 40508/36. Canto Virgen En este trabajo se ha estudiado la interacción entre el primer de PVA y el canto de madera planteándose como futuro estudiar la diferente adhesión a tablero de partículas y de densidad media (DM) de cantos de madera virgen con distintas cantidades de PVA y observar cómo afecta la diferente cantidad de PVA a la adhesión con un adhesivo termofusible de EVA. 4. CONCLUSIONES 1mm Papel cara 1 - Los cantos de madera natural comerciales llevan un primer de papel preencolado con PVA. Papel cara 2 1mm - En cada tipo de canto comercial recubierto de cola de PVA existe un diferente nivel de recubrimiento, es decir aparece una cantidad diferente de fibras de papel expuestas al adhesivo termofusible. 1mm Figura 6. Micrografías SEM de canto de madera virgen y del papel preencolado (ambas caras). Dado que la superficie que se expone al adhesivo termofusible de EVA es la cola de PVA, y las cantidades de PVA son diferentes en los distintos cantos, se planteó estudiar como sistema modelo cómo afecta la cantidad de cola PVA aplicada sobre el canto de madera virgen. El experimento se diseñó aplicando distintas cantidades de cola de PVA sobre canto virgen mediante varillas de distinto gramaje. Se aplicaron cantidades de PVA equivalentes a un espesor de 50 µm, 100 µm y 150 µm. Las micrografías SEM que se han obtenido se muestran en la Figura 7. Canto Virgen CV+50µm 1 mm CV+100µm 1 mm - Lo que se expone a la adhesión con el adhesivo termofusible para la unión con el tablero es la cola de PVA. - Hasta 150 micras de grosor de PVA no se recubre totalmente la matriz del canto virgen de madera. - Se está estudiando cómo afecta la diferente cantidad de PVA en la adhesión a tablero con un adhesivo termofusible de EVA. 5. AGRADECIMIENTOS Este trabajo de investigación ha sido posible gracias a la financiación concedida al CETEM en el proyecto ENCCANTO (DEX-560550-2008-20) por parte del Ministerio de Ciencia e Innovación a través del Programa Nacional de Proyectos de Desarrollo Experimental-2008. 6. REFERENCIAS [1] CV+150µ [2] 1 mm 1 mm Figura 7. Micrografías SEM de canto virgen y canto virgen con distintas cantidades de PVA. [3] En la Figura 7 se muestra que al ir aumentando la cantidad de PVA aplicado al canto de madera virgen, van desapareciendo los surcos de los vasos de la madera virgen rellenándose de adhesivo. XI CONGRESO NACIONAL DE MATERIALES [4] Ortiz Gutiérrez J, “El encolado de cantos con colas termofusibles”, Asociación de Investigación de las Industrias de la Madera. www.infomadera.net Navarro Bañon, M. V; Melero Muñoz, F J; Azorín Soriano, C; Sanz Perpiñan J.M; Donate Robles, J; Martín Martínez J.M. , “Adhesivos termofusibles en base EVA para encolado de cantos en el sector del mueble y la madera. Problemas de delaminación de encolados”, Plásticos Modernos, 2009 (641). Norma UNE 53.433 “Tableros de partículas de madera melaminizados. Especificaciones y Métodos de Ensayo. Martín Martínez J.M., “Adhesivos. Volumen I”, Red CYTED VIII.D. Universidad de Alicante. 2001. 281 6. POLíMEROS PREPARACIÓN Y CARACTERIZACIÓN DE MEZCLAS TERNARIAS BASADAS EN POLI(ETILÉN TEREFTALATO), POLI(AMINO ÉTER) Y POLI(HIDROXI ÉTER DE BISFENOL A) A. Granado, J. I. Eguiazábal, J. Nazábal Dpto. de Ciencia y Tecnología de Polímeros e Instituto de Materiales Poliméricos POLYMAT, Facultad de Química, UPV/EHU, Paseo Manuel de Lardizábal, 3, 20018 Donostia-San Sebastián [email protected] Resumen: La incorporación de cantidades minoritarias de poli(hidroxi éter de bisfenol A) (fenoxi) produce un descenso en la temperatura de fusión (Tm) del poli(etilén tereftalato) (PET) que permite la obtención de mezclas con una resina de poli(amino éter) (PAE), superando las dificultades derivadas de las diferencias en las condiciones de procesado idóneas para cada uno de los polímeros. A pesar de la aparente miscibilidad que se deduce tras la obtención de un único pico de Tg en el análisis mecánico-dinámico, el análisis morfológico muestra la naturaleza bifásica de las mezclas ternarias. La buena adhesión entre fases y el pequeño tamaño de partícula dispersa, junto con la mayor orientación de las mezclas ternarias con respecto a los polímeros puros, justifican el favorable comportamiento a rotura de las mezclas, así como el sinergismo observado en las propiedades mecánicas de baja deformación. Palabras clave: mezclas poliméricas, procesado, compatibilidad, propiedades mecánicas. 1. INTRODUCCIÓN El PET es un polímero de notable importancia en los sectores del envasado y el embalaje [1]. Sus propiedades pueden ser optimizadas para estas aplicaciones por mezclado con una resina PAE, que presenta, entre otras, excelentes propiedades barrera frente a los gases. Sin embargo, las limitaciones en cuanto a las condiciones de procesado idóneas para cada uno de los polímeros, dificultan la obtención de estas mezclas potencialmente interesantes en el ámbito aplicado. Una modificación adecuada por medio de un compatibilizador puede solventar dicho problema permitiendo la preparación de las mezclas. En cualquier caso la elección del agente compatibilizador debe venir determinada por una mínima miscibilidad, o al menos compatibilidad, del mismo con los componentes de la mezcla básica a fin de evitar efectos negativos en las propiedades finales. En este trabajo se estudian las mezclas PET/PAE con contenidos mayoritarios de PET, previamente modificado mediante la adición de 20% de fenoxi a fin de posibilitar su procesado. La elección de la resina fenoxi se realizó en base a su similitud química con la PAE y su posibilidad de interacción/reacción con el PET [2,3]. 2. PARTE EXPERIMENTAL Los polímeros empleados, PET, PAE y fenoxi, son polímeros comerciales suministrados respectivamente por Brilén S.A., Dow Chemical y Union Carbide. El PET y la resina fenoxi fueron inicialmente mezclados en composición 80/20 en una extrusora bihusillo a 260ºC. La premezcla obtenida fue a su vez mezclada con la resina PAE, en contenidos hasta 40%, en un XI CONGRESO NACIONAL DE MATERIALES proceso directo de moldeo por inyección a 240ºC. Se estudió el comportamiento de fases, la morfología y las propiedades mecánicas de las mezclas mediante análisis mecánico-dinámico (DMA) y calorimetría diferencial de barrido (DSC), microscopía electrónica de barrido (SEM) y ensayos de tracción respectivamente. 3. RESULTADOS Y DISCUSIÓN El estudio preliminar de las mezclas binarias PETfenoxi y PAE-fenoxi reveló la parcial miscibilidad entre el PET y la resina fenoxi, que daba lugar a un descenso en la Tm del PET de aproximadamente 6ºC, así como a una favorable dispersión y elevada adhesión interfacial. Por otro lado, la compatibilidad entre el PAE y la resina fenoxi se vio reflejada igualmente en la homogeneidad y elevada adhesión entre las fases así como en la linealidad de las propiedades mecánicas de sus mezclas. Comportamiento de fases El comportamiento de fases de las mezclas ternarias PET-fenoxi/PAE se estudió mediante análisis mecánicodinámico debido a la cercanía de las Tg´s de la matriz PET-fenoxi (77ºC) y el PAE (73ºC). Como se puede observar en la Figura 1, todas las mezclas muestran un único pico de tan , similar al de la matriz, cuya temperatura tiende a disminuir al aumentar el contenido de PAE. La presencia de un único pico en las mezclas es en principio representativa de miscibilidad, que aparentemente se veía corroborada por la ausencia de reacciones entre los componentes, comprobada por espectroscopía infrarroja (FTIR), junto con el descenso de la Tm y la cristalinidad del PET en las mezclas. Sin embargo, y dada la cercanía de ambas Tg´s, la naturalezamono fásica o bifásica de las mezclas debió ser confirmada por SEM. 282 6. POLíMEROS 80-20/0 Tan δ 80-20/10 80-20/20 80-20/30 80-20/40 PAE 0 20 40 60 Temperatura (ºC) 80 100 (b) Figura 1. Barridos mecánico-dinámicos de las mezclas PET-fenoxi/PAE. Figura 2. Superficies de rotura criogénica de las mezclas PET-fenoxi/PAE con 10% (a) y 30% (b) de PAE. Morfología El estudio morfológico mostró la naturaleza bifásica de las mezclas ternarias. La morfología de las mezclas con 10% PAE (Figura 2a) era similar a la de la matriz PETfenoxi. Como se puede observar, las partículas de PAE son difícilmente visibles, presentan un pequeño tamaño (1 m aproximadamente) y están homogéneamente distribuidas y fuertemente adheridas a la matriz. Al aumentar el contenido de PAE (30%, Figura 2b), aumenta el número y tamaño de las partículas dispersas, aunque se mantienen las elevadas homogeneidad y adhesión interfacial. Además, se observan subpartículas ocluidas dentro de las partículas de PAE, características de mezclas con baja tensión interfacial entre los componentes [4]. La elevada compatibilidad deducida del análisis morfológico concuerda con los resultados del estudio térmico discutidos anteriormente. (a) XI CONGRESO NACIONAL DE MATERIALES Propiedades mecánicas Las propiedades mecánicas de baja deformación, módulo de Young y tensión de rendición, de las mezclas y del PET puro como referencia, se presentan en la Tabla 1. Como se puede observar, la adición de un contenido minoritario de fenoxi aumenta ligeramente la rigidez del PET. Las mezclas ternarias, por su parte, presentan en ambas propiedades una ligera desviación positiva con respecto a la linealidad entre los valores de la matriz PET-fenoxi y de la PAE pura. Tabla 1. Módulo de Young y tensión de rendición de las mezclas PET-fenoxi/PAE. PET-fenoxi/PAE PET 80-20/0 80-20/10 80-20/20 80-20/30 80-20/40 0/100 Módulo de Young (MPa) 1990 ± 70 2290 ± 30 2410 ± 50 2460 ± 40 2480 ± 80 2560 ± 40 2700 ± 25 Tensión de rendición (MPa) 49,8 ± 0,6 52,7 ± 0,2 56,2 ± 0,3 56,5 ± 0,7 57,3 ± 0,7 57,8 ± 0,4 57,6 ± 0,2 Para explicar el comportamiento del módulo elástico y la tensión de rendición se descarta la influencia de parámetros que pueden afectar a estas propiedades como la cristalinidad [5] que disminuye o el volumen libre [6] que sigue una tendencia lineal al aumentar el contenido de PAE. Sin embargo, la mayor orientación de las mezclas con respecto a la matriz PET-fenoxi y el PAE, reflejada en los valores de birrefringencia representados en la Figura 3, parece ser responsable de las ligeras desviaciones positivas observadas en estas propiedades. 283 6. POLíMEROS 1,6 4. CONCLUSIONES Birrefringencia (x10 3) 1,4 1,2 1,0 0,8 0,6 0,4 0,2 0,0 0 20 40 % PAE 60 80 100 Figura 3. Birrefringencia de las mezclas PETfenoxi/PAE. La ductilidad de las mezclas se muestra en la Figura 4. Como se puede observar, la adición de fenoxi al PET prácticamente no modifica su ductilidad. Sin embargo, la ductilidad de las mezclas ternarias disminuye al aumentar el contenido de PAE, aunque independientemente del contenido de este polímero las mezclas presentan una naturaleza claramente dúctil, con deformaciones a rotura superiores al 50%. 250 Ductilidad (%) 200 150 5. AGRADECIMIENTOS Los autores agradecen la financiación proporcionada por el Gobierno Vasco (Proyecto GIC07/48-IT-234-07). A. Granado agradece la ayuda otorgada por la Universidad del País Vasco para la realización de este trabajo. 6. REFERENCIAS 100 [1] 50 0 La adición de 20% de fenoxi reduce la Tm del PET en una medida suficiente para permitir el procesado de sus mezclas con contenidos minoritarios de PAE. La aparente miscibilidad de las mezclas ternarias, deducida de la presencia de un único pico en los ensayos mecánico-dinámicos, no se corrobora en el estudio morfológico. Sin embargo, y a pesar de la naturaleza bifásica de las mezclas, el pequeño tamaño de partícula dispersa, la baja tensión interfacial y la elevada adhesión entre fases ponen de manifiesto la elevada compatibilidad entre los componentes y justifican sus buenas propiedades a rotura. Por otro lado, el aumento de la orientación de las mezclas con respecto a los componentes puros parece ser responsable del ligero sinergismo observado en las propiedades mecánicas de baja deformación. [2] 0 20 40 % PAE 60 80 100 Figura 4. Ductilidad de las mezclas PET-fenoxi/PAE. Este comportamiento mecánico resulta muy favorable teniendo en cuenta que corresponde a un sistema multifásico procesado en condiciones que no resultan las más idóneas para sus componentes. La buena respuesta del sistema a altas deformaciones se atribuye a la baja tensión interfacial y elevada adhesión entre las fases, que permite una efectiva transmisión de esfuerzos entre la matriz y la fase dispersa. XI CONGRESO NACIONAL DE MATERIALES [3] [4] [5] [6] Paul, D. R., Bucknall, C. B., “Polymer Blends”, Ed. John Wiley & Sons, 2000. Cortázar, M., Eguiazábal, J. I., Iruin, J. J., Eur. Polym. J., 30, 901 (1994). Gao, G., Zhang, L., Sun, H., Chen, G., Zhang, M., Ma, R., Liu, F., J. Appl. Polym. Sci., 97, 878 (2005). Granado, A., Eguiazábal, J. I., Nazábal, J., J. Appl. Polym. Sci., 109, 3892 (2008). Hara, M., Sauer, J. A., J. Macromol. Sci. Rev. Macromol. Chem. Phys., C38, 327 (1998) Barlow, J. W., Paul, D. R., Polym. Eng. Sci., 21, 985 (1981) 284 6. POLíMEROS DEPÓSITO ELECTROASISTIDO DE MATERIALES SOL-GEL: POLÍMERO HÍBRIDO INTERPENETRANTE SÍLICE-PANI POR INSERCIÓN REACTIVA ELECTROQUÍMICA D. Salinas-Torres1, F.Montilla1, F.Huerta2, E.Morallón1 Dpto. de Química-Física e Instituto Universitario de Materiales, Universidad de Alicante, Ap. 99, E-03080, Alicante, España, [email protected] 2 Dpto. de Ingeniería Textil y Papelera, Univ. Politécnica de Valencia, E-03801, Alcoy, España 1 Resumen: En este trabajo se ha realizado un depósito electroquímico de capas porosas de sílice sobre electrodos de carbón vítreo usando la metodología sol-gel. Estas capas actúan como plantilla para el crecimiento de polianilina (PANI). La síntesis de los materiales compuestos sílice-PANI se ha realizado por inserción reactiva mediante electroxidación del monómero. Los materiales híbridos se han caracterizado mediante microscopía electrónica de barrido y mediante métodos electroquímicos (cronoamperometría y voltametría cíclica). Se han obtenido los valores de capacidad y además, se ha realizado un estudio de la cinética de polimerización del polímero conductor sobre los electrodos modificados con sílice. Palabras clave: materiales compuestos, sílice, polianilina, electrodepósito, carbón vítreo. 1. INTRODUCCIÓN Los nanomateriales híbridos están siendo objeto de un intenso estudio debido a que la combinación de materiales de diferente naturaleza puede resultar en una mejora de las características fisicoquímicas respecto a los materiales individuales (actividad catalítica, estabilidad ambiental, etc). En el caso de materiales basados en sol-gel, la sílice ha sido empleada como plantilla para el crecimiento de diversas entidades moleculares [1,2] debido a que presenta porosidad, elevada estabilidad térmica y morfología modulable. Entre los materiales que se han combinado con la sílice se encuentran los polímeros conjugados (polianilina, PEDOT, polipirrol, etc), que presentan buena conductividad eléctrica y actividad electroquímica. De este modo, se obtiene un material compuesto que combina las propiedades de ambos. El material híbrido de sílice-polímero conductor se puede sintetizar por inserción heterogénea, inserción homogénea e inserción reactiva (electroquímica). Durante los últimos años, los polímeros conjugados han sido objeto de estudio en diversos campos como el de los supercondensadores, los sensores, las baterías o los dispositivos microelectrónicos. En muchas de estas aplicaciones la morfología juega un papel importante y, por ello, la combinación con un material como la sílice abre la posibilidad de modular las propiedades del material resultante modificando las condiciones de síntesis (pH, presencia de surfactante, etc). Por otro lado, en los materiales compuestos se pueden incorporar funcionalidades químicas mediante el empleo de monómeros modificados. 2. EXPERIMENTAL La disolución sol-gel precursora se preparó mezclando 2 ml de tetraetoxisilano (TEOS) + 2.75 ml etanol + 2ml de una disolución 0.46M KCl+0.01M HCl. La mezcla se agita en un baño de ultrasonidos durante 15 minutos XI CONGRESO NACIONAL DE MATERIALES en un vial cerrado (formación de 1 fase, pH =2). En el electrodepósito de la sílice se utilizó una célula electroquímica de 3 electrodos. Como electrodo de trabajo se empleó una varilla de carbón vítreo y un hilo de Pt que actuó como contraelectrodo. Se utilizó un hilo de plata como pseudo-referencia durante la síntesis electroquímica del gel y un electrodo reversible de hidrógeno (RHE) en los demás experimentos. Sin embargo, todos los potenciales se han referido a la escala RHE. El depósito de la sílice se realizó a potencial constante de -1.2V (RHE) durante 60s. Figura 1. Voltagrama cíclico estabilizado de un electrodo de carbón vítreo en la disolución sol-gel precursora de la sílice. Una vez el electrodo de carbón vítreo se ha modificado con sílice, se introduce en una celda electroquímica de 3 electrodos que contiene 0.1M anilina hidrocloruro en 0.5M H2SO4 como electrolito. Como contraelectrodo se utiliza un hilo de Pt y como electrodo de referencia un RHE introducido en el mismo electrolito. El crecimiento 285 6. POLíMEROS de la polianilina se realizó mediante salto potenciostático a 1.05V. Los materiales híbridos han sido caracterizados mediante microscopía electrónica de barrido utilizando un equipo SEM JEOL JSM-840. 3. RESULTADOS Y DISCUSIÓN La Figura 1 muestra el voltagrama obtenido con un electrodo de carbón vítreo en la disolución sol-gel precursora. Podemos observar que para un potencial por debajo de -0.6V se observa una corriente de reducción importante, consecuencia de la generación de hidrógeno. Por tanto, durante la aplicación de un potencial de reducción de -1.2V constante, se produce un aumento del pH en las proximidades del electrodo desde un valor de pH=2 (en el que los coloides se encuentran dispersos) hasta valores entre 4 y 7. Este aumento de pH provoca que los coloides dispersos se agreguen quedando depositados sobre la superficie del electrodo. La Figura 2 muestra las cronoamperometrías obtenidas durante el crecimiento potenciostático de polianilina para un electrodo de carbón vítreo modificado con sílice (SiO2/GC) y para un electrodo sin modificar (GC). Podemos observar que la cinética de crecimiento de la PANI en el electrodo sin modificar (GC) es diferente al electrodo modificado con sílice (SiO2/GC). El electrodo sin modificar presenta una cinética muy rápida como muestra el crecimiento exponencial de la corriente. La forma de la curva de crecimiento indica la existencia de una cinética autocatalítica en la que la PANI ya formada cataliza la oxidación de los monómeros de anilina que se van incorporando a la cadena. Figura 2. Cronoamperogramas en una disolución 0.1M anilina hidrocloruro + 0.5M H2SO4. Ei= +0V (RHE), Ef = +1.05V (RHE), Qf(GC) = 5.67 mC, Qf(GC/Sílice) = 72.6 mC. XI CONGRESO NACIONAL DE MATERIALES Figura 3. Esquema de la evolución del crecimiento de PANI en el interior de la sílice. Por otra parte, para el electrodo modificado con sílice, se observa un régimen inicial de crecimiento lento en las zonas a y b y un crecimiento exponencial en las zonas c y d (Figura 2). En los instantes iniciales de polimerización existe un confinamiento de la PANI en el interior de la sílice, viéndose la cinética ralentizada por la dificultad que tienen los monómeros para difundir a través de los poros de la sílice. La forma de crecimiento cambia a partir de un tiempo crítico (TC) convirtiéndose en autocatalítico [3] como se observa en las zonas c y d. Este cambio en la tendencia está relacionado con la emersión del polímero conductor, PANI, sobre los poros de la sílice (Figura 3). La Figura 4 muestra las imágenes SEM del electrodo modificado con sílice en que se aprecian coloides de un tamaño que oscila entre 100 y 400 nm. Figura 4. Micrografía SEM de un electrodo GC/Sílice. 286 6. POLíMEROS En la Figura 5 se muestra la imagen SEM de un electrodo modificado con sílice sobre el que se ha crecido polianilina. En esta figura se aprecian aglomerados globulares de PANI sobre la superficie de la sílice. Debido a que la sílice no es conductora, la PANI sólo puede haber emergido a través de los poros como se indica en el esquema de la Figura 3. La Figura 7 muestra la variación de la intensidad del pico de oxidación a 0.4V, correspondiente al primer proceso de oxidación de la polianilina, con la velocidad de barrido para GC/PANI y GC/sílice-PANI. En el caso del electrodo no modificado con sílice (GC/PANI), la tendencia es lineal, como era de esperar para un proceso capacitativo. Sin embargo, para un electrodo modificado con sílice aparecen dos zonas claramente diferenciadas. La zona comprendida entre 10 y 200 mVs-1 y la zona entre 500 y 2000 mVs-1. Esta desviación que observamos a velocidades altas parece ser debida a los problemas de difusión que tienen los aniones del electrolito para compensar la carga en el proceso de oxidación del polímero, apoyando así la hipótesis de que la PANI está confinada en el interior de la matriz de sílice. Figura 5. Micrografía SEM de GC/Sílice-PANI. Los electrodos modificados con sílice y sin modificar donde se ha crecido polianilina (GC/Sílice-PANI y GC/PANI) se han caracterizado mediante voltametría cíclica. La Figura 6 muestra los perfiles voltamétricos de ambos electrodos en 0.5M H2SO4 y para una misma cantidad de polianilina depositada. Se observa que las características electroquímicas de la PANI, sus procesos de dopado y desdopado, no se ven modificados aunque el polímero esté confinado en el interior de los poros de la sílice. Esto nos indica que el área electroactiva expuesta a la disolución es mayor para el electrodo modificado con sílice. Figura 6. Voltagramas cíclicos de los electrodos GC/sílice-PANI y GC/PANI en 0.5M H2SO4. XI CONGRESO NACIONAL DE MATERIALES Figura 7. Evolución de la intensidad de corriente a +0.4V con la velocidad de barrido para GC/PANI y GC/Sílice-PANI. A partir de las pendientes de las rectas I(A) frente (mVs-1) se puede calcular la capacidad de cada material. Además, a partir de la carga voltamétrica se puede determinar la masa de PANI depositada, suponiendo que por cada monómero de anilina oxidada interviene un electrón. De esta forma se pueden obtener los valores de capacidad específica (referida al área geométrica) y másica (referida a masa de PANI) para electrodos modificados con sílice y sin modificar. Así, la Tabla 1 muestra los valores de capacidad para la misma masa de polianilina para tiempos de depósito inferiores al TC y para ambos electrodos (modificados con sílice y sin modificar). En la Figura 8 se compara los valores de capacidad másica para ambos tipos de electrodos. Podemos ver que, para pequeñas cantidades de PANI depositada, la capacidad obtenida es muy similar. Esto parece indicar que el número de núcleos de crecimiento es equivalente en ambos electrodos y que no hay un efecto claro de la capa de sílice. 287 6. POLíMEROS Tabla 1. Masa de polianilina depositada, capacidad másica y capacidad específica de difentes electrodos GC/Sílice-PANI (Tipo A) y GC/PANI (Tipo B). Tipo PANI /µg C específica/mF.cm-2 C másica/F.g-1 A1 0.678 4 463 B1 0.678 5 509 A2 1.355 10 497 B2 1.355 9 475 A3 2.711 19 481 B3 2.711 17 435 A4 5.413 38 491 B4 5.413 22 290 A5 10.501 56 373 B5 10.501 34 227 A6 15.274 86 395 B6 15.274 51 234 A7 19.092 102 377 B7 19.092 53 195 Sin embargo, cuando se incrementa la masa de PANI depositada, el valor de capacidad para GC/Sílice-PANI es mayor que para GC/PANI. Además, la capacidad másica de este último material disminuye mucho más rápidamente que para el electrodo con presencia de sílice. Este resultado puede deberse a que conforme crecen las cadenas de polianilina sobre el electrodo de GC, se produce el colapso y provoca una disminución del área electroactiva de la PANI. Sin embargo, la presencia de las capas porosas de sílice evita parcialmente el colapso entre cadenas, además hace que la polianilina sea más porosa aumentando el área electroactiva, y por tanto, la capacidad disminuye más lentamente manteniéndose en un valor elevado. 4. CONCLUSIONES Se han preparado capas de sílice sobre electrodos de carbón vítreo mediante métodos electroquímicos. La sílice sintetizada electroquímicamente se ha utilizado como molde para el crecimiento de polianilina mediante inserción reactiva. Sobre substratos GC/sílice se observan dos regímenes de crecimiento para la polianilina. En el primero ocurre un fenómeno de inducción, mientras que es en el segundo cuando se observa el crecimiento autocatalítico del polímero. Este segundo régimen es similar al observado para el crecimiento de polianilina sobre un electrodo de carbón vítreo sin recubrir con sílice. Los valores de capacidad másica y específica son mayores en los materiales híbridos de Sílice/PANI que en la PANI, al parecer, por el efecto de crecimiento interpenetrante de la PANI a través de los poros de la sílice. Estos valores indican el aumento de área superficial en contacto con la disolución al actuar la sílice como plantilla, evitando el colapso de las cadenas de polianilina y dando lugar a un polímero más poroso. 5. AGRADECIEMIENTOS Los autores agradecen el apoyo financiero del MICINN y FEDER (MAT2007-60621).F.M. agradece al MICINN la financiación de su contrato Ramón y Cajal. 6. REFERENCIAS [1] Figura 8. Evolución de la capacidad másica para GC/PANI y GC/Silica-PANI respecto cantidad de PANI depositada. XI CONGRESO NACIONAL DE MATERIALES [2] [3] Walcarius, A.; Mandler, D.; Cox, J. A.; Collinson, M.; Lev, O.; J. Mater. Chem., Y1 - 2005/// 2005, 15 (35-36), 3663-3689. Collinson, M.M.;.Accounts of Chemical Research, 2007, 40 (9), 777-783. Montilla, F.; Cotarelo, M.A.; Morallon, E. J. Mater. Chem., 2009, 19 (2), 305-310. 288 6. POLíMEROS OPTIMIZACIÓN DE LAS PROPIEDADES ADHESIVAS EN UNIONES DE POLIETILENO TRATADO CON PLASMA ATMOSFÉRICO O. Fenollar1, L. Sánchez1, D. García1, J.F. Balart1, V. Fombuena1 1 Instituto de Tecnología de Materiales (ITM), Universidad Politécnica de Valencia, Plaza Ferrandiz y Carbonell s/n, 03801, Alcoy, Alicante, [email protected] Resumen: Industrialmente los principales métodos para mejorar este comportamiento marcadamente hidrofóbico de las poliolefinas debido a su carácter no polar, se basan en la modificación de la superficie. El polietileno de baja densidad presenta valores muy bajos de energía superficial, por debajo de 30 mJ.m-2, que dificulta el proceso de adhesión durante el conformado de este tipo de materiales. Los polímeros tratados con plasma atmosférico presentan un aumento de sus propiedades de humectabilidad ya que el proceso se caracteriza por modificar la funcionalidad superficial, aportándole un carácter hidrofílico. Después del tratamiento de plasma atmosférico, la superficie del polímero sufre un fenómeno de funcionalización por la formación de varios grupos polares que contienen oxígeno, como grupos ester, alcohol, cetona, hidroperóxido, peróxido, ácido carboxílico,… Estos radicales libres formados en la superficie tratada del polímero pueden reaccionar con especies reactivas presentes en la atmósfera modificando químicamente la superficie lo que mejora sus propiedades adhesivas. Palabras clave: plasma atmosférico, humectabilidad, adhesión, hidrofílico, olefinas. 1. INTRODUCCIÓN El comportamiento químico o la naturaleza química de la superficie de los sustratos sólidos afecta directamente a su comportamiento en muchas de sus aplicaciones al servicio de la industria. Este fenómeno ha suscitado un creciente interés dentro de la comunidad científica, tanto a nivel académico como industrial, en el desarrollo de nuevas tecnologías o procesos que modifiquen las superficies de los sustratos sólidos sin alterar sus propiedades generales. En las últimas décadas se han investigado distintas tecnologías de modificación superficial de sustratos sólidos mediante tecnologías de tipo químico, térmico o eléctrico. Uno de los tratamientos más interesantes es el basado en la tecnología de plasma, ya que permite modificaciones superficiales de sólidos en condiciones medioambientalmente correctas al no emitir ningún tipo de residuos dañinos, además de ser fácilmente adaptable a los procesados de materiales a nivel industrial. Una de las poliolefinas de mayor demanda es el Polietileno de Baja Densidad LDPE, debido la óptima combinación de propiedades/procesado/precio que presenta como buena resistencia térmica y química, buena resistencia al impacto, flexibilidad de diseño y de acabado, muy buena procesabilidad. Pero uno de los mayores defectos frente a solicitaciones tecnológicas es la pobre adherencia que presenta en uniones con otros materiales, debido a sus inherentes estructuras químicas, lo que conlleva a la necesidad de un pretratamiento del polímero para ser recubierto, laminado, unido…. Industrialmente los principales métodos para mejorar este comportamiento marcadamente hidrofóbico de las poliolefinas debido a su carácter no polar, se basan en la modificación química de la superficie. Hay que tener en cuenta que el LDPE presenta valores muy bajos de energía superficial, por debajo de 30 mJ.m-2, que XI CONGRESO NACIONAL DE MATERIALES dificulta el proceso de adhesión durante el conformado de este tipo de materiales. Uno de los tratamientos más interesantes a nivel industrial, que permite aumentar las propiedades de adhesión del polímero al aumentar su energía superficial, es el tratamiento de plasma atmosférico. Los polímeros tratados con plasma atmosférico presentan un aumento de sus propiedades de humectabilidad ya que el proceso se caracteriza por modificar la funcionalidad superficial del polímero, aumentando su energía superficial (s) aportándole un carácter hidrofílico. Este efecto se debe a la formación de radicales libre sobre la superficie polimérica tratada., debido a la interacción de las cadenas del polímero con las especies ionizadas que ha creado el tratamiento con plasma atmosférico y que proceden del aire. Estos radicales libres formados en la superficie tratada del polímero pueden reaccionar con especies reactivas presentes en la atmósfera modificando químicamente la superficie y por lo tanto su comportamiento hidrofóbico inicial lo que mejora sus propiedades. [1,4] 2. EXPERIMENTAL 2.1. Materiales Las planchas poliméricas utilizadas en este estudio son de Polietileno de Baja Densidad ALCUDIA PE-019 suministrado por Repsol YPF (Madrid, España), en forma de granza sin aditivos, especialmente indicado para moldeo por inyección debido a su baja viscosidad, con un Indice de fluidez (190ºC y 2.16 kg) de 20 g/10min y densidad de 919 kg/m3. las planchas de LDPE se inyectaron en una inyectora industrial suministrada por Mateu-Solé, mod. 270/5 (Mateu-Solé S.A., Barcelona, España). Las dimensiones de las probetas inyectadas en forma de lámina son 160x60x2 mm. 289 6. POLíMEROS El adhesivo utilizado en la caracterización de a uniones LDPE/LDPE es un un poliuretano monocomponente de alta densidad comercializado por Kefren: Poliuretano 801 Blanco. 2.2. Tratamiento de plasma atmosférico Las planchas de polietileno de baja densidad fueron expuestas al tratamiento superficial de plasma atmosférico. El generador de plasma opera a 50/60 Hz , 230 voltios y 16 A. El equipo ha sido suministrado por PLASMATREAT GmbH, mod. FG 3001 (Steinhagen, Alemania). Todas las muestras se tratan con plasma atmosférico a 6, 10, 14 y 20 mm de distancia tobera/sustrato y a velocidades de pasada desde 100 a 1000 mm/s. 2.3. Instrumental Para evaluar la humectabilidad se realizan las medidas de ángulos de contacto mediante el goniómetro Easydrop Standard, mod. FM 40 (KRÜSS GmbH Instruments, Hamburg, Alemania). Con este equipo es posible evaluar de forma cuantitativa el carácter hidrofílico de ls laminas de LDPE tratadas con plasma atmosférico. A partir de estos datos, se aplica el método de Owens-Wendt para calcular las enregias superficiales, ya que es un métode sencillo que además prmite evaluar las componentes polar y dispersiva de la energía superficial. La expression utilizada, gráficamente representa una ecuación del tipo y = a + bx, y es la siguiente: ( ) γ + θ γ = γ γ ( ) γ ( ) + γ In this equation, θ is the contact angle, l is the surface tension of the liquid, and γs is the surface tension of the solid or surface free energy. The terms with the subscripts d and p refer to the dispersive and polar component respectively. Surface morphology of low density polyethylene sheets treated with atmospheric plasma were evaluated with microscopio electrónico de barrido SEM, suministrado por FEI, modelo PHENOM (FEI Company, Eindhoven, The Netherlands), con un voltaje de aceleración de 5KV. Los cambios en la topografia superficial de las superficies tratadas es evaluado mediante microscopía de fuerza atómica (AFM) usando un microscopio Multimode AFM con un controlador Nanoscope IIIa ADCS (Veeco Metrology Group, Cambridge, Inglaterra). y un cantilever de silicio (NanoWorld Pointprobe ® NCH) con una fuerza constante de 42 N·m-1 y una frecuencia de resonancia de 320kHz. En el análisis de las imágenes se evaluó la rugosidad (Rrms) en una dimensión de 20m x 20m. Los ensayos de pelado en T para la evaluación de la resistencia de las uniones LDPE/LDPE, se realizan en una máquina universal de ensayos IBERTEST ELIB 30 (S.A.E. Ibertest, Madrid, España) a una velocidad de 300 mm/min a temperatura ambiente y con la célula de XI CONGRESO NACIONAL DE MATERIALES carga de 5 kN, aplicando los criterios de la norma UNEEN 1895. 3. RESULTADOS Y DISCUSIÓN La acción del plasma atmosférico sobre las láminas de LDPE estudiadas provoca la funcionalización de la superficie del polímero por la interacción de las especies ionizadas del aire que genera el plasma, aumentando la humectabilidad de las mismas, pero dicho aumento es función de las condiciones de aplicación del tratamiento de modificación superficial. Por este motivo, en primer lugar, se optimizan los parámetros de procesado con plasma atmosférico: velocidad de pasada y distancia tobera/sustrato, mediante el análisis de la variación de los ángulos de contacto como principal indicador de la hidrofilidad o hidrofobicidad de la superficie de los sólidos. Las medidas de los ángulos de contacto sobre láminas de LDPE tratado con plasma atmosférico a diferentes distancias de la tobera a la superficie polimérica, y para distintas velocidades de pasada, utilizando diferentes líquidos de medida que permiten el estudio de los cambios de humectabilidad del LDPE en función de las condiciones de aplicación del plasma. Hay que tener en cuenta que la lámina de LDPE sin tratamiento presenta un fuerte carácter hidrofóbico cuantificado por los altos valores de ángulos de contacto obtenidos con los cuatro líquidos de medida utilizados. El estudio de los datos obtenidos permite establecer de forma general cómo cualquiera que sea el líquido de medida utilizado, los ángulos de contacto alcanzan los mínimos valores en condiciones de baja velocidad de aplicación del tratamiento y poca distancia entre la superficie polimérica a tratar y la tobera del plasma atmosférico. Mediante la aplicación del método de Owens-Wendt se calculan los valores de energía superficial de las diferentes muestras obtenidas, a partir de los valores de ángulos de contacto anteriormente citados, como parámetro cuantificador del comportamiento frente a humectabilidad de los sólidos. La figura 1 muestra la variación de las energías superficiales (s) de las superficies tratadas de polietileno de baja densidad como función de la velocidad de pasada y de la distancia tobera/sustrato. Para condiciones de procesado a pequeña distancia (6 mm) entre la boquilla del plasma y la superficie del LDPE se observa un importante aumento de la energía superficial para el rango de bajas velocidades de pasada (100, 200 y 300 mm s-1) de aplicación del plasma, ya que pasa de 27.44 mJ m-2 de la muestra sin tratar a valores de entre [63, 62 mJ m-2] para estas velocidades, lo que supone un incremento del 127%. Gráficamente se observa como el aumento de la velocidad de pasada en la aplicación del plasma atmosférico hace disminuir de forma considerable los valores de la energía superficial, que para velocidades altas (900, 1000 mm s-1) son de 34 mJ m-2, pero comparativamente con la muestra sin tratar supone un aumento de alrededor de 25%. Si se aumenta la distancia entre la tobera y la superficie de la muestra, 20 mm, este mismo efecto anterior se ve menos favorecido, ya que para las condiciones de más 290 6. POLíMEROS baja velocidad de pasada (100 mm s-1) el aumento de la energía superficial respecto a la muestra sin tratar es sólo del 42%, que todavía es menor para 200 mm s-1, 25%, y a partir de 600 mm s-1 los valores de la energía superficial son de la misma magnitud que los que presenta la muestra sin tratar, como se observa en la gráfica anterior. De forma general se observa que la modificación superficial de las láminas de LDPE mediante la aplicación de plasma atmosférico, permite una óptima funcionalización de la superficie del polímero en condiciones de baja velocidad de pasada y poca distancia tobera/sustrato. 85 0n m 0nm Figura 3. Imagen AFM en 2D y 3D de la superficie del LDPE tratado a una distancia de 6mm y a una velocidad de pasada de 100mm/s. (Escala 20 µm x µm 20) La agresividad del tratamiento se ve acentuada con la menor distancia entre tobera/substrato y por otra parte con la menor velocidad de tratamiento, ya que el tiempo de permanencia de la muestra sobre el foco de plasma es mayor. Esto se corrobora con la elevada rugosidad media alcanzada con una muestra a 6mm de distancia y una velocidad de 100 mm/min: 123.64 nm. Por otra parte se ve la diferencia de agresividad del proceso si se compara con el valor obtenido por una muestra tratada a 20mm y una velocidad de 1000mm/min: 24.92 nm. La pérdida de masa sufrida por los substratos viene a afirmar lo observado mediante la técnica de AFM. Una de las técnicas más exactas en la cuantificación del fenómeno de microetching en la superficie de las muestras tratadas con plasma es la microscopía de fuerza atómica (AFM). La figura 2 y 3 muestra comparativamente los perfiles de rugosidad para muestras no tratadas y tratadas con plasma atmosférico para una distancia de 6 mm y 100 mm s-1 velocidad de pasada. La superficie de la muestra sin tratamiento se caracteriza por presentar un perfil de rugosidad suave, sin picos, mientras que el perfil de la muestra tratada presenta picos más marcados, indicadores de una alta rugosidad superficial provocada por el efecto de la abrasión del plasma sobre la superficie polimérica. Para velocidades de pasada mayores, el perfil de la rugosidad presenta menor densidad de picos y menos pronunciados. 8 50 nm 0nm Figura 2. Imagen AFM en 2D y 3D de la superficie del LDPE sin tratamiento. (Escala 20 µm x µm 20) XI CONGRESO NACIONAL DE MATERIALES Para comprobar la mejoría en las propiedades de adhesión tras el proceso de plasma atmosférico se han realizado ensayos de pelado en T de las muestras tratadas en función de las condiciones del tratamiento, es decir, diferentes velocidades de paso y diferentes distancias tobera/sustrato polimérico. 180 6mm 10mm 14mm 20mm 160 Fuerza máxima de pelado (N) Figura 1. Variación de la energía superficiale (s) sobre polietileno de baja densidad tratado con plasma atmosférico en función de la velocidad de pasada y de la distancia tobera/sustrato. 140 120 100 80 60 40 0 200 400 600 800 1000 velocidad tratamiento (mm/min) Figura 4. Variación de la fuerza máxima de pelado (N) en uniones adhesivas LDPE/LDPE tratado con plasma atmosférico en función de la velocidad de pasada y de la distancia tobera/sustrato. En la figura 4, se observa que para distintas velocidades de pasada y alta distancia boquilla/substrato, se obtienen las menores resistencias de adhesión, producidas entre las superficies de las muestras tratadas mediante plasma atmosférico y el adhesivo. Por el contrario, se obtiene un mayor efecto del tratamiento de plasma atmosférico, para velocidades bajas y poca distancia boquilla/substrato, en 291 6. POLíMEROS concreto los mejores resultados se obtienen para una altura boquilla/substrato de 6 mm y velocidades de pasada de 100 a 300 mm/s. Para ver los mecanismos de adhesión en las intercaras substrato/adhesivo y poder analizar la causa de la variación de resistencias obtenidas, se observa macroscópica y microscópicamente las superficies de fractura de las muestras ensayadas. El aspecto visual de las muestras es diferente en función de la velocidad de pasada o la distancia a la que han sido tratadas las muestras. La morfología de las superficies de fractura en función de las condiciones de tratamiento se observan en la figura 5. El aspecto visual de las muestras es diferente en función de la velocidad o la distancia a la que han sido tratadas las muestras. misma. La segunda fotografía, muestra el detalle de la rotura del poliuretano de una muestra tratada a una velocidad de 100mm/s y 6 mm. En ella se puede observar formaciones circulares y rugosas que surgen como motivo del agarre del adhesivo a ambas superficies, indicativas de mayor adhesión, y correspondientes a rotura cohesiva del propio adhesivo. Estas formaciones circulares corresponden a la rotura de la estructura interna del propio poliuretano y corresponden a las celdillas propias de su carácter elastomérico como se puede observar con mayor detalle en la figura 7. A medida que la velocidad del tratamiento aumenta, esta morfología de rotura del PU, se va sustituyendo por zonas de deslizamiento del adhesivo, o zonas donde el adhesivo no se ha adherido correctamente al sustrato. Figura 7. Tipos de morfologías de las superficies de fractura de las uniones LDPE/LDPE. (x500) 4. CONCLUSIONES Figura 5. Fotografía que muestra los diferentes aspectos morfológicos de las muestras tratadas a una distancia de 6mm y diferentes velocidades de paso, comparadas con una muestra sin tratamiento. En la serie analizada se observan dos tipos de morfologías claramente diferenciadas y que se repiten en todas las series estudiadas: una de aspecto liso, uniforme y homogéneo; y otra más rugosa e irregular, que se pueden ver con mayor detalle en la figura 6. El polietileno de baja densidad tratado con plasma atmosférico presentan un interesante aumento de sus propiedades de humectabilidad ya que el proceso permite la modificación superficial del polímero, aumentando su energía superficial (s) aportándole un carácter hidrofílico, que mejora de forma óptima sus propiedades adhesivas. La utilización de los parámetros óptimos del tratamiento de plasma atmosférico: altura boquilla/substrato de 6 mm y velocidades de pasada bajas, de 100 mm/s a 300 mm/s, permiten la obtención de las mayores resistencias a la rotura de las muestras de LDPE ensayadas a pelado, con fracturas de tipo cohesivo. 5. AGRADECIMIENTOS Los autores agradecen al “Ministerio de Ciencia y Tecnología”, Ref: DPI2007-66849-C02-02 y Generalitat Valenciana FPA/2010/027 & ACOMP/2010 por la financiación recibida. (1) (2) Figura 6. Detalle de las morfologías de fractura observadas en las uniones en T: 1) muestra sin tratar, 2) muestra tratada a 100mm/s y 6 mm. (x16) En la primera fotografía se observa el aspecto superficial liso del adhesivo PU en una muestra sin tratar, al no presentar ningún tipo de adherencia sobre la superficie polimérica y ser totalmente despegado de la XI CONGRESO NACIONAL DE MATERIALES 6. REFERENCIAS [1] [2] [3] [4] M. R. Sanchis, V. Blanes, M. Blanes, D. Garcia and R. Balart, Eur. Polym. J. 42, 1558-1568 (2006). M. R. Sanchis, O. Calvo, O. Fenollar, D. Garcia and R. Balart, J. Appl. Polym. Sci. 105, 10771085 (2007). Z. Fang, Y. C. Qiu and H. Wang, Plasma Sci. Technol. 6, 2576-2580 (2004). O. J. Kwon, S. W. Myung, C. S. Lee and H. S. Choi, J. Colloid Interface Sci. 295, 409-416 (2006). 292