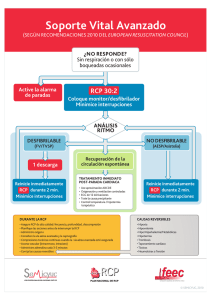
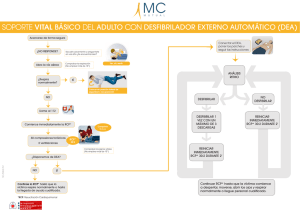
anexo i lista del titular de la autorizacion de comercializacion
Anuncio

ANEXO I LISTA DEL TITULAR DE LA AUTORIZACION DE COMERCIALIZACION, NOMBRE DE FANTASIA, DOSIS, FORMA FARMACÉUTICA, VIA DE ADMINISTRACION, , PRESENTACIÓN Y TAMAÑO DEL ENVASE DEL MEDICAMENTO EN LOS ESTADOS MIEMBROS 1 Estado Miembro Titular de la Autorización de Comercialización Nombre de fantasía Beriate P Dosis Forma farmacéutica Vía de administración Envase Volumen de llenado Tamaño del envase AUSTRIA Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania 250 IU Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 250 IU Disolvente: 2,5 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 500 IU Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 500 IU Disolvente: 5 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* AUSTRIA Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 1000 IU Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 1000 IU Disolvente: 10 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* ALEMANIA Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 250 IU Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 250 IU Disolvente: 2,5 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* ALEMANIA Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 500 IU Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 500 IU Disolvente: 5 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* ALEMANIA Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 1000 IU Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 1000 IU Disolvente: 10 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* ITALIA Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Beriate P 250 IU Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para AUSTRIA Vía intravenosa Polvo: Vial (vidrio) Polvo: 250 IU Disolvente: 2,5 ml 1 vial + 1 vial + 2 Estado Miembro Titular de la Autorización de Comercialización Nombre de fantasía Dosis Alemania Forma farmacéutica Vía de administración Envase Volumen de llenado Tamaño del envase Disolvente: Vial (vidrio) (100 IU/ml) 1 set de dispositivos de administración* Vía intravenosa Polvo: Vial (vidriovidrio) Disolvente: Vial (vidriovidrio) Polvo: 500 IU Disolvente: 5 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 1000 IU Disolvente: 10 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 250 IU Disolvente: 2,5 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 500 IU Disolvente: 5 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 1000 IU Disolvente: 10 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* Vía intravenosa Polvo: Vial (vidrio) Polvo: 250 IU Disolvente: 2,5 ml 1 vial + 1 vial + solución inyectable o perfusión ITALIA Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 500 IU ITALIA Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 1000 IU PORTUGAL Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 250 IU PORTUGAL Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 500 IU PORTUGAL Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 1000 IU ESPAÑA Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Beriate P 250 IU Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para 3 Estado Miembro Titular de la Autorización de Comercialización Nombre de fantasía Dosis Alemania ESPAÑA Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 500 IU ESPAÑA Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 1000 IU SUECIA Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 250 IU SUECIA Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 500 IU SUECIA Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 1000 IU REINO UNIDO Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 250 IU Forma farmacéutica solución inyectable o perfusión Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para solución inyectable o percusión Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para solución inyectable o 4 Vía de administración Envase Volumen de llenado Tamaño del envase Disolvente: Vial (vidrio) (100 IU/ml) 1 set de dispositivos de administración* Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 500 IU Disolvente: 5 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 1000 IU Disolvente: 10 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 250 IU Disolvente: 2,5 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 500 IU Disolvente: 5 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 1000 IU Disolvente: 10 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 250 IU Disolvente: 2,5 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* Estado Miembro Titular de la Autorización de Comercialización Nombre de fantasía Dosis REINO UNIDO Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 500 IU REINO UNIDO Aventis Behring GmbH, P.O. Box 1230, 35002 Marburg, Alemania Beriate P 1000 IU Forma farmacéutica perfusión Polvo y disolvente para solución inyectable o perfusión Polvo y disolvente para solución inyectable o perfusión *1 trasvasador *1 filtro de un solo uso 5 Vía de administración Envase Volumen de llenado Tamaño del envase Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 500 IU Disolvente: 5 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* Vía intravenosa Polvo: Vial (vidrio) Disolvente: Vial (vidrio) Polvo: 1000 IU Disolvente: 10 ml (100 IU/ml) 1 vial + 1 vial + 1 set de dispositivos de administración* ANEXO II CONCLUSIONES CIENTÍFICAS Y MOTIVOS DE LA REVISIÓN DEL DICTAMEN DE 18 FEBRERO 2003 PRESENTADAS POR LA EMEA 5 RESUMEN GENERAL DE LA EVALUACIÓN CIENTÍFICA DE BERIATE P A. Resumen de la conclusión científica del CPMP respecto al dictamen adoptado sobre la remisión (17 de octubre de 2002) Sobre la base de los motivos de la remisión, los aspectos considerados por el CPMP fueron: 1. La advertencia sobre los agentes transmisibles en la Sección 4.4 del RCP. 2. Si había justificación para desviarse del texto de la advertencia sobre los agentes transmisibles en la Sección 4.4 del RCP básico para productos que contienen factor VIII derivado de plasma humano (CPMP/BPWG/1619/99, 29 de junio de 2000), y añadir el siguiente texto sobre las pruebas NAT/PCR: “Para reducir el número de donaciones potencialmente infecciosas durante el “período de latencia” (serológicamente no reactivo), se analiza todo el plasma para la detección de material genómico del VHB, VHC y VIH-1 (mediante la técnica de reacción en cadena de la polimerasa, PCR), evitando así la entrada de plasma reactivo positivo en los procesos de producción.” 3. La modificación del RCP para equipararlo al RCP básico antes mencionado. El CPMP adoptó su dictamen el 17 de octubre de 2002 (véase el Apéndice 1). El CPMP concluyó que el texto propuesto por el titular de la autorización de comercialización para la advertencia sobre los agentes transmisibles en la Sección 4.4 del RCP no era aceptable. El CPMP consideró que los argumentos presentados por el titular de la autorización de comercialización no justificaban una desviación del texto del actual RCP básico. El RCP debe contener información relevante para que el que prescribe el medicamento pueda hacer un uso seguro y eficaz del mismo. Por consiguiente, el CPMP recomendó la modificación del RCP en línea con la advertencia sobre los agentes transmisibles contenida en la Sección 4.4 del RCP básico para productos que contienen factor VIII derivado de plasma humano (CPMP/BPWG/1619/99, 29 de junio de 2000). Además, el CPMP recomendó otras modificaciones específicas del RCP para equipararlo al RCP básico. B. Resumen de la evaluación científica del recurso sobre Beriate B (Opinión 18 Febrero 2003) 1. Ámbito de la evaluación El titular de la autorización de comercialización comunicó el 25 de octubre de 2002 su intención de recurrir el dictamen y el 20 de diciembre de 2002 remitió los motivos de dicho recurso. El 22 de enero de 2003 y el 19 de febrero de 2003 presentó alegaciones verbales ante el CPMP. Los informes de evaluación del ponente y el ponente adjunto se distribuyeron el 31 de enero y el 3 de febrero, respectivamente. El recurso fue considerado por los grupos de trabajo BPWG y BWP en las reuniones de 6-7 de febrero de 2002 y 10-12 de febrero de 2003, respectivamente. Los motivos del recurso interpuesto por el titular de la autorización de comercialización se adjuntan (Apéndice 2) al dictamen del CPMP del 18 de Febrero. La moción del recurso interpuesto por dicho titular fue: “El dictamen del CPMP y el rechazo de la solicitud del Apelante incumplen tanto el RCP básico como la Directiva 2001/83/CE. El rechazo del idioma en el que se presenta el etiquetado del producto carece de todo fundamento legal. Además, dicho rechazo contraviene el concepto del RCP básico del CPMP de incluir toda la información clínicamente relevante • al no permitir que el Apelante incluya información completa y científicamente sólida respecto a las pruebas PCR, 6 • al dar a entender que la seguridad viral de los productos derivados de plasma está garantizada, entre otras medidas, por las pruebas PCR para un único virus (VHC), y al considerar que esa información es suficiente, necesaria y adecuada. • Por consiguiente, el dictamen parece basarse en una decisión arbitraria. El CPMP obliga al Apelante a proporcionar a los profesionales sanitarios y a los pacientes información incompleta y confusa y, por tanto, infringe su derecho a ser debidamente informados sobre el medicamento descrito en el RCP correspondiente. Así pues, el Apelante solicita al CPMP que revoque su dictamen con fecha 17 de octubre de 2002 y apruebe el siguiente texto o uno equivalente para el etiquetado del producto: “Para reducir el número de donaciones potencialmente infecciosas durante el “período de latencia” (serológicamente no reactivo), se analiza todo el plasma para la detección de material genómico del VHB, VHC y VIH-1 (mediante el método de reacción en cadena de la polimerasa, PCR), evitando así la entrada de plasma reactivo positivo en los procesos de producción.” 2. Fundamento legal y reglamentario del RCP y función de los RCP básicos El CPMP ha tenido muy en cuenta el fundamento legal y reglamentario del RCP y la función de los RCP básicos en su evaluación científica de la remisión y durante el recurso. Dada la importancia que estos aspectos han tenido en la evaluación, se reproduce a continuación. La Notificación a los Solicitantes publicada por la DG Empresa de la Comisión Europea contiene una directriz sobre el RCPa en la que se explica lo siguiente sobre el RCP : “De conformidad con el apartado a del artículo 4 de la Directiva 65/65/CEEb, modificada, la solicitud tiene que ir acompañada de una propuesta del resumen de las características del producto (RCP). La Parte I B corresponde a dicha propuesta. Además, el apartado b del artículo 4 de la Directiva 65/65/CEEc requiere que el contenido sea aprobado por la autoridad competente. Así pues, el RCP constituye una parte intrínseca y esencial de la autorización de comercialización. El RCP contiene la información aprobada sobre el medicamento basada en el proceso de evaluación. Es la declaración definitiva entre la autoridad competente y el titular de la autorización de comercialización y constituye el fundamento común de la comunicación entre las autoridades competentes de todos los Estados miembros. Como tal, su contenido no puede modificarse salvo con la aprobación de la autoridad competente original.” En conclusión, el contenido del RCP debe ser aprobado por las autoridades reguladoras. Además, la directriz explica la finalidad del RCP : “El RCP constituye la base de la información para que los profesionales sanitarios sepan como utilizar el medicamento con seguridad y eficacia.” En ello se fundamenta el requisito de que el RCP debe contener “información necesaria para el médico y el paciente”. La directriz contiene una lista de aspectos relevantes para cada sección del RCP. La remisión actual con arreglo al artículo 31d hace referencia al texto de la Sección 4.4. (Advertencias y precauciones especiales de empleo). Como indica el propio nombre de esta sección, todas las cuestiones tratadas en ella tienen que estar relacionadas con información que sea clínicamente importante para médicos y pacientes. En la directriz se ofrecen ejemplos de este tipo de información. Finalmente, la directriz establece que «Siempre que exista, se tendrá en cuenta la directriz sobre el a Directriz sobre el Resumen de las Características del Producto, Revisión final 0, diciembre de 1999. b Artículo 11 de la Directiva 2001/83/CE. c Artículo 21 de la Directiva 2001/83/CE. d Corresponde al artículo 12 de la Directiva 75/319/CEE, para remisiones presentadas antes del 18 de diciembre de 2001. 7 RCP de un área terapéutica (por ejemplo, antibióticos) o de un grupo farmacológico (por ejemplo, benzodiacepinas) específicos». Así pues, la función del RCP básico es proporcionar directrices. Las directrices no son legalmente vinculantes, pero representan la forma mejor y más apropiada de cumplir con una obligación establecida en la legislación comunitaria. En consecuencia, las directrices pueden incumplirse por razones científicas válidas. No obstante, el contenido del RCP ha de ser aprobado por las autoridades reguladoras. El CPMP adoptó el resumen de las características del producto (RCP) básico revisado para los productos que contienen FVIII derivado de plasma en junio de 2000 (CPMP/BPWG/1619/99). La finalidad del RCP básico es proporcionar un formato común para los RCP de ciertas clases de medicamentos con el fin de facilitar la armonización de los RCP para productos comercializados en la Unión Europea y la concordancia de los RCP establecidos a través de procedimientos nacionales o comunitarios para esos productos. La Sección 4.4 de los RCP básicos para medicamentos derivados de plasma contiene un texto armonizado de advertencia sobre los agentes transmisibles. Esta sección puede modificarse en el caso de que el solicitante pueda demostrar que algunas advertencias no son válidas para sus productos. 3. Fundamentos científicos Antes de considerar las cuestiones que se plantean en este recurso, es importante establecer los fundamentos científicos de: • La seguridad viral de los medicamentos derivados de plasma y cómo se consigue dicha seguridad • La finalidad de las pruebas NAT para la detección de material genómico viral. 3.1 Seguridad viral de los medicamentos derivados de plasma y cómo se consigue dicha seguridad El reconocimiento a mediados del decenio de 1980 de que los medicamentos derivados de plasma, sobre todo los concentrados de factores de la coagulación, habían causado la transmisión generalizada del virus de la inmunodeficiencia humana (VIH) y la hepatitis no A no B (conocida ahora como hepatitis C) trajo consigo importantes cambios en los procesos de producción, con la adopción de medidas específicas para inactivar/eliminar estos y otros virus transmitidos por la sangre. Sin embargo, a principios del decenio de 1990 se produjeron algunos casos aislados de transmisión de virus con envoltura (virus de la hepatitis B (VHB) y C (VHC)) y del virus sin envoltura de la hepatitis A (VHA) a través de medicamentos derivados de plasma. Como consecuencia de ello, se revisaron los requisitos de seguridad viral y se incrementó la eficacia de los procesos de inactivación/eliminación de los virus. A mediados del decenio de 1990 se revisó asimismo la directriz del CPMP sobre validación viral1 y sobre medicamentos derivados del plasma2. La seguridad viral actual de los medicamentos derivados de plasma fue examinada recientemente por el CPMP y sus correspondientes grupos de trabajo (BPWG y BWP)3 y se evaluó asimismo con motivo de la reciente Declaración del CPMP sobre los donantes remunerados y no remunerados: seguridad y suministro de los medicamentos derivados de plasma4. Ninguna medida es capaz por sí sola de garantizar la seguridad viral de los medicamentos derivados de plasma, exigiendo ésta la adopción de una serie de medidas complementarias que deben considerarse conjuntamente para evaluar la seguridad global de cada producto individual. Dichas medidas son: • Buenas prácticas de fabricación • Selección/cualificación de donantes individuales 8 • • • Consideración de la epidemiología de los virus que se transmiten por la sangre en la población de donantes, incluidos los subtipos virales específicos. Análisis de las donaciones individuales y las mezclas de plasma para detectar marcadores de la infección con virus conocidos, considerando también la especificidad, sensibilidad y robustez de los métodos de ensayo y su capacidad para detectar subtipos o variantes de virus. Y, muy importante, la aplicación de procesos de producción validados, capaces de inactivar o eliminar diversos virus. Esas medidas están sujetas a control reglamentario según establece la Directiva 2001/83/CE. Dicho control incluye inspecciones de los centros de extracción y producción. Además, muchos Estados miembros de la UE disponen de un sistema de pruebas para la liberación de mezclas de plasma o medicamentos derivados de plasma realizadas por los Laboratorios Oficiales de Control de Medicamentos (LOCM). El riesgo de enfermedades infecciosas debidas a la transmisión de agentes infecciosos, entre ellos patógenos de naturaleza desconocida, se reduce con la aplicación de este conjunto de medidas, pero no puede descartarse del todo. Los medicamentos actuales derivados de plasma ofrecen una seguridad viral excelente con respecto a los virus con envoltura, entre ellos VIH, VHB y VHC. Se han establecido métodos para la inactivación/eliminación del VHA, un virus sin envoltura, en algunos productos que contienen factores de la coagulación. El CPMP ha recomendado la adopción de métodos eficaces para la inactivación/eliminación del VHA en todos los productos que contienen factores de la coagulación2,3. Se han notificado casos de infecciones por el parvovirus B19 asociados a la administración de factores de la coagulación y plasma tratado con disolventes o detergentes. La infección por el parvovirus B19 rara vez constituye un problema médico grave y se pueden identificar los grupos que corren mayor riesgo de contraer la infección. El parvovirus B19, un virus sin envoltura, es difícil de inactivar o eliminar con los métodos actuales. El CPMP ha recomendado la introducción de etapas de inactivación/eliminación en el proceso de fabricación o medidas complementarias tales como el uso de técnicas de amplificación de ácidos nucleicos (NAT) para mejorar la seguridad viral con respecto al parvovirus B19 como medida prioritaria para los factores de la coagulación y el plasma tratado con disolventes o detergentes, puesto que parece ser que transmiten el parvovirus B19, y para la inmunoglobulina anti-D, porque se utiliza en un grupo de pacientes de alto riesgo3. Según se establece en la Nota de orientación sobre los medicamentos derivados de plasma2, un objetivo para todos los medicamentos derivados de plasma consiste en la adopción de métodos eficaces para la inactivación/eliminación de un gran número de virus con distintas características físicoquímicas. De esta forma podrían tenerse ciertas garantías de la eficacia de los métodos frente a virus que actualmente no se conocen o que están empezando a conocerse. 3.2 Finalidad de las pruebas NAT para la detección de material genómico viral La tecnología de amplificación de ácidos nucleicos (NAT) es una técnica sensible capaz de detectar genomas virales incluso antes de que las pruebas serológicas den resultados positivos (“período de latencia”) en una persona infectada. Por consiguiente, ofrecen la posibilidad de reducir más aún la carga viral a que puede enfrentarse un proceso de fabricación. El CPMP había recomendado que se realizaran pruebas NAT para la detección del ARN del VHC en todos los medicamentos derivados de plasma ya desde julio de 19995 y esa recomendación se convirtió en obligación tras su inclusión en la Farmacopea Europea. Este requisito se introdujo después de un período de consultas internas y públicas y exigió a los Laboratorios Oficiales de Control de Medicamentos la realización de ensayos de aptitud antes de su aplicación formal. Con respecto a la realización de la prueba, la norma de la Farmacopea estipula que una mezcla no reactiva para los procesos de producción es una mezcla que da resultado negativo en un análisis capaz de detectar concentraciones de 100 UI/ml de ARN del VHC. 9 El CPMP explicó en su recomendación sobre la realización de pruebas NAT para detectar ARN del VHC5 que “La utilización de técnicas NAT para la detección de ácido nucleico viral es un “avance” en el proceso de asegurar la calidad de los medicamentos derivados del plasma.” Explicó también que la seguridad viral de los medicamentos derivados de plasma con respecto a la hepatitis C estaba ya asegurada con la selección de los donantes, la detección de anticuerpos frente a la hepatitis C en donaciones individuales y materiales de partida y la incorporación de etapas eficaces para la inactivación/eliminación de virus en los procesos de producción. El CPMP confirmó que no existían motivos científicos ni problemas de seguridad que justificaran la retirada de los lotes de productos liberados antes de julio de 1999 que se habían fabricado con mezclas de plasma no sometidas a pruebas NAT para la detección de ARN del VHC. En el Seminario de la EMEA celebrado en septiembre de 2000e sobre la seguridad viral de los productos derivados de plasma se habló de las pruebas NAT para la detección de VHC, VIH y VHB, dedicando una especial atención a los virus sin envoltura 3. La PPTA informó sobre la experiencia de las pruebas NAT realizadas a más de 10 millones de donaciones, en las que se obtuvieron unas tasas de detección de donaciones reactivas de 8,5, 1,3 y 0,4 por 100.000 donaciones, respectivamente para el VHC, el VHB y el VIH. (Los datos incluyen donaciones de donantes potenciales y una o más donaciones positivas por donante). Según la EPFA/EBA (European Blood Alliance), los datos epidemiológicos de sus poblaciones de donantes hacían esperar aproximadamente una donación positiva al NAT y negativa a los anticuerpos por cada 1.000.000 donaciones de donantes repetidos para el VHC y el VHB, y 0,22 donaciones positivas al NAT y negativas a los anticuerpos por cada 1.000.000 donaciones de donantes repetidos para el VIH. La incidencia de donantes positivos al ARN del VHC y negativos a los anticuerpos detectados en minimezclas fue algo menor de lo que preveían los cálculos del riesgo. Tras considerar la información presentada en el Seminario celebrado en septiembre de 2000, el CPMP y los correspondientes grupos de trabajo concluyeron que3: “Ya se realizan pruebas NAT para la detección del VHC en las mezclas de plasma. La necesidad de realizar otras pruebas NAT depende de una serie de consideraciones, tales como la situación epidemiológica de los donantes de sangre o plasma, la duración de la fase de latencia y la carga viral, así como la capacidad del proceso de producción de inactivar o eliminar los virus. Desde la perspectiva reguladora, se considera que los sistemas actuales empleados para garantizar la seguridad viral de los medicamentos derivados de plasma con respecto al VIH y el VHB son adecuados y que las pruebas NAT para detectar el VIH y el VHB en mezclas de plasma no son necesarias actualmente. Sin embargo, se reconoce que, en determinadas circunstancias, las pruebas NAT para detectar VIH y VHB podrían contribuir a la selección de la población de donantes.” El CPMP consideró también las pruebas NAT para la detección de VHA y parvovirus B19 y concluyó que3: “Debido a que las infecciones pueden transmitirse incluso con niveles muy bajos de viremia, se considera actualmente que las pruebas NAT realizadas en mezclas de plasma para la detección de ARN del VHA tienen una utilidad limitada para mejorar la seguridad de los productos.” e El Seminario fue presidido por el Presidente del BWP y contó con la asistencia de miembros y expertos de los grupos BWP/BPWG, representantes de la Comisión Europea, el Departamento Europeo para la Calidad de los Medicamentos (EDQM), asociaciones de fabricantes europeos de medicamentos derivados de plasma (PPTA (Asociación de Terapéutica de Proteínas Plasmáticas) y EPFA (Asociación Europea de Fraccionamiento del Plasma), y representantes del Consorcio Europeo de la Hemofilia (EHC) y la Asociación Europea de Pacientes con Inmunodeficiencia Primaria (EPPIC). La lista completa de participantes puede encontrarse en el informe de la reunión publicado en el sitio web de la EMEA. 10 “Las pruebas NAT de minimezclas para detectar parvovirus B19 pueden emplearse para reducir la carga viral de las mezclas utilizadas en fabricación. Se considera como una medida complementaria de las etapas de inactivación/eliminación en el proceso de producción, pero todavía no se ha demostrado el efecto que tienen estas medidas complementarias en la seguridad global de cada producto.” 4. Evaluación de las cuestiones que se plantean en el recurso interpuesto por la empresa 4.1 ¿Por qué se incluyen las pruebas NAT para el VHC en la Sección 4.4 del actual RCP básico para los medicamentos que contienen factor VIII ? La empresa alega que la mención de las pruebas NAT para el VHC en el texto actual del RCP básico demuestra que el CPMP considera que es información clínicamente importante. Tal como se establece en el dictamen del CPMP sobre la remisión (EMEA/CPMP/5008/02), el motivo de mencionar las pruebas NAT en el RCP básico es que se trata de pruebas obligatorias y necesarias para todos los medicamentos derivados de plasma que se citan en el RCP básico. En el momento de aprobarse el RCP básico actual para los productos que contienen factor VIII derivado de plasma, algunas empresas realizaban otras pruebas (ya fueran NAT u otros análisis) de las donaciones individuales o las mezclas de plasma que no se incluyeron como texto opcional en el RCP básico. La empresa ha alegado también que la obligación de realizar pruebas NAT para la detección de VHC no significa necesariamente que deban mencionarse en el RCP. El CPMP está de acuerdo con la empresa. La Directriz sobre el resumen de las características del producto6 dice que “Los solicitantes deben mantener la integridad de todas las secciones del documento incluyendo sólo información en cada sección que se corresponda con su encabezamiento.” Puesto que la información sobre las medidas de obligado cumplimiento no constituye una precaución ni una advertencia, el texto de la Sección 4.4 del RCP básico para los productos que contienen factor VIII derivado de plasma humano debe modificarse para excluir todo lo que no sea ni una advertencia ni una precaución. Este análisis es consecuente con la revisión que está realizando el CPMP del texto actual de las advertencias, que se describía en el dictamen del CPMP de octubre de 2002 y en el informe de evaluación sobre la remisión de Beriate P: «El CPMP revisa constantemente sus directrices, incluidos los RCP básicos para los medicamentos derivados de plasma, y actualiza los textos según lo considera necesario. Así pues, en paralelo con esta remisión, el CPMP ha considerado si el texto actual de la advertencia sobre los agentes transmisibles en el RCP básico es óptimo. El texto del RCP básico actual permite la diferenciación de los productos en función de la eficacia de las medidas adoptadas contra el VHA y el parvovirus B19, siempre que dicha información sea necesaria para el que prescriba el medicamento. La opinión provisional del CPMP es que la advertencia podría mejorarse suprimiendo la referencia a las pruebas obligatorias (que ya se ha publicado en otro lugarf) y centrándose en los otros riesgos potenciales. Se ha creado un grupo de trabajo del CPMP para mejorar el texto de la advertencia sobre los agentes transmisibles para todos los medicamentos derivados de plasma y el trabajo no ha terminado todavía. Su objetivo es publicar una directriz sobre el texto de las advertencias que se incluirá en los RCP y los prospectos. Como se hace habitualmente, habrá una consulta pública para conocer las opiniones de las partes interesadas sobre la propuesta de directriz antes de su publicación definitiva.” La finalidad del nuevo texto es proporcionar una clara indicación de si las medidas de seguridad en su conjunto pueden considerarse una garantía de la seguridad del producto frente a virus específicos o si sigue existiendo un riesgo. El texto se ha modificado basándose en la filosofía de que lo importante es la seguridad viral del propio producto, no una lista de medidas individuales. Por ejemplo, este tipo de lista puede llevar al usuario a creer erróneamente que un producto es seguro frente a un virus específico aunque no sea ese el caso. Un producto fabricado a partir de una mezcla de plasma cuya carga viral se reduce por debajo del límite establecido mediante pruebas NAT para el ADN del f Las pruebas obligatorias de donaciones y mezclas de plasma se han publicado en la monografía de la Farmacopea Europea sobre plasma humano para fraccionamiento. . 11 parvovirus B19 podrá seguir siendo capaz de transmitir el virus si el proceso de producción es incapaz de inactivar o eliminar la carga viral por debajo de ese límite. Sería contradictorio que se permitiera ampliar la descripción de las pruebas NAT para hacer referencia al VIH y al VHB en la Sección 4.4 del RCP para Beriate P, cuando al mismo tiempo se está considerando la posibilidad de no mencionar ni siquiera las pruebas obligatorias. 4.2 ¿Por qué el CPMP diferencia entre las pruebas NAT según sean obligatorias o voluntarias? Como se decía antes, el CPMP está de acuerdo con la empresa en que el hecho de que la prueba del VHC sea obligatoria no significa que deba mencionarse en el RCP. El CPMP tiene la intención de suprimir dicha mención en la Sección 4.4 del RCP básico. Las pruebas NAT para el VHC constituyen un requisito obligatorio para todos los medicamentos derivados de plasma y la inclusión de esta información en los RCP no supone hacer distinción entre productos. Por el contrario, la mención en la Sección 4.4 (“Advertencias y precauciones especiales de empleo”) de pruebas voluntarias como son las pruebas NAT para VIH y VHB, que se realizan en algunos productos pero no en otros, daría a entender que los productos sometidos a esas pruebas son más seguros que los otros. Pero eso no es cierto, como se explica más adelante (Sección 4.4). La inclusión de esa información tendría, por tanto, un elemento de carácter publicitario totalmente inaceptable en un RCP y no sería útil para la “educación sanitaria” según se establece en el artículo 62 de la Directiva 2001/83/CE. Además, al no ser obligatorias las pruebas NAT para el VIH y el VHB, no existe un límite inferior de detección oficialmente establecido y exigido para el VHC. Por consiguiente, el significado de que una mezcla de plasma ha sido sometida a pruebas para la detección de VIH y VHB (mediante PCR) podría ser distinto para productos de diferentes fabricantes, puesto que dependería de la sensibilidad del método utilizado y del valor límite establecido para definir la mezcla no reactiva. En las alegaciones verbales presentadas en febrero de 2003, la empresa ha propuesto un nuevo texto para la Sección 4.4 (las partes suprimidas del texto del RCP básico se indican en negrita y las adiciones, en cursiva): “Cuando se administran medicamentos derivados de la sangre o plasma humanos, no se puede excluir totalmente la aparición de enfermedades debidas a la transmisión de agentes infecciosos. Esto también se refiere a la posible transmisión de patógenos de naturaleza desconocida. Sin embargo, el riesgo de transmisión de agentes infecciosos se reduce por: - la selección de donantes mediante un reconocimiento médico y la detección de AgHBs y anticuerpos frente a VIH y VHC en las donaciones individuales y en las mezclas de plasma. - el análisis de material genómico del VHC en las mezclas de plasma. - la vigilancia epidemiológica de los centros de extracción de sangre y plasma en Austria, Alemania y Estados Unidos. - la reducción de donaciones potencialmente infecciosas durante el período de latencia mediante el análisis de mezclas de plasma para la detección de material genómico de VHC, VHB y VIH-1, evitando con ello la entrada de plasma reactivo en los procesos de producción. - Los procedimientos de inactivación/eliminación incorporados al proceso de producción y validados utilizando virus modelo. Estos procedimientos se consideran efectivos para VIH, VHC<, VHA>, < parvovirus B19>, y VHB. <Los procedimientos de inactivación/eliminación viral pueden tener una utilidad limitada frente a virus sin envoltura como <VHA > <y/o> parvovirus B19.> Se recomienda la vacunación apropiada (hepatitis A y B) para los pacientes que reciban concentrados plasmáticos de factor VIII. 12 La infección por parvovirus B19 puede ser grave para la mujer embarazada (infección fetal) y para sujetos con inmunodeficiencia o con producción aumentada de hematíes (por ejemplo, con anemia hemolítica).” El CPMP considera que el texto revisado no resuelve sus objeciones al texto propuesto en la moción de recurso, como se expone en este dictamen. 4.3 ¿Puede incluirse información sobre otras pruebas NAT en el RCP como información farmacéutica ? Otra posibilidad considerada por el CPMP fue la inclusión de las pruebas NAT como información farmacéutica en la Sección 2 del RCP “Composición cualitativa y cuantitativa”. Sin embargo, el CPMP concluye que ello supondría también proporcionar información aislada sobre un único aspecto del proceso de fabricación de algunos productos. La finalidad del RCP no es facilitar datos técnicos sobre los procesos de producción. Los artículos 11 y 62 de la Directiva 2001/83/CE, que especifican la información que deben contener los RCP y los prospectos respectivamente, no mencionan los procesos de producción. Además, ello daría pie a la inclusión de otro tipo de información irrelevante para el uso clínico apropiado del producto en los RCP de los medicamentos derivados de plasma y de otros medicamentos. En las alegaciones verbales presentadas el 18 de febrero, la empresa propuso el texto siguiente para la Sección 2 “Composición cualitativa y cuantitativa”: “El producto se prepara con plasma humano procedente de Austria, Alemania y Estados Unidos. El riesgo de transmisión de agentes infecciosos se reduce por: - la selección de donantes mediante un reconocimiento médico y la detección de AgHBs y anticuerpos frente a VIH y VHC en las donaciones individuales y en las mezclas de plasma. - la vigilancia epidemiológica de los centros de extracción de sangre y plasma en Austria, Alemania y Estados Unidos. - la reducción de donaciones potencialmente infecciosas durante el período de latencia mediante el análisis de las mezclas de plasma para la detección de material genómico de VHC, VHB y VIH-1, evitando con ello la entrada de plasma reactiva en los procesos de producción. - Los procedimientos de inactivación/eliminación incorporados al proceso de producción y validados utilizando virus modelo. Estos procedimientos se consideran efectivos para VIH, VHC<, VHA>, < parvovirus B19>, y VHB.” El CPMP no consideró aceptable la propuesta porque proporciona sólo información selectiva sobre los procesos de producción para medicamentos derivados de plasma y, tal como se expuso antes, la finalidad del RCP no es facilitar información sobre la fabricación, puesto que ello daría pie a la inclusión de otro tipo de información. Además, aunque se haya transladado a una sección sobre información farmacéutica, la introducción del texto dice así: “El riesgo de transmisión de agentes infecciosos se reduce por:”. Por consiguiente, sigue existiendo un vínculo con la seguridad y se da a entender que los productos sometidos a pruebas NAT para la detección de VHB y VIH-1 son más seguros que los demás. 4.4 ¿Puede mejorar la seguridad de los productos con las pruebas NAT para la detección de VIH y VHB, en comparación con los productos no sometidos a esas pruebas ? Como se explica en el apartado 3. Fundamentos científicos, la seguridad viral se consigue con una combinación de diferentes factores, que han de considerarse conjuntamente para poder evaluar la seguridad global de cada producto. Por ejemplo, es posible que un producto deba someterse a pruebas NAT adicionales, mientras que otro tenga un proceso de producción con mayor capacidad de inactivación/eliminación viral. Para poder comparar productos han de tenerse en cuenta todos los factores que influyen en el margen de seguridad conseguido. 13 Todos los medicamentos actuales derivados del plasma ofrecen una excelente seguridad viral frente a los virus con envoltura como el VIH, el VHB y el VHC y las pruebas NAT no deben utilizarse como única información para diferenciar un producto de otro. Las autoridades reguladoras no autorizarían un medicamento derivado de plasma si no consideraran garantizada su seguridad frente a los virus con envoltura. La seguridad de los productos actuales se refleja en la mención que se hace en el recurso de la empresa a la información publicada sobre los últimos casos ocurridos de transmisión de VIH en 1989/90, de VHC en 1993/94 y de VHB en 1994. En cada uno de estos casos estaba implicado un producto específico. Los medicamentos derivados de plasma pueden diferir en su seguridad frente a la hepatitis A y el parvovirus B19. En consecuencia, el texto actual de las advertencias en el RCP básico permite la diferenciación de los productos en función de la eficacia de las medidas adoptadas contra el VHA y el parvovirus B19. Esa información es útil para el que prescribe el medicamento. El texto seguirá mejorándose con la revisión continua del texto de las advertencias. 4.5 ¿Son las pruebas NAT para el VIH-1 y el VHB útiles en el caso de Beriate P? Como se expone en el apartado 3. Fundamentos científicos, el CPMP reconoce que, en determinadas circunstancias, las pruebas NAT para el VIH y el VHB pueden contribuir a la selección de la población de donantes. Las poblaciones de donantes difieren en cuanto a la epidemiología de los diferentes virus que se transmiten por la sangre y el beneficio relativo de las pruebas NAT depende de ello. La empresa afirmó que la frecuencia de donaciones positivas en las pruebas NAT es de 12,8, 1,6 y <0,1 por 100.000 donaciones para el VHC, el VHB y el VIH-1 respectivamente. Partiendo de esta información, puede calcularse que, si no se realizaran pruebas NAT, aproximadamente la totalidad de las mezclas de plasma contendría una donación positiva para el ARN del VHC, una de cada 10 mezclas contendría una donación positiva para el ADN del VHB y una de cada 100 mezclas contendría una donación positiva para el ARN del VIH-1. (La empresa calcula igualmente que aproximadamente 1,6 mezclas de cada 10 contendrían una donación positiva para el VHB). En tales circunstancias, la realización de pruebas NAT es una medida razonable para esta empresa. Además, la empresa extrae plasma principalmente de donantes para plasmaféresis, que donan con más frecuencia que los donantes de sangre y que pueden hacer varias donaciones antes de que ocurra la seroconversión. Las pruebas NAT permiten a la empresa detectar antes a los donantes infectados y evitar que sigan donando. Se hace también un seguimiento retrospectivo para identificar las donaciones infectadas en las reservas existentes. Debe señalarse que, pese a todo ello, las mezclas utilizadas en la fabricación podrían seguir conteniendo virus en niveles inferiores al límite de detección de los métodos utilizados o genotipos variantes que pasaran inadvertidos con el método de detección. En las alegaciones verbales de febrero de 2003, la empresa presentó datos que comparan los resultados de las pruebas NAT de Aventis Behring con los resultados de las pruebas NAT de la Cruz Roja alemana. Estos últimos indican una menor frecuencia de donaciones positivas para el VHC y el VHB en las pruebas NAT de los donantes de la Cruz Roja alemana (véase la Tabla 1). 14 Tabla 1: Comparación de los resultados de las pruebas NAT de Aventis Behring y la Cruz Roja alemana VHC Aventis Behring Cruz Roja alemana 12,8 0,9 NAT positivos por 100.000 donaciones VHB 1,6 <0,1 0,14 0,1 VIH La empresa admite que, con una menor frecuencia de donaciones positivas en las pruebas NAT, el riesgo de que una mezcla contenga una donación positiva es menor. Sin embargo, si bien en el caso de Aventis Behring es posible detectar y retirar del proceso de producción las donaciones que en las pruebas NAT dan resultados superiores al valor límite, en las empresas que no hagan pruebas NAT esas donaciones entrarán en el proceso de producción. La conclusión de la empresa es que «las pruebas NAT eliminan las mezclas positivas con independencia de la prevalencia y son una alternativa preferible a no realizar ninguna prueba.» El CPMP considera que las alegaciones de la empresa coinciden con la opinión del CPMP que se expone en el apartado 3. Fundamentos científicos La seguridad global de los medicamentos derivados de plasma frente a un virus específico puede garantizarse con métodos capaces de reducir al mínimo la carga viral en las mezclas utilizadas en la fabricación, siendo estos métodos diferentes en cada empresa, y sobre todo con la capacidad del proceso de fabricación de inactivar/eliminar virus residuales en la mezcla. El éxito de estos métodos complementarios se confirma por la excelente seguridad viral de los medicamentos actuales derivados de plasma frente a los virus VIH, VHB y VHC (véase la Sección 4.4). La empresa alega que el aumento del margen de seguridad frente a los tres virus (VIH, VHB y VHC) es equivalente cuando sus pruebas NAT se comparan con la sensibilidad exigida por las pruebas NAT obligatorias para el VHC. Este argumento no se considera justificado. La incidencia de infecciones, el período de latencia y la carga viral durante el período de latencia en el caso de infección por los virus VIH-1 y VHB son diferentes de los de la infección por el VHC. La empresa no tuvo en cuenta todos los factores en sus cálculos. Cuando los datos se analizan de nuevo tomando en consideración la frecuencia de donaciones durante el período de latencia, la reducción del riesgo es entre 100 y 1000 veces menor para el VIH y el VHB que para el VHC. La empresa argumenta que los datos epidemiológicos no son relevantes para los cálculos del riesgo ni se tuvieron en cuenta cuando se introdujo el requisito de realizar pruebas de seguridad viral a los componentes sanguíneos utilizados para transfusiones (componentes lábiles de la sangre), al calcular los márgenes de seguridad viral de un producto de acuerdo con la directriz ICH o al recomendar en 1999 las pruebas NAT para el VHC. El CPMP no considera válidos estos argumentos. Aunque quede fuera del ámbito de esta remisión (véase más adelante), los datos epidemiológicos sí se tuvieron en cuenta al considerar el valor añadido de una prueba adicional de seguridad viral exigida a los componentes sanguíneos utilizados para transfusiones. La directriz ICH se refiere a los productos biotecnológicos obtenidos mediante un proceso cerrado a partir de líneas celulares caracterizadas y no contempla los medicamentos derivados de plasma. Finalmente, el hecho de que el documento del CPMP en el que se recomiendan las pruebas NAT5 no haga referencia a la epidemiología no significa que ésta se ignorara a la hora de hacer la recomendación. Como se expone en el apartado 3. Fundamentos científicos, la utilidad de las pruebas NAT depende de una serie de consideraciones, entre ellas la situación epidemiológica de los donantes de sangre/plasma, la duración de la fase de latencia y la carga viral, así como la capacidad del proceso de fabricación para inactivar o eliminar los virus. Cuando el CPMP recomendó las pruebas NAT para el VHC, tuvo en cuenta la incidencia de infecciones, el período de latencia más largo y la mayor carga viral durante el período de latencia del VHC, comparado con el VIH y el VHB. La utilidad de las pruebas NAT para el VHC se refleja también en los datos de Aventis Behring descritos antes, según los cuales, si no realizaran esas pruebas NAT, prácticamente todas las mezclas tendrían una donación 15 positiva al VHC. No obstante, en el momento de recomendarse las pruebas NAT para la detección del ARN del VHC, la seguridad viral de los productos derivados del plasma con respecto al VHC estaba ya asegurada por la selección de los donantes, las pruebas de seguridad viral de las donaciones individuales y la incorporación de medidas eficaces para la inactivación/eliminación del virus durante el proceso de fabricación. Por consiguiente, en la recomendación del CPMP5 se explica que la utilización de técnicas NAT para la detección del ácido nucleico viral es un “avance” en el proceso de asegurar la calidad de los medicamentos derivados del plasma.” La empresa alega que la recomendación en Estados Unidos7 de realizar pruebas NAT para el VIH-1 en los medicamentos derivados de plasma fuente (plasma extraído mediante plasmaféresis) es otra evidencia que cuestiona la posición del CPMP. Éste observó que el proyecto de directriz se basa también en consideraciones epidemiológicas: «Según un informe reciente, los donantes de plasma remunerados tienen una mayor probabilidad de donar unidades potencialmente infecciosas (Ref. 3). No obstante, las iniciativas emprendidas por la industria de plasma fuente, como las pruebas NAT bajo IND, reducen considerablemente el riesgo de que estas unidades estén presentes en las mezclas utilizadas para fabricación.» La empresa alega además que el requisito obligatorio en Japón de realizar pruebas NAT a mezclas de plasma para la detección de material genómico de VIH-1, VHC y VHB utilizadas en procesos de producción es otra evidencia que cuestiona la posición del CPMP. El CPMP no considera que ese argumento sea válido, puesto que la epidemiología de las infecciones virales varía según la región geográfica, siendo éste uno de los factores que tienen en cuenta las autoridades de otras regiones geográficas. En las alegaciones verbales presentadas en febrero de 2003, la empresa utilizó el requisito de realizar pruebas NAT a los componentes sanguíneos utilizados en transfusiones (como hematíes, plaquetas y plasma) en algunos Estados miembros como argumento para aplicar normas similares al plasma utilizado para fraccionamiento. El CPMP considera que el requisito de realizar pruebas NAT a los componentes sanguíneos no es relevante para la remisión actual, puesto que los componentes de la sangre no suelen someterse a procedimientos de inactivación/eliminación. 4.6 ¿Sería útil para médicos y pacientes que se incluyera información sobre las pruebas NAT para el VIH-1 y el VHB? El CPMP entiende que los usuarios desean información sobre las medidas adoptadas para garantizar la seguridad viral de los productos derivados de plasma. No obstante, esta información tiene que permitir a médicos y pacientes valorar la contribución relativa de las medidas a la seguridad global del producto. Por consiguiente, el hecho de proporcionar información aislada sobre las pruebas NAT hechas voluntariamente por una empresa puede confundir a los usuarios y ejercer un efecto promocional injustificado e inaceptable. La evaluación de la seguridad viral es una cuestión compleja que no puede expresarse simplemente con factores de reducción o listas de métodos analíticos sofisticados. Esa información es insuficiente para que las autoridades reguladoras puedan evaluar la seguridad viral de un producto y, en consecuencia, sería también insuficiente para los usuarios. La información importante que debe proporcionarse a los profesionales sanitarios en el RCP y a los pacientes en el prospecto es si la suma de todas las medidas adoptadas se considera suficiente para garantizar la seguridad global del producto frente a virus específicos o si existen riesgos residuales. No obstante, la declaración de haber realizado una prueba en particular (como pruebas NAT para la detección del VHB y el VIH-1) no proporciona información completa y precisa salvo en el contexto de una información mucho más detallada sobre la seguridad general del producto y, de hecho, puede inducir a error. Para que la información sea completa y precisa, tendrían que declararse también otras medidas adoptadas que contribuyen a la seguridad del producto (como la vigilancia epidemiológica de la población de donantes, la prevalencia de subtipos virales específicos en la población de donantes, el análisis de donaciones individuales y mezclas de plasma para detectar marcadores de infección por 16 virus conocidos, considerando la especificidad, sensibilidad y robustez de los métodos analíticos y su capacidad de detectar subtipos o variantes virales durante el proceso de fabricación y su eficacia para eliminar toda posible infectividad residual, la validez de los virus modelo elegidos para validar el proceso de producción. etc.). Tratar de proporcionar toda esta información en el RCP no sólo sería una tarea enorme, sino también imposible. Los RCP habrían de ampliarse enormemente, con el efecto negativo que ello tendría en la transmisión clara de información importante para garantizar el uso seguro y eficaz de los productos. Otra dificultad es que, en la actualidad, los datos epidemiológicos de las poblaciones de donantes no se expresan de manera uniforme en los expedientes, motivo por el cual los datos de los diferentes fabricantes no son estrictamente comparables. (Pese a esta dificultad, las autoridades reguladoras pueden hacer comparaciones generales de los datos epidemiológicos durante la evaluación de la seguridad viral de los medicamentos derivados de plasma.) Esta cuestión está siendo considerada actualmente por el BWP8. Al no existir una presentación uniforme de los datos, la inclusión de datos epidemiológicos en el RCP no permitiría la comparación precisa de las diferentes poblaciones de donantes. No existe una definición aceptada en toda la UE de lo que son una incidencia y una prevalencia “grandes” o “pequeñas”. Además, estos datos tendrían que ser actualizados con frecuencia como consecuencia de los cambios de las poblaciones de donantes o de su situación epidemiológica. Aunque son datos facilitados periódicamente a las autoridades reguladoras, su inclusión en un RCP sería imposible. Volviendo al texto específico propuesto por la empresa para el RCP del Beriate P: “Para reducir el número de donaciones potencialmente infecciosas durante el “período de latencia” (serológicamente no reactivo), se analiza todo el plasma para la detección de material genómico del VHB, VHC y VIH-1 (mediante el método de reacción en cadena de la polimerasa, PCR), evitando así la entrada de plasma reactivo positivo en los procesos de producción.” Al no ser obligatorias las pruebas, tampoco se han establecido a escala comunitaria los valores límite para las pruebas NAT del VHB y el VIH-1, como se hizo para la prueba NAT del VHC (después de realizar los correspondientes ensayos de aptitud). El requisito de una prueba NAT obligatoria para la detección del ARN del VHC se introdujo después de un período de consultas internas y públicas y exigió a los Laboratorios Oficiales de Control de Medicamentos la realización de ensayos de aptitud antes de su aplicación formal. La mención de las pruebas para la detección de ARN del VHC en el RCP está vinculada a una norma de la Farmacopea que estipula que una mezcla no reactiva para los procesos de producción es una mezcla que da resultado negativo en un análisis capaz de detectar valores de 100 UI/ml de ARN del VHC. La mención de pruebas NAT para el VIH y el VHB no facilita información sobre el valor límite utilizado para definir la mezcla no reactiva (o plasma reactivo positivo) y puede tener un significado diferente según la empresa. En las alegaciones verbales presentadas en febrero de 2003, la empresa preguntó si las pruebas NAT para el VHC se definen con precisión, puesto que la validación se basa en el genotipo 3 y existen diferencias de sensibilidad importantes para otros genotipos. El CPMP considera que existe una norma específica de la Farmacopea Europea y directrices sobre validación para el VHCg. La empresa se refirió también a las dificultades que han tenido los LOCM para la liberación de lotes cuando estos laboratorios utilizan métodos más sensibles que los establecidos en la norma de la Farmacopea. El CPMP considera que éste es un problema operativo para la liberación de los lotes por los LOCM que guarda relación con la forma de interpretar el límite y procesar el resultado; pero no cuestiona la existencia de una norma específica de la Farmacopea Europea. Finalmente, la empresa recuerda que la sensibilidad de las pruebas para el VIH y el VHB en 4 laboratorios importantes es similar. El CPMP g Farmacopea Europea, Cuarta edición, 2002 (4.3): Plasma humano para fraccionamiento (01/2002:0853 corregida), 2.6.21 Técnicas de amplificación de ácidos nucleicos y validación de las técnicas de amplificación de ácidos nucleicos (NAT) para la detección del ARN de virus de la hepatitis C (VHC) en mezclas de plasma: Directrices. 17 considera que la comparación de métodos entre distintas empresas no tiene el mismo peso que una norma específica de la Farmacopea. Además, el texto propuesto por la empresa en la moción de recurso es confuso. Cuando escribe “se analiza todo el plasma”, puede entenderse que todas las donaciones individuales se someten a pruebas NAT. En realidad, las pruebas se hacen en mezclas piloto de alícuotas procedentes de unas 1.200 donaciones. Cuando escribe “evitando así la entrada de plasma positivo (reactivo) en los procesos de producción”, puede entenderse que sólo se procesa plasma negativo, cuando en realidad puede seguir procesándose plasma con un contenido viral inferior al valor límite de la prueba NAT. Las propuestas presentadas por la empresa en las alegaciones verbales de febrero (véanse las Secciones 4.2 y 4.3) corrigen estas frases confusas del texto, pero no resuelven las otras objeciones que se expresan en este dictamen. Fuera de Europa, la advertencia exigida por la FDA como parte de la información sobre los productos que contienen factor VIII, no incluye detalles sobre medidas individuales9: «Puesto que este producto contiene sangre humana, puede conllevar cierto riesgo de transmisión de agentes infecciosos, como virus y, en teoría, el agente responsable de la enfermedad de CreutzfeldtJakob.» Los médicos estadounidenses reciben información detallada sobre las etapas de fabricación y los estudios de validación viral, así como sobre los resultados de los ensayos clínicos. Se observa una diferencia general en el enfoque europeo y estadounidense de la información sobre los productos. No obstante, hasta esa información tan detallada sería insuficiente para que las autoridades reguladoras pudieran evaluar la seguridad viral de un producto y emitir un juicio sobre los márgenes relativos de seguridad entre productos. En consecuencia, sería también información insuficiente para los usuarios. Una de las conclusiones de la compañía en las alegaciones verbales de febrero de 2003 fue que la aceptación de las propuestas presentadas contribuiría a la armonización internacional. En el debate mantenido con la empresa con ocasión de las alegaciones verbales presentadas en febrero, ésta declaró que el grado de detalle de la información facilitada en Japón está en un punto medio entre Estados Unidos y Europa. El CPMP no considera que la aceptación de cualquiera de las propuestas de la empresa vaya a contribuir a la armonización internacional, puesto que los enfoques adoptados por estas 3 regiones son diferentes. En conclusión, la seguridad viral se consigue con una combinación de diferentes factores, que han de considerarse conjuntamente para poder evaluar la seguridad global de cada producto. Como orientación importante para las futuras revisiones de los RCP básicos, el CPMP tiene la intención de publicar una Declaración sobre la evaluación de la seguridad viral de los medicamentos derivados de plasma, considerando también la función de las pruebas NAT y la base para las alegaciones contenidas en el RCP. 4.7 ¿Qué formas existen de proporcionar información a los usuarios sobre todas las medidas que contribuyen a garantizar la calidad y la seguridad? Existen dos formas de proporcionar información relacionada con los agentes transmisibles: 1. Indicar cuándo se considera garantizada la seguridad y cuándo siguen existiendo riesgos potenciales. 2. Proporcionar información sobre todas las medidas que contribuyen a garantizar la calidad y la seguridad. El documento apropiado para facilitar información que sirva a la primera finalidad es el RCP. Este precisa una descripción concisa, centrada en los posibles riesgos residuales y las precauciones que pueden ser necesarias. Esta opinión es consecuente con el concepto general del RCP, en el que no se incluye la información detallada sobre la calidad, la seguridad y la eficacia que se remite y evalúa para obtener y mantener una autorización de comercialización. El RCP no es el documento apropiado para proporcionar información que sirva al segundo propósito, porque no es la información que necesitan los profesionales sanitarios para hacer un uso seguro y eficaz del medicamento. Además, la inclusión 18 de información detallada sobre todas las medidas adoptadas sobrecargaría el RCP y podría influir negativamente en la transmisión de información importante para la seguridad. El CPMP considera que un informe público de evaluación es el documento apropiado para facilitar información sobre todas las medidas adoptadas para garantizar la seguridad de un producto individual frente a los agentes transmisibles y, lo que es muy importante, información sobre su evaluación independiente por parte de las autoridades reguladoras competentes. En el caso de medicamentos evaluados por el procedimiento centralizado, esta información está ya contenida en el informe público europeo de evaluación (EPAR). Se acepta que hasta la fecha sólo se han autorizado 2 medicamentos derivados de plasma por el procedimiento centralizado. Sin embargo, este hecho no justifica la inclusión en el RCP de información aislada sobre pruebas NAT voluntarias, que podría ser engañosa y contener un elemento de carácter publicitario. El argumento de la empresa insiste en la importancia de ampliar los informes públicos de evaluación a los productos autorizados por procedimientos nacionales. Como parte de la revisión continua de la legislación farmacéutica, propone la publicación de EPAR también para productos evaluados por el procedimiento de reconocimiento mutuo, como Beriate P. En el caso de los productos evaluados por procedimientos nacionales, la decisión de publicar o no informes públicos de evaluación recae en las autoridades nacionales competentes. Además, cuando se remiten productos específicos al CPMP para que adopte un dictamen, como en este caso, se publica en el sitio web de la EMEA la información resumida del dictamen sobre la remisión. Aunque la herramienta para realizar una evaluación independiente de productos específicos es el informe público de evaluación, el CPMP puede y debe proporcionar información sobre los factores que contribuyen a la seguridad viral en su sitio web1-5,8. Además, en el Seminario de la EMEA celebrado en septiembre de 2000 sobre la seguridad viral de los medicamentos derivados de plasma, con especial atención a los virus sin envoltura, participaron representantes de las asociaciones de paciente europeos con hemofilia e inmunodeficiencia primaria (EHC y EPPIC)3. El BPWG invita periódicamente a las organizaciones relevantes de médicos y pacientes a los debates sobre la seguridad y la eficacia de los medicamentos derivados de plasma, en los que también se trata del contenido del RCP básico. Este procedimiento habitual se seguirá en la próxima consulta pública sobre el texto revisado de las advertencias para los RCP y los prospectos. Además, el CPMP pretende publicar una Declaración sobre la evaluación de la seguridad viral de los medicamentos derivados de plasma. Su finalidad será explicar, de una manera clara y sencilla para los profesionales sanitarios y los pacientes, los factores que han de tenerse en cuenta para evaluar la seguridad viral de los medicamentos derivados de plasma, como la utilidad de las pruebas NAT, y para determinar el contenido del RCP. Esta declaración será la continuación lógica de la publicada el año pasado por el CPMP sobre los donantes remunerados y no remunerados: seguridad y suministro de los medicamentos derivados de plasma4. 4.8 ¿Apoyan los Estados miembros de la UE la directriz del CPMP sobre el texto de la advertencia relativa a los agentes transmisibles? Los RCP básicos para medicamentos derivados de plasma son una herramienta para armonizar mejor la información sobre estos productos en la UE. La empresa ha alegado que la mayoría de los Estados miembros (8/15) hacen caso omiso de la directriz del CPMP y permiten que en se haga referencia a pruebas NAT para otros virus sus RCP nacionales. La empresa cita, en el Anexo 1 de la documentación de su recurso, casos en los que se ha permitido la referencia a otras pruebas NAT diferentes a la obligatoria para el VHC. La lista de productos presentados por la empresa en cuyos RCP se hace referencia a pruebas NAT es engañosa a primera vista. Sin embargo, como puede observarse en la Tabla 2, cuando la información proporcionada por la empresa se analiza más a fondo, revela una situación diferente. 19 Tabla 2: Análisis de la información contenida en el Anexo 1 de la documentación del recurso de la empresa País Número de productos cuyos RCP hacen referencia a otras pruebas NAT para el VIH y el VHB Austria Bélgica Dinamarca Finlandia Francia Alemaniac) Grecia Irlanda Italia Luxemburgo Países Bajos España Suecia Portugal Reino Unido 16 0 5 0 0 32 6 0 1 1 0 0 0 0 4 Número de productos cuyos RCP hacen referencia a otras pruebas NAT para el VIH y el VHC, con fecha posterior a la del RCP básico correspondiente al productoa) 6 0 1 0 0 6 2 0 0 0 0 0 0 0 2 Número de productos que contienen FVIII cuyos RCP hacen referencia a otras pruebas NAT para el VIH y el VHB, con fecha posterior al RCP básico 3b) 0 0 0 0 0 0 0 0 0 0 0 0 0 1b) a) No se consideran los RCP básicos antiguos, publicados en 1992, que no han sido revisados posteriormente. b) Un ejemplo en Austria es el Haemate P y otro en el Reino Unido, Haemate P, un producto de Behring. c) En Alemania, las pruebas NAT para el VIH y el VHB se mencionan en la información facilitada a los profesionales sanitarios alemanes (Fachinformation) en una sección titulada «Notas adicionales», que está permitida por la legislación nacional (Arzneimittelgesetz). Aventis Behring ha declarado durante las alegaciones verbales presentadas en febrero de 2003 que las pruebas NAT se mencionan en la sección de advertencias y precauciones de algunos RCP actualizados para equipararlos a los RCP básicos. Esta remisión se refiere a la inclusión de información adicional sobre pruebas NAT para el VIH y el VHB. Sin contar los casos en que se mencionan pruebas NAT para otros virus, 7 de los 15 Estados miembros tienen productos cuyos RCP mencionan las pruebas NAT para el VIH y el VHB (columna 1 de la Tabla 2). Muchos de estos casos corresponden a RCP anteriores a la entrada en vigor de los RCP básicos actuales o a productos que no tienen todavía un RCP básico. Considerando que todas las empresas de la PPTA están realizando voluntariamente pruebas NAT para el VIH y el VHB, se observa que el número de productos cuyos RCP actualizados mencionan las pruebas NAT para el VIH y el VHB es pequeño (columna 2 de la Tabla 2). En el caso del factor VIII, el objeto de este recurso, sólo 2 Estados miembros tienen RCP actualizados después de la entrada en vigor del RCP básico que mencionen las pruebas NAT para el VIH y el VHB (columna 3 de la Tabla 2). En el único caso del Reino Unido, se trata de otro producto de Aventis Behring, la empresa implicada en esta remisión. Ese mismo producto se corresponde también con 1 de los 3 casos de Austria. En las alegaciones verbales presentadas en febrero, la empresa declaró que, al haber autorizado varios Estados miembros la mención de otras pruebas NAT para virus distintos al VHC, la desestimación de este recurso supondría una discriminación contra Aventis Behring. El CPMP considera que el análisis de la situación de otros productos que contienen factor VIII derivado de plasma no justifica esa supuesta discriminación. De hecho, si se permitieran otras pruebas NAT para el factor VIII de Aventis Behring, sería una discriminación contra otros productos con RCP actualizados para equipararlos al RCP básico. Este análisis demuestra la utilidad de la directriz sobre el RCP básico y el respaldo que ha obtenido de los Estados miembros. No obstante, los Estados miembros podrían encontrar dificultades si los titulares de las autorizaciones de comercialización se negaran a actualizar sus RCP en línea con la última directriz durante los procedimientos nacionales. 20 Además, el texto acordado para el RCP básico, el rechazo de la variación de Beriate P por el procedimiento de reconocimiento mutuo y el dictamen del CPMP sobre la remisión de Beriate P indican que los expertos de los Estados miembros respaldan la directriz del CPMP. De igual modo, tampoco se mencionan otras pruebas NAT para la detección del ADN de VIH-1 y el ADN de VHB en los RCP de los medicamentos derivados del plasma autorizados por el procedimiento centralizado. La revisión continua de la advertencia sobre los agentes transmisibles culminará con la publicación de una directriz aplicable a todos los medicamentos derivados de plasma y contribuirá así a la armonización de todos los medicamentos de este tipo. La empresa argumenta también que Bélgica publicó recientemente un Real Decreto que obliga a realizar pruebas NAT para el ARN del VIH-1, además de las pruebas NAT para el ARN del VHC. El CPMP recuerda que el Real Decreto belga se refiere principalmente a los componentes sanguíneos lábiles (hematíes, plaquetas, etc.) que, al contrario que los medicamentos derivados de plasma, no suelen someterse a procedimientos de inactivación/eliminación viral. Los hemoderivados estables constituyen una excepción a este Real Decreto. En las Secciones 4.5 y 4.6 pueden encontrarse las consideraciones del CPMP sobre los argumentos expuestos por la empresa respecto a la situación en Estados Unidos y Japón. 5. Respuesta del CPMP a la moción de recurso de la empresa Moción: “El dictamen del CPMP y el rechazo de la solicitud del Apelante incumplen tanto el RCP básico como la Directiva 2001/83/CE. El rechazo del idioma en el que se presenta el etiquetado del producto carece de todo fundamento legal. Además, dicho rechazo contraviene el concepto del RCP básico del CPMP de incluir toda la información clínicamente relevante.” Respuesta del CPMP: Según se establece en la directriz sobre el resumen de las características del producto6: «El RCP constituye la información básica facilitada a los profesionales de la salud sobre cómo utilizar el medicamento con seguridad y eficacia.» En ello se fundamenta el requisito de que el RCP contenga «información necesaria para el médico y el paciente». La directriz contiene una lista de aspectos que son relevantes para cada sección del RCP. La remisión actual se refiere al texto de la sección 4.4 (Advertencias y precauciones especiales de empleo). Como indica el propio nombre de esta sección, todas las cuestiones tratadas en ella deben estar relacionadas con información que sea clínicamente importante para médicos y pacientes. Moción: • “al no permitir que el Apelante incluya información completa y científicamente sólida sobre las pruebas PCR.” Respuesta del CPMP: La finalidad del RCP no es facilitar datos técnicos sobre los procesos de producción. Los artículos 11 y 62 de la Directiva 2001/83/CE que especifican la información que deben contener los RCP y los prospectos, respectivamente, no mencionan los procesos de producción. Además, ello daría pie a la inclusión de otro tipo de información irrelevante para el uso clínico apropiado del producto en los RCP de los medicamentos derivados de plasma y de otros medicamentos. Moción: • “al dar a entender que la seguridad viral de los productos derivados de plasma está garantizada, entre otras medidas, por las pruebas PCR para un único virus (VHC), • y al considerar que esa información es suficiente, necesaria y adecuada.” 21 Respuesta del CPMP: La empresa ha alegado que la mención de las pruebas NAT para el VHC en el texto actual del RCP básico demuestra que el CPMP considera que es información clínicamente importante. Tal como se establece en el dictamen del CPMP sobre la remisión (EMEA/CPMP/5008/02), el motivo de mencionar las pruebas NAT en el RCP básico es por tratarse de una de las pruebas obligatorias y necesarias para todos los medicamentos derivados de plasma. La empresa ha alegado también que la obligación de realizar pruebas NAT para la detección de VHC no significa necesariamente que deba mencionarse en el RCP. El CPMP está de acuerdo con la empresa. La Directriz sobre el resumen de las características del producto6 dice que “Los solicitantes deben mantener la integridad de todas las secciones del documento, incluyendo sólo información en cada sección que se corresponda con su encabezamiento.” Puesto que la información sobre las medidas de obligado cumplimiento no constituye una precaución ni una advertencia, el texto de la Sección 4.4 del RCP básico para los productos que contienen factor VIII derivado de plasma humano debe modificarse para excluir todo lo que no sea ni una advertencia ni una precaución. Este análisis es consecuente con la revisión que está realizando el CPMP del texto actual de las advertencias, que se describía en el dictamen del CPMP de octubre de 2002 y en el informe de evaluación sobre la remisión de Beriate P: «El CPMP revisa constantemente sus directrices, incluidos los RCP básicos para los medicamentos derivados de plasma, y actualiza los textos según lo considera necesario. Así pues, en paralelo con esta remisión, el CPMP ha considerado si el texto actual de la advertencia sobre los agentes transmisibles en el RCP básico es óptimo. El texto del RCP básico actual permite la diferenciación de los productos en función de la eficacia de las medidas adoptadas contra el VHA y el parvovirus B19, siempre que dicha información sea necesaria para el que prescriba el medicamento. La opinión provisional del CPMP es que la advertencia podría mejorarse suprimiendo la referencia a las pruebas obligatorias (que ya se ha publicado en otro lugarh) y centrándose en los otros riesgos potenciales.” Sería contradictorio que se permitiera ampliar la descripción de las pruebas NAT para hacer referencia al VIH y al VHB en la Sección 4.4 del RCP para Beriate P, cuando al mismo tiempo se está considerando la posibilidad de no mencionar ni siquiera las pruebas obligatorias. Moción: “Por consiguiente, el dictamen parece basarse en una decisión arbitraria. El CPMP obliga al Apelante a proporcionar a los profesionales sanitarios y a los pacientes información incompleta y confusa y, por tanto, infringe su derecho a ser debidamente informados sobre el medicamento descrito en el RCP correspondiente.” Respuesta del CPMP: Como se señaló antes, el CPMP está de acuerdo con la empresa en que el hecho de que la prueba del VHC sea obligatoria no significa que deba mencionarse en el RCP. El CPMP tiene la intención de suprimir dicha mención en la Sección 4.4 del RCP básico. Las pruebas NAT para el VHC constituyen un requisito obligatorio para todos los medicamentos derivados de plasma y la inclusión de esta información en los RCP no suponer hacer distinción entre productos. Por el contrario, la mención en la Sección 4.4 (“Advertencias y precauciones especiales de empleo”) de pruebas voluntarias como son las pruebas NAT para VIH y VHB, que se realizan en algunos productos pero no en otros, daría a entender que los productos sometidos a esas pruebas son más seguros que los otros. Pero eso no es cierto, como se explica más adelante (Sección 4.4). La inclusión de esa información tendría, por tanto, un elemento de carácter publicitario totalmente inaceptable en un RCP y no sería útil para la “educación sanitaria” según se establece en el artículo 62 de la Directiva 2001/83/CE. h Las pruebas obligatorias de donaciones y mezclas de plasma se han publicado en la monografía de la Farmacopea Europea sobre plasma humano para fraccionamiento. . 22 Además, al no ser obligatorias las pruebas NAT para el VIH y el VHB, no existe un límite inferior de detección oficialmente establecido y exigido para el VHC. Por consiguiente, la declaración de que una mezcla de plasma ha sido sometida a pruebas para la detección de VIH y VHB no facilita información sobre el valor límite inferior utilizado para definir una mezcla no reactiva (o un plasma reactivo positivo) y podría tener un significado diferente según el fabricante. Al estar muy avanzado ya el trabajo de revisión de la advertencia sobre los agentes transmisibles que se incluirá en los RCP de los medicamentos derivados de plasma, el CPMP recomienda que las autorizaciones de comercialización puedan revisarse conforme al resumen de las características del producto que se incluye en el Anexo III del dictamen. Por consiguiente, el CPMP recomienda la revisión del dictamen que adoptó el 17 de octubre de 2002, en el que recomendaba que las autorizaciones de comercialización debían revisarse. De esta forma, el titular de la autorización de comercialización podrá elegir entre armonizar el texto del RCP básico actual o esperar a la revisión de la advertencia sobre los agentes transmisibles. 6. Conclusión 6.1 La seguridad viral se consigue con una combinación de diferentes factores, que han de considerarse conjuntamente para poder evaluar la seguridad global de cada producto. La evaluación de la seguridad viral es una cuestión compleja que no puede expresarse simplemente con factores de reducción o listas de sofisticados métodos analíticos. Tratar de proporcionar toda esta información en el RCP no sólo sería una tarea enorme, sino también imposible. Los RCP tendrían que ampliarse enormemente, con el efecto negativo que ello tendría en la transmisión clara de información importante para garantizar el uso seguro y eficaz de los productos. Además, esa información seguiría siendo insuficiente para que las autoridades reguladoras pudieran evaluar la seguridad viral de un producto y, en consecuencia, sería también insuficiente para los usuarios. 6.2 Las pruebas NAT realizadas a mezclas de plasma constituyen sólo uno más de los diferentes factores que contribuyen a la seguridad global de un producto. Se reconoce la utilidad de las pruebas NAT para detectar VHB y VIH-1 y contribuir al control de la contaminación en los materiales de partida, como parte de la estrategia de fabricación y el epidemiología de las poblaciones donantes en esa empresa en particular. Sin embargo, las pruebas NAT para el HIV1 y el HBV no deben mencionarse en la Sección 4.4 («Advertencias y precauciones especiales de empleo») del RCP porque la información aislada sobre pruebas NAT voluntarias induciría a error, al dar a entender que los productos sometidos a esas pruebas son más seguros que los que no se someten a ellas, e introducir así un elemento de carácter publicitario. La inclusión de esa información podría fomentar alegaciones publicitarias injustificadas. Además, el RCP no tiene como finalidad facilitar información detallada sobre los procesos de fabricación. 6.3 Todos los medicamentos actuales derivados de plasma ofrecen una excelente seguridad viral respecto a los virus con envoltura, entre ellos el VIH, el VHB y el VHC, y las pruebas NAT no deben utilizarse como la única información para diferenciar un producto de otro. En lo que sí pueden diferir los productos es en su seguridad respecto a la hepatitis A y el parvovirus B19. En consecuencia, el texto actual de las advertencias en el RCP básico permite diferenciar a los productos según la efectividad de las medidas adoptadas contra el VHA y el parvovirus B19. Este tipo de información es útil para el que prescribe los medicamentos. El texto se optimizará próximamente con la revisión que se está realizando de las advertencias. 6.4 Los usuarios deben recibir información exhaustiva sobre la seguridad de los productos y no información parcial. Los informes públicos de evaluación constituyen la herramienta apropiada para facilitar información sobre todas las medidas adoptadas para garantizar la seguridad de un producto en particular frente a los agentes transmisibles y, lo que es muy importante, información sobre su evaluación independiente por parte de las autoridades reguladoras competentes. En el caso de medicamentos evaluados por el procedimiento centralizado, esta 23 información está ya contenida en el informe público europeo de evaluación (EPAR). Como parte de la revisión continua de la legislación farmacéutica, se propone la publicación de EPAR también para productos evaluados por el procedimiento de reconocimiento mutuo. En el caso de los productos evaluados por procedimientos nacionales, la decisión de publicar o no informes públicos de evaluación recae en las autoridades nacionales competentes. Además, cuando se remiten productos específicos al CPMP para que adopte un dictamen, como en este caso, se publica en el sitio web de la EMEA información resumida del dictamen sobre la remisión. 6.5 Aunque la herramienta para realizar una evaluación independiente de productos individuales es el informe público de evaluación, el CPMP puede proporcionar y proporciona información objetiva sobre los factores que contribuyen a la seguridad viral en su sitio web. Además, representantes de las asociaciones relevantes de pacientes y médicos participan activamente en los seminarios y reuniones de la EMEA sobre agentes transmisibles y, más generalmente, sobre la seguridad y eficacia de los medicamentos derivados de plasma. Este procedimiento habitual se seguirá en la próxima consulta pública sobre el texto revisado de la advertencia sobre los agentes transmisibles en los RCP y los prospectos. Además, el CPMP tiene la intención de publicar una Declaración sobre la evaluación de la seguridad viral de los medicamentos derivados de plasma. Su finalidad será explicar a los profesionales sanitarios y a los pacientes los factores que tienen que tener en cuenta para evaluar la seguridad viral de los medicamentos derivados del plasma, entre ellos la utilidad de las pruebas NAT y el contenido del RCP. Referencias 1. Nota de orientación del CPMP sobre los estudios de validación de virus : Diseño, contribución e interpretación de los estudios para validar la inactivación y eliminación de virus, CPMP/BWP/268/95, 14 de febrero de 1996. 2. Nota de orientación del CPMP sobre CPMP/BWP/269/95, 25 de enero de 2001. 3. Seminario de la EMEA sobre la seguridad viral de los medicamentos derivados de plasma, con especial atención a los virus sin envoltura , CPMP/BWP/BPWG/4080/00, 28 de marzo de 2001, Anexo: Conclusiones y recomendaciones del Grupo de trabajo «Biotecnología» (BWP) y el Grupo de trabajo ad hoc sobre hemoderivados (BPWG), CPMP/BWP/BPWG/93/01, 28 de marzo de 2001. 4. Declaración del CPMP sobre los donantes remunerados y no remunerados: seguridad y suministro de los medicamentos derivados del plasma, EMEA/CPMP/BWP/1818/02, 30 de mayo de 2002. 5. Nota de orientación sobre la «Utilización de NAT para la detección de ARN del virus de la hepatitis C en mezclas de plasma» (CPMP/BWP/390/97), 24 de marzo de 1998. 6. Directriz sobre el resumen de las características del producto, diciembre de 1999, Normas que regulan los medicamentos en la Comunidad Europea, Volumen 2A y 2B, Notificación a los solicitantes. 7. Proyecto de Directriz de la FDA para la industria, Determinación de ácidos nucleicos en mezclas de plasma fuente para conseguir una reducción apropiada del riesgo de transmisión de VIH-1 y VHC, diciembre de 2001. 8. Informe del seminario de la EMEA sobre EMEA/CPMP/BWP/1737/02, Diciembre 2002. 24 los medicamentos el archivo derivados maestro de de plasma, plasma, 9. Directriz de la FDA para la industria sobre la revisión de las medidas preventivas para reducir el posible riesgo de transmisión de la ECJ y los vectores de la ECJ a través de la sangre y los medicamentos derivados de plasma, enero de 2002. 25 C. MOTIVOS DE LA REVISIÓN DEL DICTAMEN DE 18 DE FEBRERO DE 2003 El 14 de abril de 2003, la Comisión Europea devolvió a la EMEA el Dictamen Final del CPMP con fecha 18 de febrero de 2003 solicitando aclaraciones sobre dicho dictamen y la recomendación según la cual «podrán revisarse las autorizaciones de comercialización, de conformidad con el resumen de las características del producto que figura en el Anexo III del Dictamen». La Comisión Europea, en su carta a la EMEA, decía lo siguiente: «El texto da a entender que las autoridades nacionales pueden elegir entre modificar o no las autorizaciones nacionales de comercialización de Beriate P. Teniendo en cuenta que el objetivo del artículo 31 de la Directiva 2001/83/CE es alcanzar una decisión armonizada en el ámbito comunitario, la Comisión no puede adoptar una decisión basada en dicha declaración.» La Comisión Europea solicitó aclaraciones sobre la evaluación final del CPMP en ese sentido. En respuesta a la carta antes mencionada de la Comisión Europea, el CPMP aclara que no pretendía dar a entender que las autoridades nacionales pueden elegir entre modificar o no las autorizaciones nacionales de comercialización de Beriate P. Su intención fue la que se describe en el Anexo II del Dictamen Final revisado del CPMP (Sección B.5., Respuesta del CPMP al recurso presentado por la compañía). Por consiguiente, el CPMP, teniendo en cuenta la carta antes mencionada de la Comisión Europea, recomienda que su Dictamen Final de 18 de febrero sea revisado para suprimir la recomendación «podrán revisarse las autorizaciones de comercialización, de conformidad con el resumen de las características del producto que figura en el Anexo III del Dictamen». Además, el CPMP se declara conforme con la decisión adoptada en el procedimiento de reconocimiento mutuo de rechazar la solicitud de variación en las condiciones de las autorizaciones de comercialización de Beriate P (SE/H/135/01-03/W16) que proponía incluir una mención a las pruebas NAT para el VIH y el VHB en la Sección 4.4 (Advertencias y precauciones especiales de empleo) del RCP. En consecuencia, los resúmenes de las características del producto siguen siendo los mismos autorizados por el procedimiento de reconocimiento mutuo. Los fundamentos científicos de la conclusión del CPMP se exponen en el informe de evaluación de la remisión que se adjunta y en el Anexo II del Dictamen Final revisado del CPMP e incluyen los siguientes aspectos: • El RCP constituye la base de la información para que los profesionales sanitarios sepan como utilizar el medicamento con seguridad y eficacia. La Sección 4.4 (Advertencias y precauciones especiales de empleo) debe contener información clínicamente relevante para el que prescriba el medicamento y para el paciente. • La finalidad del RCP no es facilitar datos técnicos sobre los procesos de producción. • El motivo de mencionar las pruebas NAT para el VHC en el texto actual de la advertencia sobre los agentes transmisibles contenida en el RCP básico es que se trata de una de las pruebas obligatorias para todos los medicamentos derivados del plasma. Las pruebas NAT para el VIH y el VHB no son pruebas obligatorias y algunos fabricantes, pero no todos, las realizan de manera voluntaria. • El CPMP tiene ya muy avanzado el trabajo de revisión de la advertencia para transmitir un mensaje clínico más claro sobre los riesgos residuales. La revisión no mencionará prueba específica alguna, pero indicará si las medidas generales adoptadas se consideran eficaces contra los virus especificados. • La seguridad viral se consigue mediante una combinación de factores diferentes, que han de considerarse conjuntamente para evaluar la seguridad global de cada producto individual. (Por ejemplo, un producto puede someterse a pruebas NAT adicionales, mientras que otro puede proceder de un proceso de fabricación con mayor capacidad de inactivación o destrucción). El margen de seguridad conseguido ha de tener en cuenta todos los factores para poder comparar los 26 • productos. Todos los medicamentos actuales derivados del plasma ofrecen una excelente seguridad viral con respecto a los virus VIH, VHB y VHC y no existe razón alguna para hacer este tipo de distinciones entre productos en el RCP. La inclusión en la Sección 4.4 (Advertencias y precauciones especiales de empleo) de pruebas voluntarias, como las pruebas NAT para el VIH y el VHB, que se realizan en algunos productos pero no en otros, daría a entender que los productos sometidos a esas pruebas son más seguros que los otros. Sin embargo, como se explicaba antes, no es éste el caso. Esta revisión no altera las conclusiones científicas del CPMP respecto al recurso, sino sólo su forma de expresión en el dictamen final revisado. Conclusión El CPMP, tras haber considerado la solicitud de aclaración de la Comisión, recomienda que su dictamen final de 18 de febrero sea revisado para suprimir la recomendación de «podrán revisarse las autorizaciones de comercialización, de conformidad con el resumen de las características del producto que figura en el Anexo III del Dictamen». Asimismo, el CPMP se declara conforme con la decisión adoptada en el procedimiento de reconocimiento mutuo de rechazar la solicitud de variación de las condiciones de las autorizaciones de comercialización de Beriate P (SE/H/135/01-03/W16), que propone incluir una mención a las pruebas NAT para el VIH y el VHB en la Sección 4.4 (Advertencias y precauciones especiales de empleo) del RCP. En consecuencia, los resúmenes de las características del producto siguen siendo los mismos autorizados por el procedimiento de reconocimiento mutuo. 27