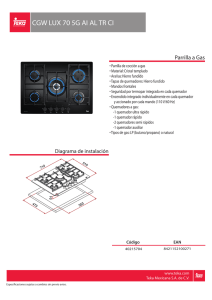
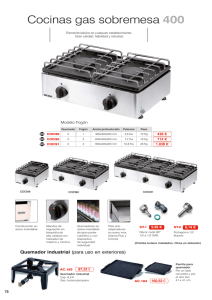
manual de prácticas del espectrofotómetro de absorción atómica
Anuncio

UNIVERSIDAD VERACRUZANA FACULTAD DE CIENCIAS QUÍMICAS QUÍMICA INDUSTRIAL TRABAJO TEÓRICO PRÁCTICO EDUCATIVO MANUAL DE PRÁCTICAS DEL ESPECTROFOTÓMETRO DE ABSORCIÓN ATÓMICA GBC 932 AA PRESENTA MAYRA DEL SOCORRO MÁRQUEZ MARTÍNEZ DIRECTOR M.C. MARISOL CASTILLO MORALES ORIZABA, VER. JULIO, 2009 INDICE GENERAL Pág. INDICE I INDICE DE FIGURAS III INDICE DE CUADROS V INDICE Pág. 1. INTRODUCCIÓN 1 1.1 Justificación 3 1.2 Objetivo General 4 1.3 Objetivo Particular 4 2. MARCO TEÓRICO 4 2.1. Principios de Absorción Atómica 6 2.1.1 Espectros Ópticos 6 2.2. Fundamento 7 2.2.1 Instrumentación en Espectroscopia de Absorción Atómica 8 2.2.2 Métodos de Introducción de la muestra 9 2.2.3 Producción de la nube atómica (Sistema Nebulizador-Quemador) 10 1 2.2.4 Sistema Óptico 11 2.2.5 Fuente de luz o radiación primaria 12 2.2.6 Sistema de preajuste óptico 13 2.2.7 Monocromador 14 2.2.8 Detector 15 3. MÉTODOS 16 Practica num. 1 Conocimiento General de un Espectrofotómetro de 17 Absorción Atómica Practica num. 2 Conocimiento del Manejo del Software 31 Practica num. 3 Calibración 52 Practica num. 4 Elaboración de una Curva de Calibración 55 Practica num. 5 Determinar calcio (Ca), sodio (Na), potasio (K), magnesio 60 (Mg) en muestras de propóleo Practica num. 6 Determinación de potasio (K) en muestras de plátano 73 Practica num. 7 Determinación de potasio (K) en muestras de piña y 77 corazón deshidratado. Practica num. 8 Determinación de calcio (Ca), sodio (Na), potasio (K), 83 magnesio (Mg) en muestras de higuerilla. Practica num. 9 Determinar calcio (Ca), sodio (Na), potasio (K), magnesio 96 (Mg) en muestras de gigantón Practica num. 10 Determinar plomo (Pb) y cadmio (Cd) en muestras de 104 pescado. Practica num. 11 Determinación de plomo (Pb) y cadmio (Cd) en muestras 112 2 de agua residual. Practica num. 12 determinar calcio (Ca), sodio (Na), potasio (K), magnesio 123 (Mg) en muestras de colorín. Practica num. 13 Determinación de mercurio (Hg) en muestra de pescado 136 por medio del generador de hidruros. 4. COMENTARIOS 148 5. ANEXOS 150 ANEXO 1 151 ANEXO 2 152 ANEXO 3 153 ANEXO 4 155 ANEXO 5 157 ANEXO 6 160 6. BIBLIOGRAFIA 177 3 INDICE DE FIGURAS Figura Nombre de Figura Pág. 1 Espectros Ópticos 6 2 Espectro continuo de la luz blanca 7 3 Espectro de emisión de vapores de Li 7 4 Espectro de absorción de vapores de Li 7 5 Proceso de la flama 8 6 Componentes del Sistema Óptico 12 7 Lámpara de Cátodo Hueco 13 8 Función del monocromador 15 9 Partes del Instrumento de Absorción Atómica 17 10 Quemador aire-acetileno 18 11 Quemador N2O-Acetileno 19 12 Botón liberador del quemador 20 13 Cámara de Rociado 21 14 Nebulizador removido 24 15 Soquet 29 16 Lámpara de Cátodo Hueco 29 17 Colocación del Soquet 30 18 Colocación del soquet con la lámpara de cátodo hueco 30 4 19 Programa GBC Avanta Ver 1.32 31 20 Ventana principal del programa GBC Avanta 1.32 32 21 Ayuda 32 22 Configuración del Instrumento: Modelo 35 23 Propiedades del Método del Sistema 35 24 Método 36 25 Libro del Método 36 26 Tabla Periódica 36 27 Añadir un elemento a lámpara 37 28 Pagina del Instrumento 37 29 Modo de Medición 38 30 Página de Calibración 39 31 Página de Estándares 39 32 Página de Calidad 40 33 Página de Flama 41 34 Muestras 41 35 Tabla de Muestras 42 36 Análisis 43 37 Análisis Único 43 38 Resultados 44 39 Página de Resultados 45 5 40 Resultados 45 41 Página del Reporte 46 42 Panel de Optimización del Flujo de Gases 47 43 Mensaje del Orden Cero No Encontrado 48 44 Baja Señal de la HCL 49 45 Panel de Medidores de Servicio 51 46 Operación del Instrumento 138 47 Componentes del Instrumento 139 48 Mezcle de Gases 140 49 Componentes del Instrumento 140 50 Accesorio HG 141 6 INDICE DE TABLAS Tablas Nombre de las Tablas Pág. 1 Temperatura de los Gases 11 2 Información general de como usar otros módulos para desarrollar un 33 análisis 3 Clave y peso de muestras de propóleo 65 4 Clave y peso de muestras de plátano 74 5 Clave y peso de muestras de Piña. 79 6 Clave y peso de muestras de Higuerilla. 85 7 Clave y peso de muestras de gigantón. 98 8 Clave y peso de muestras de pescado. 108 9 Clave y volumen de muestras de agua residual. 116 10 Clave y peso de muestras de colorín 125 11 Componentes del Instrumento 141 12 Clave y peso de muestras de pescado 145 7 1. INTRODUCCION 8 Los métodos instrumentales pueden detectar sólo rara vez especies químicas a niveles de microgramos, muchos métodos instrumentales son sorprendentemente sensibles. Entre ellos se encuentra Absorción Atómica consisten en transformar la muestra en átomos en estado de vapor (atomización) y medir la radiación electromagnética absorbida o emitida por dichos átomos. México requieren de nuevas estrategias en la formación de profesionistas competitivos que impulsen el avance económico, social y político del país, por lo que se tiene el desafío de fortalecer el objetivo fundamental y de encontrar un equilibrio entre la tarea que implica la inserción en la comunidad internacional y la atención a las circunstancias primordiales del país, entre la búsqueda del conocimiento por si mismo y la atención a necesidades sociales; entre fomentar capacidades genéricas o desarrollar conocimientos específicos, entre responder a demandas del empleador o adelantarse y descubrir anticipadamente el mundo futuro del trabajo que probablemente se sustentara mas en el autoempleo. Es por ello que la Universidad Veracruzana ha tenido que implementar un nuevo modelo educativo con un enfoque basado en el aprendizaje para promover que cambie el paradigma tradicional de la educación centrada en aprendizaje; para promover el desarrollo de habilidades y la apropiación de valores para formar profesionales de alta calidad que respondan eficientemente a las necesidades del país, basando la educación en competencias es resultado de análisis instrumental forma parte importante en la formación de profesores del área química y requieren fortalecer competencia y habilidades para que realicen análisis que son difíciles o imposibles por los métodos clásicos. 9 1.1 JUSTIFICACION La espectroscopia de Absorción Atómica es una de las técnicas analíticas más importantes en donde se realizan análisis o investigaciones químicas. La mayoría de las técnicas espectroscópicas se utilizan para el estudio y caracterización de moléculas o iones, la espectroscopía de absorción atómica se usa casi exclusivamente para el análisis de átomos. Por consiguiente, la técnica resulta casi insuperable como método de análisis elemental de metales. En principio, puede utilizarse para la identificación y la determinación cuantitativa de todos los elementos de la tabla periódica. Es muy importante para el alumno universitario conozca esta técnica fundamental con los conocimientos teóricos y que este familiarizado con el manejo adecuado del equipo. El campo de trabajo para el químico es muy amplio sin embargo hoy en día es muy común encontrar un sin número de industrias cementeras, control ambiental, alimentarias, etc. u otros organismos dedicados a la investigación cuyo principal requisito es que el químico este capacitado en esta técnica de absorción atómica. La técnica de Absorción Atómica ofrece un resultado confiable, reproducible y exacto que garantiza los análisis. Este trabajo pretende dar una referencia básica a los usuarios de la técnica de absorción atómica 10 1.2 OBJETIVO GENERAL Implementar un trabajo teórico-practico educativo sobre la las técnicas de Absorción Atómica asi como la utilización, que permitan al estudiante la adquisición de conocimientos para el desarrollo de habilidades que de soporte a la Experiencia Educativa (EE) de Análisis Instrumental, de tal manera que el estudiante lo haga mas competitivo y este acorde con el modelo educativo flexible. 1.3 OBJETIVO PARTICULAR Determinar e identificar los minerales y metales pesados presentes en agua, alimentos, plantas, mediante espectroscopia de absorción atómica. • Establecer la metodología que permita al estudiante el conocimiento básico del equipo de Absorción Atómica. • Implementar metodología básica para el análisis de agua en los analitos de Cadmio (Cd), Plomo (Pb), Mercurio (Hg). • Implementar metodología básica para el análisis de alimentos: Determinar potasio (K) en Piña (Ananas comosus) y Plátano Implementar técnicas que utilicen el generador de hidruros como Mercurio (Hg) en pescado Determinar los siguientes minerales: Sodio (Na), Magnesio (Mg), Calcio (Ca), Potasio (K) en Higuerilla (Ricinus communis L.) Gigantón, Colorín (Eritrina coralloides), Propóleo (americano) 11 2. MARCO TEORICO 12 2.1. Principios de Absorción Atómica La espectroscopia por absorción atómica puede definirse como un método analítico para la determinación cuantitativa de elementos, para llevarlo a cabo es necesario producir una nube atómica partiendo de una solución en la cual se encuentra el elemento a determinar cuyos átomos fundamentales están en la nube atómica mencionada, simultáneamente se hace pasar a través de esta nube atómica una luz con la longitud de onda correspondiente a la línea de resonancia del elemento a cuantificar midiendo la absorbancia. Los demás elementos presentes en la nube atómica no interfieren en la cuantificación, ya que ellos absorben en otras longitudes de onda. (19) Figura 1. Espectros Ópticos (8) 2.1.1 Espectros Ópticos Cuando se hace pasar la radiación emitida por un cuerpo caliente a través de un prisma óptico, se descompone en distintas radiaciones electromagnéticas dependiendo de su distinta longitud de onda (los distintos colores de la luz visible, radiaciones infrarrojas y ultravioleta) dando lugar a un espectro óptico. Todas las radiaciones obtenidas impresionan las películas fotográficas y así pueden ser registradas. Cada cuerpo caliente da origen a un espectro diferente ya que esta depende de la propia naturaleza del foco. (8) Los espectros pueden ser de emisión y absorción. A su vez ambos se clasifican en continuos y discontinuos: Espectros de Emisión Son aquellos que se obtienen al descomponer las radiaciones emitidas por un cuerpo previamente excitado. (8) 13 Los espectros de emisión continuos se obtienen al pasar las radiaciones de cualquier sólido incandescente por un prisma. Todos los sólidos a la misma Temperatura producen espectros de emisión iguales. Figura 2. Espectro continúo de la luz blanca (8) Los espectros de emisión discontinuos se obtienen al pasar la luz de vapor o gas exitado. Las radiaciones emitidas son características de los átomos exitados. (8) Figura 3. Espectro de emisión de vapores de Li (8) Espectros de Absorción son los espectros resultantes de intercalar una determinada sustancia entre una fuente de luz y un prisma. (8) Los espectros de absorción continuos se obtienen al intercalar el sólido entre el foco de radiación y el prisma. Así, por ejemplo, si intercalamos un vidrio de color azul quedan absorbidas todas las radiaciones menos el azul. (8) Los espectros de absorción discontinuos se producen al intercalar vapor o gas entre la fuente de radiación y el prisma. Se observan bandas o rayas situadas a la misma longitud de onda que los espectros de emisión de esos vapores o gases. (8) Figura 4. Espectro de absorción de vapores de Li (8) 14 2.2. Fundamento La ley de Beer establece que la absorción de una muestra, es directamente proporcional a la concentración de las especies absorbentes bajo ciertas condiciones experimentales dadas. (19) La ecuación que la define es: A=abc Donde: A = es la absorbancia. a = es el coeficiente de absortividad. b = es la longitud del paso de la luz ocupado por celda de absorción. c = es la concentración de las especies absorbentes. En la espectroscopía de absorción atómica, el quemador hace las veces de una celda como las utilizadas en los métodos de colorimetría. (19) Si se gráfica la absorción de soluciones de concentración conocida, contra su concentración, se obtiene una curva de calibración. A partir de esta curva, se puede medir la absorbancia de las soluciones problema e interpolar dicha absorbancia en las curvas de calibración para obtener la concentración de las soluciones. (19) Para obtener un valor real de concentración en absorción atómica se asegurara de estar trabajando en el rango lineal de la curva normal, para ello, nos indican el rango y las condiciones de trabajo para cada elemento. (19) 2.2.1 Instrumentación en Espectroscopia de Absorción Atómica En esta metodología se utiliza un atomizador de llama en el que la disolución de la muestra es nebulizada mediante un flujo de gas oxidante, mezclado con el gas combustible y se transporta a una llama donde se produce la atomización, una serie compleja de procesos encadenados tiene lugar en la flama. (19) 15 Figura 5. Proceso de la flama (2) El primero es la desolvatación en el que se evapora el disolvente hasta producir un aerosol molecular sólido finamente dividido. (19) El segundo la disociación de la mayoría de estas moléculas produce un gas atómico. La mayoría de los átomos así formados se ionizan originando cationes y electrones, indudablemente se producen también otras moléculas y átomos de la llama como resultado de las interacciones del gas combustible con el gas oxidante y con las distintas especies de la muestra. (19) El tercero es una fracción de las moléculas, átomos e iones también se excitan por el calor de la llama produciéndose así espectros de emisión moleculares, atómicos e iónicos. Como ya se mencionó, produciéndose tantos procesos complejos resulta comprensible que la atomización sea la etapa más crítica en la espectroscopia de llama y por ello sea ésta la que limita la precisión de dichos métodos. Esta técnica tiene la ventaja de ser bastante precisa y más económica que otras ya mencionadas. (19) 2.2.2 Métodos de Introducción de la muestra La introducción de la muestra se considera como el punto clave de la espectroscopia atómica, ya que en muchos casos es la etapa limitante de la exactitud, precisión y límites de detección de las medidas de los espectrofotómetros atómicos. Siendo el objetivo del sistema de introducción de la muestra transferir una parte reproducible y representativa de la muestra al atomizador. (19) 16 A continuación se mencionan dos de los métodos más empleados. • Generador de Hidruros: Proporciona un método para introducción de muestras que contienen arsénico, antimonio, estaño, mercurio, selenio, bismuto, dicho procedimiento mejora los límites de detección de éstos elementos. (19) • Atomización Electrotérmica (horno de grafito): aparecen en los setentas y proporcionan generalmente mayor sensibilidad, debido a que toda la muestra se atomiza en un periodo muy corto y el tiempo de permanencia de los átomos en el camino óptico. Para su funcionamiento unos cuantos microlitros de muestra se evaporan primero a baja temperatura y posteriormente se calcinan a una temperatura más elevada dentro de un tubo de grafito, el cual es calentado eléctricamente, tras la calcinación la corriente se incrementa rápidamente a varios cientos de amperios lo que eleva la temperatura de 2000 a 3000°C; la atomización de la muestra se produce en un periodo de tiempo de pocos milisegundos a segundos, en estas condiciones se mide la absorción de las partículas atomizadas en la zona situada inmediatamente por encima de la superficie calentada. Ofrece la ventaja de su elevada sensibilidad para pequeños volúmenes de muestra, en general se emplean volúmenes de muestra entre 0.5 y 10 microlitros. En estas condiciones los límites de detección absolutos se encuentran normalmente de 1010 – 10-13g de analito. Se puede considerar que entre sus desventajas se encuentra el ser una técnica lenta, requiriéndose habitualmente varios minutos por elemento. Por lo anteriormente señalado esta técnica se aplica solo cuando la atomización con flama proporcionan límites de detección inadecuados. (19) Este mecanismo cuenta con un aspersor que toma la muestra y la envía a una cámara donde es nebulizada en pequeñísimas gotas. Finalmente pasa a un quemador con flama lo suficientemente caliente para producir el vapor atómico por el cual pasará la luz. (19) 17 2.2.3 Producción de la Nube Atómica (Sistema Nebulizador-Quemador) El sistema nebulizador – quemador. Cuenta con un mecanismo de aspersión que succiona la muestra convirtiéndola en un aerosol haciéndola pasar a través de una cámara donde es impactada varias veces. De ahí pasa a una cámara de mezcla por la cual entran el gas oxidante y el gas combustible. (19) Una vez realizada la mezcla, pasa al quemador donde está encendida la flama para que se produzca el vapor atómico. (19) Dependiendo de la temperatura de la flama a utilizar se usará un quemador distinto. Los tipos de flama más utilizados son: Cuadro 1. Temperatura de los Gases (19) OXIDANTE COMBUSTIBLE TEMPERATURA Aire propano 2200 ºC Aire acetileno 2450 ºC oxido nitroso N2O acetileno 3200 ºC . La mezcla más utilizada es la de aire – acetileno, la de óxido nitroso – acetileno se utiliza principalmente cuando se requieren altas temperaturas que rompan compuestos termoestables que podrían dar una falsa lectura al estar parte de los elementos en forma de compuestos y no formar parte de la nube atómica. (19) El quemador aire – acetileno mide aproximadamente diez centímetros, mientras que el de óxido nitroso – acetileno mide cinco centímetros. (19) ‘Es muy importante mantener siempre muy limpio el sistema nebulizador – quemador para que haya una buena producción de vapor atómico y para que no existan impurezas en el quemador que podrían alterar el análisis. (19) 18 2.2.4 Sistema Óptico Un sistema detector: Capaz de realizar la absorbancia de la línea de resonancia correspondiente. (19) Es el cual se elijan las distintas longitudes de onda y se emita la luz que incidirá sobre el vapor atómico. (19) El sistema óptico de un espectrofotómetro de absorción atómica que consiste principalmente en: • Fuente de luz o radiación primaria • Sistema de preajuste óptico • Monocromador Divergencia Rendija Abertura Rendija Monocromador Rendija Detector Tubo Muestra Figura 6. Componentes del Sistema Óptico (9) 2.2.5 Fuente de luz o radiación primaria En la mayoría de los equipos de absorción atómica, se utiliza como fuente de radiación primaria las lámparas de cátodo hueco, que genera un espectro atómico lineal característico del elemento a cuantificar. (19) 19 La lámpara de cátodo hueco es un cilindro de vidrio llenada de un gas, que es generalmente argón o neón, al extremo se encuentra una ventana transparente a la radiación emitida por el cátodo que también se encuentra dentro de la chaqueta de vidrio. (19) El proceso de emisión del espectro atómico, sucede cuando se aplica una diferencia de potencial entre el cátodo y el ánodo, entonces algunos de los átomos del gas de relleno se ionizan. Los iones cargados positivamente se aceleran a través del campo eléctrico y colisionan con el cátodo cargado negativamente, desalojando así algunos de los átomos de la superficie del cátodo; los átomos desalojados son excitados debido a las subsecuentes colisiones que sufren con los iones de gas emitiendo el correspondiente espectro atómico al regresar a su estado fundamental. (19) Las lámparas de cátodo hueco pueden operar en un intervalo más ó menos amplio pero es necesario escoger el miliamperaje apropiado para cada lámpara y análisis a realizar. Es la fuente de líneas espectrales característica para cada elemento. La lámpara de cátodo hueco es la más comúnmente empleada. Su rendimiento promedio es de 1000 h/trabajo, si el sistema óptico es deficiente, se deberán utilizar intensidades de corriente más elevadas, que terminan por desgastar la señal de emisión y acortar la vida útil de la lámpara. (19) Figura 7. Lámpara de Cátodo Hueco (9) Como la descarga de la lámpara es la media estadística de todas las micro descargas producidas por la excitación de los átomos desalojados del cátodo, la señal emitida no es perfectamente estable presentando pequeñas variaciones o ruido, el cual se manifiesta en 20 el parpadeo de la señal. Por ello debe haber un mínimo de intensidad de corriente, debajo del cual el ruido es muy grande por lo que el valor escogido de la corriente debe ser mayor para minimizar el efecto, pero no tan alto que provoque el ensanchamiento de las líneas de resonancia. Se recomienda que el ruido sea menor al 0.2% y que la fluctuación de la intensidad sea menor al 5% por hora. (19) 2.2.6 Sistema de preajuste óptico En cualquier espectrofotómetro de un solo haz, la luz que llega al transmisor es proporcional a la transmisión de la muestra que se encuentra en el camino óptico del rayo, por ello, es necesario llevar a cabo cuando la muestra está presente y cuando no lo está, para que pueda obtenerse la absorbancia de la muestra, por ello es muy importante que la intensidad de la luz de la fuente principal sea lo más estable posible. (19) Las pequeñas variaciones que puede sufrir la intensidad de la lámpara pueden resolverse utilizando un sistema de preajuste óptico de doble haz. En la mayoría de estos sistemas, el rayo de luz proveniente de la fuente cae a un espejo rotatorio antes de pasar a través de la flama, lo que trae como consecuencia que el rayo se vea dirigido alternativamente a través de la flama y por fuera de ella. Ya en el detector ambas señales son comparadas. (19) 2.2.7 Monocromador La función del monocromador es la de seleccionar la línea de resonancia de trabajo de las otras líneas que están cerca de ella. Por lo tanto el monocromador debe ser capaz de separar las líneas de resonancia de cualquier espectro que llegue a la ventanilla de entrada independientemente de la complejidad de éste. (19) El elemento dispersante del sistema monocromador puede ser un prisma o una rejilla de reflexión. La diferencia entre los dos sistemas estriba principalmente en que la dispersión dada por el prima varía con la frecuencia, siendo mayor en la región ultravioleta. (19) 21 En el caso de las rejillas, la dispersión es prácticamente constante a través de todo el espectro. Estas consisten en una superficie aluminizada de alto poder de reflexión que contiene un gran número de canales paralelos muy finos. (19) Absorción atómica existe una fuente independiente de luz monocromática, específica para cada elemento a analizar y que se hace pasar a través del vapor de átomos, midiéndose posteriormente la radiación absorbida. (19) Figura 8. Función del monocromador (26) 2.2.8 Detector Es casi universal y el uso de un sistema fotomultiplicador para la detección en los espectrofotómetros de absorción atómica. En este sistema se tiene una determinado de superficie fotoemitiva conectada a potenciales mayores en forma sucesivas. (19) Los electrones liberados en la primera superficie por el impacto de la radiación incidente, son acelerados por la diferencia de potencial existente hacia la segunda superficie, donde parte de la energía cinética adquirida es empleada en la liberación de más electrones, éstos a su vez son acelerados hacia la superficie siguiente. La señal inicial llega a ser multiplicada hasta un millón de veces. (19) 22 3. METODOS 23 PRACTICA NUM. 1 CONOCIMIENTO GENERAL DE UN ESPECTROFOTOMETRO DE ABSORCIÓN ATÓMICA OBJETIVO Que el alumno tenga un conocimiento general del equipo de Absorción Atómica. EQUIPO Nombre: Espectrofotómetro de Absorción Atómica. Marca: GBC Modelo: Serie 932 AA Guarda Flama Lámpara de Cátodo Cámara de Espreado Interruptor Principal Panel de Control Caja de Gases Perillas de Ajuste del Quemador Figura 9. Partes del Instrumento de Absorción Atómica 24 Componentes Principales Guarda Flama Es una ventana de vidrio duro localizada enfrente de la sección del quemador. El panel de vidrio esta montado atrás de una rejilla de fuerte metal que tiene bisagras sobre el lado izquierdo sobre la sección del quemador. El guarda flama debe permanecer cerrado todo el tiempo cuando esta operando la flama. Remoción • Abra parcialmente el guarda flama • Levante el lado izquierdo un poco hasta que las orejeras libre los agujeros guía de la pared del comportamiento del quemador. • Remuévalo facilitándolo hacia la derecha. Mantenimiento • Lavar con jabón y agua y dejar escurrir. • Invierta este procedimiento de remoción para volverlo a colocar. Quemador A) Quemador de aire-acetileno tiene una ranura larga (10cm), cuenta con una llave de seguridad, esta consiste en que si la llave no esta colocada en su lugar el equipo no se encenderá. Figura 10. Quemador aire-acetileno 25 B) Quemador N2O-Acetileno tiene una ranura corta (5cm), cuenta con una llave de seguridad, esta consiste en que si la llave no esta colocada en su lugar el equipo no se encenderá. Figura 11. Quemador N2O-Acetileno C) Rotación del Quemador Para máxima sensibilidad el quemador debe ser alineado con el haz óptico. Para reducir la sensibilidad sin diluir las muestras, es posible rotando el quemador. Para rotar el quemador: • Inserte la herramienta de rotación del quemador en el interior del orificio en la esquina superior izquierda del quemador. • Rote el quemador hasta el ángulo deseado. D) Remoción • Apague la flama. • Espere unos minutos para que el quemador se enfrié. • Abra la puerta del compartimiento del quemador. • Desconecte la llave del quemador. • Presione abajo el botón de liberación del quemador en el costado izquierdo de la cámara de rociado. 26 Botón liberador del quemador Figura 12. Botón liberador del quemador • Rote el quemador a las manecillas del reloj tanto como corra. • Levante el quemador verticalmente fuera de la cámara de rociado. Colocación Invierta el procedimiento anterior. Mantenimiento Los quemadores que se utilizan son hechos de titanio, con una bayoneta de montado en alto grado de acero inoxidable. Titanio ofrece excelente resistencia aun amplio rango de químico pero es un metal blando. No use cualquier objeto de metal para limpiar la ranura del quemador. Los quemadores pueden llegar a obstruirse parcialmente durante su uso por depósitos de carbono o sales. Remoción Para limpiar la ranura del quemador • Remueva el quemador. • Inserte una tarjeta de limpiado del quemador dentro de la ranura. • Remueva cualquier depósito de la ranura usando un movimiento hacia fuera. Si un depósito duro de sal esta presente, lave el quemador en un chorro de agua caliente. Inspeccione regularmente los empaques del quemador. 27 Colocación y Posición del Quemador Ajuste primero el ángulo del quemador. Ajuste la posición horizontal del quemador, revise el ángulo. Tome nota de la posición vertical. Baje el quemador hasta que un pico sea alcanzado. En la mayoría de las aplicaciones aire-acetileno no debes necesitar bajar más de 1 cm. aproximadamente. Sistema de Atomización por Flama El sistema de atomización por flama comprende el quemador, el nebulizador, el ensamble cámara de rociado/trampa de liquido. Esta sección describe cada uno de los componentes del sistema de atomización por flama y su mantenimiento, por favor es aconsejable no tocar si no domina el procedimiento. Cámara de rociado Nebulizador Montaje de Quemador Capilar de Muestra Tornillo de Sujeción Cable de Enclaves Figura 13. Cámara de Rociado Remoción • Apague la flama. • Abra la puerta del compartimiento del quemador. 28 • Remueva la llave del quemador. • Desconecte el cable de la cámara de rociado. • Usando la llave de 9/16” provista, desconecte la línea del oxidante (azul) del nebulizador y la línea del combustible (roja) del lado derecho de la cámara de rociado. • Desconecte la línea del drenaje de la trampa de liquido (tubería flexible de PVC transparente). • Desatornille el tornillo de sujeción de la cámara de rociado localizado bajo el cuerpo de esta. • Remueva la cámara de rociado levantándola verticalmente fuera del instrumento. Colocación Invierta el procedimiento para colocar la cámara de rociado. Asegúrese que las líneas de los gases estén correctamente conectadas (Línea azul al nebulizador, roja al cuerpo de la cámara de rociado) y que las tuercas están apretadas. Asegúrese que el quemador, nebulizador y tapón de alivio están apropiadamente instalados y asegurados. Tapón de alivio de presión El tapón de alivio de presión esta localizado en la pared derecha de la cámara de rociado. Removiéndola directamente. Para colocarla, presione firmemente el tapón colocándolo y rotando al mismo tiempo. • Inspeccione el empaque del tapón de alivio de presión regularmente. Trampa de Líquido La trampa de liquido esta sujeta a la pared izquierda de la cámara de rociado. Esta provista con dos tapones: • El tapón ventilado esta localizado en la parte superior. • El tapón no ventilado (o tapón de drenado) esta en la parte inferior. 29 Drenando la trampa de líquido • Remueva el ensamble cámara de rociado/trampa de liquido del instrumento como se mostró arriba. • Manteniendo la cámara de rociado sobre el contenedor de deshechos del instrumento, desatornille el tapón de PTFE para drenado de la trampa de líquido localizado en la parte inferior de esta. • Permita fluir cualquier líquido fuera de la trampa de líquido. • Coloque el tapón de drenado y apriételo firmemente (No use pinzas, un apriete firme con los dedos es el apropiado) Llenado de la trampa de líquido Nunca intente encender la flama sin llenar la trampa de líquido. Llene la trampa de liquido con el mismo liquido empleado para preparar las muestras. Algo del líquido puede fluir fuera por el drenaje en el proceso. Asegúrese que la tubería de drenaje esta conectada y corra hacia el contenedor de deshechos. • Desatornille el tapón ventilado en la parte superior de la trampa de líquido. • Usando una pizeta, sirva algo del solvente de preparación de las muestras al interior de la trampa de líquido hasta que algo del solvente influya hacia fuera por la tubería de drenaje. • Coloque nuevamente el tapón ventilado. Limpiando la cámara de rociado No sumerja la cámara de rociado dentro de agua o solventes • Remueva el quemador, nebulizador y tapón de alivio de presión. • Drene la trampa de líquido. • Limpie las superficies interiores usando una fuente solución de detergente para laboratorio y un pequeño cepillo (una escobetilla es conveniente). • Enjuague bajo una corriente de agua. El mejor curso es tener agua corriendo hacia el interior del nebulizador abierto, y hacia fuera a través del quemador. • Limpie las superficies externas usando un algodón húmedo. 30 Nebulizador El cuerpo del nebulizador esta atornillado dentro de un soporte de polipropileno que es asegurado a la cámara de rociado. Una esfera de impacto es usada para formar una neblina fina. El ensamble del capilar se atornilla dentro del cuerpo Remoción • Apague la flama • Usando la llave de “9/16” proveída con el instrumento, desconecte la línea del oxidante “azul” del nebulizador. • Remueva las dos perillas atornillables que sujetan la placa de retención del nebulizador al cuerpo de la cámara de rociado. • Remueva la placa de retención • Sujete el cuerpo del nebulizador firmemente retire el nebulizador de la cámara de rociado. Soporte Ensamble del Capilar Cuerpo Esfera de Impacto Figura 14. Nebulizador removido Colocación Invierta este procedimiento para colocar el nebulizador. Asegúrese que la perrilla de seguridad de la placa de retención esté apretada. 31 Mantenimiento • Inspeccione los empaques del nebulizador regularmente. • Las únicas partes usadas servibles son la esfera de impacto y el capilar. No intente remover cualquier otra parte del nebulizador si no domina el instrumento. • Si el nebulizador comienza a obstruirse intente lo siguiente en este orden. Remplacé el capilar de muestra de teflón (PTFE). Note que partículas externas pueden fácilmente depositarse en las uniones del capilar. Limpie el ensamble del capilar. Remueva el cuerpo del nebulizador del soporte, después remueva el ensamble del capilar del cuerpo. Limpiando el ensamble del capilar • Desconecte el capilar de la muestra de PTFE del nebulizador. • Remueva el ensamble del capilar desatornillándolo del cuerpo del nebulizador. • Inserte el alambre de limpiado del capilar dentro del extremo trasero del capilar, después empújelo completamente para limpiar cualquier obstrucción. • Cuando coloque el ensamble del capilar tenga cuidado de no trasroscarlo. Inserte el capilar, después gire en sentido contrario a las manecillas del reloj hasta sentir el final de la cuerda. Solo después gírelo suavemente en sentido de las manecillas del reloj. • Evite cualquier esfuerzo. Toma del Nebulizador Si el instrumento no ha sido optimizado con anterioridad, puede ser prudente envolverlo primero completamente en sentido de las manecillas del reloj. Enciendo la flama y aspire el estándar. Rote la perilla del nebulizador y observe el medidor de absorbancia en la pantalla. De la posición completamente en el sentido de las manecillas del reloj, el nebulizador puede presentar uno o tres picos de absorbancia. Ajuste la toma al pico de mayor absorbancia, o al pico que obtenga el menor ruido. 32 La flama Aire-Acetileno Antes de encender la flama, asegúrese que: • La cámara de rociado debe estar apropiadamente instalada con el nebulizador, el tapón de alivio de presión y el quemador deben estar colocados en su lugar y asegurados. • La trampa de líquido este llena. • La puerta del compartimiento del quemador este cerrada. • El ventilador del extractor de humos encendido. • Ambas válvulas de los gases (Auxiliar y Combustible) estén completamente cerradas (envuelta completamente en sentido a las manecillas del reloj). • Portar los lentes de seguridad. Encendido de la flama • Encienda los suministros de aire y acetileno. • En el panel frontal de la caja de gases automática gire la perilla del combustible en sentido de las manecillas del reloj cerca de 2 vueltas. • Presione el botón “AIR-ACET”. • Presione el botón “IGNITION” por 3 segundos. La flama debe encender y permanecer así. Apagando la flama Presione el botón “AIR-ACET”. La flama se apagara automáticamente. La flama N2O-Acetileno Antes de encender la flama, asegúrese que: • La cámara de rociado debe estar apropiadamente instalada con el nebulizador, el tapón de alivio de presión y el quemador deben estar colocados en su lugar y asegurados. • La trampa de líquido este llena. 33 • La puerta del compartimiento del quemador este cerrada. • El ventilador del extractor de humos encendido. • Ambas válvulas de los gases (Auxiliar y Combustible) estén completamente cerradas (envuelta completamente en sentido a las manecillas del reloj). • Portar los lentes de seguridad. • El quemador de N2O-Acetileno este en su lugar. Encendiendo la flama • Encienda los suministros de aire, N2O y acetileno. • Presione el botón “N2O-ACET”. • Presione el botón de ignición “IGNITION” por 3 segundos. La flama aire-acetileno enciende primero y es convertida automáticamente a la flama N2Oacetileno. La perilla del combustible puede ser ajustada disminuyendo si es requerido, dependiendo de la aplicación. La flama se mantendrá encendida hasta que sea apagada como se muestra a continuación. Apagando la flama • Presione el botón “N2O-ACET”. • La flama automáticamente se convertirá a la de aire-acetileno y se apagara. También puedes convertir la flama de regreso al aire-acetileno presionando el botón “AIR-ACET”. Optimizando la Flama Aire-Acetileno Preparación • Disponer de un estándar del elemento a ser analizado. • Una lámpara de cátodo hueco del mismo elemento. • Una tarjeta blanca. Preajuste del Quemador • Instale la lámpara y optimícela. 34 • Asegúrese que la cámara de rociado este apropiadamente instalada. • Dibuje una línea sobre una tarjeta blanca como se muestra. • Coloque la tarjeta sobre el quemador con el extremo de la línea vertical sobre la ranura del quemador. Optimizando la Flama N2O-Acetileno Use el mismo método como para la flama aire-acetileno con las siguientes precauciones: Seguridad Una flama N2O-acetileno puede retroceder si el flujo del combustible es bajo. La caja de gases automática esta diseñada para asegurar un flujo de seguridad mínimo. Con la caja de gases manual, observe las siguientes precauciones: Si es posible use un flujo de combustible de al menos 5 unidades. Si necesitas usar una flama oxidante con un bajo flujo de combustible, asegura que un teñido rosa de 5mm este siempre presente en la base de la flama. El teñido es solo visible si no estas aspirando muestra. Por lo tanto: • Puedes reducir el flujo del combustible, o incrementar la toma del nebulizador solo aspirando fuera de la muestra por que estas operaciones pueden hacer que la flama se empobrezca. Necesitas observar el teñido rosa. • Es seguro incrementar el flujo del combustible, o para reducir la toma del nebulizador mientras este aspirando la muestra porque estas operaciones estarán enriqueciendo la flama. Flujo del Combustible Ajuste el flujo del combustible para una absorbancia máxima. Si tiene duda, use una flama delgada como en la mayoría de los caso esta reducirá el ruido. Algunos elementos semejantes al Sílice (Si) no producirá absorbancia sin el uso de una flama reductora, y el quemador necesita una colocación más baja. Para otros elementos semejantes necesitas iniciar el proceso de optimización con una flama rica. 35 Lámparas de Cátodo Hueco • Se inserta la lámpara en el soquet 1 o 2. Figura 15. Soquet • En la pantalla aparecerá una ventanilla preguntando en que soquet se encuentra y después de haber le indicado en donde esta se encenderá la lámpara como se muestra en la siguiente figura. 16 Figura 16. Lámpara de Cátodo Hueco • Después de que la lámpara esta encendida se introduce dentro del equipo como se muestra en las figuras siguientes. 36 Figura 17. Colocación del Soquet Figura 18. Colocación del soquet con la lámpara de cátodo hueco. Una vez que la lámpara sea insertada, el símbolo del elemento activo y el numero del cátodo (1 o 2). En la lámpara mostrada abajo, el elemento Cu es encontrado en el cátodo No 1. CONCLUSIONES Esta práctica es de carácter demostrativa para que el alumno conozca las funciones y características de los componentes del equipo de Espectrofotómetro de Absorción Atómica GBC 932 AA. 37 PRACTICA NUM. 2 CONOCIMIENTO DEL MANEJO DEL SOFTWARE OBJETIVO Que el alumno aprenda a utilizar el software así como llevar acabo el manejo del mismo. Activación del Software • Encender la computadora donde se encuentra instalado el Software del instrumento de absorción atómica. • Dar doble Clic en el programa de GBC Avanta Ver 1.32 como se muestra en la siguiente figura 20. Programa GBC Avanta Ver 1.32 Figura 19. Programa GBC Avanta Ver 1.32 • Presione el interruptor principal de la esquina inferior derecha del instrumento como se muestra en la figura • Espere hasta que el programa GBC Avanta Ver 1.32 este listo. Ventana Principal del programa GBC Avanta Ver 1.32 La ventana principal abre automáticamente cuando el programa es activado. La ventana principal es dividida en seis áreas como se muestra en la figura de abajo. 38 Barra de Titulo Barra de Menú Principal Barra de Inicio Barra de Sistema Barra de Módulos Área Principal de la Pantalla Figura 20. Ventana principal del programa GBC Avanta 1.32 El Sistema de Ayuda Figura 21. Ayuda Este manual provee solo información general a cerca del programa. Para información detallada, Dar un clic al icono de ayuda (Help). El sistema de ayuda es sensible al contexto y provee detalles de la página abierta actualmente. Para acceder al sistema de ayuda por tabla de contenido, índice y búsqueda, toque la opción Help en la barra del menú principal. 39 Módulos del Programa El programa esta dividido lógicamente en seis módulos. Un modulo es abierto dando clic en el icono en la barra de módulos en el costado izquierdo de la pantalla. Note que la segunda opción del menú principal se despliega el nombre del modulo actualmente abierto. Una vez activado el modulo, la información ingresada por el operador o el resultado del análisis puede ser almacenado en un archivo. El nombre del archivo tiene una extensión que es característica del modulo usado. Cuadro 2. Información general de como usar otros módulos para desarrollar un análisis. Modulo Información Almacenada Extensión del Archivo Método ( Method) Elemento .mth Parámetros de Instrumento. Tipo de calibración (concentración, adición de estándares, etc.) Posición del estándar en el automuestreador, si es usado. Parámetros de Calidad Parámetros de la flama u Horno Muestras ( Samples) Lista de muestras de orden de analizar. .sam Cuando se debe desarrollar una calibración, reescalar, muestra control. Para cada muestra, etiqueta y factores de disolución donde aplique. Posición de la muestra en la gradilla del automuestreador si es usado. Análisis (Análisis) Análisis Único (Single) : .anl Archivos del método, muestras resultados a se usados. Análisis Múltiple ( Múltiple) : Una tabla en que cada línea (conocida como una 40 partida de análisis) es un análisis simple puede ser salvada como un archivo de análisis. Resultados (Results) Los resultados del análisis. .res Un archivo de resultados puede incluir los resultados de un análisis o de varios. Reporte Que información y que formato será usado para (Report) el reporte Instrumento El tipo de sistema usado ( flama u horno) (Instrument) Cualquier accesorio usado (automuestreador, .rep .ins generador de hidruros, etc.) Opciones y configuraciones del instrumento. Configuración del Instrumento El modelo del instrumento y accesorios pueden ser seleccionados de la caja de dialogo “Hardware Setup” en el modulo del instrumento. • Haga clic en el icono de modulo de instrumento. • Sobre el menú principal, haga clic “Instrument/Properties/Configuration” • Haga clic en el boton “Hardware Setup” Modelo El tipo de instrumento es seleccionado de fábrica y ajustado a 932,933, Prospector. Este no debe ser cambiado. 41 Figura 22. Configuración del Instrumento: Modelo Accesorios En esta tarjeta se encuentra los accesorios que fueron comprados con el instrumento. Esto hará disponible a estos accesorios en otras áreas como las propiedades del método. Figura 23. Propiedades del Método del Sistema 42 Método de Flama Figura 26. Método • Haga Clic en la pantalla de la computadora el botón del método (Method). Figura 25. Libro del Método • Haga clic en la pestaña descripción (description). • Seleccione la flecha verde • En la tabla periódica desplegada seleccione el elemento correspondiente a la cercana al campo Elemento (element). lámpara que tienes instalada. Figura 26 Tabla Periódica 43 • La caja de dialogo para añadir un elemento a la lámpara “Add an Element to the Lamp” es automáticamente desplegada si el elemento aun no a sido registrado. Figura 27. Añadir un elemento a lámpara • Esperar unos minutos para que el instrumento complete la rutina de optimización. El mensaje de instrumento listo “Instrument ready” se mostrara en la barra de estado inferior de la pantalla. Instrumento Figura 28. Pagina del Instrumento La corriente de la lámpara, longitud de onda y ancho de ranura son automáticamente ajustadas a los valores predeterminados. El campo de la longitud de onda ofrece alternativas, longitudes de onda menos sensibles están en un listado desplegable. La 44 altura de la ranura predeterminada a normal para métodos de flama. Habilite la corrección de fondo si es requerida. Medición Figura 29. Modo de Medición • Seleccione el modo de medición (Integración “Integration”, Altura del Pico “Peak Height”, Área del Pico “Peak Area”, Promedio de la corrida “Running Mean”). Para un método por flama, Integración o Promedio de la corrida son las opciones más comunes. • Ingrese el numero de replicas y el tiempo de integración, o la precisión requerida si seleccionas promedio de la corrida. 45 Calibración Figura 30. Página de Calibración • Seleccione el modo de calibración (Calibration Mode) en el listado del menú desplegable. Las opciones son un número reducido de métodos de curvas de calibración, adición de estándares o estándares de ruptura. El campo predetermina a Concentración. • Si se habilita auto salvar el método después de calibrar (AutoSave Method After Cal), el programa actualizara los resultados de la calibración como parte del archivo del método automáticamente cada vez que la calibración sea completada durante la corrida del análisis. Estándares Figura 31. Página de Estándares 46 Agregar estándares dando un clic al icono para ingresar las concentraciones de los estándares utilice el teclado. Calidad Figura 32 Página de Calidad • Esta pagina se activa si se habilita la revisión del rango de concentración “Concentration Range Checking”. Después ingresar una concentración mínima y máxima para muestras, y la acción requerida si una muestra se sale de estos limites. Una muestra de control “Check Sample” es una muestra de concentración conocida usada para revisar el método. Con un sistema completamente automatizado una opción de porción de recobro “Spike Recovery” es también disponible. 47 Flama Figura 33. Página de Flama • Usar esta página para grabar el tipo de flama y condiciones. Haga clic en el botón de Optimizar “Optimize” para optimizar los parámetros de la flama. Modulo de Muestras Figura 34. Muestras • Toque el botón de muestras para abrir el modulo de muestras como se muestra la figura 37 de abajo. • Inicialmente, la tabla de muestras incluye una sola llamada “Sample 1” Si quieres correr una calibración primero • Haga clic y sostenga la línea “Sample 1” hasta que un menú secundario sea desplegado. • Dar un clic en propiedades. 48 • En la caja de dialogo propiedades de medición “Measurement Properties” desplegada, dando un clic a la flecha abajo para continuar al campo del tipo de medición “Measurement Type” • Activar con un clic “Calibration” en el listado desplegado que aparece. Para agregar muestras al final del listado, dar un clic en el botón de agregar renglón “Add Rows” . Las etiquetas de las muestras se auto incrementarán en uno a partir de la primera (Sample 1 en este caso). Figura 35. Tabla de Muestras • Cada renglón de la tabla de muestras es conocida como una medición. Una medición puede ser una muestra (Sample), una calibración completa (Calibration), una reescalada (Rescale), una muestra de recobro “Spike Sample” o una muestra blanco “Sample Blank”. • Puedes cambiar el tipo de medición de cualquier renglón usando la caja de dialogo de propiedades de medición como se mostró arriba. • Puedes renombrar cualquier etiqueta de la muestra realizando un doble clic sobre el renglón, después usando el teclado para escribir sobre él. 49 Modulo del Análisis Figura 36. Análisis Toque el botón del análisis para abrir el modulo del análisis como se muestra abajo. Para cambiar entre un análisis único y múltiple, toque “Analysis” en el menú principal y seleccione o deseleccione múltiples análisis “Multiple Analysis" del menú desplegable mostrado. Análisis único Figura 37. Análisis Único Archivos • Seleccioné el archivo del método, muestras y resultados usando el botón del explorador en el área de archivos “Files”. • El archivo de resultados necesita ser creado en el modulo de resultados antes de seleccionarlo aquí. Secuencia • Se correr la tabla de muestras entera, o parte de ella solo seleccionando la primera y la ultima medición. 50 Modulo de Resultados Figura 38. Resultados Creando un Archivo de Resultados en Blanco • Haga clic en el botón de resultados “Result”. • Sobre la barra del sistema, dar clic en el icono de Nuevo “New”. La página de un archivo de resultados nueva será mostrada. • Ingrese un nombre valido en el campo para nombrar el archivo “File name”. • Haga clic en el botón Crear “Create”. Editando los resultados Si el modulo de resultados esta abierto mientras se realizac un análisis, los resultados son automáticamente escritos en la pantalla conforme el análisis progresa. Se observa los resultados pero estos no pueden ser editados antes de que el análisis finalice. Cualquier analisis concluido de resultados puede ser visto abriéndolo y viéndolo en el modulo de resultados. La pantalla de resultados esta dividida en tres áreas principales como se muestra abajo: • La tabla de Resultados (Results). • El panel de Replicas (Replicates). • El panel de Gráficos (Graphics). 51 Tabla de Resultados Replicas Grafica Figura 39 Página de Resultados Haga clic en cualquier parte de la tabla de resultados. Las replicas para esta partida son desplegadas en el área de replicas. Cualquier replica puede ser borrada o editada si el resultado buscado es erróneo (por ejemplo si la muestra analizada se salió durante la lectura). Cualquier edición de resultados se desplegara e imprimirá con un asterisco (*) una vez hechos. El panel de gráficos muestra las señales gráficamente para el resultado seleccionado. Si la calibración completa es seleccionada, esta mostrara la grafica de calibración. Modulo del Reporte Figura 40. Resultados Haga clic en el icono de Reporte para abrir el modulo. 52 Los reportes no pueden ser impresos a partir de este modulo. La mayoría de los reportes son impresos del modulo de resultados. El modulo de reporte es usado para decidir que incluir en tu reporte. La página del reporte incluye cuatro tarjetas como se muestra abajo. Figura 41. Página del Reporte Elemento Único Seleccionar el elemento que deseas incluir en los reportes de elemento único haga un clic seleccionando la caja de activación apropiada. Las posibles opciones se muestran en la figura 43. Encabezado del Reporte • Incluir un encabezado para cada página de tu reporte seleccionar la caja de activación para incluir el encabezado del reporte “Include Report Header” en esta tarjeta. • Escriba su encabezado del reporte dentro de la caja provista. • Pruebas de Desempeño Exportación durante la corrida • Durante una corrida en tiempo real los datos pueden ser exportados a los siguientes destinos. 53 • Archivo • Cualquier puerto serie disponible. Alineación del Quemador • Ajuste el quemador usando los ajustes horizontal y vertical, y la rotación del quemador, posicione el quemador de tal manera que la ranura éste exactamente paralela al haz y cerca de 1 cm. abajo de éste. • Abra el modulo del método “Method”, después seleccione la pestaña de Flama “Flame” y el botón de Optimizar “Optimise”. Esto abrirá el panel de optimización de gases “Gas Flow Optimisation” mostrada abajo. Figura 42. Panel de Optimización del Flujo de Gases • El medidor de absorbancia debe mostrar cero. De otro modo ejecute un cero al instrumento tocando el botón “Perform Instrument zero”. • Lentamente eleve el quemador hasta que la absorbancia del medidor comience a mostrar lecturas positivas. Esto es debido a que el quemador esta parcialmente obstruyendo el haz. • Baje el quemador de tal manera que la lectura de absorbancia regrese a cero. 54 El quemador comenzara en esta posición para optimizar una flama aire-acetileno. Para aplicaciones N2O éste deberá estar típicamente 1cm o tal vez menos. En cualquier caso, asegúrese que el quemador nunca se coloque más alto de tal manera que no presente interferencia con el haz. Deje el panel de optimización del flujo de gases abierta como esta, aunque se usara para el procedimiento de optimización descrito a continuación. Optimizando la Flama aire-acetileno • Instale y optimice la lámpara de cátodo hueco. • Coloque el quemador. Deje el panel de optimización de flujos abierto. • Encienda la flama • Ajuste la toma del nebulizador. • Ajuste la posición horizontal y ángulo del quemador. • Ajuste la altura del quemador. • Ajuste el flujo del combustible. • Si es necesario revise la toma del nebulizador, la altura del quemador y flujo del combustible hasta que tenga el resultado satisfactorio buscado. Situaciones que se pueden presentar en el equipo Orden Cero No Encontrado Figura 43. Mensaje del Orden Cero No Encontrado Este mensaje puede ser desplegado rápidamente después de energizar el instrumento. Esto indica que la luz no ha alcanzado el detector. Las causas pueden ser: 55 • Falla la lámpara de cátodo hueco, o no esta instalada. • Selección de la lámpara incorrecta. • Selección incorrecta de la posición en la tabla de elementos de la lámpara. Baja Señal de la Lámpara de Cátodo Hueco Después de encontrar el orden cero el instrumento se va a optimizar en la longitud de onda seleccionada para el elemento. El siguiente mensaje puede ser mostrado después: Figura 44. Baja Señal de la HCL Las posibles causas son: • Lámpara incorrecta • Lámpara HC pobremente alineada • Corriente de la lámpara HC baja • Lámpara defectuosa, o sucia la ventana de la lámpara • Haz de referencia parcialmente obstruida • Ventanas ópticas ensuciadas (especialmente para longitudes de onda menores a 250nm). Dificultades de Ignición Antes de encender la flama, asegúrese que: • La cámara de rociado este apropiadamente instalada con el nebulizador, el tapón de alivio de presión y quemador en su lugar y asegurados. • La trampa de líquido llenada. 56 • La puerta del compartimiento de muestra cerrada. • El ventilador de extracción encendido. • Ambas válvulas del gas (“auxiliary” y “fuel”) estén completamente cerradas (enroscadas completamente en sentido de las manecillas del reloj). • Colócate tus lentes de seguridad. • El quemador instalado sea el correcto. Condiciones para Ignición Para obtener una ignición, dos condiciones deben ser completamente satisfactorias: • El filamento de la bujía de ignición debe de encandecer. • Acetileno debe pasar sobre el filamento de la bujía. La operación del filamento de la bujía esta enclavado a parámetros críticos como se muestran a continuación. Caja de Gases Automática En la caja de gases automática la ignición esta también enclavada a las presiones de los gases. Por ello no es posible revisar la operación de la bujía separadamente. Tenga listo al instrumento para la ignición Presione el botón de ignición. Si la flama no ignita cuando todos los enclaves han sido revisados: • Si el ignitor falla para producir una flama, la línea del acetileno puede necesitar ser cargada. Intente incrementando el flujo a cerca de 3 unidades de flujo. • Si el ignitor trabaja, pero la flama principal no enciende, el flujo del combustible es muy bajo. Incremente el flujo del combustible a cerca de 2 unidades. 57 Panel de Medidores de Servicio El panel de medidores de servicio es muy usado para diagnosticar fallas del sistema fotométrico y para probar lámparas corriendo un barrido en longitud de onda. Este también puede ser usado para alinear lámparas y el cabezal. Para abrir los medidores de servicio: • Toque el botón Instrumento • Sobre el menú principal, toque “Instruments / Service Options / Service Meters” Figura 45. Panel de Medidores de Servicio La imagen superior muestra el estado de los medidores de servicio cuando el instrumento ha sido optimizado usando una lámpara de Zn. La señal D2 puede bajar para otras lámparas, y será muy baja para cualquier longitud de onda arriba de 450nm, ajustando con la máxima corriente D2. Esto es normal y no debe ser tratado. CONCLUSIONES El alumno conocerá el manejo básico del software Avanta Ver 1.32 pero será necesario con el uso rutinario del mismo. 58 PRACTICA NUM. 3 CALIBRACIÓN OBJETIVO Que el alumno tenga la seguridad de que el equipo este funcionado correctamente antes de iniciar el análisis. FUNDAMENTO Una verificación de calibración periódica es para demostrar de que no han cambiado las condiciones del instrumento en una forma significativa. La verificación debe realizarse cada vez que se utilice el espectrofotómetro. (22) Cuando se calibra un instrumento se debe tener una razonable certeza de que este responda de igual manera a los patrones al igual que la muestra, aunque estas tengan una matriz relativamente diferente. Si estas diferencias son relativamente grandes, puede llegar a invalidar el proceso de calibración. Es necesario estar completamente seguro de que el calibrado es valido antes de utilizarlo para obtener el valor de concentración. Importancia de la calibración. (22) El envejecimiento de los componentes, los cambios de temperatura y el estrés mecánico que soporta los equipos deterioran lentamente sus funciones. Cuando esto sucede, los ensayos y las medidas aumentar su intervalo de incertidumbre y se refleja tanto en el diseño como en la calidad del producto final ya que durante el proceso de producción es importante que cada parte sea realizada con calidad para obtener los resultados deseados. (22) MATERIAL Ver Anexo 1 Limpieza del material. • Pipeta Volumétrica de 5 mL • Pipeta graduada de 5 mL 59 • Matraz aforado de 100 mL • Probeta de 100 mL • Pizeta • Perilla REACTIVOS Ver Anexo 5 Seguridad • Solución Estándar de Cobre (Cu) de 1000 mg/L • Acido Nítrico (HNO3) Concentración. • Agua desionizada o tridestilada. EQUIPO Ver Anexo 6 Métodos analíticos • Espectrofotométrica de Absorción Atómica • Lámpara de Cátodo Hueco de Cobre (Cu) PREPARACION DE SOLUCIONES Para la realización la verificación se debe preparar una solución de cobre (Cu) de 5 mg/L y preparar un blanco de acido nítrico (HNO3) Preparación de la Solución de Cobre 5 mg/L • Medir una alícuota de 5mL del estándar de Cobre (Cu) de 1000 mg/L en pipeta volumétrica y llevarlo a un matraz aforado de 100 mL. • Adicionar 2 mL de acido nítrico (HNO3) con pipeta graduada. • Llevar al aforo con agua desionizada o tridestilada. • Cuidar el aforo. 60 Preparación del Blanco • Medir una alícuota de acido nítrico (HNO3) conc. Con pipeta graduada, llevar a la probeta graduada. • Aforar a 100 mL con agua desionizada o tridestilada. • Cuidar el aforo. DESARROLLO EXPERIMENTAL Ver Anexo 2 Desarrollo de análisis • Encender el equipo y conectar la lámpara de Cobre (Cu). • Alinear la lámpara hasta obtener la máxima energía. • Seleccionar el ancho de banda espectral óptimo, el cual depende de cada elemento en particular. • Seleccionar la longitud de onda para el metal de interés. • Optimizar la longitud de onda ajustándola hasta obtener la máxima energía. • Esperar de 10 min a 20 min para que se estabilice el equipo, una vez encendida la lámpara. • Encender la flama. Permitir que el sistema alcance el equilibrio de temperatura. • Aspirar un blanco (matriz libre de analitos a la cual se le agregan todos los reactivos en los mismos volúmenes y proporciones usadas en el procesamiento de la muestra). El blanco en absorbancia debe de ser 0.0000 • Aspirar una disolución estándar de Cobre de 5 mg/L, • Buscar una absorbancia de 0.6 a 0.7 unidades de absorbancia • Si en caso de que no se obtenga la absorbancia mencionada, ajustar la velocidad de flujo del nebulizador hasta obtener la máxima sensibilidad, así como ajustar el quemador horizontal y verticalmente hasta obtener la máxima respuesta. CONCLUSIONES Al concluir esta practica el alumno conocerá los pasos a seguir en la calibración del Espectrofotómetro de Absorción Atómica, y se considera cuando cumpla los parámetros de absorbancia de 0.6 a 0.7 el coeficiente de correlación R2 mayor o igual 0.9500. 61 PRACTICA NUM. 4 ELABORACION DE UNA CURVA DE CALIBRACION OBJETIVO Que el alumno aprenda a elaborar una curva de calibración para el desarrollo del análisis en muestras. FUNDAMENTO Es una curva que se construye a partir de diferentes concentraciones de un analito y su respuesta frente a una reacción. Este gráfico tiende a ser lineal, mientras progrese según la ley de Lambert Beer. (22) Factores que determinan la calidad de una calibración son: La precisión de las medidas estimadas através de la repetibilidad y la reproducibilidad de las medidas. La repetibilidad se evalúa através del calculo de la desviación estándar relativa (RSD%) de la medida de los patrones de calibrado. (22) Exactitud de los patrones el valor de la concentración o masa designado a cada patrón trae aparejado un error pequeño si es preparado a partir de reactivos puros (grado analítico) con estequiométria bien definida este error en lo general se desprecia, frente al error de las medidas de las señales producidas por el instrumento. (22) MATERIAL Ver Anexo 1 Limpieza de material • Pipeta Volumétrica de 10, 5, 2, 3, 1 mL • Pipeta graduada de 10 mL • Matraz aforado de 100 mL 62 • Pizeta • Perilla • Frascos de boca ancha • Etiquetas REACTIVOS Ver Anexo 5 Seguridad • Solución Estándar de Cobre (Cu) de 1000 mg/L • Acido Nítrico (HNO3) Conc. • Agua desionizada o tridestilada. EQUIPO Ver Anexo 6 Métodos analíticos • Espectrofotométrica de Absorción Atómica • Lámpara de Cátodo Hueco de Cobre (Cu) DESARROLLO EXPERIMENTAL Calcular los volúmenes que necesitamos de la solución estándar para la elaboración de la curva. Para la preparación utilizaremos un estándar de Cobre (Cu) de 1000 mg/L de esta solución tomaremos una alícuota para preparar una concentración de 100 mg/L y de esta partiremos para preparar los demás puntos para la curva. Nota: Cuidar de no contaminar el estándar de Cobre (Cu) de 1000 mg/L Calculo para la preparación de los puntos de la curva. Concentración Teórica = mL de alícuota x Concentración del Estándar Deseada Aforo 100 mL 63 Calculo para la preparación de la solución Cobre 100 mg/L: Concentración Teórica = 10 mL x 1000 mg/L Deseada 100 mL = 100 mg/L Preparación de la solución de Cobre 100 mg/L: Tomar una alícuota de 10 ml del estándar de Cobre (Cu) de 1000 mg/L, llevarlo a un matraz aforado de 100 mL, igualar la matriz agregándole 2 mL de Acido Nítrico (HNO3) concentrado y llevarlo al aforo. Transvasar a un frasco y etiquetar. Calculo para la preparación de la solución de Cobre 6 mg/L: Concentración Teórica = 6 mL x 100 mg/L Deseada 100 mL = 6 mg/L Preparación de la solución de Cobre 6 mg/L: Tomar una alícuota de 6 ml del estándar de Cobre (Cu) de 1000 mg/L, llevarlo a un matraz aforado de 100 mL, igualar la matriz agregándole 2 mL de Acido Nítrico (HNO3) conc y llevarlo al aforo. Transvasar a un frasco y etiquetar. Calculo para la preparación de la solución de Cobre 4 mg/L: Concentración Teórica = 4 mL x 100 mg/L Deseada 100 mL = 4 mg/L Preparación de la solución de Cobre 4 mg/L: Tomar una alícuota de 4 ml del estándar de Cobre (Cu) de 1000 mg/L, llevarlo a un matraz aforado de 100 mL, igualar la matriz agregándole 2 mL de Acido Nítrico (HNO3) conc y llevarlo al aforo. Transvasar a un frasco y etiquetar. 64 Calculo para la preparación de la solución de Cobre 3 mg/L: Concentración Teórica = 3 mL x 100 mg/L Deseada 100 mL = 3 mg/L Preparación de la solución de Cobre 3 mg/L: Tomar una alícuota de 3 ml del estándar de Cobre (Cu) de 1000 mg/L, llevarlo a un matraz aforado de 100 mL, igualar la matriz agregándole 2 mL de Acido Nítrico (HNO3) conc y llevarlo al aforo. Transvasar a un frasco y etiquetar. Calculo para la preparación de la solución de Cobre 2 mg/L: Concentración Teórica = 2 mL x 100 mg/L Deseada 100 mL = 2 mg/L Preparación de la solución de Cobre 2 mg/L: Tomar una alícuota de 2 ml del estándar de Cobre (Cu) de 1000 mg/L, llevarlo a un matraz aforado de 100 mL, igualar la matriz agregándole 2 mL de Acido Nítrico (HNO3) conc y llevarlo al aforo. Transvasar a un frasco y etiquetar. Calculo para la preparación de la solución de Cobre 1 mg/L: Concentración Teórica = 1 mL x 100 mg/L Deseada 100 mL = 1 mg/L Preparación de la solución de Cobre 1 mg/L: Tomar una alícuota de 1 ml del estándar de Cobre (Cu) de 1000 mg/L, llevarlo a un matraz aforado de 100 mL, igualar la matriz agregándole 2 mL de Acido Nítrico (HNO3) conc y llevarlo al aforo. Transvasar a un frasco y etiquetar. 65 Después de la preparación de la curva, se analiza o se guarda en refrigeración. Ver Anexo 2 Desarrollo de análisis Se realiza el análisis como se muestra en la Práctica Num. 3 CONCLUSIONES Para la cuantificación de cualquier mineral es necesario la preparación de soluciones de diferentes concentraciones, con el echo de construir una curva de calibración que cumpla con una linealidad (R2 mayor o igual a 0.95) y una reproducibilidad antes de analizar las muestras. 66 67 PRACTICA NUM. 5 DETERMINAR CALCIO (Ca), SODIO (Na), POTASIO (K), MAGNESIO (Mg) EN MUESTRAS DE PROPOLEO OBJETIVO Que el alumno realice análisis de minerales en cualquier tipo de muestras como lo es el propóleo. FUNDAMENTO El propóleo es una sustancia resinosa utilizada por las abejas para cubrir y proteger la colmena. Las abejas obtienen esta sustancia a partir de las yemas y cortezas de algunos árboles. (21) El propóleo es rico en bioflavonoides y aceites esenciales, además de contener oligoelementos, vitaminas y aminoácidos. (21) El termino propóleo proviene del griego Propolis que significa “defensa de la ciudad” (Proantes de Polis-ciudad, lo cual se traduce como defensas antes de la ciudad o Defensor de la ciudad). (21) Gracias a la acción antibiótica del propóleo, que protege de la actividad de virus y bacterias, la colmena es uno de los lugares mas estériles conocidos en la naturaleza. El propóleo contiene una gran variedad de elementos: Aminoácidos, vitaminas, minerales, etc. Entre todos estos compuestos destacan los bioflavonoides. El propóleo en estado bruto contiene 500 veces más bioflavonoides que las naranjas, los cuales son considerados hoy en día beneficiosos en estados de convalecencia. No obstante, estudios científicos llevados a cabo por diversos investigadores en todo el mundo han demostrado que el efecto del propóleo se consigue gracias a la acción sinérgica de todos sus componentes. Por su composición y propiedades suele recomendarse en caso de afecciones respiratorias recurrentes o en cualquier situación en la que las defensas del organismo están bajas. (21) 68 MATERIAL Ver Anexo 1 Limpieza del material. • 8 Matraz microkjeldhal • Pipeta Volumétrica 1, 2, 4, 5 10 • Pipeta graduada de 10 mL • 5 Matraz aforado de 100 mL • Probeta de 100 mL • Pizeta • Papel filtro • Perilla • 8 Frascos de boca ancha • Etiquetas REACTIVOS Ver Anexo 5 Seguridad • Solución Estándar de Calcio (Ca), Sodio (Na), Potasio (k), Magnesio (Mg) de 1000 mg/L • Acido Nítrico (HNO3) Concentrado. • Agua desionizada o tridestilada. EQUIPO Ver Anexo 6 Métodos analíticos • Microdigestor • Espectrofotométrica de Absorción Atómica • Lámpara de Cátodo Hueco de Calcio (Ca), Sodio (Na), Potasio (K), Magnesio (Mg). 69 DESARROLLO EXPERIMENTAL Ver Anexo 4 Interferencias y Anexo 3 Técnicas de muestreo. Para la digestión de la muestra se realiza de acuerdo a la norma NOM-117-SSA1-1994 ya que no hay una norma específica para esta muestra, así como también hay diferentes tipos de digestión para su desarrollo. Se realiza de acuerdo al material que se tiene. En este caso se realiza por vía húmeda. (15) Digestión por vía húmeda. • Pesar con precisión de ± 0,1 mg, una cantidad apropiada de muestra. Para la determinación por el método de absorción por flama pesar como máximo 40 g de jugo o bebida, 20 g de alimentos que contengan del 50 al 75% de agua y 10 g de alimentos sólidos o semisólidos. Limite el contenido de grasa o aceite a un máximo de 4 g y el total de materia orgánica a 5 g. • Añadir 10 ml de ácido nítrico concentrado y dejar reposar toda la noche o iniciar directamente la digestión. • Usar matraz de microkjeldhal o matraz conectado al sistema de refrigerantes. • Calentar suavemente. • Digerir la muestra 3 horas o más tiempo si es necesario (algunas muestras requieren la adición de mayor cantidad de ácido nítrico) hasta la aparición del color traslúcido, si queda ámbar, adicionar peróxido de hidrógeno gota a gota con agitación continua (reacción exotérmica). • Enfriar. • Recuperar, filtrar y llevar a un volumen conocido en matraz volumétrico. • Correr un blanco de reactivos. • Leer en el aparato de elección (espectrómetro de absorción atómica por flama). (15) 70 Digestión por vía seca. • Pesar con precisión de ± 0,1 mg, una cantidad apropiada de muestra. • Para la determinación por el método de absorción por flama pesar como máximo 40 g de jugo o bebida, 20 g de alimentos que contengan del 50 al 75% de agua y 10 g de alimentos sólidos y semisólidos. Limite el contenido de grasa o aceite a un máximo de 4 g y el total de materia orgánica a 5 g. • Añadir 10 ml de ácido nítrico concentrado y dejar reposar toda la noche o iniciar directamente la digestión. En productos con alta concentración de proteínas adicionar una solución de nitrato de magnesio al 7,0% p/v y mezclar completamente, llevar a sequedad aproximadamente durante 6 horas en estufa a una temperatura de 90 a 95ºC. • Colocar la muestra en una mufla y elevar la temperatura lentamente de 2 a 4ºC por minuto hasta 350°C. Mantener la temperatura hasta que cesen los humos. • Elevar gradualmente la temperatura de 500 a 550ºC para evitar que la muestra se incinere y mantener esa temperatura durante 16 horas o toda la noche. • Apagar la mufla y dejar enfriar. • Un segundo paso de calcinación puede ser requerido para remover algunos residuos de carbón, mediante el siguiente procedimiento: • Lavar las paredes del crisol con 2 ml de ácido nítrico al 50%. Colocar la muestra en una placa de calentamiento puesta a 120ºC para remover el exceso de ácido. Colocar la muestra en una mufla fría y elevar la temperatura gradualmente de 500 a 550ºC, manteniéndola por el tiempo necesario. Repetir este procedimiento cuantas veces sea necesario hasta que quede libre de carbón remanente. • Disolver las cenizas completamente en 5 ml de ácido clorhídrico 1N, transferir la muestra disuelta a un tubo de propileno o a un matraz de volumen conocido, enjuagar el crisol con dos alícuotas de 5 ml de ácido clorhídrico 1 N y transferir al mismo tubo o matraz para obtener un volumen de 15 ml en el primero y llevar al aforo en el segundo, tapar y mezclar, si existe presencia de partículas o materia insoluble, filtrar en papel Whatman No. 2, antes de la determinación. • Correr un blanco de reactivos. • Leer en el aparato de elección (espectrómetro de absorción atómica: flama). (15) 71 Después de la digestión se preparan las curvas para cada analito, como se muestra en la Práctica Num. 4 Ver Anexo 2 Desarrollo del análisis. Para el análisis de Calcio se realiza por Oxido Nitroso, así que se cambia de quemador como se muestra en la pagina 20 y se utiliza la flama con Oxido Nitroso-Acetileno ver la paginas 26, 27. DESARROLLO DE LOS CALCULOS Método de cálculo. Interpolar los valores de absorbancia o altura de pico de la muestra analizada en la curva de calibración y obtener los mg/kg del elemento en la muestra y realizar los cálculos empleando la siguiente fórmula: (16) mg/ kg = AxB C en donde: A = Concentración en mg/kg de la muestra a interpolar en la curva de calibración. B = Volumen final al que se llevó la muestra (ml). C = Peso de la muestra (g) o volumen de la muestra (ml) en el caso de agua. Cuadro 3. Clave y peso de muestras de propóleo Clave de Muestra W = Peso de Muestra Jalacingo 0.809 gr Tenango 0.732 gr Campo Grande 0.812 gr Nevería 0.940 gr 72 Cálculos de Potasio (K) Jalacingo = 10.63272 mg/L x 100 mL = 1314.3040 mg/L de K 0.809 gr Tenango = 6.35871 mg/L x 100 mL = 868.6762 mg/L de K 0.732 gr Campo grande = 4.99172 mg/L x 100 mL = 614.7438 mg/L de K 0.812 gr Nevería = 12.38090 mg/L x 100 mL = 1317.1170 mg/L de K 0.940 gr Cálculos de Magnesio (Mg) Jalacingo = 3.77899 mg/L x 100 mL = 467.1186 mg/L de Mg 0.809 gr Tenango = 26.29313 mg/L x 100 mL = 3591.9576 mg/L de Mg 0.732 gr Campo grande = 3.99633 mg/L x 100 mL = 492.1588 mg/L de Mg 0.812 gr 73 Nevería = 18.00709 mg/L x 100 mL = 1915.6478 mg/L de Mg 0.940 gr Cálculos de Sodio (Na) Jalacingo = 18.67718 mg/L x 100 mL = 2308.6749 mg/L de Na 0.809 gr Tenango = 16.70930 mg/L x 100 mL = 2282.6912 mg/L de Na 0.732 gr Campo grande = 12.94869 mg/L x 100 mL = 1594.6662 mg/L de Na 0.812 gr Nevería = 5.51976 mg/L x 100 mL = 587.2085 mg/L de Na 0.940 gr Cálculos de Calcio (Ca) Jalacingo = 5.23902 mg/L x 100 mL = 647.5920 mg/L de Ca 0.809 gr Tenango = 31.54016 mg/L x 100 mL = 4308.7650 mg/L de Ca 0.732 gr 74 Campo grande = 17.49314 mg/L x 100 mL = 2154.3275 mg/L de Ca 0.812 gr Nevería = 29.47782 mg/L x 100 mL = 3135.9382 mg/L de Ca 0.940 gr CONCLUSIONES Se toman muestras identificadas por el lugar donde se muestrean, que son de la región. El análisis de distintas muestras de propóleo presentaron niveles de 600 ppm a 1300 ppm de potasio; de 400 ppm a 3500 ppm de magnesio; y de 500 ppm a 2300 ppm de sodio, y de 600 ppm a 4500 ppm de calcio, lo q representa que los productos de origen natural tiene una variación de los productos q lo rodean, el propóleo es un producto que recoge las abejas de las resinas de la vegetales o de los árboles de la región. 75 76 77 78 79 PRACTICA NUM. 6 DETERMINACION DE POTASIO (K) EN MUESTRAS DE PLATANO OBJETIVO Que el alumno realice análisis de minerales en muestras de alimentos. FUNDAMENTO El plátano es una fruta que ofrece un alto contenido de potasio, y por ello es muy recomendado por médicos cuando pacientes tienen niveles bajos de potasio. Un plátano contiene 600mg de este mineral. Adicionalmente contiene vitamina A, vitamina C, riboflavina, niacina, vitamina B6 y acido fólico. El comer plátanos ayuda a combatir enfermedades como: anemia, presión alta, problemas digestivos, entre otros y es para quienes buscan alimentos que contengan mucho potasio. (18) MATERIAL Ver Anexo 1 Limpieza de material. • 6 Matraz microkjeldhal • Pipeta Volumétrica 1, 2, 5 10 mL • Pipeta graduada de 10 mL • 5 Matraz aforado de 100 mL • Probeta de 100 mL • Pizeta • Papel filtro • Perilla • 6 Frascos de boca ancha • Etiquetas 80 REACTIVOS Ver Anexo 5 Seguridad • Solución Estándar de Potasio de 1000 mg/L • Acido Nítrico (HNO3) Concentrado • Agua desionizada o tridestilada. EQUIPO Ver Anexo 6 Métodos analíticos • Microdigestor • Espectrofotométrica de Absorción Atómica • Lámpara de Cátodo Hueco de Potasio (K). DESARROLLO EXPERIMENTAL Ver Anexo 4 Interferencias, Técnicas de toma de muestra en Anexo 3 y desarrollo de análisis en Anexo 2 Para la digestión de la muestra se realiza de acuerdo a la norma NOM-117-SSA1-1994 ya que no hay una norma específica para esta muestra. En este caso se realiza por vía húmeda como se muestra en la Practica Num. 5 Después de la digestión se preparan las curvas para cada analito, como se muestra en la Práctica Num. 4 CALCULOS Se realizan los cálculos utilizando la formula de la Practica Num. 5 Cuadro 4. Clave y peso de muestras de plátano Clave de Muestra W = Peso de Muestra Plátano Violeta 2.1314 gr Plátano Morado 2.2392 gr Plátano Naranjado 2.2449 gr 81 Cálculos de Potasio (K) Plátano Violeta = 114.8815 mg/L x 100 mL = 5389.9549 mg/L de K 2.1314 gr Plátano Morado = 115.8846 mg/L x 100 mL = 5175.2679 mg/L de K 2.2392 gr Plátano Naranjado = 121.9432 mg/L x 100 mL = 5432.0103 mg/L de K 2.2449 gr CONCLUSIONES Los niveles de potasio que se encontraron en los plátanos de musa ornata fueron de 5000 ppm por lo q resulta una fuente importante de potasio que podría utilizarse como alimento. 82 83 PRACTICA NUM. 7 DETERMINACION DE POTASIO (K) EN MUESTRAS DE PIÑA Y CORAZON DESHIDRATADO OBJETIVO Que el alumno realice análisis de minerales en muestra de alimentos. FUNDAMENTO Nombre científico y familia Ananas comosus. pertenece a la familia de las Bromeliáceas (Bromeliaceae). Esta familia comprende unas 1.400 especies de plantas, casi todas herbáceas y de hoja perenne y con flores muy llamativas. Algunas de ellas, como es el caso de la piña tropical, producen enzimas proteolíticas, es decir, sustancias capaces de facilitar la digestión de las proteínas. Todas las especies de esta extensa familia se crían en la América tropical. (5) Esta fruta tiene un contenido de agua muy alto, por lo que su valor calórico es bajo. Bien madurado, el ananás contiene alrededor del 11% de hidratos de carbono simples o de absorción rápida. Su contenido en azúcares y en principios activos se duplica en las últimas semanas de maduración, por lo que los frutos recolectados prematuramente resultan ácidos y pobres en nutrientes. En cuanto a minerales, destacan en cantidad el potasio, magnesio, cobre y manganeso. Las vitaminas más abundantes de la piña son la vitamina C y, en menor cantidad, la tiamina o B1 y la B6 o piridoxina. (5) Los componentes no nutritivos de la piña son los más significativos desde el punto de vista dietético: • Su contenido en fibra es considerable. • Contiene una enzima, la bromelina o bromelaína, similar a las enzimas digestivas, que ayuda a digerir las proteínas. 84 • Los ácidos cítrico y málico son los responsables de su sabor ácido y como ocurre en los cítricos, el primero potencia la acción de la vitamina C. (5) MATERIAL Ver Anexo 1 Limpieza de material. • 6 Matraz microkjeldhal • Pipeta Volumétrica 1, 2, 4, 5 10 • Pipeta graduada de 10 mL • 5 Matraz aforado de 100 mL • Probeta de 100 mL • Pizeta • Papel filtro • Perilla • 6 Frascos de boca ancha • Etiquetas REACTIVOS Ver Anexo 5 Seguridad • Solución Estándar de Potasio (k) de 1000 mg/L • Acido Nítrico (HNO3) Conc. • Agua desionizada o tridestilada. EQUIPO Ver Anexo 6 Métodos analíticos • Microdigestor • Espectrofotométrica de Absorción Atómica • Lámpara de Cátodo Hueco de Potasio (K). 85 DESARROLLO EXPERIMENTAL Ver Anexo 4 Interferencias, Técnicas de muestreo en Anexo 3 y Desarrollo de análisis en Anexo 2 Para la digestión de la muestra se realiza de acuerdo a la norma NOM-117-SSA1-1994 ya que no hay una norma específica para esta muestra. En este caso se realiza por vía húmeda como se muestra en la Practica Num. 5 Después de la digestión se preparan las curvas para cada analito, como se muestra en la Práctica Num. 4 CALCULOS Se realizan los cálculos como la Practica Num. 5 Cuadro 5. Clave y peso de muestras de Piña. Clave de Muestra W = Peso de Muestra CA 1 0.0550 gr CA 2 0.0517 gr CA 3 0.0547 gr CO 1 0.0495 gr CO 2 0.0332 gr CO 3 0.0345 gr Cálculos de Potasio (K) CA 1 = 15.86496 mg/L x 100 mL = 28845.381 mg/L de K 0.0550 gr CA 2 = 21.92887 mg/L x 100 mL = 42415.609 mg/L de K 0.0517 gr 86 CA 3 = 18.14120 mg/L x 100 mL = 33164.899 mg/L de K 0.0547 gr CO 1 = 9.00888 mg/L x 100 mL = 18199.757 mg/L de K 0.0495 gr CO 2 = 71.60112 mg/L x 100 mL = 215666.02 mg/L de K 0.0332 gr CO 3 = 122.04768 mg/L x 100mL = 353761.39 mg/L de K 0.0345 gr CONCLUSIONES Los resultados obtenidos que el corazón deshidratado de la piña es una fuente rica de potasio con valores que van de 18000 ppm a 40000 ppm de potasio. 87 88 89 PRACTICA NUM. 8 DETERMINACION DE CALCIO (Ca), SODIO (Na), POTASIO (K), MAGNESIO (Mg) EN MUESTRAS DE HIGUERILLA OBJETIVO Que el alumno realice análisis de minerales en cualquier tipo de semillas como esta muestra. FUNDAMENTO La higuerilla o ricino, corresponde a Ricinus communis L., familia de las Eupharbiaceae. Planta originaria de África tropical (Abisinia) y posiblemente de la india; y se a extendido en los climas cálidos de todo el mundo, es de la familia Euforbiaceae, que según sus variedades se presenta como árbol, arbusto o hierba. La planta procede de África pero se ha hecho silvestre en muchas regiones cálidas de nuestro país, incluido el estado de Morelos, donde nace, crece y se reproduce en todos los predios baldíos de las ciudades y poblados así como a la orilla de las carreteras. (23) Planta herbáceo alta a veces algo arbustiva, de color verde claro a azul grisáceo, en ocasiones rojiza mide hasta 6 metros de alto, su tallo es engrosado y ramificado, sus hojas son lamina casi orbicular de 10 a 60 centímetros de diámetro profundamente palmatilobada, las divisiones ovado-oblongas a lanceoladas, agudas o acuminadas, borde irregular dentado-glanduloso; pecíolo tan largo o mas largo que la lamina. Glándulas entre la lamina y el pecíolo y sus flores rojas son masculinas con un perianto de 6 a 12 mm de largo, el de las flores femeninas de 4 a 8 milímetros de largo, ovario densamente cubierto por largos tubérculos blandos, que parecen pelos gruesos, frutos y semillas es una cápsula subglobosa, de uno 1.5 a 2.5 cm de largo, con espinas cortas y gruesas; semillas elipsoides, algo aplanadas, de 10 a 17 mm de largo, lisas brillantes, frecuentemente jaspiadas de café y gris carunculadas. (23) 90 Sus semillas contienen aceite fijo (oleum ricini) en porcentajes del 35 al 55 % principalmente constituido por los glicéridos de los ácidos ricinoleico, iso-recinoleico. Etc; también ricina y ricinina, la primera es una fitotoxina sumamente venenosa, por vía endovenosa y menor por vía oral, aun que esta última vía puede ocasionar la muerte; su actividad desaparece por acción del calor moderado; el segundo es un alcaloide de fórmula C8H8N2O2. (23) MATERIAL Ver Anexo 1 Limpieza de material • 4 Matraz microkjeldhal • Pipeta Volumétrica 1, 5, 10, 20 mL • Pipeta graduada de 10 mL • 5 Matraz aforado de 100 mL • Probeta de 100 mL • Pizeta • Papel filtro • Perilla • 4 Frascos de boca ancha • Etiquetas REACTIVOS Ver Anexo 5 Seguridad • Solución Estándar de Calcio (Ca), Sodio (Na), Potasio (k), Magnesio (Mg) de 1000 mg/L • Acido Nítrico (HNO3) Concentrado. • Agua desionizada o tridestilada. EQUIPO Ver Anexo 6 Métodos analíticos 91 • Microdigestor • Espectrofotométrica de Absorción Atómica • Lámpara de Cátodo Hueco de Calcio (Ca), Sodio (Na), Potasio (K), Magnesio (Mg). DESARROLLO EXPERIMENTAL Ver Anexo 4 Interferencias, Técnicas de toma de muestra en Anexo 3 y Desarrollo de análisis en Anexo 2 Para la digestión de la muestra se realiza de acuerdo a la norma NOM-117-SSA1-1994 ya que no hay una norma específica para esta muestra. En este caso se realiza por vía húmeda como se muestra en la Practica Num. 5 Después de la digestión se preparan las curvas para cada analito, como se muestra en la Práctica Num. 4 CALCULOS Se realizan los cálculos como la Practica Num. 5 Cuadro 6. Clave y peso de muestras de Higuerilla. Clave de Muestra W = Peso de Muestra Semilla 1.053 gr Cáscara 1.104 gr Cáscara c/semilla 1.003 gr 92 Cálculos de Magnesio (Mg) Semilla = 4.10603 mg/L x 100mL = 389.9363 mg/L de Mg 1.053 gr Cáscara = 1.24440 mg/L x 100mL = 112.71739 mg/L de Mg 1.104gr Cáscara c/semilla = 2.34165 mg/L x 100mL = 233.4646 mg/L de Mg 1.003 gr Cálculos de Potasio (K) Semilla = 4.15235 mg/L x 100 mL = 394.3352 mg/L de K 1.053 gr Cáscara = 2.53864 mg/L x 100 mL = 229.9275 mg/L de K 1.104 gr Cáscara c/semilla = 1.81738 mg/L x 100mL = 181.1944 mg/L de K 1.003 gr Cálculos de Sodio (Na) Semilla = 0.40473 mg/L x 100 mL = 38.435897 mg/L de Na 1.053 gr 93 Cáscara = 0.87874 mg/L x 100 mL = 79.596014 mg/L de Na 1.104 gr Cáscara c/semilla = 0.69067 mg/L x 100 mL = 68.860418 mg/L de Na 1.003 gr Cálculos de Calcio (Ca) Semilla = 0.50224 mg/L x 100mL = 47.696106 mg/L de Ca 1.053 gr Cáscara = 0.43185 mg/L x 100mL = 39.116847 mg/L de Ca 1.104 gr Cáscara c/semilla = 0.46977 mg/L x 100mL = 46.83649 mg/L de Ca 1.003 gr CONCLUSIONES Se toman las muestras que se encuentran en la región. El análisis se realizo con distintas muestras y presentaron niveles de 100 ppm a 400 ppm de magnesio, y de 100 ppm a00 ppm de potasio; y de 40 ppm a 80 ppm de sodio, y de 30 ppm a 40 ppm de calcio, lo q representa que los productos de origen natural tiene una variación sus productos. 94 95 96 97 98 99 100 101 102 PRACTICA NUM. 9 DETERMINAR CALCIO (Ca), SODIO (Na), POTASIO (K), MAGNESIO (Mg) EN MUESTRAS DE GIGANTON OBJETIVO Que el alumno realice análisis de minerales en cualquier tipo de plantas como esta muestra. FUNDAMENTO Thithonia diversifolia es una planta herbácea de la familia Asteracea, originaria de Centro América (Nash, 1976). Tiene un amplio rango de adaptación, tolera condiciones de acidez y baja fertilidad en el suelo. Es además una especie con buena capacidad de producción de biomasa, rápido crecimiento y baja demanda de insumos y manejo para su cultivo. Presenta características nutricionales importantes para su consideración como especie con potencial en alimentación animal. (25) Se utiliza para alimentación de cabras en un sistema de corte y acarreo en Mindanao, Filipinas. El estiércol de los animales se aplica en los callejones del cultivo. Este sistema combina los beneficios de la producción pecuaria, el ciclaje eficiente de nutrientes y la conservación de suelos. También se aprovecha para el ramoneo de ovejas y, en Luzón, algunos agricultores esparcen hojas de T. diversifolia en los estanques para ser consumida por tilapias. Adicionalmente en Indonesia y Filipinas se han realizado ensayos con resultados promisorios, al incorporar hojas de esta especie en raciones para alimentación de gallinas. (25) MATERIAL Ver Anexo 1 Limpieza de material. • 1 Matraz microkjeldhal • Pipeta Volumétrica 1, 5, 10, 20 mL 103 • Pipeta graduada de 10 mL • 5 Matraz aforado de 100 mL • Probeta de 100 mL • Pizeta • Papel filtro • Perilla • 1 Frascos de boca ancha • Etiquetas REACTIVOS Ver Anexo 5 Seguridad • Solución Estándar de Calcio (Ca), Sodio (Na), Potasio (k), Magnesio (Mg) de 1000 mg/L • Acido Nítrico (HNO3) Concentrado. • Agua desionizada o tridestilada. EQUIPO Ver Anexo 6 Métodos analíticos • Microdigestor • Espectrofotométrica de Absorción Atómica • Lámpara de Cátodo Hueco de Calcio (Ca), Sodio (Na), Potasio (K), Magnesio (Mg). DESARROLLO EXPERIMENTAL Ver Anexo 4 Interferencias, Técnicas de toma de muestra en Anexo 3 y Desarrollo de análisis en Anexo 2 Para la digestión de la muestra se realiza de acuerdo a la norma NOM-117-SSA1-1994 ya que no hay una norma específica para esta muestra. 104 En este caso se realiza por vía húmeda como se muestra en la Practica Num. 5 Después de la digestión se preparan las curvas para cada analito, como se muestra en la Práctica Num. 4 CALCULOS Se realizan los cálculos como la Practica Num. 5 Cuadro 7. Clave y peso de muestras de gigantón. Clave de Muestra W = Peso de Muestra Gigantón 1.0137 gr Cálculos de Magnesio (Mg) Gigantón= 3.07019 mg/L x 100 mL = 302.8696 mg/L de Mg 1.0137 gr Cálculos de Potasio (K) Gigantón= 3.40551 mg/L x 100 mL = 335.9485 mg/L de K 1.0137 gr Cálculos de Sodio (Na) Gigantón= 2.06013 mg/L x 100 mL = 203.2287 mg/L de Na 1.0137 gr 105 Cálculos de Calcio (Ca) Gigantón= 1.23629 mg/L x 100 mL = 1.0137 mg/L de Ca 1.0137 gr CONCLUSIONES Se toman muestras que se encuentran en la región. El análisis de distintas muestras presentaron niveles de de 300 ppm de magnesio; 335ppm de potasio; y de 200 ppm de sodio; y de 1.0 ppm de calcio, lo q representa que los productos de origen natural se encuentran diferentes minerales. 106 107 108 109 110 PRACTICA NUM. 10 DETERMINAR PLOMO (Pb) Y CADMIO (Cd) EN MUESTRAS DE PESCADO OBJETIVO Que el alumno realice análisis de metales pesados como el plomo en muestra de alimentos. FUNDAMENTO En los últimos años, el medio ambiente marino se ha ido contaminando visiblemente, como resultado de las actividades del hombre. Grandes cantidades de sustancias, algunas nocivas, llegan al ecosistema acuático y parte de ellas proceden de residuos industriales, agrícolas o domésticos. Dentro de la variable lista de contaminantes, los metales pesados en cantidades de trazas, ocupan una posición única, ya que ellos no pueden ser descompuestos posteriormente y, una vez depositados, permanecerán en el medio acuático, prácticamente sin ningún cambio cualitativo. (3) Las fuentes de los metales pesados Las fuentes principales del mercurio son las siguientes: el pescado (a causa de la contaminación de los mares); los insecticidas (que contienen normalmente uno o dos metales pesados, que se cuelan en la cadena alimentaria); el agua ‘potable’ (tenemos que suponer que el agua contiene tóxicos a menos que se haya comprobado mediante análisis lo contrario); algunos medicamentos (especialmente los que regulan la alta presión sanguínea y la vacuna contra el tétanos); y el aire contaminado por la industria y los coches (por la tecnología de combustión). Otra fuente de mercurio muy importante es el traspaso de la madre al feto a través de la placenta y al bebé a través de la leche materna por procesos hormonales. Mediante estos procesos la madre traspasa del 40 al 60% de su carga al niño. (3) 111 MATERIAL Ver Anexo 1 Limpieza de material. • 4 Matraz microkjeldhal • Pipeta Volumétrica 0.5, 1, 2, 10 • Pipeta graduada de 10 mL • 5 Matraz aforado de 100 mL • Probeta de 100 mL • Pizeta • Papel filtro • Perilla • 4 Frascos de boca ancha • Etiquetas REACTIVOS Ver Anexo 5 Seguridad • Solución Estándar de Plomo (Pb), Cadmio (Cd) de 1000 mg/L • Acido Nítrico (HNO3) Concentrado • Agua desionizada o tridestilada. EQUIPO Ver Anexo 6 Métodos analíticos. • Microdigestor • Espectrofotométrica de Absorción Atómica • Lámpara de Cátodo Hueco de Plomo (Pb), Cadmio (Cd). DESARROLLO EXPERIMENTAL Ver Anexo 4 Interferencias, técnicas de toma de muestra en Anexo 3 y desarrollo de análisis en Anexo 2 112 Para la digestión de la muestra se realiza de acuerdo a la norma NOM-027-SSA1-1993 y esta nos rige a esta NOM-117-SSA1-1994. En este caso se realiza por vía húmeda como se muestra en la Práctica Num. 5 Después de la digestión se preparan las curvas para cada analito, como se muestra en la Práctica Num. 4 CALCULOS Se realizan los cálculos como en la Practica Num. 5 Cuadro 8. Clave y peso de muestras de pescado. Clave de Muestra W = Peso de Muestra PC 1 19. 9670 gr Pch 1 20.2328 gr PCB 1 19.7582 gr PchB 1 20.0523 gr Cálculos de Plomo (Pb) PC 1 = 0.06103 mg/L x 100 mL = 0.3056 mg/L de Pb 19.9670 gr PCh 1= 0.11991 mg/L x 100 mL = 0.5926 mg/L de Pb 20.2328 gr PCB 1= 0.09698 mg/L x 100 mL = 0.4908 mg/L de Pb 19.7582 gr 113 PCHB 1 = 0.48462 mg/L x 100 mL = 2.4167 mg/L de Pb 20.0523 gr Cálculos de Cadmio (Cd) PC 1 = 0.00000 mg/L x 100 mL 19.9670 gr =No se realizan cálculos por que el instrumento no detecto nada CONCLUSIONES Los resultados obtenidos del pescado con valores que van de 0.30 ppm a 2.00 ppm de Plomo, nos indica la presencia del mismo, y para la determinación de Cadmio el equipo no detecto trazas. 114 115 116 117 118 PRACTICA NUM. 11 DETERMINACION DE PLOMO (Pb) Y CADMIO (Cd) EN MUESTRAS DE AGUA RESIDUAL OBJETIVO: Que el alumno realice la digestión de la muestra y lleve acabo su análisis. FUNDAMENTO: Los efectos de los metales que se encuentran en las aguas naturales, potables y residuales en la salud humana, pueden ir desde el intervalo de benéficos, causantes de problemas hasta tóxicos, esto es dependiendo de su concentración, por lo que su cuantificación en cuerpos de agua es importante. Algunos metales son esenciales, otros pueden afectar adversamente a los consumidores de agua, sistemas de tratamiento de aguas residuales y cuerpos receptores de agua. (11) Aguas naturales Se define como agua natural el agua cruda, subterránea, de lluvia, de tormenta, residual y superficial. (11) Aguas residuales Las aguas de composición variada provenientes de las descargas de usos municipales, industriales, comerciales, agrícolas, pecuarias, domésticos y similares, así como la mezcla de ellas. (11) Aguas potables Para uso y consumo humano: Aquella que no contiene contaminantes objetables ya sean químicos o agentes infecciosos y que no causa efectos nocivos al ser humano. (11) Estos métodos se implementaran de acuerdo a las normas mexicanas que se involucren en el tipo de análisis.NMX-AA-051-SCFI-2001 119 MATERIAL Ver Anexo 1 Limpieza de material. • 4 Matraz microkjeldhal • Pipeta Volumétrica 0.5, 1, 2, 10 • Pipeta graduada de 10 mL • 5 Matraz aforado de 100 mL • Probeta de 100 mL • Pizeta • Papel filtro • Perilla • 4 Frascos de boca ancha • Etiquetas REACTIVOS Ver Anexo 5 Seguridad • Solución Estándar de Plomo (Pb), Cadmio (Cd) de 1000 mg/L • Acido Nítrico (HNO3) Concentrado. • Agua desionizada o tridestilada. EQUIPO Ver Anexo 6 Métodos analíticos • Microdigestor • Espectrofotométrica de Absorción Atómica • Lámpara de Cátodo Hueco de Plomo (Pb), Cadmio (Cd). DESARROLLO EXPERIMENTAL Ver Anexo 4 Interferencias, Técnicas de toma de muestra en Anexo 3 y Desarrollo de análisis en Anexo 2 120 Preparación de la muestra para la determinación de metales totales por digestión en parrilla de calentamiento y en vaso abierto en aguas naturales, potables y residuales. • Homogeneizar perfectamente la muestra, verificando que no existan sólidos adheridos en el fondo del contenedor. Inmediatamente después tomar una alícuota de 50 mL a 100 mL dependiendo de la naturaleza de la muestra, y transferir a un vaso de precipitados para digestión. • Añadir 3 mL de ácido nítrico concentrado y calentar en una placa, evaporar, cuidando que no hierva, hasta aproximadamente de 2 mL a 5 mL de muestra y enfriar. • Adicionar 5 mL de ácido nítrico concentrado, cubrir con un vidrio de reloj y pasar nuevamente la muestra a la placa de calentamiento. Incrementar la temperatura de calentamiento hasta que exista reflujo de vapores. • Continuar calentando y en caso de ser necesario, agregar mayor cantidad de ácido nítrico concentrado y continuar la digestión. Cuando la digestión es completa (cuando la muestra tenga apariencia cristalina constante) retirar la muestra y enfriar. • Por cada 100 mL de volumen de disolución final adicionar 10 mL de ácido clorhídrico (1:1) y 15 mL de agua, calentar la muestra sin llegar a ebullición por espacio de 15 min para disolver precipitados o residuos resultantes de la evaporación y llevar al aforo correspondiente (de 50 mL a 100 mL). • Enfriar, lavar las paredes del vaso y vidrio de reloj con agua y filtrar para remover el material insoluble que pueda tapar el nebulizador. Ajustar el volumen basado en la concentración esperada de los analitos. • La muestra está lista para su análisis. • Las concentraciones deben reportarse como metales totales. (11) Cálculos que se realizan para una muestra Realizar las gráficas de la curvas de calibración de cada uno de los metales de acuerdo a las concentraciones esperadas de la muestra. 121 Calcular la concentración de la muestra por medio de la ecuación de la recta que se obtiene de las curvas de calibración para cada metal empleando la siguiente ecuación: Ecuación 1: Y = mX + b donde: Y es la absorbancia de la muestra ya procesada; m es la pendiente (coeficiente de absortividad), y b es la ordenada al origen. despejar X que debe ser la concentración de la muestra procesada y tomar en cuenta los factores de dilución que se realicen en cada uno de los metales según la técnica utilizada, y se debe obtener la concentración del metal en la muestra. Si se trabaja con el método de adición de estándares, obtener la gráfica, el coeficiente de correlación y el valor de la muestra sin añadir. Reporte de resultados: No se deben reportar concentraciones de elementos por debajo del límite de detección. Reportar los resultados del análisis en mg/L. (11) Cálculos que se realizan para una muestra fortificada Se debe fortificar al menos una muestra por grupo o el 10% de ellas lo que resulte mayor. La concentración añadida debe ser de aproximadamente 0,1 unidades de absorbancia. Se debe calcular el % de recuperación para el analito, de acuerdo a: (11) R = CM - C x 100 CA R = % recuperación CM = Concentración de la muestra fortificada C = Concentración de la muestra CA = Concentración equivalente de analito añadido a la muestra. 122 Si la recuperación del analito en la muestra fortificada está fuera del intervalo previamente establecido y el blanco de reactivos fortificado está correcto, puede existir un problema relacionado con la matriz de la muestra. Los datos se deben verificar por el método de las adiciones estándar. (11) CALCULOS Cuadro 9. Clave y volumen de muestras de agua residual. Clave de Muestra W = ml de Muestra 1 Orilla 100 mL 1 Orilla Fortificada 100 mL 2 Profundo 100 mL 2 Profundo Fortificada 100 mL 3 Vieja 100 mL 3 Vieja Fortificada 100 mL Cálculos de Plomo (Pb) 1 Orilla = 0.69642 mg/L x 100 mL =0.6964 mg/L de Pb 100 mL 1 Orilla = Fortificada 2 Profundo = 1.57534 mg/L – 0.69642 mg/L 1.00 mg/L 0.69299 mg/L x 100 mL X 100 % = 87.89 % de Recuperación de Pb = 0.69299 mg/L de Pb 100 mL 2 Profundo = 1.66302 mg/L – 0.69642 mg/L Fortificada 1.00 mg/L X 100 % = 96.66 % de Recuperación de Pb 123 3 Vieja = 0.73088 mg/L x 100 mL = 0.73088 mg/L de Pb 100 mL 3 Vieja = 1.71525 mg/L – 0.73088 mg/L 1.00 mg/L Fortificada X 100 % = 98.43 % de Recuperación de Pb Cálculos de Cadmio (Cd) 1 Orilla = 0.00000 mg/L x 100 mL =0.00000 mg/L de Cd 100 mL 1 Orilla = 0.50692 mg/L – 0.00000 mg/L 0.50 mg/L Fortificada 2 Profundo = 0.00176 mg/L x 100 mL X 100 % = 101.38 % de Recuperación de Cd = 0.00176 mg/L de Cd 100 mL 2 Profundo = 0.52219 mg/L – 0.00000 mg/L Fortificada 0.50 mg/L 3 Vieja = 104.43 mg/L x 100 mL X 100 % = 104.43 % de Recuperación de Cd = 0.00000 mg/L de Cd 100 mL 124 3 Vieja = Fortificada 0.51502 mg/L – 0.00000 mg/L 0.50 mg/L X 100 % = 103.00 % de Recuperación de Pb CONCLUSIONES Los resultados obtenidos en las muestras de agua residual en el análisis con valores que van de 0.600 ppm a 0.700 ppm de plomo; y en cadmio no se detectaron trazas en las muestras. El porcentaje que se obtuvo en las muestras fortificadas tienen valores del 87% a 98% en plomo. Por lo tanto se considera que los análisis obtenidos son confiables ya que es un porcentaje de recuperación aceptable. 125 126 127 128 129 PRACTICA NUM. 12 DETERMINAR CALCIO (Ca), SODIO (Na), POTASIO (K), MAGNESIO (Mg) EN MUESTRAS DE COLORIN OBJETIVO Que el alumno realice análisis minerales en cualquier tipo de plantas como esta muestra. FUNDAMENTO Erythrina coralloides, colorín, es una especie de árbol ornamental, debido a sus flores rojas y blancas en grupos, y mezcladas. Las flores son también alimento. Es nativa de Norteamérica: México, EE. UU., y de Centroamérica. Alcanza 5 m de altura, y su madera blanca se usa para tapones, y en San Luis Potosí para hacer figuras, decoración y joyería por artesanos. (27) Tiene semillas muy tóxicas, contiene eritroidina, poderoso paralizante de músculos, eritroresina: emético, coralina y ácido eritrico. Su extracto es un sustituto del curare. Son semillas elípticas, brillantes, rojo coral, con una línea saliente longitudinal en el dorso, y un hilo blanco, rodeado con un borde negro. (27) Del análisis bioquímico: 13,35 sólidos y aceites; 0,32 resina soluble en éter; 13,47 resina soluble en alcohol; 1,61 eritrococaloidina, alcaloide; 5,6 albúmina; 0,83 goma; 1,55 azúcar; 0,42 ácido orgánico; 15,87 almidón; 7,15 humedad; 39,15 materia inorgánica. (27) MATERIAL Ver Anexo 1 Limpieza de material. • 3 Matraz microkjeldhal • Pipeta Volumétrica 0.5, 1, 2,10 mL • Pipeta graduada de 10 mL 130 • 5 Matraz aforado de 100 mL • Probeta de 100 mL • Pizeta • Papel filtro • Perilla • 3 Frascos de boca ancha • Etiquetas REACTIVOS Ver Anexo 5 Seguridad • Solución Estándar de Calcio (Ca), Sodio (Na), Potasio (k), Magnesio (Mg) de 1000 mg/L • Acido Nítrico (HNO3) Concentrado. • Agua desionizada o tridestilada. EQUIPO Ver Anexo 6 Métodos analíticos • Microdigestor • Espectrofotométrica de Absorción Atómica • Lámpara de Cátodo Hueco de Calcio (Ca), Sodio (Na), Potasio (K), Magnesio (Mg). DESARROLLO EXPERIMENTAL Ver Anexo 4 Interferencias, Técnicas de toma de muestra en Anexo 3 y Desarrollo de análisis en Anexo 2 Para la digestión de la muestra se realiza de acuerdo a la norma NOM-117-SSA1-1994 ya que no hay una norma específica para esta muestra. 131 En este caso se realiza por vía húmeda como se muestra en la Practica Num. 5 Después de la digestión se preparan las curvas para cada analito, como se muestra en la Práctica Num. 4 CALCULOS Se realizan los cálculos como la Práctica Num. 5 Cuadro 10. Clave y peso de muestras de colorín Clave de Muestra W = Peso de Muestra Colorín 1 1.3120 gr Colorín 2 1.2806 gr Colorín 3 1.0211 gr Cálculos de Potasio (K) Colorin 1= 1.47750 mg/L x 100 mL = 112.6143 mg / L de K 1.3120 gr Colorin 2= 1.09072 mg/L x 100 mL = 85.1725 mg / L de K 1.2806 gr Colorin 3= 1.56615 mg/L x 100 mL = 153.3787 mg / L de K 1.0211 gr Cálculos de Sodio (Na) Colorin 1= 0.56488 mg/L x 100 mL = 43.0548 mg / L de Na 1.3120 gr 132 Colorin 2= 0.56595 mg/L x 100 mL = 44.1941 mg / L de Na 1.2806 gr Colorin 3= 0.16301 mg/L x 100 mL = 13.5717 mg / L de Na 1.0211 gr Cálculos de Magnesio (Mg) Colorin 1= 4.33979 mg/L x 100 mL = 335.2057 mg / L de Mg 1.3120 gr Colorin 2= 2.87044 mg/L x 100 mL = 224.1480 mg / L de Mg 1.2806 gr Colorin 3= 3.03944 mg/L x 100 mL = 297.6633 mg / L de Mg 1.0211 gr Cálculos de Calcio (Ca) Colorin 1= 0.16761 mg/L x 100 mL = 51.5320 mg / L de Ca 1.3120 gr Colorin 2= 0.7753 mg/L x 100 mL = 60.5419 mg / L de Ca 1.2806 gr 133 Colorin 3= 60.541933 mg/L x 100 mL = 12.2622 mg / L de Ca 1.0211 gr CONCLUSIONES Se toman muestras identificadas por el lugar donde se muestrean, que son de la región. El análisis de muestras de colorín presentaron niveles de 80 ppm a 150 ppm de potasio; y de 10 ppm a 40 ppm de sodio; de 200 ppm a 350 ppm de magnesio; y de 12 ppm a 60 ppm de calcio, lo q representa que los productos de origen natural presentan minerales en concentraciones a nivel trazas y tiene una variación de los productos por ser naturales. 134 135 136 137 138 139 140 141 142 PRACTICA NUM 13 DETERMINACION DE MERCURIO EN MUESTRA DE PESCADO POR MEDIO DEL GENERADOR DE HIDRUROS OBJETIVO Que el alumno realice análisis, utilizando el generador de hidruros a concentraciones a nivel trazas FUNDAMENTO Generador de Hidruros Es un método instrumental para análisis de muestras que contienen arsénico, antimonio, estaño, selenio, bismuto en forma de gas, dicho procedimiento mejora los límites de detección de éstos elementos de 10 a 100 veces, debido a que varias de estas especies son altamente tóxicas es de considerable importancia su determinación a concentraciones bajas (para el mercurio que se determina por la técnica de vapor frío, se deben manejar concentraciones menores a 0.001 mg/L, ya que éste es el límite permisible que establece la NOM-127-SSA1-1994 para el agua potable). El Generador de Hidruros se emplea en la técnica de vapor frío aplicable a la determinación de mercurio. (11) El arsénico en la muestra es reducido de As V a As III mediante la reacción con SnCl2 o NaBH4 y convertido a arsina (AsH3). El hidruro gaseoso formado AsH3 es arrastrado por el gas argón a la flama del quemador del aparato de absorción atómica para su determinación, registrándose la absorbancia correspondiente. (11) Los hidruros gaseosos tampoco forman métodos no estables a temperaturas altas. Una vez que forma y separa desde el liquido, el vapor de hidruro es llevado por un flujo de gas inerte (nitrógeno o argon) hacia al tubo de cuarzo caliente donde ocurre la descomposición térmica. El absorbe luz por átomos analíticos es entonces determinado de manera normal como en espectrofotómetro absorción atómica. (11) 143 En el caso de mercurio (Hg) las reacciones con cualquiera NaBH4 o SnCl2 en la presencia de acido, produce un átomo Hg vapor directo, así no requiere flama. Esto pasan con “Cerrando flujo directo” celda y analiza en la vía usual. (11) MATERIAL Ver Anexo 1 Limpieza de material. • 3 Matraz microkjeldhal • Pipeta Volumétrica 0.5, 1, 2 • Pipeta graduada de 10 mL • 5 Matraz aforado de 100 mL • Probeta de 100 mL • Pizeta • Papel filtro • Perilla • 3 Frascos de boca ancha • Etiquetas REACTIVOS Ver Anexo 5 Seguridad • Solución Estándar de Mercurio (Hg) de 1000 mg/L • Acido Clorhídrico (HCl) Concentrado. • Agua desionizada o tridestilada. • Borohidruro de Sodio • Acido Clorhídrico 3M. EQUIPO • Microdigestor • Espectrofotométrica de Absorción Atómica 144 • Generador de Hidruros • Lámpara de Cátodo Hueco de Mercurio (Hg) DESARROLLO EXPERIMENTAL Ver Anexo 4 Interferencias, Técnicas de toma de muestra en Anexo 3 y Desarrollo de análisis en Anexo 2 Conocimiento del instrumento HG Generador de Hidruros Este método es aplicable para la determinación de antimonio, arsénico, mercurio, selenio, etc. Figura 46. Operación del Instrumento El HG 3000 generador hidruros automático de flujo continuo es un sistema de generación de vapor. La solución mezclada fluye a través de una bocina de reacción cuando el metal hidruro de vapor se elimina de la utilización del liquido, entre un trasportador de gas. El hidruro se introduce en una celda de cuarzo es absorbido sobre un quemador y se calienta por una flama de aire-acetileno. 145 Figura 48. Componentes del Instrumento Figura 47. Componentes del Instrumento 146 Figura 48. Mezcle de Gases Figura 49. Componentes del Instrumento. 147 Cuadro 11. Componentes del Instrumento 1. Frasco de reactivo acido. 18. Polea de montaje de drenaje 2. Frasco de reactivos de borohidruro. 19. Tubo de desagüe 3. Entrada de capilar acido. 20. Selector de gas. 4. Entrada de capilar de borohidruro. 21. Bomba de interruptor de encendido/apagado 5. Entrada de capilar muestra. 22. Montaje del ala 6. Polea de montaje de la bomba de tubo. 23. Separador de gas-liquido 7. Bomba peristática cabeza. 24. Separador de pinza 8a. Abrazaderas de tubo de bomba. 25. Separador de tapa 8b. Abrazadera de la bomba de tubo rápido. 26 Capilar de gas inerte 9. Tubo de la bomba de acido 27. Tubo de hidruro 11. Tubo de la bomba de borohidruro. 28. Polea de mezclador 12. Bomba de muestreo de tubo. 29. Capilar de mezclador de gases. 13. Interconexión de capilar acido. 30. Tubo de silicona 14. Interconexión de capilar borohidruro. 15. Interconexión de capilar muestra. 16 Polea de mezcla. 17 Adaptador de polea de reacción. Se activa el instrumento de HG en la sección de accesorios que se encuentra en la Práctica Num. 2 en la pagina 36 Generador de Hidruros Figura 50. Accesorio HG 148 Análisis por Generador de Hidruros. En forma directa para aguas naturales y potables (transparentes) y/o después de previa digestión para aguas residuales y alimentos. • Calibrar el espectrofotómetro de absorción atómica con el aditamento generador de hidruros de acuerdo a lo indicado por el manual del fabricante. • Análisis por vapor frío. • Para el análisis con boro hidruro de sodio. • Los estándares, blanco y muestras, más un oxidante se conectan al sistema de vapor frío para ser detectados, ver manual de fabricante. • Obtener una gráfica con los valores de la curva de calibración y realizar los cálculos cuantitativos. • Analizar el lote de muestras. Preparación de Reactivos Borohidruro de Sodio Preparar unos 500 mL de borohidruro de sodio (NaBH4) de la siguiente manera. Disolver 3 g de polvo de NaBH4 y 3 g de NaOH (Reactivos de laboratorio de alto grado) en agua desionizada hasta disolver y filtrar la solución en el frasco de reactivos de borohidruro. Acido Clorhídrico 3M Llenar la otra botella con 500 mL de acido clorhídrico conc. Aproximadamente 3M Tapar con seguridad ambas botellas y en un lugar fresco en el anaquel de reactivos. Almacenar Solución es inestable y se descompone, si se almacena por más de 3-4 días. Después de la fecha de correr trasvasar la solución en un contenedor y asegurar la tapa, no sea hermético (Debido a la evolución de gas hidrogeno). 149 El acido clorhídrico puede almacenarse indefinidamente después de la fecha, decantar el acido en un contenedor sellado. Enjuague las botellas de reactivos mucho antes de almacenar en el HG 3000 frascos de reactivos del anaquel. Dicromato de potasio Preparar 100 mL al 20% g/g de dicromato de potasio (K2Cr2O7). Pesar 20 g de dicromato de potasio en un vaso de 200 mL. Agregar 50 mL de acido nítrico concentrado (HNO3) y calentar suavemente el dicromato de potasio hasta disolver (No hervir la solución). Transferir cuantitativamente esta solución a un matraz volumétrico de 100 mL y llevar al volumen con agua desionizada. Bajo al nivel Hg (Mercurio) es añadido a todas las muestras. Blanco estándar y soluciones para dar una concentración final al menos 0.05 % de Dicromato de Potasio. Operación del Equipo Generador de Hidruros HG-3000. • La presión del gas acarreador debe tener de 30 a 60 Kpa., esta presión se observa en el manómetro del tanque del argón. • Instalar la lámpara adecuada, colocar la corriente de la lámpara dependiendo del metal a analizar. • Encender el espectrofotómetro y esperar a que se estabilice. • Seleccionar la longitud de onda y el ancho de banda espectral para el elemento que va a ser determinado de acuerdo al protocolo del laboratorio o del manual del fabricante. • Alinear la lámpara a su máxima energía. • Alinear el accesorio que se va a usar para atomizar la muestra. • Ajustar el rayo de luz de la lámpara de acuerdo con las especificaciones del fabricante del equipo. • Ajustar los flujos de gas de aire y acetileno. Este ajuste no se requiere para la determinación de mercurio. 150 • Alinear la celda de cuarzo en el rayo de luz y esperar de 20 min a 30 min para su estabilización en la flama antes de iniciar el análisis; en este período, preparar las disoluciones estándar y los reactivos. • Colocar en el recipiente del reductor una disolución de borohidruro de sodio en hidróxido de sodio y conectar el recipiente al sistema según las especificaciones del fabricante del equipo. • Abrir el suministro de gas inerte y ajustar la presión de acuerdo a las especificaciones del fabricante del equipo. • Conectar el vaso de reacción al sistema generador y esperar el tiempo suficiente para que todo el aire se purgue del sistema, entonces registrar el cero en el espectrofotómetro (autocero). • Conectar el vaso de reacción que contiene el blanco de reactivos. • Purgar el sistema hasta eliminar completamente el aire, permitir la entrada de la disolución de borohidruro de sodio, hasta obtener la lectura del blanco. • Limpiar el sistema haciendo pasar agua o ácido clorhídrico diluido. • Realizar la curva de calibración con un mínimo de cuatro concentraciones y un blanco de reactivos en el intervalo lineal demostrado para cada elemento. El primer punto deberá ser igual o mayor al límite de cuantificación, y el último deberá estar dentro del intervalo lineal. Al finalizar los trabajos con el Generador de Hidruros. • Apague el Generador de Hidruros con el botón principal ON/OFF, desconecte la entrada de gas al Generador de Hidruros con la perilla GAS-ON. Enseguida desconecte las mangueras de los frascos reactivos (Borohidruro de sodio, HCl, y muestra), introduzca las tres mangueras en un vaso de precipitados con agua desionizada y lave el sistema (100 ml aproximadamente). • Vaciar las soluciones de HCl y Borohidruro de Sodio y vaciarlas en recipientes con tapa, etiquetados y ponerlos en refrigeración. • Quitar las mangueras de la bomba peristáltica para evitar su desgaste. • Desconectar la manguera que conecta la celda de cuarzo con el generador de hidruros (hacerlo con precaución). 151 Digestión por vía húmeda para la determinación de Hg. Sistema de reflujo. • Pesar con precisión de ± 0,1 mg, la cantidad apropiada de muestra, dependiendo el tipo de ésta, en un matraz de digestión y adicionar perlas de ebullición. • Conectar el matraz al sistema de reflujo y agregar poco a poco la cantidad necesaria de ácido nítrico concentrado y calentar durante media hora o hasta que no se observen cambios en la digestión. • Dejar enfriar y agregar una mezcla de ácido nítrico y ácido sulfúrico concentrados (1 + 1). • Calentar y agregar más ácido nítrico gota a gota sobre las paredes del recipiente, hasta que el color obscuro de la solución desaparezca. • Enfriar. • Si existe grasa o cera filtrar la solución. • Correr un blanco de reactivos. • Leer en el aparato de elección (espectrómetro de absorción atómica de vapor frío). CALCULOS Cuadro 12. Clave y peso de muestras de pescado Clave de Muestra W = Peso de Muestra PC 1 19. 9670 gr Pch 1 20.2328 gr PCB 1 19.7582 gr PchB 1 20.0523 gr Cálculos de Mercurio (Hg) PC 1 = 1.40005 mg/L x 100 mL = 7.0118 mg/L de Hg 19.9670 gr 152 PCh 1= 1.31723 mg/L x 100 mL = 6.5103 mg/L de Hg 20.2328 gr PCB 1= 0.19789 mg/L x 100 mL = 1.0001 mg/L de Hg 19.7582 gr PCHB 1 = 0.47039 mg/L x 100 mL = 2.3458 mg/L de Hg 20.0523 gr CONCLUSIONES Los resultados obtenidos en la muestra de pescado, con valores que van de 1.0 ppm a 7.0 ppm de mercurio, por lo cual nos indica que se encuentra el mercurio en el pescado en trazas. 153 154 4. COMENTARIOS 155 En este manual educativo teórico practico, detalla la técnica de operación para el espectrofotómetro de absorción atómica, GBC 932 AA haciéndolo comprensible e introductorio para el uso. Este manual educativo es una referencia donde proporciona los fundamentos básicos del espectrofotómetro de absorción atómica y los requisitos para el desarrollo de un método analítico. Las prácticas permiten al estudiante conocer los diferentes análisis para su determinación así como conocer en cantidades a nivel trazas y así conocer los minerales que los forman. 156 5. ANEXOS 157 ANEXO 1 1. LIMPIEZA DEL MATERIAL Todo el material usado en esta determinación debe ser exclusivo para este procedimiento. • Para el lavado del material remojar durante 1 h en una disolución de ácido nítrico al 10 % y enjuagar con agua. Los detergentes con base de amoniaco no deben usarse para la limpieza del material. Su uso debe restringirse dentro del laboratorio. • Los contenedores de las muestras deben lavarse con disolución de detergente no iónico, libre de metales, enjuagarse con agua, remojarse en ácido toda la noche y volver a enjuagarse con agua libre de metales y dejar secar (con cuidado especial para el análisis de trazas). • En los casos de que se presenten adherencias en el material debe dejarse remojando de 12 h a 24 h con HNO3 (1:5), HCl (1:5) o con agua regia (3 partes de HCl concentrado + 1 parte de HNO3 concentrado) a 70ºC, después debe ser enjuagado con agua libre de metales. • En los casos de que el material presente grasas, enjuagar con acetona y/o hexano. 158 ANEXO 2 PROCEDIMIENTO BASICO EN EL DESARROLLO DE UN ANÁLISIS Paso Acción 1 Encienda el instrumento del interruptor principal. 2 Instale la lámpara y permita que se optimice la lámpara Pagina 17 29,30,36,37 instalada. 3 Instale el quemador 18 - 21 4 Optimice el sistema 48 - 50 5 6 • Asegúrese que el quemador no bloquea el haz de luz • Optimice la Lámpara. • Alinear el quemador Organice los archivos del análisis. • Crear el archivo del Método • Crear el archivo de Muestra • Crear el archivo de Resultados • Complete el modulo de Análisis • Complete el modulo de Reporte Encienda la flama • 7 26 Caja de gases automático Optimice • Método de flama Aire-Acetileno • Método de flama Oxido Nitroso-Acetileno • Generador de Hidruros 8 Corre Análisis seleccionar el botón verde en la barra de inicio 9 Cuando el análisis haya finalizado. 10 32 - 46 • Limpiar el nebulizador con agua desionizada • Apagar la flama Apagar el instrumento. 26, 27 32 26 - 27 17 159 ANEXO 3 1. TECNICAS DE PREPARACIÓN DE MUESTRAS 1.1 Toma y Conservación de Muestras Antes de tomar una muestra es necesario conocer los analitos que se le han de determinar, así como la fracción de éstos (fracción disuelta, suspendida, total o extraíble con ácido). Esta decisión determinará en parte si se acidulará con o sin filtración, así como el tipo de digestión requerido. Durante la toma y conservación de las muestras, pueden cometerse errores debido a: a) La contaminación del dispositivo de la toma de muestras b) La limpieza impropia del recipiente colector c) La pérdida de metales por adsorción y/o precipitación en el recipiente de la muestra como consecuencia de una inapropiada acidulación de la misma. d) Condiciones inapropiadas del transporte de la muestra. 1.2 Contenedores Los mejores recipientes para las muestras son los fabricados con cuarzo o TFE. Estos recipientes son de alto costo, por lo cual son más recomendables los de polipropileno o polietileno lineal. También pueden emplearse recipientes de vidrio de borosilicato, aunque es necesario evitar el empleo de recipientes de vidrio blando tratándose de muestras que contienen metales del orden de µg/L. Las muestras para la determinación de plata deben guardarse en recipientes que no absorban la luz, solo se deben utilizar recipientes y filtros que hayan sido enjuagados con ácido. 1.3 Conservación de la muestra La conservación de las muestras debe ser inmediatamente después de la toma, acidulando con HNO3 concentrado hasta pH<2, filtrándose antes de guardarse, en lo práctico se ha visto que es suficiente 1.5 mL de HNO3 concentrado por litro de muestra. 160 Para muestras cuya capacidad reguladora es elevada, es necesario aumentar la cantidad de ácido, siendo en ocasiones necesario emplear hasta 5 mL de ácido por litro de muestra. Dadas las pequeñas magnitudes de analito a determinar es necesario utilizar HNO3 comercial de pureza elevada o prepararlo en el laboratorio por destilación. Una vez acidulada la muestra debe ser conservada en refrigeración a 4ºC para evitar un cambio de volumen ocasionado por la evaporación, así como la alteración de ésta por reacciones secundarias. En estas condiciones las muestras con concentraciones de metal de varios µg/L se mantienen estables por periodos de hasta 6 meses (excepto el mercurio cuyo límite es de 5 semanas). Tratándose de niveles de concentración de metales del orden de µg/L es recomendable analizar las muestras inmediatamente después de tomarlas. Las muestras para el análisis de mercurio se deben conservar añadiendo 2mL/L de solución de K2Cr2O7 al 20% preparado en HNO3 1+1. 161 ANEXO 4 1. INTERFERENCIAS Las que a continuación se informan son de las interferencias más comunes. Aspiración directa Interferencias químicas. Son causadas por la pérdida de absorción por saltos cuánticos de átomos en combinaciones moleculares en la flama. Este fenómeno puede ocurrir cuando la flama no está lo suficientemente caliente para disociar la molécula. La adición de lantano o estroncio a blancos, muestra y estándares disminuye esta interferencia así como la flama NO2/C2H2 ayuda a la disociación efectiva de las moléculas. Interferencia de absorción no especifica (fondo): La absorción molecular y la dispersión de la luz causadas por partículas sólidas en la flama pueden causar errores positivos. Para evitar este problema se debe utilizar corrección de fondo. Estos sólidos además de presentar una barrera física al paso de la luz de la lámpara en la flama, forman depósitos en la cabeza del quemador, sin embargo esto se puede evitar aspirando continuamente agua acidulada. Interferencias de ionización: Ocurren cuando la temperatura de la flama es lo suficientemente alta para generar la remoción de un electrón de su átomo neutral, generándose un ion con carga positiva. Este tipo de interferencias pueden controlarse generalmente con la adición de elementos fácilmente ionizables tales como Na, K y Cs en blancos, muestras y estándares. Interferencia física: 162 Están relacionadas con las diferentes propiedades existentes entre las muestras y los estándares. Las cuales pueden afectar a la aspiración y eficiencia de nebulización en el sistema de atomización. Si las soluciones presentan diferencias de viscosidad y/o tensión superficial, la eficiencia de nebulización no será igual y los resultados analíticos se ven afectados. La presencia de otros compuestos además del elemento de interés pueden afectar a los resultados analíticos. Estas interferencias pueden ser corregidas utilizando el método de adición interna (adición de estándares). Para las interferencias específicas de cada elemento en el análisis de metales por flama se recomienda ver manual del fabricante. NOTA.- Para los elementos que presenten interferencias significativas en ciertas matrices se recomienda el uso de la técnica de adición de estándares. 1.1 GENERADOR DE HIDRUROS Las interferencias en generador de hidruros se presentan por presencia de otros elementos o moléculas presentes en la muestra. Los efectos se ven reflejados en una disminución de la cantidad de hidruro formado y por lo tanto en una disminución de la señal analítica. La forma de eliminar este tipo de interferencias es modificando la concentración del ácido y/o del borohidruro de sodio. Para las interferencias específicas de cada elemento en el análisis de metales por generador de hidruros se recomienda ver manual del fabricante. 163 ANEXO 5 1. SEGURIDAD Para el muestreo se necesita tener los cuidados que se establecen en la norma mexicana NMX-AA-003. No se ha determinado la carcinogenicidad de todos los reactivos con precisión, por lo que cada sustancia química debe tratarse como potencialmente peligrosa para la salud. La exposición a estas sustancias debe reducirse al menor nivel posible. Este método puede no mencionar todas las normas de seguridad asociadas con su uso. El laboratorio es responsable de mantener un ambiente de trabajo seguro y un archivo de las normas de seguridad respecto a la exposición y manejo seguro de las substancias químicas especificadas en éste método. Debe tenerse un archivo de referencia de las hojas de información de seguridad el cual debe estar disponible a todo el personal involucrado en estos análisis. El borohidruro de sodio es una sustancia tóxica, flamable y corrosiva. Se requiere el uso de una campana de extracción, ropa de protección, lentes de seguridad y mascarilla cuando se preparan las soluciones donde las reacciones entre el disolvente y el soluto son exotérmicas, esto es, óxido de lantano en solución ácida. Se requieren iguales precauciones cuando se diluyen, ácidos fuertes, debe evitarse el contacto con la piel y vías respiratorias. Se requiere de un sistema de ventilación permanente para eliminar una gran cantidad de gases calientes y algunas veces tóxicos producidos por el quemador durante la operación del instrumento. Como el acetileno es un gas flamable deberán tomarse las precauciones adecuadas cuando se use. Para evitar explosiones nunca pase el acetileno a través de instalaciones o tuberías de cobre o aleaciones con alto contenido de cobre (latón, bronce). Si el espectrofotómetro no esta equipado con un escudo protector, el operador deberá usar lentes de seguridad para atenuar la luz ultravioleta emitida por la flama. El óxido 164 nitroso es un gas que se usa como anestésico, por lo que el lugar debe estar bien ventilado. Los gases oxidantes deben separarse de los gases reductores mediante una pared a prueba de fuego. Seguir cuidadosamente las guías de operación del fabricante del equipo para optimizar la velocidad del flujo de gas. Si no se emplean las precauciones adecuadas, puede resultar una combustión peligrosa dentro de la cámara de mezcla de los gases. Para evitar explosiones en la línea, no permitir que la presión de llegada del acetileno al instrumento exceda 1,06 kg/cm2 (15 psi). Cuando se usa óxido nitroso como oxidante, debe utilizarse una cabeza de quemador de 50,8 mm de diámetro, ya que utilizando una cabeza de 101,6 mm (4-in) ocurre un regreso de la flama. La flama de óxido nitroso debe encenderse usando primero una combinación de aire-acetileno y luego cambiar a óxido nitroso-acetileno. El óxido nitroso nunca debe pasarse a través de líneas que contengan residuos de aceites o grasas, ya que puede causar una explosión. Revisar que el tubo del desagüe de la cámara de mezcla de gas esté lleno con agua antes de comenzar cualquier análisis. Se recomienda el uso de una trampa de seguridad o de cualquier válvula. Siga las instrucciones del fabricante para mantener una presión positiva en el sello del líquido. Dada la alta toxicidad del berilio, plomo, cadmio, níquel, mercurio, antimonio, plata, bario, cromo, así como todos los pasos de preparación y digestión de las muestras, extremar las precauciones de manejo de todas las disoluciones y utilizar un cuarto bien ventilado. El arsénico, el selenio y sus correspondientes hidruros son tóxicos. Manéjese con cuidado. Los compuestos del antimonio son irritantes para la piel y las membranas de las mucosas. 165 La inhalación de los vapores de manganeso han sido reportados como tóxicos para el ser humano. 2. MANEJO DE RESIDUOS Es la responsabilidad del laboratorio cumplir con todos los reglamentos federales, estatales y locales referentes al manejo de residuos, particularmente las reglas de identificación, almacenamiento y disposición de residuos peligrosos. Cada laboratorio debe contemplar dentro de su programa de control de calidad (CC) el destino final de los residuos generados durante la determinación. Confinamiento. El laboratorio debe contar con áreas especiales, que tengan señalamientos adecuados, para almacenar temporalmente las soluciones contaminadas con metales pesados. Los desechos ácidos deben neutralizarse para poder transportarlos a su disposición final. Todas las muestras que cumplan con la norma de descarga al alcantarillado pueden descargarse en el mismo. 166 ANEXO 6 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 6. BIBLIOGRAFIA 1. http://www.alimentacion-sana.com Mayo 20,2009 2. http://arturobola.tripod.com Mayo 20,2009 3. http://www.ceniap.gov. Mayo 20, 2009 4. http://www.buap.mx Mayo 19, 2009 5. http://www.euroresidentes.com Mayo 18, 2009 6. GBC 932/933 Atomic Absorption Spectrophotometers Operation manual. Edition July, 1993 7. http://www.ecoportal.net Mayo 20, 2009 8. http://www.fao.org Mayo 20, 2009 9. http://www.institucional.us.com Mayo 20,2009 10. http://www.monografias.com Mayo 19, 2009 11. NMX-AA-051-SCFI-2001 Análisis de Agua - Determinación de metales por absorción atómica en aguas naturales, potables, residuales y residuales tratadas método de prueba 12. Norma Oficial Mexicana NOM-001-ECOL-1996, Que establece los límites máximos permisibles de contaminantes en las descargas de aguas residuales en aguas y bienes nacionales. 184 13. Norma Oficial Mexicana NOM-002-ECOL-1996, que establece los límites máximos permisibles de contaminantes en las descargas de aguas residuales a los sistemas de alcantarillado urbano o municipal. 14. Norma Oficial Mexicana NOM-027-SSA1-1993, Bienes y servicios. productos de la pesca. Pescados frescos-refrigerados y congelados. especificaciones sanitarias. 15. Norma Oficial Mexicana NOM-041-SSA1-1993, Bienes y servicios. Agua purificada envasada. Especificaciones sanitarias 16. Norma Oficial Mexicana NOM-117-SSA1-1994, Bienes y servicios. método de prueba para la determinación de cadmio, arsénico, plomo, estaño, cobre, fierro, zinc y mercurio en alimentos, agua potable y agua purificada por espectrometría de absorción atómica. 17. Norma Oficial Mexicana NOM-127-SSA1-1994, "Salud ambiental, agua para uso y consumo humano-limites permisibles de calidad y tratamientos a que debe someterse el agua para su potabilización". 18. http://www.nutriologo.com.mx Mayo 20, 2009 19. http://propiedadesfrutas.jaimaalkauzar.es Mayo 20, 2009 20. Principles of Analytical Chemistry M. Valcarcel ED. Springer 2000 21. http://www.pronara.com.mx. Mayo 18, 2009 22. http://www.quiminet.com.mx Mayo 19, 2009 23. http://www.tlahui.com Mayo 19, 2009 185 24. http://www1.us.es.com Mayo 18, 2009 25. http://www.utafoundation.org Mayo 17,2009 26. http://www.xtec.net.com Mayo 20, 2009 27. http://es.wikipedia.org Mayo 20, 2009 186