Trastornos paroxísticos no epilépticos
Anuncio

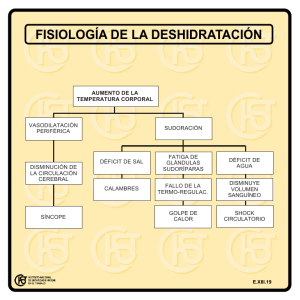
Capítulo 587 Trastornos paroxísticos no epilépticos & e587-1 Tabla 587-1 TRASTORNOS PAROXÍSTICOS NO EPILÉPTICOS SEGÚN LA EDAD DE PRESENTACIÓN EDAD Neonato Lactante Niño y adolescente PAROXISMOS GENERALIZADOS Apnea Hiperekplexia Temblor Paroxismos por dolor intenso Hiperekplexia Crisis anóxicas reflejas Espasmo del sollozo Vértigo paroxístico benigno Sobresaltos patológicos Paroxismos por dolor intenso Vértigo paroxístico benigno Sobresalto patológico Valsalva compulsivo Hemiplejía alternante de la infancia Migraña hemipléjica familiar Síncope (QT largo, vasovagal, vagovagal, ortostático, inducido por migraña) Crisis psicógenas Cataplejía Amnesia global transitoria Crisis de hiperventilación MOVIMIENTOS Y POSTURAS ANORMALES ANOMALÍAS OCULOMOTORAS Temblor Coreoatetosis paroxística distónica Paroxismo tónico de la mirada Hemiplejía alternante de la infancia Temblor Sandifer Coreoatetosis paroxística distónica Mioclonías del lactante Crisis de estremecimiento Tortícolis paroxística benigna Trastornos psicológicos Hemiplejía alternante de la infancia Jactatio capitis o movimientos estereotipados de la cabeza Reacciones medicamentosas Tics Temblor Discinesia paroxística Tortícolis paroxística benigna Ataxia episódica Trastornos psicológicos (síndrome de Munchausen por poderes, simulación) Masturbación Jactatio capitis o movimientos estereotipados de la cabeza Ataque de ira Reacciones medicamentosas Paroxismo tónico de la mirada Apraxia oculomotora Spasmus nutans o movimientos de cabeceo Síndrome de opsoclonus-mioclonus Ensimismamiento (soñar despierto) Reacciones medicamentosas TRASTORNOS DEL SUEÑO Mioclonías neonatales benignas durante el sueño Trastornos de la transición vigilia-sueño Trastornos por despertar incompleto en fase no REM Trastornos del sueño REM Narcolepsia Trastornos de la transición vigilia-sueño (sonambulismo, somniloquia) Trastornos por despertar incompleto en fase no REM Trastornos del sueño REM Narcolepsia Trastornos de la transición vigilia-sueño (sonambulismo, somniloquia) Mioclonus durante el sueño Síndrome de piernas inquietas De Obeid M, Mikati M: Expanding spectrum of paroxysmal events in children: potential mimickers of epilepsy, Pediatr Neurol 37(5):309-316, 2007. PAROXISMOS GENERALIZADOS Apnea Los episodios de apnea en el recién nacido generalmente se acompañan de bradicardia. Sin embargo, la apnea secundaria a una crisis epiléptica suele acompañarse de taquicardia. Puede haber excepciones, de manera que una apnea grave también puede producirse después de una crisis anóxica. © ELSEVIER. Fotocopiar sin autorización es un delito. Hiperekplexia y sobresaltos patológicos La hiperekplexia es un trastorno poco frecuente, esporádico o de herencia dominante, de comienzo neonatal, que se presenta como episodios aparentemente letales con rigidez tónica que precipitan una apnea y crisis convulsivas hipóxicas. La causa genética consiste en un defecto en las subunidades alfa o beta de los receptores sensibles a la estricnina-glicina. Se caracteriza por una tríada consistente en rigidez generalizada, mioclonías nocturnas y, posteriormente, un reflejo de sobresalto patológico. Se puede provocar un signo específico de diagnóstico tocando la nariz, lo que produce un reflejo de sobresalto inagotable con retracción de la cabeza. El baño, un despertar repentino y los estímulos táctiles o auditivos pueden inducir los ataques. El electroencefalograma (EEG) ictal muestra un artefacto muscular que puede confundirse con puntas. Este diagnóstico no se debe olvidar, ya que se puede producir una lesión cerebral hipóxica. La flexión repetida del cuello y de las caderas del niño (maniobra de Vigevano) puede abortar los episodios. Se puede utilizar clonazepam y, en ocasiones, otros antiepilépticos. En otros niños, después de una lesión cerebral, y en muchos pacientes con parálisis cerebral, puede observarse un reflejo de sobresalto exagerado. Esto es más frecuente que la hiperekplexia. En la enfermedad de Tay-Sachs y en otras gangliosidosis similares se produce un sobresalto exagerado al sonido, que ha sido interpretado, de manera inapropiada, como hiperacusia. continuación. A menudo los espasmos del sollozo pálidos se agravan por la anemia ferropénica. El segundo es el espasmo del sollozo cianótico. El término «espasmos del sollozo» es, en realidad, un nombre inapropiado, ya que no está relacionado con alteraciones mediadas por la voluntad o la conducta. La apnea espiratoria prolongada es la responsable de los episodios cianóticos, que resultan del cortocircuito intrapulmonar. Por otro lado, el reflejo cardíaco vagal de bradicardia es el responsable de los episodios pálidos. Un episodio comienza con un grito (a menudo un grito «silencioso» y una marcada palidez, en el caso del tipo pálido) y progresa a apnea y cianosis. Los espasmos del sollozo comienzan, por lo general, entre los 6 y 18 meses de edad. Evolutivamente pueden verse síncopes, posturas tónicas y crisis anóxicas reflejas, sobre todo en los espasmos del sollozo de tipo pálido. Los traumatismos, la ira y la frustración, sobre todo si son por sorpresa, son los desencadenantes más frecuentes. La educación y la tranquilidad de los padres es todo lo que se necesita, ya que estos episodios son, por regla general, autolimitados y desaparecen en pocos años. Sin embargo, es necesario administrar tratamiento si coexiste un déficit de hierro. Es importante la educación de los padres en primeros auxilios sobre cómo manejar los espasmos más graves. Los fármacos anticolinérgicos (p. ej., sulfato de atropina 0,01 mg/kg/24 horas en dosis divididas, con una dosis máxima diaria de 0,4 mg), la instrucción de los padres en maniobras de reanimación cardiopulmonar (RCP) básica, o el tratamiento con fármacos antiepilépticos para las crisis anóxicas recurrentes y que no responden a otras medidas pueden, en raras ocasiones, ser necesarios. Se debe enseñar a los padres a que no se obtenga una ganancia secundaria cuando se producen los episodios, ya que esto puede reforzarlos. Además, se debe preparar al niño para experiencias desagradables (como la recepción de un susto) en lugar de sorprenderle con ellas, lo que puede ayudar a limitar el número de espasmos. Valsalva compulsivo Espasmos del sollozo Este término se ha aplicado a dos tipos de espasmos. El primero es el espasmo del sollozo pálido, que es el reflejo vasovagal descrito a En los niños con retraso mental, incluido el síndrome de Rett, las maniobras de Valsalva pueden inducir convulsiones sincopales. En este caso, ocurren verdaderos espasmos del sollozo y, por lo general, e587-2 & Parte XXVII El sistema nervioso duran unos 10 segundos durante la inspiración. Algunos autores apoyan la utilización de naloxona en estos casos. Síncope vagal El síncope es una crisis anóxica que puede manifestarse como una caída súbita o como una crisis generalizada. Estas convulsiones, provocadas por una repentina falta de oxígeno al cerebro, son clínicamente similares y pueden ser erróneamente diagnosticadas como crisis generalizadas primarias. El síncope vasovagal (neurocardiogénico) es una de las causas que con mayor frecuencia se confunde con una convulsión tónico-clónica generalizada y habitualmente es provocado por el calor, la deshidratación, estar de pie durante mucho tiempo sin moverse, una ducha de agua caliente, la visión de la sangre, el dolor o el estrés repentino. La historia clínica suele ser la clave para distinguir entre el síncope y las crisis epilépticas. Inicialmente se produce palidez y sudoración seguidas de visión borrosa, sensación de mareo, náuseas, a las que sigue colapso gradual con pérdida de conciencia. Sin embargo, estos síntomas no siempre están presentes en un síncope. En el síncope vasovagal se puede observar incontinencia urinaria y alguna sacudida convulsiva, con una frecuencia del 10% y el 50% de los casos, respectivamente. También puede apreciarse una confusión postictal en algún caso. El dolor abdominal, un aura común en la epilepsia del lóbulo temporal, se produce en el síncope vasovagal, y puede ser un desencadenante o una consecuencia (crisis vagal intestinal). La mayoría de los niños con síncope vasovagal tiene un familiar de primer grado afectado. El EEG es normal y la prueba de la mesa basculante se ha utilizado con fines diagnósticos, aunque en la mayoría de los casos con una historia clínica típica no son necesarios. El vómito puede provocar un síncope vasovagal y progresar a una crisis convulsiva si la asistolia es lo suficientemente prolongada. La exposición repentina al frío en la cara o en el cuerpo también puede desencadenar un síncope vagal. El síncope también puede desencadenarse, aunque de forma rara, en relación con la tos, el trenzado de un cabello apretado, al peinarse, con la extensión forzada del cuello debido a la compresión de las vértebras, y con la flexión del cuello secundaria a un proceso estilomastoideo muy prolongado que produzca una compresión de la carótida. Las dos últimas situaciones requieren un estudio de neuroimagen (TC, RM) para un diagnóstico correcto. En la hipotensión ortostática y la intolerancia ortostática los síntomas se observan durante la bipedestación y pueden ser aliviados por el decúbito. El síndrome de taquicardia postural, cuya fisiopatología sigue siendo difícil de explicar, es una enfermedad de las adolescentes que se caracteriza por taquicardia e hipotensión con la postura de bipedestación. La insuficiencia autonómica primaria es rara en niños, y la disautonomía familiar es la única forma relativamente frecuente. La disautonomía familiar es una enfermedad común en los judíos Ashkenazi, y se caracteriza por la ausencia de secreción de lágrimas con las emociones, hiporreflexia rotuliana y ausencia de reacción de exacerbación tras la administración de histamina intradérmica. El déficit de dopamina b-hidroxilasa es una causa muy rara de insuficiencia autonómica primaria, y se caracteriza por la alteración de la eyaculación, ptosis, nicturia, paladar elevado, articulaciones hiperflexibles y unas complicaciones en el período neonatal (hipotensión, hipotonía, hipotermia). También puede observarse hipotensión en la insuficiencia suprarrenal. La prueba de la mesa basculante provoca una disminución de la tensión arterial y de la frecuencia cardíaca en pacientes con síncope vasovagal clásico. En la insuficiencia autonómica se aprecia una disminución de la tensión arterial con un cambio mínimo de la frecuencia cardíaca, y en el síndrome de taquicardia postural se observa una disminución de la tensión arterial y un aumento en la frecuencia cardíaca. El tratamiento del síncope consiste en evitar los factores precipitantes (mantener una hidratación adecuada, evitar permanecer quieto, ponerse de pie lentamente tras estar sentado, medidas de primeros auxilios, elevación de las piernas, medidas posturales) y el tratamiento de cualquier trastorno médico concomitante o subyacente que los pueda favorecer (anemia, insuficiencia suprarrenal, cardíaca, etc.). Además, los b-bloqueantes (p. ej., metoprolol a dosis inicial de 1-2 mg/kg una vez al día hasta un máximo de 6 mg/kg/día), o el tratamiento con fluorhidrocortisona (0,05-0,1 mg/día), pueden ser necesarios en algún caso seleccionado. Síncope cardíaco Los síndromes de QT largo (LQT) pueden causar síncopes de tipo «pálido» amenazantes para la vida. Acompañando a éstos pueden aparecer arritmias ventriculares, por lo general torsades de pointes o incluso fibrilación ventricular. Hay más de 10 tipos de síndromes de QT prolongado. Cuando se acompaña de sordera congénita forma parte del síndrome de Jervell y Lange-Nielson, de herencia autosómica recesiva (tipo 1, LQT 1, asociado con una mutación del canal del potasio KvLQT1). El síndrome de Romano-Ward es un síndrome autosómico dominante con penetrancia incompleta que se caracteriza por episodios de permanecer quieto como si el sujeto estuviese muerto, durante varios segundos, antes de producirse el episodio anóxico convulsivo (LQT 2 asociado con una mutación en el canal HERG del potasio). LQT 3 se asocia con una mutación en el canal del sodio SCN1A, el tipo 4 con una mutación en la proteína ankyrin, el tipo 5 (forma más leve) con la mutación KCNE1 minK, el tipo 6 con mutaciones en el gen del potasio KCNE2, el tipo 9 con mutaciones en la proteína caveolina relacionada con los canales del sodio y el tipo 10 con mutaciones SCN4B de los canales del sodio. Los tipos 7 y 8 son de especial interés debido a las manifestaciones clínicas y neurológicas asociadas. El tipo 7 (síndrome de Andersen-Tawil) está asociado con parálisis periódica, anomalías en el desarrollo del esqueleto, clinodactilia, orejas de implantación baja y micrognatia (mutaciones en el gen KCNJ2). El tipo 8 o síndrome de Timothy (con mutaciones en el gen del canal de calcio CACNA1c) se manifiesta con cardiopatía congénita, autismo, sindactilia e inmunodeficiencia. Se debe estudiar a todos los miembros de la familia de una persona afectada por un síndrome de LQT. Las personas afectadas necesitan la implantación de desfibriladores cardíacos, y a sus familias se les debe enseñar maniobras de RCP. Como regla general, a todos los niños con crisis de nueva aparición se les debería realizar un ECG para descartar el síndrome LQT, ya que puede enmascarar crisis comiciales. El síncope cardíaco es generalmente súbito, sin aparición gradual y presenta los síntomas que acompañan al síncope vagal. La estenosis aórtica puede causar un síncope repentino por ejercicio (por lo general hipertrófica) o directamente al final (por lo general valvular) y, si se sospecha, habría que realizar un ecocardiograma. Migraña hemipléjica familiar Es un tipo raro de migraña con un aura motora de debilidad. Los ataques comienzan a los 5-7 años de edad. En una persona genéticamente susceptible, los ataques se pueden precipitar por un traumatismo craneoencefálico, ejercicio físico o estrés emocional. Puede haber un déficit cerebeloso interictal (p. ej., nistagmus, ataxia). Los casos familiares y esporádicos tienen una prevalencia similar. Los 3 genes implicados en los tipos familiares son SCN1A (subunidad del canal del sodio neuronal) CACNA1A (subunidad del canal del calcio neuronal) y ATP1A2 (subunidad ATPasa del sodio y el potasio). Varios tipos de migraña hemipléjica familiar pueden presentar crisis concomitantes (p. ej., en asociación con encefalopatía mitocondrial con acidosis láctica y episodios de ictus [MELAS], epilepsia occipital y con la ataxia episódica [cap. 586.2]). Vértigo paroxístico benigno Se trata de un equivalente migrañoso común, que consiste en episodios de desequilibrio breve durante el cual el niño parece asustado. Pueden estar presentes nistagmo, sudoración, náuseas y vómitos. Los episodios remiten a los 5 años de edad. La RM cerebral y los EEG son normales, pero la prueba calórica, si se hace, puede mostrar una función vestibular anormal. Los pacientes pueden responder a la difenhidramina a la dosis de 5 mg/kg/día (máximo 300 mg/día) por vía oral, vía intramuscular, intravenosa o rectal. Síndrome de vómitos cíclicos Este síndrome es otra variante de migraña periódica que responde a los fármacos antimigrañosos o antiepilépticos. Éste y otros síndromes periódicos se han asociado con mutaciones que también pueden causar una migraña hemipléjica. Capítulo 587 Trastornos paroxísticos no epilépticos & e587-3 Síndrome de Alicia en el país de las maravillas Consiste en la experiencia episódica de distorsiones transitorias de la imagen corporal o de las imágenes visuales que, muy a menudo, constituyen un equivalente migrañoso. También puede ser un fenómeno epiléptico. Migraña inducida por síncope La migraña, por lo general la forma basilar, puede desencadenar un síncope vasovagal y, con menor frecuencia, crisis epilépticas. Una historia clínica cuidadosa del preludio migrañoso del síncope ayuda en la identificación de este fenómeno. Trastornos psicológicos Las crisis psicógenas no epilépticas son reacciones de conversión cuya sospecha clínica se basa, por lo general, en las características de los episodios (tabla 587-2). El diagnóstico se puede confirmar mediante vídeo-EEG con la captación de un episodio que permita eliminar cualquier duda residual acerca de su naturaleza, ya que suelen ocurrir en pacientes que también sufren crisis epilépticas. Las crisis psicógenas se asocian con menor frecuencia a un aumento de los niveles séricos de prolactina, 15-120 minutos después del evento, que las crisis epilépticas. Se manejan mejor en el momento agudo tanto por la certeza de su naturaleza relativamente benigna como por la realización de un tratamiento de soporte. Se necesita una evaluación psiquiátrica y un seguimiento para no enmascarar una potencial psicopatología subyacente, sobre todo en adolescentes y adultos, y para establecer un apoyo continuado puesto que las crisis psicógenas pueden persistir durante largos períodos de tiempo. La simulación y el síndrome de Munchausen por poderes son a menudo difíciles de diagnosticar, aunque en numerosas ocasiones puede resultar útil realizar un enfoque similar al de las crisis psicógenas, incluyendo un seguimiento con vídeo-EEG. MOVIMIENTOS Y POSTURAS ANÓMALOS Temblor y clonus neonatal El temblor consiste en movimientos simétricos de las extremidades hacia delante y hacia atrás, que se produce de forma espontánea o provocado por el tacto o sonidos fuertes. La supresión del movimiento tras la eliminación del estímulo o mediante la relajación de las extremidades afectadas, la ausencia de síntomas autonómicos y la clara diferencia de las dos fases (contracción rápida, relajación lenta) de actividad clónica y sacudidas mioclónicas muy rápidas indican un evento no epiléptico. Pueden ser posibles causas etiológicas la hipocalcemia, la hipoglucemia, la retirada de un fármaco y la encefalopatía hipóxico-isquémica. El clonus producido por una lesión del tracto corticoespinal se observa al final de la infancia o en la adolescencia, en estos casos puede suprimirse con el cambio de posición. Trastorno por dolor paroxístico intenso (previamente síndrome del dolor rectal familiar) Este síndrome (causado por la mutación del gen del canal de sodio SCN9A) por lo general comienza en el período neonatal o en la infancia y persiste durante toda la vida. En la mayoría de los pacientes al inicio predominan las manifestaciones autonómicas, con enrojecimiento de la piel en todos los pacientes y cambio de color arlequín y ataques tónicos. Es frecuente observar un síncope muy aparatoso con bradicardia y a veces con asistolia. Más tarde, el trastorno se caracteriza por ataques de dolor profundo insoportable tipo quemazón a menudo en la región ocular, rectal, o en áreas de la mandíbula, pero también de forma difusa en algunos casos. Los ataques son provocados por la defecación, el aire frío, el viento, la comida y las emociones. Se puede usar carbamazepina, pero la respuesta es a menudo incompleta. Tortícolis paroxística benigna de la infancia Este trastorno se presenta generalmente como episodios matutinos sin dolor y más tarde tortícolis, a menudo causada por los cambios de postura. Los ataques pueden comenzar con movimientos oculares anormales, para progresar a la permanencia en una postura anormal. Esto, por lo general, tiene una duración de minutos u horas, y a veces días. Los exámenes neurológicos, EEG y neuroimagen, entre los ataques, son normales. Afecta más a las niñas que a los niños (3:1), a menudo comienza antes de los 3 meses de edad, y remite de forma espontánea antes de cumplir los 5 años. El tratamiento médico no es Tabla 587-2 COMPARACIÓN ENTRE CRISIS GENERALIZADAS Y ALGUNOS TRASTORNOS QUE PUEDEN SIMULARLAS © ELSEVIER. Fotocopiar sin autorización es un delito. TRASTORNO PRECIPITANTES (NO APLICABLE A TODOS LOS PACIENTES) PRÓDROMOS Crisis generalizadas Privación de sueño, televisión, videojuegos, modelos visuales y estimulación lumínica Raramente irritabilidad o cambios de conducta inespecíficos Síncope: vasovagal Fatiga, estrés emocional, deshidratación Pequeños golpes en la cabeza, sustos Vómitos, deglución, derramar agua fría sobre la cara Visión borrosa, tinnitus, mareo Llanto en los espasmos del sollozo Síncope con crisis anóxicas reflejas Síncope: vagovagal (también trigémino-vagal) Síncope: ortostático Hiperekplexia Bipedestación, baño, despertar Estímulos auditivos y táctiles Ninguno Cardíaco Ejercicio físico Ninguno Psicógeno Sugestión, estrés Ninguno SÍNTOMAS ICTALES SÍNTOMAS POSTICTALES Habitualmente 2-3 minutos La conciencia puede estar conservada si se producen crisis atónicas o en algunas crisis tónicas Movimientos bilaterales sincrónicos Mordedura de lengua Pérdida de conciencia: segundos Aspecto pálido en la crisis anóxica refleja Retraso en la recuperación con un período postictal Rigidez tónica, cianosis si grave, inducidos por toques en la nariz, no agotable, sobresaltos Pérdida de conciencia: a menudo de sólo pocos segundos Palidez cutánea Comienzo gradual Movimientos asíncronos de agitación de las extremidades que varían entre los ataques No traumatismo, ojos cerrados Puede responder a la sugestión durante la «pérdida de conciencia» Habitualmente >2-3 minutos Dependiendo de la gravedad puede haber un período postictal De Obeid M, Mikati M: Expanding spectrum of paroxysmal events in children: potential mimickers of epilepsy, Pediatr Neurol 37(5):309-316, 2007. Recuperación rápida con ausencia de período postictal Raramente No período postictal e587-4 & Parte XXVII El sistema nervioso necesario. Se considera como un equivalente migrañoso y se puede observar asociado con migraña en algunas familias. Síndrome de Sandifer El reflujo gastroesofágico en los bebés puede causar episodios paroxísticos de rigidez generalizada y postura en opistótonos que puede acompañarse de apnea, mirada fija y sacudidas mínimas de extremidades. Los episodios suelen ocurrir 30 minutos después de la toma. Hemiplejía alternante de la infancia Consiste en ataques de hemiplejía flácida en uno o en ambos lados, con una duración de minutos a días, a partir de los primeros 18 meses de vida. Las manifestaciones más precoces incluyen nistagmo paroxístico que a menudo es monocular e ipsilateral a la hemiplejía. Con frecuencia se producen episodios distónicos y tónicos que pueden ser confundidos con convulsiones y la hemiplejía con la parálisis de Todd. Por lo general, el sueño anula los ataques y el tratamiento con flunarizina, 2,5-10 mg/día, los reduce. La mayoría de los niños en última instancia desarrolla ataxia, retraso del neurodesarrollo y coreoatetosis persistente. Discinesias paroxísticas y otros trastornos del movimiento Estos trastornos se caracterizan por ataques repentinos, que consisten en movimientos coreicos y distónicos, balísticos o mixtos (tabla 587-3). Una sensación de fatiga o debilidad limitada hacia un lado puede anunciar un ataque. La conciencia está preservada y los pacientes pueden realizar una actividad motora, como caminar, a pesar del ataque. La variabilidad en el patrón de la gravedad y la localización entre los distintos ataques también pueden ayudar a diferenciarlas de las convulsiones. La frecuencia de los ataques aumenta en la adolescencia y disminuye constantemente en la tercera década de la vida. El examen neurológico, entre los ataques, el EEG, las pruebas de laboratorio y los estudios de neuroimagen son normales. Estas discinesias a menudo responden a la fenitoína, carbamazepina, clonazepam, o a los antidopaminérgicos como el haloperidol. Las reacciones medicamentosas pueden dar lugar a movimientos anormales como la crisis oculógira con muchos antieméticos, coreoatetosis con la fenitoína, distonía y discinesias faciales con los fármacos antidopaminérgicos y los tics con la carbamazepina. Los accidentes cerebrovasculares, las lesiones focales cerebrales, los trastornos del tejido conjuntivo (p. ej., el lupus eritematoso sistémico), la vasculitis, o los trastornos metabólicos y genéticos también pueden causar trastornos del movimiento. Se han descrito mutaciones en los genes del transportador de glucosa 1 (GLUT1/SLC2AI) en pacientes con discinesia inducida por el ejercicio. Tics motores Los tics pueden ser voluntariamente controlados de forma parcial, se asocian con un impulso para realizarlos y un alivio posterior, generalmente se agravan con las emociones, y con frecuencia cambian de características a lo largo del tiempo. En los pacientes con tics que tienen el síndrome de Tourette, a menudo existe una historia familiar de tics y/o trastorno obsesivo compulsivo o rasgos de personalidad. Ataxias episódicas La ataxia episódica abarca 7 síndromes clínica y genéticamente heterogéneos, sólo dos de los cuales (los tipos 1 y 2) se han descrito en un gran número de familias. El tipo 1 está causado por mutaciones en los canales Kv1.1 de potasio dependientes de voltaje. Se caracteriza por breves episodios (de segundos a minutos de duración) de ataxia cerebelosa, y ocasionalmente crisis parciales con mioquimia interictal clínica o electrofisiológica como principal característica diagnóstica. El tipo 2 se caracteriza por ataques más largos (de minutos a horas de duración) y signos cerebelosos interictales. Está causado por mutaciones en el gen del canal del calcio voltaje-independiente CACNA1A. Mioclonías benignas neonatales, ataques de temblor y temblor mentoniano El mioclonus benigno consiste en sacudidas mioclónicas de las extremidades en estado de vigilia y, a veces, también durante el sueño. Se ha sugerido por algunos autores que estos ataques están en el mismo espectro que los ataques de temblor. Éstos se caracterizan por temblor rápido de la cabeza, los hombros y el tronco, de segundos de duración, a menudo asociados con la comida, que se repiten varias veces al día. Otros han considerado a estos ataques como una manifestación temprana del temblor esencial, puesto que a menudo presentan antecedentes familiares de temblor esencial. Los eventos clínicos de cualquiera de ellos pueden ser confundidos con los espasmos infantiles; sin embargo, los EEG ictales e interictales, la RM y el neurodesarrollo son normales. La remisión espontánea se produce, en general, al cabo de unos meses. También se ha documentado temblor del mentón hereditario, a 3 ciclos por segundo, precipitado por el estrés. Síndrome por autoanticuerpos contra el canal del potasio Este síndrome se asocia a menudo con cáncer y con deterioro neurocognitivo. Se puede manifestar con muchos trastornos del movimiento que simulan crisis epilépticas y responden a la inmunoterapia en lugar de a los fármacos antiepilépticos. Estos movimientos pueden ser coreoatetosis, distonía, mioclonías, cambios autonómicos y alucinaciones. También pueden presentar crisis epilépticas. Se ha documentado en niños mayores y es más frecuente en adultos. Tormenta autonómica (crisis diencefálica) Los episodios de hiperhidrosis, los cambios en la presión arterial, la temperatura y la inestabilidad autonómica se producen en pacientes con lesión cerebral difusa o lesión hipotalámica localizada y se han denominado tormentas autonómicas. El término «crisis diencefálicas» no se recomienda ya que no son verdaderas convulsiones. El tratamiento es difícil y ha incluido, con resultados mixtos, la clonidina, los antiepilépticos, la ciproheptadina, la morfina o la simpatectomía. Trastornos psicológicos Muchos trastornos psicológicos pueden confundirse con crisis epilépticas. Una conducta placentera similar a la masturbación puede aparecer desde la infancia, y puede consistir en movimiento de balanceo Tabla 587-3 CARACTERÍSTICAS CLÍNICAS DE ALGUNOS TRASTORNOS DEL MOVIMIENTO QUE PUEDEN SIMULAR CRISIS EPILÉPTICAS PRÓDROMOS Tortícolis paroxística benigna de la infancia Coreoatetosis paroxística distónica (discinesia de Mount y Reback) Coreoatetosis paroxística cinesiogénica (discinesia de Kertesz) Distonía paroxística inducida por ejercicio (discinesia de Lance) FACTORES PRECIPITANTES Palidez, irritabilidad, vómitos, ataxia Cambios en la postura Sensación de fatiga o debilidad limitada a un lado Mareo, hormigueo y entumecimiento, se puede observar en la discinesia de Kertesz Alcohol, café, té, chocolate, fatiga, hambre, estrés Movimientos abruptos o de sobresalto Ejercicio continuo De Obeid M, Mikati M: Expanding spectrum of paroxysmal events in children: potential mimickers of epilepsy, Pediatr Neurol 37(5):309–316, 2007. DURACIÓN Y FRECUENCIA DE LOS ATAQUES Pocos minutos, días, o raramente semanas 2 minutos hasta 6 horas 4/día hasta 1/mes <1 minuto 100/día hasta 1/mes 5-30 minutos 1/día hasta 2/mes Capítulo 587 Trastornos paroxísticos no epilépticos & e587-5 rítmico en una posición de sentado o acostado, o flexión y aducción rítmica de la cadera. La masturbación puede presentarse en niñas de 2-3 años y se asocia a menudo con la sudoración, respiración irregular y gruñidos, pero sin pérdida de conciencia. Ocasionalmente se asocia con abuso infantil o con otras psicopatologías. Las estereotipias o movimientos repetitivos que son más complejos que los tics y que no cambian no aparecen ni desaparecen como los tics (p. ej., pequeños golpes en la cabeza, movimientos de balanceo de la cabeza y del cuerpo y movimientos de aleteo de las manos), por lo general ocurren en niños con daño neurológico. Los ataques de pánico y ansiedad se han descrito, a veces, en niños y pueden ser clínicamente indistinguibles de las crisis epilépticas verdaderas, y por tanto requieren de una monitorización de vídeo-EEG. Los ataques de ira, por lo general, ocurren en pacientes con trastorno de la personalidad y no son convulsiones. Los episodios de hiperventilación pueden ser precipitados por ansiedad y se asocian con mareos, hormigueo y, a veces, espasmo carpopedal. La amnesia global transitoria consiste en la pérdida aislada de memoria a corto plazo de minutos a horas que se produce sobre todo en los ancianos. La etiología puede estar relacionada con epilepsia, trastornos vasculares, o con drogas. Ensimismamiento y mirada perdida ANOMALÍAS OCULOMOTORAS Las mioclonías neonatales durante el sueño consisten en movimientos repetitivos y rítmicos, por lo general bilaterales, con participación de los miembros superiores e inferiores durante el sueño no REM, a veces imitando crisis clónicas. Un balanceo lento (1 Hz) del niño en una dirección cefalocaudal es una prueba diagnóstica específica que reproduce el mioclonus. La falta de cambios autonómicos, la aparición sólo en el sueño y la supresión por despertares pueden ayudar a diferenciar estos eventos de las crisis epilépticas. La remisión es espontánea a los 2-3 meses de edad. En niños mayores y adultos, las mioclonías durante el sueño consisten en sacudidas mioclónicas al azar de las extremidades. Paroxismo tónico de la mirada vertical de la infancia Por lo general, comienza antes de los 3 meses de edad y consiste en ataques prolongados (horas o días) de desviación de la mirada continua o episódica hacia arriba, conservándose los movimientos oculares horizontales. Se puede observar un nistagmo con la fase rápida hacia abajo con la mirada hacia el suelo. Los síntomas disminuyen o se alivian con el sueño, se exacerban con la fatiga y las infecciones, y remiten espontáneamente después de unos años. Hasta un 50% de los pacientes puede tener retraso psicomotor y del lenguaje. Los estudios de laboratorio y de neuroimagen y los exámenes neuropsicológicos son, por lo general, inespecíficos. El tratamiento con dosis bajas de levodopa/carbidopa puede ser útil. Apraxia oculomotora e intrusiones sacádicas © ELSEVIER. Fotocopiar sin autorización es un delito. Se afectan los movimientos sacádicos de los ojos. La cabeza se gira de forma súbita para compensar el deterioro de la mirada lateral y simula convulsiones. Este trastorno puede ser idiopático (apraxia oculomotora de Cogan) o puede ocurrir en el contexto de la ataxiatelangiectasia o en enfermedades lisosomales. Se han implicado defectos genéticos en los mecanismos de reparación del ADN en, por lo menos, 4 trastornos de ataxia espinocerebelosa que están acompañados de apraxia oculomotora. Se cree que en estos trastornos ocurre una pérdida selectiva de células de Purkinje necesarias para inhibir a las neuronas que regulan los movimientos rápidos de los ojos e iniciar el movimiento sacádico del ojo. Las intrusiones sacádicas son movimientos conjugados de los ojos, involuntarios y repentinos, desde la posición ocular deseada, y que no son necesariamente patológicos. Spasmus nutans o movimientos de cabeceo Este trastorno se presenta con una tríada consistente en nistagmo, inclinación de la cabeza y movimientos de cabeceo de la misma. Si se produce una fluctuación diurna los síntomas pueden simular una crisis epiléptica. Se debe realizar una RM cerebral, puesto que la tríada se ha asociado con masas en el quiasma óptico y en el tercer ventrículo. También se debe descartar una retinopatía. La remisión se produce antes de los 5 años de edad. Síndrome opsoclonus mioclonus Los llamados «ojos danzantes» se refieren a los movimientos oculares continuos, al azar, irregulares y conjugados que pueden fluctuar en intensidad. Por lo general, se acompañan de mioclonías y ataxia («pies danzantes»). La encefalitis y el neuroblastoma son causas posibles. El tratamiento consiste en tratar la causa subyacente, pero la corticotropina (ACTH), los corticoides y el clonazepam pueden ser necesarios. El rituximab se ha estudiado y en ensayos preliminares se ha visto que puede ser también eficaz. La mirada perdida o mirada fija puede ser una manifestación de una crisis de ausencia, aunque debe diferenciarse de la actitud de soñar despierto o ensimismamiento, del comportamiento de mirada fija por fatiga o de una falta de atención. Los episodios de mirada fija sólo en ciertos ambientes (p. ej., el colegio) es poco probable que sean una convulsión. Además, la sensibilidad al tacto y la falta de interrupción del juego caracterizan los episodios de mirada fija no epiléptica. TRASTORNOS DEL SUEÑO Los eventos paroxísticos no epilépticos relacionados con el sueño son más frecuentes en los pacientes epilépticos que en la población general, lo que hace que su diagnóstico sea difícil. La semiología, el calendario de eventos, el vídeo-EEG y la polisomnografía ayudan a distinguir entre los eventos epilépticos y los no epilépticos. Las parasomnias ocurren típicamente menos de una o dos veces por la noche, pero si los episodios son más frecuentes sugieren crisis epilépticas. Mioclonus benigno durante el sueño y mioclonías neonatales durante el sueño Trastornos por despertar incompleto en la fase no REM Los despertares confusionales nocturnos breves que se producen 1-2 horas después del inicio del sueño, en la fase 4 del sueño, son normales en los niños. Estos episodios pueden variar desde la masticación, la sedestación y la murmuración hasta el sonambulismo agitado, y suelen durar 10-15 minutos. Los terrores nocturnos también ocurren pocas horas después del inicio del sueño, en la fase 3 o 4 del mismo, la mayoría entre los 2-7 años de edad y, sobre todo, en varones. El niño grita, parece aterrorizado, tiene las pupilas dilatadas, taquicardia, taquipnea, falta de respuesta, agitación y actitud furiosa que se incrementan al intentar consolarle, es difícil de despertar y puede tener alguna o ninguna vocalización. En los niños mayores con terrores nocturnos puede haber una etiología psicológica subyacente. El diagnóstico se basa en la historia clínica. Sin embargo, en raras ocasiones, se necesita una monitorización con vídeo-EEG. A veces, el uso de diazepam antes de acostarse (0,2-0,3 mg/kg) o clonazepam (0,01 mg/kg) puede ayudar a controlar el problema mientras que se investigan los factores psicológicos. El síndrome de piernas inquietas puede provocar disestesias dolorosas en las piernas que causan despertares nocturnos e insomnio. Puede ser de etiología genética o asociado con la deficiencia de hierro, enfermedad sistémica, o con algunas drogas. El tratamiento se basa en actuar sobre la causa subyacente y, si es necesario, en el uso de fármacos dopaminérgicos como la levodopa/carbidopa, o antiepilépticos como la gabapentina. Trastornos del sueño REM Las pesadillas y la parálisis del sueño son trastornos frecuentes. A diferencia de los terrores nocturnos, las pesadillas tienden a ocurrir más tarde durante la noche y el niño recuerda el episodio. Trastornos de la transición del sueño Los movimientos estereotipados de la cabeza (jactatio capitis) durante la noche y los movimientos de balanceo de la cabeza y del cuerpo ocurren a menudo en lactantes y en niños pequeños mientras tratan de conciliar el sueño. Generalmente remiten de forma espontánea a la edad de 5 años. No precisan un tratamiento específico. e587-6 & Parte XXVII El sistema nervioso Síndrome de narcolepsia-cataplejía BIBLIOGRAFÍA La narcolepsia se caracteriza por somnolencia diurna excesiva, cataplejía (pérdida súbita del tono muscular), parálisis del sueño, alucinaciones hipnagógicas y trastornos del sueño durante la noche. La pérdida del tono se produce en respuesta a las emociones fuertes y progresa de forma cefalocaudal. En la cataplejía no hay afectación del nivel de conciencia. La pérdida selectiva de las neuronas secretoras de hipocretina en el hipotálamo da lugar a este trastorno. El hecho de que DQB1*0602 sea un alelo HLA predisponente identificado en el 85-95% de los pacientes con narcolepsia-cataplejía sugiere que la pérdida neuronal se produzca por un trastorno autoinmune. El diagnóstico se basa en la prueba de latencias múltiples del sueño y el tratamiento consiste en siestas programadas, anfetaminas, metilfenidato, antidepresivos tricíclicos y el asesoramiento acerca de las precauciones en el trabajo y la conducción. Bakker MJ, van Dijk JG, van den Maagdenberg AM, et al: Startle syndromes, Lancet Neurol 5:513-524, 2006. Crompton DE, Berkovic SF: The borderland of epilepsy: clinical and molecular features of phenomena that mimic epileptic seizures, Lancet Neurol 8(4):370-381, 2009. Dauvilliers Y, Arnulf I, Mignot E: Narcolepsy with cataplexy, Lancet 369:499-511, 2007. DiMario FJ Jr: Paroxysmal nonepileptic events of childhood, Semin Pediatr Neurol 13:208-221, 2006. Fertleman CR, Ferrie CD, Aicardi J, et al: Paroxysmal extreme pain disorder (previously familial rectal pain syndrome), Neurology 69: 586-595, 2007. Tan KM, Lennon VA, Klein CJ, et al: Clinical spectrum of voltage-gated potassium channel autoimmunity, Neurology 70:1883-1890, 2008. Webster G, Berul CI: Congenital long-QT syndromes: a clinical and genetic update from infancy through adulthood, Trends Cardiovasc Med 18:216-224, 2008.