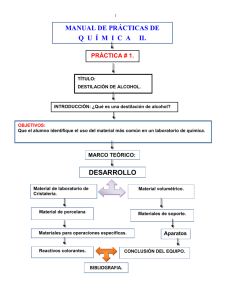
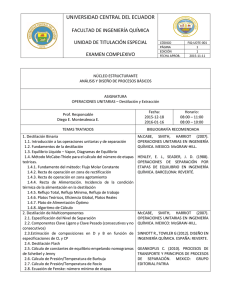
UNIVERSIDAD VERACRUZANA
Anuncio

UNIVERSIDAD VERACRUZANA FACULTAD DE CIENCIAS QUIMICAS “OPTIMIZACIÓN DE COSTOS PARA LA SEPARACIÓN DE UNA MEZCLA AZEOTRÓPICA MEDIANTE SIMULACIÓN CON ASPEN PLUS” TESIS: PARA APROBAR EL EXAMEN DEMOSTRATIVO DE LA EXPERIENCIA RECEPCIONAL DEL PROGRAMA DE INGENIERÍA QUÍMICA PRESENTA: NANCY AURIE MENDIOLA VICENCIO DIRECTOR: M.C. CARLOS ANTONIO MÁRQUEZ VERA ASESOR: DR. SERGIO NATAN GÓNZALEZ ROCHA POZA RICA, VER. DICIEMBRE 2013 CONTENIDO CONTENIDO PÁGINAS RESUMEN ............................................................................................................ I INTRODUCCIÓN .................................................................................................. II JUSTIFICACIÓN ................................................................................................. IV OBJETIVO GENERAL ......................................................................................... VI OBJETIVOS ESPECIFICOS ....................................................................................VI HIPÓTESIS ........................................................................................................ VII CAPITULO I: MARCO TEÓRICO I.1. Generalidades ................................................................................................ 1 I.2 Mezclas ideales ............................................................................................... 2 I.2.1 Ley de Raoult .................................................................................................... 2 I.2.2 Mezclas Azeotrópicas........................................................................................ 3 I.3 Separación de Mezclas ..................................................................................... 4 I.4 Operaciones de Separación ............................................................................. 6 I.4.1 Destilación Extractiva ........................................................................................ 6 I.4.1.1. Aplicaciones industriales ............................................................................ 9 I.4.2 Extracción líquido-líquido ................................................................................ 10 I.4.2.1 Aplicaciones de utilidad............................................................................. 11 I.5 Selección de mezcla azeotrópica ..................................................................... 14 I.6 Simulación en Aspen Plus ............................................................................... 17 I.7 Estimación de Costos de inversión del Proceso ................................................ 18 I.7.1 Método de Guthrie ........................................................................................... 19 I.7.2 Procedimiento ................................................................................................. 20 CAPÍTULO II: METODOLOGÍA II.1 Selección y simulación de la mezcla azeotrópica ............................................. 21 II.2 Diseño del diagrama y simulación de la separación método no convencional ..... 24 II.3 Diseño del diagrama y simulación de la separación método convencional .......... 32 CONTENIDO CAPITULO III: RESULTADOS III.1 Resultados método de destilación extractiva primera columna en el proceso ..... 41 III.1.1 Resultados en la segunda columna en el proceso de destilación extractiva .. 46 III.1.2 Resultados en la tercera columna en el proceso de destilación extractiva ..... 48 III.1.3 Cantidad de agua necesaria para reducir la temperatura del disolvente ........ 50 III.2 Resultados en el método de la extracción L-L columna RadFrac ...................... 52 III.2.1 Cantidad de agua necesaria para reducir la temperatura del disolvente ........ 54 III.2.2 Cantidad de disolvente para en la extracción L-L .......................................... 55 III.3 Optimización de costos del proceso. .............................................................. 55 III.3.1 Optimización de costos del disolvente .......................................................... 56 III.3.2 Optimización de costos de equipo. ................................................................ 57 III.4 Comparación de métodos en costos generales ............................................... 59 III.4.1 Comparación en la eficiencia de los métodos ................................................ 59 III.5 Costos del material utilizando método de Guthrie en la destilación extractiva ..... 60 III.5.1 Especificaciones para la primera columna ..................................................... 61 III.5.1.1 Determinación de costos del material para la primera columna .............. 62 III.5.2 Especificaciones para la segunda columna ................................................... 64 III.5.2.1 Determinación de costos del material para la segunda columna ............. 65 III.5.3 Especificaciones para la tercera columna ...................................................... 66 III.5.3.1 Determinación de costos del material para la tercera columna ............... 67 III.6 Costos del material utilizando método de Guthrie en la extracción L- L .............. 69 III.6.1 Especificaciones para la única columna RadFrac del proceso ...................... 69 III.6.1.1 Determinación de costos material única columna RadFrac del proceso .. 70 III.6.2 Especificaciones para la columna de extracción L-L...................................... 72 III.6.2.1 Determinación del costo del material para la columna de extracción L-L 73 III.6.3 Comparación en los costos de operación y material adquiridos .................... 73 CONCLUSIONES Y RECOMENDACIONES ......................................................... 75 REFERENCIA BIBLIOGRAFICA .......................................................................... 76 ANEXOS............................................................................................................ 78 CONTENIDO INDICE DE FIGURAS I.1 Esquema clásico del proceso de destilacion extractiva para separacion de un sistema binario............................................................................................................ 76 I.2 Ejemplo de una destilación extractiva ................................................................... 10 II.1 Datos de equilibrio para la mezcla etanol-agua ..................................................... 21 II.2 Datos de equilibrio para la mezcla etanol-agua predecidos por NRTL .................. 22 II.3 Equipo de separacion Flash2 ................................................................................ 22 II.4 Condiciones en la corriente de alimentación ......................................................... 23 II.5 Acceso al tipo de análisis deseado con los datos de equilibrio.............................. 23 II.6 Diagrama de proceso para la serparacón de la mezcla azeotrópica etanol-agua por medio del método de destilación extractiva ................................................................ 25 II.7 Condiciones en la corriente de alimentación de la mezcla azeotrópica ................. 26 II.8 Condiciones en la corriente de alimentación del disolvente en destilación ext. ..... 26 II.9 Configuración de operación para la primera columna .......................................... 27 II.10 Configuración de operación para la segunda columna ........................................ 27 II.11 Etapa de alimentación en las corrientes de alimentación .................................... 28 II.12 Configuración de operación para la columna de recuperación del disolvente ..... 28 II.13 Configuración en el condensador de la tercera columna ..................................... 28 II.14 Condiciones de operación para el intercambiador de calor en destilación ext ..... 29 II.15 Condiciones iniciales en el divisor para el método de destilación extractiva ........ 29 II.16 Simulación de la separación de la mezcla azeotrópica sin reflujo agregado ....... 30 II.17 Configuración final del divisor ............................................................................. 30 II.18 Configuración final en la corriente de alimentación de disolvente ....................... 31 II.19 Simulación final con cantidad de disolvente fijo en la corriente de desahogo y en la retroalimentación por el método de destilación extractiva ....................................... 31 II.20 Curva de equilibrio obtenida por medio de las ecuaciones de estado PENG-ROB, WILSON, VANLAAR, NRTL y UNIQUAC .................................................................... 33 II.21 Curva de equilibrio predicha por la ecuación CHAO-SEA ................................... 33 II.22 Curva de equilibrio predicha por la ecuación UNIFAC ........................................ 33 CONTENIDO II.23 Diseño del diagrama para la separación de la mezcla azeotrópica por el método convencional .............................................................................................................. 34 II.24 Selección del ortoxileno como disolvente ............................................................ 34 II.25 Configuración de alimentación para la corriente de la mezcla ............................. 35 II.26 Configuración de alimentación para la corriente del disolvente ........................... 35 II.27 Número de etapas en la columna Extract ........................................................... 36 II.28 Selección del refinado y el extracto como productos en la extracción ................. 36 II.29 Número de etapas para las corrientes de alimentación y de los productos ......... 36 II.30 Configuración en la rectificación del método de extracción L-L ........................... 37 II.31 Etapa de alimentación para el extracto ............................................................... 37 II.32 Presión de operación en el condensador de la columna RadFrac en el método de la extracción L-L ......................................................................................................... 37 II.33 Configuración inicial del divisor en el método de la extracción L- L ..................... 38 II.34 Condiciones de operación para el intercambiador de calor en extracción L-L .... 38 II.35 Simulación sin reflujo de disolvente método de la extracción L-L ....................... 39 II.36 Configuración final del divisor ............................................................................. 39 II.37 Configuración final en la corriente de reposición del disolvente extracción L-L ... 40 II.38 Simulación final por el método de la extracción L-L ............................................ 40 III.1 Menú para hacer visibles las cargas térmicas en las simulaciones ...................... 41 III.2 Primera columna en el proceso de destilación extractiva ..................................... 42 III.3 Estados de la materia: Temperatura vs Entalpia .................................................. 43 III.4 Condiciones de alimentación en el Flash2 para conocer las entalpias ................. 44 III.5 Condiciones de operación para obtener un líquido saturado ................................ 44 III.6 Condiciones de operación para obtener un vapor saturado ................................. 44 III.7 Diagrama de interpretación de transferencia de energía en el condensador de la primera columna del método de destilación extractiva ................................................ 45 III.8 Segunda columna en el proceso de destilación extractiva ................................... 46 III.9 Diagrama de interpretación de transferencia de energía en el condensador de la segunda columna del método de destilación extractiva .............................................. 48 III.10 Tercera columna en el proceso de destilación extractiva ................................... 49 CONTENIDO III.11 Diagrama de interpretación de transferencia de energía en el condensador de la tercera columna del método de destilación extractiva ................................................. 50 III.12 Intercambiador de calor requerido en el proceso ............................................... 50 III.13 Diagrama de interpretación de transferencia de energía en el intercambiador de calor del método de destilación extractiva................................................................... 51 III.14 Única columna RadFrac en el método de extracción L-L ................................... 52 III.15 Diagrama de interpretación de transferencia de energía en el condensador de la columna del método extracción L-L ............................................................................ 53 III.16 Intercambiador de calor en el método de la extacción L-L ................................. 54 III.17 Diagrama de interpretación de transferencia de energía en el intercambiador de calor del método de la extracción L-L ......................................................................... 55 INDICE DE TABLAS I.1 Lista de algunos ejemplos de mezclas azeotrópicas con su respectivo disolvente para la separación de la misma por el método de destilación extractiva ..................... 16 I.2 Nivel de presición, el cual puede variar desde un 40% para una estimación preliminar y hasta un 3% para una estimación detallada ............................................ 18 I.3 Factores de módulos ............................................................................................ 20 III.1 Resultados predichos por Aspen en el Liquído saturado...................................... 43 III.2 Condiciones en la primera columna del método de destilación extractiva ............ 45 III.3 Condiciones en la segunda columna del método de destilación extractiva........... 47 III.4 Condiciones en la tercera columna del método de destilación extractiva ............. 49 III.5 Condiciones del disolvente en el Intercambiador de calordestilación ext. ............ 51 III.6 Condiciones en la única columna del método extacción L-L ............................... 53 III.7 Condiciones del disolvente en el Intercambiador de calor .................................... 54 III.8 Optimización de costos de disolvente mediante ................................................... 57 III.9 Optimización de costos de equipo a partir de la modificación del S y la R .......... 58 III.10 Comparación de las condiciones del etanol como producto principal ................ 60 III.11 Costo total de operación en los métodos del proceso ........................................ 74 III.12 Costo de material adquirido en cada método del proceso .................................. 74 RESUMEN RESUMEN La separación de las sustancias en una mezcla es un importante proceso para los que estudian el área de la química y en muchas industrias. La comparación de separación entre métodos convencionales y no convencionales mediante la simulación ASPEN PLUS, es una práctica por la cual se puede determinar, el proceso favorable para una mezcla. Como se presenta en el caso de la mezcla azeotrópica etanol-agua la cual requiere ser separada para la obtención de etanol con una pureza de un 99% en peso de alcohol, debido a que la principal fuente de energía que dispone la humanidad son los combustibles fósiles, en especial el petróleo y carbón, cuyas reservas se están agotando rápidamente. Cuando el etanol se encuentra a elevadas purezas, puede ser utilizado como aditivo para la gasolina, ganando explosividad y reduciendo importantemente las emisiones contaminantes en la combustión. La mezcla etanol-agua es sometida a diversas simulaciones con el fin de obtener su separación y conseguir un alcohol con purezas muy elevadas. El primer método de separación, es el método de la destilación extractiva el cual consta de tres columnas de rectificación, utilizando al etilenglicol como disolvente ya que si se utiliza un disolvente más caro, este método podría dejar de ser el más indicado, por su alta cantidad de disolvente retroalimentado. El segundo método empleado es el de la extracción L-L, la cual consta de una columna de rectificación y una columna de extracción, utilizando al ortoxileno como disolvente. I INTRODUCCIÓN INTRODUCCIÓN La separación de mezclas azeotrópicas es una tarea muy usual en la industria química; en ella nos encontramos con una inmensa variedad de mezclas, y todas ellas han sido clasificadas por el diferente comportamiento de sus componentes presentes. Uno de los procesos más importantes e indispensables de la ingeniería química es la separación de componentes mezclados, y actualmente existen métodos convencionales de separación para todo tipo de mezclas, como la destilación que es la operación de separar, comúnmente mediante calor, los diferentes componentes líquidos de una mezcla, aprovechando los diferentes puntos de ebullición (temperaturas de ebullición) de cada una de las sustancias a separar o la extracción líquido-líquido que consiste en la separación de los componentes de una mezcla líquida, por contacto con otro líquido, inmiscible con ella o parcialmente inmiscible y que disuelve preferentemente a uno de los constituyentes. Pero la separación de componentes que tienen aproximadamente las mismas temperaturas de ebullición es difícil de alcanzar su separación por destilación simple aun cuando las mezclas sean ideales, y una separación completa a menudo resulta imposible debido a la formación de azeotrópos. En los casos de las mezclas azeotrópicas suelen utilizarse métodos no convencionales que son más complejos y por lo tanto en ocasiones más costosos, como una destilación extractiva. Una modalidad de destilación es la llamada destilación extractiva la cual se trata de un método de rectificación de multicomponentes. A una mezcla binaria que es difícil o imposible de separar por los métodos ordinarios, se le agrega un tercer componente, un agente másico de separación (AMS), conocido como disolvente, el cual altera la volatilidad relativa de los componentes originales II INTRODUCCIÓN y permite, de esa forma, la separación. Este disolvente agregado es de baja volatilidad, no evaporándose de modo apreciable durante todo el proceso. Una aplicación de la destilación extractiva es la producción de etanol, para el cual el desarrollo de tecnologías que permitan la producción por diferentes métodos de deshidratación y consigan la obtención de altos niveles de pureza, convierte a este como uno de los compuestos más atractivos ya que proviene de una materia prima biológica renovable, no es corrosivo, ni tóxico y, además, mejora el desempeño de las gasolinas. El principal problema de la producción de bioetanol anhidro es el alto costo energético que conlleva su separación, ya que durante la etapa de fermentación se obtienen grandes cantidades de caldo de fermentación con bajas concentraciones de alcohol (entre 5-12 % en peso) por lo que es necesario eliminar el exceso de agua (Quintero et al. 2007). A través de los métodos convencionales de destilación es posible obtener etanol con concentraciones cercanas a la composición azeotrópica (95.57 % en peso); sin embargo, para que el etanol pueda ser mezclado con la gasolina es necesario que su contenido de agua sea muy bajo cercano al 99 % en peso de etanol, con el fin de evitar formación de 2 fases líquidas en la mezcla (Quintero et al. 2007). Debido a que los procesos de separación antes mencionados son muchas veces complejos, se recomienda el empleo de herramientas computacionales especializados en el área, en la cual se encuentra Aspen Plus, que es una herramienta de proceso líder en el mercado de modelado para el diseño conceptual, la optimización y supervisión del rendimiento para la industria química, polímeros, productos químicos especiales, metales y minerales, y las industrias de carbón. Aspen Plus es un elemento central de aspenONE AspenTech ® aplicaciones de ingeniería. Ahora que se conocen las herramientas y los diferentes métodos, se realizará la simulación de la separación de una mezcla azeotrópica industrial comparando los métodos convencionales y no convencionales, debido a que aún no se ha encontrado un método convencional lo suficientemente bueno para dicha separación. III JUSTIFICACIÓN JUSTIFICACIÓN La principal fuente energética que dispone la humanidad son los combustibles fósiles, en especial el petróleo y carbón, cuyas reservas se están agotando rápidamente. La utilización de estos combustibles contamina el ambiente con gases como el dióxido de carbono, óxidos de nitrógeno, los óxidos de azufre (SOx) y material particulado (hollín), entre otros (Bobadilla, 1997). Con la finalidad de encontrar fuentes alternativas se ha encontrado que el bioetanol es un compuesto químico atractivo (Quintero et al. 2007), el cual se produce por la fermentación de los azúcares contenidos en la materia orgánica de las plantas. En este proceso se obtiene el alcohol hidratado, con un contenido aproximado del 88 % al 95 % en peso de agua, que tras ser deshidratado se puede utilizar como combustible. El bioetanol mezclado con la gasolina produce un biocombustible de alto poder energético con características muy similares a la gasolina pero con una muy importante reducción de emisiones contaminantes en los motores tradicionales de combustión (Becerra, 2008), por lo tanto se requiere la separación de dicha mezcla. La separación de las sustancias en una mezcla es importante para los que estudian el área de la química y en muchas industrias, dado que la mayor parte de los materiales, son obtenidos de productos naturales o preparados en el laboratorio, y la mayoría son mezclas de sustancias, por lo tanto en el diseño de columnas de destilación para la separación de mezclas no ideales, métodos como McCabe Thiele o Ponchon Savarit, se vuelven obsoletos. Los procesos de separación simples usados en el laboratorio son los mismos que los de las industrias. IV JUSTIFICACIÓN Cada uno de ellos tiene una enorme importancia práctica por lo que su análisis riguroso permite la propuesta de nuevas tecnologías para lograr la separación de mezclas no ideales. Hay que hacer mención especial al proceso de destilación, muy empleado en la industria química por su efectividad y versatilidad para combinarse con otras operaciones y llevar acabo separaciones más complejas. Debido a que los cálculos rigurosos para los procesos de separación no ideal son muy extensos y complejos, el uso de Aspen Plus se convierte en una herramienta ideal para el tipo de proceso ya que este permite el diseño conceptual, la optimización mediante la aplicación de la Ingeniería Económica y supervisión del rendimiento para la industria química, polímeros, productos químicos especiales, metales y minerales, y las industrias de carbón. El presente trabajo se propone determinar la separación de la mezcla no ideal etanol-agua. Para esto se hace necesario evaluar la posibilidad de elegir un nuevo disolvente de extracción que tenga un mayor grado de inmiscibilidad con el etanol que el agua, que no forme mezcla azeotrópica heterogénea con él y que, además, tenga una mayor temperatura de ebullición. Este nuevo agente extractivo debe disminuir su consumo global en la etapa de extracción y facilitar las separación del etanol en las posteriores etapas de destilación, con la consiguiente optimización de los costos del proceso. V OBJETIVOS OBJETIVO GENERAL Determinar la configuración y condiciones de proceso óptimas para la separación de la mezcla azeotrópica etanol-agua empleando métodos convencionales y no convencionales mediante la Simulación con Aspen Plus OBJETIVOS ESPECIFICOS Seleccionar una mezcla azeotrópica en la cual uno de los componentes sea útil, rentable y óptimo (etanol-agua). Comparar el equilibrio de bases de fases de la mezcla azeotrópica mediante los resultados obtenidos por Aspen Plus Realizar la Simulación Aspen Plus utilizando el método de separación Destilación Extractiva Reducir los consumos energéticos de equipo y solventes procurando no modificar la eficiencia del proceso en la destilación extractiva Realizar la Simulación Aspen Plus utilizando el método de separación Extracción L-L Reducir los consumos energéticos de equipo y solventes procurando no modificar la eficiencia del proceso en la Extracción L-L Determinar los costos de servicio, de materias primas y solventes Comparar los costos de operación y equipos entre la destilación extractiva y la extracción L-L VI HIPÓTESIS HIPÓTESIS Mediante la aplicación de métodos convencionales y no convencionales asociados a un proceso de simulación de tipo computacional Aspen Plus, es posible determinar el análisis y la optimización de costos de un proceso de separación. VII I MARCO TEÓRICO CAPITULO I MARCO TEÓRICO I.1. Generalidades La principal fuente energética que dispone la humanidad son los combustibles fósiles, en especial el petróleo y carbón, cuyas reservas se están agotando rápidamente (Cassedy, 1990). A nivel mundial, aproximadamente 90 % de la energía consumida proviene de fuentes no renovables, por lo que estos recursos fósiles se están agotando aceleradamente y su tasa de disminución es cada vez mayor. Por lo anterior, desde hace algunos años, distintas naciones han incursionado en la búsqueda de fuentes alternas de energía (Becerra, 2008). Dentro de las fuentes alternativas para la producción de energía se ha encontrado que el etanol presenta características favorables, el etanol (alcohol etílico, alcohol de grano) es un líquido claro, incoloro con un olor característico y agradable. En una disolución acuosa, tiene un sabor algo dulce, pero en soluciones más concentradas tiene un sabor ardiente. El etanol, CH3-CH2-OH, es un alcohol, un grupo de compuestos químicos cuyas moléculas contienen un agrupo hidroxilo. – OH, en condiciones de enlace a un átomo de carbono. El etanol se solidifica a 114.1°C, hierve a 78.5°C, y tiene una densidad de 0.789 g/ml a 20°C (Shakhashiri, 2009). El etanol es una droga psicoactiva y una de las más antiguas drogas recreativas. Mejor conocido como el tipo de alcohol encontrado en bebidas alcohólicas, también se utiliza en termómetros, como un disolvente y como combustible. La obtención del etanol se debe a la fermentación de azucares, en este proceso se obtiene un alcohol diluido en agua. El agua como compuesto químico habitualmente se piensa que es el agua natural que conocemos es un compuesto químico de fórmula H2O, pero no es así, debido a su gran capacidad como disolvente toda el agua que se encuentra en la naturaleza contiene diferentes cantidades de diversas sustancias en solución y hasta en suspensión, lo que corresponde a una mezcla. El agua es, quizá el compuesto químico más 1 I MARCO TEÓRICO importante en las actividades del hombre y también más versátil, ya que como reactivo químico funciona como ácido, alcalino, agente oxidante y agente reductor; siendo a su vez el disolvente universal. I.2 Mezclas ideales Una mezcla es una combinación de dos o más sustancias en la cual las sustancias conservan sus propiedades características. Las mezclas pueden ser homogéneas (la composición de la mezcla es la misma en toda la solución) o heterogéneas (su composición no es uniforme). Cualquier mezcla ya sea homogénea o heterogénea, se puede formar y separar en sus componentes puros por medios físicos sin cambiar la identidad de dichos componentes. Después de la separación no habrá ocurrido cambio alguno en las propiedades y composición de los componentes de la mezcla (Chang 1999). Por lo tanto, se llama mezcla ideal a aquélla en la que no hay interacción entre sus partes y por tanto cualquier magnitud conservativa y aditiva será igual a la media ponderada de los valores que corresponden a cada especie en su estado estándar de referencia en esas condiciones de P y T (para gases el estado de gas ideal puro, para disolventes el de su fase pura, y para solutos el de dilución infinita), aunque aquí se va a usar el símbolo * para designar cualquiera de estos estados estándar. I.2.1 Ley de Raoult Cuando se habla de mezclas ideales, a estas se les pueden determinar los datos para la construcción de los diagramas de ebullición a partir de las tensiones de vapor de los componentes puros. Este comportamiento ideal se presenta en mezclas cuyos constituyentes muestran gran semejanza química, y se aproximan a este comportamiento las mezclas cuyos componentes tienen iguales presiones críticas. Estas disoluciones obedecen la ley de Raoult, según la cual “la presión de vapor de cada componente es igual al producto de la fracción molar de dicho componente en la fase liquida por la tensión de vapor del componente puro a la misma temperatura”. 2 I MARCO TEÓRICO Sin embargo, cuando la concentración de un componente, expresada en fracción molar se aproxima a la unidad, su comportamiento se aproxima al previsto por la ley de Raoult; por tanto, en disoluciones muy concentradas, esta ley se puede aplicar como limite al componente que se halla en mayor proporción (Ocon 1968). Si se trata de disoluciones diluidas, y para el componente que se encuentra en menor proporción, se puede aplicar la ley de Henry, según la cual “la presión de un componente en el vapor es proporcional a su concentración en el líquido”. Comprobando el intervalo de aplicabilidad de estas leyes, podemos decir que la ley de Raoult es aplicable al disolvente, y la de Henry es aplicable al soluto. I.2.2 Mezclas Azeotrópicas Azeotrópo o mezcla azeotrópica es una mezcla líquida de dos o más sustancias que se comporta como una sustancia única, en el hecho que el vapor producido por la evaporación parcial del líquido tiene la misma composición que el líquido. La mezcla en ebullición constante muestra un punto máximo o mínimo de ebullición, comparado con el de otras mezclas de las mismas sustancias. Un azeotrópo, puede hervir a una temperatura superior, intermedia o inferior a la de los constituyentes de la mezcla, permaneciendo el líquido con la misma composición inicial, al igual que el vapor. Un ejemplo es la mezcla de etanol y agua, que forma un azeotrópo para una concentración del 96% en peso de alcohol, que hierve a una temperatura de 78,2 °C. Cuando las desviaciones de la ley de Raoult son suficientemente grandes, el diagrama obtenido por los datos de las tensiones de vapor sufre una modificación y aparece un máximo (en el caso de desviaciones positivas o un mínimo (si las desviaciones son negativas). La mezcla recibe el nombre de azeotrópica, ya que proviene del griego azeotrópos que significa hervir sin cambio. Son más abundantes las mezclas que presentan el máximo en la presión de vapor (que corresponde a un mínimo en la temperatura de ebullición), y en este caso decimos 3 I MARCO TEÓRICO que la mezcla presenta un azeotrópo mínimo cuya composición corresponde al mínimo que aparece en la curva de ebullición para una presión total determinada. En estas mezclas, las curvas de composición del líquido y del vapor son tangentes en el punto de azeotropismo. Las mezclas cuya concentración es menor que la correspondiente al azeotrópo dan lugar en la ebullición a un vapor más rico en el componente más volátil que el líquido de partida, mientras que las mezclas de concentración superior a aquella dan un vapor más pobre en componente más volátil que el líquido de partida. Naturalmente, si se trata de una mezcla cuya composición sea la del azeotrópo dará lugar a un vapor de la misma composición, comportándose como si se tratara de un componente puro; por tanto, las mezclas de esta estructura no pueden separarse por el método de destilación simple (Ocon 1968). La composición del azeotrópo varia con la presión total, siendo posible en muchos casos eliminar el azeotrópo o al menos desplazarlo hacia el lugar que nos interese por variación adecuada de la presión. I.3 Separación de Mezclas La separación de mezclas es una práctica muy común en la industria de la química fina. Los procesos de destilación y extracción líquido-líquido se emplean generalmente dentro de las ramas farmacéutica y biotecnológica para la recuperación de disolventes utilizados en la síntesis química o bioquímica y en los procesos de separación y purificación del producto final (Acosta, 2010). Tal es el caso en la recuperación del alcohol etílico producido a partir de diferentes materias primas se hace prácticamente de la misma manera. La concentración de alcohol en los caldos de cultivo resultantes de los procesos de fermentación oscila entre 5 y 12 % en peso de etanol, por lo que es necesario concentrar el etanol hasta valores mayores a 99 % en peso (Uyazan, 2004) y obtener así alcohol anhidro (deshidratado), que es el utilizado en calidad de aditivo para la gasolina, ya que la presencia de agua en el etanol puede con llevar a fallas durante la combustión en el motor. Estas mezclas por destilación no son factibles económicamente debido a la elevada cantidad de calor necesaria para la vaporización de los compuestos involucrados, lo que significa mayor energía de calentamiento. 4 I MARCO TEÓRICO Por otra parte, la extracción con disolventes resulta más viable que la destilación cuando presenta componentes que tienen una baja volatilidad relativa o son relativamente no volátiles. Otra aplicación la ha encontrado en aquellos compuestos que son sensibles al calor como hormonas, antibióticos, productos bioactivos naturales, etc. Esta técnica ha encontrado gran utilización en la separación de metales y nuevas sustancias encuentran aplicaciones como agentes de extracción (Valer, 2009). La separación de mezclas azeotrópicas en fracciones de elevada pureza constituye uno de los problemas técnicos y económicos más importantes y retadores de la industria de procesos químicos (Vásquez, 2006). Sin embargo, la necesidad de obtener alcohol deshidratado con el fin de ser usado como aditivo oxigenante de la gasolina, ha impuesto una serie de retos a la industria y a los centros de investigación, a fin de reducir los costos energéticos de la recuperación de etanol cumpliendo los correspondientes estándares de calidad. Adicionalmente, el impacto ambiental que presentan algunas tecnologías tradicionales de separación ha hecho que la investigación sobre esquemas alternativos y no convencionales sea mucho más intensa (Montoya et al., 2005). Por otro lado, la diversidad de alternativas tecnológicas para la producción de alcohol carburante ha hecho crucial el análisis del proceso global, a la par del diseño y desarrollo de cada una de las operaciones que lo integran. Dentro de las nuevas tendencias de investigación y desarrollo en esta área se cuenta la integración del proceso con miras a develar las muy complejas interacciones entre las diferentes etapas del proceso productivo (Lynd, 1996). El desarrollo de procesos integrados permitirá una reducción sustancial de los costos de producción y el incremento de la competitividad del bioetanol frente a la gasolina. De otro lado, la integración de procesos es una condición indispensable para optimizar el proceso de producción de etanol, de tal manera que se consideren como objetivos no sólo la minimización de los costos productivos o la maximización de diferentes indicadores financieros, sino también el mejoramiento de los índices de desempeño ambiental de este proceso (Sánchez, 2005). 5 I MARCO TEÓRICO I.4 Operaciones de Separación Sería prácticamente imposible estudiar el número casi infinito de procesos químicos que se llevan a cabo en la industria diariamente, si no hubiera un punto en común a todos ellos. Afortunadamente, esta conexión existe. Cualquier proceso que se pueda diseñar consta de una serie de operaciones físicas y químicas que, en algunos casos son específicas del proceso considerado, pero en otros, son operaciones comunes e iguales para varios procesos. Cada una de estas operaciones es una operación unitaria. Este concepto fue introducido en 1915 por el profesor Little, del Massachussets Institute of Technology (M.I.T.). La definición dada entonces, fue la siguiente: “... todo proceso químico conducido en cualquier escala puede descomponerse en una serie ordenada de lo que pudieran llamarse operaciones unitarias, como pulverización, secado, cristalización, filtración, evaporación, destilación, extracción, etc. El número de estas operaciones básicas no es muy grande, y generalmente sólo unas cuantas de entre ellas intervienen en un proceso determinado” (Marcilla 1998). I.4.1 Destilación Extractiva Con el nombre de destilación se entiende la separación de los componentes de una mezcla liquida por vaporización parcial de la misma, de tal manera que la composición del vapor obtenido sea distinta de la composición del líquido de partida resultando distinta también la composición del líquido residual. La destilación es una de la operaciones básicas más importantes de la industria química y permite separar los componentes de una mezcla liquida al estado de sustancia puras (Ocon 1968). La destilación es la operación de separar, comúnmente mediante calor, los diferentes componentes líquidos de una mezcla, aprovechando los diferentes puntos de ebullición (temperaturas de ebullición) de cada una de las sustancias a separar. Una modalidad de destilación es la llamada destilación extractiva. Se trata de un método de rectificación de multicomponentes. A una mezcla binaria que es difícil o imposible de separar por los métodos ordinarios, se le agrega un tercer componente, un agente másico de separación (AMS), conocido como disolvente, el cual altera la volatilidad relativa de los 6 I MARCO TEÓRICO componentes originales y permite, de esa forma, la separación. Este disolvente agregado es de baja volatilidad, no evaporándose de modo apreciable durante todo el proceso. La destilación extractiva se usa en la industria petroquímica y química para la separación de sistemas de puntos de ebullición cercanos, críticos, o azeotrópicos, para los cuales la destilación sencilla con una única alimentación es demasiada cara o imposible. La figura I.1 ilustra el esquema clásico de un proceso de destilación extractiva para la separación de un sistema binario. La configuración consiste en una columna extractiva de doble alimentación y en una columna de recuperación de disolvente. Los componentes A y B pueden tener una volatilidad relativa baja o formar un azeotrópo de mínimo punto de ebullición. El disolvente se introduce en la columna de extracción a una elevada concentración unos platos por debajo del condensador, pero por encima del plato de alimentación. Debido a que el disolvente se elige no volátil, permanece a una concentración relativamente elevada en la fase líquida a lo largo de las distintas secciones de la columna, por debajo del plato de alimentación de disolvente. Figura I.1 Esquema clásico de un proceso de destilación extractiva para la separación de un sistema binario Uno de los componentes, A (no necesariamente el componente más volátil de la mezcla original), se extrae como una corriente de destilado esencialmente pura. 7 I MARCO TEÓRICO Debido a que el disolvente es no volátil, como mucho unos pocos platos situados por encima del plato de alimentación del disolvente son suficientes para separar el disolvente del destilado. El producto de cola, consistente en B y el disolvente, se envía a la columna de recuperación. El destilado procedente de la columna de recuperación es B puro, y el disolvente-producto de cola se recicla de vuelta a la columna extractiva. La destilación extractiva trabaja mediante el aprovechamiento de las mejoras inducidas por el disolvente o las moderaciones de las no idealidades de la fase líquida de los componentes que van a ser separados. El disolvente modifica, selectivamente, los coeficientes de actividad de los componentes que van a ser separados. Para lograr esto, es necesaria una alta concentración de disolvente. Varios aspectos son esenciales respecto al disolvente: Alta selectividad, o habilidad para alterar de tal modo que el equilibrio vapor-líquido de la mezcla original permita su fácil separación utilizando pequeñas cantidades del disolvente. Baja volatilidad, con el fin de prevenir la evaporación del disolvente con el producto principal y manteniendo una concentración elevada en la fase líquida. Separabilidad. El disolvente debe poder separarse con facilidad de la mezcla a la cual se adicionó; en particular, no debe formar azeótropos con las sustancias originales. Además es importante considerar el costo, toxicidad, carácter corrosivo, estabilidad química, punto de congelamiento y viscosidad. La columna de destilación extractiva debe ser una columna de doble alimentación, con el disolvente alimentado por encima de la alimentación primaria. 8 I MARCO TEÓRICO I.4.1.1. Aplicaciones industriales La destilación extractiva es generalmente sólo aplicable a sistemas en los que los componentes que van a ser separados contienen uno o más grupos funcionales diferentes. En un método antieconómico normalmente para la separación de estereoisómeros, homólogos, o isómeros homólogos o estructurales que contienen los mismos puntos funcionales, a menos que las diferencias en las estructuras también contribuyan a significativas diferencias en la polaridad, el momento dipolar o el carácter hidrofóbico. Uno de los ejemplos de la destilación extractiva es el caso de la separación de tolueno de hidrocarburos parafínicos (Figura I.2). Ambos compuestos tienen pesos moleculares muy parecidos y es muy difícil separarlos debido a su volatilidad relativamente baja. A pesar de eso, es necesario recuperar el tolueno a partir de ciertas mezclas de hidrocarburos del petróleo. Como hidrocarburo parafínico se toma el isooctano (punto de ebullición 93ºC). El isooctano es más volátil que el tolueno (punto de ebullición 110,8 ºC). En presencia de fenol (punto de ebullición 181,4 ºC) la volatilidad relativa del isooctano aumenta, de forma que la separación del tolueno es relativamente sencilla con aproximadamente 83 % en mol de fenol en el líquido. El diagrama de flujo es el que se muestra en la figura: La mezcla binaria se introduce más o menos en el centro de la torre de destilación extractiva (1); el fenol, como disolvente, se introduce cerca de la parte superior, a fin de que posea concentraciones elevadas en la mayoría de los platos en la torre. En estas condiciones, el isooctano se destila fácilmente como producto principal, mientras que el tolueno y el fenol se separan como residuo. Aunque el fenol tiene un punto de ebullición relativamente elevado, su presión de vapor es suficiente para que pueda evitarse su presencia en el producto principal. La sección de recuperación del disolvente en la torre, que puede ser relativamente corta, sirve para separar el fenol del isooctano. EI residuo de la torre debe rectificarse en una torre auxiliar (2), para separar el tolueno del fenol que se recircula; esta es una separación relativamente sencilla. En la práctica, el hidrocarburo parafínico es una mezcla y no isooctano puro, pero el principio de la separación es el mismo. 9 I MARCO TEÓRICO Un proceso de este tipo depende de la diferencia del alejamiento del ideal entre el disolvente y los componentes de la mezcla binaria que se va a separar. En el ejemplo dado, tanto el tolueno como el isooctano por separado forman soluciones líquidas no ideales con el fenol, pero la no idealidad es mayor con el isooctano que con el tolueno. Por lo tanto, con las tres sustancias presentes, el tolueno y el isooctano se comportan como una mezcla no ideal y su volatilidad relativa se vuelve más alta. Las consideraciones de este tipo forman las bases para la elección de un disolvente para la destilación extractiva (Treybal 1988). Figura I.2 Ejemplo de una destilación extractiva (Robert E. Treybal) I.4.2 Extracción líquido-líquido El estudio experimental del equilibrio de fases líquido-líquido de sistemas multicomponentes es de gran importancia para los procesos industriales de extracción en fase liquida, ya que se requiere que los disolventes elegidos sean no tóxicos para el proceso, más eficientes, más baratos, menos corrosivos y más selectivos. Por esta razón es indispensable contar con datos experimentales confiables de equilibrio de fases líquido-líquido de los compuestos de interés. La extracción líquida, llamada algunas veces extracción con disolventes, es la separación de los componentes de una solución líquida por contacto con otro líquido insoluble. Si las sustancias que componen la solución original se distribuyen de manera distinta entre las dos fases líquidas, se puede lograr cierto 10 I MARCO TEÓRICO grado de separación, que puede incrementarse mediante el uso de contactos múltiples o su equivalente en la forma de la absorción de gases y la destilación. Por ejemplo, si una solución de ácido acético en agua se agita con un líquido como acetato de etilo, parte del ácido, pero relativamente poca agua, entrará en la fase éster. Puesto que las densidades de la capa acuosa y la del éster son diferentes en el equilibrio, se separarán al cesar la agitación; también se pueden separar por decantación. Puesto que ahora la relación de ácido a agua en la capa de éster es diferente de la relación de la solución original y distinta también de la relación de la solución acuosa residual, se ha logrado cierto grado de separación. Este es un ejemplo de contacto por etapas; puede llevarse a cabo en lotes o en forma continua. El agua residual puede extraerse repetidamente con más éster para reducir más aún el contenido de ácido, o se puede re arreglar una cascada a contracorriente de etapas. Otra posibilidad es utilizar algún tipo de aparato de contacto continuo a contracorriente, en donde no se tienen etapas discretas. La utilización del reflujo, como en la destilación, puede mejorar más aún la separación final. En todas las operaciones de este tipo, la solución que se va a extraer se llama alimentación y disolvente el líquido con el cual se pone en contacto la alimentación. El producto de la operación rico en disolvente se llama extracto; el líquido residual de donde se separó el soluto es el refinado. En procesos más complicados se pueden utilizar dos disolventes para separar los componentes de una alimentación. Por ejemplo, una mezcla de ácido p- y o-nitrobenzoico puede separarse distribuyendo los ácidos entre cloroformo y agua, que son líquidos insolubles. El cloroformo disuelve preferencialmente al isómero para y el agua al isómero orto. A esto se le llama extracción con doble disolvente o fraccionada (Treybal 1988). I.4.2.1 Aplicaciones de utilidad Las aplicaciones de la extracción líquida se clasifican en varias categorías: aquellas aplicaciones en que la extracción esta en competencia directa con otros 11 I MARCO TEÓRICO métodos de separación y aquellas aplicaciones en que es el único método adecuado. En competencia con otras operaciones de transferencia de masa, aquí, los costos relativos son importantes. La destilación y la evaporación son métodos directos de separación; los productos obtenidos están formados básicamente de sustancias puras. Por otra parte, la extracción líquida produce nuevas soluciones, que a su vez deben separarse, frecuentemente por destilación o evaporación. Por ejemplo, es difícil separar, por destilación, al ácido acético de una solución diluida con agua; en cambio, puede separarse con relativa facilidad mediante la extracción con un disolvente adecuado y la destilación posterior del extracto. En particular, para las soluciones más diluidas en las cuales el agua debe evaporarse por destilación, la extracción es más económica; especialmente, porque el calor de evaporación de la mayoría de los disolventes orgánicos es sustancialmente menor que el del agua. La extracción también puede resultar aconsejable como alternativa frente a la destilación al alto vacío, a temperaturas muy bajas, para evitar la descomposición térmica. Por ejemplo, los ácidos grasos de cadena larga pueden separarse de los aceites vegetales mediante destilación al alto vacío, pero se separan en forma más económica por extracción con propano líquido. El Tántalo y el Niobio se pueden separar mediante una tediosa cristalización fraccionada de los fluoruros dobles con potasio; en contraste, su separación es bastante sencilla por extracción líquida de las soluciones de ácido fluorhídrico con metilisobutilcetona (MIK). Como un sustituto de métodos químicos (Los métodos químicos consumen reactivos y con frecuencia conducen a una costosa eliminación de los subproductos químicos), la extracción líquida, que no provoca gastos químicos o eliminación de subproductos, puede ser menos costosa. La separación de metales como uranio-vanadio, hafnio-zirconio, tungstenomolibdeno y los productos de fisión de los procesos de energía atómica, se llevan a cabo más económicamente por extracción liquida. Aun los metales menos costosos como cobre y sustancias químicas inorgánicas como ácido fosfórico, ácido bórico y similares, se pueden purificar de manera económica mediante 12 I MARCO TEÓRICO extracción líquida, a pesar de que el costo de recuperación del disolvente debe incluirse en las cuentas finales. En la destilación, en donde la fase vapor se crea a partir del líquido por adición de calor, el vapor y el líquido están compuestos necesariamente de las mismas sustancias; por lo tanto, son muy similares químicamente. Entonces, las separaciones producidas dependen de las presiones de vapor de las sustancias. En contraste, en el caso de la extracción líquida, los componentes principales de las dos fases son muy distintos químicamente; por esto, son posibles las separaciones de acuerdo con el tipo químico. Por ejemplo, los hidrocarburos aromáticos y parafínicos de aproximado peso molecular no se pueden separar por destilación, ya que sus presiones de vapor son casi iguales; sin embargo, pueden separarse fácilmente por extracción con distintos disolventes, como dióxido de azufre líquido, di etilenglicol o sulfolano. (Es importante observar que la destilación extractiva también es útil en estas operaciones, pero es simplemente la extracción de la fase vapor con un disolvente, mientras que la destilación líquida es la extracción de la fase líquida. Con frecuencia, los mismos disolventes son útiles en los dos casos. Muchos productos farmacéuticos -penicilina, por ejemplo, se producen en mezclas tan complejas que sólo la extracción líquida es un método adecuado de separación (Treybal 1988). La clave de un proceso eficaz reside en la posibilidad de disponer de un disolvente adecuado. Además de no toxico, barato y fácilmente recuperable, un buen disolvente deberá ser relativamente inmiscible con solvente de los componentes de la alimentación y poseer diferente densidad. Ha de tener una afinidad muy grande para el soluto, del cual será fácilmente separable por destilación, cristalización u otros medios. Los modelos de solución utilizados para la correlación de los datos experimentales de los equilibrios de fases líquidolíquido estudiados con UNIQUAC (Universal Quasichemical Activity Coefficients) y NRTL (Non Random Two Liquids). Estos modelos se consideran los más adecuados para la correlación de datos de equilibrio de fases líquido-líquido (Aspen Plus 2006). 13 I MARCO TEÓRICO I.5 Selección de mezcla azeotrópica Se han encontrado miles de ejemplos de mezclas azeotrópicas con punto azeotrópico mínimo. Una de las más importantes es la mezcla etanol-agua, el cual a 1 atm aparece a 95.57 % en peso de etanol y se encuentra a 78.2 °C, este punto azeotrópico desaparece a presiones de 70 mmHg (Treybal 1988). Para esta mezcla se encuentran diferentes métodos no convencionales de separación, como por ejemplo la destilación a bajas presiones, la cual hace uso del cambio en el equilibrio de fases a presiones inferiores a la atmosférica, lo que conlleva a la desaparición del azeotrópo por debajo de los 6 kPa. Pero para obtener un producto de alta pureza es necesario utilizar torres con gran número de etapas (por encima de 40) y con altas relaciones de reflujo, con elevados costos de capital y energéticos debido al mantenimiento del vacío en columnas con gran cantidad de platos. En lugar de la destilación por vacío se emplea la destilación azeotrópica, que consiste en la adición de un tercer componente a la mezcla etanol-agua y se forman nuevos azeotrópos que facilitan la separación en esquemas tecnológicos que involucran dos o tres columnas de destilación. Entre las sustancias (llamadas arrastradores) que se agregan a las mezclas de etanol-agua resultantes del proceso de obtención de alcohol se encuentra el benceno, el tolueno y el pentano. El proceso consiste de una columna de deshidratación (columna azeotrópica) que se alimenta con una mezcla cercana al 90 % de peso de etano. A esta columna se le agrega en el plato superior el benceno, mientras de la parte inferior alcohol anhídrido con una concentración de agua menor al 1 %. El vapor de salida de la parte superior de la columna, con una composición igual o cercana a la del azeotrópo ternario, se condensa y se lleva a un separador en donde la fracción 14 I MARCO TEÓRICO rica en agua se alimenta a una pequeña columna de lavado (columna despojadora) para la regeneración del arrastrador, mientras la otra fracción se recircula como reflujo a la parte superior de la columna azeotrópica. Sin embargo el uso de benceno no es deseable debido a sus propiedades carcinogénicas, por lo que se ha propuesto la destilación extractiva, en donde la tercera sustancia que se agrega (denominada disolvente) modifica la volatilidad relativa de los componentes de la mezcla etanol-agua sin formar nuevos azeotrópos, facilitando así la separación. El disolvente debe ser de baja volatilidad a escoger a voluntad. Para que su separación en la tercera torre de destilación, donde se recupera, sea mucho más fácil. Como disolvente se ha usado tradicionalmente etilenglicol pero se debe de cuidar la presión y temperatura del proceso ya que el etilenglicol realiza con mayor eficiencia la separación a 80 °C pero se descompone a trabajos mayores de 0.2 atm y se oxida a los 150 °C (Quintero, 2007), se indica que bajo condiciones de operación específicas, la destilación extractiva para la obtención de etanol anhidro puede ser competitiva energéticamente en comparación con la destilación azeotrópica, lo cual ha sido corroborado por Montoya y Quintero et al. (2005) mediante simulación. Se puede apreciar en la tabla I.1 la cual contiene una lista de los de algunos ejemplos de mezclas azeotrópicas, indicando el tipo de azeotrópo en ellas, el disolvente favorables para su separación y una breve reseña del sistema. 15 I MARCO TEÓRICO Tabla I.1 Lista de algunos ejemplos de mezclas azeotrópicas con su respectivo disolvente para la separación de la misma por el método de destilación extractiva SISTEMA Etanol-agua TIPO Azeotrópo mínimo Teb BencenoAzeotrópo ciclohexano mínimo Teb Acetato de Azeotrópo etilo-etanol mínimo Teb THF-agua Azeotrópo mínimo Teb AcetonaAzeotrópo metanol mínimo Teb IsopropenoAzeotrópo pentano mínimo Teb Piridina-agua Azeotrópo mínimo Teb Acetato de Azeotrópo metilo-metanol mínimo Teb DISOLVENTE RESEÑA de Etilen-glicol, sales Alternativa a la destilación de acetato para azeotrópica, destilación por procesos con sal variación de presión de Anilina de Esteres altos alcoholes aromáticos de Propilen-glicol o Proceso similar para otros sistemas alcohol-ester Alternativa a la destilación por variación de presión de Agua, anilina, Etilen-glicol de Furfural, DMF, acetonitrilo de Bisfenol de Etilen-glicol, monometil éter Elemento de un sistema de recuperación como alternativa a la producción de acetato de metilo por destilación reactiva; alternativa a la destilación azeotrópica por variación de presión Acido nítrico- Máximo Teb nitrato de Los procesos de ácidos agua magnesio para sulfúrico dependen procesos salinos fuertemente de la curvatura del limite Isómeros de Teb cercanos y Anilina-fenol heptanoazeotrópos de tolueno mínimo Teb Acetato de Teb cercanos Fenol, aromáticos Alternativa a la destilación vinilo-acetato simple de etilo PropanoTeb cercanos Acrilonitrilo Alternativa a la destilación propileno simple EtanolTeb cercanos Benzoato de Alternativa a la destilación isopropanol metilo. simple 16 I MARCO TEÓRICO I.6 Simulación en Aspen Plus La simulación es una técnica muy poderosa y ampliamente usada en las ciencias para analizar y estudiar sistemas complejos. En Investigaciones se formularon modelos que se resolvían en forma analítica. En casi todos estos modelos la meta era determinar soluciones óptimas. La simulación permite al analista imitar la realidad, así como, analizar metas múltiples simultáneamente; es una herramienta muy buena porque proporciona una descripción más adecuada de la realidad. Los beneficios que se obtienen al utilizar simulación son ahorros económicos para la empresa, ahorro en cuanto a tiempo e incremento de la productividad. Un análisis puede efectuarse manualmente, pero cuando se vuelve complicado es necesario utilizar una herramienta de computadora como una hoja de cálculo, no obstante esta presenta una limitante pues no tiene la habilidad de incluir la aleatoriedad que ocurre en un modelo, ni las interdependencias de los recursos y las entidades que se mueven en el. El análisis estático utiliza parámetros promedio, como consecuencia, el analista puede llegar a conclusiones erróneas, sobre estimando o subestimando una determinada situación, mientras que la simulación proporciona una descripción adecuada de la realidad. Las técnicas de optimización como la programación lineal, la programación por metas y la dinámica son útiles, pero se encuentran limitadas a alcanzar solo una meta excluyendo otras metas que pueden tener relevancia. En conclusión la simulación ofrece muchas más ventajas frente a otras herramientas pues permite al analista imitar la realidad, incluyendo la aleatoriedad que ocurre basado en una distribución de probabilidad identificada apropiadamente, o una distribución tomada de los datos. Examina múltiples metas simultáneamente, analiza el desempeño de un sistema, con respecto a varios factores, proporcionando información a múltiples disciplinas en el sistema en estudio permitiendo determinar un mejor sistema y una mejor estrategia. Además ofrece beneficios económicos como: inversiones, trabajos y contrataciones innecesarias, incrementa la productividad, la satisfacción del cliente, mejora la toma de decisiones y ahorro en los tiempos de análisis. 17 I MARCO TEÓRICO I.7 Estimación de Costos de inversión del Proceso Cuando una tecnología es bien conocida, puede tenerse acceso a su costo de inversión a través de fuentes especializadas o del licenciador de la tecnología. Cuando el proceso está en desarrollo a escala laboratorio, esta estimación generalmente no está disponible. Es necesario en estos casos tener alguna estimación razonable del potencial económico del proceso en desarrollo, aun cuando esta estimación no sea completamente precisa. Una estimación aceptable en un momento dado puede servir para discriminar alternativas, o para evitar esfuerzos de tiempo y dinero en proyectos que no ofrezcan un buen potencial económico. Tabla I.2 Nivel de precisión, el cual puede variar desde un 40 % para una estimación preliminar y hasta un 3 % para una estimación detallada. PRECISIÓN DE ESTIMACIÓN DE INVERSIONES TIPOS DE ESTIMACIÓN Orden de magnitud PRECISIÓN BASE GENERAL PROBABLE % Información previa sobre costos 40 similares Estudio vía factores de estimación Estudio preliminar Conocimiento de un diagrama de 25 flujo Datos suficientes para la 12 preparación de un presupuesto Definitivo (Control del proyecto) Detallado (Firma constructora) Datos detallados, pero diagramas 6 incompletos Diagramas y especificaciones 3 completas En general, cualquier método de estimación requiere primero de un diagrama de flujo que muestre los principales componentes de equipos y sus dimensiones. El proceso de estimación tiene dos pasos generales: a) La estimación del costo base de las unidades de equipo Y b) El uso de factores de experiencia para incluir los accesorios adicionales para la operación de esos equipos de procesos. 18 I MARCO TEÓRICO I.7.1 Método de Guthrie Guthrie publicó en 1969 una de la mejores recopilaciones que se tengan sobre estimaciones de costos. Para el manejo de esta información, se divide la planta en módulos. Esta técnica se usa para estimar el costo de una unidad o planta instalada. Para una estimación actual de algún equipo en particular, se recomienda consultar la revista Chemical Engineering, donde se publican con frecuencia métodos de estimación de inversiones de diferentes tipos de equipos de procesos. Aunque el trabajo de Guthrie incluye la posibilidad de estimar módulos como edificios, oficinas administrativas, terrenos y desarrollo del lugar, los módulos de equipo de proceso son los que representan el mayor interés y utilidad en este caso. Los módulos de equipo consisten de una combinación de varios elementos de costos, tales como: Costo de equipo (fob) Mano de obra directa en campo Costo directo de material y mano de obra Costos indirectos Costo del módulo desnudo Costo del módulo total La estimación del costo de un módulo de equipo de proceso representa el costo de la construcción del equipo (intercambiador de calor, bomba, columna, etc.) y el costo del material, mano de obra e indirectos necesarios para instalar el equipo en un circuito de proceso químico. El método comienza con la estimación de un costo base en función de alguna dimensión del equipo. Ese costo base implica acero al carbón como material de construcción, una geometría base de equipo, una presión de operación moderada y un año base de 1968. Ese costo debe corregirse luego al incorporar los datos del material de construcción, geometría, presión y año para la estimación del equipo deseado. Otra parte del ajuste implica el uso de factores de módulo, aplicables al costo base para luego corregir ese valor por el efecto de las características de la unidad deseada. 19 I MARCO TEÓRICO Tabla I.3 Factores de módulos (pueden verse conceptualmente como una especie de factores de Lang modificados) FACTORES DE MÓDULOS Unidad Factor del Módulo Hornos de procesos 2.30 Calentadores de fuego directo 2.30 Intercambiadores de calor 3.39 Enfriadores de aire 2.54 Recipientes verticales 4.34 Recipientes horizontales 3.29 Bombas 3.48 Compresores 3.21 I.7.2 Procedimiento Guthrie presenta en su trabajo varios caminos alternativos cuyo punto final es la estimación del costo de un módulo del proceso. Una de las maneras más simples de procesar la información es la siguiente: 1) Obtener el costo base para una geometría base, acero al carbón y 1968, 2) Ajustar el costo base por efecto de la geometría requerida para el equipo y material de construcción. Se obtiene el costo , que implica que no se incluyen costos de transporte y por la base de datos usados este costo se aplica al año 1968. 3) Para obtener el costo del módulo: - Usar el factor del módulo desnudo: - Sumar diferencia entre la unidad deseada y la base ( - ) Ajustar el costo hacia el año deseado usando índices de costos como los de Chemical Engineering. Añadir contingencias. Guthrie recomienda usar un factor de 15%: 20 II METODOLOGÍA CAPÍTULO II METODOLOGÍA II.1 Selección y simulación de la mezcla azeotrópica Dentro de las mezclas empleadas en la industria química se observó que la mezcla etanol-agua tiene gran importancia económica ya que se emplea en la producción de etanol anhídrido el cual es utilizado como aditivo para las gasolinas pero se requiere de su pureza en un mínimo de 99 % en peso de etanol (Caicedo, 2004). Esta mezcla forma un azeotrópo aproximadamente al 95.57 % en peso de etanol lo cual representa un problema económico. En base a esto, se eligió como mezcla azeotrópica a estudiar. Se encontró que la ecuación de estado que describe el equilibrio líquido vapor para la mezcla etanol-agua es NRTL (Quintero, 2007), con el fin de demostrar que el simulador reproduce los datos experimentales de forma correcta, se compararon los resultados experimentales (figura II.1) contra los obtenidos en el simulador (figura II.2) observándose el mismo comportamiento. Figura II.1 Datos de equilibrio para la mezcla etanol agua (Quintero 2007) 21 II METODOLOGÍA Figura II.2 Datos de equilibrio para la mezcla etanol - agua predichos por NRTL (mediante simulador Aspen Plus) La simulación se realiza mediante el empleo de un separador Flash2. Esta es una herramienta fundamental en la simulación y trabaja como un separador de una entrada con dos salidas (Figura II.3), se seleccionan los componentes y la ecuación de estado la cual predice de forma correcta los resultados y por la corriente de entrada de flujo se consideró con una concentración equimolar, a 25 °C, un flujo de 100 lbmol/hr y una presión de 14.7psi (Figura II.4). Figura II.3 Equipo de separación Flash2 22 II METODOLOGÍA Figura II.4 Condiciones en la corriente de alimentación Dichas curvas de equilibrio pueden ser vistas accediendo en el menú TOOLS, ANALYSIS, PROPERTY, BINARY que se encuentra en la barra de menús (Figura II.5), dicha acción nos permite seleccionar YX como tipo de análisis y en base a que componente es considerada la composición, la cual normalmente es considerada en base al componente más volátil. Figura II.5 Acceso al tipo de análisis deseado con los datos de equilibrio 23 II METODOLOGÍA II.2 Diseño del diagrama y simulación de la separación por el método no convencional (destilación extractiva) El método empleado fue destilación extractiva debido a que según lo reportado en bibliografía muestra mayores rendimientos (Quintero, 2007). Para esta simulación se seleccionó un disolvente de separación, recordando que este debe de ser miscible en la mezcla para que pueda modificar, selectivamente y considerablemente los coeficientes de actividad de los componentes que van a ser separados en este caso etanol-agua, sin formar nuevos azeótropos. Además el disolvente agregado debe de tener una baja volatilidad para que su recuperación en la tercera torre sea fácil, además de un punto de ebullición muy alto. Para lograr la separación, es necesaria una alta concentración de disolvente. El etilenglicol es la mejor opción (Tabla I.1) ya que es un solvente muy miscible con ambos componentes y esto permite la separación de la mezcla original (Morrison 1990), además de tener un punto de ebullición muy alto (197.08°C). En base a esto se construyó un diagrama de procesos en ASPEN PLUS el cual consta de tres columnas de rectificación RadFrac dicha columna realiza trabajos rigurosamente en sistemas de 2 o 3 fases de fraccionamiento en columnas individuales. La primera realiza una operación de rectificación esto permite acercarse al azeotrópo, por lo que se puede realizar una mejor separación ya que ese es el punto donde el disolvente debe de alterar las volatilidades, la segunda columna realiza la destilación extractiva mediante el procedimiento ya explicado y la última mediante otra rectificación recupera el disolvente el cual recircula por todo el sistema por unidad de tiempo. Esta simulación se obtuvo mediante el diseño de un diagrama de procesos y el uso de tres Columnas RadFrac, un Intercambiador de calor Heater, un Mezclador Mixer y un Divisor FSplit (Figura II.6). 24 II METODOLOGÍA Figura II.6 Diagrama de procesos para la separación de la mezcla azeotrópica etanol-agua por medio del método de destilación extractiva Una vez diseñado el diagrama de procesos para la separación se toma en consideración las condiciones de entrada para la mezcla y para el disolvente realizando la primera simulación sin reflujo alguno, por lo tanto en la salida de desahogo aproximadamente se obtendrá todo el etilenglicol agregado. Para la simulación de separación se seleccionaron los componentes y la ecuación de estado ya estudiada para este método. Considerando una composición de 0.0505 en fracción mol de etanol ya que aproximadamente esa es la concentración a la que se obtiene el etanol por métodos de fermentación de caldos (0.1197 en fracción masa), una temperatura ambiente de 25 °C o 77°F, la cantidad de flujo de alimentación de la mezcla es de 1400.2 Kg/hr y la presión en la corriente de alimentación de la mezcla etanol-agua es de 14.7 psi (Figura II.7). La corriente del etilenglicol que entra por el mezclador Mixer debe de estar presente en gran cantidad, por lo que es necesario agregar 17900Kg a una temperatura de 25 °C y una presión de entrada de 14.7 psi (Figura II.8). 25 II METODOLOGÍA Figura II.7 Condiciones en la corriente de alimentación de la mezcla azeotrópica Figura II.8 Condiciones en la corriente de alimentación del disolvente en el método de destilación extractiva. La configuración de la primera columna consta de 24 etapas o platos, este número de etapas permite estar lo más cerca posible del azeotrópo. Cuenta con un condensador total, esto asegura que todo el producto se convertirá en un líquido con una concentración de etanol en el punto azeotrópico. La mezcla contiene 167.63 Kg de etanol pero no es posibles recuperar todo el etanol, por lo tanto se calcula la cantidad de destilado mediante una tanteo de simulaciones llegando a la conclusión de que la cantidad de destilado en la columna de acercamiento al azeotrópo es de 153.2 Kg/hr; la torre cuenta con una relación de reflujo de 100 ya que se probaron varias cantidades de reflujo hasta llegar a esta que es la más favorable (Figura II.9). Y la presión de operación en el condensador es de 0.2 atm. 26 II METODOLOGÍA Figura II.9 Configuración de operación para la primera columna La segunda columna RadFrac realiza la destilación extractiva y esto hace necesario un mayor número de etapas que una rectificación común además de tener dos corrientes de alimentación, esta columna trabaja con 32 etapas. El destilado es totalmente condensado y esta corriente es el principal producto de la separación obteniendo un alcohol con purezas de más de 99% en peso de etanol. La cantidad de flujo que se obtiene por el destilado es de 3.1635Kmol/hr. Este valor es la cantidad aproximada de etanol que se encuentra en la mezcla ya procesada y es obtenido mediante los resultados producidos en la rectificación de la columna anterior. La columna ejerce una relación de reflujo de 100 (Figura II.10) y las corrientes de alimentación de la mezcla azeotrópica y del disolvente entran en las etapas 16 y 15 respectivamente (Figura II.11), con una presión de operación en el condensador de 0.2 atm. Figura II.10 configuración en la segunda columna 27 II METODOLOGÍA Figura II.11 Etapa de alimentación en las corrientes de alimentación La tercera columna RadFrac es una rectificación simple la cual permite recuperar el disolvente por el fondo de la columna, separándola de los residuos de la mezcla inicial. Cuenta con 21 etapas las cuales son en menor número comparadas con la columna anterior, el tipo de condensador que tiene la columna es uno total, ya que a pesar de que este es el final del proceso de separación el producto obtenido por el domo de la torre sigue siendo de gran interés, aunque es muy poca cantidad (0.809lbmol/hr) este sigue teniendo trazas de etanol las cuales pueden ser recuperadas después. La relación de reflujo de es de 100 (Figura II.12) y la presión de operación para el condensador de 0.2 atm (Figura II.13). Figura II.12 Configuración de operación para la columna de recuperación del disolvente Figura II.13 Configuración en el condensador de la tercera columna. 28 II METODOLOGÍA Uno de los servicios más importantes en el proceso es el intercambiador de calor Heater, el cual permite que el etilenglicol siempre al momento de entrar a la columna de destilación extractiva se mantenga a 80 °C, esto es muy indispensable ya que ese es el punto en el que el disolvente realiza mejor la separación además de controlar su temperatura, porque si el disolvente alcanza los 150 °C o si trabaja con presiones superiores a 0.2 atm (Quintero, 2007) este se descompone (Figura II.14). Figura II.14 Condiciones de operación para el intercambiador de calor en el método de destilación extractiva La operación del Divisor FSplit, determina la cantidad de disolvente que regresa a la columna de destilación extractiva. Pero en el proceso el disolvente ha perdido pureza por haberse mezclado con la mezcla azeotrópica en la segunda columna, por ello se somete a una rectificación con el residuo del agua-etanol, para poder determinar la cantidad correcta que debe regresar primero se debe saber cuánta es la cantidad de disolvente impuro que sale por el fondo de la tercera columna. Para poder determinar esto es necesario escoger la corriente de desahogo e indicar que la fracción de salida sea de cero (Figura II.15). Figura II.15 Condiciones iniciales en el divisor para el método de destilación extractiva Una vez establecidas todas las condiciones de operaciones se realiza la simulación (Figura II.16) donde que la corriente de retorno del disolvente no presenta alguna variacion y que los resultados son correctos. 29 II METODOLOGÍA Corriente de retorno Corriente de desahogo Resultados correctos Figura II.16 Simulación de la separación de la mezcla azeotrópica sin reflujo agregado. Como ahora se sabe que por la corriente de desahogo la cantidad de disolvente impuro es de 17900.2106 Kg/hr se realiza la operación de reflujo editando la configuración del divisor descontando el flujo de la corriente de desahogo en pequeñas porciones, la cantidad que es retirada también debe de ser excluida de la alimentación del etilenglicol fresco. Llegando al término de la simulación con resultados correctos y determinando que por cada hora se deben de retirar del sistema 17897.165 Kg (Figura II.17) de etilenglicol contaminado y por lo tanto por cada hora se debe de añadir 2.835 Kg (Figura II.18) de etilenglicol fresco; en la última simulación, por el método de destilación extractiva se puede observar que se han obtenido resultados favorables y que las corrientes de desahogo y retorno tienen actividad (Figura II.19). Figura II.17 Configuración final del divisor 30 II METODOLOGÍA Figura II.18 Configuración final en la corriente de alimentación de disolvente Te mper a ture ( K) Q Duty ( c a l/se c) Figura II.19 Simulación final con cantidad de disolvente fijo en la corriente de desahogo y en la retroalimentación por el método de destilación extractiva. 31 II METODOLOGÍA II.3 Diseño del diagrama y simulación de la separación por el método convencional (extracción L-L) La operación unitaria seleccionada para realizar la separación de la mezcla etanolagua por el método convencional fue la extracción líquido-líquido. Para dicho método en la selección del disolvente de separación se tomaron en cuenta las dos cualidades más importantes en la extracción L-L; el disolvente de separación debe de ser inmiscible con el solvente de la mezcla original para este caso el agua y no debe de formar azeotrópo con el soluto de la mezcla o sea el etanol. El agua es el solvente que forma el mayor número de inmiscibilidades con otros solventes, por lo tanto además de tomarse en cuenta la inmiscibilidad también se consideró la solubilidad de los solventes en el agua. De los solventes que mostraron mejor inmiscibilidad y una muy poca solubilidad con el agua fue el Xileno. Así que se decide tomar al Ortoxileno como disolvente de separación por ser el xileno con el más alto punto de ebullición (144.43 °C), lo que facilitara su recuperación en la columna de rectificación. Por lo tanto la mezcla etanol-ortoxileno se convertirá en el extracto y el agua en el refinado para la extracción líquido-líquido. Debido a que se no conocen los datos de equilibrio para el sistema etanolortoxileno los cuales son necesarios para comprobar que el extracto no forma azeotrópo y además será sometida a una rectificación para la recuperación de ambos componentes se realizaron diversas simulaciones considerando las 8 ecuaciones de estado de mayor importancia para procesos químicos como son PENG-ROB, SRK, WILSON, VANLAAR, CHAO-SEA, UNIFAC incluidas NRTL (Non Random Two Liquids) y UNIQUAC (Universal Quasi-Chemical Activity Coefficients) ya que son las dos ecuaciones de estado más favorables para la extracción L-L (Aspen Plus 2007). Para dichas simulaciones se empleó un separador Flash2, obteniendo estos resultados (Figura II.28; Figura II.29; Figura II.30). 32 II METODOLOGÍA Figura II.20 Curva de equilibrio obtenida por medio de las ecuaciones de estado PENG-ROB, SRK, WILSON, VANLAAR, NRTL Y UNIQUAC. Figura II.21 Curva de equilibrio predicha por la ecuación CHAO-SEA Figura II.22 Curva de equilibrio predicha por la ecuación UNIFAC 33 II METODOLOGÍA Para la simulación de la extracción L-L se diseñó un diagrama (Figura II.23) en el cual se utilizó una Columna Extract que nos permite resolver sistemas de extracción a contra corriente con un solvente líquido, una Columna RadFrac, un Intercambiador de calor Heater, un Mezclador y un Divisor. Figura II.23 Diseño del diagrama para la separación de la mezcla azeotrópica por el método convencional (extracción L-L) Como se puede apreciar en las curvas de equilibrio dos de las ocho ecuaciones predicen la formación de un azeotrópo y el resto nos permitió visualizar la misma curva sin formación de azeotrópo. Por lo tanto se decide tomar a NRTL como la ecuación de estado que nos permite resolver el sistema por dicho método, y como disolvente se escoge al orto-xileno (Figura II.24). Figura II.24 Selección del ortoxileno como disolvente 34 II METODOLOGÍA Para la corriente alimentación se considera una mezcla con las mismas condiciones a la simulación anterior, una composición de 0.0505 en fracción mol de etanol. Se toman 1400.2 Kg/hr como flujo de alimentación para la mezcla etanol-agua, a una temperatura ambiente de 77 °F ó 25 °C con una presión de 14.7 psi (Figura II.25). Y por la alimentación del mezclador son necesarios 3040 Kg/hr de ortoxileno como alimentación inicial del proceso a una temperatura de 77 °F y una presión de 14.7 psi (Figura II.26). Figura II.25 Configuración de alimentación para la corriente de la mezcla Figura II.26 Configuración de alimentación para la corriente del disolvente Como condiciones de operación en la columna Extract se consideraron 20 etapas mediante un proceso adiabático (Figura II.27), a diferencia de la destilación, en la extracción se obtienen dos productos, el refinado el cual para este caso es el agua, y el extracto, en el cual se forma la nueva mezcla de etanol-ortoxileno (Figura II.28) que después será sometida a una rectificación para la obtención del producto de interés y para la recuperación del disolvente. 35 II METODOLOGÍA Además otra diferencia entre estos métodos es que en la destilación se puede seleccionar la etapa de alimentación y en la extracción las entradas tanto como las salidas siempre son por el domo y el fondo de la columna (Figura II.29). Figura II.27 Número de etapas en la columna Extract Figura II.28 Selección del refinado y el extracto como productos en la extracción Figura II.29 Número de etapas para las corrientes de alimentación y de los productos 36 II METODOLOGÍA Una vez culminada la operación de extracción L-L, el extracto que ahora se ha convertido en la nueva mezcla de interés es sometido a una rectificación en una columna RacFrac para la recuperación del disolvente y del producto principal. Considerando 25 etapas para una mejor pureza, el tipo de condensador debe de ser total debido a que como producto final se requiere un líquido saturado, una cantidad de destilado total de 172.5 Kg/hr porque aunque la cantidad de etanol en la mezcla sea de 167.63 Kg/hr el proceso de extracción y rectificación no son suficientes para eliminar el exceso de agua, la relación de reflujo para esta columna es de 50 (Figura II.30). La corriente de alimentación es ingresada en el plato número 13 (Figura II.31) y la presión de operación para el condensador es de 14.7 psi (Figura II.32). Figura II.30 Configuración en la rectificación del método de extracción L-L Figura II.31 Etapa de alimentación para el extracto del proceso anterior Figura II.32 Presión de operación en el condensador de la columna RadFrac en el método de la extracción L-L 37 II METODOLOGÍA De igual forma, como en la destilación extractiva aún no se conoce la cantidad ideal de disolvente para reflujo, es necesario realizar el mismo procedimiento configurando por primera y única ocasión al divisor con la opción de Split Fraction de 0 para la corriente de retorno y así poder conocer la cantidad de disolvente contaminado que abandona el proceso (Figura II.33). Figura II.33 Configuración inicial del divisor en el método de la extracción L-L Como la corriente de reflujo regresa al mezclador junto con la corriente de disolvente puro es necesario enfriar el disolvente contaminado. El intercambiador de calor que se encuentra entre el divisor y el mezclador tiene la función de disminuir la temperatura del xileno ya utilizado a los 25°C que es la misma temperatura que el disolvente puro; el enfriamiento del disolvente contaminado se realiza a la presión de 1atm (Figura II.34). Figura II.34 Condiciones de operación para el intercambiador de calor en el método de la extracción L-L Se realiza la simulación una vez que ya se han terminado de configurar todos los servicios. Pero el resultado es diferente a la simulación anterior ya que en esta ocasión ASPEN reproduce unos resultados correctos pero indica una advertencia en el proceso, esto se debe porque todo el disolvente sale por la corriente de desahogo, por lo tanto el intercambiador de calor no recibe flujo alguno esto indica que el simulador no solo considera las condiciones de operación sino que además toma en cuenta el diseño del proceso, esto se puede apreciar en la figura II.35. 38 II METODOLOGÍA Corriente de desahogo con su respectiva T Corriente de retorno con su respectiva T Figura II.35 Simulación sin reflujo de disolvente, resultados favorables con advertencia en el método de la extracción L-L Una vez que ya se conoce la cantidad de disolvente contaminado que sale al final del proceso, se edita la configuración del divisor cambiando la opción de Split Fraction por Flow, en esta opción se agrega el flujo de salida y se empieza a descontar la cantidad de disolvente contaminado en la corriente de desahogo y al mismo tiempo, se excluye esa misma cantidad por la corriente de alimentación. Ese procedimiento se realiza hasta llegar al punto en el que es necesario retirar 2987.553 kg/hr de ortoxileno contaminado (Figura II.36) y solo es necesario añadir 0.7835 Kg/hr de disolvente fresco (Figura II.37). Figura II.36 Configuración final del divisor 39 II METODOLOGÍA Figura II.37 Configuración final para la corriente de reposición del disolvente en el método de la extracción L-L Una vez que se encuentran activados todos los servicios y que ya se conoce la cantidad ideal de salida de disolvente contaminado y al mismo tiempo la cantidad ideal de disolvente puro para relleno se realiza la simulación final obteniendo resultados favorables para dicho proceso (Figura II. 38). Figura II. 38 Simulación final por el método de la extracción L-L 40 III RESULTADOS CAPITULO III RESULTADOS Una vez realizadas todas las simulaciones se determina la cantidad de vapor que se requiere en cada re-hervidor de las columnas RadFrac, la cantidad de agua necesaria en los condensadores y los costos de agentes másicos (etilenglicol y ortoxileno). Para este análisis se utilizan los resultados que ASPEN predijo utilizando la carga térmica del condensador y del re-hervidor, estas cargas se pueden apreciar accediendo desde la barra de menús al menú TOOLS, OPTIONS y se desplegara una ventana secundaria y en la pestaña de Results View se selecciona Heat/Work y Temperatura (Figura III.1). Figura III.1 Menú para hacer visibles las cargas térmicas en las simulaciones III.1 Resultados del método de destilación extractiva primera columna en el proceso En la primera columna de este proceso y ya editadas las opciones en los resultados, se obtiene que la energía en el condensador necesaria es de Qc=1134874 cal/s y la energía en el re-hervidor es de Qr=1147025 cal/s. Además la corriente del domo de la columna tiene una temperatura de 315 K, la corriente del fondo tiene una temperatura de 332K y la corriente de alimentación de la mezcla la cual se encuentra a una temperatura ambiente (Figura III.2). 41 III RESULTADOS Figura III.2 Primera columna en el proceso de destilación extractiva En el re-hervidor de esta columna necesitamos un vapor de 352K ya que el incremento de temperatura ideal para todos los sistemas es ΔT=20°C. Para conocer la cantidad de vapor necesaria se tienen que encontrar la temperatura ideal del vapor (Cengel 1996) y ubicar la temperatura ideal de vapor a 352K. A la temperatura de 352 K el vapor del agua tiene una (Occon), por lo tanto la diferencia y de estas es , siendo este el calor latente ( ) cedido. Para el re-hervidor de esta columna, la cantidad de vapor requerido es: ( ) En la torre de RadFrac se obtiene un producto en punto de rocío, el cual pasa a través de un condensador y este entrega el mismo componente pero en su punto de burbuja o punto de ebullición. La temperatura en la que se obtiene el líquido es a la misma que se produce el vapor, ya que es una mezcla prácticamente pura y la única diferencia entre ellos es su entalpia (Figura III.3). 42 III RESULTADOS Figura III.3 Estados de la materia; Temperatura VS Entalpia Debido a un balance de energía se puede determinar que la entalpia perdida por la mezcla es la misma que gana el líquido de enfriamiento, donde normalmente se utiliza el agua en este proceso, dependiendo de la temperatura que se desea alcanzar. En este caso el cambio de entalpia no es más que la cantidad de energía necesaria para cambiar de estado a la materia, cambiando un vapor saturado a un líquido saturado. Por lo tanto la diferencia de entalpia en la mezcla es igual a la cantidad de entalpia necesaria del agua. Para conocer la entalpia del producto en la torre es necesario ingresar a los resultados predichos por Aspen. En los resultados se pueden apreciar el caudal de salida, la temperatura que ya antes se conocía, se puede observar que el producto es completamente líquido además de poder conocer las unidades de la entalpia en este sistema (Tabla III.1) Tabla III.1 Resultados predichos por ASPEN en el líquido saturado Condiciones del líquido saturado Flujo total (kg/h) 153.1998 Temperatura (K) 315.4175 Fracción líquido 1 Entalpia (cal/s) -63 855.01 43 III RESULTADOS Ahora solo es necesario conocer la entalpia del vapor saturado y así poder conocer la diferencia de entalpias para la mezcla, para obtener estos resultados requeridos es necesario realizar una simulación más. Como equipo se utiliza un separador Flash2, un flujo másico de mezcla de 153.200295 Kg/hr que es la cantidad del destilado, una presión de entrada de 1 atm, a una temperatura de 25 °C y las concentraciones del etanol en el líquido resultante que es de 0.92345 en fracción masa (Figura III.4). En la configuración del separador se adaptan 0.2 atm ya que es la presión de operación en el sistema y se especifica como resultado un producto con una fracción de vapor en 0 ya que esto es un líquido saturado (Figura III.5). Al terminar la simulación se edita la configuración del separador cambiando la fracción vapor de 0 a 1 y esto es un vapor saturado (Figura III.6). Figura III.4 Condiciones de alimentación en el Flash2 para conocer las entalpias de la mezcla Figura III.5 Condiciones de operación Figura III.6 Condiciones de operación para obtener un líquido saturado. para obtener un vapor saturado. 44 III RESULTADOS Una vez realizadas ambas simulaciones se observan los resultados, y así podemos conocer la diferencia de entalpia entre el vapor (Tabla III.2) y el líquido. Además se puede apreciar que los resultados en el líquido saturado son los mismos que en el proceso; y se hace notar que los resultados en la temperatura, como ya se había mencionado, se mantiene constante. Tabla III.2 Condiciones en la primera columna del método de destilación extractiva Propiedades Condiciones del Condiciones del líquido saturado vapor saturado Flujo total (kg/h) 153.1998 153.1998 Temperatura (K) 315.4175 315.4178 Fracción líquido 1 0 Fracción vapor 0 1 Entalpia (cal/s) -63 855.01 -54 237.39 La diferencia de entalpias determina el calor latente, y se cálcula: ( ) ( ) ( ) Ahora se requiere calcular la cantidad de agua para lograr la diferencia de energía. Para este cálculo se diseña un diagrama (Figura III.7) en el cual se interpreta la transferencia de energía de la mezcla al agua que se encuentra a un temperatura ambiente. Figura III.7 Diagrama de interpretación de transferencia de energía en el condensador de la primera columna del método de destilación extractiva 45 III RESULTADOS La cantidad de agua necesaria se calcula: ( )( ) Por lo tanto como resultado final se concluye que la primera columna RadFrac del proceso no convencional requiere de 2.07765 Kg/s de vapor de agua a 352K en el re-hervidor y 1.37395 Kg/s de agua a 298K en el condensador. III.1.1 Resultados en la segunda columna en el proceso de destilación extractiva En la segunda columna de este proceso se puede observar que la energía necesaria en el condensador es de Qc=-904684 cal/s y la energía en el re-hervidor es de Qr=1127942 cal/s. Además la corriente del domo de la columna tiene una temperatura de 315 K, la corriente del fondo tiene una temperatura de 422 K, la corriente de alimentación superior es el flujo del etilenglicol el cual se presenta a 80 °C y la corriente de alimentación inferior es el producto de la primera columna que se encuentra a 315 K (Figura III.8). Figura III.8 Segunda columna en el proceso de destilación extractiva 46 III RESULTADOS Para poder realizar la separación del componente principal es requerido ingresar un vapor de agua de 442K. A la temperatura de 442K el vapor del agua tiene una y (Occon), por lo tanto la diferencia de estas es , siendo este el calor latente ( ) cedido. Para el re- hervidor de esta columna, la cantidad de vapor requerido esta temperatura es: ( ) Para el cálculo de la cantidad de agua necesaria para condensar el producto principal se obtienen las características en los productos tanto como en el vapor saturado como en el líquido saturado (Tabla III.3). Tabla III.3 Condiciones en la 2a columna del método de destilación extractiva Propiedades Condiciones del Condiciones del líquido saturado vapor saturado Flujo total (kg/h) 145.6317 145.6317 Temperatura (K) 315.4702 315.4704 Fracción líquido 1 0 Fracción vapor 0 1 Entalpia (cal/s) -57 771.64 -49 072.77 La diferencia de entalpias determina el calor latente, y se cálcula: ( ) ( ) ( ) Para este cálculo se diseña un diagrama (Figura III.9) en el cual se interpreta la transferencia de energía del producto principal al agua que se encuentra a una temperatura ambiente. 47 III RESULTADOS Figura III.9 Diagrama de interpretación de transferencia de energía en el condensador de la segunda columna del método de destilación extractiva La cantidad de agua necesaria se calcula: ( )( ) Por lo tanto como resultado final concluimos que la segunda columna RadFrac del proceso no convencional requiere de 2.3015 kg/s de vapor de agua a 442K en el re-hervidor y 1.242695 kg/s de agua a 25°C en el condensador. Además en esta columna se requieren 17900Kg de etilenglicol, pero esta es solo una carga inicial de disolvente en el proceso ya que después en esta misma columna es necesario añadir 2.835 kg/h. III.1.2 Resultados en la tercera columna en el proceso de destilación extractiva En la tercera columna de este proceso se puede observar que la energía necesaria en el condensador es de Qc=-68791 cal/s y la energía en el re-hervidor es de Qr=72434cal/s. Además la corriente del domo de la columna tiene una temperatura de 322 K, la corriente del fondo tiene una temperatura de 423 K, la corriente del fondo de la columna anterior es el flujo de la nueva mezcla aguaetilenglicol la cual se encuentra a 422 K y es el producto del fondo de la segunda columna (Figura III.10). 48 III RESULTADOS Figura III.10 Tercera columna en el proceso de destilación extractiva Para poder realizar la recuperación del disolvente es requerido ingresar un vapor de agua de 443K. A la temperatura de 443K el vapor del agua tiene una y (Occon), por lo tanto la diferencia de estas es , siendo este el calor latente ( ) cedido. Para el re- hervidor de esta columna, la cantidad de vapor requerido esta temperatura es: ( ) Para el cálculo de la cantidad de agua necesaria para condensar el agua residual se obtienen las características en los productos tanto como en el vapor saturado y como en el líquido saturado (Tabla III.4). Tabla III.4 Condiciones en la 3a columna del método de destilación extractiva Propiedades Flujo total (kg/h) Temperatura (K) Fracción líquido Fracción vapor Entalpia (cal/s) Condiciones del líquido saturado 10.20059 322.4079 1 0 -7 379.147 Condiciones del vapor saturado 10.20059 329.4537 0 1 -6 943.814 La diferencia de entalpias determina el calor latente, y se cálcula: ( ) ( ) ( ) 49 III RESULTADOS Para este cálculo se diseña un diagrama (Figura III.11) en el cual se interpreta la transferencia de energía de la mezcla al agua que se encuentra a un temperatura ambiente. Figura III.11 Diagrama de interpretación de transferencia de energía en el condensador de la tercera columna del método de destilación extractiva La cantidad de agua necesaria se calcula: ( )( ) Por lo tanto como resultado final se concluye que la tercera columna RadFrac del proceso no convencional requiere de 0.14803 kg/s de vapor de agua a 431K en el re-boiler y de 0.02291 kg/s de agua a 25°C en el condensador. III.1.3 Cantidad de agua necesaria para reducir la temperatura del disolvente El intercambiador de calor es muy indispensable en el proceso. Este permite reducir la temperatura del disolvente, en la corriente que es proveniente del mezclador y que se encuentra a 423K y es entregada por la corriente que ingresa a la segunda columna del proceso y esta se encuentra a 353K. Se puede observar que la cantidad de energía retirada es Q=-226901cal/s (Figura III.12). Figura III.12 Intercambiador de calor requerido en el proceso. 50 III RESULTADOS Para el cálculo de la cantidad de agua necesaria para enfriar el disolvente requerido en el reflujo se obtienen las características en la entrada del intercambiador y en la salida del mismo (Tabla III.5). Tabla III.5 Condiciones del disolvente en el intercambiador de calor del método de destilación extractiva Propiedades Flujo total (kg/h) Temperatura (K) Entalpia (cal/s) Presión (atm) Disolvente caliente Disolvente frio 17 900 422.9959 -8.3901 x 106 0.2 17 900 353.9959 -8.6170 x 106 0.2 Para este cálculo se diseña un diagrama (Figura III.13) en el cual se interpreta la transferencia de energía del disolvente al agua que se encuentra a una temperatura ambiente. Figura III.13 Diagrama de interpretación de transferencia de energía en el intercambiador de calor del método de destilación extractiva La diferencia de entalpias determina el calor latente, y se cálcula: ( ) La cantidad de agua necesaria se calcula: ( )( ) Se concluye que en el intercambiador de calor del proceso no convencional se requiere de 9.076 kg/s de agua a 25°C para poder enfriar el disolvente. 51 III RESULTADOS III.2 Resultados en el método de la extracción L-L columna RadFrac En la única columna RadFrac del proceso de separación por el método convencional se puede observar que la energía necesaria en el condensador es de Qc=-475451cal/s y la energía en el re-boiler es de Qr=525139cal/s. Además la corriente del domo de la columna tiene una temperatura de 351K, la corriente del fondo tiene una temperatura de 417K y la corriente proveniente de la torre de extracción L-L se encuentra a 296K (Figura III.14). Figura III.14 Única columna RadFrac en el método de la extracción L-L Para poder realizar la separación de la mezcla es requerido ingresar un vapor de agua de 437K. A la temperatura de 437K el vapor del agua tiene una y estas es (Occon), por lo tanto la diferencia de , siendo este el calor latente ( ) cedido. Por lo tanto, para el re-hervidor de esta columna la cantidad de vapor requerido a esta temperatura es: ( ) Para el cálculo de la cantidad de agua necesaria para condensar el producto principal se obtienen las características en los productos tanto como en el vapor saturado como en el líquido saturado (Tabla III.6). 52 III RESULTADOS Tabla III.6 Condiciones en la única columna del método de extracción L-L Propiedades Flujo total (kg/h) Condiciones del líquido saturado 172.5 Condiciones del vapor saturado 172.5 Temperatura (K) Fracción líquido Fracción vapor Entalpia (cal/s) 351.3386 1 0 -70 069.31 315.4703 0 1 -60 093.52 La diferencia de entalpias determina el calor latente, y se cálcula: ( ) ( ) ( ) Para este cálculo se diseña un diagrama (Figura III.15) en el cual se interpreta la transferencia de energía del producto principal al agua que se encuentra a una temperatura ambiente. Figura III.15 Diagrama de interpretación de transferencia de energía en el condensador de la columna del método de la extracción L-L La cantidad de agua necesaria se calcula: ( )( ) Por lo tanto como resultado final se concluye que la única columna RadFrac del proceso convencional requiere de 1.0630 kg/s de vapor de agua a 437K en el rehervidor y de 0.5868 kg/s de agua a 25°C en el condensador. 53 III RESULTADOS III.2.1 Cantidad de agua necesaria para reducir la temperatura del disolvente El intercambiador de calor es muy indispensable en el proceso. Este permite reducir la temperatura del disolvente, en la corriente que proviene del divisor y que se encuentra a 417 K y es entregada por la corriente que ingresa al mezclador del proceso y que se encuentra a 298 K. Se puede observar que la cantidad de energía retirada es Q=- 47.379 Kcal/s (Figura III.16). Figura III.16 Intercambiador de calor en el método de la extracción L-L Para el cálculo de la cantidad de agua necesaria para enfriar el disolvente requerido en el reflujo se obtienen las características en el disolvente de entrada y salida (Tabla III.7) del intercambiador de calor. Tabla III.7 Condiciones del disolvente en el intercambiador de calor Propiedades Disolvente caliente Disolvente frio Flujo total (kg/h) 3038.952 3038.952 Temperatura (K) 417.4535 298.15 Entalpia (cal/s) 1815.411 - 45 563.64 Presión (atm) 1 1 Para este cálculo se diseña un diagrama (Figura III.17) en el cual se puede interpretar la transferencia de energía del disolvente al agua que se encuentra a una temperatura ambiente. 54 III RESULTADOS Figura III.17 Diagrama de interpretación de transferencia de energía en el intercambiador de calor del método de la extracción L-L La diferencia de entalpias determina el calor latente, y se cálcula: ( ) La cantidad de agua necesaria se calcula: ( )( ) Se concluye que en el intercambiador de calor del proceso convencional se requiere de 1.8951 kg/s de agua a 25 °C para poder enfriar el disolvente. III.2.2 Cantidad de disolvente para en la extracción L-L Para la columna de extracción líquido-líquido se requieren 3040Kg de ortoxileno como cantidad de disolvente de inicio, pero una vez que el proceso comienza con a recircular solo es necesario añadir 0.7835 kg de ortoxileno por cada hora. III.3 Optimización de costos del proceso. Optimizar implica lograr un mejor resultado posible del proceso mediante el aprovechamiento al máximo de sus potencialidades, tomando en cuenta ciertos factores importantes como lo son: tiempo, costo, calidad y eficiencia, es decir, hacer más con menos. Dado que el objetivo primordial es la optimización de costos (de servicio, procesos de operación y equipo) se deduce que la 55 III RESULTADOS optimización de costos es la obtención de un buen resultado aprovechando todos los elementos disponibles que intervienen en el proceso, permitiendo conocer el mejor precio disponible y contar con los datos acerca de áreas que requieren atención en cuanto a pérdidas…etc., para la toma de una decisión, en este caso la elección de un mejor proceso. III.3.1 Optimización de costos del disolvente La operación del Divisor FSplit, determina la cantidad de disolvente que regresa a la columna de destilación extractiva. Pero en el proceso el disolvente ha perdido pureza por haberse mezclado con la mezcla azeotrópica en la segunda columna y se sometió a una rectificación con el residuo del agua y etanol, para poder determinar la cantidad correcta que debe regresar primero se debe saber cuánta es la cantidad de disolvente impuro que sale por el fondo de la tercera columna. Para poder determinar esto es necesario escoger la corriente de desahogo e indicar que la fracción de salida sea de uno. Una vez establecidas todas las condiciones de operaciones se realizó la simulación, determinando que la corriente de retorno del disolvente no presenta alguna variacion y que los resultados son correctos. Se sabe que por la corriente de desahogo la cantidad de disolvente impuro es de 17900.2106 Kg/hr se realiza la operación de reflujo editando la configuración del divisor descontando el flujo de la corriente de desahogo en pequeñas porciones, la cantidad que es retirada también debe de ser excluida de la alimentación del etilenglicol fresco (Tabla III.8) 56 III RESULTADOS Tabla III.8 Optimización de costos de disolvente mediante la variacion de alimentación del disolvente (etilenglicol) y en el Split Optimización de costos de disolvente en la destilación Extractiva Alimentación del Disolvente en Moles de etanol puro disolvente (kg/h) el Split (kg/h) en el destilado 17 930.33 17 900.2 0 8 914.835 8 985.165 0.5019 4 914.835 12 985.165 0.7253 914.835 16 985.165 0.9488 514.835 17 385.165 0.9711 214.835 17 685.165 0.9879 14.835 17 885.165 0.9991 4.835 17 895.165 0.9996 2.835 17 897.165 0.9998 Llegando al término de la simulación con resultados correctos y quedando como conclusión que por cada hora se deben de retirar del circuito 2.835 kg de etilenglicol contaminado y por lo tanto por cada hora se debe de añadir 2.835kg de etilenglicol fresco. Y en la última simulación por el método de la destilación extractiva se observaron resultados favorables dado que las corrientes de desahogo y retorno tienen actividad. Por lo tanto, la cantidad de disolvente a utilizar en el proceso es el mínimo, cumpliendo así con los aspectos que determinan la optimización de costo siendo un disolvente de gran calidad, a un mínimo gasto en determinado tiempo y con una gran eficiencia. III.3.2 Optimización de costos de equipo. Para la optimización de costos de equipo se toma como base estándar las especificaciones (número de platos y relación de reflujo) ya establecidas en los equipos (columnas, intercambiadores de calor, etc.), las cuales se modificaran de manera proporcional (Tabla III.9); ya sea aumentando o disminuyendo el valor de 57 III RESULTADOS dichas especificaciones tomando en cuenta la eficiencia en el proceso (cantidad de etanol en el destilado). Tabla III.9 Optimización de costos de equipo a partir de la modificación del S y la relación de reflujo. Optimización de costos de Equipo en la Destilación Extractiva con una alimentación de etanol de 70.7101 kg/h 1a Columna 2a Columna 3a Columna Cantidad de etanol S Relación S de reflujo Relación S de reflujo Relación destilado (kg/h) Eficiencia de reflujo 24 100 32 100 21 100 145.4509 0.9985 25 105 33 105 22 105 145.4509 0.9985 27 113 34 113 20 95 145.5318 0.9992 28 117 . . 18 86 145.5318 0.9992 . . . . 16 76 145.5318 0.9992 . . . . 14 67 145.5368 0.9998 28 117 34 113 12 57 145.5368 0.9998 La tabla anterior muestra los resultados obtenidos por la simulación, en ella se puede observar el comportamiento de las modificaciones realizadas en cada una de las columnas. La modificación del número de platos y la relación de reflujo determinaron la cantidad de etanol puro en el destilado; nótese que en la segunda columna no se realizaron demasiadas modificaciones, dado que los resultados se mantuvieron constantes. Por otro lado, al aumentar ambas especificaciones en la primera y tercera columna no hubo cambio alguno en la cantidad de destilado, mientras que al disminuir las especificaciones en ambas, tanto la eficiencia como la cantidad de destilado fueron aumentando de manera proporcional a pequeña escala, cumpliendo así con los aspectos que determinan la optimización de costo de equipo, es decir, con un mínimo de equipo es posible llevar a cabo un proceso de calidad y eficiencia en la destilación extractiva. 58 III RESULTADOS III.4 Comparación de métodos en costos generales Una vez calculadas las cantidades totales de los insumos y servicios es necesario comparar entre métodos los costos de operación considerando también las condiciones del producto principal. Los costos de los disolventes son de: 2.77 dollar/kg de etilenglicol (Ballester Productos Químicos S.A 2013) y el costo del ortoxileno es de 3 dollar/kg (Poveda 2000). Se considera un vapor estándar debido a que los precios de tal varían con respecto a la ciudad y se considera un vapor con una temperatura aun mayor a la máxima requerida en todos los procesos el costo de este vapor a los 164°C es de 0.121 dollar/kg (CONAE, 2013), el costo de agua para enfriamiento es de 0.285dollar/tonelada (CONAGUA San Luis Potosí 2012). Como característica principal en el producto se considera su composición en fracción masa ya que anteriormente en este trabajo se mencionó que la concentración mínima requerida para el producto pueda ser ocupado con aditivo para la gasolina es de 0.99 en fracción masa, la cual se logró alcanzar en uno de los dos métodos. También es necesario tomar en cuenta la cantidad de producto obtenido y la eficiencia del proceso para poder calcular el costo aproximado de un kilogramo de etanol. III.4.1 Comparación en la eficiencia de los métodos La eficiencia de los diferentes métodos es aquella que como su nombre lo menciona, demuestra la eficiencia del proceso, multiplicado la cantidad de etanol obtenido por la composición obtenida (Tabla III.10) y dividiéndola por la cantidad de etanol disponible en la mezcla inicial la cual para todos los casos es 167.6382 Kg/hr. 59 III RESULTADOS Tabla III.10 Comparación de las condiciones del etanol como producto principal entre los dos método. Propiedades Método No convencional Método convencional (Destilación extractiva) (Extracción L-L) Fracción mol Etanol 0.9998 0.9472 Agua 1.2139 x 10-3 0.05243 Solvente 8.5999 x 10-35 (etanol) 4.8676 x 10-4 (o-xileno) Flujo total (kmol/h) 3.163500 3.899428 Flujo total (kg/h) 145.6317 172.5 Temperatura (K) 315.4702 351.3385 En ese caso la eficiencia para cada método se obtiene que de la siguiente manera: ( ) ( ) III.5 Costos del material utilizando método de Guthrie en la destilación extractiva (método no convencional) Cuando una tecnología es bien conocida, puede tenerse acceso a su costo de inversión a través de fuentes especializadas o del licenciador de la tecnología. Cuando el proceso está en desarrollo a escala laboratorio, esta estimación generalmente no está disponible. Es necesario en estos casos tener alguna estimación razonable del potencial económico del proceso en desarrollo, aun cuando esta estimación no sea completamente precisa. 60 III RESULTADOS Una estimación aceptable en un momento dado puede servir para discriminar alternativas, o para evitar esfuerzos de tiempo y dinero en proyectos que no ofrezcan un buen potencial económico. A partir de los balances de materia realizados y mediante la implementación de las operaciones unitarias tomando como base principal el simulador Aspen, es posible determinar los parámetros que delimitaran los costos del proceso y los servicios que implican la optimización de costos mediante la aplicación del método de Guthrie. III.5.1 Especificaciones para la primera columna a) Determinación del diámetro de la columna (Dc): ( ) ( ) ( )( [( [( ( )( ) ) )( )( )( )( )] )( )( )( )( )( )] b) Número de platos (S): c) Altura de la columna (Hc), considerando ( ) ( ) d) Área de la columna: ( ( ) ) ( ( ) ) 61 III RESULTADOS Despejando A de la ecuación se tiene que: )( ( ) III.5.1.1 Determinación de costos del material para la primera columna Una vez determinadas las especificaciones de la columna se procede a calcular los costos de los materiales a utilizar, tales como: Costo del intercambiador de calor: a) El costo base (Cb) de unidad de es de $ 3500 b) Los factores de ajuste son: c) El costo ajustado es (Cfob): ( ) [ ( ) ] [ ( )( )] d) Para intercambiadores de calor, el factor del módulo desnudo( ) e) El costo de una unidad en base acero al carbón, presiones inferiores a 150 psi, reboiler Kettle y en 1968 es: ( ) ( ) f) El costo de la unidad deseada se obtiene sumando al rubro anterior la diferencia entre ( ) [ , lo cual da lugar al módulo desnudo ajustado: ( )] [ ( )] Este valor representa el costo de la unidad deseada en 1968, año base de la información usada. Para estimar el costo de la unidad en el año 2000, se emplean los índices de costos de Chemical Engineering. Por lo tanto ( ) ( ) ( ) 62 III RESULTADOS Añadiendo 15% de contingencias se obtiene el costo deseado: ( ) ( ) ( ) Costo del recipiente de proceso: a) El Cb de una unidad de es de $ 32 500 b) Los factores de ajuste son: ( ) c) [ [( ] ) ( ) ( )] d) Para recipientes verticales, el factor del módulo desnudo ( ) e) ( ) ( ( ) f) [ ( ) ) ( ( )] ) ( ) Añadiendo 15% de contingencias se obtiene el costo deseado: ( ) ( ) ( ) Costo de los platos: a) El Cb de una unidad de es de $ 15 000 b) Los factores de ajuste son: c) ( ) [ ( [ )] ( )] d) Para recipientes horizontales, el factor del módulo desnudo ( ) e) ( ) ( ) f) ( ) ( [ ) ( ( ) )] ( ) 63 III RESULTADOS Añadiendo 15% de contingencias se obtiene el costo deseado: ( ) ( ) ( ) III.5.2 Especificaciones para la segunda columna a) Determinación del diámetro de la columna (Dc): ( ) [( ( ( ) ) )( )( )( )( )( )( )] b) Número de platos (S): c) Altura de la columna (Hc), considerando ( ) ( ) d) Área de la columna: ( ( ) ) ( ( ) ) Despejando A de la ecuación se tiene que: ( )( ) 64 III RESULTADOS III.5.2.1 Determinación de costos del material para la segunda columna Una vez determinadas las especificaciones de la columna se procede a calcular los costos de los materiales a utilizar, tales como: Costo del intercambiador de calor: a) El Cb de una unidad de es de $ 2800 b) Los factores de ajuste son: c) ( ) [ ( ) [ ] ( )( )] d) Para intercambiadores de calor, el factor del módulo desnudo( ) e) El costo de una unidad en base acero al carbón, presiones inferiores a 150 psi, reboiler Kettle y en 1968 es: ( ) ( ) f) ( [ ) ( )] [ ( )] Por lo tanto ( ) ( ) ( ) Añadiendo 15% de contingencias se obtiene el costo deseado: ( ) ( ) ( ) Costo del recipiente de proceso: a) El Cb de una unidad de es de $ 35 000 b) Los factores de ajuste son: c) ( ) [ ] [( ) ( ) ( )] d) Para recipientes verticales, el factor del módulo desnudo ( ) e) f) ( ) ( ) ( [ ( ) )] 65 III RESULTADOS ( ) ( ) ( ) Añadiendo 15% de contingencias se obtiene el costo deseado: ( ) ( ) ( ) Costo de los platos: a) El Cb de una unidad de es de $ 16 500 b) Los factores de ajuste son: ( ) c) [ ( [ )] ( )] d) Para recipientes horizontales, el factor del módulo desnudo ( ) ( ) e) ( ) f) ( [ ( ) ) ( ( )] ) ( ) Añadiendo 15% de contingencias se obtiene el costo deseado: ( ) ( ) ( ) III.5.3 Especificaciones para la tercera columna a) Determinación del diámetro de la columna (Dc): ( ) [( ( ( ) )( ) )( )( )( )( )( )] 66 III RESULTADOS b) Número de platos (S): c) Altura de la columna (Hc), considerando ( ) ( ) d) Área de la columna: ( ( ) ) ( ) ( ) Despejando A de la ecuación se tiene que: )( ( ) III.5.3.1 Determinación de costos del material para la tercera columna Una vez determinadas las especificaciones de la columna se procede a calcular los costos de los materiales a utilizar, tales como: Costo del intercambiador de calor: a) El Cb de una unidad de es de $ 325 b) Los factores de ajuste son: c) ( ) [ ( ) [ ] ( )( )] d) Para intercambiadores de calor, el factor del módulo desnudo( ) e) f) ( ) ( ) ( [ ) ( )] 67 III RESULTADOS Por lo tanto ( ) ( ) ( ) Añadiendo 15% de contingencias se obtiene el costo deseado: ( ) ( ) ( ) Costo del recipiente de proceso: a) El Cb de una unidad de es de $ 6 000 b) Los factores de ajuste son: ( ) c) [ ] [( ) ( ) ( )] d) Para recipientes verticales, el factor del módulo desnudo ( ) e) ( ) ( ( ) f) [ ( ) ) ( ( )] ) ( ) Añadiendo 15% de contingencias se obtiene el costo deseado: ( ) ( ) ( ) Costo de los platos: a) El Cb de una unidad de es de $ 750 b) Los factores de ajuste son: c) ( ) [ ( )] [ ( )] d) Para recipientes horizontales, el factor del módulo desnudo ( ) e) f) ( ) ( ) ( [ ( ) )] 68 III RESULTADOS ( ) ( ) ( ) Añadiendo 15% de contingencias se obtiene el costo deseado: ( ) ( ) ( ) III.6 Costos del material utilizando método de Guthrie en la extracción L-L (método convencional) III.6.1 Especificaciones para la única columna RadFrac del proceso a) Determinación del diámetro de la columna (Dc): ( ) [( ( ) ( ) )( )( )( )( )( )( )] b) Número de platos (S): c) Altura de la columna (Hc), considerando ( ) ( ) d) Área de la columna: ( ( ) ) ( ( ) ) 69 III RESULTADOS Despejando A de la ecuación se tiene que: )( ( ) III.6.1.1 Determinación de costos del material para la única columna RadFrac del proceso Una vez determinadas las especificaciones de la columna se procede a calcular los costos de los materiales a utilizar, tales como: Costo del intercambiador de calor: a) El Cb de una unidad de es de $ 2 200 b) Los factores de ajuste son: c) ( ) [ ( ) [ ] ( )( )] d) Para intercambiadores de calor, el factor del módulo desnudo( ) e) f) ( ) ( ) ( [ ) ( )] Por lo tanto ( ) ( ) ( ) Añadiendo 15% de contingencias se obtiene el costo deseado: ( ) ( ) ( ) Costo del recipiente de proceso: a) El Cb de una unidad de es de $ 12 000 b) Los factores de ajuste son: 70 III RESULTADOS c) ( ) [ [( ] ) ( ) ( )] d) Para recipientes verticales, el factor del módulo desnudo ( ) e) ( ) ( ( ) f) [ ( ) ) ( ( )] ) ( ) Añadiendo 15% de contingencias se obtiene el costo deseado: ( ) ( ) ( ) Costo de los platos: a) El Cb de una unidad de es de $ 3 000 b) Los factores de ajuste son: c) ( ) [ ( )] [ ( )] d) Para recipientes horizontales, el factor del módulo desnudo ( ) e) ( ) ( ) f) ( [ ( ) ) ( ( )] ) ( ) Añadiendo 15% de contingencias se obtiene el costo deseado: ( ) ( ) ( ) 71 III RESULTADOS III.6.2 Especificaciones para la columna de extracción L-L Suponiendo una velocidad de flujo de la alimentación del solvente de 500 y 750 gal/min respectivamente. Asumiendo un tiempo de residencia ( una capacidad de ) de 5 min, con se determina el diámetro y altura de la columna de extracción. a) Diámetro y altura de la columna asumiendo que ( ) )( ( ( ( √ √ ) ) ) ( ) } b) Altura de la columna: ( ) 72 III RESULTADOS III.6.2.1 Determinación del costo del material para la columna de extracción L-L Costo del recipiente de proceso: a) El Cb de una unidad de es de $ 8 000 b) Los factores de ajuste son: c) ( ) [ [( ] ) ( ) ( )] d) Para recipientes verticales, el factor del módulo desnudo ( ) e) ( ) ( ) f) ( [ ( ) ) ( ( )] ) ( ) Añadiendo 15% de contingencias se obtiene el costo deseado: ( ) ( ) ( ) III.6.3 Comparación en los costos de operación y material adquiridos en cada método En la comparación de costos de procesos solo se toman en cuenta los costos de servicios e insumos como la cantidad total de vapor debido que se utiliza el mismo costo para todas las temperaturas, la cantidad de agua para enfriamiento y la cantidad de disolvente en la reposición. La suma de todos los costos es el costo total de operación. En las siguientes tablas se pueden observar los costos de disolvente (Tabla III.11) y el costo operación (Tabla III.12), sin embargo, en el costo de operación no se incluye el costo del disolvente, ya que este solo es una inversión inicial, el costo que en realidad interesa es el costo del disolvente retroalimentado, ya que este se añade cada hora del proceso. 73 III RESULTADOS Tabla III.11 Costo total de operación en los métodos del proceso Costo total de Operación en el Proceso Método No Convencional Método Convencional (Destilación Extractiva) (Extracción L-L) Cantidad Precio Precio Cantidad Precio Precio total requerida unitario total requerida unitario (dólar/h) (kg/h) (dólar/kg) (dólar/h) (kg/h) (dólar/kg) 17 900 2.77 3 040 3.81 11 582.4 2.835 2.77 7.8530 0.7835 3.81 2.9851 42 175.998 2.85 E-4 12.0202 8 934.84 2.85 E-4 2.5464 16 297.848 0.121 1972.04 3 826.8 0.121 463.0428 Disolvente de inicio “no incluido en el costo del 49 583 proceso Disolvente reposición Agua total enfriam. Vapor total generación de calor Costo total operación (dólar/h) 1 991.9132 468.5743 Tabla III.12 Costos de material adquirido en cada método del proceso Costos generados Método No Convencional Método Convencional (en dólares) (Destilación Extractiva) (Extracción L-L) Costo del intercambiador $ 130 890.6574 $ 42 031.40624 $ 1 271 513.218 $ 345 989.9913 $ 499 875.772 $ 60 867.13325 Total $ 1 902 279.647 $ 448 888.5308 Total (más 10% déficit) $ 2 092 507.612 $ 493 777.3839 de calor Costo del recipiente del proceso (Columna) Costo de los platos de la columna 74 CONCLUSIONES CONCLUSIONES Y RECOMENDACIONES Se concluye que el método de destilación extractiva es el menos eficiente de los dos procesos, así como también es el más alto en costos (de material y operación), por lo tanto el costo de un kg de etanol es muy elevado. Sin embargo el proceso logra una pureza muy elevada, deduciendo que este método no es el indicado para la mezcla etanol-agua; sin embargo si es un buen método para la separación de mezcla azeotrópicas. Por otra parte el método de la extracción L-L es el más eficiente de los dos, su costo (de material y operación) es el más económico por lo tanto el costo del etanol producido es el más bajo, dado que la concentración con la que se obtiene el alcohol es menor, por lo tanto, es poco viable la obtención de etanol como aditivo para la gasolina , pero si se llegase a encontrar un disolvente más miscible y menos soluble en el agua, puede ser que este método sea el más competente. También se sugiere comprobar los datos de equilibrio de la mezcla etanolortoxileno mediante un método experimental. Uno de los aspectos de mayor importancia desarrollados durante el proceso de la simulación fueron: la modificación de los platos de las columnas tomando en cuenta la relación de reflujo, la cual consistió en el aumento o disminución en el número de platos y en la relación de reflujo, cantidad de etanol tomando siempre en cuenta la en el destilado. Esto permitió determinar la eficiencia del proceso, el análisis previo a la optimización de los costos y la concentración de etanol obtenida en el destilado; y la modificación del solvente a partir del cual se determinó la pureza del etanol, el costo del solvente inicial y el costo de solvente durante el proceso. De acuerdo a la metodología utilizada en cada proceso, fue posible determinar la configuración, las condiciones de proceso y los costos tanto de operación como de material óptimas para la separación de dicha mezcla mediante el Simulador Aspen; logrando cumplir con los objetivos propuestos, concluyendo teóricamente que si de optimizar costos en la separación de una mezcla azeotrópica se trata, el Método Convencional (Extracción L-L) es el más viable. 75 BIBLIOGRAFÍA REFERENCIA BIBLIOGRAFICA Quintero, Montoya, Sánchez y Cardona (2007). EVALUACIÓN DE LA DESHIDRATACIÓN DE ALCOHOL CARBURANTE MEDIANTE SIMULACIÓN DE PROCESOS [versión electrónica] Facultad de Ciencias Agropecuarias Vol. 5 No. 2 Agosto 2007, pp. 72-83 Mejía, Espinal, y Mondragón (2006). ESTUDIO DEL AZEOTRÓPO ETANOL - AGUA. CARACTERIZACIÓN MOLECULAR DE DÍMEROS DE ETANOL, HETERODÍMEROS Y HETEROTRÍMEROS DE ETANOL-AGUA [versión electrónica] Energética, núm. 36, diciembre, 2006, pp. 5-18 Becerra (2008). LA INDUSTRIA DEL ETANOL EN MÉXICO [versión electrónica] Economía unam vol. 6 núm. 16, 2008, pp. 82-98 Shakhashiri (n.d.). CHEMICAL OF THE WEEK. Revised: 5 Feb 2009 www.scifun.org R. Chang. (1999). Química (6ta ed.). McGraw-Hill, México. Ocon y Tojo, Problemas de Ingeniería Química, Ed. Aguilar, España, 1968. José Luis Moreno Frigols, Ramón García Doménech, Gerardo M. Introducción a la fisicoquímica. Publi. Universitat Valencia, 2007 Acosta, Rodríguez, Lodeiro y Nuevas (2010) SEPARACION DE LOS COMPONENTES DE LA MEZCLA AZEOTRÓPICA ACETONA-N-HEXANO MEDIANTE UN PROCESO COMBINADO: EXTRACCION LÍQUIDOLÍQUIDO Y DESTILACION DISCONTINUA [versión electrónica] Ciencias Químicas, vol. 41, núm. 1, enero-abril, 2010, pp. 37-42 Uyazán, Gil, Aguilar, Rodríguez y Caicedo (2004) DESHIDRATACION DEL ETANOL [versión electrónica] Ingeniería e Investigación, núm. 003, diciembre, 2004 pp. 49-59 Robert E. Treybal, OPERACIONES DE TRANSFERENCIA DE MASA (2da ed.) 1988 76 BIBLIOGRAFÍA Fabrizzio Valer Gómez (2009) EXTRACCION LÍQUIDO-LÍQUIDO; DISEÑO Y ANALISIS DEL FUNCIONAMIENTO DEL EQUIPO Escuela Profesional de Ingeniería Química Laboratorio de Control de Procesos Vásquez, Ruiz, Arango, Caicedo, Sánchez, Ríos y Restrepo (2007) PRODUCCIÓN DE ETANOL ABSOLUTO POR DESTILACIÓN EXTRACTIVA COMBINADA CON EFECTO SALINO [versión electrónica] Dyna, Año 74, Núm. 151, pp. 53-59. Medellín, Marzo de 2007. ISSN 00127353 Jiménez Gutiérrez, Arturo. ”Estimación de costos de Inversión” DISEÑO DE PROCESOS EN INGENIERIA QUÍMICA. Editorial Reverté S.A. Pp. 38- 51.1968 Guthrie, K.M. Capital Cost Estimation, Chemical Engineering. Pp.114124.1969. Seader J.D. y Henley, Ernest J. Separation Process Principles. Editorial John Wiley y Sons, Inc. Segunda Edición. Pp. 123 – 129. A. Marcilla Gomis (1998) INTRODUCCIÓN A LAS OPERACIONES DE SEPARACIÓN CÁLCULO POR ETAPAS DE EQUILIBRIO Publicaciones Universidad de Alicante Morrison, R. T. y Boyd, R. N.: QUÍMICA ORGÁNICA. Addison-Wesley Iberoamericana, S. A. Wilmington. Delaware, E.U.A., 1990. Perry Robert, H. Perry´s Chemical engineers` handbook. 7ªEdición. Ed. McGraw -Hill 1999. Warren L. McCabe, Julian C. Smith, Peter Harriott. Operaciones unitarias en ingeniería química. 7ª Edición. Ed. McGraw -Hill 2007. Rosa María Jiménez Olmos (2008) USO EFICIENTE DE LA ENERGIA EN CALDERAS. Seminario “Uso eficiente de la energía en calderas y sistemas de refrigeración”, CONAE. Gabriel Poveda Ramos (2000) TAMAÑO OPTIMO DE PLANTAS INDUSTRIALES [versión electrónica] Dyna, año 2000, Nro. 130, pp. 21-30. Medellín 77 ANEXOS ANEXOS Tablas y gráficas que determinan la optimización de costos de equipo por el método de Guthrie. I ANEXOS II ANEXOS Costo base de recipientes de proceso III