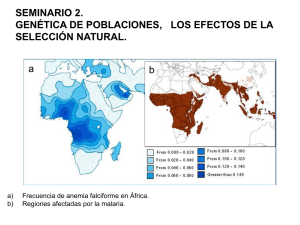
Hipotesis y Objetivos - Universidad de Granada
Anuncio

Universidad de Granada Instituto de Biotecnología Facultad de Medicina Departamento de Fisiología Determinación de los haplotipos de anemia falciforme y su correlación con los niveles de estrés oxidativo en pacientes de 6 meses a 15 años de edad en Panamá Iryna Rusanova Granada, 2010 Editor: Editorial de la Universidad de Granada Autor: Iryna Rusanova D.L.: GR 830-2011 ISBN: 978-84-694-0210-8 D. DARÍO ACUÑA CASTROVIEJO, Catedrático de Fisiología de la Facultad de Medicina de la Universidad de Granada, CERTIFICA QUE Dña Iryna Rusanova, Licenciada en Bioquímica, ha realizado bajo su dirección y en el Instituto de Biotecnología de la Universidad de Granada, el trabajo titulado: “Determinación de los haplotipos de anemia falciforme y su correlación con los niveles de estrés oxidativo en pacientes de 6 meses a 15 años de edad en Panamá”, reuniendo el mismo las condiciones necesarias para optar al grado de Doctor. Granada, 20 de octubre de 2010 Vº Bº Director La interesada Darío Acuña Castroviejo Iryna Rusanova Dña. GERMAINE ESCAMES, Profesora Titular Fisiología de la Facultad de Medicina de la Universidad de Granada, CERTIFICA QUE Dña Iryna Rusanova, Licenciada en Bioquímica, ha realizado bajo su dirección y en el Instituto de Biotecnología de la Universidad de Granada, el trabajo titulado: “Determinación de los haplotipos de anemia falciforme y su correlación con los niveles de estrés oxidativo en pacientes de 6 meses a 15 años de edad en Panamá”, reuniendo el mismo las condiciones necesarias para optar al grado de Doctor. Granada, 20 de octubre de 2010 Vº Bº Director Germaine Escames La interesada Iryna Rusanova Beca y proyectos de investigación que han financiado este estudio: Este trabajo se ha realizado en el Laboratorio del Grupo de Investigación CTS101: Comunicación Intercelular, del Instituto de Biotecnología de la Universidad de Granada, en el Centro de Investigación Biomédica, Parque Tecnológico de Ciencias de la Salud, Granada, y el Hospital del Niño de Panamá. 1. Becas disfrutadas Beca SENACYT-IFARHU. 2. Proyectos de Investigación que han financiado la investigación: 1. Financiación del Grupo de Investigación CTS-101: Comunicación Intercelular. 2. Secretaría Nacional de Cencia, Tecnología e Innovación (SENACYT) de Panamá, proyecto FID08-124. 3. Publicaciones científicas: Autores: Rusanova I, Escames G, Cossio G, de Borace R, Chahbouni M, López LC, Díez T, Acuña-Castroviejo D Título: Oxidative stress status, clinical outcome, and -globin haplotypes in sickle cell anemic pediatric patients Ref. Revista: Eur J Haematol 2010, in press: doi: 10.1111/j.1600-0609.2010.01528.x. Financiación: CTS-101 - FID08-124 Áreas: Hematology Posición/Áreas: 35/61 F. Impacto: 2.345 PMDI: 20846340 Clave: artículo Autores: Rusanova I, Cossio G, Moreno B, Perea J, de Borace R, Perea M, Escames G, Acuña-Castroviejo D Título: β-Globin Gene Cluster Haplotypes in Sickle Cell Patients From Panamá Ref. Revista: Am J Hum Biol 2010, submitted: Financiación: CTS-101 - FID08-124 Áreas: Biology Posición/Áreas: 23/76 F. Impacto: 2.121 PMDI: Clave: artículo 4. Congresos: Comunicación oral en el XIII Congreso Nacional de Ciencia y Tecnología de APANAC. Tema: ―Estrés Oxidativo, clínica y los haplotipos del custer globínico Beta en los pacientes pediátricos panameños con Anemia Falciforme‖ Presentado el día viernes 8 de octubre, en la sala 102, en el horario de 14:00 a 14:15 horas en el Centro de Convenciones de la Ciudad del Saber, Panamá, República de Panamá. Comunicación en forma de poster titulada: ―BIOMARCADORES DEL ESTRÉS OXIDATIVO, MANIFESTACIONES CLÍNICAS Y LOS HAPLOTIPOS DEL CLUSTER GLOBÍNICO BETA EN LOS PACIENTES PEDIÁTRICOS PANAMEÑOS CON ANEMIA DREPANOCÍTICA―, dentro de la LII Reunión Nacional de la Sociedad Española de Hematología y Hemoterapia (SEHH) y XXVI Congreso Nacional de la Sociedad Española de Trombosis y Hemostasia (SETH), a celebrar en Las Palmas de Gran Canaria, los días 28 al 30 de octubre de 2010. Nº DE PÓSTER: PO-24. “Hay dos formas de vivir la vida. Una es pensar que nada es un milagro. La otra es pensar que todo es un milagro.” Albert Einstein “Elige un trabajo que te guste y no tendrás que trabajar ni un día de tu vida. “ Confucio AGRADECIMIENTOS Desde que inicié este programa de Doctorado en Biotecnología impartido entre las dos universidades: Universidad de Panamá y Universidad de Granada, puedo mirar hacía atrás y decir que se ha recorrido un camino muy interesante, lleno de experiencias. Quiero agradecer en primer lugar a la Universidad de Panamá por haber abierto este programa, por las clases, ricas en conocimientos impartidos por distintos profesores. Quiero dar un agradecimiento especial al Doctor Tomas Díez como coordinador de este programa y persona que me apoyó mucho en este camino. Este tipo de experiencias en primer lugar abren camino para conocer gente, las necesidades del país, y en este sentido quiero agradecer la suerte de haber conocido a la Dra. Gradys Cossio, pediatra- genetista del Hospital del Niño, quién me presentó la problemática de salud que existe en Panamá y la necesidad de realizar estudios científicos. Sin este enlace entre el hospital y la universidad, no sería posible hacer investigaciones biomédicas aplicadas, tan necesarias para los países como Panamá. Gracias a todos los directivos del Hospital por abrirnos puertas y poder conseguir las muestras de sangre requeridas para este estudio. Gracias a la doctora Gradys Cossio por su positivismo y gran espíritu de luchadora constante por mejorar las condiciones de la investigación en el hospital. Sin el apoyo del Doctor Darío Acuña- Castroviejo de la Universidad de Granada no sería posible hacer este estudio completo y novedoso. Gracias, Doctor Acuña, por depositar su confianza en mí, por compartir sus conocimientos, por ofrecerme una oportunidad de aprender las técnicas y poder realizar las determinaciones completas en su laboratorio. Agradezco a la Doctora Germaine Escames por su don de gente, su amabilidad y apoyo en los momentos difíciles. Estos aspectos son sumamente valiosos cuando uno se encuentra frente a nuevos retos. Muchas gracias, Germaine, por tu paciencia, de todo mi corazón. Sin la sonrisa y un trato tan amable de todos los miembros del laboratorio este trabajo de día a día no sería nada fácil. Muchísimas gracias a Aracely, Ana, Anilla, Carolina, Carmen, José Antonio, Francis, José Carlos, Mariam, Miquel, Alberto, Laura y Luís Carlos. Agradezco a mi esposo Antonio, por estar a mi lado, brindándome ánimo y confianza. Gracias, mi amor, por saber escucharme y apoyarme en los momentos difíciles. Nada en mi vida sería posible sin el apoyo de mis padres, muchas gracias, papá y mamá por darme sus consejos, ánimo y esperanza. Sin ustedes no podría lograr muchas cosas en mi vida. Finalmente, agradezco a la Secretaría Nacional de Ciencia y Tecnología de Panamá por haber financiado una parte de este proyecto que corresponde al estudio de haplotipos del cluster beta en los pacientes panameños con anemia falciforme. -Abreviaturas- Iryna Rusanova ABREVIATURAS ADN: ácido desoxirribonucleico NADPH: nicotinamida adenina ARN: ácido ribonucleico dinucleótido fosfato BH4: tetrahidrobiopterina NOS: óxido nítrico sintasa b-NFkB O2-•: anión superóxido Ca2+: calcio ONOO-: peroxinitritos ELAM-: molécula de adhesión PCR: reacción en cadena de la endotelial de leucocitos 1 polimerasa eNOS: óxido nítrico sintasa endotelial RFLP: fragmentos de restricción de GPx: glutation peroxidasa longitud variable GRd: glutation reductasa RNS: especies reactivas de nitrógeno GSH: glutation reducido ROS: especies reactivas de oxígeno GSSG: glutation oxidado SOD: superóxido dismutasa H2O2: peróxido de hidrógeno SCD: sickle cell disease (anemia de HO•: radical hidroxilo células falciforme, inglés) IL: interleuquina TNF-: factor de necrosis tumoral iNOS: óxido nítrico sintasa inducible XO: xantina oxidasa LPO: peroxidación lipídica -Índice- Iryna Rusanova ÍNDICE Introducción 1. Anemia falciforme……………………………………………………..….. 2 1.1. Historia……………………………………………………..……. 2 1.2. Epidemiología…………………………………………….…..….. 4 1.3. Diagnóstico…………………………………………………...….. 7 1.4. Clínica de la enfermedad……………………………………...….10 1.5. Fisiopatología de la anemia falciforme………………………..….12 1.5.1. Fisiología de la hemoglobina ……………………….…12 1.5.2. Polimorfismos de genes del cluster beta………….........15 1.5.3. Otros polimorfismos de un solo nucleótido (SNPs)…….17 1.5.4. La variaciones en la expresión de la hemoglobina fetal...19 1.5.5. Niveles del estrés oxidativo asociado a la hemólisis…….20 1.5.6. Reducción en la biodisponibilidad de óxido nítrico……..21 2. Haplotipos de anemia falciforme……………………………………………24 3. Anemia falciforme y estrés oxidativo………………………………………..29 3.1. Radicales libres y enzimas involucradas en su producción…………29 3.2. Sistemas antioxidantes………………………………………………32 3.3. Fuentes de radicales libres en la anemia falciforme……………….. 37 3.4. Consecuencias del estrés oxidativo en la anemia falciforme……….39 4. Estrategias terapéuticas en la anemia falciforme…………………………..39 4.1. Terapias tradicionales………………………………………………..39 4.2. Terapias alternativas…………………………………………………40 Hipótesis y Objetivos…………………………………………. 44 Material y métodos…………………………………………… 48 1. Pacientes y preparación de las muestras……………………………….….. 50 2. Métodos analíticos……………………………………………………………52 2.1. Estrés oxidativo…………………………………………………… 52 2.1.1. Determinación de la concentración de proteínas: método de Lówry……………………………………………………….. 53 Iryna Rusanova 2.1.2. Determinación de glutatión oxidado y glutatión reducido……..53 2.1.3. Determinación de la actividad enzimática de la glutatión peroxidasa (GPx)………………………………………….…………..55 2.1.4. Determinación de la actividad enzimática de la glutatión reductasa (GRd)………………………………………….……………56 2.1.5. Lipoperoxidación……………………………………………….57 2.1.6. Determinación de nitritos……...………………………………..58 2.1.7. Determinación de la actividad enzimática de la superóxido dismutasa……………………………………………………………...60 2.2. Parametros hematológicos………………………………………….62 3. Análisis de AND……………………………………………………………… 63 3.1. Extracción de AND………………………………………………….63 3.2. Análisis de haplotipos con la técnica de RFLP……………………..64 3.2.1. PCR……………………………………………………..64 3.2.2. RFLP……………………………………………………64 3.2.3. Electroforesis……………………………………………65 4. Análisis estadístico………………………………………………………….. 69 RESULTADOS……………………………………………………………………….72 1. Haplotipos del cluster globínico beta………………………………………. 74 1.1. Datos generales de los pacientes y del grupo control……………….74 1.2. Haplotipos del cluster globínico beta………………………………..75 1.3. Distribución geográfica de los haplotipos identificados…………….79 2. Correlación de los haplotipos de anemia falciforme identificados en pacientes atendidos en el Hospital del Niño de Panamá con el fenotipo clínico de la enfermedad (leve, moderada y severa) y los datos hematológicos………………………………………………………………….80 Iryna Rusanova 2.1. Análisis de fenotipo clínico de acuerdo a los haplotipos del cluster globínico beta…………………………………………………………81 2.2. Valores hematológicos de acuerdo a los haplotipos determinados..82 2.3. Análisis de los valores hematológicos de acuerdo a las manifestaciones clínicas de los pacientes con anemia falciforme……82 3. Valoración del estrés oxidativo en el grupo de enfermos y el grupo control…………………………………………………………………………84 4. Correlaciones entre el estrés oxidativo y los haplotipos del cluster globínico beta, manifestaciones clínicas y los niveles de HbF expresados en los pacientes con anemia falciforme…………………………………………….88 4.1. Resultados del estrés oxidativo de acuerdo a los grupos haplotípicos………………………………………………………….88 4.2. La relación entre los biomarcadores del estrés oxidativo y el fenotipo clínico de anemia falciforme en la población panameña…………...90 4.3. La relación entre los biomarcadores del estrés oxidativo y la expresión de HbF en los pacientes falcémicos de Panamá…………………….92 DISCUSIÓN DE LOS RESULTADOS……………………………………………...94 1. Haplotipos del cluster globínico beta………………………………………. 96 1.1. Datos generales de los pacientes y del grupo control………………..96 1.2. Haplotipos del cluster globínico beta…………………...…………...96 2. Correlación de los haplotipos de anemia falciforme identificados en pacientes atendidos en el Hospital del Niño de Panamá con el fenotipo clínico de la enfermedad (leve, moderada y severa) y los datos hematológicos……………………………………………………………… 100 3. Valoración del estrés oxidativo en el grupo de enfermos y el grupo control……………………………………………………………………….. 103 4. Correlación del estrés oxidativo con los haplotipos, fenotipo clínico de la enfermedad y los niveles de HbF expresados……………………………... 108 CONCLUSIONES…………………………………………………………………...114 BIBLIOGRAFÍA…………………………………………………………………….119 2 -Introducción- Introducción 1. ANEMIA FALCIFORME. 1.1. Historia. La primera observación de eritrocitos falciformes se realizó en 1910 por James Herrick en un frotis de sangre periférica de un estudiante negro originario de la Grenada, el cual murió a los 32 años a causa de neumonía. Debido a la forma anormal de los eritrocitos, Herrick por primera vez utilizó el término de ―enfermedad falciforme‖. Sin embargo, tuvieron que pasar 15 años antes de que esta enfermedad fuera considerada como primaria y no debida a una infección u otra condición (Savitt y colbs., 1989). En el año 1917 Víctor Emnel por primera vez usó el término ―anemia de células falciformes‖, para establecer una entidad clínica definida, sobre la base del descubrimiento de Herrick y referiendose a la peculiar y alargada forma de las células falciformes. Hann y Guillespie, en 1927, descubrieron que la deformación de los eritrocitos estaba relacionada con el estado de oxigenación de la hemoglobina (Hb) en el desempeño de su función, y que se podía provocar esta forma falciforme saturando de dióxido de carbono una suspensión de eritrocitos. En 1947, el norteamericano James Neel, en el artículo de revisión ―The Clinical Detection of the Genetic Carriers of Inherited Disease‖ publicado en la revista Medicine (Neel, 1992), postuló la hipótesis de herencia autosómica recesiva para la anemia falciforme. Dos años después, James Neel confirmó la hipótesis en la revista Science (Nell, 1949) tras estudiar 21 familias de niños con anemia falciforme, de manera que los individuos enfermos eran homocigóticos recesivos y los individuos aparentemente sanos, que presentaban glóbulos rojos falciformes y normales, eran heterocigóticos y portadores de la enfermedad. Por lo tanto, en cada pareja de padres portadores existe un 25% de probabilidades de que nazca un hijo con anemia falciforme independientemente del sexo (Chang y colbs., 1995). Una gran aportación en el entendimiento de la base molecular de anemia falciforme fue hecha por Pauling LC, Premio Nobel de Química en 1954. Pero años antes, en 1949, Pauling, junto con Harvey Itano, S. J. Singer e Ibert Wells, publicó en la revista Science la primera prueba de la relación entre una enfermedad humana y un cambio en una proteína específica (Pauling y colbs., 1949). Utilizando la 2 Introducción electroforesis, demostraron que la hemoglobina de los pacientes con anemia falciforme tiene la carga eléctrica diferente a la de las moléculas de personas sanas. Afirmó que la enfermedad puede ser provocada por alteración en una molécula protéica, de manera que la herencia podía influir en las mutaciones de dicha proteína, marcando así el inicio de la genética molecular. Poco después, en el año 1959, Vernon M. Ingram (Ingram, 1959) demostró que la hemoglobina S es el resultado de una mutación en el codón 6 del gen de la globina β en la que la base adenina es sustituida por la timina (GAG GTG) lo que ocasiona que el ácido glutámico en la posición 6 de la globina sea sustituido por la valina. Como esta sustitución se lleva a cabo en una posición superficial de la molécula de hemoglobina y la carga eléctrica es diferente, la movilidad electroforética de la HbS es menor que la de la hemoglobina normal, pudiéndose ser fácilmente separada por electroforesis. El test de Murayama (Murayama M y Nalbandian RM, 1973) muestra que una solución concentrada de HbS desoxigenada se convierte en gel, mientras que la Hb adulta (HbA) desoxigenada sigue siendo soluble. Además, desde aquella época la patología es considerada como una enfermedad relacionada con el rasgo racial, ya que en su mayoría afecta a la raza negra. Drepanocitosis (del griego drepanon=hoz, y kytos= células) es otro término que determina la presencia en sangre de hematíes en forma de media luna; y es sinónimo al término anemia falciforme. La hemoglobinopatía S, o drepanocitosis, o SCD (sickle cell disease en inglés). Puede existir bajo 4 formas diferentes: Forma heterocigota o rasgo falciforme (HbAS). Aparece cuando la mutación se encuentra en uno de los dos alelos que codifica para la proteína globina β (βA/ βS). En este caso, el paciente tiene un 30-40% de HbS y no presenta manifestaciones clínicas. Forma homocigota o anemia de células falciforme (HbSS). Aparece cuando la mutación afecta a los 2 alelos del gen correspondiente a la cadena β (βS/βS). En estos casos, prácticamente toda la Hb (75-95%) es HbS, siendo el resto (5-15%) la fetal (HbF). Presenta una variedad de síntomas clínicos, desde ligeros hasta muy graves. Forma doble heterocigota HbS-talasemia (HbS-Tal). Aparece cuando en el mismo paciente coexisten 2 alelos anormales, uno para la HbS y otro para la β3 Introducción talasemia (βS/βtal). Si la síntesis a nivel del gen talasémico es nula (β0-tal), la cantidad de HbS será prácticamente la misma que en el estado homocigoto (70-90%). Si, por el contrario, sólo presenta una disminución en el gen talasémico (β+- tal), se observa la coexistencia de HbA (10-30%), HbS (60-85%), y una pequeña proporción de HbF (5%). No son tan graves como las formas de herencia tipo homocigoto HbSS y predominan en el área mediterránea más que en la raza negra. Forma doble heterocigota HbS-HbC (HbSC). Se debe a la coexistencia de 2 alelos anormales. Un alelo codifica la síntesis de HbS y el otro, la síntesis de HbC (βS/βC). No existe HbA mientras que existen cantidades similares de HbS y HbC (50%). La expresión clínica suele ser menos severa. Otras formas de asociación de HbS con distintas hemoglobinopatías (Ángeles, G-Philadelphia, HbO Arab, etc) pueden tener una variabilidad clínica diversa. 1.2. Epidemiología. La anemia falciforme y la beta-talasemia son las dos de las enfermedades genéticas más frecuentes del mundo. Según los datos de la Organización Mundial de la Salud (OMS) se estima, que un 7% de la población mundial porta genes de las hemoglobinopatías, y el 50% de estas hemoglobinopatías viene dado por la anemia falciforme o drepanocitosis (Cao y colbs., 1998). Cada año nacen alrededor de 300.000-400.000 niños con drepanocitosis, y la prevalencia de esta enfermedad oscila entre 0,1/1.000 en los países no indémicos, hasta 20/1.000 en algunas partes del continente Africano (Weatherall and Clegg, 2001). En todo el mundo, hay más portadores de talasemias que de anemia falciforme, pero la elevada frecuencia del gen de la drepanocitosis en ciertas áreas da lugar a elevadas tasas de recién nacidos afectados por esta enfermedad. La mayor concentración del gen para la drepanocitosis se da en la población negra de África ecuatorial, donde hay grupos con el gen que afecta hasta a un 40% de la población (Chang y colbs., 1995), y es aún más frecuente en Benín (Nigeria) y África oeste central (Lukens y colbs., 1993). También se presenta en países de la cuenca mediterránea, como Turquía, Grecia, Italia, España y Oriente Medio, así como en los países árabes e India Oriental. En 4 Introducción América se da en los Estados Unidos (en personas de origen africano o afroamericano) y en el Caribe, América Central y del Sur (Brasil) (Lukens y colbs., 1993). Esta distribución se debe a que la población con el rasgo falciforme es resistente a la malaria, una enfermedad transmitida por un parásito, el Plasmodium falciparum, que vive en el interior de los hematíes durante una etapa de su ciclo vital. En zonas donde la malaria ha sido históricamente endémica, ha favorecido a la población poseedora del rasgo falciforme, aumentando la prevalencia del alelo HbS (Thomas y colbs., 2001). El gen de la drepanocitosis se ha hecho frecuente en África porque el rasgo heterocigoto confiere cierta resistencia al paludismo por Plasmodium falciparum durante la infancia, favoreciendo así la supervivencia del huésped y la consiguiente transmisión del gen de la hemoglobina anormal. Aunque la presencia de un único gen anormal puede proteger del paludismo, la herencia de dos genes anormales produce anemia falciforme y no confiere la mencionada protección, por lo que el paludismo constituye una importante causa de enfermedad y muerte en niños con anemia drepanocítica (Föller y colbs., 2009). La anemia falciforme tiene importantes repercusiones en la salud pública. Sus efectos en la salud humana se pueden evaluar en función de la mortalidad infantil. La proporción de niños afectados que sobreviven más allá de los cinco años es cada vez mayor, pero aun muchos niños corren el riesgo de muerte prematura. Cuando el impacto en la salud se mide en función de la mortalidad de los menores de cinco años, la anemia falciforme es la causa de la muerte de un 5% de este segmento de población en el continente africano (Modella and Darlison, 2008); de más de un 9% en África occidental y de hasta un 16% en determinados países como Nigeria. El 24% de la población es portadora del gen mutante, y la prevalencia de la anemia falciforme es de aproximadamente 20 casos por cada 1.000 nacidos. Esto significa que sólo en Nigeria nacen anualmente unos 150.000 niños con esta hemoglobinopatía (Magaña y colbs., 2002). La detección masiva de anemia falciforme en Estados Unidos surgió en el año 1975. Un seguimiento en la evaluación de estos niños y la administración temprana de penicilina redujo notablemente la morbilidad y la mortalidad en los niños diagnosticados. La supervivencia media estimada en los Estados Unidos de América en 1994 era de 42 años para los hombres y 48 años para las mujeres, mientras que en 5 Introducción el año 2001 era de 53 años para los hombres y 58,5 años para las mujeres. En Jamaica, donde existe una gran migración del continente africano, la mayor mortalidad se registra entre los 6 y los 12 meses de vida, que es cuando fallece el 10% de los pacientes, pese a la considerable experiencia en el diagnóstico y tratamiento de la enfermedad y a la ausencia de paludismo. En África subsahariana la mitad de los pacientes con anemia falciforme mueren antes de los cinco años, generalmente por infecciones como el paludismo y la sepsis neumocócica, o por la propia anemia. En general, en los últimos años, la esperanza de vida ha mejorado notablemente estimándose que el 85% de los niños con anemia falciforme sobreviven hasta los 18 años. La anemia falciforme representa un importante problema para la salud pública (Roberts and de Montalambert, 2007). Solo en Estados Unidos entre los años 1989 y 1992 se registraron 75.000 hospitalizaciones por año. En 1996 estas hospitalizaciones supusieron un coste de $475 millones de dólares (Ashley-Koch y colbs., 2000). Según los estudios de la Organización Panamericana de la Salud (OPS) en el año 2004, la prevalencia de HbS en algunos países latinoamericanos es importante, aunque las muestras de individuos estudiados varían entre 150 en Guatemala hasta más de 20.000 en Brasil (Tabla 1). Tabla 1. Prevalencia de HbS en algunos países latinoamericanos, según los datos de la OPS, 2004. País No. De Individuos % Brasil 21372 6,2 Colombia 1184 11,9 Costa Rica 1830 8,1 Cuba 6996 6,1 Guatemala 150 18,3 México - 11,2 Venezuela 2132 8,7 Panamá 7604 16,0 En Panamá, según algunos estudios realizados, existe un 8 % de la población afectada por esta enfermedad (Schroeder y colbs., 1900). Sin embargo, no hay datos 6 Introducción claros actualizados y publicados sobre la prevalencia de drepanocitosis en la República de Panamá. Sólo recientemente y gracias a la aprobación de la ley nacional de tamizaje neonatal en el año 2007, se va a poder a realizar un diagnóstico masivo y determinar la prevalencia de anemia falciforme. 1.3. Diagnóstico. Las técnicas para el diagnóstico de anemia falciforme han evolucionado al igual que nuestro conocimiento sobre esta enfermedad. Existen varias técnicas de laboratorio que permiten hacer una detección temprana de drepanocitosis y que luego deben ser comprobados con las técnicas de biología molecular. La HbS resulta de la sustitución de un aminoácido con carga (ácido glutámico), situado en la sexta posición de la cadena ß, por otro neutro (valina). Esta modificación de la carga superficial de la hemoglobina disminuye la solubilidad de la Hb, especialmente en su estado reducido (deoxi-Hb), y facilita la formación de agregados fibrilares o polímeros de molécula de Hb que alteran profundamente la morfología eritrocitaria y aumentan su rigidez. Las técnicas que se utilizan para el diagnóstico de drepanocitosis son las siguientes: 1. Inducción de drepanocitosis: En un medio carente de oxígeno (O2) la hemoglobina S se hace menos soluble y forma agregados cristalinos con la consiguiente formación de drepanocitos. Se coloca una gota de sangre en un portaobjetos, se le agrega metabisulfito de sodio 2%, que es un reactivo consumidor de O2, se tapa con un cubreobjetos y se sellan los bordes con petrolato; en pocos minutos se puede observar el fenómeno (Winterbourn, 1974). 2. Prueba de solubilidad de la hemoglobina: Aquí también ocurre una desoxigenación de la hemoglobina S, pero utilizando sustancias como el dithionato sódico, que hace que la hemoglobina S se haga insoluble y precipite, observándose turbia la solución (Huntsman y colbs., 1970). Esta prueba es más fácil y rápida de realizar, pero puede dar falsos negativos en los pacientes con anemia severa, a menos que se realice junto al hematocrito, y en casos de hemoglobina F aumentada. Los 7 Introducción falsos positivos pueden estar dados por otras causas de turbidez como en las disglobulinemias (Surve y colbs., 2000). Figura 1. Inducción de drepanocitos en metabisulfito de sodio (aumento 100x). 3. Electroforesis de hemoglobina: Prueba diagnóstica principal que involucra separación de las diferentes clases de hemoglobina por movilidad que difiere según su carga molecular y tamaño. Sin embargo, algunas hemoglobinas migran juntas en un medio alcalino (acetato de celulosa) (Figura 2) y es necesario repetirla a un pH ácido (agar citrato) en que pueden migrar diferente (Figura 3). Por ejemplo, la hemoglobina C y la A2 migran juntas en acetato de celulosa (Kohn J, 1969), pero separadas en el agar citrato y, en este medio, la hemoglobina F se separa mejor. Entre las técnicas complementarias utilizadas para identificar hemoglobinas anormales o electroforéticamente ―silenciosas‖, se menciona la isoelectrofocalización como el método de elección por su rapidez y facilidad, además de la seguridad de los resultados (Dacie JV y Lewis SM, 1991). Figura 2. Electroforesis de Hb en acetato de celulosa a un pH 8.6, para mostrar el sitio de migración de diferentes Hb. 4. Figura 3. Electroforesis de Hb en agar citrato a un pH 6,2, para comparar con la electroforesis en acetato de celulosa. HPLC (cromatografía líquida de alta presión) es un método automatizado de alta precisión que permite detectar distintas moléculas de hemoglobina (F, A, S, C, E 8 Introducción y D). Es suficiente contar con la muestra de sangre en forma de frotis en papel filtro. Además, esta técnica es un método útil para la cuantificación inicial de HbS y F en el seguimiento de los tratamientos (Surve y colbs., 2000). 5. Diagnóstico molecular: basado en el uso de las enzimas de restricción específicas que son capaces de detectar el cambio de un sólo nucleótido en la cadena de ADN. La digestión con la enzima de restricción DdeI es para confirmación del genotipo de la mutación A→T en el codón 6 (6 GAG→GTG), que permite salir de la duda en caso de la talasemia beta, o HbD que puede alterar el análisis de los haplotipos. La enzima reconoce el sitio 5’-C•TNAG-3’. Si la muestra tiene la mutación S, la enzima no es capaz de realizar el corte (figuras 4 y 5) (Varawalla, 1992). Figura 4. La enzima DdeI corta las secuencias en posiciones 27, 40, 62, 107, 488, 576. El nucleótido rojo indica el sitio mutado. A: CCT-GAG-GAG S: CCT-GTG-GAG DdeI Pro –Val – Glu 5 6 7 5' 3' -/- = SS 308 pb +/+ = AA Figura 5. Uso de la enzima DdeI para confirmar el diagnóstico HbS. 9 Introducción 1.4. Clínica de la enfermedad. Las manifestaciones clínicas de la anemia drepanocítica clásica han sido bien definidas desde hace mucho tiempo y se han caracterizado por un conjunto de signos y síntomas própios de una anemia hemolítica crónica grave, con incremento de la susceptibilidad a las infecciones, retraso del crecimiento y desarrollo, crisis de dolor por oclusión vascular, cuadro torácico agudo, síndrome de secuestro esplénico, daño tisular, trastornos del sistema nervioso central, crisis aplásicas y hemolíticas y asplenia funcional (Rodgers y Schetcher, 1991). La anemia falciforme tiene un amplio espectro de manifestaciones clínicas. La mayoría de los afectados presentan anemia crónica con una hemoglobinemia de alrededor de 8 g/dl. De acuerdo al estudio cooperativo de SCD, se definen los siguientes síntomas severos de anemia falciforme (Inati y colbs., 2003): Accidente cerebro vascular: síndrome neurológico agudo vascular debido a la oclusión o hemorragia. Los síntomas o signos neurológicos se prolongan más de 24 horas. Síndrome torácico agudo: presencia de un nuevo infiltrado pulmonar en asociación con una enfermedad aguda del tracto respiratorio. Crisis de dolor: dolor en brazos y piernas, espalda, abdomen, pecho o cabeza que dura al menos 2 horas, y que requiere atención médica. Existen dos tipos de crisis: ―crisis dolorosa o infártica‖, que se caracteriza por dolor óseo intenso y fiebre en la que no se producen cambios en la concentración de Hb; y ―crisis de secuestro‖ que suele afectar a los lactantes y niños pequeños en la que se produce un descenso rápido de la concentración de Hb en sangre y puede producir ―crisis hemolítica‖ que se caracteriza por una disminución de Hb e ictericia intensa (Ballas, 1991). Dactilitis: sensibilidad o dolor con o sin hinchazón de las manos y pies. Secuestro esplénico: atribuido a la ampliación esplénica de al menos 2 cm por debajo del margen costal y una disminución de los niveles de hematocrito por debajo del 20%. 10 Introducción La clínica de los pacientes con anemia falciforme varía inmensamente a pesar de que existe la misma mutación genética. Algunos autores clasifican los cuadros clínicos como grave o severo, moderado y suave, dependiendo de la presencia o ausencia de los síntomas descritos anteriormente. Los distintos subfenotipos de la clínica de anemia falciforme fueron sugeridos por Ballas en 1991, quién notó que los pacientes con frecuentes crisis de dolor tenían una evolución distinta a los que tenían ulceraciones en los píes (Ballas, 1991). La variedad clínica de anemia falciforme sigue siendo el rompecabezas para los científicos y clínicos, y se busca encontrar la explicación de ello en la fisiopatología de esta enfermedad. La situación de anemia crónica produce daños, muchas veces irreversibles a distintos órganos. Órganos afectados: Bazo: las células falciformes tienen una vida media más corta y, por tanto, son atrapados en el bazo, para ser destruidos. El secuestro esplénico es causado por el atrapamiento de las células rojas en la circulación del bazo, causando disminuciones en los niveles de Hb y un rápido aumento del órgano. Inmediatamente se requiere tratamiento con transfusiones, y un 50% requiere esplenectomía. Sistema nervioso central: se producen pequeños infartos que pueden ser clínicamente silenciosos, pero que a largo plazo producen defectos cognitivos que pueden ser detectados con pruebas neuro-psiquiátricas. Pacientes adultos pueden presentar aneurismas y hemorragias cerebrales. La mayoría de los pacientes con daños cerebrales requieren terapia de transfusión, y en el caso de los niños, trasplante de la médula ósea. Huesos y articulaciones: los problemas óseos se inician en la infancia. En los huesos y articulaciones se producen episodios de vaso oclusión, que conllevan a que se desarrollen anormalidades en las vertebras. La necrosis en los hombros y las caderas causan episodios de dolor que pueden requerir una intervención quirúrgica. La enfermedad puede conllevar a que se desarrollen las infecciones de las articulaciones, que acaba con la colocación de prótesis. Ojos: curiosamente, los pacientes que llevan un cuadro clínico más suave y tienen valores de hemoglobina más altos, son los que más sufren problemas oftalmológicos. La mayoría de estos daños se revelan a una edad adulta y pueden 11 Introducción consistir en hemorragias oculares, desprendimiento de la retina y oclusiones de la arteria retinal central (Claster y Vichinsky, 2003). Sistema genitourinario: los problemas del aparato genitourinario son bastante frecuentes en pacientes con SCD. Se desarrolla esclerosis glomerular, que se manifiesta con proteinuria, y más de 5% de los pacientes desarrollan daño renal crónico. Otro gran problema es el priapismo: dolorosa y persistente erección debido a la obstrucción vaso-oclusiva de drenajes venosos de pene. Aproximadamente el 89% de los hombres desarrollan algún episodio de priapismo, que si llegan ser prolongados pueden resultar en impotencia (Claster y Vichinsky, 2003). Sistema pulmonar: el síndrome torácico agudo está en segundo lugar como causa de admisión hospitalaria. La situación se agrava por infecciones y/o embolias de grasa en las arterias pulmonares. Se desarrollan daños crónicos pulmonares que resultan en hipoxia y fibrosis pulmonar. Uno de cada tres pacientes desarrolla hipertensión pulmonar y esto disminuye significativamente la esperanza de vida de los pacientes. La principal causa de muerte en mayores de cinco años suele ser el síndrome torácico agudo, con fiebre, dolor torácico y leucocitosis. Se ha indicado que hoy en día la supervivencia de los pacientes con anemia falciforme guarda relación con la frecuencia de estas crisis (Claster y Vichinsky, 2003). 1.5. Fisiopatología de la anemia falciforme. 1.5.1. Fisiología de la hemoglobina. La Hb es una molécula compleja presente en los eritrocitos; su función es transportar el oxígeno a todos los tejidos del organismo. Se integra con cuatro cadenas peptídicas (globinas), cada una unida a una molécula de protoporfirina ferrosa (grupo hem), sitio donde se fija y se desplaza el oxígeno. En un hematíe hay alrededor de 280 millones de moléculas de Hb, cada una con un peso molecular de 64.458 Dalton, forma elipsoide y dimensiones 64x55x60 Aº. 12 Introducción Figura 6. Estructura del grupo hemo de Hb, de las cuatro cadenas globinicas que forman un tetrámero conjugado, y del cromosoma 11, donde se codifica la estructura de las cadenas globinicas , , , . Existen varias formas de hemoglobinas normales: más de 95% de la hemoglobina del adulto y de los niños mayores de 7 meses es la HbA1, compuesta por dos cadenas α y dos cadenas (α2 2). La HbA2 es la hemoglobina que se encuentra en menor concentración en los adultos (2,5%) y está compuesta por dos cadenas α y dos cadenas . Se representa como α2 2. La HbF es el componente principal de la Hb de recién nacido. Su concentración disminuye en los primeros meses de vida y está constituida por dos cadenas α y dos . Las hemoglobinas Gower I (z2ε2), Gower II (2ε2) y Portland (2z2) son embrionarias y sólo aparecen durante el primer trimestre de gestación (Hoppe y colbs., 2004). La Hb Gower II es la más importante y alcanza entre 50% y 60% de toda la Hb embrionaria (Hardison, 1996). Al parecer, las cadenas polipeptídicas épsilon () y zeta (z) se sintetizan en el saco vitelino, y la Hb embrionaria tiene mucha mayor afinidad por el oxígeno. Se han descrito más de 400 variantes anormales de hemoglobinas, que difieren entre sí en uno o más nucleótidos, lo que puede traducirse en cambio de aminoácidos que repercute en la función de la proteína. Las alteraciones en la estructura de Hb se denominan hemoglobinopatías. Existen varias hemoglobinopatías: las más comunes son α- y - talasemia, que se caracterizan por deficiencia en la síntesis de una de las 13 Introducción dos globinas, y hemoglobinopatías estructurales producidas por una mutación puntual en la estructura de la proteína. La alteración en la hemoglobina S (HbS) es la más frecuente a nivel mundial, y conlleva a la producción de anemia hemolítica crónica (Peñalosa-Espinosa y cols., 2008). Entre otras hemoglobinopatías estructurales podemos mencionar las de HbC y HbE. La HbC se caracteriza por la sustitución del ácido glutámico de la posición 6 de la cadena beta por lisina. Es una hemoglobinopatía propia de África occidental, pero puede encontrarse con cierta frecuencia en España. El estado homocigoto (CC) se caracteriza por una ligera anemia hemolítica crónica con esplenomegalia. El estado heterocigoto (AC) no produce trastorno alguno. Aunque la HbC tiende a polimerizarse en condiciones de hipoxia, no produce crisis vaso oclusivas como las de la HbS (Peñalosa-Espinosa y cols., 2008). La HbE (26 GluLys) es más frecuente en el sur de Asia y se han hecho estudios que sugieren que esta variante de Hb también ofrece cierta protección contra la malaria (Steinberg, 1995). La anemia falciforme es la primera enfermedad congénita en la cuál fue identificado el error genético específico. Durante muchas décadas fue considerado que la única causa de vaso-oclusión era la acumulación de eritrocitos falciformes en los capilares que conllevaba a los daños orgánicos. El concepto cambió a finales del siglo XX debido a las evidencias de interacción entre las células sanguíneas circulantes y el endotelio, aumento de la adhesión celular, detección de los factores de inflamación, biomarcadores de estrés oxidativo elevados y cambios en el equilibrio del sistema de vasoconstricción-vasodilatación. Finalmente, Hebbel fue el primer científico quién sugirió la presencia de la disfunción endotelial generalizada que afecta el curso clínico de la enfermedad (Hebbel y colbs., 1985; Kaul y colbs., 1989). La disfunción endotelial se define como un estado patológico caracterizado por el desequilibrio entre la producción de factores vasodilatadores y vasoconstrictores, entre las propiedades procoagulantes y anticoagulantes y por alteración de la permeabilidad vascular. Entre los factores involucrados en la disfunción endotelial se consideran los siguientes: hemolisis activado por el estrés oxidativo elevado, biología de la producción de óxido nítrico (NO), adhesión celular e inflamación (Mack y Kato, 2006). 14 Introducción La vaso-oclusión, responsable de las crisis de dolor y daño multiorgánico, se relaciona con la inflamación, un aumentado estrés oxidativo y la disfunción endotelial. Además, se producen cambios en la disponibilidad de sustratos y cofactores para la enzima óxido nítrico sintasa, que conlleva a la disminución de biodisponibilidad de óxido nítrico y el aumento en la formación de las especies reactivas de nitrógeno. Otros estudios también indican que aumenta la respuesta inflamatoria microvascular, del mismo modo que durante la ateroesclerosis e hipertensión (Mack y Kato, 2006). Se produce un desequilibrio en el funcionamiento de las células endoteliales que conlleva al aumento de vasoconstricción y adhesión celular. 1.5.2. Polimorfismos de genes del clúster globínico beta. Los aspectos genéticos más estudiados son los cambios de un solo nucleótido o SNPs en el bloque o clúster de genes beta, denominados haplotipos del clúster beta. El clúster globínico beta (HBB, gene ID: 3043), se localiza en cromosoma 11 (11p15.5) y contiene alrededor de 45 kb, incluyendo cinco genes funcionales y un pseudogen (ver figura 7). El orden de ubicación de los genes en la dirección desde terminal 5' hacía terminal 3' es: épsilon, gamma G, gamma A, beta 1 pseudogen, delta, beta. Todos estos genes tienen una estructura similar (dos exones y tres intrones) que lleva a concluir que se derivan de un solo gen ancestral. Los genes de las globinas son regulados durante el desarrollo y se expresan en los momentos específicos de la vida humana. El gen épsilon y los dos gamma se expresan en las etapas embrionaria y fetal, en los tejidos eritroides, mientras que los genes delta y beta se expresan en la vida adulta. Las cadenas globinicas , , , están compuestos por 146 aminoácidos y poseen homología estructural. De la misma forma, las cadenas z y α son productos de genes localizados en el extremo 5 ׳del cromosoma 16, y están compuestos de 141 aminoácidos. El análisis del ADN de diversos individuos en distintas poblaciones ha puesto de manifiesto que existen mutaciones productoras de polipéptidos cuya secuencia de aminoácidos está alterada (por ejemplo, variantes de la Hb), y también diferencias en las secuencias nucleotídicas que no alteran la función del gen. Estas variantes ―silenciosas‖ se definen como polimorfismos del ADN. Se ha calculado que aproximadamente una de cada 100 bases es polimórfica, ello significa que en esa 15 Introducción posición puede haber nucleótidos distintos, y se ha demostrado que estas variaciones se presentan casi exclusivamente en las regiones adyacentes a los genes y en los intrones, mientras que las secuencias de los exones se mantienen prácticamente constantes. La introducción de técnicas de biología molecular, entre ellas, el uso de enzimas de restricción obtenidas de bacteriófagos específicos, dió un nuevo impulso para el estudio de la HbS al encontrar que existen distintos patrones de digestión o restricción dentro del clúster beta y que además, se relaciona con sus orígenes geográficos. Las técnicas de biología molecular hacen posible la determinación de los distintos haplotipos del clúster S a través de los 11 sitios polimórficos (Nagel y Fabry, 1984). La presencia o ausencia del sitio polimórfico produce los llamados fragmentos de restricción de longitud variable (en inglés, restriction fragment length polymorphisms, o RFLP). Ciertas enzimas ―cortadoras‖ o de restricción tienen afinidad exclusiva por los distintos puntos polimórficos en la cadena de ADN y por lo tanto se usan para dividirla en sus diferentes RFLP. Cuando se habla de haplotipos se hace alusión a los diversos patrones o fragmentos polimórficos de ADN a lo largo de una misma región cromosómica (Chang y colbs., 1995). La combinación o patrón aleatorio de los sitios de restricción polimórficos en un cromosoma es lo que define el haplotipo de un gen. El clúster beta se divide en clúster 5' y 3'. Figura 7: Esquema de cluster globínico beta. Desde la primera descripción de estos fragmentos por Kan y Dozy (Kan y Dozy, 1980), se ha demostrado que al menos hay 5 tipos de HbS: SS-Benín, SS-Bantú (CAR, República Central Africana), SS-Senegal, SS- Camerún, SS-Árabe-Hindú (Peñuela, 16 Introducción 2005; Ofori-Acquah y colbs., 2002). Otros autores prefieren hacer la clasificación en seis haplotipos, separando el haplotipo Árabe-Hindú en Arabé-Saudi y Asiático-Índio (Rodríguez y Saénz, 1998). La importancia de conocer los haplotipos radica en que los de tipo Bantú y Benín acusan una gravedad mayor que los de tipo Senegal, Arabé-saudi y Asiáticoindio, en los cuales los pacientes pueden presentar síntomas diversos desde el segundo mes de vida, razón por la que requieren mayor y mejor atención médica continua (Peñalosa-Espinosa y colbs., 2008). Por ejemplo, se ha visto que los neonatos con haplotipo Bantú (CAR) en su estado homocigoto tienen un mayor riesgo para desarrollar accidentes cerebrovasculares (Schacter y colbs., 1988). También hay estudios donde no se encuentra la correlación entre la gravedad clínica de la enfermedad y los haplotipos (Inati y colbs., 2003). 1.5.3. Otros polimorfismos de un solo nucleótido (SNPs). Recientemente se han hecho estudios que asocian polimorfismos de un solo nucleótido (SNPs) de varios genes con la gravedad clínica de la anemia falciforme. El uso de técnicas modernas de biología molecular (cDNA microarrays, análisis de polimorfismos, proteómica) permiten identificar polimorfismos (SNPs) de los genes que potencialmente pueden influir en diferentes aspectos de la fisiopatología de la anemia falciforme. En la tabla 2 citamos algunos de los genes posiblemente involucrados en las manifestaciones clínicas de la anemia falciforme. Como podemos observar, casi todos estos genes están relacionados con la inflamación, proliferación celular, reactividad vascular y apoptosis, lo cuál apoya la idea que drepanocitosis es una enfermedad que se caracteriza por la disfunción endotelial e inflamación (Kutlar, 2007). 17 Introducción Tabla.2. Genes que posiblemente impactan en la fisiopatología de anemia falciforme. Gen estudiado Asociado a Referencia SNPs en los genes de fosfodiesterasa 7 Aumento (PDE7), proteína asociada a producción de HbF en la Kutlar, 2007 microtubulo7 (MAP7), factor de biogénesis peroxisomal7 (PEX7) y proteína kinasa 5 mitogeno-activada (MAP3K5). Receptor de Interleucina-4 (IL-4R, Accidente cerebro- Kutlar, 2007 S503P), factor de necrosis tumoral vascular (TNFα,-308G) y receptor - adrenergico-2 (ADRBP, Q27E). Óxido nítrico sintasa 2 (NOS2A, T- Hipertensión Sharan y colbs., 786C) y factor de crecimiento de pulmonar, priapismo, 2004; receptor beta tipo II (TGFBR2). úlceras en los pies, Koch síndrome Ashleyy colbs, torácico 2005. agudo. Factor de crecimiento de receptor beta Coagulación, adhesión Ashley-Koch tipo III (TGFBR3), celular, priapismo Aquaporina (AQP1) colbs, y 2004, Elliott y colbs., 2007. Gen reductasa methylenetetrahydrofolato Necrosis avascular (MYHFR), la mutación Milner y colbs., 1991 C677T Receptor de Interleucina-4 (IL-4R, Accidente S503P), factor de necrosis tumoral vascular (TNFα,-308G), receptor beta cerebro- Hoppe y colbs., 2004, Hoppe y colbs., 2007. adrenérgico (ADRB2), molécula de adhesión celular vascular (VCAM1,1594), receptor de lipoproteína de baja densidad LDLR. 18 Introducción 1.5.4. Las variaciones en la expresión de la hemoglobina fetal. La hemoglobina fetal tiene mayor afinidad por el oxigeno e inhibe la polimerización de HbS, permitiendo llevar cantidades suficientes de oxígeno a los tejidos periféricos. La concentración de HbF al nacer es de 80% pero disminuye al cumplir los seis meses de vida. Varios factores influyen en las variaciones de HbF en los pacientes SCD: edad, sexo, cantidad de las globinas alfa, haplotipos del cluster globínico beta y locus ligado al cromosoma X, que regula la producción de los eritrocitos F (FCP). Cada una de estas variables presenta asociación con los niveles de HbF, pero la participación de FCP es de mayor prevalencia (40%) (Chang y colbs., 1995). En los pacientes con anemia falciforme se ha observado que los niveles elevados de hemoglobina fetal y los haplotipos Senegal y Árabe/Hindú se asocian con el cuadro clínico menos grave (Ruiz Cruz y colbs., 2003). También, Nagel y colaboradores (Nagel y colbs., 1991) demostraron que la presencia de por lo menos un cromosoma con haplotipo Senegal, a diferencia de otros haplotipos africanos, se asocia con altos niveles de HbF y un curso clínico mas leve. Los altos niveles de HbF se relacionan a su vez con el polimorfismo en la región promotora del gen G (-158 (CT)) que regula la expresión de HbF (Lu y Steinberg, 1996). Sin embargo, otros estudios sugieren que los niveles de hemoglobina fetal son importantes, pero no son los únicos factores que afectan la gravedad de la enfermedad. Por ejemplo, se encontraron altos niveles de HbF en pacientes con haplotipos CAR en el Líbano que no parecen mejorar la severidad de síntomas, lo que sugiere que existen otros factores y que los niveles de HbF no son los principales determinantes de la gravedad de esta enfermedad (Inati y colbs., 2003). La utilización de hidroxiurea es uno de los tratamientos más aceptados hasta ahora. Se piensa que el principal mecanismo de acción es el aumento de los niveles de HbF. Hidroxiurea es un citotóxico que causa la regeneración eritroide y también su metabolismo da lugar a un aumento en Guanilil ciclasa soluble dependiente de óxido nítrico, con una elevación de cGMP que aumenta la expresión de gen de globina gamma (Cokic y colbs., 2003). A pesar su amplio uso en la práctica médica, no todos los pacientes respondan al tratamiento con hidroxiurea. Además, se han encontrado SNPs que participan en el metabolismo de hidroxiurea y en la respuesta a 19 Introducción la misma (Steinberg, 2005). La predicción de cómo un individuo responde al tratamiento con hidroxiurea, podría ayudar a una mejor selección de los pacientes para este fin y reducir al mismo tiempo los efectos por su toxicidad. Desafortunadamente, a pesar de algunas pistas interesantes, no es posible predecir actualmente la respuesta al tratamiento con esta molécula. 1.5.5. Niveles del estrés oxidativo asociados a la hemólisis. Los glóbulos rojos de los pacientes anémicos tienen una membrana mucho más frágil y son mucho más vulnerables a producir hemolisis. La hemolisis supone un aumento de la destrucción prematura de los eritrocitos. Si la vida media de los eritrocitos normales es alrededor de 120 días, en un individuo con SCD los eritrocitos tienen una vida media entre 4 y 21 días. Antes se pensaba que la hemolisis se debía al contenido de HbS polimerizada y adherida a la membrana que la hace ser más rígida. Hoy día se ha demostrado que estos eritrocitos se encuentran en condiciones de alto estrés oxidativo, que provoca daño en la membrana celular. Parece ser que la hemolisis es uno de los factores implicados en la heterogeneidad de las complicaciones de esta enfermedad. Evidencias recientes demuestran que los pacientes con las complicaciones agudas, tales como hipertensión pulmonar y sistémica (Morris y colbs., 2005), priapismo (Nolan y colbs., 2005), ulceraciones cutáneas (Nolan y colbs., 2006) y riesgo de muerte repentina en los niños, se caracterizan por altos niveles de hemolisis, una vasculopatía proliferativa y disfunción en el sistema vasomotor (Morris y colbs., 2005). Se puede estimar la hemolisis a través del índice de reticulocitos. La otra forma de medir la hemolisis es a través de lactato deshidrogenasa (LDH), arginasa I y ratio arginina/ornitina, en el plasma. A cambio, los pacientes con las manifestaciones clínicas clásicas de la enfermedad, que incluyen crisis de dolor y síndrome torácico agudo, están más asociados con la patogénesis que involucra vaso-oclusión, infartos e inflamación (Platt y colbs., 1994, Kaul y Hebbel, 2000). La patogénesis de la hemolisis incluye daños en la membrana debido al estrés oxidativo. La producción de los radicales libres de oxigeno (ROS), disminución en la 20 Introducción efectividad del sistema antioxidante, peroxidación de lípidos y proteínas de las membranas eritrocitarias conllevan a la salida del contenido hacia el espacio extracelular. El estrés oxidativo a su vez provoca activación de la iNOS (Escames G, 2006) y VEGF (factor de crecimiento endotelial vascular), que a su vez potencia producción de las especies reactivas de nitrógeno, vasoconstricción y la producción de la hipertensión arterial (Kato, 2007; Aslan, 2007). Por tanto, la posibilidad de estudiar los niveles del estrés oxidativo como un biomarcador de estado y avance de anemia falciforme y su posible relación con los haplotipos, tiene una significancia clínica (Kato, 2007; Aslan, 2007; Wood y Granger, 2007). Además, las ROS pueden desactivar directamente a la proteína NOS endotelial (Schulz y cols., 2004), responsable de producir NO, un importante vasodilatador. Los eritrocitos poseen un sistema de defensa complejo y eficiente en el cual se incluye el sistema glutatión y las enzimas como la superóxido dismutasa (SOD), la catalasa (CAT), la glucosa-6-fosfato deshidrogenasa (Glu-6PDH) y la NADH o NADPH metahemoglobina reductasa. Se ha demostrado que en los eritrocitos SS se generan cantidades excesivas de ROS debido a la presencia de la HbS inestable y la oxidación espontánea del hierro en el grupo hemo. Por otra parte, la vaso-oclusión que ocurre en la drepanocitosis provoca daños por isquemia-reperfusión y activación de procesos inflamatorios tisulares que aumentan la producción y liberación de ROS, así como la expresión de la óxido nítrico sintasa inducible (iNOS) (Aslan y cols., 2003; Schulz y cols., 2004), que contribuye a la producción de las especies reactivas de nitrógeno (RNS). En los estudios con eritrocitos se ha demostrado que los niveles de anión superóxido (O2-•), radical hidroxilo (OH•) y peroxidación lipídica son aproximadamente 2 veces mayores en pacientes con drepanocitosis que en donantes voluntarios (Ren, 2008). Igualmente se encontró una disminución en la producción o liberación de óxido nítrico durante las crisis vasooclusivas (Schulz y cols., 2004). 1.5.6. Reducción en la biodisponibilidad de óxido nítrico. Al NO se atribuye una serie de funciones importantes que participan en el mantenimiento del buen funcionamiento vascular. A parte de ser un vasodilatador, 21 Introducción inhibe la agregación plaquetaria y la adhesión de los leucocitos, la proliferación y la migración de las células del músculo liso, y tiene efectos antioxidantes y antiinflamatorios (Mack y Kato, 2006). El NO liberado del endotelio difunde hacia las células musculares adyacentes, donde reacciona con hierro ferroso del grupo hemo de guanilato ciclasa aumentando la síntesis de guanosín monofosfato cíclico (cGMP) a través de guanosín trifosfato (GTP). A su vez, el cGMP activa la proteín kinasa G (PKG), que estimula el bombeo activo de Ca++ hacía sus depósitos, disminuyendo los niveles de Ca++ intracelular y, por tanto, produciendo una relajación muscular, vasodilatación y aumento del flujo sanguíneo local (Ignarro y colbs., 1987; Lincoln y Cornwell, 1993). El mismo mecanismo señalizado por el sistema cGMP/PKG a su vez inhibe la agregación plaquetaria (Radomski y colbs., 1987). La reducción de Ca++ intracelular en plaquetas disminuye la expresión de P-selectina y suprime la expresión de glicoproteína IIb/IIIa, requerida para las uniones de fibrinógeno y la activación de coagulación (Loscalzo, 2001). El NO también es un potente antagonista de la inflamación (De Caterina y colbs., 1995). En parte este efecto se debe a que NO induce la expresión de IBα, un importante inhibidor del factor NF-B (Peng y colbs., 1995). A su vez NF-B, controla la expresión de varios genes involucrados en la inflamación y en la respuesta inmune, tales como VCAM-1, ICAM-1, y E-selectina. Actualmente se estudian las posibilidades de uso de NO para la prevención de adhesión de las células rojas al endotelio. Por ejemplo, se ha demostrado que los donadores exógenos de L-arginina y NO previenen la acción protrombótica causada por la inhibición de la eNOS en las vénulas (Broeders y colbs., 1998). El NO se sintetiza a partir de la L-arginina mediante la acción de la NOS y casi enseguida adquiere un electrón no apareado, transformandose en radical de óxido nítrico (NO) (Ghafourifar and Cadenas, 2005). En el medio extracelular NO reacciona con el oxígeno y agua, formando aniones de nitratos (NO3-) y nitritos (NO2), los cuáles conjuntamente se denominan como NOx. Se conocen tres isoformas de la NOS cuyos nombres se deben al sitio donde primeramente fueron encontradas: NOS neuronal (nNOS o NOS 1), inducible (iNOS o NOS 2), endotelial (eNOS o NOS 3). La eNOS se expresa de forma constitutiva, se encuentra en las células endoteliales, plaquetas y el cerebro y su actividad es calcio-calmodulina dependiente (Gladwin y Kato, 2008). La inhibición de la eNOS conlleva a un efecto 22 Introducción vasoconstrictor y provoca efectos secundarios graves, como la hipertensión pulmonar, una de las causas de crisis y de muerte de individuos con anemia falciforme. Esta hipótesis se apoya en algunos estudios que demuestran que aumentando la actividad eNOS se mejora vasodilatación, mientras, en ratones SCD con eNOS inhibida se produce una mayor vasoconstricción (Nath y colbs., 2000). La iNOS se induce ante determinados estímulos, es capaz de sintetizar NO• en cantidades mil veces superiores a las normales y es calcio-calmodulina independiente. En ciertos procesos como la inflamación, hipoxia y el estrés oxidativo, la actividad y la expresión de la iNOS aumentan (Crespo y colbs., 1999; Sullivan y colbs., 2008). El NO• reacciona con O2•-, produciendo grandes cantidades de peroxinitritos (ONOO-), tan tóxicos como el HO• (Cadenas y colbs., 1996). También se encuentra un incremento de esta enzima en los pacientes hipertensos (Escames y colbs., 2004). Se ha demostrado que la inhibición de la producción de los radicales libres mejora la clínica de estas patologías (Crespo y colbs., 1999; Escames y colbs., 2004, López y colbs., 2008). La expresión de la iNOS está aumentada en los riñones y el hígado de los pacientes y/o ratones SCD (Mack y Kato, 2006; Diwan y colbs., 2002; Aslan y colbs., 2003). Desde los años noventa, los datos producidos en el laboratorio y con animales de experimentación proporcionaron gran información para comenzar a establecer los efectos de los eritrocitos falciformes y la hemolisis crónica sobre la disfunción endotelial y el metabolismo de NO•. Por ej., French y colbs. (1997) demostraron que la administración de las drogas inhibidoras de la eNOS en ratas provoca cambios en el endotelio cerebral, disminuye el flujo sanguíneo cerebral y aumenta la mortalidad. En los ratones SCD la reducción de concentración de NO• se asocia con el aumento en la degradación de fibrina y en la producción de trombos (Ballas y colbs., 1988). Las tres causas más importantes de la disminución de biodisponibilidad de NO• son: disminución de L-arginina en el plasma, consumo de NO• mediante la hemoglobina extracelular (Reiter y colbs., 2002) y a través de su reacción con ROS (Mack y Kato, 2006). Una de las consecuencias de la hemolisis es la liberación de la arginasa, que a su vez degrada la L-arginina, precursor necesario para la síntesis de NO•. Se ha observado que la actividad de la arginasa es dos veces mayor en pacientes con 23 Introducción hipertensión pulmonar, dando como resultado un bajo ratio arginina/ ornitina, y una menor disponibilidad de NO• (Morris y colbs., 2005). Figura 8. Efectos de la hemolisis sobre la biodisponibilidad de óxido nítrico (Gladwin y Kato, 2008). El NO• reacciona con la desoxiHb y la oxiHb formando nitrosilHb (Hb(Fe 2+ )- NO) y metHb, respectivamente (Gibson y Roughton, 1957). Las constantes de estas reacciones están en el orden de 3-5 x107M -1·S -1, que significa una vida media de 1 µseg para el NO•. Con esta vida tan corta, la concentración de NO• no sería suficiente para activar a la guanilato ciclasa y no produciría sus conocidos efectos de vasodilatación (Peñuela, 2005; Herold y colbs., 2001). La reducción de biodisponibilidad de NO• puede predisponer a la crisis vasooclusiva y/o los procesos inflamatorios, ya que NO• inhibe la formación de las moléculas inflamatorias: VCAM-1, ICAM-1 y ELAM-1 (Rother y colbs., 2005). Es más, en los ratones con otras anemias, por ejemplo, con anemia hemolítica alloinmune, se ha visto que se desarrolla la hipertensión pulmonar asociada a una disminución en vasodilatación causada por una menor biodisponibilidad de NO• (Dweik y colbs., 1998). 2. HAPLOTIPOS DE ANEMIA FALCIFORME. Los haplotipos que más se asocian con los diversos aspectos clínicos son los haplotipos del extremo 5 ׳del clúster beta. 24 Introducción Figura 9. El patrón de digestión del clúster con distintas enzimas de restricción (Zago y colbs., 2000). Los primeros estudios de distribución geográfica de los β haplotipos demostraron que la mutación ocurrió independientemente, al menos en tres ocasiones en África, en la Península Arábica y la India Central (El-Hazmi y colbs., 1999; Ruiz y colbs., 2003; Ofori-Acquah y colbs., 2004). En estos grupos étnicos existen varios haplotipos, pero hay un haplotipo más predominante que es el que se denomina de acuerdo al sitio. Por ejemplo, el haplotipo Senegal es más común en Senegal, donde también está presente el haplotipo Benín. El haplotipo Benín es mas común (92%) en Yoruba, Nigeria, pero también es común en Jamaica (aprox. 71%), aunque Bantú y Senegal también están presentes en Jamaica. La distribución geográfica de los s haplotipos clásicos se atribuye a los sitios de origenes independientes (Hanchard y colbs., 2007). La mutación βS apareció hace aproximadamente 30.000 años, es mucho más reciente que la separación de los genes β y α (hace mas de 450 millones de años atrás) y está relacionada con la aparición del sedentarismo humano y seleccionada bajo la presión ejercida por la malaria (Rodríguez y Saénz, 1998). Se ha demostrado que los haplotipos s clásicos son altamente conservados a lo largo de cientos de bases (Hanchard y colbs. 2007), pero también existen otros haplotipos menos frecuentes. Basándonos en el uso de las cinco enzimas de restricción dentro del clúster , tales como ε-HincII, G-HindIII, A-HindIII, 5'ψ-HincII, 3'ψ25 Introducción HincII, podemos distinguir los haplotipos 5' mas comunes, denominados de acuerdo al sitio de su origen, y otros haplotipos encontrados, pero menos frecuentes (Shimitzu y colbs., 2001). Tabla 3. Patrón de digestión de los haplotipos 5' del clúster globínico beta. Haplotipo Benin Bantu (CAR) Senegal Cameron ArabeHindu 1 2 3 Haplotipo 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 ε-Hinc + + + + ε-Hinc + + + + + + + G-HindIII + + + + G-HindIII + + + + + + + + + + + + A-HindIII + + A-HindIII + + + + + + - 5'ψ-HincII 3'ψ-HincII + + + + + + 5'ψ-HincII + 3'ψ-HincII + + + + + + + + + + + + + + + + + + + + - El signo mas (+) significa corte de una región específica de ADN con la enzima de restricción mencionada, y el signo menos (-) significa no corte. Se ha observado que el principal beneficio conferido por algunos de estos haplotipos en relación con la gravedad de la drepanocitosis se explica en términos de su relación directa con las concentraciones sintéticas de Hb fetal, que suele considerarse ―antifalciforme‖ porque no copolimeriza con la HbS, a la que torna más soluble. Los diferentes polimorfismos del gen de la HbS estimulan una mayor o 26 Introducción menor síntesis de HbF, además, modifican la proporción de las cadenas G y A de la HbF en el adulto drepanocítico (Powars, 1991; Rodríguez y colbs., 1994; Powars y colbs.). Los haplotipos Sen y Árabe-hindú se asocian con mayor producción de las cadenas G, y por ende mayor porcentaje de la HbF. En el neonato y en el niño de pocos meses hay más cadenas G que cadenas A (razón 6:4). En la medida en que se inactiva la síntesis de HbF en años posteriores, se activa un mecanismo genético que favorece un descenso más acelerado de la producción de cadenas G en relación con cadenas A (Powars y colbs., 1990; Nagel, 1991), por lo que después del cuarto o quinto mes de edad la razón G:A empieza a ser 4:6, como en el adulto (Nagel, 1991). Los haplotipos Senegal y Árabe-hindú se asocian con mayores concentraciones de HbF y con la conservación de razón G:A del período neonatal normal, 6:4, lo que posiblemente contribuye a la clínica más leve. Además, los estudios del clúster globínico beta permiten hacer conclusiones de orígenes antropológicos de las poblaciones. En las Américas, los haplotipos βs permiten entender mejor las raíces ancestrales africanas de las poblaciones de raza negra. Por ejemplo, varios estudios hechos en América Central apuntan que allí predenomina el haplotipo CAR. Tabla 4. Los parámetros de HbF, ratio G: A y la clínica de acuerdo al haplotipo del clúster beta. Haplotipo Benín (Ben) Conc. de HbF Intermedia 5-15% Ratio G: A 1:1 Clínica Gravedad intermedia, más leve que CAR, las principales complicaciones son uremia y la insuficiencia o disfunción de órganos importantes, tales como cerebro, riñón e hígado. De estos pacientes, 90% presentan crisis de osteonecrosis importantes después de los 20 o 25 años de edad. Ubicación geográfica Occidente centroafricano 27 Introducción Haplotipo Conc. HbF Rep. Centroafricana (CAR) o Bantú Senegal (Sen) Camerún (Cam) Arabéhindú de Ratio G: A Clínica Ubicación geográfica Muy baja, <5% Muy baja África central Altas, 20% 6:4 Intermedia , 5-15% Altas, 20% 1:1 La clínica más grave, presenta daño en alguno de sus órganos vitales en los primeros 40 años de vida. Más leve, por su parte, se vincula con un menor número de episodios ―drepanocitémicos‖, de síndrome torácico agudo y de infarto esplénico y óseo. Curso clínico más leve. 6:4 Curso clínico inocuo. más África atlántica Zona de Camerún India, países árabe En la zona Mediterránea el haplotipo más frecuente es el Benín, que llegó a Grecia y Sicilia por medio de las invasiones musulmanas y a Portugal, por el tráfico de los esclavos (Rodríguez y Sáenz, 1998). Los haplotipos βs árabe-saudí y el indio o asiático han tenido un origen distinto del de los haplotipos africanos, estando estrechamente relacionados entre sí por las corrientes migratorias que se establecieron entre territorios cercanos al río Indo (cuna de la civilización de Harappa en el subcontinente indio) y pueblos originarios del Oasis del Este en la península arábica, y de manera más concreta entre indios, árabes chiítas e iraníes. Fue aparentemente a orillas del río sagrado de la India donde se originó la mutación asiática de la HbS, lo que sugiere, a diferencia de lo sucedido en África, que el origen del gen βs en la India fue de carácter unicéntrico (Labie y colbs., 1989). Por ejemplo, los estudios en la población iraní demostraron que el haplotipo que más pre denomina es el Árabe-hindú. En segundo lugar se encuentra haplotipo Bantú A2 (Rahimi y colbs., 2003). Desde los primeros análisis de haplotipo beta en los años ochenta, se han hecho varios estudios epidemiológicos que relacionan distintos haplotipos con el curso clínico de la enfermedad. Por ejemplo, Powars, en varios estudios hechos en África y América, encontró que el haplotipo CAR incrementa el riesgo de desarrollar complicaciones desde una edad temprana, además se relaciona con la 28 Introducción microvasculopatía que lleva a los daños orgánicos durante tres primeras décadas de la vida, y sugiere aplicar una terapia de trasplante desde una edad temprana (Powars e Hiti, 1993). El haplotipo Benín tiene síntomas menos severos que el CAR pero más severos que el haplotipo Senegal (Powars e Hiti, 1993; Powars, 1991). Los CAR/CAR tienen mayor riesgo de padecer accidente cerebro vascular que el resto de los haplotipos (Sarnaik y Ballas, 2001). 3. ANEMIA FALCIFORME Y ESTRES OXIDATIVO. 3.1. Radicales libres y enzimas involucrados en su producción. Los radicales libres son producidos en el organismo como parte de sus funciones bioquímicas y fisiológicas. El radical libre se define como una especie química cargada con uno o más electrones desapareados en su orbital electrónico externo y que es capaz de existir independientemente (Halliwell y Gutteridge, 1989). Pero el peligro de contener una gran cantidad de radicales libres consiste en que estas especies son sumamente inestables y reaccionan con otras moléculas a su alrededor, provocando paso de electrones libres y alterando el funcionamiento correcto de estas moléculas. La formación de los radicales libres en el organismo es inevitable (López y colbs., 2008), pero estas especies no causan daño oxidativo en condiciones normales debido a que la célula está prevista de un efectivo sistema antioxidante. Los antioxidantes se definen como aquellas sustancias que a bajas concentraciones respecto a las de un sustrato oxidable, retardan o previenen la oxidación. Este proceso consiste, básicamente, en el hecho de que un antioxidante cede un electrón al radical libre, neutralizándolo, y transformándolo en una molécula inocua. Existen fuentes externas e internas de los radicales libres; al igual que existen dos tipos de antioxidantes: los exógenos tomados en la dieta y los endógenos, dotados por el propio sistema biológico. Otra forma de clasificar a los agentes antioxidantes es de acuerdo al nivel de su acción: primario, secundario y terciario (ver Tabla 5). 29 Introducción Tabla 5. Tipos de antioxidantes. Primarios Neutralizan los RL recién formados Secundarios Capturan los RL evitando la reacción en cadena Terciarios Reparan las biomoléculas dañadas por los RL Glutation peroxidasa (GPx), glutation reductasa (GRd), glutation S- transferasa, catalasa (CAT), superóxido dismutasa (SOD) Melatonina, vitamina E, C, betacaroteno, ácido úrico, estrógenos. Ej.; enzimas reparadoras de ADN (endonucleasas, exonucleasas), la metionina sulfóxido reductasa. El grupo de ROS incluye la formación de anión superóxido O2-•, peróxido de hidrógeno (H2O2) y radical hidroxilo (HO•): O2 + e- O2-• O2-•+ e- + 2H+ H2O2 H2O2 + e- 2OH• O2- + H2O2 → OH- + OH• + O2 Entre las RNS se incluyen los radicales libres óxido nítrico (NO•), dióxido de nitrógeno (NO2•) y el potente oxidante peroxinitrito (ONOO-). Entre los enzimas que contribuyen a la formación de los radicales libres podemos destacar el papel de NADPH oxidasa, xantina oxidasa e iNOS. Las enzimas intracelulares tales como, SOD y CAT, al igual que el sistema de glutatión, actúan como la primera línea de defensa frente a los radicales libres (Bradley y Nathan, 1984). Sólo cuando se produce un desequilibrio entre sustancias prooxidantes y antioxidantes a favor de las primeras, el resultado es un daño oxidativo que puede afectar varias moléculas alterando su funcionamiento y llevando el organismo entero a un envejecimiento prematuro, enfermedades cardiovasculares, cáncer, diabetes, afecciones a nivel genético y otras patologías. Este daño oxidativo producido por un desequilibrio entre la producción de radicales libres y el funcionamiento del sistema antioxidante, se denomina estrés oxidativo. 30 Introducción Factores prooxidantes: Externos Internos: NADPH oxidasa Xantina oxidasa eNOS Factores antioxidantes: Sistema glutation Enzimas: SOD, CAT, GPx Figura 10. Balance de procesos prooxidantes y antioxidantes que debe existir en el organismo. La NADPH oxidasa cataliza la reducción de oxígeno a O2-•, lo que resulta en la producción de H2O2. Este enzima se encuentra en la membrana de los leucocitos polimorfonucleares, es mejor caracterizada en los neutrófilos, y se activa en condiciones de inflamación, en respuesta a la presencia del organismo microbiano (Market y colbs., 1984; Elsbach y Weiss, 1985). La xantina oxidorreductasa (XOR) es un molibdenoflavoenzima complejo, que existe en dos formas funcionalmente distintas: xantina deshidrogenasa (XDH), que produce NADH y urato; y puede ser transformada en xantina oxidasa (XO), oxígeno dependiente, que origina radical anión superóxido y/o peróxido de hidrógeno y urato. En condiciones normales la XDH representa 80% de la XOR funcional in vivo, y la XO constituye un 20% restantes. Pero en las condiciones de injuria por isquemia- reperfusión la enzima XO aumenta notablemente su actividad y contribuye a la formación de ROS (McCord, 1985; Chambers y colbs., 1985). Tanto la XDH, como la XO catalizan la degradación de la hipoxantina (HX) en xantina (X), y subsecuentemente en urato. Sin embargo, sólo XO produce O2•- en el último paso de esta reacción (McCord, 1985), ver figura 11. 31 Introducción Adenosina AMP Adenosina Xantino deshidrogenasa (i) Inosina Xantino oxidasa (a) ATP Acido úrico Hipoxantina Xantina O2-• H2O2 Figura 11. Esquema de producción de RL por la enzima xantino oxidasa. El enzima iNOS participa en la producción de RNS en condiciones de inflamación y el estrés oxidativo (Escames y colbs., 2004). 3.2. Sistemas antioxidantes. Superoxido dismutasa (SOD) La SOD fue la primera enzima de la cuál se conoció que actuaba sobre un radical libre y fue descubierta en 1968 (McCord y Fridovich, 1969). La familia de las superóxido dismutasas está integrada por tres tipos de métalo enzimas, Cu/Zn-SOD, Mn-SOD, y Fe-SOD. La Cu/Zn-SOD es la más abundante, se encuentra en la fracción soluble celular, y en los líquidos extracelulares. La SOD dependiente de Mn++ se encuentra en procariotas y eucariotas y se localiza en la matriz mitocondial. En las bacterias se ha descrito una tercera forma de la SOD, dependiente de Fe++. 32 Introducción La SOD neutraliza el anión superóxido, convirtiéndolo en agua oxigenada, por lo cual forma una primera línea de defensa ante la producción de ROS generados en numerosos procesos oxidativos: O2•- + O2•- + 2H+ → H2O2 + O2 La actividad de la enzima aumenta en situaciones de estrés oxidativo prolongado, normalizándose sus valores cuando la producción de ROS disminuye. No obstante, el agua oxigenada producida por la SOD es, a su vez, fuente de ROS, como el radical hidroxilo, mucho más agresivo que el radical superóxido. Por ello, es muy importante disponer de otros sistemas de defensa, tales como la catalasa, para evitar que el agua oxigenada se transforme en radicales hidroxilo. Catalasa (CAT) La catalasa se una proteína compuesta de cuatro subunidades, cada una de las cuáles contiene grupo heme y la molécula de NADPH. En los mamíferos la CAT se sintetiza en la mayoría de las células. A nivel subcelular, el 80% de la actividad de la CAT se localiza en los peroxisomas y el 20% restante en el citosol. Catalasa elimina el peróxido de hidrógeno sin necesidad de un agente reductor: 2H2O2 →2H2O + O2 Sistema del Glutatión El glutatión (L--glutamyl-L-cysteinylglycine) es un tripéptido involucrado en la defensa antioxidante de casi todas las células. Su capacidad antioxidante se debe a la presencia de un residuo de cisteína (grupo tiol –SH), capaz de formar el puente de disulfuro entre las dos moléculas de glutatión, proceso acompañado con la liberación de dos electrones y dos protones. El glutatión puede existir en su forma reducida (GSH) y oxidada (GSSG) o unido a las proteínas. Cuando la forma reducida de glutatión pasa a su forma oxidada, se liberan protones y dos electrones, los cuáles pueden utilizarse para 33 Introducción reducir el efecto dañino de los radicales libres. El enzima que participa en este proceso es la glutatión peroxidasa (GPx), y a su vez, la vía de recuperación del glutatión reducido a partir del GSSG es catalizada por el enzima glutatión reductasa (GRd). Este último es dependiente del NADPH proveniente del ciclo de las pentosas fosfato. GSSG + NADPH + H + → 2 GSH + NADP+ Las enzimas GRd y GPx juntos forman la maquinaria antioxidante dependiente de glutatión. Ambas enzimas se encuentran tanto en el citosol, como en la mitocondria, aun que predomina el componente citosólico (Kim y colbs., 2003). El sistema de glutatión es un indicador de la funcionalidad celular y puede ser medido a través del ratio GSH/GSSG. En condiciones normales la forma reducida debe predominar la forma oxidada y el ratio oscila entre 1 y 10 (Pastore y colbs., 2003). Figura 12. Sistema del glutation. En algunas patologías el estrés oxidativo tiene una contribución significativa en su desarrollo, como en la enfermedad de Parkinson, Alzheimer, artritis reumatoide y el fenómeno llamado "paradoja del oxígeno" que se presenta posterior a un fenómeno de isquemia, durante la reperfusión, cuando el aporte de oxígeno no disminuye el daño sino que lo incrementa participando en reacciones de oxidación de proteínas y 34 Introducción lipoperoxidación. (Halliwell y Gutteridge, 1999). El fenómeno isquemia-reperfusión es bastante común en los pacientes con anemia falciforme. Según los datos epidemiológicos, los niveles de hemolisis crónica se asocian, con las complicaciones, tales como la hipertensión pulmonar y las ulceraciones cutáneas (Wood y colbs., 2008). Los últimos estudios demuestran que una característica común de la anemia falciforme es la hemólisis crónica provocada por el estrés oxidativo. Sobre la participación del estrés oxidativo en anemia falciforme se han hecho varios estudios en animales que revelan, por ejemplo, que los ratones con sobreexpresión de la SOD, tienen una atenuada adhesión de las células sanguíneas en las paredes de los vasos sanguíneos (Wood y colbs., 2005). Por otro lado, la utilización de allopurinol, inhibidor de XO, y por lo tanto, de ROS, mejora la dilatación arteriolar y reduce la acumulación de leucocitos en las vénulas (Kaul y colbs., 2004; Kaul y colbs., 2006). Al exponer los eritrocitos normales a un tratamiento con phenazin methosulfato, un fuerte productor de radicales libres, aparecen las características propias de eritrocitos SS, tales como, rigidez de las membranas, inestabilidad mecánica, lisis y el tiempo de vida más corto (Hebbel y colbs., 1990). Existen varias causas del estrés oxidativo en anemia falciforme. Por un lado, la hemólisis de las células falciforme conlleva a la liberación de la Hb y la arginasa. El grupo hemo de la Hb liberada se oxida y el mismo Fe en su estado elemental contribuye a la formación de ROS a través de la reacción de Fenton. Por otro lado, la vasooclusión intermitente provoca daños por isquemia-reperfusión y activación de los procesos inflamatorios tisulares que aumentan la producción de radicales libres activando la iNOS (Aslan y colbs., 2003; Schulz y colbs., 2004), y otras enzimas prooxidantes, como la XO y la NADPH oxidasa. 3.3. Fuentes de los radicales libres en la anemia falciforme. 1. Autooxidación de la hemoglobina falcémica Hebbel y colbs. (1988) por primera vez demostraron que la HbS tiene una velocidad de autooxidación mucho más alta que la de HbA, en presencia de oxígeno. 35 Introducción Por lo tanto, es implicada en una mayor producción de superóxido y radical hidroxilo detectados en los pacientes con SCD (Hebbel y colbs., 1988). La autooxidación de Hb conduce a la formación de superóxido y subsecuentemente, a la formación de peróxido (reacción de Fenton): Fe2+ + O2 Fe3+ + O2-• O2-•+ O2-• + 2H+ H2O2 2. Xantina oxidasa Se ha demostrado, que la enzima XO tiene una mayor actividad en el endotelio aórtico (Kaul y colbs., 2006) y si los ratones SCD son tratados con allopurinol, inhibidor de XO, y por lo tanto de las ROS, mejora la dilatación arteriolar y se reduce la acumulación de leucocitos (Kaul y colbs., 2004). Las fuentes principales de XO son dos: células endoteliales y el hígado. Se propone que en condiciones de la crisis vaso-oclusiva el hígado libera la XO soluble, cuál se une a la superficie de las células endoteliales de los vasos sanguíneos de distintos órganos (Aslan y olbs., 2001). Se ha observado que la actividad de la XO en plasma de los pacientes con SCD es significativamente mayor que en el grupo control (Aslan y colbs., 2001). 3. NADPH oxidasa En los pacientes con SCD la cantidad de leucocitos está aumentada, y los leucocitos contienen la enzima NADPH oxidasa, implicada en la producción del radical superóxido. Además, estudios recientes demuestran que los aniones superóxido producidos por la NADPH oxidasa endotelial están implicados en la adhesión leucocitaria y plaquetaria en las vénulas del cerebro de ratones SCD (Wood y colbs., 2005). 4. Otras fuentes 36 Introducción La enzima eNOS está compuesta por dos subunidades monoméricas: oxygenasa y reductasa, trabaja en presencia de oxigeno, y requiere de cofactores NADPH y tetrahidropterina (BH4). En condiciones normales, la eNOS transforma la L-arginina en NO, produciendo pequeñas cantidades de NO necesarias para mantener homeostasis vascular. En ausencia o baja concentración de la BH4, la enzima NOS se desacopla y en vez de producir NO, puede producir superóxido y sus derivados (H2O2, OH•, ONOO-) (Aslan y colbs., 2003). Esta situación se ha observado en los estudios de la diabetes, hipertensión y ateroesclerosis. Pero hay estudios que apuntan que una situación similar se presenta en el caso de anemia falciforme también. Las experimentaciones en los ratones SCD en condiciones de hipoxiareperfusión estimulada, han demostrado una adhesión anormal de las células rojas a las paredes de las vénulas del cerebro, y esta situación fue normalizada con una deleción genética de la eNOS, o con tratamiento con eNOS inhibidor- L-NAME (Wood y colbs., 2006). En condiciones de hiperhomocistenemia, presente en SCD, se promueve la formación de dimetilarginina asimétrica (ADMA), que compite con la L-arginina como sustrato de la NOS. En los ratones transgénicos SCD bajo estas condiciones la eNOS se desacopla y se evidencia un aumento en las cantidades de ROS y RNS (Forstermann y Munzel, 2006). 3.4. Consecuencias del estrés oxidativo en la anemia falciforme. 1. Hemolisis: La membrana es uno de los componentes que más se afecta en condiciones de sobreproducción de ROS, se produce la lipoperoxidación, la membrana se hace más rígida e inestable a cualquier toque mecánico. 2. Adhesión de las células rojas: En los estudios de cultivos de células endoteliales de venas del cordón umbilical humano de los pacientes con SCD, se ha observado un aumento de concentración de ácido tiobarbiturico, activación de NFkB y una expresión elevada de las moléculas de adhesión celular. La adhesión se disminuye luego de tratar las células con la SOD, catalasa y antioxidante probucol (Sultana y colbs., 1998). 37 Introducción 3. Adhesión de las células plaquetarias y los leucocitos: La adhesión plaquetaria en microcirculación de los ratones SCD es 4-5 veces mayor que en los ratones normales, hecho que se relaciona con el aumento en la expresión de Pselectina en condiciones del estrés oxidativo, lo que a su vez motiva el reclutamiento de las plaquetas y los leucocitos (Wood y colbs., 2004). Además, se ha demostrado que en los ratones con deficiencia genética de NADPH oxidasa (gp92phox) y sobreexpresión de SOD citosólica (SOD1), la adhesión de los leucocitos disminuye (Wood y colbs., 2005). Figura 13. Consecuencias del estrés oxidativo en anemia falciforme. Entonces, la enfermedad de células falciforme es compleja, y el proceso de oclusión de los vasos sanguíneos y los daños en los órganos internos puede ser 38 Introducción explicado a través de los procesos de adhesión celular, inflamación y un desequilibrio en la función vaso motora. 4. ESTRATEGIAS TERAPÉUTICAS EN LA ANEMIA FALCIFORME. 4.1. Terapias tradicionales. Hidroxiurea: La hidroxiurea es una droga citotóxica y citostática que la ―Food and Drug Adminitration‖ (FDA) estadounidense ha aprobado como tratamiento preventivo de anemia falciforme. Varios estudios indican que su uso reduce la frecuencia de las crisis vaso-oclusivas, complicaciones pulmonares, disminuye la necesidad de las transfusiones y reduce la mortalidad en los pacientes SCD (Charanhe y colbs., 1995; Lima y colbs., 1997). Otra ventaja es su bajo coste que permite utilizarla de una manera bastante generalizada. El mecanismo de acción de hidroxiurea no está muy claro. Por un lado, se ha demostrado que ésta induce la síntesis de la HbF, que tiene un efecto beneficioso en el proceso de oxigenación de los tejidos. Pero por otro lado, también se sugiere que la hidroxiurea es un agente que reduce la actividad de mieloperoxidasa (Saleh y colbs., 1999), libera el NO• y estabiliza la eNOS (Cokic y colbs., 2007). Algunos estudios confirman la capacidad de hidroxiurea de reaccionar con la oxihemoglobina, deoxyhemoglobina y metahemoglobina para liberar NO• in vitro e in vivo (Pacelli y colbs., 1996; Kim-Shapiro y colbs., 1999; Nahavandi y colbs., 2000). Sin embargo, no todos los pacientes responden al tratamiento con hidroxiurea. Además, su uso prolongado puede incrementar el riesgo de los defectos hereditarios y de leucemia, según los datos de National Toxicology Program, 2007. Transfusiones crónicas: Las transfusiones de sangre mejoran notablemente el estado de un paciente crítico, siendo una terapia alternativa que evita daños mayores a los órganos vitales. Sin embargo, siempre queda algún porcentaje de células falciformes en el organismo, y el proceso patológico continua. Así, los niveles elevados de la Hb plasmática y de recuento de reticulocitos disminuyen, pero no se normalizan completamente (Tancabelic y colbs., 1999; Lezcano y colbs., 2006). Además, el acceso a las transfusiones sanguíneas es limitado, especialmente en países 39 Introducción africanos. Las transfusiones frecuentes conllevan a que se desarrolle riesgo de alloinmunización eritrocitaria, sobrecarga de hierro y las infecciones. Trasplante de células madre hematopoyéticas: Es un tipo de tratamiento curativo pero la desventaja está en que es difícil de conseguir donante HLA compatible y que no tenga rasgo falciforme, el trasplante es más efectivo en los niños que en los adultos, es difícil predecir el curso que va a tomar el paciente luego de la operación, y las complicaciones aún son bastante frecuentes. Si se evalúan esos tres tratamientos tradicionales, se puede decir que son menos que satisfactorios, que se requieren nuevas opciones para disminuir el ratio riesgobeneficio. En Francia actualmente se exploran primeros ensayos para la terapia génica, que sí podría ser un tratamiento definitivo, pero aun no está claro si el gen correctivo puede tener una expresión efectiva y a largo plazo, si es un procedimiento seguro y cuál es el perfil de su coste. 4.2. Terapias alternativas. Inhalación de óxido nítrico: La inhalación de óxido nítrico puede ser una terapia beneficiosa para los pacientes SCD, como lo indican varios estudios. Por ejemplo, puede usarse para tratar síndrome torácico agudo, aumentar la tensión arterial de oxigeno, reducir la tensión arterial pulmonar, e inclusive, el tiempo de hospitalización (Montero-Huerta y colbs., 2006; Opperty colbs., 2004). Además, se han visto efectos a nivel sistémico, debido a que al inactivar la Hb plasmática mediante su unión a NO, disminuye el consumo del NO arterial y se restaura su biodisponibilidad (Reiter y colbs., 2002), lo que a su vez permite efectuar la vasodilatación, aliviar los dolores asociados con la vaso oclusión (Weiner y colbs., 2003) y mejorar la tensión arterial de oxígeno (Zuzak y colbs., 2003). Terapia con quelantes de hierro y aclaradores de hem/Hb: El 99% de hierro en el cuerpo se encapsula dentro de la Hb eritrocitaria o en las proteínas de almacenamiento (transferrina, lactoferrina, y ferritina). Si el Fe2+ es liberado de la célula y es oxidado, se desencadenan los eventos relacionados con los daños de la membrana mediante la lipoperoxidación. Se ha investigado que el uso del quelante de 40 Introducción hierro sérico, deferiprona, disminuye los daños membranales (Shalev y colbs., 1995), y el uso de la desferoxamina, quelante intracelular, atenúa la adhesión de los eritrocitos en las vénulas del cerebro de los ratones SCD (Wood y colbs., 2005), y puede ser usado para prevenir los daños asociados con las transfusiones. La haptoglobina es una glicoproteína fijadora de la Hb. El complejo de hemoglobina-haptoglobina se depura en cuestión de minutos por el sistema mononuclear-fagocítico. En los pacientes con hemólisis significativa, sea intravascular o extravascular, es característico un descenso de la haptoglobina sérica. Si la cantidad de hemoglobina liberada supera la capacidad de la fijación de haptoglobina del plasma, la hemoglobina libre restante contribuye a la formación de los radicales libres o atraviesa los glomérulos, produciendo hemogloglobinuria. Por lo tanto, las estrategias para aumentar las concentraciones de haptoglobina pueden tener utilidad en la SCD y los pacientes talasémicos cuya capacidad de unión de la transferrina con hemoglobina está afectada por el exceso de la hemólisis (Gladwin y colbs., 2004). Participación de NOS: Este tipo de terapia se enfoca a mejorar el funcionamiento de la enzima eNOS desacoplada, suministrándole el sustrato o su cofactor. La suplementación alimenticia con L-arginina da resultados dudosos, quizás por el hecho que en el plasma la arginasa se encarga de metabolizarla. La inactivación de la arginasa con su inhibidor NG-hidroxi-L-arginina protege la L-arginina endógena y exógena y da mejores resultados. Experimentalmente se ha visto, que el suplemento de BH4 o su precursor sepiapterina, puede restaurar la síntesis de NO y su rol en la homeostasis vascular en los ratones (Schmidt y Alp, 2007). Se han hecho estudios que confirman que la inhibición de la relajación vascular en los ratones SCD se restaura totalmente por la acción de los donantes de NO, como es el sodio nitroprusiato (SNP). Este hecho se debe a que SNP se metaboliza por las células del músculo liso (Shalev y colbs., 1995), que a su vez excluye el efecto negativo del mismo en la producción de RNS y es efectivo debido a que actúa directamente sobre guanilato ciclasa, provocando la relajación (Kowaluk y colbs., 1992). Drogas para combatir ROS: Este tipo de terapia está enfocado hacía el uso de los agentes químicos que disminuyen la producción endógena de los radicales libres. La sulfasalazina es un potente anti-inflamatorio que inhibe el factor de transcripción 41 Introducción -NFkB necesario para la producción de las enzimas que participan en la formación de las ROS y de moléculas inflamatorias, incluyendo citokinas, quimiokinas y moléculas de adhesión. En modelos experimentales con ratones SCD se demostró que sulfasalazina reduce la adhesión celular (Kaul y colbs., 2004). Además, se demostró que tiene propiedades antioxidantes y previene eventos que conllevan a la disfunción endotelial (MacDermott, 2000). Otra droga antioxidante que se ha demostrado que tiene efectos favorables para la vasodilatación, es la L-4F, análogo de la ApoA-1, (Ou y colbs., 2003). Los estudios preliminares indican que en los pacientes SCD con hipertensión pulmonar y resistencia vascular a vasodilatación acetilcolina-dependiente, hay niveles reducidos de ApoA-1 (Yuditskaya y colbs., 2005). Terapia combinada: El uso de algunas de estas estrategias terapéuticas por separado en los pacientes con SCD puede tener limitaciones importantes. Por ejemplo, las terapias que aumentan la biodisponibilidad de NO (suplemento de L-arginina, la inhibición de la arginasa, donantes de NO) en un entorno de aumento de estrés oxidativo pueden producir la lesión nitrosativa a través de especies nitrosativas muy reactivas (ONOO-) (Wood y colbs., 2006; Ferdinandy y Schulz, 2001). Del mismo modo, las estrategias que restablezcan funcionamiento de la eNOS a través de la disponibilidad del cofactor sólo alimentan oxidación si la xantina oxidasa, NADPH oxidasa o la autooxidación de HBS son principales contribuyentes al medio oxidativo. Así que una estrategia combinada que tome en cuenta desacoplamiento de la eNOS, la inhibición de la arginasa, la captación de la hemoglobina libre o su inactivación, eliminación de ROS y donadores de NO, puede ser mas efectiva. Finalmente, el énfasis tradicional del fenómeno de la formación de eritrocitos falciformes y oclusión de los vasos sanguíneos en la SCD debe ser equilibrado con una apreciación de que esta hemoglobinopatia se manifiesta en gran medida como una vasculopatía generalizada (Aslan y Canatan, 2008). Las estrategias terapéuticas destinadas a contrarrestar la disfunción endotelial, alteraciones inflamatorias y estrés oxidativo pueden reducir la frecuencia de las hospitalizaciones, de las transfusiones, y de la incidencia de dolor y la aparición del síndrome torácico agudo y la hipertensión pulmonar en pacientes con SCD. 42 43 Hipótesis y Objetivos -Hipótesis y Objetivos- 44 Hipótesis y Objetivos 45 Hipótesis y Objetivos Para contribuir al conocimiento sobre la implicación del daño oxidativo en la fisiopatología de la anemia falciforme, hemos evaluado y compararado los niveles del estrés oxidativo entre el grupo de los pacientes y el grupo control, y analizado los biomarcadores de estrés oxidativo de acuerdo a la expresión de HbF en dichos pacientes. Nuestra hipótesis es que el grado de estrés oxidativo podría usarse como biomarcador del estado y avance clínico de la enfermedad y que, en consecuencia, pueda tener relación con los haplotipos del cluster globínico beta. En el caso que exista una correlación directa entre la expresión de los biomarcadores de estrés oxidativo y el avance clínico, se podrá pensar en buscar un tratamiento que disminuya el estrés oxidativo, y mejore el funcionamiento endotelial, ofreciendo una alternativa relativamente económica para mejorar la calidad de vida de los pacientes pediátricos. En este sentido, una intervención en la edad temprana de los pacientes podría tener mayores posibilidades de éxito. Objetivos: 1. Diagnosticar los haplotipos del cluster globínico beta en 100 pacientes pediátricos panameños y analizar su distribución geográfica y prevalencia en comparación con otros países latinoamericanos. 2. Correlacionar los haplotipos de anemia falciforme encontrados en los pacientes panameños con el fenotipo clínico de la enfermedad (leve, moderado y severo) y los datos hematológicos. 3. Comparar el grado de estrés oxidativo, de acuerdo a los biomarcadores seleccionados, entre el grupo de enfermos y el grupo control. 4. Evaluar la correlación de los niveles de estrés oxidativo de la sangre de los pacientes con el haplotipo diagnosticado. 5. Correlacionar los niveles de estrés oxidativo que presentan estos pacientes con el fenotipo clínico de la enfermedad y la expresión de la HbF. 46 47 -Materiales y Métodos48 49 Material y Métodos 1. PACIENTES Y PREPARACIÓN DE LAS MUESTRAS. Para este estudio se cuenta con el consentimiento informado aprobado por el Comité Nacional de Bioética de Panamá, siguiendo las normas de ética establecidas para trabajos de investigación en humanos por la Organización Mundial de la Salud (OMS) y la Declaración de Helsinki ratificada por la 29th World Medical Assembly, Tokio 1975, en su última revisión en 2008. Los niños incluídos en el estudio ya están diagnosticados previamente como enfermos falcémicos y se citan con sus familiares una vez a la semana para escuchar charlas sobre la enfermedad y los cuidados que se debe tener en cuenta. Posterior a la charla programada, se les explica el objetivo de este estudio y entre aquellos que quieren participar en el mismo, se escogen al azar los donantes. A lo largo de un mes se repite el mismo procedimiento una vez por semana hasta llegar a obtener las 100 muestras necesarias para este estudio. El mismo procedimiento se utiliza en un colegio de la localidad, para obtener las 40 muestras de sangre de niños aparentemente sanos. Criterios de inclusión: Niños HbSS, HbSF y HbSC de ambos sexos con el diagnostico confirmado a través de la prueba de electroforesis. Edad de 6 meses hasta los 15 años. Pacientes cuyos padres o tutor legal han firmado el consentimiento informado. Criterios de exclusión: Pacientes mayores de 15 años. Pacientes falcémicos sin diagnóstico confirmado. Pacientes que reciben tratamiento con hidroxiurea en el momento de este estudio. La determinación del fenotipo hemoglobínico. El fenotipo hemoglobínico se determina por electroforesis a pH 8,6 en acetato de celulosa (Lehmann y colbs., 1974). 50 Material y Métodos Las muestras de sangre se extraen mediante la punción venosa periférica en horas de la mañana en el laboratorio especializado de hematología del Hospital del Niño de Panamá. La sangre se distribuye en los tubos con EDTA-K3 como anticoagulante. La cantidad de tubos varía entre 2 y 3 dependiendo de la edad del niño y, a veces, por las dificultades para extraer la sangre debido al comportamiento del niño pequeño. La sangre se extrae de una forma cuidadosa, evitando la lisis de los hematíes, y los tubos se colocan verticalmente en una gradilla situada en la nevera a 4 ºC. A continuación, uno de los tubos se utiliza para realizar el análisis hematológico y para separar aproximadamente 500 l de sangre total para una posterior extracción de ADN y análisis de los patrones de restricción con las enzimas de digestión. El resto de los tubos se centrifuga durante 10 minutos a 5.000 g y 4 ºC. Tras la centrifugación, se hacen las alícuotas de plasma; y los hematíes se lavan dos veces con una solución salina isotónica (NaCl 0,9%), centrifugando cada vez a 5.000 g durante 10 min a 4 ºC. El plasma y los hematíes se alicuotan para una posterior determinación de la peroxidación lipídica (LPO) y de nitritos y nitratos (NOx) en el plasma; del glutatión reducido (GSH), glutatión oxidado (GSSG), las actividades de la glutatión peroxidasa (GPx), glutatión reductasa (GRd) y superoxido dismutasa (SOD) en los hematíes. Obtención de sangre por punción venosa periférica 500 l ST para la extracción de ADN Centrifugar 5000 g, 15׳, 4ºC PLASMA PCR LPO Nitritos GLOBULOS ROJOS 2 lavados 2,5 ml sol. NaCl 0.9%, 5000g, 5׳, 4ºC RFLPHaplotipos GSSG, GSH, GPx, GRd, SOD, Hb Figura 14. Preparación de las muestras para el estudio. 51 Material y Métodos Las alícuotas de plasma y glóbulos rojos se guardan a -80 ºC y posteriormente se trasladan a España, conservando la cadena de frío. Es importante destacar que no se hizo ninguna manipulación hasta el día en que se realizan las determinaciones bioquímicas. 2. MÉTODOS ANALÍTICOS. 2.1. Estrés oxidativo. 2.1.1. Determinación de la concentración de proteínas: método de Lówry. Todos los resultados obtenidos en este trabajo se normalizan por las proteínas de la muestra de hematíes tratados. Para calcular la concentración de proteínas se utiliza el método de Lowry (Lowry y colbs., 1951) con las modificaciones del método de Biuret (Layne y colbs., 1957). Se trata de una técnica colorimétrica con dos reacciones complementarias: a) Reacción de Biuret: es específica de los grupos amino de los enlaces peptídicos e implica la formación de un complejo coordinado coloreado de estos grupos con el cobre en medio alcalino. La misión del pH alcalino es facilitar el desplegamiento de las proteínas globulares en medio acuoso, por tanto, actúa como un agente desnaturalizante para que los iones de cobre puedan llegar hasta los grupo amino de los enlaces peptídicos. b) Reducción del reactivo de fenol: es específica de grupos reductores como el fenol. La reducción del reactivo de fenol se produce por el efecto de ciertos radicales aromáticos de los aminoácidos y de los complejos de Biuret. Este reactivo actúa como un indicador redox que al reducirse adquiere un color azul. La intensidad de color dependerá de la cantidad de proteínas presentes en la muestra, y puede cuantificarse midiendo la absorbancia a 650 nm. 52 Material y Métodos Complejo Proteínas-Cu2+ AA red. Complejo Proteínas-Cu2+ AA oxid. Reactivo Folin oxid. AMARILLO Reactivo Folin red. AZUL Protocolo: La medición de la concentración de proteínas totales en la muestra se realiza en placas de 96 pocillos. Para hacer la recta patrón se utiliza albúmina sérica como estándar (disuelta en TRIS 20 mM) a concentraciones entre 0,05-0,6 mg/ml. En primer lugar, se pone en los pocillos 50 l de blanco, patrones o muestras. A continuación se añade 200 l de reactivo de Lowry (carbonato disódico al 2% en NaOH 0,1 M, tartrato de sodio-potasio al 1% y sulfato cúprico al 0,5%, la proporción de estas soluciones es 98:1:1) y se deja reposar durante 10 minutos en agitación (agitador de placas: 1296-001 Delphia Plateshake). Finalmente, la reacción se revela con 50 l de reactivo de fenol diluido 1:10 con agua destilada. Se incuba durante 30 minutos a temperatura ambiente y se mide la absorbancia a 650 nm en un espectrofotómetro de placa (Bio-Tek Power-Wave, Microplate Scanning Spectrophotometer). Todas las determinaciones se realizan por duplicado. 2.1.2. Determinación de glutation oxidado (GSSG) y glutation reducido (GSH) en hematíes. Glutation oxidado y reducido son determinados mediante el ensayo fluorimétrico (Hissin y Hilf, 1976). Medición de GSSG: 53 Material y Métodos 1. La muestra descongelada de hematíes se hemoliza en tampón de hemólisis (10 mM fosfato sódico, 1mM EDTA-Na2, pH 6,25) en proporción 1:1. Posteriormente se realiza la desproteinización de la muestra añadiendo la misma cantidad de ácido tricloracético al 10%. Centrifugando a 20.000 g durante 15 minutos a 4 ºC, se obtiene el sobrenadante que se utiliza para la medición de GSSG. 2. Se incuban 200 l del sobrenadante con 80 l de solución N-etilmaleimida (NEM, 5 mg/ml en agua destilada) durante 40 minutos a temperatura ambiente. La NEM previene la oxidación del GSH presente en la muestra a GSSG. 3. Tras la incubación, se añaden 720 l de NaOH 0,1 N. 4. Se añaden a los pocillos de una microplaca 30 l de las muestras correspondientes con NEM y NaOH, 160 l de NaOH y 10 l de solución oftalaldehído (OPT, 1 mg/ml). El OPT reacciona con el GSSG a pH alcalino, emitiendo fluorescencia en proporción directa a la cantidad de GSSG presente. 5. Se incuba 15 minutos a temperatura ambiente y se lee la fluorescencia en un fluorímetro de placas (FLx 800 Microplate Fluorescence Reader, Bio-Tek Instruments, Inc). 6. Para cuantificar GSSG se compara la fluorescencia emitida por las muestras con las de una curva de disoluciones GSSG estándar (0,1 a 1,63 nmol/ml). 7. Los valores de GSSG se expresan en mol/g proteínas totales. Medición de GSH: 1. La muestra descongelada de hematíes se hemoliza en tampón de hemólisis (10 mM fosfato sódico, 1mM EDTA-Na2, pH 6,25) en proporción 1:20. Posteriormente se realiza la desproteinización de la muestra añadiendo la misma cantidad de ácido tricloracético al 10%. Centrifugando a 20.000 g durante 15 minutos a 4 ºC, se obtiene el sobrenadante que se utiliza para la medición de GSSG. 2. 10 l de sobrenadante se incuba con 10 l de solución ophtaldehído-etanol (1 mg/ml) y 180 l de buffer fosfato (100 mM fosfato sódico, 5 mM EDTA-Na2, pH 8.0) durante 20 minutos a temperatura ambiente. 54 Material y Métodos 3. Se mide la fluorescencia de las muestras en un lector de fluorimetría (FLx 800 Microplate Fluorescence Reader, Bio-Tek Instruments, Inc). 4. Para cuantificar el GSH se compara la fluorescencia emitida por las muestras con las de una curva de disoluciones GSH estándar (0,2 a 3,25 nmol/ml). 5. Los valores de GSH se expresan en mol/g proteínas totales. 2.1.3. Determinación de la actividad enzimática de la glutation peroxidasa (GPx). Existen dos tipos de GPx, una dependiente de selenio (Se-GPx) y otra independiente de selenio (iSe-GPx). Ambos tipos pueden usar hidroperóxidos orgánicos (cumeno hidroperóxido) como substratos, pero solo la Se-GPx es capaz de utilizar la forma inorgánica (H2O2) (Lawrence y Burk, 1976). Se utiliza la reacción acoplada con GRd para reducir GSSG producido por la reducción de peróxidos orgánicos por medio de GPx, e hidroperóxido cuménico como substrato. (Jaskot y colbs., 1983; Dolphin y colbs., 1989). El método empleado consiste en una determinación espectrofotométrica indirecta de consumo de NADPH, midiendo decremento de la absorbancia en función de tiempo a una longitud de onda de 340 nm. Se tienen lugar las siguientes reacciones: GPx R-OOH + 2 GSH R-OH + GSSG GRd GSSG + NADPH + H+ 2 GSH + NADP+ Protocolo: 1. La muestra descongelada de hematíes se hemoliza en tampón de hemólisis (10 mM fosfato sódico, 1mM EDTA-Na2, pH 6,25) en proporción 1:20, se deja cinco minutos en reposo y a continuación se centrifuga a 20.000 g, 10 min, 4ºC. 2. 10 l de muestra procesada se agrega a 240 l de la solución de trabajo (tampón catalizado) que contiene tampón fosfato 100 mM, EDTA-Na2 1 mM, pH 7,5, 4 mM azida sódica, 60 mM GSH, 20 mM NADPH y 0,5 U/ml GRd. 55 Material y Métodos 3. Paralelamente se sirvan 10 l de muestra procesada con 240 l de la solución de tampón no catalizado (tampón fosfato 100 mM, EDTA-Na2 1 mM, pH 7,5). 4. Después de la incubación a temperatura ambiente durante 5 minutos, la reacción inicia agregando 10 l de hidroperóxido cumenico disuelto en el tampón no catalizado (0,3%). 5. La absorbancia se mide a 340 nm durante tres minutos en un espectrofotómetro de placa (Bio-Tek Power-Wavex Microplate Scanning Spectrophotometer). En todas las determinaciones se resta la oxidación no enzimática del NADPH. 6. La actividad de la GPx se expresa en mol/ min/ g proteínas totales. 2.1.4. Determinación de la actividad enzimática de la glutation reductasa (GRd). La medida de la actividad de la GRd se realiza siguiendo el método de Jaskot (Jaskot y colbs., 1983). Este método se basa en el mismo principio que el anterior, es decir, cuantificar la oxidación del NADPH en el tiempo como consecuencia del proceso de reducción del GSSG. La reacción que tiene lugar es la siguiente: GRd GSSG + NADPH + H+ 2 GSH + NADP+ Una unidad de la actividad de la GRd se define como la cantidad de enzima necesaria para reducir un mol de GSSG durante un minuto a un pH=7,6 y temperatura 25 ºC, y se ha comprobado que para la reducción de una molécula de GSSG se requiere una molécula de NADPH. Protocolo: 1. Los hematíes descongelados de hemolisan en tampón de hemólisis (10 mM fosfato sódico, 1mM EDTA-Na2, pH 6,25) en proporción 1:20, se dejan cinco minutos en reposo y a continuación se centrifugan a 20.000 g, 10 min, 4 ºC. 2. 35 l de muestra se añaden a 465 l de una solución extemporánea de trabajo que contiene también el tampón fosfato 10 mM pH 7,5 y 2,5 mM GSSG (30 mg en 20 ml de tampón fosfato). 3. Tras una incubación a 37 ºC durante cinco minutos, se añaden 8.5 l de NADPH 12 mM para disparar la reacción y la oxidación del NADPH se mide durante tres 56 Material y Métodos minutos a 340 nm en un espectrofotómetro de cubeta (UV1603 Shimadzu spectrophotometer). 4. En todas las determinaciones se resta la oxidación no enzimática del NADPH sustituyendo los 465 l de solución de trabajo por tampón fosfato 10 mM, pH 7,5. 5. La actividad de la GRd se expresa en mol NADP oxidado/ min/ g proteínas totales. 2.1.5. Determinación de la peroxidación lipídica. La medición de lipoperoxidación se hace en base a las determinaciones de malonildialdehido (MDA) y 4-hidroxialkenal (4-HDA) que proveen un índice conveniente de la peroxidación lipídica. Los peróxidos lipídicos son inestables y se descomponen formando compuestos carbonilos. Los ácidos grasos poliinsaturados forman malonildialdehido (MDA) y 4-hidroxialkenal (4-HDA) en condiciones de oxidación (Esterbauer y Cheeseman, 1990; Botsoglou y colbs., 1994). Se utiliza el kit BioXytech LPO-568 (Cayman Chemical, Ann Arbor, MI, USA). El kit contiene un reactivo cromógeno (N-metil-2-fenilindol) que reacciona con MDA y 4-HDA a 45 ºC formulándose un cromóforo estable con máximo de absorbancia a 586 nm. Para una determinación simultanea de los dos biomarcadores (MDA y 4HDA), se agrega ácido metasulfónico como un solvente ácido. La incubación se realiza a 45 ºC durante 40 minutos agitando. En calidad del estándar no se usa MDA directamente, debido a que éste es muy inestable. El kit ofrece solución de 1,1,3,3- tetrametoxypropano (TMOP) en Tris-HCl, un acetal, que 57 Material y Métodos durante la incubación ácida a 45 ºC se hidroliza formando MDA. Las concentraciones de los patrones utilizados son de 0,625; 1,25; 2,5; 5; 10 y 15 M/ml. Los resultados se expresados en nmol MDA+4HDA/ ml plasma. La absorbancia se lee en el espectrofotómetro (lector de placa Power Wave x Bio-Tek Instruments, Inc) a una longitud de onda de 586 nm. La relación entre la absorbancia y el valor de concentración del estándar es lineal y permite que el programa calcule el valor de (MDA + 4HDA) de la muestra basándose en la siguiente fórmula: (MDA+ 4HDA) = (A586 –b)/a x df, Donde: A586 es la absorbancia neta de la muestra a 586 nm, a= coeficiente de regresión (pendiente) b = factor de intercepción df = factor de dilución de la muestra. 2.1.6. Determinación de nitritos. La molécula de NO•, producida endógenamente, es muy inestable y reacciona rápidamente con el agua produciendo los nitritos. A su vez, los nitritos se oxidan fácilmente a nitratos en solución acuosa y a pH 7,4, lo que ocurre normalmente en el organismo en proporción variable. Por lo tanto, la determinación de los nitritos nos proporciona una información indirecta sobre los niveles de NO• en el organismo. La concentración de nitritos presentes en la muestra se determina mediante una técnica colorimétrica basada en la reacción de Griess (Green y colbs., 1982). El reactivo de Griess está formado por naftil-etilen-diamina (NEDA) y sulfanilamida, se combina con los nitritos para formar un compuesto nitrogenado coloreado. La intensidad del color púrpura producido es proporcional a la cantidad del cromógeno formado y, en consecuencia, a la cantidad de nitritos existentes según la ley de Lambert-Berr (Green y colbs., 1982; Verdon y colbs., 1995). Para determinar los nitratos, éstos son tratados previamente con las enzimas nitrato reductasa (NRd), que reduce los nitratos a nitritos a expensas del NADPH, y glucosa 6-fosfato deshidrogenasa (GP6DH), que recicla el NADPH a partir del 58 Material y Métodos NADP+ producido, evitando que la reacción de la NRd se detenga por falta de los cofactores. Estas reacciones se esquematizan de la siguiente manera: NR a) NO3- + NADPH + H+ NO2- + NADP+ + H G6PDH b) H2O + NADP + G6P 6-GP + NADPH + H+ + Protocolo: 1. Las muestras de plasma descongeladas se centrifugan a 4.000 g, 4 ºC, durante 5 min. Posteriormente el sobrenadante se desproteiniza añadiendo a 130 l de muestra, 26 l de ácido sulfosalicílico 6%. 2. Se mezcla y se incuba durante 30 min a temperatura ambiente, agitando cada 5 min. 3. Se centrifuga 15 min a 10.000 g y 4 ºC, cuando termina, se sacan 150 l del sobrenadante en tubos nuevos, sobre hielo. 4. En un tubo eppendorff se mezclan los siguientes reactivos para preparar el medio de reacción: Glucosa-6-fosfato deshidrogenasa (G6PDH) (50 mU/muestra…… 2 l Glucosa-6-fosfato (G6P) (750 M/muestra)…………….……...3,75l Nitrato reductasa (NR) (30 mU/muestra)………………………… 12 l Tampón fosfato (14 mM, pH = 7,4)……………………………18,25 l 5. En una microplaca se dispensan: 50 l de muestra 4 l de NaOH 36 l de medio de reacción 10 l de NADPH. 6. Agitar 15 minutos, incubar hasta 60 min adicionales, tapando con papel aluminio. 7. Se añaden 100 l de reactivo de GRIESS y se incuba 10 minutos adicionales a temperatura ambiente. 59 Material y Métodos 8. La absorbancia se mide a 540 nm en un espectrofotómetro de placa (Bio-Tek Power-Wavex Microplate Scanning Spectrophotometer), frente a una curva estándar construida con nitrito sódico (2-20 nmol/ml) disuelto en agua. 9. La concentración de nitritos se expresa en nmol/ ml plasma. 2.1.7. Determinación de la actividad enzimática de la superoxido dismutasa. El método utilizado para la determinación de SOD eritrocitaria dependiente de cobre y zinc, (Cu/Zn/-SOD), se basa en el hecho de que la epinefrina (adrenalina) en medio alcalino experimenta una auto-oxidación, generando anión superóxido (Misra y Fridovich, 1972), según el siguiente esquema: RH3- + O2 RH2 + O2•- + H+ Donde RH3- es adrenalina antes de autooxidarse. A su vez el anión superóxido O2•- actúa con la propia adrenalina transformándola en adrenocromo RH3•. En presencia de la SOD, se reduce la producción de adrenocromo, ya que la enzima neutraliza el anión superóxido: RH3 - + O2•- + 2H+ RH 3• + H2O2 SOD H2O2 La epinefrina es estable en medio ácido, pero al incrementar el pH tiene tendencia a oxidarse. Las condiciones de pH=10,2 y temperatura 30 ºC son las más óptimas para producir mayor cantidad de adrenocromo. El método está dirigido a determinar específicamente la Cu/Zn-SOD, y la mezcla de cloroformo-etanol se utiliza para inactivar las Mn-SOD y Fe-SOD. La reacción de autooxidación de epinefrina a adrenocromo se realiza midiendo la absorbancia del adrenocromo a 480 nm durante 10 minutos en condiciones de ausencia y presencia de la SOD a distintas concentraciones de la muestra analizada. Para valorar la acción de la SOD calculamos la dosis inhibitoria 50% (ID50) del hemolizado, es decir, la cantidad de proteínas capaces de inhibir en un 50% la producción de adrenocromo. Por tanto, una unidad de enzima (U) se define como la 60 Material y Métodos cantidad de la proteína total equivalente, capaz de inhibir en un 50% la producción de adrenocromo durante la incubación, y se expresa en U/g proteínas totales. Protocolo: 1. Se prepara tampón de carbonato Na2CO3 /NaHCO3 50 mM, EDTA-Na2 0,1 mM, pH 10,2. 2. Los 19,9 mg de epinefrina se diluyen en 10 ml de HCl 1 mM. 3. Se produce la hemolisis de hematíes en agua fría en una proporción de 1:4. 4. La extracción de la SOD se realiza mezclando 1 ml del hemolizado con 1,6 ml de la mezcla etanol: cloroformo (6,25:3,75), se procede a centrifugar a 3,000 rpm, 4 ºC, durante 5 minutos. 5. El sobrenadante se guarda. 6. Se procede a realizar distintas diluciones del sobrenadante (SOD): Dilución Sobrenadante SOD H2O bidistilada (ul) 1:50 10 490 1:25 20 480 3:50 30 470 2:25 40 460 7. Las determinaciones se realizan en una microplaca, dispensando 250 l de tampón Na2CO3 /NaHCO3 50 mM, EDTA-Na2 0,1 mM, pH 10,2; 30 l de distintas diluciones de la muestra, 20 l de epinefrina diluida que se agrega con una pipeta multicanales justamente antes de realizar la lectura de las absorbancias cada 60 seg durante 10 minutos. En calidad de blanco se usan 250 l de la solución tampón Na2CO3 /NaHCO3 50 mM, EDTA-Na2 0,1 mM, pH 10,2, 30 l de H2O2 y 20 L de HCl 1 mM. 8. Se imprimen las curvas de los resultados y se realizan los cálculos. 61 Material y Métodos Los cálculos se hacen encontrando los valores de la absorbancia en el rango de tiempo, donde la gráfica tiene una máxima pendiente y es aproximadamente lineal. Se calcula el ratio de la diferencia de las absorbancias presentadas y el rango de tiempo. Utilizando el análisis de regresión lineal o logarítmica, se calcula el porcentaje del incremento en condiciones de distintas concentraciones de la muestra y, finalmente, el cálculo de la actividad de la SOD se expresa en relación al contenido de proteína total en la muestra (U/ mg proteínas totales). 2.2. Parametros hematológicos. El análisis hematológico se realiza utilizando el contador electrónico Coulter Count T890 en el Hospital del Niño de Panamá. Se miden valores de hemoglobina (Hb), hematocrito (Hcto), volumen corpuscular medio (VCM), concentración de hemoglobina corpuscular media (CHCM), hemoglobina corpuscular media (HCM) y reticulocitos. La cuantificación de la hemoglobina fetal (HbF) se realiza por el método de desnaturalización alcalina (Betke y colbs., 1959). 62 Material y Métodos 3. ANÁLISIS DE ADN. 3.1. Extracción de ADN. El ADN se obtiene de la sangre extraída en los tubos con EDTA-K3, utilizando el kid Pure LinkTM Genomic DNA Kits de Invitrogen. Se usan 200 l de sangre total fresca y se realiza el paso de lísis celular y la degradación de la fracción proteica añadiendo 20 l de proteíncinasa K, 20 l de RNAsa A y 200 l de Lysis/Binding Buffer del kit, con su posterior incubación a 55 ºC. Para la precipitación de ADN se agregan 200 l del etanol al lisado y luego se procede al paso de purificación de ADN, el cuál se realiza primero, uniendo el ADN a las columnas PureLink Spin mediante la centrifugación, y luego, a través de su lavado y elución con los Wash Buffers de estas columnas a los tubos de recolección. El ADN extraído se conserva a -20 ºC. Las cantidades de ADN extraído se miden con Espectrofotometro Biomate Thermo, y se llega a preparar las diluciones de ADN de las muestras a una concentración de 0,3 g/ml aproximadamente. 3.2. Análisis de haplotipos con la técnica de RFLP. Antes de realizar la identificación de los sitios polimórficos propuestos para este estudio, se procede a la confirmación molecular del diagnóstico de la presencia del gen S de la anemia falciforme. Para esto el fragmento específico del exón que contiene codón 6 del gen beta de 308 pb se amplifica con los iniciadores 5'GGGCTGAGGGTTTGAAGTCC-3' y 5'-CCACTTCATCCACATTCACC-3', se pone en contacto con la enzima de restricción DdeI y los productos de la digestión se analizan en el gel de agarosa al 2%, la ausencia de sitio de corte nos confirma que el alelo en estudio está mutado, es decir, contiene la mutación S. La identificación de los sitios polimórficos para la construcción de haplotipos se realiza en los siete fragmentos del bloque de genes globínicos , en los sitios específicos, amplificados por RCP de acuerdo a lo descrito por Varawalla y colaboradores (Varawalla y colbs. 1992). La amplificación de cada uno de los segmentos de ADN donde se localizan los sitios polimórficos se lleva a cabo mediante 63 Material y Métodos la utilización de dos iniciadores específicos (primers). Los sitios polimórficos en el gen globínico se identificarán mediante la utilización de endonucleasas de restricción y a partir de los productos amplificados mediante PCR. Las sustituciones nucleotídicas neutras o polimorfismos, pueden introducir o remover sitios de corte para endonucleasas de restricción (Antonarakis y colbs., 1982) y permiten realizar el diagnostico de haplotipos. El patrón de digestión de los productos amplificados con las enzimas de restricción se observa en un gel de agarosa al 2%. 3.2.1. PCR. Todos los oligos usados para este estudio se resuspenden en agua bidistilada hasta llegar a una concentración de trabajo de 10 M. Para cada reacción de PCR se utilizan 2 l de ADN genómico en 25 l de la mezcla de PCR, compuesta por: buffer 1x (KCl 50 mM, Tris-HCl 20 mM); MgCl2 1,5 mM, dNTPs 200 M, oligo sentido y antisentido 10 pmol de cada uno, y la enzima Taq polimerasa (Invitrogen) 1 Unidad). El protocolo de PCR para todos los fragmentos se basa en propuesto por Varawalla (Varawalla, 1992) y consiste en la fase inicial de desnaturalización de un ciclo a 95 ºC durante 3 minutos. Luego se realizan 30 ciclos consecutivos de amplificación: 95 ºC durante 1 minuto, 55 ºC durante 1 minuto, 72 ºC durante 1 minuto, y la última fase a 72 ºC durante 3 minutos. Para los fragmentos HindIII-G, HindIII-A, AvaIII-, HinfI- la temperatura que se utiliza en la fase de amplificación es de 60 ºC y la cantidad de ciclos de amplificación requeridos para HindIII es de 35. 3.2.2. RFLP. Los productos de amplificación se digirieren con las enzimas de restricción correspondientes en concentración de 1 Unidad de enzima por cada 1 g de ADN, en condiciones de 37 ºC durante al menos 2 horas o toda la noche. Las condiciones de digestión se dan en presencia del buffer comercial para cada una de las enzimas y, en el caso de la enzima HincII se usa la solución de BSA (albúmina de suero bovina). Para la digestión con HindIII la cantidad mínima de la enzima para optimizar los resultados es de 5 U. 64 Material y Métodos 3.2.3. Electroforesis. Los productos de PCR y de la digestión son visualizados en el gel de agarosa 2% preparado en buffer TBE 1x (EDTA-Na2 0,2 mM, Tris 8,9 mM, ácido bórico 8,9 mM). En cada pocillo se sirven 5 l de amplificado y 4 l de colorante Gel Loading Solution, dejando correr las muestras en el aparato de electroforesis en condiciones de 100V y 20-30 mA, durante 2 horas aproximadamente. La determinación del tamaño de las bandas separadas se realiza frente a los marcadores correspondientes (AMRESCO), servidos en cada gel en relación de 6 l de marcador y 3 l de colorante. En el caso de los productos de digestión con las enzimas de restricción, se usan 20 l del digerido y 4 l del colorante. La visualización se realiza por coloración con bromuro de etidio 1 g/ml y se denota con los signos más (presencia) o menos (ausencia) (+/-) la presencia o ausencia de los sitios de restricción. (Salazar R. y colbs., 2006). El patrón de digestión para los cinco haplotipos 5' está esquematiado en la tabla 6. Tabla. 6. Patrones de digestión de los fragmentos de ADN con las enzimas de restricción de acuerdo a los haplotipos del cluster beta. Bantu Benin Senegal Camerun Arabehindú εHincII γGHindIII γ AHindIII 5ώβHincII 3ώβHincII -AvaII -HinfI + + + + + + - + + + + + + + + + + + + + + - En la tabla 7 detallamos los oligos usados y los fragmentos producto de digestión con las enzimas de restricción utilizadas. 65 Material y Métodos Tabla 7. Tamaños de los fragmentos amplificados con los oligos utilizados y digeridos con las enzimas de restricción correspondientes. Sitio de restricción Oligos usados Posición de la restricción Tamaño del fragmen to Ausencia de sitio de restricción (-) Presenci a de sitio de restricción (+) ε-HincII 5’´-TCTCTGTTTGATGACAAATTC-3’ 890 pb corriente arriba del inicio del gen ε Segundo intron del gen G 760 bp 760 bp 446 bp 314 bp 328 bp 328 bp 91 y 237 bp Segundo intron del gen A 635 bp 635 bp 308 y 327 bp Segundo intron del gen ψ 794 bp 794 bp 104 y 690 bp 28 kb corriente abajo del gen ψ Segundo intron del gen 350 pb corriente abajo del gen 914 bp 914 bp 435 y 479 bp 328 bp 328 bp 100 y 228 bp 638 bp 336 y 302-el 123 y 213 bp 5’-AGTCATTGGTCAAGGCTGACC-3’ 5’ AGTGCTGCAAGAAGAACAACTACC 3’ G- 5’CTCTGCATCATGGGCAGTGAGCTC 3’ HindIII 5’ ATGCTGCTAATGCTTCATTAC 3’ A- 5’ TCATGTGTGATCTCTCTCAGCAG 3’ HindIII 5’ TCCTATCCATTACTGTTCCTTGAA 3’ 5'ψ- 5’ ATTGTCTTATTCTAGAGACGATTT 3’ HincII 5’ GTACTCATACTTTAAGTCCTAACT 3’ 3'ψ- 5’TAACGCAAAGATTATTTCTGGTCTCT3’ HincII -AvaII 5’GTGGTCTACCCTTGGACCCAGAGG3’ -HinfI 5’-AGTTAGAGGCTTGATTTGGAGG-3’ 5’TTCGTCTGTTTCCCATTCTAAACT 3’ 5’-GTTAAGGTGGTTGATGGTAAC-3’ fragment o constante REACTIVOS: Enzimas de restricción (como una unidad se define la cantidad de enzima requerida para digerir 1 g de ADN durante 1 hora a temperatura 37 ºC, si el volumen total de la reacción es de 50 l): 66 Material y Métodos AvaII, (Bio Labs, New Ingland), 10,000 unidades, 10,000 U/ml, recombinante, sitio de reconocimiento: 5'. . . G•G W C C . . . 3' 3'. . . C C W G•G . . . 5' HindIII, (Bio Labs, New Ingland), 50,000 unidades, 20,000 U/ml, recombinante, sitio de reconocimiento: 5'. . . A•A G C T T . . . 3' 3'. . . T T C G A•A . . . 5' HincII, (Bio Labs, New Ingland), 5,000 unidades, 10,000 U/ml, recombinante, sitio de reconocimiento: 5'. . . G T Y•R A C . . . 3' 3'. . . C A R•Y T G . . . 5' HinfI, (Bio Labs, New Ingland), 5,000 unidades, 10,000 U/ml, recombinante, sitio de reconocimiento: 5'. . . G•A N T C . . . 3' 3'. . . C T N A•G . . . 5' DdeI, (Bio Labs, New Ingland), 1,000 unidades, 10,000 U/ml, recombinante, sitio de reconocimiento: 5'. . . C•T N A G . . . 3' 3'. . . G A N T•C . . . 5' La enzima utilizada para PCR es la Taq DNA polymerase, Invitrogen, 5 U/l. Para la preparación del gel se utilizaron los siguientes: Agarosa UltraPureAgarose, (gel strength 1%1.200 g/cm3), Invitrogen. Gel Loading Solution (Sigma), contiene los colorantes azul de bromofenol y azul de xilencianol. Marcadores de las bandas de ADN: X174 Market Hae III digest, Sigma, 477 ug/ml, revela 11 fragmentos de rango 72 – 1.353 bp; y Marcador AMRESCO de 100 bp y de 1 kb. 67 Material y Métodos Marcador de 100 bp separado en el gel de agarosa 2% -TAE buffer, tenido con marcador SYBR Green I Nucleic Acid Stain. Marcador de 1 Kp separado en el gel de agarosa 0.75% -TAE buffer, tenido con bromuro de etidio. Tabla 8. Características de las regiones amplificadas. Sitio Temperatura Enzima Fragmentos 60ºC Pares de bases 760 bp 5'ε HincII G 60ºC 328 bp HindIII A 60ºC 635 bp HindIII 5'ψ 55ºC 794 bp HincII 3'ψ 55ºC 914 bp HincII 60ºC 328 bp AvaII 60ºC 638 bp HinfI 446 bp 314 bp 91 y 237 bp 308 y 327 bp 104 y 690 bp 435 y 479 bp 100 y 228 bp 123 y 213 y 302 bp 68 Material y Métodos 4. ANÁLISIS ESTADÍSTICO. Se utiliza estadística descriptiva para el análisis de los datos mediante los programas GraphPad Inc. Versión 3.05, 32 bit para Win 95/NT y Slide Write plus para Windows Versión 7.0. Los datos son expresados como la media ± error estándar medio de las determinaciones por duplicado de las muestras dentro del mismo grupo. Para comparar la media entre los diferentes grupos se ha usado un análisis de la t de Student, considerando un valor de p menor de 0,05 como estadísticamente significativo. Las frecuencias genotípicas se estiman mediante contéo manual, mientras las inferencias haplotípicas, equilibrio de Hardy-Weinberg y variabilidades intrapoblacionales se obtienen mediante el programa ARLEQUIN 3.1. (Excoffier et al., 2005). 69 Resultados 70 -Resultados- 71 Resultados 72 Resultados 1. HAPLOTIPOS DEL CLUSTER GLOBÍNICO BETA. 1.1. Datos generales de los pacientes y del grupo control. Las 100 muestras de sangre de los niños de 6 meses a 15 años de edad son diagnosticadas previamente como enfermos de anemia falciforme (HbS). Los datos electroforéticos revelan que el 95 % de ellos son homocigotos HbS/HbS (SS), y el 5% son doble heterocigotos HbS/HbC (SC). En este estudio la presencia de la mutación falciforme se confirma con la técnica de biología molecular. Para ello el fragmento específico de 308 pb se amplifica con PCR y se digiere con la enzima de restricción DdeI. Todos los pacientes presentan patrón de digestión -/- que confirma el diagnóstico y todos los sujetos se incluyen en el estudio. En calidad del grupo control se utilizan 40 muestras de niños aparentemente sanos de 9 a 12 años de edad. A estas muestras se les aplica la prueba de la solubilidad de la Hb y la de electroforesis, resultando todas las muestras negativas para la anemia falciforme. Ambas extracciones son tomadas en horas matutinas, por el personal técnico especializado del Hospital del Niño de Panamá. En la tabla 9 observamos los datos generales y las características hematológicas de los dos grupos: Tabla 9. Datos generales y características hematológicas de los pacientes de anemia falciforme y del grupo control Grupo Control (n=40) 11,27 ± 0,28 Grupo Pacientes (n=100) 7,77 ± 0,42*** 23/17 52/48 13,45 ± 0,2 8,3 ± 0,14*** 39 ± 4 25 ± 0,4*** HbF, % <2 14,5 ± 1,8*** VCM, fl 90 ± 6 91 ± 1.1 CHCM, g/dL 34 ± 2 33,6 ± 0,2 1,1 ± 0,6 11,2 ± 0,5*** Edad, años Sexo, niños/niñas Hb, g/dL Hematocrito, % Reticulocitos, % Los datos se expresan como media ± error medio estándar; VCM- volumen corpuscular medio, CHCM- concentración de hemoglobina corpuscular media, *** P < 0.001 vs. control. 73 Resultados Como podemos observar, los valores de Hb total, HbF, hematocrito y % de los reticulocitos difieren significativamente entre ambos grupos (P < ,0001). 1.2. Haplotipos del clúster globínico beta. Las frecuencias haplotípicas de los 200 cromosomas analizados y su patrón de restricción con los 7 enzimas usados, están presentadas en la Tabla 10. Tabla 10. Las frecuencias haplotípicas del cluster globínico β encontradas en la población de los niños panameños. Sitios polimórficos Haplotipos εHincII γHindIII γHindIII 5´βHincII 3´βHincII βAvaII βHinfI N1 H. Fr.2 Bantu (Hp7) - + - - - + + 102 0.510 Benin (Hp4) - - - - + + + 60 0.300 Senegal (Hp3) Cameroon (Hp2) Arabe- Hin. (Hp10) Haplotipos atipicos Hp5 - + - + + + + 17 0.085 - + + - + + - 8 0.040 + + - + + + - 2 0.010 - + - - + + + 5 0.025 Hp1 + - - - - + + 5 0.025 Hp11 + - - - + + + 1 0.005 1 2 G A Número de cromosomas. Frecuencias haplotípicas. Las combinaciones haplotípicas encontradas son las siguientes: 39% de los pacientes presentan haplotipo Bantú, le sigen 22% con el haplotipo Benin, 6% con Senegal, 4% con Camerún, 1% Árabe-Hindú, 2% atípicos Hp14/Hp14, y 1 paciente con el haplotipo Hp5/Hp5. En el estado heterocigoto encontramos 15% Bantú/Benin, 5% Bantú/Senegal, 3% Bantú/Hp5, 1% Bantú/Hp14, 1% Benin/Hp11 (ver Tabla 11). 74 Resultados Tabla 11. Genotipos y haplotipos del cluster beta identificados en 100 pacientes panameños con anemia falciforme. Combinación haplotípica Genotipo según Número de Frequencia electroforesis pacientes (n=100) Bantu / Bantu SS 39 0,39 Benin / Benin SS 22 0,22 Senegal / Senegal SS 6 0,06 Cameroon / Cameroon SS 4 0,04 Arab-Indian / Arab-Indian SS 1 0,01 Bantu / Benin SS 15 0,015 Bantu / Senegal SS 5 0,05 Bantu /Hp5 SC 3 0,03 Bantu /Hp14 SS 1 0,01 Benin / Hp11 SC 1 0,01 Atipico Hp14/Hp14 SS 2 0,02 Atipico Hp5 / Hp5 SC 1 0,01 Homocigotos Heterozygous Atipicos Entre todos los pacientes, el 75% de los diagnosticados presentan patrón homocigoto y el 25% son heterocigotos para los sitios de restricción analizados. La distribución porcentual de los resultados de frecuencias genotípicas del clúster globínico beta se presenta en la figura 15. Dentro del grupo de los pacientes HbSC encontramos que tres de ellos presentan haplotipo Bantú/Hp5, un paciente es Benin/Hp11 y uno es homocigoto atípico Hp5/Hp5. La frecuencia del alelo HbC (β6GluLys) en nuestra población es de 2,5%, y este porcentaje es algo menor que las frecuencias publicadas en otros países (Ingram, 2004). Pero para determinar las frecuencias de HbC en Panamá, nosotros sugerimos que se hagan más estudios y, preferiblemente, en pacientes capturados mediante diagnóstico neonatal masivo. 75 Resultados Figura 15. Distribución porcentual de las frecuencias haplotípicas del clúster globínico beta. De acuerdo a los resultados obtenidos mediante la técnica utilizada, nosotros encontramos que 11 de los 200 cromosomas analizados presentan haplotipo atípico (5,5%): Bantu/Hp5 (3 casos), Bantú/Hp14 (1 caso), Benin/Hp11 (1 caso), Hp14/Hp14 (2 casos) y doble homocigoto Hp5/Hp5 (1 caso). Para todos los locus estudiados la frecuencia genotípica esperada es significativamente mayor que la frecuencia genotípica observada (P < ,0001). Los patrones de digestión para cada uno de los sitios de restricción realizados por RFLP’s correspondientes al cluster globínico de los extremos 5' y 3' se ilustran en las fotos tomadas durante el estudio (ver figuras 16 y 17). 76 Resultados HincII 3’ + + + - + - + - HincII 5’ + + + - M 914pb 479pb 435pb 794 pb HindIIIG HindIII A M -/- +/- +/+ +/- M 323pb 308 pb 235pb 98 pb Figura 16. Patrones de digestión con las enzimas HincII 3’ψ, HincII 5’ψ, HindIII A, HindIII G. AvaII + / - Patron +/+ HinfI +/- -/- P M +/+ +/- M 328 pb 227 pb 336 pb 301 pb 101 pb 213 pb 123 pb Figura 17. Patrones de digestión con las enzimas AvaII y HinfI . 77 Resultados 1.3. Distribución geográfica de los haplotipos identificados. La distribución geográfica de los haplotipos se realizó de acuerdo a las siguientes regiones: A- Región Metropolitana de Panamá y Colón. B- Provincias Centrales (Herrera, Los Santos, Veraguas, Coclé). C- Provincias del Este del país (Darién). Resulta que en la región Metropolitana de Panamá y Colón predomina el haplotipo Bantú (53,80% de los cromosomas), le siguen Benín (31,64%), y Senegal (9,50%). Del mismo modo, el haplotipo predominante en las Provincias Centrales es Bantú (42,5%). Además hay una importante presencia del alelo Benin (25%) y otros haplotipos atipicos. En la muestra analizada tenemos presente sólo un paciente de la provincia del Este de Panamá y es diagnosticado como Senegal (ver tabla 12, figura 18). Tabla 12: Distribución geográfica de los haplotipos diagnosticados en Panamá. Haplotipo Región Provincias Provincia de Metropolitana Centrales Este n, (%) n, (%) n, (%) Bantú 85 (53,80%) 17 (42,5%) 0 Benin 50 (31,64%) 10 (25%) 0 Senegal 15 (9,50%) 0 2 (100%) Camerún 4 (2,53%) 4 (10%) 0 Árabe-Hindú 2 (1,26%) 0 0 Otros 2 (1,26%) 9 (22,5%) 0 Total de 158 40 2 haplotipos n- número de cromosomas estudiados 78 Resultados 53 % Bantú 31 % Benin 9 % Senegal 42 % Bantú 25 % Benín 10 % Camerún Senegal Figura 18. Los haplotipos del cluster globínico beta determinados en las tres Regiones de Panamá. 2. CORRELACIÓN DE LOS HAPLOTIPOS DE ANEMIA FALCIFORME IDENTIFICADOS EN PACIENTES ATENDIDOS EN EL HOSPITAL DEL NIÑO DE PANAMÁ CON EL FENOTIPO CLÍNICO DE LA ENFERMEDAD (LEVE, MODERADO Y SEVERO) Y LOS DATOS HEMATOLÓGICOS. En primer lugar definimos los criterios utilizados para asignar el fenotipo clínico y dividir a los pacientes en los siguientes tres grupos: I- fenotipo leve, sólo presenta la crisis vasooclusiva (CVO) y con una baja frecuencia. II- fenotipo moderado, tiene la crisis vasooclusiva frecuente, más otro tipo de manifestación clínica, como dactilitis, neumonía, secuestro esplénico. III- fenotipo severo, presenta enfermedad cerebro vascular, accidente cerebro vascular o daño al órgano, además de las CVO. 79 Resultados 2.1. Análisis del fenotipo clínico de acuerdo a los haplotipos del cluster globínico beta. Como puede observarse en la tabla 13, todos los pacientes con el haplotipo Senegal presentan un cuadro clínico leve. En cambio, el 54,55% de niños con el haplotipo BEN/BEN y el 33,32% de CAR/CAR tienen el fenotipo moderado y severo. El haplotipo Camerún se encuentra en segundo lugar en cantidad de pacientes con el fenotipo leve (75,00%), y sólo un paciente diagnosticado como Árabe-Hindú tiene la clínica favorable. El haplotipo heterocigoto CAR/SEN también presenta clínica bastante favorable (80,00% es con fenotipo leve y el 20,00% es moderado). Tabla 13. Distribución de los haplotipos diagnosticados de acuerdo al fenotipo clínico Haplotipos diagnosticados n (%) Fenotipo clínico Leve Bantu (CAR/ CAR) Benin (BEN/ BEN) Senegal (SEN/ SEN) n=39 n=22 n=6 26/39 10/22 6/6 (66,67 (45,45 %) (100 %) Camerún (CAM/ CAM) n=4 Bantu/ Senegal (CAR/SEN Otros n=1 Bantu/ Benin (CAR/ BEN) n=15 n=5 n=8 3/4 1/1 10/15 4/5 5/8 (75 %) (100 (66,67 %) (80 %) (62,50 %) Moderado ÁrabHindu %) 6/39 10/22 (15,38 (45,45 %) 0/6 1/4 0/1 (25 %) %) 3/15 1/5 3/8 (20 %) (20 %) (37,50 %) Severo %) 7/39 2/22 (17,94 (9,10 %) 0/6 0/3 0/1 2/15 0/5 0/8 (13,33 %) %) n = número de casos 80 Resultados 2.2. Valores hematológicos de acuerdo a los haplotipos determinados. Los datos hematológicos de acuerdo a los diferentes haplotipos se presentan en la Tabla 14. Se muestra una diferencia estadística significativa de los valores de HbF entre los haplotipos homocigotos Bantú y Senegal en su estado homocigoto (P 0,01). El recuento de reticulocitos de los haplotipos Bantú y Benin tiene tendencia al incremento, además, el haplotipo Bantú presenta recuento reticulocitario significativamente más alto en comparación con el haplotipo Camerún (P 0,05). El resto de los valores hematológicos no difieren significativamente. 2.3. Análisis de los valores hematológicos de acuerdo a las manifestaciones clínicas de los pacientes con anemia falciforme. Cuando los pacientes fueron clasificados de acuerdo a su fenotipo clínico, hallamos que altos niveles de HbF se comparten entre los pacientes con el fenotipo leve, mientras que los niveles más bajos de HbF se mantienen en los pacientes portadores de un fenotipo severo (Tabla 15). 81 Resultados Tabla 14. Valores hematológicos de acuerdo a los haplotipos encontrados. Valores de hematología Haplotipos Bantú Benín Senegal Camerún Árabe- Bantú/Benin Bantú/Senegal (CAR/CAR) (BEN/BEN) (SEN/SEN) /Camerún Hindú (CAR/BEN) (CAR/SEN) (n=39) (n=22) (n=6) (n=4) (n=1) (n=15) (n=5) Grupo Valores Pacientes de referencia Hb total g/dl 7.43 ± 0.25 7.41 ± 0.40 8.04 ± 0.34 7.79 ± 0.32 7.74 7.83 ± 0.26 7.99 ± 0.34 8.32 ± 0.14 Hb fetal 10.65 ± 3.02 15.63 ± 2.39 28.62 ± 5.18 11.02 ± 3.08 23.2 16.04 ± 5.82 8.88 ± 1.85 15.39 ± 1.21 12,516,5 <2 Hematocrito % VCM (fl) 24.47 ± 064 25.24 ± 1.08 25.98 ± 1.05 27.00 ± 1.05 19.2 23.24 ± 1.05 25.04 ± 0.38 25.02 ± 0.42 35- 43 91.78 ± 1.79 89.06 ± 2.08 98.78 ± 3.81 94.33 ± 6.17 74 93.15 ± 2.61 100.64 ± 5.04 91.06 ± 1.11 84- 96 34.00 ± 0.18 33.55 ± 0.38 32.23 ± 0.97 34.70 ± 0.02 33 33.63 ± 0.08 33.75 ± 0.64 33.62 ± 0.16 32- 36 12.31 ± 1.23 11.62 ± 0.82 10.1 ± 1.78 6.63 ± 1.74 8 11.16 ± 1.59 13.75 ± 1.78 11.16 ± 0.54 0,5- 1,8 CHCM (g/dL) Reticulocitos (%) Los datos se expresan como media ± error medio estándar. Ver las abreviaturas en la Tabla 9. P < 0.01 vs. Bantú P < 0.05 vs. Bantú. 82 Resultados Tabla 15. Valores hematológicos de acuerdo al fenotipo clínico de la enfermedad Hb (g/dL) Leve 8,33 ± 0,16 Fenotipo clínico Moderado 8,12 ± 0,27 Severo 8,60 ± 0,59 HbF (%) 18,48 ± 2,68*** 12,73 ± 2,53* 5,85 ± 1,40 Hematocrito (%) 24,92 ± 0,40 24,45 ± 0,88 25,54 ± 1,42 VCM (fl) 90,43 ± 1,42 90,35 ± 1,89 98,07 ± 4,22 CHCM (g/dL) 33,55 ± 0,27 33,58 ± 0,24 33,80 ± 0,15 Reticulocitos (%) 11,40 ± 0,76 11,81 ± 0,87 9,32 ± 0,45 Los datos de expresan como media ± error medio estándar. Las abreviaciones ver en la Tabla 9. *P < 0.05 y ***P < 0.001 vs. fenotipo severo. 3. VALORACIÓN DEL ESTRÉS OXIDATIVO EN EL GRUPO DE ENFERMOS Y EL GRUPO CONTROL. Realizamos determinaciones de los biomarcadores seleccionados en el grupo control y en las muestras de los niños con anemia falciforme. En la tabla 16 se presentan los valores estadísticos de los siete biomarcadores del estrés oxidativo de las muestras analizadas. Los niveles plasmáticos significativamente elevados de LPO (P < 0,001) y NOx (P < 0,05), en el grupo de los enfermos en comparación con los del grupo control, reflejan un daño oxidativo a nivel celular. El estado redox intracelular se estudió mediante el sistema de glutatión y la actividad de la enzima SOD. El ratio GSSG/GSH, considerado como un biomarcador del daño oxidativo intracelular, se encuentra significativamente elevado en los pacientes falcémicos (P < 0,001), básicamente, debido al incremento de los niveles de GSSG. Las actividades de las enzimas del ciclo de glutatión, GPx y GRd, también son alteradas, de modo que la actividad de GPx está elevada y la de GRd está disminuida (P < 0,001). La actividad de la SOD está significativamente reducida en el grupo de los enfermos (P < 0,05). 83 Resultados Tabla 16. Resultados de los biomarcadores de estrés oxidativo en los niños sanos versus los niños enfermos. Biomarcador LPO, nmol/ml NOx, nmol/ml GSSG, mol/g prot. total GSH, mol/g prot. total GSSG/GSH GRd, mol/min/g prot. total GPx, mol/min/g prot. total SOD, U/mg prot. total Valores en los niños sanos 5,40 ± 0,30 Valores en los niños con anemia falciforme 7,84 ± 0,36 *** 30,00 ± 2,47 36,10 ± 1,81* 0,38 ± 0,02 0,63 ± 0,02 *** 2,54 ± 0,15 3,08 ± 0,08 ** 0,12 ± 0,01 0,21 ± 0,01 *** 1,28 ± 0,09 0,76 ± 0,07 *** 15,15 ± 1,06 37,93 ± 1,33 *** 817,45 ± 69,47 676,59 ± 34,58 * *P < 0,05; **P < 0,01, y ***P < 0,001 vs. grupo control. 84 Resultados Fig.19. LPO (MDA + 4-HDA) en plasma de niños sanos y niños con anemia falciforme. *** P < 0,001 versus control. Fig.21. Niveles de GSSG en el grupo de niños sanos y niños con anemia falciforme. *** P < 0,001 versus control. Fig.20. Niveles de nitritos en el grupo de niños sanos y niños con anemia falciforme. *P < 0,05 versus control. Fig.22. Niveles de GSH en el grupo de niños sanos y niños con anemia falciforme. ** P < 0,01 versus control. 85 Resultados Fig.23. Ratio de GSSG/GSH en el grupo de niños sanos y niños con anemia falciforme. *** P < 0,001 versus control. Fig.25. Actividad de GPx en el grupo de niños sanos y el grupo de falcémicos. *** P < 0,001 versus control. Fig.24. Actividad de SOD en el grupo de niños sanos y el grupo de falcémicos. * P < 0,05 versus control. Fig.26. Actividad de GRd en el grupo de niños sanos y el grupo de falcémicos. *** P < 0,001 versus control. 86 Resultados Tomando en cuenta que la edad media de los pacientes con anemia falciforme es significativamente distinta a la edad media del grupo control, nosotros analizamos el estado del estrés oxidativo en los pacientes divididos entre los dos subgrupos: niños menores de 6 años y niños mayores de 6 años. Las diferencias estadísticas entre los dos grupos no han sido encontradas (Ver Tabla 17). Table 17. Biomarcadores del estrés oxidativo en los pacientes divididos en dos grupos según la edad. Edad < 6 años (n = 39) Edad 6 años (n = 61) LPO, nmol/ml 7.67 ± 0.54 7.95 ± 0.49 NOx, nmol/ml 32.0 ± 2.47 34.10 ± 1.81 GSSG, mol/g proteína 0.59 ± 0.02 0.63 ± 0.02 GSH, mol/g proteína 3.28 ± 0.36 3.01 ± 0.09 GSSG/GSH 0.17 ± 0.01 0.20 ± 0.01 GRd, mol/min/g proteína 0.87 ± 0.14 0.67 ± 0.07 GPx, mol/min/g proteína 36.81 ± 2.04 38.72 ± 1.81 798.75 ± 72.23 691.08 ± 48.75 SOD, U/mg proteína 87 Resultados 4. CORRELACIONES HAPLOTIPOS DEL DEL MANIFESTACIONES ESTRÉS CLUSTER CLÍNICAS Y OXIDATIVO CON GLOBÍNICO LOS NIVELES LOS BETA, DE HbF EXPRESADOS EN LOS PACIENTES PANAMEÑOS CON ANEMIA FALCIFORME. 4.1. Resultados del estrés oxidativo de acuerdo a los grupos haplotípicos. Con el objetivo análisar una posible relación entre distintos haplotipos determinados y los biomarcadores del estrés oxidativo, hemos dividido a todos los pacientes en tres grupos haplotípicos, de acuerdo a Inati y colb. (Inati, 2003). Esta división está basada en que las enzimas cortan ciertos sitios de ADN de manera no aleatorina, como es el caso de la enzima HincII que corta un sitio específico en el gen 5'ψ solo para los haplotipos Senegal y Árabe-hindú. Otro sitio importante es el 3'ψ que se corta (+) con la enzima HincII en el caso de Benín y no se pude cortar (-) en el caso de Bantú. El mecanismo de producción de estas asociaciones no aleatorias dentro de ciertas regiones de ADN puede explicarse por la selección de ADN en condiciones de la presión ejercida y/o por diferentes frecuencias de recombinación dentro de estas regiones (Antonarakis y colbs., 1985). La división utilizada es la siguiente: Grupo I: los pacientes asociados al haplotipo Benín en su estado homocigoto. Grupo II: los pacientes que tienen por lo menos un cromosoma Bantú. Grupo III: incluye pacientes Árabe/hindú y los Sen/n, donde n - es cualquier otro haplotipo, excepto Bantú. La tabla 18 presenta los valores de biomarcadores de estrés oxidativo en los grupos haplotípicos. Se observa que el grupo III, donde están incluidos la mayoría de los pacientes con la clínica leve, tiene actividades de la GRd (P < 0,01) y de la SOD (P < 0,05) más altas que el grupo I. La actividad GRd del grupo II es significativamente más alta en comparación con el grupo I (P < 0,05). Además, hay diferencias significativas en los valores de la actividad de la SOD entre los grupos II y III (P < 0,05). La actividad de la SOD del grupo III es más alta. Con relación a los 88 Resultados niveles de LPO observamos una tendencia al incremento en el grupo I, donde se encuentra a mayoría de los pacientes con la clínica desfavorable. Tabla 18. Los biomarcadores del estrés oxidativo de acuerdo a los grupos haplotípicos. GRUPO I GRUPO II GRUPO III Pacientes LPO, nmol/ml 9,03 ± 0,84 7,54 ± 0,44 7,67 ± 1,53 7,84 ± 0,36 NOx, nmol/ml 38,05 ± 3,15 35,54 ± 2,63 32,97 ± 3,14 36,10 ± 1,81 GSSG, mol/g 0,61± 0,01 0,67 ± 0,02 0,61 ± 0,02 0,63 ± 0,02 3,06 ± 0,22 3,06 ± 0,11 3,26 ± 0,28 3,08 ± 0,08 GSSG/GSH 0,20 ± 0,01 0,22 ± 0,01 0,19 ± 0,01 0,21 ± 0,01 GRd, mol/min/g 0,54 ± 0,08 0,81 ± 0,11 * 0,93 ± 0,17 ** 0,74 ± 0,06 37,47 ± 2,30 37,68 ± 1,96 42,67 ± 5,38 37,93 ± 1,33 670,85 ± 64,01 620,03 ± 44,50 831,55 ± 106,52 676,59 ± 34,58 proteína GSH, mol/g proteína proteína GPx, mol/min/g proteína SOD, U/mg proteína * Grupo I: los pacientes con haplotipo BEN/BEN; Grupo II: los pacientes CAR/n, donde n es cualquier otro haplotipo; Grupo III: incluye paciente Árabe/Hindú y los Sen/n, donde n - es cualquier otro haplotipo, excepto Bantú. *P <0.05 vs. Grupo I, **P < 0.01 vs. Grupo I, P < 0.05 vs. Grupo II. 89 Resultados 4.2. La relación entre los biomarcadores del estrés oxidativo y el fenotipo clínico de la anemia falciforme en la población panameña. La relación entre el estado del estrés oxidativo y las características fenotípicas de la enfermedad se presenta en la Tabla 19. Los niveles de LPO son significativamente mayores en el grupo de los pacientes con manifestación clínica severa en comparación con aquellos que presentan manifestaciones clínicas leves y frente al promedio de todos los enfermos (P < 0,05, figura 27). Además, el grupo con la clínica severa tiene las actividades de la GRd (P < 0,01) y de la SOD (P < 0,05) más bajas (figuras 28 y 29). Tabla 19. Biomarcadores de estrés oxidativo de acuerdo al fenotipo clínico. Leve Moderado Severo Enfermos LPO, nmol/ml 7,56 ± 0,45 8,03 ± 0,65 10,31 ± 1,41* 7,89 ± 0,36 NOx, nmol/ml 33,22 ± 2,05 35,27 ± 2,29 38,16 ± 9,79 36,10 ± 1,81 GSSG, mol/g protein GSH, mol/g protein GSSG/GSH 0,62 ± 0,03 0,61 ± 0,06 0,67 ± 0,03 0,65 ± 0,02 3,06 ± 0,09 3,05 ± 0,14 3,62 ± 0,41 3,12 ± 0,08 0,21 ± 0,01 0,22 ± 0,01 0,18 ± 0,01 0,21 ± 0,01 GRd, mol/min/g protein GPx, mol/min/g protein SOD, U/mg protein 0,79 ± 0,09 0,78 ± 0,12 0,48 ± 0,02** 0,76 ± 0,07 38,36 ± 1,58 35,02 ± 2,01 43,21 ± 9,11 37,93 ± 1,35 758,45 ± 54,08 759,37 ± 64,06 468,03 ± 65,62* § 676,59 ±34,58 *P < 0,05 y **P < 0,01 vs. fenotipo leve., §P<0,05 vs. fenotipo moderado y P < 0,05 vs. grupo enfermos. 90 Resultados Fig.27. Niveles de lipoperoxidación encontrados en diferentes fenotipos clínicos (leve, moderado, severo). *P < 0,05 versus cuadro leve. P < 0,05 vs. Pacientes. Fig. 28. Actividades de SOD encontradas en diferentes fenotipos clínicos (leve, moderado, severo). P<0,05 vs. fenotipo leve, §P<0,05 vs. fenotipo moderado y P < 0,05 vs. grupo enfermos. Fig. 29. Actividades de la GRd encontradas en diferentes fenotipos clínicos (leve, moderado, severo). **P < 0,05 versus cuadro leve. 91 Resultados 4.3. La relación entre los biomarcadores del estrés oxidativo y la expresión de HbF en los pacientes falcémicos de Panamá. Al comparar los valores hematológicos entre diferentes haplotipos del cluster globinico beta y entre los fenotipos clínicos de la enfermedad, nos damos cuenta que la característica común, estadísticamente significativa, es el valor de HbF. Suponiendo, que su expresión puede estar relacionada con la gravedad de la enfermedad y/o los niveles del estrés oxidativo, tomamos la decisión de comparar los siete biomarcadores del estrés oxidativo entre los tres grupos de pacientes (Grupo A: pacientes que tienen niveles de HbF menores a 5%; Grupo B: HbF entre 5 y 15 %; y Grupo C: HbF mayor a 15%) (Inati y colbs., 2003). Los niveles altos de HbF se asocian con niveles de LPO más bajos (P < 0,05, vs. Grupo A, ver tabla 20). Además, se observa que a medida que aumentan los valores de HbF, se tiene tendencia al incremento de NOx y de la actividad de GRd. Tabla 20. Biomarcadores del estrés oxidative de acuerdo a los niveles de HbF. HbF < 5 5 < HbF < 15 HbF 15 LPO, nmol/ml 11,22 ± 0,72 9,51 ± 0,68 7,91 ± 1,06* NOx, nmol/ml 37,92 ± 2,01 39,25 ± 3,39 42,57 ± 5,57 GSSG, mol/g protein 0,64 ± 0,05 0,55 ± 0,03 0,56 ± 0,04 GSH, mol/g protein 3,28 ± 0,14 3,28 ± 0,09 3,30 ± 0,15 GSSG/GSH 0,19 ± 0,01 0,17 ± 0,01 0,17 ± 0,01 GRd, mol/min/g protein 0,64 ± 0,12 0,71 ± 0,09 0,81 ± 0,11 GPx, mol/min/g protein 38,36 ± 1,58 35,02 ± 2,01 43,21 ± 9,11 877,38 ± 140,25 686,53 ± 72,89 821,71 ± 91,84 SOD, U/mg protein *P < 0,05 vs. grupo con HbF < 5. 92 93 -Discusión de los resultados- 94 95 Discusión de los resultados 1. HAPLOTIPOS DEL CLUSTER GLOBÍNICO BETA. 1.1. Datos generales de los pacientes y del grupo control. Para la realización de este estudio en una población pediátrica de Panamá, se logra obtener 100 muestras de sangre de los niños diagnosticados de anemia de células falciforme, que representa alrededor de 40% del total de la población atendida por el Hospital del Niño de Panamá. Entre los 100 pacientes, existe una representación de ambos sexos (52 niños y 48 niñas) y su edad promedio es de 7,77 ± 0,42 años. Para obtener valores de referencia de una población pediátrica del mismo país, se cuenta con las 40 muestras de sangre de los niños sanos, de ambos sexos (23 niños y 17 niñas) y la edad promedio 11,27 ± 0,82 años. Los estudios en niños falcémicos muestran que los valores de hemoglobina pueden variar alrededor de 8,09 ± 4,03 g/dl (Fernández-García y González, 2006). En nuestro estudio el valor promedio de la Hb en el grupo de los enfermos es de 8,31 ± 0,14 g/dL y está dentro del rango correspondiente a esta patología hematológica. Este valor es significativamente menor (P < 0,0001) que el promedio de la Hb del grupo control. Una acelerada destrucción de los glóbulos rojos afectados también explica los valores bajos de hematocrito y altos del % de recuento reticulocitario. 1.2. Haplotipos del cluster globínico beta. De las cien muestras diagnosticadas con la técnica de RFLP, se encontró que el 75% de los pacientes son homocigotos y los 25% son heterocigotos para los sitios de restricción analizados, siendo haplotipo Bantú/Bantú el predominante (39%), le siguen haplotipo Benín/Benín (22%), Senegal/Senegal (6%), Camerún/Camerún (4%) y un paciente con haplotipo Árabe-Hindú/Árabe-Hindú. En el grupo heterocigoto (ver resultados en la tabla 12), también predominan alelos Bantú y Benín. Con estos resultados se confirma el origen afro-antillano de la mutación de las células falciformes en Panamá. La enfermedad de anemia falciforme oscila entre el 8% y el 16% en la población panameña (Schroeder et al., 1990; OMS, 2006). Sin embargo, no hay 96 Discusión de los resultados suficientes datos actualizados y publicados sobre la prevalencia de la enfermedad de células falciformes en la República de Panamá. Sólo recientemente, y gracias a la aprobación de la ley tamizaje neonatal en el 2007, se va a poder realizar un diagnóstico masivo y determinar la prevalencia de la anemia de células falciformes en el país. El porcentaje de haplotipos atípicos en los pacientes panameños es de 5,5% y es similar al señalado previamente en otras poblaciones (Zago, 2001). Estos resultados sugieren que el cluster clobínico β es dinámico y que los haplotipos son producidos por diversos mecanismos genéticos, incluyendo la recombinación. La República de Panamá es un país que se caracteriza por un alto porcentaje de mezclas entre distintas razas. La migración africana está relacionada con dos momentos históricos: la traída de los esclavos africanos a partir del siglo XVI destinado a las plantaciones de la costa tropical del Pacífico sudamericano, y la actividad relacionada con la construcción del Canal de Panamá a finales del siglo XIX, inicio del siglo XX. La cantidad exacta de los inmigrantes es difícil de estimar debido a la falta de registros escritos pero, por ejemplo, en la segunda mitad del siglo XVIII, se estima que los esclavos en el Istmo de Panamá formaban el 7% de la población (Conte-Porras and Castillero, 1998). La situación de la llegada de los esclavos permaneció hasta el año 1821, cuando se prohibió la esclavitud (la ley de libertad de vientres). Pero la presencia de la población de origen africano permaneció en el país. Además, el mismo año comienzan a llegar los contingentes de antillanos, jornaleros libres para la construcción del ferrocarril de Panamá. Ellos constituyen el antecedente de llegadas más importante que se producen durante la construcción del Canal de Panamá, sobre todo desde el 1881 hasta el 1889 y entre 1904 y 1914. En el período francés de la construcción del Canal de Panamá (1881-1889) la población se duplicó debido a la llegada de los trabajadores del continente africano. Durante el período norteamericano de la construcción del Canal de Panamá (19041914), de los 60.000 trabajadores extranjeros traídos a propósito, aproximadamente un tercio eran blancos y dos tercios, negros. Llegaron los migrantes de Cartagena, Venezuela, Cuba, Barbados y Santa Lucía. Algunos africanos llegan de Senegal. Europa representa alrededor de 26,3% de trabajadores, de los cuales más de 80% eran españoles y el resto, italianos. El impacto de las obras del canal en la formación de la población de Panamá, sobre todo en el siglo XX, es considerable. Así, en 1911, uno de cada cuatro habitantes 97 Discusión de los resultados del Istmo, incluyendo la Zona del Canal, es extranjero. En la Región Metropolitana, más de la mitad de la población ha nacido en el exterior, de la cuál una proporción importante en las Antillas y es de origen afroamericano. La presencia afro-antillana en Panamá se confirma con los hallazgos de haplotipos, que en su gran mayoría son de origen africano (Bantú, Benín, Senegal). Siendo el Hospital del Niño un centro de referencia en atención a la población pediátrica de la mayor parte del país, podemos hacer una estimación importante de la distribución geográfica de los haplotipos encontrados. Para esto hemos observado que en la población estudiada existe una representación de las tres regiones: Región Metropolitana (provincias de Panamá y Colón y la Zona del Canal de Panamá), las Provincias Centrales (Herrera, Los Santos, Veraguas, Coclé), y sólo un paciente de Darién (provincia del Este del país). No observamos muchas diferencias en las prevalencias de haplotipos más comunes (Bantú y Benín) entre la Región Metropolitana y las Provincias Centrales. La presencia de muchas mezclas entre las personas de distintos orígenes, permite estimar una alta presencia de haplotipos en su estado heterocigoto. La llegada de los trabajadores de Senegal para la construcción del Canal de Panamá, estima su relativamente alta presencia (9,5%) en la Ciudad de Panamá, donde actualmente se dedican al comercio en distintas áreas de la economía panameña. Además, la presencia de haplotipo de origen árabe-hindú, se puede explicar por la presencia de los inmigrantes árabes en la ciudad. Los estudios de anemia falciforme en América del Norte y del Sur, y en el área de Caribe, demuestran, en su mayoría, la prevalencia de los haplotipos de origen africano, y mayoritariamente, de Benín (Currat M, y colbs, 2002). En algunos países la prevalencia de haplotipos depende de la región, por ejemplo, en el estado Sucre de Venezuela predomina haplotipo Benín (Salazar y colbs., 2006), pero en el resto del país el más frecuente es Bantú (32,2%) (Arends, y colbs, 2007). En Brasil predomina el haplotipo Bantú (Zago y colbs, 1992), al igual que en la Costa Chica de México (Magaña, 2005). En este estudio nosotros encontramos que el haplotipo Bantú es el más frecuente en Panamá. Una situación similar se ha presentado en otros países, tales como Costa Noreste de Venezuela, Brasil y el norte de la Región Costa Chica de México (Vívenes de Lugo et al., 2003; Lemos Cardoso Farías y Guerreiro, 2006; Magaña et al., 98 Discusión de los resultados 2002). En los Estados Unidos, Trinidad y Jamaica, el haplotipo predominante es Benin, y es el resultado del tráfico de africanos a estas regiones durante el comercio británico de esclavos del Atlántico (Wainscoat et al., 1983; Schroeder et al., 1989; JonesLecointe et al., 2008) (Tabla 21). Si comparamos las prevalencias de los haplotipos encontrados en Panamá con los países vecinos, podemos darnos cuenta que, por ejemplo, en la Costa Atlántica de Costa Rica el haplotipo más frecuente es Benín (73.3%) (Rodríguez Romero et al., 1998). En Colombia predomina haplotipo Bantú (55.5%), le siguen Benín (34.8%) y Senegal (4.3%) (Cuéllar-Ambrosi F, y colbs., 2000). Nuestro estudio confirma la prevalencia de Bantú y Benín en la población panameña y una cercanía en los porcentajes de los mismos con los encontrados en Colombia, lo que sugiere un origen común de las poblaciones de Panamá y Colombia. Tabla 21. Distribución de los βs haplotipos en algunos países americanos s haplotipos Costa Noreste de Venezuelaa Cubab Brazil Nortec Benin 36.0 51.0 21.8 Bantu 50.0 41.0 66.0 Senegal 2.0 8.0 10.9 a Vívenes de Lugo y colbs., 2003 b Muniz y colbs., 1995 c Lemos Cardoso y Farias Guerreiro, 2006 d Magaña y colbs., 2002 e Rodríquez Romero y colbs., 1998 f Cuéllar-Ambrosi y colbs., 2000 g Jones-Lecointe y colbs., 2008 h Datos del presente estudio. Mexico (Costa Chica)d Costa Ricae Colombiaf 18.2 78.8 0.0 73.3 6.7 20 34.8 55.5 4.3 Trinidad Panama g h 61.8 17.3 8.5 30.0 51.0 8.5 99 Discusión de los resultados 2. CORRELACIÓN DE LOS HAPLOTIPOS DE ANEMIA FALCIFORME IDENTIFICADOS EN PACIENTES PEDIÁTRICOS PANAMEÑOS CON EL FENOTIPO CLÍNICO DE LA ENFERMEDAD (LEVE, MODERADO Y SEVERO) Y LOS DATOS HEMATOLÓGICOS. Hemos realizado correlaciones de los haplotipos con los tres niveles de gravedad de la enfermedad de células falciforme, de acuerdo a los criterios descritos en el apartado de los resultados. En el grupo con fenotipo leve encontramos un 100% presencia de los pacientes con haplotipo Senegal siguiéndoles los haplotipos Camerún (75%), Bantú (66,67%) y Benín (45,45%). El único paciente con haplotipo Árabehindú, tiene un cuadro clínico leve. Si sumamos los pacientes con los fenotipos severo y moderado, el total es liderado por el haplotipo Benín, le siguen Bantú y Camerún. El mayor porcentaje del fenotipo severo (17.94%) es representado por CAR/CAR. Estos datos en parte concuerdan con los publicados en la literatura, donde el fenotipo más severo se le otorga a Bantú, le siguen Benín, Senegal y Camerún. En nuestro estudio parece ser que el haplotipo Benín es el que tiene un mayor número de pacientes con la clínica moderada y severa. Pero hay que tener en cuenta que es un estudio de la población pediátrica, donde por un lado, pueden existir casos de niños que aún no han llegado a la edad en la que se revelan las complicaciones clínicas de la enfermedad. Por otro lado, casi todos los informes científicos anteriores se han hecho con las poblaciones de adultos enfermos. Los estudios de Kulosik y colbs. (1987), Nagel y colbs. (1991), y Steinberg y colbs. (1995) demuestran que diferentes haplotipos del cluster globínico βs influyen en la severidad clínica y los valores hematológicos de la enfermedad. En nuestro estudio encontramos las diferencias significativas de HbF y del % de reticulocitos. Hemos realizado análisis estadístico de los datos hematológicos tomados en cuenta para este estudio: Hb, HbF, hematocrito, VCM, CHCM, % de reticulocitos. El primer paso al valorar la analítica de un niño con sospecha de anemia es comparar sus niveles de Hb y Hcto con las cifras normales correspondientes para su edad y sexo (Tabla 22). Otro aspecto importante es valorar los índices eritrocitarios. El volumen corpuscular medio (VCM) se calcula como: Hematocrito (%) x10/núm. eritrocitos (mln/mm3), este índice valora el tamaño del hematíe y permite clasificar la anemia en microcítica, normocítica o macrocítica. La anemia falciforme por lo regular se 100 Discusión de los resultados encuentra en el grupo de anemias normociticas, lo cuál se confirma en este estudio (VCM = 91,06 ± 1,11). La concentración de la hemoglobina corpuscular media (CHCM) se calcula como Hbx100/Hematocrito y valora en contenido de Hb en los eritrocitos. Este valor también se encuentra dentro del rango de referencia (ver Tabla 14). Tabla 22: Valores normales de Hb, Hcto, VCM (Fernández-García y González, 2006). Edad Hb (g/dl), media Hematocrito (g/dl), media VCM (m3) Recién nacido 16,5 51 108 6 meses- 2 años 12,5 35 96 2-4 años 12,5 38 79 5-7 años 13,0 39 81 8-11 años 13,5 40 83 Mujer 13,5 41 85 Varón 14,0 43 84 12-14 años Un hallazgo importante del estudio es que el haplotipo CAR/CAR tiene valores de HbF significativamente menores que el haplotipo SEN/SEN (P < 0,01), ver tabla 14. Estos resultados corresponden a los de la mayoría de las publicaciones (Powars, 1991b; Lu y Steinberg, 1996). Nagel et al. (Nagel et al., 1991) que demuestran que la presencia de un solo cromosoma con el haplotipo Senegal, en comparación con otras combinaciones de haplotipos africanos, se asocia con un mayor nivel de HbF y un menor daño a los órganos. Nuestros resultados nos llevan a pensar que el haplotipo Bantú, siendo en nuestro caso el haplotipo con una clínica desfavorecida, se caracteriza por un menor valor de HbF, en comparación con el haplotipo Senegal. Estos datos corresponden con la mayoría de los presentes en la literatura. Por ejemplo, Steinberg y colaboradores informan que en los estudios hechos en la población adulta e independientemente del sexo, los individuos con haplotipo Senegal homocigoto o en combinación BEN/SEN, 101 Discusión de los resultados tienen niveles de HbF significativamente mayores que el resto de los grupos (Steinberg y colbs., 1995). En nuestro estudio no contamos con los pacientes BEN/SEN, pero una mayor producción de la HbF se ha observado no solamente en haplotipo Senegal, sino también en el paciente con haplotipo Árabe-hindú (23,2%). Se ha identificado una asociación entre la presencia del alelo Senegal y el polimorfismo 158(CT) en el promotor del gen G (Lu y Steinberg, 1996). A su vez, la HbF inhibe la polimerización de la HbS y está relacionada con las manifestaciones clínicas más suaves (Marotta y colbs., 1977). El recuento de reticulocitos nos evalúa la producción eritrocitaria: Reticulocitos (%) = [Número de Reticulocitos / Número de Glóbulos Rojos] x 100. El recuento de reticulocitos es un reflejo de la actividad reciente de la médula ósea e indirectamente de la hemólisis. Si la médula responde adecuadamente al incremento de la demanda de los hematíes, ésta permite una liberación temprana de los glóbulos rojos inmaduros (reticulocitos) y se observa un aumento en su recuento. Si un paciente sufre hemorragias, el número de reticulocitos aumentará unos días después para compensar la pérdida de los hematíes. Si un paciente sufre hemorragias crónicas, el número de reticulocitos quedará aumentado de forma permanente, ya que la médula está tratando de mantener la demanda constante de glóbulos rojos. Se ha observado que los pacientes drepanocíticos con un cuadro clínico más leve, muestran un aumento de supervivencia de células con HbF debido a una disminución de la concentración de Hb corpuscular media (CHCM) y a un menor grado de hemólisis, con el consiguiente incremento de las concentraciones de Hb y del hematócrito y con un menor % de los reticulocitos (Adekile, 1994). En nuestro estudio se observó un elevado recuento de reticulocitos en el caso de los pacientes con haplotipos Benín y Bantú, siendo inclusive que Bantú tiene estos valores significativamente mayores que Camerún en su estado homocigoto (P < 0,05). Algunos autores relacionan altos niveles de reticulocitos con una elevada tasa de hemólisis. Nuestros datos sugieren que los niños con haplotipos Bantú y Benín en su estado homocigoto son más propensos a los fallos hemolíticos y una elevada destrucción de los glóbulos rojos en comparación con los pacientes Camerún en su estado homocigoto. 102 Discusión de los resultados 3. VALORACIÓN DEL ESTRÉS OXIDATIVO EN EL GRUPO DE ENFERMOS Y EL GRUPO CONTROL. Varios estudios evidencian la participación del estrés oxidativo en la fisiopatología de la anemia falciforme (Hebbel y colbs, 1982; Lachant y colbs., 1983; Ren y colbs., 2008). Una excesiva producción de anión superóxido, peróxido de hidrógeno y radical hidroxilo, junto con una menor actividad de las enzimas antioxidantes, conlleva a la producción del daño oxidativo. En este trabajo hemos evaluado el estado del estrés oxidativo en los pacientes a través de las mediciones de siete biomarcadores en el plasma (LPO, NOx) y en los eritrocitos (GSSG, GSH, GRd, GPx, SOD). Encontramos que la LPO media del grupo de los pacientes presenta un 45% de incremento en comparación con el grupo control (ver figura 19). Estos resultados reflejan que los pacientes con anemia falciforme tienen una sobreproducción de los radicales libres de oxigeno (Lachant y colbs., 1983; Hebbel y colbs., 1990), que dañan la membrana celular y contribuyen a la hemólisis. Los eritrocitos poseen un sistema enzimático muy específico capaz de neutralizar los radicales libres. La primera línea de antioxidantes enzimáticos es la Cu/Zn superóxido dismutasa (SOD), que cataliza dismutación del anión superóxido (O2•-) a peróxido (H2O2), con su posterior conversión en H2O por medio de la catalasa y GPx. En el presente estudio se ha encontrado que los enfermos tienen actividad de la SOD significativamente más baja en comparación con el grupo control (P < 0,05). Es posible que esta disminución sea secundaria a la inactivación de la enzima por los radicales libres (Ren y colbs., 2008). Para evaluar el estado del sistema antioxidante del organismo, hemos realizado una evaluación completa del sistema de glutation: GSH, GSSG, GRd y GPx. En este trabajo resulta que los niños con anemia falciforme presentan la actividad de la GRd más baja (P<0,001, figura 26) y de GPx mas alta (P < 0,001, figura 25), con un valor del cociente GSSG/GSH más alto (P < 0,001, figura 23), en comparación con el grupo control. Los niveles de GSH pueden variar dependiendo de varios factores, tales como disponibilidad de sustrato para la síntesis endógena de GSH, el reciclaje de GSSG por medio de la GRd o un aumento en el consumo de GSH en condiciones del estrés 103 Discusión de los resultados oxidativo. La mayoría de los estudios de GSH en los pacientes adultos con anemia falciforme muestran una disminución de este biomarcador (Reid M y colbs., 2006). Con relación a la síntesis de GSH, existen ciertas contradicciones. Por ejemplo, Morris y colbs. (2008) indican que existe una disminución en la biodisponibilidad de glutamina necesaria para la formación de glutamato, un sustrato para la síntesis endógena de GSH. Por otro lado, Reid y colbs. (2006) publicaron un estudio donde confirman que la síntesis de GSH en pacientes con anemia falciforme era un 57% mayor que en el grupo control, lo que puede indicar que bajos niveles de GSH se deben no a la disminución de la síntesis, sino al aumento de consumo de GSH. Por otro lado, la síntesis endógena puede estar condicionada por los niveles de cisteína, un aminoácido requerido para la formación endógena de GSH (Reid y colbs., 2006). El GSH es el cofactor fundamental de la glutation peroxidasa. Constituye además el principal agente reductor de los eritrocitos y su síntesis requiere de dos reacciones: 1. Ácido glutámico + cisteína γ-glutamil-cisteína 2. γ-glutamil-cisteína + glicina GSH La primera reacción está catalizada por la γ-glutamil-cisteína sintetasa y la segunda por la glutation sintetasa. El proceso que evita la oxidación de la Hb requiere la oxidación del glutation reducido (GSH) a glutation oxidado (GSSG) por la enzima glutation peroxidasa. Para ello, es necesario un sistema que mantenga un suministro continuo de GSH. Este sistema está representado por la glutation reductasa, que cataliza la reducción de GSSG a GSH, con la participación del NADPH, producido en la vía de las pentosas fosfato por medio de la glucosa-6-fosfato-deshidrogenasa (Peñuela, 2005). A pesar, que al principio se pensaba que los pacientes drepanocíticos tienen bajos niveles de la glucosa-6-fosfato-deshidrogenasa, un amplio estudio de pacientes con HbSS y grupo control (HbAA) en Congo, ha demostrado que no existe ninguna relación biológica entre la mutación de la deficiencia de glucosa-6 fosfato deshidrogenasa y la mutación HbS (Bouanga y colbs., 1998). Estos datos parecen indicar que en la población pediátrica con anemia falciforme existe una elevación en los niveles de GSH debido a que se incrementa su síntesis endógena. Pero en condiciones del estrés oxidativo elevado se afecta la actividad de la enzima GRd y su actividad baja significativamente. Esta deficiencia en la actividad de 104 Discusión de los resultados GRd, encontrada en nuestros resultados, confirma algunos estudios previos (Lachant y colbs, 1983). El GSH juega un papel importante en la protección contra el estrés oxidativo: elimina los radicales libres y regenera antioxidantes como GPx, glutation transferasa, y vitaminas C y E (Masella y colbs. 2005). Pero para poder evaluar mejor el daño oxidativo de un organismo, se usa el cociente GSSG/GSH que a su vez es un indicador importante del estado redox intracelular y sus oscilaciones determinan el funcionamiento celular. En condiciones fisiológicas el estado redox se mantiene dentro de un rango permisible, algo similar al sistema biológico de regulación del pH. El ratio GSSG/GSH actúa como un potente amortiguador frente al daño oxidativo (Dröge, 2002). Bajo un elevado estrés oxidativo, aumentan los niveles de GSSG, que a su vez se relaciona con el aumento de los puentes de disulfuro en las proteínas que contienen el grupo thiol. Si estos cambios se producen en proteínas funcionales, tales como los receptores, enzimas, o factores de transcripción, se altera no solo su estructura sino también el funcionamiento. Por tal motivo podemos estimar que el GSSG actúa como una molécula de señalización no específica. Una de las enzimas antioxidantes que se activa en condiciones del estrés oxidativo elevado es la glutation peroxidasa (Elosua A y colbs., 2005). En este estudio se observa un incremento de la actividad de GPx en más de dos veces, comparando con los niños sanos (figura 25), confirmando otros estudios previos (Aslan y colbs., 2001; Manfredini y colbs., 2008). Nuestros resultados sugieren que una alta producción de los radicales libres acompañada por una disminución de la actividad de la GRd y un aumento de la actividad de GPx en los niños con anemia falciforme, conllevan al aumento del cociente GSSG/GSH, cambiando el estado redox en los eritrocitos (Droge y colbs., 1994). Podemos concluir que los pacientes pediátricos con anemia falciforme tienen una elevada producción de ROS, con una disminución de la actividad antioxidante de las enzimas SOD y GRd. El incremento del cociente GSSG/GSH activa la enzima GPx y finalmente, se produce daño a las biomoléculas, incluyendo un aumento de la LPO (Hebbel y colbs., 1982; Misra y Fridovich, 1972, ver figura 30). 105 Discusión de los resultados Fig. 30. Eventos dentro del eritrocito SS. No sólo el estrés oxidativo es responsable del daño a las macromoléculas. En nuestro estudio, el grupo de pacientes de anemia falciforme presenta elevados niveles de NOx en comparación con el grupo control (P < 0,05, figura 20). Estos datos suponen un aumento del la producción de RNS y por tanto, de estrés nitrosativo, que confirman algunos datos previos (Díaz-Da-Motta y colbs, 1996). La biodisponibilidad del NO se mantiene a través el balance entre su producción por las isoenzimas iNOS y eNOS, y su consumo. Este balance se encuentra interrumpido en el caso de la enfermedad de células falciformes (Wood y 106 Discusión de los resultados Granger, 2007). Los niveles de NOx son una medida indirecta de la actividad de la NOS. En condiciones del estrés oxidativo elevado y hemólisis estos niveles aumentan por distintas razones (figura 40). Por un lado, la hemoglobina liberada al plasma se oxida espontáneamente consumiendo NO y de esta forma contribuye a la disminución de su biodisponibilidad (Reiter y colbs., 2002) (ver figura 40). Fig. 40. Producción de los NOx en situación de la hemolisis en anemia falciforme y aumento en el consumo de NO•. Las enzimas eNOS desacoplada, xantin oxidasa y NADPH oxidasa producen superóxido (O2•-), éste reacciona con NO• formando peroxinitritos (ONOO-). La Hb liberada durante la hemolisis consume el NO• formando nitratos. Por otro lado, los radicales libres a través de la activación de la vía inflamatoria del NF-kB inducen la expresión de la iNOS, responsable del aumento de los niveles de NO y la producción de RNS (Aslan y colbs., 2003; Schulz y colbs., 2004). El O2•reacciona con NO• formando peroxinitritos (ONOO-), altamente pro-oxidantes, que pueden inactivar proteínas por nitrosilación, y pueden descomponerse en radical hidroxilo que a su vez origina daño al ADN y LPO (Carr et al., 2000): NO• + O2•- → ONOOLos estudios en los modelos animales con isquemia/reperfusión cardíaca, hepática y renal demostraron un incremento en la expresión de la iNOS y una elevación de los niveles de NO2- (Lee y colbs., 2001). Se ha visto que el aumento de NOx, derivado de 107 Discusión de los resultados la inducción de la iNOS en neutrofilos, junto a una disminución en la formación de GSH, da lugar a la formación de peroxinitritos citotóxicos (Aslan y Canatan, 2008). En humanos, niveles elevados de ROS contribuyen a que el NO reaccione con O2produciendo peroxinitritos que, al reaccionar con lípidos, producen lípidos nitrogenados (LNO2, L(O)NO2). Como consecuencia de ello, se deteriora el funcionamiento vascular e incrementa el daño inflamatorio de las paredes de vasos sanguíneos. Igualmente, varios estudios demuestran que los defectos en la producción de NO se asocian con varias enfermedades cardiovasculares, incluyendo ateroesclerosis, hipertensión, diabetes, obesidad, hiperlipidemias, y SCD (Simionescu y Simionesci, 1986). La biodisponibilidad de NO y su efecto sobre la vasodilatación es difícil de evaluar sin conocer la actividad de la eNOS. 4. CORRELACIONES DEL ESTRÉS OXIDATIVO CON LOS HAPLOTIPOS DEL CLUSTER GLOBÍNICO BETA, MANIFESTACIONES CLÍNICAS Y LOS NIVELES DE HbF EXPRESADOS EN LOS PACIENTES PANAMEÑOS CON ANEMIA FALCIFORME. El daño oxidativo ocurre en condiciones de producción elevada de ROS/RNS y/o la disminución de la actividad de los sistemas antioxidantes endógenos y exógenos. Las diferencias individuales en el equilibrio entre los factores prooxidantes y antioxidantes pueden contribuir a la heterogeneidad clínica de la enfermedad. Al agrupar a los pacientes de acuerdo al patrón de los haplotipos diagnosticados, se ha encontrado que aquellos pacientes que tienen aunque sea un alelo Senegal y no llevan el alelo Bantú (Grupo III), presentan las actividades de GRd y de la SOD más elevados que los pacientes CAR/CAR (grupo I, ver Tabla 18). Es relevante subrayar que la mayoría de los pacientes del Grupo III tienen clínica más suave. Esto puede indicar que, en condiciones del estrés oxidativo la enzima antioxidante GRd está produciendo una mayor cantidad de GSH, y la enzima SOD pueda que se relaciona con una mejor protección frente al daño oxidativo. Al realizar análisis del resto de los biomarcadores del estrés oxidativo entre distintos grupos haplotípicos, podemos señalar que el grupo I (CAR/CAR) tiene una 108 Discusión de los resultados tendencia a la elevación de los niveles de LPO y de NOx, en comparación con el resto de los grupos haplotípicos. Existen otros estudios que relacionan niveles de NOx con el estado del paciente. López y colbs. (2003) encontraron que los adultos sanos y los adultos SCD pero en su estado controlado, sin crisis de dolor, tienen niveles similares de NOx, mientras que los pacientes con las crisis y dolor fuerte tienen niveles de NOx más bajos que aquellos enfermos que tienen sensación de dolor mucho menor. Esta observación posiblemente se explica por el hecho de que en condiciones de una menor producción de NO•, hay un aumento de la isquemia tisular y de la sensación de dolor. En pacientes pediátricos, Morris y colaboradores (2005) observaron que los niños controlados tienen valores similares de arginina y NOx que los niños normales. En cambio, cuando se presenta la crisis de dolor, los niveles de NOx y de arginina disminuyen significativamente. Los mismos autores informaron que tras el suplemento oral con arginina, los niveles de NOx en el plasma de pacientes aumentan. Todos estos datos sugieren que los niveles de arginina y NOx fluctúan durante las crisis de dolor y el estado clínico del paciente. La eliminación de NO• a su vez aumenta expresión de la molécula vascular de adhesión celular (VCAM-1), producción de endotelina-1 y citoquinas inflamatorias (De Caterina y colbs., 1995; Rybicki y Benjamin LJ, 1998; Lin y colbs., 2001). Por todo lo expuesto anteriormente, es posible pensar que puedan existir biomarcadores de estrés oxidativo que se relacionen con las manifestaciones clínicas en los enfermos con anemia falciforme. Al analizar los biomarcadores del estrés oxidativo entre los tres fenotipos clínicos (leve, moderado y severo), podemos resaltar que los pacientes con la clínica menos favorecida presentan unos niveles de LPO significativamente más elevados, y las actividades de la GRd y SOD más bajas (P < 0,01 y P < 0,05, respectivamente), en comparación con el grupo de los pacientes que tienen la clínica más leve. Hay estudios que indican que los pacientes con una mayor gravedad clínica tienen bajos niveles de actividad de la SOD. Y que la acumulación de H2O2, la activación de NF-kB y la adhesión leucocitaria en ratones SCD, disminuyen al tratarlos con análogo de catalasa (polinitroxyl albumina) (Sultana y colbs., 1998) y antioxidantes probucol (Mahaseth y colbs., 2005). Estos datos refuerzan nuestros 109 Discusión de los resultados resultados con relación a una baja actividad de la SOD en el grupo de pacientes con clínica severa. Teniendo en cuenta que la mayoría de los pacientes con la clínica grave tienen haplotipo CAR/CAR o CAR/BEN, y al contrario, la mayor parte de los pacientes con la clínica leve es de tipo SEN/n, nuestros resultados concuerdan en parte con las evaluaciones de los biomarcadores del estrés oxidativo para los grupos haplotípicos. Basándonos en el hecho que los niveles de HbF son significativamente diferentes entre distintos haplotipos y entre diferentes fenotipos clínicos de la enfermedad (ver tablas 14 y 15), nosotros estudiamos los biomarcadores del estrés oxidativo entre los grupos de pacientes que difieren en las expresiones de HbF. Encontramos resultados que concuerdan en parte con los hallazgos hechos en los modelos animales. Por ejemplo, Dasgupta y colbs. (2010) hicieron estudios en los ratones con anemia falciforme y genéticamente modificados de acuerdo a distintos niveles de expresión de HbF. Los autores hallaron que aquellos ratones que expresaban una mayor cantidad de HbF (mayor a 40%), presentan menores niveles de LPO y mayores valores de GSH y de las actividades de las enzimas antioxidantes SOD, GPx y catalasa, comparándolos con el grupo que tenía menor expresión de HbF (menor de 1%). Nuestros resultados en parte confirman la función protectora de HbF en la población pediátrica con anemia falciforme. En efecto, los pacientes con altos niveles de HbF presentan menores valores de LPO; a cambio, los NOx y la actividad de GRd tienden a elevarse (ver tabla 20) en este grupo de pacientes, confirmando en parte la hipótesis del grupo de Kaul (Dasgupta y colbs., 2010). El aspecto central en la fisiopatología de anemia de células falciforme es la polimerización de la HbS y drepanocitosis intravascular, cuyas consecuencias son la oclusión de los vasos sanguíneos, daños por isquemia-reperfusión, la hemólisis de los hematíes, la disminución en la biodisponibilidad de NO y el estrés oxidativo/nitrosativo (Fabry y Nagel, 1982; Hebbel y colbs., 2004; Persons y colbs., 2003; Dasgupta y colbs., 2010). El aumento de la expresión de HbF puede estar relacionado con los haplotípos del cluster globínico beta, y conducir a la reducción del estrés oxidativo y aumento de la biodisponibilidad del óxido nítrico, sustancia antiinflamatoria y anti-oclusiva. Al disminuir el estrés oxidativo, se reduce la hemólisis y a su vez, disminuye la liberación de la arginasa y el consumo de la arginina, el sustrato de la NOS. Por otro 110 Discusión de los resultados lado, la reducción de la hemólisis contribuye a que disminuya la liberación de hemoglobina libre al espacio extracelular, lo que a su vez contribuye al estrés oxidativo. En conjunto, estos acontecimientos conducen al incremento de la biodisponibilidad de NO y una disminución del daño causado por el estrés oxidativo (Morris y colbs., 2008; Dasgupta y colbs., 2010). Los estudios en los ratones demuestran que a una mayor expresión de los genes de la HbF, existe una inversa relación entre el estrés oxidativo medido a través de LPO (es menor) y la biodisponibilidad del óxido nítrico medido a través de los metabolitos NOx (es mayor). A su vez el daño a los órganos es menor en aquellos ratones que expresan mayores cantidades de HbF. Por el contrario, elevados niveles de estrés oxidativo hacen fallar a los sistemas antioxidantes que a su vez contribuye aun más al proceso de hemolisis y múltiples daños orgánicos (Dasgupta y colbs., 2006). Considerando que la generación de HbS es un evento monogénico, producto de la expresión de los genes G y A, el fenotipo de la anemia falciforme es multigénico (Kutlar, 2007). Otros genes, aparte del locus globínico , participan en los eventos patológicos que a su ves pueden mejorar o agravar las manifestaciones clínicas (Morris y colbs., 2005). Por lo tanto, la pregunta es: ¿el nivel de estrés oxidativo podría ser empleado para predecir el estado clínico de los pacientes? Nosotros observamos que el grupo III de los pacientes, con alelos Senegal y Arabe-Hindú, tienen mayores actividades de la GRd y de la SOD, lo que sugiere que los pacientes de este grupo (todos ellos con fenotipo leve), tienen una mejor defensa antioxidante que los demás. Por otra parte, los pacientes con manifestaciones clínicas graves tienen valores más altos de LPO, y actividades más bajas de la GRd y de la SOD. La participación del estrés oxidativo relacionado con hemólisis y la expresión en la enfermedad de HbF SCD puede influir en diferentes subfenotipos clínicos. Por ejemplo, algunos autores sugieren que altos niveles de HbF reducen la incidencia de algunas subfenotipos de la enfermedad de células falciformes, como la osteonecrosis, el síndrome torácico agudo y los episodios dolorosos agudos. Sin embargo, alto nivel de HbF no se ha asociado con la protección frente a la hipertensión pulmonar, derrame cerebral o priapismo (5). Los agentes que disminuyen la hemólisis o restablecen la biodisponibilidad de NO pueden ser potencialmente útiles para reducir la incidencia y severidad de la enfermedad de células falciformes. 111 Discusión de los resultados En conclusión, los resultados de este estudio apoyan la existencia de un importante estrés oxidativo en pacientes de anemia falciforme y sugieren que al menos tres marcadores redox, LPO, GRd y SOD, están relacionados con la clínica de estos apcientes. Estos datos, junto a la relación entre HbF elevada y LPO bajo, y entre HbF elevada y elevada actividad GRd y niveles de NOx, indcian que el uso de marcadores de estrés oxidativo/nitrosativo como éstos puede ser importante en la evaluación del estado y evolución clínica de los pacientes. Asimismo, el uso de terapias para mejorar su estado redox y aumentar la biodisponibilidad del óxido nítrico podrían ser beneficioso para mejorar la función vascular y reducir la severidad de la anemia drepanocítica. 112 113 -CONCLUSIONES114 Conclusiones 115 Conclusiones 1. Este estudio evalúa por primera vez los haplotipos del cluster beta en pacientes pediátricos panameños de anemia falciforme. Como resultado, se ha encontrado un 75% de los haplotipos homocigotos y un 25% heterocigotos. Se confirma la presencia de las raíces afro-antillanas en el bagaje genético del país y una cercanía en los porcentajes de los haplotipos del cluster globínico beta con los encontrados en Colombia, lo que sugiere un origen común de las poblaciones de Panamá y Colombia. 2. La presencia de por lo menos un alelo Senegal está relacionada con la clínica más leve y valores de HbF significativamente más elevados en comparación con el resto de los haplotipos hallados. El haplotipo Bantú, por el contrario, presenta valores significativamente menores de HbF y el recuento de reticulocitos más elevado, indicando un mayor grado de hemólisis intravascular. 3. Este estudio ha permitido obtener y publicar por primera vez los valores de referencia de los biomarcadores de estrés oxidativo en niños panameños sanos prebúberes. 4. Los pacientes con anemia falciforme presentan valores significativamente más altos de los marcadores de estrés oxidativo extra e intracelulares que los niños controles, indicando un mayor estrés oxidativo/nitrosativo en los primeros. 5. Se obtiene por primera vez una relación entre los valores de estrés oxidativo y los distintos haplotipos de anemia falciforme identificados en Panamá. La presencia del alelo Senegal y haplotipo Árabe-Hindú, con actividades más altas de los enzimas antioxidantes GRd y SOD, se traduce en un fenotipo leve. Por el contrario, los pacientes con manifestaciones clínicas graves tienen valores más altos de LPO, y actividades más bajas de GRd y SOD. 6. El grupo de pacientes con altos niveles de HbF, que presentan una clínica más leve, tienen menor estrés oxidtivo y una tendencia al aumento de los niveles de NOx y de la actividad de GRd. Es decir, estos pacientes están mejor protegidos frente al daño oxidativo. 116 Conclusiones 7. Los resultados confirman que el estrés oxidativo está relacionado con la hemólisis y la expresión en la enfermedad de HbF, y puede influir en diferentes subfenotipos clínicos. Por lo tanto, el uso de marcadores de estrés oxidativo, sobre todo LPO, GRd y SOD, puede ser importante en la evaluación del estado y evolución clínica de los pacientes pediátricos con anemia falciforme, y sugiere que las terapias para mejorar el estado redox serán beneficiosas para mejorar la función vascular y reducir la severidad de anemia drepanocítica. 117 118 -BIBLIOGRAFÍA- 119 120 Bibliografía Adekile AD. Factors associated with the distribution and severity of sickle cell disease in Africa. Sphere 1994; 17: 1–5. Alexander N, Higgs D, Dover G, Serjeant GR. Are there clinical phenotypes of homozygous sickle cell disease? Br J Haematol 2004;126:606-611. American Sickle Cell Anemia Association. Sickle cell anemia and antisickling agents then and now." Current Medical Chemistry 2001. Antonarakis SE, Boehm CD, Giardina PJ, and Kazazian HH, Nonrandom association of polymorphic restriction sites in the β-globin gene cluster, Proc Natl Acad Sci U S A. 1982; 79: 137–141. Arendes A. Hemoglobinopatías en Venezuela. Interciencia. 2007; 32(8): 516-521. Ashley-Koch AE, De Castro L, Lennon-Graham F, Jonassaint J, Jackson TL, Price J, Galloway J, Jones S, Randall E, Eckman J, Orringer EP, Vance JM, Telen MJ. Clinical and genetic profiles of the aging sickle cell patient. Abstract presented at the 28th Annual Meeting of the National Sickle Cell Disease Program, Cincinnati, 2005. Ashley-Koch A, DeCastro L, Lennon-Graham F, Jonassaint J, Jackson TL, Price J, Galloway J, Ataga K,Orringer EP, Vance JM, Telen MJ. Genetic polymorphisms associated with the risk of pulmonary hypertension and proteinuria in sickle cell disease. Blood 2004; 104:464-475. Ashley-Koch A, et al, Sickle Hemoglobin (Hb S) Allele and Sickle Cell Disease. Am J Epidemiology 2000; 151(1): 839-845. Aslan M, Ryan TM, Adler B et al. Oxygen radical inhibition of nitric oxidedependent vascular function in sickle cell disease. Proc. Natl Acad. Sci. USA 2001; 98: 15 215–20. 121 Bibliografía Aslan M, Ryan TM, Townes TM, Coward L, Kirk MC, Barnes S y col. Nitric oxidedependent generation of reactive species in sickle cell disease. Actin tyrosine induces defective cytoskeletal polymerization. J Biol Chem. 2003; 278: 125-130. Aslan M. Redox-Dependent Impairment of Vascular Function in Sickle Cell Disease. Free Radic Biol Med. 2007; 43(11): 1469–1483. Aslan Mutay, Canatan Duran, Modulation of redox pathways in neutrophils from sickle cell disease patients, Experimental Hematology, 2008; 36: 1535-1544. Ballas SK, Larner J, Smith ED, Surrey S, Schwartz E, Rappaport EF. Rheologic predictors of the severity of the painful sickle cell crisis. Blood 1988; 72: 1216-23. Ballas SK. Sickle cell anemia with few painful crises is characterized by decreased red cell deformability and increased number of dense cells. Am J Hematol. 1991; 36: 122–130. Bernini L. Rapid determination of hemoglobin A2 by DEAE-cellulose chromatograkhy. Biochem Genet 1969; 2: 305-8. Betke K, Martin H, Schilicht I. Estimation small percentages of foetal haemoglobin. Nature 1959; 184:1877-8. Botsoglou, N.A. et al (1994) Rapid, Sensitive, and Specific Thiobarbituric Acid Method for Measuring Lipid Peroxidation in Animal Tissue, Food and Feedstuff Samples; J. Agric. Food Chem. 42, 1931-1937. Bradley AA, Nathan CF: Glutathione metabolism as a determinant of therapeutic efficiency. Cancer Res 1984, 44: 4224-4232. 122 Bibliografía Broeders MA, Tangelder GJ, Slaaf DW, Reneman RS, oude Egbrink MG. Endogenous nitric oxide protects against thromboembolism in venules but not in arterioles. Arterioscler Thromb Vasc Biol 1998; 18:139–145. Cao, A, Galanillo, R, and Rosarelli MC. Prenatal diagnosis and screening of the haemoglobinopathies in Sickle Cell Disease and Thalassaemia. Rodgers C.P. Ed. 1998; 11(1): 215-238. Cadenas E, Poderoso JJ, Antunes F, Boveris A, Lizasoain I, Moro MA, et al. Nitric oxide and peroxynitrite exert distinct eff ects on mitochondrial respiration which are differentially blocked by glutathione or glucose. Biochem J 1996; 314: 877-80. Carr, A., McCall, M. R., & Frei, B. Oxidation of LDL by myeloperoxidase and reactive nitrogen species-reaction pathways and antioxidant protection. Arterioscl. Thromb. Vasc. Biol. 2000; 20: 1716–1723. Ghafourifar, P. and Cadenas, E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci. 2005; 26, 190–195. Chambers DE, Parks DA, Patterson G, Roy R, McCord JM, Yoshida S, Parmley LF, Downey JM. Xanthine oxidase as a source of free radical damage in myocardial ischemia. J Mol Cell Cardiol. 1985; 17(2):145-52. Chang YC, Smith KD, Moore RD, Serjeant GR, Dover GJ. An Analysis of Fetal Hemoglobin Variation in Sickle Cell Disease: The Relative Contributions of the X-Linked Factor, @-Globin Haplotypes, a-Globin Gene Number, Gender, and Age. Blood 1995; 85(4): 1111-1117. Chang YC, Smith KD, Moore RD, Serjeant GR, Dover GJ. An analysis of Fetal Hemoglobin Variation in Sickle Cell Disease: The Relative Contribof the X-lked Factor, beta-globin Haplotypes, -Gene Number, Gender and Age. Blood 1995; 85(4): 1111-1117. 123 Bibliografía Charache, S.; Terrin, M. L.; Moore, R. D.; Dover, G. J.; Barton, F. B. Eckert, S. V.; McMahon, R. P.; Bonds, D. R. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N. Engl. J. Med.1995; 332:1317–1322. Cho CS, Kato GJ, Yang SH, Bae SW, Lee JS, Gladwin MT, Rhee SG. Hydroxyurea-induced expression of glutathione peroxidase 1 in red blood cells of individuals with sickle cell anemia. Antioxid Redox Signal. 2010;13:1-11. Claster S, Vichinsky EP. Managing sickle cell disease. British Medical Journal. 2003; 327 (15):1151-65. Cokic VP, Smith RD, Belestin-Cokic BB, Njoroge JM, Miller JL, Gladwin MT, Schechter AN. Hydroxyurea induces fetal hemoglobin by the nitric oxide-dependent activation of soluble guanylyl cyclase. J. Clin. Invest. 2003; 111:231-239. Cokic, V. P.; Beleslin-Cokic, B. B.; Noguchi, C. T.; Schechter, A. N. Hydroxyurea increases eNOS protein levels through inhibition of proteasome activity. Nitric Oxide 2007; 16:371–378. Cokic VP, Andric SA, Stojilkovic SS, Noguchi CT, Schechter AN. Hydroxyurea nitrosylates and activates soluble guanylyl cyclase in human erythroid cells. Blood. 2008;111(3):1117-23. Cosby K, Partovi, K. S.; Crawford, J. H.; Patel, R. P.; Reiter, C. D.; Martyr, S.; Yang, B. K.; Waclawiw, M. A.; Zalos, G.; Xu, X.; Huang, K. T.; Shields, H.; KimShapiro, D. B.; Schechter, A. N.; Cannon III, R. O.; Gladwin, M. T. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat. Med. 2003; 9:1498–1505. Crespo E, Macias M, Pozo D, Escames G, Martin M, Vives F, Guerrero JM, AcuñaCastroviejo D. Melatonin inhibits expression of the inducible no Synthase II in liver and lung and prevents Endotoxemia in lipopolysaccharide-induced multiple Organ dysfunction syndrome in rats. FASEB J. 1999; 13(12):1537-46. 124 Bibliografía Cuéllar-Ambrosi F., Mondragón MC, Figueroa M, Préhu C, Galactéros F, RuízLinares A, Sickle Cell Anemia and -globin Gene Haplotypes in Colombia. Hemoglobin 2000; 24:221-225. Dacie JV, Lewis SM. Practical heamatology, 7th edn. London: Churchill Livingstone. 1991: 227-57. Dasgupta T, Hebbel RP, Kaul DK. Protective effect of arginine on oxidative stress in transgenic sickle mouse models. Free Radic Biol Med. 2006:41(12): 1771-80. Dasgupta T, Fabry ME, Kaul DK. Antisickling property of fetal hemoglobin enhances nitric oxide bioavailability and ameliorates organ oxidative stress in transgenic-knockout sickle mice. Am J Physiol Regul Integr Comp Physiol 2010;298: 394-402. De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA Jr, Shin WS, Liao JK. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest 1995; 96:60–68. Dias-Da-Motta P, Arruda VR, Muscara MN, et al: The release of nitric oxide and superoxide anion by neutrophils and mononuclear cells from patients with sickle cell anemia. Br J Haematol 1996; 93:333–340. Diwan BA, Gladwin MT, Noguchi CT, Ward JM, Fitzhugh AL, Buzard GS. Renal pathology in hemizygous sickle cell mice. Toxicol. Pathol. 2002; 30:254–62. Dolphin D, Poulson R, Avramovic O, Glutathione: Chemical, Biochemical and Metabolic Aspects. J. Wiley and Sons. 1989, Vols. A and B. Dröge W, Schulze-Osthoff K, Mihm S, Galter D, Schenk H, Eck HP, Roth S, and Gmunder H. Functions of glutathione and glutathione disulfide in immunology and immunopathology. FASEB J 1994; 8:1131–1138. 125 Bibliografía Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002; 82: 47–95. Eaton WA, Hofrichter J. Sickle cell hemoglobin polymerization. Adv Protein Chem 1990;40:63-279. El-Hazmi de MAF, A.S. Warsy N, Bashir A, Beshlawi IR, Hussain S, Temtamy F. Haplotypes del gene del b-globin como factores pronósticos en enfermedad de la célula de hoz. Cell Molec Biol, 1999, 5:1154-1158. Elliott L, Ashley-Koch AE, De Castro L, Jonassaint J, Price J, Ataga KI, Levesque MC, Brice Weinberg J, Eckman JR, Orringer EP, Vance JM, Telen MJ. Genetic polymorphisms with priapism in sickle cell disease. Br J Haematol. 2007; 137(3): 262-7. Elosua A., Shields R., Nielsen D. Effects of a maximal graded exercise test on glutathione as a market of acute oxidative stress. J of Cardiopulmonary Rehabilitation 2005; 25:215-219. Elsbach P, Weiss J. Oxygen-dependent and oxygen-independent mechanisms of microbicidal activity of neutrophils, Immunol Lett 1985; 11:159-63. Escames G, Khaldy H, León J, Gonzalez L, Acuña-Castroviejo D. Changes in iNOS activity, oxidative stress and melatonin levels in hypertensive patients treated with lacidipine. J of Hypertension 2004, 22:626-635. Escames G, López LC, Ortiz F, Ros E, Acuña-Castroviejo D. Age-dependent lipopolysaccharide-induced Inos expression and multiorgan failure in rats: effects of melatonin treatment. Exp Gerontol. 2006; 41(11): 1165-73. Esterbauer H, Cheeseman KH, Determination of aldehydric LPO products: malonaldehyde and 4-hydroxynonenal. Methods Enzymol 1990; 186:407-421. 126 Bibliografía Ferdinandy P, Schulz R. Peroxynitrite: toxic or protective in the heart? Circ. Res. 2001; 88:12–13. Fernández-García N, González A. Anemia en la infancia. Anemia ferropénica. Bol Pediatr. 2006; 46:311-317. Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation. 2006; 113:1708–14. Föller M, Bobbala D, Koka S, Huber SM, Gulbins E and Lang F. Suicide for Survival -Death of Infected Erythrocytes as a Host Mechanism to Survive Malaria. Cellular Physiology and Biochemistry 2009; 24: 133-140. French JA, Kenny D, Scott JP. Mechanisms of stroke in sickle cell disease: sickle erythrocytes decrease cerebral blood flow in rats after nitric oxide synthase inhibition. Blood 1997; 89:4591–4599. Frenett PS, Sickle cell vaso-occlusion: multistep and multicellular paradigm. Current Opinion in Hematology 2002, 9:101–106. Fujii S, Kaneko T. Selenium, glutathione peroxidase and oxidative hemolysis in sickle cell disease. Acta Haematol 1992;87:105-106. Gibson Q, Roughton FJW. The kinetics and equilibria of the reactions of nitric oxide with sheep hemoglobin. J Physiol (Lond) 1957; 136:507–526. Gladwin MT, and Kato GJ. Hemolysis-associated hypercoagulability in sickle cell disease: the plot (and blood) thickens! Hematológica. 2008; 93(1):115-123. Gladwin MT, Shelhamer JH, Ognibene FP. Nitric oxide donor properties of hydroxyurea in patients with sickle cell disease. Br J Haematol. 2002; 116(2):43644. 127 Bibliografía Gladwin MT, Kato GJ. Cardiopulmonary complications of sickle cell disease: role of nitric oxide and hemolytic anemia. Hematology Am Soc Hematol Educ Program 2005: 51–57. Gladwin MT, Sachdev V, Jison ML, Shizukuda Y, Plehn JF, Minter K, Brown B, Coles WA, Nichols JS, Ernst I, Hunter LA, Blackwelder WC, Schechter AN, Rodgers GP, Castro O, Ognibene FP. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease. N. Engl. J. Med. 2004; 350:886–895. Green LC, Ruiz de Luzuriaga K, Wagner DA. Nitrate biosíntesis in man. Proc Natl Acad Sci USA 1982; 78(12):7764-8. Halliwell B, Gutteridge JMC. Antioxidant defences. In: Halliwell B, Gutteridge JMC, editors. Free Radicals in Biology and Medicine. 3rd ed. Oxford: Clarendon Press; 1999; 200-216. Halliwell B; Gutteridge JM. Free radical in biology and medicine, 2nd ed. Oxford: Clarendon Press, 1989. Hammerman SI, Klings ES, Hendra KP, et al: Endothelial cell nitric oxide production in acute chest syndrome. Am J Physiol 1999; 277:1579–1592. Hanchard N, Elzein A, Trafford C, Rockett K, Pinder M, Jallow M, Harding R, Kwiatkowski D, and McKenzie C. Classical sickle beta-globin haplotypes exhibit a high degree of long-range haplotype similarity in African and Afro-Caribbean populations. BMC Genetics 2007; 8:52. Hardison R. A brief history of hemoglobins: plant, animal, protest and bacteria. Proc Natl Acad Sci USA. 1996; 93: 5675-5679. Hasegawa, H.; Sawabe, K.; Nakanishi, N.; Wakasugi, O. K. Delivery of exogenous tetrahydrobiopterin (BH4) to cells of target organs: role of salvage pathway and 128 Bibliografía uptake of its precursor in effective elevation of tissue BH4. Mol. Genet. Metab. 2005; 86:2–10. Haynes JJr, Obiako B, King JA, Hester RB, Ofori-Acquah S. Activated neutrophilmediated sickle red blood cell adhesion to lung vascular endothelium: role of phosphatidylserine-exposed sickle red blood cells. Am J Physiol Heart Circ Physiol 2006;291: 1679-85. Hebbel RP, Leung A, Mohandas N. Oxidation-induced changes in microrheologic properties of the red blood cell membrane. Blood. 1990; 76: 1015–20. Hebbel RP, Morgan WT, Eaton JW, Hedlund BE. Accelerated autoxidation and heme loss due to instability of sickle hemoglobin. Proc. Natl Acad. Sci. USA. 1988; 85: 237–41. Hebbel RP, Schwartz RS, Mohandas N. The adhesive sickle erythrocyte: Cause and consequence of abnormal interactions with endothelium, monocytes/macrophages and model membranes. Clin Haematol. 1985; 14:141–161. Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. 2004; 11(2):129-51. Herold S, Exner M, Nauser T. Kinetic and mechanistic studies of the NO-mediated oxidation of oxymyoglobin and oxyhemoglobin. Biochemistry 2001; 40:3385–3395. Hissin PJ and Hilf R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal Biochem 1976; 74: 214-226. Hoppe C, Klitz W, Cheng S, Apple R, Steiner L, Robles L, Girard T, Vichinsky E, Styleset L. Gene interactions and stroke risk in children with sickle cell anemia. Blood 2004; 103(6):2391-6. 129 Bibliografía Hoppe C, Klitz W, D'Harlingue K, Cheng S, Grow M, Steiner L, Noble J, Adams R, Styles L; Stroke Prevention Trial in Sickle Cell Anemia Investigators. Stroke 2007; 38(8):2241-6. Huntsman RG, Burclay GPT, Canning DM, Yawson GI. A rapid whole blood solubility test to differentiate the sickle-cell trait from Sickle-cell anaemia. J Clin Path. 1970; 23: 781-783. Ignarro LJ, Byrns RE, Buga GM, Wood KS. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circ Res. 1987; 61(6):866–879. Inati A, Taher A, Bou Alawi W, Koussa S, Kaspa H, Shbaklo H, Zalloua PA. Globin gene cluster haplotypes and HbF levels are not the only modulators of sickle cell disease in Lebanon. Eur J Haematol. 2003; 70: 79–83. Ingram VM. Abnormal human haemoglobins. III. The chemical difference between normal and sickle cell haemoglobins. Biochim Biophys Acta. 1959; 36: 402-11. Jaskot RH, Charlet EG, Grose EC, Grady MA, Roycroft JH, An automatic analysis of glutathione peroxidasa, glutathione-S-transferase, and reductase activity in animal tissue. J Anal Toxicol 1983; 7:86-8. Jean Claude Bouanga, René Mouélé, Claude Préhu ,Henri Wajcman, Josué Feingold, Frédéric Galactéros, Glucose-6-Phosphate Dehydrogenase Deficiency and Homozygous Sickle Cell Disease in Congo. Hum Hered 1998; 48:192–197. Kan YW, Dozy AM. Evolution of the hemoglobinS and C genes in world populations. Science 1980; 209:388–391. Kato GJ, McGowan VR, Machado RF, et al. Lactate dehydrogenase as a biomarker of hemolysis-associated nitric oxide resistance, priapism, leg 130 Bibliografía ulceration, pulmonary hypertension and death in patients with sickle cell disease. Blood 2006; 107: 2279–2285. Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: Reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007; 21:37–47. Kaul DK, Fabry ME, Nagel RL. Microvascular sites and characteristics of sickle cell adhesion to vascular endothelium in shear flow conditions: Pathophysiological implications. Proc Natl Acad Sci USA 1989; 86:3356–3360. Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J. Clin. Invest. 2000; 106:411–420. Kaul DK, Liu XD, Choong S, Belcher JD, Vercellotti GM, Hebbel RP. Antiinflammatory therapy ameliorates leukocyte adhesion and microvascular flow abnormalities in transgenic sickle mice. Am. J. Physiol. Heart Circ. Physiol. 2004; 287:293–301. Kaul DK, Liu XD, Zhang X, Ma L, Hsia CJ, Nagel RL. Inhibition of sickle red cell adhesion and vaso-occlusion in the microcirculation by antioxidants. Am. J. Physiol. Heart Circ. Physiol. 2006; 291:167–75. Khan BV, Harrison DG, Olbrych MT, et al. Nitric oxide regulates vascular cell adhesion molecule 1 gene expression and redox sensitive transcriptional events in human vascular endothelial cells. Proc Natl Acad Sci USA 1996;93:9114-9119. Kiessling K, Roberts N, Gibson JS, Ellory JC. A comparison in normal individuals and sickle cell patients of reduced glutathione precursors and their transport between plasma and red cells. Hematol J 2000;1: 243-249. 131 Bibliografía Kim HG, Hong SJ, Park HJ et al. Age-related changes in the activity of antioxidant and redox enzymes in rats, Mol. Cells 2003; 16:78-284. Kim-Shapiro, D. B.; King, S. B.; Shields, H.; Kolibash, C. P.; Gravatt, W. L.; Ballas, S. K. The reaction of deoxy-sickle cell hemoglobin with hydroxyurea. Biochim. Biophys. Acta 1999; 1428:381–387. Kohn J. Separation of haemoglobins on cellulose acetate. J Clin Pathol. 1969;22:109-111. Kowaluk EA, Seth P, Fung HL. Metabolic activation of sodium nitroprusside to nitric oxide in vascular smooth muscle. J Pharmacol Exp Ther 1992; 262:916–922. Kulozik AE, Wainscoat JS, Serjeant GR, Kar BC, Al-Awamy B, Essan GJ, Falusi AG, Haque SK, Hilali AM, Kate S, et al. Geographical survey of beta S-globin gene haplotypes: evidence for an independent Asian origin of the sickle-cell mutation. Am J Hum Genet 1986;39:239-244. Kutlar A. Sickle Cell Disease: A Multigenic Perspective of a Single Gene Disorder. Hemoglobin 2007; 31(2): 209-224. Labie D, Srinvas R, Dunda O. Haplotypes in tribal bearing the sickle gene: evidence for the unicentric origin of the βs mutation and the unicentric origin of tribal populations in India. Hum Biol 1989; 61: 479–491. Lachant NA, Davidson WD, Tanaka KR, Impaired pentose phosphate shunt function in sickle cell disease: a potential mechanism for increased Heinz body formation and membrane lipid peroxidation. Am J Hematol, 1983; 15(1): 1-13. Lawrence RA, Burk RF. Glutathione peroxidase activity in selenium-deficient rat liver. Biochem Biophys Res Commun, 1976; 71(4):952-8. 132 Bibliografía Lee VG, Johnson ML, Baust J, Laubach VE, Watkins SC, Billiar TR. The roles of iNOS in liver ischemia-reperfusion injury. Shock 2001; 16:355–360. Lezcano, N. E.; Odo, N.; Kutlar, A.; Brambilla, D.; Adams, R. J. Regular transfusion lowers plasma free hemoglobin in children with sickle-cell disease at risk for stroke. Stroke. 2006; 37:1424–1426. Lima, C. S.; Arruda, V. R.; Costa, F. F.; Saad, S. T. Minimal doses of hydroxyurea for sickle cell disease. Braz. J. Med. Biol. Res. 1997; 30:933–940. Lin G, Macdonald RL, Marton LS, et al: Hemoglobin increases endothelin-1 in endothelial cells by decreasing nitric oxide. Biochem Biophys Res Commun 2001; 280:824–830. Lincoln TM, Cornwell TL. Intracellular cyclic GMP receptor proteins. FASEB J 1993; 7:328–338. Lopez BL, Kreshak AA, Morris CR, et al. L-arginine levels are diminished in adult acute vaso-occlusive sickle cell crisis in the emergency department. Br J Haematol. 2003; 120:532–534. López A, García JA, Escames G, et al. Melatonin protects the mitocondria from oxidative damage reducing oxygen consumption, membrane potencial and superoxide anion production. J. Pineal Res. 2008; 19-23. Lopez BL, Barnett J, Ballas SK, Christopher TA, Davis-Moon L, Ma X. Nitric oxide metabolite levels in acute vaso-occlusive sickle-cell crisis. Acad Emerg Med 1996;3:1098-1103. Lopez BL, Davis-Moon L, Ballas SK, Ma XL. Sequential nitric oxide measurements during the emergency department treatment of acute vasoocclusive sickle cell crisis. Am J Hematol 2000;64:151-9. 133 Bibliografía Loscalzo J. Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circ Res 2001; 88:756–762. Lu ZH, Steinberg MH, Fetal hemoglobin in sickle cell anemia: relation to regulatory sequences cis to the beta-globin gene. Multicenter Study of Hydroxyurea. Blood. 1996; 87(4):1604-11. Lukens JN. Hemoglobinopathies, S, C, D, E and o associated diseases. Hematology clinic. 1993; 38. MacDermott, R. P. Progress in understanding the mechanisms of action of 5aminosalicylic acid. Am. J. Gastroenterol. 2000; 95:3343–3345. Mack KA, Kato GJ. Sickle cell disease and nitric oxide: A paradigm shift? Int J Biochem Cell Biol. 2006; 38(8): 1237-1243. Magaña MT, Ongay Z, Tagle J, Bentura G, Cobián JG, Perea FJ, Casas- Castañeda M, Sánchez-López IJ, Ibarra B. Analysis of S and A Genes in a Mexican Population with African Roots. Blood Cells, Molecules and Diseases. 2002; 28(1): 1-6. Mahaseth, H.; Vercellotti, G. M.; Welch, T. E.; Bowlin, P. R.; Sonbol, K. M.; Hsia, C. J.; Li, M.; Bischof, J. C.; Hebbel, R. P.; Belcher, J. D. Polynitroxyl albumin inhibits inflammation and vasoocclusion in transgenic sickle mice. J. Lab. Clin. Med. 2005; 145: 204–211. Manfredini V, Lazzaretti LL, Griebeler IH, Santin AP, Brandão VD, Wagner S, Castro SM, Peralba Mdo C, Benfato MS. Blood antioxidant parameters in sickle cell anemia patients in steady state. J Natl Med Assoc. 2008 Aug; 100(8):897-902. Market M, Andrew PC, Babiar BM. Measurement of superoxide production by human neutrophils. Methods Enzimol 1984; 105: 358-65. 134 Bibliografía Marotta CA, Forget BG, Cohne-Solal M, Wilson JT, Weissman beta-globin messenger RNA. I. Nucleotide sequences SM. Human derived from complementary RNA. J Biol Chem 1977; 252: 5019–5031. Marvin Reid, Asha Badaloo, Terrence Forrester, and Farook Jahoor. In vivo rates of erythrocyte glutathione synthesis in adults with sickle cell disease. Am J Physiol Endocrinol Metab 2006; 291: E73–E79. Masella R, Di Benedetto R, Vari R, Filesi C, Giovannini C. Novel mechanisms of natural antioxidant compounds in biological systems: Involvement of glutathione and glutathione-related enzymes. J. Nutr. Biochem. 2005; 16: 577–586. Mathias Currat et al., Molecular Analysis of the b-Globin Gene Cluster in the Niokholo Mandenka Population Reveals a Recent Origin of the bS Senegal Mutation, Am. J. Hum. Genet. 70:207–223, 2002. Mayer B, Hemmens B. Biosynthesis and action of nitric oxide in mammalian cells. Trends Biochem Sci 1997; 22:477–481. McCord JM, Fridovich I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J Biol Chem 1969; 244:6049-55. McCord JM. Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med 1985; 312:159-63. Milner PF, Kraus AP, Sebes JI, Sleeper LA, Dukes KA, Embury SH, Bellevue R, Koshy M, Mohr JW, Smith J. Sickle cell disease as a cause of osteonecrosis of the femoral head. N Eng J Med. 1991; 325(21):1476–1481. Misra HP, Fridovich I, The Generation of Superoxide Radical during the Autoxidation of Hemoglobin, J Biological Chemistry, 1972, 247(21): 6960-62. 135 Bibliografía Misra HP, Fridovich I. The role of super oxide anion in the auto oxidation of epinephrine and a simple assay for super oxide dismutase. J Biol Chem 1972; 247:3170-5. Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480-7. Montero-Huerta, P.; Hess, D. R.; Head, C. A. Inhaled nitric oxide for treatment of sickle cell stroke. Anesthesiology. 2006; 105:619–621. Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, Hazen SL, Vichinsky EP, Morris Jr. SM, Gladwin MT. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. J Am Med Assoc. 2005; 294:81–90. Morris CR, Suh JH, Hagar W, Larkin S, Bland DA, Steinberg MH, Vichinsky EP, Shigenaga M, Ames B, Kuypers FA and Klings ES. Erythrocyte glutamine depletion, altered redox environment, and pulmonary hypertension in sickle cell disease. Blood 2008; 111:402-410. Murayama M, Nalbandian RM. Sickle Cell Hemoglobin: Molecular Basis of Sickling Phenomenon Theory And Therapy. Critical Reviews in Biochemistry and Molecular Biology. 1973; 1 (4): 461-499. Nagel RL, Fabry ME. Sickle cell anemia as a miltigenetic disease: new insights into the mechanism of painful crisis. Prog Clin Biol Res. 1984;165: 93-102. Nagel RL, Fabry ME, Pagnier J, et al. Hematologically and genetically distinct forms of sickle cell anemia in Africa. N Engl J Med 1985;312:880-884. Nagel RL. Severity, pathobiology, epistatic effects, and genetic markers in sickle cell anemia. Semin Haematol 1991; 28:180–201. 136 Bibliografía Nahavandi, M.; Wyche, M. Q.; Perlin, E.; Tavakkoli, F.; Castro, O. Nitric oxide metabolites in sickle cell anemia patients after oral administration of hydroxyurea, hemoglobinopathy. Hematology. 2000; 5:335–339. Nath KA, Shah V, Haggard JJ et al. Mechanisms of vascular instability in a transgenic mouse model of sickle cell disease. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000; 279: R1949–55. National Toxicology Program—Center for the Evaluation of Risks to Human Reproduction U. S. D. o. H. a. H. S. White Paper on Hydroxyurea Risk:Benefit Oct 2007. In: NTP-CERHR expert panel report on the reproductive and developmental toxicity of hydroxyurea. Nell JV. James Van Gundia Neel 1915. The Clinical Detection of the Genetic Carriers of Inherited Disease 1947. Medicine (Baltimore). 1992; 71(2): 96-101. Neel JV. The inheritance of Sickle Cell Anemia. Science. 1949; 110(2846): 64-66. Nolan, VG, Adewoye A, Baldwin C, Wang L, Ma Q, Wyszynski DF, Farrell JJ, Sebastiani P, Farrer LA, Steinberg MH. Sickle cell leg ulcers: associations with haemolysis and SNPs in Klotho, TEK and genes of the TGF-beta/BMP pathway. Br. J. Haematol. 2006; 133: 570–578. Nolan, VG, Wyszynski DF, Farrer LA, Steinberg MH. Hemolysis-associated priapism in sickle cell disease. Blood.2005; 106:3264–3267. Ofori-Acquah SF, Lalloz MR, Layton M. Localisation of cis Regulatory Elements at the β-Globin Locus: Analysis of Hybrid Haplotype Chromosomes. Dentistry and Biomedical Sciences, London, United Kingdom. 2002. 137 Bibliografía Ofori-Acquah SF, Lalloz MR, Serjeant G, Layton DM. Dominant influence of gamma-globin promoter polymorphisms on fetal haemoglobin expression in sickle cell disease. Cell Molec Biology. 2004; 50(1):35-42. Oppert M, Jorres A, Barckow D, Eckardt KU, Frei U, Kaisers U. Inhaled nitric oxide for ARDS due to sickle cell disease. Swiss Med. Wkly. 2004; 134:165–167. Ou, J.; Ou, Z.; Jones, D. W.; Holzhauer, S.; Hatoum, O. A.; Ackerman, A. W.; Weihrauch, D. W.; Gutterman, D. D.; Guice, K.; Oldham, K. T.; Hillery, C. A.; Pritchard Jr., K. A. L-4F, an apolipoprotein A-1 mimetic, dramatically improves vasodilation in hypercholesterolemia and sickle cell disease. Circulation. 2003; 107: 2337–2341. Pacelli, R.; Taira, J.; Cook, J. A.; Wink, D. A.; Krishna, M. C. Hydroxyurea reacts with heme proteins to generate nitric oxide. Lancet. 1996; 347:900. Pagnier J, Mears JG, Dunda-Belkhodja OD, Schaefer-Rego KE, Beldjord C, Nagel RL, and Labie D. Evidence for the multicentric origin of the sickle cell hemoglobin gene in Africa. Proc Natl Acad Sci USA 1984;81:1771-1773. Pastore A, Federici G, et al. Analysis of glutathione: implication in redox and detoxification. Clinica Chimica Acta 2003; 333: 19-39. Pauling L, Itano HA, et al. Sickle cell anemia a molecular disease. Science. 1949; 110(2865): 543-8. Peng HB, Libby P, Liao JK. Induction and stabilization of I kappa B alpha by nitric oxide mediates inhibition of NF-kappa B. J Biol Chem 1995; 270:14214–14219. Peñalosa-Espinosa RI, Buentello-Malo L, Hernández-Maya MA, Nieva-García B, Lisker-Yurkowitzki R, Salamanca-Gómez F. Frecuencia de la hemoglobina S en cinco poblaciones mexicanas y su importancia en la salud pública. Salud Pública Mex. 2008; 50:325-329. 138 Bibliografía Peñúela OA. Hemoglobina: una molécula modelo para el investigador: Colomb Med. 2005; 36(3): 215-225. Persons DA, Hargrove PW, Allay ER, Hanawa H, Nienhuis AW. The degree of phenotypic correction of murine beta -thalassemia intermedia following lentiviralmediated transfer of a human gamma-globin gene is influenced by chromosomal position effects and vector copy number. Blood. 2003; 101(6): 2175-83. Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro Steinberg MH, Klug PP. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N. Engl. J. Med. 1994; 330:1639–1644. Powars D, Chan L, Schroeder WA. The variable expression of sickle cell disease is genetically determined. Sem Hematol 1990; 27:360–370. Powars D, Hiti A. Sickle cell anemia. Beta s gene cluster haplotypes as genetic markers for severe disease expression. Am J Dis Child. 1993; 147(11):1197-202. Powars DR. Beta s-gene-cluster haplotypes in sickle cell anemia. Clinical and hematologic features. Hematol Oncol Clin North Am. 1991; 5(3):475-93. Powars DR. Sickle cell anemia: s-gene cluster haplotypes as prognostic indicators of vital organ failure. Semin Hematol 1991; 28: 202–208. Radomski MW, Palmer RM, Moncada S. The role of nitric oxide and cGMP in platelet adhesion to vascular endothelium. Biochem Biophys Res Commun 1987; 148:1482. Rahimi Z, Karimi M, Haghshenass M, and Merat A. B-globin gene cluster haplotypes in sickle cell patients from southwest Iran. American Journal of Hematology. 2003; 74:156–160. 139 Bibliografía Reid M, Badaloo A, Forrester T, Jahoor F In vivo rates of erythrocyte glutathione synthesis in adults with sickle cell disease. Am J Physiol Endocrinol Metab 2006;291:73-79. Reiter C.D., Gladwin M.T. An emerging role for nitric oxide in sickle cell disease vascular homeostasis and therapy. Curr. Opin. Hematol. 2003; 10: 99-107. Reiter, C. D.; Wang, X.; Tanus-Santos, J. E.; Hogg, N.; Cannon III, R. O.; Schechter, A. N.; Gladwin, M. T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002; 8:1383–1389. Ren H, Chebreneskel K, Okpala I, Lee A, Ibegbulam O, Crawford M. Patients with sickle cell disease have reduced blood antioxidant protection. Int J Vitam Nutr Res. 2008; 78(3): 139-48. Roberts I, de Montalembert M. Sickle cell disease as a paradigm of immigration hematology: new challenges for hematologists in Europe.Haematologica. 2007;92(7):865-71. Rodgers GP, Schetcher AN. Molecular pathology of the hemoglobin structure. En: Hoffman R, Benz E, Shattil S, Fume B, Cohen H. Haematology: basic principles and practice. New York: Churchill-Livingstone. 1991; 441–470. Rodríguez RW, Saénz RG. Haplotipos de la hemoglobina S: importancia epidemiológica, antropológica y clínica. Public Health. 1998; 3(1). Rodríguez WE, Sáenz GF, Josafovska O, Nagel RL. The African ports of origin of individuals of African descent in Costa Rica: the use of s-gene cluster haplotypes. Rev Invest Clin 1994; (supl):245. Rother Russell P, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin, a novel mechanism of human disease. JAMA. 2005; 293(13): 1653-63. 140 Bibliografía Ruiz Cruz ED, Maya AH, García BN, Azuara JG, Hernández Carvajal C, Salamanca Goméz F, Peñalosa Espinosa RI. Anemia de células falciformes y niveles de hemoglobina fetal, Med IMSS 2003; 41 (4): 299-303. Rybicki AC, Benjamin LJ: Increased levels of endothelin-1 in plasma of sickle cell anemia patients. Blood 1998; 92:2594–2596. Sáenz GF, Chaves MA, Quintana E. Las hemoglobinopatías en Costa Rica: aspectos históricos, culturales y epidemiológicos. Rev Costarricense Cien Med 1989; 7: 95106. Salazar Lugo R, González M, Castro-Guerra D, Arends A. Haplotipos de los genes βA y βS globina en la población de Campoma, Estado Sucre, Venezuela. Acta Bioquím Clín Latinoam 2006; 40 (2): 205-11. Saleh, A. W.; Hillen, H. F.; Duits, A. J. Levels of endothelial, neutrophil and platelet-specific factors in sickle cell anemia patients during hydroxyurea therapy. Acta Haematol.1999; 102:31–37. Sarnaik SA, Ballas SK. Molecular characteristics of pediatric patients with sickle cell anemia and stroke. Am J Hematol. 2001; 67(3):179-82. Savitt TL, Golgberg MF. Herrick´s 1910 case report of sickle cell anaemia. The rest of the story. JAMA. 1989; 261(2): 266-71. Schacter, L.; Warth, J. A.; Gordon, E. M.; Prasad, A.; Klein, B. L. Altered amount and activity of superoxide dismutase in sickle cell anemia. FASEB J. 1988; 2:237– 243. Schmidt, T. S.; Alp, N. J. Mechanisms for the role of tetrahydrobiopterin in endothelial function and vascular disease. Clin. Sci. (Lond.). 2007; 113:47–63. 141 Bibliografía Schroeder WA, Munger ES, Powars DR. Sickle cell anemia, genetic variations and the trade to the United States. J African History 1990; 31:163–180. Schulz E, Anter E, Keaney JF Jr. Oxidative stress, antioxidants, and endothelial function. Curr Med Chem. 2004; 11(9):1093-104. Setty BN, Betal SG, Zhang J, Stuart MJ. Heme induces endothelial tissue factor expression: potential role in hemostatic activation in patients with hemolytic anemia. J Thromb Haemost. 2008; 6(12):2202-9. Shalev O, Repka T, Goldfarb A, Grinberg L, Abrahamov A, Olivieri, NF, Rachmilewitz EA; Hebbel RP. Deferiprone (L1) chelates pathologic iron deposits from membranes of intact thalassemic and sickle red blood cells both in vitro and in vivo. Blood. 1995; 86:2008–2013. Sharan K, Surrey S, Ballas S, Borowski M, Devoto M, Wang KF, Sandler E, Keller M. Association of T–786C eNOS gene polymorphism with increased susceptibility to acute chest syndrome in females with sickle cell disease. Br J Haematol. 2004; 124(2): 240–243. Shimizua K, Hashimotoa T, Hariharab S, Tajimac K, Sonodad S, Zaninovice V. ß-Globin Gene Haplotype Characteristics of Colombian Amerinds in South America, Hum Hered 2001;51:54–63. Simionescu M, Simionescu N. Functions of the endothelial cell surface. Annu. Rev. Physiol.1986; 48:279–293. Stamler JS, Jia L, Eu JP, et al: Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science. 1997; 276:2034–2037. Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, Orringer E, Bellevue R, Olivieri N, Eckman J, Varma M, Ramirez G, Adler B, Smith W, Carlos T, Ataga K, DeCastro L, Bigelow C, Saunthararajah Y, Telfer M, Vichinsky E, 142 Bibliografía Claster S, Shurin S, Bridges K, Waclawiw M, Bonds D, Terrin M. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. J Am Med Assoc. 2003;289(13):1645-51. Steinberg MH, Hsu H, Nagel RL, Milner PF, Adams JG, Benjamin L, Fryd S, Gillette P, Gilman J, Josifovska O, et Gender and haplotype effects upon hematological manifestations of adult sickle cell anemia. Am J Hematol. 1995 Mar; 48(3):175-81. Steinberg MH. Genetic modulation of sickle cell anemia. Soc Exp Biol Med. 1995; 209: 1-13. Steinberg MH. Predicting clinical severity in sickle cell anemia. British Journal of Hematology. 2005; 129:465–481. Stevens MC, Maude GH, Beckford M, Grandison Y, Mason K, Taylor B, Serjeant BE, Higgs DR, Teal H, Weatherall DJ, Alpha thalassemia and the hematology of homozygous sickle cell disease in childhood. Blood. 1986; 67(2):411-4. Sullivan KJ, Kissoon N, Gauger C. Nitric oxide metabolism and acute chest syndrome of sickle cell anemia. Pediatr Crit Care Med. 2008; 9:159-168. Sultana C, Shen Y, Rattan V, Johnson C, Kalra VK. Interaction of sickle erythrocytes with endothelial cells in the presence of endothelial cell conditioned medium induces oxidant stress leading to transendothelial migration of monocytes. Blood 1998; 92: 3924–35. Surve RR, Mukherjee MB, Kate SL, Nagtilak SB, Wadia M, Tamankar AA, Ghosh K, Colah RB, Mohanty. Detection of the S gene: an evaluation of the solubility test against automated chromatography end haemoglobin electrophoresis. British J of Biomedical Science, 2000; 57: 292-294. 143 Bibliografía Tancabelic J, Sheth S, Paik M, Piomelli S. Serum transferrin receptor as a marker of erythropoiesis suppression in patients on chronic transfusion. Am. J. Hematol. 1999; 60: 121–125. Thomas SL, Egee S, Lapaix F, Kaestner L, Staines HM, Ellory JC: Malaria parasite Plasmodium gallinaceum up-regulates host red blood cell channels. FEBS Lett 2001; 500: 45-51. Valko M, Leibfritz D, Moncol J, Cronin M, Mazur M, Telser J. Free radicals and antioxidants in normal physiological function and human disease. The International J of Bioquem. And Cell Biology. 2007; 39:44-84. Varawalla NY, Fitches AC, Old JM, Analysis of -globin gene haplotypes in Asian Indians: origin and spread of -thalassaemia on the Indian subcontinent. Hum Genet, 1992; 90:443-449. Venters Jr., H. D.; Bonilla, L. E.; Jensen, T.; Garner, H. P.; Bordayo, E. Z.; Najarian, M. M.; Ala, T. A.; Mason, R. P.; Frey II, W. H. Heme from Alzheimer's brain inhibits muscarinic receptor binding via thiyl radical generation. Brain Res. 1997; 764:93–100. Verdon CP, Burton CA, Prior RL. Sample pretreatment with nitrate reductase and glucose-6-phosphate dehydrogenase quantitatively reduces nitrate while avoiding interference by NADP+ when the Griess reaction is used to assay for nitrite. Anal Biochem 1995; 224(2):502-8. Vilachá D, Salazar R., Patrón clínico y hematológico de pacientes con drepanocitosis o diagnóstico presuntivo de talasemia, Revista de Investigación Clínica, vol 58, No. 2; 2006, 94-100. Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79(8):704-12. 144 Bibliografía Weiner, D. L.; Hibberd, P. L.; Betit, P.; Cooper, A. B.; Botelho, C. A.; Brugnara, C. Preliminary assessment of inhaled nitric oxide for acute vaso-occlusive crisis in pediatric patients with sickle cell disease. JAMA. 2003; 289:1136–1142. Winterbourn CC and Carrell RW. Studies of hemoglobin denaturation and Heinz body formation in the unstable hemoglobins. J. Clin. Invest. 1974; 54: 678-689. Wood KC, Hebbel RP, Granger DN. Endothelial cell P-selectin mediates a proinflammatory and prothrombogenic phenotype in cerebral venules of sickle cell transgenic mice. Am. J. Physiol. Heart Circ. Physiol. 2004; 286: 1608–14. Wood KC, Granger K. Sickle cell disease: role of reactive oxygen and nitrogen metabolites. Clinical and Experimental Pharmacology and Physiology. 2007; 34 (9): 926-932. Wood KC, Hebbel RP, Granger DN. Endothelial cell NADPH oxidase mediates the cerebral microvascular dysfunction in sickle cell transgenic mice. FASEB J. 2005; 19: 989–91. Wood KC, Hebbel RP, Lefer DJ, Granger DN. Critical role of endothelial cellderived nitric oxide synthase in sickle cell disease-induced microvascular dysfunction. Free Radic. Biol. Med. 2006; 40:1443–1453. Wood KC, Hsu LL, Gladwin MT, Sickle cell disease vasculopathy: A state of nitric oxide resistance. Free Radical Biology and Medicine 2008, 44:1506-1528. World Health Organization, 59th World Health Assembly, A59/9. Sickle Cell Anemia: Report of the Secretariat 2006. Yuditskaya, S.; Hoehn, G.; Suffredini, A. F.; Xu, X.; Munson, P.; Gladwin, M. T.; Kato, G. J. Proteomic identification of dysregulated apolipoprotein expression in 145 Bibliografía pulmonary hypertension secondary to sickle cell disease. Society for Free Radical Biology and Medicine. 2005. Zago MA, Silva WA Jr, Dalle B, Gualandro S, Hutz MH, Lapoumeroulie C, Tavella MH, Araujo AG, Krieger JE, Elion J, Krishnamoorthy R. Atypical beta(s) haplotypes are generated by diverse genetic mechanisms: PMID: 2000, Feb; 63(2):79-84. Zuzak KJ, Gladwin, M. T.; Cannon III, R. O.; Levin, I. W. Imaging hemoglobin oxygen saturation in sickle cell disease patients using noninvasive visible reflectance hyperspectral techniques: effects of nitric oxide. Am. J. Physiol., Heart Circ. Physiol. 2003; 285:1183–1189. 146