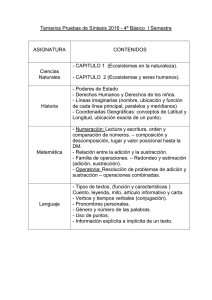
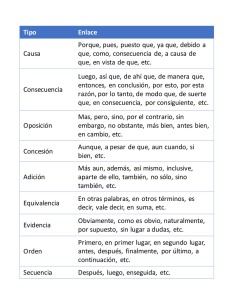
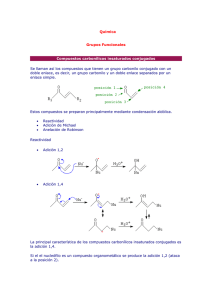
Reacciones de adición conjugada organocatalítica - Academica-e
Anuncio

Universidad Pública de Navarra Máster en Química Sintética e Industrial Trabajo fin de máster Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Presentado por: Álvaro Astiz Erro Línea de investigación: Organocatálisis enantioselectiva Director: Jesús Razkin Lizarraga Ciudad: Pamplona Fecha: Septiembre 2014 2 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Índice 1. Introducción .................................................................................................................. 11 1.1 Quiralidad .................................................................................................................... 11 1.2 Estrategias para la obtención de compuestos enantioméricamente puros ............... 12 1.3 Modos de activación de los organocatalizadores ....................................................... 17 1.4 Importancia y ventajas de las -hidroxicetonas en síntesis asimétrica ...................... 20 1.5 Precedentes en catálisis asimétrica con -hidroxienonas .......................................... 24 1.6 Importancia de las benzofuran-2(3H)-onas ................................................................ 28 2. Objetivos generales ....................................................................................................... 29 3. Resultados y discusión .................................................................................................. 35 4. Conclusiones.................................................................................................................. 45 5. Parte experimental ........................................................................................................ 47 5.1 Técnicas experimentales ............................................................................................. 47 5.2 Reacciones................................................................................................................... 49 5.3 Selección de espectros de RMN .................................................................................. 60 5.4 Selección de cromatogramas de HPLC ........................................................................ 67 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 3 4 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 5 6 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Abreviaturas y acrónimos Listado de símbolos, abreviaturas y acrónimos empleados en esta memoria. * Centro de simetría AcEt Acetato de etilo Ar Arilo/Aromático BB Base de Brønsted Bn Bencilo DCE Dicloroetano DBU 1,8-Diazabiciclo[5.4.0]undec-7-eno ee Exceso enantiomérico Enant. Enantiómero EPC Enantiomerically Pure Compounds (Compuestos enantioméricamente puros) HPLC Cromatografía líquida de alta resolución Hex Hexano iPr Isopropilo Isóm. Isómero L Ligando M Metal Me Metilo n.r. No reacciona Ph Fenilo Rdto. Rendimiento químico RMN Resonancia magnética nuclear T Temperatura t Tiempo THF Tetrahidrofurano TLC Cromatografía en capa fina UV Ultravioleta Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 7 8 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Resumen El presente proyecto se enmarca dentro del área denominada “organocatálisis” y tiene como objetivo explorar y comparar la eficiencia de catalizadores en reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona sobre plantillas de ’hidroxienonas. Se ha llevado a cabo un estudio para optimizar el exceso enantiomérico obtenido en dicho tipo de reacciones, utilizando diferentes compuestos que actúan como bases de Brønsted en cantidades catalíticas, y modificando las estructuras de las plantillas de las hidroxienonas, así como las condiciones de reacción. Este tipo de reacciones han sido poco exploradas utilizando métodos organocatalíticos y los correspondientes resultados, además de aportar luz sobre el comportamiento de dichos catalizadores y sustratos, proporcionan aductos de gran valor sintético. Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 9 10 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 1. Introducción 1.1 Quiralidad La palabra quiral fue introducida por William Thomson (Lord Kelvin) en 1894 para designar objetos que no son superponibles con su imagen especular. Aplicado a la química orgánica, podemos decir que una molécula es quiral cuando ella y su imagen en un espejo no son superponibles. La quiralidad está a menudo asociada a la presencia de carbonos asimétricos. Un carbono asimétrico es aquel que se une a cuatro sustituyentes diferentes. Un ejemplo de carbono asimétrico lo tenemos en la molécula de bromocloroyodometano (figura 1). El carbono está unido a bromo, cloro, yodo e hidrógeno, cuatro sustituyentes diferentes que lo convierten en quiral o asimétrico. La molécula y su imagen en un espejo son diferentes, ningún giro permite superponerlas. Figura 1. Quiralidad de la molécula de bromocloroyodometano. Ambas moléculas son estereoisómeros y se distinguen con la notación R y S. Los estereoisómeros que se relacionan entre sí como un objeto y su imagen especular no superponible se denominan enantiómeros. Existen además moléculas quirales portadoras de otros elementos de quiralidad como ejes y planos. Estas moléculas quirales se diferencian de las aquirales en que tienen actividad óptica, es decir, desvían el plano en el que vibra la luz polarizada; una de las formas lo desvía a la derecha y la otra a la izquierda. Por lo demás, ambas formas poseen casi idénticas propiedades. Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 11 La quiralidad tiene una gran importancia en la vida1. La mayor parte de los compuestos de importancia biológica fundamental son quirales y, además, solo se encuentran como un enantiómero en la naturaleza. Los seres vivos estamos compuestos por moléculas quirales como las proteínas, los ácidos nucleicos o los hidratos de carbono y las funciones biológicas de las que depende el curso de la vida se basan en el reconocimiento entre estas moléculas. Es en este reconocimiento donde la quiralidad de las moléculas puede ser determinante. De hecho, cada uno de los enantiómeros puede presentar propiedades biológicas diferentes. Por ejemplo, el (R)-propanolol se emplea como anticonceptivo mientras que su enantiómero, de configuración (S), actúa como antidepresivo. Por el contrario, la síntesis de moléculas en el laboratorio suele conducir a la obtención de mezclas racémicas. Dado que los enantiómeros poseen propiedades biológicas diferentes, la demanda de compuestos quirales enantioméricamente puros (EPC) por parte de diversas industrias ha crecido considerablemente en los últimos años. 1.2 Estrategias para la obtención de compuestos enantioméricamente puros Para lograr la síntesis de compuestos enantioméricamente puros, existen tres metodologías2 (figura 2): resolución de racematos, empleo de fuentes quirales naturales y síntesis asimétrica. Figura 2. Metodologías para la obtención de compuestos enantiopuros. 1 a) M. Avalos, R. Babiano, P. Cintas, J. L. Jiménez, J. Palacios, Thtrahedron: Asymmetry, 2000, 11, 28452874; b) A. Guijarro, M. Yus, The Origin of Chirallity in the Molecules of Life: A Revision from Awareness to the Current Theories and Perspectives of this Unsolved Problem, 2008, RSC Publishing, Cambridge (UK) 2 Para más información sobre estas metodologías, ver: G. Beck, Synlett., 2002, 6, 637-650. 12 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas En primer lugar, la resolución de mezclas racémicas3 es una estrategia ampliamente utilizada a escala industrial. Se hace reaccionar la mezcla racémica con un agente de resolución quiral para dar diastereómeros. Posteriormente, estos son separados por técnicas convencionales (cristalización, cromatografía…). Finalmente, se elimina el agente de resolución para obtener uno de los enantiómeros con un rendimiento máximo del 50%. Dos técnicas de separación más modernas son la resolución cromatográfica con fases estacionarias quirales y la resolución cinética. La primera de ellas consiste en la diferente afinidad que presentan los enantiómeros con la fase quiral; la segunda se basa en la diferencia de velocidad de reacción de cada uno de los enantiómeros con un agente quiral. Otra metodología para la obtención de productos enantioméricamente puros es el empleo de compuestos quirales naturales, o también denominado “chiral-pool”4. Estos compuestos actúan como plantillas quirales determinando la configuración de los nuevos elementos estereogénicos. Una última estrategia para la obtención de compuestos enantiopuros es la síntesis asimétrica5, que consiste en la obtención de compuestos quirales a partir de compuestos no quirales. En el proceso se lleva a cabo la transformación de un sustrato aquiral en un producto quiral con la consiguiente generación de uno o más elementos estereogénicos de forma estereoselectiva. La información quiral puede provenir de auxiliares quirales, de enzimas o de catalizadores quirales. En el uso de auxiliares quirales6, se produce una unión covalente entre el sustrato y el auxiliar. La función del auxiliar consiste en controlar la estereoselectividad de la reacción dando lugar a una proporción de isómeros. Las mezclas diastereoméricas pueden enriquecerse en uno de los diastereómeros mediante procedimientos físicos y dar lugar a los aductos estrictamente enantiopuros tras la escisión del auxiliar quiral, que se recupera y reutiliza como fuente promotora de quiralidad. En la figura 3 se muestran algunos de los auxiliares quirales más representativos: 3 Para una revisión actualizada de los procesos de resolución, véase: N.G. Anderson, Org. Proc. Res. Dep., 2005, 9, 800-813. 4 a) S. Hanessian, Pure Appl. Chem., 1993, 65, 1189-1204; b) K. C. Nicolaou, E. J. Sorensen, Classics in Total Synthesis, 1996, Wiley-VCH; c) K. C. Nicolaou, E. J. Sorensen, Classics in Total Synthesis II, 2003, Wiley-VCH. 5 Libros generales sobre síntesis asimétrica: a) M. Christmann, S. Bräse, Asymmetric Synthesis- The Essentials, Wiley-VHC, 2007. b) D. Enders, K-E. Jaeger, Asymmetric Synthesis with Chemical and Biological Methods, Wiley-VHC, 2007. 6 Para información sobre auxiliares quirales, ver: a) D. A. Evans, G. Helmchen, M. Rueping, Asymmetric Synthesis-The Essentials (Part I. Chiral Auxiliaries in Asymmetric Synthesis), Wiley-VCH, 2007. b) L. A. Paquette, Handbook of Reagents for Organic Synthesis: Chiral Reagents for Asymmetric Synthesis, Wiley, 2003. c) J. Seyden-Penne, Chiral Auxiliaries and Ligands in Asymmetric Synthesis, Wiley, 1995. Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 13 Enders, 1976 Evans, 1981 Oppolzer, 1989 Figura 3. Auxiliares quirales representativos. Los auxiliares quirales pueden ser productos naturales o sintéticos. Entre los primeros cabe destacar derivados de terpenos, aminoácidos y carbohidratos7. Un fragmento estructural que ha demostrado su eficacia como elemento de información quiral es el de las -hidroxicetonas, descritas inicialmente por Hearhcock8 y Masamune9, dada su facilidad para coordinarse con metales (figura 4): Figura 4. Síntesis estereoselectiva de ácidos carboxílicos -alquil-β-hidroxi sustituidos desarrollada por Heathcock. El empleo de auxiliares quirales proporciona alto grado de estereoselectividad. Sin embargo, presenta dos desventajas: el uso de cantidades estequiométricas del auxiliar quiral y la necesidad de alargar la secuencia de síntesis en dos etapas (anclaje y escisión del auxiliar). 7 Revisiones significativas: Terpenos: a) W. Oppolzer, Tetrahedron, 1987, 43, 1969-2004; b) B. H. Kim, D. P. Curran, Tetrahedron, 1993, 49, 293-318. Aminoácidos: c) J. S. Johnson, D. A. Evans, Acc. Chem. Res. 2000, 33, 325-335. Carbohidratos: d) R. J. Ferrier, Carbohydr. Chem., 2003, 34, 338-366; e) S. Knauer, B. Kranke, H. Kunz, Curr. Org Chem. 2004, 8, 1739-1761. 8 N. A. Van Draanen, S. Arseniyadis, M. T. Crimmins, C. H. Heathcock, J. Org. Chem., 1991, 56, 2499-2506 y referencias allí citadas. 9 a) A. Abiko, J.-F. L, D. C. Buske, S. Moriyama, S. Masamune, J. Am. Chem. Soc., 1999, 121, 7168-7169; b) A. Abiko, T. Inoue, H. Furuno, H. Schwalde, C. Fieres, S. Masamune, J. Am. Chem. Soc., 2001, 123, 46054607. 14 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Pero sin duda, el método de mayor interés dentro de la síntesis asimétrica es la catálisis asimétrica. Hace uso de catalizadores quirales enantiopuros que son empleados en cantidades subestequiométricas, lo que supone una gran ventaja sobre otros métodos tradicionales. El catalizador interacciona de forma reversible con el sustrato formando una especie de “bolsillo quiral” en el estado de transición, favoreciendo así la formación de un enantiómero frente a otro. Dependiendo del catalizador utilizado en la reacción, hay tres campos principales: biocatálisis, catálisis metálica y organocatálisis (figura 2). La biocatálisis consiste en el empleo de enzimas o células para sintetizar los compuestos quirales. Las enzimas son las proteínas responsables de la regulación de las transformaciones químicas a nivel celular y su eficacia química y quimioselectividad es excepcional. Por lo tanto, se trata de una alternativa muy eficaz para la preparación de compuestos ópticamente activos. La única limitación es su alto grado de especificidad, que restringe su versatilidad. La catálisis metálica10 emplea metales complejados a ligandos orgánicos quirales. Consiste en la reactividad y capacidad coordinante de los metales de transición sumado a la estereoespecificidad de los ligandos orgánicos al formar complejos. Esta estrategia de síntesis es muy utilizada en la industria11 gracias a la diferente reactividad química de las especies organometálicas en combinación con la amplia variedad estructural del ligando orgánico. En las últimas décadas, la catálisis metálica ha experimentado un crecimiento realmente considerable. No obstante, aunque los catalizadores metálicos tienen una gran importancia en la industria, muestran dos limitaciones importantes como son la necesidad de atmósferas inertes y la incompatibilidad de los metales con sistemas biológicos debido a su toxicidad. 10 Para revisiones generales sobre catálisis organometálica, ver: a) R. Noyori, Asymmetric Catalysis in Organic synthesis, 1994, Wiley, New York (EEUU); b) E. N. Jacobsen, A. Pfaltz, H. Yamamoto, Comprehensive Asymmetric Catalysis, 1999, Springer, (vol. I-III), Berlín (Alemania); c) I. Ojima, Catalytic a Asymmetric synthesis, 2000, 2 edición, Wiley-VCH, New York (EEUU); d) número especial "Catalytic Asymmetric Synthesis”, Acc. Chem. Res., 2000, 33, 323-440; e) J. A. Ma, D. Cahard, Angew. Chem. Int. a Ed., 2004, 43, 4566-4583; f) M. Beller, C. Bolm, Transition Metals for Organic Synthesis, 2004, 2 edición, Wiley-VCH, Weinheim (Alemania); g) J.-A. Ma, D. Cahard, Angew. Chem. Int. Ed., 2004, 43, 4566-4583; h) M. Christmann, S. Bräse, Asymmetric Synthesis: The Essentials, M. Shibasaki, S. Matsunaga, "Part II, Metal Catalyzed Asymmetric Shynthesis", 2007, Wiley-VCH, Weinheim (Alemania). 11 H. U. Blazer, E. Schmidt, Asymmetric Catalysis on Industrial Scale, 2004, Willey-VHC, Weinheim (Alemania). Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 15 Por ello, en los últimos años ha cobrado un considerable protagonismo la organocatálisis12. Se define como la aceleración de una reacción orgánica promovida por moléculas orgánicas de pequeño tamaño sin la participación de elementos metálicos en el ciclo catalítico13. Esta estrategia de síntesis tiene tres beneficios principales que se resumen en: 1. Las moléculas orgánicas son generalmente insensibles al oxígeno y la humedad de la atmósfera, por lo que no es necesario tomar medidas o cuidados especiales. 2. Una amplia variedad de reactivos orgánicos como aminoácidos, carbohidratos o hidroxiácidos son accesibles a partir de fuentes naturales como un único enantiómero, lo cual conlleva que organocatalizadores simples basados en estas moléculas sean baratos y fácilmente accesibles en cantidades adecuadas tanto para llevar a cabo reacciones a pequeña escala como para reacciones a escala industrial. 3. La mayoría de las moléculas en las que están basados estos catalizadores son inocuas, aumentando así la seguridad a la hora de trabajar con ellas. En la última década, y especialmente en los últimos años, la catálisis en general y la organocatálisis en particular, han experimentado un crecimiento exponencial, lo que se refleja en una extensa bibliografía en la materia que se encuentra recopilada en numerosos artículos de revisión. Así, en el año 2003 una serie de sistemas, tanto metálicos como organocatalíticos, eran ya considerados como “catalizadores privilegiados”14 (figura 5) por su efectividad, versatilidad y tolerancia de sustrato. Precisamente, es en este campo concreto de la organocatálisis donde se enmarcará el presente trabajo de investigación. 12 Referencias generales sobre organocatálisis: a) K. Mikami, M. Lautens, New Frontiers in Assymetric Catalysis, G. Lelais, D. W. C. MacMillan "cap. 11: History and Perspective of Chiral Organic Catalysts", 2007, Wiley Inter-Science, New Jersey (EEUU); b) M. Christmann, S. Bräse, Asymmetric Synthesis: The Essentials, S. V. Ley, "Part III, Asymetric Organocatalysis", 2007, Wiley-VCH, Weinheim (Alemania); c) M. J. Gaunt, C. C. C. Johansson, A. McNally, N. T. Vo, Drug Discovery Today, 2007, 12, 8-27; d) P. H.-Y. Cheong, C. Y. Legault, J. M. Um, N. Çelebi-Ölçüm, K. N. Houk, Chem. Rev., 2011, 111, 5042-5137; e) F. Giacalone, M. Gruttadauria, P. Agrigento, R. Noto, Chem. Soc. Rev., 2012, 41, 2406-2447; f) B. List, Asymmetric Organocatalysis 1, Lewis Base and Acitd Catalysts, Science of Synthesis, 2012, Thieme, Stuttgart (Alemania); g) K. Maruoka, Asymmetric Organocatalysis 2, Brønsted Base and Acid Catalysi.s, and Additional Topics: Science of Synthesis, 2012, Thieme, Stuttgart, (Alemania); i) R. Rios, Stereoselective Organocatalysis: Bond Formation Methodologies and Activation Modes, 2013, Wiley, Jon Willey and Sons (New Jersey, EEUU). 13 P. I. Dalko, L. Moisan, Angew. Chem. Int. Ed., 2004, 43, 5138-5175. 14 a) T.P. Yoon, E. N. Jacobsen, Science, 2003, 299, 1691-1693; b) A. Pfaltz, W. J. Drury III, Proc. Nat. Acad. Sci. USA, 2004, 101, 5723-5726; c) M. Shibasaki, S. Matsunaga, Privileged Chiral Ligands and Catalysts, 2001, 295-332, Ed. Q.-L. Zhou, Wiley-VCH, Weinheim, Alemania. 16 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Figura 5. Ligandos privilegiados y algunas reacciones catalíticas en las que han sido empleados. 1.3 Modos de activación de los organocatalizadores Los organocatalizadores cumplen dos funciones: por un lado activan al nucleófilo o al electrófilo (en algunos casos incluso ambos, denominándose en estos casos catalizadores bifuncionales15) y por otro controlan la aproximación del electrófilo o nucleófilo determinante para la estereoselectividad de la reacción. Los organocatalizadores pueden considerarse versiones minimizadas de enzimas no metálicas y su mecanismo de acción se puede clasificar atendiendo a las categorías y mecanismos de catálisis enzimática. La clasificación más extendida es aquella relacionada con el tipo de interacción que se da entre el catalizador y el sustrato. Se distingue entre catálisis covalente y no covalente. 15 Para revisiones sobre organocatálisis bifuncional, ver: a) P. S. Bhadury, B. A. Song, S. Yang, D. Y. Hu, W. Xue, Curr. Org. Synth., 2009, 6, 380-399. b) S. J. Connon, Chem. Commun., 2008, 2499-2510. Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 17 1.3.1 Catálisis covalente La catálisis covalente incluye aquellos procesos que requieren la unión covalente entre el catalizador y el sustrato en el ciclo catalítico para formar el complejo activado. Estas reacciones son promovidas por bases de Lewis (en general aminas). Algunos ejemplos típicos son los aminoácidos, péptidos, alcaloides y moléculas sintéticas que presentan nitrógeno, azufre o fósforo en su estructura. 1.3.2 Catálisis no covalente La catálisis no covalente agrupa a los procesos en los que la interacción entre el catalizador y los sustratos se realiza a través de interacciones más débiles como enlaces de hidrógeno (ácidos de Brønsted) o catálisis por formación de pares iónicos. La catálisis por enlace de hidrógeno16 o ácidos de Brønsted se establece, generalmente, entre un sustrato que contiene grupos coordinantes y un ácido de Brønsted quiral que actúa como catalizador. La formación de estos enlaces entre el catalizador y el eletrófilo de una reacción provoca un aumento del carácter electrófilo de este último, facilitando así el ataque del nucleófilo. Según la fortaleza del enlace de hidrógeno y el número de interacciones que pueden establecerse, se pueden distinguir activaciones monodentadas o bidentadas17, e incluso formación de pares iónicos cuando hay protonación neta del sustrato por acción del ácido de Brønsted (figura 6): Figura 6. Modos de activación de un sustrato electrofílico por enlace de hidrógeno. 16 Para revisiones sobre catálisis asimétrica mediante enlace de hidrógeno, ver: a) M. S. Taylor, E. N. Jacobsen, Angew. Chem. Int. Ed., 2006, 45, 1520-1543; b) T. Akiyama, J. Itoh, K. Fichibe, Adv. Synth. Catal., 2006, 348, 999-1010; c) A. G. Doyle, E. N. Jacobsen, Chem. Rev., 2007, 107, 57135743; d) X. Yu, W. Wang, Chem. Asian. J., 2008, 3, 516-532; e) Z. Zhang, P. R. Scheriner, Chem. Soc. Rev., 2009, 38, 1187-1198; f) P. M. Pihko, Hydrogen Bonding in Organic Synthesis, 2009, Wiley-VCH, Weinheim (Alemania); g) Y. Sohtome, K. Nagasawa, Synlett, 2010, 1-22; h) S. Schenker, A. Zamfir, M. Freund, S. B. Tsogoeva, Eur. 1. Org. Chem., 2011, 2209-2222. 17 P.M. Pihko, Angew. Chem. Int. Ed., 2004, 43, 2062-2064. 18 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Las estructuras más representativas que se han descrito para este tipo de catálisis son el TADDOL18, derivados de ureas y tioureas19, escuaramidas20, derivados de binaftol21 o ácidos fosfóricos22 (figura 7): Figura 7. Ácidos de Brønsted quirales representativos. Otro tipo de catálisis no covalente es la promovida por bases de Brønsted23. Las bases de Brønsted retiran un protón de la especie pro-nucleófila para convertirla en una especie con mayor nucleofilia, creando simultáneamente un entorno quiral a través de la formación de un par iónico. La dificultad para conocer la direccionalidad de las interacciones electrostáticas entre los pares iónicos resultantes dificulta la predicción de la estereoinducción del proceso. Este problema puede minimizarse combinando el carácter básico del catalizador con un grupo donador de enlaces de hidrógeno pudiendo así anclar tanto el nucleófilo como el electrófilo en el estado de transición. De este modo, se obtiene un catalizador más activo, ya que activa a los dos sustratos. Además, este tipo de catálisis bifuncional puede proporcionar un mayor control estereoquímico y se considera catálisis básica siempre que la activación del nucleófilo sea la etapa crucial del transcurso del proceso. Este tipo de bases son las que van a ser utilizadas como catalizadores a lo largo de este trabajo. 18 Para el primer ejemplo organocatalítico y enantioselectivo empleando TADDOL como catalizador, ver: Y. Huang, A. K. Unni, A. N. Thadani, V. H. Rawal, Nature, 2003, 424, 146. 19 Para revisiones sobre organocatálisis empleando derivados de tioureas ver: a) Y. Takemoto, Org. Biomol. Chem., 2005, 3, 4299-4306; b) S. J. Connon, Chem. Eur. J., 2006, 12, 5418-5427; c) H. Miyabe, Y. Takemoto, Bull. Chem. Soc. Jpn., 2008, 81, 785-795; d) S. 3. Connon, Synlett., 2009, 3, 354-376; e) Z. Zhang, P. R. Schreiner, Chem. Soc. Rev., 2009, 38, 1187-1198; f) W.-Y. Siau, J. Wang, Catal. Sci. Technol., 2011, 1, 1298-1310. 20 Para revisiones sobre escuaramidas, ver: a) J. Alemán, A. Parra, H. Jiang, K. A. Jørgensen, Chem. Eur. J., 2011, 17, 6890-6889; b) R. I. Storer, C. Aciro L. H. Jones. Chem. Soc. Rev., 2011, 40, 2330-2346. 21 Para revisiones generales sobre el uso de binaftol y derivados, ver: a) Y. Chen, S. Yekta, A. K. Yudin, Chem. Rev., 2003, 103, 3155-3211; b) P. Kočovský, S. Vyskočil, M. Smrčina, Chem. Rev., 2003, 103, 32133245; c) G. Bringmann, A. J. P. Mortimer, P. A. Keller, M. J. Gresser, J. Garner, M. Breuning, Angew. Chem. Int. Ed., 2005, 44, 5384-5427; d) J. M. Brunel, Chem. Rev., 2005, 105, 857-897; e) J. M. Brunel, Chem. Rev., 2007, 107, PR1-PR45; f) M. Terada, Chem. Commun., 2008, 4097-4112. 22 a) S. J. Connon, Angew. Chem. Int. Ed. 2006, 45, 3909-3912; b) ref. 72f, c) M. Terada, Curr. Org. Chem., 2011, 15, 2227-2256; d) M. Terada, Synthesis, 2012, 1929-1982. 23 Para revisiones con bases de Brønsted, ver: a) M. S. Taylor, E. N. Jacobsen, Angew. Chem. Int. Ed., 2006, 45, 1520-1543; b) C. Palomo, M. Oiarbide, R. López, Chem. Soc. Rev., 2009, 38, 632-653; c) A. Ting, J. M. Goss, N. T. McDougal, S. E. Schaus, Top. Curr. Chem. 2010, 291, 145-200. Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 19 1.4 Importancia y ventajas de las -hidroxicetonas en síntesis asimétrica Hoy en día, la síntesis de moléculas asimétricas con formación de enlaces C-C o C-X sigue teniendo especial interés para ensayos biológicos o investigación en ciencia de los materiales. El diseño de un catalizador eficiente24, tanto química como esteroquímicamente, es una tÁrea complicada que requiere, además de un diseño preciso de su estructura, un cuidado riguroso de las condiciones de reacción. Por tanto, un cambio mínimo en las proximidades del sitio de reacción del sustrato puede alterar la reactividad y la estereoquímica de la reacción. Esta idea es la que ha llevado a sintetizar plantillas que faciliten la acción del catalizador para llevar a cabo la reacción. Los sustratos de tipo acilo (figura 8) se encuentran entre las plantillas preferidas por dos razones: 1. Se pueden formar nuevos enlaces de manera estereoselectivamente en las posiciones y β respecto del grupo carbonilo. 2. Existe gran variedad de derivados de acilo o productos relacionados por transformación de grupo funcional, lo que nos da gran versatilidad. Reactividad en (enolato/enamina) Reactividad en (sustitución de un grupo X) Reactividad en β (adición conjugada o cicloadición Figura 8. Concepto general de una plantilla tipo acilo. 24 Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz, H. Yamamoto, Springer, Heidelberg, 1999, vol. I-II. 20 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas De las muchas plantillas de tipo acilo que se han probado, las N-acil oxazolidinonas (figura 9) han demostrado destacar respecto del resto, tras el trabajo desarrollado por el grupo de Evans en Harvard25. Inducción estereocontrolada por el catalizador. Inducción estereocontrolada por el sustrato. Figura 9. Caso de ejemplo: de oxazolidinona como auxiliar quiral a oxazolidinona como plantilla aquiral. En los últimos años, se ha descubierto que las - hidroxicetonas, y más concretamente las ’-hidroxienonas, sus análogos insaturados, son plantillas aquirales efectivas26 para la catálisis asimétrica (catálisis metálica y organocatálisis). Es conveniente hacer un análisis de las características estereoelectrónicas fundamentales de las ’-hidroxienonas (así como de sus derivados análogos ’alcóxidos y ’-sililóxidos) para entender su importancia en catálisis asimétrica. En primer lugar, podemos destacar la existencia de patrones de coordinación con metales o protones. La buena coordinación entre el sustrato y el catalizador es considerada el elemento clave para la activación y el control estereoquímico en las reacciones enantioselectivas. Así pues, la mayoría de plantillas aquirales de tipo acilo obedecen a uno de los tres modelos descritos en la figura 10: Sustratos bidentados (estereodiscriminación efectiva) Sustratos monodentados (mala estereodiscriminación) Figura 10. Patrones de coordinación metal-sustrato para plantillas de tipo acilo ,β-insaturadas (monodentadas y bidentadas). 25 26 J. S. Johnson, D. A. Evans, Acc. Chem. Res., 2000, 33, 325. C. Palomo, M. Oiarbide, J. M. García, Chem. Soc. Rev., 2012, 41, 4150-4164. Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 21 En la figura 10 C tenemos un sistema coordinado entre el carbonilo y un ácido de Lewis donde, en principio, las dos geometrías pueden coexistir en equilibrio mediante un sustrato monodentado. Solo unos pocos catalizadores metálicos han sido desarrollados que sean capaces de lograr un control estereoquímico efectivo con sustratos monodentados27. Sin embargo, los sustratos bidentados pueden inducir a la coordinación por dos puntos de anclaje, como se observa en la figura 11 A y B. Esto conduce a una conformación del estado de transición menos flexible y, por lo general, con mejor enantioselectividad. En segundo lugar, dichas ’-hidroxienonas, y análogos, coordinan de forma bidentada y pueden formar quelatos metálicos de unión 1,4 de tipo B (figura 10) con centros metálicos de carácter oxofílico o con protones. Estos quelatos muestran un modelo de geometría diferente en comparación con el modelo típico de quelación de metal 1,5 propuesto para las reacciones de N-acil oxazolidinonas con sus respectivas plantillas bidentadas. Esto se explicaría debido a que las y β-hidroxicetonas libres también forman ciclos intramoleculares de 5 y 6 miembros por puentes de hidrógeno. Una característica de las quelaciones 1,4 (anillos de 5 miembros) en las ’hidroxienonas es que la conformación más probable es la planar con los dos enlaces CO eclipsados, permitiendo el enlace por puente de hidrógeno intramolecular (figura 11). Así por ejemplo, se pudo predecir la preferencia conformacional del grupo enona por la forma s-cis antes que por la s-trans para el estado de transición en el caso de las reacciones de Diels-Alder28. s-trans (preferido en el estado fundamental) s-cis (preferido en el estado de transición) Modelo de actuación para las reacciones catalizadas por metal Figura 11. Conformaciones más probables y modelos de quelación para las -hidroxienonas. 27 a) D. Liu, E. Canales, E. J. Corey, J. Am. Chem. Soc., 2007, 129, 1498 y referencias dentro del texto; b) J. N. Payette, Yamamoto, J. Am. Chem. Soc., 2007, 129, 9536 y referencias dentro del texto. Para más ejemplos sobre organocatálisis, ver: c) A. B. Northrup, D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 2458. 28 a) W. Choy, L. A. Reed III, S. Masamune, J. Org. Chem., 1983, 48, 1139; b) S. Masamune, L. A. Reed III, J. T. Davis, W. Choy, J. Org. Chem., 1983, 48, 4441; c) B. Satammen, U. Berlange, R. Kindermann, M. Kaiser, B. Günther, W. S. Sheldrick, P. Welzel, W. R. Roth, J. Org. Chem., 1992, 57, 6566. 22 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Por otra parte, cabe señalar que podría darse la situación de que existiese competencia con una reacción intramolecular de ciclación de Michael. Sin embargo, este es un proceso no favorable termodinámicamente según las reglas de Baldwin29, por lo que no es posible que se forme el producto ciclado mostrado en la figura 12: Figura 12. Posible producto secundario por reacción de Michael intramolecular. Además de estas tres útiles características estereoelectrónicas, otra gran ventaja de las ’-hidroxienonas es su facilidad para ser preparadas a partir de reactivos más sencillos. Algunos de los principales métodos son (figura 13): a) Condensación aldólica del 3-hidroxi-3-metil-2-butanona (comercial) y aldehído. b) Olefinación de Horner-Wadsworth-Emmons partiendo del β-fosfonato correspondiente. c) Adición nucleófila del correspondiente metoxialeno lítico a acetona y posterior hidrólisis suave. d) Reacción de Heck. e) Metátesis cruzada con catalizador de Grubbs. 29 J. E. Baldwin, T. C. Thomas, L. I. Kruse, L. Silberman, J. Org. Chem., 1977, 42, 3846. Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 23 Figura 13. Métodos de preparación de las -hidroxienonas. Finalmente, la importancia y aplicabilidad de las ’-hidroxienonas queda demostrada por el número de reacciones catalíticas enantioselectivas en las que se ha descrito su participación. Algunos de los más destacados serían26: a) Reacciones de Diels-Alder. b) Reacciones de cicloadición con nitronas. c) Adición conjugada de carbamatos. d) Alquilaciones de Friedel-Crafts. e) Adición conjugada de carbonos nucleófilos estabilizados. f) Adición conjugada de radicales. g) Adición conjugada de reactivos dialquilcinc. 1.5 Precedentes en catálisis asimétrica con hidroxienonas Lo compuestos carbonílicos ,β-insaturados son considerados buenos aceptores en reacciones de Michael y, entre ellos, los aductos resultantes de la adición conjugada de un reactivo nucleófilo a una ’-hidroxienona tienen variedad de aplicaciones, por lo que son de gran importancia. Además, estos compuestos pueden ser fácilmente transformados posteriormente en diferentes grupos funcionales, lo que les hace muy versátiles. El mecanismo de activación electrófilo más común es la coordinación del 24 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas grupo carbonilo con un ácido de Lewis (catálisis metálica) o con una especie donadora de puentes de hidrógeno (organocatálisis). Como ya se trató en el punto 1.4, para conseguir una mejor activación del sustrato y poder llevar a cabo reacciones de adición estereocontroladas, se prefiere el uso de sustratos con dos puntos de coordinación, introduciendo un grupo X en una posición próxima al carbonilo (figura 14 B): A (monodentado) B (bidentado) Tiene un sitio extra donador de electrones Figura 14. Modelos establecidos de plantillas. Siguiendo este concepto de coordinación de las plantillas bidentadas, podemos estudiar las reacciones de adición conjugada catalizadas por bases de Brønsted (BB). El sustrato bidentado actúa como doble aceptor de puentes de hidrógeno (figura 15), al contrario que en las plantillas monodentadas, donde solo es posible que se produzca la coordinación por el oxígeno carbonílico: A (monodentado) B (bidentado) Figura 15. Modelo de coordinación en reacciones de adición conjugada catalizadas por bases de Brønsted (BB). Este tipo de bases de Brønsted, que serán las que se utilizarán en este trabajo, presentan grandes ventajas, no solo porque se encuentran fácilmente disponibles en el mercado, sino también porque el reactivo nucleófilo no necesita por lo general ser activado en un paso anterior23,30. Sin embargo, la reactividad suficiente para formar enlaces carbono-carbono a través de reacciones de adición conjugada catalizada por bases de Brønsted solo se da con algunos reactivos nucleófilos (en concreto los compuestos 1,3-dicarbonilo) o electrófilos (especialmente los nitroalcanos), mientras 30 Para más información sobre bases de Brønsted: “Asymmetric Organocatalysis 2, Brønsted Base and Acid Catalysis, and Additional Topics”: Science of Synthesis; K. Maruoka, Ed.; Thieme: Stuttgart, 2012. Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 25 que en muchos otros casos, como es el caso de los ésteres ,β-insaturados, existe poca reactividad o bajo control estereoquímico. Respecto a los compuestos 1,3-dicarbonilo, los grupos de Ito31, Shibasaki32, Sodeoka33 y Jacobsen34, entre otros35, han descrito las reacciones de adición conjugada catalizadas por metal entre 1,3-dicetonas, β-cetoésteres y -aril cianoésteres a acroleína (propenal) o a vinilcetonas (principalmente metil vinil cetona), estos últimos dos funcionando como aceptores de Michael. Además, también se han estudiado las adiciones conjugadas de compuestos carbonílicos catalizadas por bases quirales de Brønsted, posteriormente al trabajo pionero de Wynberg36. Deng ha descrito las adiciones conjugadas de compuestos β-dicarbonilo sustituidos en y -aril cianoésteres a acroleína o metil vinil cetona, catalizado por un catalizador bifuncional basado en Cinchona37,38. Jørgensen documentó la reacción entre β-cetoésteres cíclicos tanto con acroleína como con metil vinil cetona usando también un catalizador basado en Cinchona39. Más recientemente, Rodríguez y Constantieux40 extendieron el uso de bases de Brønsted como catalizadores a β-cetoamidas cíclicas como nucleófilos frente a metil vinil cetona. A pesar de estos logros, la realización de adiciones conjugadas catalizadas por bases de Brønsted con sustratos más reacios tales como compuestos carbonílicos enolizables menos reactivos sigue siendo un reto. De hecho, solo se ha descrito en tres casos concretos: 1. Dixon41 describió la adición conjugada de β-cetoésteres cíclicos a tioacrilato de naftilo y N-acriloil-pirrol, usando un alcaloide de Cinchona como base de Brønsted bifuncional, obteniéndose un alto grado de enantioselectividad. 31 M. Sawamura, H. Hamashima, Y. Ito, J. Am. Chem. Soc. 1992, 114, 8295–8296. a) H. Sasai, T. Arai, M. Shibasaki, J. Am. Chem. Soc. 1994, 116, 1571–1572; b) H. Sasai, E. Emori, T. Arai, M. Shibasaki, Tetrahedron Lett. 1996, 37, 5561–5564. 33 Y. Hamashima, D. Hotta, M. Sodeoka, J. Am. Chem. Soc. 2002, 124, 11 240–11241. 34 M. S. Taylor, D. N. Zalatan, A. M. Lerchner, E. N. Jacobsen, J. Am. Chem. Soc. 2005, 127, 1313–1317. 35 Para una revisión de adiciones conjugadas catalizadas por metal para la obtención de todos los carbonos estereogénicos, ver: C. Hawner, A. Alexakis, Chem. Commun. 2010, 46, 7295–7306. 36 a) H. Wynberg, R. Helder Tetrahedron Lett. 1975, 4057–4060; b) K. Hermann, H. Wynberg, J. Org. Chem. 1979, 44, 2238–2244. 37 a) F. Wu, H. Li, R. Hong, L. Deng, Angew. Chem. Int. Ed. 2006, 45, 947–950; b) F. Wu, H. Li, R. Hong, J. Khan, X. Liu, L. Deng, Angew. Chem. Int. Ed. 2006, 45, 4301–4305. 38 Para revisiones de catalizadores basados en Cinchona, ver: a) Cinchona Alkaloids in Synthesis and Catalysis; C. E. Song, Ed.; Wiley-VCH: Weinheim, 2009; b) T. Marcelli, H. Hiemstra, Synthesis 2010, 1229– 1279; c) E. M. O. Yeboah, S. O. Yeboah, G. S. Sing, Tetrahedron 2011, 67, 1725–1762; 39 a) S. Brandes, B. Niess, M. Bella, A. Prieto, J. Overgaard, K. A. Jørgensen, Chem. Eur. J. 2006, 12, 6039– 6052; b) M. Bell, K. Frisch, K. A. Jørgensen, J. Org. Chem. 2006, 71, 5407–5410. 40 M. M. Sanchez Duque, O. Baslé, N. Isambert, A. Gaudel-Siri, Y. Génisson, J.-C. Plaquevent, J. Rodríguez, T. Constantieux, Org. Lett. 2011, 13, 3296–3299. 41 C. L. Rigby, D. J. Dixon, Chem. Commun. 2008, 3798–3800. 32 26 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 2. Bartoli y Melchiorre42 usaron cetoésteres acíclicos como nucleófilos y maleimidas como aceptores de Michael, demostrando que el rendimiento y la selectividad disminuían. 3. El grupo de Jørgensen también ha probado con acil-fosfonatos ,βinsaturados43, resultando ser sustitutos eficaces de los enoatos en reacciones con nucleófilos reactivos, incluyendo azlactonas (compuestos derivados de 5(4H)-oxazolona, que poseen un grupo ilideno) y compuestos 1,3-dicarbonilo. Trabajos anteriores de estos laboratorios sobre adiciones conjugadas y cicloadiciones de ’-hidroxienonas catalizadas por metales, así como reacciones Diels-Alder de ’hidroxienonas quirales con catálisis ácida, mostraron que es crítica la capacidad del carbonilo tanto para el enlace 1,4-metal como para el 1,4-hidrógeno (figura 16)44: Catálisis metálica enantioselectiva Catálisis por ácido de Brønsted diastereoselectiva Figura 16. Trabajos previos con -hidroxienonas. En el trabajo de Palomo45 se propone que la capacidad de formación del enlace de hidrógeno puede ser aprovechada mucho mejor si se combina con una transferencia de protón46, tal y como ocurre en las reacciones de Michael catalizadas por bases de Brønsted (figura 17). En concreto, los modelos del estado de transición del sustrato hidroxienona actuando con dos puntos de enlace de hidrógeno pueden clasificarse en dos: modelo donador/aceptor y modelo aceptor/aceptor. 42 G. Bartoli, M. Bosco, A. Carlone, A. Cavalli, M. Locatelli, A. Mazzanti, P. Ricci, L. Sambri, P. Melchiorre, Angew. Chem. Int. Ed. 2006, 45, 4966 –4970. 43 H. Jiang, M. W. Paixão, D. Monge, K. A. Jørgensen, J. Am. Chem. Soc. 2010, 132, 2775–2783. 44 Para pruebas espectroscópicas del enlace intramolecular de hidrógeno en las ’-hidroxicetonas, ver: a) L. Joris, P. von R. Scheleyer, J. Am. Chem. Soc. 1968, 90, 4599–4611; b) T. Cho, I. Kida, J. Ninomiya, S.-i. Ikawa, J. Chem. Soc., Faraday Trans. 1994, 90, 103–107. 45 E. Badiola, B. Fiser, E. Gómez-Bengoa, A. Mielgo, I. Olaizola, I. Urruzuno, J. M. García, J. M. Odriozola, J. Razkin, M. Oiarbide, C. Palomo, “Development of Enoyl Templates with Diverting H-Bonding Capabilities For Use in Organocatalysis: Enantioselective Direct Michael Addition to ’-Hydroxy Enones Catalyzed by Brønsted Bases Forming Quaternary Stereocenters”, 2014, enviado. 46 Para revisiones de catálisis asimétrica directa en transformaciones con transferencia de protón, ver: a) N. Kumagai, M. Shibasaki, Angew. Chem. Int. Ed. 2011, 50, 4760–4772; b) Y. Yamashita, T. Tsubogo, S. Kobayashi, Chem.Sci. 2012, 3, 967–975. Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 27 Figura 17. Catálisis asociativa entre base de Brønsted y enlace de hidrógeno (X=O, NR’’). Es en este tipo de estructuras en las que se va a basar el estudio concreto de este trabajo y donde se pretende profundizar y aportar más luz en el proceso de catálisis. 1.6 Importancia de las benzofuran-2(3H)-onas Por otra parte, dentro de las muy diversas posibilidades de adición a las hidroxienonas, en este trabajo se ha optado por estudiar la adición de benzofuranonas. Concretamente, las lactonas de tipo benzofuran-2(3H)-onas (figura 18) con sustituyente en la posición 3, que al igual que sus derivados, han recibido un especial interés por ser estructuras con un carbono cuaternario en la posición 3 y que aparecen en variedad de compuestos. Además, estos compuestos muestran actividad biológica: desde propiedades antioxidantes hasta aplicación en terapia anticancerígena, por lo que pueden servir como principios activos en medicamentos47. Debido a su importante actividad, estos productos naturales han despertado el interés en su síntesis total.48 Figura 18. Estructura general de las benzofuran-2(3H)-onas con sustituyente en la posición 3. 47 a) S. A. Adediran, D. Vabaret, B. Doruillat, R. F. Pratt, M. Wakselman, Bioorg. Med. Chem. 2001, 9, 1175-1183; b) E. K. Panisheva, L. M. Alekseeva, M. I. Evstratova, S. S. Kiselev, V. G. Granik, Pharm. Chem. J. 2007, 41, 549-553. 48 a) E. Vedejs, M. A. Zajac, Org. Lett. 2001, 3, 2451; b) P. J. Gross, E. Hartmann, M. Nieger, S. Brase, J. Org. Chem. 2010, 75, 229. 28 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 2. Objetivos generales El objetivo principal de este proyecto es el estudio de las reacciones de adición de Michael entre -hidroxienonas benzofuran-2(3H)-onas), y nucleófilos (centrándose especialmente en catalizadas por bases de Brønsted en reacciones enantioselectivas de formación de enlaces carbono-carbono. Se pretende estudiar y optimizar la reacción con el fin de obtener los mayores excesos enantioméricos posibles obteniendo nuevos productos de posible interés sintético. Inicialmente, a modo de pruebas preliminares, se probaron las adiciones de un nitroderivado y un cianoderivado sobre las mencionadas -hidroxienonas, para posteriormente centrarme en las benzofuranonas, objeto de este trabajo. Así, en primer lugar, se probó la adición de Michael de 2-nitropropilbenceno (2) a 4bencil-4-hidroxi-5-fenilpent-1-en-3-ona (1a). La reacción de adición se muestra en la figura 19: Figura 19. Obtención del producto 3. Los tres catalizadores empleados para poder obtener enantioselección en el producto han sido el C1, C2 y C3 (figura 20): Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 29 Figura 20. Catalizadores C1, C2 y C3. Para ello, inicialmente hubo que preparar los reactivos 1 y 2. Se siguió el procedimiento de síntesis mostrado en los esquemas de las figuras 21 y 22: Figura 21 Esquema de síntesis del reactivo 1a. Figura 22. Esquema de síntesis del reactivo 2. 30 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas También, a modo de prueba inicial, se llevó a cabo la adición de Michael de 2-ciano-2fenilacetato de tert-butilo (9) a 2-hydroxi-2-metil-7-fenilhept-4-en-3-ona (8). La reacción de adición se muestra en la figura 23: Figura 23. Obtención del producto 10. El catalizador seleccionado en este caso ha sido el C4 (figura 24): Figura 24. Catalizador C4. Previamente, se llevó a cabo la síntesis de los reactivos 8 y 9. Se siguió el procedimiento de síntesis mostrado en los esquemas de las figuras 25 y 26: Figura 25. Síntesis del reactivo 8. Figura 26. Síntesis del reactivo 9. Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 31 Finalmente, tras mostrar estas dos reacciones de trabajo de prueba, el trabajo realmente se centra en la adición de Michael de 3-fenilbenzofuran-2(3H)-ona (15) a 1hidroxibut-3-en-2-ona, con 2 sustituyentes R en la posición ’ (1). La reacción de adición se muestra en la figura 27: Figura 27. Obtención del producto 16. La reacción se estudió a diferentes temperaturas (0 ºC, -40 ºC y -78 ºC) en dos disolventes (diclorometano y tolueno), comparando diferentes catalizadores y diferentes estructuras de la ’-hidroxienona. Se utilizaron cuatro sustratos disustituidos en posición ’ como se muestra en la figura 28: Figura 28. Sustratos de partida del reactivo 1 variando el sustituyente R. Para la síntesis de los reactivos 1a, 1b, 1c y 1d se siguió el mismo procedimiento ya mostrado en la figura 21. Por otra parte, la síntesis del reactivo 15 se desarrolló según el esquema de síntesis mostrado a continuación (figura 29): Figura 29. Obtención del producto 15. 32 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Y respecto a los catalizadores empleados en la adición, en la figura 30 se muestran las estructuras de todos ellos: C4, C5, C6, C7, C8, C9, C10, C11 y C12. C10 C11 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 33 C12 Figura 30. Catalizadores C4, C5, C6, C7, C8, C9, C10, C11 y C12. Estos catalizadores fueron previamente sintetizados por el Departamento de Química Orgánica I de la Universidad del País Vasco y el Departamento de Química Aplicada de Universidad Pública de Navarra, o adquiridos comercialmente. 3. Resultados y discusión A continuación se detallan los resultados experimentales obtenidos en cada caso. En la reacción de adición del nitroderivado 2 sobre la hidroxienona 1a, se forma un carbono asimétrico en la posición 6 de la molécula 3, que da lugar a dos enantiómeros (R y S) (figura 31): Figura 31. Enantiómeros R y S para el producto 3. En la siguiente tabla (tabla 1) se muestran las proporciones de enantiómeros (determinadas por cromatografía líquida de alta resolución (HPLC)) para cada catalizador (C1, C2, C3). La reacción se llevó a cabo en CHCl3 utilizando un exceso del nitroderivado proporción 5:1 a una temperatura de 20 ºC y utilizando un 20% de catalizador. La base empleada para obtener la mezcla racémica, necesaria como referencia para el HPLC, fue trietilamina. Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 35 # Catalizador T, ºC t, h Rdto., % Enant. 1, % Enant. 2, % ee, % 1 Trietilamina 20 5 89 - - - 2 C1 18 71 86 14 72 3 C2 76 87 13 74 4 C3 60 81 19 62 Tabla 1. Reacción de adición del nitroderivado 2 (0,5 mmol) sobre la hidroxienona 1a (0,1 mmol) para los catalizadores C1, C2, C3 (0,02 mmol). Se ha logrado un exceso enantiomérico no muy alto. El mejor de los resultados (74% ee) se obtuvo con el catalizador C2, aun siendo este insuficiente para ser considerado un catalizador eficiente. No obstante, esta reacción no fue considerada un objetivo principal de este proyecto, sino más bien una forma de aproximación e introducción a la organocatálisis y al trabajo en el laboratorio, por lo que no se siguió estudiando. En la reacción de adición del cianoderivado (9) sobre la hidroxienona (8) se forman 4 estereoisómeros (figura 32): 36 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Figura 32. Los 4 estereoisómeros obtenidos para el producto 10. En la tabla 2 se muestran las proporciones de los 4 estereoisómeros (determinadas por cromatografía líquida de alta resolución (HPLC)) para el catalizador C4. La reacción se llevó a cabo en DCE a una temperatura de 50 ºC, utilizando un exceso de la hidroxienona en proporción 3:1 y con un 10% de catalizador. La base empleada para obtener la mezcla racémica, necesaria como referencia para el HPLC, fue trietilamina. # Catalizador T, ºC t, h Rdto., % Isóm. 1, % 5 Trietilamina 50 5 91 - - - - - 18 90 0,33 2,03 2,34 95,30 96 6 C4 Isóm. 2, Isóm. 3, % % Isóm. 4, ee, % % Tabla 2. Reacción de adición del cianoderivado 9 (0,1 mmol) sobre la hidroxienona 8 (0,3 mmol) para el catalizador C4 (0,01 mmol). Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 37 Esta reacción ya estaba siendo estudiada y optimizada en mi laboratorio y simplemente se colaboró con el grupo de trabajo, lo que explica el buen resultado obtenido en esta prueba puntual concreta. De nuevo, al igual que en la reacción anterior, esta reacción fue una forma de aproximación e introducción a la organocatálisis con ’hidroxienonas. Sin embargo, como ya se ha comentado, el objetivo principal de este proyecto ha estado centrado en la adición de 3-fenilbenzofuran-2(3H)-ona (15) a ’-hidroxienonas, con 2 sustituyentes R en la posición ’ (1). Esta reacción da lugar a 2 enantiómeros R y S que se forman en la posición 3 de la benzofuran-2(3H)-ona (figura 33): Figura 33. Enantiómeros R y S obtenidos para el producto 16. A continuación, en las tablas 3, 4, 5 y 6 se muestran las proporciones de cada enantiómero (determinadas por cromatografía líquida de alta resolución (HPLC)) para los catalizadores C4, C5, C6, C7, C8, C9, C10, C11 y C12 en los dos disolventes (tolueno y diclorometano), para tres temperaturas diferentes (0 ºC, -40 ºC y -78 ºC). Como es lógico, todas las combinaciones posibles no se han llevado a término, sino que se ha ido realizando una discriminación objetiva conforme se iban obteniendo resultados. La reacción se llevó a cabo utilizando un exceso de la hidroxienona en proporción 2:1 y con un 20% de catalizador. La tabla 3 muestra los resultados obtenidos con la ’-hidroxienona 1b que presenta dos metilos en ’ 38 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas # Catalizador Disolvente T, ºC t, h Rdto., % Enant. 1, % Enant. 2, % ee, % 7 Trietilamina CH2Cl2 0 ºC 18 73 - - - 8 C4 68 36 64 29 9 -40 ºC 48 56 37 63 26 10 -78 ºC 72 58 38 62 24 0 ºC 18 64 38 62 24 12 -40 ºC 48 71 35 65 31 13 -78 ºC 72 66 41 59 17 0 ºC 18 72 46 54 8 11 Tolueno 14 C5 CH2Cl2 15 C6 Tolueno 70 43 57 13 16 CH2Cl2 68 42 58 16 17 Tolueno 73 47 53 6 CH2Cl2 66 45 55 9 18 C7 19 -40 ºC 48 57 44 56 11 20 -78 ºC 72 61 44 56 13 0 ºC 18 63 45 55 10 -40 ºC 48 70 44 56 12 0 ºC 18 75 36 64 28 24 -40 ºC 48 59 35 65 31 25 -78 ºC 72 68 35 65 30 0 ºC 18 66 39 61 21 -40 ºC 48 58 34 66 33 0 ºC 18 73 54 46 7 62 50 50 0 21 Tolueno 22 23 C8 26 CH2Cl2 Tolueno 27 28 29 C9 CH2Cl2 Tolueno Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 39 Rdto., % Enant. 1, % Enant. 2, % ee, % CH2Cl2 70 66 34 32 Tolueno 72 61 39 22 CH2Cl2 69 49 51 2 Tolueno 57 49 51 3 CH2Cl2 74 67 33 34 Tolueno 65 59 41 18 # Catalizador Disolvente 30 C10 31 32 C11 33 34 C12 35 T, ºC t, h Tabla 3. Reacción de adición de la benzofuranona 15 (0,1 mmol) sobre la hidroxienona 1b (0,2 mmol) para los catalizadores C4, C5, C6, C7, C8, C9, C10, C11 y C12 (0,02 mmol), en dos disolventes (CH2Cl2 y tolueno), para tres temperaturas diferentes (0 ºC, -40 ºC y -78 ºC). Del mismo modo, en la tabla 4 se muestran las proporciones de cada enantiómero obtenidas con los catalizadores C4 y C8 en diclorometano partiendo del sustrato 1a (R=Bn), que presenta dos grupos bencilo en posición ’. # Catalizador Disolvente T, ºC t, h Rdto., % Enant. 1, % Enant. 2, % ee, % 36 Trietilamina CH2Cl2 0 ºC 18 71 - - - 37 C4 68 36 64 28 38 -40 ºC 48 72 25 75 50 39 -78 ºC 72 74 60 40 20 0 ºC 18 67 48 52 4 -40 ºC 48 61 50 50 1 40 C8 41 Tabla 4. Reacción de adición de la benzofuranona 15 (0,1 mmol) sobre la hidroxienona 1a (0,2 mmol) para los catalizadores C4 y C8 (0,02 mmol), en diclorometano, para tres temperaturas diferentes (0 ºC, -40 ºC y -78 ºC). 40 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas El estudio continua con un nuevo sustituyente en ’: grupo fenilo (sustrato 1c) (R=Ph). Así, en la tabla 5 se muestran los resultados obtenidos con los catalizadores C4 y C8. # Catalizado r Disolvente T, ºC t, h Rdto., % Enant. 1, % Enant. 2, % ee, % 42 Trietilamina CH2Cl2 0 ºC 18 75 - - - 43 C4 65 58 42 16 44 -40 ºC 48 73 54 46 7 45 -78 ºC 72 n.r. - - - 0 ºC 18 69 66 34 32 47 -40 ºC 48 61 66 34 33 48 -78 ºC 72 n.r. - - - 46 C8 Tabla 5. Reacción de adición de la benzofuranona 15 (0,1 mmol) sobre la hidroxienona 1c (0,2 mmol) para los catalizadores C4 y C8 (0,02 mmol), en diclorometano, para tres temperaturas diferentes (0 ºC, -40 ºC y -78 ºC). Por último, se llevó a cabo el mismo procedimiento con el último sustituyente. En la tabla 6 se muestran las proporciones de cada enantiómero para los catalizadores C4 y C8 con el sustrato 1d (R=iPr). Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 41 # Catalizador Disolvente T, ºC t, h Rdto., % Enant. 1, % Enant. 2, % ee, % 49 Trietilamina CH2Cl2 0 ºC 18 68 - - - 50 C4 63 51 49 2 51 -40 ºC 48 70 53 47 6 52 -78 ºC 72 n.r. - - - 0 ºC 18 66 56 44 12 54 -40 ºC 48 72 51 49 2 55 -78 ºC 72 n.r. - - - 53 C8 Tabla 6. Reacción de adición de la benzofuranona 15 (0,1 mmol) sobre la hidroxienona 1d (0,2 mmol) para los catalizadores C4 y C8 (0,02 mmol), en diclorometano, para tres temperaturas diferentes (0 ºC, -40 ºC y -78 ºC). Los resultados obtenidos, en términos de exceso enantiomérico, no son buenos. Como puede apreciarse en las tablas 3-5, los máximos valores obtenidos se sitúan alrededor de un exceso del 30% ee, siendo el mejor resultado el obtenido para el caso de la ’hidroxienona con sustituyentes bencilo (1a), utilizando el catalizador C4 a -40 ºC en diclorometano (tabla 3, entrada 38). Desafortunadamente, este resultado no pudo ser mejorado bajando la temperatura. El catalizador que ha dado lugar a una mejor enantioselectividad ha sido la escuaramida49,50 C4 con un 50% ee para el caso del sustrato dibencilado (1a). Este resultado concuerda con los muy altos niveles de estereoselección obtenidos con dicho catalizador en las pruebas preliminares de adición de Michael sobre ’-hidroxienonas con cianoderivados (9) (tabla 2, entrada 6). 49 (a) J. P. Malerich; K. Hagihara; V. R. Rawal, J. Am. Chem. Soc. 2008, 130, 14416–14417; (b) Y. Zhu, J. P. Malerich, V. H. Rawal, Angew. Chem. Int. Ed. 2010, 49, 153–156. Para revisions de catálisis con escuaramidas, ver: (c) R. I. Storer, C. Aciro, L. H. Jones, Chem. Soc. Rev. 2011, 40, 2330–2346. (d) J. Alemán, A. Parra, H. Jiang, K. A. Jørgensen, Chem. Eur. J. 2011, 17, 6890–6899. 50 W. Yang, D.-M. Du, Org. Lett. 2010, 12, 5450–5453. 42 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Los bajos resultados obtenidos en el caso de la ’-hidroxienona (1) con la benzofuran2(3H)-ona (9) obligaron a buscar nuevos catalizadores que se coordinaran mejor con las estructuras de estos sustratos. Así, se investigaron otros 8 catalizadores (C5-C12). Cabe señalar que los catalizadores C1, C2 y C3 que habían dado resultados razonablemente buenos en la adición del cianoderivado (2) sobre la ’-hidroxienona 1b (tabla 1, pruebas preliminares), también fueron estudiados en esta adición que nos incumbe dando igualmente pobres resultados (pruebas realizadas paralelamente en Departamento de Química Aplicada de la Universidad de Navarra). En cuanto al sustrato utilizado en esta adición de Michael, la ’-hidroxienona (1), señalar que el mejor resultado se obtiene en el caso del dibencilado 1a, con un exceso enantiomérico del 50% a -40 ºC con el catalizador C4 (tabla 3, entrada 38). Los valores de exceso enantiomérico se mantienen alrededor del 30% con los sustituyentes metilo y fenilo, y disminuyen mucho para el caso del isopropilo. Como era de esperar, la estructura del sustrato influye en el resultado de la estereoselectividad. Los pobres valores alcanzados en el caso del metilo, nos llevaron a probar otros sustituyentes, aunque solamente se consiguió mejorar en el caso del bencilo (1b). En vista de los resultados obtenidos, y dado que la duración de mi periodo de investigación en el laboratorio limita el número de experimentos realizados, se propone continuar el estudio de esta reacción incidiendo en los siguientes aspectos: a) Estructura de los sustratos: i. Modificar la estructura de la ’-hidroxienona (1) introduciendo diferentes sustituyentes en ’. ii. Modificar el grupo hidroxilo derivatizandolo con grupos sililo. iii. Introducir nuevos sustituyentes en la posición 3 de la benzofuranona. b) Catalizadores. Probar nuevos catalizadores que permitan una interacción más efectiva con los sustratos. c) Condiciones de reacción. Probar más disolventes, diferentes proporciones de hidroxienona/benzofuranona, o variar la cantidad de catalizador utilizada. Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 43 44 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 4. Conclusiones a) Se ha estudiado la reacción de adición de Michael de benzofuran-2(3H)-onas a ’-hidroxienonas para diferentes sustratos, utilizando varios catalizadores que actúan como base de Brønsted. Se han obtenido excesos enantioméricos alrededor del 30% ee para hidroxienonas con sustituyentes metilo y fenilo, y del 50% ee para el caso de sustituyentes bencilo. b) En este trabajo no se han podido repetir los buenos resultados obtenidos simultáneamente en nuestro grupo de investigación con los mismos o similares catalizadores en otras adiciones sobre ’-hidroxienonas. c) Se hace necesario seguir buscando otras estructuras de los sustratos que den lugar a mejores resultados, y también un catalizador que se ajuste mejor a la estructura de la ’-hidroxienona utilizada en este caso y permita establecer interacciones catalizador-sustrato que dirijan mejor el resultado estereoquímico de la adición. Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 45 46 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 5. Parte experimental 5.1 Técnicas experimentales 5.1.1 Resonancia Magnética Nuclear (RMN) Los espectros de 1H-RMN y 13C-RMN Avance III 400 MHz (125 MHz para fueron registrados en el espectrómetro Bruker 13C). El disolvente empleado fue cloroformo deuterado (CDCl3) salvo que se especifique lo contrario. Los valores de los desplazamientos químicos se expresan en unidades δ (ppm) respecto a la señal interna del CHCl3 residual (δ = 7,26 ppm para 1H y δ = 77,0 para 13C). 5.1.2 Cromatografía El seguimiento de las reacciones y de las cromatografías en columna se efectuó por Cromatografía en Capa Fina (TLC) utilizando gel de sílice soportado sobre placas de aluminio (Panreac 60-200 UV). Fueron reveladas bajo luz ultravioleta de 254 nm o por calefacción tras contacto con una solución reveladora de p-anisaldehído y ácido sulfúrico en etanol. La purificación de productos mediante cromatografía de columna se realizó bajo presión, empleando gel de sílice Merck 230-400 mesh (0,040-0,063 mm) como fase estacionaria y mezclas de hexano/acetato de etilo en proporciones adecuadas como eluyente. Para la medida del exceso enantiomérico y de la relación diastereomérica se hizo uso de la Cromatografía Líquida de Alta Resolución (HPLC) de forma analítica. Se empleó el cromatógrafo Jasco MD2010 equipado con detector UV de haz de diodos (DAD) y monocromador. Las columnas de fase estacionaria quiral fueron la IA y AD3 de la marca Daicel Chiralpak. Para la fase móvil se usaron disolventes (hexano e isopropanol) de pureza válida para HPLC. Las muestras se prepararon disolviendo 2,0 mg de compuesto en 2 mL de isopropanol y se filtraron previamente a la inyección con filtros de 0,20 μm de poro. Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 47 5.1.3 Reactivos y disolventes Los reactivos comerciales empleados (Aldrich, Merck, Sigma) fueron utilizados sin purificación previa, salvo que se indique lo contrario. Los reactivos se almacenaron según las especificaciones de la casa comercial. Los disolventes empleados como eluyentes en cromatografía en columna (hexano y acetato de etilo) fueron purificados por destilación. Tanto el THF como el éter anhidro fueron obtenidos haciéndolos pasar a través de columnas de alúmina activada. 5.1.4 Varios Las microdestilaciones fueron llevadas a cabo en un destilador de bolas Kugelröhr Büchi B-580. En todas las reacciones fue empleada agitación magnética. Las reacciones se llevaron a cabo en atmósfera inerte, por tanto fueron preparadas con material de vidrio previamente flameado y en atmósfera de nitrógeno. 48 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 5.2 Reacciones Preparación del (2-nitropropil)benceno (2) En un matraz previamente secado y bajo atmósfera de nitrógeno se introdujo NaBH4 (2g, 52 mmol), 40 mL de dioxano y 12,5 mL de etanol, calentándose hasta una temperatura estable de 30 ºC. Se preparó una mezcla del nitroalqueno (3,92 g, 24 mmol) y 40 mL y de dioxano y se fueron añadiendo gota a gota durante 30 minutos. Pasado este tiempo, la mezcla se diluyó con agua (50 mL) y se enfrió en baño de hielo y agua. El exceso de NaBH4 se eliminó con ácido acético (50%) hasta que desaparezca la efervescencia. Por medio de rotavapor se evaporó el disolvente y a continuación se llevó a cabo una extracción por triplicado añadiendo dicolorometano. La fase orgánica se lavó con agua y NaCl y fue secada con sulfato de magnesio. Volvió a ser introducida en el rotavapor y finalmente el producto fue destilado a vacío (180 ºC y 0,6 mbar), obteniéndose un líquido amarillo con un rendimiento del 75% (2,99 g). 1H NMR (400 MHz, CDCl3), δ (ppm): 7,25–7,40 (m, 5H), 4,71-4,88 (sx, 1H), 2,98-3,40 (m, 2H), 1,59 (d, 3H). Preparación del 1-metoxipropa-1,2-dieno (5) En un matraz previamente secado y bajo atmósfera de nitrógeno se introdujo 3metoxiprop-1-ino (4) (18,18 g, 259,4 mmol) y se fue añadiendo gota a gota durante 40 minutos tert-butóxido de potasio (2,91 g, 25,94 mmol). Durante la adición, el matraz permaneció conectado a un equipo de destilación y sobre una placa calefactora. Cuando los vapores que emanan del matraz alcanzan una temperatura entre 43 y 51ºC, la mezcla del matraz comienza a destilar el producto puro (5), que se recoge en un segundo matraz introducido en baño de hielo. La destilación dura 1 hora y media aproximadamente. Se obtuvo un rendimiento del 49% (8,93 g). 1H NMR (400 MHz, CDCl3), δ (ppm): 6,75 (t, 1H), 5,48 (d, 2H), 3,41 (s, 3H). Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 49 Preparación de la 4-bencil-4-hidroxi-5-fenilpent-1-en-3-ona (1a) En un matraz de 500 mL previamente secado y bajo atmósfera de nitrógeno, con embudo de adición previamente flameado, se depositó 1-metoxipropa-1,2-dieno (5) (100 mmol, 7 g) y se disolvieron con 180 mL de éter etílico seco. La mezcla se enfrío hasta -40 ºC en baño termostático. Se añadieron 44 mL de una disolución 2,5 M de butillitio disuelto en hexano (110 mmol) gota a gota durante 35 minutos, tomando la mezcla un color amarillento. Tras la adición, se tuvo 10 minutos a -40 ºC para obtener 1-metoxipropa-1,2-dien-1-ilo de litio (6). A continuación se añadió 21,62 mL de 1,3difenilpropan-2-ona seca (110 mmol) disuelta en 90 mL de éter etílico seco. La adición fue gota a gota durante 15 minutos. La mezcla se dejó a -40 ºC durante 30 minutos y fue adquiriendo un color lechoso y una textura espesa como resultado de la formación de 2-benzil-3-metoxi-1-fenilpenta-3,4-dien-2-ol (7a). Se añadieron 200 mL de agua y se retiró del baño termostático hasta alcanzar temperatura ambiente. En un embudo de decantación se llevaron a cabo 3 extracciones con 120 mL de éter cada una, se secó la fase orgánica con sulfato sódico y se evaporó el disolvente en el rotavapor obteniéndose un aceite amarillento (25,3 g). Este producto crudo se añadió gota a gota sobre una disolución de H2SO4 (5%) y se mantuvo a temperatura ambiente durante 10 horas. La mezcla fue saturada con NaCl sólido y a continuación se procedió a extraer el producto en un embudo de extracción usando 5 porciones de 100 mL de éter etílico. La fase orgánica resultante se lavó con una disolución saturada de NaCl, se secó con sulfato sódico, fue filtrada y finalmente se evaporó el disolvente en rotavapor, obteniéndose un aceite amarillo (24 g). Este crudo fue purificado por destilación a alto vacío con trampa fría en nitrógeno líquido. Se recogieron 22,2 g del producto final (1a), dando un rendimiento del 76%. 1H NMR (400 MHz, CDCl3), δ (ppm): 7,12-7,28 (m, 10H), 6,83-6,98 (m, 2H), 6,32 (d, 1H), 5,31 (s, 1H), 2,25 (s, 2H). 50 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Obtención del producto 2-bencil-2-hidroxi-6-metil-6-nitro-1,7- difenilheptan-3-ona (3) En un vial previamente secado y bajo atmósfera de nitrógeno se introdujeron 0,0266 g (0,1 mmol) de 4-bencil-4-hidroxi-5-fenilpent-1-en-3-ona (1a), 0,0824 g (0,5 mmol) de (2-nitropropil)benceno (2), 0,02 mmol del catalizador correspondiente a cada reacción (ver tabla 1) y 0,4 mL de cloroformo. Se cerró el vial y se dejó en agitación a la temperatura y durante el tiempo indicado para cada experimento en la tabla 1. Al finalizar el tiempo de reacción, comprobando siempre el avance de la reacción con cromatografía en capa fina, se detuvo la reacción añadiendo unas gotas de HCl 1 N. Seguidamente se añadió diclorometano y se lavó la fase orgánica añadiendo HCl 1 N. Este proceso se repitió 3 veces. La fase orgánica se secó con sulfato de magnesio y el disolvente se eliminó en el rotavapor. Se procedió a la purificación mediante cromatografía de columna (Hex/AcEt 10/1). 1H NMR (400 MHz, CDCl3), δ (ppm): 7,21–7,38 (m, 15H), 4,78 (s, 1H), 3,01-3,38 (q, 6 H), 1,55 (s, 3H). Preparación de la 2-hidroxi-2-metil-7-fenilhept-4-en-3-ona (8) En un matraz de 3 bocas previamente secado y bajo atmósfera de nitrógeno conectado a un embudo de adición con n-butillitio (disolución 2,5 M en hexano, 80 mL, 200 mmol), y se añadió THF seco (200 mL) y diisopropilamina (28 mL, 200 mmol). La disolución fue enfriada a -78 ºC y se fue dejando caer gota a gota el n-butil-litio. Después de la adición, la mezcla permaneció en agitación a -78 ºC durante una hora antes de añadir gota a gota 3-hidroxi-3-metil-2-butanona (11) (8,8 mL, 80 mmol). Tras completar la adición, se añadió a la mezcla gota a gota el correspondiente aldehído (Ph(CH2)2CHO) (240 mmol, 3 equiv.) en THF (30 mL). Permaneció -78 ºC con Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 51 agitación durante una hora y se siguió el avance de la reacción por cromatografía en capa fina. Después de 3 horas, la reacción se paró con NH4Cl saturado y a continuación la fase orgánica fue extraída con diclorometano. Las fases orgánicas se juntaron, se secaron con sulfato de magnesio y se concentraron. El producto crudo fue purificado por columna cromatográfica. Se mezcló CeCl3 · 7 H2O (1,5 mmol por cada 1 mmol del sustrato aldólico) y NaI (1,5 mmol por cada 1 mmol del sustrato aldólico), se añadieron sucesivamente al producto (12), en CH3CN (10 mL/mmol) y la mezcla estuvo a reflujo y con agitación durante 3 horas. Después de enfriarse a temperatura ambiente, la reacción se diluyó con éter (200 mL) y se lavó con HCl 0,5 M (50 mL). Esta fase orgánica fue extraída con EtOAc (3 x 100 mL) y las extracciones de EtOAc se juntaron. Posteriormente, se lavó con NaHSO3 al 20% (30 mL), NaHCO3 (30 mL) y disolución saturada de NaCl (30 mL), y se secó con sulfato de magnesio. Se evaporó el disolvente a baja presión en rotavapor para dar 7,7 g del producto (8) (Rdto.: 42%), que fue purificado en columna cromatográfica (Hex/AcEt 9:1). 1H NMR (500 MHz, CDCl3), δ (ppm): 7,28-7,21 (m, 6H), 6.41 (d,1H, J=15,5 Hz), 3,98 (s, 1H), 2,8 (t, 2H, J=8Hz), 2,59 ( m, 2H), 1,34 (s, 6H); 13C NMR (125 MHz, CDCl3), δ (ppm): 202,3, 149,4, 140,6, 128,6, 128,5, 126,3, 123,1, 75,3, 34,5, 34,3, 26,3. Preparación del 2-ciano-2-fenilacetato de tert-butilo (9) Una disolución del cianoderivado (13) (10 mmol) disuelta en 10 mL de THF se añadió gota a gota a una disolución de LDA (25 mmol) disuelta en 40 mL de THF a -78 ºC. La mezcla se mantuvo en agitación a -78 ºC durante 60 minutos y a continuación se dejó otros 60 minutos a temperatura ambiente. De nuevo, la mezcla se enfrió a -78 ºC y se añadió una disolución de dicarbonato de di-tert-butilo (12 mmol) en 10 mL de THF con una jeringuilla. Después de permanecer en agitación a -78 ºC durante 5 horas, la reacción fue parada con cloruro de amonio saturado y se extrajo con dietil éter. El éter se lavó con HCl al 10% y disolución saturada de NaCl, y se secó con sulfato de magnesio. El disolvente se eliminó a baja presión en rotavapor y el crudo resultante se purificó en columna cromatográfica (Hex/AcEt 20:1) para dar 2,11 g del producto (9) (Rdto.: 81%). 1H NMR (400 MHz, CDCl3), δ (ppm): 7,53–7,29 (m, 5H), 4,61 (s, 1H), 1,44 (s, 9H). 52 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Obtención del producto 2-ciano-6-hidroxi-6-metil-5-oxo-2-fenil-3-(2- feniletil)-heptanoato de tert-butilo (10) En un vial previamente secado y bajo atmósfera de nitrógeno se introdujo una mezcla de 2-hidroxi-2-metil-7-fenil-hept-4-en-3-ona (8) (65,5 mg, 0,3 mmol), 2-ciano-2fenilacetato de tert-butilo (9) (21,7 mg, 0,1 mmol) y el catalizador C4 (6,31 mg, 0,01 mmol) en DCE anhidro (0,4 mL). Fue calentado hasta 50 ºC y se dejó en agitación durante 18 h. Después de diluir en diclorometano, la fase orgánica se lavó 2 veces con HCl 1 N y secado con sulfato de sodio y el disolvente se eliminó en el rotavapor. Se procedió a la purificación mediante cromatografía de columna (Hex/AcEt 20:1) para dar 39 mg del producto (10) (Rdto.: 90%). 1H NMR (400 MHz, CDCl3), δ (ppm): 7,56–7,54 (m, 2H), 7,42–7,36 (m, 3H), 7,20–7,10 (m, 3H), 6,92–6,90 (m, 2H), 3,60 (brs, 1H), 3,40–3,35 (m, 1H), 2,95 (dd, J = 18.8 Hz y 7.6 Hz, 1H), 2,83 (dd, J = 18,8 Hz y 2,6 Hz, 1H), 2,45–2,38 (m, 1H), 2,21–2,12 (m, 1H), 1,60–1,46 (m, 2H), 1,43 (s, 3H), 1,41 (s, 3H), 1,37 (s, 9H); 13C NMR (100 MHz, CDCl3) δ (ppm): 211,6, 166,0, 141,12, 133,4, 129,1, 129,0, 128,3, 128,1, 126,4, 125,9, 117,7, 84,7, 76,4, 60,7, 39,5, 39,2, 33,7, 33,4, 27,4, 26,8, 26,7. Preparación de la 4-hidroxi-4-metilpent-1-en-3-ona (1b) Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 53 En un matraz de 500 mL previamente secado y bajo atmósfera de nitrógeno, con embudo de adición previamente flameado, se depositó 1-metoxipropa-1,2-dieno (5) (100 mmol, 7 g) y se disolvieron con 180 mL de éter etílico seco. La mezcla se enfrío hasta -40 ºC en baño termostático. Se añadieron 44 mL de una disolución 2,5 M de butillitio disuelto en hexano (110 mmol) gota a gota durante 35 minutos, tomando la mezcla un color amarillento. Tras la adición, se tuvo 10 minutos a -40 ºC para obtener 1-metoxipropa-1,2-dien-1-ilo de litio (6). A continuación se añadió 8,08 mL de acetona seca (110 mmol) disuelta en 90 mL de éter etílico seco. La adición fue gota a gota y durante 15 minutos. La mezcla se dejó a -40 ºC durante 30 minutos y fue adquiriendo un color lechoso y una textura espesa como resultado de la formación de 3-metoxi-2metilpenta-3,4-dien-2-ol (7b). Se añadieron 200 mL de agua y se retiró del baño termostático hasta alcanzar temperatura ambiente. En un embudo de decantación se llevaron a cabo 3 extracciones con 120 mL de éter cada una, se secó la fase orgánica con sulfato sódico y se evaporó el disolvente en el rotavapor obteniéndose un aceite amarillento (13,3 g). Este producto crudo se añadió gota a gota sobre una disolución de H2SO4 (5%) y se enfrió a 0 ºC durante una hora y media. Pasado este tiempo, se llevó a temperatura ambiente. Una vez atemperado, la mezcla fue saturada con NaCl sólido y a continuación se procedió a extraer el producto en un embudo de extracción usando 5 porciones de 100 mL de éter etílico. La fase orgánica resultante se lavó con una disolución saturada de NaCl, se secó con sulfato sódico, fue filtrada y finalmente se evaporó el disolvente en rotavapor, obteniéndose un aceite amarillo (12,3 g). Este crudo fue purificado por destilación a alto vacío con trampa fría en nitrógeno líquido. Se recogieron 11,1 g del producto final (1b), dando un rendimiento del 97%. 1H NMR (400 MHz, CDCl3), δ (ppm): 7,05-7,39 (m, 9H), 3,46 (s, 4H), 1,51 (s, 6H). Preparación de la 1-hidroxi-1,1-difenilbut-3-en-2-ona (1c) 54 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas En un matraz de 500 mL previamente secado y bajo atmósfera de nitrógeno, con embudo de adición previamente flameado, se depositó 1-metoxipropa-1,2-dieno (5) (100 mmol, 7 g) y se disolvieron con 180 mL de éter etílico seco. La mezcla se enfrío hasta -40 ºC en baño termostático. Se añadieron 44 mL de una disolución 2,5 M de butillitio disuelto en hexano (110 mmol) gota a gota durante 35 minutos, tomando la mezcla un color amarillento. Tras la adición, se tuvo 10 minutos a -40 ºC para obtener 1-metoxipropa-1,2-dien-1-ilo de litio (6). A continuación se añadió 18,05 mL de benzofenona seca (110 mmol) disuelta en 90 mL de éter etílico seco. La adición fue gota a gota durante 15 minutos. La mezcla se dejó a -40 ºC durante 30 minutos y fue adquiriendo un color lechoso y una textura espesa como resultado de la formación de 2metoxi-1,1-difenilbuta-2,3-dien-1-ol (7c). Se añadieron 200 mL de agua y se retiró del baño termostático hasta alcanzar temperatura ambiente. En un embudo de decantación se llevaron a cabo 3 extracciones con 120 mL de éter cada una, se secó con sulfato sódico y se evaporó el disolvente en el rotavapor obteniéndose un aceite amarillento (22,9 g). Este producto crudo se añadió gota a gota sobre una disolución de H 2SO4 (5%) y se enfrió a 0 ºC durante una hora y media. Pasado este tiempo, se llevó a temperatura ambiente. Una vez atemperado, la mezcla fue saturada con NaCl sólido y a continuación se procedió a extraer el producto en un embudo de extracción usando 5 porciones de 100 mL de éter etílico. La fase orgánica resultante se lavó con una disolución saturada de NaCl, se secó con sulfato sódico, fue filtrada y finalmente se evaporó el disolvente en rotavapor, obteniéndose un aceite amarillo (21,2 g). Este crudo fue purificado por destilación a alto vacío con trampa fría en nitrógeno líquido. Se recogieron 19,1 g del producto final (1c), dando un rendimiento del 72%. 1H NMR (400 MHz, CDCl3), δ (ppm): 7,2-7,4 (m, 10H), 6,4-6,7 (m, 2H), 5,7 (d, 1H). Preparación de la 4-hidroxi-4-isobutil-6-metilhept-1-en-3-ona (1d) Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 55 En un matraz de 500 mL previamente secado y bajo atmósfera de nitrógeno, con embudo de adición previamente flameado, se depositó 1-metoxipropa-1,2-dieno (5) (100 mmol, 7 g) y se disolvieron con 180 mL de éter etílico seco. La mezcla se enfrío hasta -40 ºC en baño termostático. Se añadieron 44 mL de una disolución 2,5 M de butillitio disuelto en hexano (110 mmol) gota a gota durante 35 minutos, tomando la mezcla un color amarillento. Tras la adición, se tuvo 10 minutos a -40 ºC para obtener 1-metoxipropa-1,2-dien-1-ilo de litio (6). A continuación se añadió 19,40 mL de 2,6dimetilheptan-4-ona seca (110 mmol) disuelta en 90 mL de éter etílico seco. La adición fue gota a gota durante 15 minutos. La mezcla se dejó a -40 ºC durante 30 minutos y fue adquiriendo un color lechoso y una textura espesa como resultado de la formación de 4-isobutil-3-metoxi-6-metilhepta-1,2-dien-4-ol (7d). Se añadieron 200 mL de agua y se retiró del baño termostático hasta alcanzar temperatura ambiente. En un embudo de decantación se llevaron a cabo 3 extracciones con 120 mL de éter cada una, se secó con sulfato sódico y se evaporó el disolvente en el rotavapor obteniéndose un aceite amarillento (21,4 g). Este producto crudo se añadió gota a gota sobre una disolución de H2SO4 (5%) y se enfrió a 0 ºC durante una hora y media. Pasado este tiempo, se llevó a temperatura ambiente. Una vez atemperado, la mezcla fue saturada con NaCl sólido y a continuación se procedió a extraer el producto en un embudo de extracción usando 5 porciones de 100 mL de éter etílico. La fase orgánica resultante se lavó con una disolución saturada de NaCl, se secó con sulfato sódico, fue filtrada y finalmente se evaporó el disolvente en rotavapor, obteniéndose un aceite amarillo (20 g). Este crudo fue purificado por destilación a alto vacío con trampa fría en nitrógeno líquido. Se recogieron 18,3 g del producto final (1d), dando un rendimiento del 84%. 1H NMR (400 MHz, CDCl3), δ (ppm): 6,23-6,47 (m, 2H), 6,12-6,22 (d, 1H), 3,78 (s, 1H), 1,56 (q, 2H), 0,80 (d, 12H); 13C NMR (100 MHz, CDCl3) δ (ppm): 204,2, 133,6, 118,7, 115,4, 54,6, 33,0. Preparación de la 3-fenilbenzofuran-2(3H)-ona (15) 56 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas La 3-fenilbenzofuran-2(3H)-ona (2c) fue preparada usando una modificación del procedimiento de Bistrzycki y Flatau51. Se mezcló fenol finamente pulverizado (26,5 g, 0,28 mol) con ácido mandélico (30,45 g, 0,20 mol) y se enfrió en hielo. A esta mezcla se añadieron 80 mL de una disolución de ácido sulfúrico al 70%. La mezcla anterior fue agitada a 0 ºC hasta que toda la mezcla se disolvió y entonces calentada hasta 115ºC durante 45 minutos. La mezcla fue enfriada hasta temperatura ambiente, y fue vertida sobre 400 mL de una mezcla agua-hielo, y extraída con 3 porciones de 100 mL de cloruro de metileno. Se juntaron las fases orgánicas y se lavaron con 100 mL de una disolución saturada de bicarbonato de sodio. A continuación se secó con sulfato de magnesio. Al evaporar el disolvente a baja presión en rotavapor, se obtuvieron cristales blancos que fueron recristalizados con etanol al 95% para dar 11,1 g (26%) de 3fenilbenzofuran-2(3H)-ona (15). 1H NMR (400 MHz, CDCl3), δ (ppm): 7,14-7,41 (m, 9H), 4,88 (s, 1H); 13C NMR (100 MHz, CDCl3) δ (ppm): 176,3, 154,7, 135,2, 123,8-129,6, 111,0. Obtención de la 3-(4-hidroxi-4-metil-3-oxopentil)-3-fenilbenzofuran- 2(3H)-ona (16b) En un vial previamente secado y bajo atmósfera de nitrógeno se introdujeron 22,8 mg (0,2 mmol) de 4-hidroxi-4-metilpent-1-en-3-ona (1b), 21 mg (0,1 mmol) de 3fenilbenzofuran-2(3H)-ona (15), 0,02 mmol del catalizador correspondiente a cada reacción y 0,4 mL del disolvente, dependiendo de la reacción: diclorometano o tolueno (ver tabla 3). Se cerró el vial y se dejó en agitación a la temperatura y durante el tiempo indicado para cada experimento en la tabla 3. Al finalizar el tiempo de reacción, comprobando siempre el avance de la reacción con cromatografía en capa fina, se detuvo la reacción añadiendo unas gotas de HCl 1 N. Seguidamente se añadió diclorometano y se lavó añadiendo HCl 1 N. Este proceso se repitió 3 veces. La fase orgánica se secó con sulfato de magnesio y el disolvente se eliminó en el rotavapor. Se procedió a la purificación mediante cromatografía de columna (Hex/AcEt 10:1). 51 A. Bistrzycki, J. Flatau, Chem. Ber., 1895, 28, 989. Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 57 1H NMR (400 MHz, CDCl3), δ (ppm): 7,12-7,38 (m, 9H), 3,40 (s, 1H), 2,36 (s, 4H), 1,15 (s, 6H); 13C NMR (100 MHz, CDCl3) δ (ppm): 213,0, 181,5, 157,3, 144,8, 131,4-136,2, 118,1, 96,3, 66,2, 45,0, 44,2, 38,0. Obtención de la 3-(4-bencil-4-hidroxi-3-oxo-5-fenilpentil)-3- fenilbenzofuran-2(3H)-ona (1a) En un vial previamente secado y bajo atmósfera de nitrógeno se introdujeron 53,2 mg (0,2 mmol) de 4-bencil-4-hidroxi-5-fenilpent-1-en-3-ona (1a), 21 mg (0,1 mmol) de 3fenilbenzofuran-2(3H)-ona (15), 0,02 mmol del experimento correspondiente a cada reacción y 0,4 mL de diclorometano. Se cerró el vial y se dejó en agitación a la temperatura y durante el tiempo indicado para cada catalizador en la tabla 4. Al finalizar el tiempo de reacción, comprobando siempre el avance de la reacción con cromatografía en capa fina, se detuvo la reacción añadiendo unas gotas de HCl 1 N. Seguidamente se añadió diclorometano y se lavó añadiendo HCl 1 N. Este proceso se repitió 3 veces. La fase orgánica se secó con sulfato de magnesio y el disolvente se eliminó en el rotavapor. Se procedió a la purificación mediante cromatografía de columna (Hex/AcEt 20:1). 1H NMR (400 MHz, CDCl3), δ (ppm): 6,95 (m, 19H), 5,22 (s, 1H), 2,70 (s, 4H), 1,97 (s, 4H); 13C NMR (100 MHz, CDCl3) δ (ppm): 213,2, 178,3, 154,1, 138,4, 135,0, 125,1-131,7, 112,6, 83,4, 55,4, 45,3, 34,6, 32,8. Obtención de la 3-(4-hidroxi-3-oxo-4,4-difenilbutil)-3-fenilbenzofuran2(3H)-ona (1c) 58 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas En un vial previamente secado y bajo atmósfera de nitrógeno se introdujeron 47,6 mg (0,2 mmol) de 1-hidroxi-1,1-difenilbut-3-en-2-ona (1c), 21 mg (0,1 mmol) de 3fenilbenzofuran-2(3H)-ona (15), 0,02 mmol del catalizador correspondiente a cada reacción y 0,4 mL de diclorometano. Se cerró el vial y se dejó en agitación a la temperatura y durante el tiempo indicado para cada experimento en la tabla 5. Al finalizar el tiempo de reacción, comprobando siempre el avance de la reacción con cromatografía en capa fina, se detuvo la reacción añadiendo unas gotas de HCl 1 N. Seguidamente se añadió diclorometano y se lavó añadiendo HCl 1 N. Este proceso se repitió 3 veces. La fase orgánica se secó con sulfato de magnesio y el disolvente se eliminó en el rotavapor. Se procedió a la purificación mediante cromatografía de columna (Hex/AcEt 20:1). 1H NMR (400 MHz, CDCl3), δ (ppm): 7,60-7,73 (m, 10H), 6,44 (s, 1H), 6,00-6,29 (m, 2H), 5,53 (d, 1H); 13C NMR (100 MHz, CDCl3) δ (ppm): 210,2, 152,4, 140,5, 138,4, 123,3-128,7, 110,5. Obtención de la 3-(4-hidroxi-3-oxo-4,4-difenilbutil)-3-fenilbenzofuran2(3H)-ona (1d) En un vial previamente secado y bajo atmósfera de nitrógeno se introdujeron 39,66 mg (0,2 mmol) de 4-hidroxi-4-isobutil-6-metilhept-1-en-3-ona (1d), 21 mg (0,1 mmol) de 3-fenilbenzofuran-2(3H)-ona (15), 0,02 mmol del catalizador correspondiente a cada reacción y 0,4 mL de diclorometano. Se cerró el vial y se dejó en agitación a la temperatura y durante el tiempo indicado para cada experimento en la tabla 6. Al finalizar el tiempo de reacción, comprobando siempre el avance de la reacción con cromatografía en capa fina, se detuvo la reacción añadiendo unas gotas de HCl 1 N. Seguidamente se añadió diclorometano y se lavó añadiendo HCl 1 N. Este proceso se repitió 3 veces. La fase orgánica se secó con sulfato de magnesio y el disolvente se eliminó en el rotavapor. Se procedió a la purificación mediante cromatografía de columna (Hex/AcEt 10:1). 1H NMR (400 MHz, CDCl3), δ (ppm): 7,23-7,36 (m, 7H), 7,23-7,36 (d, 2H), 4,78 (s, 1H), 2,47 (s, 4H), 2,84-2,96 (sp, 2H), 0,76 (d, 12H). Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 59 5.3 Selección de espectros de RMN 60 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 61 62 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 63 64 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 65 66 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 5.4 Selección de cromatogramas de HPLC PDA 207.0 nm, Daicel Chiralpak IA 90 : 10 hex : ipr, f: 0.5 mL/min Processed Channel Descr.: PDA 207.0 nm #1 - Racémico Tiempo de retención 23.739 27.375 Area 10756910 10797867 % Área 49.90 50.10 0.30 Altura 336005 289730 27.375 23.739 1 2 AU 0.20 0.10 0.00 22.00 23.00 24.00 25.00 26.00 Minutes 27.00 28.00 29.00 #2 1 2 Tiempo de retención 23.530 27.127 Área 27646086 4676530 % Área 85.53 14.47 Altura 853390 126572 23.530 1.20 1.00 AU 0.80 0.60 27.127 0.40 0.20 0.00 16.00 18.00 20.00 22.00 24.00 26.00 28.00 Minutes 30.00 32.00 34.00 36.00 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 67 #3 1 2 Tiempo de retención 23.576 27.202 % Área 87.27 12.73 Altura 1041331 135569 23.576 1.40 Área 34560100 5039040 1.20 AU 1.00 0.80 27.202 0.60 0.40 0.20 0.00 21.00 22.00 23.00 24.00 25.00 26.00 Minutes 27.00 28.00 29.00 30.00 #4 1 2 Tiempo de retención 23.533 27.057 Área 58022913 13545446 % Área 81.07 18.93 Altura 1503154 376361 23.533 2.00 1.80 1.60 1.40 AU 1.20 1.00 27.057 0.80 0.60 0.40 0.20 0.00 23.00 24.00 25.00 26.00 27.00 28.00 Minutes 68 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas PDA 204.5 nm, Daicel Chiralpak AD3 90 : 10 hex : ipr, f: 0.5 mL/min Processed Channel Descr.: PDA 209.0 nm #5 - Racémico Tiempo de retención 22.714 23.544 24.485 26.024 1 2 3 4 Área 24775154 25841902 46819083 47133939 1.60 AU 1.00 23.544 22.714 1.40 1.20 Altura 926316 917248 1434780 1301938 26.024 24.485 1.80 % Área 17.14 17.88 32.39 32.60 0.80 0.60 0.40 0.20 0.00 20.00 22.00 24.00 26.00 28.00 30.00 Minutes #6 1 2 3 4 Tiempo de retención 21.886 22.607 23.481 24.758 Área 210126 1304069 1503226 61188267 % Área 0.33 2.03 2.34 95.30 Altura 9214 55276 56833 1669002 24.758 2.20 2.00 1.80 1.60 AU 1.40 1.20 1.00 0.80 0.00 20.00 22.00 23.481 0.20 22.607 0.40 21.886 0.60 24.00 26.00 28.00 30.00 Minutes Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 69 PDA 204.5 nm, Daicel Chiralpak IA 90 : 10 hex : ipr, f: 0.5 mL/min Processed Channel Descr.: PDA 207.0 nm #7 - Racémico 1 2 Tiempo de retención 30.675 35.080 Altura 1411994 1144307 35.080 1.50 AU % Área 50.85 49.15 30.675 2.00 Área 67728175 65452517 1.00 0.50 0.00 28.00 30.00 32.00 34.00 Minutes 36.00 38.00 40.00 #8 1 2 Tiempo de retención 23.810 27.267 Área 14516787 26099931 % Área 35.74 64.26 Altura 408127 609636 27.267 1.00 23.810 AU 0.80 0.60 0.40 0.20 0.00 20.00 70 22.00 24.00 26.00 Minutes 28.00 30.00 32.00 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas #9 1 2 Tiempo de retención 20.819 23.532 Área 32333094 54781580 20.819 AU 1.50 Altura 1020424 1378871 23.532 2.00 % Área 37.12 62.88 1.00 0.50 0.00 18.00 20.00 22.00 Minutes 24.00 26.00 % Área 38.03 61.97 Altura 1255509 1607305 28.00 #10 1 2 Tiempo de retención 26.176 29.756 Área 52912412 86226259 26.176 AU 2.00 29.756 2.50 1.50 1.00 0.50 0.00 24.00 26.00 28.00 Minutes 30.00 32.00 #11 Tiempo de retención 23.386 26.714 Área 23580906 38532012 % Área 37.96 62.04 23.386 1.20 1.00 AU 0.80 Altura 656373 877561 26.714 1 2 0.60 0.40 0.20 0.00 22.00 24.00 26.00 28.00 30.00 Minutes Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 71 #12 Tiempo de retención 25.962 29.408 Área 42457354 79957075 % Área 34.68 65.32 25.962 2.00 AU 1.50 Altura 1093874 1549215 29.408 1 2 1.00 0.50 0.00 24.00 26.00 28.00 Minutes 30.00 32.00 34.00 #13 25.272 2.50 2.00 AU Área 72406777 102827044 % Área 41.32 58.68 Altura 1688009 1797040 28.557 Tiempo de retención 25.272 28.557 1 2 1.50 1.00 0.50 0.00 24.00 26.00 28.00 Minutes 30.00 32.00 #14 1 2 Tiempo de retención 29.658 34.064 Área 46019079 54540811 % Área 45.76 54.24 Altura 965191 950402 AU 1.00 34.064 29.658 1.50 0.50 0.00 28.00 72 29.00 30.00 31.00 32.00 33.00 Minutes 34.00 35.00 36.00 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas #15 20.708 2.50 2.00 AU Área 48157297 62907810 % Área 43.36 56.64 Altura 1459174 1555341 23.405 Tiempo de retención 20.708 23.405 1 2 1.50 1.00 0.50 0.00 18.00 20.00 22.00 Minutes 24.00 26.00 #16 Área 36506634 50786793 % Área 41.82 58.18 28.339 1.20 Tiempo de retención 28.339 32.448 1.00 Altura 831766 927377 32.448 1 2 AU 0.80 0.60 0.40 0.20 0.00 27.00 28.00 29.00 30.00 31.00 Minutes 32.00 33.00 34.00 #17 1 2 Tiempo de retención 20.283 22.963 Área 35438065 40211309 % Área 46.85 53.15 Altura 1150682 1116866 AU 1.50 22.963 20.283 2.00 1.00 0.50 0.00 20.00 21.00 22.00 Minutes 23.00 24.00 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 73 #18 % Área 45.44 54.56 Altura 825949 829657 26.675 1.20 Área 32609465 39154245 30.575 Tiempo de retención 26.675 30.575 1 2 1.00 AU 0.80 0.60 0.40 0.20 0.00 26.00 27.00 28.00 29.00 Minutes 30.00 31.00 32.00 #19 Tiempo de retención 20.501 23.183 20.501 2.50 2.00 AU Área 53500739 67258704 % Área 44.30 55.70 Altura 1587600 1639988 23.183 1 2 1.50 1.00 0.50 0.00 16.00 18.00 20.00 22.00 Minutes 24.00 26.00 28.00 #20 1 2 Tiempo de retención 25.595 29.056 Área 65433855 84149437 % Área 43.74 56.26 Altura 1554806 1593806 25.595 29.056 2.50 2.00 AU 1.50 1.00 0.50 0.00 24.00 74 26.00 28.00 Minutes 30.00 32.00 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas #21 Área 3602577 4440476 % Área 44.79 55.21 0.15 Altura 110311 111377 25.246 Tiempo de retención 22.288 25.246 22.288 1 2 AU 0.10 0.05 0.00 18.00 20.00 22.00 24.00 Minutes 26.00 28.00 30.00 #22 2.00 1.50 AU Área 62553053 79673587 % Área 43.98 56.02 Altura 1508161 1541159 29.099 Tiempo de retención 25.629 29.099 25.629 1 2 1.00 0.50 0.00 22.00 24.00 26.00 28.00 Minutes 30.00 32.00 34.00 #23 Tiempo de retención 27.051 30.863 Área 62892468 110681855 27.051 2.50 AU 2.00 1.50 % Área 36.23 63.77 Altura 1474309 1840112 30.863 1 2 1.00 0.50 0.00 26.00 28.00 30.00 Minutes 32.00 34.00 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 75 #24 1 2 Tiempo de retención 20.144 22.683 Área 38244725 72351656 % Área 34.58 65.42 Altura 1242557 1746236 24.00 26.00 % Área 35.04 64.96 Altura 1488036 1835606 20.144 22.683 3.00 AU 2.00 1.00 0.00 18.00 20.00 22.00 Minutes 28.00 #25 Área 57769514 107086845 28.164 Tiempo de retención 24.875 28.164 1 2 24.875 2.50 AU 2.00 1.50 1.00 0.50 0.00 20.00 22.00 24.00 26.00 28.00 30.00 Minutes 32.00 34.00 36.00 38.00 #26 2.50 2.00 AU Área 62622409 96662231 % Área 39.31 60.69 Altura 1763236 1891313 22.540 Tiempo de retención 20.015 22.540 20.015 1 2 1.50 1.00 0.50 0.00 16.00 76 18.00 20.00 22.00 Minutes 24.00 26.00 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas #27 Tiempo de retención 25.557 28.858 Área 51222659 101101699 25.557 2.50 AU 2.00 1.50 % Área 33.63 66.37 Altura 1319696 1784638 28.858 1 2 1.00 0.50 0.00 22.00 24.00 26.00 28.00 Minutes 30.00 32.00 34.00 #28 1 2 Tiempo de retención 34.673 39.992 AU 2.00 % Área 53.68 46.32 Altura 1459493 1073233 39.992 34.673 2.50 Área 82213902 70929063 1.50 1.00 0.50 0.00 32.00 34.00 36.00 38.00 Minutes 40.00 42.00 44.00 #29 Área 65056745 65558998 Altura 1844876 1658828 AU 2.00 % Área 49.81 50.19 22.456 Tiempo de retención 19.799 22.456 19.799 1 2 1.00 0.00 14.00 16.00 18.00 20.00 Minutes 22.00 24.00 26.00 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 77 #30 1 2 Tiempo de retención 31.558 36.394 0.10 Altura 80225 34968 36.394 0.08 AU % Área 65.81 34.19 31.558 0.12 Área 3818187 1983924 0.06 0.04 0.02 0.00 30.00 31.00 32.00 33.00 34.00 Minutes 35.00 36.00 37.00 38.00 #31 Tiempo de retención 21.950 24.941 0.20 % Área 60.82 39.18 Altura 148890 81833 24.941 0.15 AU Área 4891105 3151331 21.950 1 2 0.10 0.05 0.00 20.00 21.00 22.00 23.00 24.00 Minutes 25.00 26.00 27.00 #32 1 2 Tiempo de retención 30.675 35.080 AU 1.50 % Área 50.85 49.15 Altura 1411994 1144307 35.080 30.675 2.00 Área 67728175 65452517 1.00 0.50 0.00 28.00 78 30.00 32.00 34.00 Minutes 36.00 38.00 40.00 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas #33 1 2 Tiempo de retención 19.748 22.296 % Área 48.57 51.43 Altura 2045825 1874834 22.296 19.748 3.00 Área 74566198 78950790 AU 2.00 1.00 0.00 16.00 18.00 20.00 22.00 Minutes 24.00 26.00 28.00 #34 Tiempo de retención 29.574 34.089 Área 87616564 43460220 % Área 66.84 33.16 Altura 1723029 809850 29.574 1 2 2.00 AU 34.089 1.50 1.00 0.50 0.00 26.00 28.00 30.00 32.00 34.00 Minutes 36.00 38.00 40.00 42.00 #35 Tiempo de retención 21.702 24.648 Área 64104614 44649445 % Área 58.94 41.06 AU 2.00 Altura 1789115 1144252 24.648 21.702 1 2 1.00 0.00 20.00 21.00 22.00 23.00 24.00 Minutes 25.00 26.00 27.00 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 79 PDA 204.5 nm, Daicel Chiralpak IA 90 : 10 hex : ipr, f: 0.5 mL/min Processed Channel Descr.: PDA 205.6 nm #36 - Racémico Tiempo de retención 25.202 34.899 % Área 49.92 50.08 1.50 Altura 1462732 1012987 34.899 2.00 AU Área 66912886 67118985 25.202 1 2 1.00 0.50 0.00 22.00 24.00 26.00 28.00 30.00 32.00 Minutes 34.00 36.00 38.00 40.00 #37 Tiempo de retención 20.417 30.317 % Área 35.95 64.05 Altura 1294247 1261386 30.317 1.50 AU Área 52429563 93426135 20.417 1 2 1.00 0.50 0.00 15.00 80 20.00 25.00 Minutes 30.00 35.00 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas #38 Tiempo de retención 20.472 30.625 Área 8511988 25571556 % Área 24.97 75.03 Altura 270956 472335 30.625 1 2 0.60 20.472 0.50 AU 0.40 0.30 0.20 0.10 0.00 20.00 22.00 24.00 26.00 Minutes 28.00 30.00 32.00 #40 Tiempo de retención 20.346 30.206 % Área 47.86 52.14 1.50 Altura 1458495 972837 30.206 2.00 AU Área 52204115 56878904 20.346 1 2 1.00 0.50 0.00 16.00 18.00 20.00 22.00 24.00 26.00 28.00 Minutes 30.00 32.00 34.00 36.00 #41 1.60 Área 79042198 77904776 % Área 50.36 49.64 1.40 1.20 AU Altura 1460179 1053309 32.788 1.80 Tiempo de retención 26.084 32.788 26.084 1 2 1.00 0.80 0.60 0.40 0.20 0.00 26.00 28.00 30.00 Minutes 32.00 34.00 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 81 PDA 204.5 nm, Daicel Chiralpak IA 90 : 10 hex : ipr, f: 0.5 mL/min Processed Channel Descr.: PDA 214.4 nm #42 - Racémico Tiempo de retención 30.911 38.797 1 2 % Área 46.83 53.17 2.00 Altura 1706837 1498542 38.797 30.911 2.50 Área 103739781 117804182 AU 1.50 1.00 0.50 0.00 28.00 30.00 32.00 34.00 36.00 38.00 Minutes 40.00 42.00 44.00 #43 1 2 Tiempo de retención 32.342 40.566 20.00 25.00 Área 106752947 76785340 2.00 1.50 40.566 AU Altura 1904887 979893 32.342 2.50 % Área 58.16 41.84 1.00 0.50 0.00 82 30.00 35.00 Minutes 40.00 45.00 50.00 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas #44 Tiempo de retención 26.717 34.297 1 2 Altura 2858414 2019783 34.297 3.00 AU % Área 53.62 46.38 26.717 4.00 Área 165590527 143253912 2.00 1.00 0.00 26.00 28.00 30.00 32.00 Minutes 34.00 36.00 38.00 #46 Tiempo de retención 33.144 42.953 1 2 Área 84797047 43957398 % Área 65.86 34.14 Altura 1494486 551011 33.144 2.00 1.80 1.60 1.40 1.00 42.953 AU 1.20 0.80 0.60 0.40 0.20 0.00 26.00 28.00 30.00 32.00 34.00 36.00 38.00 40.00 42.00 44.00 46.00 48.00 Minutes #47 Tiempo de retención 27.000 34.915 3.00 % Área 66.44 33.56 Altura 2787467 1130164 2.00 34.915 AU Área 143032751 72240766 27.000 1 2 1.00 0.00 24.00 26.00 28.00 30.00 32.00 Minutes 34.00 36.00 38.00 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 83 PDA 204.5 nm, Daicel Chiralpak IA 90 : 10 hex : ipr, f: 0.5 mL/min Processed Channel Descr.: PDA 208.4 nm #49 - Racémico 1 2 Tiempo de retención 6.988 8.032 Altura 2831347 2670617 8.032 3.00 AU % Área 49.32 50.68 6.988 4.00 Área 34808944 35774861 2.00 1.00 0.00 6.60 6.80 7.00 7.20 7.40 7.60 7.80 Minutes 8.00 8.20 8.40 8.60 #50 1 2 Tiempo de retención 7.464 8.465 Área 6056041 5874990 % Área 50.76 49.24 Altura 532999 447207 0.60 8.465 7.464 0.70 AU 0.50 0.40 0.30 0.20 0.10 0.00 6.00 84 6.50 7.00 7.50 8.00 Minutes 8.50 9.00 9.50 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas #51 Tiempo de retención 6.925 7.977 % Área 53.05 46.95 Altura 2637730 2224460 7.977 3.00 AU Área 31231639 27635865 6.925 1 2 2.00 1.00 0.00 6.00 6.50 7.00 7.50 Minutes 8.00 8.50 9.00 #53 1 2 Tiempo de retención 7.435 8.602 % Área 55.98 44.02 Altura 1515015 1001763 7.435 1.80 Área 18216356 14323996 1.60 8.602 1.40 AU 1.20 1.00 0.80 0.60 0.40 0.20 0.00 6.00 7.00 8.00 Minutes 9.00 10.00 11.00 #54 3.00 AU Área 45055127 43679735 % Área 50.78 49.22 Altura 2945456 2786864 7.983 Tiempo de retención 6.944 7.983 6.944 1 2 2.00 1.00 0.00 5.00 6.00 7.00 8.00 Minutes 9.00 10.00 11.00 Reacciones de adición conjugada organocatalítica de benzofuran-2(3H)-ona a ’-hidroxienonas 85