Mujer de 21 an˜os con fiebre, artralgias y leucocitosis
Anuncio

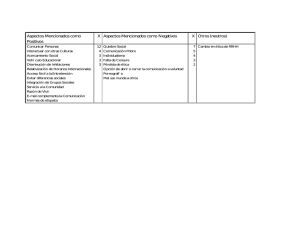
Documento descargado de http://www.elsevier.es el 20/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. Med Clin (Barc). 2011;136(1):31–37 www.elsevier.es/medicinaclinica Conferencia clinicopatológica-MIR Mujer de 21 años con fiebre, artralgias y leucocitosis A 21 year-old woman with fever, arthralgia and leucocitosis Juan Muñoz-Ortego a,*, John Lara b y José Tomás Navarro Ferrando c a b c Servicio de Reumatologı´a, Parc de Salut MAR, Barcelona, España Servicio de Anatomı´a Patológica, Hospital Universitari Germans Trias i Pujol, Badalona, Barcelona, España Servicio de Hematologı´a, Institut Català d’Oncologia, Hospital Universitari Germans Trias i Pujol, Badalona, Barcelona, España I N F O R M A C I Ó N D E L A R T Í C U L O Historia del artı´culo: Recibido el 30 de junio de 2010 Aceptado el 5 de julio de 2010 Mujer de 21 años de edad, natural de Marruecos, residente en Badalona desde hace un año, sin alergias conocidas ni hábitos tóxicos, que ingresó trasladada desde otro centro para estudio de fiebre, artralgias y leucocitosis. Una semana antes del ingreso, encontrándose previamente bien, presentó fiebre diaria de 39 C8, tos seca, odinofagia y artromialgias. Acudió a urgencias de otro centro, donde, tras practicarle una radiografı́a de tórax que fue informada como normal, se le prescribió amoxicilina e ibuprofeno y fue remitida a domicilio. Dos dı́as después, por persistencia de los sı́ntomas, la paciente volvió a urgencias, donde en un hemograma se comprobó: leucocitos 14 x 109/L (neutrófilos 86%; bandas 2%). En la exploración fı́sica la paciente estaba normotensa y eupneica. La faringe estaba hiperémica. No se evidenciaron adenopatı́as. No existı́an signos menı́ngeos. La auscultación cardiorrespiratoria fue normal y no se palparon visceromegalias. No se evidenció artritis. Se decidió su ingresó. Los hemocultivos y urocultivos fueron negativos. En el electrocardiograma se comprobó ritmo sinusal sin signos de sobrecarga o hipertrofia. La ecografı́a abdominal y ginecológica fue normal y las serologı́as para virus de Epstein-Barr (VEB), citomegalovirus (CVM), Brucella, Salmonella, virus de la inmunodeficiencia humana (VIH) y Toxoplasma fueron negativas. El aspirado nasofarı́ngeo para gripe A fue negativo. Las pruebas de autoinmunidad, que incluyeron anticuerpos antinucleares (ANA), anticuerpos contra el citoplasma de los neutrófilos (ANCA) y factor reumatoide (FR), fueron negativas. Se practicó un ecocardiograma en el que no se visualizaron alteraciones de las válvulas ni derrame pericárdico. Se * Autor para correspondencia. Correo electrónico: [email protected] (J. Muñoz-Ortego). practicó una tomografı́a computarizada (TC) torácica-abdominal, que también resultó normal. La paciente persistı́a febril, con dos picos febriles diarios. Para ampliar el estudio se le practicó un ecocardiograma transesofágico que resultó normal, ası́ como serologı́as para Rickettsia, Borrelia, estudio de gota gruesa y estudio de fiebre mediterránea familiar, resultando todos ellos negativos o normales. La paciente fue trasladada al Hospital Germans Trias i Pujol. En el momento de su ingreso la temperatura axilar era de 37 8C, la frecuencia cardı́aca de 96 latidos/minuto y la presión arterial de 97/68 mmHg, con una saturación de oxı́geno del 99%. En la exploración fı́sica se evidenció una paciente consciente y orientada, con un exantema evanescente asalmonado en el torso. La auscultación cardiorrespiratoria era normal. No se evidenciaron visceromegalias. No habı́a signos menı́ngeos y se pudo comprobar dolor articular a la movilización en articulaciones de las manos, muñecas, codos, rodillas y tobillos. En las pruebas de laboratorio destacaba: velocidad de sedimentación globular (VSG) 108 mm en la primera hora, leucocitos 14,6 x 109/L (neutrófilos 83%, linfocitos 15%, monocitos 2%), hemoglobina 11, 8 g/dL, plaquetas 443 x 109/L, actividad de protrombina del 83%, fibrinógeno 747 mg/dL, tiempo de tromboplastina parcial activado (TTPA) 29 seg, glucemia 76 mg/dL, proteı́nas 73 g/L, urea 10 mg/dL, creatinina 0,42 mg/dL, uricemia 2,3 mg/dL, Na 138 mmol/L, K 4,1 mmol/L, bilirrubina 0,39 mg/dL, fosfatasa alcalina (FA) 73 U/L (25120), aspartato-aminotransferasa (AST) 40 U/L (valores normales 531), alanino-aminotransferasa (ALT) 38 U/l (valores normales 5-31), gammaglutamiltranspeptidasa (GGT) 32 U/L (valores normales 6-50), lactatodeshidrogenasa (LDH) 562 U/L (valores normales 240-480) y triglicéridos 107 mg/dL. El sedimento de orina fue normal y la determinación de proteinuria fue negativa. La proteı́na C reactiva (PCR) fue de 167 mg/dL (valores normales 0-30) y los ANA, ANCA y FR eran negativos. Los marcadores tumorales antı́geno 0025-7753/$ – see front matter ß 2010 Elsevier España, S.L. Todos los derechos reservados. doi:10.1016/j.medcli.2010.07.003 Documento descargado de http://www.elsevier.es el 20/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. 32 J. Muñoz-Ortego et al / Med Clin (Barc). 2011;136(1):31–37 carcinoembrionario, alfafetoproteı́na y beta-2 microglobulina eran normales. La IgG frente a parvovirus fue positiva, mientras que la IGM resultó negativa. La serologı́a luética fue negativa. La concentración plasmática de ferritina resultó de 6.540 ng /ml (valores normales 15-90). Se inició tratamiento con glucocorticoides a dosis de 1 mg/kg/ dı́a con mejorı́a inicial en las primeras 24 horas. Sin embargo, 48 horas después la enferma empeoró, presentando deterioro del estado general, además de fiebre y exantema. En la analı́tica destacaba: VSG 59, leucocitos 7,6 x 109/L (neutrófilos 53%, linfocitos 31,5%, monocitos 0,90%, eosinófilos 0,2%, basófilos 0%), hemoglobina 11,9 g/dL, plaquetas 204 x 109/L, fibrinógeno 218 mg/ dL, ferritina 43.600 ng/ml, bilirrubina 5,4 mg/dL, FA 197 U/L, AST 1.422 U/L, ALT 982 U/L, LDH 6.114 U/L, GGT 713 U/L y PCR 48 mg/l. Se practicaron pruebas diagnósticas. Diagnóstico diferencial Dr. Juan Muñoz Ortego. En primer lugar, para discutir las etiologı́as más probables del caso clı́nico tomaremos como referencia los signos y sı́ntomas guı́a iniciales: la fiebre con leucocitosis y neutrofilia, las artralgias, el exantema evanescente asalmonado y la hiperferritinemia. Posteriormente analizaremos las causas del cuadro de hepatitis tras recibir terapia inmunodepresora. Inicialmente debemos pensar en una etiologı́a infecciosa, fundamentalmente vı́rica, aunque también hay que tener en consideración la bacteriana y la protozoaria. El primer grupo de agentes infecciosos a considerar es el de los virus, en especial los hepatotropos. En nuestro caso, las serologı́as para VEB, CMV, VIH, IgM para parvovirus, y el frotis nasofarı́ngeo para virus Influenza A fueron negativos. No obstante, serı́a de gran interés el cribado para los virus de las hepatitis A, B y C, herpes simple, varicela-zóster y Coxsackie, pues durante el curso de su historia natural pueden causar el cuadro clı́nico inicial de la paciente1. El grupo de serologı́as solicitadas para enfermedades bacterianas fueron Rickettsia, Borrelia, Brucella, Salmonella y Treponema. En este punto es importante resaltar que hubiera sido conveniente realizar más serologı́as complementarias, como son las de leptospirosis, Yersinia y Coxiella, ası́ como frotis farı́ngeo en busca del estreptococo betahemolı́tico. Del mismo modo que los virus, todas estas entidades pueden presentar cuadros febriles con artromialgias, leucocitosis y exantemas durante el curso de su historia natural2. En contra de esta etiologı́a bacteriana tenemos la negatividad de los hemocultivos, los urinocultivos y las pruebas de imagen, en especial de la TC tóraco-abdominal, en la que no se visualizaron colecciones o abscesos, ası́ como el ecocardiograma transesofágico, que no objetivó signos de endocarditis. Tampoco se realizaron la prueba intradérmica de tuberculina o un estudio de quantiferón en sangre a fin de descartar la tuberculosis, enfermedad prevalente en el norte de África y con una incidencia en aumento en nuestro medio. En el caso de positividad se deberı́a de haber buscado activamente en fluidos mediante cultivos o reacción en cadena de la polimerasa para el bacilo de Koch. A pesar de todo, recordemos que la TC tóracoabdominal era normal y el cuadro clı́nico no es muy sugestivo de esta entidad3. Las enfermedades protozoarias, en especial las causadas por Toxoplasma y Plasmodium, fueron debidamente estudiadas y rechazadas. Otro grupo de enfermedades a considerar son las neoplásicas. Las de mayor interés en este caso son la enfermedad de Hodgkin, los linfomas no hodgkinianos, los sı́ndromes mieloproliferativos y las neoplasias sólidas, sobre todo las de pulmón y de mama, que pueden cursar como un sı́ndrome paraneoplásico similar al cuadro clı́nico de nuestra paciente4. Todas estas enfermedades fueron razonablemente descartadas mediante exploración fı́sica, pruebas de imagen y de laboratorio. El estudio de la médula ósea mediante una biopsia-aspirado serı́a una prueba complementaria que debiera de valorarse si la sospecha fuera muy alta. En este punto comentaremos las causas principales de hiperferritinemia, pues deberı́amos tener en cuenta los cuadros de citólisis hepáticas y los cuadros hematológicos con activación macrofágica. En un trabajo, Ramı́rez et al5 analizaron 269 determinaciones de 135 pacientes con valores de ferritina sérica mayor de 2.000 ng/ml y observaron que las enfermedades hematológicas representaban el grupo más numeroso, en concreto la leucemia mieloblástica aguda seguida de la leucemia linfoblástica aguda, la leucemia mieloide crónica y la aplasia medular. Otras entidades frecuentes fueron las hepatitis vı́ricas, la insuficiencia renal crónica, las infecciones sistémicas y la enfermedad de Still. Respecto a este último trastorno, la media de valor de ferritina sérica fue estadı́sticamente superior al resto de las enfermedades previamente nombradas. La determinación de ferritina glucosilada debiera de tomarse en consideración como prueba complementaria a realizar, pues un valor inferior al 20% sugiere una enfermedad de Still, mientras que valores muy bajos se detectan en sı́ndromes de activación macrofágica causados sobre todo por sepsis graves o neoplasias6. El tercer grupo de entidades que debemos considerar es el de las enfermedades sistémicas. La enfermedad de Still del adulto (ESA) es considerada una enfermedad reumática e inflamatoria de etiologı́a desconocida, caracterizada por la asociación de fiebre intermitente, exantema evanescente, artralgias y leucocitosis con neutrofilia. Si aplicamos los criterios diagnósticos más aceptados, los de Yamaguchi et al7, con una sensibilidad del 96,2% y una especificidad del 92,1%, o los de Frautel et al8, con una sensibilidad del 80,6% y una especificidad del 98,5%, nos percatamos de que nuestra paciente puede ser clasificada como tal (tabla 1). Se ha de reseñar que en los criterios de Yamaguchi et al, a diferencia de los de Fautrel et al, la ESA es considerada una enfermedad por exclusión de todas las entidades que hemos expuesto previamente y de las enfermedades sistémicas que comentaremos a continuación. El grupo de las vasculitis también debiera tenerse en consideración, en especial la poliarteritis nodosa. Si tenemos en cuenta los criterios de clasificación de la American College of Rheumatology9, en nuestro caso no se nos refiere pérdida de peso, livedo reticularis, disestesias, hipertensión arterial ni afectación renal. La paciente presentaba mialgias y serı́a de interés realizar serologı́as para virus de la hepatitis B, por su relación, y evaluar la necesidad de realizar una angiografı́a de vasos mesentéricos. En el caso de observar aneurismas u oclusiones arteriales se deberı́a valorar realizar biopsia de las arterias de pequeño o mediano tamaño. La negatividad de los ANCA y la caracterı́stica evanescencia de la lesión cutánea de nuestra paciente son elementos en contra de otras vasculitis, como la poliangeı́tis microscópica o las vasculitis cutáneas. El sı́ndrome de Sweet, o dermatosis neutrofı́lica febril aguda, es una entidad caracterizada por el comienzo repentino de fiebre, leucocitosis y pápulas y placas bien delimitadas y eritematosas10. En contra de esta posibilidad hay que destacar la edad de presentación, pues suele ser en torno a los 50 años, y las caracterı́sticas de la lesión cutánea, pues acostumbran a ser dolorosas. No obstante, la realización de una biopsia cutánea debiera tenerse en cuenta con el fin de objetivar la lesión histopatológica más especı́fica de esta entidad, un infiltrado neutrofı́lico denso con una disposición perivascular. Los sı́ndromes autoinflamatorios familiares, también conocidos como sı́ndromes hereditarios de fiebre periódica, forman un grupo Documento descargado de http://www.elsevier.es el 20/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. J. Muñoz-Ortego et al / Med Clin (Barc). 2011;136(1):31–37 33 Tabla 1 Criterios de clasificación de la enfermedad de Still del adulto Yamaguchi et al Fautrel et al Criterios mayores Fiebre de 39 8C o más, de duración superior a una semana* Artralgias de 2 o más semanas de duración Rash tı́pico cutáneo: maculopapular, no pruriginoso, asalmonado y concomitante con picos febriles* Leucocitosis (> 10,000/mm3) con granulocitos > 80%* Criterios mayores Fiebre 39 8C* Artralgias* Eritema evanescente* Faringitis* Neutrofilia > 80%* Fracción glucosilada de la ferritina 20% Criterios menores Odinofagia* Linfadenopatı́a y/o esplenomegalia Alteración de la biologı́a hepática Factor reumatoide negativo y anticuerpos antinucleares negativos* Criterios menores Leucocitosis (> 10,000/mm3)* Rash tı́pico cutáneo* Criterios de exclusión Infecciones Neoplasias Otras enfermedades sistémicas Criterios de exclusión No especifica Diagnóstico 5 criterios, al menos 2 mayores Diagnóstico 4 mayores o 3 mayores y 2 menores * Criterios presentes en nuestra paciente. de trastornos con un fenotipo común de episodios febriles crónicos recurrentes acompañados de diarrea, dolor abdominal, exantema y artralgias con intervalos asintomáticos11. Suelen debutar en la infancia, aunque su diagnóstico puede realizarse en el adulto. En contra de estos sı́ndromes figuran la falta de adenopatı́as y de un cuadro clı́nico abdominal, ya sea en forma de dolor o alteración del ritmo deposicional. En nuestra paciente se realizó estudio de fiebre mediterránea familiar, pero habrı́amos de considerar realizar estudio genético para el sı́ndrome de hiperinmunoglobulina D y el sı́ndrome de fiebre periódica asociado al receptor del factor de necrosis tumoral (TNF). Hay otro grupo de enfermedades sistémicas a tener en cuenta, pero ya en un segundo plano. A favor de la polimiositis y la dermatomiositis tan solo encontramos una LDH discretamente elevada; no obstante, no presenta debilidad proximal ni las lesiones tı́picas como son el rash en heliotropo, las pápulas de Gottron ni el signo de la V. Serı́a aconsejable la determinación de enzimas musculares, como son la creatincinasa y la aldolasa, y, en el caso de positividad, deberı́a valorarse la realización de un electromiograma y posterior biopsia muscular si procediera12. Respecto al lupus eritematoso sistémico, la negatividad de los ANA va en contra y tampoco presenta alteraciones hematológicas, renales ni viscerales compatibles. Al referirnos a las lesiones cutáneas, tampoco presenta las más especı́ficas, como son el eritema facial, la fotosensibilidad ni el lupus discoide. Serı́a de interés solicitar la determinación de anticuerpos antifosfolı́pidos, anti-Sm y anti-ADN, pues hasta un 3% de pacientes con este proceso puede presentar ANA negativos13. Si tomamos como referencia el cuadro de poliartralgias y el dolor a la movilización articular, hay que tener en consideración las artritis reactivas, como la postestreptocócica, por lo que un frotis nasofarı́ngeo para identificar el estreptococo betahemolı́tico podrı́a haber sido de utilidad. La artritis reumatoide seronegativa es otra entidad que debe tenerse en cuenta, no obstante, no se refleja la rigidez articular matutina y en la descripción de la sintomatologı́a no se resalta una clara artritis. Serı́a de utilidad la realización de radiologı́as y ecografı́as articulares con el fı́n de objetivar erosiones o sinovitis activa, respectivamente. También añadirı́a la determinación de anticuerpos antipéptido cı́clico citrulinado, ya que su sensibilidad y especificidad para su diagnóstico es mayor14. Llegados a este punto, se nos informa que la paciente recibió tratamiento con glucocorticoides a dosis de 1 mg/kg/dı́a con una mejorı́a inicial, pero que en 48 horas empeoró, presentando deterioro del estado general, además de fiebre y exantema. En este momento nos muestran parámetros compatibles con una hepatopatı́a aguda, no obstante la leucocitosis y la neutrofilia, ası́ como la VSG y la PCR, disminuyeron tras la infusión de glucocorticoides. Por otro lado, los valores de ferritina y de citólisis hepática se elevaron claramente. En la literatura están descritas varias series de pacientes con ESA15,16, donde las alteraciones hepáticas son muy frecuentes, hasta un 60%, normalmente de intensidad leve o moderada. Se han implicado mecanismos relacionados con el uso de antiinflamatorios no esteroides (AINE) y metotrexato. Sin embargo, en las series descritas, la elevación de transaminasas no es una contraindicación para usar los fármacos descritos, ya que este tipo de hepatopatı́a suele responder correctamente a glucocorticoides o añadiendo otros inmunodepresores como el mismo metotrexato. En algunos casos, la terapéutica biológica con anakinra, inhibidor de interleucina 1, ayuda a controlar la sintomatologı́a17. A pesar de todo, hay descritos pacientes cuya insuficiencia hepática aguda llega a requerir trasplante hepático18. En nuestro caso, en primer lugar debemos de realizar el diagnóstico diferencial de la hepatopatı́a con las infecciones por los virus de las hepatitis A, B, C y D, ası́ como los virus herpes simple tipo 1 y 2, pues, como ya comentamos, no se realizaron previamente y podrı́an ser la causa del cuadro clı́nico. Habrı́amos de solicitar las siguientes pruebas: anticuerpos frente al VHA de tipo IgM, antı́geno de superficie del VHB (HBsAg), anticuerpos anticore del VHB (anti-HBc) de tipo IgM, ADN del VHB, anticuerpos frente al VHC, ARN del VHC y ADN del VHS. En el caso de positividad para VHB deberı́amos de tener en consideración una coinfección con el virus D. A favor de esta etiologı́a reside el hecho de haber presentado el cuadro de citólisis tras el indicio de la glucocorticoterapia y pudiera ser que la paciente fuera una portadora de VHB latente que se hubiera reactivado al iniciar tratamiento inmunodepresor19. No obstante, recordemos que el diagnóstico de ESA es un diagnóstico por exclusión y habrı́amos de considerar los virus hepatotropos, en especial el VHB, como una de las etiologı́as probables de todo el cuadro clı́nico20. En segundo lugar debemos de considerar el sı́ndrome de reactivación hemofagocı́tico (SRH), un subtipo de la linfohistiocitosis hemofagocı́tica (LH), proceso sistémico caracterizado por la activación y la proliferación de linfocitos T y macrófagos en la Documento descargado de http://www.elsevier.es el 20/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. 34 J. Muñoz-Ortego et al / Med Clin (Barc). 2011;136(1):31–37 médula ósea, el sistema reticuloendotelial y el sistema nervioso. Ha sido descrito como complicación de la ESA y suele presentarse con fiebre, hepatoesplenomegalia, pancitopenia, alteración de la biologı́a hepática, hiperferritinemia e hipertrigliceridemia21. El sı́ndrome de activación macrofágica (SAM), descrito como complicación de enfermedades reumáticas, sobre todo de la forma sistémica de la artritis idiopática juvenil (AIJ) y del lupus eritematoso sistémico, se caracteriza por un cuadro similar al SRH22. Recientemente, algunos autores consideran que SRH, SAM, AIJ y ESA son el espectro de un mismo trastorno, con presentaciones clı́nicas diferenciales y que deben ser clasificadas dependiendo de su gravedad, pero que esencialmente comparten la misma patogenia, marcada por una intensa reacción inmunológica asociada a una cascada de liberación de citotocinas y defectos de apoptosis que perpetúan la agresión inmune23. Atendiendo a los últimos criterios diagnósticos para LH, no objetivamos en nuestro caso citopenias, hipertrigliceridemia, hipofibrinoginemia ni esplenomegalia, y serı́a de interés comprobar una elevación del receptor soluble de interleucina 2 (IL-2) por encima de 2.400 U/L y la disminución de actividad de las células NK en el laboratorio24. Habrı́a que valorar, dependiendo de la evolución de la paciente y una vez descartadas las otras entidades, en especial las hepatitis vı́ricas, la realización de un aspirado de médula ósea y una biopsia hepática a fin de objetivar hemofagocitos. En tercer lugar, debemos de considerar las causas tóxicomedicamentosas. No se nos refiere ingesta alcohólica ni la relación AST/ALT es > 2, por lo que esta etiologı́a se excluye razonablemente. Respecto a los fármacos, debemos recordar que tomó ibuprofeno y amoxicilina durante 2 dı́as, sin mejorı́a, y la reacción de hipersensibilidad deberı́a haber acontecido poco tiempo después, con lo que la cronologı́a de los hechos va en contra. Además, en el caso de la amoxicilina, el patrón de afectación hepática suele ser más colostásico que citolı́tico, suele haber eosinofilia y el tratamiento con glucocorticoides suele mejorar la clı́nica25. Otras etiologı́as a considerar en un segundo término son los sı́ndromes mieloproliferativos, pues, como ya comentamos, son una de las principales causas de cuadros de hiperferritinemia y pueden desarrollar una hepatitis5. En contra de estos tastornos hematológicos encontramos la normalidad en las tres series del hemograma y una TC en la que no se evidencian adenopatı́as, masas ni visceromegalias. La realización de un frotis de sangre periférica y el aspirado de médula ósea podrı́a ser de utilidad si quisiéramos profundizar en esta etiologı́a. Entre los trastornos hereditarios que cursan con hepatopatı́a deberı́amos destacar el déficit de alfa1-antitripsina y la enfermedad de Wilson. Respecto al primer proceso, no se encontró afectación pulmonar en la TC y la determinación sanguı́nea de alfa1-antitripsina serı́a de utilidad. Está descrita la enfermedad de Wilson como presentación en forma de hepatitis fulminante, sin tener que presentar afectación neurológica. Un examen con lámpara de hendidura a fin de observar los anillos de KayserFleischer y la determinación de ceruloplasmina baja y cupruria elevada serı́an de utilidad26. Respecto a la hepatitis autoinmune, la determinación de anticuerpos antimúsculo liso y antimicrosomales tipo 1 pudiera ser de interés, aunque en contra de esta etiologı́a fue la negatividad en los ANA y, sobre todo, que el cuadro se inició tras el tratamiento con glucocorticoides, ya que estos fármacos son considerados la primera lı́nea terapéutica27. La hemodinamia de la paciente fue estable en todo momento, sin presentar cuadros de insuficiencia cardı́aca, shock o hipotensión grave, por lo que no nos hace pensar en un origen isquémico del cuadro. La realización de una ecografı́a-doppler abdominal podrı́a ser de utilidad a fin de excluir las enfermedades vasculares hepáticas, a saber, la trombosis portal o enfermedad de Budd-Chiari. También nos ayudarı́a para descartar obstrucción de la vı́a biliar aguda, aunque recordemos que las primeras ecografı́a y TC fueron normales, y en este caso una colangiorresonancia serı́a la prueba de elección a contemplar28. No obstante, estas entidades suelen cursar con dolor abdominal agudo y exploración fı́sica abdominal patológica, que en nuestro caso no se refiere. En conclusión, mi impresión diagnóstica es que nos encontramos ante una paciente con una ESA, excluidas razonablemente las etiologı́as infecciosas, neoplásicas y sistémicas que hemos comentado. Toda la evolución clı́nica se podrı́a explicar, como ya hemos comentado, por el mismo proceso, con un SRH como episodio final. Pero, dado que carecemos de ciertos datos clı́nicos, no podemos descartar que fuera portadora de un virus hepatotropo, como pudiera ser el VHB, y que se reactivara tras el inicio de terapia inmunodepresora, provocando una hepatitis aguda. Por ello, creo que un aspirado de médula ósea y una biopsia hepática serı́an las pruebas diagnósticas a realizar si las serologı́as para hepatitis fueran negativas. Diagnóstico clı´nico Enfermedad de Still del adulto, hepatitis aguda o sı́ndrome hemofagocı́tico. Discusión anatomopatológica Dr. John Lara. En el departamento de Anatomı́a Patológica se recibieron cuatro biopsias, dos de ellas correspondı́an a muestras en sacabocados de la erupción cutánea, otra era una punción hepática y la última, una biopsia de médula ósea. En la primera biopsia de piel, la epidermis no mostraba alteraciones histológicas importantes, mientras que en la dermis superficial llamaba la atención la presencia de infiltrado inflamatorio mixto de neutrófilos y linfocitos rodeando a los vasos capilares, sin evidencia de leucocitoclasia, necrosis fibrinoide o extravasación hemática (fig. 1). En cuanto al segundo punch cutáneo, únicamente presentaba cambios urticariales con edema dérmico difuso, leve infiltrado linfocitario perivascular y algunos mastocitos y eosinófilos que podı́an apreciarse mejor con la tinción de Giemsa (fig. 2). El cilindro hepático poseı́a una arquitectura conservada. Los [(Figura_1)TD$IG]lobulillos mostraban necrosis focal con pequeños agregados Figura 1. Punch cutáneo mostrando un infiltrado perivascular superficial de linfocitos y neutrófilos (Hematoxilina-eosina, x100). Documento descargado de http://www.elsevier.es el 20/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. [(Figura_2)TD$IG] [(Figura_4)TD$IG] J. Muñoz-Ortego et al / Med Clin (Barc). 2011;136(1):31–37 35 Figura 2. La tinción histoquı́mica de May-Grünwald-Giemsa ayuda a poner de manifiesto la presencia de numerosos mastocitos y eosinófilos en el infiltrado perivascular (Giemsa, x400). Figura 4. Médula ósea hipercelular con disminución relativa de la celularidad grasa (Hematoxilina-eosina, x25). linfohistiocitarios y cambios regenerativos con algunas figuras de mitosis. Además, se identificaban algunos megacariocitos (fig. 3) indicativos de hematopoyesis extramedular. Ası́ pues, los hallazgos histológicos encontrados en las diferentes biopsias analizadas son poco especı́ficos y deben ser valorados en el contexto clı́nico. La médula ósea era moderadamente hipercelular y de aspecto reactivo, con una representación de las tres series hematopoyéticas dentro de los lı́mites de la normalidad, pero habı́a algunos macrófagos con fenómenos de hemofagocitosis (fig. 4), por lo que agradecerı́a al hematólogo, con más experiencia en este cuadro, que ampliara la discusión. Dr. José Tomás Navarro Ferrando. En el aspirado de médula ósea se observó un celularidad global abundante con disminución de la grasa medular. Se encontraban presentes las tres series hematopoyéticas. La relación mieloide/eritroide se encontraba ligeramente aumentada y la serie granulocı́tica presentaba un aumento de la granulación y una ligera eosinofilia. Entre la celularidad hematopoyética se observaron numerosos macrófagos, muchos de los cuales tenı́an fenómeno de hemofagocitosis, es decir, mostraban elementos precursores de la hematopoyesis en sus citoplasmas (fig. 5). La LH es una entidad que comprende dos trastornos diferentes: la LH familiar y la LH secundaria o SRH. La primera es una enfermedad autosómica recesiva con una incidencia estimada de 1:50.000 nacimientos vivos. Se han descrito mutaciones en varios genes, como el que codifica la perforina (PRF)29, el gen UNC13D (17q25)30 y el gen STX11 en el cromosoma 6q2431, que codifica la sintaxina 11. Es una enfermedad que aparece tı́picamente durante la infancia, con un curso fatal y una mediana de supervivencia inferior a 2 meses si no se instaura tratamiento. El SRH es una entidad clı́nico-patológica que se caracteriza por fiebre, hepatoesplenomegalia, citopenias, cifra de ferritina muy elevada, junto a la proliferación de histiocitos en la médula ósea, bazo, ganglios linfáticos, hı́gado y lı́quido cefalorraquı́deo. Otros hallazgos frecuentes son hipertrigliceridemia, coagulopatı́a con hipofibrinogenemia, elevación de las transaminasas y sı́ntomas neurológicos. La fisiopatologı́a está ligada directamente a la activación y la proliferación incontrolada de linfocitos T y macrófagos que dan lugar a la hiperproducción de diferentes citocinas. El SRH puede aparecer en pacientes con neoplasias, infecciones y enfermedades autoinmunes. Entre estas últimas, las más frecuentes son el lupus eritematoso sistémico, la artritis reumatoide, la dermatomiositis y la ESA. El tratamiento debe incluir el control de la enfermedad de base y se debe acompañar de un tratamiento inmunodepresor con glucocorticoides, etopósido y ciclosporina A32. Los criterios diagnósticos de la LH son los establecidos por la Histiocyte Society32. A los 5 criterios iniciales establecidos en 1991, se añadieron otros 3 en 2004 (tabla 2). Se deben cumplir 5 de los 8 criterios, aunque los pacientes con un diagnóstico molecular no necesitarı́an cumplirlos. El SRH se asocia a la ESA con una frecuencia estimada del 15%. Resulta difı́cil distinguir entre la sintomatologı́a de la ESA y la del SRH, y se sugiere que la presencia de coagulación intravascular diseminada, hipertrigliceridemia y el descenso de haptoglobina, junto a la presencia de citopenias, deberı́an hacer sospechar un SRH cuando se presentan en un individuo con ESA33. Se han postulado diferentes fármacos como desencadenantes del SRH en la ESA, como los AINE, las sales de oro, la penicilamina o la sulfasalazina. El tratamiento debe incluir dosis altas de glucocorticoides que pueden combinarse con inmunoglobulinas [(Figura_3)TD$IG] Figura 3. Biopsia hepática con presencia de grandes megacariocitos en los sinusoides, rodeados de hepatocitos de aspecto normal (Hematoxilina-eosina, x400). Documento descargado de http://www.elsevier.es el 20/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. [(Figura_5)TD$IG] J. Muñoz-Ortego et al / Med Clin (Barc). 2011;136(1):31–37 36 Figura 5. Imágenes del aspirado de médula ósea que muestran el fenómeno de hemofagocitosis (May-Grünwald-Giemsa, x1.000). A) Macrófago fagocitando un polimorfonuclear neutrófilo. B) Macrófago con un cayado y un linfocito fagocitados. En el extremo superior izquierdo se observa un magacariocito con emperipolesis. C) Imagen de un macrófago con restos celulares en su citoplasma, rodeado por precursores de la serie eritroide y granulocı́tica. D) Imagen de un macrófago que ha fagocitado varios eritroblastos maduros. intravenosas. En los casos resitentes, la ciclosporina A o el infliximab pueden ser beneficiosos. Dr. Agustı́n Urrutia. La serologı́a de hepatitis (IgM del virus A, HBsAg y anticuerpos frente al virus C) realizada cuando la paciente empeoró resultó negativa. Apoyan los hallazgos histológicos el diagnóstico de enfermedad de Still? Dr. Lara. Aunque histológicamente los hallazgos son poco especı́ficos, concuerdan con los descritos en la literatura en pacientes con esta enfermedad34–37. Dr. Agustı́n Urrutia. Se observó hemofagocitosis en alguna de las biopsias? Dr. Lara. No, las muestras fueron revisadas exhaustivamente tras conocerse los hallazgos hematológicos y ninguna de ellas mostraba hemofagocitosis. Probablemente se debe a que el extendido citológico permite estudiar elementos celulares completos e identificar más fácilmente estructuras fagocitadas dentro de la membrana de una célula. Por el contrario, en la histologı́a se estudian secciones de unas pocas micras que solo permiten apreciar la fagocitosis cuando es más abundante. Diagnóstico anatomopatológico Enfermedad de Still del adulto. Sı́ndrome hemofagocı́tico. Editores de la Conferencia Clinicopatológica-MIR Editor: Agustı́n Urrutia de Diego Editor asociado: Marı́a Teresa Fernández Figueras Celebrada en el Hospital Universitari Germans Trias i Pujol, Badalona, el 18 de febrero de 2010. Conflicto de intereses ? ? Los autores declaran no tener ningún conflicto de intereses. Tabla 2 Criterios diagnósticos de la linfohistiocitosis hemofagocı́tica A. Criterios diagnósticos iniciales Fiebre Esplenomegalia Citopenias ( 2 de las 3 lı́neas en la sangre periférica): Hemoglobina < 90 g/L (recién nacidos < 4 semanas: hemoglobina < 100 g/l) Plaquetas < 100 x 109/L Neutrófilos < 1,0 x 109/L Hipertrigliceridemia y/o hipofibrinogenemia Triglicéridos 3,0 mmol/L ( 265 mg/dL) Fibrinógeno 1,5 g/L Hemofagocitosis en la médula ósea, bazo o ganglios linfáticos B. Nuevos criterios diagnósticos Actividad de células NK baja o ausente Ferritina 500 mg/L CD25 soluble (receptor de IL-2 soluble) 2.400 U/mL IL-2: interleucina 2. Bibliografı́a 1. Ausina V, Urrutia A. Infecciones bacterianas. In: Rozman C, Cardellach F, editors. Farreras-Rozman Medicina Interna. 16a ed, Elsevier: Barcelona; 2009. p. 2259– 391. 2. Ausina V, Urrutia A. Infecciones vı́ricas. In: Rozman C, Cardellach F, editors. Farreras-Rozman Medicina Interna. 16a ed, Elsevier: Barcelona; 2009. p. 2490– 573. 3. Timothy J. Infecciones micobacterianas y fúngicas. In: Harris E, Budd R, Firestein G, Genovese M, Sergent J, editors. Kelley. Tratado de Reumatologı́a. Madrid: Elsevier; 2006. p. 1660–74. 4. Chakravarty E, Genovese M. Sı́ndromes musculoesqueléticos en las neoplasias malignas. In: Harris E, Budd R, Firestein G, Genovese M, Sergent J, editors. Kelley. Tratado de Reumatologı́a. Madrid: Elsevier; 2006. p. 1769–86. 5. Ramı́rez C, Rubio C, Fernández R, Aguilera C, Espejo I, Fuentesa F. Significado clı́nico de los valores elevados de ferritina sérica. Med Clin (Barc). 2004;122:532–4. 6. Fardet L, Coppo P, Kettaneh A, Dehoux M, Cabane J, Lambotte O. Low glycosylated ferritin: A good marker for the diagnosis of hemophagocytic syndrome. Arthritis Rheum. 2008;58:1521–7. Documento descargado de http://www.elsevier.es el 20/11/2016. Copia para uso personal, se prohíbe la transmisión de este documento por cualquier medio o formato. J. Muñoz-Ortego et al / Med Clin (Barc). 2011;136(1):31–37 7. Yamaguchi M, Ohta A, Tsunematsu T, Kasukawa R, Mizushima Y, Kashiwagi H, et al. Preliminary criteria for classification of adult Still’s disease. J Rheumatol. 1992;19:424–30. 8. Fautrel B, Zing E, Golmard JL, Le Moel G, Bissery A, Rioux C, et al. Proposal for a new set of classification criteria for adult-onset still disease. Medicine (Baltimore). 2002;81:194–200. 9. Lightfoot R, Michel B, Bloch D, Hunder G, Zvaifler N, McShane D, et al. The American College of Rheumatology 1990 criteria of the classification of Polyarteritis Nodosa. Arthritis Rheum. 1990;33:1088–93. 10. Ginarte M, Toribio J. Sı́ndrome de Sweet. Med Clin (Barc). 2009;133:31–5. 11. Drenth J, Meer J. Hereditary periodic fever. N Engl J Med. 2001;345:1748–57. 12. Bohan A, Peter JB. Polymiositis and dermatomyositis. N Engl J Med. 1975; 292:344–407. 13. Reichlin M. ANA negative systemic erythematosus sera revisited serologically. Lupus. 2000;9:116–9. 14. Venrooij J, Beers J, Pruijn G. Anti-CCP Antibody, a Marker for the Early Detection of Rheumatoid Arthritis. Ann N Y Acad Sci. 2008;1143:268–85. 15. Andres E, Kurtz J, Perrin A, Pflumio F, Ruellan A, Goichot B, et al. Retrospective monocentric study of 17 patients with adult Still’s disease, with special focus on liver abnormalities. Hepatogastroenterology. 2003;50:192–5. 16. Zhu G, Liu G, Liu Y, Xie Q, Shi G. Liver abnormalities in adult onset Still’s disease: a retrospective study of 77 Chinese patients. J Clin Rheumatol. 2009;6:284–8. 17. Mylona E, Golfinopoulou S, Samarkos M, Fanourgiakis P, Papadakos V, Skoutelis A. Acute hepatitis in adult Still’s disease during corticosteroid treatment successfully treated with anakinra. Clin Rheumatol. 2008;27:659–61. 18. Taccone F, Lucidi V, Donckier V, Bourgeois N, Decaux G, Vandergheynst F. Fulminant hepatitis requiring MARS and liver transplantation in a patient with Still’s disease. Eur J Intern Med. 2008;19:26–8. 19. Hoofnagle J. Reactivation of hepatitis B. Hepatology. 2009;5:156–65. 20. Liang TJ. Hepatitis B: the virus and disease. Hepatology. 2009;49:13–21. 21. Hot A, Toh ML, Coppéré B, Perard L, Madoux MH, Mausservey C, et al. Reactive hemophagocytic syndrome in adult-onset Still disease: clinical features and long-term outcome: a case-control study of 8 patients. Medicine (Baltimore). 2010;89:37–46. 22. Tristano AG. Macrophage activation syndrome: A frequent but under-diagnosed complication associated with rheumatic diseases. Med Sci Monit. 2008;14:27–36. 23. Félix F, Leal L, Fontenele J. Cloak and dagger: the case for adult onset still disease and hemophagocytic lymphohistiocytosis. Rheumatol Int. 2009;29:973–4. 37 24. Henter J, Horne A, Aricó M, Egeler R, Filipovich A, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31. 25. Chang CY, Schiano TD. Review article: drug hepatotoxicity. Aliment Pharmacol Ther. 2007;25:1135–51. 26. Roberts E, Schilsky M. Diagnosis and treatment of Wilson disease: an update. Hepatology. 2008;47:2089–111. 27. Granito A, Muratori P, Ferri S, Pappas G, Quarneti C, Lenzi M, et al. Diagnosis and therapy of autoimmune hepatitis. Mini Rev Med Chem. 2009;9:847–60. 28. Lee WM. Etiologies of acute liver failure. Semin Liver Dis. 2008;28:142–52. 29. Stepp SE, Dufourq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. 1999;286:1957–9. 30. Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, et al. Munc 13-4 is essential for cytolytic granules fusion and it is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 2003;115:461–73. 31. Zur Stadt U, Beutel K, Kolberg S, Schneppenheim R, Kabisch H, Janka G, et al. Mutation spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular and functional analyses of PRF1, UNC13D, STX11, and RAB27A. Hum Mutat. 2006;27:62–8. 32. Henter J-I, Horne AC, Aricó M, Egeler M, Filipovich AH, Imashuku S, et al. HLH2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124–31. 33. Hot A, Toh M-L, Coppéré B, Perard L, Madoux MH, Mausservey C, et al. Reactive hemophagocytic syndrome in adult-onset Still disease: clinical features and long-term outcome: a case-control study of 8 patients. Medicine (Baltimore). 2010;89:37–49. 34. Wolgamot G, Yoo J, Hurst S, Gardner G, Olerud J, Argenyi Z. Unique histopathologic findings in a patient with adult-onset Still disease. Am J Dermatopathol. 2007;29:194–6. 35. Kieffer C, Cribier B, Lipsker D. Neutrophilic urticarial dermatosis: a variant of neutrophilic urticaria strongly associated with systemic disease. Report of 9 new cases and review of the literature. Medicine (Baltimore). 2009;88: 23–31. 36. Kakar S, Kamath PS, Burgart LJ. Sinusoidal dilatation and congestion in liver biopsy: is it always due to venous outflow impairment? Arch Pathol Lab Med. 2004;128:901–4. 37. Min JK, Cho CS, Kim HY, Oh EJ. Bone marrow findings in patients with adult Still’s disease. Scand J Rheumatol. 2003;32:119–21.