Hiperpituitarismo, talla alta y exceso de crecimiento
Anuncio

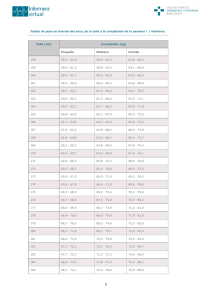
Capítulo 554 Hiperpituitarismo, talla alta y exceso de crecimiento & e554-1 La hipersecreción primaria de hormonas hipofisarias por un adenoma es poco frecuente en la infancia. El adenoma diagnosticado con más frecuencia en la infancia es el prolactinoma, seguido del corticotropinoma y del somatotropinoma, que secretan prolactina, corticotropina y hormona del crecimiento, respectivamente. Se han descrito unos pocos casos de tirotropinomas en niños y adolescentes. No se han publicado casos pediátricos de gonadotropinoma. Los hamartomas hipotalámicos que secretan un exceso de hormona liberadora de gonadotropinas no son una causa infrecuente de pubertad precoz. La naturaleza monoclonal de la mayoría de los adenomas hipofisarios implica que la mayoría se origina a raíz de un suceso clonal en una única célula. Se sospecha que algunos tumores hipofisarios se deben a la estimulación con hormonas liberadoras hipotalámicas, y en otros casos, como en el síndrome de McCune-Albright (SMA), el tumor está causado por mutaciones activadoras del gen GNAS1 que codifica la subunidad a de Gsa, una proteína que une guanil nucleótidos. La presentación clínica suele depender de la hormona hipofisaria que se hipersecreta. Son frecuentes alteraciones de la regulación del crecimiento y/o de la maduración sexual debido a una hipersecreción hormonal o a una compresión local por el tumor. El SMA asocia también displasia fibrosa poliostótica y manchas café con leche de diferente localización. TALLA ALTA La distribución normal de la altura predice que el 2,3% de la población tendrá una talla 2 DE (97,7%) por encima de la media. La aceptación social e incluso el deseo de altura («altismo») hacen que la talla alta sea un motivo de consulta poco frecuente. En Norteamérica es excepcionalmente infrecuente que los niños y hombres busquen atención médica por una talla excesiva, aunque en Europa es algo más habitual. Incluso en las niñas y las mujeres, la talla alta se ha convertido en algo socialmente más aceptado, aunque las niñas continuarían acudiendo a su médico con el deseo de frenar su velocidad de crecimiento. © ELSEVIER. Fotocopiar sin autorización es un delito. Diagnóstico diferencial La tabla 554-1 enumera las causas de talla alta en la infancia y la adolescencia. De éstas, la variante normal, familiar o constitucional de talla alta es con diferencia la causa más frecuente. De forma casi invariable, se puede obtener una historia familiar de talla alta y no existe ninguna patología orgánica. El niño suele ser más alto que sus pares durante la infancia y disfruta de una salud excelente. Los padres del adolescente con talla alta constitucional podrían entristecerse por su propia adolescencia como jóvenes altos. La exploración física no muestra alteraciones y las pruebas de laboratorio, si se obtienen, son negativas. Otras características de los síndromes de hipercrecimiento se recogen en la tabla 554-2. El síndrome de Klinefelter (síndrome XXY) es una alteración relativamente frecuente (1:500-1.000 recién nacidos vivos varones) asociada a talla alta, dificultades de aprendizaje (incluyendo necesidad de logopedia), ginecomastia y una disminución en la relación entre los segmentos superior e inferior del cuerpo. Los niños afectados pueden tener hipotonía, clinodactilia e hipertelorismo. Los testículos son invariablemente pequeños aunque la producción de andrógenos por las células de Leydig suele situarse en el límite bajo de la normalidad. La espermatogénesis y la función de las células de Sertoli son deficientes, por lo que existe infertilidad. Pueden encontrarse otras alteraciones genitales como un pene relativamente pequeño, hipospadias y criptorquidia. El síndrome XYY se asocia con talla alta, problemas en el desarrollo motor y del lenguaje y posible comportamiento antisocial. El síndrome de Marfan es un trastorno del tejido conjuntivo autosómico dominante que consiste en talla alta, aracnodactilia, extremidades finas, gran envergadura y una disminución en la relación entre los segmentos superior e inferior del cuerpo (cap. 693). Pueden presentar otras alteraciones como trastornos oculares (p. ej., subluxación del cristalino), hipotonía, cifoescoliosis, malformaciones de las válvulas cardíacas y dilatación de la raíz aórtica. Tabla 554-1 DIAGNÓSTICO DIFERENCIAL DE LA TALLA ALTA Y LOS SÍNDROMES DE HIPERCRECIMIENTO HIPERCRECIMIENTO FETAL Diabetes mellitus materna Gigantismo cerebral (síndrome de Sotos) Síndrome de Weaver Síndrome de Beckwith-Wiedemann Otros síndromes por exceso de IGF-2 HIPERCRECIMIENTO POSTNATAL QUE PRODUCE TALLA ALTA EN LA INFANCIA Talla alta familiar (constitucional) Gigantismo cerebral Síndrome de Beckwith-Wiedemann Obesidad exógena Secreción excesiva de GH (gigantismo hipofisario) Síndrome de McCune-Albright o MEN asociado con exceso de secreción de GH Pubertad precoz Síndrome de Marfan Síndrome de Klinefelter (XXY) Síndromes por exceso de SHOX Síndrome de Weaver Síndrome de X frágil Homocistinuria XYY Hipertiroidismo HIPERCRECIMIENTO POSTNATAL QUE PRODUCE TALLA ALTA ADULTA Talla alta familiar (constitucional) Déficit de andrógenos o estrógenos, o resistencia a los estrógenos (en varones) Feminización testicular Déficit o resistencia a ACTH o cortisol Secreción excesiva de GH (gigantismo hipofisario) Síndrome de Marfan Síndrome de Klinefelter (XXY) XYY ACTH, corticotropina; GH, hormona del crecimiento; IGF, factor de crecimiento similar a insulina; MEN, neoplasia endocrina múltiple. La homocistinuria es un error congénito del metabolismo de los aminoácidos autosómico recesivo producido por la deficiencia de la enzima cistationina sintetasa. Se caracteriza por retraso mental si no se trata, y muchas de sus características clínicas recuerdan al síndrome de Marfan, en especial las manifestaciones oculares (cap. 79). El hipertiroidismo en los adolescentes se asocia a un crecimiento rápido, pero la talla adulta es normal. Casi siempre está causado por una enfermedad de Graves y es mucho más frecuente en las niñas (cap. 562). La obesidad exógena es un trastorno habitual en la adolescencia y puede asociarse a un crecimiento lineal rápido y a un comienzo precoz de la pubertad. La talla adulta suele ser normal. El propósito de la evaluación diagnóstica de la talla alta es distinguir las habituales variantes constitucionales normales de las poco frecuentes entidades patológicas. A menudo, cuando la historia sugiere una talla alta familiar y la exploración física es completamente normal, no está indicado realizar pruebas de laboratorio. Es interesante obtener la edad ósea mediante una radiografía para predecir la talla adulta, lo que sirve como base para comunicarse con la familia y para la toma de decisiones. Si la historia sugiere alguno de los trastornos previamente mencionados o la exploración física muestra alguna alteración, deben obtenerse pruebas complementarias adicionales. El factor de crecimiento similar a la insulina 1 (IGF-1) y la proteína transportadora de IGF-3 (IGFBP-3) son excelentes pruebas de cribado del exceso de GH y pueden confirmarse con una prueba de supresión con glucosa. La comprobación analítica de un exceso de GH obliga a realizar una evaluación de la hipófisis mediante RM. El análisis cromosómico es útil en los niños varones, en especial cuando la relación entre los segmentos superior e inferior del cuerpo está disminuida o cuando existen alteraciones del desarrollo, para descartar un síndrome de Klinefelter. Si se sospecha un síndrome de Marfan o una homocistinuria a partir de la exploración física, el paciente debe ser remitido al cardiólogo y al oftalmólogo. Las pruebas de función tiroidea son útiles para diagnosticar o descartar un hipertiroidismo cuando se sospecha este trastorno. e554-2 & Parte XXVI Sistema endocrino Tabla 554-2 SÍNDROMES GENÉTICOS DE HIPERCRECIMIENTO SÍNDROMES GENÉTICOS CARACTERÍSTICAS CLÍNICAS Síndrome de BeckwithWiedemann* Hipoglucemia, macroglosia, pliegues auriculares, onfalocele o hernia umbilical, hemihiperplasia Síndrome de Perlman* Macrosomía, facies peculiar Nefroblastosis Facies tosca, macroglosia, surco central en labio inferior, mamilas supernumerarias Síndrome de SimpsonGolabi-Behmel* Síndrome de Sotos Síndrome PTEN hamartoma (Bannayan-RuvalcabaRiley) Síndrome de Weaver Síndrome de Marfan tipo I Síndrome de Marfan tipo II o síndrome de Loeys-Dietz Síndrome de Beals Homocistinuria Síndrome de Lujan Aneuploidía de cromosomas sexuales Klinefelter 47XXY 47XYY, 47XXX Anomalías autosómicas Tetrasomía 12p en mosaico*, pat 11pdup, 4pdub, 22q13del, 15q26-qter dup Facies típica (cara fina y alargada, frente ancha) Problemas de alimentación Hipotonía Macrocefalia (>percentil 97) habitualmente progresiva desde el nacimiento, hipotonía, piel pigmentada, máculas pigmentadas en pene, lipomas Frente ancha, hipertelorismo, micrognatia, filtrum largo, campodactilia, pulpejos de los dedos prominentes Facies característica, aracnodactilia, escoliosis, pectus carinatum o excavatum, dilatación de la raíz aórtica, luxación del cristalino Habito marfanoide, dilatación raíz aórtica, disección aórtica, vasculopatía INCIDENCIA DE MALIGNIDAD (%) ETIOLOGÍA 7,5 Rara autosómica recesiva 7,5 Recesivo ligado al X (mutaciones de glipican-3) 4 Normalmente mutación o deleción de novo dominante de NSD1 Raros casos familiares Mutación esporádica o autosómica dominante de PTEN Desconocida 5-6 Artrogriposis distal congénita, hélix plegado Habito marfanoide Retraso del desarrollo Luxación del cristalino Habito marfanoide con discapacidad intelectual PRUEBAS COMPLEMENTARIAS Y MANEJO ECO cardíaca, renal FISH y/o MLPA de cromosoma 11p, estudios de metilación Vigilancia de tumores ECO cerebral (ACC), cardíaca (coartación), renal ECO cardíaca, renal Rayos X columna (segmentación vertebral anómala) Vigilancia de tumores ECO cardíaca, renal Vigilar desarrollo ECO cerebral, cardíaca y renal Vigilar desarrollo Rara, desconocida ECO cerebral, cardíaca y renal Autosómica dominante fibrilina-1 (FBN1) Examen y seguimiento oftalmológico ECO y seguimiento cardiológico Vigilar escoliosis Autosómica dominante, alteración de la vía del TGF-b y genes TGFBR1 y TGFBR2 Autosómica dominante fibrilina 2 (FBN2) Autosómica recesiva Mutación de la cistationina b sintetasa (CBS) Recesivo ligado al X gen MED12 Examen oftalmológico habitualmente normal ECO y seguimiento cardiológico Vigilar escoliosis Examen oftalmológico y ECO cardíaca habitualmente normales Cribado metabólico en orina Examen oftalmológico Vigilar desarrollo Examen oftalmológico habitualmente normal ECO cardíaca habitualmente normal Tratamiento sustitutivo con andrógenos desde la pubertad en el síndrome de Klinefelter Vigilar desarrollo Vigilar desarrollo Talla alta, testículos pequeños, ginecomastia Talla alta dificultades de aprendizaje Hipercrecimiento congénito o talla alta en la infancia con discapacidad intelectual *El hipercrecimiento suele estar presente al nacimiento. ACC, agenesia de cuerpo calloso; aFP, a-fetoproteína; ECO, ecografía; FISH, hibridación fluorescente in situ; MLPA, amplificación de ligasa de múltiples sondas; PTEN, fosfatasa y homólogo de la tensina; TGF, factor de crecimiento transformante; TGFBR, receptor b del factor de crecimiento transformante. De Verge CF, Mowat D: Overgrowth, Arch Dis Child 95:458–463, 2010. La pubertad precoz, ya sea central (aumento de la secreción de gonadotropinas) o periférica (aumento de la secreción de andrógenos, estrógenos o de ambos), produce un crecimiento lineal acelerado durante la infancia, que simula el estirón puberal. Debido a que la maduración esquelética también se encuentra avanzada, la talla adulta suele comprometerse. La evaluación diagnóstica y el tratamiento de la pubertad precoz se exponen en el capítulo 556. Aunque la pubertad retrasada puede asociarse con una talla baja durante la infancia, como en el retraso constitucional, el retraso del desarrollo puberal y de la maduración sexual completa puede conllevar un crecimiento sostenido durante la edad adulta, con una talla final alta. El caso de una talla alta con epífisis abiertas como consecuencia de una mutación del receptor de estrógenos en un varón con una maduración sexual masculina normal subraya el papel fundamental de los estrógenos en la estimulación de la fusión de las epífisis y la terminación del crecimiento esquelético normal. El déficit de aromatasa produce una talla alta por un mecanismo similar. Además, la resistencia a los andrógenos se asocia con una talla alta en las niñas, lo que demuestra el papel de los andrógenos en este proceso. Tratamiento Tranquilizar a la familia y a los pacientes es la clave en el tratamiento de la variante normal de talla alta. El uso de la edad ósea para predecir la talla adulta podría proporcionarles cierta seguridad, como lo harían conversaciones de apoyo sobre la aceptabilidad social de este trastorno. Aunque existe un tratamiento para las niñas y los niños con crecimiento excesivo, su uso debería restringirse a los pacientes con una predicción de talla adulta >3-4 DE por encima de la media (79 pulgadas o 200 cm en los niños, 73 pulgadas © ELSEVIER. Fotocopiar sin autorización es un delito. Capítulo 554 Hiperpituitarismo, talla alta y exceso de crecimiento & e554-3 o 185 cm en las niñas), con una afectación psicosocial significativa. Para la familia que cree firmemente en el tratamiento, debe considerarse hacer una prueba con esteroides sexuales. Los esteroides sexuales se han usado en el tratamiento de la talla alta para acelerar la pubertad y promover la fusión de las epífisis; por ello tienen un beneficio escaso cuando se administran en la pubertad tardía. El tratamiento se inicia preferiblemente antes de la pubertad o al comienzo de la misma. En los niños varones el tratamiento debería comenzar antes de alcanzar una edad ósea de 14 años. Aunque la testosterona puede darse como tratamiento de la talla alta constitucional en niños, rara vez se emplea. En la práctica clínica se usa enantato de testosterona a dosis de 500 mg por vía intramuscular cada 2 semanas durante 6 meses. En las niñas, los estrógenos orales a diferentes dosis se han empleado con éxito para reducir la talla prevista en 5-10 cm de media. Esto es un resultado directo de los efectos conocidos de los esteroides sexuales en la estimulación de la fusión epifisaria, por lo que el tratamiento debe comenzar antes de que la edad ósea haya alcanzado los 12 años. El etinilestradiol oral en dosis de 0,15-0,5 mg/ día hasta que se produzca la interrupción del crecimiento se ha utilizado con éxito en niñas. Si es necesario, se puede añadir un progestágeno después de un año sólo con estrógenos. Los efectos secundarios a corto plazo incluyen patología benigna de mama, colelitiasis, hipertensión, irregularidades menstruales, ganancia de peso, náuseas, dolores en las extremidades, galactorrea y trombosis. La reducción de la fertilidad en la vida adulta puede ser una complicación a largo plazo. La falta de una amplia experiencia con esta forma de tratamiento y los riesgos del tratamiento con estrógenos para la talla alta deben sopesarse y discutirse cuidadosamente con la familia antes de comenzar la terapia. El mecanismo de acción de los estrógenos implica efectos indirectos sobre el eje hormona del crecimiento-IGF-1 así como su efecto directo sobre las epífisis. Los estrógenos median la fusión de las epífisis tanto en las niñas como en los niños. En niñas prepúberes, la talla adulta se reduce en hasta 5-10 cm con respecto a las predicciones pretratamiento. Cuando se inicia el tratamiento tras el comienzo de la pubertad, la disminución de la talla adulta no será tan importante. El tratamiento de los niños varones con talla alta es incluso más problemático. Los estrógenos serían probablemente más eficaces para acelerar la fusión de las epífisis, pero obviamente no son deseables en los niños. Los andrógenos también aceleran la maduración ósea, presumiblemente mediante su aromatización a estrógenos, pero se asocian con virilización. En los niños con síndrome de Klinefelter, el tratamiento con testosterona puede iniciarse durante la pubertad para facilitar el desarrollo de los caracteres sexuales secundarios, y este tratamiento puede disminuir la talla final adulta. Los prolactinomas no deben confundirse con la hiperprolactinemia y la hiperplasia hipofisaria que pueden producirse en los pacientes con hipotiroidismo primario, que se trata fácilmente con hormona tiroidea (cap. 559). Las elevaciones moderadas (<200 ng/ml) de la prolactina se asocian también con varios fármacos (fundamentalmente antipsicóticos), con disfunción del tallo hipofisario, como puede ocurrir en el craneofaringioma, y con otros trastornos benignos. En la mayoría de los pacientes esto puede tratarse de forma efectiva con el agonista de la dopamina bromocriptina o el de larga acción cabergolina. Cuando el tratamiento con agonistas de la dopamina no ha sido efectivo para disminuir los niveles plasmáticos de prolactina o el tamaño del adenoma, y cuando persisten durante el tratamiento los síntomas o signos secundarios a la hiperprolactinemia o al tamaño del adenoma, debe considerarse la cirugía transesfenoidal. PROLACTINOMA En las personas jóvenes con epífisis abiertas, la hiperproducción de GH produce gigantismo; en las personas con epífisis cerradas, el resultado es la acromegalia. Con frecuencia, en el gigantismo se aprecian características de la acromegalia, incluso en niños y adolescentes. Tras el cierre de las epífisis, los rasgos acromegálicos se hacen más evidentes. El gigantismo hipofisario es extremadamente infrecuente y su causa suele ser un adenoma hipofisario, pero se ha observado gigantismo en un niño de 2,5 años de edad con un tumor hipotalámico que presumiblemente secretaba GHRH. Otros tumores, como los que forman parte de los síndromes MEN, en especial los pancreáticos, producen acromegalia por secreción de grandes cantidades de GHRH con hiperplasia secundaria de las células somatotropas. La manifestación clínica principal del gigantismo es la aceleración del crecimiento longitudinal secundaria al exceso de GH. Las manifestaciones habituales son los rasgos faciales toscos y unas manos y pies grandes. En los niños pequeños el rápido crecimiento de la cabeza puede preceder al crecimiento lineal. Algunos pacientes tienen problemas visuales y de comportamiento. En la mayoría de los casos descritos, el crecimiento anómalo se hizo evidente en la pubertad, pero el trastorno se había establecido tan pronto como en el período neonatal en un niño y a los 21 meses de edad en otro. Los gigantes rara vez crecen por encima de los 2,44 m. Los adenomas hipofisarios secretores de prolactina son los tumores hipofisarios más frecuentes en la adolescencia. Con la llegada de la RM se están detectando más tumores de este tipo, sobre todo microadenomas (<1 cm de diámetro). Las manifestaciones más frecuentes al inicio son la cefalea, la amenorrea primaria o secundaria y la galactorrea espontánea o tras estimulación. El trastorno afecta al doble de niñas que de niños; la mayoría de los pacientes ha tenido una pubertad normal antes del inicio de los síntomas. Sólo unos pocos pacientes tienen una pubertad retrasada. En algunas familias con neoplasia endocrina múltiple (MEN) de tipo I, los prolactinomas son las manifestaciones iniciales durante la adolescencia. Los niveles de prolactina pueden estar elevados de forma leve (40-50 ng/ml) o importante (10.000-15.000 ng/ml). La mayoría de los prolactinomas en los niños son grandes (macroadenomas), causan aumento de tamaño de la silla turca y, en algunos casos, provocan defectos del campo visual. Aproximadamente el 30% de los pacientes con macroadenomas desarrolla hipopituitarismo, en especial déficit de GH. De forma alternativa, los adenomas secretores de prolactina también pueden ser positivos en la tinción para GH y/o TSH y secretar una cantidad excesiva de estas hormonas. CORTICOTROPINOMA El corticotropinoma es muy infrecuente en niños, y su pico de incidencia se sitúa a los 14 años. La enfermedad de Cushing se define específicamente por un adenoma hipofisario secretor de ACTH que estimula una producción y secreción excesivas de cortisol. Es más frecuente que las causas adrenales de síndrome del Cushing, excepto en los niños más pequeños, en los que los carcinomas adrenales, aunque raros, son la causa más frecuente de esta enfermedad. Los adenomas que provocan enfermedad de Cushing son significativamente menores que todos los demás tipos de adenomas en el momento de su presentación. El indicador más sensible del exceso de secreción de glucocorticoides en la edad pediátrica es el retraso del crecimiento, que suele preceder a las demás manifestaciones. Los pacientes presentan una ganancia de peso que suele ser centrípeta más que generalizada. La interrupción de la pubertad, la hipertensión, el cansancio y la depresión también son frecuentes. La localización del microadenoma suele ser determinada por RM y la toma de muestras bilaterales del seno petroso inferior. La cirugía transesfenoidal es el tratamiento de elección de la enfermedad de Cushing en los niños. Se han descrito tasas de remisión inicial del 70-98% de los pacientes y tasas de éxito a largo plazo del 50-98%. Es frecuente observar una insuficiencia suprarrenal transitoria residual tras la cirugía, que puede durar hasta 30 meses. La radioterapia hipofisaria se usa si los niveles de cortisol permanecen elevados y/o los niveles de ACTH continúan siendo detectables. SECRECIÓN EXCESIVA DE HORMONA DEL CRECIMIENTO Y GIGANTISMO HIPOFISARIO e554-4 & Parte XXVI Sistema endocrino Las características de la acromegalia consisten principalmente en un agrandamiento de las partes distales del cuerpo, pero las manifestaciones del crecimiento anómalo implican todas las regiones del cuerpo. El perímetro craneal aumenta, la nariz se ensancha y la lengua con frecuencia aumenta de tamaño, los rasgos faciales se vuelven toscos. La mandíbula crece de forma excesiva y los dientes se separan. Los defectos del campo visual y las alteraciones neurológicas son frecuentes; los signos de aumento de la presión intracraneal aparecen más tarde. Los dedos de las manos y los pies crecen sobre todo en grosor. Puede haber cifosis dorsal. El cansancio y la laxitud son síntomas precoces. Los niveles de GH están elevados y ocasionalmente superan los 100 ng/ml. De forma típica no existe supresión de los niveles de GH por la hiperglucemia en una prueba de sobrecarga de glucosa. Los niveles de IGF-1 y de IGFBP-3 están elevados de forma constante en la acromegalia, mientras que otros factores de crecimiento no lo están. El gigantismo es infrecuente, con sólo varios centenares de casos documentados hasta la fecha. A diferencia del inicio insidioso de la acromegalia en adultos, la presentación del gigantismo es habitualmente dramática. La masa tumoral por sí misma puede causar cefalea, alteraciones visuales debido a compresión del nervio óptico e hipopituitarismo. Cerca del 50% de los pacientes también tiene una importante hiperprolactinemia como consecuencia de adenomas plurihormonales que secretan GH y prolactina. Esto se debe a que las mamosomatotropas son el tipo más frecuente de células secretoras de GH implicadas en el gigantismo infantil. Los tumores hipofisarios secretores de GH son típicamente adenomas eosinofílicos o cromofóbicos. Los adenomas pueden comprometer otras funciones de la hipófisis anterior por el crecimiento o la degeneración quística. La secreción de gonadotropinas, tirotropina o corticotropina puede estar afectada. Puede haber un retraso de la maduración sexual o un hipogonadismo. Cuando la hipersecreción de GH se acompaña de déficit de gonadotropinas, el crecimiento lineal acelerado puede persistir durante décadas. En algunos casos el tumor se extiende por fuera de la silla turca, invadiendo el hueso esfenoides, los nervios ópticos y el cerebro. En los pacientes pediátricos los tumores secretores de GH suelen ser más invasivos o agresivos que en los adultos. La etiología es incierta, aunque los estudios en acromegálicos han sugerido que muchos casos pueden deberse a mutaciones que generan una activación constitutiva de las proteínas G con una actividad GTPasa reducida. El incremento resultante del adenosina monofosfato cíclico intracelular en la hipófisis lleva a una secreción aumentada de GH. El síndrome de McCune-Albright (SMA), que también puede estar causado por mutaciones que producen proteínas G activadas de forma constitutiva, puede asimismo presentar tumores somatotropos y exceso de secreción de GH. Alrededor del 20% de los pacientes con gigantismo son aquellos con SMA (que consiste habitualmente en la tríada de pubertad precoz, manchas café con leche y displasia fibrosa). Los tumores secretores de GH también se han descrito en la adenomatosis endocrina múltiple y asociados con neurofibromatosis, esclerosis tuberosa y el complejo de Carney. En el SMA se han encontrado mutaciones activadoras de las proteínas estimuladoras Gsa en las lesiones hipofisarias y se cree que son las responsables de los otros adenomas glandulares observados también en este síndrome. Asimismo se han identificado mutaciones puntuales somáticas de la proteína Gsa en las células somatotropas hasta en un 40% de los adenomas hipofisarios esporádicos secretores de GH. Diagnóstico La prueba de referencia para el diagnóstico de exceso de GH es la no supresión de los niveles plasmáticos de GH hasta <5 ng/dl tras una sobrecarga oral con 1,75 g/kg (máximo 75 g) de glucosa. Esta prueba mide la capacidad de IGF-1 de suprimir la secreción de GH, ya que la sobrecarga de glucosa estimula la secreción de insulina, lo que produce supresión de IGFBP-1, provocando un incremento agudo de los niveles de IGF-1 libre. El incremento de IGF-1 libre suprime la secreción de GH en 30-90 minutos. Esta prueba puede proporcionar resultados anómalos en los pacientes diabéticos. Una medición aislada de GH es inadecuada porque la GH se secreta de una forma pulsátil. Por tanto, el uso de una medición al azar de GH puede dar resultados falsos positivos y falsos negativos. La medición de la concentración plasmática de IGF-1 es una prueba sensible de cribado para detectar el exceso de GH. Se ha demostrado una excelente correlación lineal dosisrespuesta entre los niveles plasmáticos de IGF-1 y la secreción media de GH en 24 horas. Una concentración elevada de IGF-1 en un paciente con una sospecha clínica adecuada indica casi siempre un exceso de GH. Puede surgir confusión al evaluar adolescentes sanos, porque durante la pubertad la concentración de IGF-1 es significativamente más alta que durante la edad adulta; los niveles de IGF-1 deben ajustarse por edad y sexo. Los niveles plasmáticos de IGFBP-3 son un marcador sensible de las elevaciones de GH y pueden estar aumentados aunque las concentraciones de IGF-1 sean normales. Si los hallazgos de laboratorio sugieren un exceso de GH, debe buscarse la presencia de un adenoma hipofisario con una RM cerebral. En casos excepcionales no se identifica una masa hipofisaria. Esto podría deberse a un microadenoma hipofisario oculto o a una producción ectópica de GHRH o GH. La TC es aceptable cuando no se dispone de RM. Tratamiento Los objetivos del tratamiento son extirpar o reducir el volumen de la masa hipofisaria, restaurar los patrones de secreción y los niveles de GH, normalizar la concentración de IGF-1 y de IGFBP-3, mantener la secreción hipofisaria normal de otras hormonas y prevenir la recidiva de la enfermedad. Para los adenomas hipofisarios bien circunscritos, la cirugía transesfenoidal es el tratamiento de elección y puede ser curativa. El tumor debe extirparse completamente. La probabilidad de curación quirúrgica depende en gran medida de la experiencia del cirujano, así como del tamaño y la extensión de la masa. Las mediciones intraoperatorias de GH pueden mejorar los resultados de la resección tumoral. La cirugía transesfenoidal para la resección de tumores es tan segura en niños como en adultos. Algunas veces puede ser necesario un abordaje transcraneal. El objetivo inicial del tratamiento es normalizar los niveles de GH. Las concentraciones de GH (<1 ng/ml 2 horas después de una sobrecarga de glucosa) y de IGF-1 (intervalo normal ajustado por edad) son las mejores exploraciones para definir una curación bioquímica. Si la secreción de GH no se normaliza mediante cirugía, las opciones incluyen la radioterapia hipofisaria y el tratamiento médico. La radioterapia estereotáxica se recomienda si la hipersecreción de GH no se normaliza mediante la cirugía. La irradiación evita el crecimiento tumoral en 99% de los pacientes. La principal desventaja es el retraso en la eficacia para disminuir los niveles de GH. La GH se reduce alrededor de un 50% respecto a la concentración inicial en 2 años, un 75% en 5 años, y se acerca al 90% en 15 años. Es previsible que se produzca un hipopituitarismo, que ocurre en el 40-50% de los pacientes 10 años después de la radioterapia. La cirugía no puede curar un número significativo de pacientes y, por tanto, el tratamiento médico tiene un importante papel en el tratamiento de los pacientes que tienen un exceso de GH. El tratamiento es efectivo y bien tolerado con análogos de acción larga de la somatostatina y con agonistas de la dopamina, así como con los nuevos antagonistas de la GH. Los análogos de la somatostatina han demostrado ser muy eficaces en el tratamiento de los pacientes con exceso de GH. La octreotida suprime la GH hasta <2,5 ng/ml en el 65% de los pacientes con acromegalia y normaliza los niveles de IGF-1 en el 70%. Los efectos de la octreotida se mantienen bien en el tiempo. También produce reducción del volumen tumoral, aunque suele ser modesta. Se puede obtener una supresión mantenida de la GH mediante una bomba de infusión subcutánea continua de octreotida en niños en edad puberal con gigantismo hipofisario. Las formulaciones de acción prolongada, incluida la octreotida de acción prolongada y la lanreotida, producen supresión mantenida de GH e IGF-1 en pacientes acromegálicos con inyecciones depot intramusculares Capítulo 554 Hiperpituitarismo, talla alta y exceso de crecimiento & e554-5 mensuales o bisemanales. Las preparaciones de liberación mantenida no han sido evaluadas oficialmente en niños. En la población pediátrica se han empleado inyecciones de octreotida a dosis de 1-40 mg/kg/24 h. Para los casos con hipersecreción de GH y de prolactina deben considerarse los agonistas dopaminérgicos, como la bromocriptina, que se une a los receptores dopaminérgicos hipofisarios de tipo 2 (D2) y suprime la secreción de GH, aunque su mecanismo de acción exacto sigue sin estar claro. Los niveles de prolactina se suprimen con frecuencia de forma adecuada; los niveles de GH y de IGF-1 rara vez se normalizan con esta modalidad de tratamiento. Menos del 20% de los pacientes alcanza niveles de GH <5 ng/ml y <10% consigue la normalización de los niveles de IGF-1. La reducción del volumen tumoral se produce en una minoría de los pacientes. La bromocriptina puede emplearse como tratamiento médico adyuvante para el exceso de GH. Su eficacia puede sumarse a la de la octreotida. Otros agonistas de la dopamina, como la pergolida y la cabergolina, ofrecen una mayor eficacia, definida por la normalización de la concentración de IGF-1 y una reducción del volumen tumoral. La dosis de bromocriptina varía entre 10-60 mg/24 horas vía oral divididos en cuatro dosis. Sólo una pequeña parte de los pacientes se beneficia de dosis >20 mg/24 h. Se ha demostrado que la utilización durante largos períodos de tiempo en niños es segura, pero los efectos secundarios pueden incluir náuseas, vómitos, dolor abdominal, arritmias, congestión nasal, hipotensión ortostática, alteraciones del sueño y cansancio. El tratamiento con octreotida de acción prolongada y cabergolina es prometedor en adultos con acromegalia; sin embargo la experiencia en pediatría es muy limitada. El pegvisomant es un antagonista del receptor de GH que compite con la GH endógena por la unión con el receptor de GH. Suprime de forma eficaz los niveles de GH y de IGF-1 en pacientes con acromegalia debida a tumores hipofisarios así como a hipersecreción ectópica de GHRH. La normalización de los niveles de IGF-1 se produce hasta en el 90% de los pacientes tratados diariamente con este fármaco durante 3 meses o más. La dosis en el adulto es de 10-40 mg vía subcutánea una vez al día, aunque protocolos de 3 veces por semana también han sido muy efectivos. Deben monitorizarse los niveles de IGF-1 y las enzimas hepáticas. El tratamiento combinado con análogos de la somatostatina e inyecciones semanales de pegvisomant también ha mostrado eficacia. La experiencia pediátrica es muy limitada. © ELSEVIER. Fotocopiar sin autorización es un delito. SÍNDROME DE SOTOS (GIGANTISMO CEREBRAL) Los niños con gigantismo cerebral (también conocido como síndrome de Sotos) se encuentran por encima del percentil 90 tanto para la longitud como para el peso al nacimiento; también pueden tener macrocefalia en ese momento. El gen NSD1 (receptor nuclear de unión al dominio de proteína-1) es responsable de este síndrome. Aunque se caracteriza por un crecimiento rápido, no hay pruebas de que el síndrome de Sotos esté causado por una alteración endocrina. Se ha sugerido un defecto hipotalámico como causa, pero no se ha podido demostrar ninguno funcionalmente o en las autopsias. El crecimiento es llamativamente rápido; al año de vida, los lactantes afectados tienen una talla superior al percentil 97. El crecimiento acelerado continúa durante los primeros 4-5 años y después vuelve a la normalidad (fig. 554-1). La pubertad suele producirse en el momento esperado, pero puede adelantarse ligeramente. La talla adulta se encuentra generalmente en el límite alto de la normalidad. El síndrome se caracteriza por una cabeza grande (macrocefalia) y dolicocefálica, una frente y una mandíbula prominentes, hipertelorismo, orientación antimongoloide de las fisuras palpebrales, paladar ojival y unas manos y unos pies grandes con engrosamiento del tejido subcutáneo. También se observa torpeza y la marcha tosca, y los niños afectados suelen tener grandes dificultades para los deportes, para aprender a montar en bicicleta y para otras tareas que requieran coordinación. La mayoría de los pacientes presenta cierto grado de retraso del desarrollo; en algunos niños afectados pueden predominar los déficits perceptivos. La maduración ósea es [(Figura_1)TD$IG] Figura 554-1 Gigantismo cerebral (síndrome de Sotos) en un niño de 8 años. La talla correspondía a un niño de 12 años de edad y la edad ósea era de 12 años. El CI era 60. El electroencefalograma estaba alterado. Nótese la prominencia de la frente y la mandíbula, y las manos y los pies de gran tamaño. El desarrollo sexual era consecuente con la edad cronológica. Los resultados del estudio hormonal eran normales. La talla final adulta fue 208 cm; su desarrollo sexual fue normal. Utiliza zapatos de la talla 54. habitualmente compatible con la talla del paciente, aunque se ha descrito la edad ósea avanzada. Las GH, el IGF-1 y otros estudios endocrinológicos suelen ser normales; no existe ningún marcador bioquímico o radiológico característico de este síndrome. Las alteraciones en los electroencefalogramas son frecuentes; los estudios de imagen suelen mostrar un sistema ventricular dilatado. Los pacientes afectados pueden presentar un mayor riesgo de desarrollar neoplasias, particularmente carcinoma hepático, aunque también se han descrito tumores de Wilms, ováricos y parotídeos. HIPERCRECIMIENTO FETAL Y NEONATAL La diabetes materna es la causa más frecuente de niños grandes para la edad gestacional (GEG). Incluso en ausencia de síntomas clínicos o de antecedentes familiares, el nacimiento de un niño de GEG indica la evaluación de una posible diabetes materna (o gestacional). Se ha descrito un grupo de trastornos asociados con un crecimiento excesivo tanto somático como de órganos específicos, conocido de forma conjunta como síndromes de hipercrecimiento. Parece que estos trastornos están causados por una producción y disponibilidad excesivas de IGF-2 codificado por el gen Igf2. El síndrome mejor descrito de este grupo es el síndrome de BeckwithWiedemann (SBW), que es un síndrome malformativo con hipercrecimiento que ocurre con una incidencia de 1:14.000 nacimientos. Se manifiesta como un síndrome de hipercrecimiento fetal en el que la hipertrofia domina el cuadro clínico. Los niños afectados son característicamente macrosómicos, con macroglosia, hepatoesplenomegalia, nefromegalia y onfalocele. También presentan hipoglucemia secundaria a una hiperinsulinemia derivada de una hiperplasia de las células b del páncreas. Los niños con SBW están predispuestos a un grupo específico de neoplasias infantiles constituido por el tumor de Wilms y el carcinoma cortical suprarrenal. La e554-6 & Parte XXVI Sistema endocrino expresión excesiva de IGF-2 en el SBW puede estar causada por un número de alteraciones genéticas entre las que se incluyen la duplicación genética, la pérdida de heterocigosidad y la relajación o la pérdida de impronta del gen Igf2. Varias líneas de investigación han localizado improntas genéticas implicadas en el SBW y los tumores infantiles asociados en el cromosoma 11p. Éstos comprenden, además del Igf2, el gen H19, que está implicado en la supresión del Igf2, así como el gen WT-1 (el gen del tumor de Wilms). Las mutaciones en GPC3, un gen glipicano (que codifica un receptor de membrana que neutraliza el IGF-2), causan el síndrome de hipercrecimiento de Simpson-Golabi-Behmel, relacionado con el anterior. BIBLIOGRAFÍA Acharya SV, Gopal RA, Bandgar TR, et al: Clinical profile and long term follow up of children and adolescents with prolactinomas, Pituitary 12:186-189, 2009. Bademci G: Pitfalls in the management of Cushing’s disease, J Clin Neurosci 14:401-408, 2007. Bulun SE: Aromatase deficiency and estrogen resistance: from molecular genetics to clinic, Semin Reprod Med 18:31-39, 2000. Carel JC, Blumberg J, Bougeard-Julien M, et al: Long-acting lanreotide in adolescent girls with constitutional tall stature, Horm Res 71:228-236, 2009. Chahal HS, Stals K, Unterlander M, et al: AIP mutation in pituitary adenomas in the 18th century and today, N Engl J Med 364 (l):43-50, 2011. Drop SL, De Waal WJ, De Muinck Keizer-Schrama SM: Sex steroid treatment of constitutionally tall stature, Endocr Rev 19:540-558, 1998. Faravelli F: NSD1 mutations in Sotos syndrome, Am J Med Genet C Semin Med Genet 137C:24-31, 2005. Feenstra J, de Herder WW, ten Have SM, et al: Combined therapy with somatostatin analogues and weekly pegvisomant in active acromegaly, Lancet 365:1644-1646, 2005. Horvath A, Stratakis CA: Clinical and molecular genetics of acromegaly: MEN1, Carney complex, McCune-Albright syndrome, familial acromegaly and genetic defects in sporadic tumors, Rev Endocr Metab Disord 9:1-11, 2008. Lania A, Spada A: G-protein and signalling in pituitary tumours, Horm Res 71(Suppl 2):95-100, 2009. Leventopoulos G, Kitsiou-Tzeli S, et al: A clinical study of Sotos syndrome patients with review of the literature, Pediatr Neurol 40:357-364, 2009. Melmed S, Colao A, Barkan A, et al: Guidelines for acromegaly management: an update, J Clin Endocrinol Metab 94:1509-1517, 2009. Melmed S, Cook D, Schopohl J, et al: Rapid and sustained reduction of serum growth hormone and insulin-like growth factor-1 in patients with acromegaly receiving lanreotide Autogel therapy: a randomized, placebo- controlled, multicenter study with a 52 week open extension, Pituitary 13:18-28, 2010. Misra M, Cord J, Prabhakaran R, et al: Growth hormone suppression after an oral glucose load in children, J Clin Endocrinol Metab 92:4623-4629, 2007. Morison IM, Becroft DM, Taniguchi T, et al: Somatic overgrowth associated with overexpression of insulin-like growth factor II, Nat Med 2:311-316, 1996. Narayanaswamy V, Rettig KR, Bhowmick SK: Excessive growth, Clin Pediatr (Phila) 47:705-708, 2008. Prevedello DM, Pouratian N, Sherman J, et al: Management of Cushing’s disease: outcome in patients with microadenoma detected on pituitary magnetic resonance imaging, J Neurosurg 109: 751-759, 2008. Reddy R, Hope S, Wass J: A cromegaly, BMJ 341:c4189, 2010. Reeve AE: Role of genomic imprinting in Wilms’ tumour and overgrowth disorders, Med Pediatr Oncol 27:470-475, 1996. Riccio A, Sparago A, Verde G, et al: Inherited and sporadic epimutations at the IGF2-H19 locus in Beckwith-Wiedemann syndrome and Wilms’ tumor, Endocr Dev 14:1-9, 2009. Rosenfield RL, Lipton RB, Drum ML: Thelarche, pubarche, and menarche attainment in children with normal and elevated body mass index, Pediatrics 123:84-88, 2009. Sacks DA: Etiology, detection, and management of fetal macrosomia in pregnancies complicated by diabetes mellitus, Clin Obstet Gynecol 50:980-989, 2007. Simm PJ, Bajpai A, Russo VC, et al: Estrogens and growth, Pediatr Endocrinol Rev 6:32-41, 2008. Socin HV, Chanson P, Delemer B, et al: The changing spectrum of TSHsecreting pituitary adenomas: diagnosis and management in 43 patients, Eur J Endocrinol 148:433-442, 2003. Sotos JF: Overgrowth: genetic syndromes and other disorders associated with overgrowth, Clin Pediatr 36:157-170, 1997. Sotos JF, Argente J: Overgrowth disorders associated with tall stature, Adv Pediatr 55:213-254, 2008. Tajima T, Tsubaki J, Ishizu K, et al: Case study of a 15-year-old boy with McCune-Albright syndrome combined with pituitary gigantism: effect of octreotide-long acting release (LAR) and cabergoline therapy, Endocr J 55:595-599, 2008. Topaloglu AK, Sansaricq C, Snyderman SE: Influence of metabolic control on growth in homocystinuria due to cystathionine B-synthase deficiency, Pediatr Res 49:796-798, 2001. Venn A, Bruinsma F, Werther G, et al: Oestrogen treatment to reduce the adult height of tall girls: long-term effects on fertility, Lancet 364:1513-1518, 2004. Verge CF, Mowat D: Overgrowth, Arch Dis Child 95:458-463, 2010. Zeger MP, Zinn AR, Lahlou N, et al: Effect of ascertainment and genetic features on the phenotype of Klinefelter syndrome, J Pediatr 152:716-722, 2008.