Epilepsia mioclónica progresiva
Anuncio

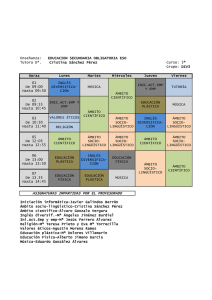
A. VISTORTE, ET AL ORIGINAL Epilepsia mioclónica progresiva: caracterización clínica de 18 pacientes A. Vistorte, N. Sardiñas, E.M. Esteban, J. Vargas-Díaz, L. Novoa-López, E. Rojas-Massippe, E.M. Pestana PROGRESSIVE MYOCLONIC EPILEPSY: THE CLINICAL CHARACTERISTICS OF 18 PATIENTS Summary. Introduction. The term progressive myoclonic epilepsy (PME) includes a groups of heterogeneous conditions, with genetic causes, characterized by having different types of seizures, basically myoclonic, and other neurological findings due to a progressive lesion of the central nervous system. Objective. To demonstrate the aetiology and clinico-encephalographic changes seen in patients with PME. Patients and methods. A retrospective, descriptive study was done of patients attended for PME in the Instituto de Neurología y Neurocirugía de Cuba between 1990 and 1995. Eighteen patients were included. All were interviewed and had a physical examination, EEG and the specific complementary tests for each aetiology. Results. There was a predominance of neural ceroid lipofuschinosis in 10 patients (55.5%), and in 9 of these the illness started before the age of 9 years. The second most frequent condition was myoclonic epilepsy with red-torn fibres (16.6%) and Unverricht-Lundborg disease (16.6%). The latter began in late childhood or adolescence. The most marked clinical characteristics were epilepsy, which was difficult to control and intellectual deterioration in 100%, followed by cerebellar signs in 88.8%. Myoclonias were the commonest type of seizures (94.4%) and many children presented with prior tonic-clonic seizures (88.8%). Conclusion. Response to treatment was poor but the best results were obtained using valproate either alone or associated with benzodiazepines [REV NEUROL 1999; 29: 102-4]. Key words. Ceroid lipofuschinosis. Intellectual deterioration. Myoclonias. Progressive myoclonic epilepsy. Valproate. INTRODUCCIÓN La epilepsia constituye una de las enfermedades neurológicas más atendidas en las consultas de Neuropediatría. Alrededor del 2% de la población sufre de epilepsia y aproximadamente las tres cuartas partes debutan en la infancia. Por otra parte, es probable que entre un 30 y un 40% de estos casos no logren un control adecuado de sus crisis; dentro de este grupo se encuentra el síndrome de epilepsia mioclónica progresiva (EMP) [1,2]. Este término agrupa a un conjunto de entidades clínica y etiológicamente heterogéneas provocadas por alteraciones genéticas, y que se caracterizan por presentar diferentes tipos de crisis epilépticas, principalmente mioclónicas, deterioro intelectual y otras manifestaciones clínicas, predominantemente cerebelosas, que originan una lesión progresiva del sistema nervioso central (SNC) [1,2]. Las causas más frecuentes y que constituyen la mayoría de los casos de EMP son [3-7]: – Enfermedad de Unverricht-Lundborg – Enfermedad de Lafora – Lipofuscinosis ceroide neural en sus distintas variedades: – Infantil precoz o enfermedad de Santavuori-Haltia – Infantil tardía o enfermedad de Jansky-Bielschowsky – Juvenil o enfermedad de Spielmeyer-Vogt – Adulta o enfermedad Kufs – Sialidosis – Encefalopatía mitocondrial y EMP. El diagnóstico etiológico de estas entidades resulta fundamental pues permite seleccionar el tratamiento más apropiado, asesorar desde el punto de vista genético al paciente y a sus familiares y predecir adecuadamente el pronóstico en cada caso. La motivación fundamental de la presente investigación ha sido describir la etiología y las características clínicas y electroencefalográficas de los pacientes diagnosticados de EMP en el Instituto de Neurología y Neurocirugía (INN) de Cuba. PACIENTES Y MÉTODOS Existen otros cuadros que, con mucha menos frecuencia, pueden cursar con EMP; entre ellos se encuentran la distrofia neuroaxonal, la encefalopatía sensible a la biotina, el síndrome de insuficiencia renal y las mioclonías [2,5,8]. Se ha realizado un estudio retrospectivo y descriptivo de todos los pacientes diagnosticados como EMP que debutaron en edades comprendidas entre 1 y 25 años, y que fueron estudiados en el INN en el período 1990-1995. La muestra se seleccionó a partir del registro hospitalario de 823 pacientes que se diagnosticaron como epilépticos en ese período en el INN. Los criterios de selección utilizados fueron: evidencias clínicas de epilepsia mioclónica de difícil control, acompañada por otros tipos clínicos de crisis, deterioro psicomotor o intelectual, manifestaciones neurológicas de localización difusa en el SNC, sobre todo de tipo cerebeloso, e identificación etiológica definida de alguno de los procesos que se han descrito como causa del síndrome de EMP [5]. La muestra quedó conformada por 18 pacientes que cumplían estrictamente los criterios diagnósticos de EMP. En cada caso se registraron los datos de edad en el momento del diagnóstico, sexo, tipo de crisis epiléptica al inicio de la enfermedad, manifestaciones clínicas neurológicas y de otros sistemas afectados, exámenes complementarios más importantes realizados para confirmar el diagnóstico y sus resultados, incluyendo el EEG del cual se recogió el tipo de alteración paroxística, la actividad de base y la presencia de respuesta fotosensible. Respecto al tratamiento, se registraron los fármacos antiepilépticos utilizados y la respuesta a los mismos. Los datos se incorporaron a una base de datos y se procesaron mediante el sistema estadístico SPSS utilizando la distribución de frecuencias. Recibido: 22.03.99. Aceptado tras revisión externa sin modificaciones: 02.05.99. RESULTADOS Servicio de Neurología Infantil. Instituto de Neurología y Neurocirugía. La Habana, Cuba. Al analizar nuestra casuística, encontramos que el grupo de edades de entre 5 y 9 años fue el más nutrido, ya que en él debutaron 9 casos, lo que representa un 50% del total. En cuanto a la etiología, 6 de ellos se correspondían con lipofuscinosis ceroide neural (LFC), variedad Spielmeyer-Vogt. Los restantes correspondían a dos niños con enfermedad de Unverricht-Lundborg y uno con enfermedad de Lafora. El 22,2% de los casos debutaron entre Correspondencia: Dr. Norberto Sardiñas Hernández. Servicio de Neurología Infantil. Instituto de Neurología y Neurocirugía. 29 y D, Vedado. CP 10400 La Habana, Cuba. E-mail: [email protected] 1999, REVISTA DE NEUROLOGÍA 102 REV NEUROL 1999; 29 (2): 102-104 EPILEPSIA MIOCLÓNICA PROGRESIVA Tabla I. Relación entre la edad de presentación y la etiología de la epilepsia mioclónica progresiva. Tabla II. Epilepsia mioclónica progresiva: manifestaciones clínicas asociadas más frecuentemente. Edad Etiología Manifestaciones clínicas 1-4 años LFC 3 16,6 MERRF 1 5,5 LFC 6 33,3 U-Lundborg 2 11,1 Enf. de Lafora 1 5,5 LFC 1 5,5 MERRF 1 5,5 U-Lundborg 1 5,5 15-19 años MERRF 1 5,5 5,5 20-24 años Mioclonía y fallo renal 1 5,5 5,5 18 100,0 100,0 5-9 años 10-14 años N.º de casos Total % Total % 22,1 50,0 16,6 los 0 y 4 años de edad, y en este grupo predominó también la LFC, variedad Jansky-Bielschowsky (Tabla I). En cuanto al sexo, el mayor número de casos correspondió al sexo masculino con 11 niños (71%). Entre las manifestaciones neurológicas, las más frecuentes fueron la epilepsia (100%), seguida de las manifestaciones cerebelosas en 16 pacientes (88,8%) y en 9 casos se detectaron trastornos oftalmológicos (Tabla II). Es de destacar que los 18 pacientes estudiados presentaron deterioro intelectual. Con relación al tipo de crisis (Tabla III), las más frecuentes fueron las mioclónicas en 17 casos (94,4%), y las tonicoclónicas generalizadas en 16 (88,8%). En la tabla IV se muestran las alteraciones del EEG en los pacientes con EMP. El 100% de los pacientes mostró algún tipo de alteración, siendo las más frecuentes los paroxismos focales en 17 pacientes (94%) y el sufrimiento cortical global en 16 (88,8%). Se realizaron diversos estudios para comprobar el diagnóstico etiológico específico de las EMP. Entre ellos, los potenciales evocados visuales (PEV), realizados a 10 pacientes (55,5%), mostraron las alteraciones correspondientes a las LFC. Los potenciales somatosensoriales (PESS) fueron patológicos en 4 niños (22,2%). El electromiograma (EMG) mostró alteraciones en los 3 niños (16,6%) portadores del síndrome de MERRF (del inglés, Myoclonus Epilepsy and Ragged-Red Fibers). Las biopsias realizadas (piel, músculo, conjuntiva y nervio) fueron patológicas en 13 (72,2%) de los casos, y se observaron alteraciones en relación con la entidad nosológica que presentaban estos pacientes. Respecto a la terapéutica antiepiléptica empleada, se administró valproato de sodio en 11 de los casos (61,1%), clonacepam en 7 (38,8%), fenobarbital en 6 pacientes (33,3%), carbamacepina y fenitoína en 5 casos (27,8%) y otros medicamentos en 13 (72,2). En todos los pacientes (100%) se utilizó politerapia. En nuestra serie sólo 3 (16,6%) pacientes lograron la supresión parcial de las crisis. El diagnóstico etiológico en estos casos fue: uno con EMP y fallo renal, otro padecía una enfermedad de Unverricht-Lundborg y el último era portador de un síndrome MERRF. DISCUSIÓN Como se desprende de nuestra serie, existe un franco predominio de comienzo de los síntomas de la enfermedad antes de los 9 años de edad y las LFC son las entidades más frecuentes, tal y como se informa en la literatura [2]. Queremos destacar que la mayoría de los autores señalan las sialidosis dentro de las cinco primeras causas de EMP, pero en nuestra serie no tuvimos ningún niño REV NEUROL 1999; 29 (2): 102-104 N.º de casos % Epilepsia de difícil control 18 100,0 Deterioro intelectual 18 100,0 Trastornos cerebelosos 16 88,8 Fallo visual progresivo 7 38,8 Hipoacusia 1 5,5 Tabla III. Tipos de crisis más frecuentes en las epilepsias mioclónicas progresivas. Tipo de crisis N.º de casos % Mioclónicas 17 94,4 Tonicoclónicas generalizadas 16 88,8 Parciales 5 27,8 Crisis de ausencia 1 5,5 Tabla IV. Manifestaciones electroencefalográficas en las epilepsias mioclónicas progresivas. EEG N.º de casos % Paroxístico focal 17 94,0 Sufrimiento cortical global 16 88,8 Ausencia de ritmo de base 7 38,8 Fotosensibilidad 5 27,7 Paroxístico generalizado 1 5,5 portador de esta entidad y sí un caso de EMP y fallo renal que se describe como menos frecuente [8]. En cuanto al sexo, el predominio del sexo masculino creemos que es puramente casual ya que no existe predilección por el sexo en ninguno de estos cuadros [5]. Además de las crisis epilépticas, se asociaron otras manifestaciones neurológicas; de ellas, las más frecuentes fueron las cerebelosas y en algunos casos se detectaron trastornos oftalmológicos, lo que se explica por la mayor frecuencia de LFC que incluye en su cuadro clínico fallo visual progresivo. Sólo en un caso de LFC no se observaron las alteraciones típicas del fondo de ojo, si bien estudios neuroftalmológicos (ERG, PEV) y la biopsia de conjuntiva corroboraron el diagnóstico en este paciente. La mayoría de los autores revisados coinciden en señalar la presencia de las mencionadas alteraciones oftalmológicas en estas entidades [9-11]. Todos los pacientes estudiados presentaron deterioro intelectual, tal como aparece descrito en la literatura [12]. Con relación al tipo de crisis, queremos señalar que en el momento del diagnóstico un niño no presentaba crisis mioclónicas pero posteriormente, durante la evolución de su enfermedad, éstas aparecieron. Destaca que la mayoría de los pacientes debutaron con crisis tonicoclónicas generalizadas, antes de las mioclonías, dato que no difiere de la literatura revisada [1-3]. El EEG mostró alteraciones en todos los casos. Las más frecuentes fueron los signos de sufrimiento cortical global y los 103 A. VISTORTE, ET AL elementos paroxísticos localizados. En todos los casos de enfermedad de Unverricht-Lundborg se presentó fotosensibilidad, como se ha descrito; sin embargo, a pesar de que predominaron las LFC, en ninguno de nuestros pacientes se comprobó el fenómeno de Pampiglioni positivo, hecho descrito con relativa frecuencia en esta entidad, fundamentalmente en la variedad de Jansky-Bielschowsky [13]. En nuestra casuística el medicamento más utilizado fue el valproato de sodio, solo o asociado a benzodiacepinas. Estos fármacos constituyen hasta el momento la mejor opción terapéutica [14,15], ya que no contamos con medicamentos de nueva generación como la zonizamida que se preconiza en la actualidad para el control de crisis mioclónicas. En nuestra serie sólo 3 pacientes lograron la supresión parcial de las crisis. Ello demuestra que estas entidades son generalmente refractarias al tratamiento, por lo que podemos englobarlas dentro del grupo de epilepsias de difícil control [16]. CONCLUSIONES 1. La mayoría de los casos estudiados debutaron en edad pediátrica, fundamentalmente en el grupo de 5 a 9 años. 2. La etiología predominante en las EMP fue la lipofuscinosis ceroide neuronal. 3. El sexo más afectado fue el masculino. 4. La epilepsia de difícil control, el deterioro intelectual y las manifestaciones de tipo cerebeloso fueron las manifestaciones clínicas más frecuentemente observadas. 5. Las crisis epilépticas predominantes fueron las mioclónicas y las tonicoclónicas generalizadas. 6. El EEG se comportó con cambios de tipo paroxístico focal y signos de sufrimiento cortical global, principalmente. 7. Los medicamentos más utilizados fueron, de forma combinada, el valproato de sodio y las benzodiacepinas. 8. No existió control de las crisis de forma significativa en la mayoría de los pacientes. BIBLIOGRAFÍA 1. Delgado-Escueta AV, Serratosa JM, Liu A, Weissbecker K, Medina 10. Berkovic SF, Carpenter S, Evans A. Myoclonus epilepsy and raggedMT, Gee M, et al. Progress in mapping human epilepsy genes. Epilepred fibres (MERRF). A clinical, pathological, biochemical, magnetic sia 1994; 35 (Suppl 1): S529-40. resonance spectrographic positron emission tomography study. Brain 2. Berkovic SF, Anderman F, Carpenter F, Wolfe LF. Progressive myo1989; 112: 1231-60. clonus epilepsies: specific causes and diagnostic. N Engl J Med 1986; 11. Jarvela D, Schleutrer J, Haataja L. Infantile neuronal ceroid lipofuscinosis 15: 296-305. (CLNI) maps to the short arm of chromosome 1. Genomic 1991; 9: 170-3. 3. Aicardi J. Epilepsy in children. New York: Raven Press; 1996. 12. Anderman F, Berkovic S, Carpenter S, Wolfe LS. The Ramsay Hunt syn4. Malafosse A, Lehesjoki A, Genton P. Identical genetic locus for Baltic drome is no longer a useful diagnostic entity. Mov Disord 1989; 4: 3-7. and Mediterranean myoclonus. Lancet 1992; 339: 1080-1. 13. Pampiglioni G, Harden A. So-called neuronal ceroid lipofuscinosis: 5. Roger G, Genton P, Bureau M, Dravet Ch. Progressive myoclonus epilepneurophysiological studies in 60 children. J Neurol Neurosurg Psychisies in childhood and adolescence. London: John Libbey; 1992. p. 13-23. atry 1977; 40: 323-30. 6. Ponsford S, Pye IF, Elliot EJ. Posterior proximal discharge: an aid to 14. Lavanainen M, Hilberg JJ. Valproate and clonazepam in the treatment early diagnosis in Lafora disease. J R Soc Med 1993; 86: 597-9. of severe progressive myoclonus epilepsy. Arch Neurol 1982; 38: 7. Kaufaman MA, Dwark AJ, Wilson NJ, Jahn S. Late-onset Lafora’s dis236-8. ease with typical intraneuronal inclusions. Neurology 1993; 43: 1246-8. 15. Obeso JA, Artieda J, Rothwell JC, Day B, Thompson P, et al. The 8. Anderman E, Anderman F, Carpenter S. Action- myoclonus-renal treatment of severe action myoclonus. Brain 1989; 112: 765-77. failure syndrome: a previously unrecognized neurological disorder 16. Delgado-Escueta AV, Serratosa JM, Medina MT. Myoclonic seizures unmarked by advances in Nephrology. Adv Neurol 1986; 43: 87-103. and progressive myoclonus epilepsy syndromes. In Wyllie E, ed. The 9. Marsden CD, Obeso AJ. Viewpoints in the Ramsay Hunt syndrome. treatment of epilepsy. Principles and practice. 2 ed. Baltimore: WilMov Disord 1989; 4: 6-12. liams & Wilkins; 1997. p. 467-83. EPILEPSIA MIOCLÓNICA PROGRESIVA: CARACTERIZACIÓN CLÍNICA DE 18 PACIENTES Resumen. Introducción. El término epilepsia mioclónica progresiva (EMP) agrupa a un conjunto de entidades heterogéneas, de causas genéticas, caracterizadas por presentar diferentes tipos de crisis, fundamentalmente mioclónicas, y otras manifestaciones neurológicas que traducen lesión progresiva del sistema nervioso central. Objetivo. Mostrar la etiología y las manifestaciones clínicoelectroencefalográficas de los pacientes con EMP. Pacientes y métodos. Se realizó un estudio descriptivo retrospectivo de los pacientes atendidos por EMP en el Instituto de Neurología y Neurocirugía de Cuba durante el período comprendido entre 1990 y 1995. La muestra quedó constituida por 18 pacientes. A todos se les realizó interrogatorio, examen físico, EEG y exámenes complementarios específicos de cada etiología. Resultados. Predominaron las lipofuscinosis ceroideas neurales en 10 pacientes (55,5%), con debut de la enfermedad antes de los 9 años en 9 de ellos. Le siguieron en frecuencia la epilepsia mioclónica con fibras rojo-rasgadas (16,6%) y la enfermedad de Unverricht-Lundborg (16,6%), esta última con debut en la infancia tardía y la adolescencia. Las manifestaciones clínicas más prominentes fueron la epilepsia de difícil control y el deterioro intelectual en el 100%, seguido de manifestaciones cerebelosas en el 88,8%. Las mioclonías fueron el tipo de crisis más frecuente (94,4%) y un gran número de niños presentaron crisis tonicoclónicas precediendo a éstas (88,8%). Conclusión. La respuesta al tratamiento fue pobre y los mejores resultados se obtuvieron con el valproato solo o asociado a benzodiacepinas [REV NEUROL 1999; 29: 102-4]. Palabras clave. Deterioro intelectual. Epilepsia mioclónica progresiva. Lipofuscinosis ceroide. Mioclonías. Valproato. 104 EPILEPSIA MIOCLÓNICA PROGRESSIVA: CARACTERIZAÇÃO CLÍNICA DE 18 DOENTES Resumo. Introdução. O termo epilepsia mioclónica progressiva (EMP) engloba um conjunto de entidades heterogéneas, de causas genéticas, caracterizadas por apresentar diferentes tipos de crises, sobretudo mioclónicas, e outras manifestações neurológicas que traduzem lesão progressiva do sistema nervoso central. Objectivo. Mostrar a etiologia e as manifestações clínico-electroencefalográficas dos doentes com EMP. Doentes e métodos. Realizou-se um estudo descritivo, retrospectivo dos doentes observados por EMP no Instituto de Neurología y Neurocirurgía de Cuba durante o período compreendido entre 1990 e 1995. A amostra ficou constituída por 18 doentes. Foi efectuado a todos, anamnese, exame objectivo, EEG e exames complementares específicos para cada etiologia. Resultados. Predominaram as lipofuscinoses ceroideias neuronais em 10 doentes (55,5%), com início da doença antes dos 9 anos em 9 deles. Seguiram, em frequência, a epilepsia mioclónica com fibras vermelhas rasgadas (16,6%) e a doença de Unverricht-Lundborg (16,6%), esta última com início na infância tardia e adolescência. As manifestações clínicas mais proeminentes foram a epilepsia de dificil controlo e a deterioração intelectual em 100%, seguida de manifestações cerebelosas em 88,8%. As mioclonias foram o tipo de crise mais frequente (94,4%). Um grande número de crianças apresentaram crises tónico-clónicas precedendo as crises mioclónicas (88,8%). Conclusão. A resposta ao tratamento foi escassa e os melhores resultados foram obtidos com o valproato em monoterapia, ou associado a benzodiazepinas [REV NEUROL 1999; 29: 102-4]. Palavras chave. Deterioração intelectual. Epilepsia mioclónica progressiva. Lipofuscinose ceroide. Mioclonias. Valproato. REV NEUROL 1999; 29 (2): 102-104