Síndrome de regresión caudal: Revisión de 12 casos en Teletón
Anuncio

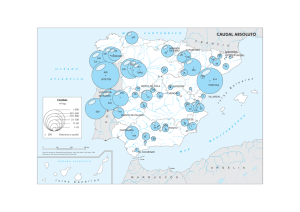
Rehabil. integral 2009; 4 (2): 100-106 Artículo de Revisión Síndrome de regresión caudal: Revisión de 12 casos en Teletón Santiago DANIELA GARCÍA P1 y VERÓNICA MARÍN V1. Residentes de Medicina Física y rehabilitación Hospital Clínico Universidad de Chile. Teletón-Santiago. 1 Recibido: 14 octubre 2009 Aceptado: 23 noviembre 2009 Correspondencia a: Verónica Marín P. [email protected] ABSTRACT Caudal regresion sindrome: A review of 12 cases in TeletónSantiago Caudal regression syndrome is a group of conditions with one thing in common: malformation of the distal spine. The degree of bony abnormality often determines clinical repercussions. The syndrome is associated to joint deformity, neurological impairment and visceral anomalies. The object of this article is to describe and analyze the population of children with caudal regression syndrome attending Teleton-Santiago. Key words: Caudal regresion, sacral agenesis, lumbar spine, orthopedic malformation, visceral anomalies. RESUMEN El síndrome de regresión caudal es un conjunto de patologías que tienen como nexo la malformación de la columna vertebral baja. El grado de compromiso óseo va a determinar la repercusión clínica en el paciente. Este síndrome se asocia a diversas deformidades articulares, alteraciones neurológicas y malformaciones viscerales. El objetivo de este artículo es describir y analizar la población de niños con diagnóstico de síndrome de regresión caudal que asisten a Teletón Santiago. Palabras clave: Regresión caudal, agenesia sacra, columna lumbosacra, deformaciones ortopédicas, malformaciones viscerales. Introducción El síndrome de regresión caudal engloba un espectro amplio de malformaciones de la columna vertebral inferior1. Varía desde una agenesia de la columna lumbosacra hasta casos muy severos como la sirenomelia, con fusión de las extremidades inferiores, alteraciones en la morfología genitourinaria y agenesia renal bilateral2. Esta última patología, conocida también 100 como los niños sirena, es incompatible con la vida3,4. Afortunadamente, la gran mayoría de los pacientes se concentra en las agenesias sacras con columna lumbar normal. El cuadro de agenesia de columna inferior fue descrito por primera vez en 1852 por Zur A. Hohl. Desde entonces se han recopilado más de doscientos casos en la literatura5. En 1957 Williams utiliza por primera vez el término “agenesia sacra”, que muchas veces se emplea SÍNDROME DE REGRESIÓN CAUDAL: REVISIÓN DE 12 CASOS EN TELETÓN-SANTIAGO como sinónimo a pesar de su diferencia6. Otros sinónimos usados son displasia caudal o agenesia caudal. No es hasta 1961 que se introduce el término de regresión caudal para incluir todas estas diversas patologías1. Este aporte se lo debemos a Duhamel quien utilizó la siguiente definición para describir el síndrome: “Espectro de malformaciones que van desde aplasia coccígea asintomática aislada hasta ausencia sacra, lumbar y de vértebras torácicas con graves déficit neurológicos asociados y malformaciones de otros órganos y sistemas”7. La incidencia mundial de este síndrome varía de 1/7 500 a 1/60 000 nacidos vivos dependiendo de la fuente que lo reporte8. Pero si se toma en consideración sólo la población de madres con diabetes pregestacional o gestacional, esta cifra aumenta a 1/350 nacimientos9. Otros autores hablan que hasta un 1% de los nacimientos en madres diabéticas puede presentar este tipo de patologías9. La presentación suele ser esporádica con bajo riesgo de recurrencia, pero también se han descrito casos de herencia autosómica recesiva o incluso dominante10. También se han estudiado diversos factores de riesgo que pudiesen estar influyendo en la aparición del síndrome. Dentro de estos destacan ser hijo de madre diabética, el uso de insulina durante el embarazo, la hipertermia durante la gestación, el uso de sales de litio, la hipoperfusión fetal, y el déficit de ácido fólico o vitaminas1,2,10. La etiopatogenia de este síndrome aun no está completamente dilucidada, pero se cree que su presentación podría deberse a una alteración de la neurulación secundaria, o en los casos más severos, también de la primaria11. Recordemos que la neurulación primaria es el proceso por el que se originan la placa y tubos neurales y concluye con la formación de la médula y canal neural hasta la unión lumbosacra1. La neurulación secundaria completa los segmentos caudales pendientes. Es por esto que la noxa debe ocurrir antes de la cuarta semana de gestación, pero su mecanismo de acción es aun desconocido. En el último tiempo han aparecido artículos que sugieren que la persistencia de la arteria vitelina podría influir en la aparición del síndrome al desviar la irrigación de la porción caudal del embrión12,13. Rehabil. integral 2009; 4 (2): 100-106 Consecuencia del gran abanico de malformaciones incluidas en el término de regresión caudal, los hallazgos al examen físico pueden ser muy variables. En los casos más leves podría no encontrarse nada visible o sólo un pliegue interglúteo corto con un discreto aplanamiento glúteo. En estos pacientes es útil preguntar por antecedentes de infección urinaria a repetición, constipación o escoliosis6. Otros signos que se pueden observar en las presentaciones más severas son estatura corta por reducción del tren inferior, agenesia variable de la columna caudal, cifoescoliosis con inestabilidad espino-pélvica, alteraciones costales, contractura en flexión de la rodilla por pterigion poplíteo, flexo de caderas asociado a displasia o luxación, y malformaciones de los pies donde lo más frecuente es el equinovaro2,14. Generalmente, se acompaña de compromiso de la médula espinal que se traduce en una falta de fuerza muscular bajo el nivel de la última vértebra intacta. El compromiso sensitivo suele ser más inconstante y partir en un nivel más bajo que el motor14. Por otra parte, hasta en 75% de los casos se puede asociar una alteración de la columna cervical como la inestabilidad atlantoaxoidea, por lo que se debe siempre incluir un estudio de esta zona15. Como se describió anteriormente, al síndrome de regresión caudal también se puede asociar con alteraciones en otros órganos. Se ha observado que mientras mayor es la deformidad a nivel del esqueleto, mayores son las malformaciones viscerales asociadas6. Las alteraciones más frecuentes se encuentran a nivel del aparato urogenital, donde encabezan la lista la hidronefrosis y la vejiga neurogénica16. También se puede encontrar agenesia renal, fimosis o hermafroditismo10. En otros sistemas podemos encontrar diversas malformaciones como ano imperforado, atresia colorrectal, distonía del esfínter anal, fisura labio palatina, cardiopatías congénitas, o alteraciones en los grandes vasos9,14. El diagnóstico se puede realizar en el período prenatal utilizando la ecografía transvaginal. Esta tiene una alta resolución a partir de la semana 17 de gestación y el hallazgo clásico que se espera encontrar es el feto en “posición de Buda” o “posición de rana” que se observa cuando las extremidades inferiores están en 101 D. GARCÍA P. y V. MARÍN V. triple flexión8. Otras alteraciones que se pueden detectar son la interrupción abrupta de la columna vertebral, oligohidroamnios o agenesia renal9. Si se sospecha el síndrome en el período postnatal el estudio de elección es la resonancia magnética. La literatura describe varias alteraciones que pueden observarse en este examen, dentro de las que destacan la disrafia oculta, presente hasta en un 35% de los casos; el cono medular globoso, identificado en la mitad de los pacientes; la separación neta de las raíces anteriores y posteriores de la cola de caballo, el sacro ausente, los huesos iliacos fusionados, y la expansión del canal ependimario5. Al realizar una búsqueda bibliográfica, se encontraron muy pocos estudios nacionales e internacionales que mencionen y analicen el síndrome de regresión caudal17. Se planteó como objetivo, describir y analizar los pacientes portadores de síndrome de regresión caudal que acuden a Teletón- Santiago. Siendo este último un centro de referencia para la población de niños con discapacidad a nivel regional, parece el lugar indicado para buscar y analizar las características de este polimorfo síndrome y comparar esos resultados con los encontrados en la literatura. Materiales y Método Se exploró la base de datos computarizada de Teletón Santiago, introduciendo el término “agenesia sacra”. Se obtuvo una lista de 16 pacientes activos, es decir, con al menos un control médico durante el último año. Cinco de ellos se encontraban viviendo fuera de la Región Metropolitana, lo que impidió acceder a sus fichas. De los 11 pacientes restantes, una ficha no se pudo revisar por dificultades administrativas. Se sumaron dos casos más que no estaban consignados en la base de datos, pero que fueron señalados por sus médicos tratantes. La descripción de los resultados se realizó a partir de estas 12 fichas médicas obtenidas. Una vez concluida la revisión, se confeccionó una base de datos con toda la información extraída utilizando el programa Excel. Dentro de los datos recolectados se incluyeron los antecedentes maternos y perinatales, las mal102 formaciones ortopédicas presentes, las malformaciones viscerales asociadas, la presencia de vejiga o intestino neurogénicos, edad de comienzo y abandono de marcha, uso de órtesis y ayudas técnicas y los rangos articulares de cadera y rodilla. Se obtuvieron porcentajes y se compararon con literatura internacional. Resultados La población estudiada presentó una edad promedio de 10,8 años (21 meses a 20 años). Se observó una proporción equitativa de hombres y mujeres. Del total de casos, en el 42% se encontraron antecedentes maternos de diabetes. Todas las madres eran insulino-requirentes, menos una que se manejaba con dieta. Además, en un 25% de ellos se pesquisaron antecedentes familiares de malformaciones congénitas (Figura 1). Al analizar el período prenatal de estos pacientes, destaca que en 10/12 casos existieron complicaciones del embarazo de diversa índole. Los problemas más frecuentes fueron la amenaza de aborto y el síndrome hipertensivo. Consecuencia de lo anterior, el embarazo llegó a término en sólo 58% de los casos, siendo la vía de este vaginal en casi la mitad de los niños (Figura 2). En cuanto a su diagnóstico, el 100% de los pacientes analizados presentó agenesia parcial o total del sacro comprobada con estudios imaginológicos. Si analizamos las malformaciones ortopédicas asociadas al síndrome de regresión caudal, se observa que la más frecuente de ellas fue la deformación de los pies (75%). Al desagregar, se encontró un paciente con pie Figura 1. Patología materna en sujetos con regresión caudal. Rehabil. integral 2009; 4 (2): 100-106 SÍNDROME DE REGRESIÓN CAUDAL: REVISIÓN DE 12 CASOS EN TELETÓN-SANTIAGO Figura 2. Patología del embarazo en sujetos con regresión caudal. cavo bilateral; el resto de ellos se dividen en igual proporción de pies talos y equinovaros. En cuanto a otros problemas ortopédicos destaca que un 25% de los niños tenía displasia de cadera y se encontró un caso con luxación de cadera (Tabla 1). En las malformaciones viscerales, se detectó que 10/ 12 pacientes presentaban algún tipo de alteración morfológica. Al subdividir, el 50% tuvo algún grado de malformación anorrectal, un tercio se asoció a disrafia o mielomeningocele, 2 pacientes presentaron situs inverso, y un paciente tenía labio leporino (Tabla 1). Si consideramos el funcionamiento vesical de estos pacientes, observamos que el 100% de ellos fue portador de una vejiga neurogénica. 11/12 pacientes padecían de intestino neurogénico. Cabe destacar que el paciente que no presentaba intestino neurogénico tenía una colostomía permanente por una agenesia de colon. Analizando el examen físico de rangos articulares se observa que más del 50% de los pacientes conservó sus valores normales al ser examinado hasta su último control médico consignado en la ficha. Dentro de las alteraciones descritas destacan 3 pacientes con un flexo progresivo de rodillas y caderas de muy difícil manejo, que no se logró corregir ni siquiera tras cirugía. Como último análisis se intentó establecer el pronóstico de ambulación de estos pacientes. Cabe resaltar que el 83,4% de los niños logró una marcha, al menos asistida, en algún momento de su desarrollo. La edad promedio en que se alcanzó este hito fue a los 2 años y 3 meses, con una variabilidad entre 14 meses y 7 años. Incluso 4 de los 12 pacientes lograron alcanzar Rehabil. integral 2009; 4 (2): 100-106 Tabla 1. Malformaciones encontradas en niños con síndrome de regresión caudal de Teletón- Santiago Malformaciones n (%) Agenesia sacra parcial o total 12 (100,0) Malformación anorrectal 6 (50,0) Mielomeningocele/disrafia 5 (41,7) Pie equino varo bilateral 3 (25,0) Pie talo bilateral 3 (25,0) Displasia de cadera 2 (16,6) Situs inverso 2 (16,6) Luxación de cadera 1 (8,3) Pie equino varo + pie talo 1 (8,3) Pie cavo 1 (8,3) Agenesia vena cava inferior 1 (8,3) Agenesia renal unilateral 1 (8,3) Riñón en herradura 1 (8,3) Labio leporino 1 (8,3) una marcha sin ayudas técnicas a una edad promedio de 3 años y 3 meses. Sin embargo, de los 10 pacientes que deambulaban un 20% terminó por abandonar la marcha en algún momento de su seguimiento (Figura 3). Discusión El síndrome de regresión caudal agrupa diversos cuadros clínicos de muy baja incidencia. Dada la alta heterogeneidad de las patologías involucradas, el pronóstico y manejo también son muy variables. Se han publicado diversos 103 D. GARCÍA P. y V. MARÍN V. Figura 3. Tipo de marcha en sujetos con regresión caudal. case report de esta patología, pero no encontramos artículos que analicen una población y ninguno de ellos a nivel nacional. Es por esto que nos parece que nuestro análisis de los niños con síndrome de regresión caudal que asisten a Teletón Santiago es un gran aporte al estudio y descripción de este poco frecuente, pero interesante síndrome. Al comparar los resultados obtenidos en este estudio con los publicados en la literatura científica se puede observar que se reafirman muchos de los conceptos entregados por ésta. En la muestra estudiada, efectivamente se respalda que la diabetes materna es un factor de riesgo importante para presentar este tipo de patología; sin embargo, los datos publicados hablan que un 16% de los niños portadores del síndrome serían hijos de madres diabéticas1,5,9 nosotros encontramos una cifra bastante mayor. Más del 40% de nuestros pacientes tenía el antecedente presente, lo que sugiere que tal vez este factor de riesgo tenga un peso mayor en nuestro medio. Además, la gran mayoría de los pacientes estudiados presentaron complicaciones de diversa índole en el período prenatal. La más frecuente de ellas, fue la amenaza de aborto. No encontramos artículos que describan este tema, por lo que no podemos determinar si este mayor grado de complicaciones es consecuencia directa del síndrome. Nos parece un tema interesante a profundizar ya que si efectivamente hay problemas prenatales asociados, las madres debiesen ser derivadas en forma precoz a un policlínico 104 especializado en embarazos de alto riesgo una vez que se haya realizado el diagnóstico de regresión caudal. Al analizar las malformaciones viscerales se aprecia que éstas también siguen la línea de lo expuesto en la literatura. Prácticamente el 100% de los niños son portadores de vejiga e intestino neurogénico, por lo que el estudio y manejo de estas patologías debe realizarse inmediatamente después de confirmado el diagnóstico de regresión caudal y no esperar a que estas se presenten con síntomas o complicaciones graves. La muestra estudiada presentó múltiples problemas ortopédicos, todos ellos dentro de lo descrito como parte del síndrome. A pesar de que el 50% de los pacientes lograba mantener sus rangos articulares en el tiempo, la mayoría de ellos perdía la habilidad de caminar con el trascurso de los años. Sería de gran utilidad contar con algún tipo de clasificación que nos ayude a conocer mejor el pronóstico de cada uno de estos pacientes en forma precoz. Revisando la literatura encontramos que hay varios tipos que clasificaciones que han intentado subdividir a los pacientes con síndrome de regresión caudal. La primera de ellas fue descrita en 1978 por Renshaw1. Esta clasificación intenta subdividir los pacientes según el grado de agenesia sacra observada en la radiografía. Los pacientes tipo 1 sólo tienen agenesia sacra unilateral, en los tipo 2 hay una agenesia bilateral, en los tipo 3 se agrega agenesia de la columna lumbar y los huesos iliacos se articulan con la última vértebra existente, el tipo 4 es el más severo donde la última vértebra sólo “reposa” sobre los huesos iliacos que están fusionados18. En 1994 Bollini y Jouve crearon una nueva clasificación que incorpora la existencia y tipo de problemas ortopédicos que se pueden observar según las alteraciones radiológicas del paciente19. El grupo 1 incluye a los pacientes con agenesia sacra total más agenesia torácicolumbar; en estos niños las alteraciones ortopédicas asociadas son el flexo de cadera y de rodilla, presencia de pterigion poplíteo, e inestabilidad espinopélvica. El grupo 2 son los pacientes con disgenesia de las articulaciones sacroiliacas y pueden presentar una escoliosis de grado variable. El grupo 3 sólo tiene una agenesia parcial del sacro por lo que sólo hay riesgo de displasia Rehabil. integral 2009; 4 (2): 100-106 SÍNDROME DE REGRESIÓN CAUDAL: REVISIÓN DE 12 CASOS EN TELETÓN-SANTIAGO de caderas y deformidades del pie. Hace tan sólo tres años, Vergara H14, propuso una nueva clasificación en la que se intenta definir el pronóstico ambulatorio de este tipo de pacientes. El tipo I corresponde a la agenesia parcial unilateral de sacro, que se subdivide en tipo Ia si es que no hay evolución a la escoliosis, y en tipo Ib si es que hay escoliosis. El tipo II incluye la agenesia total de sacro y también se subdivide en dos grupos. El tipo IIa es cuando no hay traslación de la columna sobre los huesos iliacos ni escoliosis. En tipo IIb sí hay traslación y/o deformidad de la columna. En cuanto al pronóstico ambulatorio, el autor reportó a grandes rasgos que los pacientes tipo Ia suelen tener marcha comunitaria sin requerir uso de órtesis, los IIa suelen utilizar órtesis, y los IIb requieren del uso de silla de ruedas para movilizarse. Todas las clasificaciones antes expuestas nos parecen de gran utilidad para ser aplicadas en el manejo de los pacientes con diagnóstico de síndrome de regresión caudal, ya sea para conocer mejor el pronóstico de cada niño, como para orientar el tratamiento. Este último puede diferir mucho de paciente a paciente según el nivel de compromiso. Es por esto que el manejo de estos niños debe ser siempre multidisciplinario e individual para cada caso. Aunque prácticamente todos los pacientes requerirán de un gran apoyo ortopédico para manejar las múltiples deformidades articulares y un buen control urológico para manejar la vejiga neurogénica9. Siempre es importante insistir en el fortalecimiento del musculo cuádriceps para intentar contrarrestar en lo posible el flexo de rodilla. Pero muchas veces no queda más alternativa que optar por soluciones de tipo quirúrgicas como la liberación de tejidos blandos, la osteotomía supracondílea de fémur, la estabilización de pelvis y columna, o en casos más drásticos, la desarticulación de rodilla o cadera10. No olvidar nunca que estos pacientes suelen tener un coeficiente intelectual completamente normal por lo que deben ser incluidos desde pequeños en cualquier toma de decisiones que comprometan su futuro. Con este artículo hemos querido sacar a relucir un síndrome de rara frecuencia y pocas veces considerado como diagnóstico por los médicos tratantes. La precocidad del diagnósRehabil. integral 2009; 4 (2): 100-106 tico y el manejo apropiado tienen una repercusión importante en la calidad de vida de estos pacientes. Es por esto que el conocimiento de esta patología y sus variadas presentaciones deben estar al alcance de los especialistas que trabajan con niños. Agradecimientos Agradecemos la dedicación y ayuda incondicional de la Dra. María Antonieta Blanco, fisiatra de Teletón Santiago. Referencias 1.- Ramírez N, Pabón C, Mendoza A, Maldonado C, Mamani F, Mamani N. Síndrome de regresión caudal. Presentación de un caso. Ciencia e Invest Médica Estudiantil Latinoamérica 2005; 10: 69-72. 2.- Hortelano M, Palencia J, García J, Reig C, Herrera M, Romero M, et al. Síndrome de regresión caudal. Rev Neurología 1998; 27: 613-15. 3.- Bracho V, Tovar J, Rodríguez M, Moreno B. Sirenomielia. Estudio de cinco casos y revisión de la literatura. VITAE Acad Biom Dig 2005; 24. Disponible en: www. caibco.ucv.ve. [revisado el 10 de abril 2008]. 4.- Balakumar K. Case control: Sirenomelia-early second trimester antenatal ultrasonographic diagnosis. Obst and Gynec Radiology 2007; 17: 273-4. 5.- Romero C, Intruvini S, Couto J, Massaro M, Meli F. Síndrome de regresión caudal. Presentación característica en RM. Diagnóstico Journal 2000; 9 (97). Disponible en: www.diagnostico.com.ar. [revisado el 22 de abril 2008]. 6.- García E, Olmos M, Beguiristáin J. Defecto vertebral múltiple con otras anomalías acompañantes. An Esp Pediatr 2001; 54: 409-10. 7.- Duhamel B. From de mermaid to anal imperforation: the syndrome of caudal regression. Arch Dis Child 1961; 30: 152-5. 8.- Fayyaz A, Ilyas M, Iqbal O. Pre-natal diagnosis of caudal regression syndrome. J College Phys Surgeons Pakistan 2007; 17: 425-6. 9.- Aslan H, Yanik H, Celikaslan N, Yildirim G, Ceylan Y. Prenatal diagnosis of caudal regression syndrome: a case report. BMC Pregnancy and Childbirth 2001; 1: 8. 10.- Loera R, Rodríguez I, Rodríguez R, Delgado C, Cruz A. Agenesia lumbosacra. Med Univ 2007, 9: 38-41. 11.- Stierkorb E, Hentschel J, Schneider G, Gortner L, 105 D. GARCÍA P. y V. MARÍN V. Rohrer T. Sonographic diagnosis of caudal regression syndrome. Ultraachall Med 2007; 28: 521-4. 12.- Duesterhoeft S, Ernst L, Siebert J, Kapur R. Five cases of caual regression with an aberrant abdominal umbilical artery: Furher support for a caudal regression- sirenomelia spectrum. Am J Med Genetics Part A 2007; 143A: 3175-84. 13.- Hentschel J, Stierkorb E, Schneider G, Goedde S, Siemer S, Gortner L. Caudal regression sequence: vascular origin? J Perinatol 2006; 26: 445-7. 14.- Vergara H, Cardoso A, Rosales M, Orellana C. Agenesis lumbosacra: Tratamiento y propuesta de nueva clasificación. Acta Ortop Mex 2005; 19: 6-12. 15.- Thiryayi W, Alakandy L, Leach P, Cowie R. Craniocervical instability in an infant with partial sacral agenesis. 106 Acta Neurochir 2007; 149: 623-7. 16.- Torre M, Bufa P, Jasonni V, Cama A. Long-term urologic outcome in patients with caudal regression syndrome, compared with meningomyelocele and spinal cord lipoma. J Pediatr Surgery 2008; 43: 530-3. 17.- Naze J, Ramírez R. Malformaciones congénitas en los hijos de madres diabéticas, Rev Méd Chile 2000; 128 (9): 1045-52. 18.- Renshaw T. Sacral agenesis. J Bone Joint Surg Am 1978; 60: 373-83. 19.- Bollini G, Jouve J, Cottalorda J. Malformations congénitales de la ceinture pelvienne. Encycl Méd Chir (Elsvier, Paris-France), Appareil locomoteur 1994; 15: 225-310. Rehabil. integral 2009; 4 (2): 100-106