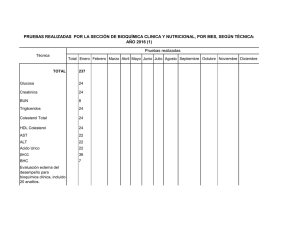
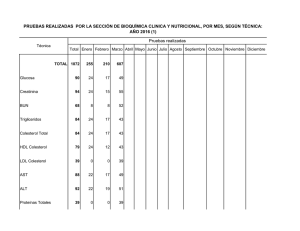
Revista ABA 71-2-PC.indd - Asociación Bioquímica Argentina
Anuncio

VOL 71 - Nº 2 - 2007 Ciudad de Bs. As. Argentina ISSN 1515-6761 Bioquímica y Patología Clínica Revista de la Asociación Bioquímica Argentina - Volumen 71 - Nº 2 - 2007 Bioquímica y Patología Clínica Actinomucor elegans asociado a infección en humanos. Primeros registros mundiales. 50 µm Revista de la Asociación Bioquímica Argentina Incorporada al Latindex y a la Red de Revistas Científicas de América Latina y el Caribe, España y Portugal (REDALYC) 50 µm EDITORIAL Actinomucor elegans asociado a infección en humanos. Primeros registros mundiales. La Asociación Bioquímica Argentina, y por extensión, los bioquímicos argentinos –por quienes existe y a quienes pertenece y se debe - tienen hoy un motivo más de alegría. El órgano científico de la Asociación, Bioquímica y Patología Clínica – ByPC, ha recibido una honrosa distinción. En efecto, por medio de la Resolución 1071/07 del Directorio del Consejo Nacional de Investigaciones Cientificas y Tecnicas - CONICET – se ha incorporado a Bioquímica y Patología Clínica – ByPC al Núcleo Básico de Revistas Científicas y Tecnológicas Argentinas, en el que se nominan a los órganos científicos considerados de excelencia por dicho Consejo. En esta Resolución se hace lugar a las consideraciones del Comité Asesor del Núcleo Básico de Revistas Científicas Argentinas. Debemos hacer notar que para que esas recomendaciones se produzcan la Revista debe cumplir con estrictas exigencias científicas, estructurales y editoriales que las justifiquen. La Revista de la Asociación Bioquímica Argentina, hoy Bioquímica y Patología Clínica – ByPC, vio la luz en 1935, cuando los creadores de la ABA, en 1934 comprendieron la necesidad de un órgano científico que permitiera la publicación y expresión de la actividad científica de los bioquímicos clínicos e investigadores argentinos, transformándose así en pionera de las publicaciones bioquímicas científicas en nuestro país. Sus páginas dieron así cabida al trabajo de varias generaciones de bioquímicos argentinos, incluyendo a los más destacados exponentes de nuestra ciencia, como nuestro Premio Nobel, el Dr. Luis Federico Leloir. Desde esos años, ha aparecido en forma ininterrumpida hasta la actualidad, a pesar de las dificultades económicas y restricciones presupuestarias que ha sufrido y sufre la ABA. Esto ha sido posible porque todas las conducciones desde 1934 hasta la actualidad han priorizado la educación permanente del bioquímico como el fin supremo de nuestra Asociación, y su razón de existir. Desde hace unos años, una característica saliente de ByPC, es la publicación en la portada de fotografías producto de trabajos de bioquímicos argentinos, con su correspondiente explicación en la primera página de la Revista. Debemos hacer notar que dichas fotografías no son copias de otras publicaciones, sino verdaderos originales con un gran valor de consulta y a la vez de exposición de la excelencia de nuestros bioquímicos. La Asociación Bioquímica Argentina, desde 1934 y hasta la actualidad, ha fomentado el continuo perfeccionamiento del bioquímico a través de sus Cursos, Congresos, Jornadas, Conferencias y en lo que hoy nos ocupa, su Revista Bioquímica y Patología Clínica – ByPC. Su tarea es silenciosa y ardua, pues todos quienes tienen algún grado de responsabilidad en su conducción o participan de las Divisiones o Comisiones de la Asociación, lo hacen, como establecen sus estatutos, en forma absolutamente honoraria, siguiendo el ejemplo de sus creadores. Recientemente, el decidido apoyo de empresas editoriales, profesionales y comerciales, a quienes quedamos profundamente agradecidos, ha permitido asegurar la continuidad de nuestra Revista. En forma muy especial deseo agradecer la infatigable tarea de la Lic. Ana María Flores, Coordinadora del Área de Publicaciones Científicas del CAICYT – CONICET, labor que cristalizó en la honrosa inclusión de ByPC en el Núcleo Básico de publicaciones argentinas de excelencia. Quiero también agradecer la inestimable colaboración de las Dras. Raquel Osatinsky (anterior Directora de la Revista) y Patricia Indaburu, en la difícil tarea de la corrección y compaginación de los trabajos, así como la de los profesionales bioquímicos, investigadores y científicos que colaboran con sus arbitrajes en la selección de los trabajos a publicar. Asimismo, mi agradecimiento al apoyo incondicional de todos los miembros de la Comisión Directiva de la Asociación, en especial a su Presidente, Dra. Silvia Morilla. Su colaboración con la Revista fue siempre un factor decisivo que ha permitido sortear muchas dificultades que se han presentado en su accionar. Resulta insoslayable destacar el trabajo tesonero y eficiente del Dr. Aníbal Bagnarelli, que fue uno de los pilares que, con su férreo espíritu organizativo, llegó a concretar todos los pasos administrativos y requisitos indispensables para la gestión de la Revista. Su compromiso merece mi especial reconocimiento. La distinción conferida a nuestra Revista nos enorgullece y emociona, pero no es solo un reconocimiento a sus setenta y dos años de trayectoria, sino también el premio a la excelencia de los bioquímicos argentinos y de otros países, quienes con su aporte científico han llevado a lo que es hoy Bioquímica y Patología Clínica – ByPC, Revista de la Asociación Bioquímica Argentina. Néstor Litwin Director Bioquímica y Patología Clínica – ByPC Asociación Bioquímica Argentina VOL 71 - Nº 2 - 2007 Ciudad de Bs. As. Argentina ISSN 1515-6761 Bioquímica y Patología Clínica Revista de la Asociación Bioquímica Argentina Incorporada al Latindex y a la Red de Revistas Científicas de América Latina y el Caribe, España y Portugal (REDALYC) SUMARIO 8 EDITORIAL. Un honroso reconocimiento a la Revista Bioquímica y Patología Clínica - ByPC, órgano científico de la Asociación Bioquímica Argentina. 13 36 Gestión del Laboratorio de Análisis Clínicos Implementación de la documentación según normas internacionales. Dra. Graciela Briozzo. 37 40 Evolución de los pacientes neutropénicos en nuestro HospitalPaula Bermúdez1. Juan Tentoni, Alejandra Larregina, Nélida Polini. 41 46 Hipotiroidismo congénito en los inicios de la pesquisa neonatal en el sector público de la ciudad de Posadas. Alvaro Errasti, María Marinela Tibolla, Nora Graciela Márquez, Ana Lía Albrekt, Sandra Steffens. 47 51 Identificación fenotípica de micobacterias. Belén Imperiale, Nora Morcillo, Amelia Bernardelli. 52 54 Valores de referencia del hemograma en embarazadas, con tecnología actual. Canalejo Katia, Juan Tentoni, Aixalá Mónica, Jelen Ana María. 55 59 Monitoreo de depósitos de hierro elevados mediante protoporfirina eritrocitaria. Silvia Haydee Langini, Marta Mabel Lardo, Silvana Fleischman, Alberto Lazarowski, María Esther Rio. 60 66 ¿Que es la electroforesis capilar?. Raquel Osatinsky. 67 75 Resúmenes de las comunicaciones libres aprobadas en el IV Congreso y 8º Encuentro Bioquímico 2007. XXX Reunion del Capítulo Argentino de la Sociedad Latinoamericana de Nutrición (CASLAN) FOTO DE TAPA: Actinomucor elegans asociado a infección en humanos. Primeros registros mundiales. Solo existe en la literatura un caso informado de infección en humanos por Actinomucor elegans por nuestro grupo en 2001 (1). El segundo caso fue registrado también en nuestro laboratorio (datos no publicados). Las imágenes corresponden a la observación microscópica de A. elegans, (993313) aislado en 1999 a partir de material mucopurulento obtenido de senos paranasales de una niña de 11 años con sinusitis y lagrimeo ocular, sin señales de otra enfermedad subyacente (Figura 1) (1). En el año 2000, se identificó otro aislamiento de A. elegans (00291) proveniente de una lesión cutánea de un paciente inmunocomprometido (Figura 2). Figura 2. Esporangios globosos que nacen de un esporangióforo ramificado. Magnificación, 62 x. Figura 1. Esporangio terminal roto y separado de la columela, con algunas esporangiosporas espinosas sueltas y tres esporangios globosos, hialinos o subhialino de menor tamaño unidos al esporangióforo. Magnificación, 125x. 50 µm nentes de los profesionales del laboratorio en el campo de la micología, para lograr la identificación de estos microorganismos poco comunes. 50 µm En ambos casos el examen directo del material clínico mostró la presencia de hifas hialinas sin septos (cenocíticas). El género Actinomucor pertenece al orden de los Mucorales, incluye dos especies, A. elegans y A. taiwanensis, que difieren entre sí por el tamaño de sus esporangiosporas, por la capacidad de crecer en el medio de Czapek y el desarrollo a 37 ºC. Su morfología microscópica es similar a Absidia, Rhizopus y Rhizomucor, diferenciándose de estos por las características del esporangio, el limitado desarrollo de los estolones y las temperaturas de crecimiento (2). Actinomucor elegans fue descrito por primera vez por Schostakowish en 1898. La especie se aísla frecuentemente de heces de herbívoros y vegetación en descomposición. Nuevos hongos oportunistas surgen como agentes de infecciones o colonizaciones en una amplia variedad de pacientes. Esto requiere una capacitación y actualización perma- Autores: Refojo, Nicolás Abrantes, Ruben Hevia, Alejandra Lugar de trabajo: Laboratorio de Hongos Miceliales. Departamento Micología. INEI. ANLIS “Dr. Carlos G. Malbrán” Dirección Postal: Nicolas Refojo Avenida Vélez Sarsfield 563 1281 Ciudad Autónoma de Buenos Aires. Argentina TE/FAX 43025066 [email protected] 1.- Davel G, Featherston P, Fernandez A, Abrantes R, Canteros C, Rodero L, Sztern C, Perrotta D. Maxillary sinusitis caused by Actinomucor elegans. J Clin Microbiol. 2001; 39(2): 740-742. 2.- Benjamin CR, Hesseltine CW. The genus Actinomucor. Mycologia. 1957; 49 240–249. VOL 71 - Nº 2 - 2007 Ciudad de Bs. As. Argentina ISSN 1515-6761 Bioquímica y Patología Clínica REVISTA DE LA ASOCIACION BIOQUIMICA ARGENTINA Venezuela 1823 - Piso 3 - CP (1096) Buenos Aires - Argentina Tel/ fax: 4384-7415 - Tel: 4381-2907 - e-mail: [email protected] - www.aba-online.org.ar ASOCIACION BIOQUIMICA ARGENTINA Fundada el 3 de septiembre de 1934 Registro Nacional de Derechos de Autor Nº 034772 Publicación cuatrimestral COMISION DIRECTIVA COMITE EDITORIAL Presidente Dra. Silvia Morilla Vicepresidente Dr. Anibal E. Bagnarelli Secretaria Dra. María Ruggiero Tesorero Dra. Estela Meyer Vocales Titulares Dra. Mabel Villareal - Dra. Silvia de Pardo Dr. Néstor Litwin Director Dr. Néstor Litwin Secretaria Dra. Raquel Osatinsky Redacción Dra. Patricia Indaburu Dr. Gabriel Carballo COMISION REVISORA DE CUENTAS Dra. Mónica Aixalá Dr. Juan Antonio Barcat Dra. Alicia Blanco Dra. María de la Paz Castresana Dr. Sergio Damilano Dra. Ana Di Martino Dr. Luis Gamek Dr. Emilio Labal Dra. Graciela Lucero Dr. Alejandro C. Paladini Dra. Juana María Pasquini Dr. Marco Pizzolato Dra. Elvira Roehr Dr. Edgardo Poskus Dra. Liliana Roquel Dr. Otmaro Roses Dra. Sandra Rozenthal Dr. Modesto Rubio Dra. Nora Slobodianik Dra. Marysia Szefner Dra. Graciela Valdéz Dra. Patricia Sorroche Titulares Dr. Abel Pallares - Dra. Isabel de Simone Suplentes Dra. Verónica Natoli - Dra. Vanina Berardi Dr. Alberto Villagra COMISIONES INTERNAS Actividades Culturales Dr. Alberto Villagra - Dra. Liliana Paz Dra. Silvia Morilla Acreditación de Laboratorios Dra. Silvia de Pardo - Dra. Mabel Villareal - Dr. Mariglino - Dra. Silvia Morilla Dr. Alberto Villagra Certificación Profesional Dr. Edgardo Poskus Dr. Anibal E. Bagnarelli - Dr. Néstor Litwin Cursos y Conferencias Dra. Raquel Osatinsky - Dra. Verónica Natoli - Dr. Alberto Villagra - Dra. Vanina Berardi- Dra. María Ruggiero - Dra. Gladys Oviedo - Dra. Estela Meyer Premios y Distinciones Dra. Juana M. Pasquini - Dr. Edgardo Poskus - Dr. Modesto Rubio - Dr. Néstor Litwin - Dra. Alicia Blanco COMITE ASESOR CIENTIFICO REGLAMENTO DE PUBLICACIONES DE BIOQUIMICA Y PATOLOGIA CLINICA, REVISTA DE LA ASOCIACION BIOQUIMICA ARGENTINA Bioquímica y Patología Clínica (ByPC), Revista de la Asociación Bioquímica Argentina publica artículos relacionados con aplicaciones de la bioquímica clínica en todas sus especialidades en el campo asistencial y de investigación clínica humana, como así también en bioquímica animal y vegetal. Los trabajos enviados a la Revista ByPC no deben haber sido publicados en otra revista u órgano de difusión científica nacional o extranjero y los autores deben informar si han sido presentados previamente en carácter de comunicaciones libres. Los manuscritos serán enviados a un jurado de la especialidad del Comité Científico Asesor de la Revista, quienes podrán convocar a otros profesionales de relevantes méritos. La resolución del jurado será remitida a la Comisión Revista ByPC dentro de los 30 días de recibido. El dictamen del jurado que será reservado, deberá fundamentarse de modo explicito. La aceptación o rechazo del trabajo será comunicada a los autores por la Comisión Revista ByPC, mencionando los motivos del mismo. REQUERIMIENTOS GENERALES Imprimir el manuscrito en papel blanco de 80 g/m2 - A4 ( 21 x 29.7 cm) en Word-Window, folios a simple faz, letra fuente Arial, tamaño de la fuente 12 e interlineado 1.5 cm. Margen izquierdo 2.5 cm. y margen derecho 1.0 cm en hojas numeradas en el extremo superior derecho, comen-zando con la pagina del Titulo. Se debe adjuntar diskette con el archivo del trabajo en formato Word. Los trabajos podrán ser enviados por email y la información adicional del trabajo como ser, fotos de color, gráficos etc podrán ser enviados por correo postal. De cualquier manera el correcto envío-recepción del trabajo queda bajo exclusiva responsabilidad de los autores. A.TRABAJO ORIGINAL Las siguientes secciones del trabajo original serán confeccionadas en folios separados: 1 | Título del trabajo 2 | Resumen en español e inglés 3 | Texto 4 | Agradecimientos 5 | Referencias bibliográficas 6 | Cada una de las tablas 7 | Figuras y leyendas 1 | Título: indicar: a. Título del trabajo. b. Nombre, Apellido y Título profesional de cada autor. c. Institución/es en la/s cual/es el trabajo fue llevado a cabo. d. Dirección postal y de email del autor al que deberá ser dirigida la correspondencia 2 | Resumen : en español e inglés Se utilizarán hasta un máximo de 150 palabras para definir el problema, delinear el diseño experimental y comentar resultados y conclusiones. 3 | Texto: Deberá constar de: a. Introducción: definir la razón del estudio, la naturaleza del problema, su relación con trabajos previos y el objetivo del trabajo. b. Métodos: describir sujetos experimentales, equipamiento, reactivos y procedimiento utilizado, incluyendo marcas registradas cuando corresponda y referencias al utilizar métodos establecidos. lndicar consideraciones éticas que correspondan si seres humanos han sido involucrados en el estudio. c. Resultados:presentar en secuencia lógica los datos generados utilizando apropiadamente tablas o figuras sin duplicación. Indicar los procedimientos estadísticos utilizados y sus referencias si corresponde. d. Discusión: indicar las conclusiones que se extraen ubicándolas críticamente en el contexto de la experiencia anterior. Distinguir claramente entre nueva información y hallazgos previos, así como las especulaciones de los hechos. Se podrán identificar nuevos problemas emergentes del trabajo así como las nuevas hipótesis generadas por el mismo. 4 | Agradecimientos: Indicar asistencia o ayuda científica, técnica, financiera y obsequios. 5 | Referencias Bibliográficas: Las referencias deberán ser numeradas por orden de aparición en el trabajo, citando en la forma más completa posible las que sean estrictamente relevantes a los fines del trabajo. Las abreviaturas utilizadas corresponden a la lista de revistas indexadas publicadas anualmente en el número de enero del Index Medicus. El formato de presentación será: a. Publicación en revistas: Gainer AL, Stinson RA. Evidence that alkaline phosphatase from human neutrophils is the same gene product as the liver/kidney/bone isoenzyme. Clin Chim Acta 1982; 123: 11-17. b. Publicación en libros: Wright LA. Diagnostic Clinical Toxicology. En: Gornall AG, ed. Applied Biochemistry of Clinical Disorders. Pp. 378-88. Hagerstown: Harper & Row, 1980. 6 | Tablas: Tipear cada tabla a doble espacio en hojas separadas, numerándolas consecutivamente en números arábigos y dando un encabezamiento descriptivo y con notas al pie aclarando las abreviaturas. 7 | Figuras y leyendas: Enviar un juego de figuras de alta calidad con símbolos de tamaño sufi-ciente para que permanezcan legibles al reducirlas para su publicación. B.COMUNICACIONES BREVES Seguirán las normas explicadas para los trabajos originales, con la diferencia que el TEXTO (punto 3 de Trabajos Originales) deberá tener una extensión no mayor de 3 páginas. C.ACTUALIZACIONES,EDITORIALES Y COMENTARIOS Las actualizaciones, editoriales y comentarios serán usualmente solicitados por el Comité Editorial de la Revista de la Asociación Bioquímica Argentina a autores considerados expertos en el campo, disciplina o especialidad en cuestión. Sin embargo serán consideradas para su publicación las enviadas espontáneamente. Deberán seguir los lineamientos expuestos para la publicación de trabajos originales a diferencia que su texto no necesitará contar con introducción, métodos, resultados y discusión, debiendo contener, referencias bibliográficas completas y actualizadas a los fines del tema tratado. D.TRABAJOS DE LAS RESIDENCIAS BIOQUIMICAS Serán bienvenidos los trabajos originales o actualizaciones bibliográficas de temas producidos por residentes bioquímicos, a condición que integre el plantel de autores un profesional asesor acreditado en el campo, disciplina o especialidad considerada REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 13 | 36 Pág | 13 Gestión del laboratorio de análisis bioquímicos - clínicos. Implementación de la documentación según normas internacionales. Dra. Graciela Briozzo. Bioquímica Tesis de Maestría Maestría en Gerencia y Administración de Sistemas y Servicios de Salud Universidad Favaloro Enviar Correspondencia a: e-mail: [email protected] RESUMEN Con el fin de poder desarrollar un Sistema de Gestión de la Calidad (SGC) eficaz en cualquier empresa de salud, debe elaborarse la documentación necesaria, que además de ser imprescindible para el ordenamiento interno de todos los procesos, es de suma utilidad para la preparación de la acreditación de los servicios, para la presentación ante las auditorías y para la capacitación permanente del recurso humano. Dado que todos los laboratorios de análisis clínicos deben poseer documentos de diferentes tipos (y son pocos los que los poseen en su totalidad), se presenta en este trabajo un modelo de sistema de documentación que contiene los documentos requeridos en la actualidad por los organismos nacionales e internacionales y se formulan directivas y sugerencias para el control de dicha documentación. La documentación del SGC permite la identificación y gestión de procesos y actividades relacionadas con un enfoque integral y el desarrollo de una cantidad de documentos necesarios para la efectiva planificación (planes de gestión), operación (planes operativos), incluyendo elementos organizativos, inventariales, control y mejora continua del SGC y sus procesos. El modelo propuesto de “Sistema de Gestión de la Documentación”, permite conocer qué procesos implica y la mejor manera de llevarlos a cabo para que sirva de soporte al SGC. Es de referencia para que en cualquier servicio de laboratorio clínico pueda ser implantada la gestión documental. Un sistema de calidad que funcione correctamente es crítico para el desarrollo y la prestación de servicios de los laboratorios de análisis clínicos, mientras que un sistema de documentación aplicable y consolidado representa una fuerte herramienta para el futuro crecimiento de la estructura. Ambos sistemas son complejos y están en mutua dependencia, constituyendo su relación armoniosa la clave del éxito en la gestión. Palabras Clave | Sistema de Gestión de la Calidad, Sistema Documentario, Laboratorio Clínico. SUMMARY Revista ByPC. Incorporada al Latindex. ISSN 1515-6761 Código Bibliográfico: RByPC Trabajo Recibido: 20-03-07 Aceptado: 11-05-07 In order to develop an effective Quality Management System (QMS) into a Health Organisation, it is essential to elaborate all the mandatory documentation, which, besides critical for ruling internal processes, is very useful to make Services ready for Accreditation, to fulfil Auditory and for permanent qualification of Human Resource. According to the fact that every Clinical Laboratory must hold a wide variety of documents (and only a few fill up this subject), this paper describes a standard documentary scheme pattern that includes both national and international organisations documentation requirements and provide directions and suggestions for its control. The Documentation of a QMS allows identification and management of processes and activities with a broad scope and the development of many documents necessary for an effective planification (management tactics), operation (operative tactics), including organization issues, inventory, control and permanent upgrading of QMS and its processes. The proposed scheme of “Management Documentation System” shows the involved processes and the best way to accomplish them as the basis for QMS. Is a reference to any Clinical Laboratory to meet the needs of a Documentary Management. The accurate operation of the Quality System is critical for development and provision of Clinical Laboratory services, while a suitable and well-built Documentary System represents a strong tool for structure future expansion. Both systems are complex and interdependent, being its mutual relation the key for a successful Management. Key words | Quality Management System, Documentary System, Clinical Laboratory. Gestión del Laboratorio de Análisis Clínicos Implementación de la documentación según normas internacionales 1.INTRODUCCION La década de los noventa ha sido, indudablemente, una de las más importantes para proponer nuevos enfoques para la gestión, en este campo se han experimentado profundos cambios, tanto en los ambientes en los que se desarrollan estas actividades dentro de las empresas como en las teorías y enfoques que tratan de explicar la manera en que éstas deben desempeñarse para adaptarse con éxito a la nueva realidad. Dadas las características actuales de la globalización de los mercados, para el abordaje concerniente a los Servicios de Salud, cada país, debe adoptar esquemas y requerimientos conformes a las normas y estándares de validez y aceptación internacional para el gerenciamiento de sus organizaciones. La acreditación de los Servicios de Salud tiene como propósitos mejorar la calidad de los productos, servicios, sistemas y personas, generar confianza en las actividades de las entidades acreditadas, aportar transparencia a los mercados, facilitar el comercio y participar en el mejoramiento de la calidad de vida de la población. La aceptación y confianza en los análisis del laboratorio por una segunda parte puede conseguirse demostrando la competencia a través de la acreditación de los procedimientos de análisis por una tercera parte (organismos de acreditación nacionales e internacionales). El concepto “acreditación” se define en la Guía 2:1996 de la Organización Internacional de Normalización /Comisión Electrotécnica Internacional (ISO/IEC) como el “Procedimiento mediante el cual un organismo con autoridad reconoce formalmente que un organismo o persona es competente para desarrollar determinados trabajos”. La acreditación de los laboratorios clínicos en su sentido más amplio tiene cada vez más importancia como instrumento de gestión y como medio para el aseguramiento de los resultados (1). Es el reconocimiento de haber cumplido las normas requeridas para llevar a cabo una función propia y la puerta que abre la posibilidad de competir en un mercado de mayor tamaño. De igual modo, cuando se intenta normatizar los sistemas de calidad de los laboratorios de análisis bioquímicos - clínicos, deben tenerse en cuenta las características peculiares de este tipo de servicio para lograr una normativa precisa, congruente y armónica que refleje lo más acabadamente dichos Sistemas de Calidad. Los laboratorios clínicos, a diferencia de otros laboratorios de análisis, tienen características particulares en relación a su contacto con los pacientes: • Consideraciones preanalíticas hacia los pacientes relacionadas con la extracción, preparación, identificación y transporte de muestras • Consideraciones postanalíticas hacia el personal sanitario en relación a la validación, información, interpretación y asesoramiento. Además hay observaciones de seguridad, ética y prevención de enfermedades. Para asegurar que se aplica un sistema de procedimientos normatizado a fin de controlar y mejorar la calidad de un servicio, proceso o producto, se requiere una documentación de soporte, completa y consensuada que describa los procedimientos adecuados que representen las mejores prácticas. Con el fin de poder desarrollar un Sistema de Gestión de la Calidad eficaz en cualquier empresa de salud, debe elaborarse toda la documentación necesaria, que además de ser imprescindible para el ordenamiento interno de todos los procesos, es de suma utilidad para la preparación de la acreditación de los servicios, para la presentación ante las auditorías y para la capacitación permanente del recurso Humano (2). Dado que todos los laboratorios de análisisbioquímicos Clínicos deben poseer documentos de diferentes tipos (y son pocos los que los poseen en su totalidad), se presenta en este trabajo un modelo de sistema de documentación que contiene los documentos requeridos en la actualidad por los organismos nacionales e internacionales y se formulan directivas y sugerencias para el control de dicha documentación. 2.JUSTIFICACIÓN En el área de la gestión en salud, los ciudadanos cuentan cada vez con mayor información acerca de las políticas generales de salud y su financiación, los métodos de recaudación de impuestos y el uso de los recursos públicos. Esto motiva que los contribuyentes sean más críticos y conocedores de sus derechos en el momento de la evaluación de la atención recibida, exigiendo mejor organización y gerenciamiento de los servicios de salud a los cuales acuden. No existe duda de la necesidad de una estrategia sólida y bien articulada para el cumplimiento de las normas de una organización. En el caso de las empresas de salud, esta estrategia constituye un gran desafío para todos los que tratamos de implementar acciones tendientes a lograr el mejor desempeño de nuestros servicios, haciéndolos más accesibles, solidarios, éticos y equitativos. La información y su consecuencia, la documentación, para que pueda utilizarse y genere ventajas competitivas debe tener tres características básicas: completa, confiable y oportuna. Además debe emplearse para establecer relaciones con clientes, colaboradores, proveedores; realizar procesos en la organización y crear productos/ servicios con alto grado de valor. La documentación se convierte así, en una parte importante de la gestión del conocimiento entendida como el establecimiento de vínculos entre las personas y la información para lograr ventajas competitivas y añadir valor más allá del almacenamiento y manipulación de datos. La naturaleza y extensión de la documentación debe satisfacer los siguientes requisitos: (ISO 9004:2000 (3). Pág | 15 REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 13 | 36 • • • • Contractuales Legales Reglamentarios Necesidades y expectativas de clientes y otras partes interesadas. Y deben ser apropiadas para otras partes de la organización. El éxito en la implantación y mejora de los sistemas de calidad depende en gran parte de la correcta fluidez de la información, de la adecuada selección, de su distribución y de la disponibilidad para el destinatario y por estas razones la organización de la documentación de los servicios de salud se ha convertido en uno de los temas cruciales de la gestión. La documentación de los laboratorios de análisis bioquímicos - clínicos, ha sido tradicionalmente confeccionada según el mejor parecer y conocimientos de sus directivos y hasta el momento actual, en la mayoría de los servicios, la documentación existente es frecuentemente escasa, a veces desorganizada y otras, incompleta. En nuestro medio es habitual que los laboratorios no posean la documentación completa sobre por qué hacer, qué hacer, cómo hacerlo y los registros que demuestran el cumplimiento de lo expresado en la documentación. Para dar sustento práctico a las afirmaciones realizadas anteriormente, he llevado a cabo una encuesta a un conjunto de 40 laboratorios de análisis bioquímicos - clínicos (estatales y privados) acerca de la existencia y grado de complejidad de la documentación que poseen (Anexo I). Las razones anteriormente mencionadas proveen la justificación necesaria para el presente trabajo, dado que se propone una metodología para elaborar un sistema documental de acuerdo al ISO/TR 10013: 2001(E) (4), implementarlo según los requerimientos de las normas ISO 9000:2000 (5) y que sea controlado de acuerdo a las directivas de la norma IRAM-ISO 15189:2003 (6). Además, dado que todos los laboratorios clínicos que aspiren a la acreditación, deberán cumplir con los requisitos de algunas de estas normas, el trabajo está elaborado para instruir y asistir a los laboratorios de análisis clínicos para conseguir de manera ordenada y lógica, la documentación necesaria que agilice tanto la gestión como la implementación y control de los documentos. En la medida en que seamos capaces de asegurar que los procesos de las instituciones de salud estén formalizados, documentados, difundidos y aplicados en forma correcta, la calidad de los mismos estará ligada a su ejecución, y el control permitirá la corrección de las desviaciones en tiempo real, obteniéndose un servicio personalizado no obstante la normalización. 3.OBJETIVOS 3.A-OBJETIVO GENERAL Este trabajo tiene el propósito, dentro de sus objetivos, de servir como guía para los laboratorios que deseen acreditar por organismos nacionales e internacionales. Para ello, se plantean elementos de juicio concretos a nivel de la conducción que permitan desarrollar políticas contribuyentes a fortalecer la documentación del sistema de gestión de la calidad para poder acceder a futuras acreditaciones. 3.B-OBJETIVOS ESPECÍFICOS 1) Elaboración de un modelo de sistema de documentación del sistema de gestión de la calidad para los Laboratorios de análisis bioquímicos - clínicos tomando como base: a) Reporte técnico ISO/TR 10013: 2001(E). ”Guía para la documentación del sistema de gestión de la calidad”. b) La Norma ISO 9001:2000 “Sistema de Gestión de la Calidad. Requisitos”.Punto 4.2 “Requisitos de la documentación”. c) La norma ISO 9004:2000 “sistemas de gestión de la calidad. Directrices para la mejora del desempeño”. Punto 4.2 “Documentación”. 2) Elaboración del modelo del manual de la calidad del laboratorio y directivas y sugerencias para el control de toda la documentación para los laboratorios de análisis bioquímicos - clínicos de acuerdo a la norma IRAM-ISO 15189: 2003 “Laboratorios bioquímicos - Clínicos. Requisitos para la calidad y la competencia”. 4.HIPÓTESIS La organización, implementación y control de un sistema de gestión de la documentación para los laboratorios de análisis bioquímicos - clínicos permitirá una mejor organización y gestión de los mismos y cumplir con los requerimientos documentarios para acceder a la acreditación de los laboratorios (7,8). 5.LIMITACIONES Y DELIMITACIONES La presente propuesta ha sido asumida como un medio, dado que cada laboratorio adaptará los criterios de la propuesta a sus reales necesidades y a los recursos humanos y materiales de los que dispone. El tema “documentación” será enfocado desde una perspectiva conceptual multidimensional, analizándolo en su totalidad a fin de elaborar un modelo de sistema de confección, implementación y control de la documentación que sea soporte del sistema de gestión de la calidad, de utilidad tanto para los laboratorios con pequeño número de prestaciones como los servicios que poseen gran demanda, dado que los modelos brindan un marco conceptual tanto para el diagnóstico organizacional como para la gestión de la calidad. Un modelo es el producto de metodologías que aplican criterios estructurados y enfoques rigurosos, tiene un sustento práctico y permite la evaluación basada en evidencias Gestión del Laboratorio de Análisis Clínicos Implementación de la documentación según normas internacionales fácticas y concretas. Las experiencias de aplicación de los modelos indican que en todos los casos se genera un aprendizaje que siempre conduce a buenos resultados. La investigación implicará la búsqueda de datos y materiales, actualizar conocimientos y emplear la creatividad para el ordenamiento por etapas, para obtener una propuesta de sistema de documentación completo y accesible. Decidí volcar en este estudio mis conocimientos y experiencia de varios años de trabajo en el laboratorio de análisis bioquímicos - clínicos junto con la revisión bibliográfica adaptada a la realidad de nuestro medio. Considero el presente proyecto viable y accesible por las obtención de datos de fuentes primarias y por la coherencia entre el resultado a producir y las condiciones esenciales para su realización (técnicas y personales). Los costos que acarreará este estudio corresponden al tiempo personal, viáticos y material de estudio y son los esperados para una investigación documental bibliográfica. La especialización necesaria para llevar a cabo este proyecto, está fundamentada en el trabajo de investigación, en los cursos correspondientes al tema y en la búsqueda de las normas ISO internacionales referentes a la documentación necesaria según el tipo de laboratorio, para hacerlos acreedores a la confección de los documentos para la acreditación tanto a nivel nacional como internacional. 6.MARCO DE REFERENCIA Cada unidad de servicio de una institución de salud es un centro de archivo en sí mismo para el tratamiento y custodia de la documentación que le compete, sea ésta de carácter científico o de gestión y es su función recoger, tratar, conservar, administrar y facilitar la información que poseen. Para asegurarse de que se aplica un sistema de procedimientos normalizados para controlar la calidad de un producto o un servicio, se requiere una documentación completa que describa los procedimientos adecuados. De la misma forma que las organizaciones, los documentos son construcciones dinámicas y necesitan ser actualizados permanentemente para validarse como instrumentos útiles. Visualizar en forma sencilla un tema complejo, tal como la documentación de los laboratorios de análisis bioquímicos - clínicos, exige un trabajo de análisis, reflexión y síntesis. Algunos laboratorios pioneros en el tema han tomado como referencia la norma ISO 17025: 1999, IRAM 301: 2000 (9), que es una norma para los laboratorios en general (laboratorios de ensayo y calibración), que no habla de procesos y que no se refiere específicamente a los laboratorios de análisis bioquímicos - clínicos, o las normas de la serie ISO 9000 que también adolecen de falta de especificidad para los laboratorios de análisis bioquímicos clínicos, aunque los beneficios de su implementación son claros para el desarrollo del sistema de gestión de la calidad. El trabajo posee tres marcos de referencia propiamente dichos: • Para la elaboración e implementación de la documentación: la Norma ISO 9001:2000 “Sistema de Gestión de la Calidad. Requisitos” , la Norma ISO 9004:2000 “Sistemas de Gestión de la Calidad. Directrices para la mejora del desempeño” y el Reporte Técnico ISO/TR 10013: 2001(E) “Guía para la documentación del Sistema de Gestión de la Calidad”. Las dos Normas ISO mencionadas en primer término son conocidas como Sistemas de Estándares Genéricos de Gestión. Son sistemas de gestión porque proveen de un modelo para ordenar y operar una organización. Son genéricos porque las mismas normas pueden ser aplicadas a cualquier tipo de empresa cualquiera sea su complejidad, tamaño, rubro o sector. El Reporte técnico ISO/TR 10013: 2001(E) “Guía para la documentación del Sistema de Gestión de la Calidad” provee las líneas directrices para el desarrollo de la documentación de los sistemas de gestión de la calidad. • Para la confección del modelo del Manual de la Calidad y el control de la documentación: la Norma ISO 15189:2000. Versión original en inglés “Medical Laboratories. Particular requirements for quality and competence”. En este punto la razón para utilizar la Norma ISO 15189 se debe a la necesidad de consideraciones especiales en cuanto a las relaciones de los laboratorios de análisis clínicos con los pacientes, en particular para las etapas pre y postanalíticas, que son las fases clave en el contacto del laboratorio con sus clientes externos. Esta norma es específica para los laboratorios clínicos y posee principalmente dos componentes: la parte de gestión y la parte técnica. Lo referente de gestión corresponde a los requisitos para la certificación del Sistema de Calidad, mientras que la parte técnica describe los requisitos para el personal, equipos, instalaciones, procedimientos, garantía de calidad e informes. Este soporte teórico, convenientemente elaborado, permitirá la consecución de los objetivos planteados. 7.DESARROLLO 7.1-DOCUMENTOS Y ARCHIVOS Los nuevos tiempos exigen nuevos modelos y paradigmas que reemplacen o mejoren los existentes y en ese aspecto, el conocimiento ocupa un lugar preponderante en su consideración e importancia como factor productivo. El interés por la forma en que las empresas adquieren, generan, almacenan y expanden información y conocimientos para desarrollar nuevos productos o servicios, ha pasado a ser prioritario, tanto desde una perspectiva académica como desde el punto de vista práctico. En este contexto, las transformaciones tan aceleradas que está viviendo la sociedad apuntan a que cada vez se agrupen esfuerzos para lograr objetivos y metas comunes, situación que también repercute en los centros de información REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 13 | 36 documental y en los archivos. Las sociedades se han convertido en “sociedades de la información” (Cornella, 1994) y todas las organizaciones son sistemas de procesamiento de información (Kaye, 1996). El concepto de documento varía sustancialmente según se lo analice desde los puntos de vista de los profesionales de las distintas ciencias relacionadas con el mismo: historiadores, diplomáticos, juristas, bibliotecarios o documentalistas. En un sentido muy amplio y genérico, un documento es un objeto tangible producto de la actividad humana, que sirve de fuente de conocimiento y que demuestra o prueba algo. También puede definirse como el testimonio de la actividad del hombre fijado en un soporte perdurable que contiene información.(10) Un documento de archivo es un instrumento fehaciente, que da fe de un hecho y que prueba o justifica la certeza o verdad de una cosa (11), tanto sea de naturaleza jurídica como administrativa. Es esencial su relación con la entidad productora, así como las demás circunstancias que condicionan sus características y su finalidad. “Un documento de archivo es el testimonio material de un hecho o acto realizado en el ejercicio de sus funciones por personas físicas o jurídicas, públicas o privadas, de acuerdo con unas características de tipo material y formal” (12). Los documentos de archivo son garantía de que el hecho relatado es verdadero, poseen valor testimonial científico y pueden ser utilizados como prueba y como fuente de datos, dado que reflejan las actividades del hombre, producto y testimonio de una gestión. La autenticidad es una de las características fundamentales de los documentos de archivo, y debe ser siempre probada científicamente. Para establecer una clara relación de archivo con Institución de Salud, puede definirse según Robert Bautier “Un depósito de archivo es ante todo, como un establecimiento de carácter científico, encargado de funciones administrativas”, lo cual representa las dos caras de una misma institución. La razón última de la conservación documental es siempre una razón práctica y utilitaria, ya sea con fines jurídicos, administrativos o de investigación y su principal función es comunicar de la información contenida en los archivos. 7.2-INFORMACIÓN Y GESTIÓN. ARCHIVOS INSTITUCIONALES La gestión de la información es el proceso que se encarga de suministrar los recursos necesarios (input) para la toma de decisiones en todos los niveles, así como para mejorar los procesos, productos y servicios (output) de una organización. Las instituciones deben conseguir, procesar, usar y comunicar información, tanto interna como externa, en sus procesos de planificación, dirección y toma de decisiones (Kaye, 1996). La información es la representación activa y dinámica de la realidad que ella misma nos proporciona, en un contexto mediante Pág | 17 datos que se extraen de hechos ocurridos o percibidos (13). Sus objetivos son: • Obtener y diseminar el conocimiento para reducir la incertidumbre. • Proporcionar alternativas que impulsen a tomar decisiones. • Resolver problemas en base a datos útiles y oportunos • Confirmar los datos. • Estar al servicio de la organización. • Garantizar la coherencia de formas y expresiones a partir de una única fuente de datos. Sus valores son: • Sincronía con los hechos. • Anticipación a los problemas. • Oportunidad resultante de la sincronía y anticipación. • Confianza de la relación entre el costo de su obtención y su valor. Sus atributos son: • Finalidad deseada. • Formato deseado de presentación y circulación. • Equilibrio entre su redundancia y su eficiencia. • Densidad o equilibrio entre el exceso y el grado óptimo de información. • Frecuencia con que se necesita. • Rapidez requerida en la explotación. La información es un mensaje significativo que se transmite de la fuente a los usuarios, es la expresión material del conocimiento con fines de uso y para resolver determinados problemas. Cuando se habla de organización es casi imposible no hablar de información y de su resultante: el conocimiento, el cual a través del tiempo ha comenzado a convertirse en un bien público. Los archivos institucionales están constituidos por un conjunto de documentos que integran todos los archivos de una institución, pública o privada y constan de tres elementos básicos: • La institución productora, como marco de producción y acumulación de la información. • El fondo documental, como suma de esa información. • La sistematización de la organización de ese fondo documental, para la puesta al servicio de los clientes externos e internos. En el modelo europeo de excelencia en la gestión de la Fundación Europea para la Gestión de la Calidad (EFQM) la administración de la información tiene peso como agente facilitador en la obtención de resultados y es parte fundamental en la innovación y aprendizaje. El éxito en la implantación y mejora de los sistemas de calidad depende en gran parte de la correcta fluidez de la información, de la adecuada selección, de su distribución y de la disponibilidad para el destinatario y es por estas razones que la organización de la documentación de los Servicios de Salud se ha convertido en uno de los temas cruciales de la gestión. Gestión del Laboratorio de Análisis Clínicos Implementación de la documentación según normas internacionales 7.3-DOCUMENTACIÓN DE UN SISTEMA DE GESTIÓN DE LA CALIDAD (SGC) La implementación del Sistema de Gestión de la Calidad del Laboratorio Clínico tiene su soporte en el Sistema Documental, dado que en el mismo no solamente se expresa la forma de operar el servicio, sino toda la información que posibilita el desarrollo de cada uno de los procesos operativos y la toma de decisiones para la gestión. La estructura de la documentación es una combinatoria de los procesos del servicio y de los apartados de la norma aplicable. Debe responder al tamaño del servicio, a la complejidad de las actividades que desarrolla y a la interrelación entre sus procesos y el grado de preparación de su Recurso Humano. La documentación facilita la comprensión de los procesos y nivela los conocimientos del personal. Los procesos son la parte menos variable de la asistencia sanitaria, aún cuando cambie su contenido y los criterios del enfoque sobre el proceso son más fácilmente traducibles a resultados que los enfoques estructurales. Desde esta perspectiva, la descripción tanto de la estructura como de los procesos documentales facilita la distribución, el mantenimiento y comprensión de la documentación, disminuyendo la incertidumbre y ayudando a actualizar los conocimientos. La documentación del Sistema de Calidad permite la identificación y gestión de procesos y actividades relacionadas con un enfoque integral y el desarrollo de una cantidad de documentos necesarios para la efectiva planificación (planes de gestión), operación (planes operativos), incluyendo elementos organizativos, inventariales, control y mejora continua del SGC y sus procesos. La Figura 1 muestra la ubicación de la Documentación dentro del Sistema de la Calidad. Figura 1. Ubicación de la Documentación dentro del Sistema de Calidad Sistema de la Calidad Establecer Implementar Mantener Documentación Comunicada Comprendida Disponible Implementada La familia de normas 9000-2000 requiere que el Sistema de Gestión de la Calidad de una organización debe estar documentado. La documentación del SGC debe alinearse con: a) las actividades globales del laboratorio b) las actividades seleccionadas del servicio relativas a: • Los productos • Los procesos • Los contratos • Las regulaciones vigentes • Las exigencias propias del servicio • La satisfacción de los clientes ¿Qué es un documento? Un documento es la información y su soporte (IRAM-ISO 9000:2000). Son evidencias escritas objetivas del Sistema de Calidad que permiten la trazabilidad de los resultados. Donde trazabilidad significa: “aptitud de reconstruir la historia, la utilización o la localización de un producto por medio de identificaciones registradas”. También se puede definir “documento” como: a) Recopilación estructurada de información para la comprensión humana. b) Resumen del resultado de una tarea. c) Formalización de un conocimiento obtenido. En la práctica un documento es una entidad que tiene autores, lectores, se almacena en algún soporte y cumple con una finalidad. En este contexto el término “documento” puede significar declaraciones de la política, procedimientos, especificaciones, tablas de calibración, gráficos, manuales, pósters, software, dibujos, planos, etc. Pueden estar en diversos medios tales como papel o soportes electrónicos y pueden ser digitales, analógicos, fotográficos o escritos. La documentación del Sistema de Calidad de los laboratorios está constituida por manuales, procedimientos generales, procedimientos específicos, instrucciones operativas, formularios, hojas de trabajo y protocolos. La documentación provee evidencia de que: • Los procesos y operaciones técnicas están definidos. • Los procedimientos han sido aprobados. • Los cambios están bajo control. • Los requisitos han sido cumplidos. • La organización está ordenada y balanceada. La documentación provee soporte para la mejora del sistema: • Permite detectar un cambio. • Permite evaluar el efecto de un cambio. • Permite establecer el estado de mejora alcanzada. • Provee una base para la mejora continua. • Provee un marco de referencia para las relaciones con los proveedores. • Provee de base para los procesos de Auditoría. La documentación provee la base para la capacitación: • Facilita la capacitación del personal en adiestramiento. Pág | 19 REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 13 | 36 • Promueve un equilibrio entre aptitud y capacitación del personal y el grado de detalle de la documentación. • Ayuda a los empleados a comprender su rol dentro del laboratorio así como el propósito e importancia de su tarea. • Permite el mutuo entendimiento entre empleados y directivos. • Si no se documenta, la organización no sabe concretamente como trabaja. ¿Para qué se documenta? • Proveer confianza en cada una de las actividades que se desarrollan en el laboratorio y saber que están consensuadas por los responsables de la calidad y que se cumplen siempre y del mismo modo. • Disminuir errores sistemáticos mediante la comparación con una referencia externa establecida. • Conocer las variaciones de los indicadores. • Proveer entrenamiento y guía para el personal. • Capacitar al personal que ingresa. • Facilitar la comunicación del laboratorio con otros servicios relacionados. • Conocer la calidad de la asistencia brindada. • Comparar las actividades del servicio con patrones de desempeño que se desean alcanzar. • Planificar las actividades futuras. 7.4-ESTRUCTURA DE LA DOCUMENTACIÓN DE UN SISTEMA DE GESTIÓN DE LA CALIDAD (SGC) La preparación de esta documentación es posiblemente la parte más extensa y la que más tiempo requiere de todo el proceso de acreditación del laboratorio, pero debido a su naturaleza comprobatoria de varias normas, es esencial que se haga de una manera sistemática y estructurada. La estructura de la documentación del Sistema de Gestión de la Calidad se alinea con los procesos del laboratorio y los puntos de la norma aplicable a dicho sistema. Debido a la amplia variedad de documentos involucrados, es útil establecer una “jerarquía de la documentación” según el ISO/TR (Technical Report) 10013: 2001(E). El ISO/TR 10013:2001(E) provee una guía para asistir a una organización que pretende desarrollar y mantener la documentación necesaria para asegurar su Sistema de Gestión de Calidad, hecho a medida de sus necesidades específicas mediante la adopción del Enfoque por Procesos. La Figura 2 muestra la Pirámide de Calidad de una organización. ISO 10013:2001(E). Notas: 1. El número de niveles puede ser ajustado a las necesidades de la organización. 2. Los formularios pueden ser aplicables a todos los niveles jerárquicos. 3. El desarrollo de una jerarquía depende de las circunstancias de la organización. Figura 2. Pirámide del Sistema de Calidad según ISO 10013:2001 Política de Calidad y Objetivos Manual de la Calidad Generación de Procedimientos generales Registros y Instrucciones de trabajo Datos Por encima de la pirámide y fuera de ella, están las Políticas de Calidad y los Objetivos de la Organización. Una política puede definirse como “la declaración de principios de una organización para seguir un procedimiento de acción en particular”. Un laboratorio clínico puede tener diseñada una política general en la cual se incluyan todos los aspectos del trabajo del laboratorio o tener una serie de políticas separadas relacionadas con los diferentes aspectos del trabajo. Las políticas pueden estar relacionadas con la gestión, la garantía de la calidad, los métodos del laboratorio, etc y los procedimientos necesarios que se realizan en esas mismas áreas. El primer nivel lo constituye el Manual de la Calidad de la organización. Es un documento que sirve para introducir las políticas y campo de actuación de los servicios del laboratorio, así como para crear una estructura que permita organizar su documentación. Es obligatorio para todo laboratorio que desee certificar su Sistema de Calidad. El segundo nivel en la jerarquía está constituido por los Procedimientos. Los procedimientos son “Una manera especificada de llevar a cabo una actividad” (IRAM-ISO 8402:1994) (12) o “Forma especificada para llevar a cabo una actividad o un proceso” (IRAM-ISO 9000:2000). A menudo se denominan PNT (procedimientos normalizados de trabajo) o POES (procedimientos operativos estándar). Son la manera práctica en que las políticas se trasladan a la acción. El tercer nivel de la documentación son las Instrucciones de trabajo. Estas son las instrucciones prácticas del día a día y pueden estar incluidas en un procedimiento o pueden estar citadas en el procedimiento y escritas separadamente. Cada una de estas jerarquías es una fuente de generación de datos y registros, los cuales siguen en importancia documental a las instrucciones de trabajo y que generalmente se asientan en formularios. Un registro es un formulario que contiene alguna información. Los registros deben hacerse en formularios que tengan un formato adecuado y aprobado y su soporte no debe ser necesariamente papel sino cualquiera que provea evidencia de lo actuado. Gestión del Laboratorio de Análisis Clínicos Implementación de la documentación según normas internacionales Es importante que se registre cualquier información o dato, como por ejemplo los de ingreso y resultados de los pacientes, los resultados de los controles de calidad interno y externo o los resultados de una inspección o auditoría. Esta documentación “revisable”, operativa al nivel de los procesos, constituye la columna vertebral del Sistema Documental de los laboratorios y provee en todos los casos evidencias del funcionamiento del sistema. 7.5-ELABORACIÓN DE UN SISTEMA DE DOCUMENTACIÓN PARA LOS LABORATORIOS DE ANÁLISIS CLÍNICOS Previo a la implantación de un Sistema de Gestión de la Documentación, la Dirección del Laboratorio deberá tener la plena convicción de que el Sistema Documental es crucial para el logro de la calidad asistencial y una necesidad insoslayable para permanecer en el mercado. El Sistema Documental de un Laboratorio de Análisis Clínicos debe ser apropiado para el ambiente que opera, basándose en un análisis objetivo de las necesidades de documentación y no ser meramente un instrumento burocrático que desmotive la creatividad y la iniciativa. Una recomendación para que el Laboratorio no posea un sistema documental burocrático es que toda la documentación del Sistema de Calidad debe relacionarse con el riesgo que acarrearía la falta de determinado documento. La documentación del Sistema de Calidad debe identificar y desarrollar los procesos y actividades, relacionándolos con un enfoque sistémico a fin de elaborar una serie de documentos necesarios para la planificación, operación y mejora continua del sistema y sus procesos. La consulta de la documentación de los laboratorios facilita la aplicación de un procedimiento de trazabilidad (14) para la evaluación de resultados, consistencia de la producción y análisis de no conformidades. La trazabilidad de los archivos permite reconstruir certeramente la información disponible a lo largo de cualquier proceso practicado sobre una muestra de un paciente en particular, independientemente del grado de procesamiento de esa muestra o aún con el resultado analítico ya emitido. Existe amplio consenso acerca de la importancia de la documentación como soporte del Sistema de Gestión de la Calidad, pero existe una tendencia a limitar esta cuestión solamente a cuál es la mejor manera de elaborar un Manual de la Calidad y un Manual de Procedimientos. Si bien estos aspectos son importantes, es esencial la visión del Sistema Documental como un todo que abarca tanto los documentos como los pasos necesarios para garantizar que el sistema funcione como un conjunto armonioso, que esté bajo control y sea una herramienta eficaz para la administración del laboratorio. 7.6-MODELO DE SISTEMA DE GESTIÓN DE LA DOCUMENTACIÓN A continuación se describe el modelo propuesto de “Sistema de Gestión de la Documentación”, a fin de conocer qué procesos implica y la mejor manera de llevarlos a cabo para que sirva de soporte al Sistema de Gestión de la Calidad. Será de referencia para que en cualquier servicio de laboratorio clínico pueda ser implantada la Gestión Documental. El modelo está basado en el Enfoque por Procesos, por medio del cual se lograron identificar eslabones críticos de los procesos de la documentación y proponer los pasos para implementarlos. Enunciado de cada proceso principal seguido del desarrollo de cada uno: a) Determinar qué tipo de documentación debe existir en el servicio para garantizar que los procesos se ejecuten en condiciones controladas. b) Estado actual de la documentación del laboratorio c) Diseño del Sistema de Documentación. d) Definición de la autoridad y responsabilidad para la elaboración de la documentos en cada nivel. e) Definición del plan de elaboración de la documentación f) Determinación de la circulación o flujo de la documentación. g) Definición del cronograma de implantación. h) Estimación de los costos que acarreará el proceso en general. a) Establecimiento de qué tipo de documentación debe existir en el servicio para garantizar que los procesos se ejecuten en condiciones controladas Se propone a las autoridades del servicio el estudio e interpretación de las Normas ISO 9001:2000, 9004:2000 y el ISO/TR (Technical Report) 10013:2001(E) para la confección de la documentación y la Norma ISO 15189:2000 para el control de la misma. Deben identificarse los requisitos de las normas relacionados con los procesos del laboratorio, y aplicando el sentido común, determinar la extensión de la documentación de acuerdo a: • Tamaño del laboratorio. • Menú de prestaciones. • Cantidad de prestaciones. • Complejidad de los procesos y sus relaciones. • Competencia y preparación del Recurso Humano. La Documentación del Sistema de Gestión de la Calidad de acuerdo al ISO/TR 10013:2001(E) incluye: • Declaración documentada de la: - Política de la Calidad. - Objetivos de la Calidad. • Manual de Calidad. • Procedimientos documentados. REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 13 | 36 • • • • • Instrucciones de trabajo. Formularios. Planes de la Calidad. Especificaciones Documentos Externos. Registros. En el modelo propuesto, tanto las Políticas de Calidad como los Objetivos de Calidad forman parte de la Introducción del Manual de la Calidad (6). b) Estado actual de la documentación del laboratorio Debe investigarse la situación de la documentación existente en el laboratorio comparando lo que se dispone con los requisitos determinados en el punto anterior. Para relevar la documentación deberá conocerse: • Qué documentos existen y responden a la norma. • Qué prácticas se efectúan y no están documentadas. • Qué actividades no se hacen y se deberían hacer. En general, es difícil encontrar laboratorios que no posean algún grado más o menos complejo de documentación. En este caso, debe analizarse la utilidad de los documentos existentes a fin de adaptarlos a los nuevos requerimientos, pudiéndose así acortar significativamente el tiempo de implementación del Sistema Documental. Debe hacerse un listado de los documentos existentes y analizarlos para establecer su utilidad. Este estudio es conocido como “Análisis Gap” y es muy útil para determinar en qué aspectos dichos documentos deberían ser completados o corregidos. c) Diseño del Sistema de Documentación Establecer las pautas y elementos generales necesarios que abarcará el Sistema Documental. Se propone el modelo de la pirámide ISO 10013:2001(E), donde se ubica en el nivel más alto el Manual de la Calidad, en el segundo nivel los procedimientos, expuestos en el Manual de Procedimientos y en el tercer nivel, instrucciones de trabajo, registros, especificaciones y otros documentos. Por encima de la pirámide y fuera de ella, están las Políticas de Calidad y los Objetivos de la Organización. La Dirección del laboratorio debe considerar los requisitos legales, reglamentarios y normativos del Sector Salud, la información atinente a las necesidades y expectativas de los pacientes, médicos y otras partes interesadas, tales como los administradores y los proveedores. Es importante establecer las relaciones que los documentos guardan entre sí, para poder diseñar una documentación que contemple los posibles efectos de las interacciones entre ellos. En algunos casos conviene confrontar el diseño propuesto con la documentación existente en otros servicios que cumplen iguales funciones (benchmarking) para tomar ideas y adaptarlas al proyecto en estudio. Pág | 21 El laboratorio debe definir la estructura y formato de los procedimientos documentados (impresos o en soporte informático) en cuanto al texto, diagramas de flujo, tablas o alguna combinación de los anteriores y deben contar con una identificación única. d) Definición de la autoridad y responsabilidad para la elaboración de la documentos en cada nivel El proyecto para la implantación del Sistema Documental debe tener un líder (que no sea externo al laboratorio), que será el representante de la Dirección y el nexo con los integrantes de la tarea operativa propiamente dicha. En general, esta persona pertenece a los mandos medios del servicio y está capacitada tanto para coordinar las tareas como para efectuar comentarios o correcciones sobre la preparación o elaboración de los documentos. Es muy importante que tanto el proyecto de documentación como su implementación esté conducido por personal del propio laboratorio y no que la tarea sea encargada a un asesor externo al servicio, dado que al no pertenecer al plantel permanente no vive los problemas diarios de la organización. En este punto debe decidirse quienes elaborarán, quienes revisarán y quienes aprobarán los documentos acorde a los conocimientos, el nivel jerárquico y el organigrama vigente. Debe comprometerse a todo el personal que sea posible en la elaboración, revisión y modificación de los borradores o versiones preliminares de los documentos de acuerdo a su capacitación y entrenamiento, dado que quien mejor conoce una tarea es aquél que la realiza habitualmente. También debe contemplarse la necesidad de capacitación del personal involucrado en la confección de los documentos, proveyéndolo además de otras fuentes documentales accesorias y los recursos de asesoramiento y materiales disponibles para llevarla a cabo. e) Definición del plan de elaboración de la documentación Se definirán los procesos operativos de confección de cada grupo de documentos según los criterios y alcances de la norma a aplicar, así como también un orden de prioridad de elaboración y plazos para el inicio y la terminación para cada documento. Para esto se procederá a crear un procedimiento técnico para la elaboración de los documentos, los cuales tendrán un responsable de la confección, un responsable de la revisión y/o corrección y un responsable de la aprobación definitiva del documento en cada nivel. Con la finalidad de hacer más ágil esta etapa, es conveniente que la revisión y aprobación de los documentos se realice a medida que éstos se vayan confeccionando. El enfoque del plan de elaboración puede ser personalizado (el documento lo elaborará una sola persona ) o en equipo. En este último caso, el número de integrantes variará de acuerdo a la importancia y extensión del documento y deberán realizarse varias reuniones de consenso y discusión antes de la elaboración propiamente dicha. Gestión del Laboratorio de Análisis Clínicos Implementación de la documentación según normas internacionales f) Determinación de la circulación o flujo de la documentación Deberá organizarse el circuito de la documentación desde su aprobación por la autoridad competente hasta el momento en el que el documento sale de circulación por reemplazo por una versión más actualizada (revisión del documento) o porque ya no es más útil. Debe preverse que los documentos sean accesibles a las personas autorizadas y que se encuentren en todo momento en el lugar requerido para su consulta. Ciclo de vida de un documento: 1) Creación o generación: El autor o autores elaboran el contenido del documento, el cual estará mejor diseñado por la persona que habitualmente realiza la tarea o la que está familiarizada con la actividad en particular. 2) Revisión: Se revisa y pueden agregarse comentarios o efectuar alguna sugerencia en cuanto al contenido para mejorar su facilidad de uso, lo cual hace a su funcionalidad. La revisión del documento debe ser hecha preferentemente por la persona que emplea ese documento y proporcionarle un estilo similar acorde a toda la documentación. 3) Aprobación: Hasta el momento de su aprobación el documento se considera no controlado y es susceptible de cambios para perfeccionarlo. Luego se controla, aprueba y autoriza la distribución, previa identificación adecuada. 4) Distribución y conservación Se edita, imprime y se aseguran los medios para almacenarlo con seguridad. 5) Baja: El documento sale de circulación y pasa a un archivo de “Documentos Obsoletos”. Actualmente ya existe un número apreciable de laboratorios que aplican medios electrónicos como soporte de la documentación, los cuales tienen las siguientes ventajas: • El personal autorizado tiene acceso todo el tiempo a la misma información actualizada. • Facilidad para efectuar controles, cambios y distribución de los documentos. • Facilidad de impresión y copia. • Acceso a documentos antiguos. • Facilidad para el retiro de documentos obsoletos. Los documentos conservados en soporte informático deben almacenarse de dos formas: en un soporte para la consulta habitual y en otro guardado como reserva, la cual debe ser accesible y consultable durante todo el tiempo de vigencia del documento. El laboratorio debe garantizar la confidencialidad de los datos de los pacientes o toda otra información relativa a ellos, mediante la preservación de los bancos de datos en el caso de soporte informático o de los libros de registros en caso de soporte en papel (copia dura). El acceso a los datos archivados debe estar debidamente reglamentado y restringido según nivel de responsabilidades para asegurar la confidencialidad de la información. En algunas ocasiones, la distribución de documentos tales como el Manual de Calidad y objetivos de calidad, pueden trascender el ámbito del laboratorio, para ser difundidos externamente, ya sea a organismos de acreditación o laboratorios derivantes. En la Figura 3 puede analizarse el diagrama de flujo propuesto para la emisión de la documentación. Figura 3. Flujograma de emisión de la documentación Responsable de la Calidad Elaboración del documento ajustes posibles Responsable de la comprobación No aprueba Si notificación Distribución del documento copias Responsable de la distribución g) Definición del cronograma de implantación Para efectuar esta tarea deberán considerarse las características propias del laboratorio en cuanto a su organización y los recursos materiales y humanos para una efectiva implementación. La documentación aprobada debe ser distribuida a los sectores a medida que se realiza su aprobación. La implementación es conveniente que sea en etapas, encargando a los empleados tareas que, a nuestro juicio, sean capaces de realizar con cierta autonomía (bit size pieces), propiciando el “poder hacer”. En el caso frecuente en que se presenten dificultades para la implantación de algún procedimiento, deben efectuarse las medidas correctivas apropiadas para el caso en el tiempo más corto posible. Debe mantenerse el sistema adecuado a las necesidades operativas y de gestión del servicio para garantizar su utilidad a través del tiempo. Esto se logra mediante auditorías internas documentales, las cuales identificarán las oportunidades para mejorar algún aspecto del Sistema Documental. h) Control de la documentación Debe establecerse un procedimiento para el control de todos los documentos del Sistema de Calidad, tanto los generados internamente como de fuentes externas, asegurando REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 13 | 36 la disponibilidad y adecuación y previniendo el uso de los documentos obsoletos. i) Estimación de los costos que acarreará el proceso en general Los costos a considerar incluyen los correspondientes a los materiales e insumos que serán utilizados y los de capacitación del Recurso Humano involucrado (horas- hombre). 7.7-EL MANUAL DE CALIDAD. MODELO PARA SU ELABORACIÓN. La Gestión de la Calidad (planificación, organización, control y mejora) en todo servicio, sobrepasa los límites del problema técnico por afectar a la estructura organizativa del mismo. Por esta razón debe establecerse un Sistema de Calidad que asegure, externa e internamente, el cumplimiento de la Política de Calidad formulada por la Dirección (en nuestro caso la Jefatura del Laboratorio). Dicho sistema se almacena en un documento, denominado Manual de Calidad, que recopila y ordena todo lo referente a la organización y documentación, relativas a las políticas y procedimientos de la función Calidad, definiendo el perfil de servicio que el laboratorio pretende proveer y el compromiso del laboratorio con la buena práctica profesional. Es un documento ejecutivo único para cada organización que sirve para introducir las políticas y campo de actuación de los servicios del laboratorio, así como para crear una estructura para organizar la documentación. Proporciona un documento permanente de referencia que permite al Recurso Humano comprender el compromiso de la Dirección con la Política de la Calidad y usarlo para enfocar sus actividades. Es obligatorio y punto de partida para la evaluación de todo laboratorio que desee certificar su Sistema de Calidad. Aún cuando no se busque el reconocimiento formal, la elaboración del Manual de Calidad será un procedimiento edificante que conducirá a una mejor práctica, asegurando que la dirección sepa lo que sucede en el laboratorio y que el personal tenga un marco de referencia confiable. Es un instrumento para la comunicación de la responsabilidad que tiene el servicio con respecto a la calidad. Propósitos del Manual de la Calidad: • Comunicar las Políticas de Calidad a fin de que puedan ser aplicadas. • Definir la competencia del laboratorio y la continuidad de las operaciones independientemente de la rotación del personal. • Describir y facilitar actividades, siendo patrón de referencia para evaluar los procedimientos. • Documentar el Sistema de Calidad, otorgando evidencias de la capacidad operativa del servicio. • Capacitar al Recurso Humano, contribuyendo a la formación interna y a la normalización de su comportamiento. Pág | 23 • Exhibir el Sistema de Calidad ante requerimientos externos de inspecciones o auditorías. • Establecer los procedimientos necesarios para cumplir con requerimientos legales establecidos, tanto referentes al Derecho propiamente dicho como a la contratación de servicios o la compra de material o equipos. Cada grupo directivo de los laboratorios elegirá las características del Manual de Calidad que elaborará, tales como la extensión y complejidad. Su redacción consiste en la revisión sucesiva de los distintos capítulos de la norma aplicable para adecuar las actividades del laboratorio a sus requerimientos. Debe tener un formato material definido, de buen aspecto formal, consistente en: • Portada Es la primera página del manual, en la cual se lo enuncia. Debe contener el nombre y dirección del laboratorio, número de copia y forma de distribución (copia controlada o no controlada). • Índice de Capítulos y Secciones Es aconsejable que todos los capítulos guarden la misma estructura y que definan el objetivo del capítulo, actividades, personas o recursos afectados, documentos empleados como base en su elaboración y la descripción de actividades para el cumplimiento de los objetivos. • Control de modificaciones Esta página debe tener un registro completo de las modificaciones efectuadas en el Manual de Calidad, proporcionando evidencias de que se mantiene al día y que se han emitido nuevas páginas con las modificaciones. • Paginación Que incluya una codificación del número de revisión. El manual puede incluir las políticas, gestión y los procedimientos, o solamente las políticas y el Sistema de Gestión y hacer referencia al Manual de Procedimientos y otros documentos en los Anexos. El Manual de Calidad puede considerarse también como la introducción a los procedimientos que deben prepararse como resultado de las políticas definidas en el mismo. Una vez que la conducción del laboratorio ha tomado la decisión de elaborar el Manual de Calidad, la tarea se delega al Responsable de Calidad, para coordinar su confección, quien trabajará con un grupo abocado a tal proyecto. De este modo se involucra al personal y se fomenta el sentimiento de pertenencia al servicio. Las recomendaciones que propongo para la preparación de la redacción del Manual de Calidad son: • Designar un responsable (que conozca bien la operativa del laboratorio). • Acordar el contenido con todas las funciones involucradas. Gestión del Laboratorio de Análisis Clínicos Implementación de la documentación según normas internacionales • Describir el Sistema de Calidad sin incluir detalles técnicos o específicos • Describir efectivamente lo realizado, no lo que debería hacerse. • Seguir una lógica en las etapas, redactando con estilo simple y directo. • Referirse a funciones, no a personas • Acordar su contenido con todas las funciones intervinientes. • Diagramar el contenido para facilitar su actualización (hojas intercambiables, ordenado y de fácil acceso a las personas autorizadas). Todo el personal debe ser instruido sobre el uso y la aplicación del Manual de Calidad, todos sus documentos de referencia y el requerimiento de su implementación. La responsabilidad del manual en todo aspecto (conservación, revisión, circulación, etc) está bajo la órbita del Responsable de Calidad del laboratorio, que es el representante de la jefatura respecto de la calidad. Luego de analizar las normas disponibles para la confección de manuales de calidad, especialmente la IRAM-ISO 15189:2003, considero que es necesario adaptar esas directivas a la realidad de los laboratorios de nuestro medio, a fin de hacer más simple la confección sin dejar de lado ningún aspecto esencial de los contenidos. Propongo el siguiente ordenamiento con tres secciones generales: 1. Introducción 2. Manual de Calidad 3. El laboratorio 1. Introducción Esta sección contiene la Política de Calidad y los Objetivos de Calidad del servicio. La Política de Calidad es redactada por las máximas jerarquías de los servicios, es revisada en forma periódica por los sectores afectados y es aprobada por la dirección. La definición de la Política de Calidad es propia de cada servicio y debe poseer los siguientes atributos: a) Ser coherente con las políticas de la organización a la cual pertenece. b) Ser clara y objetiva. c) Ser comprendida, aplicada y mantenida en todos los niveles del laboratorio. d) Establecer los medios para alcanzar y medir los objetivos de la calidad declarados. e) Asignar responsabilidades para su instrumentación. f) Estar refrendada por la jefatura, afirmando su compromiso con la calidad suministrada. Ejemplo de Política de Calidad : “La Política de Calidad para nuestro laboratorio de Análisis Clínicos es alcanzar dentro del ámbito de trabajo niveles de calidad competitivos y un hábito de mejorar diariamente nuestra labor para lograr la satisfacción de nuestros clientes (pacientes y demás integrantes del equipo de salud) tendiendo a eliminar o corregir cualquier causa de error en nuestro trabajo cotidiano para mejorar la productividad y eficiencia a través de la mejora de la Calidad” (14). La planificación estratégica del servicio y la política de la calidad proporcionan un marco de referencia para el establecimiento de los Objetivos de la Calidad. La Dirección establece estos objetivos para mejorar el desempeño del laboratorio (4). Los objetivos de calidad son metas de calidad definidas y cuantificadas, base para la planificación de resultados. Sirven para unificar criterios, motivar, actuar y comparar resultados. En un servicio existen dos niveles de Objetivos de Calidad: a) A nivel dirección: son objetivos de mejora y de gestión. b) A niveles inferiores: son objetivos de control y tecnológicos. Ejemplo de objetivos de calidad: “Objetivos de la política de calidad de nuestro laboratorio de análisis bioquímicos - clínicos: b. Proporcionar información clínicamente útil a través de los análisis de las muestras de los pacientes teniendo en cuenta los recursos destinados a tal fin. c. Los datos que se proporcionen deben ser confiables y las incertidumbres deben estar de acuerdo con las necesidades clínicas y las normas técnicas apropiadas para nuestra profesión. d. Capacitar al recurso humano en políticas de calidad comprometiendo a todo el personal en la realización de acciones concretas para llevarlas a cabo. e. Mejorar la productividad y eficiencia a través de la Política de Calidad. f. Informar y concientizar a todo el equipo de salud sobre las acciones del laboratorio. g. Incorporar nuevas técnicas analíticas de acuerdo a la demanda necesaria y justificada del Equipo de Salud “(15). 2. Manual de Calidad 2.1 Objeto Describir el Sistema de Calidad del laboratorio. 2.2 Alcance Describe qué actividades regirá el manual. Puede referirse a todas las actividades del laboratorio o solamente a algunas. 2.3 Referencias Se basará en la Norma ISO 15189 versión 2000 (original en inglés). Como advertencia, esta normativa aclara que “aunque el documento no fue pensado como una guía para la acreditación, podría ser usada a tales propósitos por gobiernos, profesionales u otra autoridad. El documento se en- REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 13 | 36 foca en los rasgos y requerimientos característicos de los laboratorios clínicos”. 2.4 Definiciones A los fines de la Norma ISO 15189 han sido aplicadas más de 100 definiciones, las cuales no serán expuestas en este trabajo y que pueden encontrarse en el texto original de la norma. 2.5 Responsabilidades Define quienes serán los responsables tanto de la confección como del mantenimiento actualizado del manual. 3. El Laboratorio 3.1 Introducción En este capítulo se “presenta” al laboratorio, haciendo una breve reseña de su historia, ubicación geográfica y las características físicas del servicio. Deben especificarse las características de la población que asiste, así como la zona geográfica de cobertura de los servicios que el laboratorio brinda. 3.2 Requisitos de Gestión 3.2.1 Organización y Gestión La forma en que el laboratorio se organiza y gestiona afectará no solamente la calidad del producto final sino que tendrá un marcado efecto sobre el personal que trabaja en él. La Gestión del laboratorio debe seguir algunos principios fundamentales: • Clara demarcación de las responsabilidades • Líneas jerárquicas bien definidas Líneas de comunicación bien establecidas y usadas apropiadamente • Funciones asumidas por cada nivel 3.2.2 Sistema de Gestión de la Calidad Las políticas, procesos, programas, procedimientos e instrucciones deben estar documentadas y en conocimiento del personal relevante. La jefatura asegura que los documentos son comprendidos e implementados. La Jefatura del Laboratorio debe impartir las directivas para la confección de indicadores que evalúen sistemáticamente su contribución al cuidado de los pacientes. Asimismo debe asegurar la participación del laboratorio en actividades de mejoramiento de la calidad que involucren áreas relevantes para el cuidado de los pacientes. Es aconsejable que estas actividades comprendan: a) Sistema de control de calidad interno b) Sistema de control de calidad externo En cada uno de estos controles el personal participante deberá estar familiarizado en el manejo de los Documentos de Calidad y la implementación de los procedimientos. Debe establecerse e implementarse un programa que regu- Pág | 25 larmente controle y demuestre la adecuada calibración y funcionamiento de los instrumentos, reactivos y sistemas analíticos, así como también un programa preventivo de mantenimiento y calibración el cual, como mínimo debe seguir las recomendaciones de los fabricantes. La Jefatura del laboratorio debe designar un responsable técnico, encargado de la Función Calidad, el cual será el representante de la Dirección del laboratorio para los temas concernientes a la calidad. Tendrá acceso directo a la Jefatura, dispondrá de recursos para la calidad y autoridad para efectuar correcciones. 3.2.3 Control de documentos El laboratorio debe poseer procedimientos para controlar toda la información y documentos que forman su Documentación de Calidad. Nota: en este contexto, “documento” es cualquier información o instrucciones incluyendo manifiestos de políticas, libros de texto, procedimientos, especificaciones tablas de calibración, intervalos de referencia biológicos y sus orígenes, gráficos, posters, noticias, memoranda, software, planes, documentos de origen externo tal como regulaciones, estándares y procedimientos de examen. Estarán conservados en un medio apropiado, el cual puede o no incluir papel. El Responsable de Calidad se encargará de la confección de un determinado número de copias y determinará a quién serán entregadas. La copia controlada mencionada se guarda en un archivo previa aprobación por el Jefe del Laboratorio. Los documentos se revisarán anualmente y aquellos que deban salir de circulación se archivarán en un archivo destinado a tal fin llamado “Archivo de Documentos Obsoletos”. 3.2.4 Consulta con laboratorios de referencia El Laboratorio deberá poseer un procedimiento efectivo documentado para evaluar y seleccionar y registrar Laboratorios de Consulta competentes a fin de proveer segundas opiniones para disciplinas tales como Inmunoserología, Análisis Hormonales, Química Clínica, Microbiología o Hemocitología. Los arreglos y/o contratos con los Laboratorios Consultores se revisarán anualmente para asegurar que: a) Los requerimientos y procedimientos son adecuadamente definidos, documentados y comprendidos. b) El Laboratorio de Consulta tiene la capacidad de cumplir con los requerimientos y c) La selección de procedimientos es adecuada para el uso pretendido. 3.2.5 Servicios externos y suministros La Administración del Laboratorio debe contar con un sistema de control para la aceptación o rechazo de insumos que responda a sus políticas y procedimientos. Es recomendable que todo insumo a ser usado por primera vez en el Laboratorio pase previamente por las fases de prue- Gestión del Laboratorio de Análisis Clínicos Implementación de la documentación según normas internacionales ba, control y aceptación o rechazo, siendo muy útiles en estas etapas los sistemas de control de calidad del laboratorio. El equipamiento adquirido y los insumos consumibles que puedan afectar la calidad del servicio no serán puestos en uso hasta que demuestren haber cumplido con las especificaciones estándar de operabilidad de los procedimientos correspondientes. En estos casos es conveniente solicitar al fabricante/ proveedor los resultados documentados de todos los controles o verificaciones efectuadas a sus productos. Debe existir un inventario actualizado y móvil de todos los insumos que están siendo utilizados en un momento dado. El mismo incluirá el registro de los números de lote de los reactivos más importantes, materiales de control y calibradores; la fecha de recepción por el laboratorio, la fecha de vencimiento y la fecha en la que el material es puesto en uso. 3.2.6 Identificación y control de no conformidades El laboratorio debe poseer un sistema de detección y procedimientos a implementar cuando descubra que algún aspecto de sus exámenes no concuerda con sus propios procedimientos o con los requerimientos del manejo del Sistema de Calidad. Nota: los exámenes o actividades no conformes ocurren en muy diferentes áreas y pueden ser identificadas de muchas maneras. Estas incluyen disconformidades de los médicos y/ o pacientes, indicaciones originadas por el análisis de los controles de calidad, calibraciones de instrumentos, chequeo de materiales consumibles, comentarios del personal, revisiones del manejo del laboratorio y auditorías internas y externas. Si se determina la existencia de una “no conformidad”, deben implementarse rápidamente procedimientos para identificar, documentar y eliminar la causa(s) de esas no conformidades. 3.2.7 Acciones correctivas Los procedimientos para las acciones correctivas incluirán un proceso de investigación para determinar la(s) causa(s) subyacentes del problema, teniendo en cuenta que la acción correctiva deberá ser apropiada a la magnitud del problema y relacionada con los riesgos posibles. 3.2.8 Servicios de asesoramiento y resolución de quejas El Laboratorio debe poner a disposición de los profesionales de la institución el listado de prácticas que se realizan y sus frecuencias así como tipo de muestra y cantidad requerida para cada ensayo. De ser necesario el Jefe de cada Sector asesorará al demandante sobre la interpretación de los resultados emitidos. El servicio debe poseer procedimientos para la resolución de quejas recibidas pos los médicos, pacientes u otros. Se llevará en libro foliado un registro de quejas y sus acciones correctivas bajo el nombre de “Libro de Quejas y Sugerencias”. 3.2.9 Proceso de Mejora Continua Todos los procedimientos operacionales deberán ser sistemáticamente revisados a intervalos regulares, tal como está definido en el Sistema de Calidad, a fin de identificar alguna fuente de no conformidad u otras oportunidades de mejorar el Sistema de Manejo de la Calidad o las prácticas técnicas. Se desarrollarán y documentarán planes para el mejoramiento y los mismos serán implementados. 3.2.10 Registros técnicos y de calidad El Laboratorio implementará procedimientos para la identificación, recolección, acceso, almacenamiento, mantenimiento y seguridad de los registros técnicos y de calidad, los cuales serán legibles y expuestos de manera fácilmente comprensible. Estarán en un lugar apropiado para prevenir daño, pérdida, deterioro o acceso no autorizado, existiendo un tiempo de retención para cada registro de acuerdo a su validez e importancia. Se proponen los siguientes registros: 1. Formularios de pedido 2. Insertos y/ o manuales del instrumental 3. Insertos de las técnicas analíticas 4. Manuales de factores de conversión 5. Catálogos de productos 6. Listas de precios de insumos y equipamiento 7. Registros de Control de Calidad Externo e Interno 8. Libro de Quejas 9. Registro de Estadística 10. Manual de Bioseguridad 11. Libro de presentismo del personal 12. Libro de Registro de Accidentes de Trabajo 3.2.11 Auditorías internas El Responsable de Calidad organizará auditorías internas a intervalos establecidos en las distintas áreas de trabajo fin de verificar que todas las operaciones cumplan con los requerimientos del Sistema de Calidad del Laboratorio. Estas auditorías se efectuarán especialmente en las áreas críticas para el cuidado de los pacientes. Las auditorías son llevadas a cabo por personal competente designado por el Responsable de Calidad con acuerdo del Jefe del Laboratorio. El personal no audita sus propias actividades. 3.2.12 Revisión por la gerencia La Gerencia del Laboratorio revisa anualmente el Sistema del Gerenciamiento de la Calidad del Laboratorio y de todos sus servicios incluyendo las actividades de examen y asesoramiento a fin de asegurar que continúan siendo apropiados y efectivos como soporte para el cuidado de los pacientes y también para introducir cualquier cambio necesario o mejora en los servicios dentro de un tiempo acordado. Los resultados de esta revisión son incorporados a una planifi- REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 13 | 36 Pág | 27 cación que incluye metas, objetivos y planes de acción para el año venidero. también un estricto régimen de confidencialidad respecto de todos los datos o resultados obtenidos. 3.3 Requerimientos Técnicos 3.3.1 Personal El Laboratorio debe poseer un plan de organización (organigrama) con descripciones de las políticas para el personal y descripciones de trabajo que definan las calificaciones y tareas de todo el Recurso Humano, manteniendo registros con datos relevantes de las calificaciones educacionales y profesionales, entrenamiento, experiencia y competencia de todo el personal. 3.3.2 Comodidades y condiciones ambientales El Laboratorio deberá poseer un espacio asignado de manera tal de poder desarrollar sus tareas sin comprometer la calidad del trabajo, de los procedimientos de Control de Calidad, la seguridad del personal y el servicio del cuidado de los pacientes. Las instalaciones deberán ser adecuadas para la atención de los pacientes y para soportar las actividades del Laboratorio y estarán mantenidas en condiciones confiables y funcionales. Las mismas provisiones deben asegurarse para los lugares de recolección primaria de muestras, en especial con respecto al confort y privacidad de los pacientes a fin de que no se invaliden o afecten adversamente la calidad de las mediciones. El Laboratorio estará diseñado para ser eficiente en su operación, optimizar el confort de sus ocupantes y minimizar el riesgo de accidentes o enfermedades ocupacionales. Los pacientes, empleados y visitantes estarán protegidos de los riesgos conocidos y deberá estar controlado el acceso y uso de áreas donde se pueda afectar la calidad de las determinaciones. Deberá existir adecuado espacio y condiciones para el almacenamiento de muestras, reactivos, manuales de operación e insumos, archivos, documentos, registros y resultados, mientras que las áreas de trabajo poseerán las condiciones de higiene y seguridad ambiental apropiadas y el almacenamiento y descarte de materiales peligrosos se realizará según normas vigentes. La documentación propuesta incluye: • Título habilitante con matrícula. • Referencias de empleos previos. • Descripciones de su trabajo. • Evaluación de competencia. • Estado de inmunización. • El Laboratorio es dirigido por una persona(s) con responsabilidad ejecutiva que tiene(n) la competencia para asumir responsabilidades, dependiendo de los servicios provistos: médicos, científicos y técnicos. • Nota: “Competencia” se entiende como el resultado de la educación académica de grado, posgrado y continuada así como el entrenamiento y la experiencia de varios años en el laboratorio clínico. • Las responsabilidades del Jefe del Laboratorio o sus designados incluyen asuntos científicos, consultivos, organizativos, administrativos y educacionales y estarán en concordancia con los servicios ofrecidos. El Jefe del Laboratorio autorizará al personal a desempeñar tareas particulares tales como muestreo, exámenes u operación de algún tipo de equipamiento particular, incluyendo el uso de computadoras utilizadas en el sistema informático del Laboratorio. La Dirección del Laboratorio debe asegurar la competencia del personal que: • Opera equipos específicos • Realiza ensayos o calibraciones • Evalúa los resultados obtenidos • Valida con su firma los protocolos emitidos Deberán existir políticas definidas en cuanto al uso del sistema de computación: quiénes tendrán acceso solamente a los datos de los pacientes y quiénes estarán autorizados a ingresar y/o corregir resultados de pacientes, corregir protocolos o modificar programas de computación, los cuales estarán adecuadamente protegidos para prevenir el acceso, alteración o destrucción por usuarios casuales o no autorizados. Se propone la existencia obligatoria de un Programa de Educación Continua para el personal en todos los niveles que incluya entrenamiento específico en el aseguramiento de la calidad de los servicios ofrecidos a los usuarios, así como 3.3.3 Equipamiento Nota: para esta sub-cláusula se incluye como equipamiento del laboratorio a los instrumentos, materiales de referencia, consumibles, reactivos y sistemas analíticos. El Laboratorio estará provisto con todo el equipamiento necesario para la provisión de servicios (incluida toma de muestras primarias y preparación, almacenamiento, transporte y procesamiento de muestras). Las autoridades del laboratorio deben establecer un programa para monitorear regularmente los aparatos y exhibir una calibración y funcionamiento adecuado de los instrumentos, reactivos y sistemas analíticos, así como un programa documentado de mantenimiento preventivo que cumpla con las recomendaciones de los fabricantes. Cada ítem del equipamiento estará unívocamente rotulado para su identificación. Los registros podrán incluir: a) Identidad del equipamiento. b) Nombre del fabricante, identificación y número de serie. c) Persona a contactar con el fabricante y número de teléfono. d) Fecha de recepción y de puesta en servicio. Gestión del Laboratorio de Análisis Clínicos Implementación de la documentación según normas internacionales e) Ubicación dentro del Laboratorio (de acuerdo con las recomendaciones del fabricante). f) Condición a la recepción (nuevo, usado, reacondicionado). g) Planes de mantenimiento. Estos registros serán mantenidos durante todo el período útil del equipamiento. El equipamiento debe ser operado solamente por personal autorizado y tanto el manual como las instrucciones de uso estarán a disposición del personal del laboratorio. Es aconsejable que las computadoras o equipos automáticos que se usan para la recolección, procesamiento, registro, reporte o almacenamiento de datos de análisis cumplan con lo siguiente: a) El software, incluido el de los aparatos deberá estar documentado y validado como apropiado para el uso requerido. b) Los procedimientos estarán establecidos e implementados para proteger en todo momento la integridad de los datos y c) Las computadoras y equipos automáticos estarán mantenidos para asegurar su correcto funcionamiento y provistos de las condiciones ambientales y de operación necesarias para mantener la integridad de los datos. El equipamiento, incluyendo hardware, software, materiales de referencia, consumibles, reactivos y sistemas analíticos deberán estar resguardados de ajustes o manoseos que puedan invalidar los resultados de los exámenes. 3.3.4 Proceso Pre-analítico El Laboratorio requerirá de los demandantes una Solicitud de Análisis que debe contener los siguientes datos: a) Identificación unívoca del paciente 1) Apellido y nombre. 2) HC o Registro Ambulatorio o Cama. b) Examen requerido. c) Información clínica relevante o diagnóstico presuntivo en algunos casos. d) Firma y sello del demandante. e) Fecha de la Solicitud de Pedido. Las instrucciones específicas para la correcta recolección y manipuleo de muestras primarias (MP) estarán implementadas y documentadas por la Jefatura del Laboratorio y deberán estar disponibles para aquellas personas responsables de dicha recolección. Estas instrucciones estarán contenidas en el “Manual para recolección de Muestras Primarias”. El laboratorio debe controlar el acarreo de muestras hacia su planta física: a) Dentro de un tiempo apropiado para su procesamiento. b) Dentro de un rango de temperatura especificado en el “Manual de Recolección de Muestra Primarias” y con los preservantes adecuados para asegurar la integridad de las muestras. c) De modo que se brinde seguridad para el acarreador, el público en general y el receptor en el Laboratorio. El laboratorio poseerá un sistema para la recepción, rotulado, procesamiento y reporte de muestras recibidas y calificadas como “urgentes” que incluya un rotulado especial con indicación del servicio solicitante, procedimientos para el inmediato procesamiento e informe y comunicación con el demandante vía intercomunicador, así como una política respecto a los requerimientos verbales sobre los resultados de los pacientes. Las muestras se almacenarán por un tiempo dado y en condiciones que aseguren la estabilidad de las propiedades de las mismas para permitir repeticiones luego de ser informado el resultado. 3.3.5 Proceso analítico Todos los procesos analíticos desarrollados en el Laboratorio, incluidos los de selección/toma de muestra deberán responder a las necesidades de los usuarios y serán apropiados para los exámenes. Todas las operaciones estarán validadas por procedimientos documentados que confirmen que los procesos son adecuados para el uso que se les requiere. Los métodos y procedimientos que se han seleccionado deberán haber sido evaluados y haberse encontrado satisfactorios antes de utilizarse para los exámenes del laboratorio. Las instrucciones de uso (insertos) provistas por los fabricantes serán usadas como parte de los procedimientos en caso de que contribuyan a hacer más claros los mismos, mientras que todo cambio estará debidamente documentado e información adicional se hallará agregada en caso necesario y cada nueva versión de equipos de diagnóstico que implique cambios en los reactivos o el procedimiento se controlará para su desempeño. Se propone que además de la identificación del documento, consten los siguientes ítems: a) Propósito del examen. b) Principio del procedimiento. c) Especificaciones de desempeño (linealidad, precisión, exactitud, límite de detección, intervalo de medida, sensibilidad y especificidad analítica). d) Tipo de muestra primaria incluyendo envase y aditivos. e) Equipo y reactivos requeridos. f) Procedimientos de calibración. g) Proceso de validación. h) Procedimientos de Control de Calidad. i) Interferencias y reacciones cruzadas. j) Procedimientos para efectuar los cálculos. REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 13 | 36 k) Intervalo de referencia biológico. l) Valores de alerta o críticos. m) Interpretación analítica y clínica de los datos. n) Precauciones de seguridad. o) Fuentes potenciales de variabilidad. El Jefe del Laboratorio será responsable de asegurar que la descripción de los procedimientos de los exámenes es completa, sencilla y ha sido exhaustivamente revisada. En caso de modificación de un Procedimiento Operativo que implique cambios en la interpretación de los resultados, los usuarios serán avisados previamente a la introducción de esos cambios mediante notas o aclaraciones en el protocolo de resultados. 3.3.6 Aseguramiento de la calidad de los Procesos Operativos El Laboratorio debe obligatoriamente poseer un procedimiento de Control de Calidad Interno apropiado a la calidad de los resultados que emite. Este Sistema de Control de Calidad provee al personal de información clara y fácilmente comprensible sobre la cual tomar decisiones, prestando especial atención a la supresión de errores en los procesos de manipuleo de muestras, pedidos, exámenes, informes, etc. Debe determinarse la incertidumbre de sus medidas y sus componentes tales como la preparación de muestras, calibradores, materiales de referencia, equipamiento usado, condiciones ambientales, condiciones de muestra, cambio de operador, etc. Siempre que sea posible es conveniente la participación en programas de Evaluación Externa de Calidad, los cuales proveen muestras similares a las de los pacientes en variedad y complejidad y los resultados son un reflejo completo del proceso de examen. El Responsable de Calidad del laboratorio debe inspeccionar los resultados del Programa Externo de Calidad y participar en la implementación de acciones correctivas cuando sea necesario. 3.3.7 Proceso post-analítico e informe de los resultados La Jefatura del Laboratorio es responsable del formato de los informes, previa discusión con los demandantes, compartiendo la responsabilidad de que los informes lleguen a ellos en tiempo y forma. Se propone que los informes del Laboratorio incluyan: a) Identificación clara del examen. b) Identificación del laboratorio que emite el resultado. c) Identificación unívoca del paciente. d) Fecha de la recolección de muestra primaria. e) Resultados del examen incluyendo unidades de medida si correspondieren. f) Intervalo de referencia biológico. g) Comentarios sobre la calidad o propiedad de la muestra primaria que puedan comprometer el resultado. Pág | 29 h) Interpretación de los resultados, si corresponde o es oportuna. h) Identificación de la persona que autoriza la emisión del reporte. El laboratorio deberá poseer procedimientos para el inmediato aviso al médico (u otro personal responsable del cuidado del paciente) cuando los resultados de los exámenes sean “de alerta” o “críticos”; dejando asentada la hora del aviso y a quién se dirigió el mismo. La Jefatura del Laboratorio establecerá la tardanza para cada uno de los exámenes y la misma estará de acuerdo a las necesidades clínicas; de igual modo deberán existir mecanismos de aviso cuando una demora en la entrega de un dato pueda comprometer el cuidado del paciente. El laboratorio poseerá procedimientos de distribución de informes que incluyen a quién se le entregan los resultados y de qué modo. Los resultados emitidos verbalmente serán seguidos de un reporte escrito adecuado y no se comunicarán resultados por teléfono, salvo excepciones debidamente justificadas y autorizadas por algún jefe. 3.3.8 Alteraciones y enmiendas de informes Todas las alteraciones de los registros deben indicar fecha y persona que efectúa los cambios. Si ocurriese un error en un informe, el mismo deberá ser rehecho, no debiendo ser borrado, enmendado ni tachado. 3.4 Anexos 3.4.1 Anexo I. Organigrama del laboratorio 3.4.2 Anexo II. Formulario de control de documentos 3.4.3 Manuales de Procedimientos Operativos 3.4.4 Manual de Bioseguridad 3.4.5 Manual de recopilación de formularios, planillas y protocolos 7.8-EL MANUAL DE PROCEDIMIENTOS. MODELO DE ELABORACIÓN DE UN PROCEDIMIENTO. El Manual de Procedimientos del laboratorio describe el conjunto organizado de los procedimientos operativos del servicio, que están referidos a la parte más importante de sus actividades: las relacionadas directamente con los pacientes. Todas las actividades que se realizan en el laboratorio constituyen una cadena de procesos donde el resultado de una actividad es el punto de partida para la siguiente. Es por esta razón que el Manual de Procedimientos debe reflejar acabadamente el desempeño global de los procesos del servicio. Para decidir qué procedimientos y qué documentación pueden ser necesarias para cubrir adecuadamente todas las actividades de los procesos operativos, es necesario considerar los atributos que están asociados a cada paso. Estos atributos pueden plantearse como una serie de pre- Gestión del Laboratorio de Análisis Clínicos Implementación de la documentación según normas internacionales guntas: “¿Qué implica este paso y porqué se realiza?”, “¿Dónde y cuándo se realiza?” y finalmente “¿Quién y cómo lo realiza?”. El Manual de Procedimientos debe tener un índice y considerarse como un anexo del Manual de la Calidad o constituir un documento separado. También debe tener una copia maestra y un número conocido de copias. En algunos laboratorios, el Manual de Procedimientos puede ser algo voluminoso, por lo cual, por razones prácticas, recomiendo su división por secciones o áreas del laboratorio, tales como Química Clínica, Hemocitología, Hormonas, Inmunoserología, Microbiología, etc, de acuerdo a la cantidad y extensión de los procedimientos de cada sector. Además de los procedimientos para la fase analítica del laboratorio, el Manual de Procedimientos podrá contener también procedimientos pre y post-analíticos. El método que considero más práctico para la elaboración del Manual de Procedimientos es incluir progresivamente los documentos hasta alcanzar la totalidad de los procedimientos de todas las actividades desarrolladas. Un procedimiento es un registro escrito de cómo un proceso particular debe darse en una organización. “Una forma especificada para llevar a cabo una actividad o un proceso”. (ISO 9000:2000). Los procedimientos pueden abarcar una o varias áreas o funciones y resaltan la coordinación, las responsabilidades y la comunicación entre sectores. ¿Cuándo es necesario un procedimiento escrito? 1. Cuando sea requisito obligatorio de una norma de aplicación. 2. Cuando, por su importancia o complejidad sea conveniente que las tareas estén documentadas apropiadamente a fin de no poner en riesgo el Sistema de Calidad. Un procedimiento operativo estándar (POE) es un documento que contiene: • El objeto y alcance de una actividad • Qué debe hacerse • Quién debe hacerlo • Cómo, cuándo y cómo • Qué materiales, equipos y documentos de registro y control deben utilizarse Los procedimientos pueden describir gran variedad de procesos y son: • De conducción • De comunicación y coordinación • Técnicos • De emergencia y alarma Los procedimientos documentados deben contar con la información necesaria y una identificación única y constan básicamente de los siguientes elementos: • Encabezado Logotipo y nombre del laboratorio y sección que aplica el POE. Título descriptivo Firma de quien lo elabora (día, mes y año) Firma de quien lo revisa (día, mes y año) Firma de quien lo autoriza (día, mes y año) Vigencia Fecha de revisión Código Número de página y número total de páginas del documento. • Objetivo Expresar en forma clara y concisa el propósito del procedimiento que se intenta elaborar. • Alcance Mención de la unidad operativa en la que se aplicará el procedimiento y su campo de aplicación. • Personal afectado Describe quién operará el procedimiento en cuanto a su calificación, preparación o destrezas. • Definiciones y abreviaturas Se indica el significado de los principales términos de importancia utilizados en el procedimiento, así como también las abreviaturas empleadas. • Referencias Se enumeran los documentos que dan soporte al procedimiento. • Responsabilidades Se enuncia la responsabilidad y autoridad de los destinatarios del procedimiento y sus interrelaciones con otros procesos. • Desarrollo Se efectúa una descripción secuencial de las instrucciones operativas. Debe ser claro y sin ambigüedades. Es recomendable usar diagramas de flujo o gráficos para describir los mecanismos de medición y seguimiento de los procesos. El nivel de detalle varía dependiendo de la complejidad de las actividades y del nivel de capacitación y destreza del personal involucrado. En procedimientos operativos de métodos de ensayo o medición este punto describe además: principios básicos, materiales (equipos y reactivos), técnica, especificaciones (criterios de aceptación y rechazo), cálculos, estadística y las condiciones de validez del método de ensayo. • Condiciones de seguridad Indica las condiciones y medidas de seguridad a tener en cuenta para la ejecución segura del POE. • Registros Se mencionan planillas y/ o protocolos en los cuales deben registrarse los datos o mediciones que involucran al procedimiento. Cada procedimiento debe tener adjuntos todos los documentos complementarios, tales como planillas, protocolos, etc, que contribuyan al esclarecimiento del mismo. Las planillas o protocolos deben asociarse a cada proced- Pág | 31 REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 13 | 36 imiento para asegurar que se ha registrado toda la información necesaria para: controles/ auditorías, no conformidades o controversias y para el desempeño del recurso humano. Deben incluir la fecha de los ensayos, las iniciales de las personas que los realizan, los resultados, la declaración inequívoca de aceptación o rechazo y la fecha y firma del responsable designado. • Anexos Contienen información suplementaria o de soporte y pueden estar constituidos por tablas, gráficos, formularios o flujogramas. Los procedimientos analíticos, pueden en ocasiones ser documentos algo extensos, por lo tanto, es útil elaborar un resumen de una o dos páginas con los pasos sobresalientes, o confeccionando un diagrama de flujo (flujograma) donde se describan esquemáticamente las etapas del procedimiento. Cuando un documento está en uso, y ocasionalmente se descubren pequeños errores de escritura o es necesario hacer correcciones menores, es útil adjuntar en la parte posterior del procedimiento un formulario de procedimiento de enmienda, donde se mantiene un registro de tales cambios, evitando un número excesivo de enmiendas. Es importante señalar que el documento perteneciente a un procedimiento estará mejor preparado o diseñado por la persona que normalmente realiza el procedimiento o la que está familiarizada con la actividad en particular. Por eso es recomendable que el primer bosquejo de un procedimiento operativo sea hecho por quien realiza la tarea, para luego consultarlo y corregirlo, consensuando su aceptabilidad con el Responsable de Calidad. En el Anexo II se incluyen, a modo de ejemplos, los procedimientos: a) Procedimiento de preparación de la paciente para la Prueba de Tolerancia a la Glucosa de 75 gramos. b) Procedimiento para la determinación de Glucosa en suero. c) Procedimiento para compra de insumos de laboratorio para la Sección Bioquímica Clínica. 7.9 INSTRUCCIONES DE TRABAJO Y REGISTROS TÉCNICOS Las actividades diarias de los laboratorios producen gran cantidad de información necesaria tanto para la atención de los pacientes como para las tareas de gestión. Las instrucciones de trabajo y los registros técnicos que de ellas proceden, constituyen el tercer nivel de la documentación. Estos documentos deben estar expuestos de manera fácilmente comprensible y principalmente abarcan: 1. Solicitudes de análisis 2. Insertos y/o manuales del instrumental 3. Protocolos de procedimientos diarios 4. Manuales de factores de conversión 5. Registro de corridas analíticas y controles 6. Registros de control y aseguramiento de la calidad 7. Registros de Control de Calidad Externo e Interno 8. Hojas de trabajo 9. Registros de mantenimiento de equipos 10. Registros de inspección 11. En algunos casos fotografías, portaobjetos teñidos y todo otro registro que se considere de utilidad. Los instructivos de trabajo son documentos que especifican el modo en que se debe realizar una tarea día a día en un área o por personas establecidas, destacando los pormenores operativos para que no se ocasionen errores. Las instrucciones de trabajo indican como debe realizarse una tarea dentro de un procedimiento. Responden a la pregunta: ¿Cómo hacerlo? Los registros son evidencia documentada de que el sistema se ha implementado tal como se lo describe (funcionamiento real del sistema). Responden a la pregunta: ¿Cómo probar lo que se hace? Un registro constituye el asiento de un hecho vital o de salud, es individual, continuo y permanente y de obligatorio por su importancia legal (12). En general un registro debe incluir los siguientes datos: • Procedimiento utilizado • Identificación del operador • Todo otro factor que deba ser controlado para asegurar la validez de los resultados. El laboratorio debe establecer y mantener procedimientos para la identificación, acceso, archivo, conservación y eliminación de los registros. En caso de registros electrónicos, debe prevenirse el acceso o la modificación no autorizados. Deberán estar en lugar apropiado para prevenir daño, pérdida, deterioro o acceso no autorizado, existiendo un tiempo de retención para cada registro de acuerdo a su validez e importancia. Con respecto al control de los registros, no recomiendo una codificación dado que estos registros provienen de planillas internas creadas en el mismo laboratorio. El Anexo III contiene el ejemplo de un Procedimiento de Registro de las Inspecciones sobre el instrumental. 8.CONCLUSIONES He presentado en este trabajo los siguientes conceptos fundamentales: • La importancia de la implementación del Sistema de Gestión de la Calidad en los laboratorios clínicos • La posibilidad para los laboratorios de aplicar, implementar y adecuar a Normas Nacionales y/o Internacionales Gestión del Laboratorio de Análisis Clínicos Implementación de la documentación según normas internacionales • La especialización y la diferenciación por sus características especiales, de la Gestión de Calidad en el laboratorio clínico • El rol fundamental e imprescindible de la documentación tanto en forma como en contenido como soporte necesario de Sistema de Calidad • La aplicación de las normas pertinentes para la elaboración de un modelo de Sistema Documental adaptable a cualquier servicio de laboratorio clínico. v La documentación garantiza el soporte del Sistema de Gestión de la Calidad, dado que en ella se plasman no solamente las formas de operar de la organización sino toda la información que permite el desarrollo de todos los procesos y la toma de decisiones. La aplicación extensiva del Sistema Documental de los laboratorios de análisis clínicos traerá aparejados los siguientes beneficios para los servicios que la implementen: • Mejora de la calidad de los servicios brindados • Acceso garantizado a la acreditación • Mejora de las condiciones contractuales frente a otros prestadores • Mejora del rendimiento de la inversión reduciendo costos • Mejora de la situación legal frente a juicios al ajustarse a normativas • Mejora de la orientación y servicio al paciente y médico. • Mejora de las condiciones laborales y de bioseguridad. • Incorporación de conceptos ético-profesionales a la actividad asistencial. Un Sistema de Calidad que funcione correctamente es crítico para el desarrollo y la prestación de servicios de los Laboratorios de Análisis Clínicos, mientras que un Sistema de Documentación aplicable y consolidado representa la herramienta para el futuro crecimiento de la estructura. Ambos sistemas son complejos y están en mutua dependencia, constituyendo su relación armoniosa la clave del éxito en la Gestión. 1. ¿Tiene el laboratorio definida y declarada su Política de Calidad? a) SI b) NO 2. ¿Posee el laboratorio un Sistema de Gestión de la Calidad documentado? a) SI b) NO 3. ¿Se cumple? a) SI b) NO 4. ¿Posee el laboratorio un Manual de la Calidad? a) SI b) NO 5. Posee el laboratorio un Manual de Procedimientos o alternativamente un conjunto de procedimientos individuales de análisis? a) SI b) NO 6. ¿Incluye todos los procedimientos? a) SI b) NO En la Tabla 1 se exponen los resultados de la Encuesta Tabla 1 N: 40 Pregunta N° SI NO 1 10 30 2 9 31 3 8 32 4 15 25 5 35 5 6 30 10 ANEXO I Encuesta para los Laboratorios de Análisis Clínicos Existencia y estado de la documentación La finalidad de la encuesta fue conocer la existencia y el grado de completitud de la documentación de un grupo de laboratorios de Análisis Clínicos (que fueron utilizados como muestra) a fin de tener un panorama de cuál sería el aporte de una propuesta de modelo para la confección y el control de dicha documentación. La muestra para la encuesta consistió en 40 laboratorios de Análisis Clínicos, de diversas dimensiones y complejidad, ubicados en Capital Federal y Conurbano Bonaerense. ANEXO II Ejemplos de Procedimientos PROCEDIMIENTO TÉCNICO- ADMINISTRATIVO Procedimiento de atención de la paciente para P 75 o P 50 Objetivo: Conocer el procedimiento para efectuar la Prueba de Tolerancia Oral a la Glucosa con ingestión de 75 g o de 50 g de Glucosa (de acuerdo al pedido médico). Pág | 33 REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 13 | 36 Alcance: Todas las pacientes y muestras a las que se les solicita ese estudio. Personal afectado: Recepcionistas: Reciben el pedido y dan las instrucciones. Extraccionistas: Toman la muestra. Profesionales y técnicos: Efectúan las determinaciones. Definiciones y abreviaturas: P 75: Prueba de tolerancia oral a la glucosa con 75 g de dextrosa. P 50: Prueba de tolerancia oral a la glucosa con 50 g de dextrosa. Dextrosa: glucosa La ejecución de estas pruebas puede realizarse en cualquier momento del embarazo, pero lo ideal es realizarlas entre las 24 y 26 semanas de gestación y luego de las 32 semanas (por la mayor influencia de las hormonas placentarias). Valores mayores a los establecidos como límite determinan continuar con el siguiente paso diagnóstico, que es la Prueba de sobrecarga oral con 100 g de glucosa. Este procedimiento abarca: • Horario de Recepción y Toma de Muestras • Atención personal o telefónica de las consultas sobre condiciones de concurrencia. • Verificación del cumplimiento de ambas pautas. Referencias: Indicaciones de Estudios Carpeta de Instrucciones para Prácticas Generales y Especiales. Bibliografía al respecto en Biblioteca del Laboratorio. Responsabilidades: Elabora: Jefe de Bioquímica Clínica. Revisa: Jefe de Hemocitología. Aprueba: Jefe del Laboratorio. Desarrollo: a. Cuando la paciente concurre a solicitar turno, la recepcionista le explica en qué consiste la prueba, la duración de la misma y le entrega un instructivo donde consta en qué condiciones y qué elementos deben traer el día de la prueba. (Ver Anexo I del procedimiento). b. El día del estudio la paciente concurre a la Mesa de Entrada del laboratorio a las 7.00 hs., se la recepciona inmediatamente (sin hacer fila) y se procede al ingreso del pedido en el Libro de Ingreso de Pacientes de la Mesa de Entrada. c. En la Recepción se le entrega un número de orden (los números de estas pruebas son los primeros otorgados cada día) y se indica a la paciente la fecha de retiro del resultado consignando por escrito la misma en el talón adjunto. d. Se envía a la paciente al sector “Toma de muestra”, donde la extraccionista para un mejor ordenamiento de los pacientes y horarios de tomas de muestra, consigna los datos de la paciente en una planilla rotulada “Planilla de Curvas” (ver Anexo II de este procedimiento) y extrae la muestra de ayuno. La metódica para la extracción de sangre figura en un procedimiento aparte. e. Se le administra a la paciente una solución dextrosada de 75 o 50 gramos de glucosa uso oral (según el caso) disuelta en 400 ml de agua a temperatura ambiente y aromatizada con unas gotas de jugo de limón, indicándole que esta solución debe ser ingerida dentro de los siguientes 10 minutos. f. Se indica a la paciente que debe permanecer sentada en reposo en el lugar destinado para ese fin hasta la finalización de la prueba excepto durante las extracciones de sangre. g. Se efectúa luego una extracción de sangre: en el caso de la P 75 a las 2 horas de la toma de la muestra de ayuno y en el caso de la P 50 a la hora, luego de lo cual se da por finalizada la prueba y se le comunica a la paciente que puede retirarse. h. En caso de que existiera alguna objeción al pedido dentro del ámbito administrativo, se considera cada caso en particular y se consigna la conducta seguida en la correspondiente solicitud. i. En caso de que existiera alguna objeción al pedido dentro del ámbito “Toma de Muestra”, se procede de igual forma. j. Si durante el desarrollo de la prueba la paciente sufriera algún tipo de malestar o descompensación, se debe llamar sin demora a la Guardia de Obstetricia (Interno 21) a fin de que un médico concurra al laboratorio para asistir la emergencia. En estos casos se da por finalizada la prueba. Anexo I Instrucciones impartidas a la paciente previas a la realización de la prueba • No se requiere dieta previa pero sí una alimentación regular. • Concurrir con ayuno de aproximadamente 8 horas. • Traer una taza para tomar la solución. • Traer 2 limones para acompañar la ingestión de la glucosa. • Calcular permanencia en el laboratorio de aproximadamente 3 horas. Gestión del Laboratorio de Análisis Clínicos Implementación de la documentación según normas internacionales Desarrollo: La determinación de glucosa se lleva a cabo principalmente para contribuir al diagnóstico y tratamiento de diabetes mellitus, hipoglucemia y enfermedades de los sistemas renal, tiroideo e hipofisiario. La glucosa oxidasa cataliza la oxidación de glucosa a ácido glucónico y agua oxigenada, la cual es cuantificada luego por reacción con 4-aminofenazona y peroxidasa obteniéndose una quinonimina roja cuantificable a 505 nm. Diagrama de flujo del proceso Paciente concurre a Mesa de Entrada Recepcionista Verificación de Solicitud Médica Se le otorga el número Talón para retirar resultado Extraccionista Aclaraciones en solicitud Toma demuestra de ayuno Ingestiónde la sol. de glucosa Paciente en reposo Toma demuestra a la hora o 2 hs. Paciente se retira PROCEDIMIENTO DE LABORATORIO Procedimiento para la determinación de Glucosa Objetivo: conocer el procedimiento para la determinación cuantitativa de Glucosa en suero. Alcance: todas las muestras de suero a las que se les realiza Glucosa. Personal afectado: personal profesional y técnico del sector Química Clínica. Definiciones y abreviaturas: Glu: Glucosa Li: Litio Na: Sodio K: Potasio Referencias: insertos de los Equipos Sera-Pak Plus y Wiener y bibliografía que figura en los mismos. Responsabilidades: Elabora: personal del Sector Química Clínica Revisa: Jefe de Sección Bioquímica Clínica. Aprueba: Jefe del Laboratorio. Recolección y almacenamiento de muestras: La muestra recomendada es suero no hemolizado. Podrán utilizarse plasmas que contengan oxalato de K, fluoruro de Na, heparina de Li o EDTA. No se requiere ningún aditivo o conservante especial si el contacto con las células rojas no excede los 90 minutos. Procesamos las muestras dentro de la media hora de su extracción. Debe prestarse atención a las muestras hemolizadas, ictéricas y lipémicas. Los niveles de glucosa disminuyen en sangre entera a razón de 7% por hora a temperatura ambiente. En suero hemolizado no separado, la glucosa es estable hasta 48 hs. cuando es refrigerada. La adición de fluoruro de Na a la sangre entera estabiliza la glucosa hasta 48 horas cuando se mantiene refrigerada. Se adjuntan insertos explicativos de los equipos de reactivos en uso: Glicemia enzimática AA línea líquida Wiener (presentación 1 x 1000 ml) y Sera-Pak Plus (presentación 6 x 100 ml), los cuales utilizamos alternativamente de acuerdo a costos. Se emplea un procedimiento automatizado: analizadores automáticos Technicon RA-XT y Opera cuyas programaciones se adjuntan. Calibración: • Set Point Bayer Cat. 08174(R2) Control de Calidad Interno: • Seracheck N Cat. 6656 Bayer • Seracheck A Cat. 6657 Bayer • Ocasionalmente Qualitrol HSN Cat. 1.10937 Merck. Control de Calidad Externo: • Programas “Buenos Aires” (C.E.M.I.C.) y Prevecal. Criterio de dilución de la muestra: • Datos crudos por encima de 400 mg/dl. Se efectúa dilución al ½ con solución fisiológica. Criterio de repetición de datos: • Se repite todo dato igual o superior a 200 mg/dl. Valores de Referencia: • 70-110 mg/dl. Precaución: • Evitar el contacto del reactivo con la piel, ojos y ropa. La toxicidad no ha sido establecida. Pág | 35 REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 13 | 36 PROCEDIMIENTO ADMINISTRATIVO Procedimiento de compra de insumos de laboratorio para la Sección Bioquímica Clínica Objetivo: Conocer el procedimiento para la adquisición de insumos de laboratorio para la Sección Bioquímica Clínica. Alcance: Todos los insumos de laboratorio para Bioquímica Clínica que se proveen directamente por compra. Personal afectado: Jefe del Laboratorio y Jefe de Sección Bioquímica Clínica. Definiciones y abreviaturas: “Directamente por compra”: significa que el laboratorio, además de generar la solicitud de pedido, asesora las ofertas en cuanto a su aceptabilidad. En este caso la mercadería ingresa al laboratorio directamente desde el proveedor sin ningún servicio intermediario (ejemplo: Farmacia). El procedimiento de compra de insumos es general para todas las secciones del laboratorio, pero a pesar de ello, en este procedimiento existen algunas particularidades que hacen oportuna una descripción separada. A.R.D.: Administrador de Recursos Desconcentrados. Referencias: Decreto 5720 del G. C. B. A. (establece y regula los procedimientos de compra). Manual de Calidad del Laboratorio. de todas las operaciones y las correspondientes conclusiones. Dicho informe es revisado por el Jefe de Sección y aprobado por el Jefe del Laboratorio para pasar luego a ser archivado en un archivo llamado “Pruebas y Controles de Insumos Nuevos”. De esta manera, todo insumo a ser usado por primera vez en la Sección Bioquímica Clínica pasa previamente por estas fases de prueba, control y aceptación o rechazo, siendo muy útiles en estas etapas los Sistemas de Control de Calidad del Laboratorio. El equipamiento adquirido y los insumos consumibles que puedan afectar la calidad del servicio no son puestos en uso hasta que demuestran haber cumplido con las especificaciones estándar de los procedimientos correspondientes. En estos casos es conveniente solicitarle al fabricante/ proveedor los resultados documentados de todos los controles o verificaciones efectuadas a sus productos. La Sección Bioquímica Clínica posee un inventario actualizado y móvil de todos los insumos que están siendo utilizados en un momento dado. El mismo incluye el registro de los números de lote de los reactivos más importantes, materiales de control y calibradores; la fecha de recepción por el Laboratorio y la fecha en la que el material es puesto en uso. Este inventario figura en los “Cuadernos de Stock”. Circuito de adquisición de insumos Secc. Bioq. Clínica Dirección Administrativa Responsabilidades: Elabora: Jefe de Bioquímica Clínica. Revisa y aprueba: Jefe del Laboratorio. Desarrollo: Para realizar observaciones y mediciones, el laboratorio requiere, además de los equipos de medición, de reactivos, calibradores, materiales de control y todos los otros insumos que le permiten desempeñar su tarea. El abastecimiento bien organizado es un requisito esencial para el uso óptimo de los insumos, para su mejor rendimiento y para evitar el desperdicio de materiales vencidos. La Administración del Laboratorio cuenta con un sistema de control para la aceptación o rechazo de insumos que responde a sus políticas y procedimientos. Antes de la adquisición de cada insumo de Bioquímica Clínica, el mismo es probado en cuanto a su desempeño analítico por el responsable del área de su aplicación, el cual, en consenso con el Jefe de Sección, efectúa las pruebas o controles necesarios para conocer la propiedad del insumo y procede a su aceptación o rechazo. En cualquier caso, el operador que efectúa los ensayos de prueba eleva al Jefe de Sección un informe escrito detallado Stock Consumo mensual Demanda promedio Listado de precios Pedido Director A.R.D. Programación Compras Toma conocimiento Autoriza Firma Afecta elgasto Realiza el trámitede compra Trámite de compra Original proveedor 2 copias carpeta Copia Programación Copia Servicio Programación Afecta provisorio Tesorería Copia Tesorería Recepción de Documentación Listado de facturas recibidas por orden de fecha de recepción Gestión del Laboratorio de Análisis Clínicos Implementación de la documentación según normas internacionales ANEXO III EJEMPLO DE REGISTRO Planilla diaria de Registro de Pacientes Prueba de Tolerancia a la Glucosa MES: N° DIA: PACIENTE HORA P 75 P 100 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 BIBLIOGRAFÍA 1.- Burnett D. BSc, PhD. Acreditación del Laboratorio Clínico. Editorial Reverté, 1998. 2.- Briozzo G, Perego M. Etapas Pre y post-analíticas en el Laboratorio de Análisis Clínicos. Mejora continua de la calidad. Agosto 2004. 3.- ISO/TR 10013: 2001E. ”Guía para la documentación del Sistema de Gestión de la Calidad”. 4.- Norma ISO 9004:2000. Sistemas de Gestión de Calidad. Directrices para la mejora del desempeño. 5.- Norma ISO 9001:2000. Sistemas de Gestión de Calidad. Requisitos. 6.- Norma ISO 15189:2000. “Medical Laboratories. Particular requirements for quality and competence”. 7.- Reale E. Metodología de la Investigación. Universidad Fa- valoro, 2005. 8.- Reale E. La Tesis y su desarrollo. Universidad Favaloro, 2005. 9.- Norma ISO 17025:1999, IRAM 301:2000. Requerimientos generales para la competencia de los laboratorios de ensayo y calibración. 10.- Núñez Contreras L. “Concepto de documento”. Archivística. Estudios básicos. Sevilla: Diputación Provincial, 1983, 31. 11.- Fuster Ruiz F. Archivística, Archivo, Documento de archivo. Necesidad de clarificar los conceptos. Anales de Documentación. 1999, 2:103-120. 12.- Rezzónico R. “Módulo de Gestión de la información clínica y gerencial”. Maestría en Gerencia y Administración de Sistemas y Servicios de Salud. 2° año. Universidad Favaloro. 13.- Diccionario de Terminología Archivística. Comisión de Terminología de la Dirección de Asuntos Estatales. (Madrid. Ministerio de Cultura, 1993). 14.- Norma IRAM-ISO 8402:1994. 15.- Briozzo G, Perego MC. “Manual de Calidad”. Laboratorio Central. Hospital Materno-Infantil “Ramón Sardá”. G.C.B.A. Año 2002. Presentado en exposición oral de Dra. Briozzo. “Primer Encuentro Mercosur de Calidad en Organizaciones de Salud”. Organizado por la Asociación Médica Argentina y el IRAM. Buenos Aires. 24, 25 y 26 de junio de 2002. REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 37 | 40 Pág | 37 Evolución de los pacientes neutropénicos en nuestro Hospital Paula Bermúdez1 Juan Tentoni2 Alejandra Larregina3 Nélida Polini4 1 Dra. en Bioquímica 2 Dr. en Bioquímica 3 Médica. Especialista en Hematología y Hemoterapia. 4 Dra. en Bioquímica. Especialista en Hemostasia. Lugar de Trabajo: Unidad de Hematología y Hemoterapia. Hospital Municipal de Agudos “Dr. Leónidas Lucero”. Ciudad de Bahía Blanca. Enviar Correspondencia a: Dra. Paula Bermúdez Amancay 601. B8002 GRG - Bahía Blanca [email protected] Revista ByPC. Incorporada al Latindex. ISSN 1515-6761 Código Bibliográfico: RByPC Trabajo Recibido: 03-07-07 Aceptado: 06-08-07 RESUMEN Las neutropenias son insuficiencias medulares parciales que recaen sobre la serie granulocítica. Se definen por un recuento absoluto de neutrófilos (RAN) inferior a 1,5 x109/L. El objetivo fue evaluar en nuestra unidad, la evolución de los pacientes con neutropenia. Se revisaron las historias clínicas durante cinco años (2001-2005). Se incluyeron 93 pacientes con edades entre 4 y 89 años. El RAN osciló entre 0.02 y 1,50 x 109/L. Se observaron 204 episodios, de los cuales 103 fueron febriles: 36 sin foco apreciable. Se aislaron gérmenes en 37 episodios. Para el tratamiento se usaron antibióticos, antivirales, antimicóticos y factor estimulante de colonias. La mortalidad global fue del 77% y para las neutropenias febriles del 25%. La neutropenia de cualquier etiología constituye un desafío diagnóstico. Dadas las características de nuestros pacientes y la flora hospitalaria encontrada consideramos importante realizar un trabajo interdisciplinario para disminuir la morbilidad y mortalidad de estos pacientes. Palabras claves | neutropenia, neutropenia febril, recuento absoluto de neutrófilos. SUMMARY Neutropenias are partial insuffiencies of the bone marrow on the granulocitic linage, defined by an absolute neutrophil count (ANC) lower than 1,5 x 109/L. We studied the evolution of this particular group in our Unit. We checked medical files during a five years period (2001-2005) and found 93 neutropenic patients whose ages varied among 4 and 89 years. The ANC range was 0,02-1,50 x 109/L in 204 episodes, 103 of them registered fever: 36 with unknown origin. Pathogenic microorganisms were isolated in37 events. Theraphy included the use of antibiotics, antifungical, antiviruses and colony granulocitic stimulating factors. Global mortality arose 77% and 25% in patients with febrile neutropenia. Despite its cause, neutropenias represents a medical challenge. According with the microorganism isolated and our patients particularities, we consider imperative to study this population with the commitment of all the hospital staff to decrease the mortality and morbidity. Keywords | neutropenias, febrile neutropenias, absolut recount of neutrophil. Evolución de los pacientes neutropénicos en nuestro Hospital INTRODUCCION Los neutrófilos son granulocitos polimorfonucleares. Poseen funciones fagocíticas y bactericidas. Constituyen la primera línea de defensa inespecífica del organismo contra las infecciones junto con la integridad de la piel y mucosas (1,2). A causa de trastornos en la producción; distribución y recambio; enfermedades infecciosas y toxicidad por la administración de drogas, el paciente puede presentar un recuento absoluto de neutrófilos (RAN) inferior a lo normal conocido como neutropenia (3). Las neutropenias y su expresión extrema, la agranulocitosis, son insuficiencias medulares parciales que recaen sobre los granulocitos y sus precursores. Se definen por RAN un recuento absoluto de neutrófilos inferior a 1,5 x 109/L y a 0,5 x109/l respectivamente (4). El objetivo de nuestro trabajo fue evaluar en nuestra unidad, la evolución de los pacientes con neutropenia, su causa, duración, tratamiento y gérmenes aislados. MÉTODOS Se realizó la revisión de las historias clínicas de todos los pacientes atendidos en la Unidad de Hematología y Hemoterapia del Hospital Municipal de Agudos Dr. Leónidas Lucero de la ciudad de Bahía Blanca, en el período comprendido entre enero de 2001 y diciembre de 2005. Se seleccionaron aquellos pacientes que presentaron algún evento de neutropenia. De las historias clínicas seleccionadas se tomaron en cuenta las siguientes variables: nombre, número de historia clínica, fecha del evento, sexo, edad, enfermedad de base, duración de la neutropenia, recuento absoluto de neutrófilos, foco identificado, germen aislado, presencia de fiebre y tratamiento instituido, con las que se confeccionó una plantilla Excel. Los recuentos de leucocitos se realizaron en cámara de Neubauer y la fórmula leucocitaria fue observada por un doctor en bioquímica experimentado en hematología. La tipificación de gérmenes aislados se realizó en el laboratorio de bacteriología del Laboratorio Central de nuestro hospital. El procesamiento estadístico de los datos nominales recavados en este estudio retrospectivo incluyó la representación gráfica de porcentajes, histogramas y sectores circulares (5, 6, 7) eventos de neutropenia. El mayor porcentaje de ellos estaba asociado con la quimioterapia, mientras que los restantes estuvieron relacionados a la enfermedad de base (leucemia, aplasia medular, anemia megaloblástica, mielodisplasia, infección por HIV, entre otros),con un mínimo porcentaje de otras causas. Figura 1. Episodios de neutropenias quimioterapia enfermedad de base danantizol sepsis agranulocitosis tóxica En el 66% de los eventos se indicó factor estimulante de colonias granulocíticas como tratamiento de la neutropenia. Hubo un 50% de los eventos de neutropenia febril, de los cuales el 65% tuvo un foco identificado. Los focos hallados y los gérmenes aislados en cada caso se muestran en las figuras 2 y 3. Figura 2. Focos infecciosos presentes Infecciones urinarias celulitis infecciones orofaríngeas endocarditis peritonitis candidiasis neumonías sepsis colitis del neutropénico infecciones asociadas a cateter acceso hepático infecciones perianales Figura 3. Gérmenes aislados 20 18 16 14 12 10 6 4 2 ag ala ct iae Ca lbi ca ns Kp ne um on ia e S vir ida ns S Ec oli ae ru gin os a S p io ge ne s Sp ne um on iae Sa lm on ell as p P ur eu s pid er m idi s 0 Sa Se incluyeron 93 pacientes, 47 mujeres y 46 varones con un rango de edades comprendido entre 4 y 89 años, siendo la mediana de 59 años. Como puede observarse en la figura 1, se detectaron 204 8 Se RESULTADOS Pág | 39 REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 37 | 40 Los pacientes con neutropenia febril con foco, recibieron tratamiento antibiótico, antimicótico y/o antiviral según el germen aislado. En la Tabla 1 se enumeran las drgas empleadas. En los casos en que no se identificó el foco se utilizó el esquema de tratamiento empírico de nuestro hospital con una cefalosporina y un aminoglucósido (figura 4). Tabla 1. Drogas empleadas en el tratamiento Droga Número de pacientes Amikacina 16 Amoxicilina 8 Amoxicilina clavulanico 4 Ampicilina 7 Anfotericina 4 Cefalexina 2 Cefalotina 7 Cefazolina 8 Ceftacidima 18 Ceftriaxona 9 Ciprofloxacina 10 Claritromicina 1 Clindamicina 2 Fluconazol 1 Gentamicina 21 Metronidazol 1 Nistatina 6 Ornidazol 2 Rifampicina 1 Roxitromicina 2 Vancomicina 1 DISCUSIÓN La neutropenia febril es una entidad común en el manejo de los pacientes onco-hematológicos (8). La posibilidad de infecciones aumenta en aquellos enfermos con neutropenia severa (menos de 100 neutrófilos) (9) Según la literatura entre el 30 y el 60% de los pacientes neutropénicos febriles tienen un foco definido (9,10); en nuestra población este porcentaje fue del 65% lo que coincide con lo descripto. (figura 1). Por otra parte es importante destacar que el porcentaje de recuperación de gérmenes en nuestro laboratorio fue del 35%. Para el hematólogo la neutropenia de cualquier etiología constituye un desafío diagnóstico. Una de las decisiones más importantes con respecto a los pacientes inmunocomprometidos es determinar si la fiebre requiere una evaluación urgente, y el inicio temprano y empírico de tratamiento antimicrobiano (11,12). Como vemos, la neutropenia febril, es una de las mas temidas por su alto índice de mortalidad y en general, el manejo ha mejorado dramáticamente la sobrevida de los enfermos, con una mortalidad del 90% en la década del 70 a menos del 10% a fines de los 90 (13); en nuestra población la mortalidad fue del 25%. Concluimos con los datos recabados que sigue siendo muy importante realizar un trabajo interdisciplinario, conocer muy bien las características de la población con que trabajamos, la flora microbiana de cada institución, para de esta manera disminuir la morbilidad y mortalidad, de estos enfermos. REFERENCIAS BIBLIOGRÁFICAS Figura 4. Tratamiento Antibióticos Antiviral Antimicóticos Factores estimulantes de colonias La mortalidad global de nuestros pacientes fue del 77%, donde solo un 25% correspondió a aquellos pacientes que cursaban un cuadro de neutropenia febril. 1.- Florensa L, Woessner S. Capítulo 1: Hematopoyesis. Morfología de los elementos formes de la sangre y órganos hematopoyéticos. En: Hematología clínica. 5° edición. Elsevier España. Madrid. España. 2006; 1-32. 2.- Trevani A, Zwirner N, Geffner J, Fainboim L. Capítulo 3: Inmunidad innata: neutrófilos, macrófagos y células natural killer. En: Introducción a la Inmunología Humana. 5° edición. Editorial Médica Panamericana. Buenos Aires. Argentina. 2005; pp: 60-63. 3.- Rizzardini C, Espinoza X. Urgencias oncológicas. Rev Per Elec (en línea) 2005;2:25-32 4.- Pizzo PA. Current Concepts: Fever in Inmunocompromised Patients. N Engl J of Med 1999; 341:893-900. 5.- Dawson-Saunders B, Trapp RG. Capítulo 3: Exploración y presentación de datos. En: Bioestadística médica. 2º edición. Ediciones El Manual Moderno, SA de CV, México DF, 1997; pp 25-35. 6.- Macchi RL. Capítulo 4: Resumen de datos nominales. En: Introducción a la estadística en Ciencias de la Salud. Editorial Médica Panamericana. Buenos Aires. Argentina. 2001; pp 19-24. 7.- Steel RGD, Torrie JH. Capítulo 2: Observaciones. En: Bio- Evolución de los pacientes neutropénicos en nuestro Hospital estadística: principios y procedimientos. Segunda edición. Editorial McGraw-Hill. México. 1985; pp: 7-36. 8.- Torres HA, Bodey GP, Rolson KVI, Kantarjian HM, Raad II, Kontoyiannis Dp. Infections in patients with aplastic anemia: Experience at a Tertiary Care Cancer Center. Cancer 2003; 98:86-93. 9.- Hughes WT, Armstrong D, Bodey GP, Bow EJ, Brown AE, Pizzo PA. Guidelines for the use of antimicrobial agents in neutropenic patients with cancer. Clin Infect Dis. 2002; 34: 730-751. 10.- Pizzo PA. The compromised host. En: Goldman L, Bennet JC, eds. Cecil textbook of medicine. 2000;1569-1581. 21st ed Philadelphia: W.B. Saunders. 11.- Pizzo PA. Fever in inmunocompromised patients. N Engl J Med 1999; 341(12):893-900. 12.- Pizzo PA. Management of fever in patients with cancer and treatment-induced neutropenia. N Engl J Med. 1993;328: 1323-1332. 13.- Klastersky J, Zinner SH, Calandra T, Gaya H, Glauser MP, Meunier F, et al. Empiric patients therapy for febrile granulocytopenic cancer patients: Lessons from four EORTC trials. Eur J Can Clin Oncol 1988; 24 Suppl1;35-45. REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 41 | 46 Pág | 41 Hipotiroidismo congénito en los inicios de la pesquisa neonatal en el sector público de la ciudad de Posadas Alvaro Errasti1 María Marinela Tibolla2 Nora Graciela Márquez3 Ana Lía Albrekt4 Sandra Steffens5 1 Bioquímico 2 Bioquímica Especialista 3 Bioquímica Especialista 4 Bioquímica 5 Bioquímica Lugar de Trabajo: Facultad de Ciencias Exactas, Químicas y Naturales. Universidad Nacional de Misiones. Hospital Central Provincial Dr. Ramón Madariaga de la Ciudad de Posadas. Enviar Correspondencia a: Alvaro Errasti “Módulo de Farmacia y Bioquímica” Mariano Moreno y López Torres, Posadas, Misiones, CP 3300. Tel: 03752-427687 e-mail: [email protected] Revista ByPC. Incorporada al Latindex. ISSN 1515-6761 Código Bibliográfico: RByPC Trabajo Recibido: 05-06-07 Aceptado: 10-08-07 RESUMEN El Hipotiroidismo Congénito (HC) es una enfermedad producida por falla de la glándula tiroides del niño. La detección precoz de HC se realiza mediante el Programa de Pesquisa Neonatal (PPN), constituido en nuestra provincia desde Enero del 2006 en el Sector Público, cubriendo de esta manera, a todos los Recién Nacidos vivos del Hospital Dr. Ramón Madariaga de la Ciudad de Posadas. El objetivo de este Trabajo fue brindar un análisis epidemiológico descriptivo sobre la acción del PPN y calcular la Incidencia de HC en el Sector Público. La cobertura del PPN sobre los Recién Nacidos vivos fue del 84,94%, la Incidencia calculada fue de 1:1146. Los casos detectados fueron tres. Desde su inicio el programa ha demostrado un mejoramiento y, si bien, el intervalo de tiempo y la población tomada fue pequeña, este estudio es un valioso apoyo para la optimización del PPN y posteriores análisis epidemiológicos. Palabras claves | Hipotiroidismo congénito - Pesquisa Neonatal - Análisis Epidemiológico. SUMMARY The Congenital Hypothyroidism (CH) is a child thyroid gland fault produced disease. The CH precocious detection through the Newborn Screening Program (NSP) was constituted in the public sector of our province since January of 2006, covering, in this way, all the Dr. Ramón Madariaga Hospital of Posadas City living newborns. This work objective was to offer a descriptive epidemiological analysis about NSP and calculate the CH incidence on the public sector. The NSP cover on the newborns living was 84.94 %, the calculated incidence 1:1146. The detected cases were three. Since its beginning, an improvement of the program has demonstrated and, even the time interval and the selected population were small, this study is a valuable support for the NSP optimization and to subsequent epidemiological analysis. Keywords | Congenital Hypothyroidism – Newborn Screening – Epidemiological Analysis. Hipotiroidismo congénito en los inicios de la pesquisa neonatal en el sector público de la ciudad de Posadas INTRODUCCION El tiroides es una glándula de secreción endocrina situada inmediatamente por debajo de la laringe y a ambos lados y por delante de la tráquea. Elabora dos hormonas importantes, la Tiroxina y la Triyodotironina, T4 y T3 respectivamente, también secreta Calcitonina, una hormona importante para el metabolismo del calcio. Las funciones mas importantes de T3 y T4 son, un aumento generalizado de la actividad funcional de todo el organismo, dando como resultado un aumento importante de la actividad metabólica celular; por otra parte promueve el crecimiento y desarrollo del cerebro durante la vida fetal, y en los primeros años de la vida posnatal favoreciendo la ramificación y mielinización de las células neuronales del Sistema Nervioso Central. La regulación en la secreción del tiroides se encuentra definida por la Hormona Estimulante de Tiroides (TSH), hormona hipofisiaria que actúa de la siguiente manera: cuando el nivel de T3 y T4 baja en sangre, la hipófisis lo detecta y aumenta la producción de TSH, que estimula el tiroides para que produzca y libere mas hormonas tiroideas; cuando el nivel de hormonas tiroideas es alto, la hipófisis se frena, baja la TSH en sangre y el tiroides ralentiza su actividad. Las enfermedades del tiroides abarcan las alteraciones relacionadas con la liberación excesiva de hormona tiroidea (hipertiroidismo), otras vinculadas con una deficiencia de hormona tiroidea (hipotiroidismo), y lesiones tumorales. (1) Cualquier trastorno funcional o estructural que interfiera en la producción de concentraciones adecuadas de T3 y T4 origina hipotiroidismo. Este trastorno puede ser primario, si se debe a una anomalía intrínseca del tiroides, o secundario si se origina por un trastorno hipotalámico o hipofisiario (2,3) El Hipotiroidismo Congénito (HC) es una enfermedad producida como consecuencia de la falla de la glándula tiroides del niño, que se puede deber a la alteración embriológica del desarrollo de ésta (atireosis, ectopía o hipoplasia), o a la alteración en la hormonogénesis, debido a defecto enzimático que suele ser en general familiar y cursar con bocio. Ambas formas conllevan a una hipofunción tiroidea definitiva, y la terapia sustitutiva con T4 es de por vida. Por lo tanto, estos Hipotiroidismos Congénitos son permanentes. Existen otras formas de hipotiroidismo, los llamados transitorios. Su frecuencia no esta bien determinada, generalmente se relacionan con la administración de drogas antitiroideas a madres hipertiroideas, afectas de Enfermedad de Graves Basedow, o por el pasaje transplacentario de anticuerpos bloqueantes anti-receptor de TSH al feto. (4) Los bebes sin tiroides o con problemas en la formación de las hormonas tiroideas, en el momento del nacimiento son absolutamente normales, ya que se han desarrollado con la ayuda de las hormonas tiroideas de la madre. Los signos de HC se van a desarrollar a partir de la primera semana de vida pero no van a ser evidentes hasta el segundo o tercer mes, y para entonces se ha producido ya un deterioro mental que puede ser muy significativo. La clínica mas típica es la de un bebe con ictericia prolongada, constipación, hernia umbilical o caída tardía de cordón, macroglosia, letargo, motilidad disminuida, llanto ronco y poco vigoroso, entre otros menos específicos. (5) El HC se detecta con la determinación de TSH en el recién nacido mediante la Pesquisa Neonatal (PN), que comprende una batería de pruebas bioquímicas de “screening” para ciertos desórdenes metabólicos o errores innatos del metabolismo, que se practican a los recién nacidos, a partir de las 48 horas y hasta los 7 (siete) días de vida. La enfermedad en cuestión es la causa de discapacidad mental profunda prevenible más frecuente. Su tratamiento consiste en la toma diaria de un comprimido de hormona tiroidea. En nuestra provincia no hay trabajos que documenten datos epidemiológicos sobre HC, con lo que se hace difícil saber con que prevalencia e incidencia se presenta. El HC es un defecto metabólico. Este tipo de enfermedades puede ser de tipo congénito o hereditario, carecen de síntomas específicos tempranos y si no se tratan precozmente pueden provocar discapacidades físicas e intelectuales, e incluso la muerte. Hasta la fecha se han descrito mas de 4000 defectos metabólicos, de los cuales aproximadamente 70 conducen a un daño cerebral leve o severo, disponiendo 50 de ellos de tratamiento efectivo. La detección de estos últimos cobra importancia por la posibilidad de recuperación del individuo afectado a una calidad de vida cuasi-normal. El diagnóstico y tratamiento temprano asegura el desarrollo mental, psicomotor y crecimiento normal de los niños afectados. Se sabe que la prevalencia mundial de HC es de 1:2000 a 1:3000 RN, sin embargo se han descrito variaciones en la frecuencia, tanto geográficas como poblacionales. Como se puede observar, el principio de esta PN es detectar enfermedades congénitas o hereditarias, que no tienen manifestaciones clínicas en los primeros meses de vida y que pueden producir daños severos e irreversibles, pero que si se detectan a tiempo, tienen tratamiento efectivo. Por lo expresado, la PN es una gran herramienta de la medicina preventiva.(6-11) En nuestro país existen tres leyes nacionales que convierten el test en obligatorio: 23413 (de 1986), la 23874 (de 1990) y la 24438 (de 1994). Hacen referencia a tres enfermedades: HC, Hiperfenilalaninemia (PKU), y Fibrosis Quística (FQ). Sin embargo, si bien la ley es Nacional, Argentina es un país federal, esto es, las Provincias se adhieren a las leyes que quieren, tienen derecho a elegir. El Estado Nacional no tiene injerencia, salvo el caso que lanzara un Programa Nacional de PN. (12) Una de las restantes dos pruebas que componen el Screening o PN básico es la determinación de Fenilalanina (Fal) para la detección de un grupo de afecciones hereditarias denominadas “Hiperfenilalaninemias”, que se caracterizan porque su inconveniente es no poder transformar el ami- Pág | 43 REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 41 | 46 noácido esencial Fal a Tirosina, de tal forma que esa sustancia, de amplia presencia en los alimentos, se deposita hasta niveles tóxicos en sangre, llevando a la producción de cetoacidosis. Puede llevar a daño cerebral severo en caso de no recibir tratamiento adecuado, el cual es una dieta con bajo contenido de Fal. Las Hiperfenilalaninemias pueden ser: transitoria benigna, maligna o la forma más comúnmente conocida, la Fenilcetonuria clásica. (13) La Fibrosis Quística (FQ) es una enfermedad genética, que afecta los canales de conductancia del cloruro por deficiencia de la Proteína Reguladora de la Conductancia Transmembrana (CFTR), haciendo más viscosas todas las secreciones de las glándulas exocrinas. El daño principal se produce en los pulmones y el páncreas; su tratamiento precoz mejora la calidad y duración de vida. Se detecta mediante la determinación de Tripsina (Tripsina Inmuno Reactiva) neonatal en la PN. El tratamiento es específico para cada caso; en cuanto al costo, se encuentra vinculado a las Leyes Provinciales establecidas. En nuestra provincia, la PN es obligatoria para las determinaciones de TSH y Fal desde 2004, según ley provincial Nº 4100. En el año 2005 se modificó la ley para incluir dentro de la PN la determinación de Tripsina y, de esta forma, desde Enero de 2006 se conformó el Programa de Pesquisa Neonatal (PPN) en el Sector Público, cubriendo totalmente de esta manera a los bebes nacidos vivos en el Hospital Dr. Ramón Madariaga de la ciudad de Posadas. sólida, placas de tiras con pocillos revestidos previamente con anticuerpos monoclonales Anti Cadena Beta de la TSH. Las muestras, calibradores y el control en manchas de sangre seca se eluyeron con un conjugado Anti TSH Humana (Carnero)/Fosfatasa Alcalina. Este eluato se depositó en los pocillos de las tiras, permitiendo la formación del complejo anticuerpo/TSH/anticuerpo-enzima. Se realizó un lavado posterior, eliminando así los componentes no fijados. Se añadió un sustrato fluorogénico (4-metilumbeliferil fosfato) en los pocillos de las tiras, que resultó hidrolizado por acción de la enzima del conjugado. Por último, se midió la intensidad de la fluorescencia emitida, que es proporcional a la concentración de TSH presente en la muestra. Figura 1. Tarjeta de Guthrie modelo. MATERIALES Y MÉTODOS Se tomó como población para este estudio a los Recién Nacidos vivos del Hospital Dr. Ramón Madariaga de la ciudad de Posadas, a quienes se les realizó la PN, en el período comprendido entre Enero-Noviembre de 2006, lapso de tiempo durante el cual se encuentra instituido el PPN. Las muestras se obtuvieron mediante punción del talón o punción venosa entre las 24 hs. y los 7 días de vida. La sangre se depositó en papel de filtro Schleicher & Schuell 903 (Tarjeta de Guthrie), diseñados específicamente para tal fin, provistos de 6 (seis) círculos de 13 mm de diámetro, de acuerdo con las técnicas internacionalmente aceptadas. (Fig.1). En el diseño de esta tarjeta se incluyó un espacio destinado al registro de los siguientes datos: identificación del paciente, de la madre, del centro asistencial de origen donde se produjo el nacimiento; fecha de nacimiento y primera ingesta de leche, y otras informaciones de interés médico. Posteriormente las mismas fueron enviadas al Laboratorio de PN para su evaluación y procesamiento bioquímico, que consistió en la cuantificación de la TSH mediante el método Ultra Micro ELISA (UMELISA-kit comercial). El UMELISA TSH neonatal es un ensayo heterogéneo inmunoenzimático tipo sándwich, en el cual se utiliza como fase Los valores de TSH menores al punto de corte (15μUI/ml), se consideraron normales. Los valores mayores a éste, se consideraron como sospechosos, recitándose a los pacientes para una nueva toma de muestra antes del día 21 de vida, para posterior confirmación mediante perfil tiroideo (TSH, T4total y T4libre). Los valores elevados de TSH y disminuidos de T4 confirmaron los diagnósticos de HC. Es importante señalar que al momento de la recitación se les suministró a los pacientes el tratamiento pertinente, que consistió en dosis de Levotiroxina, evitando así la posible tardanza, si se trataba de un caso positivo. Se realizó un estudio descriptivo y retrospectivo sobre los ca- Hipotiroidismo congénito en los inicios de la pesquisa neonatal en el sector público de la ciudad de Posadas sos positivos para HC detectados mediante la PN. Para el mismo, se obtuvo el número de Nacidos Vivos durante el período estudiado de los registros de la Sección Partos del Hospital Dr. Ramón Madariaga. Para la carga de estos datos y realización de los gráficos, se utilizó el programa Microsoft Excel. Figura 4. Observación de la frecuencia renacimientos distribuidos según las Semanas de Gestación. DISTRIBUCION POR SEMANAS DE GESTACION Frecuencia RESULTADOS 600 En el período comprendido entre Enero-Noviembre 2006, el número de nacidos vivos fue de 3440, con una distribución por sexos de: 500 400 300 200 Figura 2. Observación de la distribución por sexos durante el período Ene-Nov 2006. DISTRIBUCION POR SEXO Masculino 50,25% Femenino 49,75% El rango de peso al nacer que predominó fue de 2500-3500gr., con un mínimo de 910gr., y un máximo de 6700gr. (Fig. 3). Figura 3. Distribución por Peso de los Recién Nacidos vivos. DISTRIBUCION POR PESO EN GRAMOS Frecuencia 600 100 0 sem sem sem 8 sem 9 sem 0 sem 1 sem 2 sem 3 4 3 4 4 3 0 1-3 4 5- 37 5 2 3 3 Semanas de Gestación Fig. 4: Observación de la frecuencia renacimientos distribuidos según las Semanas de Gestación El número de tamizajes en dicho período fue de 2922, obteniéndose así una cobertura del 84,94% del total de Recién Nacidos vivos en el Hosp. Dr. Ramón Madariaga. Se define Incidencia de una enfermedad como el número de casos nuevos de una enfermedad durante un período de tiempo, que se desarrollan en una población sana, pero en riesgo de contraer la misma. (14) En esta enfermedad el riesgo a contraerla lo poseen todos los recién nacidos vivos, pues no hay otro factor que influya mas que el de nacer, ya que, teóricamente no se tienen evidencias de deficiencia de yodo en nuestra región. La incidencia de casos positivos de HC calculada para nuestra ciudad es de 1:1146 Recién Nacidos vivos. Del 100% de muestras tamizadas: • El 0,3% de ellas (9 muestras), fueron mal tomadas (papel filtro sobresaturado de sangre, gotas de sangre insuficiente, etc.), solicitándose nuevas muestras. (Fig.5). 500 400 Figura 5. Invalid Specimens 300 200 100 0 0 gr gr gr gr 00 450 00 00 00 5 5 5 5 1 4 2 3 1 11de 350 900 150 250 yor a m Rangos de Peso (gr.) El rango de edad de gestación mas frecuente fue de 37-40 semanas, con mínimo de 26 y máximo de 42 semanas. (Fig. 4). REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 41 | 46 Fig. 5: Muestras rechazadas por diferentes motivos: 1. muestra insuficiente; 2. muestra desgastada; 3. muestra no secada antes del envío; 4. muestra sobresaturada; 5. muestra diluida, descolorida o contaminada; 6. muestra con anillos de suero; 7. muestra coagulada; 8. muestra sin sangre. Fuente: www.marchofdines.com/files/neonatal.pdf. • Otro 0,3% de ellas (9 muestras) correspondió a resultados sospechosos, al dar un valor de TSH mayor a 15μUI/ml. De éstas: • 2 (dos) no se pudieron recitar para la toma de una segunda muestra. • A las 7 (siete) restantes se las pudo localizar, realizándoles las pruebas de confirmación, de las cuales: • 4 (cuatro), resultaron con valores dentro de los rangos normales. • 3 (tres), acabaron siendo confirmadas como HC. El tiempo promedio empleado para confirmar y tratar los tres casos detectados fue de 39 ± 20 días, con un rango de 12-63 días. Hasta el momento no se han realizado las pruebas para detectar la etiología de ninguno de los tres casos. (Fig. 6). Figura 6. Tardanza del PPN en confirmar diagnostico de los casos positivos. DIAS ENTRE Dx Y Tto Días 28 / 07 /2006 70 60 25 / 07 /2006 DIAS 50 40 30 26 / 10 /2 006 20 10 1 0 1 2 3 CASOS Por último, de estos 3 (tres) casos, 2 (dos) eran de sexo femenino (66,6%), mientras que el restante era masculino (33,3%). CONCLUSIÓN El Peso y las Semanas de Gestación no tienen asociación con el riesgo para contraer HC, pero, si la toma de muestra se realiza sobre un neonato pretérmino, es probable que el valor de su TSH esté elevado, dando lugar a potenciales falsos positivos. Para evitar estos sucesos es que se recomienda una segunda toma de muestra a las 2 (dos) semanas de vida, con el objeto de reevaluar al bebe. Pág | 45 El porcentaje de cobertura estimado es elevado respecto al del interior de nuestra provincia, dado que la divulgación del PPN está en sus comienzos. Sumado a esto, no debemos descartar la dificultad que acarrea el envío de las muestras tomadas por otros laboratorios del interior de Misiones al central, sito en la ciudad de Posadas. La Dirección Materno-Infantil del Ministerio de Salud de la Nación establece una cobertura de aplicación en el orden Nacional de aproximadamente un 75%. En Cuba y en la ciudad de Navarra, España, existen programas donde el porcentaje de cobertura es del orden del 97-99%. Estos últimos son PPN establecidos desde hace ya 15-20 años, que utilizan sangre de cordón umbilical para la detección de TSH neonatal, elevando el valor de corte de la TSH, medida por la técnica utilizada. El problema con esta muestra es que las hiperfenilalaninemias no se detectan antes de las 48 horas, lo que hace imposible su práctica en nuestro medio. La Incidencia calculada es elevada con respecto a la Incidencia mundial, según literatura (1:2000 a 1:3000), como así también lo es frente a publicaciones pertenecientes a otros lugares (1:2778 en Ciudad Autónoma de Buenos Aires, período 2000-2002 ; 1:3571 en México, período 20012002).(15,16) Sin embargo, para hablar con certeza de la verdadera frecuencia de la patología tendríamos que contar con una cobertura total, es decir, practicarles la PN a todos los Recién Nacidos sin excepción y con cero fallas en localización de casos sospechosos, hecho que aun no se puede alcanzar. Una posible explicación a la alta incidencia de HC calculada para el Sector Público de nuestra ciudad es que existe la probabilidad de obtener varios casos juntos, como ninguno en un determinado lapso de tiempo, siendo el de este estudio, un período considerablemente corto como para brindar una descripción precisa. Se debe continuar el seguimiento epidemiológico, ya sea para evaluar nuestra población como para optimizar el PPN. Por tal motivo este estudio ofrece una base sobre la cual poder apoyarse en trabajos posteriores. Las muestras rechazadas por haber sido mal tomadas correspondieron al intervalo de tiempo Enero-Marzo, lapso en que se dio inicio al PPN, lo que refleja la falta de entrenamiento y/o capacitación de los extraccionistas para la técnica elegida (punción de talón en Tarjeta de Guthrie). Sin embargo, otros trabajos muestran un porcentaje mas elevado de muestras mal tomadas (1,85% México, 2001-2002; 2,7% Colombia, 2001). (17) En cuanto a las muestras que no pudieron ser ubicadas para su recitación, fue debido a la falta de datos sobre la identificación del paciente. Este hecho ocurrió, ya sea por el mal relleno de las tarjetas, que como hemos mencionado, tienen un espacio para identificación del paciente; o por cambios de domicilio o número de teléfono. Los resultados elevados de TSH, los cuales normalizaron sus valores en la segunda determinación, se debieron a que la primera toma de muestra se efectuó antes de las 48 Hipotiroidismo congénito en los inicios de la pesquisa neonatal en el sector público de la ciudad de Posadas horas de vida. La muestra se aconseja obtenerla el tercer día de vida, no antes de las 48 horas, por la elevación fisiológica de la TSH, que podría dar lugar a falsos positivos, ni mas tarde del quinto día, con objeto de no retrasar mas el tratamiento. El curso de tiempo transcurrido entre la detección y la respectiva confirmación y tratamiento de cada caso positivo de HC ha ido en progresivo descenso, lo que se traduce en una optimización del PPN, del primero al tercer caso y, lo que es mas importante en un pronóstico muchísimo mas favorable para los niños. El tiempo de espera entre la detección y la confirmación del resultado obtenido, y por tanto, diagnóstico de la patología, se ha demostrado que descendió notoriamente, llegando al tiempo óptimo de 12 días. Lo mismo sucedió con el número de muestras rechazadas por mala toma. Esto atestigua del buen trabajo multidisciplinario desarrollado por todos los Profesionales intervinientes, tanto en la actividad técnica como en la social, considerando las históricas falencias de estos servicios de PN en la respuesta a las situaciones de recitado; y que además avala la optimización del servicio, debido a que el imprescindible acortamiento de ese tiempo de espera redunda en la aplicación del tratamiento (mayor precocidad) y por ello, en la calidad de vida del paciente. La preponderancia del género femenino sobre el masculino en los casos de HC ocurrida en nuestra ciudad se asemeja a la literatura consultada (70% niñas-20% niños, Navarra, 1997; 90% niñas-10% niños, Ciudad Autónoma de Buenos Aires, 2001). REFERENCIAS BIBLIOGRÁFICAS 1.- Robbins, S.; Cotran, R.; Kumar, V.; Collins, T. “El Sistema Endocrino”. En: Patología Estructural y Funcional. Pag. 1174-1178. Ed. Mc Graw Hill. Interamericana, 6° ed., Madrid. 1999. 2.- Greenspan, F.; Gardner, D. “Endocrinología Básica y Clínica”. Editorial Manual Moderno. 5ta. ed. México DF., 2003. 3.- Albarrán Jara, A. “Endocrinología”. Editorial Médica Panamericana SA. 1°ed. Madrid. 2003. 4.- Oyarzábal, M.; Chueca, M.; Elso, J.; Sola, A.; y la comisión de tiroides de la SEEP. “Screening Neonatal del Hipotiroidismo Congénito: Resultados del Programa en Navarra”. Rev. Anales del Sistema Sanitario de Navarra. Navarra, 1998; Vol.21, N°3. 5.- Cattani, A.”Transtornos Tiroideos Neonatales”. Boletín de la Escuela de Medicina de la Universidad Católica de Chile. Vol.29 6.- Fisher, D. A. “Second International Conference on Neonatal Thyroid Screening”: Progress report. J. Pediatr 1983; 102: 653-654. 7.- Toublanc, J. E. “Comparision of Epidemiological data on Congenital Hypothyroidism in Europe with those of other Parts in the World”. Horm Res. 1992; Vol.38: 230-235. 8.- Klein, R. Z.; Mitchell, M. L. “Hypothyroidism in Infants and Children”. Neonatal Screening. In: Braverman, L.; Utiger, R., (eds). Werner and Inglar´s. The Thyroid. New York. 984-994. Lippincott-Raven Press. Philadelphia; 1996. 9.- Klett, M. “Epidemiology of Congenital Hypothyroidism”. Exp. Clin. Endocrinol Diabetes 1997; 105 Suppl 4: 19-23. 10.- Güell GR. “Hipotiroidismo”. En: Caparrós CHM. Introducción a la Endocrinología Pediátrica. 66-79. Editorial científico-técnica. Ciudad de La Habana 1987. 11.- Marrero N, Frómeta A, Coto R, Villegas L. “Medición de TSH, T4 y Phe en muestras de sangre del cordón umbilical en papel de filtro: impacto en el tamizaje neonatal”. Biomed 2000;20:30-41. 12.- Borrajo, G. “Males Congénitos”. Rev. Fundación Bioquímica Argentina. 13.- Chiesa, A; Papendieck, L; Prieto, L; Bergadá, C. “Hiperfenilalaninemias: Fisiopatología y Pesquisa”. Rev Hosp. Niños Buenos Aires. Vol. XXXVI-N°157. Junio de 1994. 14.- Fernandez Pita, S.; Pértigas Díaz, S.; Valdez Cañedo, F. “Medidas de Frecuencia de Enfermedad: Incidencia y Prevalencia”. www.fisterra.com. Atención Primaria en la Red. 15.- “Resultados Estadísticos del Programa de Pesquisa Neonatal de la Ciudad Autónoma de Buenos Aires”. Publicación de la Red de Pesquisa Neonatal Coordinación Redes de Salud Secretaría de Salud Gobierno de la Ciudad Autónoma de Buenos Aires. 29 y 30 de noviembre 2002. Buenos Aires. 16.- Vela-Amieva, M.; Gamboa-Cardiel, S.; Perez-Andrade, M.; Ortiz-Cortés, J. “Epidemiología del Hipotiroidismo Congénito en México”. Salud Pública Mex 2004; 46: 141-148. 17.- De Bernal, M.; Caldas, M.; Bonilla, R.; Chamorro, G.; Matallana, A. “Tamización para Hipotiroidismo Congénito en Cali y Constitución de un Centro Piloto de Referencia para la Identificación Temprana de la Enfermedad”. Rev. Colombia Médica, Vol. 34 N°1, 2003. Pág | 47 REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 47 | 51 MONOGRAFÍA Identificación fenotípica de micobacterias Belén Imperiale1 Nora Morcillo1 Amelia Bernardelli3 1 Bioquímico 2 Bioquímica Especialista 3 Bioquímica Especialista 4 Bioquímica 5 Bioquímica Lugar de Trabajo: 1 Laboratorio de Referencia del Programa de Control de la Tuberculosis de la Provincia de Buenos Aires, Hospital Dr. Antonio A. Cetrángolo. 2 Laboratorio de Referencia en Tuberculosis Bovina (OIE), Laboratorio de Referencia en Paratuberculosis (OIE) DILAB -Servicio Nacional de Sanidad y Calidad Agroalimentaria (SENASA) Revista ByPC. Incorporada al Latindex. ISSN 1515-6761 Código Bibliográfico: RByPC Trabajo Recibido: 14-05-07 Aceptado: 31-07-07 La tuberculosis (TB) es una enfermedad infecciosa producida por Mycobacterium tuberculosis. Se estima que alrededor de un tercio de la población mundial se encuentra infectada con dicha micobacteria, y que dos millones de personas mueren anualmente en todo el mundo por esta enfermedad.1 En la Argentina la TB es considerada una enfermedad endémica. Con la epidemia del HIV/SIDA ha habido un aumento importante en el número de enfermos de TB. 2 La infección con el HIV, no sólo ha llevado a un aumento de la incidencia de TB, sino que también produjo un aumento de enfermedades producidas por las micobacterias no tuberculosas (NTM), llamadas micobacteriosis.3 Este hecho lleva frecuentemente a la necesidad de la identificación micobacteriana. Dentro del Complejo M. tuberculosis se encuentran las siguientes especies: M. tuberculosis, M. bovis, M. bovis BCG, M. africanum, M. microti, M. caprae, M. canetti y M. pinnipedii. 4 El M. bovis es el agente etiológico de la TB bovina, pero puede también producir enfermedad en el hombre, convirtiendo a la enfermedad en una zoonosis. Por tal motivo, es necesario contar con herramientas adecuadas para diferenciar M. bovis de M. tuberculosis en aquellos casos donde el enfermo en estudio posea características epidemiológicas compatibles con una tuberculosis causada por M. bovis. Una de las causas por la que es importante saber si la enfermedad esta causada por M. tuberculosis o M. bovis es por el tratamiento. M. bovis es intrínsecamente resistente a pirazinamida, una de las principales drogas utilizadas en el tratamiento de la TB causada por M. tuberculosis. 5 Las micobacterias no tuberculosas son microorganismos ambientales que pueden producir desde colonización hasta enfermedad en el huésped susceptible. Las personas están habitualmente en contacto con las micobacterias ambientales, las cuales son comúnmente ingeridas, inhaladas o hasta pueden estar en la piel. Según su patogenicidad y virulencia, las micobacterias se clasifican en tres grupos. El primero incluye a los patógenos estrictos altamente virulentos. Estos no son encontrados en el medio ambiente. Algunos ejemplos de este grupo son el complejo M. tuberculosis, M. leprae. En el segundo grupo se encuentran las micobacterias potencialmente patógenas, de virulencia intermedia. La mayoría se encuentran en la naturaleza y se convierten en patógenas en circunstancias especiales del huésped, es decir se comportan como oportunistas. Como ejemplo de este grupo se encuentra el complejo Mycobacterium avium-intracellulare. Pertenecen al tercer grupo las especies normalmente saprófitas, avirulentas, que son no patógenas o lo son sólo excepcionalmente. Como ejemplo se encuentran M. gordonae, M. fortuitum. Otra clasificación de las micobacterias es la que fue descrita por Runyon, y se basa en caracteres como el tiempo de crecimiento y la cromogenicidad de las colonias obtenidas en cultivo. 6 Los grupos son: I. Fotocromógenas: son de crecimiento lento. Colonias no pigmentadas en la oscuridad, los cultivos jóvenes adquieren color amarillo al exponerlos a la luz. II. Escotocromógenas: Son de crecimiento lento. Colonias pigmentadas, amarillentas o anaranjadas, aunque desarrollen en la oscuridad. III. No cromógenas: Crecimiento lento. Colonias no cromógenas generalmente. En algunos casos los cultivos viejos adquieren color amarillento. IV. De crecimiento rápido: Desarrollan colonias en los medios de cultivo en menos de una semana. Identificación fenotípica de micobacterias Las enfermedades causadas por micobacterias se diagnostican generalmente por exámenes bacteriológicos, baciloscopía y cultivo. A través de la baciloscopía no es posible identificar la especie causal y solo se informa presencia de bacilos ácido-alcohol resistentes, en cambio el cultivo permite continuar con el proceso de identificación micobacteriana. En el caso de diagnosticar a un paciente con TB o con micobacteriosis el comportamiento clínico debe ser completamente distinto, ya que la implicancia epidemiológica y el tratamiento eficaz deben ser diferentes en ambos casos. Hasta el presente no ha sido demostrada la transmisión persona a persona, especialmente por vía aerógena, de las NTM. Además, la resistencia intrínseca de la mayoría de es- Identificación a partir de aislamientos obtenidos en cultivos sólidos en base de huevos. Velocidad de crecimiento Rápida (< 7 días) Pigmentado No Pigmentado Fotocromógena / Escotocromógena Nitrato Reductasa Catalasa SC Catalasa 68ºC + Nitrato Reductasa - + Catalasa SC Catalasa 68ºC - +/- + M. smegmatis Arilsulfatasa (3 y 14 días) - -/+ M. phlei M. vaccae SC: Semicuantitativa Crecimiento en ClNa 5% - M. chelonae + M. chelonae + M. fortuitum M. peregrinum (M. fortuitum se colorea con el verde de malaquita) Pág | 49 REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 47 | 51 tas micobacterias a los agentes activos contra M. tuberculosis hace imprescindible incorporar otros fármacos al régimen terapéutico adecuado. Debido a estos motivos es que surge la imperiosa necesidad de efectuar una correcta identificación micobacteriana cuando el caso clínico lo requiera. dad. Los métodos genotípicos son mucho más rápidos, también son más costosos y complejos, quedando solamente disponibles para laboratorios de alta complejidad. Además estos métodos no reemplazan a los fenotípicos, sino que aportan datos complementarios para la correcta identificación micobacteriana. En presencia de un aislamiento clínico de una NTM, se deben considerar las características del mismo y la frecuencia del aislamiento. En los pacientes no infectados por el HIV es necesario aislar la micobacteria por lo menos tres veces en un cultivo abundante y relacionarlo con el estado clínico de la persona para definir la micobacteriosis. En cambio, en los pacientes infectados por el HIV o con alguna otra inmunosupresión severa, un único aislamiento de una NTM aporta un dato relevante para el diagnóstico de una micobacteriosis oportunista como causa de enfermedad. Las micobacterias más frecuentemente aisladas de pacientes atendidos en el Laboratorio de Referencia del Programa de Control de TB del Hospital Dr. Antonio Cetrángolo, resultaron ser M. gordonae en pacientes no infectados con el HIV y M. avium en pacientes infectados con HIV. Sobre la base de estos hallazgos clínicos y a la posibilidad de nuestro laboratorio de realizar las pruebas fenotípicas de identificación, se presenta el siguiente algoritmo destinado a la rápida y correcta aproximación de la identificación de las principales NTM encontradas en la población que concurre a nuestro servicio de salud. El algoritmo esta basado en la observación de las características culturales de las micobacterias y en las pruebas bioquímicas que revelan la presencia de enzimas asociadas a alguna vía metabólica de estos microorganismos. 6, 7 Algunas veces se utiliza la sensibilidad o resistencia a antibióticos como indicación de la especie bacteriana. Existen distintos métodos para la identificación de micobacterias: los fenotípicos y los genotípicos. Las pruebas fenotípicas constituyen el principal método para identificar micobacterias en laboratorios clínicos, especialmente en establecimientos de medianos o bajos recursos y compleji- Cultivos en medio sólido Características culturales: colonias secas, rugosas, no pigmentadas, de crecimiento lento, mayor de 15 días Probable complejo M. tuberculosis Niacina - TCH: sensible Pirazinamidasa: negativa M. bovis + M. tuberculosis TCH: resistente Pirazinamidasa: positiva Identificación fenotípica de micobacterias Cultivos en medio líquido Ziehl-Neelsen Formación de cuerdas + - Probable complejo M. tuberculosis Probable NTM Niacina - TCH: sensible Pirazinamidasa: negativa M. bovis + M. tuberculosis La observación microscópica es el primer control que se debe realizar de todo cultivo que se quiera identificar. Mediante la coloración de Ziehl-Neelsen se puede observar la presencia de microorganismos ácido alcohol resistentes, su morfología y tamaño. Es importante también la observación de las características culturales de las colonias del aislamiento obtenido. Este trabajo fue realizado como producto del curso “Clasificación Fenotípica de Micobacterias concretado en el Departamento de Micobacterias, DILAB-SENASA, Martínez –Pcia. de Bs. As.,9 al 11 de octubre de 2006. BIBLIOGRAFÍA 1.- The WHO/IUATLD Global Project on Anti-tuberculosis Drug Resistance Surveillance: Anti-tuberculosis drug resistance in the world, report number 2 volume. Geneva; 2004. 2.- Instituto Nacional de Enfermedades Respiratorias Emilio TCH: sensible Pirazinamidasa: positiva Coni. Tuberculosis Notificación de casos de tuberculosis en Argentina. Situación nacional y por provincias. Año 2003. PRO.TB.44/03 3.- Pitchenik, A. E., Fertel D. Medical management of AIDS patients: tuberculosis and nontuberculous mycobacterial diseases. Med. Clin. North Am. 1992; 76: 121-171 4.- “Practical Handbook for the phenotypic and genotypic identification of mycobacteria”. Cardoso Leao, S., Martin A., Mejía G., et al. 2004. 5.- World Health Organization (WHO). Emerging and another communicable diseases, surveillance and control. 1996; WHO/EMC/ZOO/96.4 6.- Centro Panamericano de Zoonosis (CEPANZO, OPS/OMS). Nota técnica número 11 “Bacteriología de la tuberculosis”. Kantor I. 1979. 7.- Material bibliográfico del curso “Clasificación Fenotípica de Micobacterias”.DILAB- SENASA, 9-11 de octubre de 2006. Pág | 51 REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 47 | 51 Identificación a partir de aislamientos obtenidos en cultivos sólidos en base de huevos Velocidad de crecimiento Lenta (>7 días, no M. tuberculosis) No Pigmentado Pigmentado Fotocromógena Catalasa SC Catalasa SC Escotocromógena Catalasa 68ºC Catalasa SC Catalasa 68ºC - Catalasa 68ºC + Nitrato Reductasa + + - Reducción de Telurito (3 y 9 días) Reducción de Telurito (3 y 9 días) Nitrato Reductasa - - Nitrato Reductasa Red. Telurito (3 y 9 días) + - M. kansasii M. gordonae + + + Red. Telurito 3 días 9 días - Complejo M. aviumintracellulare + M. scrofulaceum PRA M. avium M. intracellulare Valores de referencia del hemograma en embarazadas, con tecnología actual Valores de referencia del hemograma en embarazadas, con tecnología actual Katia Canalejo1 Juan Tentoni2 Mónica Aixalá3 Ana María Jelen4 1 Bioquímica Especialista en Hematología 2 Bioquímica Especialista en Hematología 3 Bioquímica 4 Bioquímica Lugar de Trabajo: Instituto de investigaciones Hematológicas Academia Nacional de Medicina 1 Pacheco de Melo 3081 Ciudad A. de Buenos Aires. Hospital Vélez Sarsfield 2, Calderón de la Barca 1550 Ciudad A. de Buenos Aires. Enviar Correspondencia a: Katia Canalejo Laguna 1120 dpto 2 1407 - Capital Federal [email protected] Revista ByPC. Incorporada al Latindex. ISSN 1515-6761 Código Bibliográfico: RByPC Trabajo Recibido: 22-03-07 Aceptado: 18-06-07 RESUMEN Se procesaron 435 muestras de embarazadas clínicamente sanas con la finalidad de obtener valores hematológicos de referencia utilizando un contador con tecnología actual. Los resultados, media y desvío estándar de cada parámetro hallado, se presentan agrupados por trimestres de gestación, lo cual permite observar las variaciones a lo largo del embarazo. Han sido significativos la disminución de hemoglobina y el aumento de leucocitos a expensas de neutrófilos y monocitos. Cualquier mínimo cambio en los valores hematimétricos de una embarazada, fuera de lo esperado fisiológicamente para cada trimestre, debe ser considerado en cuanto a magnitud y tiempo de gesta. Esto puede sugerir la presencia de alguna patología. SUMMARY 435 blood samples of clinically healthy pregnancies were examined by means of a state of the art cell-counter in order to establish the haematological reference data. The results, with means and standard deviations of each parameter measured, were presented according to each trimester of gestation, thus allowing the observation of the variation of this data throughout the pregnancy. The lowering of haemoglobin and rise of leukocytes levels at the expense of neutrophils and monocytes were significant. Other than that physiologically expected for each trimester, the magnitude and timing of any minimum haematimetric values change during pregnancy should be taken into account. This may indicate the presence of some pathology. Pág | 53 REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 52 | 54 INTRODUCCION RESULTADOS El análisis completo del hemograma, incluido el recuento diferencial de leucocitos, es usado en el seguimiento clínico de las embarazadas. Por lo tanto, tener valores de referencia correctos, es esencial para la interpretación de los resultados (1). La mayoría de los contadores hematológicos usados hoy en día, en centros asistenciales de alta complejidad o de referencia, tienen incorporaciones tecnológicas de avanzada. Por ejemplo: al método tradicional para el recuento de células, denominado impedancia o resistencia eléctrica se le han agregado algunas modificaciones técnicas tales como Editing (circuito revisor de pulsos que en combinación con un transductor Von Behrens reduce la recirculación de células) y Focusing (enfoque hidrodinámico, método que controla la orientación y el número de células que pasan a través de la apertura)(2). Esta combinación de tecnologías proporciona un alto grado de precisión en los resultados, tanto en muestras normales como patológicas. El recuento diferencial de leucocitos se realiza por citometría de alta resolución por flujo enfocado, que permite identificar cinco subtipos de células blancas en estado natural. Por todo lo mencionado anteriormente se hace necesario establecer los valores de referencia para estos equipos (3). Se procesaron 455 muestras de embarazadas clínicamente sanas, en un contador hematológico multiparamétrico totalmente automatizado, diseñado para el recuento diferencial de poblaciones celulares sanguíneas (4). El tamaño muestral es adecuado para considerar los resultados hallados como valores de referencia (5). Los mismos se presentan en las Tablas 1 (serie eritroide y plaquetas) y 2 (serie leucocitaria con el recuento diferencial correspondiente expresado en valores absolutos). En ambas tablas se muestran los valores obtenidos en cada trimestre de gestación para poder evaluar lo que sucede a lo largo del embarazo normal (6). OBJETIVO Actualizar los valores de referencia del hemograma en los tres trimestres del embarazo con las nuevas tecnologías MATERIAL Y METODOS Población: se estudiaron 455 embarazadas con hemoglobinas mayores de 11gr/dL, en edades comprendidas entre 18 y 40años, clínicamente sanas, que no tomaban ningún suplemento vitamínico ni hierro. Las embarazadas otorgaron su consentimiento oral para el estudio. Muestras: se obtuvieron muestras anticoaguladas con EDTAK3 y los hemogramas fueron realizados en un autoanalizador hematológico Cell Dyn 3700 (tecnología: impedancia electrónica, citometría de alta resolución por flujo enfocado). El sistema de medición se basa en impedancia volumétrica para el recuento de hematíes, plaquetas y leucocitos, tecnología laser para el recuento y diferenciación de leucocitos y método espectrofotométrico basado en la obtención de cianmetahemoglobina para la medición de la concentración de la hemoglobina. Tabla 1. Serie eritroide y plaquetas: Valores promedio de referencia en cada trimestre del embarazo Primer Trimestre Segundo Trimestre Tercer Trimestre Parámetros Media ± DE Media ± DE Media ± DE RBC (x 10 /L) (a) 4,18 ± 0,27 4,09 ± 0,31 4,09 ± 0,26 Hb (g/dL) (b, c) 12,3 ± 0,66 12,1 ± 0,70 11,9 ± 0,65 Hto (L/L) (a) 0,37 ± 0,02 0,36 ± 0,02 0,36 ± 0,02 VCM (fL) 89,4 ± 3,2 90,0 ± 3,6 89,7 ± 4,2 HCM (pg) 29,5 ± 1,3 29,6 ± 1,4 29,1 ± 1,6 ADE (%) 15,0 ± 1,1 15,2 ± 1,0 15,2 ± 1,2 Plaq (x 109/L) 244 ± 50 245 ± 47 244 ± 50 12 RBC: Recuento de hematíes, Hb: Hemoglobina, Hto: Hematocrito, VCM: Volumen corpuscular medio, HCM: Hemoglobina corpuscular media, ADE: Amplitud de distribución eritrocitaria, Plaq: Plaquetas a- p<0,01entre primero y tercer trimestre b- p<0,001 entre primero y tercer trimestre c- p<0,01 entre primero y segundo trimestre Tabla 2. Serie leucocitaria: Valores promedio de referencia en cada trimestre del embarazo. Primer Trimestre Segundo Trimestre Tercer Trimestre Parámetros Media ± DE Media ± DE Media ± DE Leucocitos (x109/L) (a) 8,77 ± 1,99 9,34 ± 2,10 9,70 ± 2,30 Neutrófilos (x109/L) (a) 5,83 ± 1,73 6,55 ± 1,96 6,58 ± 1,83 Linfocitos (x10 /L) 2,11±0,58 2,09±0,53 2,25±0,68 Monocitos (x10 /L) (a) 0,57±0,19 0,60±0,19 0,66±0,24 Eosinófilos (x10 /L) 0,20±0,13 0,24±0,23 0,21±0,17 Basófilos (x10 /L) 0,05±0,02 0,06±0,03 0,06±0,03 9 9 9 ESTADÍSTICA Acopio de datos en planilla excel Funciones estadísticas utilizadas: media y un desvío estándar 9 a- p<0,01 entre primero y tercer trimestre b- p<0,001 entre primero y tercer trimestre Valores de referencia del hemograma en embarazadas, con tecnología actual CONCLUSIONES Se establecieron los valores de referencia del hemograma de embarazadas en cada trimestre de gestación. El recuento de los eritrocitos, la concentración de la hemoglobina y el hematocrito tuvieron descenso significativo entre el primer y el tercer trimestre (p < 0,01). Esto se relaciona con la mayor expansión plasmática con respecto a la de la masa globular, que se produce durante la gestación. La hemoglobina del primero al segundo trimestre disminuyó un 2,2% (p<0,01), del segundo al tercero, el 1,5% y del primero al tercero, el 3,4% (p<0,001) (7). Los índices hematimétricos volumen corpuscular medio (VCM), hemoglobina corpuscular media (HCM), concentración de hemoglobina corpuscular media (CHCM) y ADE (amplitud del tamaño celular) no tuvieron variación significativa entre los trimestres (8). El recuento de plaquetas no sufrió cambios significativos del primero al tercer trimestre de gesta. (9). Se encontró un aumento significativo en el recuento de leucocitos (p<0.001), siendo este aumento a expensas de neutrófilos (p<0.001) y monocitos (p<0.001). En los linfocitos, eosinófilos y basófilos no se encontró ninguna variación cuantitativa. Hay que tener en cuenta las diferencias encontradas en los distintos parámetros, de un trimestre al siguiente. Cualquier mínimo cambio en la hematimetría de una embarazada debe ser considerado en cuanto a magnitud y tiempo de gesta. Cualquier modificación fuera de la variación normal esperada para cada trimestre y, de acuerdo al analito, puede indicar la presencia de alguna patología (10, 11). BIBLIOGRAFÍA 1.- Zarama Marquez FA, Cruz Mejía R, Buitron García R. Perfil hematólogico durante el embarazo. Ginecol Obstet Mex, 2002; 70(3): 136-40 2.- Van den Bossche J, Devreese K, Malfait R, Van de Vyvere M ,Wauters A, Neels H. Reference values using different haematology analysers. Clin Chen Lab Med 2000; 40 (1): 69-73 3.- Vives Corrons, J.Ll, Besson I. Los valores de referencia en hematología. Sangre 1992; 38 (1): 63-65 4.- Bessman JD. Automated Blood Counts and differentials: A Practical Guide.Baltimore : John Hopkins University Press, 1986. 5.- National Committee for Clinical Laboratory Standards (NCCLS). How to define, determine, and utilize reference intervals in the clinical laboratory; Proposed Guideline Vol 12, nº 2, 1992. 6.- Per Bergsjo. Haemoglobin concentration in pregnant women. Acta Obstet Scand 1996; 75: 241-244 7.- Canalejo K, Casella A, Bucci M, Cappanera P, Jelen A, Aixala M. Anemia microcítica: ferrropenia como causa de anemia en el embarazo. B y PC 2000; 64 (2):17-24 8.- Artaza JR, Carbia CD, Ceballo MF, Diaz NB. Indice de distribución de glóbulos rojos (RDW): su aplicación en la caracterización de anemias microcíticas e hipocrómicas. Medicina 1999;59: 17-22 9.- Tygart SG, McRoyan DK, Spinnato JA, McRoyan CJ, Kitay DZ. Longitudinal study of platelet indices during normal pregnancy. Am J Obstet Gynecol 1986 Apr;154(4):883-7. 10.- Marti-Carvajal A, Pena –Marti G, Comunian G, Muñoz S. Prevalence of anemia during pregnancy: results of Valencia (Venezuela) anemia during pregnancy study. Arch Latinoam Nutr. 2002 Mar; 52(1): 5-11 11.- Van Hove L, Schisano T, Brace L. Anemia Diagnosis, Classification, and Monitoring-Laboratory Hematology 2000; 6: 93-108. REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 55 | 59 Pág | 55 Monitoreo de depósitos de hierro elevados mediante protoporfirina eritrocitaria Silvia Haydee Langini1 Marta Mabel Lardo2 Silvana Fleischman3 Alberto Lazarowski4 María Esther Rio5 1 Doctora en Bioquímica, UBA 2 Bioquímica, Especialista en Bioquímica Clínica. Area Hematología, UBA 3 Bioquímica y Farmacéutica, UBA 4 Doctor en Bioquímica, UBA 5 Dra. en Farmacia y Bioquímica, UBA Lugar de Trabajo: Cátedra de Nutrición y Sección Hematología. Departamento de Bioquímica Clínica. Facultad de Farmacia y Bioquímica. UBA. Buenos aires, Argentina Enviar Correspondencia a: Dra Silvia H LANGINI Cátedra de Nutrición. Facultad de Farmacia y Bioquímica (UBA) Junín 956 2do piso (1113) Capital Federal Tel.: 4964-8242 Fax: 4964-8243 [email protected] Revista ByPC. Incorporada al Latindex. ISSN 1515-6761 Código Bibliográfico: RByPC Trabajo Recibido: 11-04-07 Aceptado: 12-07-07 RESUMEN Ferritina sérica (FS)>200 μg/L se relaciona con mayor riesgo cardiovascular. Como alternativa, se estudió la protoporfirina eritrocitaria (PE) para estimar FS. En sangre entera de dadores voluntarios: varones (V) (n=183; 19-64 años) y mujeres (M) (n=75; 18-59 años) se determinó: Hematocrito (Hto) (contador hematológico MAXM de Coulter) y PE (Piomelli) -μg PE/dL eritrocitos -(PE/Hto); en suero, Proteína C Reactiva (PCR) (PCR-látex, Wiener lab) y FS (IMMULITE Ferritin, DPC). Para el análisis de la relación PE vs FS se incorporaron datos de FS y PE de niños sanos (N) (n=51,<3 años) de estudios previos. PE vs FS forman una curva del tipo Log-Log: Log PE/Hto= a. Log FS + b (p<0.0001); en la intersección de N con V y M, PE/Hto=21 se corresponde con FS= 630 μg/L, valor correspondiente a sobrecarga de Hierro (Fe). Se propone el uso de PE, para el monitoreo de elevados depósitos de Fe en estudios epidemiológicos. SUMMARY Iron (Fe) overload is proposed as a factor increasing cardiovascular risk. Erythrocyte protoporphyrin (EP) was studied as an alternative test for serum ferritin (SF), usually used to assess Fe stores. Blood samples from volunteer donors: men (V) (n=183; 19-64 y) and women (M) (n=75; 18-59 y) were analyzed for Hematocrit (electronic counter MAXM de Coulter ) and EP by Piomelli’s (μg EP/dL erythrocytes); in serum, C-Reactive Protein (PCR) (PCR-latex, Wiener lab) and SF (IMMULITE Ferritin, DPC) were determined. EP and SF data from healthy children (N) (n=51,<3 y) were taken from previous studies. EP correlated inversely to SF in a Log-Log type curve: Log EP = a. LogSF + b (p<0.0001). The lines intersect at EP = 21 μg /dL erythrocytes and SF = 630 μg/L compatible with Fe overload. These results suggest EP as a screening non invasive test for the assessment of high Fe stores in adult population. Monitoreo de depósitos de hierro elevados mediante protoporfirina eritrocitaria INTRODUCCION Los problemas relacionados con el estado nutricional respecto del Hierro (Fe) no se limitan a la deficiencia sino que incluyen también la sobrecarga que, si bien es mucho menos prevalente, ha sido asociada con aumento del riesgo de padecer enfermedades crónicas (1). Esta sobrecarga ha emergido desde la década de 1980 en los países industrializados como el desorden clínico más importante en el balance de Fe en poblaciones adultas (2); al respecto se ha propuesto el exceso de Fe como una hipótesis alternativa de riesgo cardiovascular (3,4). En nuestro país, los hábitos alimentarios incluyen elevado consumo poblacional de carne vacuna (5), la que además de aportar Fe de elevada biodisponibilidad, es un alimento que tiene la capacidad de incrementar la biodisponibilidad del Fe poco disponible presente en otros alimentos (6). Si bien la mayoría de los cuadros por sobrecarga se deben a procesos que llevan a su acumulación patológica en el organismo (7,8), el consumo de cantidades excesivas de Fe de alta biodisponibilidad podría ser también considerada entre las posibles causas nutricionales que ponen a los grupos vulnerables, particularmente el hombre adulto y la mujer postmenopáusica, en situación de riesgo de sufrir sobrecarga de Fe, especialmente cuando se asocia con el consumo prolongado de alimentos fortificados o suplementos dietarios. Al respecto, un estudio longitudinal realizado por Snowdon y col en California, entre 1960 y 1980 sobre el hábito de consumir carne, indicó que el riesgo de padecer enfermedad coronaria isquémica era 3 veces mayor en hombres adultos jóvenes que consumían carne diariamente respecto de los que no lo hacían (9). En este punto es conveniente recordar que la carne es el alimento básico en la dieta de los argentinos. Para detectar la deficiencia de Fe se dispone de una profusa metodología que abarca varios tipos de indicadores capaces de medir diferentes etapas del proceso (10); la situación es más compleja cuando se trata de medir el exceso. Así, la ferritina sérica es considerada tradicionalmente el indicador de elección para evaluar la magnitud de los depósitos de Fe corporal en individuos sanos; sin embargo, en presencia de afecciones inflamatorias crónicas o procesos patológicos que ocasionan destrucción tisular, la ferritina sérica aumenta considerablemente sus niveles, por ser un reactivo de la fase aguda, y puede alcanzar valores falsamente elevados compatibles con una posible sobrecarga (10). Por otra parte, el elevado costo de los equipos comerciales basados en técnicas de enzimoinmunoensayo, así como la forma en que su determinación puede ser afectada por deficiencias en el proceso de conservación de las muestras, son factores que contribuyen a limitar su aplicación, particularmente cuando se trata de estudios epidemiológicos. Es por estos motivos que resulta de interés la estandari- zación de un indicador alternativo de la ferritina sérica que permita monitorear el exceso de Fe con un costo relativamente bajo, en especial cuando se realizan estudios a nivel poblacional. En este sentido, la determinación de protoporfirina eritrocitaria en sangre entera reúne características que alientan su estudio con este fin ya que la determinación puede ser realizada en un laboratorio de mediana complejidad utilizando una muestra de fácil acceso y conservación. MÉTODOS Se estudió la relación entre protoporfirina eritrocitaria (PE) y ferritina sérica (FS) en muestras de sangre entera obtenidas de adultos (>18 años) clínicamente sanos: varones (V) (n= 183) y mujeres (M) (n=75), 9 de ellas postmenopausicas, que concurrieron voluntariamente a donar sangre al Servicio de Hemoterapia del Hospital de Clínicas José de San Martín (UBA) entre abril y agosto de 2005. En sangre entera se determinó PE por el método fluorométrico de Piomelli (11) y hematocrito (Hto) (contador hematológico MAXM de Coulter) a los fines de expresar los resultados como μg PE/dL eritrocitos (PE/Hto). En los sueros se determinó Proteína C Reactiva (PCR) (PCR-látex, Wiener lab) para descartar FS falsamente elevada (10), y en los que resultaron PCR negativos, se midió FS por ensayo inmunométrico (IMMULITE Ferritin, DPC). Un valor de PE<70 μg/dL eritrocitos surgido de las encuestas nutricionales realizadas en población de USA (NHANES II) (12) ha sido considerado compatible con adecuación respecto del Fe en el adulto por el International Anaemia Consultative Group (INACG) (13). Los valores de FS < 15 μg/L son indicativos de bajos depósitos de Fe mientras que aquellos >200 μg/L para las mujeres en edad fértil y >400 μg/L para los varones se consideraron como posibles indicadores de depósitos elevados de Fe en adultos (14). Para estandarizar un indicador bioquímico de estado nutricional se necesita disponer de un amplio espectro de datos, entre los muy bajos y los muy altos del indicador usado como referencia (en este caso la FS) tal que la distribución de los valores correspondientes a la relación Y (en este caso PE) vs FS permita identificar la función matemática a la que dicha relación se ajusta; sin embargo, en este caso particular es difícil encontrar adultos sanos con valores de FS < 15 μg/L. En los niños, en cambio, durante la etapa de crecimiento, tales valores pueden considerarse normales. Por tal motivo, con el fin de completar la zona de FS bajas, se incluyeron para el análisis de la relación entre ambos indicadores datos de FS y PE de niños (N) (n=51) antropometricamente normales, sin patologías diagnosticadas que habían participado de estudios previos 1 (15). De esta Piazza N, Dupraz H, Zago LB, Rio ME. Efecto protector de la carne sobre los depósitos de hierro. Presentado al 33 Congreso Argentino de Pediatría. Sociedad Argentina de Pediatría. Mar del Plata, octubre 1-4, 2003. 1 Pág | 57 REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 55 | 59 manera el total de datos de PE y FS incluidos en el análisis fueron 309. Las características de los grupos estudiados se indican en la Tabla 1. Figura 1. Relación entre protoporfirina eritrocitaria y ferritina sérica 180 Tabla 1. Características de los grupos estudiados 160 Niños Varones Mujeres Grupo nº Edad (años)* Niños 51 < 3 (6 a 35 meses) Varones 183 34 ± 10 (19-64) Mujeres# 75 35 ± 11 (18-59) ug PE/dL eritrocitos 140 120 100 80 60 40 20 * promedio ± desvío estándard y rangos entre paréntesis # nueve mujeres con edad > 50 años RESULTADOS Los valores promedio ± desvío estandard y rangos de PE/ Hto (μg PE/dL eritrocitos) y ferritina sérica (μg/L) correspondientes a las muestras de sangre de los individuos estudiados se presentan en la Tabla 2. Tabla 2. Valores promedio, desvío estándar y rangos (entre paréntesis) de protoporfirina eritrocitaria y ferritina sérica en los grupos estudiados Grupo μg PE/ dL eritrocitos Ferritina sérica (μg/L) Niños 61.4 ± 33.8 a (15-170) 54 ± 58 c (1-270) Varones 23.7 ± 8.7 b (9.4 -84) 192 ± 131 d (26.4 -706) Mujeres 27.3 ± 8.0 b (11.4 -49.6) 87 ± 117 c (4.6 -828) a,b) p< 0.001 c,d) p<0.001 La relación entre PE/Hto y FS se muestra en la (Fig. 1). Al graficar los datos individuales de PE/Hto vs FS de los grupos N, V y M se obtuvo una relación inversa no lineal, cuyos puntos se agruparon en una curva del tipo hipérbola X . Y = K. En un segundo nivel de análisis se verificó que los datos se ajustaban a una curva del tipo Log-Log descomponible en rectas cuyas ecuaciones y valores estadísticos fueron: en el grupo N, PE/Hto vs FS, y = -0.2845x +2.1308; r=0.769; p<0.0001, en los grupos V y M, y = -0.0505x+1.4732; r =0.141, p=0.0233. La pendiente cercana a 0 obtenida en los adultos fue el reflejo de una PE constante en el rango de valores de FS entre 4.6-828 μg/L (Fig. 2). 0 100 200 300 400 500 600 700 800 900 FS (ug/L) Figura 2. Relación Log-Log entre protoporfirina eritrocitaria y ferritina sérica Niños Varones & Mujeres 2,5 Log PE/Hto (ug/dL eritrocitos) Para analizar los resultados, las medias se compararon utilizando el test estadístico ANOVA de un criterio considerándose significativo un p<0.05 y como test a posteriori el test de Tukey-Kramer (GraphPad InStat Version 3.01, 1998. GraphPad Software Inc.); para el análisis de las correlaciones entre ambos indicadores se usó el programa Microsoft Excel 2000. 0 Niños y = -0,2845x + 2,1308 r=0,769; p<0,0001 2 1,5 1 Varones & mujeres y = -0,0505x + 1,4732 r=0,141; p=0,0233 0,5 0 0 0,5 1 1,5 2 2,5 3 3,5 Log FS (ug/L) DISCUSIÓN En la dieta argentina, la carne es el principal aportador de Fe. De acuerdo con las Hojas de Balance de FAO, la disponibilidad de carne vacuna para el consumo en Argentina en el año 2003 era 54.7 Kg/habitante/año (5). Esto representa un consumo /día /habitante de ~150 g carne vacuna y ~3.0 mg Fe, del cual un elevado porcentaje es Fe hemínico de alta biodisponibilidad, que ejerce además un efecto facilitador sobre la absorción del Fe no hemínico presente en otros alimentos (6). Esta característica de la dieta argentina se ha visto reflejada en diversos estudios de nuestro grupo de trabajo. Así, en 1993 en hombres adultos sanos del área metropolitana de Buenos Aires, se encontró que el 15 % presentaba valores de FS> 300 μg/L (16); en otro iniciado en 2004, se observó una clara tendencia de la PE a valores más bajos que los ya bajos que se habían observado en 1993 (17). En un grupo de embarazadas clínicamente sanas de bajo nivel socioeconómico asistidas en el Hospital Paroissien (La Matanza, Pcia de Buenos Aires, 1994-1996) se constató un consumo promedio de carne de 152 g/día, que sugiere una biodisponibilidad del Fe superior al 15% y explica que los valores de PE fueran compatibles con adecuada eritropoyesis Monitoreo de depósitos de hierro elevados mediante protoporfirina eritrocitaria en el 93.5% de ellas (18). Es importante también destacar resultados recientes de la Encuesta Nacional de Nutrición y Salud llevada a cabo en Argentina entre 2004 y 2006 (19), según la cual la concentración media de FS en mujeres de 10-49 años de edad fue de 54.6 μg/L, (valores mínimo y máximo de 0.4 y 785 μg/L) siendo el valor de la mediana 38.6 μg/L. Por lo tanto, en todos los casos mencionados se observa corrimiento hacia valores adecuados o elevados de depósitos de Fe, lo que también ya se había percibido en un trabajo previo en niños en edad escolar que consumían carne habitualmente (20). El presente estudio corrobora tal situación respecto del Fe. En 4.4% de los varones los valores de FS estuvieron entre 300-400 μg/L, y en 8.2% la FS fue > 400 μg/L, de lo cual resulta un 12.6% de varones con valores compatibles con depósitos de Fe elevados, comparable a los datos de 1993. En las mujeres, la FS estuvo entre 200-400 μg/L en 5 de ellas (7.5%) y un solo caso superó los 800 μg/L; entre las postmenopausicas ninguna presentó FS>400 μg/L. Los valores de PE/Hto fueron compatibles con adecuación respecto del Fe en todas las mujeres estudiadas y en un solo varón fue levemente superior al punto de corte. La PE es el precursor inmediato en la síntesis del grupo hemo, que normalmente está en baja concentración en el eritrocito maduro; cuando no se dispone de suficiente Fe para completar la eritropoyesis la concentración de PE aumenta e, inversamente, se refleja en valores muy bajos cuando el organismo dispone de suficiente Fe de depósito (21). La Fig. 1 muestra la relación inversa entre la disponibilidad de Fe para la síntesis de hemoglobina, evaluada por la PE, y el tamaño de los depósitos de Fe estimados a partir de los datos de FS. Tal como se esperaba se observaron valores de PE más elevados en los niños que en los adultos, siendo las diferencias altamente significativas (p<0.001) (Tabla 2). Este comportamiento era el esperado ya que es sabido que, la PE disminuye en función de la edad, desde los primeros años de vida hacia la adultez. Este hecho se verificó en individuos entre 6 meses y 74 años en la encuesta nutricional Second National Health and Nutritional Examination Survey (NHANES II) llevada a cabo en EE.UU entre los años 1976 y 1980 (12) y, llevó a postular que dicho comportamiento sería atribuible a cambios fisiológicos producidos como consecuencia del crecimiento normal (22). En este aspecto, los resultados de un trabajo de tesis en el que se estudió el comportamiento hematológico en niños desnutridos y anémicos y los efectos de la velocidad de crecimiento durante la recuperación nutricional, permitieron postular que los valores más elevados de PE observados en los niños sugieren que al acelerar la velocidad de ganancia de peso (VGP), existe una deficiencia transitoria de Fe intraeritrocitario, que eleva las concentraciones de PE determinando una relación particular entre ésta y la FS según la VGP desarrollada, tanto sean los niños desnutridos en recuperación, cuanto adecuadamente nutridos en etapa de crecimiento activo (23). Tradicionalmente, la PE ha sido estandarizada para iden- tificar la deficiencia de Fe, siendo el valor de PE/Hto <70 μg/dL eritrocitos compatible con adecuación respecto del Fe (12,13). En cambio, no se ha fijado un punto de corte inferior por debajo del cual existe posibilidad de exceso de Fe. Por ello, en este trabajo, hemos encarado el estudio de la estandarización de la PE como posible indicador de riesgo de elevados depósitos de Fe en el adulto. Para ello, se analizó la curva hipérbola de la Fig. 1 transformándola en una curva Log-Log tal como se muestra en la Fig. 2, obteniéndose así dos rectas en cuyo punto de intersección la FS es igual a 630 μg/L y la PE es igual a 21 μg PE/dL eritrocitos. Por lo tanto, la inclusión de los datos de los niños en el estudio permitió extrapolar un valor de PE que estaría reflejando depósitos elevados de Fe. En el laboratorio clínico de rutina es habitual que valores de FS> 200 μg/L para las mujeres en edad fértil y >400 μg/L en los varones, junto a porcentajes de saturación de transferrina (%ST) >50% ó >60% para mujeres y varones, respectivamente sean considerados indicativos de probable exceso de Fe (10). Si bien el % ST es un indicador útil para el rastreo de la sobrecarga de Fe, el método requiere un volumen importante de muestra y resulta laborioso porque se debe trabajar con material adecuadamente lavado y reactivos de pureza pro-análisis, para evitar la presencia de Fe contaminante que invalide los resultados (24). Contrariamente, la medición de la PE puede realizarse, mediante métodos simples de extracción que liberan la protoporfirina del Zn, con el que está quelada, mediante una extracción ácida, utilizando un pequeño volumen de sangre entera (20 μL), y efectuando la lectura por fluorometría (11). Asimismo, en la década del 70, se desarrolló el hematofluorómetro, que dispone de un sistema óptico especialmente diseñado para la medición de la fluorescencia emitida por la zinc protoporfirina (ZnPP) presente en una gota de sangre entera oxigenada. Esta técnica provee resultados inmediatos; sin embargo, los métodos por extracción presentan ciertas ventajas con respecto al hematofluorómetro, particularmente en lo referente a la estandarización y estabilidad de las muestras. No obstante, Blumberg y col. han demostrado que existe una excelente correlación lineal (r=0.99) entre los resultados de EP y ZnPP dado que � 90% de la PE se encuentra como ZnPP (25). Si bien el requisito de disponer de un fluorómetro o hematofluorómetro podría ser una desventaja en estudios clínicos de rutina, por tratarse de equipos costosos, no lo sería si se considera la aplicación de este indicador a nivel masivo, como lo son las encuestas nutricionales. Un ejemplo de esto han sido las encuestas nutricionales realizadas en la población norteamericana (NHANES II), en las cuales el número de individuos en quienes se aplicó este indicador fue más de 3 veces mayor que en los que se utilizó la medición de la FS (12). Es conocido que el organismo humano carece de mecanismos que le permitan excretar el exceso de Fe. Cuando la carga de Fe corporal sobrepasa la capacidad de su almacenamiento seguro, es decir unido a proteínas, se produce daño REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 55 | 59 tisular y alteraciones funcionales (14). Está bien establecido que el Fe libre es altamente tóxico y puede participar en la generación de radicales libres e intermediarios reactivos del oxígeno responsables de la peroxidación de lípidos de membrana, lo que conduce a injuria celular y daño severo en distintos órganos y podrían constituir un factor de riesgo para el desarrollo de enfermedades degenerativas como cáncer, cataratas, ateroesclerosis, entre otras. No obstante, los estudios observacionales no han sido suficientes como para establecer una relación causa-efecto, por lo que, por ejemplo, la relación entre estado nutricional del Fe y ateroesclerosis continua siendo un tema de debate en la literatura, sin haber aún consenso al respecto (3,4,14,26). A pesar de ello, las tendencias observadas indican que no es aconsejable pasar por alto ningún indicio sobre el aumento del almacenamiento de Fe como factor de riesgo. Sobre esta base es que proponemos el uso de la protoporfirina eritrocitaria para el monitoreo de elevados depósitos de Fe en estudios epidemiológicos, por la simplicidad de la determinación y fácil conservación de la muestra y cuyo costo es significativamente menor que el de la ferritina sérica. AGRADECIMIENTOS Los autores agradecen al Servicio de Hemoterapia del Hospital de Clínicas José de San Martín (UBA), por la provisión de las muestras. Parcialmente financiado por CONICET (PIP N° 02306) y por UBACyT 2004-2007, Urgencia Social B703. BIBLIOGRAFÍA 1.- Bothwell TH. Overview and mechanisms of iron regulation. Nutr Rev 1995; 53: 237-245. 2.- Skikne B, Cook J. Screening test for iron overload. Am J Clin Nutr 1987; 46: 840-843. 3.- Sullivan J. Iron and the sex difference in heart disease risk. Lancet 1981; 317: 1293-1294. 4.- Yuan XM, Li W. The iron hypothesis of atherosclerosis and its clinical impact. Ann Med 2003; 35: 578-591. 5.- Organización de las Naciones Unidas para la Agricultura y la Alimentación (FAO). Hoja de Balance de Alimentos, Argentina año 2003. FAOSTAT. FAO Dirección de estadística 2006. http://www.faostat.fao.org/ 6.- Hallberg L. Bioavailability of dietary iron in man. Ann Rev Nutr 1981; 1: 123-147. 7.- Halliday JW. Hemochromatosis and iron needs. Nutr Rev 1998; 56: S30-S37. 8.- Orsilles MA. Hemocromatosis hereditaria: genética y diagnóstico. Acta Bioquímica Clínica Latinoamericana 2000; XXXIV: 339-350. 9.- Snowdon DA, Phillips RL, Fraser GE. Meat consumption and fatal ischemic heart disease. Prev Med 1984; 13: 490-500. 10.- Gibson R. Assessment of iron status. En: Gibson RS, ed. Pág | 59 Principles of Nutritional Assessment. Pp. 349-76. New York – Oxford: Oxford University Press,1990. 11.- Piomelli S. A micromethod for free erythrocyte porphyrins: The FEP test. J Lab Clin Med 1973; 81: 932-940. 12.- Expert Scientific Working Group. Summary of a report on assessment of the iron nutritional status of the United States population. Am J Clin Nutr 1985; 42:1318-1330. 13.- Sauberlich H. Laboratory test for the assessment of nutritional status. I. Wolinsky, ed. Boca Raton, FL: CRC press, 1999. 14.- Yip R. Hierro. En: Bowman BA, Russell RM, eds. Conocimientos Actuales sobre Nutrición. Pp 340-59. Washington, D.C: OPS e Instituto Internacional de Ciencias de la Vida, 2003. 15.- Piazza N, Dupraz H, Zago LB, Rio ME. 2002. Datos no publicados sujetos a cláusula de confidencialidad. 16.- Langini SH, Godoy MF, Fleischman S, Rio ME, Pita Martín de Portela ML. Estado nutricional con respecto al hierro en adultos del área metropolitana de Buenos Aires. Acta Bioquímica Clínica Latinoamericana 1996; XXX: 221-230. 17.- Langini S, Fleischman S, Portela ML de, Río ME. Desarrollo de un sistema para controlar el programa nacional de fortificación con hierro. Estudio piloto en adultos de Buenos Aires. Revista FABICIB 2005; 9: 233-238. 18.- Pita Martin de Portela ML, Langini SH, Fleischman S, Lopez LB, Garcia M, Ortega Soler CR. Indicadores bioquímicos e ingesta de hierro en un grupo de gestantes del Gran Buenos Aires. Medicina 1998; 58:194-196. 19.- Encuesta Nacional de Nutrición y Salud. Documento de resultados 2006. Ministerio de Salud, Presidencia de la Nación., Argentina. 20.- Portela ML, Rodriguez P, Weisstaub A, Gaón D, Río ME. Análisis de los valores de protoporfirina eritrocitaria libre en escolares de una zona urbana de Buenos Aires. Acta Bioquímica Clínica Latinoamericana 1992; XXIV: 329-334. 21.- White A, Handler P, Smith EL, Hill RL, Lehman IR. Principios de Bioquímica. Libros McGraw - Hill de México ,S.A de C.V, 1983. 22.- Yip R, Johnson C, Dallman PR. Age -related changes in laboratory values used in the diagnosis of anemia and iron deficiency. Am J Clin Nutr 1984; 39:427-436. 23.- Morasso MC. Comportamiento hematológico en niños desnutridos y anémicos. Efectos de la velocidad de crecimiento. Tesis doctoral, 1981 Universidad Nacional de La Plata, Argentina. 24.- Lynch SR. Iron deficiency anaemia. En: Sandler MJ, Strain JS, Caballero B, eds. Encyclopedia of Human Nutrition. Vol I, Pp 81-85. Londres, Academic Press, 2000. 25.- Blumberg WE, Eisenger J, Lamola AA, Zuckerman DM. The hematofluorometer. Clin Chem 1977; 23: 270-274. 26.- Ramakrishna G, Rooke TW, Cooper LT. Iron and peripheral arterial disease: revisiting the iron hypothesis in a different light. Vasc Med 2003; 8: 203-210. ¿Que es la electroforesis capilar? ¿Que es la electroforesis capilar? Raquel Osatinsky * Bioquímica – Especialista en Química Clínica: Orientación Proteínas Lugar de Trabajo: MANLAB - Diagnóstico Bioquimico M. T. de Alvear 2263 [email protected] Enviar Correspondencia a: Av. Corrientes 2818 - Piso10 - Dto. “D” 1193 – Ciudad A. de BS.As e- mail : [email protected]. RESUMEN La electroforesis capilar (CE) es una técnica de separación basada en la diferente velocidad de migración de las distintas especies cargadas bajo la acción de un campo eléctrico. La separación se lleva a cabo en un capilar de sílice fundida de diámetro muy pequeño ( 10-200 µm). El uso de estos capilares tiene múltiples ventajas: a) los capilares son anticonvectivos en sí mismos, por lo tanto, no es necesaria la utilización de un gel soporte como medio; b) el calor generado al pasar la corriente eléctrica (efecto Joule), que daría lugar a problemas de gradientes de temperatura no uniformes ( cambios locales de viscosidad ), es fuertemente reducido, ya que la disipación de calor es muy efectiva; c) pueden aplicarse altos voltajes consiguiéndose una reducción del tiempo de análisis y altas eficiencias. Se la considera una técnica intermedia entre la clásica electroforesis de zona (ZE) y la cromatografía (HPLC) líquida.Utiliza el principio de la electroforesis capilar en solución libre. La separación por CE es muy rápida, se realiza en menos de 5 minutos, con una reproducibilidad que muestran un coeficiente de variación (CVs) < 2%, muchos investigadores consideran que su sensibilidad es 10 veces mayor que la de la cromatografía líquida de alta performance. Se conocen varios sistemas de separación por ésta tecnología. La electroforesis capilar de zona(CZE); el isoelectroenfoque capilar (CIEF); la isotacoforesis capilar ( CITF) y la cromatografía micelar electrocinética (MECC). Desde 1990 el avance en ésta campo fue vertiginoso, ya que podemos incluir diagnósticos estudiando DNA, dficiencias enzimáticas, estudios de drogas de abuso en orina , estudios de esteroides y en cuando a el labortorio clínicoa, métodos para separasión de protéinas séricas, urinarias , variantes de hemoglobina, crioglobulinas, líquido céfalo raquídeo, proteinas reactantes de la fase aguda y muchas variantes más. SUMMARY Revista ByPC. Incorporada al Latindex. ISSN 1515-6761 Código Bibliográfico: RByPC Trabajo Recibido: 20-07-07 Aceptado: 08-08-07 The process of electrophoresis is defined as the differential movement or migration of ions by attraction or repulsion in an electric field. In practical terms, a positive ( anode) and negative (cathode) electrode are placed in a solution containing ions. Then, when a voltage is applied across de electrodes, solute ions of different charge, i.e. anions (negative) and cations (positive), will move through the solution towards the electrode of opposite charge. Capillary electrophoresis, then is the technique of performing electrophoresis in buffer-filled, narrow-bore capillaries, normally from 25 to 100 µm in internal diameter. The ends of a capillary are placed in separate buffer reservoirs, each containing a electrode connected to a high-voltage power supply. Capillary electrophoresis is very suited to automation, and the arrangement of comercial CE instruments will seen familiar to those with knowledge of modern HPLC. A vitally important feature of CE is the bulk flow of liquid through the capillry. This is called the electroosmotic flow. An uncoated fused-silica capillary tube is typically used por CE. The surface of the inside of the tube has ionisable silanol groups, which are in contac with the buffer during CE. These silanol groups readily dissociate, giving the capillary wall a negative charge. Therefore, when the capillary is filled with buffer, the negatively charged capillary wall attracts positively charged ions fron the buffer solution, creating an electrical double layer and a potential difference. REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 60 | 66 Pág | 61 INTRODUCCION La electroforesis capilar (CE) es una técnica de separación basada en la diferente velocidad de migración de las distintas especies cargadas bajo la acción de un campo eléctrico. La separación se lleva a cabo en un capilar de sílice fundida de diámetro muy pequeño ( 10-200 μm). El uso de estos capilares tiene múltiples ventajas: a) los capilares son anticonvectivos en sí mismos, por lo tanto, no es necesaria la utilización de un gel soporte como medio; b) el calor generado al pasar la corriente eléctrica (efecto Joule), que daría lugar a problemas de gradientes de temperatura no uniformes ( cambios locales de viscosidad ), es fuertemente reducido, ya que la disipación de calor es muy efectiva; c) pueden aplicarse altos voltajes consiguiéndose una reducción del tiempo de análisis y altas eficiencias. (1-2-3) Se la considera una técnica intermedia entre la clásica electroforesis de zona (ZE) y la cromatografía (HPLC) líquida.(45) Utiliza el principio de la electroforesis capilar en solución libre. J. Gras en el Capítulo 1 de su libro Proteínas Plasmáticas (1983) dice: “Electroforesis libre.- Los grandes avances obtenidos en el estudio de las proteínas plasmáticas fueron conseguidos gracias a la electroforesis libre. Los elementos técnicos más fundamentales para la misma están constituídos por la célula con electrodos y los dispositivos ópticos que permiten visualizar el fenómeno. Célula y electrodos: Desde las primeras determinaciones de movilidad electroforética llevados a cabo por Nernst y por Hardy, el tubo en U utilizado fue perfeccionado incesantemente por numerosos investigadores, entre ellos por Tiselius, quien llego en 1937 a idear un tipo que ha sido desde entonces la base del utilizado en todos los aparatos de electroforesis analítica libre y mediante el cual el método adquiere todo su desarrollo actual”. Arne Tiselius, Bioquímico sueco, fue Premio Nobel de Química 1948. aisló e identificó por electroforesis proteínas de la sangre y de la leche, además obtuvo por absorción la separación de diversos aminoácidos. (6) La separación por CE es muy rápida, se realiza en menos de 5 minutos, con una reproducibilidad que muestran un coeficiente de variación (CVs) < 2%, muchos investigadores consideran que su sensibilidad es 10 veces mayor que la de la cromatografía líquida de alta performance.(7-8-9) Desde 1937, luego de la primera publicación de Tiselius, se multiplicaron las investigaciones sobre el tema, mejorando el equipo , los capilares empleados, aplicando metodos como el isoelectroenfoque sobre éste sistema y en los últimos tiempos su aplicación en bioquímica clínica. Se conocen varios sistemas de separación por ésta tecnología. La electroforesis capilar de zona(CZE); el isoelectroenfoque capilar (CIEF); la isotacoforesis capilar ( CITF) y la cromatografía micelar electrocinética (MECC). Desde 1990 el avance en ésta campo fue vertiginoso, ya que podemos incluir diagnósticos estudiando DNA, dficiencias enzimáticas, estudios de drogas de abuso en orina , estudios de esteroides y en cuando a el labortorio clínicoa, métodos para separasión Figura 1a. Tubo en U de Tiselius (J. Gras Proteinas Plasmáticas pag. 31 Ed. JMS 1983) Figura 1b. Tubo de Tiselius acoplado a portaelectrodos y baño termo regulador. (J.Gras Proteínas Plasmáticas- pag- 32. Ed. JMS 1983) de protéinas séricas, urinarias , variantes de hemoglobina, crioglobulinas, líquido céfalo raquídeo, proteinas reactantes de la fase aguda y muchas variantes más. (10-11-12) ¿Que es la electroforesis capilar? FUNDAMENTOS DESCRIPCIÓN DE UN EQUIPO La electroforesis capilar es la modalidad más utilizada por su simplicidad operacional y versatilidad. Requiere pequeños volúmenes de muestra por lo que ha despertado un gran interés en diferentes campos, tal como lo demuestra el incremento de publicaciones realizadas en el área de la bioquímica y biotecnología, industria farmaceútica, análisis clínicos y medio ambiente. (13) El capilar y los viales se llenan con una solución buffer. La muestra está formada por un conjunto de aniones y cationes que se introduce dentro del sistema ocupando una única zona. Al someterlo a la influencia de un campo eléctrico, migran hacia el electrodo correspondiente, estableciéndose un movimiento de iones que forman parte del sistema. Permite la separación de moléculas cargadas en función de su movilidad electroforética, en un buffer a un pH determinado, según el punto isoeléctrico (pI) de la molécula y de un flujo electrosmótico (EOF) más o menos importante. El EOF es el flujo del líquido en el interior del capilar originado por la carga eléctrica negativa existente en la pared interna del capilar. En el caso de los capilares de sílice fundida ésta superficie de carga es generado por la ionización de los grupos silanol. La carga de la superficie interna del capilar atrae hacia si iones positivos que forman una capa adyacente fija por adsorción ( capa de Stern ), y una capa difusa y móvil, también positiva ( capa de Gouy – Chapman ). Los iones de la capa difusa experimentan una fuerza paralela a la superficie y migran hacia el cátodo al aplicarse una diferencia de potencial entre los extremos del capilar. Éstos iones, al estar sulfatados, generan un movimiento global del flúido hacia el cátodo. Éste movimiento del flúido constituye la fuerza electrosmótica (EOF). (14-15) Requiere una fuente de alto voltaje ( +/- 8000 voltios). Uno o más capilares cuyos extremos se encuentren sumergidos, junto con dos electrodos, en dos viales que contienen una solución buffer, un detector y un sistema de adquisición de datos. Los capilares son generalmente de sílice fundida, con diametros internos de +/- 50 nm, cubiertos con poliamida para darle flexibilidad y disminuir su fragilidad. El pequeño diámetro facilita la disipación del calor generado por la resistencia eléctrica del electrolito dentro del capilar. La sílice fundida es mejor conductor de calor que cualquiera de los otros materiales usados para fabricar capilares de diámetro pequeño ( teflón, pyrex). Durante las separaciones, los extremos del capilar están colocados en dos recipientes que contienen un buffer, el mismo con el que se ha llenado la columna. Junto con otras condiciones, la composición del buffer define el método de análisis. Ambos recipientes contienen el mismo buffer y su nivel debe ser el mismo en los dos para evitar cualquier tipo de flujo debido a un desequilibrio hidrostático. Al introducir el electrolito en el capilar, pueden formarse burbujas de aire, que provocan fluctuaciones en la línea de base e interrumpir la corriente eléctrica. Debe evitarse este problema.(16) Existen distintos modelos de aparatos con sistemas muy similares, variando en la cantidad de capilares que funcionan en forma simultánea. Es un método totalmente automatizado. Se conocen dos equipos en el mercado*. que emplean el principio de electroforesis capilar en solución libre, uno tiene ocho capilares que funcionan en paralelo, permitiendo realizar ocho determinaciones simultáneas y 90 en una hora y otros trabajan con seis capilares en forma simultánea.(17) Figura 2. Diagrama que muestra la dirección del flujo electrosmótico (EOF). (Jenkins MA.-Clin Apli. of CEpag.3-Human Press Inc.1999) Figura 3. Esquema de un aparato de electroforesis capilar. (Jenkins M A. Clin Aplic of CE.- pag 2.- Human Press Inc. 1999) High Voltage Detector Capilary Buffer Recorder La separación electroforética se efectúa en un capilar de díametro inferior a 100 μ m, que contiene un buffer. El uso de capilares para la separación electroforética tiene múltiples ventajas. Los capilares son anticonvectivos, razón por la que no es necesario el empleo de un gel como soporte. El calor generado por el paso de la corriente eléctrica, que daría lugar a problemas de temperatura, está muy reducido porque la disipación del calor es muy efectiva. INYECCIÓN DE LA MUESTRA Existen diferentes sistemas de inyección en la electroforesis capilar, se basan en aplicar un gradiente de voltaje ( electrocinética) o en un gradiente de presión ( hidrodinámica o hidrostática), entre los dos extremos del capilar. En todos Pág | 63 REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 60 | 66 los casos, para introducir la muestra dentro del capilar, se cambia el vial de entrada por un vial con muestra y se aplica la inyección de la misma durante 10-30 segundos. Introduciendose en el capilar un volumen de muestra del orden de los nanolitros. En la inyección hidrodinámica , la introducción de la muestra en el capilar se efectúa mediante una diferencia de presión entre los dos extremos de la columna ( es el método convencional de inyección). Existen diferentes formas de producir una presión superior al extremo del capilar en contacto con la disolución de la muestra: por flujo gravitatorio de la disolución electrolítica mediante la elevación del vial con la muestra y la aplicación de presión sobre la misma, luego por succión de la muestra aplicando vacío al vial de salida. Éste tipo de inyección no depende de la movilidad electroforética , ni de la composición de la muestra. El volumen de muestra inyectada se define por las dimensiones del capilar, la viscosidad del electrolito, la presión aplicada y el tiempo. Se debe controlar con presición la temperatura capilar porque puede verse afectada la viscosidad y variar la cantidad de muestra que se inyecta. En gran parte de los instrumentos de CE la diferencia de presión se utiliza, además de para introducir la muestra, para renovar el buffer del capilar, ya que normalmente debe cambiarse luego de un número determinado de análisis para conseguir repetibilidad en el tiempo de migración en las muestras a determinar. La inyección electrocinética se basa en que el voltaje puede causar movimientos electroforéticos y electrosmóticos. Se introduce un extremo del capilar en el vial que contiene la muestra y el otro extremo en el vial que contiene el buffer, se aplica un voltaje elevado durante un breve período de tiempo. Los compuestos se introducen en el caplilar por la acción conjunta de la migración y del efecto de la movilidad electrosmótica. Si el voltaje se aplica durante un un período de tiempo corto, la muestra se introducirá por el movimiento electroforético. Diversos investigadores demostraron que por una serie de fenómenos que produce la inyección electrocinética es menos reproducible que la inyección hidrodinámica. Los límites de detección de los compuestos estudiados mediante la inyección electrocinética son aproximadamente cinco veces más bajos que los conseguidos con la hidrodinámica. SISTEMA DE DETECCIÓN La detección se realiza directamente sobre el capilar, es decir que el mismo capilar actúa como celda de detección. El tipo de detector escogido dependerá de los analitos a determinar y, siempre que se pueda, se escogerá aquel que proporcione una sensibilidad elevada para todos los compuestos. El detector ultravioleta – visible es uno de los más empleados en la CE. La longitud de onda escogida es de 214 nm, o de 200 nm de acuerdo al equipo con el que se trabaja. Éste sistema a pesar de la estabilidad y facilidad de operación, presenta algunos incovenientes ya que opera a una única longidtud de onda. La introducción del detector de diodos en fila ha resuelto este problema permitiendo adquirir al mismo tiempo el espectro de cada uno de los analitos en un mismo análisis. Una alternativa a la detección UV-Visible es la detección de fluorescencia, que es ampliamente utilizada en bioquímica clínica. Otro tipo de detección utilizada por la CE es la electroquímica, los más utilizados han sido los conductimétricos y los amperométricos. PRINCIPIOS DE SEPARACIÓN Flujo electrosmótico (EOF).- En la CE, se desplazan por el capilar bajo la influencia de la corriente eléctrica, las moléculas de soluto y la solución buffer. Éste es un fenómeno conocido como flujo electrosmótico. El polo positivo o ánodo se encuentra en el extremo del capilar donde se realiza la inyección de la muestra. El EOF va del polo positivo al negativo, de manera que el buffer se mueve del vial que lo contiene ( el de entrada) hacia el detector, en el vial de salida. La pared del capilar está constituida por un entramado de grupos silanol , por lo tanto al trabajar con bufferes básicos se encuentra cargada negativamente con grupos sialonatos, y hace que las otras cargas positivas libres se situen cerca de las cargas negativas de la pared formando una doble capa. Al aplicar una diferencia de potencial las cargas libres (positivas y negativas) se moveran hacia los polos contrarios. Las cargas negativas de la pared no pueden moverse dando como resultado un flujo de cargas positivas hacia el polo negativo. FLUJO ELECTROFORÉTICO Bajo la acción de un campo electrico, los solutos ionicos se desplazan a través del capilar : a) por el flujo electrosmotico que arrastra todo el seno de la disolución y b) la carga propia de un soluto hace que éste muestre una movilidad por si mismo, a lo que se denomina flujo electroforético. La movilidad electroforética de un soluto es función de su carga, del radio de la molécula y de la viscosidad del medio. Cuando se introduce una muestra en el capilar y se aplica una diferencia de potencial, cada uno de sus componentes tendrá una movilidad que dependerá de las velocidades electrosmóticas y electroforéticas. (18-19) Una vez inyectada la muestra por aspiración en el o los capilares, se procede a la separación de la misma, aplicando una diferencia de potencial de varios miles de voltios (≈ 8000), en los extremos de cada capilar. La detección directa de las proteínas se lleva a cabo a 200 nm en el extremo catódico del capilar. Las proteínas separadas ( en capilares de sílice fundido) son detectadas directamente en una burbuja existente en el capilar por espectrofotometría de absorbancia. Emplean- ¿Que es la electroforesis capilar? do buffer a pH alcalino, el orden de migración de las proteínas es el siguiente: Gamma globulinas 2 globulinas 1 globulinas 2 globulinas 1 globulinas Albúmina Figura 5a. Aumento de las fracciones α1 y α2 Gamma globulina con patente oligoclonal. *** Se visualiza la curva que produce el contenido proteico de Figura 4a. Curva de CE normal * Figura 5b. Gamma globulina con patente *** oligoclonal. Alb. �1 �2 �1 �2 � Figura 4b. Curva de CE normal * Observar en fig. a y en la b los distintos picos correspondientes a las múltiples bandas homogéneas. Alb. �1 �2 � 1 �2 � Éste método tiene una sensibilidad mayor en la definición de las fracciones que presenta la curva, relacionada con lasconcentraciones de las proteínas tanto absolutas como las relativas. la muestra. La detección es directa por cuanto “no existe el paso correspondiente a la coloración y decoloración de las corridas”. La figura similar a una electroforesis en agarosa que se observa en un costado de la pantalla donde se obtienen los datos es virtual. (20-21) Como ya se mencionó los equipos de CE para su aplicación en el laboratorio bioquímico clínico separan las proteínas séricas en forma similar a las que se observan en los equipos que emplean agarosa como soporte. Debe quedar claro que el proteinograma electroforético está expresado directamente en la curva que se observa en la pantalla del monitor. La lectura de los valores se realizan a 200 nm y expresan el contenido directo de cada fracción visualizada. Tiene una sensibilidad muy grande que se ve en los detalles de la curva, sobre todo en la zona de las alfa 1, alfa 2 y gamas. Con éste método se puede observar con más frecuencia la presencia de bisalbuminemias. Respecto de las patentes oligo y monoclonales en la zonade las gamas, asi como la presencia de pequeñas bandas homogéneas son visibles conclaridad. Probablemente las pequeñas bandas homogéneas en la zona de las betas deban observarse con más detenimiento. Es un método completamente distinto a las electroforesis de zona tanto en agarosa como en acetato de celulosa Pág | 65 REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 60 | 66 y como toda innovación debe estudiarse, validarse, hacer trabajos comparativos para asegurar un resultado correcto. La gammapatías monoclonales se diagnostican por la curva y la tipificación de las mismas se realiza por inmunofijación (22) . En el sistema de CE la IF se efectúa por el método de inmunosubstracción. (23-24-25-26) Las electroforesis de hemoglobina realmente son inmejorables, ya que permite la separación y valoración precisa de las diferentes fracciones de la hemoglobina A, A2 y HbF, asi como de las difrentes hemoglobinopatías estructurales. En el caso de la presencia de una probable HbLepore la separa de una probable HbS, tambien cuando se presentan probables Hb C y/o E se puede cuantificar la fracción de la HbA2 que ninguno de los métodos de electroforesis de zona sobre agarosa lo soluciona. (27) Figura 6a. Gamma globulina con un pico de banda **** homogénea sobre patente policlonal Figura 6b. Gamma globulina con un pico correspondiente a **** un componente monoclonal, y dos pequeños picos agregados *,**,***,****, Estudios realizados con el Capillarys 2 -Sebia CONCLUSIÓN Considero que la aplicación de la electroforesis capilar en el laboratorio clínico permite contar con una herramienta técnica de gran versatilidad. Es completamente automático, tiene una gran sensibilidad, acorta el tiempo por cada análisis que realiza disponiendo el profesional a cargo emplearlo para analizar con tranquilidad los resultados y validarlos y/o realizar otras tareas dentro del laboratorio. * Los equipos mencionados son :Capillarys 2 de Sebia y Paragon 2000 CZE de Beckman. BIBLIOGRAFÍA 1.- Meunier C .-La detection et la caracterisation des composes monoclonaux difficiles dans le serum: techniques sur gel au electrophorese capillaire.-Ann Biol Clin 1999 ; 57 : 605-10. 2.- Blessum c, Joppson O, Aguzzi F, Bernon H, Bienvenu J.L´electrophorese capillaire : principe et applications au laboratoire de biologie clinique.- Ann Biol Clin 1999 ;57 :643-57. 3.- Keren D.- Capillary zone electrophoresis in the evaluation of serum protein abnormalities. Am J Clin Pathol.- 1998; 110; 248-252. 4.- Compton S W, Brownlee R G.- Capillary electrophoresis.Biotechniques.- 1988; 6:432. 5.- Deyl z, Rochlicek V, Struzinsky R.- Some rules applicable to capillary zone electrophoresis of peptides and proteins. J Liquid Chromatograf. 1989; 12: 2515. 6.- Tiselius A.- New apparatus for electrophoretic analysis of colloidal mixtures.- Trans Faraday Soc 1937; 33:524-536 7.- Herten S.- Zone broadening in electrophoresis whith special reference to high – permormance electrophoresis in capillaries: an interplay between theory and practice. Electrophoresis.-1990; 11: 665-690. 8.- Gordon M J, Huang X, Pentoney S LJr, and Zare RN.- Capillary electrophoresis. Science 1988; 242:224-228 9.- Deyl Z and Struzinsky R.- Review. Capillary zone electrophoresis: its applicability and potential in biochemical analysis. J Chromatograf. 1991; 569: 63-122. 10.- Mazzeo J R and Krull I S.- Coated capillaries and additives for the separations of proteins by capillary zone electrophoresis and capillary isoelectric focusing. Biotechniques 1991; 10: 638-645. 11.- Jenkins M A and Guerier M D.- Cappillary electrophoresis as a clinical tool. J Chromatograf. B. 1996; 682: 23-24. 12.- Lehmann R, Liebick H M and Voelter W.- Application of capillary electrophoresis in clinical chemistry: developments from prelimilary trials to routine analysis. J Cap Electrophor. 1996; 3: 89-110.13.- Ruiz Marrondo S.- Desarrollo de métodos de electrofore- ¿Que es la electroforesis capilar? sis capilar en fase micelar. Aplicación al análisis de herbicidas y sus productos de degradación.- Universidad Politécnica de Cataluña.- Tesis defendida : 2001; 2 de Octubre. 14.- Schwartz H E, Ulfeder K J, Chen F T A and Pentoeny S R. Utility of laser- induced fluorescence detection in applications of capillary electrophoresis. J Cap Electroph. 1994; 3: 89-110. 15.- Zhu M, Rodriguez R, Hansen P and Wehr T. Capillary electrophoresis under alkaline conditions.- J Chromatogrf. 1990; 516: 123- 131. 16.- Robert F, Bouilloux J P, Denoroy L..- Revues. L´electrophorese capillaire : principe et applications.- Ann Biol Clin 1991 ; 49 : 137-148 17.- Lissoir B, Wallemacq P, Maisin De.- Electrophorese des proteins seriques : compairison de la technique en capillaire du zon Capillarys (Sebia) et la electrophorese au gel d´agarose Hydrasis ( Sebia ). An Biol Clin.- 2003 ; 61 : 557-562. 18.- Gordon M J, Lee K J, Arias A A and Zare R N.- Protocol for resolving protein mixtures in capillary zone electrophoresis.-Ann Chem 1991; 63: 69-72 19.- Kun J W, Park J H, Park J W, Doh H J, Heo G S and Lee K J.- Quantitative analysis of serum proteins separated by capillary electrophoresis- Clin Chem 1993; 39: 689-692. 20.- Jonsson M and Carlson J.- Computer-sopported interpretation of protein profiles after capillary electrophoresis.Clin Chem 2002; 48: 1084-1093. 21.- Gay Bellier C, Bengunfa D, Honze P, Diddier Le Carrier, Bentakehal M, Bousquet B, Gourmel B and Le Bricon T.- Automated multicapillary electrophoresis for analysis of human serum proteins.- Clin Chem 2003; 49: 1909-191522.- Huskens Y, De Winter J, Pekelharing M and Ponzee G.Detection and identification of monoclonal gammopathies by capillary electrophoresis. Clin Chem .-1998; 44: 11841190. 23.- Narvaiza R C, Casado B, Fernandez M A, Lobeera I, Borque L A.- Inmunosustracción en fase homogénea : un nuevo procedimiento para la tipificación de componentes monoclonales en suero.- Revista de Diagnóstico Biológico 2006; 2: 88-94. 24.- Luraschi P, Infusmo I, Melotti F , Franzeni C.- Analytical variation in the measurement of serum monoclonal component by capillary electrophpresis. Clin Chim Acta 2004; 349 (1-2): 151-6 25.- Roudiere l, Boularan A M, Boncudet A, Vallat C, Cristol J P, Dupuy A M.- Evaluation of a capillary zone electrophoresis system versus a conventional agarose gel system for routine serum protein separation and monoclonal component typing. Clin Lab 2006; 52(1-2) 19-27. 26.- Terreni A, Caldieri A, Ognbene A, Salvadori B, Messeri G.Serum proteins capillary zone electrophoresis: A comparison of two systems.-Lab Med 2006; 37(4): 233-236. 27.- Clinical Applications of Capillary Electrophoresis.- Edited by Stephen M Palfrey.- Human Press Inc. 1999. Pág | 67 REVISTA BIOQUIMICA Y PATOLOGIA CLINICA VOL 71 Nº 2 2007 Trabajo: págs 67 | 75 IV Congreso y 8º Encuentro Bioquímico 2007 “Actualización y Avances en Bioquímica Clínica, Microbiología y Nutrición” XXX REUNION DEL CAPÍTULO ARGENTINO DE LA SOCIEDAD LATINOAMERICANA DE NUTRICIÓN (CASLAN) IV Jornadas de Actualización “Alimentos, Nutrición y Salud” Rosario 9,10 y 11 de agosto de 2007 COMISIÓN ORGANIZADORA SECRETARÍA CIENTÍFICA Presidente Dr. Sergio Ghersevich Vice-Presidente Dra. Laura Ramos Secretaria Dra. Sandra Arriaga Pro-secretaria Dra. Claudia Echenique Tesorero Dr. Carlos Cotorruelo Pro-tesorero Dra. Alicia Luque Dra. Marisa Biasoli Dra. Claudia Biondi Dra. Irma Bragós Dra. Nélida Noguera Dra. Gilda Revelant (CASLAN) Bioq. Darío Marinozzi (CASLAN) COMISIÓN HONORARIA SECRETARÍA DE DIFUSIÓN Dr. Miguel A. Capiello Dra. Claudia Balagué Dr. Héctor Campanini Dra. Adriana Limansky Bioq. Susana Lioi Dr. Fabián Pelusa Dra. Susana Pérez Bioq. Silvana Ramadán Bioq. Miguel Taborda Dra. María Catalina Olguín (CASLAN) Dra. Marta Posadas (CASLAN) Dr. Manuel Forgoso (CASLAN) Lic. Verónica Labourdette (CASLAN) COMISIÓN CIENTÍFICA Dra. Clara López Dra. Hortensia Magaró Dra. Angela Milani Dra. Lida Morisoli Dra. Amelia Racca Dra. Irene Rosillo Dra. Emma Sutich Dra. Juana Valverde Dr. Enrique Agulló (CASLAN) Dra. Marcela Martinelli (CASLAN) Dra. Patricia Ronayne (CASLAN) Dra. María Elena Sambucetti (CASLAN) Dra. Nora Slobodianik (CASLAN) Dra. Angela Zuleta (CASLAN) Revista ByPC. Incorporada al Latindex. ISSN 1515-6761 Código Bibliográfico: RByPC Resúmenes recibidos: 20-07-07 VOCALES Dra. Isabel Bogado Bioq. Silvia García Borrás Bioq. Lucía Bulacio Dra. Stella Daniele Bioq. Jorgelina Pérez Bioq. Patricia Ponce de León Bioq. Inés Toresani Dra. María Elena Tosello Bioq. Marisa Zingale ASOCIACIÓN BIOQUIMICA ARGENTINA Programa De Educación Continua 2do. Semestre 2007 ACTUALIZACIÓN BIOQUIMICA DE LA PATOLOGÍA Curso anual 2007 por módulos y talleres de aplicación clínica Organizado por: Asociación Bioquímica Argentina Universidad Argentina J.F.Kennedy (por convenio) MÓDULO SOBRE BIOMARCADORES ONCOLÓGICOS: del 25 de Octubre al 15 de Noviembre FECHA : 19 de Abril – 29 de Noviembre DÍA Y HORARIO: Jueves de 18:00 a 21:30 Hs. CARGA HORARIA: 325 Hs. CON EVALUACIÓN FINAL AUDITORIO: U K. Colegio Sarmiento. Sarmiento 4562 Cap. Fed. Coordinadores: Dr. Sergio Nosetto –Director Asociado del Laboratorio Bioanalítica Dra. Angela Solano – Laboratorio HRDC – Dto. de Bioquímica, Facultad de Medicina UBA.-Profesional Principal CONICETDra. Halina Grosman. – Dto. de Análisis Clínicos F.F.y B, Sección marcadores Tumorales-. DIRECTORES : Dra. Raquel Osatinsky, Dra. Silvia B. González, Dr. Emilio S. Labal COORDINADORES : Dr. Néstor Litwin – Dra. Sandra Rozental MÓDULOS: • Patología Respiratoria • Factores de riesgo coronario • Patología digestiva • Genética • Marcadores oncológicos Se pueden realizar por separado los módulos de éste curso. Quedan dos para finalizar, los interesados deben consultar en la Secretaría de la ABA, costos de los mismos. MÓDULO SOBRE GENÉTICA: del 30 de Agosto al 18 de Octubre COORDINADORA: Dra. Sandra Rozental (Jefe a Cargo del Dto. de Diagnóstico Genético, Centro Nacional de Genética Médica.-ANLIS.-Dr. Carlos G.Malbran) DOCENTES: Dres. Liliana Alba, Noemí Buzzalino, Roberto Coco, Lilian Furforo, Marta Gallego, Maria Ester Móllica Temas a desarrollar: • Defectos congénitos; concepto, etiología, impacto y detección. • Estructura del material genético. Cariotipo. Mutaciones • Anomalías cromosómicas., Técnicas de identificación cromosómica. Citogenética molecular. • Técnicas de biología molecular para el diagnóstico de enfermedades genéticas. • Diagnóstico prenatal de enfermedades genéticas • Genética y cáncer. • Asesoramiento genético y aspectos bioéticos. • Taller con problemas para la integración del módulo. Temas a desarrollar: • Biología del cáncer. Aspectos biológicos de los procesos oncológicos• Marcadores tumorales: Clasificación – Conceptos básicos. Aspectos dinámicos – Valoración en diferentes etapas clínicas. • Los diversos tipos de tumores y marcadores asociados. • Biología molecular oncológica. • Cáncer hereditario. • Utilización clínica de los marcadores. • Garantía de calidad en oncología. • Medicina basada en la evidencia. Guías de consenso Internacionales. Evaluación del curso: Los alumnos realizaran una presentación oral en Power Point eligiendo un tema desarrollado en los módulos. La modalidad será por grupos con la participación de todos los integrantes. 22-11-2007 Presentación por los alumnos 29-11-2007 Presentación por los alumnos ARANCELES Socios ABA y Egresados de la UK: $ 90.- por mes No socios ABA: $ 180.- por mes Nota: Los alumnos que opten por cursar módulos individuales acreditaran 50 Hs. por módulo Secretaría de la Asociación Bioquímica Argentina: Venezuela 1823 Piso 3 1096 – Ciudad de Buenos Aires. e-mail: [email protected]. WEB: www.aba-online.org.ar TELEFAX (011)4384-7415-TEL (011) 4381-2907. Horario: Lunes a Viernes de 15:00 a 19:00 Hs. ASOCIACIÓN BIOQUIMICA ARGENTINA Programa De Educación Continua 2do. Semestre 2007 CURSO TALLER- GESTIÓN DE COSTOS EN EL LABORATORIO LABORATORIO SEGÚN DIRECTRICES DE ANALISIS CLINCOS PARA DE LA NORMA ISO 15189 LA TOMA DE DECISIONES Directores: Dres. Silvia Depardo, Mabel Villarreal, Gabriel Migliarino Coordinación: Dra. Silvia Morilla Duración: desde el 16 de abril a agosto de 2007 Días y horarios: lunes de 18.30 Hs a 22 Hs Carga Horaria: 350 horas Modalidad: teórico práctico con evaluación y monografía final Auditorio: Asociación Bioquímica Argentina Aranceles del curso: Socios ABA $ 90 por mes- No socios $ 180 por mes Este curso se puede realizar por módulos Independientes con evaluación final al término de los mismos Docentes: Mabel Villarreal - Silvia Depardo (AGOSTO) Carga horaria: 50 hs Arancel: Socios ABA: $100 No socios: $200 DIRECTOR: Dr. Juan Carlos Galli. Gerente de Laboratorio Centros Médicos: Dr. Stamboulian Fecha: 5 al 26 de Setiembre de 18 a 21:30 Evaluación final Carga Horaria: 52 Hs. AUDITORIO : CEI. Centros Médicos Dr. Stamboulian. Scalabrini Ortiz 676.Cap. Fed ARANCELES: Socios ABA: $ 90, No socios ABA: $ 180 Temario a desarrollar en cuatro sesiones. • • • • • • • • • • • • • Formación de auditores ISO 15189 Impacto de la formación de los mismos en la eficacia del Sistema de Gestión Auditorias internas. Norma ISO 19011 Calificación de Auditores. Gestión de un programa de auditoria: objetivos y extensión. Responsabilidades, recursos, procedimientos, registros. Seguimiento y revisión del programa Actividades de la auditoria • • • • • • • • • • • • Conceptos básicos de análisis de costos. Definiciones de términos utilizados habitualmente. Costos directos de mano de obra y de insumos. Concepto de costos fijos y variables. Importancia de cada uno. Costos indirectos y su asignación al producto final. Concepto de índices o “drivers”. Diferentes sistemas de costeo. Ventajas y desventajas. Elección de un sistema de costeo. Sistema de cuentas contables. Costos reales y estándar. Diferentes aplicaciones. Concepto de punto de equilibrio (Break-even). Análisis conceptual y matemático del punto de equilibrio. Costos fijos y variables en el análisis del punto de equilibrio. Factores que afectan la incorporación de equipamiento. Cálculo de costos por práctica en equipos automatizados. Determinación de precio de venta por estudio. La influencia de los factores del mercado. Concepto y aplicación del “costo marginal”. Relación entre costo – volumen – ganancia. Estrategias en la fijación de precio de venta. Decisión “hacer o derivar” Costos en el sector privado y en el sector público. Formas de retribución, “fee for service”, capitación, módulos, etc. Estrategias de estudio de costos global del sistema. Ejercicios de aplicación. “La ciencia pura que ha sido el pilar de la práctica de laboratorio a través de la historia, no puede seguir siendo el único sostén, hoy la ciencia económica debe ser una parte integral de la práctica de laboratorio en aquellos laboratorios que intenten sobrevivir en el entorno desafiante de negocios de hoy en día” Eleanor Travers, M.D. Washington D.C. Secretaría de la Asociación Bioquímica Argentina: Venezuela 1823 Piso 3 1096 – Ciudad de Buenos Aires. e-mail: [email protected]. WEB: www.aba-online.org.ar TELEFAX (011)4384-7415-TEL (011) 4381-2907. Horario: Lunes a Viernes de 15:00 a 19:00 Hs. ASOCIACIÓN BIOQUIMICA ARGENTINA Programa De Educación Continua 2do. Semestre 2007 HERRAMIENTAS METODOLOGICAS PARA LOS DESAFÍOS DEL BIOQUIMICO EN SU DESEMPEÑO Directoras: Dra. Marysia Szefner y Dra. María José Rial Coordinador: Dra. María Cristina Cañadas Docentes: Dra. Gloria Califano, Dra. Claudia Ferrario, Dr. Pablo Duran, Dra. Vivian Bóxer Auditorio: Asociación Bioquímica Argentina Fecha: a partir del miércoles 12 de septiembre Encuentros semanales: 12 Día y Horario: miércoles de 18,00 a 21,30 h Carga horaria: 260 h Evaluación final Cada alumno deberá desarrollar un trabajo de investigación que tenga en cuenta los aportes teóricos prácticos incorporados Temario a desarrollar: • Diseño de diferentes tipos de estudios: ventajas y limitaciones. Errores potenciales de los estudios epidemiológicos • Selección del tipo de diseño de investigación más adecuado al objetivo del estudio • Validez interna y externa de los estudios .Interpretación Extrapolación. Normalidad y anormalidad • Discriminación diagnóstica de las pruebas Variabilidad de una prueba • Selección de una prueba estadística • Niveles de evidencia Dirigido a: El curso está dirigido a los bioquímicos que desean desarrollar trabajos de investigación clínica dentro de instituciones asistenciales así como a aquellos que tienen interés en incorporar estas herramientas a su práctica profesional. Metodología El curso combina conferencias participativas, exposiciones y talleres con provisión de materiales de lectura .y ejercicios de aprendizaje basado en análisis de casos y resolución de problemas desde la perspectiva de los conocimientos apropiados. , .Se tiende al desarrollo de habilidades y actitudes concretas y para que el aprendizaje pueda transferirse a la práctica laboral; el diseño de los ejercicios individuales y grupales está basado en problemas reales Objetivo: Desarrollar las capacidades del bioquímico para: • Participar activamente en la elaboración de los planes y desarrollo de la investigación clínica • Elaborar protocolos de investigación clínica • Evaluar y validar los procedimientos de diagnóstico de laboratorio • Efectuar el análisis estadístico y utilizar programas informáticos en la investigación clínica. • Evaluar fuentes de información y utilizar correctamente la información científica Aranceles: Socios ABA: $ 90.- por mes No socios ABA: $ 180 por mes Secretaría de la Asociación Bioquímica Argentina: Venezuela 1823 Piso 3 1096 – Ciudad de Buenos Aires. e-mail: [email protected]. WEB: www.aba-online.org.ar TELEFAX (011)4384-7415-TEL (011) 4381-2907. Horario: Lunes a Viernes de 15:00 a 19:00 Hs.