Ámonio - R-Biopharm AG
Anuncio

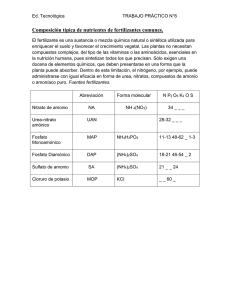
Ámonio BOEHRINGER MANNHEIM / R-BIOPHARM Enzymatic BioAnalysis/Food Analysis Método UV Para uso in vitro solamente Para la determinación de Amonio en alimentos y otros materiales y para la determinación de nitrógeno luego de digestión por Kjeldahl (ver Pto. 12.2) Cat. No. 10 112 732 035 Test-Combinado para 50 determinaciones Principio (Ref. A1) En presencia de glutamato deshidrogenasa (GIDH) y nicotinamida adenina dinucleótido reducido (NADH), el amonio reacciona con 2oxoglutarato a L-glutamato con oxidación del NADH. + 2-oxoglutarato + NADH + NH4 ⎯ GlDH → L-glutamato + NAD + H2O + La cantidad de NADH oxidado en la reacción anterior es estequiométrica con la cantidad de amonio y se mide por medio de su absorbancia a 334, 340 ó 365 nm. El ensayo combinado contiene: 1. Botella 1 con aprox. 60 ml de solución de: buffer trietanolamina, pH 8.0; 2-oxoglutarato, 150 mg. 2. Botella 2 con 50 tabletas cada una contiene NADH, aprox. 0.4 mg 3. Botella 3 con aprox. 1.2 ml de solución de glutamato deshidrogenasa, aprox 1000 U 4. Botella 4 control de amonio para control del ensayo (la medición del control no es necesaria para el cálculo de los resultados). Usar la solución control sin diluir. (Vto.: ver etiqueta) Preparación de las soluciones 1. Usar el contenido de la Botella 1 sin diluir. 2. Disolver una tableta de la Botella 2 con un ml de solución de la Botella 1 en un tubo de reacción para cada ensayo (blanco y muestras) dependiendo del número de determinaciones. Usar una pinza para tomar las tabletas de la Botella 2. Esto resulta en la mezcla de reacción 2*. 3. Usar el contenido de la Botella 3 sin diluir. Estabilidad de los reactivos La Solución 1 es estable a 2-8°C (ver etiqueta). Llevar la solución 1 a 20-25°C antes de su uso. Las tabletas 2 son estables a 2-8ºC (ver etiqueta) La mezcla de reacción 2 es estable por 3 días a 2-8ºC. Llevar la mezcla de reacción 2 a 20-25ºC antes de su uso. El contenido de la botella 3 es estable a 2-8°C (ver etiqueta). Procedimiento 1 Long. de Onda : 2 Cubeta de Vidrio : Temperatura: Volumen final: Leer contra: de 340 nm, Hg 365 nm ó Hg 334 nm 1.00 cm paso de luz 20-25°C 3.020 ml blanco de aire (sin cubeta en el paso Solución de Muestra: luz) ó contra agua. 3 0.2-8 μg amonio /ensayo (en 0.1002.000 ml volumen muestra) 1 La absorción máxima de NADH es a 340 nm. En espectrofotómetros las mediciones se toman a la máxima absorción, si se utilizan fotómetros espectrales equipados con lámpara de vapor de mercurio, las mediciones se toman a 365 nm ó 334 nm. 2 Si se desea se pueden utilizar cubetas descartables en lugar de cubetas de vidrio. 3 Ver las instrucciones para el rendimiento del ensayo. Almacenar a 2-8° C Esta traducción en español has sido realizada por R−Biopharm Latinoamérica con el objeto de ayudar a los usuarios a entender el procedimiento, pero no se actualizan regularmente por lo que no pueden remplazar las instrucciones en inglés. Las instrucciones de uso que son válidas, son aquellas incluidas en cada kit en Alemán y en Inglés, dado que están escritas e impresas por el fabricante (Roche). Refiérase siempre a las instrucciones incluidas en cada kit. Pipetear en la cubeta Blanco Muestra mezcla de reacción 2 1,000 ml 1,000 ml sol. Muestra 0,100 ml agua bidestilada 2,000 ml 1,900 ml Mezclar ***, leer las absorbancias de las soluciones (A1) luego de aprox. 5 min. a 20-25ºC. Iniciar la reacción con: solución 3 0,020 ml 0,020 ml Mezclar ***,esperar a que finalice la reacción (aprox. 20 min.) leer las absorbancias de las soluciones (A2). Si la reacción no se ha detenido luego de 20 min. continuar leyendo las absorbancias a intervalos de 2 min. hasta que el aumento de las mismas sea constante. * Para simplificar el desarrollo del ensayo es posible pipetear directamente 1.000 ml de la solución 1 en la cubeta y agregarle 1 tableta de la botella 2. Luego de la disolución de la tableta con la ayuda de la espátula, continuar como se describe en el procedimiento. La diferencia de volumen (ca. 1%, aumento del volumen por una tableta en 3.020 ml de volumen del ensayo) tiene que tomarse en cuenta en el cálculo, multiplicando el resultado por 1.01. ** Enjuagar el tip de la pipeta en la solución de muestra antes de dispensarla. *** Por ejemplo con espátula plástica ó por agitación suave luego de cerrar la cubeta con Parafilm (trademark of the American Can Company, Greenwich, Ct., USA) Si la absorbancia A2 disminuye constantemente, extrapolar la misma al tiempo de la adición de la solución 3 (GIDH). Determinar la diferencia de absorbancias (A1-A2) para ambos, el blanco y las muestras. Sustraer la diferencia de absorbancias del blanco de las diferencias de absorbancias de las muestras. ΔA = (A—A2)muestra – (A1 – A2)blanco Las diferencias en las medidas de absorbancia deben, como regla, ser al menos de 0.100 unidades de absorbancia para obtener suficiente precisión en los resultados (ver “Instrucciones para el rendimiento del ensayo” y “Sensibilidad y límite de detección”, punto 4). Cálculos De acuerdo a la ecuación general del cálculo de las concentraciones: V x MW c = ------------------------- x ∆A (g/l) ε x d x v x 1000 V v MW d ε = volumen final (ml) = volumen de muestra (ml) = peso molecular de la sustancia a ensayar (g/mol) = paso de luz (cm) = coeficiente de extinción del NADH a: -1 -1 340 nm = 6.3 (l x mmol x cm ) -1 -1 Hg 365 nm = 3.4 (l x mmol x cm ) -1 -1 Hg 334 nm = 6.18 (l x mmol x cm ) Corresponde para amonio 3.020 x 17.03 0.5143 c = ----------------------------------- x ∆A = ------------- x ∆A (g amonio / l ε x 1.00 x 0.100 x 1000 ε sol. muestra) Si la muestra se ha diluido durante la preparación, los resultados deben multiplicarse por el factor de dilución F. 2013-10 1/4 Cuando se analizan muestras sólidas ó semisólidas que se pesan para la preparación de la muestra, los resultados se calculan a partir de la cantidad pesada: c amonio (g/l de sol. ) Contenido amonio = ------------------------------------------- x 100 (g / 100g) peso muestra en g/l sol. 1. Instrucciones para el funcionamiento del kit La cantidad de amonio presente en el ensayo debe ser entre 0.2 ug y 8 ug. Para obtener una diferencia de absorbancias suficiente, la solución de muestra debe diluirse para dar una concentración de amonio entre 0.01 y 0.08 g/l. Tabla de dilución Cantidad estimada de amonio por litro Dilución con agua Dilución Factor F < 0,08 g - 1 0,08-0,8 g 1+9 10 0,8-8,0 g 1 + 99 100 Si la medida de la diferencia de absorbancias (∆A) es muy pequeña (ej. < 0.100), debe prepararse nuevamente la solución de muestra (pesar mas cantidad de muestra ó diluir menos la muestra) ó el volumen de muestra que se pipetea en la cubeta puede aumentarse hasta 2.000 ml. El volumen de agua agregada debe, en ese caso, disminuirse para obtener el mismo volumen final en el ensayo para la muestra y el blanco. El nuevo volumen de muestra v debe tomarse en cuenta en los cálculos. 2. Información técnica 2.1 Usar solamente agua bidestilada fresca para el ensayo. 2.2 Trabajar en una atmósfera libre de amonio (prohibido fumar en el laboratorio). 3. Especificidad (Ref. 1) El método es específico para amonio. En el análisis de sulfato de amonio comercial se obtienen valores aprox. del 100%. 4. Sensibilidad y límite de detección (Ref. 1.2) La mínima diferencia de absorbancia para el procedimiento es de 0.005 unidades. Esto corresponde a un volumen máximo de muestra v = 2.000 ml y medición a 340 de una concentración de amonio de 0.02 mg/l de solución de muestra (si el v = 0.100 ml, esto corresponde a 0.4 mg/l de solución de muestra). El límite de detección de 0.08 mg/l deriva de una diferencia de absorbancia de 0.020 (medida a 340 nm) y de un volumen máximo de muestra de v = 2.000 ml. 5. Linealidad La linealidad de la determinación existe desde aprox. 0.2 ug de amonio / ensayo (0.08 mg amonio/ l de solución de muestra; volumen de muestra v = 2.000 ml) hasta 8 ug de amonio / ensayo (0.08 g amonio/ l de solución de muestra; volumen de muestra v = 0.100 ml). 6. Precisión En una determinación por duplicado de una única solución de muestra, se puede obtener una diferencia de 0.005 a 0.010 unidades de absorbancia. Con un volumen de muestra de v = 0.100 ml y medido a 340 nm, esto corresponde a una concentración aprox. de amonio de 0.4-0.8 mg/l. (Si la muestra se diluye durante la preparación de la misma, el resultado se debe multiplicar por el factor de dilución F. Si la muestra se pesa para su preparación, ej. usando 1 g muestra/100 ml = 10 g/l, se puede obtener una diferencia de 0.004-0.01g/100 g). En la literatura se han publicado los siguientes datos: CV = 1.6 % (plasma) CV = 0.88-1.16 % (soluciones de Cloruro de amonio) CV = 0.34 % (solución de cloruro de amonio) CV = 0.36-0.96 % (muestras de carne) (Ref. 1.1) (Ref. 1.2) (Ref. 3.2) 7. Interferencias y fuentes de error Durante la precipitación de proteínas con ácido perclórico que se lleva a cabo en alimentos se pueden obtener, ocasionalmente, fragmentos proteicos. Estos fragmentos se mantienen en solución y gradualmente forman amonio en buffer alcalino, generando reacciones “creep”. La formación de amonio es muy baja y puede calcularse y diferenciarse del contenido de amonio de la muestra por extrapolación de la absorbancia A2 al tiempo de adición de la solución 3 (GIDH). Los ingredientes comunes de los alimentos no interfieren con la determinación de amonio. Solamente altas concentraciones de taninos en los jugos de fruta pueden causar una inhibición de la reacción de la GIDH. Por ello los jugos de fruta deben tratarse con PVPP. Dado que altas concentraciones de metales pesados causan turbidez, ello también produce dificultad en la determinación de amonio. En la mayoría de los casos las altas concentraciones de iones metálicos pueden removerse como hidróxidos por alcalinización de la solución de muestra (pH > 7.5). El tiosulfato de sodio agregado ocasionalmente al agua de las piletas de natación, no interfiere en el ensayo hasta 1 mg/ensayo. 8. Reconocimiento de interferencia durante el procedimiento del ensayo 8.1 Si la conversión de amonio se ha completado en el tiempo estipulado en “Procedimiento” se puede concluir, en general, que no han ocurrido interferencias. 8.2 Al completarse la reacción, la determinación puede reiniciarse con el agregado de amonio (cloruro de amonio, sulfato de amonio, cualitativa ó cuantitativamente): si la absorbancia se altera posteriormente al agregado del material estándar, esto también es una indicación que no ha ocurrido interferencia. 8.3 Se pueden reconocer errores del operador ó interferencia en la determinación por la presencia de sustancias contenidas en la muestra, realizando una doble determinación utilizando dos volúmenes diferentes de muestra (ej. 0.100 ml y 0.200 ml): las diferencias en las mediciones de las absorbancias deben ser proporcionales a los volúmenes de muestra utilizados. Cuando se analizan muestras sólidas, se recomienda que se pesen diferentes cantidades (ej. 1g y 2g) en recipientes de 100 ml. Las diferencias de absorbancias medidas y los pesos de la muestra utilizados deben ser proporcionales para volúmenes idénticos. 8.4 Se pueden reconocer posibles interferencias causadas por sustancias contenidas en la muestra utilizando un estándar interno como control: además de las determinaciones de la muestra, el blanco y el estándar, se lleva a cabo otra determinación con la muestra y el control juntos en el mismo ensayo. La recuperación se puede calcular de la diferencia de absorbancias medidas. 8.5 Posibles pérdidas durante la determinación se puede reconocer por medio de ensayos de recuperación: la muestra debe prepararse y analizarse con y sin el agregado de material estándar. Dicha adición debe recuperarse cuantitativamente dentro del rango del error del método. 9. Riesgo de los reactivos Los reactivos utilizados en la determinación de amonio no son materiales riesgosos de acuerdo a las Regulaciones de Sustancias Peligrosas, las Leyes Químicas ó la Regulación 67/548/EEC de la CEE y subsecuentes Guías de alteraciones, suplementos y adaptaciones. Aún así, deben cumplirse todas las medidas de seguridad generales que se aplican a todas las sustancias químicas. Luego de su utilización, los reactivos deben desecharse con el descarte del laboratorio, pero deben observarse siempre las regulaciones locales. El material de empaque puede desecharse en recipientes destinados al reciclado. 10. Información general sobre la preparación de muestra Al llevar a cabo el ensayo: Usar muestras líquidas límpidas, incoloras y prácticamente neutras directamente ó luego de diluirlas de acuerdo a la tabla de dilución, y usar un volumen hasta 2.000 ml; Filtrar soluciones turbias; Desgasear muestras que contengan dióxido de carbono (ej. por filtración); Ajustar muestras ácidas a pH 7-8 por el agregado de una solución de hidróxido de sodio ó de potasio; Ajustar muestras ácidas y levemente coloreadas a pH 7-8 por el agregado de una solución de hidróxido de sodio ó de potasio y posterior incubación durante 15 min.; Tratar las muestras “altamente coloreadas” que se usan sin diluir ó en grandes volúmenes con polivinilpolipirrolidona (PVPP) – (ej. 2.55 g/100 ml); Muela ú homogeneice las muestras sólidas ó semisólidas, extraer con agua ó disolver en agua y filtrar si es necesario; Desproteinizar muestras que contengan proteínas con ácido perclórico ó ácido tricloroacético; 2013-10 2/4 Extraer muestras que contengan grasa con agua caliente (la temperatura de extracción debe ser mayor que el punto de fusión de la grasa involucrada). Enfriar para permitir que la grasa se separe, colocar el recipiente en baño de hielo por 15 min y filtrar; Romper emulsiones con ácido tricloroacético; Nota importante La clarificación de Carrez no debe utilizarse en la preparación de la muestra para la determinación de amonio debido a la baja tasa de recuperación (absorción de amonio). 11. Ejemplos de aplicación Determinación de amonio en jugos de fruta Agregar 0.5-1.0 g (!) de polivinilpolipirrolidona húmeda (PVPP) a 10 ml de jugo de fruta (jugos claros, turbios ó coloreados) – cuando aumenta el volumen de muestra, neutralizar, si es necesario, y llevar a 20 ml con agua en un recipiente. Agitar por 1 min. con agitador magnético. Filtrar la solución de muestra inmediatamente y usar para el ensayo. Determinación de amonio en agua (piletas de natación) (Ref. 3.1, 3.4) Filtrar la muestra de agua si es necesario, y usar la muestra clara para el ensayo. Diluir la muestra de acuerdo a la tabla de dilución ó usar hasta un volumen de muestra de v = 2.000 ml para el ensayo. Determinación de amonio en leche Mezclar 1 ml de leche con 4 ml de ácido tricloroacético (0.3 M). Luego de aprox. 5 min. centrifugar para separar el precipitado. Decantar el sobrenadante y neutralizar con KOH (10 M) (el factor de dilución puede despreciarse debido a la alta concentración del KOH), filtrar y usar 1.000-2.000 ml de solución de muestra para el ensayo. Determinación de amonio en productos panificados Pesar aprox. 10 g de muestra en homogeneizador, agregar aprox. 20 ml de ácido perclórico (1 M) y homogeneizar por 2 min. Proceder según “carne y productos cárnicos”. Usar como máximo 1.000 ml para el ensayo. Determinación de amonio en carne, productos cárnicos (Ref. 3.2, 3.3) y queso Pesar aprox. 5 g de muestra homogeneizada (de una muestra de 100 g, que ha sido molida y homogeneizada en un homogeneizador) en un recipiente, agregar 20 ml de ácido perclórico (1 M) y homogeneizar por 2 min. Transferir el contenido cuantitativamente con 40 ml de agua. Ajustar el pH a 7.0 (< 7.5) primero con hidróxido de potasio (5 M) y luego exactamente con hidróxido de potasio (2 M). Transferir el contenido cuantitativamente en un recipiente de 100 ml, llevar a volumen con agua, teniendo en cuenta que la fase lipídica se encuentre sobre el nivel y la fase acuosa en encuentre en el nivel. Para la separación de la grasa y para la precipitación del perclorato de potasio, refrigerar por 20 min. Luego filtrar. Descartar los primeros mililitros. Usar la solución clara, posiblemente algo turbia para el ensayo, diluyendo si es necesario de acuerdo a la tabla de dilución. Calcular la cantidad de amonio de acuerdo a la fórmula anteriormente detallada, pero debe multiplicarse por el factor de desplazamiento del volumen K = 0.98. Determinación de amonio en pastillas de orozuz y de sales de amonio Pesar 1 g de muestra molida y homogeneizada en un recipiente de 100 ml, agregar aprox. 60 ml de agua a 60 – 70ºC y agitar con agitador magnético por 10 min. hasta que el material esté completamente disuelto. Ajustar a 20-25ºC, llevar a volumen con agua, mezclar y filtrar. Descartar los primeros mililitros, usar el filtrado claro (generalmente) para el ensayo, luego de diluirlo de acuerdo a la tabla de dilución si es necesario. M) ó con hidróxido de sodio diluido (1 M) en el caso de fertilizantes acídicos. Calentar a 60-70ºC con agitador magnético termostatizado por aprox. 10 min. Dejar enfriar, transferir cuantitativamente a un recipiente de 100 ml y llevar a volumen con agua. Mezclar la solución y filtrar si es necesario. Usar 0.100 ml de la solución clara diluida, si es necesario, para el ensayo. 12.2 Determinación de nitrógeno luego de digestión por Kjeldahl La determinación de nitrógeno total se puede obtener por medio de la determinación de amonio en una muestra mineralizada de acuerdo al método de Kjeldahl. Normalmente, las muestras se incineran (ácido sulfúrico). El amonio, originado del nitrógeno, se determina de acuerdo a el siguiente procedimiento: Pesar 2 g de muestra molida y homogeneizada en un recipiente de Kjeldahl de 100 ml, agregar 20 ml de ácido sulfúrico (gravedad específica = 1.84 g/ml) y aproximadamente 30 mg de muestra catalítica (ej. de acuerdo a Wieninger) ó un tableta de Kjeldahl, calentar por aprox. 2-3 h hasta que la muestra se haya desintegrado (solución amarilla ó verde amarilla). Dejar que la muestra enfríe y con cuidado (usar anteojos protectores) transferir el material cuantitativamente en un recipiente con 600 ml de agua fría (con baño de hielo), agitando todo el tiempo (agitador magnético). Neutralizar con aprox. 60 ml de KOH (10 M) (pH 6-8). Transferir la solución neutralizada cuantitativamente a un recipiente de 1 l, llevar a volumen con agua y mezclar.. SI es necesario, filtrar la mezcla (en general es necesario después de la desintegración con las pastillas de Kjeldahl), descartar los primeros mililitros. Usar la solución diluida, si es necesario, para el ensayo. Cálculo Contenido de nitrógeno de la muestra (en %) = ∆A × V × MW × 100 = --------------------------------------------------------ε × d × v × 1000 × cantidad pesada [g] ∆A × 3.020 × 14.01 × 100 = -----------------------------------------------------------------ε × 1000 × 0.100 × 1000 × cantidad pesada [g] 12.3 Determinación de amonio en muestras de fermentación y medio de cultivo de células Colocar la muestra (luego de centrifugar, si es necesario) en un baño de agua a 80ºC por 15 min. para detener las reacciones enzimáticas. Centrifugar y usar el sobrenadante (diluido de acuerdo a la tabla de dilución si es necesario) para el ensayo. Alternativamente se puede desproteinizar con ácido perclórico. Ver los ejemplos mencionados anteriormente. Homogeneizar el medio gelatinoso de agar con agua y tratar como se describe. Referencias 1.1 da Fonseca-Wollheim, F., Bergmeyer, H. U. & Gutmann, I. (1974) in Methoden der enzymatischen Analyse (Bergmeyer, H. U. Hrsg.) 3. Aufl., Bd. 2, S. 1850-1853, Verlag Chemie, Weinheim and (1974) in Methods of Enzymatic Analysis (Bergmeyer, H. U. ed.) 2nd ed., vol. 4, pp. 1802-1806, Verlag Chemie, Weinheim/Academic Press, Inc., New York and London 1.2 Bergmeyer, H. U. & Beutler, H.-O. (1985) in Methods of Enzymatic Analysis (Bergmeyer, H. U., ed.) 3rd ed., vol. VIII, pp. 454-461, Verlag Chemie, Weinheim, Deerfield Beach/ Florida, Basel 2.1 Untersuchung von Papieren, Kartons und Pappen für die Lebensmittelverpackungen (gem. Empfehlungen XXXVI der Kunststoffkommission des Bundesgesundheitsamtes) Kapitel 8 (Methoden) Pkt. 3.4.2 (Ammoniak) 2.2 Brautechnische Analysenmethoden, Band III, S. 597-599 (1982), Methodensammlung der MitteleuropÑischen Brautechnischen Analysenkommission (MEBAK), herausgegeben von F. Drawert im Selbstverlag der MEBAK, Freising 3.1 Höpner, Th. (1977) Enzymatische Methoden in der Wasseranalytik - Mîglichkeiten und renzen, Vom Wasser 49, 173-182 3.2 Gerhardt, U. & Quang, T. D. (1979) Methoden zur Ammoniakbestimmung in Fleisch und Fleischerzeugnissen, Fleischwirtschaft 59, 946-948 3.3 Barchietto, G., Cantoni, C., Frigerio, R. & Provera, D. (1984) Esame comparativo dei prodotti di autolisi nella carne di maiale (Azoto non proteico, Urea, Ammoniaca), Conservazione degli Alimenti 3, 12-17 3.4 Bartels, U. (1991) Die enzymatische Bestimmung von Ammonium im Niederschlagswasser, CLB Chemie in Labor und Biotechnik 42, 377-382 3.5 Cheuk, W. L. & Finne, G. (1984) Enzymatic determination of urea and ammonia in refrigerated seafood products, J.Agric.Food Chem. 32, 14-18 12. Otras aplicaciones El método también puede ser utilizado para analizar fertilizantes, fármacos, cosméticos, papel (Ref. 2.1) y en investigación para análisis de muestras biológicas. Para detalles de muestreo, tratamiento y estabilidad de los reactivos ver Ref. 1.1-1.2. Ejemplos 12.1 Determinación de amonio en fertilizantes Moler aprox. 10 g de muestra y mezclar. Pesar aprox. 100 mg de material homogeneizado en un recipiente de 100 ml y agregar aprox. 50 a 60 ml de agua. Ajustar el pH 7-8 con ácido clorhídrico diluido (1 2013-10 3/4 . Solución control del ensayo de Amonio (Botella 4) Concentración *: ver etiqueta en frasco El control del ensayo de amonio es una solución acuosa estabilizada de amonio. Sirve como control del ensayo enzimático para la determinación de amonio en alimentos y otros materiales. Aplicaciones 1 Adición de la solución control de amonio a la mezcla de reacción: En vez de la solución de muestra utilizar la solución control en el ensayo. 2. Reinicio de la reacción cuantitativamente: Una vez finalizada la reacción con la solución de muestra y tomada la medida de A2, agregar 0.050 ml de solución control a la mezcla del ensayo. Leer la absorbancia A3 luego de finalizada la reacción (aprox. 20 min.). Calcular la concentración de la diferencia (A2-A3) de acuerdo a la ecuación general del cálculo de la concentración. Aunque hay una dilución de la mezcla del ensayo por el agregado de la solución control, los resultados casi no difieren del dato estipulado en la etiqueta. 3. Estándar interno: La solución control del ensayo puede utilizarse como estándar interno para controlar el correcto funcionamiento del ensayo (errores groseros) y para ver si la solución de muestra está libre de sustancias interferentes. Pipetear en la Muestra + Blanco Muestra Estándar cubeta estándar sol. Reacción 2 1,000 ml 1,000 ml 1,000 ml 1,000 ml sol. Muestra - sol. Control - 0,100 ml - 0,050 ml - 0,100 ml 0,050 ml agua bidestilada 2,000 ml 1,900 ml 1,900 ml 1,900 ml Mezclar y leer las absorbancias de las soluciones (A1) luego de aprox. 5 min. Continuar como se describe en "Procedimiento". Seguir las instrucciones dadas bajo "Instrucciones para el funcionamiento del ensayo" y las notas al pié. La recuperación del estándar se calcula de acuerdo a la siguiente fórmula: 2 x ∆A muestra + estándar - ∆A muestra recuperación = ----------------------------------------------- x 100 (%) ∆A estándar 2013-10 4/4