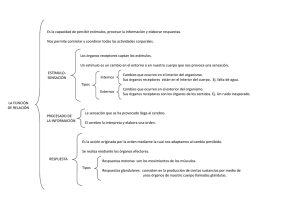
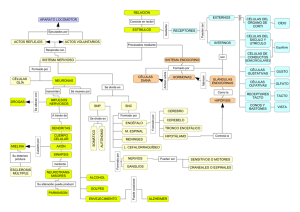
pdf Papel de los receptores de kainato en la regulación de la
Anuncio

Universidad Autónoma de Madrid Papel de los receptores de kainato en la regulación de la transmisión sináptica gabaérgica Antonio Rodríguez Moreno Tesis de Doctorado Facultad de Ciencias Director: Dr. Juan Lerma Gómez 2000 ÍNDICE INTRODUCCIÓN....................................................................................................................1 1 1. RECEPTORES IONOTRÓPICOS DE GLUTAMATO..................................... 2 . 4 1.1. RECEPTORES DE NMDA...............................................................................................4 5 1.2. RECEPTORES DE AMPA...............................................................................................5 6 1.3. RECEPTORES DE KAINATO.......................................................................................6 7 1.3.1. Biología Molecular de los receptores de kainato........................................ A. La familia de subunidades de los receptores de kainato: GluR5-7, KA1 y KA2.............................................................................7....7 B. Distribución de las subunidades de los receptores de kainato............................... 7 C. Diversidad estructural................................................................................................ 8 10 1.3.2. Propiedades funcionales de los receptores de kainato............................ A. Propiedades biofísicas. Propiedades de los receptores 10 de kainato a nivel de canal único........................................................................10 B. Propiedades de activación y de desensibilización 11 de los receptores de kainato..................................................................................11 C. Propiedades farmacológicas de los receptores de kainato...................................... 13 13 C.1. Agonistas......................................................................................................13 C.2. Antagonistas.......... ....................................................................................... 14 1515 D. Papeles fisiológicos de los receptores de kainato.................................... OBJETIVOS...............................................................................................................................17 17 MATERIALES Y MÉTODOS.......................................................................................18 18 1. CULTIVOS NEURONALES...........................................................................................18 18 Microcultivos de neuronas........................................................................................................ 18 2. RODAJAS DE CEREBRO...............................................................................................18 18 19 3.REGISTROS ELECTROFISIOLÓGICOS...............................................................1 3.1. Cultivos...................................................................................................................................19 19 21 3.2. Rodajas....................................................................................................................................21 4. ANÁLISIS DE LOS REGISTROS................................................................................2 23 24 5. SOLUCIONES........................................................................................................................24 24 5.1. Cultivos....................................................................................................................................24 5.2. Rodajas.......................................................................................................................................... 24 6. EXPERIMENTOS IN VIVO................................................................................................. 25 RESULTADOS................................................................................................................................ 26 1. La activación de los receptores de kainato produce una depresión de la transmisión sináptica GABAérgica en el hipocampo de rata................26 1.1. Kainato produce una depresión de la transmisión GABAérgica................................................................................................................................. 1.2. La modulación de la transmisión GABAérgica por kainato es de origen presináptico..................................................................................................................... 1.3. El efecto de kainato no se debe a la activación de los receptores metabotrópicos de glutamato.................................................................................................. 1.4. CNQX antagoniza el efecto de kainato de forma dosis-dependiente.......................... 1.5. Kainato no actúa sobre otro tipo de receptores descritos que regulan la liberación de neurotransmisor..................................................................... 1.6. El agonista endógeno de los receptores de kainato, glutamato, ejerce el mismo efecto que kainato sobre las eIPSCs....................................................... 1.7. Kainato deprime la transmisión GABAérgica en la región CA1 del hipocampo in vivo...................................................................................................................... 26 29 31 32 32 33 35 2. La modulación de la liberación de GABA por los receptores de kainato involucra una función metabotrópica.................................................. 37 2.1. La activación de una proteína G sensible a toxina pertúsica y de proteína kinasa C son necesarias para que se produzca la disminución de liberación de GABA mediada por la activación de receptores de kainato......................................................................................................... 2.2. Kainato activa una población particularde PKC............................................................. 2.3. La acción de kainato es independiente de la actividad del receptor como canal iónico............................................................................................. 2.4. AMPA no activa una cascada similar................................................................................ 2.5. La activación de los receptores metabotrópicos de tipo I no ocluye la acción de kainato............................................................................................. 2.6. La activación de los receptores de kainato modula la señal de calcio..................... 37 40 42 43 43 44 3. Las interneuronas de hipocampo presentan dos poblaciones de receptores de kainato con mecanismos de señalización diferentes....................................................................................................... 46 3.1. El aumento de actividad espontánea y el descenso de amplitud de las respuestas evocadas son efectos producidos por la activación de receptores de kainato diferentes...................................................................... 46 4. La modulación de la liberación de GABA mediada por la activación de los receptores de kainato presinápticos es un fenómeno común a diferentes zonas del cerebro 4.1. Hipocampo................................................................................................................................ 4.2. Cerebelo..................................................................................................................................... 4.3. Neocorteza................................................................................................................................ DISCUSIÓN................................................................................................................................. 1. La acción de kainato es presináptica.................................................................................... 2. La curva dosis-respuesta para el efecto de kainato y de glutamato presenta forma de campana................................................................................................... 3. La depresión de las corrientes inhibidoras mediada por activación de receptores de kainato requiere una cascada metabotrópica........................................... 4. ¿Son las proteínas exocitóticas los efectores de una población específica de PKC?.................................................................................................................... 5. En las interneuronas de hipocampo coexisten las dos poblaciones de receptores de kainato con sistemas de señalización diferentes........................................................................................................ 6. La modulación de la liberación de GABA mediada por la activación de receptores de kainato presinápticos es un fenómeno común a diferentes áreas del cerebro.................................................................................................... 7. Relevancia fisiológica del bloqueo de la liberación de GABA mediada por receptores de kainato............................................................................................................... 53 53 54 56 58 59 60 60 61 63 65 66 CONCLUSIONES................................................................................................................... 68 BIBLIOGRAFÍA..................................................................................................................... 69 ABREVIATURAS Y ACRÓNIMOS. AMPA: ácido α-amino-3-hidroxi-5-metil-4-isoxazolepropiónico AMPc: adenosín monofosfato cíclico ATP: adenosín trifosfato ATPA: ácido (RS)-α-amino-3-hidroxi-5-tert-butil-4-isoxazolepropiónico BIS: bisindolilmaleimida Ca2+: ion calcio CA: Cornus ammonis (nombre de las subcapas del hipocampo) CNQX: 6-ciano-7-nitorquinoxalín-2,3-diona CV: Coeficiente de Variación D-AP5: ácido D-2-amino-5-fosfopentanoico DNA: DRG: ácido desoxirribonucleico ganglios de la raíz dorsal EEM: error estándar de la media EGTA: ácido etilenglicol-bis-(β-aminetiléter)-N, N, N’, N’-tetracético. eIPSCs: corrientes postsinápticas inhibidoras evocadas EPSPs: potenciales postsinápticos excitadores de campo GABA: ácido γ-aminobutírico GTP: guanosín trifosfato HEK: Human embryonic kidney cells (células embrionarias de riñón humano) HEPES: N-(2-hidroxietil) piperacina-N-(2-ácido etanosulfónico) Hz: Hercio iGluR: receptor de glutamato ionotrópico IP3: inositoltrifosfato K+: ion sodio KBPs: kainate binding proteins (proteínas que unen kainato) KDa: Kilodalton LTD: long Term Depression (Depresión de larga duración) LTP: long Term Potentiation (Potenciación de larga duración) MΩ: megaohmios Mg2+ : ion magnesio mGluR: receptor de glutamato metabotrópico i mIPSCs: corrientes postsinápticas inhibidoras miniatura mM: milimolar mOsm: miliosmoles mRNA: ácido ribonucleico mensajero ms: milisegundo mV: milivoltio + Na : ion sodio NMDA: N-metil-D-aspartato pA: picoamperio PKA: proteína kinasa A PKC: proteína kinasa C PLC: fosfolipasa C pS: picosiemens PTx: toxina pertúsica sIPSCs: corrientes postsinápticas inhibidoras espontáneas SNC: Sistema Nervioso Central TEA: tetraetilamonio TTX: tetrodotoxina µM: micromolar ii Introducción INTRODUCCIÓN. Han pasado 46 años desde que se demostró por primera vez que la aplicación de glutamato monosódico en la corteza cerebral inducía depolarizaciones masivas (Hayashi, 1954). A esta observación puntual indicando un posible papel del glutamato como agente excitador del Sistema Nervioso Central (SNC), le siguieron estudios electrofisiológicos más detallados demostrando efectos depolarizantes tanto del glutamato como del aspartato en neuronas aisladas del SNC de vertebrados y que éstos eran el resultado de un aumento de la conductancia de la membrana a sodio. En la década de los 70 se realizaron los trabajos demostrativos de que el glutamato cumplía la mayor parte de los criterios para ser considerado neurotransmisor en el SNC de los mamíferos. La presencia de receptores para glutamato en las membranas neuronales (Evans et al., 1979), la liberación de glutamato por las terminales nerviosas tras la excitación (Hamberger et al., 1979), así como la existencia de sistemas de transporte específicos de alta afinidad tanto en células gliales como en terminales nerviosas (Logan y Snyder, 1972), apoyaron fuertemente la hipótesis de que el glutamato era el neurotransmisor excitador por excelencia en el SNC. Además, el conocimiento de la existencia de rutas metabólicas biosintéticas y degradativas de glutamato en neuronas así como la presencia de un transportador dependiente de ATP implicado en el almacenaje de glutamato en vesículas sinápticas, igualmente sirvieron para consolidar esta conclusión. En la actualidad, está bien establecido que el glutamato es el agente neurotransmisor usado en la mayoría de las sinapsis excitadoras del Sistema Nervioso Central de los mamíferos. Además de la función del glutamato como mediador de la transmisión sináptica, este aminoácido participa durante la formación del sistema nervioso en procesos de crecimiento y maduración neuronal, en la formación y eliminación de sinapsis y, en determinadas áreas y de forma dependiente de actividad, en la formación de patrones precisos de conectividad sináptica. Igualmente desencadena cambios duraderos en la eficacia sináptica, fenómenos conocidos como LTP (potenciación de larga duración, “longterm potentiation”) y LTD (depresión de larga duración, “long-term depression”), que se consideran el correlato celular de los procesos de aprendizaje y formación de la memoria. 1 Introducción Además, alteraciones de la neurotransmisión glutamatérgica están implicadas en el daño neuronal observado después de episodios de isquemia y de hipoglucemia, así como en la etiología de una serie de estados neurológicos patológicos que incluyen la epilepsia y las enfermedades de Alzheimer, de Parkinson, la corea de Huntington y la esclerosis lateral amiotrófica. El glutamato realiza sus funciones mediante la activación de varias moléculas receptoras. Los receptores que el glutamato activa se han dividido en dos familias: receptores metabotrópicos y receptores ionotrópicos. Los receptores metabotrópicos (mGluRs) están acoplados a proteínas G (proteínas que unen GTP) y regulan la producción de mensajeros intracelulares (Pin y Duvoisin, 1995). Esta superfamilia está formada por 8 genes diferentes (mGluR1-8), cada uno de los cuales da lugar a distintos mRNAs por mecanismos de ayuste (splicing) alternativo. Según el nivel de conservación de su secuencia aminoacídica estos receptores se han subdividido en tres grupos (tipos I, II y III) (Nakanishi, 1992), con dos posibles mecanismos de transducción (adenilato-ciclasa y fosfolipasa C). Estos tres grupos presentan también propiedades farmacológicas propias. Por el contrario, los receptores ionotrópicos de glutamato (iGluRs) forman un canal catiónico con diferente selectividad iónica según el tipo de receptor, siendo permeables a sodio (Na+), potasio (K+) y, en ocasiones, a calcio (Ca2+) (Nakanishi, 1992; Hollmann y Heinemann, 1994). 1. RECEPTORES IONOTRÓPICOS DE GLUTAMATO. Se estima que en el SNC de los mamíferos, el glutamato es el neurotransmisor empleado en el 90 % de las sinapsis excitadoras rápidas, donde actúa sobre receptores ionotrópicos (iGluR). En función del agonista que produce la activación de éstos con mayor afinidad o selectividad, los receptores ionotrópicos de glutamato se han clasificado en tres tipos: receptores de NMDA (ácido N-metil-D-aspártico), receptores de AMPA (ácido α-amino-3-hidroxi-5-metil-4-isoxazolepropiónico) y receptores de kainato. Desde el punto de vista molecular, los receptores ionotrópicos de glutamato son proteínas integrales de membrana formadas, probablemente, por cuatro subunidades que 2 Introducción GLUTAMATO NMDA EXTRACELULAR Zn GLU AMPA KAINATO mGluR I, II, III GLY PA Mg G INTRACELULAR NR2A NR2B SUBUNIDADES NR1+ NR2C NR2D GluR5-7 KA1, KA2 GluR1-4 AdC PLC IONOTRÓPICOS mGluR1-8 METABOTRÓPICOS Figura 1. Los receptores de glutamato se dividen en ionotrópicos (canales iónicos) y metabotrópicos (acoplados a proteínas G). Los receptores ionotrópicos se subdividen en función de su activación selectiva por los agonistas NMDA, AMPA y kainato. Por su parte, los metabotrópicos se subdividen con respecto a la homología de sus secuencias en los grupos I, II y III. En la figura se incluyen también los nombres de las subunidades conocidas a la fecha para cada uno de los diferentes tipos de receptor y, en el receptor de NMDA, se indican los diferentes sitios de modulación que se conocen. AdC, adenilato ciclasa; PLC, fosfolipasa C; PA, poliaminas. A NH2 Sitio R/G flip/flop Glu 1 Sitio Q/R/N Sitio I/V Sitio Y/C 2 3 4 B NH 2 C Canal Iónico Glu 1 2 3 4 5 6 7 COOH COOH M2 Figura 2. Topología de los receptores de glutamato. A) Los tres tipos de receptores ionotrópicos poseen tres dominos transmembranales (1, 3 y 4) y un segmento hidrofóbico que se introduce en la membrana de manera incompleta (2) formando la pared del canal iónico. El sitio de unión del glutamato está formado por aminoácidos de la región N-terminal y del asa entre el tercero y el cuarto dominios transmembranales. También se muestra la posición de los diferentes sitios modificados por la edición y el procesamiento o ayuste alternativo de los RNA mensajeros que codifican estos receptores y que se discuten en el texto. B) Los receptores metabotrópicos poseen siete dominios transmembranales análogos a los observados en otras familias de receptores acoplados a proteínas G. El sitio de unión de glutamato está formado por aminoácidos del segmento N-terminal exclusivamente. C) La disposición en el plano de la membrana de las subunidades que conforman un receptor ionotrópico revela que se trata de un tetrámero en el que el segmento hidrofóbico M2 se ubica hacia el centro de la molécula formando el poro del canal. 3 Introducción forman un tetrámero (Rosenmund et al., 1998). Estas subunidades son cadenas polipeptídicas de aproximadamente 900 aminoácidos (≈100 KDa), las cuales están formadas por tres dominios transmembranales (M1, M3 y M4) y un dominio intramembranal (M2); con el extremo carboxiterminal intracelular y el aminoterminal situado en el lado extracelular. Se han identificado 28 subunidades distintas, codificadas por, al menos, 17 genes diferentes. Tal variedad se genera porque los RNAs pueden ser modificados mediante mecanismos de ayuste alternativo. Además, se conocen variantes generadas por edición del RNA, lo que incrementa el número de isoformas. 1.1. RECEPTORES DE NMDA. De los tres tipos de receptores ionotrópicos de glutamato, el mejor conocido es el receptor de NMDA, debido sobre todo, a la rápida disponibilidad de ligandos selectivos, como el agonista NMDA (N-metil-D-aspartato) y los antagonistas D-AP5, MK801 o ketamina. Los receptores de NMDA son canales catiónicos que permiten el paso a su través de Na+, K+ y Ca2+. Funcionalmente, se caracterizan por poseer una alta permeabilidad a Ca2+ (Ascher y Nowak, 1988), presentar dependencia de voltaje debida al bloqueo del canal por concentraciones fisiológicas de magnesio (Mg2+)(Nowak et al., 1984; Mayer y Westbrook, 1987), un tiempo de apertura largo (Nowak et al., 1984) y una cinética de activación y de deactivación relativamente lenta (Lester et al., 1990). Además de los sitios de unión para el agonista, el receptor de NMDA presenta diversos lugares de regulación alostérica, que son diana para compuestos tanto endógenos como exógenos. Estos sitios de unión incluyen: un sitio de alta afinidad para glicina (Johnson y Ascher, 1987; Klecner y Dingledine, 1998; Lerma et al., 1990); un sitio localizado en la luz del canal donde se unen el Mg2+ y moléculas como las fenciclidinas (PCP), el MK801, TCP o ketamina (Macdonald et al., 1991; Lerma et al., 1991); además de un sitio adicional en el que actúa Zn2+ inhibiendo las respuestas inducidas por el agonista de forma independiente de voltaje (Peters et al., 1987; Westbrook y Mayer, 1987). Finalmente, es conocido que las poliaminas espermina y espermidina potencian de forma alostérica el receptor de NMDA (Lerma, 1992). Igualmente, se ha descrito que los receptores de NMDA son sensibles a altas concentraciones extracelulares de H+ (i.e. son sensibles al pH) (Traynelis y Cull-Candi, 1990) y son susceptibles a los estados de 4 Introducción oxidación-reducción (Aizenman et al., 1989). A nivel estructural estos receptores están formados por, al menos, dos tipos de subunidades: NR1 (Moriyoshi et al., 1991) y NR2 (Monyer et al., 1992; Meguro et al., 1992). Esta última consta de cuatro isoformas, NR2A, NR2B, NR2C y NR2D. La subunidad NR1 presenta 8 variantes generadas por ayuste alternativo (Sugihara et al., 1992; Nakanishi et al., 1992), que tienen diferentes propiedades farmacológicas. Expresando en sistemas heterólogos la subunidad NR1 con cualquiera de las cuatro subunidades NR2, se obtienen receptores de NMDA con características funcionales similares a los nativos. La subunidad NR1 parece ser el constituyente común de los receptores de NMDA, puesto que ninguna de las cuatro subunidades NR2 es capaz de generar receptores funcionales por sí misma. Dependiendo de la subunidad NR2 que se ensamble, los receptores heteroméricos NR1-NR2 tienen características propias. La subunidad NR1 se encuentra expresada en prácticamente todas las neuronas (Moriyoshi et al., 1991) mientras que las distintas subunidades NR2 muestran patrones de expresión espacio-temporales característicos en el cerebro y en la médula espinal, sufriendo alteraciones durante el desarrollo (Watanabe et al., 1992; Monyer et al., 1994). Los receptores de NMDA están involucrados en procesos fisiológicos complejos del SNC. Además de participar en la transmisión normal de la información, la activación de los receptores de NMDA se requiere para la generación de algunas formas de LTP (Nicoll et al., 1988). Igualmente, la alta permeabilidad a calcio de los receptores de NMDA hace que estos jueguen un importante papel en procesos de muerte celular por excitotoxicidad. 1.2. RECEPTORES DE AMPA. La familia de los receptores de AMPA está formada por cuatro subunidades que presentan una homología entre ellas del 68 al 75 %. Se denominan GluR1-4 o GluRA-D, según el criterio seguido por el grupo que las clonó. Cada una de las cuatro subunidades presenta dos variantes generadas por procesamiento alternativo de los mRNAs, denominadas “flip” o “flop”. Un determinante molecular importante para las propiedades de canal de los receptores de AMPA es el aminoácido situado en la posición 586 (sitio Q/R). En tres de las cuatro subunidades del receptor de AMPA (GluR1, 3 y 4) esta posición está ocupada por una glutamina (Q). Los canales formados por cualquiera de estas 5 Introducción subunidades presentan una marcada rectificación entrante: dejan pasar más corriente en sentido entrante (potenciales de membrana negativos) que en sentido saliente (potenciales de membrana positivos) y son significativamente permeables a calcio (Hollman et al., 1991; Verdoorn et al., 1991; Burnashev et al., 1992). Por el contrario, la subunidad GluR2 presenta una arginina (R) en esta posición. Este cambio hace que estos receptores presenten rectificación saliente (dejan pasar más corriente a potenciales de membrana positivos que negativos) y no permeen calcio. La subunidad GluR2 es fenotípicamente dominante, determinando el comportamiento del canal. De forma contraria a los receptores de NMDA, que poseen varios sitios de modulación, los receptores de AMPA carecen, aparentemente, de esta compleja farmacología. En 1990 se describió que la sustancia Aniracetam potenciaba las respuestas de quisqualato a través de los receptores de AMPA (Ito et al., 1990). Investigaciones posteriores demostraron que una familia de compuestos, las benzotiodiacidas, eran capaces de reducir la desensibilización de los receptores de AMPA. En este sentido, los diazóxidos, clínicamente usados como antidepresivos, son un orden de magnitud más potentes que el aniracetan. El compuesto más efectivo de esta familia es la ciclotiacida que reduce notablemente la desensibilización rápida de los receptores de AMPA, aumentando las corrientes postsinápticas (Yamada et al., 1993; Patneau et al., 1993; Trussell et al, 1993). Los receptores de AMPA se distribuyen por todo el cerebro, existiendo cambios de expresión según la etapa del desarrollo y el tipo de subunidad. De forma general, estos receptores se encuentran altamente expresados en el hipocampo y en las capas superficiales de la corteza. Por el contrario, las capas profundas de la corteza y el caudado/putamen expresan niveles intermedios. Además de participar en la transmisión rápida de la información sináptica, estos receptores también han sido involucrados en algunas formas de plasticidad sináptica. Asimismo, una excesiva entrada de calcio a su través en condiciones patológicas también contribuye a la muerte de las neuronas. 1.3. RECEPTORES DE KAINATO. El receptor de kainato es el componente del sistema de señalización de glutamato que 6 Introducción se ha mostrado más elusivo a los investigadores. La falta de herramientas farmacológicas específicas ha impedido la detección de este tipo de receptores en neuronas del Sistema Nervioso Central (SNC), así como la determinación de sus papeles fisiológicos. Hasta la clonación de las subunidades que forman los receptores de kainato, la evidencia de su existencia como receptores independientes era inexistente y sólo recientemente se han definido los procesos en los que estos receptores están involucrados (Lerma, 1997a). 1.3.1. Biología molecular de los receptores de kainato. A. La familia de subunidades de receptores de kainato: GluR5-7, KA1 y KA2. La familia de subunidades que puede contribuir a la formación de receptores de kainato nativos se puede dividir en dos subfamilias. Una primera incluye las subunidades GluR5, GluR6 y GluR7 y muestran entre sí un grado de homología del 75-80 %. Todas estas subunidades generan canales funcionales homoméricos, conteniendo lugares de baja afinidad para la unión de kainato. La otra subfamilia la constituyen las subunidades KA1 y KA2, las cuales, aunque no forman canales homoméricos por sí mismas, generan lugares de alta afinidad para la unión de kainato cuando se expresan en células de mamíferos. Mientras que las secuencias aminoacídicas de estas subunidades muestran un grado de homología entre sí del 70 %, sólo presentan un 45 % de homología con las de la otra subfamilia. De forma similar a las subunidades de otros receptores de glutamato, las subunidades de los receptores de kainato se componen de aproximadamente 900 aminoácidos (≈100 KDa) y tienen una topología de membrana similar a la descrita para las subunidades de los receptores de AMPA y de NMDA. B. Distribución de las subunidades del receptor de kainato. Estudios de hibridación in situ han revelado que los receptores de kainato se encuentran ampliamente distribuidos en el sistema nervioso. Sin embargo, los patrones de expresión de las distintas subunidades son bastante heterogéneos (Wisden y Seeburg, 1993; Bahn et al, 1994). Así, el tránscrito de GluR5 se encuentra presente sobre todo en neuronas de los ganglios de la raíz dorsal (DRG), el subiculum, el núcleo septal, el córtex piriforme y cingulado así como en las células de Purkinje del cerebelo (Bettler et al., 1990); GluR6 es abundante en las células granulares del cerebelo, en el giro dentado y en la región CA3 del hipocampo, al igual que en el estriado. La subunidad GluR7 está presente con bajos niveles en el cerebro pero, se expresa en particular en las capas profundas del córtex cerebral, el 7 Introducción estriado, y en las neuronas inhibidoras de la capa molecular del cerebelo. KA1 se restringe de forma casi exclusiva a la región CA3 del hipocampo, aunque también se expresa en bajos niveles en el giro dentado, en la amígdala y en el córtex entorrinal (Werner et al., 1991). Por el contrario, el mensajero de KA2 se encuentra prácticamente en todos los núcleos del sistema nervioso. Aunque las diferentes subunidades de los receptores de kainato están ya presentes a nivel embrionario, la expresión emerge durante el período postnatal. Así, la cantidad de mRNA de GluR5 alcanza un pico entre P0 y P5, y luego comienza a declinar hasta los valores adultos. Esto ocurre también con otras subunidades. C. Diversidad estructural. Los receptores de kainato tienen la misma topología transmembrana y la misma estequiometría que los receptores de AMPA y de NMDA. Es decir, son tetrámeros en los cuales cada monómero lleva su propio lugar de unión y contribuye con una secuencia de aminoácidos específica a la formación del lumen del canal, llamado segmento M2. Ese segmento está compuesto por residuos hidrofóbicos que entran en la membrana formando una estructura en forma de horquilla. Además de esta secuencia hidrofóbica, cada subunidad posee tres segmentos transmembrana (M1, M3 y M4) distribuidos de tal forma que el dominio N-terminal de la proteína se sitúa extracelularmente y la región C-terminal está localizada intracelularmente (Hollmann et al., 1994; Roche et al., 1994; Taverna et al., 1994; Wo y Oswald, 1994; Bennet y Dingledine, 1995). Por analogía con los datos disponibles para los receptores de AMPA, se supone que el sitio de unión a glutamato en los receptores de kainato está formado por residuos distribuidos entre el dominio Nterminal y el lazo extracelular existente entre los segmentos M3 y M4 (Stern-Bach et al., 1994). Sin embargo, la estructura real del bolsillo de unión a kainato debe presentar diferencias significativas entre los distintos receptores de glutamato, que podrían ayudar a explicar, entre otras cosas, porqué los receptores de AMPA pueden unir kainato con alta afinidad mientras que algunos receptores de kainato no son capaces de unir AMPA. Algunas subunidades de los receptores de kainato se presentan como isoformas generadas por ayuste alternativo (e.g. Sommer y Seeburg, 1992). La subunidad GluR5 fue inicialmente descrita en dos formas moleculares que difieren en la presencia (GluR5-1) o 8 Introducción ausencia (GluR5-2) de un fragmento de 15 aminoácidos en el extremo N-terminal (Bettler et al., 1990). Análisis adicionales revelaron la existencia de dos formas moleculares adicionales para GluR2 (Sommer et al., 1992), que difireren en regiones del extremo carboxiterminal, por lo que la subunidad GluR5-2 puede presentar 3 regiones o dominios C-terminal diferentes. Estas isoformas se denominaron GluR5-2a (la más corta), GluR-2b y GluR5-2c (la más larga). Se desconoce si estas isoformas presentan el mismo comportamiento farmacológico, puesto que es la subunidad GluR5-2a la que se ha estudiado más, debido a que es la más eficiente a la hora de expresarse cuando se transfecta en células de mamíferos (Sommer et al., 1992). La existencia de ayuste o procesamiento aternativo no se ha descrito para las subunidades GluR6, KA1 y KA2. Por el contrario, recientemente se ha descrito que la subunidad GluR7 existe en dos variantes generadas por ayuste alternativo del extremo C-terminal, denominadas GluR7a y GluR7b. (Schiffer et al., 1997). Aunque se ha demostrado que las variantes por ayuste alternativo de otros receptores de glutamato son fundamentales para la función del receptor, las implicaciones funcionales de estas variaciones de los receptores de kainato todavía están por determinar. Se podría hipotetizar que estas variantes con diferentes secuencias C-terminales podrían interaccionar con diferentes proteínas del citoesqueleto dotando al receptor de un mecanismo de interacción diferencial con distintos sistemas de señalización (e.g. Nishimune et al., 1998; Osten et al., 1998; Ehlers et al., 1996). De forma similar a otros muchos receptores de neurotransmisores (ver Swope et al., 1992 para revisión), las subunidades de los receptores de kainato contienen sitios consenso de fosforilación para diversas proteínas kinasas. Así, un residuo de serina en la posición 684 parece ser el principal sitio de fosforilación para la proteína kinasa A (PKA). Se dispone de algunas subunidades clonadas de pollo, sapo o peces que no forman canales funcionales ni receptores heteroméricos con otras subunidades cuando se expresan en sistemas recombinantes. Estas subunidades han sido designadas proteínas que unen kainato (KBP, kainate-binding proteins), (ver Henley, 1994 para revisión). La edición de los mRNA es una fuente de heterogeneidad molecular para 9 Introducción determinadas proteínas (Cattaneo, 1991; ver Sommer y Seeburg, 1992 para revisión). Las subunidades GluR5 y GluR6, pueden sufrir edición postranscripcional del mRNA en un sitio localizado en la posición 590 (GluR6) o 591 (GluR5), conocido como sitio glutamina/arginina (Q/R). Es conocido que las propiedades de rectificación de los receptores de kainato están controladas exclusivamente por el sitio Q/R, de forma análoga a lo observado para los receptores de AMPA (Herb et al., 1992, Egebjerg y Heinemann, 1993; Köler et al., 1993). La versión no editada codifica para una glutamina (Q) y muestra una clara rectificación entrante, mientras que la versión editada codifica para una arginina (R) en esta posición y no rectifica (Egebjerg y Heinemann, 1993). GluR6 puede sufrir también edición en dos sitios adicionales localizados en el primer dominio transmembrana, donde una isoleucina puede ser sustituida por una valina (sitio I/V) y una tirosina puede ser sustituida por una cisteína (sitio Y/C). La edición del RNA en estas posiciones tiene consecuencias funcionales en los canales GluR6 homoméricos. Se ha visto que la permeabilidad a calcio de los canales GluR6 es modulada por el sitio Q/R, cuando los sitios I/V y Y/C del primer dominio transmembrana están editados (Khöler et al., 1993). El alcance de la edición es regulado durante el desarrollo. A diferencia de la subunidad GluR2 de los receptores de AMPA, una proporción significativa de subunidades de receptores de kainato no editadas está presentes tanto en el cerebro embrionario como en el adulto (Paschen et al., 1997; Bernard y Khrestchatisky, 1994). Igualmente, se ha observado la edición parcial de las subunidades GluR5 en el sitio Q/R también en neuronas individuales de hipocampo de rata adulta (Mackler y Eberwine, 1993). Aproximadamente el 60 % de los mRNAs de GluR5 se encuentran editados en tejido adulto, mientras que para GluR6 la proporción encontrada es del 80 %. 1.3.2. Propiedades funcionales de los receptores de kainato. A. Propiedades biofísicas. Propiedades de los receptores de kainato a nivel de canal único. Los receptores homoméricos editados GluR6(R) y GluR5(R) presentan una conductancia unitaria del orden de femtosiemens. La edición del sitio Q/R altera de forma drástica la conductancia de canal único, en el sentido que la forma no editada (Q) presenta una conductancia unitaria mucho mayor. La combinación de las subunidades GluR5 (R) o GluR6 (R) con KA2 produce receptores heteroméricos que tienen 2-3 veces mayor 10 Introducción conductancia que sus respectivos homoméricos en la forma R, pero muestran menor afinidad por su agonista (Howe, 1996; Swanson et al., 1996). Sin embargo, GluR6(Q)/KA2 presenta características de canal único que son indiferenciables de las que presentan los receptores homoméricos GluR6 (Q), aunque la coexpresión de KA2 y GluR5 (Q) acorta la duración de las respuestas cuando se compara con los canales homoméricos (Swanson et al., 1996). Este efecto está de acuerdo con el hecho de que los mRNAs de las subunidades KA1 y KA2 no son susceptibles de ser editados y ambos llevan una Q en el lugar Q/R. En células embrionarias de riñón (HEK) que expresan la forma Q de GluR5 o de GluR6 en receptores homoméricos (o heteroméricos con KA2), se realizaron registros de corrientes en la configuración de parches de membrana escindidos, que permitieron resolver tres niveles de subconductancias (Swanson et al., 1996). Recientemente, se ha descubierto que la corriente de canal único media de los receptores de glutamato también depende de cuántos de los sitios de unión para agonista están ocupados (Rosemund et al., 1998) mientras el receptor no entre en estado desensibilizado. B. Propiedades de activación y de desensibilización de los receptores de kainato. Un sello de identidad de los receptores de kainato es que en presencia continuada del agonista, la corriente que fluye a través de los canales activados decae rápidamente. Ello se debe a la desensibilización del receptor. En neuronas de hipocampo en cultivo el desarrollo de la desensibilización sigue una curva exponencial única (Lerma et al., 1993; Paternain et al., 1998), aunque también se ha observado procesos que se ajustan mejor a una exponencial doble (Lerma et al., 1993; Wilding y Huettner, 1997). La conclusión que permiten alcanzar los datos observados por algunos autores en diferentes preparaciones es que la desensibilización de los receptores de kainato es un proceso rápido y muy marcado. La desensibilización del receptor sigue una exponencial con una constante de tiempo de 11-13 ms tanto en neuronas de hipocampo como en canales GluR6 recombinantes (Lerma et al, 1993; Paternain et al, 1998). La velocidad de desensiblización estimada para receptores GluR6 en parches de membrana escindidos es más rápida (5-8 ms; Heckmann et al., 1996; Traynelis y Wahl, 1997), pero no muy diferente a los valores medidos en condiciones de célula completa. 11 Introducción Por el contrario, la recuperación de la desensibilización de los receptores de kainato es muy lenta. La recuperación total de la respuesta inducida por glutamato en neuronas de hipocampo se obtiene después de 15 segundos de una aplicación inicial, aunque la velocidad de recuperación depende del agonista usado para desensibilizar el receptor (Paternain et al, 1998). Es necesario esperar 1 minuto para recuperar la amplitud inicial en células de hipocampo, cuando se usa kainato para desensibilizar el receptor. En general la recuperación de la desensibilización sigue una exponencial simple, con una constante de tiempo que depende del tipo de subunidades que componen el receptor. Por ejemplo, los receptores homoméricos GluR5 cuando son desensibilizados con (S)-5-IW se recuperan en 2.5 minutos, mientras que los receptores heteroméricos GluR5/KA2 se recuperan de la desensibilización con una constante de tiempo de 12 segundos (Swanson et al. 1998). Independientemente del tipo de subunidad estudiada, estos datos implican que los receptores de kainato tienden a estar largos períodos de tiempo en el estado desensibilizado y que este estado puede ser considerado como un estado absorbente ya que el equilibrio está casi completamente desplazado en este sentido. El análisis de las curvas dosisrespuesta para el agonista endógeno, ya sea para receptores nativos o recombinantes, permiten concluir que los receptores de kainato no tienen una alta afinidad por glutamato. Sin embargo, en términos de desensibilización en el estado estacionario, los receptores de kainato son muy sensibles a la concentración ambiente de agonista. Estos receptores son aproximadamente dos órdenes de magnitud más sensibles al agonista para su desensibilización que para su activación. El cálculo de la desensibilización en el estado estacionario para los receptores de kainato expresados por las neuronas de hipocampo en cultivo da un valor de IC50, (i.e. la concentración de agonista a la cual el 50 % de los receptores se desensibilizan), de aproximadamente 3 µM para glutamato (Paternain et al., 1998). Esto significa que a concentraciones de glutamato incapaces de producir activación, los receptores están desensibilizados. Lo mismo ocurre cuando se usa kainato como agonista. Esta diferencia de sensibilidad entre activación y desensibilización también se encontró en receptores GluR6 recombinantes (Heckmann et al., 1996; Paternain et al., 1998). Curiosamente, los receptores GluR6 recombinantes son más sensibles a la desensibilización por glutamato (IC1/2 de 0.3 µM) que los nativos. Por el contrario, son menos sensibles a la activación por 12 Introducción glutamato (EC50 de 500-700 µM). C. Propiedades farmacológicas de los receptores de kainato. El principal obstáculo para el estudio de los receptores de kainato ha sido la falta de agonistas y antagonistas específicos. El desarrollo de agonistas y antagonistas específicos de los receptores de NMDA ha permitido determinar numerosos procesos en los cuáles estos receptores están involucrados. Sin embargo, la separación funcional de los receptores nativos de kainato y de AMPA en neuronas ha sido dificultosa ya que ambos receptores se activan por la misma colección de agonistas, y durante mucho tiempo se denominaron genéricamente receptores de tipo No-NMDA. Por ejemplo, el AMPA carece de efecto sobre los receptores de kainato recombinantes (Egebjerg et al., 1991; Herb et al., 1992) dado que es completamente inactivo sobre GluR6 y GluR7 y presenta una EC50 de 3 mM para GluR5 (Sommer et al., 1992). De forma similar, AMPA no activa los receptores de kainato nativos en cultivos de neuronas de hipocampo, y sólo a altas concentraciones a los presentes en células de los ganglios de la raíz dorsal (DRG) de la médula espinal (Huettner, 1990; Lerma et al., 1993; Wong et al., 1994). Sin embargo, kainato, aunque con menor afinidad que para los receptores de kainato, activa todos los tipos de receptores de AMPA a dosis relativamente bajas. C.1. Agonistas. La afinidad de kainato por sus receptores varía según la composición de subunidades de los mismos. Los valores de EC50 calculados para las distintas subunidades en sistemas recombinantes corresponden a 33.6 µM para GluR5 (Sommer et al., 1992), 299 µM para GluR6 (Paternain et al., 1998) y >1 mM para GluR7. Una situación similar se encontró cuando se usó glutamato como agonista (631 µM para GluR5, 270-762 µM para GluR6 y 5.9 mM para GluR7). Estos datos nos indican que los receptores de kainato no muestran alta afinidad ni por kainato ni por glutamato. Además de kainato y glutamato, se han encontrado algunas moléculas capaces de activar los receptores de kainato con cierto grado de especifidad. El domoato, una toxina que se aisló de mejillones contaminados con algas, tiene 20-25 veces mayor preferencia por los receptores de kainato que por los de AMPA en células de DRG y en receptores GluR5 recombinantes (Huettner et al., 1990; Sommer et al., 1992), aunque es inactivo sobre 13 Introducción GluR7 (Schiffer et al., 1997). El ácido (RS)-α-amino-3-hidroxi-5-tert-butil-4-isoxazolepropiónico (ATPA), un derivado de AMPA, ha sido descrito como agonista selectivo de receptores recombinantes GluR5. La EC50 para ATPA es 0.6 µM para receptores nativos en neuronas de DRG y de 2.1 µM para receptores GluR5 recombinantes, un orden de magnitud mayor que kainato (Clarke et al., 1997). Al mismo tiempo su afinidad por los receptores de AMPA es unas 500 veces más baja. El derivado de willardina, (S)-5-iodo-wilardina, muestra una selectividad de unas 130 veces mayor para los receptores de kainato que para los de AMPA. Su EC50 es 140 nM. C.2. Antagonistas. La búsqueda de antagonistas de los receptores de kainato no ha sido tan exitosa como la búsqueda de agonistas. La primera generación de antagonistas de los receptores de glutamato de tipo No-NMDA, las quinoxalinodionas (CNQX; DNQX y NBQX), son incapaces de discriminar entre los receptores de AMPA y los de kainato. El compuesto denominado NS102, otro antagonista de receptores de tipo No-NMDA, fue descrito como un producto que, en membranas de corteza, inhibe completamente la unión de kainato (Johansen et al., 1993). Además, NS102 antagoniza parcialmente las respuestas generadas por activación de receptores GluR6 en células embrionarias de riñón (HEK) con un IC50 de 2.2 µM (Verdoorn et al., 1994). Sin embargo, NS102 también es activo sobre los receptores de AMPA, aunque de forma menos potente. Por ello, esta droga se consideró ineficaz para separar las actividades mediadas por ambos receptores (Paternain et al., 1996; Wilding y Huettner, 1996). El desarrollo de las 2,3-benzodiazepinas como antagonistas de los receptores de AMPA (ver Vizi et al., 1996 para revisión), y la posterior demostración de que estos compuestos son selectivos para estos receptores (Paternain et al., 1995; Wilding y Huettner, 1995), ha sido crucial para el entendimiento de la función de los receptores de kainato. En particular GYKI 53655 (LY300168 según el código de Elli Lilly) es un antagonista no competitivo de los receptores de AMPA, capaz de bloquear completamente estos receptores a una concentración de 100 µM. A esta concentración, no ejerce 14 Introducción antagonismo sobre los receptores de kainato, y por ello ha sido usado para desenmascarar las corrientes de kainato en neuronas maduras (Paternain et al., 1995; Wilding y Huettner, 1997). Aunque GYKI 53655 es la mezcla racémica, presenta una IC50 de ∼1µM para antagonizar los receptores de AMPA (Paternain et al., 1995; Wilding y Huettner, 1997), siendo el isómero activo, LY303070, más potente. Recientemente, se ha desarrollado una nueva serie de compuestos que muestran una diferente sensibilidad para los receptores de AMPA y de kainato. LY293558 inhibe GluR5 pero no GluR6. Sin embargo, este compuesto también antagoniza las respuestas mediadas por los receptores de AMPA (Bleakman et al., 1996). Otro derivado, LY294486, muestra mayor selectividad para GluR5, y más recientemente se ha demostrado que el compuesto LY382884 es específico para antagonizar la unión a los receptores GluR5, no presentando afinidad significativa por las subunidades de los receptores de AMPA GluR1,2,3 ni por las de kainato GluR6, GluR7 y KA2 (Simmons et al., 1998). 1.3.3. Papeles fisiológicos de los receptores de kainato. La primera evidencia acerca de la existencia de receptores de kainato se obtuvo en el sistema nervioso periférico. Los primeros estudios mostraron que los nervios periféricos de la raíz dorsal de ratas inmaduras, específicamente las fibras de tipo C, se depolarizaban con kainato, un efecto que luego se observó que se debía a la inactivación de canales de sodio dependientes de voltaje (Davies et al., 1979; Agrawal y Evans, 1986). Estudios posteriores demostraron que la aplicación de kainato en células disociadas de los DRG inducía respuestas que se desensibilizaban rápidamente (Huettner et al., 1990), un efecto sorprendente puesto que, el conocido hasta ese momento, se correspondía con una respuesta a kainato sostenida debido a la activación de los receptores de AMPA por kainato. La posterior demostración de que los receptores de kainato producían respuestas desensibilizantes cuando se expresaban en sistemas heterólogos así como la localización de subunidades de receptores de kainato en células de DRG, condujeron a la aceptación de que las respuestas observadas se debían, efectivamente, a la activación de receptores de glutamato de tipo kainato. Este constituyó la primera evidencia de la presencia de receptores de kainato específicos. La disponibilidad del GYKI53655 como antagonista selectivo de los receptores de 15 Introducción AMPA, hizo posible el estudio de la participación de los receptores de kainato en la transmisión sináptica excitadora. Los primeros experimentos en cultivos de neuronas de hipocampo indicaban que, al contrario que los de AMPA, estos receptores no participaban en la transmisión sináptica excitadora. Aunque se demostró que las neuronas estudiadas tenían receptores de kainato, no fue posible provocar corriente postsináptica alguna en presencia de antagonistas de los receptores de AMPA y de NMDA (Lerma et al., 1997b). Sin embargo, estudios posteriores en rodajas de hipocampo han permitido identificar una serie de sinapsis donde los receptores de kainato parecen mediar una fracción pequeña de la corriente sináptica. En este sentido, se ha demostrado que se pueden inducir corrientes excitadoras postsinápticas (EPSCs) con características farmacológicas propias de los receptores de kainato en las sinapsis formadas por la fibras musgosas y las neuronas piramidales de CA3 (Castillo et al., 1997; Vignes y Collingridge, 1997). Sin embargo, estas respuestas sólo se observan en circunstancias de actividad presináptica repetitiva y podrían reflejar disposición extrasináptica de los receptores. La corriente inducida por estos trenes de estímulos está ausente en ratones a los que se les ha anulado el gen GluR6 (Mulle et al., 1998), lo que indica que los receptores de kainato que contienen la subunidad GluR6 participan en la transmisión sináptica a nivel fibra musgosa-neurona de CA3. Además del hipocampo, se ha identificado la presencia de receptores postsinápticos de kainato en neuronas de la amígdala lateral (Li y Rogawski, 1998), en neuronas del asta dorsal de la médula espinal (Li et al., 1999) (posiblemente implicados en nocicepción), en algunas células bipolares de la retina (DeVries y Schwartz, 1999) y en el córtex cerebral (Kidd e Isaac, 1999). 16 Objetivos OBJETIVOS. Teniendo en cuenta la disponibilidad del antagonista específico de los receptores de AMPA, GYKI53655, se planteó determinar el papel de los receptores de kainato en la fisiología cerebral. Para ello, en la presente tesis se han abordado los siguientes objetivos generales: 1. Determinar el papel de los receptores de kainato en la modulación de la transmisión sináptica inhibidora en el hipocampo. 2. Determinar el/los mecanismos mediante los cuáles estos receptores interfieren con la transmisión inhibidora. 17 Materiales y Métodos MATERIALES Y MÉTODOS. 1. CULTIVOS NEURONALES. Microcultivos de neuronas. Las neuronas de hipocampo se cultivaron en condiciones de microcultivo (Fotografía 1), según el protocolo descrito por Bekkers y Stevens (1991) con algunas modificaciones (Lerma et al., 1997b). Las células se disociaron de hipocampos aislados de embriones de 1718 días de gestación y se sembraron a una densidad de 100 células/mm2 en placas de Petri de 35 mm (Nunc). Cuarenta y ocho horas antes de comenzar el cultivo, el fondo de las placas de Petri fue recubierto con una fina capa de agarosa estéril (0.2 %) que se dejó secar en el interior de un incubador a 37 ºC. Antes de comenzar la disociación, se pulverizó sobre la agarosa una solución que contuvo poli-D-lisina (100 µg/ml) y laminina (16 µg/ml), con el objeto de producir múltiples microgotas de sustrato permisivo. La pulverización se ajustó para obtener islas con un diámetro de aproximadamente 100 µm, con una densidad de 30-50 gotas/mm2. El medio de cultivo usado fue Neurobasal con suplemento B27 al 4 %, (ambos de Gibco) (Brewer et al., 1993) al que se añadió glutamina (0.5 mM), mercaptoetanol (25 µM) (Grill y Pixley, 1993), penicilina (100 unidades/ml) y estreptomicina (100 mg/ml). En los días de cultivo 4, 7, 14 y 21 un 40 % del medio de cultivo de cada placa se reemplazó con medio fresco. Tras una semana de cultivo, la distribución aleatoria de las neuronas en las placas permitió encontrar numerosas islas ocupadas por una sóla neurona así como microgotas conteniendo dos o más células. En aquellas gotas que sólo albergaban una neurona, esta al crecer y desarrollarse termina estableciendo contactos sinápticos con ella misma (autapsis) y en aquellas gotas en las cuáles se encuentran dos neuronas estas establecen contacto entre sí y así se obtuvieron conexiones heterosinápticas. 2. RODAJAS DE CEREBRO. Las rodajas de hipocampo fueron preparadas de acuerdo con los procedimientos descritos (Stuart et al., 1993), a partir de ratas Wistar de 14-16 días de edad. Brevemente, tras sacrificar el animal por decapitación, el cerebro fue sumergido en una solución fría 18 Materiales y Métodos conteniendo (en mM) 124 NaCl, 2.69 KCl, 1.25 KH2PO4, 2 MgSO4, 1.8 CaCl2, 26 NaHCO3 y 10 glucosa (300 mOsm). El cerebro completo conteniendo los dos hipocampos se posicionó sobre la platina de un vibratomo (Word Precision Instruments, VSLM1) y se seccionó para obtener rodajas transversales de 350 µm. Las rodajas fueron transferidas a una cámara de incubación conteniendo la solución descrita que se mantuvo oxigenada continuamente (95 % O2, 5 % CO2, pH=7.4). Así se mantuvieron a temperatura ambiente durante, al menos, una hora antes de ser usadas para los registros electrofisiológicos. Las rodajas de cerebelo fueron obtenidas usando igual metodología que en el caso de las rodajas de hipocampo, salvo que en este caso se posiciona sobre la platina del vibratomo sólo el cerebelo que ha sido previamente separado del resto del cerebro, el cual se orientó de forma que al seccionar se obtuvieron rodajas sagitales del mismo. Las rodajas de corteza se obtuvieron usando igual metodología y soluciones que las descritas para la obtención de rodajas de hipocampo. 3. REGISTROS ELECTROFISIOLÓGICOS. 3.1. Cultivos. Se hicieron registros de actividad sináptica entre pares de neuronas interconectadas entre sí y también en neuronas que establecen conexiones sinápticas consigo mismas. En los pares de neuronas, una de ellas se utilizó como célula presináptica y la otra como neurona postsináptica, de forma que se hizo disparar la neurona presináptica (aplicándole pulsos depolarizantes) y las corrientes así evocadas se registraron en la célula postsináptica. En el caso de las conexiones autápticas la misma neurona se usó como pre y postsináptica. Los registros se obtuvieron usando las técnica de patch clamp en su configuración de célula completa (whole cell) en condiciones de fijación de voltaje (Hamill et al., 1981) para la célula postsináptica, para lo cual se usó un amplificador Axopatch-200A y en condiciones de fijación de corriente para la célula presináptica, para lo cual se usó un amplificador Axoclamp-2A. Para el caso de los registros obtenidos de conexiones autápticas se usó un amplificador Axopatch-200A y se obtuvieron los registros en condiciones de fijación de corriente. 19 Materiales y Métodos Fotografía 1. Dos neuronas creciendo en microcultivo. Estas neuronas establecen contactos sinápticos entre sí y consigo mismas. La imagen corresponde a neuronas de un cultivo de aproximadamente 21 días. 20 Materiales y Métodos Las micropipetas se fabricaron a partir de capilares de borosilicato en un estirador de pipetas horizontal (Sutter P-81), y tuvieron una resistencia de 2.5-5 MΩ cuando se llenaron con la solución interna. Con el fin de minimizar los cambios en el potencial de punta del electrodo cuando se intercambiaron las soluciones, el baño se conectó al amplificador mediante un puente salino de agar (2 %; NaCl 135 mM) y mediante un hilo de plata previamente clorurado en una solución de ácido clorhídrico 1 N. Las respuestas fueron supervisadas continuamente con un osciloscopio (Tektronix TDS 310). Todos los experimentos se llevaron a cabo a temperatura ambiente (22-25 ºC). Las señales electrofisiológicas se filtraron a 1-2 KHz, se digitalizaron mediante un convertidor analógico-digital (Lab-Master TM-40, Axon Instruments, USA) y se almacenaron en un ordenador personal usando el programa de adquisión de datos Clampex (versión 5.5.1, pClamp 5.6, Axon Instruments, USA). El programa Clampex igualmente permitió generar protocolos de estimulación que fueron usados para imponer cambios de voltaje a las células a través del amplificador. 3.2. Rodajas. Las rodajas se transfirieron a una cámara de registro, montada sobre un microscopio (Axioscop, Zeiss) y se mantuvieron en posición mediante una rejilla de hilos de nylon con un armazón de platino. Esta cámara se perfundió a razón de 3-4 ml/min con una solución externa igual a la descrita para la obtención de las rodajas. Las células de las rodajas se visualizaron por videomicroscopía infrarroja de contraste interdiferencial (IR-DIC) (Stuart et al., 1993) a través de un objetivo de inmersión en agua 40x. Las soluciones se aplicaron por gravedad cambiando entre cuatro líneas de perfusión mediante una electroválvula de multiples vías. Todos los experimentos se llevaron a cabo a temperatura ambiente (22-25 ºC). En el hipocampo, las corrientes inhibidoras postsinápticas (IPSCs) se indujeron mediante pulsos eléctricos aplicados con un electrodo bipolar colocado en el stratum oriens, a 50-150 µm del lugar de registro. Los electrodos se fabricaron con capilares de borosilicato presentando una resistencia de 5-10 MΩ cuando se llenaron con la solución interna. Las corrientes se registraron en el soma de neuronas de la capa CA1 o CA3 en la configuración de célula completa tras el establecimiento de sellos >1 GΩ. 21 Materiales y Métodos Registro Estimulación CA1 St. Oriens St. Radiatum CA3 Figura 3. Diseño experimental para la obtención de registros de las células de la región CA1 del hipocampo. Como se observa en la figura, el electrodo de estimulación se coloca en el stratum oriens y la pipeta de registro en las neuronas de la capa CA1 del hipocampo. Para obtener respuestas inhibidoras al registrar el soma de células de Purkinje se estimuló (con un electrodo igual al descrito para hipocampo) en la zona de la capa molecular cerca de la capa de células de Purkinje. Para obtener respuestas inhibidoras en las células de los núcleos profundos del cerebelo se estimuló en la zona de los terminales axónicos de las células de Purkinje y se registró en el soma de las células de los núcleos profundos del cerebelo. En las rodajas de corteza, se obtuvieron registros de los somas de la neuronas piramidales situadas en la capa V, estimulando las interneuronas situadas en las proximidades de estas neuronas piramidales. En todas los casos usó un amplificador Axopatch 200A (Axon Instruments). La resistencia de acceso fue monitorizada continuamente durante los registros, los cuales fueron rechazados si durante un experimento sufrió un cambió >15 %. Las señales electrofisiológicas se filtraron a 1-2 KHz, se digitalizaron y se almacenaron en el disco duro de un ordenador personal. Las corrientes elementales (miniatura, mIPSCs) fueron registradas durante periodos de 180-360 segundos bajo cada situación experimental en presencia de tetrodotoxina (TTX; 0.522 Materiales y Métodos 1µM). Estas respuestas miniatura fueron adquiridas usando el programa Axotape 2.0 (Axon Instruments) y almacenados en el disco duro del ordenador. 4. ANÁLISIS DE LOS REGISTROS. Los registros se analizaron con el programa Clampfit, versión 6.02 (Axon Instruments, USA). Las respuestas miniatura se analizaron, además de con Clampfit, con Fetchan 6.02 y con el programa de análisis de corrientes elementales Snap, diseñado en el Instituto Cajal por el Dr. Imanol Martínez-Padrón. El análisis estadístico se realizó usando los programas SigmaPlot y Origin, versiones 2.0 y 3.5, respectivamente. El grado de inhibición (o de incremento) de la amplitud de las respuestas se calculó como [1-(IPSCtest /IPSCcontrol)] ∗100. Donde, IPSCtest representa la amplitud de las corrientes (en pA) tras aplicar kainato, glutamato o cualquier otra sustancia, según el caso, a las células en cultivo o a las rodajas y IPSCcontrol representa la amplitud (en pA) de las corrientes antes de aplicar alguna sustancia a las células cultivadas o a las rodajas. El Coeficiente de Variación libre de ruido (CV) de las corrientes inhibidoras se calculó como: CV = σ 2 ( IPSC ) − σ 2 ( ruido) amplitud IPSC donde σ2(IPSC) y σ2(ruido) son la varianza de las respuestas de corriente y la varianza del ruido basal, respectivamente. La razón de CV en ambas situaciones (CVR) se obtuvo para cada neurona como CVka/CVcontrol. La gráfica comparando la variación en la amplitud media de las IPSCs (M) con respecto al estadístico 1/CV2 que refleja el cambio en la varianza de la amplitud de las respuestas, se construyó como se describe en Bekkers y Stevens (1990) y Malinow y Tsien (1990; ver también Siegelbaum y Kandel, 1991 para una mayor explicación). Las corrientes miniaturas fueron analizadas tras ser detectadas usando un programa de detección. Este calculó la primera derivada de la corriente registrada, teniendo en cuenta sólo 23 Materiales y Métodos aquellas respuestas que sobrepasan un umbral aleatorio, (típicamente 1.5 veces la amplitud del ruido; i.e. 8-12 pA). En algunos casos, la detección se hizo bajo control visual. Con el fin de comprobar si las condiciones de registro influenciaron las variaciones en la amplitud espontánea de las mIPSCs, se representaron tanto el tiempo de subida frente al tiempo de caída de los eventos registrados en cada célula. Estos dos parámetros no mostraron ningún tipo de correlación significativa por lo que se conluyó que el filtro electrotónico no fue una fuente importante de variación. La frecuencia de mIPSCs fue calculada mediante la exponencial de las distribuciones cumulativas tanto de los intervalos como de las amplitudes. Las distribuciones obtenidas tras la aplicación de kainato o de glutamato se compararon estadísticamente con las obtenidas en condiciones control usando el test de KolmogorovSmirnov. 5. SOLUCIONES. 5.1. Cultivos. Solución extracelular. La solución externa usada consistió (en mM) en: 165 NaCl, 2.5 KCl, 1.8 CaCl2, 2 MgCl2, 20 Glucosa y 10 HEPES. La solución se ajustó a un pH de 7.4 con NaOH. La osmolaridad, medida con un osmómetro de presión de vapor (Wescor 5500), se ajustó a 320330 mOsm mediante la adición de sacarosa, cuando fue necesario. Solución intracelular. Las pipetas de registro se llenaron con la siguiente solución (en mM): 130 K-metanosulfonato, 20 CsCl, 0.5 CaCl2, 5 MgCl2, 4 mM ATP-Mg, 10 EGTA y 10 HEPES. Esta solución se ajustó a un pH de 7.3 con KOH. La osmolaridad se ajustó a 315320 mOsm, siempre entre 5 y 10 mOsm inferior a la solución externa. 5.2 Rodajas. Soluciones extracelulares. Las solución externa usada en la mayoría de los experimentos estuvo compuesta por (en mM): 124 NaCl, 2.69 KCl, 1.25 KH2PO4, 2 MgSO4, 1.8 CaCl2, 26 NaHCO3 y 10 glucosa (300 mOsm) (1). 24 Materiales y Métodos En los experimentos en los que el calcio extracelular se varió, los componentes fueron los mismos, salvo que las concentraciones de CaCl2 se ajustaron a 0.5 o 10 mM. En algunos experimentos la concentración de Na+ se modificó, ajustándose a 60 mM, 37.5 y, en algún caso, 0 mM, de forma que la solución (1) se modificó para obtener estas condiciones, la osmolaridad se ajustó a 300 mOsm usando N-metil-Glucamina y HCl. Los agonistas o antagonistas que se emplearon se prepararon con estas soluciones a partir de soluciones madre altamente concentradas. Soluciones intracelulares. En los experimentos realizados en condiciones de fijación de voltaje (voltaje clamp) la solución interna usada estuvo compuesta por (en mM): 120 CsCl, 20 QX-314, 8 NaCl, 1 MgCl2, 0.2 CaCl2, 10 HEPES y 2 EGTA (pH=7.3, 287 mOsm). En los experimentos realizados en condiciones de fijación de corriente, la solución interna usada estuvo compuesta por (en mM): K-gluconato 120, 8 NaCl, 1 MgCl2, 0.2 CaCl2, 10 HEPES y 2 EGTA (pH=7.3). 6. EXPERIMENTOS IN VIVO. En colaboración con el Dr. Oscar Herreras del Departamento de Investigación del Hospital Ramón y Cajal, se realizaron algunos experimentos in vivo con el fin de comprobar el efecto de kainato sobre la excitabilidad hipocámpica en el circuito intacto. Los experimentos se realizaron en ratas Sprague-Dawley de 200-250 g de peso, anestesiadas con uretano (1-2 g/kg), cuya temperatura se mantuvo a 37 ºC usando una manta eléctrica. Los métodos quirúrgicos y estereotáxicos usados han sido descritos previamente por el grupo (ver Largo et al., 1996). Brevemente, se implantó un electrodo bipolar concéntrico en el lado homolateral de CA3 para estimular ortodrómicamente las colaterales de Schaffer y un electrodo de registro en la capa piramidal de CA1 con el fin de registrar el potencial de campo. El estímulo consistió en pulsos de 0.1 ms a una frecuencia de 0.1 Hz y a una intensidad que fue supramáxima para evocar una espiga poblacional. Igualmente, se implantó una sonda de microdiálisis, (diámetro externo, 220 µm; longitud 0.8-1 mm) manufacturada de la forma descrita (Herreras et al., 1994; Largo et al., 1996), la cual se usó para introducir kainato (3 µM) en el fluido extracelular. La sonda se bajó hasta una posición en la que toda la extensión dorso-ventral de CA1 quedó expuesta a la diálisis. 25 Resultados RESULTADOS. 1. La activación de los receptores de kainato produce una depresión de la transmisión sináptica GABAérgica en el hipocampo de rata. 1.1. Kainato produce una depresión de la transmisión GABAérgica. En microcultivos de neuronas hipocámpicas y en condiciones de bloqueo de los receptores de AMPA (con GYKI 53655, 100 µM) y de NMDA (con D-AP5, 50 µM), la aplicación de bajas concentraciones de kainato provocó una disminución de las respuestas inhibidoras postsinápticas (IPSCs) (Figura 4). Una concentración de kainato de 0.6 µM produjo una disminución del 43.0 ± 11.5 % (n=5). En nuestras condiciones de microcultivo, sólo el 25 % de los pares de células estaban conectados sinápticamente y menos del 20 % de estas conexiones eran inhibidoras. Por lo tanto, este modelo no permitió hacer un análisis exhaustivo del efecto de kainato sobre la inhibición sináptica. Sin embargo, se registraron un total de 6 pares, uno de los cuales fue descartado debido a que las respuestas inhibidoras mostraban una marcada disminución de la amplitud con el tiempo (rundown) que hizo imposible cualquier tipo de cuantificación. En el resto de los casos, se observó de forma clara un aumento en el número de fallos en la liberación de neurotransmisor (se genera un potencial de acción en la célula presináptica pero no se produce liberación de neurotransmisor) y en dos de estos pares se observó un aumento en la latencia de las IPSCs en presencia de kainato. En dos casos adicionales se evaluó el efecto de 0.6 µM de kainato en IPSCs autápticas (obtenidas en neuronas que establecen conexiones sinápticas consigo mismas). Kainato no tuvo ningún efecto en uno de estos casos, en el cual se comprobó la falta de receptores de kainato en la membrana, pero disminuyó la amplitud de las IPSCs en el otro. Para descartar una posible interacción del GABA con su receptor se examinó la respuesta a 100 µM GABA en presencia y en ausencia de kainato en neuronas de hipocampo en cultivo. La magnitud de las respuestas mediadas por GABA fue similar en ambas condiciones indicando que la acción de kainato sobre las IPSCs no se debe a una interacción del kainato con los receptores de GABA postsinápticos. Con el interés de extender estos resultados a modelos experimentales más fisiológicos, se pasó a realizar el estudio en rodajas de hipocampo de rata, las cuales retienen en gran medida las conexiones sinápticas originales. Para ello, las interneuronas GABAérgicas se estimularon con un electrodo emplazado en el stratum oriens del área CA1 y se registraron las corrientes 26 Resultados A Dos células en una isla Post Pre B Control Bicuculina 100 pA 40 ms C Control Kainato 0.6 µM Lavado 100 pA 40 ms Vm=-30 mV Figura 4. Kainato inhibe las eIPSCs mediadas por GABA en microcultivos de neuronas de hipocampo. En una microisla se registraron dos neuronas conectadas entre sí (A) y se generó un potencial de acción en una de ellas para evocar una respuesta sináptica en la otra. B) Las corrientes postsinápticas fueron sensibles a bicuculina (100 µM). Los registros son el promedio de 10 respuestas. C) La aplicación de kainato redujo de forma reversible la transmisión inhibidora, aumentando el número de fallos. Nótese, que durante la perfusión de kainato, la latencia de las eIPSCs aumenta ligeramente. Resultados similares se obtuvieron en 4 pares adicionales de células. inhibidoras en células piramidales de CA1 usando la técnica de patch-clamp en su configuración de célula completa. Estas células fueron identificadas visualmente mediante microscopía de infrarrojos. Para evitar la activación de los otros receptores ionotrópicos de glutamato, se incluyeron en el líquido de perfusión, de manera sistemática, el antagonista específico no competitivo de receptores de AMPA, GYKI 53655 (100 µM) y el antagonista de los receptores de NMDA, D-AP5 (50 µM). Debido a que la concentración de Cl- en la pipeta coincidió con la del líquido de perfusión (e.g. igual concentración de Cl- en el interior y en el exterior celular) la apertura de los canales de Cl- determinó la salida de este ion al exterior celular, observándose como corrientes entrantes. Las IPSCs así registradas se abolieron completamente en presencia de bicuculina, el antagonista específico de los receptores de GABA del tipo A, lo que demostró que sólo son responsables de las mismas receptores de este tipo. 27 Resultados Los estímulos se aplicaron a una frecuencia de 0.1-0.2 Hz (un estímulo cada 10 ó cada 5 segundos) y tras la estabilización de la amplitud de las eIPSCs se adicionó kainato a la cámara de registro a diferentes concentraciones (0.3-300 µM). Como muestra la figura 5A, durante la perfusión de kainato la amplitud de las eIPSCs decreció, recuperándose la amplitud original tras la retirada del kainato en la mayoría de los casos. Las concentraciones más efectivas de kainato fueron 0.3-10 µM (fig.5B), concentraciones mayores o menores a éstas fueron menos eficaces para disminuir las eIPSCs. De esta forma, la curva dosis-respuesta presentó forma de campana. Control Control Kainato 0.3 µM B Lavado 50 pA Kainato 10 µM Lavado 50 pA Control Kainato 200 µM Lavado Inhibicion de eIPSCs (%) A IC1/2=0.37 µM 80 EC50=20 µM 60 40 Incluso a bajas concentraciones,100 kainato indujo una corriente de membrana en dirección 20 pA 1 10 100 1000 entrante, indicando que tuvo un efecto depolarizante. Sin0.1 embargo, este efecto fue variable de 100 ms (µM)de kainato usada. célula a célula y no se observó una correlación evidente con laKainato concentración Figura 5. Efecto de kainato sobre la transmisión sináptica inhibidora en la capa CA1 del hipocampo en rodajas de cerebro de rata. A) Las corrientes sinápticas inhibidoras (IPSCs) fueron registradas en neuronas piramidales de CA1 y provocadas por estimulación del stratum oriens. A -60 mV de potencial de membrana, las IPSCs fueron entrantes debido a la concentración simétrica de cloro. B) Curva dosis-respuesta para la inhibición de las IPSCs inducida por kainato. El grado de inhibición se calculó como [1(IPSCKA/IPSCControl]·100 y se representó gráficamente frente a la concentración de kainato. Cada punto corresponde a un experimento. La línea continua representa la curva dosisrespuesta en estado estacionario para los receptores de kainato con parámetros determinados en células cultivadas de hipocampo. Ésta se calculó de acuerdo con la expresión: I= [KA] 1 + ni ( IC1 / 2 ) ni 1 +C (EC50 )na ⋅ 1 + [KA]n a con los siguientes parámetros: EC50= 20 µM; IC1/2= 0.37 µM; ni= 0.8 and na= 1.2. La curva resultante fue escalada para ajustar con los valores máximos, añadiendo un 25 % de “offset”. 28 Resultados Incluso a bajas concentraciones, kainato indujo una corriente de membrana en dirección entrante, indicando que tuvo un efecto depolarizante. Sin embargo, este efecto fue variable de célula a célula y no se observó una correlación evidente con la concentración de kainato usada. Por ejemplo, a 0.5, 10 y a 200 µM, la amplitud fue de 23 ± 3.4, 27 ± 5.4 y de 30 ± 3.5 pA, respectivamente, presentando un valor medio para las células estudiadas de 31.9 ± 3.1 pA (n=54). 1.2. La modulación de la transmisión GABAérgica por kainato es de origen presináptico. Para determinar si la modulación de las respuestas inhibidoras GABAérgicas se producía a través de un mecanismo de acción pre o postsináptico se usaron varias aproximaciones experimentales basadas en el análisis cuántico de la liberación de neurotransmisor. Dado que tanto en células microcultivadas como en rodajas de hipocampo, kainato produjo un aumento en el número de fallos en la transmisión sináptica, se cuantificó la proporción de éstos representándose frente al grado de inhibición de las eIPSCs observado, independientemente de la concentración usada de kainato. Para este análisis se seleccionaron 12 de las 54 células estudiadas en las rodajas, células en las que los fallos en la transmisión sináptica se pudieron identificar inequívocamente. Como muestra la figura 6B, el número de fallos se correlacionó bien con el grado de inhibición de las eIPSCs inducido por kainato. Ello sugiere que un cambio en la fiabilidad sináptica podría ser la causa de la reducción de las eIPSCs. Para comprobar si realmente un origen presináptico podía explicar estos cambios, se calculó el coeficiente de variación (CV) de las respuestas sinápticas tanto en condiciones control como en presencia de kainato, calculándose la razón entre ambos valores. Este valor, fue >1 en 48 de las 54 células analizadas (1.82 ± 0.1), lo cual según está establecido, señala un cambio en los parámetros de liberación como la explicación más probable para la reducción de las eIPSCs. Esta conclusión fue también evidente cuando se representó la variación de la amplitud media de las eIPSCs (M) frente al cambio en el estadístico 1/CV2 (el inverso del cuadrado del coeficiente de variación), que denota la varianza de las eIPSCs (Fig. 6C). Se ha establecido que un cambio en M no acompañado de un cambio correspondiente en la varianza denota que sólo el contenido cuántico (q) sufre modificación durante un tratamiento dado, indicando por tanto, un mecanismo postsináptico (Figura 6C, línea punteada) como causa de la modificación de la transmisión. Por el contrario, un cambio en la amplitud de las respuestas 29 Resultados acompañado por un cambio en el coeficiente de variación (i.e. en el estadísitico 1/CV2), sugiere una variación en los parámetros de liberación (n ó p), lo que indica fuertemente un origen presináptico como fuente de la variación (línea discontínua en la figura 6C; ver revisión de Thompson y Deuchars, 1995). Como se puede observar en la figura 6C, la reducción en la amplitud de las eIPSCs producida por kainato se vió acompañada de un cambio paralelo en 1/CV2, lo que indica un mecanismo de acción presináptico para kainato. Kainato 0.3 µM Control A C 1.5 r=0.93 85 r=0.7 Postsináptico 2 1.0 1/CV Inhibición de eIPSCs (%) B 65 0.5 45 Presináptico 25 0 25 50 75 100 Aumento de Fallos (%) 0.0 0.5 1.0 1.5 M Figura 6. Kainato activó un mecanismo presináptico para inhibir la transmisión GABAérgica. A) Kainato produce una reducción de la amplitud de las eIPSCs y un aumento en el número de fallos. B) El aumento en el número de fallos se correlacionó con el grado de disminución de las eIPSCs, independientemente de la concentración de kainato usada. Sólo se computaron las neuronas en las que los fallos pudieron determinarse de forma segura (n=12). Las líneas discontinuas corresponden al intervalo de confianza de la regresión del 95 %. C) Las amplitudes medias y su coeficiente de variación (CV) se midieron durante la aplicación de kainato y se normalizaron con sus respectivos valores control en cada célula. La variación proporcional de la amplitud (M) vs. la variacion proporcional en 1/CV2 en 49 neuronas se representó independientemente de la concentración usada de kainato. Nótese, que los datos experimentales siguen la relación predicha para una acción presináptica (línea discontinua) más que para una acción postsináptica (línea punteada). Por claridad, la recta de regresión (r=0.7, p<0.01) no se muestra, pero fue indistinguible de la diagonal. Está bien establecido que la frecuencia de las corrientes postsinápticas miniatura (mIPSCs) puede ser, en determinadas condiciones, un buen indicador de los cambios producidos en la probabilidad de liberación por efecto de alguna sustancia. Para determinar con 30 Resultados una tercera aproximación si las reducciones de las eIPSCs inducidas por kainato poseen origen presináptico, se estudiaron las corrientes miniatura (mIPSCs) antes, durante y después de la adición de kainato 3-10 µM a la rodaja. Para ello, se incluyó tetrodotoxina a una concentración de 0.5-1 µM para evitar la generación de potenciales de acción. De las 38 células estudiadas, en 29 de ellas se observó un descenso en la frecuencia de las mIPSCs (39.0 ± 3.8 %); en 3 células el kainato no tuvo efecto notable y en las 6 restantes se produjo un aumento en la frecuencia (17.3 ± 4.0 %). En conjunto la disminución media fue del 28.4 ± 4.8 % (n=38 células de 24 rodajas). Por el contrario, la amplitud de los mIPSCs no sufrió variación significativa. La figura 7 muestra un ejemplo representativo en una neurona piramidal de CA1. Estos datos también son congruentes con un modo de acción presináptico para kainato. Kainato 3 µM Recuperación 50 pA 4s C 1.0 0.8 Control 0.6 Kainato 3 µM 0.4 0.2 0.0 0 40 80 Amplitud (pA) 120 Probabilidad Acumulativa B Control Probabilidad Acumulativa A 1.0 0.8 Control 0.6 Kainato 3 µM 0.4 0.2 0.0 0 2 4 6 8 10 Intervalo (s) Figura 7. Efecto de kainato sobre las corrientes inhibidoras GABAérgicas postsinápticas miniatura (mIPSCs). Las respuestas miniatura se registraron en presencia de TTX 0.5-1 µM. A) Registros de las mIPSCs antes, durante y después de la aplicación de kainato. B) Distribución cumulativa de amplitud de las mIPSCs antes y durante la aplicación de kainato. Como se puede observar, no hay ningún cambio en la amplitud de las respuestas durante la aplicación de kainato. C) Distribución cumulativa de intervalos entre miniaturas antes y durante la aplicación de kainato 3 µM. Se puede observar que el intervalo de tiempo que pasa entre la ocurrencia de dos miniaturas aumenta cuando se aplica kainato a las rodajas. 1.3. El efecto de kainato no se debe a la activación de los receptores metabotrópicos de glutamato. Con el fin de descartar que un aumento en la concentración extracelular de glutamato inducido por la adición de kainato a la rodaja, y la subsecuente activación de los receptores de glutamato metabotrópicos, fuera el origen de las acciones observadas, se ensayó el efecto de kainato en presencia de inhibidores de los receptores de glutamato metabotrópicos. Esta 31 Resultados posibilidad se descartó dado que en presencia de MPPG y MCPG (1.5 mM cada uno de ellos), los efectos de kainato permanecieron inalterados. 1.4. CNQX antagoniza el efecto de kainato de forma dosis-dependiente. Aunque CNQX está considerado como el antagonista prototípico de los receptores de AMPA, también bloquea a los receptores de kainato. Dado que en nuestras condiciones experimentales los receptores de AMPA se encuentran bloqueados con GYKI 53655, a todos los efectos se puede usar CNQX como antagonista de los receptores de kainato. Como se puede observar en la figura 8A, cuando se aplicó kainato 10 µM en presencia de CNQX, éste fue menos efectivo en reducir la amplitud de las eIPSCs, efecto que fue dosis-dependiente. Estos resultados sugirieron que la reducción de la amplitud de las eIPSCs por kainato se debió, Inhibición de las eIPSCs (%) 100 80 60 * 40 * 20 0 KA 10 µM ** Reducción de frec. mIPSCs (%) por kainato 3 µM efectivamente, a la activación de receptores de glutamato de tipo kainato. + CNQX 25 µM 50 µM 100 µM 60 45 30 15 0 Control Cóctel Figura 8. Propiedades farmacológicas de la acción de kainato sobre las IPSCs. A) La acción inhibidora de kainato fue antagonizada de forma dosis-dependiente por CNQX. (* p ≤ 0.05; ** p ≤ 0.01, test t de Student). Las barras representan la media ± EEM de 6 (control), 3 (CNQX 25 y 100 µM) ó 2 (CNQX 50 µM) experimentos. B) Kainato fue igualmente efectivo en reducir la frecuencia de las mIPSCs aún en presencia de un cóctel de antagonistas de otros receptores. Este consistió en: MCPG (1.5 mM); MPPG (1.5 mM), Naloxona (100 µM), Atropina (50 µM), 2-OH-Saclofén (100 µM), DPCPX (0.1 µM), D-AP5 (50 µM) y GYKI 53655 (100 µM). 1.5. Kainato no actúa sobre otro tipo de receptores descritos que regulan la liberación de neurotransmisor. Para descartar la posibilidad de que un mensajero indeterminado, liberado tras la depolarización neuronal y/o glial fuera la causa del efecto descrito, al activar algún receptor 32 Resultados presináptico, kainato se aplicó en presencia de un cóctel de antagonistas compuesto por: MCPG y MPPG (1.5 mM cada uno), naloxona (100 µM), atropina (50 µM), hidroxisaclofén (50-150 µM) y DPCPX (0.1 µM). Sin embargo, el efecto de kainato sobre las mIPSCs estuvo presente aún en presencia de este cóctel (Fig. 8B), con lo que se pueden descartar los receptores metabotrópicos de glutamato, de opiáceos, muscarínicos, de GABAB y A1 de adenosina como mediadores de estos efectos. La somatostatina, aplicada al baño, no tuvo efecto alguno sobre las eIPSCs, con lo cual también se puede descartar que tras la adición de kainato a la rodaja ésta sea liberada y deprima las corrientes inhibidoras GABAérgicas. 1.6. El agonista endógeno de los receptores de kainato, glutamato, ejerce el mismo efecto que kainato sobre las eIPSCs. Para verificar si el agonista endógeno de los receptores de kainato, glutamato, era capaz de actuar sobre los receptores de kainato para regular la liberación de GABA, este agonista se aplicó a las rodajas en presencia de una mezcla de bloqueantes compuesto por MCPG y MPPG (1.5 mM cada uno), GYKI 53655 (100 µM) y D-AP5 (200 µM) para prevenir la activación de cualquier tipo de receptor de glutamato excepto la de los de kainato. Al igual que el kainato, el glutamato redujo la amplitud de las corrientes inhibidoras presentando una curva dosis-respuesta en forma de campana (Figura 9). Los datos experimentales se ajustaron bien a la curva dosis-respuesta para glutamato calculada en condiciones de estado estacionario a partir de los parámetros de activación e inactivación obtenidos en neuronas en cultivo. Al igual que ocurrió cuando se aplicó kainato, el coeficiente de variación (medido como 1/CV2) fluctuó de manera paralela a la variación de la media (M) entre las condiciones test y control. Ello sugirió de nuevo una localización presináptica para los receptores que median este efecto. Igualmente en presencia de glutamato, la frecuencia de las mIPSCs disminuyó ligera pero significativamente (14.0 ± 4.0 %, n=5, Glutamato 30 µM), sin que se produjera cambio alguno en la amplitud de las mismos. El efecto de glutamato sobre las eIPSCs (42.2 ± 6.1% de reducción, glutamato 10 µM) fue antagonizado por CNQX (100 µM; Fig. 10B) ya que en presencia de este antagonista, glutamato produjo una depresión de la amplitud de las eIPSCs de un 5.9 ± 12.3 % (n=4). 33 Resultados A Control Glutamato 3 µM B Recuper. 80 Inhibición eIPSCs (%) 200 pA 100 µM 200 pA 1 mM EC50 =300 µM IC1/2= 2.5 µM 60 40 20 0 0.1 100 pA 100 ms 1 10 100 1000 Glutamato (µM) A eIPSCs Glu Control 50 pA 100 ms + Cóctel B Glu Control 20 pA 100 ms Reduccion IPSCs (%) Figura 9. Efecto de glutamato sobre las eIPSCs. La aplicación de glutamato en las condiciones de estado estacionario del receptor, reduce la transmisión inhibidora en neuronas de CA1 de acuerdo con la ventana predicha según las propiedades de activación e inactivación de los receptores de kainato. A) Corrientes sinápticas inhibidoras (evocadas al estimular en el stratum oriens del hipocampo) registradas en rodajas en neuronas piramidales de CA1 antes, durante y después de la aplicación de glutamato. B) Curva dosis-respuesta. Los puntos rellenos corresponden a los valores promedio ± EEM, mientras que los círculos no rellenos corresponden a valores individuales. La línea continua representa la curva dosis-respuesta teórica para las condiciones de equilibrio del receptor, calculada con los parámetros que se indican. 60 40 20 * 0 Glu + CNQX 10 µM Cóctel 100 µM Figura 10. Propiedades farmacológicas del efecto de glutamato sobre las eIPSCs. A), B) La acción depresiva de glutamato (10 µM) sobre las eIPSCs se mantuvo en presencia de un cóctel de antagonistas de otros receptores (ver figura 8) y fue antagonizada por CNQX. 34 Resultados Finalmente, el efecto de glutamato sobre la amplitud de las eIPSCs persistió en presencia del cóctel anteriormente descrito (Fig. 10) (MCPG, MPPG, naloxona, atropina, 2-ohsaclofén, DPCPX, APV y GYKI 53655). 1.7. Kainato deprime la transmisión GABAérgica en la región CA1 del hipocampo in vivo. De los resultados anteriores, obtenidos in vitro, es evidente que los receptores de kainato regulan la liberación de GABA. Estudios realizados en colaboración con el laboratorio del Dr. Herreras, del Departamento de Investigación del Hospital “Ramón y Cajal”, indicaron que este resultado también era el observado en experimentos in vivo. En este sentido, se hicieron experimentos en el hipocampo de ratas anestesiadas, en las que se implantaron cánulas de diálisis. Se registraron potenciales de campo extracelulares en la capa piramidal de CA1, evocados por estímulos eléctricos en las colaterales de Schaffer (Figura 11A). En estos registros, la onda negativa representa los potenciales de acción sincrónicos de una gran población de neuronas (espiga poblacional). Se ha demostrado que la amplitud de la espiga poblacional es proporcional al número de células que dispara al mismo tiempo. La estimulación por pares de pulsos sirve para medir la efectividad de la inhibición GABAérgica mediante la comparación de la respuesta al segundo pulso (test) con la respuesta al primero (condicionada). Estos 2 pulsos fueron separados 40 ms, y las respuestas de campo se promediaron cada 5 ensayos. En este intervalo, predomina la activación inhibidora mediada por receptores de tipo GABAA. Este experimento se repitió en 5 animales con resultados similares (Fig. 11). La efectividad de la inhibición fue drásticamente reducida cuando se introdujo kainato 3 µM en la cánula de diálisis. Se eligió una concentración baja de kainato para evitar la activación de los receptores de AMPA (se estima que la concentración que se alcanza extracelularmente es aproximadamente 1/10 de la concentración perfundida, Herreras et al., 1994). Incluso a esta baja concentración de kainato, una espiga poblacional, ausente en los controles, apareció claramente en respuesta al pulso test, aumentando con el tiempo de perfusión. Este fenómeno fue reversible tras retirar el kainato del líquido de perfusión (Figuras 11A y 11B). Por el contrario, no se observó efecto alguno sobre la amplitud de la espiga inducida por la respuesta condicionada, lo cual fue indicativo de que a estas concentraciones, kainato no produjo una depolarización significativa. Además, en dos animales se hicieron registros sobre la capa sináptica (stratum radiatum), no observándose un efecto significativo sobre los potenciales postsinápticos de campo excitadores (EPSPs). De acuerdo con su acción sobre la inhibición sináptica, durante la exposición a kainato, en el electroencefalograma hipocámpico apareció un 35 Resultados patrón de actividad similar al de los estados epilépticos (Fig. 11C), siendo evidentes la presencia de espigas epilépticas en la actividad electroencefalográfica (Fig. 11D inferior). Es conocido que este tipo de actividad aparece en vivo y/o en rodajas después de exponer el tejido A B 40 ms Ringer Kainato 3 µM t=1' t=3' t=8' t=18' Espiga Poblacional (mV) a sustancias convulsantes que bloquean la inhibición mediada por GABA. 12 Cond. 8 4 Test 0 C Kainato 3 µM 0 25 50 75 100 125 Tiempo de perfusión (min) t=60' 2 mV 5 mV 1s 2 mV 20 ms Figura 11. Efecto de bajas concentraciones de kainato sobre la inhibición GABAérgica en experimentos llevados a cabo in vivo en el hipocampo. A) Para testar la inhibición recurrente, se registraron potenciales de campo en la capa piramidal de CA1 evocados por pares de pulsos en la ruta de las colaterales de Schaffer. Los estímulos fueron separados 40 ms. Nótese la ausencia de espiga poblacional (la señal negativa puntiaguda) en respuesta al segundo estímulo (test) en condiciones control. Esta espiga poblacional se desarrolla progresivamente tras la inclusión de kainato en el lugar de perfusión. A tiempos de exposición prolongados aparecieron dobles espigas en el potencial de campo (inferior), lo cual es una indicación de actividad epiléptica. B) Evolución de la amplitud de las espigas poblacionales evocadas por el primer (condicionada, Cond.) y el segundo (Test) estímulo. La respuesta test fue aumentada de forma reversible mientras que la primera espiga se mantuvo constante a lo largo del experimento. C) Kainato indujo actividad epiléptica en el hipocampo, caracterizada por la presencia de espigas interictales. El recuadro representa un registro expandido de 1 segundo. 36 Resultados 2. La modulación de la liberación de GABA por los receptores de kainato involucra una función metabotrópica. 2.1. La activación de una proteína G sensible a toxina pertúsica y de proteína kinasa C son necesarias para que se produzca la disminución de liberación de GABA mediada por la activación de receptores de kainato. Una vez determinado que la activación de los receptores de kainato produce una depresión de la transmisión inhibidora GABAérgica y que estos receptores deben estar localizados en los terminales presinápticos de los contactos inhibidores, se intentó determinar el mecanismo mediante el cual se produce este efecto de disminuir la liberación del GABA. Para determinar si los efectos de kainato sobre la liberación de GABA involucraban la activación de una proteína G, se analizó, en primer lugar, la capacidad de este agonista para deprimir las eIPSCs mediadas por GABA en rodajas que previamente habían sido incubadas con toxina pertúsica (PTx) (5 µg/ml, 3-4 horas a 37 ºC). Al igual que en anteriores experimentos, la activación de los receptores de AMPA por kainato se evitó añadiendo GYKI 53655 (100 µM) en todos las experimentos y la activación de los receptores de NMDA se evitó incluyendo D-AP5 (50 µM). En las rodajas tratadas con PTx, las eIPSCs presentaron cinéticas de activación y de deactivación similares a las observadas en las rodajas sin tratar. En estas rodajas, kainato fue incapaz de alterar la amplitud de las respuestas inhibidoras (Figura 12). Kainato (3 µM) produjo una reducción de la amplitud de las eIPSCs del 9.4 ± 5.3 % (n=6 neuronas de 5 rodajas diferentes) en rodajas que previamente fueron tratadas con PTx. Por el contrario, el tratamiento similar con toxina colérica (20 µg/ml) tuvo un efecto mínimo en la acción de kainato sobre la amplitud de las eIPSCs (50.6 ± 3.8 % de reducción, n=5 rodajas). Estos resultados indican que una proteína G sensible a toxina pertúsica es necesaria para que tengan lugar los efectos depresores de kainato sobre las eIPSCs en el hipocampo. Las proteínas G pueden interaccionar de forma directa con dominios estructurales de canales de calcio voltaje-dependientes (De Waard et al., 1997; Zamponi et al., 1997), siendo éste uno de los mecanismos por el que actúan algunos receptores acoplados a proteínas G (e.g. Dolphin et al., 1986; Boland y Bean, 1993). Para determinar si un mensajero soluble estaba implicado o no en el efecto de kainato, se determinaron los efectos de kainato sobre las eIPSCs tras incubar las rodajas con estaurosporina, un inhibidor de proteínas kinasas de amplio 37 Resultados espectro. A una concentración de 500 nM, estaurosporina abolió completamente la acción de kainato sobre las eIPSCs (2.0 ± 13.9 %, 3 µM de kainato; Figura 13), indicando que la activación de una proteína kinasa, más que una modulación directa de los canales de calcio por una proteína G, era responsable de la acción observada sobre la transmisión GABAérgica. A Kainato 3 µM Control Lavado 100 pA C Reducción de las eIPSCs(%) Amplitud de las eIPSCs B Control PTx 5 µg/ml 1.5 1.0 0.5 Control 0.0 0 50 Kainato 3 µM 100 150 Lavado 50 ms 60 40 20 ** 0 Control 200 Tiempo (s) PTx ChTx 5 µg/ml 20 µg/ml Figura 12. La modulación de la liberación de GABA mediada por receptores de kainato involucra una proteína G sensible a toxina pertúsica. A) Inhibición de las IPSCs por kainato. Las respuestas fueron registradas de neuronas de CA1 cuyo potencial de membrana fue mantenido a -60 mV y fueron evocadas por estimulación del stratum oriens. Los registros son el promedio de 5 respuestas consecutivas. B) Curso temporal de la amplitud de las eIPSCs antes, durante y depués de aplicar kainato en rodajas sin tratar (círculos negros) y en rodajas tratadas con toxina pertúsica (círculos abiertos). Los valores son la media+EEM de 5 respuestas consecutivas. C) Promedio del efecto de kainato 3 µM sobre la amplitud de las eIPSCs en rodajas normales (control) y en rodajas tratadas con toxinas pertúsica (PTx) o colérica (ChTx). Las barras representan la media+EEM de resultados obtenidos de 20, 6 (5 rodajas) y 5 (5 rodajas) neuronas, respectivamente. Para determinar qué kinasa estaba involucrada en esta acción, se realizaron experimentos farmacológicos con inhibidores específicos. La inhibición de la proteína kinasa dependiente de cAMP (PKA) por H-89, un inhibidor específico, al cual la membrana plasmática es permeable, aplicado a 500 nM (≅ 10 veces su IC50) no modificó el efecto de kainato sobre las eIPSCs (63.1 ± 8.3 %, n=5 neuronas de 3 rodajas; Figura 13). Por el contrario, la calfostina C, un inhibidor 38 Resultados altamente específico de proteína kinasa C (PKC), previno de forma dosis-dependiente la acción de kainato (Fig. 13B), demostrando que la activación de la PKC está involucrada en la depresión de la liberación de GABA inducida por kainato. De acuerdo con este resultado, la activación de PKC con ésteres de forbol (PDBu, 0.5 µM) ocluyó totalmente la acción inhibidora de kainato 3 µM (Figura 14). La incubación de las rodajas con Sp-cAMPS (50 µM), un análogo permeante de cAMP, activador de PKA, no afectó a la reducción de la amplitud de las eIPSCs inducida por kainato (Figura 14B), siendo ésta reducida en un 13 % (de un 63.2 % a un 50.4 ± 11.1 % en presencia de Sp-cAMPS, n=5 células de 4 rodajas). Igualmente, el bloqueo de fosfolipasa C (PLC) también atenuó en gran medida el efecto de kainato sobre la liberación de GABA (Fig. 14B). Incubando las rodajas con U-73122 (10 µM), inhibidor de la PLC, se redujo de forma significativa la atenuación de las eIPSCs inducida por kainato (21.2 ± 8.6 % de inhibición, n=7 células de 4 rodajas; cf. 63.2 % en el control), un efecto que fue específico ya que el análogo inactivo, U-73343, no ejerció acción alguna sobre la inhibición inducida por kainato (52.0 ± 8.5 % de inhibición, n=4 células de 4 rodajas; Figura 14B). B Amplitud de las eIPSCs 2.0 Control Estaurosp. 500 nM 1.5 1.0 0.5 C 0.0 0 Kainato 3 µM 100 200 Lavado 300 400 Reducción de las eIPSCs (%) A Tiempo (s) 60 40 ** 20 0 ** ** . C C sp 9 u ro M H -8 M lfo s. M lf os. a t n µ a µ M a s µ 0 E 50 0 .5 C 0 .5 C 1 Figura 13. La modulación de las amplitudes de las eIPSCs por kainato involucra la activación de una proteína kinasa C. A) Experimento representativo en el cual la acción de kainato sobre la amplitud de las eIPSCs se evaluó en ausencia (círculos negros) y en presencia de estaurosporina (círculos abiertos). Los valores son la media+EEM de 5 respuestas consecutivas. B) Efecto de inhibidores específicos a los cuales la membrana es permeable, de proteínas kinasas sobre la acción de kainato. La inhibición de las PKC por estaurosporina y por calfostina C previno el efecto de kainato (3 µM). La inhibición de la PKA por H-89 no tuvo ningún efecto. Las barras representan la media+EEM de 4, 5, 4 y 6 neuronas respectivamente. La barra horizontal rayada representa el intervalo de confianza para el decremento en amplitud de las eIPSCs inducido por kainato (3 µM) en rodajas sin tratar. **p<0.01, test t de Student. 39 Resultados B Amplitud de las eIPSCs 1.5 Reducción de las eIPSCs (%) A Control PDBu 0.5 µM 1.0 0.5 0.0 Control Kainato 3 µM Lavado 50 150 250 350 60 40 * 20 0 450 Time (s) ** S u 3 2 34 DB MP 12 73 µ M -73 µ M -cA M P 5 µ M U U 0 0. Sp 0 µ 1 10 5 Figura 14. La estimulación de la PKC ocluyó la acción de kainato. A) Experimento representativo mostrando que kainato no afecta la amplitud de las eIPSCs en presencia del éster de forbol PDBu. Los valores son la media+EEM de 5 respuestas consecutivas. B) La inhibición de fosfolipasa C, por U73122, previno el decremento en amplitud inducido por kainato. El análogo inactivo, U73343, no tuvo efecto. La activación de PKC (con PDBu), pero no la de PKA (con Sp-cAMPS) ocluyó la acción depresiva de kainato. Las barras son la media+EEM de datos obtenidos de 7, 4, 5, y 4 rodajas, respectivamente. La barra horizontal rayada representa el intervalo de confianza para el decremento de la amplitud de las eIPSCs inducido por kainato (3 µM) en rodajas sin tratar. *p<0.05; **p<0.01, test t de Student. Estos resultados indicaron que los receptores de kainato activan una proteína G acoplada a fosfolipasa C (PLC). La activación de PLC induciría la producción de diacilglicerol, el cual es sabido que es un potente activador de PKC. 2.2. Kainato activa una población particular de PKC. La activación de la PKC produce un aumento de la liberación de neurotransmisor en sinapsis tanto excitadoras (e.g. Malenka et al., 1986) como inhibidoras (e.g., Copogna et al., 1995). Sin embargo, nuestros resultados, muestran que kainato reduce la probabilidad de liberación de GABA a través de un fenómeno mediado por PKC. La explicación más sencilla consistente con estos resultados, es que existen dos poblaciones separadas de PKC y que la activación de cada una produce efectos diferentes. Para determinar si éste era o no el caso, se diseñaron experimentos de oclusión con el éster de forbol PDBu. Tras perfundir la rodaja con PDBu (0.5µM), kainato fue totalmente inefectivo en decrecer la amplitud de las eIPSCs (Figura 14A); esto es, el éster de forbol ocluyó completamente la acción de kainato. Con el fin de evitar la modificación de los receptores de GABA por los ésteres de forbol (e.g. Vaello et al., 1994), 40 Resultados se midió la frecuencia de miniaturas (mIPSCs), registrados en presencia de TTX (0.5 µM) (Fig. 15 ). PDBu triplicó la frecuencia de mIPSCs (311.9 ± 68.4 % , n=6 células). Kainato produjo un aumento adicional de la frecuencia de mIPSCs (66.0 ± 21.2 %). El efecto de PDBu fue prevenido con la inclusión de 1 µM de calfostina C (19.0 ± 19.0 de aumento, n=4), lo que demuestra que la acción general de la estimulación de PKC ocluye totalmente la acción de kainato. Cuando el experimento se realizó a la inversa, incluyendo PDBu una vez que kainato hubo realizado su efecto, la inhibición fue totalmente revertida, aumentando la frecuencia de minis hasta 166.7 ± 39.6 % del valor control (n=5). Por tanto, kainato fue incapaz de ocluir la acción del éster de forbol. Estos resultados indicaban, que los receptores de kainato tienen acceso a una población limitada y específica de PKC, cuya activación no compromete a la población total. A Control Kainato 3 µM Kainato 3 µM + PDBu 0.5 µM C 75 75 mIPSCs (#/30 s) mIPSCs (#/30 s) B 20 1 s pA 50 25 Kainato PDBu 0.5 µM 0 0 2 4 6 50 25 PDBu Kainato 3 µM 0 8 10 12 14 0 Tiempo (min) 2 4 6 8 10 12 14 Tiempo (min) Figura 15. PDBu ocluyó la acción inhibidora de kainato pero kainato no ocluyó la acción estimuladora de PDBu. A) Registros de las corrientes inhibidoras miniatura (mIPSCs), registradas en presencia de TTX (0.5 µM) antes (superior), durante la aplicación de kainato (intermedio) y durante la aplicación de kainato más PDBu (inferior). B) Un experimento representativo mostrando el efecto de PDBu y de la subsecuente inclusión de kainato (3 µM) sobre la frecuencia de mIPSC. C) En una neurona diferente, cuando el efecto de kainato sobre las mIPSC alcanzó un estado de equilibrio se añadió PDBu. Las líneas punteadas indican en cada caso el valor medio de la frecuencia de mIPSC en esta célula en particular. Más que decrecer la frecuencia de minis, kainato la aumenta en presencia del éster de forbol. Resultados similares se observaron en 5 (B) y 6 (C) neuronas. 41 Resultados 2.3. La acción de kainato es independiente de la actividad del receptor como canal iónico. Para determinar si la acción metabotrópica de kainato era dependiente o independiente de la actividad del receptor de kainato como canal, se examinó si el efecto inhibidor de kainato cambiaba cuando se modificaba la concentración extracelular de sodio. Es conocido que la mayor parte de la carga a través del canal es acarreada por este ion en la mayoría de los receptores ionotrópicos de glutamato. El sodio fue sustituido por concentraciones equimoleculares de N-metil-D-glucamina. La reducción de hasta el 60% del sodio extracelular no impidió la generación de potenciales de acción, y por tanto, se pudo estudiar la actividad sináptica evocada. En estas circunstancias, kainato fue igualmente efectivo en reducir la amplitud de las eIPSCs (Fig. 16C). Menores concentraciones de sodio (<60 mM) no permitieron la generación de potenciales de acción. A concentraciones de Na+ menores (37.5 mM; 25% de lo normal, en presencia de TTX), kainato fue igualmente efectivo en reducir la frecuencia de mIPSCs (Figura 16A). Este experimento se repitió eliminando completamente el sodio de la solución extracelular con un resultado idéntico (Fig. 16B). Estos datos indican que la inhibición de la liberación de GABA por kainato es esencialmente independiente de la función como canal del receptor. A Na extracelular = 37.5 mM B 60 IPSCs miniaturas C 80 Kainato 3 µM 30 pA Lavado 2s 40 20 0 Na+ Na+ Na+ 150 mM 37.5 mM 0 mM Reducción de amplitud (%) Control Reducción frecuencia (%) + IPSCs evocadas 60 40 20 0 Na+ 150 mM Na+ 60 mM Figura 16. La regulación de la liberación de GABA por kainato es independiente de la actividad del receptor como canal iónico. A) Registros mostrando el efecto de kainato sobre la frecuencia de mIPSCs en condiciones de bajo Na+ extracelular (25% del total). La reducción del sodio extracelular no afectó al grado de reducción en la frecuencia de mIPSC inducido por kainato (B) ni al grado de reducción de las respuestas evocadas (eIPSC) (C). Las barras son la media+EEM de los valores obtenidos de 12, 5 y 4 neuronas, respectivamente (B) y 20 y 3, respectivamente (C). 42 Resultados 2.4. AMPA no activa una cascada similar. El antagonista competitivo de los receptores de tipo AMPA/kainato, CNQX, previene de forma dosis-dependiente la acción inhibidora de kainato sobre las eIPSCs (ver Figura 8A). En los experimentos descritos, la activación de los receptores de AMPA se previno añadiendo 100 µM de GYKI 53655 a la solución de perfusión. Sin embargo, como GYKI es un antagonista no competitivo de los receptores de AMPA (Paternain et al., 1995), no impide la unión del agonista al receptor. Tras la demostración de que la subunidad del receptor de AMPA GluR1 podía producir la activación de una proteína G de forma independiente a su actividad como canal (Wang et al., 1997), todavía podría ser posible que GYKI 53655 impidiera la apertura del canal tras la unión del agonista pero no la subsecuente activación de la proteína G. Para descartar la posibilidad de que kainato estuviese produciendo su actividad moduladora por su acción como agonista de los receptores de AMPA, se determinó si la unión del agonista prototípico de los receptores de AMPA, era capaz de producir acciones similares sobre la amplitud de las eIPSCs. Para ello, se incluyó GYKI 53655 (100 µM) y ciclotiazida (50 µM) (una sustancia que elimina la desensibilización de los receptores de AMPA). En estas circunstancias, 50 µM de AMPA fue completamente inefectivo en reducir la amplitud de las eIPSCs (101.2 ± 4.7 % del valor control, n=4), pudiéndose descartar que los receptores de AMPA estén involucrados en la depresión de la transmisión GABAérgica mediada por kainato. 2.5. La activación de los receptores metabotrópicos de glutamato de tipo I no ocluye la acción de kainato. Es sabido que los receptores metabotrópicos de glutamato de tipo I están acoplados a una proteína G, la cual es capaz de activar PLC y producir IP3 y diacilglicerol (ver Conn y Pin, 1997). Con el fin de comprobar si la activación de esta ruta en los terminales inhibidores podía ocluir la acción de kainato, se estudiaron los efectos de kainato en presencia de DHPG, un agonista específico de los receptores metabotrópicos del grupo I (Schoepp et al., 1994). DHPG (100 µM) produjo un aumento de la actividad espontánea así como un cambio en la corriente de fijación. Transcurridos 5 minutos, la actividad espontánea cesó y la corriente de fijación volvió a sus valores originales, aunque la amplitud de la respuesta inhibidora evocada permaceció aumentada un 29.6 ± 13.3 %, n=4. En esta situación, la adición de kainato (3µM) produjo un decremento de las eIPSCs de igual magnitud que la de los experimentos control (Fig.17), produciéndose una reducción del 67.5 ± 4.7 % (n=4; cf. 63.2 % en el control). 43 Resultados Estos resultados demuestran que o bien no existen receptores mGluRs de tipo I en los terminales inhibidores GABAérgicos del hipocampo estudiados o que esta ruta de señalización no converge con la activada por los receptores de kainato. Inhibición eIPSCs (%) por KA 3 µM 80 Figura 17. La activación de los receptores metabotrópicos de tipo I no ocluye la acción de kainato. En presencia de DHPG 100 µM (un agonista de los receptores metabotrópicos de glutamato de tipo I) kainato fue capaz de producir una depresión de las eIPSCs de igual cuantía que en condiciones control. Las barras representan la media ± EEM de 26 neuronas en el caso control y de 5 neuronas en presencia de DHPG. 60 40 20 0 Control + DHPG 100 µM 2.6. La activación de los receptores de kainato modula la señal de calcio. Es conocido que la activación de la PKC facilita la liberación de neurotransmisor porque, entre otras acciones, potencia las corrientes de Ca2+ al fosforilar los canales responsables de esta corriente. Para que la activación de la PKC produzca una reducción de la liberación de neurotransmisor, como la encontrada en el presente trabajo para los receptores de kainato, sería necesario que la PKC interaccionara con un canal de potasio, facilitando su actividad, o con un canal de calcio, inhibiéndolo. Para comprobar la existencia de alguna relación entre los receptores de kainato y las corrientes de potasio o de calcio, se redujeron en primer lugar las corrientes de potasio añadiendo tetraetilamonio (TEA, 2.5 mM) a la rodaja. El bloqueo de los canales de potasio aumentó la liberación de neurotransmisor, observándose un aumento de la amplitud de las eIPSCs así como un enlentecimiento en la desactivación de las respuestas (Figura 18A). Sin embargo, el bloqueo de los canales de potasio no ocluyó el efecto de kainato 3 µM (54.1 ± 4.6 % de inhibición con 2.5 mM de TEA, n=8 neuronas de 6 rodajas; Fig 18). Un experimento similar se llevó a cabo en presencia de bajo (0.5 mM) y alto (10 mM) calcio. El aumento de la concentración extracelular de calcio produjo un incremento de la amplitud de las eIPSCs, indicando que, al igual que cuando se bloquearon los canales de K+, la cantidad de calcio que ingresaba en el terminal estaba aumentada. Sin embargo, en este caso la forma de la respuesta no se alteró (Fig. 19A). En presencia de Ca2+ 10 mM, kainato redujo la amplitud de las eIPSCs sólo un 21.0 ± 4.3 % (n=4), mientras que a calcio 0.5 mM, kainato fue 44 Resultados más efectivo en deprimir la transmisión GABAérgica (70.6 ± 12.3 %, n=4) que en las condiciones control (i.e 1.8 mM calcio). A Control TEA 2.5 mM Normalizadas C T 100 pA 1.5 Control TEA 1.0 0.5 0.0 100 ms C C 50 Kainato 150 Lavado 250 350 Reducción de eIPSCs (%) Amplitud de las eIPSCs B 60 40 20 0 Control 450 Tiempo (s) TEA 2.5 mM Figura 18. Efecto del bloqueo de los canales de K + sobre la acción inhibidora de kainato. A) Tetraetilamonio (TEA) aumentó la amplitud de las eIPSC y prolongó su curso temporal de decaimiento. Los registros son la media de 5 respuestas consecutivas, las cuáles están normalizadas en el panel de la derecha. B) Un experimento representativo mostrando que el bloqueo de los canales de K+ no tiene efecto sobre la acción inhibidora de kainato. C) Promedio de todos los experimentos. Las barras representan la media ± EEM de 5 (control) y 8 (TEA) neuronas. Estos experimentos indican que la estimulación de la PKC tras la activación de los receptores de kainato resulta en la fosforilación de elementos que están involucrados en la liberación de neurotransmisor inducida por calcio. 45 Resultados A Ca2+ 2 mM Ca2+ 10 mM Normalizadas 2 10 150 pA C 2+ 1.5 Reducción de eIPSCs(%) Amplitud de las eIPSCs B Ca 0.5 mM 2+ Ca 10 mM 1.0 0.5 0.0 50 ms C 50 Kainato 150 250 Lavado 350 450 Tiempo (s) 80 60 40 ** 20 0 Control Ca2+ Ca2+ 0.5 mM 10 mM Figura 19. Un aumento de la concentración extracelular de Ca2+ atenuó la reducción de las eIPSC inducida por kainato. A) Con el Ca2+ extracelular elevado se aumentó la amplitud de las eIPSCs con un efecto mínimo sobre su curso temporal. Ambos registros se representan en el panel derecho normalizados y sobreimpuestos. B) Experimento representativo mostrando la falta de efecto de kainato (3 µM) a altas pero no a bajas concentraciones de Ca2+. C) Comparación de las reducciones de amplitud de las eIPSCs obtenidas a concentraciones normales (1.8 mM), bajas (0.5 mM) y altas (10 mM) de Ca2+ extracelular. Las barras son la media ± EEM de 20, 4 y 4 neuronas respectivamente. **p<0.01, test t de Student. 3. Las interneuronas de hipocampo presentan dos poblaciones de receptores de kainato con mecanismos de señalización diferentes. 3.1. El aumento de actividad espontánea y el descenso de amplitud de las respuestas evocadas son efectos producidos por la activación de receptores de kainato diferentes. En nuestras condiciones experimentales, la aplicación de kainato fue generalmente acompañada por un aumento en la frecuencia de respuestas espontáneas (sIPSCs), debido al hecho de que kainato produjo la depolarización de las interneuronas (Fig. 20). 46 Resultados A Kainato 3 µM Control Interneurona del S.O. -65 mV B Recuperación 2s 30 mV Célula Piramidal 1s 40 pA Figura 20. Kainato depolariza las interneuronas y aumenta la frecuencia de las sIPSCs en las neuronas piramidales. A) Efecto depolarizante de kainato 3 µM en presencia de GYKI 53655 100 µM y D-APV 50 µM sobre las interneuronas del stratum oriens. B) Simultáneamente, se observa un aumento en la frecuencia de las IPSCs espontáneas en la neuronas piramidales de CA1. Estos efectos revierten tras el lavado de kainato. Para clarificar si la depresión de la transmisión inhibidora GABAérgica evocada y el aumento de actividad espontánea observado son fenómenos relacionados de forma directa o no, se buscaron agonistas de los receptores de kainato que permitieran discriminar entre ambos efectos. Para ello, se determinó el efecto de diferentes agonistas de los receptores de kainato sobre la excitabilidad de las interneuronas del stratum oriens así como sobre las eIPSCs registradas en neuronas piramidales de CA1, en condiciones de bloqueo de los receptores de NMDA y AMPA. En algunos experimentos, se usó el nuevo compuesto SYM 2206, que a 100 µM muestra especifidad como antagonista de los receptores de AMPA sobre los receptores de kainato. En los experimentos con glutamato, se evitó la activación de los receptores metabotrópicos de glutamato incluyendo MPPG y MCPG (1-1.5 mM cada uno). En estas condiciones, kainato (3 µM) deprimió la amplitud de las eIPSCs (63.2 ± 2.4 %; n=26). La acción de kainato fue mimetizada por ATPA, un derivado de AMPA descrito como agonista específico de los receptores de kainato formados por subunidades GluR5 (Clarke et al.1997). A 10 µM, ATPA causó una reducción de la amplitud de las eIPSCs del 58.5 ± 6.2 % (n=8). La actividad de ATPA fue parcialmente antagonizada por CNQX. Por el contrario, S-AMPA fue 47 Resultados inefectivo en reducir la amplitud de las eIPSCs a concentraciones suficientes para activar los receptores de AMPA (i.e. 50 µM; -0.1 ± 6.4 %, n=9), siendo altamente efectivo a concentraciones suficientemente altas para activar los receptores de kainato (92.5 ± 3.5 % de reducción a 500 µM; n=4). Todos los agonistas anteriormente descritos también aumentaron la frecuencia de sIPSCs en las células piramidales. Sin embargo, el agonista endógeno de estos receptores, glutamato, cuando se aplicó a bajas concentraciones (3-10 µM) fue capaz de inhibir la eIPSCs de forma significativa (42.2 ± 6.1 %, n=11 p<0.01, Fig. 21), una acción que, también fue prevenida por CNQX (5.9 ± 12.3 %, n=4). Sin embargo, a pesar de este efecto, glutamato no aumentó la frecuencia de sIPSCs (100.0 ± 7.0 %, n=4), indicando que, a esta concentración, glutamato no produjo la depolarización de las interneuronas. Glutamato fue activo incluso a más bajas concentraciones (3 µM, 20.0 ± 4.2 % de depresión; n=4; p<0.01), sin alterar la frecuencia de eventos espontáneos en la célula sometida a registro (93 ± 5 %; n=7) (Fig. 21a). En estas mismas neuronas, se comprobó que kainato (0.3 µM) causó el incremento de la frecuencia de sIPSCs (447 ± 103 %; n=6), deprimiendo la amplitud de las eIPSCs un 32.1 ± 4.6 % (n=3). Por el contrario, se observó que ATPA a 1 µM, era capaz de depolarizar las interneuronas del stratum oriens (11.8 ± 2.1 mV; n=4) y aumentar de forma notable su frecuencia de disparo y subsecuentemente, la frecuencia de sIPSCs en las células piramidales (1179 ± 261 %; n=7; p<0.001). Sin embargo, a esta concentración, ATPA, redujo la amplitud de las eIPSCs sólo un 16.1 ± 3.2 %; n=11; p<0.001). A concentraciones más bajas (0.3 µM), ATPA todavía fue efectivo en aumentar la frecuencia de sIPSCs (303 ± 65.6 %, n=5; p<0.001) siendo inhábil sobre el eIPSCs (4.0 ± 7.8 % de aumento de la amplitud, n=9) (Fig. 21C). La acción de AMPA fue similar a la de ATPA en cuanto a que a bajas concentraciones (i.e. 50 µM) aumentó de forma drástica la frecuencia de sIPSCs (973 ± 178 %; n=5), mientras que no tuvo ningún efecto sobre las eIPSCs (Fig. 22A,C). 48 Resultados Figura 21. A) A 3 µM el agonista endógeno de los receptores de kainato, glutamato, no aumentó la frecuencia de actividad espontánea inhibidora (sIPSCs) en neuronas piramidales, mientras que sí disminuyó la amplitud de las respuestas evocadas (eIPSCs), inducidas por estimulación del stratum oriens (los registros de la derecha son el promedio de 12 respuestas). A 10 µM, glutamato tuvo un efecto mayor sobre las respuestas evocadas sin alterar la frecuencia de sIPSCs (B). C) El agonista de los receptores de kainato de tipo GluR5, ATPA, aumentó de forma notable la frecuencia de sIPSCs (izquierda) sin afectar a la amplitud de las eIPSC (derecha). Estos resultados indican que la sensibilidad para el agonista de los receptores que depolarizan las interneuronas (i.e. aumento de la frecuencia de sIPSCs) y la de los receptores que controlan la liberación de GABA (i.e. amplitud de las eIPSCs) es diferente. Ello se ilustra también por la falta de correlación entre el aumento de sIPSCs y el grado de disminución de la amplitud de las eIPSCs inducida por diferentes agonistas de los receptores de kainato (Fig. 22B). Por tanto, se puede concluir que los receptores que median cada efecto tienen una naturaleza diferente. Para mejor sustanciar que el efecto de la activación de los receptores de kainato sobre la amplitud de las eIPSCs y sobre la depolarización de la membrana está mediada por receptores diferentes, se estudió el efecto de kainato tras incubar las rodajas con toxina pertúsica (5 µg/ml, 3-5 h a 37 ºC), estaurosporina (0.5 µM) y bisindolilmaleimida (inhibidor de la PKC) (BIS, 0.1 µM). 49 * 100 75 50 25 0 60 40 20 * 3 * 10 0.3 * 1 0 Redución de elPSCs (%) * Reducción de eIPSCs (%) * 1200 800 400 50 µM 80 60 40 20 0 100 600 1100 1600 Frecuencia de sIPSC (%) Glutamato ATPA AMPA 1300 * * 900 * * 500 200 100 100 AMPA 50 µM 2 4 0 6 8 Tiempo de Registro (min) 10 Amplitud de las eIPSC (%) C B Frecuencia de sIPSC (%) Frecuencia de sIPSC (%) A Resultados Figura 22. A) Efecto de diferentes concentraciones de glutamato, ATPA y AMPA sobre la frecuencia de sIPSCs (barras sombreadas) y sobre la amplitud de las eIPSCs (barras negras). * p<0.001, test t de student. B) La frecuencia de sIPSCs durante la aplicación de agonistas de receptores de kainato (normalizada con respecto del control) se representa frente al grado de reducción de las eIPSCs obtenido frente a cada concentración de agonista, (glutamato, ATPA, AMPA y kainato). Nótese la falta de correlación entre estos dos parámetros (r2<0.01). C) Curso temporal de la acción de AMPA (en presencia de GYKI 53655 100µM) sobre la frecuencia de sIPSC y sobre la amplitud de las eIPSCs. Cada punto representa el valor medio (±EEM; n=6) de periodos consecutivos de 30 segundos. (* p<0.001, ANOVA y test t de student). En todos los casos la acción de kainato sobre la modulación de las eIPSCs fue severamente impedida (Fig. 23 y 24A). Sin embargo, tanto kainato como ATPA fueron todavía capaces de depolarizar las interneuronas, no encontrándose diferencias ni en el grado de depolarización ni en el grado de aumento de la actividad espontánea en las rodajas tratadas y no tratadas. Todas las interneuronas del stratum oriens registradas (n=12) fueron sensibles a ATPA (1-10 µM). En promedio, 3 µM de kainato depolarizó las interneuronas un poco menos que 1 µM de ATPA (8.6 ± 1.8 mV, n=7 y 12 ± 1.8 mV, n=5, respectivamente). La frecuencia de disparo inducida por 3 µM de kainato fue de 3.9 ± 0.4 Hz, un valor que no fue alterado en rodajas tratadas con PTx (3.5 ± 0.5, n=5) o con estaurosporina (4.2 ± 1.0 Hz, n=3) (Fig. 25B). Estos resultados, indican que, a diferencia del control de la liberación de GABA, la depolarización de las interneuronas mediada por la activación de los receptores de kainato no 50 Resultados requiere un acoplamiento a una cascada de segundos mensajeros. A B Interneurona del stratum oriens Kainato Tratada con PTx Control KA 2 min KA 6 min KA 10 min Lavado 20 mV -65 mV 100 pA 100 ms 10 s Figura 23. El efecto depolarizante de kainato pero no su acción sobre las IPSCs persiste en rodajas tratadas con PTx. A) Efecto depolarizante de 3 µM de kainato sobre una interneurona del stratum oriens, en una rodaja tratada con toxina pertúsica. B) Kainato no reduce las IPSCs registradas en neuronas piramidales de CA1 de una rodaja tratada con toxina pertúsica. (5 µl/ml) durante 3-5 horas a 37 ºC antes de ser registrada. Frecuencia de sIPScs (%) Frecuencia de disparo (Hz) 80 (26) 60 1000 PTx 0 ** (7) (12) (6) No Tratada BIS 750 500 250 0 ** (6) BIS ** (11) 20 ** (4) Calfos.C 40 Estaurosp. C B No tratada Depresión de las eIPSCs inducida por kainato 3 µM (%) A 6 5 4 (3) (4) (5) 3 2 1 0 No Tratada PTx Estaurosp. Figura 24. A) El decremento de la amplitud de las eIPSCs inducido por kainato (No tratada, n=26) se abolió en rodajas tratadas con toxina pertúsica (PTx, 5 µl/ml), o con cada uno de los siguientes inhibidores de PKC, aplicados a concentraciones 10 veces su IC50: estaurosporina (Estaurosp., 0.5 µM), Calfostina C (Calfos. C, 0.5 µM), o bisindolilmaleimida (BIS, 0.1 µM). B) El tratamiento con PTx o con estaurosporina no tiene efecto sobre el aumento en la frecuencia de disparo de las interneuronas del stratum oriens inducido por 3 µM de kainato. C) La inhibición de la PKC no fue capaz de modificar el aumento de actividad espontánea (sIPSC) inducido por kainato. Los valores son la media ± EEM del número de neuronas indicado en paréntesis. ** p<0.001, test t de Student. 51 Resultados Finalmente, se pretendió determinar si la falta de efecto de kainato sobre las eIPSCs por inhibición de la PKC se podía revertir lavando el inhibidor. Para ello, se intentó registrar la misma neurona durante largos períodos de tiempo. En la neurona de CA1 mostrada en la figura 25A registrada de una rodaja tratada con estaurosporina, kainato produce un aumento en la amplitud de las eIPSCs, más que una disminución. Sin embargo, tras 60 minutos de lavado de la estaurosporina, la aplicación de kainato fue efectiva en reducir la amplitud de las eIPSCs registradas en la misma neurona (Fig. 25B). También se llevó a cabo el experimento inverso. Tras demostrar la efectividad de kainato para disminuir la amplitud de las eIPSCs en una célula de una rodaja control, ésta se incubó con estaurosporina. Como se puede ver en la figura 25D, la incubación durante 30 minutos con estaurosporina disminuye notablemente la acción de kainato para deprimir la amplitud de las eIPSCs. Estos mismos resultados fueron replicados en 4 rodajas diferentes. A Estaurosporina 0.5 µM Control KA 3 µM 200 pA 50 ms C Ringer Control KA 3 µM B Lavado de la estaurosporina (60’) Control KA 3 µM Promedio (n=10) C KA 100 pA 50 ms 200 pA 50 ms KA KA C 100 pA 50 ms D Estaurosporina 0.5 µM (30’) Control KA 3 µM Promedio (n=10) C 60 pA 50 ms Promedio (n=10) Promedio (n=10) C 40 pA 50 ms 60 pA 50 ms KA 40 pA 50 ms Figura 25. A) El efecto inhibidor de kainato sobre la amplitud de las eIPSCs registradas en neuronas de CA1 desapareció en rodajas que previamente habían sido tratadas con estaurosporina. A la izquierda se muestran respuestas individuales superpuestas y a la derecha su promedio. Esta neurona se pudo mantener estable durante el periodo en el que la estaurosporina se lavó (60 min). En esta situación, la aplicación de kainato indujo una reducción significativa de la amplitud de las eIPSCs (B). C) En una rodaja control, kainato aumentó la actividad espontánea, reduciendo la amplitud de las corrientes inhibidoras evocadas. D) Después de incubar esta misma rodaja con estaurosporina durante 30 minutos, el efecto de kainato sobre la amplitud de las eIPSCs registradas en la misma célula se eliminó. Nótese, que el aumento en la actividad espontánea (sIPSCs) persistió, y que la depolarización postsináptica, inducida por kainato, fue similar en cada caso. Estos resultados se obtuvieron en cuatro ocasiones. 52 Resultados 4. La modulación de la liberación de GABA mediada por la activación de receptores de kainato presinápticos es un fenómeno común a diferentes zonas del cerebro. Para determinar si en otras áreas del cerebro la activación de los receptores de kainato produce igualmente la modulación de la liberación de GABA, se llevaron a cabo experimentos sobre la transmisión inhibidora en otras zonas del cerebro. 4.1. Hipocampo. Se registraron las IPSCs evocadas en las neuronas piramidales de CA3 tras la estimulación de las interneuronas del stratum oriens o del stratum lucidum en presencia de GYKI 53655 y de D-AP5. La aplicación de kainato 3 µM a las rodajas de hipocampo en las que se registraron las eIPSCs en CA3 produjo un 60.2 ± 6.0 % de disminución de la amplitud media de estas respuestas (n=5) (Fig. 26), un efecto similar al observado en CA1. Igualmente, la aplicación de kainato produjo una corriente entrante postsináptica de 69.4 ± 2.1 pA. También se observó un aumento significativo en la actividad espontánea, que aumentó hasta el 1249 ± 360 % (n=5). A Control Kainato 3 µM 100 pA C 80 100 ms 1.5 60 1/CV2 Inhibición de eIPSCs (%) B Recuperación 40 20 0 1.0 0.5 0.0 0.0 Kainato 3 µM 0.5 1.0 1.5 M Figura 26. En la región CA3 de hipocampo kainato también reduce la liberación de GABA. A) Registros en los que se observa la acción depresora de kainato sobre las eIPSCs. Este efecto revierte tras el lavado de kainato. B) Cuantificación del efecto de kainato 3 µM sobre las eIPSCs registradas, en esta zona de hipocampo (n=5 células de 5 rodajas diferentes). C) El estadístico 1/CV2 varía de forma paralela a la amplitud media de las corrientes, lo que indica un mecanismo de acción presináptico. 53 Resultados El estudio del coeficiente de variación de estas respuestas indicó un mecanismo de acción presináptico como la acción más probable de kainato. Como se puede observar en la figura 26B, el punto medio se sitúa sobre la diagonal, lo que indica que la variación en la amplitud se corresponde con un cambio proporcional en los parámetros de liberación. 4.2. Cerebelo. En primer lugar se estudió el efecto de kainato sobre la transmisión sináptica inhibidora a nivel de las células de Purkinje del cerebelo. Para ello, el electrodo de estimulación se colocó en la capa molecular, cerca de la capa de las células de Purkinje, registrándose las corrientes inhibidoras en las neuronas de Purkinje en condiciones de bloqueo de los receptores de AMPA y de NMDA. La aplicación de kainato 3 µM produjo un descenso de la amplitud media de las eIPSCs de un 44.2 ± 9.3 % (n=15) (Fig. 27). También se observó la generación de una corriente entrante de 14.0 ± 5.4 pA, lo que sugiere la presencia de receptores de kainato funcionales en las células de Purkinje. Sin embargo, estos experimentos no revelaron cambio significativo en la actividad espontánea (sIPSCs) tras la aplicación de kainato (96.4 ± 5.3 %, n=8) El estudio del coeficiente de variación (Fig. 27B) indicó un mecanismo presináptico de acción, ya que que, como en el caso anterior, el punto medio se situó sobre la diagonal. En segundo lugar, se estudió el efecto de kainato sobre la liberación de GABA que tiene lugar en las sinapsis establecidas por las células de Purkinje sobre las neuronas de los núcleos profundos del cerebelo. En estas rodajas el electrodo de estimulación se colocó sobre las fibras que forman los axones de las células de Purkinje, registrándose las respuestas evocadas en las neuronas que forman dichos núcleos (DCN). Al aplicar kainato (3 µM) a estas rodajas en condiciones de bloqueo de los receptores de AMPA y NMDA, se observó un descenso de la amplitud media de las eIPSCs de un 44.0 ± 4.6 % (n=7) (Fig.28). En estas células, la aplicación de kainato produjo un cambio de la corriente de fijación de 34.3 ± 4.4 pA. Igualmente se valoró si la aplicación de kainato tuvo efecto sobre la actividad espontánea, observándose que en presencia de kainato se produjo un aumento de 4 veces en esta actividad (397.2 ± 95.1 %, n=5). Una vez más se determinó el coeficiente de variación, comprobándose que el punto medio caía sobre la diagonal de la gráfica, lo cual, sugiere un mecanismo de acción presináptico. 54 Resultados A Control Kainato 3 µM Recuperación 50 pA C 60 1.5 40 1.0 1/CV2 Inhibición de eIPSCs (%) B 100 ms 20 0.5 0.0 0 0.0 Kainato 3 µM 0.5 1.0 1.5 M Figura 27. Efecto de la activación de los receptores de kainato sobre las eIPSCs registradas de células de Purkinje de cerebelo. (A y B) Kainato (3 µM) produjo una reducción de la amplitud media de las eIPSCs. C) El estudio del cambio de 1/CV2 con respecto del cambio de la amplitud media de las corrientes sugiere un mecanismo de acción presináptico. A Control Kainato 3 µM Recuperación 50 pA 50 ms C 60 1.5 40 1.0 1/CV2 Inhibición de eIPSCs (%) B 20 0 0.5 0.0 0.0 Kainato 3 µM 0.5 1.0 1.5 M Figura 28. Efecto de la activación de los receptores de kainato sobre las eIPSCs registradas de neuronas de los núcleos profundos de cerebelo. (A y B) Kainato (3 µM) produce una reducción de la amplitud media de las eIPSCs que varió de forma paralela al cambio del estadístico 1/CV2 (C). 55 Resultados 4.3. Neocorteza. Se registraron células piramidales de la capa V de la neocorteza en rodajas preparadas de corteza suprahipocampal. Para generar IPSCs, el electrodo de estimulación se colocó en el entorno de la célula registrada. La aplicación de kainato (3 µM) produjo un ligero descenso de la amplitud media de las corrientes inhibidoras (18.2 ± 9.3 %, n=4) (Fig. 29). También se observó en estas células que kainato inducía una corriente entrante de 72.3 ± 17.1 pA y un incremento en el número de eventos espontáneos (al 814.8 ± 273.5 %, n=4). Como en casos anteriores, el estudio del coeficiente de variación indicó un mecanismo de acción presináptico como explicación más probable para estos resultados. A Control Kainato 3 µM Recuperación 50 pA 100 ms C 1.5 40 30 1/CV2 Inhibición de eIPSCs (%) B 20 10 0 1.0 0.5 0.0 0.0 Kainato 3 µM 0.5 1.0 1.5 M Figura 29. Efecto de la activación de los receptores de kainato sobre las eIPSCs registradas en neuronas piramidales de la capa V de corteza tras estimular las interneuronas adyacentes. A) Kainato (3 µM) produjo una reducción de la amplitud de las eIPSCs que fue paralela al cambio del estadístico 1/CV2 (C). B) Cuantificación del efecto de kainato 3 µM. La barra representa la media ± EEM de 4 neuronas de 4 rodajas diferentes. En la figura 30 se presenta de forma comparativa el efecto de kainato 3 µM sobre las eIPSCs en las diferentes áreas estudiadas. Nótese que al igual que en CA1 del hipocampo, no parece existir correlación alguna entre la disminución de las corrientes evocadas y el efecto producido sobre la actividad espontánea (Fig. 30C). 56 Resultados Estos resultados indican que los receptores de glutamato de tipo kainato están presentes en la parte presináptica de los contactos inhibidores en diferentes áreas del cerebro y que, al menos en estas zonas, estos receptores tienen una función similar, que es modular a la baja la liberación del neurotransmisor GABA. 80 70 60 50 40 30 20 10 0 Aumento de sIPSCs (%) Reducción de eIPSCs (%) B Reducción de eIPSCs (%) A 60 40 20 0 CA1CA3 DCN P 1000 500 0 CA1CA3 DCN P C C 0 1500 400 800 12001600 Frecuencia de sIPSCs (%) C Figura 30. Comparación del efecto de kainato 3 µM sobre las eIPSCs en diferentes áreas del cerebro. A) Kainato deprime las eIPSCs en las regiones CA1 y CA3 de hipocampo, en las células de los núcleos profundos (DCN) y de Purkinje (P) del cerebelo y en las neuronas piramidales de corteza (C) B) Efecto sobre la frecuencia de respuestas espontáneas (sIPSCs). C) Representación gráfica de la correlación entre la depresión de las corrientes evocadas y el cambio producido sobre la actividad espontánea. Como se puede observar, no existió correlación entre ambos fenómenos. 57 Discusión DISCUSIÓN. Los resultados expuestos demuestran que kainato regula la transmisión GABAérgica en el área CA1 del hipocampo reduciéndola. Esta acción es compatible con la bien conocida naturaleza convulsante y excitotóxica del ácido kaínico (Coyle, 1983). En el presente trabajo, se diseñaron experimentos en diferentes modelos experimentales, microcultivos, rodajas de cerebro e in vivo, obteniéndose en todos los casos resultados compatibles. Ello demuestra que este efecto es una acción general y no un resultado inherente a una preparación experimental en particular. Hace más de 15 años, cuando kainato empezó a usarse como agente excitotóxico, algunos autores describieron acciones de este compuesto sobre la inhibición GABAérgica. Sloviter y Damiano (1981) fueron los primeros en sugerir que kainato decrecía la inhibición en el hipocampo, un resultado que desde entonces ha sido encontrado por diferentes autores (Westbrook y Lothman, 1983; Fisher y Alger, 1984; Kehl y McLennan, 1985; Gaiarsa et al., 1994). Sin embargo, la falta de conocimiento sobre los receptores de kainato y de sus propiedades evitó alcanzar conclusiones válidas en esos momentos. Considerando que los receptores de AMPA, los otros receptores susceptibles de ser activados por kainato, fueron sistemática y completamente bloqueados por GYKI 53655 y que el bloqueo de los receptores metabotrópicos de glutamato no previno el efecto observado, se puede concluir que la acción de kainato sobre la inhibición GABAérgica es mediada por la activación de receptores de kainato. La falta de antagonistas específicos de los receptores de kainato impidió la demostración de una implicación directa de estos receptores en la inhibición de las eIPSCs. Sin embargo, en presencia de GYKI 53655, el antagonista de los receptores de AMPA y kainato, CNQX pudo ser usado como antagonista de los receptores de kainato. Como se ha descrito, este antagonista inespecífico fue capaz de inhibir la acción de kainato de forma dosis-dependiente. La posibilidad de que kainato produjera en las rodajas la liberación de algún tipo de sustancia que, al activar sus propios receptores, fuera capaz de producir el efecto observado, fue descartada dado que en presencia de un cóctel de antagonistas para bloquear los receptores posibles, kainato siguió produciendo la depresión de las corrientes inhibidoras. El 58 Discusión hecho de que glutamato, el agonista endógeno de estos receptores, tuviera un efecto similar sobre la transmisión inhibidora, permite concluir que la activación de estos receptores por el agonista endógeno produce la desinhibición de las poblaciones neuronales. Por tanto, considerando el perfil farmacológico y las propiedades de la acción de kainato sobre la función sináptica GABAérgica descritos en esta memoria, podemos concluir que la depresión de la inhibición es mediada por activación de receptores de glutamato de tipo kainato presentes en la parte presináptica de los contactos inhibidores y que éste es uno de los papeles principales que los receptores de kainato desempeñan en la fisiología cerebral. 1. La acción de kainato es presináptica. Efectivamente, kainato redujo notablemente la amplitud de las eIPSCs tanto en microcultivos como en rodajas de hipocampo. Todas las evidencias obtenidas con diferentes aproximaciones sugirien un sitio de acción presináptico para kainato. En primer lugar, se compararó la proporción de fallos en la liberación de neurotransmisor antes y durante la aplicación de kainato. Para este análisis sólo se consideraron aquellas células en las que los fallos pudieron ser determinados sin ambigüedad. El aumento en el número de éstos estuvo correlacionado con la magnitud del decremento en la amplitud de las eIPSCs. Ello sugiere un cambio en la probabilidad de liberación de neurotransmisor durante la aplicación de kainato. En neuronas microcultivadas, una situación en la cual la generación de la espiga presináptica pudo ser controlada, se observó igualmente un aumento en el número de fallos. En segundo lugar, kainato redujo la frecuencia de mIPSCs sin producir cambio alguno en la amplitud de las mismas, un resultado igualmente compatible con un decremento en la probabilidad de liberación. El análisis del coeficiente de variación de las respuestas evocadas (i.e. el estadístico CV2), se modificó proporcionalmente a la variación de la amplitud media de las respuestas, resultado igualmente demostrativo de un modo de acción presináptico para kainato. Algunos autores han tratado de determinar la distribución subcelular de los receptores de kainato, a pesar de la carencia de buenos anticuerpos. Sin embargo, ninguno de ellos ha podido concluir de forma inequívoca su localización. Aunque la señal inmunocitoquímica se ha encontrado preferentemente en la densidad postsináptica de sinapsis asimétricas, en algunos casos se ha observado marcaje en axones no mielinizados y en terminales presinápticos (Huntley et al., 1993; Petralia et al., 1994). Ello de acuerdo con nuestros 59 Discusión resultados, sugiere la existencia de receptores de kainato presinápticos. No obstante, la identificación de las subunidades de los receptores de kainato presentes en los botones sinápticos GABAérgicos requerirá estudios definitivos de microscopía electrónica del tipo de los llevados a cabo para otros receptores de glutamato (e.g. Luján et al., 1996; Shigemoto et al., 1996; Rubio y Wenthold, 1997). 2. La curva dosis-respuesta para el efecto de kainato y de glutamato presenta forma de campana. En el presente trabajo, la acción de kainato sobre las eIPSCs se evaluó en condiciones de equilibrio. Experimentos realizados en nuestro laboratorio indican la existencia de una ventana de activación por el agonista de este receptor (Paternain et al., 1998). Esta ventana surge de las propiedades de activación e inhibición de estos receptores en condiciones de estado estacionario. En las rodajas de hipocampo, la curva dosis-respuesta de la inhibición de las eIPSCs calculada tanto para kainato como para glutamato presentó forma de campana, existiendo un rango de concentraciones efectivas. Este hecho se debe a que dosis más altas o más bajas impiden que se mantenga una actividad del receptor en estado estacionario, debido a que las más bajas son incapaces de activar el receptor y las más altas lo desensibilizan rápidamente. Ello resulta en una curva dosis-respuesta teórica en forma de campana para las condiciones de funcionamiento del receptor en estado estacionario. La ventana de corriente teórica mostrada en la figura 5B como una línea continua se calculó como la fracción de canales no inactivados que pueden ser activados por cada concentración de kainato, teniendo en cuenta las curvas de activación y de inactivación para los receptores de kainato nativos determinadas previamente en neuronas de hipocampo en cultivo. Como se puede observar, los valores experimentales de la acción de kainato sobre las eIPSCs en rodajas de hipocampo ajustan razonablemente bien con los datos obtenidos en neuronas cultivadas. En este sentido, esta curva presentó un máximo a concentraciones de kainato en torno a 10 µM y de aproximadamente 100 µM para glutamato. 3. La depresión de las corrientes inhibidoras mediada por activación de receptores de kainato requiere la activación de una cascada metabotrópica. Con la aproximación farmacológica llevada a cabo, los resultados descritos muestran de forma clara que la depresión de la liberación de GABA inducida por la activación de los receptores de kainato en la zona CA1 de hipocampo resulta de la iniciación de una cascada de segundos mensajeros producida por la activación de una proteína G sensible a toxina 60 Discusión pertúsica. La ruta involucra la activación de fosfolipasa C y de PKC, constituyendo una acción metabotrópica de los receptores de kainato que es independiente de su actividad como canal iónico. Recientemente se ha mostrado en neuronas corticales, que los receptores de AMPA pueden regular los niveles de AMPc mediante la activación de una proteína Gi, aunque no existe ninguna evidencia acerca del papel fisiológico que esta interacción pudiera desempeñar. Sin embargo, estos resultados indican igualmente que, además de formar canales iónicos, los receptores ionotrópicos de glutamato pueden exhibir acción metabotrópica. En el caso de los receptores de kainato, semejante actividad afecta profundamente a la actividad neuronal ya que resulta en la modulación de la inhibición mediada por GABA. En sinaptosomas de hipocampo también se ha encontrado acoplamiento de los receptores de kainato a proteína G (Cunha et al., 1999). Si el receptor de kainato se acopla directamente a la proteína G o lo hace a través de una proteína adaptadora es difícil de predecir. Sin embargo, se ha observado que la afinidad por kainato de estos receptores en el pez dorado (carpín dorado), presenta cierta sensibilidad a PTx así como a la ADP ribosilación de una proteína de 40 KDa, que podría coincidir con los receptores de kainato de este pez (Ziegra et al, 1992). Aunque estos datos son consistentes con una interacción directa de los receptores de kainato con una proteína G sensible a toxina pertúsica, la topología de las subunidades de los receptores de kainato es, en principio, difícil de reconciliar con la estructura de los receptores de la familia de los metabotrópicos, que presentan sitios específicos de interacción con las proteínas G (ver Pin y Duvoisin, 1995). Por ello, es posible que una proteína adaptadora pueda estar implicada en transmitir la señal desde el receptor de kainato hasta la proteína G tras la unión de glutamato. 4. ¿Son las proteínas exocitóticas los efectores de una población específica de PKC? Es sabido que la activación de la PKC (p.e. con ésteres de forbol) aumenta la liberación de neurotransmisor en sinapsis inhibidoras (Capogna et al., 1995), un resultado que fue replicado en nuestras condiciones experimentales. Sin embargo, la activación de los receptores de glutamato de tipo kainato conduce a la activación de un reservorio de PKC que produce un descenso en la probabilidad de liberación de GABA. Este resultado indica la existencia de, al menos, dos poblaciones o reservorios de PKC en los terminales GABAérgicos. La estimulación de uno de ellos produciría un aumento de la liberación de transmisor mientras que el otro la inhibiría. En este sentido, la oclusión de la acción de 61 Discusión kainato por PDBu puede ser explicada por la activación de todos los reservorios de PKC por parte del éster de forbol, impidiendo cualquier tipo de activación por parte de kainato. Por el contrario, la activación por parte de kainato de una población inhibidora de PKC, cuantitativamente pequeña, no es capaz de impedir la acción de los ésteres de forbol. Por tanto, se puede hipotetizar que la ruta de señalización activada por los receptores de kainato involucra una población particular de PKC (ver Nishizuca, 1988). Es interesante hacer notar que en presencia de PDBu, más que disminuir la frecuencia de miniaturas (mIPSCs), kainato la aumentó y que esta acción fue adicional al efecto de PDBu. Este resultado podría ser esperado si se produjera una suave depolarización del terminal presináptico por un receptor acoplado a un canal catiónico (Lena y Changeux, 1997; ver también McGehee y Role, 1996; Glitsh y Marty, 1999), como sería el caso de los receptores de kainato. Otro e importante aspecto de los datos presentados es que la inhibición de la liberación de GABA inducida por kainato puede ser reducida aumentando la fuerza electromotriz de Ca2+. Esto prodría significar que la población de PKC susceptible de ser activada por los receptores de kainato interfiere con un paso en el mecanismo de liberación en el cual el Ca2+ debe de estar involucrado. En principio, la reducción en la liberación de neurotransmisor puede provenir de alguno de estos fenómenos: (1) la cantidad de calcio que entra en el terminal disminuye como consecuencia de la inhibición de los canales de calcio. (2) disminuye la sensibilidad a calcio de la maquinaria de liberación. En ambos casos, el resultado es una disminución en la liberación y en ambos casos un aumento en el gradiente químico para el calcio podría restaurar las condiciones iniciales. Sin embargo, es bien conocido que la activación de PKC produce un aumento de las corrientes a través de los canales de calcio y, por tanto, un aumento en la cantidad de neurotransmisor liberado (Swartz, 1993; Swartz et al., 1993). Aunque la activación de PKC ha sido también implicada en una inhibición de los canales de calcio inducida por la liberación de neurotransmisor (e.g.; Diversé-Pierluissi y Dunlap, 1993), la mayoría de la inactivación de canales de calcio inducida por la liberación de neurotransmisor involucra mecanismos mediados por proteínas G delimitados a la membrana plasmática, sin la participación de proteínas kinasa (revisado por Hille, 1992). Para minimizar un eventual déficit de calcio, se bloquearon los canales de potasio para prolongar la duración de los potenciales de acción y, por tanto, la duración de la entrada de calcio en los terminales. El aumento en la entrada de calcio al bloquear los canales de potasio, no fue suficiente para impedir la acción de kainato, aunque si se aumentó la liberación de neurotransmisor (Fig. 15). Sin embargo, aumentando el gradiente químico para 62 Discusión el calcio se aumentó la concentración de calcio que entró en el terminal sin cambiar la cinética de la entrada. Este proceder fue bastante efectivo en prevenir la acción inhibidora de kainato. Este resultado indica que la modulación de la liberación de GABA por kainato es consistente con un cambio mediado por PKC en la sensibilidad a calcio de las proteínas involucradas en la exocitosis, más que una acción directa de la PKC sobre canales de calcio presinápticos (e.g. Retting et al., 1997). En resumen, estos resultados proporcionan la evidencia que sustenta una actividad metabotrópica de un receptor típicamente ionotrópico con claras consecuencias fisiológicas. Los receptores de kainato parecen controlar la actividad de una población particular de PKC que influye notablemente la liberación de GABA en el hipocampo. Este hecho añade más diversidad a la función de los receptores de glutamato y abre nuevas vistas sobre el modo de operación de los receptores de glutamato de tipo ionotrópico (Fig. 31). GLUTAMATO NMDA EXTRACELULAR Zn GLU AMPA KAINATO mGluR I, II, III GLY PA Mg G INTRACELULAR NR2A NR2B SUBUNIDADES NR1+ NR2C NR2D GluR5-7 KA1, KA2 GluR1-4 AdC PLC mGluR1-8 IONOTROPICOS METABOTROPICOS Figura 31. Clasificación de los receptores de glutamato de acuerdo a su composición subunitaria, sus propiedades farmacológicas y a sus mecanismos de señalización. Los receptores de kainato y los de AMPA, además de funcionar como canales también presentan función metabotrópica. 5. En las interneuronas de hipocampo coexisten las dos poblaciones de receptores de kainato con sistemas de señalización diferentes. Tanto el agonista endógeno, glutamato, como ATPA, un agonista de receptores GluR5, y AMPA, un activador de algunos tipos de receptores de kainato, son capaces de disociar el aumento de actividad espontánea (sIPSCs) observada por la acción depolarizante 63 Discusión de los receptores de kainato de sus efectos sobre la liberación de GABA. Además, el efecto sobre la liberación de GABA pero no la acción depolarizante de kainato fue sensible al bloqueo de proteínas G o de PKC en las interneuronas de hipocampo. Estos resultados indican, de forma inequívoca, la existencia de dos poblaciones de receptores de kainato en las interneuronas del hipocampo que presentan desigual sensibilidad a agonista y que usan diferentes sistemas de señalización (Fig. 32). Estos resultados están en clara contradicción con uno de los argumentos usados por algunos autores en contra de la existencia de receptores presinápticos de kainato, fundamentados en la observación de que la estimulación repetitiva (i.e. 6 Hz) de las rutas inhibidoras disminuye la amplitud de las eIPSCs. Aunque no se muestra, hemos evaluado esta posibilidad en nuestras condiciones experimentales, no encontrándo una reducción de las eIPSCs tan marcada como la descrita (Frerking et al., 1998). Por el contrario, en nuestros experimentos, la estimulación repetitiva, provocó una reducción media de las respuestas similar a la recientemente observada en experimentos con activación sostenida de las sinapsis inhibidoras corticales (Galarreta et al., 1998). En definitiva, en nuestras condiciones experimentales, esta depresión dependiente de frecuencia de las eIPSCs es claramente insuficiente para explicar la reducción de las eIPSCs observada tras la adición de kainato al baño. De hecho, nuestros resultados demuestran que bajas concentraciones de glutamato deprimen las eIPSCs GABAérgicas en ausencia de disparos presinápticos repetitivos. Además, si el aumento de actividad espontánea fuera responsable de la depresión de las eIPSCs, algún tipo de correlación tendría que existir entre el aumento de actividad de las interneuronas y el descenso de la amplitud de las eIPSCs, independientemente del agonista usado. Sin embargo, de acuerdo con nuestras observaciones éste no fue el caso (Fig. 22) ni en el hipocampo ni en las otras estructuras examinadas (Fig. 30). La diferente sensibilidad a los diferentes agonistas de los tipos de receptores responsables de un fenómeno u otro, indica que estos receptores difieren en su composición molecular. Aunque las interneuronas del hipocampo expresan predominantemente la subunidad GluR5 de los receptores de kainato, recientemente también se ha encontrado que una importante población de interneuronas expresa la subunidad GluR6 y que estas dos subunidades (GluR5 y GluR6) pueden ensamblarse formando receptores heteroméricos con propiedades farmacológicas propias (Paternain et al., 2000). En consecuencia, la diversidad de receptores de kainato es mayor de lo que se preveía y hasta el momento no se puede 64 Discusión establecer ninguna conclusión sobre el tipo de subunidades que componen los receptores que median cada efecto. Curiosamente, la sensibilidad a glutamato de los receptores de kainato responsables de la regulación de la liberación de GABA, fue mayor que la de los que producen la depolarización somatodendrítica. Esto es lo que se esperaría para un receptor situado en un sitio que no es capaz de sensar el glutamato sináptico. La concentración real de glutamato que sensan los receptores de kainato in vivo es desconocida y probablemente limitada por la actividad de los transportadores de glutamato. Estimaciones recientes indican que ésta puede llegar a ser de 160-190 µM en el espacio extrasináptico. Este rango de concentraciones pueden ser suficientes para la activación de receptores de glutamato situados a distancia de los lugares de liberación, dado su aparentemente alta sensibilidad a glutamato. Por tanto, los dos requerimientos para que la modulación de la liberación de GABA ocurra en respuesta a la liberación sináptica de glutamato, una concentración suficiente fuera de la sinapsis y alta sensibilidad del receptor, parecen cumplirse. De hecho, un estudio reciente (Min et al., 1999), ha demostrado que la liberación sináptica de glutamato de las colaterales de Schaffer es suficiente para reducir la amplitud de las eIPSCs mediante un mecanismo cuya farmacología es compatible con la de los receptores de kainato. 6. La modulación de la liberación de GABA mediada por la activación de receptores de kainato presinápticos es un fenómeno común a diferentes áreas del cerebro. La disminución de la liberación de GABA producida por la activación de receptores de kainato situados en terminales presinápticos parece ser un fenómeno general, puesto que, se ha podido poner de manifiesto en las zonas CA1 y CA3 del hipocampo, en los núcleos profundos del cerebelo, en las sinapsis inhibidoras que se establecen sobre las células de Purkinje, así como en la capa V de corteza cerebral. Los datos experimentales también muestran que se produce una depolarización de las células registradas y un aumento variable de la actividad espontánea, no existiendo tampoco correlación entre el aumento de actividad espontánea observado y la depresión de las corrientes evocadas. Aunque no se ha estudiado sistemáticamente, existe la posibilidad de que en todos los casos los receptores de kainato situados en los terminales estén asociados a proteínas G y que tengan función metabotrópica al igual que ocurría en la zona CA1 del hipocampo. Sea como fuere, estos resultados indican que en diferentes áreas del cerebro existen receptores de glutamato de tipo kainato cuya activación produce una depresión de la transmisión sináptica inhibidora GABAérgica. 65 Discusión Receptores de Kainato somatodendríticos (Depolarización) A Receptores de kainato presinápticos (Inhibición de la liberación de neurotransmisor) ATPA>KA>Glu>AMPA KA>Glu>ATPA>>AMPA Figura 32. En las interneuronas del hipocampo coexisten dos poblaciones de receptores de kainato con sistemas de señalización diferentes y con diferente sensibilidad a agonistas. 7. Relevancia fisiológica del bloqueo de la liberación de GABA mediada por receptores de kainato. Es conocido que el glutamato puede regular no sólo su propia liberación sino también la liberación de GABA activando receptores de glutamato metabotrópicos localizados específicamente en terminales excitadores y GABAérgicos. El significado funcional exacto de este hecho aún no ha sido bien establecido. Nuestros resultados identifican otro receptor de glutamato capaz de ejercer esta misma actividad moduladora. Cabe imaginar que si las sinapsis inhibidoras y excitadoras están parcialmente integradas, el glutamato sináptico puede desbordarse (Kullmann et al., 1996) y alcanzar concentraciones lo suficientemente altas para activar receptores de kainato en los terminales GABAérgicos presinápticos. Teniendo en cuenta el dramático efecto que algunas concentraciones de glutamato tienen sobre la liberación de GABA en condiciones de estado estacionario (cuando sólo una pequeña cantidad de receptores están activos), el efecto de glutamato en el pico de activación puede tener como consecuencia la completa supresión de la función GABAérgica. Sin embargo, para este tipo de interacción computacional se requiere un cronometraje exacto de la ocurrencia tanto de la liberación de glutamato como de la invasión de la terminal inhibidora por el potencial de acción. La coincidencia temporal de señales excitadoras e inhibidoras en el hipocampo es común y, de existir este mecanismo, podría ser detectada en la dendrita de la célula postsináptica como una gran y duradera depolarización que no se daría si no existiera este tipo de interacción. Por tanto, éste podría ser un mecanismo de asegurar y/o reforzar la comunicación excitadora entre células. Para que esta posibilidad sea posible, es necesario además que los terminales excitadores e inhibidores 66 Discusión estén lo suficientemente cerca en el Sistema Nervioso Central. Se asume de forma general, que las sinapsis inhibidoras se disponen en los troncos dendríticos y en los somas. Por el contrario, las espinas dendríticas se han considerado siempre como blancos de los axones excitadores, formando sinapsis asimétricas. Sin embargo, estudios inmunocitoquímicos combinados con microscopía electrónica, han mostrado de forma convincente que las espinas dendríticas reciben igualmente axones GABAérgicos (Ribak, 1978; Hendry et al., 1983; Houser et al., 1984; revisado por DeFelipe y Fariñas, 1992). Consecuentemente la posibilidad de que los terminales GABAérgicos sensen la cantidad de glutamato sináptico liberado existe realmente. La hipótesis anterior no excluye la posibilidad de que el tono inhibidor esté bajo el control del glutamato extracelular. Los receptores de kainato presentan una ventana de concentraciones de agonista activas. Para el agonista endógeno, esta ventana presenta un máximo a 100 µM (menos de 10 veces la concentración alcanzada en la hendidura sináptica). Además, una concentración de aproximadamente 100 µM es la calculada como valor alcanzado en el fluido extracelular en condiciones isquémicas (Benveniste et al., 1984). La predicción es que a esta concentración de glutamato, la función GABAérgica puede estar seriamente comprometida, resultando probablemente excitotoxicidad. En efecto, en experimentos recientes, en actividad epiléptica y ratones a los que les falta el transportador de glutamato astrocítico, en los cuáles permanece elevada la concentración extracelular de glutamato en el fluido extracelular durante largos períodos, presentan ataques epilépticos espontáneos (Tanaka et al., 1997). En resumen, nuestros resultados demuestran que los receptores de kainato modifican la “fiabilidad” de las sinapsis GABAérgicas pudiendo modificar la “fuerza” de las conexiones inhibidoras. Este efecto se alcanza, no obstante, dentro de un preciso rango en la concentración extracelular de agonista. Concentraciones más altas o más bajas no son lo suficientemente eficientes en alterar la fiabilidad. De este modo, la excitabilidad del circuito estaría bajo el fino control de la concentración extracelular de glutamato a través de la activación transitoria o tónica de los receptores de kainato. Este hecho implica la existencia de reglas computacionales para la función de los receptores de kainato en la integración neuronal. 67 Conclusiones CONCLUSIONES. Teniendo en cuenta los resultados obtenidos, se han alcanzado las siguientes conclusiones: 1. La potente actividad epileptógena del ácido kaínico, conocida desde hace más de 15 años, se debe, al menos en parte, al efecto depresor de la inhibición GABAérgica que conlleva la activación de los receptores de kainato presinápticos. 2. Los receptores de kainato presentan dos tipos de señalización celular independientes, uno asociado a su actividad como canal iónico; el otro asociado a la activación de una proteína G que dispara una cascada de segundos mensajeros, constituyendo ésta la primera evidencia fisiológica de una función metabotrópica para los receptores de glutamato de tipo kainato. 3. Los dos tipos de receptores de kainato coexisten en las interneuronas del hipocampo. Una población se localiza en el compartimento somatodendrítico y su activación provoca la depolarización de la membrana; la otra se localiza en la parte presináptica de las sinapsis inhibidoras y su activación produce la disminución de la liberación de GABA a través de un mecanismo que involucra la activación de la proteína kinasa C. 68 Bibliografía BIBLIOGRAFÍA. Agrawal, S. G., and Evans, R. H. (1986). The primary afferent depolarizing action of kainate in the rat. Br. J. Pharmacol. 87: 345-355. Aizenman, E., Lipton, S. A., and Loring, S. A. (1989). Selective modulation of NMDA responses by reduction and oxidation. Neuron 2: 1257-1263. Arshavsky, V., and Pugh, E.N., Jr. (1998). Lifetime regulation of G protein–effector complex: emerging importance of RGS proteins. Neuron 20: 11–14. Ascher, P. and Nowak, L. (1988). The role of divalent cations in the N-methyl-Daspartate responses of mouse central neurones in culture. J. Physiol. 399: 247-266. Bekkers, J. M. and Stevens, C. F. (1990). Presynaptic mechanism for long-term potentiation in the hippocampus. Nature 346: 724-729. Bekkers, J. M. and Stevens, C. F. (1991). Excitatory and inhibitory autaptic currents in isolated hippocampal neurons mainatined in cell culture. Proc. Natl. Acad. Sci. U. S. A. 88:7834-7838. Bennett, J. A., and Dingledine, R. (1995). Topology profile for a glutamate receptor: three transmembrane domains and a channel-lining reentrant membrane loop. Neuron 14: 373-384. Benveniste, H., Drejer, J. Schousboe, A. and Diemer N. H. (1984). Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J. Neurochem. 43: 1369-1374. Bernard, A., and Khrestchatisky, M. (1994). Assessing the extent of RNA editing in the TMII regions of GluR5 and GluR6 kainate receptors during the rat brain development. J. Neurochem. 62: 2057-2060. Bettler, B., Boulter, J., Hermans-Borgmeyer, I., O'Shea-Greenfield, A., Deneris, E. S., Moll, C., Borgmeyer, U., Hollmann, M. and Heinemann, S. F. (1990). Cloning of a novel glutamate receptor subunit, GluR5: expression in the nervous system during development. Neuron 5: 583-595. Bleakman, D.,. Schoepp, D. D., Ballyk, B., Bufton, H., Sharpe, E. F. Thomas, K., Ornstein, P. L. and Kamboj, R.K. (1996). Pharmacological discrimination of GluR5 and GluR6 kainate receptor subtypes by (3S,4aR,6R,8aR)-6-[2-(1(2)H-tetrazole-5yl)ethyl]decahydroisoquinoline-3 carboxylic-acid. Mol. Pharmacol. 49: 581-585. Boland, L.M., and Bean, B.P. (1993). Modulation of N-type calcium channels in bullfrog sympathetic neurons by luteinizing hormone-releasing hormone: kinetics and voltage dependence. J. Neurosci.13: 516–533. Brewer, G. J., Torricelli, J. R., Evege, E. K. and Price, P. J. (1993). Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J. Neurosci. Res. 35: 567-576. 69 Bibliografía Burnashev, N. Monyer, H., Seeburg, P. H. and Sakmann, B. (1992). Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron 8: 189-198. Capogna, M., Gähwiler, B.H., and Thompson, S.M. (1995). Presynaptic enhancement of inhibitory synaptic transmission by protein kinases A and C in the rat hippocampus in vitro. J. Neurosci. 15: 1249– 1260. Castillo, P. E., Malenka, R. C. and Nicoll, R. A. (1997). Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature 388: 182-186. Cattaneo, R. (1991). Different types of messenger RNA editing. Annu. Rev. Genet. 25: 71-88. Clarke, V. R. J., Ballyk, B. A., Hoo, K. H., Mandelzys, A., Pellizzari, A., Bath, C. P., Thomas, J., Sharpe, E. F., Davies, C. H., Ornstein, P. L., Schoepp, D. D., Kamboj, R. K., Collingridge, G. L., Lodge, D. and Bleakman, D. (1997). A hippocampal GluR5 kainate receptor regulating inhibitory synaptic transmission. Nature 389: 599-603. Conn, P.J., and Pin, J.-P. (1997). Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 37: 205–237. Coyle, J. T. (1983). Neurotoxic actions of kainic acid. J. Neurochem. 41: 1-11. Cunha, R. A., Malva, J.O. and Ribeiro. (1999). Kainate receptors coupled to G(i)/G(o) proteins in the rat hippocampus. Mol. Pharmacol. 56: 429-433. Davies, J., Evans, R. H., Francis, A. A., and Watkins, J. C. (1979). Excitatory amino acid receptors and synaptic excitation in the mammalian central nervous system. J. Physiol. (Paris) 75: 641-654. DeFelipe, J. and Fariñas, I. (1992). The pyramidal neuron of the cerebral cortex: morphological and chemical characteristics of the synaptic inputs. Prog. Neurobiol. 39: 563-607. DeVries, S. H., and Schwartz, E. A. (1999). Kainate receptors mediate synaptic transmission between cones and 'Off' bipolar cells in a mammalian retina. Nature 397: 157-160. De Waard, M., Liu, H.L., Walker, D., Scott, V.E.S., Gurnett, C.A. and Campbell, K.P. (1997). Direct binding of G protein complex to voltage-dependent calcium channels. Nature 385: 446–450. Diversé-Pierluissi, M., and Dunlap, K. (1993). Distinct, convergent second messenger pathways modulate neuronal calcium currents. Neuron 10: 753–760. Dolphin, A.C., Forda, S.R., and Scott, R.H. (1986). Calcium-dependent currents in cultured rat dorsal root ganglion neurones are inhibited by an adenosine analog. J. Physiol. (Lond.) 373: 47–61. Egebjerg, J., Bettler, B., Hermans-Borgmeyer, I. and Heinemann, S. F. (1991). Cloning 70 Bibliografía of a cDNA for a glutamate receptor subunit activated by kainate but not by AMPA. Nature 351: 745-748. Egebjerg, J., and Heinemann, S. F. (1993). Ca2+ permeability of unedited and edited versions of the kainate selective glutamate receptor GluR6. Proc. Natl. Acad. Sci. U. S. A. 90: 755-759. Ehlers, M. D., Zhang, S., Bernhadt, J. P., and Huganir, R.L. (1996). Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell 84: 745-755. Evans, R. H., Francis, A. A., Hunt, Y., Oakes, D. J. and Watkins, J. C. (1979). Antagonism of excitatory amino acid-induced responses and of synaptic in isolated spinal cord of the frog. Br. J. Pharmacol. 67: 591-603. Fisher R. S., and Alger, B. E. (1984). Electrophysiological mechanisms of kainic acidinduced epileptiform activity in the rat hippocampal slice. J. Neurosci. 4: 1312-1323. Frerking, M., Malenka, R. C. and Nicoll, R. A. (1998). Synaptic activation of kainate receptors on hippocampal interneurons. Nat. Neurosci. 1: 479-486. Gaiarsa, J. L., Zagrean, L. and Ben-Ari, Y. (1994). Neonatal irradiation prevents the formation of hippocampal mossy fibers and the epileptic action of kainate on rat CA3 pyramidal neurons. J. Neurophysiol. 71: 204-215. Galarreta, M. and Hestrin, S. (1998). Frecuency-dependent synaptic depression and the balance of excitation and inhibition in the neocortex. Nat. Neurosci. 1, 587-594. Glits, M. and Marty, A. (1999). Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J. Neurosci.19 (2): 511-519. Hamberger, A. C., Chiang, G.H., Nylen, E. S., Scheff, S.W. and Cotman, C. W. (1979). Glutamate as a CNS transmitter. I. Evaluation of glucosa and glutamine as precusor for the synthesis of preferentially released glutamate. Brain Res. 168: 513-530. Hammill, O. P., Marty, A., Neher, E., Sakmann, B. and Sigworth, F. J. (1981). Improved patch-clamp techniques for high resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 391: 85-100. Hayashi, T. (1954). Effects of sodium glutamate on the nervous system. Keio. J. Med. 3: 183-192. Heckmann, M., Bufler, J., Franke, C. and J. Dudel. (1996) Kinetics of homomeric GluR6 glutamate receptor channels. Biophys. J. 71: 1743-1750. Hendry, S. H. C., Houser, C. R., Jones, E. G. and Vaughn, J. E. (1983). Synaptic organization of immunocytochemically identified GABA neurons in the monkey sensory-motor cortex. J. Neurocytol. 12: 639-660. Herb, A., Burnashev, N., Werner, P., Sakmann, B., Wisden, W. and P. H. Seeburg (1992). The KA-2 subunit of excitatory amino acid receptors shows widespread expression in brain and forms ion channels with distantly related subunits. Neuron 8: 775-785. Herreras, O., Largo, C., Ibarz, J.M., Somjen, G.G. and Martín del Río, R. (1994). Role of neuronal synchronizing mechanisms in the propagation of spreading depression in the in 71 Bibliografía vivo hippocampus. J. Neurosci. 14: 7087-7098. Hille, B. (1992). G protein-coupled mechanisms and nervous signaling. Neuron 9: 187195. Hollmann M, and Heinemann, S. (1994). Cloned glutamate receptors. Annu. Rev. Neurosci. 17: 31-108. Hollmann, M., Hartley, M. and Heinemann, S. F. (1991). Ca+2 permeability of KAAMPA-gated glutamate receptor channels depends on subunit composition. Science 252: 851-853. Houser, C. R., Vaughn, J. E., Hendry, S. H. C., Jones, E. D., and Peters, A. (1984). GABA neurons in the cerebral cortex. In: The Cerebral Cortex, Volume Two: Functional Properties of Cortical Cells, E. D. Jones and A. Peters, eds. (New York: Plenun Press), pp. 63-89. Huettner, J. E. (1990). Glutamate receptor channels in DRG neurons: activation by kainate and quisqualate and blockade of desensitization by Con A. Neuron 5: 255-266. Huntley, G. W., Rogers, S. W., Moran, T., Janssen, W., Archin, N., Vickers, J.C., Cauley, K., Heinemann, S. F. and J. H. Morrison. (1993). Selective distribution of kainate receptor subunit immunoreactivity in monkey neocortex revealed by a monoclonal antibody that recognizes glutamate receptor subunits GluR5/6/7. J. Neurosci. 13: 2965-2981. Ito, I., Tanabe, S., Kohda, A. and H. Sugiyama (1990). Allosteric potenciation of quicualate receptors by a nootropic drug aniracetam. J. Physiol. 424: 533-543. Johansen, T. H., Drejer, T., Wätjen, F., and E. Ø. Nielsen (1993). A novel non-NMDA receptor antagonist shows selective displacement of low-affinity 3H-kainate binding, Eur. J. Pharmacol. 246: 195-204. Johnson, J. W. and Ascher, P. (1987). Glycine potenciates NMDA response in cultures mouse brain neurons. Nature 325: 529-531. Kehl, S. J., and McLennan, H. (1985). A pharmacological characterization of chlorideand potassium-dependent inhibition in the CA3 region of the rat hippocampus in vitro. Exp. Brain Res. 60: 309-317. Kidd, F. L., and Isaac, J. T. R. (1999). Developmental and activity-dependent regulation of kainate receptors at thalamocortical synapses. Nature 400: 569-573. Kleckner, N. W. and Dingledine, R. (1988). Requirement for glycine in activation of NMDA-receptors expressed in Xenopus oocytes. Science 241: 835-837. Köhler, M., Burnashev, N., Sakmann, B. and Seeburg, P.H. (1993). Determinants of Ca2+ permeability in both TM1 and TM2 of high affinity kainate receptor channels: diversity by RNA editing. Neuron 10: 491-500. Kullman, D. M., Erdemli, G., and Aztely, F. (1996). LTP of AMPA and NMDA 72 Bibliografía receptor-mediated signals: evidence for presynaptic expression and extrasynaptic glutamate spill-over. Neuron 17: 461-474. Largo, C., Cuevas, P., Somjen, G.G., Martín del Río, and Herreras, O. (1996). The effect of depressing glial function on rat brain in situ on ion homeostasis, synaptic transmission and neurona survival. J.Neurosci. 16: 1219-1229. Lena, C., and Changeux, J. P. (1997). Role of Ca2+ ions in nicotinic facilitation of GABA release in mouse thalamus. J. Neurosci. 17: 576–585. Lerma J, Zukin R. S., and Bennett, M. V. (1990). Glycine decreases desensitization of Nmethyl-Daspartate (NMDA) receptors expressed in Xenopus oocytes and is required for NMDA responses. Proc. Natl. Acad. Sci. USA. 87: 2354-2358. Lerma, J., Zukin, R. S. and Bennett, M. V. (1991). Interaction of Mg2+ and phencyclidine in use-dependent block of NMDA channels. Neurosci. Lett. 123: 187191. Lerma, J. (1992). Spermine regulates N-Methyl-D-aspartate receptor desensitization. Neuron 8: 343-352. Lerma, J. (1997a). Kainate reveals its targets. Neuron 19: 1155-1158. Lerma, J. (1998). Kainate receptors: an interplay between excitatory and inhibitory synapses. FEBS Lett. 430: 100-104. Lerma, J. Kainate receptors. (1999). In: Handbook of Experimental Pharmacology. Ionotropic glutamate receptors in the CNS, edited by P. Jonas and H. Monyer. Berlin: Springer 141: 275-307. Lerma, J., Morales, M., Vicente, M. A. and Herreras, O. (1997b). Glutamate receptors of the kainate type and synaptic transmission. Trends Neurosci. 20: 9-12. Lerma, J., Paternain, A. V., Naranjo, J. R. and Mellström, B. (1993). Functional kainate-selective glutamate receptors in cultured hippocampal neurons. Proc. Natl. Acad. Sci. U. S. A. 90: 11688-11692. Lester, R. A., Clements, J. D., Westbrook, G, L. and Jahr, C. E. (1990). Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature 346: 565-567. Li, H., and Rogawski, M. A. (1998). GluR5 kainate receptor mediated synaptic transmission in rat basolateral amygdala in vitro. Neuropharmacology 37: 1279-1286. Logan, W. J. and Snyder, S. H. (1972). High affinity uptake systems for glycine, glutamic and aspartic acids in synaptosomes of rat central nervous tissues. Brain Res. 42: 413-431. Luján, R., Nusser, Z., Roberts, J. D. B., Shigemoto, R. and Somoyi, P. (1996). Perisynaptic location of metabotopic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur. J. Neurosci. 8: 1488-1500. MacDonald, J. F., Bartlett, M. C., Mody, I., Papahill, P., Reynolds, J. N., Salter, M. W., Schneiderman, J. H. and Pennefather, P. S. (1991). Actions of ketamine, phencyclidine 73 Bibliografía and MK-801 on NMDA receptor currents in cultured mouse hippocampal neurones. J. Physiol. (Lond) 432: 483-508. Mackler, S. A., and Eberwine, J. H. (1993). Diversity of glutamate receptor subunit mRNA expression within live hippocampal CA1 neurons. Mol. Pharmacol. 44: 308315. McGehee, D.S., and Role, L.W. (1996). Presynaptic ionotropic receptors. Curr. Opin. Neurobiol. 6: 342– 349. Malenka, R.C., Madison, D.V., and Nicoll, R.A. (1986). Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature 321: 175–177. Malinow, R. and Tsien, R. W. (1990). Presynaptic enhacement shown by whole-cell recordings of long-term potentiation. Nature 346: 177-180. Mayer, M. L. and Westbrook. G. L. (1987). Permeation and block of N-methyl-D-aspartatic acid receptor channels by divalent cations in mouse cultured central neurones. J. Physiol., Lond. 394: 501-527. Meguro, H., Mori, H., Araki, K., Kushiya, E., Kutsuwada, T., Yamazaki, M., Kumanishi, T., Arakawa, M., Sakimura, K. and Mishina, M. (1992). Functional characterization of a heteromeric NMDA receptor channel expressed from cloned cDNAs. Nature 357: 70-74. Min, M. Y., Meyland, Z. and Kullmann, D. M. (1999). Synaptically released glutamate reduces gamma-aminobutyric acid (GABA)ergic inhibition in the hippocampus via kainate receptors. Proc. Natl. Acad. Sci. U. S. A. 96: 9932-9937. Monyer, H., Sprengel, R., Schoepfer, R., Herb, A., Higuchi, M., Lomeli, H., Burnashev, N., Sakmann, B. and Seeburg, P. H. (1992). Heteromeric NMDA receptors: molecular and functinal distinction of subtypes. Science 256: 1217-1221. Monyer, H., Burnashev, N., Laurie, D. J., Sakmann, B., and Seeburg, P. H. (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529540. Moriyoshi, K., Masu, M., Ishii, T., Shigemoto, R., Mizuno, N., and Nakanishi, S. (1991). Molecular cloning and characterization of the rat NMDA receptor. Nature 354: 31-37. Mulle, C., Sailer, A., Pérez-Otaño, I., Dickinson-Anson, H., Castillo, P. E., Bureau, I., Maron, C., Gage, F.H., Mann, J. R., Bettler, B. and Heinemann, S. F. (1998). Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6deficient mice. Nature 392: 601-605. Nakanishi, N., Axel, R. and Shneider, N. A. (1992). Alternative splicing generates functinally distinct Nmethyl-D-aspartate receptors. Proc. Natl. Acad. Sci. USA 89: 8552-8556. Nakanishi, S. (1992). Molecular diversity of glutamate receptors and implications for brain function. Science 258: 597-603. Nishizuka, Y. (1988). The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature 334: 661–665. Nicoll, R. A., Kauer, J. A. and Malenka, R. C. (1988). The current excitement in long-term potentiation. Neuron 1: 97-103. 74 Bibliografía Nowak, L.M., Bregestovski, P., Ascher, P., Herbet, A., and Prochiantz, A. (1984). Magnesium gates glutamate-activated channels in mouse central neurones. Nature 307, 462-465. Paschen, W., Schmitt, J., Gissel, C., and Dux, E. (1997). Developmental changes of RNA editing of glutamate receptor subunits GluR5 and GluR6: in vivo versus in vitro. Dev. Brain Res. 98: 271-280. Paternain, A. V., Herrera, M. T., Nieto, M. A. and J. Lerma, J. (2000). GluR5 and GluR6 kainate receptor subunits coexist in hippocampal neurons and coassemble to form functional receptors. J. Neurosci. 20 (1): 196-205. Paternain, A. V., Morales, M. and J. Lerma, J. (1995). Selective antagonism of AMPA receptors unmasks kainate receptor-mediated responses in hippocampal neurons. Neuron 14: 185-189. Paternain, A. V., Rodríguez-Moreno, A., Villarroel, A. and J. Lerma (1998). Activation and desensitization properties of native and recombinant kainate receptors. Neuropharmacology 37: 1249-1259. Paternain, A. V., Vicente, M. A., Nielsen, E. Ø. and Lerma, J. (1996). Comparative antagonism of kainate-activated AMPA and kainate receptors in hippocampal neurons. Eur. J. Neurosci. 8: 2129-2136. Patneau, D. K., Vyklicky, L. and Mayer, M. L. (1993). Hippocampal neurons exhibit cyclothiazidesensitive rapidly desensitizing responses to kainate. J. Neuroci. 13: 3496-3509. Pemberton, K. E., Belcher, S. M., Ripellino, J. A. and Howe, J. R. (1998). High-affinity kainate-type ion channels in rat cerebellar granule cells. J. Physiol. 510: 401-420. Peters, S., Koh, J., Choi, D. W. (1987). Zinc selectively blocks the action of N-methyl-D-aspartate on cortical neurons. Science 236: 589-593. Petralia, R. S., Wang, Y. X. and Wenthold, R. J. (1994). Histological and ultrastructural localization of the kainate receptor subunits, KA2 and GluR6/7, in the rat nervous system using selective antipeptide antibodies. J. Comp. Neurol. 349: 85-110. Pin, J. P. and Duvoisin, R. (1995). The metabotropic glutamate receptors: structure and function. Neuropharmacology 34: 1-26. Pocock, J. M., Murphie, H. M., and Nicholls, D. G. (1988). Kainic Acid inhibits the synaptosomal plasma membrane glutamate carrier and allows glutamate leakage from cytoplasm but does not affect glutamate exocytosis. J. Neurochem. 50: 745-751. Represa, A., Tremblay, E. and Ben-Ari, Y. (1987). Kainate binding sites in the hippocampal mossy fibers: localization and plasticity. Neuroscience 20: 739-748. Retting, J., Heiemann, C., Ashery, U., Sheng, Z.-H., Yokoyama, C.T., Catteral, W.A., and Neher, E. (1997). Alteration of Ca 21 dependence of neurotransmitter release by disruption of Ca 21 channel/syntaxin interaction. J. Neurosci. 17: 6647–6656. Ribak, C. E. (1978). Aspinous and sparsely spinous stellate neurons in the visual cortex of rate contain glutamic acid descarboxylase. J. Neurocytol. 7: 461-478. 75 Bibliografía Roche, K. W., Raymond, L. A., Blackstone, C. and Huganir, R. L. (1994). Transmembrane topology of the glutamate receptor subunit GluR6. J. Biol. Chem. 269: 11679-11682. Rosenmund, C., Stern-Bach, Y. and Stevens, C. F. (1998). The tetrameric structure of a glutamate receptor channel. Science 280: 1596-1599. Rubio, M. E. and Wenthold, R. J. (1997). Glutamate receptors are selectively targeted to postsynaptic sites in neurons. Neuron 18: 939-950. Schiffer, H. H., Swanson, G. T. and Heinemann, S. F. (1997). Rat GluR7 and a carboxy-terminal splice variant, GluR7b, are functional kainate receptor subunits with a low sensitivity to glutamate. Neuron 19: 1141-1146. Schoepp, D.D., Goldsworthy, J., Johnson, B.G., Salhoff, C.R., and Baker, S.R. (1994). 3,5-Dihydroxyphenylglycine is a highly selective agonist for phosphoinositide linked metabotropic glutamate receptors in the rat hippocampus. J. Neurochem. 63: 769–772. Shigemoto, R., Kulik, A., Roberts, J. D. B., Ohishi, H., Nusser, Z., Kaneko, T. And Somoyi, P. (1996). Target-specific concentration of a metabotropic glutamate receptor in the presynaptic active zone. Nature 381: 523-525. Siegel, S. J., Janssen, W. G., Tullai, J. W., Rogers, S. W., Moran, T., Heinemann, S. F. and Morrison, J. H. (1995). Distribution of the excitatory amino acid receptor subunits GluR2(4) in monkey hippocampus and colocalization with subunits GluR5-7 and NMDAR1. J. Neurosci. 15: 2707-2719. Siegelbaum, S. A. and Kandel, E. R. (1991). Learning-related synaptic plasticity: LTP and LTD. Curr. Opin. Neurobiol. 1: 113-120. Simmons, R. M., Li, D. L., Hoo, K. H., Deverill, M., Ornstein, P. L. and Iyengar, S. (1998). Kainate GluR5 receptor subtype mediates the nociceptive response to formalin in the rat. Neuropharmacology 37: 25-36. Sloviter, R. S., and Damiano, B. P. (1981). On the relationship between kainic acidinduced epileptiform activity and hippocampal neuronal damage. Neuropharmacology 20: 1001-1011. Sommer, B. and Seeburg, H. (1992). Glutamate receptors channels: novel properties and new clones. TIPS 13: 291-296. Sommer, B., Keinanen, K., Verdoorn, T. A., Wisden, W., Burnashev, N., Herb, A., Kohler, M., Takagi, T., Sakmann, B., Seeburg, P. H. (1990). Flip and flop: a cellspecific functional switch in glutamate-operated channels of the CNS. Science 249: 1580-1585. Sommer, B., N. Burnashev, T. A. Verdoorn, K. Keinänen, B. Sakmann, and P. H. Seeburg (1992). A glutamate receptor channel with high affinity for domoate and kainate. EMBO J. 11: 1651-1656. 76 Bibliografía Stern-Bach. Y., Bettler, B., Hartley, M., Sheppard, P.O., O'Hara, P. J. and Heinemann, S. F. (1994). Agonist selectivity of glutamate receptors is specified by two domains structurally related to bacterial amino acid-binding proteins. Neuron 13: 1345-1357. Stuart, G.J., Dodt, H.U. and Sakmann, B. (1993). Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflügers Arch. 423: 511–518. Sugihara, H., Moriyoshi, K., Ishii, T., Masu, M. and Nakanishi, S. (1992). Structure and properties of seven isoforms of the NMDA receptor generated by alternative splicing. Biochem. Biophys. Res. Comm. 185: 826-832. Swanson, G. T, Feldmeyer, D., Kaneda, M. and Cull-Candy, S. G. (1996). Effect of RNA editing and subunit co-assembly single-channel properties of recombinant kainate receptors. J. Physiol. 492: 129-142. Swartz, K.J. (1993). Modulation of Ca2+ channels by protein kinase C in rat central and peripheral neurons: disruption of G protein-mediated inhibition. Neuron 11: 305–320. Swartz, K.J., Merrit, A., Bean, B.P., and Lovinger, D.M. (1993). PKC modulates glutamate receptor inhibition of Ca 21 channels and synaptic transmission. Nature 361: 165–168. Tanaka, K., Waase, K., Manabe, T., Yamada, K., Watanabe, M., Takahashi, K., Iwama, H., Nishikawa, T., Ichihara, N., Kikuchi, T.(1997). Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science 276:1699-1702. Taverna, F. A., Wang, L. Y., MacDonald, J. F. and Hampson, D. R. (1994). A transmembrane model for an ionotropic glutamate receptor predicted on the basis of the location of asparagine-linked oligosaccharides. J. Biol. Chem. 269: 14159-14164. Thompson, A. M., and Deuchars, J. (1995). Diverse pre- and postsynaptic properties of fast excitatory synapses. In: Excitatory Amino Acids and Synaptic Transmission. H. Wheal and A. Thompson, eds. (London: Academic Press), 145-172. Trussell, L. O., Zhang, S. and Raman, I. M. (1993). Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron 10:1185-1196. Traynelis, S. F. and Cull-Candy, S. G. (1990). Proton inhibition of N-methyl-D aspartate receptors in cerebellar neurons. Nature 345: 347-350. Traynelis, S. F., and Wahl, P. (1997). Control of rat GluR6 glutamate receptor open probability by protein kinase A and calcineurin. J. Physiol. 503: 513-531. Tsunoda, S., Sierralt, J., Sun, Y., Bodner, R., Suzuki, E., Becker, A., Socolich, M., and Zuker, C.S. (1997). A multivalent PDZ-domain protein assembles signalling complexes in a Gprotein–coupled cascade. Nature 388: 241–249. Vaello, M.L., Ruiz-Gómez, A., Lerma, J., and Mayor, F., Jr. (1994). Modulation of inhibitory glycine receptors by phosphorylation by protein kinase C and cAMP77 Bibliografía dependent protein kinases. J. Biol.Chem. 269: 2002–2008. Verdoorn, T. A., Burnashev, N., Monyer, H., Seeburg, P. H. and Sakmann, B. (1991). Structural determinants of ion flow through recombinant glutamate receptor channel. FEBS Lett. 308: 1715-1718. Vignes, M., and Collingridge, G. L. (1997).The synaptic activation of kainate receptors. Nature 388: 179-182. Vizi, E. S., Mike, A. and Tarnawa, I. (1996). 2,3-Benzodiazepines (GYKI 52466 and analogs): negative allosteric modulators of AMPA receptors. CNS Drug Rev. 2: 91-126. Wang, Y., Small, D. L., Stanimirovic, D. B., Morley, P. and Durkin, J. P. (1997). AMPA receptor-mediated regulation of a Gi-protein in cortical neurons. Nature 389: 502-504. Watanabe, M., Inoue, Y., Sakimura, K., Mishina, M. (1992). Developmental changes in distribution of NMDA receptor channel subunit mRNAs. Neuroreport 3: 1138-1140. Werner, P., Voigt, M., Keinänen, K., Wisden, W. and Seeburg, P. H. (1991). Cloning of a putative high-affinity kainate receptor expressed predominantly in hippocampal CA3 cells. Nature 351: 742-744. Westbrook, G. L., and Lothman, E. W. (1983). Cellular and synaptic basis of kainic acid-induced hippocampal epileptiform activity. Brain Res. 273: 97-109. Westbrook, G. L. and Mayer, M. L. (1987). Micromolar concentrations of Zn+2 antagonize NMDA and GABA responses of hippocampal neurons. Nature 328, 640643. Wilding, T. J., and Huettner, J. E. (1995). Differential antagonism of α-amino-3hydroxy-5-methyl-4-isoxazolepropionic acid-preferring and kainate-preferring receptors by 2,3-benzodiazepines. Mol. Pharmacol. 47: 582-587. Wilding, T. J., and Huettner, J. E. (1996). Antagonist pharmacology of kainate- and αamino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-preferring receptors. Mol. Pharmacol. 49: 540-546. Wilding, T. J., and Huettner, J. E. (1997). Activation and desensitization of hippocampal kainate receptors. J. Neurosci. 17: 2713-2721. Willard, J. M. and Oswald, R. E. (1992). Interaction of the frog brain kainate receptor expressed in chinese hamster ovary cells with a GTP-binding protein. J. Biol. Chem. 267: 19112-19116. Wo, Z. G., and Oswald, R. E. (1994). Transmembrane topology of two kainate receptor subunits revealed by N-glycosylation. Proc. Natl. Acad. Sci. U. S. A. 91: 7154-7158. Wong, L. A., Mayer, M. L., Jane, D. E. and Watkins, J. C. (1994). Willardines differentiate agonist binding sites for kainate- versus AMPA-preferring glutamate receptors in DRG and hippocampal neurons. J. Neurosci. 14: 3881-3897. 78 Bibliografía Yamada, K. A. and Tang, C. M. (1993). Benzothiadiazides inhibit rapid glutamate receptor desensitization and enhance glutamatergic synaptic currents. J. Neurosci. 13: 3904-3915. Ziegra, C. J., Willard, J. M. and Oswald, R. E. (1992). Coupling of a purified goldfish brain kainate receptor with a pertussis toxin-sensitive G protein. Proc. Natl. Acad. Sci. U. S. A. 89: 4134-4138. 79