validación de sistemas aumotáticos (mes)
Anuncio
")
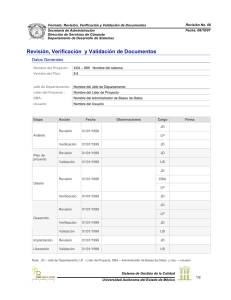
_________________________________________________________________________________________ VALIDACIÓN DE SISTEMAS AUMOTÁTICOS (MES) Oriol Argemi BASF Labiana _________________________________________________________________________________________ 1. INTRODUCCIÓN -Breve historia de las GAMP 1982-1985: Inicio de la regulación de las normativas sobre los sistemas computerizados 1983: Blue Book: Aproximación a una inspección de sistemas informáticos, mediante el “life-cycle validation” 1984: PMA/FDA creación comité para el estudio de las validaciones de los sistemas informáticos. 1987: FDA: Desarrollo Actividades de software 1990: TGA Australian Code of GMP 1991: EEC: Directiva 91/356/EEC 1995: PDA: Validation of computer related Systems 1996: ISPE. UK GAMP Forum. Supplier guide for validation of Automation Systems in Pharmaceutical Manufacturing. 1996: GMA-NAMUR: Automatisierungtechnishe Praxis 1997: 21 CFR Part 11 Electronic Signatures and Electronic Records, Federal Register, 62 Desde hace mas de 30 años, la industria química y especialmente la industria farmacéutica y alimentaria han estado sometidas a un constante proceso de introducción de nuevas tecnologías. Formando parte de estas nuevas tecnologías se ha desarrollado lo que en estos momentos es una industria de las más potentes y avanzadas. La industria del software. Esta industria cada vez incide mas en el corazón de la industria tradicional, implantando sus nuevos conceptos en el control de cualquier proceso. Es a raíz de este boom hacia finales de los años 70, en donde las autoridades empiezan a preocuparse realmente de estos sistemas de control, y su importancia en el producto final. Si desde las GMP se pide que cualquier proceso debe estar validado, y por lo tanto documentado para tener una garantía del producto final, se debe de elaborar algún sistema para tener esta certeza Conferencia de la Sección Española de ISA 1999 en cuanto a los elementos que inciden de forma sustancial en el producto. De las primeras preguntas que empiezan a salir, una de las más representativas de todas es: “¿Que controles deben tener los sistemas de control?”. En el año 1983 sale una primera guía de inspección para las inspecciones de software de la FDA. En los años siguientes, la industria farmacéutica y alimentaria de los EEUU se ve envuelta en el problema de las no-conformidades. Mientras tanto, la industria del software se pone en marcha para hacer sus programas mas seguros y acordes a los requerimientos de la administración, y es en este momento que la industria químicofarmacéutica se sitúa al frente de estos desarrollos. Los estudios y nuevas leyes duran una decena de años, en donde Europa, Japón y Australia se sitúan al nivel de la administración americana. Todos estos estudios dan sus frutos en varios documentos, interrelacionados entre sí, desde el 1995 hasta la actualidad, en donde se exponen los principios básicos para poder decir si un sistema de control de proceso es adecuado o no lo es. Estos principios quedan reflejados en lo que hoy viene a llamarse: VALIDACIÓN DE SISTEMAS APLICACIONES AUTOMÁTICAS. Y ¿En qué nivel se encuentra la automatización dentro del sistema “Quality Assurance”? -Definición de los niveles de garantía del producto, y su relación con los sistemas informáticos. Cuando se marca lo que debe abarcar el sistema de aseguramiento de un producto, en primer lograr se contempla el producto en sí mismo, en segundo lugar viene el proceso de obtención de este producto, y en tercer lugar ya viene el control de este proceso. Después vienen todos los chequeos y controles realizados discretamente o en continuo con el proceso, y al final los análisis finales de este 1 producto. Colateralmente vendrían los sistemas que interaccionan con el mismo, como los sistemas de documentación, de mantenimiento, logística, etc., hasta encontrarnos con los sistemas de Performance del producto, esto es: Finanzas, compras, comercial. Todo esto es lo que la Garantía de calidad debe abarcar si se quiere tener un control completo del sistema. -Necesidad de la validación dentro de las (GMP) GAMP. La pregunta de la necesidad de validar un sistema de control de proceso, y muy concretamente un sistema crítico de control de proceso de fabricación de un producto (MES), queda pues contestada cuando se ve en que estadio se encuentran los automatismos dentro de este aseguramiento global del producto. Ahora que sabemos la necesidad de una validación, nos podemos concentrar en ella, para sacarle todo el jugo posible, es decir, optimizar el sistema y rentabilizarlo. Para conseguirlo, los términos más importantes a tener en cuenta dentro de una validación , son los siguientes: Life-Cycle approach Hace referencia a los pasos y etapas a seguir en el tiempo, dentro de una validación. Quality management Hace referencia a la gestión de toda la información del proceso Procedimientos Toda la documentación por la que se rige cada operación dentro del proceso. Training Hace referencia a la formación de la gente de la empresa para desarrollar correctamente este proceso. Protocolos Hace referencia a los objetivos a conseguir en cada etapa de una validación. Tests de Cualificación Hace referencia a las pruebas desarrolladas en la validación del proceso Control de cambios Es la manera de documentar cualquier cambio dentro de un ciclo de vida de un proceso Hace referencia a lo apropiados que deben ser los proveedores y sus productos Evaluaciones en continuo Es la manera de documentar y revisar que el proceso sigue funcionando correctamente, una vez ya se está produciendo. -Conceptos y validaciones aclaraciones respecto a ISO 9000 Certificaciones Si analizamos los términos antes citados, vemos que todos nos suenan dentro de lo que se llaman las normas ISO. Las normas ISO no son suficientes para decir que un sistema está validado. Estas normas nos facilitan una buena base para trabajar en la validación de un proceso, y de esta manera podemos tener una garantía de que todos los elementos implicados en la validación hablan el mismo idioma, pero la validación debe realizarse, ya que es un estudio que implica a un proceso concreto, no a un sistema de calidad general. Respecto a lo que se llama GMP y la certificación de un proceso validado, se debe decir que ninguna autoridad da un certificado de correcta validación. Porqué ? Porque la validación es un proceso en continuo que se acaba cuando se acaba el producto. Es decir, que el ciclo de vida de un proceso, solo acabará con el desmantelamiento del mismo. 2. VALIDACION DE SISTEMAS AUTOMÁTICOS -Organización de la validación (los VMP). La primera parte de una validación es saber como se organiza esta validación. Se debe crear un grupo de trabajo que defina los objetivos de la validación, y que su trabajo consista en alcanzar estos objetivos. Se deben definir responsabilidades, terminologías, formas de trabajo etc. Este grupo de trabajo debe plasmar todo esto sobre la base de documentos. Debe también establecerse un comité de validación en donde haya un control en el tiempo de estos objetivos, y si deben cambiarse. Audits Conferencia de la Sección Española de ISA 1999 las 2 Identificación de la validación Una vez marcada la organización del proyecto, debe definirse exactamente cual va a ser el alcance de la validación. Que sistemas va a implicar, colateralidades, excepciones etc. Este punto es muy importante para llevar la validación a buen puerto. Si no se tiene delimitado perfectamente el marco de la validación, va a ser imposible avanzar en ésta. Consejos prácticos: Es importante delimitar lo que se va a abarcar en una validación, contando con los recursos. Se debe volver a este punto después de saber de los recursos de que se dispone, para asegurar que la validación podrá llevarse a cabo. Tener previsto un procedimiento de control de os cambios a efectuar durante toda la validación, es decir, durante toda la vida del sistema. Planes de contingencia Procedimientos para posibles eventualidades que puedan producirse en el sistema, tanto en el periodo de preparación, como en el periodo de implementación y en el de mantenimiento. Formación continua Tener un programa de formación continuada de operarios y técnicos para incorporación poder trabajar desde un principio con las máximas garantías. Revalidaciones Plan de recursos Ver de qué recursos se dispone. Normalmente, en una validación de un sistema informático se dispone de: Director del proyecto. De él depende la gestión de este proyecto. De expertos en validaciones, que son personas habituadas en las metodologías a seguir para la consecución de cada etapa del proyecto. De expertos ingenieros para temas técnicos relacionados con cada etapa del proyecto, de garantía de calidad, que nos dará las pautas para el cumplimiento en cada paso de los sistemas de calidad de la empresa, y el mantenimiento de estos en el tiempo. Auditorias a proveedores Buscar el partner más adecuado en cada caso. De encontrar una buena colaboración con la empresa proveedora, depende en buena parte el éxito de la validación. Una vez realizados estos pasos, deben ponerse las bases para que una validación no quede en una serie de documentos, archivados y sin ninguna utilidad. Estimar periodos de revalidaciones , así como nivel y profundidad de estas Desmantelamiento Procedimientos de desmantelamiento del sistema, tanto para casos de emergencia como en caso de final de operatividad del proceso. Todos estos pasos, pueden interpretarse genéricamente para toda una planta, o bien para operaciones concretas. Normalmente las industrias prefieren referirse en estos términos cuando se trata de validar toda una planta, ya que es importante pensar en la polivalencia de los trabajadores para diferentes operaciones, en programas globales de revalidaciones para el ahorro de costes, en una gestión global de la documentación , etc... Una vez realizados estos pasos, estamos dispuesto a empezar la validación propiamente dicha del proceso. Para esto, debe preverse: - -Validation Life cycle Trabajo de evaluación en continuo Plan de validación Gestión de la documentación Responsables de gestionar el volumen de información de la que se va a disponer. Es muy importante el nivel de sintetización de esta información, ya que de esto depende en gran medida la rentabilidad de una validación. Procedimientos mantenimiento y operaciones de Responsables para crear y gestionar el mantenimiento del sistema hasta que sea obsoleto. Control de cambios Requerimientos Auditorias a proveedores Especificaciones funcionales Diseño del Hardware Diseño del Software Análisis de riesgos GPP (Good Programming practices) Inspección del software Pretests (pruebas FAT) Installation Qualification Conferencia de la Sección Española de ISA 1999 3 Operational Qualification Performance Qualification • Esquemas visuales de proceso Informes de validación • Seguridades intrínsecas • Seguridad del sistema • Integridad del sistema • Mantenimiento • Procedimientos • Etc... Control de documentos cambios y control de Para seguir un ciclo de validación, pondremos un ejemplo de validación de un sistema MES, con diferentes ejemplos de los pasos a seguir en cada uno de los procesos de validación que componen el sistema. Plan de validación Auditorias El plan de validación es el documento que especifica como debe atacarse una validación de un único sistema automático o bien varios sistemas automáticos. La industria farmacéutica está obligada a determinar valorar el grado de adecuación de los proveedores a su proyecto. Normalmente se valora la capacidad del proveedor para la realización de éste. Esquema del Plan de Validación Razones de la validación. Objetivos de la validación. Informaciones iniciales. Responsabilidades. Estructura de equipo. Formación. Procedimientos a seguir. Documentación a producir. Auditorias a proveedores. Control y seguimiento de la validación. Hitos y puntos críticos del proyecto. Documentos de apoyo a la validación Requerimientos Documento donde se hace referencia a los valores de las variables criticas del proceso. Que valores se deben conseguir en cada caso Para poder desarrollar este documento, se deben tener en cuenta los siguientes parámetros: • Situación del proveedor • Organización del proveedor • Empleados • Planificación de los trabajos • Desarrollo del proyecto • Construcción del software • Especificaciones de los tests • Finalización del trabajo • Control de procedimientos • Comunicaciones con el cliente Especificaciones funcionales • Interfase humana • Interfase de proceso Las especificaciones funcionales es el documento respuesta del proveedor al documento de requerimientos, realizado por el cliente. • Comunicaciones Diseño del Hardware • Hardware -Documento-esquema de los equipos del sistema. • Software • -Unidad procesadora, interfases etc... Instrumentación • Elementos finales • Alarmas • Secuencias • Control batch o continuo • Procesado de datos Conferencia de la Sección Española de ISA 1999 pantallas, impresoras, -Instrumentación -Sistemas SAI Diseño del software Documento correspondiente a la arquitectura del software. Con diagrama y esquemas. 4 Dentro del diseño del software podemos hablar de un diseño vertical (jerárquico), y un diseño horizontal -Standard Operating System Inspección de software Revisión del software atendiendo a los puntos críticos GMP del sistema. Es el documento en donde se hace un resumen de los bloques que contiene el software, protegiendo a los puntos críticos del sistema. -Software del sistema -Preensamblaje -Configuraciones -Especificidades Análisis de riesgos HACCP. Documento de chequeo de puntos críticos del sistema, con soluciones de protección para cada punto. Pruebas FAT Factory acceptance test. Incluye los tests de integración modular y Precualificación del sistema. En el caso de que haya desarrollos separados de software, es en este estadio de la situación en donde se deben hacer las pruebas de simulación de ensamblaje de los dos soft’s. Installation Qualification Construcción del sistema Programando el software (GPP) • (Transparencia 18) Verificación de cada uno de los componentes del sistema. Confirmación de la adecuación de estos. Verificación de inputs/outputs. Chequeo I/O. Programación modular del software Operational Qualification • Estructura del software • Convenciones • Revisiones • Formatos • Control de códigos • Redundancias • Roles de compilación Selección del programa estándar de programación, IEC 1131-3 y ISA-S88 Redundancias Estudio de posibles redundancias, y extracción Compiladores Grados de compilación apropiados. Optimización de códigos Conferencia de la Sección Española de ISA 1999 Es la respuesta lógica a las especificaciones funcionales del sistema. Son los documentos de las pruebas dinámicas del sistema, así como de las condiciones ambientales que pueden afectar al mismo. Performance Qualification Son los documentos que demuestran que la instalación cumple con los requerimientos del usuario. Informes de validación Es la documentación en donde se da respuesta a los objetivos descritos en el VP. Puede contener recomendaciones de tests adicionales, y debe incluir desde el seguimiento de la validación hasta el desmantelamiento del sistema. 3. NUEVAS PERSPECTIVAS Sistemas informáticos integrales. Los sistemas informáticos integrales, que no de integración vertical, son los sistemas que nos pueden permitir interrelacionar todo el conjunto de sistemas que se tienen para cada unidad operativa, es decir, fabricación, logística, documentación, 5 mantenimiento, etc..., extrayendo los datos necesarios a analizar, y buscando soluciones de ahorro mucho más globales. Las nuevas tendencias apuntan a un futuro próximo con una gestión informática de la documentación. Eliminar todo lo que comporte Grandes Montañas de Papel. Esto no es una buena validación, y además la documentación no asegura el cumplimiento de las GMP. Para este tema, la FDA ha sacado ya las normas en cuanto a lo que a firma electrónica se refiere. La formación es un punto crítico. Se debe formar a la gente para poder validar. COSTES Y BENEFICIOS Recordar que la validación es un proceso en continuo. Si hoy no sale perfecto, mañana, al haber aprendido, saldrá mejor. Se ha hablado de todos los pasos de una validación, sin hablar apenas de los costes, por un lado, y las rentabilidades que se pueden conseguir. En primer lugar decir que si se considera que en una validación hay demasiados papeles, esto quiere decir que esta validación no se ha llevado a cabo de la manera correcta. Así como en la programación no debe de haber redundancias, en los papeles debe ocurrir exactamente igual. Debe de estar todo lo necesario y únicamente lo necesario. El coste de una validación implica el tener una aproximación del coste de un proceso no validado. Un proceso no validado quiere decir un proceso que no se puede sacar al mercado, y por lo tanto se estima en términos de miles de millones. De todas maneras, la manera más tangible de ver la rentabilidad inmediata de una validación es comparando: • Productividades • Mantenimiento • Calidad del producto. 4. CONCLUSIONES La validación, no tiene un sentido en si misma, pero permite asegurar la calidad de los productos y servicios. Y además se puede cuantificar este valor. Las empresas deben empezar a mentalizarse en la cultura de validación. Muchas veces esto es más fácil con la contratación de algún consultor externo, que no esté tan implicado en el día a día. Conviene evitar en la medida de lo posible hacer cambios innecesarios en una validación. Esto aumenta extraordinariamente su coste. Conferencia de la Sección Española de ISA 1999 6