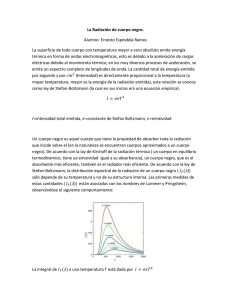
Tema 3 - OCW Usal
Anuncio

Espectrofotometría de absorción ultravioleta-visible 2 Tema 3 ESPECTROFOTOMETRIA DE ABSORCION ULTRAVIOLETA-VISIBLE La espectrofotometría de absorción en las regiones ultravioleta y visible del espectro electromagnético es, posiblemente, la más utilizada en la práctica del análisis cuantitativo de todas las técnicas espectroscópicas. Asimismo, puede resultar de utilidad como técnica auxiliar para la determinación de estructuras de especies químicas. Se basa en la absorción de radiación ultravioleta y visible por el analito, como consecuencia de lo cual se origina un estado activado que posteriormente elimina su exceso de energía en forma de calor, en un proceso que esquemáticamente puede representarse así: X + hυ —> X* —> X + calor La cantidad de calor disipado es muy pequeño, por lo que el método tiene la ventaja de originar un trastorno mínimo en el sistema que se estudia. El término generalmente empleado en la medida de la radiación absorbida es el de absorciometría, si bien, normalmente se utiliza la denominación de colorimetría cuando se trabaja en la region visible del espectro electromagnético y se emplean aparatos que utilizan filtros (ver más adelante) para seleccionar la radiación utilizada*. Por otra parte, el término espectrofotometría suele aplicarse cuando la radiación utilizada se extiende a las regiones ultravioleta e infrarroja, y además, se utilizan monocromadores en lugar de filtros, así como sistemas de detección más sensibles, como tubos fotomultiplicadores. * En sentido estricto, un colorímetro es un instrumento que compara el color de una sustancia con el de una disolución de referencia usando el ojo humano como detector, si bien, en la práctica, esta denominación suele aplicarse a muchos fotómetros de filtro con sistemas de detección eléctricos. 3 Claudio González Pérez En cuanto a la región ultravioleta, hay que indicar que la zona de mayor interés en la práctica analítica ordinaria es la denominada como ultravioleta próximo (longitud de onda entre 200 y 400 nm), pues el ultravioleta lejano o ultravioleta de vacío (de 10 a 200 nm) presenta el inconveniente de que oxígeno atmosférico absorbe en esa región, siendo necesario eliminarlo del instrumento de medida. Debido a ello, la espectrofotometría en esa región espectral no se ha desarrollado suficientemente. LEYES DE LA ABSORCION DE RADIACION Cuando un haz de radiación monocromática de una determinada longitud de onda atraviesa una capa de disolución conteniendo una especie absorbente, la potencia (energía por unidad de tiempo y unidad de área) del haz incidente Po se atenúa, disminuyendo hasta P (figura 3.1.) Se define la transmitancia, T, como la fracción de radiación incidente que consigue atravesar la muestra. Varía de 0 a 1 y puede expresarse también como porcentaje: P P x T= ; %T= 100 Po Po Un parámetro de mayor utilidad práctica es la absorbancia, A, definida como A = – log T = log Po P b Po P P–dP P db Figura 3.1. Absorción de radiación Obsérvese que cuando no hay absorción de radiación, P=Po y entonces, A=0, mientras que si se absorbe el 99 %, solo se transmite el 1%, con lo que A=2. Espectrofotometría de absorción ultravioleta-visible 4 Para una capa de disolución absorbente de espesor infinitamente pequeño, db, (Figura 3.1.) la disminución de la potencia radiante, dP, es proporcional a la propia potencia incidente, P, a la concentración de la especie absorbente, C, y al espesor db: dP = – k P C db donde k es una constante de proporcionalidad. Reagrupando la ecuación anterior e integrando, se obtiene: dP = – k C db P P dP =– P b k C db 0 Po ln P P = – k C b = 2.3 log Po Po log donde, ε = k/2.3. La expresión, P = – log T = A = ε b C Po A = ε b C se denomina ley de Beer. Es fundamental en análisis cuantitativo al relacionar la absorbancia con la concentración. La constante ε recibe el nombre de absortividad molar, cuando la concentración se expresa en moles/litro y el camino óptico, b, en centímetros. La absortividad es una propiedad característica de la sustancia absorbente y depende de la longitud de onda. Por ello, para aplicar la ley de Beer debe seleccionarse una determinada longitud de onda, y para este propósito se utiliza el espectro de absorción, siendo éste una gráfica que indica la variación de la absorbancia, o de la absortividad, con la longitud de onda. DESVIACIONES DE LA LEY DE BEER La proporcionalidad entre la absorbancia y la concentración únicamente se cumple para disoluciones muy diluidas, observándose desviaciones más o menos acusadas al aumentar la concentración (figura 3.2.). 5 Claudio González Pérez Desviaciones positivas A Desviaciones negativas C Figura 3.2. Desviaciones de la ley de Beer Las desviaciones de la ley de Beer pueden clasificarse de la forma siguiente: Reales Instrumentales Químicas Radiación no monocromática Radiación parásita Errores de lectura Dimerizaciones Influencia del equilibrio Acido-base Complejos Influencia del disolvente Influencia de la temperatura Impurezas en los reactivos Interacción entre especies absorbentes Personales DESVIACIONES REALES. En la deducción de la ley de Beer que se hizo anteriormente, no se ha considerado que la absortividad depende del índice de refracción, n, según la expresión: n ε = ε verdadero 2 2 n +2 Como, a su vez, el índice de refracción varía con la concentración, la absortividad no es rigurosamente constante para cualquier concentración, provocando desviaciones 6 Espectrofotometría de absorción ultravioleta-visible negativas. De todas formas, en la práctica, para concentraciones inferiores a 10–3 M puede prescindirse de la influencia de este factor, al ser el índice de refracción esencialmente constante. DESVIACIONES INSTRUMENTALES. Las fluctuaciones producidas en la corriente eléctrica, la inestabilidad de algunas fuentes de radiación o la respuesta no lineal del detector pueden originar el funcionamiento incorrecto de un determinado aparato. Además de éstos, pueden considerarse los siguientes factores de tipo instrumental como causas de desviaciones de la ley de Beer: a) Uso de radiación no monocromática. La deducción de la ley de Beer se hizo sobre la base de utilizar radiación monocromática, lo cual, en sentido estricto, nunca se cumple, pues en la práctica, todos los dispositivos seleccionan una banda más o menos ancha en torno a una determinada longitud de onda. La influencia de la radiación policromática sobre la ley de Beer puede mostrarse del siguiente modo: Supóngase un haz formado por las longitudes de onda λ1 y λ2 (banda M en la figura 3.3.a.). N N A A M M λ1 λ2 a) λ C b) Figura 3.3. Influencia de la radiación policromática sobre la ley de Beer. Aplicando la ley de Beer a cada una de ellas, se tiene: A1 = log P 01 –ε b C = ε 1 b C ; P 1 = P 01 10 1 P1 A2 = log P 02 –ε b C = ε 2 b C ; P 2 = P 02 10 2 P2 7 Claudio González Pérez Cuando este haz constituido por las dos radiaciones atraviesa una disolución conteniendo especies absorbentes, la potencia del haz emergente es P1+P2, mientras que la del haz incidente sobre la muestra es P01 + P02. Según esto, la absorbancia medida será: A M = log P 01 + P 02 = log P1 + P2 P 01 + P 02 P 01 10 –ε 1 b C AM = log (P01 + P02) – log (P01 10–ε1 bC + P 02 10 –ε 2 b C + P02 10– ε2b C ) En el caso particular de que ε1 = ε2 la ecuación anterior se simplifica, reduciéndose a AM = ε1 b C que es la ley de Beer. Sin embargo, cuando ε1 es distinto de ε2, la relación entre AM y la concentración deja de ser lineal. Evidentemente, cuanto mayor sea la diferencia entre ε1 y ε2, mayores serán las desviaciones de la linealidad. Por eso mismo, aún cuando la anchura de banda sea relativamente grande, si las diferencias en los valores de la absortividad son pequeños, la utilización de un haz policromático no implica diferencias significativas respecto a uno monocromático (ver Fig. 3.3.a., banda N). b) Presencia de radiación parásita. El haz de radiación que sale de un monocromador suele estar contaminado con pequeñas cantidades de radiación parásita o dispersada originada por reflexión de los distintos componentes ópticos, dispersión por partículas de polvo atmosférico, etc. Por otra parte, la propia muestra puede originar dispersiones. Con frecuencia, la radiación dispersada tiene una longitud de onda diferente respecto a la radiación principal, pudiendo además, en ocasiones, llegar al detector sin haber atravesado la muestra. Por todo ello, la absorbancia medida en presencia de radiación parásita es: AM = log P0 + Ps P + Ps donde Ps es la potencia del haz dispersado. Al aumentar la concentración de sustancias absorbentes, disminuye P, con lo que este término puede ser lo suficientemente pequeño respecto a Ps como para que la expresión anterior se transforme en: AM = log P0 + Ps P + Ps - log 0 P + Ps Ps 8 Espectrofotometría de absorción ultravioleta-visible lo cual indica la presencia de desviaciones negativas en la ley de Beer debido a este factor. c) Errores de lectura. Los errores indeterminados en la lectura de la transmitancia o absorbancia son errores que potencialmente siempre están presentes y es necesario tenerlos en cuenta. En ocasiones, pequeños errores en la lectura de la transmitancia o de la absorbancia pueden ocasionar errores grandes en la concentración cuando se opera en los extremos de la escala. Esto se ilustra en la figura 3.4. %T 1 3 2 C Figura 3.4. Errores de lectura. En 1, figura 3.4., el error absoluto cometido en la determinación de la concentración, para un cierto error de lectura de transmitancia, es pequeño, pero al ser pequeña la concentración, el error relativo puede ser grande. En 3, la incertidumbre en la determinación de la concentración es alta, y en 2, hacia la mitad de la escala, parece que se trata de la situación más favorable. Puede demostrarse, como se indica a continuación, que el mínimo error relativo se obtiene para una absorbancia de 0.434. La primera derivada de la ley de Beer es: A = ε b C = – log T ; ε b dC = – log e dT T donde dT representa el error indeterminado en la lectura de la escala de transmitancia y dC indica la incertidumbre en la concentración. Como log e = 0.434 y ε b = – log T/C, se obtiene, dC 0.434 = dT C T(log T) Derivando esta ecuación de nuevo e igualando a cero, se obtiene que la transmitancia óptima corresponde a 36.8 %, que equivale a una absorbancia de 0.434. 9 Claudio González Pérez DESVIACIONES QUIMICAS Se incluyen en este apartado toda una serie de desviaciones aparentes de la ley de Beer producidas como consecuencia de procesos químicos en los que participan las especies absorbentes. a) Influencia del equilibrio. Cuando la sustancia problema interviene o forma parte de un sistema en equilibrio con otras especies, el desplazamiento del equilibrio implica una modificación en la concentración, y, en consecuencia, en la absorbancia. Algunas situaciones que pueden producirse son las siguientes: Dimerizaciones. Cuando una disolución de dicromato potásico no tamponada se diluye, ocurre una transformación parcial en cromato, como consecuencia del equilibrio dímero-monómero: Cr2O72– + H2O <——> 2 CrO42– + 2 H+ (λmax = 350, 450 nm) (λmax = 372 nm) Acido–base. En la figura 3.5. se muestran los espectros de absorción de un indicador ácido-base. 4 A 3 1 punto isosbéstico 2 HIn <——> In– + H+ 3 rojo amarillo 2 1 4 λ Figura 3.5. Espectros de absorción de un indicador ácido-base. Es evidente que cuando las especies absorbentes están implicadas en un equilibrio ácido-base, la ley de Beer no se cumple, al menos que el pH o la fuerza iónica sean constantes, o se opere a la longitud de onda del punto isobéstico, esto es, la longitud de onda a la cual las dos especies absorbentes en equilibrio presenten la misma absortividad. En este sentido hay que mencionar que los puntos isosbésticos se toman frecuentemente como criterio para la existencia de dos especies absorbentes interconvertibles de un compuesto determinado. Complejos. Cuando se mide la absorbancia de un complejo, por ejemplo Fe(SCN)63–, para que se cumpla la ley de Beer hay que mantener constante la concentración de ligando libre, lo cual se consigue frecuentemente operando en gran exceso de ligando respecto al metal. Espectrofotometría de absorción ultravioleta-visible 10 b) Influencia del disolvente. Como consecuencia de las interacciones soluto– disolvente se originan con frecuencia desplazamientos espectrales, ensanchamientos de bandas y otros fenómenos que pueden provocar desviaciones en la ley de Beer. En este sentido, no es posible hacer predicciones de forma general. Unicamente mencionar algunos términos relacionados con los desplazamientos espectrales: desplazamiento batocrómico o desplazamiento hacia el rojo, consiste en un desplazamiento del máximo de absorción hacia longitudes de onda mayores (este efecto suele producirse en disolventes de alta constante dieléctrica). Desplazamiento hipsocrómico o desplazamiento hacia el azul, es el desplazamiento hacia longitudes de onda más cortas. c) Influencia de la temperatura. La temperatura puede influir modificando el equilibrio químico de algunos sistemas, así como, en ocasiones, dar lugar a desplazamientos batocrómicos. De todas formas, la temperatura no suele ser un factor a considerar en la mayor parte de los sistemas absorbentes sencillos. d) Presencia de impurezas en los reactivos. Muchos métodos espectrofotométricos son lo suficientemente sensibles como para detectar cantidades a nivel de trazas, por lo que la presencia de impurezas absorbentes en los mismos reactivos pueden originar errores considerables. Debido a ello, en la práctica analítica ordinaria, las medidas espectrofotométricas se llevan a cabo frente a un blanco constituido por la propia célula, el disolvente y los reactivos. En este sentido interesa que la absorbancia del blanco sea pequeña, pues si es grande, un pequeño error en su medida puede implicar un gran error relativo en el resultado final. e) Interacciones entre especies absorbentes. Cuando en una disolución existen varias especies absorbentes, la ley de Beer se cumple para cada una de ellas, si todas actúan independientemente. Sin embargo, la interacción entre ellas puede producir alteraciones en la distribución de cargas, como consecuencia de lo cual puede modificarse la energía requerida para la absorción y, en consecuencia, variaciones en la posición, forma y altura de las bandas de absorción. Por otra parte, estas alteraciones en la distribución de cargas también pueden ser originadas por la presencia de sales inertes, con el consiguiente aumento de la fuerza iónica de la disolución. Claudio González Pérez 11 f) Interacciones soluto–radiación electromagnética. Aunque en sentido estricto no son factores de tipo químico, también deben considerarse otros tipos de interacción entre la radiación y la materia, distintos de los que intervienen en el proceso de absorción. Así, la posible emisión de resonancia y la presencia de fenómenos fluorescentes y fosforescentes pueden originar desviaciones aparentes en la ley de Beer. ERRORES PERSONALES En este sentido, los mayores errores suelen cometerse por el uso inadecuado de las cubetas de absorción. Resultan de utilidad las recomendaciones siguientes: * Es necesario asegurarse de que las cubetas están perfectamente limpias, no rayadas y exentas de huellas o adherencias en las paredes por las que ha de pasar la radiación. * Las cubetas de vidrio y cuarzo pueden limpiarse con ácido nítrico o con agua regia en frío, pero no con mezcla crómica. * Una vez limpias, las cubetas deben enjuagarse con agua destilada y con varias porciones de la disolución a medir. * No deben secarse interiormente, mientras que el exterior debe secarse con papel suave, comprobando, además, que, una vez llena con la disolución problema, no contiene burbujas de aire. * Aunque se debe trabajar con cubetas idénticas para la muestra y la referencia (blanco), es buena práctica utilizar siempre la misma disolución en cada una de ellas. ABSORCION DE RADIACION Como se indicó en el capítulo anterior, cuando una molécula absorbe un fotón, se incrementa su energía, pasando a un estado excitado. En relación con esto, pueden hacerse las siguientes preguntas: ¿qué pasa con la energía absorbida? ¿en qué se invierte?, etc. Seguidamente se intentará dar respuesta a ellas. La absorción de radiación ultravioleta y visible origina transiciones entre distintos niveles de energía electrónicos, así como simultáneamente transiciones Espectrofotometría de absorción ultravioleta-visible 12 vibracionales y rotacionales, de forma que el cambio en la energía molecular de una determinada especie como consecuencia de la absorción de energía radiante puede representarse así: ∆E = ∆Eelectrónica + ∆Evibracional + ∆Erotacional + ∆Etraslacional De todos los términos que figuran en la expresión anterior, el más importante es ∆Eelectrónica, pues la diferencia de energía entre dos estados vibracionales es unas 10 veces menos, y ∆Erotacional es, a su vez, entre 10 y 100 veces menor que ∆Evibracional. Por otro lado, el último término es tan pequeño que prácticamente puede prescindirse de su influencia. Debido a lo anteriormente expuesto, suele ser de utilidad clasificar las especies absorbentes en base al tipo de transiciones electrónicas que pueden tener lugar. Asimismo, esto permite correlacionar el comportamiento espectral de una determinada especie con sus características químicas, lo cual es importante en orden a determinar estructuras moleculares y para la identificación de ciertos grupos funcionales. Tipos de transiciones electrónicas Las moléculas orgánicas, y una gran cantidad de aniones inorgánicos, contienen electrones en orbitales moleculares σ y π, así como en orbitales no enlazantes, n, por lo que cuando estas especies absorben fotones de contenido energético adecuado se pueden producir transiciones a los correspondientes orbitales moleculares antienlazantes, σ* y π*, tal como se indica en la figura 3.6., donde se muestra la estructura de Lewis para una molécula orgánica sencilla y las transiciones electrónicas correspondientes. σ* π* H C H ++ O++ σ π +n n π σ Figura 3.6. Transiciones electrónicas entre niveles de energía moleculares. 13 Claudio González Pérez Como se representa en la figura 3.6., pueden tener lugar cuatro tipos de transiciones: σ—>σ*, π—>π*, n—>σ* y n—>π*. Asimismo, también pueden producirse transiciones denominadas de transferencia de carga y de campo ligando. Seguidamente se comentarán algunas peculiaridades de cada una de las transiciones mencionadas. Transiciones σ—>σ*. Estas transiciones entre orbitales moleculares sigma enlazantes y antienlazantes implican energías relativamente grandes (ver fig. 3.6.), de forma que las bandas de absorción se observan en el ultravioleta lejano, a longitudes de onda inferiores a 200 nm (Figura 3.7.). Esta región del espectro se denomina frecuentemente ultravioleta de vacío, debido a que los constituyentes normales del aire, nitrógeno y oxígeno, absorben fuertemente a 160 y 200 nm respectivamente, por lo que los espectros en esta zona deben obtenerse operando en el "vacío". UV lejano UV próximo Visible Intensidad π —> π * transferencia de carga σ —> σ* n —> π* . n —> σ* campo ligando 200 400 λ, nm 750 Figura 3.7. Bandas de absorción originadas por diferentes transiciones electrónicas. En los hidrocarburos saturados, en los que solamente hay enlaces sencillos C–H se producen estas transiciones; así el metano presenta máximos de absorción a 125 nm y el etano a 135 nm. Desde el punto de vista analítico, estas bandas tienen poco interés, debido a dificultades de tipo instrumental. Transiciones n—>σ*. Las especies químicas saturadas que contengan átomos con pares electrónicos en orbitales no enlazantes pueden dar lugar a que se produzcan transiciones n—>σ*. Esto sucede, por ejemplo, en compuestos saturados conteniendo heteroátomos de oxígeno, azufre y halógenos. Espectrofotometría de absorción ultravioleta-visible 14 Estas transiciones requieren bastante energía, si bien en menor grado que las σ— >σ*, por lo que las bandas correspondientes se presentan fundamentalmente en el ultravioleta lejano. Por otra parte, la intensidad de las absorciones asociadas con estas bandas suelen ser de pequeña magnitud. Los máximos de absorción correspondientes a esta transiciones n—>σ* tienden a desplazarse hacia longitudes de onda menores al aumentar la electronegatividad del heteroátomo. De hecho, la mayor parte de los compuestos saturados que contienen oxígeno absorben a longitudes de onda inferiores a 200 nm, lo cual hace posible que compuestos tales como el agua, o los alcoholes, puedan utilizarse como disolventes en el ultravioleta próximo. Transiciones n—>π* y π—>π*. Estas transiciones se consideran juntas debido a que muchos grupos químicos contienen electrones en orbitales π y no enlazantes. Como se aprecia en la Figura 3.6., las transiciones π—>π* requieren la absorción de una cantidad de energía relativamente grande, por lo que suelen aparecer en el ultravioleta lejano, mientras que las n—>π* necesitan menor cantidad de energía, como consecuencia de lo cual se originan bandas en las zonas ultravioleta próximo y visible. En lo que respecta a la intensidad de la absorción, las absortividades molares de los picos asociados a las transiciones n—>π* son generalmente bajos, mientras que los correspondientes a los π—>π* son considerablemente mayores. De cualquier forma, el comportamiento mencionado puede alterarse, como se verá posteriormente, por la presencia de determinadas agrupaciones atómicas, o por el tipo de disolvente. Para que tengan lugar las transiciones consideradas en este apartado, es evidente que la especie química tendrá que contener algún grupo funcional con enlaces π, esto es, la estructura molecular deberá presentar centros absorbentes no saturados. A estos grupos de átomos responsables de la absorción en las regiones ultravioleta y visible se les denomina grupos cromóforos (por extensión, se considera que los sistemas con electrones s son cromóforos en el ultravioleta lejano). En la tabla 3.1. se muestran algunos grupos cromóforos y las transiciones implicadas en espectrofotometría ultravioleta-visible. Por otra parte, existen determinadas agrupaciones atómicas que por sí mismas no comunican color a la molécula que pertenecen, pero son capaces de reforzar la acción de un cromóforo. Estos grupos se denominan auxocromos. 15 Claudio González Pérez Tabla 3.1. Grupos cromóforos y transiciones electrónicas Grupo cromóforo Estructura Etilénico C C Acetilénico C C Carbonilo C Amida Nitro — NO2 Oxima C Ester π−> π∗ π −> π∗ π −> π∗ n −> π∗ π −> π∗ n −> π∗ π −> π∗ n −> π∗ π −> π∗ n −> π∗ O O O O C OR O C NH2 Carboxilato Transición C π −> π∗ n −> π∗ π −> π∗ n −> π∗ N Los grupos auxocromos suelen contener grupos funcionales con electrones en orbitales no enlazantes y que, en consecuencia, pueden absorber únicamente en el ultravioleta lejano, como consecuencia de transiciones n—>σ*. Sin embargo, si un auxocromo y un cromóforo se encuentran en la misma molécula, la absorción del cromóforo se desplaza hacia longitudes de onda más largas, a la vez que se produce un incremento en la intensidad de la absorción. Este efecto se atribuye normalmente a la posibilidad de que se den transiciones n—>π*. En relación con las modificaciones de las intensidades de absorción se definen los siguientes términos: efecto hipercrómico (aumento de la intensidad de la absorción) y efecto hipocrómico (disminución de la intensidad de la absorción). Cuando una molécula contiene dos o más grupos cromóforos pueden darse las siguientes posibilidades: a) Si los cromóforos están separados por dos o más enlaces sencillos se dice que están aislados, y, en estos casos, la absorción es la suma de todos los grupos cromóforos presentes. Ejemplo: CH3–CH2–CH2–CH=CH2 CH2=CH–CH2–CH2–CH=CH2 λmax (nm) 184 185 εmax 10000 20000 16 Espectrofotometría de absorción ultravioleta-visible b) Cuando los cromóforos están conjugados, esto es, separados por un enlace sencillo, se produce un desplazamiento batocrómico acompañado de un efecto hipercrómico. –C=C– –C=C–C=C– –C=C–C=C–C=C– λmax (nm) 170 220 260 εmax 16000 21000 35000 La razón de estos desplazamientos se considera que es debida a que la conjugación hace que se produzca una deslocalización de los electrones π sobre, al menos, cuatro átomos, como consecuencia de lo cual se rebaja el nivel de energía del orbital π*, con lo que al ser menor la energía requerida para las transiciones, los máximos de absorción se desplazan hacia mayores longitudes de onda, aumentando además las probabilidades de que tengan lugar las mencionadas transiciones, lo que origina un aumento simultáneo en la intensidad de la absorción. Sin embargo, cuando existe algún enlace triple, se observa una disminución de la intensidad de absorción (efecto hipocrómico) λmax ε max CH2 = CH – CH = CH 2 219 21000 CH2 = CH – C CH 219 6500 c) Cuando los cromóforos están unidos directamente, el espectro resultante normalmente es distinto del que presentan ambos cromóforos cuando están aislados, ya que en realidad se forma un nuevo cromóforo. CH2=CH2 CH2=C=CH2 λmax 170 225 εmax 10000 500 En cuanto a la influencia del disolvente, puede decirse que al aumentar la polaridad del disolvente se produce un desplazamiento hipsocrómico (hacia menor longitud de onda) de las bandas n—>π* y un desplazamiento batocrómico (hacia longitudes de onda mayores) de las bandas π—>π*. Esto puede explicarse teniendo en cuenta que el estado excitado es más polar que el estado fundamental, por lo cual, la presencia de un disolvente polar tiende a estabilizar los estados excitados, rebajando su nivel energético. De todas formas, si únicamente tuviera lugar este efecto, en ambas transiciones ocurrirían desplazamientos batocrómicos. Sin embargo, en el caso de las transiciones n—>π* se produce un aumento de solvatación del par de electrones no enlazados, lo cual rebaja la energía del orbital no enlazante en mayor medida que lo que disminuye la energía del orbital π*. En consecuencia, se produce un 17 Claudio González Pérez aumento de la energía requerida para la transición n—>π* y el consiguiente desplazamiento hipsocrómico. Bandas de transferencia de carga. Estas bandas se originan cuando se produce transferencia de electrones de una parte a otra de un sistema. Para ello es necesario que uno de sus componentes tenga características de dador de electrones y otro de aceptor. Como consecuencia de esta transferencia de electrones se origina un estado excitado que es el resultado de un proceso de oxidación-reducción interna. Así, por ejemplo, la absorción de un fotón por el complejo Fe(III)–SCN– da lugar a la transferencia de un electrón desde el tiocianato hasta uno de los orbitales del hierro, originándose una especie excitada consistente en Fe(II) y SCN neutro: Fe(III)–SCN– + hν —> Fe(II)–SCN Lo mismo que sucede con otros tipos de excitación electrónica, el electrón que ha experimentado la transferencia vuelve a su estado original, si bien, ocasionalmente puede tener lugar la disociación del estado excitado, teniendo lugar, en este caso, una reacción fotoquímica. Algunas moléculas orgánicas pueden transferencia de carga intramolecular: originar de absorción por R R C = O + hν bandas + – =C—O Las bandas de absorción originadas por estos procesos de transferencia de carga son particularmente importantes en análisis químico, ya que muchos compuestos orgánicos e inorgánicos las presentan en el ultravioleta (a veces también en el visible) y las absortividades molares suelen ser muy altas (superiores a 10000), por lo cual se obtienen límites de detección muy bajos para estas especies. Bandas de campo ligando. El color que presentan muchos iones y compuestos de metales de transición normalmente se deben a absorciones denominadas de campo ligando (junto a éstas, suelen presentarse las intensas bandas de transferencia de carga anteriormente mencionadas). Estas absorciones suelen ser de baja intensidad y 18 Espectrofotometría de absorción ultravioleta-visible aparecen en la región visible, llegando en ocasiones hasta el ultravioleta próximo y el infrarrojo próximo. El origen de las bandas de campo ligando puede explicarse del siguiente modo: los cinco orbitales d de un átomo o un ión de un elemento de transición, cuya representación se muestra en la figura 3.8., en ausencia de un campo magnético o eléctrico externo están degenerados, no siendo necesaria la absorción de radiación para promover un electrón de un orbital a otro. Como puede verse en la figura 3.8., los orbitales dx2–y2 y dz2 están orientados según los ejes de coordenadas x, y y z. Estos orbitales forman el grupo eg. Por su parte, los orbitales dxy, dyz y dxz, llamados orbitales t2g, se orientan según las bisectrices a dichos ejes. Al formarse un complejo octaédrico entre el ión metálico y el agua, o algún otro ligando, los átomos dadores de los ligandos se aproximan al ión metálico central a lo largo de los ejes, por lo que habrá una mayor repulsión entre los electrones de los ligandos y los electrones de los orbitales eg del ion central, que entre los de los ligandos y los de los orbitales t2g. z z z y x x x y d yz dxy z dxz y z y y x x d x2–y 2 dz 2 Figura 3.8. Representación esquemática de los cinco orbitales atómicos d. 19 Claudio González Pérez Como consecuencia de las repulsiones mencionadas, la aproximación de los seis átomos dadores (cargas puntuales) a lo largo de los ejes produce un campo eléctrico que elimina la degeneración existente en el grupo de orbitales d y los divide en dos grupos, los t2g con energía inferior y los eg con energía superior (figura 3.9.) * dx 2–y 2 d z2 E d xy dxz dyz dx2 –y 2 d z2 ∆oct. d xy dxz dyz Figura 3.9. Desdoblamiento de los orbitales d por efecto del campo ligando. La energía de separación entre los dos grupos, representada por ∆oct depende de las características del ión metálico (carga y número cuántico principal del orbital d) y del ligando (distribución de su densidad de carga y polarizabilidad). En este sentido, es posible ordenar los ligandos más comunes en orden creciente de ∆ (serie espectroquímica): I–<Br–<Cl–<F–<OH–<C2O42– H2O<SCN–<NH3<etilendiamina<NO2–<CN– Ejemplo: Las disoluciones acuosas de Cu2+, Cu(H2O)42+ son de color azul pálido, mientras que en medio amoniacal, Cu(NH3)42+, el color es azul violeta muy intenso. Esto es debido al aumento que experimenta el campo ligando cuando se sustituye el H2O por NH3. Las consideraciones teóricas anteriormente expuestas permiten deducir si una determinada especie química absorberá radiación electromagnética en las regiones ultravioleta y visible. Desde el punto de vista práctico es interesante poder predecir la longitud de onda aproximada a la que se producirá la absorción máxima. Para esto, se han propuesto toda una serie de reglas empíricas obtenidas de la observación de los espectros de absorción de una gran variedad de sustancias orgánicas. Las reglas que seguidamente se presentan de forma resumida** se han desarrollado para los siguientes tipos de compuestos: dienos conjugados, anillos bencénicos sustituidos, compuestos carbonílicos no saturados y ácidos, esteres, nitrilos y amidas no saturados. Cada uno de esos grupos funcionales absorbe a una cierta longitud de onda, * Cuando se forman complejos tetraédricos o con simetría cuadrada plana, se originan desdoblamientos que pueden deducirse con razonamientos similares a los utilizados para la estructura octaédrica. ** Para profundizar en el tema pueden consultarse las siguientes publicaciones: Scott, "Interpretation of the ultraviolet Spectra of Natural Products". Pergamon, Oxford (1964. H.H. Jaffe y M. Orchin, "Theory and Applications of Ultraviolet Spectroscopy", Willey, Nuw York (1962). 20 Espectrofotometría de absorción ultravioleta-visible y a este valor hay que adicionarle determinadas cantidades en función de la presencia de otras agrupaciones atómicas presentes en la molécula. En la tabla 3.2 se muestran dichas cantidades, correspondientes a algunos casos concretos. Tabla 3.2. Longitudes de onda de máxima absorción Dienos conjugados Bencenos monosutituidos Comp. carbonílicos Ac. y esteres insaturados –C=C–CO C=C–CHO 215 210 197 λ base, nm 217 203 Doble enlace adicional + 30 +45 +30 +30 +30 α +10 β +12 γ +18 α +10 β +12 γ +18 +10 α +35 β +30 γ +50 α +35 β +30 γ +50 254 Grupo alquilo +5 +20 +17 Cl, Br +5 +7 +8 — OH +7 +16 —COOH +27 +19 —NH2 +27 +26 Algunas consideraciones respecto al color Los cuerpos luminosos, como el Sol o una lámpara, emiten radiaciones en un amplio espectro que comprende muchas longitudes de onda. Aquellas longitudes de onda que están comprendidas entre unos 400 y 700 nm son capaces de afectar la retina del ojo humano y con ello dar origen a las impresiones subjetivas de la visión. Se hace mención de impresiones subjetivas, debido a que en la percepción del color influyen factores químicos, fisiológicos y físicos; de hecho, la respuesta humana al color varía de una persona a otra. Las personas que tienen una visión normal del color son capaces de correlacionar aproximadamente la longitud de onda de la radiación visible que llega al ojo con la subjetiva sensación del color, y éste, a veces se utiliza para designar ciertas porciones del espectro. 21 Claudio González Pérez Cuando incide radiación electromagnética visible sobre la materia puede ser absorbida total o parcialmente; si la absorción es total, el objeto aparece de color negro, y si se reflejan todas las radiaciones, de color blanco. Nosotros percibimos los objetos por medio de la luz emitida o reflejada, de forma que cuando la luz blanca (conteniendo todas las longitudes de onda) pasa a través de un medio que es transparente a ciertas longitudes de onda, pero que absorbe otras, para el observador ese medio aparece coloreado. Debido a que solo las radiaciones no absorbidas llegan al ojo, sus longitudes de onda indican el color del medio. Se dice que este color es complementario al color que se percibiría si la luz absorbida se pudiera detectar, debido a que la luz emitida y absorbida juntas forman la luz blanca original. Análogamente, los objetos de color que son opacos absorben algunas longitudes de onda y reflejan otras cuando son iluminadas por luz blanca. En la tabla 3.1. se indican los valores que corresponden a determinadas zonas del espectro visible. Tabla 3.1. Colores del espectro visible λ absorbida (nm) Color absorbido Color observado (complementario) 380–420 420–440 440–470 470–500 500–520 520–550 550–580 580–620 620–680 680–780 violeta azul–violeta azul verde–azulado verde amarillo–verdoso amarillo anaranjado rojo púrpura amarillo-verdoso amarillo anaranjado rojo púrpura violeta azul–violeta azul verde–azulado verde INSTRUMENTACION El instrumento que normalmente se utiliza para medir la transmitancia o la absorbancia de una muestra en función de la longitud de onda es el espectrofotómetro. Antes de pasar a describir sus componentes básicos, es conveniente indicar algunos términos relacionados con la nomenclatura utilizada a propósito de la instrumentación en los métodos ópticos de análisis. Las definiciones Espectrofotometría de absorción ultravioleta-visible 22 que se indican no pueden considerarse universales, pero sí están en razonable acuerdo con el uso popular y son las que se utilizan en las principales obras dedicadas al tema*. FOTOMETRO: Cualquier dispositivo utilizado para medir la intensidad de radiación. Normalmente se utiliza para designar un instrumento sencillo provisto de filtros para seleccionar una banda de longitudes de onda y de una fotocélula o un fototubo para medir la intensidad de radiación. ESPECTROFOTOMETRO: Instrumento más sofisticado que posee un monocromador en lugar de filtros. Además, el sistema de detección normalmente es un fotomultiplicador, más sensible que una fotocélula. COLORIMETRO: Instrumento muy simple que compara, usando el ojo humano como detector, el color de la sustancia problema con el de una disolución patrón. (El nombre de colorímetro suele aplicarse en la práctica a cualquier instrumento apropiado para medir en la region visible, y, en realidad, así se conocen muchos fotómetros de filtro comerciales). ESPECTROSCOPIO: Aparato diseñado para detectar detectar líneas espectrales a simple vista. Su aplicación está restringida al análisis cualitativo y para elementos con líneas de emisión en la zona visible del espectro. ESPECTROGRAFO: Instrumento que registra líneas espectrales sobre una placa fotográfica. ESPECTROMETRO: Denominación general que se aplica a instrumentos que poseen sistemas de detección eléctricos. Según las definiciones anteriores, un espectrofotómetro es un espectrómetro que mide fotones. Su utilización suele limitarse, en la práctica, a la región untravioleta, visible e infrarroja. * Eugene D. Olsen. "Métodos Opticos de Análisis". Ed. Reverté. Barcelona. 23 Claudio González Pérez Los componentes básicos de un espectrofotómetro son: una fuente de radiación, un monocromador, que seleccione una banda estrecha de longitudes de onda, una cubeta, o recipiente que contenga la muestra, un detector de radiación y un sistema de tratamiento y lectura de la señal detectada (figura 3.10.) espejo . Fuente de radiación Filtro o Detector monocromador Muestra Medidor o registro . Referencia Figura 3.10. Espectrofotómetro de doble haz. Fuentes de radiación Las fuentes de radiación utilizadas en espectrofotometría ultravioleta y visible deben ser continuas en una amplia zona del espectro, de intensidad elevada y ser esencialmente constante con la longitud de onda. En la zona ultravioleta y visible, la fuentes más utilizadas son de dos tipos: fuentes térmicas, basadas en la emisión de radiación por efecto de la temperatura, y fuentes cuya radiación se debe a descargas eléctricas producidas en el seno de gases. Entre las primeras, la más común es la lámpara de filamento de volframio. En condiciones ordinarias de operación, esta lámpara resulta útil entre unos 350 nm y unos 3000 nm. (figura 3.11.) volframio (- 3000ºK) deuterio visible Energia . 200 400 700 λ , nm Figura 3.11. Curvas de distribución espectral de las lámparas de deuterio y volframio. Espectrofotometría de absorción ultravioleta-visible 24 Como puede observarse en la figura 3.11., la mayor parte de la energía emitida corresponde a la zona infrarroja. La distribución de la energía depende de la temperatura del filamento, la cual depende, a su vez, del voltaje; un incremento en la temperatura de operación aumenta la energía total emitida y desplaza el máximo de intensidad hacia longitudes de onda más cortas. Sin embargo, en la práctica, esto no se utiliza para obtener mayor cantidad de radiación ultravioleta, ya que se acorta considerablemente el tiempo de vida de la lámpara. Debido a que la radiación emitida depende únicamente del voltaje suministrado, éste tiene que ser muy estable, por lo cual los instrumentos llevan incorporado un sistema para la estabilización de la corriente. Por otra parte, el calor producido por la lámpara puede constituir un problema, por lo que, con frecuencia, en el lugar donde se coloca la lámpara se instala un ventilador con objeto de evitar el calentamiento de la muestra y de los demás componentes del instrumento. Por debajo de 350 nm, la potencia de una lámpara de volframio es inadecuada, debiéndose emplear una fuente diferente. La más común es una lámpara de descarga de hidrógeno, o de deuterio. Cuando se produce una descarga eléctrica entre dos electrodos en el seno de un gas, como hidrógeno, las colisiones entre los electrones de la descarga y las moléculas gaseosas provocan la excitación electrónica, vibracional y rotacional de dichas moléculas, con lo que se obtiene un espectro de líneas que es característico del gas, siempre que la presión sea baja. Al aumentar la presión, las líneas se ensanchan, llegando a superponerse, hasta que, a presiones relativamente altas (0.2–5 mm) se produce un espectro continuo. Tanto la lámpara de hidrógeno como la de deuterio tienen un intervalo de utilización comprendido entre 175 y 350 nm (figura 3.11.). También se utilizan con la misma finalidad la lámpara de descarga de xenon y la de vapor de mercurio. Finalmente, indicar que, puesto que el vidrio absorbe fuertemente a longitudes de onda inferiores a unos 350 nm, las lámparas de ultravioleta deben utilizar ventanas de cuarzo. 25 Claudio González Pérez Filtros y monocromadores La misión de los filtros y de los monocromadores es seleccionar un haz de radiación "monocromática"*. Con este fin se utilizan los siguientes dispositivos: De corte Absorción De banda Filtros Interferencia Selección de la longitud de onda Prismas Monocromadores De transmisión Redes De reflexión Los filtros de absorción se utilizan en la región visible y se basan en la absorción selectiva de ciertas longitudes de onda. Normalmente consisten en un vidrio coloreado o una suspensión de un colorante en gelatina que se coloca entre dos placas de vidrio. Los filtros de banda (figura 3.12. A) se caracterizan por su anchura de banda (anchura a la mitad de la altura) que puede oscilar entre 30 y 250 nm. 100 %T B (filtro naranja) A (filtro verde) 50 400 anchura de banda 500 combinación de los dos filtros 600 700 λ, nm 800 Figura 3.12. Transmitancia de algunos filtros. Los filtros de corte tienen transmitancias de casi el 100 % en una zona del espectro visible, pero luego disminuye rápidamente hasta un valor de transmitancia cero (figura 3.12.B). Por combinación de diferentes filtros pueden seleccionarse bandas espectrales relativamente estrechas (figura 3.12.). * La radiación monocromática es la radiación de una sola longitud de onda. Por supuesto, es imposible producir radiación monocromática verdadera, en sentido estricto. Sin embargo, cuanto mejor sea el monocromador, tanto más estrecho será el intervalo de longitudes de onda. Espectrofotometría de absorción ultravioleta-visible 26 Los filtros de interferencia consisten en un dieléctrico transparente (frecuentemente, fluoruro cálcico o magnésico) recubierto en ambos lados con dos finas capas de plata semirreflectante. (figura 3.13.) Figura 3.13. Filtro de interferencia. Cuando un haz de radiación incide sobre este dispositivo, una fracción atraviesa la primera capa metálica, mientras que la restante se refleja. La parte que ha pasado sufre una escisión similar al llegar a la segunda capa metálica. Si la parte reflejada en la segunda interacción es de longitud de onda adecuada, se refleja, en parte, desde la cara interior de la primera capa en fase con la radiación incidente de la misma longitud de onda. El resultado es que se refuerza esa determinada longitud de onda, mientras que la mayoría de las otras, fuera de fase, sufren una interferencia destructiva. Estos filtros proporcionan anchuras de banda menores y transmitancias de pico mayores que los filtros de absorción. Se dispone de filtros de interferencia para todas las zonas de las regiones ultravioleta y visible, así como parte del infrarrojo. Un monocromador se caracteriza por producir un haz de radiación de gran pureza espectral y permitir variar, de forma continua y en un amplio intervalo, la longitud de onda de la radiación. Los componentes básicos de un monocromador son una rendija de entrada, que selecciona un haz de radiación policromática entrante, un elemento dispersante, prisma o red, que dispersa la radiación en sus longitudes de onda individuales, y una rendija de salida, que aísla la banda espectral deseada (figura 3.14.) La dispersión de radiación por un prisma se basa en el fenómeno de la refracción; esto es, el cambio de dirección que experimenta un haz de radiación al pasar de un medio a otro con distinto índice de refracción. El grado de desviación depende de la longitud de onda; así, los azules se desvían más que los rojos. 27 Claudio González Pérez El material de que está construido el prisma depende del tipo de radiación a dispersar; en la región visible se usan prismas de vidrio, mientras que en el ultravioleta es necesario usarlos de cuarzo. rendija de salida rendija de entrada λ1 λ1+λ2 λ2 . Figura 3.14. Dispersión de radiación por un prisma. Los prismas presentan las ventajas de su gran pureza espectral (no hay órdenes de dispersión superiores, como en las redes), mientras que el principal inconveniente reside en que la dispersión no es lineal; esto es, las longitudes de onda no se dispersan de manera uniforme: es mayor para las longitudes de onda más cortas. Las redes de reflexión*, que son las más utilizadas, consisten en una superficie dura, pulida, sobre la que se ha grabado un gran número de surcos paralelos y muy próximos entre sí (entre 300 y 2000 surcos por milímetro para las regiones ultravioleta y visible). Figura 3.15. Difracción de radiación por una red de reflexión. Cuando un haz de radiación incide sobre una red de reflexión con un ángulo i (figura 3.15.) se refleja según un ángulo φ, diferente del incidente. En la Figura 3.15. se muestran los haces paralelos 1 y 2. La máxima interferencia constructiva entre ambos se produce cuando la diferencia de caminos recorridos por ellos sea un múltiplo entero de la longitud de onda, y esta diferencia es AB – CD. Los segmentos AB y CD pueden expresarse en función de d y de los ángulos i y . * Tambien existen las redes de transmisión, que normalmente se construyen trazando una serie de surcos paralelos sobre una placa de vidrio, con una punta de diamante. Espectrofotometría de absorción ultravioleta-visible AB = d sen i 28 CD = – d sen φ** por lo cual, nλ = d (sen i + sen φ) donde n, número entero, se denomina orden de difracción. Según la ecuación anterior, existen distintos valores de λ para unos determinados ángulos i y φ, por lo que junto con la línea de primer orden (n=1) aparecen líneas de órdenes superiores. Ordinariamente, la línea de primer orden es la más intensa. Las líneas de órdenes superiores pueden eliminarse mediante filtros. El fenómeno de la difracción de una radiación policromática por una red se representa esquemáticamente en la figura 3.16. Para seleccionar la radiación de una determinada longitud de onda, se hace girar la red hasta hacer que la radiación que interesa coincida con la rendija de salida, eliminando los órdenes superiores mediante filtros. 3er orden 267 2º orden 400 1er orden 800 600 200 300 200 400 200 radiación incidente (200–800 nm) Figura 3.16. Difracción de una radiación policromática por una red. En cuanto a la anchura de rendija de salida debe tenerse en cuenta lo siguiente: a medida que disminuye la anchura de rendija se reduce la anchura de banda, siendo posible aumentar la resolución, pero solo hasta un cierto límite, ya que, a partir de un determinado valor, la difracción por la propia rendija comienza a ser apreciable. Además, es necesario considerar que al disminuir la anchura de rendija, disminuye también la intensidad del haz de radiación, por lo que hay que tener en cuenta la sensibilidad del detector, la cual puede limitar el estrechamiento de la rendija. ** El signo menos indica que el ángulo de reflexión, φ, cae en el lado opuesto al ángulo de incidencia, i. 29 Claudio González Pérez En resumen, y comparando con los prismas, las redes de difracción presentan las ventajas de su elevada resolución, dispersión lineal y pocas pérdidas de radiación por absorción. Posiblemente, el mayor inconveniente esté relacionado con la presencia de órdenes de difracción superiores. Recipientes para las muestras En espectrofotometría analítica, casi siempre se trabaja con disoluciones, por lo cual la mayoría de los recipientes para las muestras son celdas o cubetas para colocar líquidos en el haz del espectrómetro. Estos recipientes deben estar fabricados con un material que permita el paso de radiación de la región espectral de interés. Así, el vidrio puede emplearse entre 350 y 2000 nm, mientras que en la región ultravioleta se necesita cuarzo o sílice fundida (ambas sustancias también son transparentes en la región visible). En algunos instrumentos baratos se utilizan a veces tubos de ensayo cilíndricos como recipientes para las muestras. Es importante que estos tubos siempre se coloquen igual, para lo que se marcan en un lado, y la marca siempre se pone en la misma dirección cuando se coloca el tubo en el compartimento de cubetas del instrumento. Las celdas se deben llenar de tal forma que el haz de radiación pase a través de la disolución, con el menisco por encima del haz. Las celdas típicas para las regiones ultravioleta y visible tienen 1 cm de paso óptico, si bien existe una gran variedad en cuanto a tamaño, forma y otras peculiaridades, como se muestra en la figura 3.17. . estándar (1 cm) cilíndrica microcelda . . de flujo térmica Figura 3.17. Celdas para espectrofotometría. 2 cm 30 Espectrofotometría de absorción ultravioleta-visible Detectores Los detectores usados en espectrofotometría ultravioleta y visible son transductores que convierten la energía radiante en una señal eléctrica*. Un detector ideal deberá presentar las características siguientes: * * * * * * * Sensibilidad elevada en la región espectral de interés. Respuesta lineal para la energía radiante. Tiempo de respuesta pequeño. Utilizable en un amplio intervalo de longitudes de onda. Elevada relación señal/ruido. Mínima señal de salida en ausencia de radiación. Buena disponibilidad para la amplificación. Sin embargo, no existe el detector ideal, por lo que en la práctica, se evalúan todos los factores anteriores y se selecciona algún detector que resulte adecuado al caso. Los más utilizados son: células fotovoltaicas, fototubos y tubos fotomultiplicadores. Células fotovoltaicas. Consisten en una placa de hierro, que actúa de electrodo positivo, sobre la que se deposita una fina capa de un material semiconductor, como selenio, y éste se recubre de una capa muy fina de oro o plata, que actúa como segundo electrodo o electrodo colector (figura 3.18.) ( Plata ) Selenio Hierro ( ) Figura 3.18. Célula fotovoltaica. Cuando la radiación electromagnética incide sobre el selenio, se promocionan electrones a las bandas de conducción, haciendo que pasen electrones desde la superficie del selenio hasta el electrodo colector de plata, produciéndose un aumento de la conductividad proporcional al número de fotones que inciden sobre la superficie del semiconductor. Las células fotovoltaicas presentan las siguientes características: son sencillas de construir, relativamente baratas y no requieren una fuente de energía externa, por lo que pueden conectarse directamente a un galvanómetro o un amperímetro. En cuanto a los inconvenientes, su uso limitado a la región visible (su máxima sensibilidad * En espectrofotometría infrarroja suelen utilizarse detectores térmicos, que responden al calor. 31 Claudio González Pérez se presenta hacia los 550 mn, mientras que la respuesta a 350 y a 750 nm disminuye hasta un 10 % de la máxima), presentan dificultades para la amplificación, y fenómenos de fatiga, de forma que la corriente de salida disminuye gradualmente con el tiempo. Fototubos. Consisten en un cátodo semicilíndrico recubierto interiormente de un material fotosensible, y un ánodo, en el interior de un recipiente en el que se ha hecho el vacío (figura 3.19.). . (–) hν (+) e– . Figura 3.19. Fototubo. Cuando la radiación incide sobre el cátodo, se produce una emisión de fotoelectrones que se dirigen al ánodo, originándose una corriente que posteriormente se amplifica. La emisión de electrones depende de la naturaleza de la superficie del cátodo y de la frecuencia de la radiación. En el comercio existen fototubos que difieren en el material con el que está construida la superficie del cátodo, siendo, por tanto, diferente su respuesta a la radiación de diversas frecuencias. Muchos espectrofotómetros están provistos de detectores intercambiables que permiten mantener una buena respuesta en un amplio margen de longitudes de onda. En general, puede concluirse que los fototubos son más sensibles que las células fotovoltaicas, por la posibilidad de poder amplificar la corriente inicialmente generada. Tubos fotomultiplicadores. Este tipo de detector consiste en un cátodo fotosensible y una serie de electrodos (dínodos), cada uno a un potencial menos negativo que el que le precede (figura 3.20.) 32 Espectrofotometría de absorción ultravioleta-visible dínodos C –550 V. A –600 V. hν –500 V. A: ánodo C: càtodo Figura 3.20. Tubo fotomultiplicador. La radiación que llega al fotocátodo provoca la emisión de electrones primarios, que son acelerados hasta el primer dínodo. Al incidir en él, cada fotoelectrón origina la emisión de varios electrones adicionales; éstos, a su vez, son acelerados hasta el dínodo 2, y así sucesivamente, hasta que al final, la corriente así producida se recoge en el ánodo, se amplifica electrónicamente y se mide. Normalmente, los tubos fotomultiplicadores contienen 9 ó 10 dínodos, los cuales originan de 106 a 107 electrones por cada fotón. Este sistema de detección se caracteriza por su respuesta rápida y elevada sensibilidad. El límite de detección viene condicionado por el ruido de fondo que se origina como consecuencia de la emisión termoiónica, la cual puede reducirse enfriando. De hecho, estas corrientes pueden eliminarse virtualmente enfriando el detector a –30 ºC, si bien esto no se lleva a cabo en el trabajo espectrofotométrico ordinario. INSTRUMENTOS DE HAZ SENCILLO Y DE HAZ DOBLE Las medidas de absorción de radiación ultravioleta y visible son, por su propia naturaleza, de carácter relativo; debe de compararse, continuamente, la absorción de la muestra, conteniendo el analito, con la de una referencia o blanco conteniendo las mismas especies que la muestra, excepto el analito. En los instrumentos de haz sencillo hay solamente un haz de radiación que, después de pasar a través de la muestra llega al detector. De esta forma, para comparar la absorción del blanco, debe sustituir éste a la muestra en cada lectura, lo cual implica varios segundos entre cada medida. Alternativamente, la muestra y la referencia pueden compararse varias veces por segundo usando un instrumento de doble haz. En él, (figura 3.10.) la radiación pasa Claudio González Pérez 33 alternativamente a través de la muestra y de la referencia, lo cual se lleva a cabo mediante un motor que hace girar un espejo ("chopper") dentro y fuera de la trayectoria de la radiación. Cuando el espejo obturador intermitente no desvía el haz, éste pasa a través de la muestra y el detector mide la potencia radiante P. Cuando dicho espejo desvía el haz a través de la celda de referencia, el detector mide Po. De esta forma la radiación es desviada varias veces por segundo, y el circuito compara automáticamente P y Po para obtener la absorbancia. La operación con el sistema de doble haz presenta, fundamentalmente, las dos ventajas siguientes: la comparación muy rápida de muestra y referencia ayuda a eliminar errores debidos a fluctuaciones en la intensidad de la fuente y del detector, inestabilidad electrónica y cambios en el sistema óptico. Por otra parte, se facilita la posible automatización. APLICACIONES En principio, cualquier especie química que absorba radiación electromagnética en las regiones ultravioleta o visible es susceptible de poder ser determinada por técnicas espectrofotométricas. El mayor campo de aplicación se encuentra en el análisis cuantitativo, siendo la espectrofotometría una de las herramientas más usadas. Análisis cualitativo Los espectros de absorción ultravioleta y visible son, en general, menos útiles con fines cualitativos que los espectros en la región infrarroja, debido a que en aquellos las bandas son más anchas y, por consiguiente, menos características. La identificación de un compuesto puro requiere la comparación empírica de los detalles del espectro (máximos, mínimos y puntos de inflexión) de la sustancia problema con los del compuesto puro. Además, la intensidad de la absorción, expresada en términos de la absortividad molar, ε, se usa con frecuencia como criterio adicional para la identificación, sobre todo si ε tiene un valor elevado a determinadas longitudes de onda. Cuando se trata de identificar sustancias orgánicas, es conveniente operar en fase de vapor y a presión baja, ya que en estas condiciones, el espectro observado es Espectrofotometría de absorción ultravioleta-visible 34 debido a moléculas aisladas y, generalmente es posible resolver la estructura fina vibracional y rotacional, lo que proporciona detalles característicos para la identificación. En disolución y, sobre todo, en presencia de disolventes polares como el agua, alcoholes, ésteres y cetonas, las moléculas no se encuentran aisladas, sino solvatadas, como consecuencia de lo cual se limita la rotación. Además, los campos del disolvente pueden modificar de forma irregular los niveles de energía vibracionales y, cuando el número de moléculas es grande, los niveles de energía son poco definidos, obteniendose como resultado la aparición de una banda. Una de las pocas sustancias orgánicas con espectro de absorción característico en disolución es el benceno. Su espectro se usa frecuentemente en test de especificaciones de pureza, donde el contenido en benceno debe ser cuidadosamente controlado, por ejemplo, en alcohol absoluto o en ciclohexano. Aun cuando la identificación de un compuesto orgánico de forma inequívoca con este técnica, no es posible normalmente, el espectro de absorción en las regiones ultravioleta y visible puede ser de utilidad para la detección de ciertos grupos funcionales. Así, por ejemplo, la presencia de una banda débil entre 280 y 290 nm, que se desplaza hacia menores longitudes de onda al aumentar la polaridad del disolvente, indica la presencia de un grupo carbonilo. De cualquier forma, y para mayor seguridad, estos datos deben usarse siempre en combinación con los obtenidos a partir de espectros infrarrojos, de resonancia magnética nuclear o cualquier otra información analítica de que se disponga. En resumen, el espectro de absorción ultravioleta y visible no es una prueba infalible de la identidad de una especie química, pero sí representa otra herramienta disponible para aplicarla de forma inteligente. En cuanto a sustancias inorgánicas, casi las únicas que tienen espectro de absorción ultravioleta y visible característico son los iones trivalentes de las tierras raras. De hecho, el Ho2O3 se utiliza en ocasiones para comprobar el calibrado de la longitud de onda de algunos instrumentos. Análisis cuantitativo Ya se ha comentado que la espectrofotometría de absorción ultravioleta y visible es una de las técnicas más usadas en análisis cuantitativo. Se ha estimado que solo en el campo biosanitario, un 95 % de las determinaciones cuantitativas se llevan a cabo por espectrofotometría*. * Douglas A. Skoog y James J. Leary. "Análisis Instrumental". Ed. Mc Graw–Hill. Madrid (1993) Claudio González Pérez 35 En relación con los métodos clásicos de análisis (gravimétricos y volumétricos), los métodos espectrofotométricos son menos exactos, si bien su mayor sensibilidad (10–4–10–6 M) y rapidez los hace competir ventajosamente con aquellos en muchas ocasiones. La precisión puede considerarse aceptable, ya que, normalmente se obtienen incertidumbres relativas entre 1 y 2 %, aunque con determinadas precauciones pueden reducirse considerablemente. La base de la aplicación de los métodos espectrofotométricos al análisis cuantitativo es la ley de Beer. El procedimiento a seguir para llevar a cabo una determinación analítica consta de una serie de etapas encaminadas a establecer las condiciones de trabajo y la preparación de la curva de calibrado. a) Selección de la longitud de onda. Las medidas de absorbancia se llevan a cabo normalmente a la longitud de onda correspondiente al máximo de absorción, ya que de esta forma se obtienen máximas sensibilidades. Por otra parte, en esa zona, la curva espectral suele ser bastante plana, por lo que el cumplimiento de la ley de Beer es normalmente satisfactorio (ver anteriormente: "Uso de radiación no monocromática" en las "Desviaciones de la ley de Beer"). A veces no es posible operar a la longitud de onda del máximo de absorbancia cuando a esa longitud de onda absorbe apreciablemente alguna especie (distinta del analito) presente, incluso, en ocasiones, el propio reactivo. b) Preparación de las muestras para el análisis. Muchos compuestos orgánicos absorben en la región ultravioleta y el tratamiento de la muestra solo implica la separación de las posibles interferencias. Por otra parte, algunos elementos inorgánicos absorben intensamente en la región visible, pero solo en ciertos estados de oxidación. En estos casos, una etapa previa (además de la disolución si se trata de muestras sólidas) debe incluir un proceso redox para llevar el elemento al estado de oxidación adecuado. Así, por ejemplo, el manganeso se determina frecuentemente en forma de permanganato, previa oxidación con persulfato: 2 Mn2+ + 5 S2O82– + 8 H2O —> 2 MnO4– + 10 SO42– + 16 H+ La disolución violeta de permanganato presenta un máximo de absorbancia a 525 nm. En otras ocasiones es posible desarrollar un color por medio de reactivos orgánicos o inorgánicos. Así, por ejemplo, el Fe3+ puede determinarse mediante el Espectrofotometría de absorción ultravioleta-visible 36 color rojo de los complejos formados con SCN–; el Ni2+ por el color rosa de su quelato con dimetilglioxima, etc. En relación con estudios medioambientales, es importante la determinación de SO2 en el aire, para lo cual suele utilizarse pararosanilina como reactivo, midiendo a 560 nm la absorbancia del compuesto rojo obtenido. Asimismo, la determinación rutinaria de amoniaco, nitritos y nitratos en aguas para consumo público suele hacerse espectrofotométricamente. Para el amoniaco se utiliza el reactivo de Nessler, operando en presencia de AEDT para evitar las interferencias de Ca, Mg y Al, mientras que para el nitrito suele utilizarse la clásica reacción de diazotación con aminas aromáticas y posterior reacción con ácido naftilamino-7-sulfónico, midiendo la absorbancia a 520 nm. Por su parte, el nitrato se reduce previamente a nitrito y se determina éste por el procedimiento indicado anteriormente. Estas especias, NH3, NO3– y NO2– pueden representar peligro para la salud cuando están presentes en aguas de consumo a niveles superiores a 500, 50 y 0.1 g/mL respectivamente. La formación de especies absorbentes mediante el uso de reactivos adecuados hace necesario considerar toda una serie de factores, entre los que cabe mencionar: pH. Esta variable suele jugar un papel muy importante en las reacciones de formación de complejos. La importancia del pH y su influencia sobre los iones metálicos y los ligandos se discute detalladamente en las obras que tratan el tema correspondiente a equilibrios en disolución. Concentración de los reactivos. Previamente a cualquier nueva determinación espectrofotométrica es necesario establecer los márgenes de concentraciones adecuados de los reactivos, ya que cantidades demasiado grandes o demasiado pequeñas pueden originar desviaciones de la ley de Beer. Tiempo adecuado para la medida. Cuando la especie absorbente es estable y su formación es rápida, no es necesario controlar el tiempo al que realizar las medidas, mientras que si se incumple alguna de las premisas anteriores, esta variable será necesario tomarla en consideración. Temperatura. Determinadas reacciones requieren operar a temperatura elevada para aumentar la cinética de formación de un determinado complejo. En estas ocasiones la temperatura de trabajo deberá especificarse en el procedimiento. Orden de adición de los reactivos. A veces, es importante añadir los reactivos según una determinada secuencia. Por ejemplo, la determinación de cobalto mediante la formación del complejo entre Co(III) y nitrilotriacetato implica la formación previa del correspondiente Claudio González Pérez 37 complejo con Co(II), por lo cual el peróxido de hidrógeno (oxidante) debe añadirse en último lugar. Eliminación de interferencias. Existen muy pocas reacciones que sean totalmente específicas, por lo que en muchas ocasiones es necesario eliminar las interferencias de determinadas especies, lo cual muy frecuentemente se consigue por enmascaramiento. Extracción con disolventes orgánicos. La eliminación de sustancias interferentes puede llevarse a cabo en ocasiones mediante extracción con algún disolvente orgánico inmiscible con el agua, aunque tambien puede recurrirse a esta técnica cuando la especie absorbente es poco soluble en agua. Concentración de sales. Altas concentraciones de electrólitos influyen frecuentemente sobre la absorción de determinados compuestos, debido a la formación de asociaciones iónicas que originan desplazamientos del máximo de absorbancia. Los efectos de las variables mencionadas deben conocerse y, en consecuencia, escogerse un conjunto de condiciones de trabajo de forma que la absorbancia no sea afectada apreciablemente por pequeñas variaciones incontroladas en sus magnitudes. c) Determinación de la relación absorbancia–concentración. Una vez conocida la absorbancia de una determinada especie, en principio, se podría obtener la concentración a partir de la ley de Beer: A = ε b C. Sin embargo, como se comentó, no es prudente asumir el cumplimiento de dicha ley y utilizar un solo patrón para determinar la absortividad molar, ε. Asimismo, tampoco es conveniente basar los resultados de un análisis en un valor de la bibliografía para obtener ε. Por ello, el método normal de trabajo consiste en la preparación de un calibrado a partir de una serie de disoluciones patrón, preparadas en las mismas condiciones que la muestra y obtener los resultados por interpolación. Cuando se trata de analizar materiales complejos, es muy difícil, si no imposible, reproducir en los patrones las condiciones de las muestras. En estas ocasiones suele ser de utilidad el método de adición estándar, con objeto de contrarrestar los efectos de la matriz. Este método, en análisis espectrofotométrico, se suele aplicar de la siguiente manera: a varias alícuotas idénticas de la muestra se adicionan volúmenes variables de una disolución estándar del analito, de concentración conocida. Se añaden entonces los reactivos correspondientes al método empleado y cada disolución se enrasa a un 38 Espectrofotometría de absorción ultravioleta-visible volumen fijo, antes de medir la absorbancia*. Si se cumple la ley de Beer, los datos se representan como en la figura 3.21., a partir de la cual se obtiene el punto de corte de la recta (obtenida, si es necesario, por ajuste por mínimos cuadrados) con el eje horizontal y, a partir de ahí la concentración de la muestra original. 0.6 o A o 0.4 o 0.2o 0 5.0 10.0 V, ml 15.0 Figura 3.21. Método de adición estándar para análisis espectrofotométrico. Análisis de mezclas de sustancias absorbentes Cuando en una disolución hay varias sustancias que absorben radiación, es posible, en muchos casos, llevar a cabo su determinación sin necesidad de separar previamente los distintos componentes. Se va a considerar el caso de que la muestra esté constituida por los componentes M y N. La forma de proceder depende de los espectros de absorción de ambas especies, y pueden presentarse los siguientes casos: Espectros no superpuestos. En estas ocasiones, como se muestra en la figura 3.22, es posible encontrar algunas longitudes de onda a las que absorba solo una de las especies; λ1 para determinar M y λ2 para determinar N. A M N λ1 λ2 Figura 3.22. Análisis de mezclas. Espectros no superpuestos. * Cuando la cantidad de muestra es limitada, las adiciones estándar pueden llevarse a cabo por sucesivas adiciones de incrementos de una disolución patrón de concentración conocida a una única alícuota del problema. Las medidas de la absorbancia se hacen en el original y despues de cada adición. 39 Claudio González Pérez Espectros superpuestos parcialmente. Cuando la situación es como se muestra en la figura 3.23., la especie N no interfiere en la medida de M a la longitud de onda λ1, por lo que la determinación de esta especie puede hacerse directamente a λ1. A continuación se determina la absorbancia de M a la longitud de onda λ2 a partir de la absortividad molar (en una experiencia previa) y finalmente, se resta esta contribución de la absorbancia medida a λ2. Con ello se obtiene la absorbancia del componente N, cuya concentración se calcula posteriormente de la forma acostumbrada. M+N A M N λ2 λ1 Figura 3.23. Análisis de mezclas. Superposición parcial de espectros. Espectros completamente superpuestos. Como se indica en la figura 3.24. no es posible encontrar una longitud de onda en la que M y N absorban de forma exclusiva. M+N A N M λ1 λ2 Figura 3.24. Análisis de mezclas. Superposición completa de espectros. Espectrofotometría de absorción ultravioleta-visible 40 En este caso, el problema se resuelve midiendo las absorbancias A1 y A2 a las longitudes de onda λ1 y λ2 respectivamente, y planteando las ecuaciones siguientes: A1 = εM1 b CM + εN1 b CN (a λ1) A2 = εM2 b CM + εN2 b CN (a λ2) Esto es posible porque la ley de Beer establece que la absorbancia es una propiedad aditiva, de forma que la absorbancia total de una disolución a una longitud de onda dada es igual a la suma de las absorbancias de los componentes individuales presentes. En las ecuaciones anteriores, b representa el camino óptico y εM1, εM2, εN1 y εN2 las absortividades molares de M y N a las longitudes de onda λ1 y λ2, que deben ser determinadas previamente a partir de disoluciones patrón, o mejor, a partir de las pendientes de sus representaciones de la ley de Beer. Esta forma de proceder es válida solo si se cumple la ley de Beer y si los dos componentes se comportan independientemente uno del otro. La mayor precisión se alcanza cuando se eligen longitudes de onda en las que las diferencias entre las absortividades molares sean grandes: a λ1, εM grande y εN pequeña, y a λ2, εM pequeña y εN grande. Sin embargo, no todos los sistemas se comportan de este modo. Puede ocurrir que exista solapamiento a todas las longitudes de onda y que, para cada λ, εM>εN. Un procedimiento alternativo para analizar estas muestras consiste en un método gráfico, cuyo fundamento es el siguiente: la primera de las ecuaciones anteriores puede escribirse así: A1 ε M1 b = CM + ε N1 ε M1 CN En lugar de medir A, εM y εN solamente a dos longitudes de onda, se miden estas magnitudes a varias, para lo cual es necesario disponer de los espectros correspondientes a los componentes puros y a la mezcla desconocida. Para un cierto número de medidas, se representan gráficamente los valores de A/εM b frente a εN/εM, obteniéndose una línea recta. La pendiente de la recta permite obtener CN y la ordenada en el origen, CM. El método puede extenderse con facilidad a sistemas de tres componentes. La principal ventaja del método es que el uso de múltiples puntos a lo largo de todo el espectro, en lugar de utilizar solamente dos, permite alcanzar un grado superior de precisión y exactitud. Claudio González Pérez 41 Valoraciones fotométricas En las valoraciones convencionales, el punto de equivalencia se detecta por medio de la observación visual del cambio de color, ya sea inherente a uno de los reactivos (por ejemplo, el permanganato) o producido por un indicador. Los operadores con vista normal, en condiciones favorables, obtienen fácilmente una precisión de unas cuantas décimas por ciento. Sin embargo, es difícil o imposible obtener buenos resultados en casos donde el cambio de color es gradual, o donde los colores de las dos formas no contrastan claramente. En estos casos puede detectarse el punto final fotométricamente. En las valoraciones fotométricas se miden las cambios de absorbancia de una disolución durante la valoración. Estos cambios de absorbancia indican cambios en la concentración de alguna especie absorbente de radiación. Para llevar a cabo esta técnica, el recipiente de valoración puede colocarse directamente en el camino óptico del instrumento, si bien, esto requiere normalmente alguna modificación del compartimento de la muestra. Por ello, lo más corriente es la utilización de células de flujo. La absorbancia medida, a la longitud de onda óptima, después de cada adición de valorante, se representa frente al volumen de reactivo. Las curvas de valoración consisten, si la reacción es completa, en dos líneas rectas que se cortan en el punto final. Para las que son apreciablemente incompletas se produce una curvatura en las proximidades del punto de equivalencia. En estos casos, el punto final se obtiene por prolongación de los dos tramos rectos. En la figura 3.25. se muestra la forma de algunas curvas de valoración fotométrica. La curva a) es típica de la valoración donde solo absorbe el reactivo valorante, como en la valoración de As(III) con BrO3–—Br– leyendo la absorbancia a la longitud de onda del bromo. La curva b) es característica de sistemas donde absorbe el producto de la reacción; por ejemplo, en la valoración de Cu(II) con AEDT. Por otra parte, cuando el analito se convierte en una especie no absorbente, se obtiene la curva c). Este es el caso, por ejemplo, de la valoración de p-toluidina en butanol con ácido perclórico, midiendo a 290 nm. Cuando un analito coloreado se transforma en un producto incoloro al tratar con un reactivo valorante que también absorbe, se obtienen curvas como la d). Finalmente, las curvas e) y f) pueden representar la adición de ligandos para formar dos complejos sucesivos de diferente absortividad. Espectrofotometría de absorción ultravioleta-visible 42 Figura 3.25. Curvas de valoraciones fotométricas. En la figura 3.26. se muestra la curva de valoración fotométrica de una mezcla de Bi(III) y Cu(II) con AEDT. A Punto final del Cu Punto final del Bi vol. de AEDT Figura 3.26. Valoración de una mezcla de Bi(III) y Cu(II) con AEDT. A la longitud de onda de medida (745 nm), ninguno de los cationes ni el reactivo absorben, así como tampoco el complejo Bi(III)—AEDT. Este complejo es más estable que el de Cu(II)—AEDT y se forma en la primera parte de la valoración. Cuando se ha terminado de formar el complejo Bi(III)—AEDT, comienza la formación del complejo de Cu(II), aumentando la absorbancia hasta que éste se forma completamente. Se obtienen dos puntos finales bien definidos. 43 Claudio González Pérez Las valoraciones fotométricas presentan las siguientes ventajas sobre las convencionales: * Con ellas es posible la determinación de sustancias no absorbentes, puesto que solo es necesario que absorba alguna de estas tres especies: reactivo valorante, sustancia a valorar o producto de la reacción. * La presencia de otras sustancias que absorban a la longitud de onda a la que se mide no origina necesariamente interferencia, ya que solo importa el cambio que se observa en los valores de la absorbancia. * Los datos experimentales que realmente interesan se toman muy alejados del punto de equivalencia. De esta forma, las reacciones empleadas no necesitan tener constantes de equilibrio tan favorables como las requeridas para una valoración convencional, que depende de observaciones cerca del punto de equivalencia. Por la misma razón pueden utilizarse disoluciones más diluidas. Determinación de constantes de disociación de ácidos y bases. La determinación espectrofotométrica de constantes de disociación de ácidos y bases se basa en que el espectro de absorción de moléculas orgánicas con grupos funcionales ácidos o básicos depende del pH del medio. Para un ácido débil, – HA <—> A + H + A Ka = pK a = pH + log – H HA + HA A – Para determinar el valor de pKa es necesario conocer el pH y las concentraciones (en sentido estricto, las actividades) de las formas ácida (HA) y básica (A–). La relación [HA]/[A–] puede obtenerse espectrofotométricamente si se conocen las correspondientes absortividades molares, εHA y εA–, las cuales, a su vez, pueden obtenerse desplazando el equilibrio de forma virtualmente completa hacia la derecha y hacia la izquierda mediante la adición de un exceso de base y de ácido respectivamente. Según la ecuación anterior, cuando [HA] = [A–], pKa = pH y este valor de pH corresponde a la mitad de la curva de valoración fotométrica, esto es, al 50 % de la 44 Espectrofotometría de absorción ultravioleta-visible valoración del ácido HA. Este punto puede obtenerse representando gráficamente la absorbancia a una determinada longitud de onda frente al pH. En la figura 3.27. se han representado los espectros de absorción de un indicador ácido-base a distintos valores de pH. 615 nm A 9 8 7.5 7 6.5 6 400 500 600 λ 700 6 7 8 9 pH Figura 3.27. Espectros a varios pH y variación de la absorbancia con el pH a =625 nm El punto donde se cruzan los espectros se denomina punto isosbéstico. La presencia de un punto isobéstico evidencia que el equilibrio en cuestión implica solo dos especies absorbentes. Cuando se representa la absorbancia a 615 nm frente al pH (curva de la derecha en la figura 2.27.) se obtiene una curva en forma de S. La parte izquierda corresponde a la forma ácida del indicador, mientras que la parte superior derecha corresponde a la conversión casi total en la forma básica. Debido a que el pKa se define como el valor de pH en el cual una mitad del indicador está en la forma ácida y la otra mitad en la forma básica, dicho pKa se determina por el punto de inflexión. Determinación de la estequiometría de complejos El principal interés de la técnica espectrofotométrica para determinar la composición de complejos en disolución reside en el hecho de que pueden efectuarse medidas sin perturbar los equilibrios que se consideran. Con esta finalidad se utilizan fundamentalmente los siguientes métodos: Método de las variaciones continuas. Considérese que pueden formarse varios complejos, Claudio González Pérez 45 M + L <—> ML M + 2 L <—> ML2 ……etc pero que en ciertas condiciones predomina uno. Para determinar la estequiometría del complejo predominante por el método de las variaciones continuas, el procedimiento a seguir consiste en preparar mezclas de diferentes concentraciones de M y L, pero manteniendo constante la concentración final (en la práctica, se preparan disoluciones del catión y del ligando de concentraciones idénticas y se mezclan en distintas relaciones de volumen, pero de modo que el volumen total sea constante). La absorbancia de cada disolución se mide a una longitud de onda apropiada y se representa gráficamente la absorbancia corregida (absorbancia medida menos las absorbancias de M y L libres*) frente a la fracción molar de L. Se obtiene una gráfica donde la absorbancia máxima se corresponde con la estequiometría del complejo predominante. En la figura 3.28. se muestra la variación de la absorbancia para dos complejos de estequiometría ML2.La curvatura de las líneas experimentales es el resultado de no haberse completado la reacción de formación del complejo (de hecho, se ha desarrollado algún método para la determinación de constantes de formación de complejos, basado en la medida de las desviaciones con relación a las líneas rectas teóricas indicadas en la figura). Cuando se forma un complejo muy estable se obtiene una curva como la B, y cuando es poco estable, como la curva C. Por otra parte, la representación dará un mínimo si el complejo absorbe menos que los reactivos. Figura 3.28. Variaciones continuas para dos complejos ML2. * En muchas ocasiones, M ó L (o ambos) no absorben a la longitud de onda de interés, de manera que no es necesaria la corrección. 46 Espectrofotometría de absorción ultravioleta-visible Método de la relación molar. Este método es similar al anterior, excepto que aquí se mantiene constante la concentración de uno de los componentes (a menudo, el ión metálico) mientras que el otro se hace variar. Al representar la absorbancia del complejo frente a la relación en moles de los reactivos, se obtienen líneas rectas de diferente pendiente, produciéndose la intersección en una relación que corresponde a la estequiometría del complejo (figura 3.29.) En el complejo ML representado en la figura 3.29. el catión metálico absorbe a la longitud de onda de medida, puesto que el punto inicial tiene una absorbancia mayor que cero. Por su parte, en el complejo ML2 el ligando absorbe a la longitud de onda a la que se midió, por lo que la pendiente del segundo tramo es mayor que cero. . ML 1.2 A 1.0 0.8 . ML2 0.6 0.4 0.2 0 1 2 3 [L] /[M] Figura 3.29. Método de la relación molar. El método de la relación molar presenta sobre el de las variaciones continuas las siguientes ventajas: en primer lugar distingue mejor entre complejos de números de coordinación altos; por ejemplo, entre ML5 y ML6 (xL=0.83 y xL=0.87). Además, y en otro orden de cosas, los datos experimentales indican claramente el exceso de reactivo necesario para que el complejamiento sea total, lo cual es muy útil en procedimientos analíticos. Por otra parte, y esto puede considerarse una desventaja, el gran exceso de reactivo puede conducir a formar complejos de orden superior. 47 Claudio González Pérez ESPECTROFOTOMETRIA DE DERIVADAS La espectrofotometría de derivadas es una técnica que se conoce desde hace mucho tiempo, pero que se ha desarrollado en época relativamente reciente, sobre todo por la falta de instrumentación a un precio razonable. Actualmente, la utilización de sistemas electrónicos de diferenciación mediante el empleo de un micro ordenador acoplado en serie con el espectrofotómetro permite la representación de derivadas de primero, segundo e incluso órdenes superiores de la absorbancia frente a la longitud de onda: dA para la primera derivada dλ 2 d A 2 para la segunda derivada dλ siendo A la absorbancia y λ la longitud de onda. La primera derivada de un espectro original también puede definirse como la representación gráfica de la pendiente de la curva de absorción a cada longitud de onda. En la figura 3.30. se muestran las características del espectro de la primera y de la segunda derivada para el caso de un pico de absorción simétrico. A A 1ª derivada 2ª derivada Figura 3.30. Pico de absorbancia simétrico y su primera y segunda derivada. En el espectro de la primera o de la segunda derivada, la señal de ordenadas no es proporcional al valor de absorción, sino a la pendiente del espectro normal. Por ello, como la pendiente en el espectro normal puede tener valores positivos y negativos, el Espectrofotometría de absorción ultravioleta-visible 48 espectro de derivadas presenta valores positivos y negativos en la escala de ordenadas, o bien, máximos y mínimos, según el carácter de la pendiente. Para una sustancia absorbente, la posición de la longitud de onda y la relación entre los valores de los extremos (máximos y mínimos) representa una magnitud característica. La utilización de la espectrofotometría de derivadas presenta las siguientes ventajas: * Medida exacta de λmax. Mientras que en el espectro normal, especialmente en el caso de bandas de absorción anchas, la posición correspondiente a la longitud de onda del máximo solo puede ser fijada de manera aproximada, mientras que en la primera derivada del espectro, la posición del máximo viene definida exactamente por el paso de la curva por el valor cero. * Mejor resolución de los espectros. En el espectro normal, la estructura fina puede ser difícil de ver. En estos casos, es más conveniente emplear la segunda derivada, ya que los máximos y mínimos del espectro normal y de la segunda derivada aparecen prácticamente a las mismas longitudes de onda y la estructura fina del espectro normal se pone de manifiesto mejor que en la primera derivada. En la práctica analítica normal esto tiene gran importancia, ya sea con fines de identificación, como criterio de pureza o para control de calidad. * Determinaciones en presencia de turbidez. Las disoluciones turbias presentan en general un aumento continuo de la absorbancia hacia longitudes de onda más cortas y no ocasionan, por consiguiente, ninguna variación espectral importante en la primera y segunda derivada, al menos para un nivel de turbidez no demasiado elevado. * Determinaciones cuantitativas en presencia de dos o más componentes. La determinación cuantitativa de componentes con bandas de absorción relativamente estrechas y que estén solapadas con una banda ancha de un segundo componente es uno de los campos de aplicación más extensos de esta técnica. Cuando el componente que se desea determinar solo es visible en el espectro total por un pequeño pico o por un hombro, la utilización de métodos convencionales puede implicar grandes errores. Sin embargo, la primera o segunda derivadas de un espectro de dos componentes permite, bajo ciertas condiciones, ver claramente los diferentes compuestos y hacer determinaciones cuantitativas con errores sistemáticos muy pequeños o incluso despreciables (figura 3.31.) 49 Claudio González Pérez Espectro normal 1ª derivada a) b) Figura 3.31. Espectro normal y primera derivada. a) un componente. b) dos componentes. En la figura 3.31. se han representado los espectros normales y la primera derivada de un componente caracterizado por una pequeña banda a) y el mismo componente "oscurecido" por la presencia de otra especie con una banda de absorción más ancha. EJERCICIOS 1. Dadas las siguientes sustancias: pentano, pentanol, clorohexano, butadieno, benceno, fenol, amoniaco, butilamina, ácido acético, benzopireno, ozono, ¿Absorberán radiación visible?. ¿Cuáles absorberán radiación ultravioleta?. Justifíquelo. 2. Se determinó la absortividad molar de un compuesto químico midiendo la absorbancia de una disolución de concentración conocida utilizando una cubeta de 1 cm. Se obtuvo el valor de 2.2x104 L mol–1 cm–1. ¿Qué valor se obtendría si se utilizase una cubeta de 10 cm?. ¿Qué espesor (camino óptico) debería tener la cubeta para que la misma disolución presente una transmitancia del 8.42 %? 3. En disoluciones acuosas neutras, el fenol tiene una absortividad molar de 1.8x104.mol–1.cm–1. ¿Qué intervalo de concentraciones de fenol pueden determinarse si los valores de las absorbancias en cubetas de 10 cm deben limitarse a valores de 0.2 a 0.9 unidades de absorbancia?. 4. Se trata de determinar la concentración de Fe(III) en un agua mineral. Para ello, se pipetean alícuotas de 10 mL de la muestra en matraces aforados de 50 mL. Se adicionan a cada uno exactamente 0, 5, 10, 15 y 20 mL de una disolución patrón que 50 Espectrofotometría de absorción ultravioleta-visible contiene 11.1 p.p.m. de Fe(III), seguido de un exceso de ion tiocianato, para formar complejo rojo Fe(SCN)2+. Después de enrasar a 50 mL, las absorbancias de las cinco disoluciones medidas a 580 nm (longitud de onda de máxima absorción) fueron 0.240, 0.437, 0.621, 0.809 y 1.009 respectivamente (cubetas de 1 cm). Obtener la concentración de Fe(III) en la muestra de agua. 5. Se analizó espectrofotométricamente una mezcla de dicromato y permanganato, en medio ácido sulfúrico 1 M y utilizando cubetas de 1 cm. Para ello, se midieron las absorbancias a 440 y a 545 nm, obteniendo los valores de 0.385 y 0.653 respectivamente. Previamente, se encontró que la absorbancia de una disolución de dicromato 8.33x10–4 M era de 0.308 a 440 nm y de 0.009 a 545 nm. Análogamente, una disolución de permanganato 3.77x10–4 M presentó una absorbancia de 0.035 a 440 nm y de 0.886 a 545 nm. Calcular las concentraciones de permanganato y de dicromato en la mezcla analizada. 6. Se determinó la estequiometría de un complejo MLn por el método de las variaciones continuas y el de la relación molar, partiendo de disoluciones de M y de L 0.0100 molar. Para aplicar el primer método se mezclaron los volúmenes de cada una de ellas que se indican en la tabla, obteniendo los correspondientes valores de absorbancia. Para el método de la relación molar, se añadieron los volúmenes de L indicados en la tabla a 5.0 mL de la disolución de M, enrasando al mismo volumen en todos los casos. Variaciones continuas Relación molar mL de M mL de L A mL de L A 10.0 9.00 8.00 7.00 6.00 5.00 4.00 3.00 2.00 1.00 0.00 0.00 1.00 2.00 3.00 4.00 5.00 6.00 7.00 8.00 9.00 10.0 0.000 0.130 0.261 0.399 0.530 0.660 0.799 0.750 0.500 0.252 0.000 0.00 1.00 2.00 3.00 4.00 5.00 6.00 7.00 8.00 12.0 14.0 0.000 0.080 0.181 0.269 0.360 0.447 0,540 0.628 0.720 0.900 0.900 Obtener la estequiometría del complejo MLn.