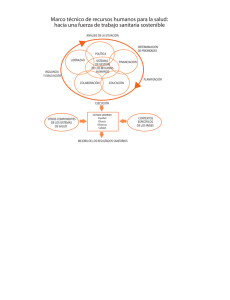
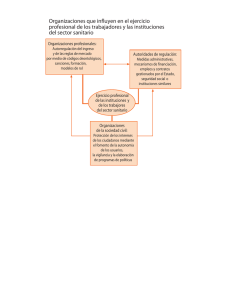
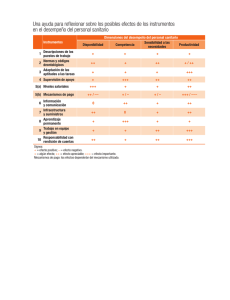
Reconocimiento de equivalencias para el registro sanitario
Anuncio

NUESTRA MISIÓN: PROTEGER TU SALUD DIRECTORIO Secretaría de Salud Dr. José Ángel Córdoba Villalobos Secretario de Salud Comisión Federal para la Protección contra Riesgos Sanitarios Lic. Miguel Ángel Toscano Velasco Comisionado Federal Dr. Roberto Mendoza Zepeda Comisionado de Autorización Sanitaria Dr. Lucio Galileo Lastra Marín Comisionado de Operación Sanitaria Dr. Juan Carlos Gallaga Comisionado de Control Analítico y Ampliación de Cobertura M en C. Rocío del Carmen Alatorre Eden-Wynter Comisionada de Evidencia y Manejo de Riesgos Mtro. Francisco Acosta Minquini Comisionado de Fomento Sanitario Lic. Manuel Ovalle Araiza Coordinador General del Sistema Federal Sanitario Lic. Erwin Roeninger Servín Secretario General 2 OBJETIVO La presente guía tiene la finalidad de proporcionar orientación para el proceso del trámite de solicitud de registro sanitario de dispositivos médicos a través del reconocimiento de equivalencias. En esta guía, encontrarán los requisitos, lineamientos y recomendaciones que se deben observar al presentar la información solicitada para evaluación en cada uno de los trámites correspondientes a los dispositivos médicos que cuentan con autorización para comercializarse en los Estados Unidos de América y Canadá, a través de las agencias regulatorias de ambos países. También se incluyen tablas y ejemplos para poder presentar las solicitudes y la documentación anexa requerida de manera correcta y ordenada, con el fin de agilizar el proceso de los trámites. 3 CONSIDERACIONES IMPORTANTES PREVIAS A LA REALIZACIÓN DEL TRÁMITE 1. ¡Clasifica tu dispositivo médico! Antes de iniciar el trámite, es importante asegurarse de contar con la clasificación con base al riesgo adecuado para el dispositivo médico que se quiere registrar, ya que: • El criterio para realizar el pago de derechos de los trámites se tomará a partir de la clasificación con base en el riesgo al que pertenece el dispositivo médico según la normatividad mexicana. ¿Cómo clasifico mis dispositivos médicos? Los dispositivos médicos se clasifican con base al riesgo en 3 clases diferentes establecidos y especificados en el artículo 83 del Reglamento de Insumos para la Salud. También se recomienda consultar en el portal de internet de la COFEPRIS en la ruta http://www.cofepris.gob.mx/wb/cfp/equivalencias_en_dispositivos_medicos el catálogo “Clasificación según riesgo sanitario y categoría” y la guía “Criterios para la clasificación con base al nivel de riesgo sanitario”, éste último también disponible en la Farmacopea de los Estados Unidos Mexicanos en el suplemento para Dispositivos Médicos. ¡Importante! Debes conocer también la clasificación del dispositivo a registrar otorgada en el país donde cuenta con autorización (EUA y Canadá), debido a que: • La clasificación del dispositivo médico ante la autoridad sanitaria extranjera determina la modalidad del trámite por el que deberá someterse el expediente ante la COFEPRIS, y por tanto establece los requisitos que serán solicitados para su registro. *La clasificación del dispositivo en México es independiente a la asignada por la autoridad sanitaria extranjera, por lo tanto, será la que se aplique al dispositivo médico en las demás consideraciones estipuladas en el marco jurídico mexicano. 2. Revisa si tu dispositivo médico puede solicitar su registro a través de éste proceso Es importante recordar que, únicamente pueden ser materia del registro sanitario por reconocimiento de equivalencias; los dispositivos médicos que cuenten con una autorización otorgada por FDA o Health Canada (EUA y Canadá), y que cuenten con permiso para su comercialización en su territorio (con excepción de los dispositivos Clase I de Health Canada). 4 3. Identifica la homoclave del trámite que quieres realizar Debes identificar cual es la homoclave correspondiente al tipo de trámite por reconocimiento de equivalencias que se desea realizar. Las homoclaves se pueden consultar en el Anexo 1 de la guía. 4. Obtén tu formato de solicitud para el trámite Puedes obtener el formato de solicitudes directamente en las ventanillas de atención de la COFEPRIS en Monterrey #33 Col. Roma, México DF., o bien vía electrónica a través de: http://www.cofepris.gob.mx/wb/cfp/solicitudes2 5. Realiza el pago de derechos correspondiente al trámite que realizarás Después de clasificar el dispositivo debes hacer el pago de derechos correspondiente para el trámite de registro sanitario. Para ello, debes requisitar el Formato SAT-5 “Declaración general de pago de derechos”, con base a la guía de llenado del formato y cubrir el monto calculado de acuerdo a la clasificación del dispositivo. El formato para el pago de derechos y la guía para el llenado del formato se encuentran disponibles vía electrónica en www.cofepris.gob.mx. 5 REQUISITOS PARA EL REGISTRO SANITARIO DE DISPOSITIVOS MÉDICOS POR RECONOCIMIENTO DE EQUIVALENCIAS A) REQUISITOS GENERALES TODAS las solicitudes de registro sanitario de dispositivos médicos por el reconocimiento de equivalencias deberán estar acompañadas por la siguiente información y documentación: I. Formato de solicitud Debe presentarse el formato de solicitudes debidamente llenado de acuerdo con la guía de llenado del formato. Esta guía puede obtenerse junto con el formato en las ventanillas de COFEPRIS o por vía electrónica a través del portal de la Comisión en la siguiente ruta: http://www.cofepris.gob.mx/wb/cfp/equivalencias_en_dispositivos_medicos * También se puede consultar el ejemplo de llenado del formato de la solicitud que se incluye al final de esta guía en el Anexo 2. II. Comprobante del pago de derechos Al realizar tu pago en el banco se te entregarán dos comprobantes de pago de derechos. Es necesario presentar ambos comprobantes y una copia simple legible junto a la solicitud y el expediente del trámite en la ventanilla de COFEPRIS. * Una vez realizada la revisión, COFEPRIS te devolverá uno de los originales sellado y se quedará con la copia y el otro comprobante original. III. Aviso de funcionamiento y de responsable sanitario: Presentar una copia simple legible del aviso de funcionamiento del establecimiento. Debe asegurarse que: • Sea expedido para el mismo establecimiento que fue especificado en el formato de solicitud. • Contenga la clave S.C.I.A.N. adecuada para el giro sanitario correspondiente. • Incluya datos y giro sanitario correspondiente para otros distribuidores, de ser el caso para aquellos establecimientos involucrados en la comercialización del dispositivo en territorio nacional. • Presente los datos del responsable sanitario del establecimiento. * En caso de que se cuente con un aviso de responsable sanitario por separado deberá anexarse una copia simple junto con el aviso de funcionamiento del establecimiento. 6 IV. Proyecto de etiqueta Debe presentarse la propuesta de etiqueta que ostentará el producto para su comercialización en el territorio nacional, con las siguientes consideraciones: • Presentado en idioma español; • Conforme a la Norma Oficial Mexicana NOM-137-SSA1-2008, “Etiquetado de Dispositivos Médicos” y demás normas aplicables.. • La información de los fabricantes, distribuidores y/o maquiladores (nacionales y extranjeros) debe coincidir con los documentos legales presentados. * Se entenderá por etiqueta a todo rótulo, inscripción, o imagen gráfica que se presente en el envase que contiene el dispositivo médico. V. Instructivo de uso/manual de operación (cuando aplique) Es necesario presentar el Instructivo de Uso para aquellos dispositivos médicos que requieren de instrucciones detalladas para su correcto funcionamiento. Debe contener la información señalada en la tabla correspondiente. * Manual de operación En el caso de equipos o instrumentos que cuenten con un manual de operación se debe presentar un ejemplar que contenga la información requerida en la siguiente tabla. INFORMACIÓN NECESARIA EN LOS INSTRUCTIVOS O MANUALES INSTRUCTIVO DE USO MANUAL DE OPERACIÓN 1 Descripción del producto. 1 Descripción del producto. 2 Listado de los componentes o partes del dispositivo. 2 Finalidad de uso. según sea el caso. 3 Componentes o partes del dispositivo médico. 3 Finalidad de uso. 4 Ensamble y desensamble, según sea el caso. 4 Condiciones de conservación y almacenamiento. 5 Operación, limpieza y mantenimiento. 5 Precauciones. 6 Calibración, cuando aplique. 6 Preparación cuando aplique. 7 Precauciones. 7 Advertencias y leyendas alusivas correspondientes. 8 Advertencias. 8 Contraindicaciones, cuando aplique. 9 Eventos Adversos, cuando aplique. 10 Para medios de contraste: Indicar vía de administración, forma farmacéutica y contenido del ingrediente activo por unidad de dosis. 7 VI. Certificado de análisis Presentar uno reciente con resultados de pruebas que confirmen las especificaciones de seguridad y eficacia del dispositivo médico emitido por el fabricante, en hoja membretada con su razón social y firmado por el responsable de calidad de la empresa extranjera. VII. Carta de representación del fabricante Si el dispositivo médico no es fabricado por una filial, subsidiaria o casa matriz del solicitante del registro, debe presentarse la carta de representación o distribución emitida por el fabricante: • Debidamente autenticada (apostillada/consularizada). • En original o copia certificada. VIII. Monografía Para que se pueda contar con la información relevante del dispositivo médico de manera completa y ordenada es necesario presentar un documento a manera de monografía, emitida por el fabricante y firmada por el responsable de calidad del dispositivo médico, conteniendo la siguiente información: 1. Nombre comercial. Nombre o Denominación Distintiva que como marca comercial le asigna el laboratorio o fabricante a sus dispositivos médicos con el fin de distinguirlos de otros similares. 2. Nombre Genérico. Nombre que describe a un dispositivo médico o grupo de dispositivos médicos que tienen características comunes, aceptado por la autoridad sanitaria. 3. Descripción del dispositivo médico y finalidad de uso. Información detallada del dispositivo y su mecanismo de acción, así como la utilidad para el que fue diseñado. 4. Esquema de la estructura del dispositivo. • En caso de productos formulados, declarar la formula cuali-cuantitativa incluyendo todos los ingredientes y su función. • Para el caso de aquellos dispositivos que no puedan definirse por medio de una formula (Equipo Médico, Prótesis, etc.) se debe presentar una descripción o diagrama de los componentes, partes y estructura. 5. Especificaciones del producto terminado. Las características físicas y funcionales que apliquen al dispositivo medico en cuestión. 6. Resumen o Diagrama de flujo del proceso de manufactura. 8 7. *Método de esterilización (Cuando aplique). En el caso de productos estériles, se debe indicar el tipo de esterilización utilizada y describir brevemente el proceso. 8. *Resumen de pruebas de atoxicidad y biocompatibilidad (Cuando aplique). 9. *Caducidad y Resumen de estudio de estabilidad (Cuando aplique). Para aquellos dispositivos médicos que por sus características y finalidad de uso ostentan una fecha de caducidad. • Debe incluir resumen y conclusiones de los estudios de estabilidad que avalen la caducidad propuesta. • Debe estar firmado por el responsable de calidad el fabricante o por el responsable sanitario. 10. Características del envase primario y secundario. Debe presentarse la descripción breve del contenedor que está en contacto directo con el dispositivo médico (envase primario). Si existen otros elementos de empaque que no estén en contacto con el dispositivo (envase secundario) deberá presentarse también su descripción. 11. Presentaciones, códigos o modelos. Los códigos que identifiquen todas las presentaciones y modelos correspondientes al dispositivo médico que se va a registrar deben incluirse en la monografía. 12. *En su caso, resumen de los estudios pre-clínicos y clínicos. 13. Referencias bibliográficas. Autor, título, revista, número, volumen, año. Y de contar con hipervínculo indicarlo. 9 B) REQUISITOS PARA DISPOSITIVOS MÉDICOS COMERCIALIZADOS EN E.U.A. I. Certificado para Gobierno Extranjero (Certificate to Foreign Government) Se debe presentar el certificado emitido por la FDA para los fabricantes de dispositivos médicos en original o una copia certificada, siendo: • Vigente al momento de presentar la solicitud. • Que avale al mismo fabricante expuesto en el formato de solicitud de registro. • Que contenga todos los códigos o modelos de los dispositivos médicos a registrar. II. Reporte de inspección del establecimiento (Establishment inspection report) Presentar el reporte de resultados de la última inspección que se haya realizado al establecimiento del fabricante del dispositivo médico por la FDA, el cual deberá mostrar: • Que el establecimiento del fabricante cumple con las buenas prácticas de manufactura. • Todas las observaciones realizadas por FDA. III. Aprobación de la FDA En el expediente presentado para la solicitud de registro sanitario se deberá incluir original o copia de la autorización de FDA para el dispositivo médico en E.U.A. Ésta debe mostrar la siguiente información: • Clasificación con base al riesgo del dispositivo. • Deberá corresponder al producto para el cual se solicita el registro sanitario en México, en cuanto a denominación y modelos, (de ser el caso). • En el caso de los dispositivos médicos Clase II debe presentarse el permiso con base a la notificación previa a la comercialización (510(k) Premarket Notification). • En el caso de los dispositivos médicos Clase III debe presentarse la aprobación previa a la comercialización (Premarket Approval). IV. Reporte de Tecno-vigilancia Presentar el resumen o comprobante del último reporte de la vigilancia posterior a la comercialización dentro de los E.U.A., que realizó la FDA al dispositivo médico que se quiere registrar en México. 10 C) REQUISITOS PARA DISPOSITIVOS MÉDICOS COMERCIALIZADOS EN CANADÁ. I. Licencia de Dispositivo Médico Presentar original o copia certificada de la licencia expedida por Health Canada (“Medical Device Licence”) en la que se aprueba el dispositivo médico a registrar. La licencia deberá contener: • Número de licencia. • Fecha de expedición. • Nombre y clasificación con base al riesgo del dispositivo médico. • Todos los códigos o modelos del dispositivo médico a registrar en México. • Nombre y dirección del fabricante. • Tipo de licencia. II. Certificado CAN/CSA-ISO 13485:03 Se debe presentar una copia simple del certificado de cumplimiento con la norma CAN/CSA-ISO 13485:03, el cual deberá: • Estar vigente al momento de presentar la solicitud. • Certificar al mismo establecimiento del fabricante que aparece en la Licencia de Dispositivo Médico y en el formato de solicitud. • Ser emitido por un tercero autorizado reconocido por Health Canada. III. Certificado ISO 17021 Se debe presentar una copia simple del certificado de cumplimiento con la norma ISO 17021 por parte del tercero autorizado que emitió el Certificado CAN/CSA-ISO 13485:03 mencionado anteriormente. Debe asegurarse que: • Esté vigente al momento de someter la solicitud. • Contenga los datos del tercero autorizado reconocido por Health Canada. IV. Autorización para tercero autorizado Presentar una copia certificada de la autorización del tercero autorizado reconocido por Health Canada, para poder certificar bajo el estándar ISO 13485:03. 11 D) PRESENTACIÓN DE DOCUMENTOS a. Debes presentar todos los documentos que acompañen a las solicitudes en español, o en caso contrario, deberán adjuntarse a los mismos su respectiva traducción al español, avalada con la firma del responsable sanitario. b. Los documentos expedidos por autoridades de otros países deberán estar autenticados y traducidos por perito traductor. *Para garantizar la autenticidad de un documento legal se debe presentar apostillado o debidamente certificado por el procedimiento legal que se aplique en el país de origen. 12 E) REQUISITOS NECESARIOS PARA CADA MODALIDAD DE SOLICITUD DE REGISTRO SANITARIO DE DISPOSITIVOS MÉDICOS REQUISITOS PARA SOLICITUD DE REGISTRO SANITARIO DE DISPOSITIVOS MÉDICOS CLASE I en FDA COFEPRIS-04-001-D DOCUMENTOS REQUERIDOS Para la solicitud de registro sanitario para dispositivos médicos con Clase I de FDA, es necesario presentar los siguientes requisitos: 1 Requisitos Generales Presentar todos los requisitos generales de acuerdo a los lineamientos anteriormente mencionados en la guía. 1. Formato de solicitud 2. Comprobante de pago de derechos 3. Aviso de funcionamiento de establecimiento y responsable sanitario 4. Proyecto de etiqueta 5. Instructivo / Manual 6. Certificado de Análisis 7. Carta de representación del fabricante 8. Monografía 2 Certificado para Gobierno extranjero 3 Último reporte de inspección del establecimiento 4 Documento de aprobación de la FDA 13 SOLICITUD DE REGISTRO SANITARIO DE DISPOSITIVOS MÉDICOS CLASE II y III en FDA COFEPRIS-04-001-E DOCUMENTOS REQUERIDOS Para la solicitud de registro sanitario para dispositivos médicos con Clase II y III de FDA, es necesario presentar los siguientes requisitos: 1 Requisitos Generales Presentar todos los requisitos generales de acuerdo a los lineamientos anteriormente mencionados en la guía. 1. Formato de solicitud 2. Comprobante de pago de derechos 3. Aviso de funcionamiento de establecimiento y responsable sanitario 4. Proyecto de etiqueta 5. Instructivo / Manual 6. Certificado de Análisis 7. Carta de representación del fabricante 8. Monografía 2 Certificado para Gobierno extranjero 3 Último reporte de inspección del establecimiento 4 Último reporte de tecnovigilancia 5 Documento de aprobación de la FDA 14 SOLICITUD DE REGISTRO SANITARIO DE DISPOSITIVOS MÉDICOS CLASE II, III y IV en HEALTH CANADA COFEPRIS-04-001-F DOCUMENTOS REQUERIDOS Para la solicitud de registro sanitario para dispositivos médicos con Clase II, III y IV de Health Canada es necesario presentar los siguientes requisitos: 1 Generales Presentar todos los requisitos generales de acuerdo a los lineamientos anteriormente mencionados en la guía. 1. Formato de solicitud 2. Comprobante de pago de derechos 3. Aviso de funcionamiento de establecimiento y responsable sanitario 4. Proyecto de etiqueta 5. Instructivo / Manual 6. Certificado de Análisis 7. Carta de representación del fabricante 8. Monografía 2 Licencia de Dispositivo Médico (Health Canada) 3 Certificado CAN/CSA-ISO 13485:03 4 Certificado ISO 17021 5 Autorización para tercero autorizado 15 SUGERENCIAS PARA LA PRESENTACIÓN DEL EXPEDIENTE 1. Llena los formatos de solicitud a máquina y de acuerdo a su guía de llenado. 2. Es conveniente llenar una lista con los documentos que presentas dentro del expediente para la solicitud de registro sanitario del dispositivo médico, de acuerdo a la modalidad del trámite a efectuar. *Podrás encontrar una lista a manera de ejemplo en el Anexo 3 de ésta guía. Se sugiere presentar dicha lista junto al expediente y disponer los documentos en el orden que ahí aparece. 3. Para agilizar la captura de los datos se sugiere presentar las presentaciones, códigos o modelos del dispositivo médico en formato electrónico contenido en un CD. 4. Anexa únicamente la documentación solicitada. 5. ¡Revisa la solicitud y todos los documentos anexos antes de realizar el trámite! Todos los expedientes presentados para los trámites de registro sanitario de dispositivos médicos deberán presentarse completos, en orden y con la información correcta, ya que de no ser así se considerará desechado el trámite. 16 REFERENCIAS 1. Ley General de Salud. 2. Ley Federal de Procedimiento Administrativo. 3. Reglamento de Insumos para la Salud (vigente al 17 de agosto del 2010). 4. Norma Oficial Mexicana NOM-137-SSA1-2008, Etiquetado de dispositivos médicos. 5. Farmacopea de los Estados Unidos Mexicanos, Suplemento para dispositivos médicos. 6. Acuerdo por el que se dan a conocer los trámites y servicios, así como los formatos que aplica la Secretaría de Salud, a través de la Comisión Federal para la Protección contra Riesgos Sanitarios, inscritos en el Registro Federal de Trámites y Servicios de la Comisión Federal de Mejora Regulatoria. Publicado en el Diario Oficial de la Federación del 19 de junio de 2009. 7. Acuerdo por el que se reconocen como equivalentes a los requisitos establecidos en los artículos 179 y 180 del Reglamento de Insumos para la Salud y a los procedimientos de evaluación técnica realizados por la Comisión Federal para la Protección contra Riesgos Sanitarios para el otorgamiento del registro de los insumos para la salud, a que se refiere el Capítulo IX del título Segundo del Reglamento de Insumos para la Salud, a los requisitos establecidos por la secciones 510(k) y 514 del Federal Food, Drug and Cosmetic Act y por el Título 21, Capítulo I, Subcapítulo H, del Code of Federal Regulations de los Estados Unidos de América, así como los establecidos por el Food and Drug Act, y las Medical Devices Regulations de Canadá para permitir la comercialización de dispositivos médicos en su territorio, y a las pruebas e inspecciones realizadas por la Food and Drug Administration de los Estados Unidos de América y por Health Canada de Canadá, para permitir la comercialización de dispositivos médicos en su territorio”, publicado en el Diario Oficial de la Federación el 26 de octubre de 2010. 17 ANEXOS Anexo 1 LISTADO DE HOMOCLAVES CORRESPONDIENTES A LOS TRÁMITES PARA REGISTROS SANITARIOS POR RECONOCIMIENTO DE EQUIVALENCIAS Anexo 2 FORMATO DE SOLICITUD DE REGISTRO SANITARIO (EJEMPLO DE LLENADO) Anexo 3 LISTA DE DOCUMENTOS NECESARIOS EN EL EXPEDIENTE 18 ANEXO 1 LISTADO DE HOMOCLAVES CORRESPONDIENTES A LOS TRÁMITES PARA REGISTROS SANITARIOS POR RECONOCIMIENTO DE EQUIVALENCIAS A) PARA REGISTRO SANITARIO DE DISPOSITIVOS MÉDICOS HOMOCLAVE COFEPRIS -04-001-D NOMBRE Solicitud Registro Sanitario de dispositivo médico MODALIDAD “D” Productos con registro Clase I en FDA. (Acuerdo de Equivalencia) HOMOCLAVE COFEPRIS -04-001-E NOMBRE Solicitud Registro Sanitario de dispositivo médico MODALIDAD “E” Productos con registro Clase II y Clase III en FDA. (Acuerdo de Equivalencia) HOMOCLAVE COFEPRIS -04-001-F NOMBRE Solicitud Registro Sanitario de dispositivo médico MODALIDAD “F” Productos con registro Clase II, Clase III y Clase IV en HC. (Acuerdo de Equivalencias) 19 ANEXO 2 FORMATO DE SOLICITUD DE REGISTRO SANITARIO (EJEMPLO DE LLENADO) COMISIÓN FEDERAL PARA LA PROTECCIÓN CONTRA RIESGOS SANITARIOS FORMATO SOLICITUDES No. DE INGRESO (USO EXCLUSIVO DE LA COFEPRIS) NO. RUPA ANTES DE LLENAR ESTE FORMATO LEA CUIDADOSAMENTE EL INSTRUCTIVO, LA GUÍA Y EL LISTADO DE DOCUMENTOS ANEXOS. LLENAR CON LETRA DE MOLDE LEGIBLE O A MÁQUINA 1.- SOLICITUD BAJA TEMPORAL DE: LICENCIA PERMISO DE IMPORTACIÓN/EXPORTACIÓN CERTIFICADO ALTA O NUEVO PRIMERA VEZ TEMPORAL PERMISO MODIFICACIÓN SUBSECUENTE DEFINITIVA MODIFICACIÓN DEPOSITO FISCAL REGISTRO NUEVO PRÓRROGA MODIFICACIÓN REVOCACIÓN PRÓRROGA OTRAS VISITAS AUTORIZACIÓN DE COMERCIALIZACIÓN E IMPORTACIÓN PARA SU COMERCIALIZACIÓN DE ORGANISMOS GENÉTICAMENTE MODIFICADOS DE PROTOCOLO MODIFICACIÓN O ENMIENDA NUEVO VISITA DE VERIFICACIÓN SANITARIA PAR PARA CERTIFICACIÓN DE BUENAS PRÁCTICAS DE FABRICACIÓN DE FÁRMACOS, MEDICAMENTOS Y OTROS INSUMOS PARA LA SALUD EN ESTABLECIMIENTOS UBICADOS EN MÉXICO Y EN EL EXTRANJERO PARA EL OTORGAMIENTO O PRORROGA DEL REGISTRO SANITARIO POR LA COMISIÓN FEDERAL PARA LA PROTECCIÓN CONTRA RIESGOS SANITARIOS OTROS HOMOCLAVE DEL TRÁMITE: NOMBRE DEL TRÁMITE: COFEPRIS-004-001-D, E ó F DEBERÁ ESCRIBIR: "SOLICITUD DE REGISTRO SANITARIO DE DISPOSITIVOS MÉDICOS" MODALIDAD DEL TRÁMITE: DEBERÁ COMPLETAR CON LA MODALIDAD DEL TRÁMITE CORRESPONDIENTE A LA HOMOCLAVE SELECCIONADA. 2.- MODIFICACIÓN DE: (solo en caso de haber seleccionado este campo en la sección 1) NÚMERO DE DOCUMENTO A MODIFICAR: DICE / CONDICIÓN AUTORIZADA DEBE DECIR / CONDICIÓN SOLICITADA SI EL ESPACIO ES INSUFICIENTE ANEXAR HOJA CON MODIFICACIONES. 3.-DATOS DEL ESTABLECIMIENTO / PROPIETARIO CLAVE (SCIAN) DESCRIPCIÓN DE SCIAN XXXXXX LA QUE INDIQUE EL AVISO DE FUNCIONAMIENTO NOMBRE DEL PROPIETARIO (PERSONA FÍSICA) O RAZÓN SOCIAL (PERSONA MORAL) R.F.C. DEBERÁ INDICAR EL NOMBRE DEL PROPIETARIO O RAZÓN SOCIAL C.U.R.P. R.F.C.DE LA EMPRESA DOMICILIO FISCAL CALLE Y NÚMERO COLONIA DELEGACIÓN O MUNICIPIO CALLE DE LA EMPRESA COLONIA DE LA EMPRESA LOCALIDAD CÓDIGO POSTAL SU LOCALIDAD (DATO OPCIONAL) DELEGACIÓN O MUNICIPIO ENTIDAD FEDERATIVA CÓDIGO POSTAL ENTIDAD FEDERATIVA ENTRE CALLE Y CALLE ENTRE QUE CALLES ENTRE QUE CALLES RAZÓN SOCIAL O DENOMINACIÓN DEL ESTABLECIMIENTO R.F.C. R.F.C.DE LA EMPRESA DEBERÁ INDICAR EL NOMBRE DEL PROPIETARIO O RAZÓN SOCIAL DOMICILIO DEL ESTABLECIMIENTO CALLE Y NÚMERO COLONIA CALLE DE LA EMPRESA COLONIA DE LA EMPRESA LOCALIDAD CÓDIGO POSTAL SU LOCALIDAD DELEGACIÓN O MUNICIPIO DELEGACIÓN O MUNICIPIO ENTIDAD FEDERATIVA CÓDIGO POSTAL ENTRE CALLE ENTIDAD FEDERATIVA Y CALLE ENTRE QUE CALLES ENTRE QUE CALLES No. DE LICENCIA SANITARIA O INDICAR SI PRESENTÓ AVISO DE FUNCIONAMIENTO. (a) RFC DEL RESPONSABLE SANITARIO O DE OPERACIÓN Y FUNCIONAMIENTO INDICAR QUE SE PRESENTÓ AVISO HORARIO DE D ACTIVIDADES: D INDICAR RFC DEL RESPONSABLE L M M J V S DE A TEL.(S) L M M J V S DE A FAX ( a ) SOLO PARA ALTA DE LICENCIA SANITARIA. TELÉFONO FÁX FECHA DE INICIO DE OPERACIONES( b ) INDICAR FECHA DÍA MES AÑO 20 INDIQUE NOMBRE COMPLETO , C.U.R.P. Y CORREO ELECTRÓNICO REPRESENTANTE LEGAL NOMBRE C.U.R.P. CORREO ELECTRÓNICO (DATO OPCIONAL) NOMBRE DEL REPRESENTANTE LEGAL PERSONA AUTORIZADA DIREECIÓN DE CORREO ELECTRÓNICO NOMBRE C.U.R.P. CORREO ELECTRÓNICO (DATO OPCIONAL) NOMBRE DE LAS PERSONAS AUTORIZADAS DIREECIÓN DE CORREO ELECTRÓNICO 4.- DATOS DEL PRODUCTO. Consultar instructivo de llenado. PRODUCTO PRODUCTO 1) NOMBRE DE LA CLASIFICACIÓN DEL PRODUCTO O SERVICIO DEBERÁ ESCRIBIR: "DISPOSITIVO MÉDICO" 2) ESPECIFICAR SUBCLASIFICACIÓN Y CLASE CON BASE EN RIESGO CONFORME A LA NORMATIVIDAD MEXICANA DENOMINACIÓN ESPECÍFICA 3) DENOMINACIÓN ESPECÍFICA DEL PRODUCTO 4) NOMBRE (MARCA COMERCIAL) O DENOMINACIÓN DISTINTIVA MARCA 5) DENOMINACIÓN COMÚN INTERNACIONAL (DCI) O DENOMINACIÓN GENÉRICA O NOMBRE CIENTÍFICO O IDENTIFICADOR ÚNICO DE LA OCDE DENOMINACIÓN GENÉRICA RECONOCIDA INTERNACIONALMENTE SÓLIDO, LÍQUIDO O GAS. 6) FORMA FARMACÉUTICA O FORMA FÍSICA 7) TIPO DE PRODUCTO MATERIA PRIMA, ADITIVO, PRODUCTO TERMINADO, PRODUCTO A GRANEL, OTROS. 8) FRACCIÓN ARANCELARIA 9) CANTIDAD DE LOTES 10) UNIDAD DE MEDIDA 11) CANTIDAD O VOLUMEN TOTAL 12) NÚMERO DE PIEZAS A FABRICAR 13) Kg. o g POR LOTE 14) No. DE PERMISO SANITARIO DE IMPORTACIÓN O EXPORTACIÓN O CLAVE ALFANUMÉRICA 15) No. REGISTRO SANITARIO 16) No. DE ACTA PRESENTACIÓN POR UNIDAD 17) PRESENTACIÓN 1 2 3 4 5 6 7 8 9 16 17 18 19 20 21 22 23 24 10 11 12 13 14 15 1 2 3 4 5 6 7 8 9 16 17 18 19 20 21 22 23 24 10 11 12 13 14 15 18) USO ESPECÍFICO O PROCESO 19) CLAVE DEL(OS) LOTE(S) 21 SEÑALAR LA FINALIDAD DE USO DEL DISPOSITIVO MÉDICO 20) INDICACIONES DE USO 21) CONCENTRACIÓN 22) INDICACIONES TERAPÉUTICAS 23) FECHA DE FABRICACIÓN 24) FECHA DE CADUCIDAD CUANDO APLIQUE 25) TEMPERATURA DE ALMACENAMIENTO 26) TEMPERATURA DE TRANSPORTE 27) MEDIO DE TRANSPORTE O ADUANA DE ENTRADA 28) IDENTIFICACIÓN DE CONTENEDORES DESCRIPCIÓN DEL ENVASE EN CONTACTO DIRECTO CON EL PRODUCTO. 29) ENVASE PRIMARIO DESCRIPCIÓN DEL EMPAQUE EN EL CUAL SE COMERCIALIZA EL INSUMO. 30) ENVASE SECUNDARIO 31) TIPO DE EMBALAJE Y No. DE UNIDADES DE EMBALAJE 32) No DE PARTIDA 33) CLAVE DEL CUADRO BÁSICO O CATÁLOGO DEL SECTOR SALUD (CBSS) EXPORTACIÓN GENÉRICO EXPORTACIÓN VENTA SECTOR SALUD GENÉRICO 34) PRESENTACIÓN DESTINADA A: SECTOR SALUD 35) FABRICACIÓN DEL PRODUCTO NACIONAL EXTRANJERO NACIONAL VENTA EXTRANJERO 36) UNIDAD DE MEDIDA DE APLICACIÓN DE LA TIGIE (UMT) 37) CANTIDAD DE UNIDAD DE MEDIDA DE APLICACIÓN DE LA TIGIE 38) TIPO DE ORGANISMO GENÉTICAMENTE MODIFICADO (OGM) SOLO UN PRODUCTO POR SOLICITUD 39) NÚMERO DE PROGRAMA IMMEX (SOLO PARA EMPRESAS QUE ESTÉN DENTRO DEL PROGRAMA PARA LA INDUSTRIA MANUFACTURERA, MAQUILADORA Y DE SERVICIOS DE EXPORTACIÓN NOTA: REPRODUCIR ESTA HOJA, TANTAS VECES COMO SEA NECESARIO CONFORME A LO ESTABLECIDO EN CADA TIPO DE TRÁMITE. 5.- INFORMACIÓN PARA CERTIFICADOS: USO DEL CERTIFICADO (PARA EXPORTACIÓN, REGISTRO, PRÓRROGA Y OTROS) PAÍS DESTINO ESPECIFICAR CARACTERÍSTICAS 6.- PROTOCOLO DE INVESTIGACIÓN TÍTULO DEL PROTOCOLO VÍA DE ADMINISTRACIÓN (Medicamentos o Dispositivos Médicos) NOMBRE DEL INVESTIGADOR PRINCIPAL NOMBRE(S) DE LA(S) INSTITUCIÓN(ES) DONDE SE REALIZARÁ LA INVESTIGACIÓN 22 DATOS DE LA OPERACIÓN: 7.- A). PARA REGISTRO (MAQUILA) NOMBRE DEL MAQUILADOR NACIONAL O EXTRANJERO (PERSONA FÍSICA) O RAZÓN SOCIAL (PERSONA MORAL) R.F.C. CALLE Y NÚMERO DELEGACIÓN O MUNICIPIO COLONIA CÓDIGO POSTAL LOCALIDAD ENTIDAD FEDERATIVA ETAPA DEL PROCESO DE FABRICACIÓN No. DE LICENCIA SANITARIA O AVISO DE FUNCIONAMIENTO R.F.C. DEL RESPONSABLE SANITARIO NOMBRE DEL RESPONSABLE SANITARIO TELÉFONO Y FAX CORREO ELECTRÓNICO 7.- B). FABRICACIÓN, DISTRIBUCIÓN O ALMACENAMIENTO DE PRODUCTOS IMPORTADOS O NACIONALES NOMBRE DEL FABRICANTE EN EL EXTRANJERO PARA PRODUCTOS DE IMPORTACIÓN (PERSONA FÍSICA) O RAZÓN SOCIAL (PERSONA MORAL) DEBERÁ INDICAR EL NOMBRE DEL PROPIETARIO O RAZÓN SOCIAL DEL FABRICANTE CALLE Y NÚMERO COLONIA LOCALIDAD CALLE DE LA EMPRESA PAÍS COLONIA DE LA EMPRESA CÓDIGO POSTAL PAIS DE LA EMPRESA SU LOCALIDAD ESTADO CÓDIGO POSTAL ENTIDAD FEDERATIVA R.F.C. (a) NOMBRE DEL PROVEEDOR O DISTRIBUIDOR (PARA INSUMOS PARA LA SALUD) DEBERÁ INDICAR EL NOMBRE DEL PROPIETARIO O RAZÓN SOCIAL CALLE Y NÚMERO R.F.C.DE LA EMPRESA DELEGACIÓN O MUNICIPIO( a ) COLONIA CALLE DE LA EMPRESA COLONIA DE LA EMPRESA LOCALIDAD ( a ) CÓDIGO POSTAL (a) SU LOCALIDAD CÓDIGO POSTAL DELEGACIÓN O MUNICIPIO ENTIDAD FEDERATIVA ( a ) ENTIDAD FEDERATIVA NOMBRE DEL ESTABLECIMIENTO QUE ACONDICIONARA O ALMACENARA LOS INSUMOS PARA LA SALUD (PERSONA FÍSICA) O RAZÓN SOCIAL (PERSONA MORAL) R.F.C. DEBERÁ INDICAR EL NOMBRE DEL PROPIETARIO O RAZÓN SOCIAL CALLE Y NÚMERO COLONIA CALLE DE LA EMPRESA COLONIA DE LA EMPRESA LOCALIDAD SU LOCALIDAD R.F.C.DE LA EMPRESA DELEGACIÓN O MUNICIPIO CÓDIGO POSTAL DELEGACIÓN O MUNICIPIO ENTIDAD FEDERATIVA ENTIDAD FEDERATIVA CÓDIGO POSTAL NOTA: EN CASO DE SER MAS DE UN FABRICANTE O DISTRIBUIDOR, REPRODUCIR EL APARTADO 8 B) EN UNA HOJA ANEXA, CUANTAS VECES SEA NECESARIO. 7.- C). IMPORTACIÓN / EXPORTACIÓN/ REGISTRO R.F.C. ( a ) NOMBRE DEL FABRICANTE CALLE Y NÚMERO COLONIA O EQUIVALENTE LOCALIDAD CÓDIGO POSTAL DELEGACIÓN O MUNICIPIO ( a ) ENTIDAD FEDERATIVA O PAÍS NOMBRE DEL PROVEEDOR O DISTRIBUIDOR R.F.C. ( a ) CALLE Y NÚMERO COLONIA O EQUIVALENTE LOCALIDAD O EQUIVALENTE CÓDIGO POSTAL DELEGACIÓN O MUNICIPIO ( a ) ENTIDAD FEDERATIVA O PAIS R.F.C. NOMBRE DEL DESTINATARIO (destino final) CALLE Y NÚMERO COLONIA O EQUIVALENTE LOCALIDAD CÓDIGO POSTAL DELEGACIÓN O MUNICIPIO ( a ) ENTIDAD FEDERATIVA O PAIS NOMBRE DEL FACTURADOR b) R.F.C. CALLE Y NÚMERO COLONIA O EQUIVALENTE LOCALIDAD O EQUIVALENTE CÓDIGO POSTAL DELEGACIÓN O MUNICIPIO ENTIDAD FEDERATIVA PAÍS DE ORIGEN PAÍS DE PROCEDENCIA PAÍS DE DESTINO ADUANA DE ENTRADA/SALIDA (Solo marque una) ( a ) SOLO CUANDO EL ESTABLECIMIENTO SEA NACIONAL. (b) SOLO PARA INSUMOS PARA LA SALUD. 23 8.- DATOS DE PUBLICIDAD MEDIO PUBLICITARIO AGENCIA (Nombre o razón social) DOMICILIO DE LA AGENCIA (CALLE, No Y LETRA, COLONIA, LOCALIDAD, C.P., TELÉFONO, CORREO ELECTRÓNICO) NÚMERO DE PRODUCTOS O TIPO DE SERVICIO DURACIÓN O TAMAÑO NOTA: SE DEBERÁ PRESENTAR UNA SOLICITUD POR CADA PROYECTO Y MEDIO PUBLICITARIO 9.- AUTORIZACIÓN DE TERCEROS B) PRUEBAS DE INTERCAMBIABILIDAD PARA MEDICAMENTOS GENÉRICOS INTERCAMBIABLES A). LABORATORIO DE PRUEBA ANÁLISIS DE ALIMENTOS, BEBIDAS Y SUPLEMENTOS ALIMENTICIOS Y PRODUCTOS DE PERFUMERIA Y BELLEZA ANÁLISIS DE MEDICAMENTOS Y DISPOSITIVOS MÉDICOS UNIDAD CLÍNICA PARA REALIZAR ESTUDIOS DE BIODISPONIBILIDAD Y/O BIOEQUIVALENCIA ANÁLISIS DE PLAGUICIDAS, FERTILIZANTES Y NUTRIENTES VEGETALES UNIDAD ANALÍTICA PARA REALIZAR ESTUDIOS DE BIODISPONIBILIDAD Y/O BIOEQUIVALENCIA UNIDAD ANALÍTICA PARA ESTUDIOS DE PERFILES DE DISOLUCIÓN OTRO (ESPECIFIQUE) C). UNIDADES DE VERIFICACIÓN. VERIFICACIÓN DE ESTABLECIMIENTOS OTRO (ESPECIFIQUE) MUESTREO DECLARO BAJO PROTESTA DECIR VERDAD QUE CUMPLO CON LOS REQUISITOS Y NORMATIVIDAD APLICABLE, SIN QUE ME EXIMAN DE QUE LA AUTORIDAD SANITARIA VERIFIQUE SU CUMPLIMIENTO, ESTO SIN PERJUICIO DE LAS SANCIONES EN QUE PUEDO INCURRIR POR FALSEDAD DE DECLARACIONES DADAS A UNA AUTORIDAD. Y ACEPTO QUE LA NOTIFICACIÓN DE ESTE TRÁMITE SE REALICE A TRAVÉS DEL CENTRO INTEGRAL DE SERVICIOS U OFICINAS EN LOS ESTADOS CORRESPONDIENTES AL SISTEMA FEDERAL SANITARIO.(Art. 35 inciso b de la Ley Federal de Procedimiento Administrativo) LOS DATOS O ANEXOS PUEDEN CONTENER INFORMACIÓN CONFIDENCIAL, ¿ESTA DE ACUERDO EN HACERLOS PUBLICOS? SI NO NOMBRE Y FIRMA DEL PROPIETARIO, O REPRESENTANTE LEGAL O RESPONSABLE SANITARIO O DE OPERACIÓN PARA CUALQUIER ACLARACIÓN, DUDA Y/O COMENTARIO CON RESPECTO A ESTE TRÁMITE, SÍRVASE LLAMAR AL SISTEMA DE ATENCIÓN TELEFÓNICA A LA CIUDADANÍA (SACTEL) A LOS TELÉFONOS 2000-2000 EN EL D.F. Y ÁREA METROPOLITANA, DEL INTERIOR DE LA REPÚBLICA SIN COSTO PARA EL USUARIO AL 01800112-0584 O DESDE ESTADOS UNIDOS Y CANADÁ AL 1-800-475-2393, O A LOS TELÉFONOS DE LA COFEPRIS EN EL D.F. DE CUALQUIER PARTE DEL PAÍS MARQUE SIN COSTO EL 01-800-033-5050 Y EN CASO DE REQUERIR EL NÚMERO DE INGRESO Y/O SEGUIMIENTO DE SU TRÁMITE ENVIADO AL ÁREA DE TRAMITACIÓN FORÁNEA MARQUE SIN COSTO AL 01-800-420-4224. 24 ANEXO 3 LISTA DE DOCUMENTOS NECESARIOS EN EL EXPEDIENTE PARA LA SOLICITUD DE REGISTRO SANITARIO DE DISPOSITIVOS MÉDICOS POR EL ACUERDO DE EQUIVALENCIA. Productos con Registro en E.U.A. Clase I por FDA COFEPRIS-04-001-D 1. Formato de Solicitud 2. Comprobante del pago de derechos 3. Aviso de funcionamiento 4. Aviso de responsable sanitario 5. Proyecto de etiqueta 6. Instructivo de uso (o Manual de Operación en su caso) 7. Monografía con la información requerida 8. Certificado de análisis 9. Certificado a gobierno extranjero 10. Reporte de inspección del establecimiento (Establishment Inspection Report) 11. Documento de aprobación del Dispositivo Médico emitido por FDA 12. Carta de representación del fabricante 25 LISTA DE DOCUMENTOS NECESARIOS EN EL EXPEDIENTE PARA LA SOLICITUD DE REGISTRO SANITARIO DE DISPOSITIVOS MÉDICOS POR EL ACUERDO DE EQUIVALENCIA Productos con Registro en E.U.A. Clase II y III por FDA COFEPRIS-04-001-E 1. Formato de Solicitud 2. Comprobante del pago de derechos 3. Aviso de funcionamiento 4. Aviso de responsable sanitario 5. Proyecto de etiqueta 6. Instructivo de uso (o Manual de Operación en su caso) 7. Monografía con la información requerida 8. Certificado de análisis 9. Certificado a gobierno extranjero 10. Reporte de inspección del establecimiento (Establishment Inspection Report) 11. Resumen del último reporte de tecnovigilancia 12. Documento de aprobación del Dispositivo Médico emitido por FDA 13. Carta de representación del fabricante 26 LISTA DE DOCUMENTOS NECESARIOS EN EL EXPEDIENTE PARA LA SOLICITUD DE REGISTRO SANITARIO DE DISPOSITIVOS MÉDICOS POR EL ACUERDO DE EQUIVALENCIA. Productos con Registro en Canadá Clase II, III y IV por HC COFEPRIS-04-001-F 1. Formato de Solicitud 2. Comprobante del pago de derechos 3. Aviso de funcionamiento 4. Aviso de responsable sanitario 5. Proyecto de etiqueta 6. Instructivo de uso (o Manual de Operación en su caso) 7. Monografía con la información requerida 8. Certificado de análisis 9. Copia certificada de licencia del dispositivo médico emitida por Health Canada 10. Certificado CAN/CSA-ISO 13485:03 del fabricante 11. Certificado ISO 17021 del tercero autorizado 12. Autorización vigente de Health Canada al tercero autorizado 13. Carta de representación del fabricante 27 www.cofepris.gob.mx NUESTRA MISIÓN: PROTEGER TU SALUD Centro de Atención Telefónica 01 800 033 50 50