Tema 1.- T Tema 1.- T ermodinámica. Conceptos previos. p
Anuncio

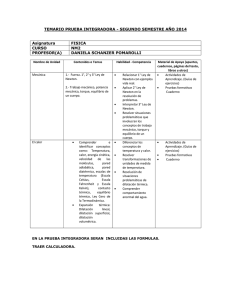
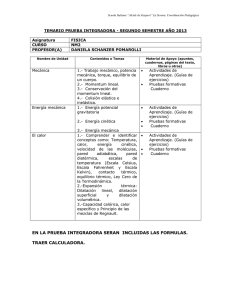
revios. rmodinámica. C onceptos pprevios. TTema ema 11..- TTeermodinámica. Conceptos §1.1.- Descripciones macro y microscópicas. Sistemas termodinámicos: Porción de materia separada del exterior por una superficie cerrada, real o imaginaria. energía masa SISTEMA Estado: Forma de estar o presentarse el Sistema Descripción del Estado: sup. real o imaginaria ENTORNO Microscópica: 6 1023 móleculas/mol 3 g.l. Mecánica Estadística Macroscópica: Se describen propiedades de conjunto o Reducido número de parámetros. Gas o Variables o coordenadas Termodinámicas independientes Variables o p, V, T coordenadas de Estado ENTORNO energía masa sup. real o imaginaria §1.2.- Clasificación de las variables termodinámicas. Externas: o Acoplamiento del sistema con su entorno o Dependen del entorno o Ej.: volumen, campo magnético externo,... Internas: o Distribución y estado de movimiento de las partículas o Tan sólo dependen indirectamente del entorno o Ej.: Presión Temperatura, energía interna,... Extensivas (X): o Dependen de la cantidad de materia o Son aditivas o Ej.: masa, volumen, energía Intensivas (Y): o NO dependen de la cantidad de materia o NO son aditivas o Son función de punto o Ej.: presión, temperatura m=m1+ m2 m1, p1, V1 , T1 V=V1+ V2 m2, p2, V2, T2, Grados de libertad del Sistema: Núm. mínimo de coordenadas independientes que lo describen. Sistema Simple: Tiene 2 g.l. Ej.: Sistema p,V,T Temperatura y dilatación 1-1/20 §1.3.- Estados de equilibrio Estado Estacionario: El valor de las variables termodinámicas permanece estacionario. Estado No-estacionario Proceso Termodinámico Estado estacionario Equilibrio Termodinámico (Imposibilidad de cambio espontáneo) El Equilibrio Termodinámico implica, además, que los parámetros intensivos independientes no varíen al pasar de un punto a otro del Sistema. Sistema simple p, V, T X extensiva V Y intensiva p Y (intensiva) p Espacio Termodinámico: 1 reversible irreversible 2 X (extensiva) V Temperatura y dilatación 1-2/20 §1.4.- Procesos e interacciones termodinámicas Los estados inicial y final son Estados de Equilibrio 1.4.a. Procesos termodinámicos Procesos reversibles (cuasiestáticos) o Los estados intermedios son estados de equilibrio Procesos irreversibles (no estáticos) o Los estados intermedios NO son estados de equilibrio Ejemplo: compresión de un gas encerrado en un cilindro provisto de un émbolo que ajusta perfectamente y que puede desplazarse sin rozamiento. Los procesos reversibles son: Invertibles mediante una modificación infinitesimal de las condiciones externas Infinitamente lentos Máximo rendimiento Irrealizables Proceso cíclico: El sistema retorna al estado inicial. Proceso infinitesimal: Implica cambios infinitesimales en las variables termodinámicas. Temperatura y dilatación Y (intensiva) p X (extensiva) V 1-3/20 §1.5.- Interacciones termodinámicas. Clasificación de los Sistemas Termodinámicos 1.5.a. Interacciones termodinámicas Naturaleza de sus paredes: móviles o deformables; permiten un intercambio energético por efectos mecánicos, que más adelante identificaremos con el trabajo. Por el contrario, las paredes fijas y rígidas impiden dicho intercambio energético. adiabáticas; impiden el intercambio energético en la forma que más adelante definiremos como calor. Las paredes que permiten el intercambio de energía en forma de calor (i.e., las no adiabáticas) se denominan paredes diatérmanas. permeables o impermeables; permiten o impiden el intercambio de materia. aislantes; no permiten ningún tipo de intercambio energético (salvo intercambios gravitacionales que podrán despreciarse cuando el sistema tenga unas dimensiones suficientemente pequeñas). Sistemas: Aislados Cerrados - Abiertos Existen tres tipos de interacciones puras, que pueden presentarse separadamente o combinadas: Interacción másica; entre los dos sistemas existe solamente intercambio de materia. La pared deberá ser permeable o semipermeable, fija y adiabática. Interacción mecánica; entre los dos sistemas se establece únicamente un intercambio energético por efectos mecánicos. La pared separadora de ambos sistemas deberá ser móvil o deformable, impermeable y adiabática. Interacción térmica; entre los dos sistemas se establece tan solo un intercambio energético por efectos no-mecánicos (calor). La pared que separa entre sí a los dos sistemas será diatérmana, fija y rígida e impermeable. 1.5.b. Clasificación de los Sistemas Termodinámicos Formas de interacción de un sistema con su entorno: Sistemas cerrados; límites impermeables al flujo de materia. Sistemas abiertos; paredes permeables o semipermeables al flujo de materia. Sistemas aislados; no permiten ningún tipo de intercambio energético (sistemas delimitados por paredes aislantes). Temperatura y dilatación 1-4/20 Atendiendo a otros aspectos. Su composición, se clasifican en monocomponentes y multicomponentes, según que estén integrados por una o varias sustancias o especies químicas. Su constitución, pueden clasificarse en homogéneos (v.g., una mezcla de dos líquidos totalmente miscibles) y heterogéneos (v.g., una mezcla de hielo y agua). §1.6.- Equilibrio Térmico. Principio Cero. Noción de Temperatura: percepción sensorial que nos permite distinguir los cuerpos fríos de los cuerpos calientes. Método operacional: La sensación térmica táctil sugiere dos hechos experimentales: Cuando dos o más cuerpos se ponen en contacto durante un tiempo suficientemente largo, la sensación táctil respecto de todos ellos es la misma (concepto de equilibrio térmico) Hay cuerpos buenos conductores del calor y otros malos conductores. Principio General de la Termodinámica: Un sistema aislado evoluciona hacia un estado de equilibrio que no podrá abandonar espontáneamente. A y B tienen la misma temperatura. Principio Cero de la Termodinámica: Si dos sistemas están en equilibrio térmico con un tercero, están en equilibrio térmico entre sí. Llamamos temperatura (T) a una magnitud que caracteriza el equilibrio térmico y, por definición, establecemos que en tal equilibrio se cumple: TA = TB = TC La temperatura caracteriza el estado interno del sistema y es independiente de la cantidad de materia del mismo (i.e., un parámetro interno e intensivo) y, en virtud de los intercambios energéticos con los otros sistemas (como consecuencia de la interacción térmica), adquiere el mismo valor en todos ellos, permaneciendo constante una vez alcanzado el equilibrio. Podemos afirmar que el Principio Cero posibilita la introducción de un nuevo parámetro termodinámico (la temperatura) que, junto con los parámetros termodinámicos externos, permite especificar el estado del sistema. Temperatura y dilatación 1-5/20 §1.7.- Repaso de escalas termométricas. Conversión Celsius-Fahrenheit: tC tF - 32 = 5 9 Ejemplo: 5 tC = (100 - 32) = 37.8 º C 9 El grado Celsius (ºC) y el kelvin (K) tiene el mismo tamaño, de modo que las diferencias de temperaturas pueden expresarse indistintamente en ºC o K. Temperatura y dilatación 1-6/20 §1.8.- Ecuaciones Térmicas de Estado. Coeficientes térmicos. Cualquier parámetro (Y) interno es función de los parámetros externos (X) y de la temperatura: Yi = Yi ( X 1 , X 2 ,...T ) Sistema simple: Y = Y ( X , T ) (2 g.l.) p = p (V , T ) gas t = t (l , T ) tensión de un hilo s = s (S ,T ) tensión superficial æ ¶Y ö÷ æ ¶Y ö÷ çç ÷ dT + X d Cambios: dY = çç ÷ çè ¶X ø÷ ç ¶T ø÷ è T X Sistema simple (p, V, T) Coef. de dilatación térmica o isobárica: a= æ ¶V / V ö÷ 1 æç ¶V ö÷ çç = ÷ ÷ çèç ÷ø V ¶T p çè ¶T ÷ø p æ ΔV / V ö÷ a = çç ÷ èç ΔT ø÷ p V = 1 + aΔT V0 sólidos: a » 10-5 K-1 líquidos: a » 10-4 K-1 gases: a » 10-3 K-1 gases ideales: a » 0.003610 K-1 Coef. piezotérmico : b= æ ¶p / p ÷ö 1 æç ¶p ö÷ ç = ÷ ÷ ç ç p èç ¶T ø÷V çè ¶T ÷øV æ Δp / p ÷ö b = çç çè ΔT ÷÷ø V p = 1 + b ΔT p0 gases: a » b » 10-3 K -1 Coef. de compresibilidad isotermo : æ ¶V / V ö÷ 1 æ ¶V ö cT = - çç ÷÷÷ = -çç ÷ çè ¶p ÷ø V çè ¶p øT T æ ΔV / V ö÷ 1 cT = -çç ÷÷ » çè Δp ø K T sólidos y líquidos: cT » 10-7 atm-1 gases: a » 10-3 K-1 gases ideales: a » 0.003610 K-1 Temperatura y dilatación 1-7/20 §1.9.- Dilatación de los sólidos A nivel microscópico, la dilatación térmica de los sólidos sugiere un aumento en la separación media entre sus átomos y moléculas constituyentes. Para una energía de vibración dada, i.e., para una temperatura de referencia (T0) dada, la separación entre los átomos cambiará periódicamente entre unos valores mínimo y máximo y, en virtud de la asimetría de la curva de energía potencial, la separación media entre los átomos (r0) será mayor que la correspondiente al equilibrio (req). dV = a dT V ìï V2 ïïln = a (T2 - T1 ) V2 = V1 ea(T2 -T1 ) ï V1 ïí ïï ΔV = a ΔT V = V0 éë1 + a (T - T0 )ùû ïï ïî V0 Dilatación lineal: = 0 ëé1 + a ¢ (T - T0 )ûù con a ¢ = a 3 a = 3a ¢ Dilatación superficial: S = S0 éë1 + a ¢¢ (T - T0 )ùû con a ¢¢ = 2a ¢ = (2 3)a Si un sólido tiene una cavidad, el volumen (o superficie) de esa cavidad aumenta, cuando el cuerpo se dilata, lo mismo que si estuviera lleno de la misma materia. Esto se cumple aunque la cavidad sea tal que el cuerpo que la rodea quede reducido a una capa delgada. Así, el volumen encerrado por un matraz aumenta como un cuerpo macizo del mismo tamaño y material. §1.10.- Esfuerzos de origen térmico Sistema de 2 g.l. Ecuación térmica de estado: s = s (l , T ) Δl l0 ΔT s E= Δl l0 a¢ = ü Δl ï = a ¢ΔT ïï ï l0 ïý a ¢ΔT = s ï Δl s E ï = ï ï l0 E ï þ s = Ea ¢ΔT Se pueden producir esfuerzos de origen térmico como consecuencia de un calentamiento no uniforme (ruptura de un recipiente de vidrio grueso al verter agua caliente en él). Temperatura y dilatación 1-8/20 §1.11.- Dilatación de los líquidos Al estudiar la dilatación de los líquidos es preciso tener en cuenta el aumento de volumen que experimenta el recipiente que los contiene. El aumento de volumen real del líquido es la suma del que experimenta el recipiente y del incremento de volumen aparente del líquido. Como consecuencia de ello, el coeficiente de dilatación (αlíq) es prácticamente igual a la suma del coeficiente de dilatación cúbica del recipiente (αrec) del coeficiente de dilatación aparente (αap) del líquido: areal = aap + arec 1.11.a. Dilatación anómala del agua La dilatación anómala del agua es de gran importancia para la vida en nuestro planeta, siendo la razón por la que los mares, lagos y ríos comienzan a helarse por la superficie, aislando el agua subyacente de nuevas pérdidas caloríficas, la cual se mantendrá casi siempre a una temperatura constante de 4 °C. Temperatura y dilatación 1-9/20 §1.12.- Dilatación de los gases Cuando cambia la temperatura de un gas, varían simultáneamente su volumen y su presión. Como tanto el volumen como la presión pueden mantenerse constantes durante un cambio en la temperatura del gas, debemos considerar por separado los efectos que los cambios de temperatura producen en el volumen y en la presión de los gases. ΔV V0 ΔT Δp p0 (V = cte) b = ΔT ( p = cte) a = V = V0 (1 + aΔT ) p = p0 (1 + aΔT ) La experiencia demuestra que los valores de los coeficientes α y coinciden aproximadamente y que no dependen de la naturaleza del gas, siendo las discrepancias tanto menores cuanto más baja sea la presión del gas. a=b = 0.003 661 K p 0 -1 = 1 1 K -1 = 273.15 T0 1.12.a. Leyes de Gay-Lussac y Charles V V0 = V µT T T0 Gases Ideales: p p0 = p µT 2ª Ley: V = cte. T T0 1ª Ley: p = cte. ì V T - T0 T0 + T - T0 T ï ï ( ) 1 1 T T a = + = + = = 0 ï ïïV0 T0 T0 T0 En efecto: í ïp T T - T0 T + T - T0 ï = 1 + b (T - T0 ) = 1 + = 0 = ï ï T0 T0 T0 ï î p0 c.q.d. c.q.d 1.12.b. Ley de Boyle-Mariotte (1661): Los volúmenes ocupados por una misma masa gaseosa son inversamente proporcionales a las presiones a la que está sometida, siempre que la temperatura se mantenga constante: Si T = cte. pv = cte pv = f (T ) A principios del siglo XIX se demostró que la ley de Boyle-Mariotte sólo tiene una validez aproximada para los gases reales, aunque estos la satisfacen tanto mejor cuanto menor es su presión. Temperatura y dilatación 1-10/20 1.12.c. Gas ideal o perfecto satisface exactamente las leyes de Gay-Lussac y de Charles obedece exactamente la ley de Boyle-Mariotte a cualquier temperatura. Aunque el gas ideal no existe, todos los gases reales a bajas presiones lo aproximan muy bien. Desde un punto de vista práctico, en las condiciones ordinarias de presión y temperatura, pueden considerarse como gases ideales los llamados gases permanentes (que no puede licuarse más que a muy bajas temperaturas), tales como el hidrógeno, el helio, el oxígeno, el nitrógeno y el aire, ... entre otros. §1.13.- Ecuación térmica de estado del gas ideal Consideremos 1 mol de gas ideal y sea v el volumen molar: Proceso AB (isocoro): p0 p¢ = T0 T m.a.m.: p0v0 pv = =R T0 T Proceso BC (isotermo): p ¢v0 = pv (cte.) Constante universal de los gases perfectos: R= 1 atm 22.41 L/mol atm.L = 0.082 05 273.15 K mol.K 1101325 Pa 22.41´10-3 m3 /mol J R= = 8.315 273.15 K mol.K Si en lugar de un mol de sustancia consideramos n moles, el volumen correspondiente será V=nv: (ecuación de Clapeyron) pV = nRT 1.13.a. Densidad de un gas perfecto Sea M la masa molecular del gas (ej. Aire, M = 28.8 g/mol) pV = nRT = m RT M r= Densidad relativa al aire: rrel = Temperatura y dilatación m pM = V RT M r = raire M aire 1-11/20 §1.14.- Mezcla de gases perfectos no reaccionantes. Ley de Dalton Consideremos una mezcla de N gases perfectos e inertes, que ocupan un volumen V a una temperatura T, y sea p la presión total de la mezcla. Designaremos por pi la presión parcial correspondiente al gas i-ésimo, definida como la que ejercería el componente i-ésimo de la mezcla si estuviese él sólo ocupando todo el volumen de la mezcla, a la temperatura a la que ésta se encuentra. piV = ni RT N N å pVi = å ni RT i=1 i = 1, 2,...N (1) (2) pV = nRT i=1 LEY DE DALTON.- La presión total de una mezcla de gases ideales no reaccionantes es la suma de las presiones parciales de sus componentes. N p = å pi i=1 Dividiendo miembro a miembro las ecuaciones (1) y (2): pi ni = = ci p n (fracción molar) pi = ci p (3) La ecuación (3) expresa que la presión parcial de cualquier componente es proporcional a su fracción molar en la mezcla, siendo la presión total la constante de proporcionalidad. Temperatura y dilatación 1-12/20 §1.15.- Calor y trabajo Hasta finales del siglo XVIII, el calor era concebido como un fluido, llamado calórico, invisible e imponderable, increable e indestructible (i.e., se conserva), capaz de mezclarse o de combinarse con los cuerpos materiales y que podía pasar de unos cuerpos a otros en virtud de la emisión de partículas calóricas. Hoy sabemos que el calor es una de las formas que puede adoptar la energía. Calor es la energía neta transmitida entre dos sistemas cerrados con un límite común, o entre un sistema cerrado y sus alrededores, en virtud de una diferencia de temperaturas y que tiene lugar sin variación de los parámetros externos, i.e., en una interacción térmica pura; Trabajo es la energía transmitida por variación de algún parámetro externo o debida a la acción de fuerzas que se desplazan. El calor y el trabajo son energías en tránsito. La estructura formal de la Termodinámica está apoyada sobre esta base y sustentada por un puente de reversibilidad formal entre calor y trabajo que instaura a aquél como agente capaz de producir trabajo, así como de ser originado por éste. El calor fluye de un punto a otro, y si el flujo cesa no se puede hablar de calor; algo parecido ocurre con el trabajo. Los cuerpos y los sistemas no tiene calor ni trabajo, solamente poseen energía. El calor y el trabajo no son funciones de estado. Convenio termodinámico de signos: Unidades de calor: Julio (J) Caloría (cal) es la cantidad de calor necesaria para elevar la temperatura de 1 gramo de agua desde 14.5 a 15.5 °C. “Equivalente mecánico de la caloría” 1 cal = 4.1868 J Con esta equivalencia es posible establecer que la constante de los gases perfectos tiene un valor de R = 1.987 cal/(mol K) 2 cal/(mol K). Se emplean corrientemente los siguientes múltiplos de la caloría: 1 kilocaloría (kcal) o caloría grande (Cal) = 1000 cal 1 Megacaloría (Mcal) o termia = 1 000 000 cal La frigoría, utilizada como unidad en la industria del frío, corresponde al desprendimiento o pérdida de 1 kcal. Temperatura y dilatación 1-13/20 §1.16.- Capacidad calorífica y calor específico El calor intercambiado entre un sistema y sus alrededores puede ponerse de manifiesto mediante dos efectos. cambia la temperatura del sistema, aunque su estado físico no se ve afectado. cambio de estado físico, pero la temperatura permanece constante. A igualdad de masa, los sistemas termodinámicos difieren unos de otros en la cantidad de calor que necesitan para que en ellos se produzca una misma variación de temperatura. Capacidad calorífica de un sistema se define como la cantidad de calor necesaria para incrementar en 1 K (o en 1 °C) su temperatura. Capacidad calorífica media de un sistema en el intervalo de temperaturas T1 a T2 es igual a la cantidad de calor (Q) que se aporta al sistema dividida por el incremento de temperatura del mismo. Capacidad calorífica del sistema a una temperatura dada. _ C= Q ΔT C= đQ dT æJö ççèç ø÷÷÷ đQ = C dT K T2 Q = ò C dT T1 Evitamos utilizar la notación Q, ya que «el calor del sistema no se incrementa»; Q significaría Q2 - Q1, lo que es inaceptable. Escribimos đQ (en lugar de dQ) para designar un «aporte» (no un «cambio») infinitesimal de calor. Calor específico (c) de una sustancia se define como la capacidad calorífica referida a la unidad de masa; representa la cantidad de calor que hay que suministrar a la unidad de masa del sistema considerado para elevar su temperatura en 1 grado (Kelvin o Celsius) a partir de una temperatura dada: c= C 1 đQ = m m dT æ J ö÷ ÷ đQ = mc dT ççç è kg ⋅ K ÷ø T2 Q = m ò c dT T1 Capacidad calorífica molar (Cm) de una sustancia se define como la capacidad calorífica referida a un mol de esa sustancia: Cm = C 1 đQ = n n dT æ J ö÷ çç đQ = nCm dT çè mol ⋅ K ÷÷ø T2 Q = n ò Cm dT T1 Relaciones: C = mc = nCm El calor específico de una sustancia varía con la temperatura; en general, el calor específico aumenta al elevarse la temperatura. Temperatura y dilatación 1-14/20 El calor específico del agua exhibe un valor mínimo de 0.99795 cal/(g.K) para la temperatura de 34.5°C, en tanto que vale 1.007 38 cal/(g K) a 0°C. Por consiguiente, el calor específico del agua varía menos del 1% respecto de su valor de 1 cal/(g K) a 15 °C, por lo que a menudo se le considera como constante. El calor específico del agua es notablemente superior al de las demás sustancias sólidas o líquidas; en consecuencia, para un mismo intercambio calorífico, la variación de temperatura que se produce en una determinada masa de agua es considerablemente menor que la que se produce en la misma masa de otra sustancia. Esto explica el poder termorregulador de las grandes masas de agua (océanos, mares, lagos, ...) que da lugar a que el clima de las zonas marítimas sea más uniforme que el de las zonas continentales. Por otra parte, la alta proporción de agua en los tejidos de los seres vivos está destinada, entre otras cosas, al establecimiento de un sistema fisiológico lo más isotermo posible. §1.17.- Calores específicos de los gases Se consideran las capacidades caloríficas molares a presión constante (Cp) y a volumen constante (CV), definidas por æ đQ ö C p = çç ÷÷÷ çè dT ø p=cte æ đQ ö CV = çç ÷÷÷ çè dT ø V =cte C p > CV La diferencia entre estas capacidades caloríficas corresponde al trabajo de dilatación que tiene lugar cuando el proceso se desarrolla a presión constante. Q p - QV = W = p Δv Con pv = RT C p ΔT - CV ΔT = R ΔT p Δv = R ΔT C p - CV = R (relación de Mayer) Las capacidades caloríficas molares a presión y a volumen constante de un mismo gas ideal difieren en una cantidad constante que es igual a la constante R de los gases perfectos [1.9867 cal/(mol.K)]. La relación de Mayer se cumple relativamente bien para los gases cuyas moléculas están constituidas por pocos átomos. Temperatura y dilatación 1-15/20 Se observa que los gases de igual atomicidad tienen calores molares cuyos valores son muy parecidos, lo que indica que los valores de las capacidades caloríficas están íntimamente relacionados con el número de átomos de las moléculas. Los valores del coeficiente adiabático g = Cp CV son prácticamente coincidentes para los gases de la misma atomicidad (1.67 para los monoatómicos, 1.40 para los diatómicos, ...). La Teoría Cinética de los Gases prevé que los valores de los calores molares de los gases mono y diatómicos son los siguientes: Temperatura y dilatación Gases g.l. Cp monoatómicos 3 5/2 R 3/2 R 5/3 = 1.67 diatómicos 5 7/2 R 5/2 R 7/5 = 1.4 --- --- --- --- --- En general f f +2 R 2 f R 2 f +2 f CV γ 1-16/20 §1.18.- Cambios de fase. Calores latentes. Concepto de fase: porción de un sistema en equilibrio que es físicamente homogénea (i.e., con propiedades intensivas uniformes) y químicamente homogénea. Un cuerpo puro en un estado de agregación determinado (sólido, líquido o vapor) constituye una fase. Cada fase de un cuerpo puro o especie química sólo puede existir entre ciertos valores límites de presión y temperatura. Cuando un sistema se presenta en fases diferentes, existen parejas de valores de presióntemperatura para los cuales tiene lugar el paso de una fase a otra. Cuando esto ocurre, decimos que se produce un cambio de fase. Experimentalmente se han establecido los resultados siguientes: a) Cuando una sustancia pura se presenta en dos fases distintas, existen pares de valores de presión-temperatura para los que hay equilibrio entre ambas. b) A una presión dada, la temperatura de equilibrio entre dos fases de una misma sustancia pura tiene un valor determinado; y recíprocamente. Con otras palabras, a una presión dada, el cambio de fase se produce siempre a la misma temperatura, que es característica de la sustancia. c) Mientras dura el cambio de fase, i.e., mientras coexisten las dos fases, si la presión permanece constante, la temperatura también permanece constante, pero es necesario intercambiar con el sistema una cierta cantidad de calor que no se invierte en modificar la temperatura del mismo, sino en cambiar la estructura física del sistema a temperatura constante. Este calor recibe el nombre de calor latente o de transformación, cuando está referido a la unidad de masa de la sustancia pura. Si no se suministra ni se sustrae calor a un sistema en el que se haya alcanzado el equilibrio entre dos fases del mismo, no se producirá cambio alguno y las proporciones relativas entre las fases coexistentes permanecerán constantes. El calor latente suele representarse por , cuando está referido a la unidad de masa de sustancia, y por L, cuando corresponde a un mol, denominándose entonces calor molar de transformación. Obviamente, la cantidad de calor puesta en juego en un proceso en el que cambian de fase una masa m de sustancia es Q=m Temperatura y dilatación 1-17/20 Cambios de fase.- Podemos comprobar fácilmente cuanto acabamos de decir estudiando como varía la temperatura de una sustancia pura en función del tiempo, cuando se le suministra calor lentamente y a ritmo constante. La gráfica de la figura corresponde a la evolución de la temperatura de un sólido cristalino (v.g, el hielo) cuando es calentado a presión constante. Temperatura y dilatación 1-18/20 Temperatura y dilatación 1-19/20 Temperatura y dilatación 1-20/20