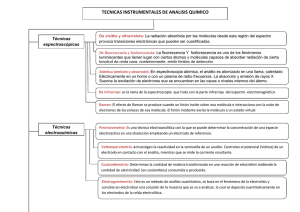
Técnicas Cromatográficas
Anuncio

Universidad Nacional Autónoma de México Facultad de Química Química Analítica Instrumental II Técnicas Cromatográficas Diciembre de 2007 Introducción a los métodos de separación Capítulo 1. Introducción a los métodos de separación 1.1 Introducción a la cromatografía En 1906, el botánico Ruso M. Tswett realizó un experimento que condujo al descubrimiento de lo que hoy conocemos como cromatografía. Colocó un extracto de pigmentos vegetales en la parte superior de una columna de vidrio rellena de carbonato de calcio (CaCO3). Al agregar éter, observó que la mezcla original se separaba en diversas bandas coloridas que descendían a través de la columna a diferentes velocidades. Un rasgo característico de la cromatografía es la presencia de dos fases; dispuestas de tal manera que mientras una permanece estacionaria dentro del sistema (fase estacionaria), la otra se desplaza a lo largo de él (fase móvil). La clave de la separación en cromatografía es que la velocidad con la que se mueve cada sustancia depende de su afinidad relativa por ambas fases (equilibrio de distribución). En el experimento de Tswett, la separación de los pigmentos vegetales se logró gracias a que cada uno de ellos tenía una afinidad diferente por las fases. En general, los componentes más afines a la fase estacionaria avanzan lentamente (más retenidos) mientras que los más afines a la fase móvil (menos retenidos) se mueven con mayor rapidez. Por consecuencia, el medio cromatográfico (columna, placa o papel) funciona como un controlador de la velocidad de cada sustancia que constituye la mezcla, logrando así su separación y mediante el uso de un detector, su caracterización química. Aunque los principios fundamentales son los mismos, se acostumbra clasificar los métodos cromatográficos según el estado físico de la fase móvil: Cromatografía líquida. La fase móvil es un disolvente o mezcla de disolventes y la fase estacionaria un sólido que interactúa con las sustancias que se desea separar (cromatografía líquido-sólido), o bien un líquido inmiscible con la fase móvil, depositado en la superficie de un sólido (cromatografía líquidolíquido). Esta forma de cromatografía puede realizarse con diferentes arreglos experimentales: en columna, en capa delgada o en papel. En el primer caso, la fase estacionaria se encuentra rellenando un tubo; en el segundo, se dispersa sobre una lámina de vidrio o aluminio formando un lecho de espesor uniforme; en la cromatografía en papel, la fase estacionaria es la solución acuosa contenida en el interior de las celdas formadas por las fibras de la celulosa, y es por tanto una forma de cromatografía líquidolíquido. Capítulo 1 1 Introducción a los métodos de separación Cromatografía de gases. En este caso la fase móvil es un gas inerte (helio o nitrógeno) y la fase estacionaria es un sólido (cromatografía gas-sólido) o un líquido “sostenido” por un sólido inerte (cromatografía gas-líquido). Este tipo de cromatografía siempre es en columna, ya que es la única manera de que la fase móvil gaseosa se mantenga fluyendo, confinada dentro del sistema. La columna puede estar rellena con la fase estacionaria, en forma semejante a la cromatografía líquida, o bien la fase estacionaria puede depositarse sobre las paredes de un tubo muy delgado (0.25mm de diámetro) y largo (hasta 100m). Este tipo de columnas se conocen como columnas capilares y proporcionan la mayor capacidad de separación. De acuerdo con el mecanismo de retención, la cromatografía se puede clasificar en los siguientes tipos: adsorción (normal, de fase reversa) permeación sobre gel partición líquido-líquido de afinidad intercambio iónico Las áreas de aplicación son muy diversas y abarcan prácticamente todas las actividades en las que interviene la química, por ejemplo: se emplea en: El análisis de drogas y fármacos en fluidos biológicos como la saliva, la sangre, la orina; Seguir la transformación de las sustancias responsables de la transmisión neurológica; Determinar la presencia de contaminantes en el medio ambiente; Descifrar la composición de los combustibles fósiles; Realizar el control de calidad de los productos químicos y farmacéuticos manufacturados; en fin, la lista de ejemplos es interminable. 1.2 Cromatografía en papel La cromatografía en papel es un proceso muy utilizado en los laboratorios para realizar análisis cualitativos ya que pese a no ser una técnica potente no requiere de ningún tipo de equipamiento. La fase estacionaria está constituida simplemente por una tira de papel de filtro. La muestra se deposita en un extremo colocando pequeñas gotas de la disolución y evaporando el disolvente. Luego el disolvente empleado como fase móvil se hace ascender por capilaridad. La separación se realiza en función de la afinidad de los solutos con las dos fases, las más solubles en agua se quedarán cerca del punto donde se aplicó la muestra, y las menos solubles en agua y más solubles en el disolvente llegarán más lejos. Las sustancias separadas se identifican mediante diversos procedimientos físicos o químicos Capítulo 1 2 Introducción a los métodos de separación La cromatografía en papel es una técnica utilizada para análisis inorgánico cualitativo, permite llevar a cabo la separación e identificación de iones, trabajando con cantidades mínimas de sustancia. Pertenece al tipo de “Cromatografía de partición” se fundamenta en que las sustancias problema, pueden tener diferentes coeficientes de reparto en dos disolventes de inmiscibilidad limitada, uno permanece fijo en la superficie del papel “fase estacionaria” generalmente en agua, la fase móvil constituida generalmente por una mezcla de disolventes parcialmente miscibles en ella. Hay varios tipos de cromatografía, la ascendente (papel hacia arriba), descendente (papel invertido), radial y de separación de zonas y sectores. Al entrar en contacto con los disolventes empieza su fase de movilidad lo que produce unas manchas características sobre el papel, generalmente, estas no son coloreadas y se revelan con una lámpara fluorescente, los contornos se marcan con lápiz. Como medida en cromatografía sobre papel se emplea el Rf (Retention factor), el cual se define como el cociente de dividir el recorrido de la sustancia por el disolvente, esto es, la distancia media desde el origen hasta el centro de la mancha (X) dividida por la distancia que media desde el origen hasta el frente del disolvente (S). 𝑅𝑓 = 𝑋 𝑆 [1] En la cromatografía en papel se utiliza como fase estacionaria una hoja de papel de celulosa de elevada pureza recubierta de una capa de agua asociada a las fibras de celulosa. La fase móvil, en la que irá disuelta la muestra, se forma por disolventes cuya naturaleza se elige en función de los componentes que se pretenden separar. 1.3 Cromatografía en capa fina La cromatografía en capa fina se basa en la preparación de una capa, uniforme, de un adsorbente mantenido sobre una placa de vidrio u otro soporte. Los requisitos esenciales son, pues, un adsorbente, placas de vidrio, un dispositivo que mantenga las placas durante la extensión, otro para Capítulo 1 3 Introducción a los métodos de separación aplicar la capa de adsorbente, y una cámara en la que se desarrollen las placas cubiertas. Es preciso también poder guardar con facilidad las placas preparadas y una estufa para activarlas. La fase móvil es líquida y la fase estacionaria consiste en un sólido. La fase estacionaria será un componente polar y el eluyente será por lo general menos polar que la fase estacionaria, de forma que los componentes que se desplacen con mayor velocidad serán los menos polares. Polaridad de los compuestos orgánicos en orden creciente: hidrocarburos < olefinas < flúor < cloro < nitro < aldehído aldehído < ester < alcohol < cetonas < aminas < ácidos < amidas La cromatografía en capa fina presenta una serie de ventajas frente a otros métodos cromatográficos (en columna, en papel, en fase gaseosa) ya que el utillaje que precisa es más simple. El tiempo que se necesita para conseguir las separaciones es mucho menor y la separación es generalmente mejor. Pueden usarse reveladores corrosivos, que sobre papel destruirían el cromatograma. El método es simple y los resultados son fácilmente reproducibles, lo que hace que sea un método adecuado para fines analíticos. Al realizar la elección del adsorbente se debe tener en cuenta el tamaño de las partículas del adsorbente, cuanto más finamente dividido esté mayor será su adhesión al soporte, aunque también se le puede añadir un adherente. En la elección del eluyente influyen varios factores: Precio. No utilizar compuestos muy volátiles. Pureza. Evitar que contengan trazas de metales No utilizar mezclas de eluyentes (catalizadores). (reproducibilidad). La elección del eluyente se realiza de forma empírica. Hay que estudiar la polaridad del componente y probar con eluyentes cada vez menos polares. Capítulo 1 4 Introducción a los métodos de separación Generalmente se utiliza como reactivo revelador yodo, el cual forma complejos coloreados con los componentes orgánicos (con tonos amarillo-marrón), pero las manchas desaparecen con el tiempo con lo que es conveniente señalar las manchas aparecidas. Otro reactivo revelador bastante utilizado es el ácido sulfúrico, que reacciona con los componentes orgánicos produciendo manchas negras. Sin embargo debe tenerse en cuenta que el tamaño de las manchas no está relacionado con la cantidad de componente separado. 1.4 Cromatografía de permeación en gel La cromatografía de permeación en gel, también conocida como cromatografía de exclusión es una variante de la CLAE que consiste en una columna cromatográfica empacada de tal manera que las partículas de dicho empacamiento tienen diferentes tamaños de poros o de redes de poros con el fin de que las moléculas de soluto sean retenidas o excluidas basándose en su forma y tamaño y no en el peso molecular. Bajo este entendido, las moléculas de mayor tamaño no caben dentro de los poros y son arrastrados alrededor de la fase estacionaria, por lo que son excluidas y eluyen en el volumen de “cama de vacío” de la fase móvil. Por el contrario, las partículas de menor tamaño eluyen hasta el final porque tiene más espacio de columna que recorrer debido a un fenómeno de difusión hacía los poros que tienen accesibles. Las moléculas de tamaños intermedios tardan diferentes tiempos en eluir, debido a la cantidad de poros por los que puedan pasar. Empaquetamientos de columna. Existen distintos tipos de empacamientos columna. Entre ellos están los polímeros macromoleculares semirígidos, los entrecruzados, las sílices rígidas ó las de “poro controlado”. Los materiales semirígidos se hinchan ligeramente se debe tener cierto cuidado en su uso ya que se limitan a una presión de 300psi. Las cuentas de poliestireno parcialmente sulfonado que tienen un tamaño usual de 5µm, son buenas para usarse con sistemas acuosos mientras los no sulfonados son compatibles con sistemas no acosos. Capítulo 1 5 Introducción a los métodos de separación Otro tipo de empacamiento hidrofílico poroso es preparado mediante una polimerización por suspensión de metacrilato de 2-hidroxietilo con dimetacrilato de etileno. Estos empacamientos resisten presiones de hasta 3000psi y pueden ser usados con sistemas acuosos y una amplia gama de disolventes orgánicos polares. Disolventes. La cromatografía de exclusión requiere un solo disolvente durante el análisis en el cual se pueda disolver la muestra. El único inconveniente que pude presentarse con los disolventes es la relación de viscosidades. Cuando la viscosidad de la muestra inyectada difiere mucho con la viscosidad de la fase móvil se pueden dar, una distorsión en los picos cromatográficos y cambios anómalos en los tiempos de retención. 1.5 Cromatografía de exclusión de iones Una variante de la cromatografía de permeación en gel o cromatografía de exclusión, es cuando se realiza para la exclusión de iones. Mediante esta modificación, se tienen separaciones que dependen de factores como tamaño de partícula de la resina, forma iónica e incluso distribución de isómeros. Esto permite separar especies que de otra forma eluirían al mismo tiempo. Las separaciones se llevan a cabo en resinas de intercambio con base en poliestireno de alta capacidad como eluyentes, ácidos minerales diluidos. Mucho del trabajo en la industria de alimentos y bebidas (vino, cerveza, jugo de frutas, etc.) o de muestras fisiológicas en donde la mayoría de los ácidos del ciclo de Krebs están presentes, puede ser hecho fácilmente mediante el uso de una columna de exclusión aniónica. El uso de las columnas de exclusión aniónica, en su modalidad con iones de calcio, se extiende a la separación de oligosacáridos usando agua como eluyente a una temperatura de 90ºC, aplicando también la separación por exclusión estérica. 1.6 Cromatografía de intercambio iónico La cromatografía de intercambio iónico se lleva a cabo con empaques de columna que tiene grupos funcionales cargados unidos a una matriz polimérica. Los grupos funcionales enlazados permanentemente y asociados con contraiones de carga opuesta. El mecanismo de retención más común es el intercambio simple de los iones de la muestra y de la fase móvil con el grupo cargado de la fase estacionaria. En el caso de empaques cargados negativamente sirven para intercambiar especies catiónicas. Los más comunes son los sulfónicos, los cuales son fuertemente ácidos y tiene las propiedades de los Capítulo 1 6 Introducción a los métodos de separación ácidos fuertes cuando está en la forma H. los empaques cargados positivamente se utilizan para el intercambio con especies aniónicas. Los más comunes son las aminas cuaternarias, las cuales son fuertemente básicas y en su forma OH presentan las propiedades de una base fuerte. Ambos grupos funcionales están totalmente disociados. Así, sus propiedades de intercambio son independientes del pH de la fase móvil. La cromatografía de intercambio iónico puede efectuarse con alguno de los varios tipos de empaques. El tipo pelicular consiste en un recubrimiento de resina, alrededor de 1-2m de espesor, sobre una cuenta de vidrio con diámetro de 30-40m. Las resinas superficialmente porosas se obtienen recubriendo cuentas de vidrio con un lecho delgado de microesferas de sílice en el que se impregna o enlaza un intercambiador de iones. Para ambos tipos de empaques la capacidad de intercambio es baja: 0.01- 0.1 meq/g. El intercambiador puede también estar enlazado a macropartículas de sílice por medio de reacciones de sililación o polimerizado dentro de los poros de un gel suficientemente poroso. El proceso primario en la cromatografía de intercambio iónico involucra la adsorción/desorción de materiales iónicos cargados en la fase móvil con una fase estacionara permanentemente cargadas (de signo contrario). Ejemplo, una resina ionizada, inicialmente en forma H, en contacto con una solución que contiene iones K+, existe un equilibrio: resina, H + K+ resina, K+ + H+ Las diferencias de retención están gobernadas esencialmente por las propiedades físicas de los iones solvatados. La fase de resina presenta preferencia por: 1. El ion de carga mayor. 2. El ion con menor radio solvatado. 3. El ion que tenga mayor polarizabilidad. Aplicaciones de intercambio catiónico: Los cationes inorgánicos se pueden determinar en alimentos, tales como los alimentos dietéticos bajos en sodio, y en muestras de orina. Aplicaciones de intercambio aniónico: Se pueden separar los ácidos HCN, carbónico, silícico y bórico de los ácidos fosfórico, sulfúrico y clorhídrico. Los metales que forman complejos cloruro y floruro. Capítulo 1 7 Introducción a los métodos de separación 1.7 Cromatografía de fluidos supercríticos La temperatura crítica de una sustancia es la temperatura por encima de la cual no puede existir en la fase líquida independientemente de la presión. La presión crítica es la presión de vapor de una sustancia a su temperatura crítica. Una sustancia a temperaturas y presiones por encima de su temperatura y de su presión crítica (punto crítico) se denomina fluido supercrítico. Los fluidos supercríticos tienen densidades, viscosidades y otras propiedades que son intermedias entre las características de esa sustancia en estado gaseoso y en estado líquido. Comparación de las propiedades de los fluidos supercríticos con las de gases y líquidos. (Los datos sólo indican el grado de magnitud). GAS FLUIDO SUPERCRÍTICO LÍQUIDO Densidad (g/cm3) (0,6-2)*10-3 0,2- 0,5 0,6- 2 Coeficiente de difusión (cm2/s) (1-4) * 10-1 10-3 – 10-4 (0,2 – 2)* 10-5 Viscosidad (g cm-1 s-1) (1-3)* 10-4 (1-3) * 10-4 (0,2- 3)* 10-2 Las propiedades seleccionadas son aquellas que son significativas para la cromatografía de gases, líquidos y fluidos supercríticos, así como la extracción de fluidos supercríticos. Una propiedad importante de los fluidos supercríticos, que está relacionada con sus densidades elevadas (de 0,2 a 0,5 g/cm3), es su notable capacidad para disolver moléculas grandes no volátiles. Por ejemplo, el dióxido de carbono supercrítico disuelve fácilmente n- alcanos que poseen entre 5 y 30 átomos de carbono. Una segunda propiedad notable de los fluidos supercríticos es que los analitos disueltos en ellos pueden ser fácilmente recuperados por el procedimiento simple de permitir que las disoluciones se equilibren con la atmósfera a temperaturas relativamente bajas. Otra ventaja de los fluidos supercríticos es que son baratos, inocuos y no son sustancias tóxicas, por lo que se pueden dejar evaporar libremente en la atmósfera sin efectos ambientales dañinos. La cromatografía de fluidos supercríticos (SFC) es una modalidad híbrida entre la cromatografía de gases y de líquidos que combina algunas características de cada una de ellas. Esta técnica es una de los tres tipos importantes de cromatografía en columna, ésta permite la separación y determinación de compuestos que no son manipulados ni por la cromatografía de gases ni por la de líquidos: compuestos no volátiles o térmicamente lábiles para los que la cromatografía de gases es inaplicable y (2) los Capítulo 1 8 Introducción a los métodos de separación compuestos que tienen grupos funcionales que no son detectables por las técnicas espectroscópicas o electroquímicas empleadas en cromatografía de líquidos. Los equipos instrumentales para la cromatografía de fluidos supercríticos son bastante parecidos en lo referente a los componentes instrumentales a los equipos de HPLC. Existen, sin embargo, dos diferencias importantes: (1) Es necesario un horno, similar para mantener la columna termostatizada y además proporcionar un control preciso de la temperatura de la fase móvil; (2) un restrictor o un dispositivo de contrapresión, que se utiliza para mantener la presión en la columna en el nivel deseado y para convertir el eluyente, de fluido supercrítico en un gas, y arrastrarlo al detector. Las variaciones de presión en cromatografía de fluidos supercríticos tienen un efecto muy marcado sobre el factor retención o de capacidad, k’. Este efecto es una consecuencia del incremento de la densidad de la fase móvil a medida que aumenta la presión. Tal incremento de la densidad origina un aumento de la capacidad disolvente de la fase móvil, lo cual acorta los tiempos de elución. La mayoría de los perfiles de presión utilizados en cromatografía de fluidos supercríticos son: presión constante (isobárica) para un período de tiempo dado seguida de un aumento lineal o asintótico de la presión hasta alcanzar el valor de la presión final. Fases estacionarias. En SFC se emplean tanto las columnas abiertas como las columnas rellenas, aunque las primeras son las más empleadas. Las columnas abiertas son similares a las columnas de sílice fundida con recubrimientos internos de varios tipos de siloxanos enlazados y de enlaces cruzados. Las columnas tienen una longitud de 10 a 20 m y un diámetro interno de 0,05 a 0.10 mm. El espesor de la película varía entre 0.05 1 micrómetro. Fases móviles. La fase móvil más utilizada en SFC es el CO2. Es un disolvente excelente par un conjunto de moléculas orgánicas no polares. Además, es una sustancia transparente en el ultravioleta, es colora, no tóxica, fácilmente disponible y muy barata cuando se compara con otras fases móviles cromatográficas. La temperatura crítica del CO2 ES 31° C y su presión crítica 72,9 atmósferas, lo cual permite jugar con una banda amplia de temperaturas y presiones sin superar las condiciones de operación de un equipo de HPLC moderno. Sustancias como etano, butano, óxido nitroso, diclorodifluorometano, éter dietílico, amoníaco y tetrahidrofurano han sido utilizadas como fases móviles de cromatografía de fluidos supercríticos. Detectores. La principal ventaja de la SFC frente al HPLC es que se pueden utilizar como en la cromatografía de gases, detectores de ionización de llama. Este detector es de respuesta universal a compuestos orgánicos, de elevada sensibilidad. Los espectrómetros de masas también se pueden Capítulo 1 9 Introducción a los métodos de separación adaptar como detectores más fácilmente para SFC que para HPLC. Otros detectores utilizados son de absorción en el ultravioleta y en el infrarrojo, de emisión de fluorescencia, termoiónico y fotométrico de llama. La cromatografía de fluidos supercríticos se ha aplicado a la separación de un amplio conjunto de sustancias, entre los que se cuentan productos naturales, fármacos, alimentos, pesticidas y herbicidas, tensoactivos, polímeros, aditivos de polímeros, combustibles fósiles y explosivos y propelentes. 1.8 Cromatografía de afinidad La cromatografía de afinidad separa las proteínas (analitos) en función de su especificidad de fijación de ligandos. Los ligandos de afinidad son moléculas poliméricas que sirven de soporte porque están unidas covalentemente sobre la columna cromatográfica o matriz inerte que debe de tener una estructura de poro abierta y estabilidad en condiciones de cambio de pH, detergentes y agentes disociantes, para así, retener y adsorber específicamente a las proteínas, la fase estacionaria es sólida. Estos ligandos se clasifican según su naturaleza química o su selectividad para la retención de analitos, esta última se clasifica en ligandos específicos (anticuerpos) y generales (como las lecitinas). Cuando las proteínas no específicas se han lavado a través de la columna, se eluye la proteína ligada con solución que contiene ligando libre. Después se introduce una nueva fase móvil que se desactiva, generalmente por el acoplamiento o alteración de los sitios activos inhibidor-ligando, para que esto pueda ser posible, se necesita un cambio en el pH, la fuerza iónica o la polaridad, debido a que estas condiciones modifican las características de los sitios activos, la finalidad de este proceso es hacer fluir a la proteína para continuar con la regeneración de la columna cromatográfica. La cromatografía de afinidad tiene la ventaja de ser altamente selectiva para la retención de proteínas afines a la columna, para ello, emplea sistemas de baja presión, columnas cortas y un campo restringido para la separación. Esquema de cromatografía de afinidad: En el primer vaso, se encuentra una mezcla de proteínas que se añaden a la columna, la cual contiene un ligando específico ligado al polímero, para la proteína de interés. En el segundo matraz se encuentra una solución de ligando, el cual se añade a una probeta para que eluya a la proteína de interés. La bureta de en medio indica que las proteínas deseadas se lavan a través de la columna. Los puntos rojos representan la proteína de interés, los negros, el ligando y las bolas beige unidas a los puntos negros, representan el ligando acoplado con el polímero. Capítulo 1 10 Introducción a los métodos de separación 1.9 Cromatografía de líquidos de alta resolución La cromatografía de líquidos de alta eficacia es la técnica analítica de separación más ampliamente utilizada. Las razones son la sensibilidad, su fácil adaptación a las determinaciones cuantitativas exactas, es ideal para la separación de especies no volátiles o termolábiles y por su gran aplicabilidad a sustancias que son de interés en la industria. Algunos ejemplos son: aminoácidos, proteínas, ácidos nucleicos, hidrocarburos, carbohidratos, fármacos, terpenoides, plaguicidas, antibióticos, esteroides, especies organometálicas y gran variedad de sustancias inorgánicas. La fase móvil es un líquido y la fase estacionaria es una columna que puede ser de acero inoxidable. 1.10 Electroforesis Los orígenes de ésta técnica se remontan a los trabajos de Tiselius en 1973, quien logró separar mezclas de proteínas (micelas cargadas eléctricamente) por su distinta movilidad en un soporte poroso al que se aplicaba un campo eléctrico. Posteriormente se ha aplicado a la separación de cualquier especie cargada o alrededor de una doble capa eléctrica. El fundamento de la técnica consiste en depositar sobre un medio poroso una mezcla de especies cargadas. Por aplicación de un campo eléctrico entre los extremos del soporte poroso las especies se separan en función de sus cargas y su movilidad iónica en ese medio. Cuanto más elevado sean el voltaje y la intensidad en menos tiempo se produce la separación; sin embargo valores altos de estas variables provocan un aumento de temperatura, con la consiguiente evaporación del disolvente y acumulación de sales del tampón, lo cual es indeseable. Para favorecer el paso de corriente se utilizan disoluciones fondo conductoras (electrolitos) que son disoluciones reguladoras en las que se empapa previamente el soporte poroso, debidamente escogidas para que no formen precipitados con las especies del problema. Como soportes se han utilizado alúmina, lana de vidrio, almidón, agar-agar, gel de sílice, etc. Capítulo 1 11 Introducción a los métodos de separación Electroforesis capilar La sección transversal del aparato de electroforesis se puede reducir se forma drástica al realizar la electroforesis en el interior de un tubo capilar de diámetro interno menor a 0.1mm y una longitud de 50cm a 1m. Es efectivo llevar de ésta forma la técnica porque la resistencia eléctrica de la disolución es tan alta que la corriente se mantiene baja, en consecuencia se pueden mantener voltajes más altos sin calentamiento excesivo. La técnica tiene gran poder de resolución y se han desarrollado métodos para llevar a cabo la electroforesis incluso en moléculas neutras no iónicas. Además, las zonas de analitos separados por electroforesis capilar se pueden detectar de igual modo que en una cromatografía en columna. El inconveniente de utilizar un capilar es que el volumen de muestra debe ser de alrededor de un nanolitro o menos, y después de la separación, cada uno de los analitos está en volúmenes inferiores al nanolitro. Con volúmenes de líquido tan pequeños, muy pocos métodos de detección se pueden usar eficazmente. Fig. 1. Diagrama de las partes básicas de un instrumento de electroforesis capilar con detección espectroscópica. La muestra penetra por la parte de la izquierda introduciendo el final del capilar en la disolución de la muestra y cargándolo, bien hidrodinámicamente, bien electrocinéticamente. Toda la unidad de la ilustración funciona a temperatura constante. El capilar está enrollado alrededor de un soporte. Durante el análisis los niveles de las dos disoluciones tampón deben ser iguales como se indica con la línea punteada) para evitar el flujo de masa debido a una diferencia de presión. 1.11 Bibliografía چ Braithwaite, A. Métodos cromatográficos, 4ª ed., ED. Chapman & Hall, Londres, 1985. Páginas 216217, 271-272. چ Burriel Marti, Felipe Lucena Conde, Siro Arribas Jimeno, Jesús Hernández Méndez, Química Analítica Cualitativa, Ed. Thompson, España, 2003 pp 321-322. چ Garrido, A., Fundamentos de química biológica. Interamericana Mc Graw-Hill. España. 1991. چ Kenneth A. Rubinson, Judith F. Rubinson, Análisis Instrumental, Ed. Prentice Hall, España 2004, pp 730-739. چ Lehninger, Albert. L. Principios de Bioquímica. Segunda Edición. Ediciones Omega. Barcelona, 1995, pp 137 Capítulo 1 12 Introducción a los métodos de separación چ McCABE / SMITH /HARRIOTT. “Operaciones Unitarias en Ingeniería Química” 1991. Cuarta Edición en Español. Editorial McGraw Hill S.A. España چ PERRY, ROBERT H. “Manual del Ingeniero Químico” 1992. Sexta Edición. Tercera Edición en Español. Editorial McGraw Hill. México چ Skoog A. Douglas. Principios de análisis instrumental, 5ª ed., Saunders Collage Publishing, Espana, 2001, págs.: 785- 791 y 831- 836. چ Willard Hobart, Métodos instrumentales de análisis. 7ª. Ed., Compañía de publicaciones Wadsworth, California, 1988, Páginas 644-650. چ http://www.javeriana.edu.co/Facultades/Ciencias/neurobioquimica/libros/celular/cromatografia.ht m چ http://www.profeonline.com/laboratorioquimico/mod_12/docs/cromatografia_de_afinidad.pdf چ http://www.textoscientificos.com/quimica/cromatografia/capa-fina چ http://es.wikipedia.org/wiki/Cromatograf%C3%ADa_en_papel http://www.monografias.com/trabajos13/sepal/sepal.shtml Capítulo 1 13 Parámetros cromatográficos Capítulo 2 Parámetros cromatográficos 2.1 Glosario Anchura de base: suele ser el intervalo de longitud de frecuencia de un pico; el intervalo pasa por un separador de banda. Banda: Es una situación ideal, es una distribución gausiana. La cantidad de compuesto que sale de cromatografico o de una columna electroforética. Coeficiente de difusión: Medida de la movilidad de una especie en unidades de cm2/s. Coeficiente de reparto: constante de equilibrio que describe la distribución de un soluto entre dos fases, para definir el coeficiente de partición solo se utiliza una forma de un soluto (kD). Coeficiente de selectividad: medida de la sensibilidad de un método para un interferente dado en comparación con su sensibilidad para el analito (KA,I). Cola: prolongación final de un pico cromatográfico, generalmente debida a la presencia de lugares muy activos en la fase estacionaria. Constante de disociación: constante de equilibrio de una reacción en la que un complejo metal- ligando se disocia para formar un ión metálico libre y un ligando (kD). Constante de equilibrio: K’ Constante que se basa en las concentraciones molares al equilibrio, su valor numérico de K’ depende de la fuerza iónica del medio. Cromatografía: separación en la que los solutos se distribuyen entre fase móvil y estacionaria. Cromatografía gaseosa: técnica cromatográfico en la que la fase móvil es un gas. Cromatograma: registro de la señal de detección en función del tiempo de elusión o del volumen. Eficiencia de la columna: medida del grado de ensanchamiento de una banda cromatográfico, se suele expresar en términos de la altura H, de los platos o del número N de platos teóricos. En la medida en que la distribución del analito dentro de la banda es gausiana, la altura de los platos está dada por la varianza dividida entre la longitud de la columna del empacado. Capítulo 2 14 Parámetros cromatográficos Ensanchamiento de banda: aumento de la anchura de base de un soluto a medida que se desplaza desde el punto de inyección al detector. Factor de capacidad: medida de la fortaleza con la que la fase estacionaria retiene en soluto dado (k’). Factor de selectividad: cociente de los factores de capacidad de dos solutos que muestra la selectividad de la columna para uno de ellos (α). Factor de separación: medida de la eficacia de una separación en lo que se refiere a la separación entre el analito y el interferente. Fase estacionaria: fase extractante que permanece en posición fija. Fase móvil: fase extractarte que se desplaza a través del sistema. Número de platos teóricos: característica de una columna cromatográfica que se emplea para medir su eficiencia. Plato teórico: medio cuantitativo para evaluar la eficacia de una columna y que consiste en tratar una columna, como si estuviera compuesta de pequeñas zonas o platos, en las que tiene lugar el reparto entre las fases móvil y estacionaria Relación de distribución: cociente que expresa la concentración total de soluto en una fase en relación con una segunda fase; en su definición participan todas las zonas del soluto (D). Resolución: separación entre dos bandas cromatográficas (R). Selectividad: Medida de la ausencia de interferencia de un método que se mide ante el cociente de selectividad del mismo. Tiempo de retención: es el tiempo entre la inyección en una columna cromatográfica y la llegada a un pico de analito al detector. Tiempo muerto: tiempo necesario para que una especie no retenida pase a través de la columna. Capítulo 2 15 Parámetros cromatográficos Un parámetro es una cantidad que puede tener distintos valores y que caracteriza un proceso, una operación o un resultado. La parametrización de datos en cromatografía, como en otros métodos facilita la tabulación y la comunicación de dichos datos. Se puede parametrizar la forma, la posición y la resolución de las bandas de una cromatografía. Estos pueden correlacionarse satisfactoriamente con descripciones de los procesos moleculares que tienen lugar durante la separación. 2.2 Constantes de Distribución Al encontrarse un soluto disperso en una fase que entra en contacto con otra, ocurre un fenómeno de distribución, en el cual dicho soluto puede tener mayor afinidad a alguna de las dos fases. De esta manera, se genera un equilibrio entre la cantidad de soluto que existe en una fase y en otra. La constante asociada es la llamada constante de distribución, que se define para un soluto A como: 𝐷𝐴 = 𝐴 𝐴 1 2 donde [A]1 y [A]2 son la concentración de A en la fase 1 y 2 respectivamente. Concretamente, en cromatografía la fase 1 es la fase estacionaria y la fase 2 es la fase móvil y el soluto A es el analito de interés. Esta constante es específica para cada compuesto con cada determinada fase estacionaria y fase móvil. 2.3 Factores de influencia en la retención La retención que ocurre del soluto en la fase estacionaria, se debe generalmente a la diferencia de polaridades entre las fases estacionaria y la móvil, promoviendo que el soluto se reparta entre ellas según su coeficiente de distribución. De esta manera, al separarse una mezcla que contenga solutos con polaridad distinta, y por lo mismo coeficientes de distribución distintos, la retención será diferente para cada uno. En la cromatografía de gases, el to, es evaluado convenientemente a partir del tiempo de retención del “pico de aire”. En la cromatografía de líquidos, cualquier compuesto no retenido puede usarse como muestra para medir el to. El tiempo muerto de la columna puede usarse para determinar el llamado tiempo de retención ajustado o corregido, t’R. t'R = tR – to Capítulo 2 16 Parámetros cromatográficos Una medida conveniente y útil de la retención del soluto está dada por el factor de capacidad (o factor de retención), k’ 𝑘´ = 𝑡´𝑟 𝑡0 FIG. 1. Cromatograma que muestra diferentes picos con tiempos de retención diferentes, así como el tiempo muerto. Los valores de k’ no tienen un significado simple en un gradiente de elución o en temperatura programada, y usualmente no son reportados cuando esos procedimientos son usados. Los valores de k’ pueden ser determinados a partir del tiempo de retención, tR. En los modos de retención de cromatografía se tiene: k’ > 0, que significa que no hay bandas eluidas antes del tiempo muerto, to. A menudo es útil expresar el tiempo de retención o el volumen en términos del factor de capacidad, k’. tR = to (1+ k’) VR = Vm (1 + k’) La retención del soluto en la fase estacionaria depende de la afinidad, y de los factores que puedan afectar a la misma. El fenómeno de adsorción depende fuertemente de la temperatura, a mayor sea ésta se fomentará la desorción, y debe por tanto controlarse dependiendo de la naturaleza del análisis. 2.4 Retención y Equilibrio en Cromatografía El soluto se encuentra en un equilibrio de distribución entre la fase móvil y la estacionaria, y dependiendo del desplazamiento de dicho equilibrio es la retención del mismo. Una separación cromatográfica típica se muestra en la figura 2. La separación está fuertemente afectada por la proximidad de bandas adyacentes en el cromatograma. Capítulo 2 17 Parámetros cromatográficos Una importante medida de la proximidad de bandas es el llamado factor de separación, α, donde dos bandas adyacentes, teniendo los valores k1 y k2: α = k2 / k1 El factor de separación, α, es usualmente identificado con la selectividad del sistema cromatográfico, es decir, la habilidad del sistema a prever diferentes tiempos de retención para dos compuestos específicos. Dicho factor debe tener un valor mayor a uno, pues esto significaría que no hay separación, pero es recomendable que sea menor a dos puesto que tiempos de retención muy largos se traducen en tiempo desperdiciado durante la operación experimental. Los valores de α depende de los dos solutos y de 1) La composición química de la fase estacionaria. 2) La composición química de la fase móvil (excepto en cromatografía de gases). 3) La temperatura. En cromatografía de fluidos supercríticos, la presión también puede afectar a α al cambiar la densidad de la fase móvil. FIG. 2. Separación de una muestra de un compuesto nitrado con 10 componentes por cromatografía de líquidos . 2.5 Eficiencia de separación de una columna Las columnas cromatográficas consisten en un número de zonas adyacentes en cada una de las cuales hay suficiente espacio para que un analito esté en equilibrio entre dos fases. Cada una de estas zonas se conoce con el nombre de plato teórico (de los que hay N en cada columna). La longitud de una columna contiene una altura del plato, H, que tiene unidades de longitud, normalmente en micras. El Capítulo 2 18 Parámetros cromatográficos valor numérico de N y H para cada columna en particular es expresado en referencia a cada analito. La altura del plato está relacionada con la anchura del pico del analito y la distancia que recorre dentro de la columna, X: 𝐻= σ2 X donde σ es un estándar de desviación de la banda gaussiana. Para los picos gaussianos simétricos la anchura de la base es igual a 4σ y la anchura del pico al punto de inflexión, ωi es igual a 2σ. Por lo tanto el valor de H puede ser calculado desde el cromatograma midiendo la anchura del pico. El número de platos teóricos en la columna entera viene dado por: 𝑁= 𝐿 𝐿∗𝑋 = 2 𝐻 σ donde L es la longitud de la columna. Si consideramos la posición del pico a X = L con el hecho de que la anchura del pico en su base es ω, obtenida de las tangentes dibujadas desde los dos puntos con más pendiente del pico, es igual a 4σ, la ecuación anterior se convierte en 𝑁= 16 ∗ 𝐿2 W2 Si en lugar de medir L y ω en unidades de longitud se hace en tiempo, la ecuación quedará: 𝑁 = 16 𝑡𝑅 𝑊 2 La medición del ancho del pico es más confiable cuando se realiza a la mitad de la altura del pico, con lo cual se puede utilizar 𝑁 = 5.545 𝑡𝑅 𝑊1/2 2 También puede hacerse en función de la altura y área del pico 𝑁 = 2𝜋 𝑡𝑅 á𝑟𝑒𝑎 2 𝑎𝑙𝑡𝑢𝑟𝑎 Es importante que tanto 𝑡𝑅 , W ó W1/2 ó á𝑟𝑒𝑎 𝑎𝑙𝑡𝑢𝑟𝑎 se expresen en las mismas unidades ya que N es adimensional. Capítulo 2 19 Parámetros cromatográficos Para que un sistema sea eficiente, se necesitan fases poco viscosas, espesores de película delgados y columnas de poco diámetro, para que se favorezca la transferencia de masa. La velocidad promedio de la fase móvil también afecta la altura del pico, por lo que hay una velocidad óptima para obtener el máximo de eficiencia. Generalmente se trabaja a flujos mayores de éste para no perder demasiado tiempo en análisis. 2.6 Procesos de ensanchamiento de banda El número de platos de una columna (eficiencia, N) es una medida del ensanchamiento de la banda cromatográfica. Cuando inyectamos un pequeño volumen de analito en una columna forma una banda estrecha en su inicio, pero a medida que el analito migra la banda se va ensanchando. La anchura de pico aumenta con la raíz cuadrada de la longitud que la banda ha recorrido. Según la ecuación de Van Deemter 𝐻= 𝐴+𝐵 𝑣+𝐶∗𝑣 A representa el término de caminos múltiples. Esto es porque las moléculas no siguen únicamente un camino, hay multicanales, que se deben a la densidad de empacado de la fase estacionaria y afecta según el tamaño y forma de la partícula. B representa la difusión axial, la cual depende de la movilidad de las moléculas en las fases. El soluto se difunde desde la zona central que es la más concentrada hacia las regiones más diluidas. En esto influye la velocidad v , pues a velocidades muy altas no se alcanza a difundir y predomina el término A. Para encontrar la velocidad óptima puede trazarse una curva de Van Deemter al graficar H en función de v. Por último, C representa la resistencia a la transferencia de masa. Es necesario éste término porque en realidad los fenómenos que ocurren están fuera del equilibrio. Esto es porque las corrientes de la fase móvil como la capa de fase estacionaria tienen una anchura determinada. Por lo que las moléculas de analito necesitan primero migrar desde el centro de ellas hasta la interfase donde ocurre la transferencia, y este retraso temporal se traduce en ensanchamiento de bandas. 2.7 Resolución La separación relativa de dos bandas a menudo se refiere a su resolución, Rs. La resolución se define como Capítulo 2 20 Parámetros cromatográficos 𝑅𝑠 = 𝑡2 − 𝑡1 1 (𝑤 + 𝑤2 ) 2 1 Aquí, t1 y t2 se refieren a los tiempos de retención (tR) de la primera y la segunda banda, y W1 y W2 son el ancho de dichas bandas. La figura 3 ilustra la separación de dos bandas adyacentes como una función de sus valores de Rs y su tamaño relativo. Se observa que la separación mejora sistemáticamente a mayores valores de Rs, y la separación es generalmente mejor para dos bandas de igual tamaño (y el mismo valor de Rs.). Esto es, mayores valores de Rs se requerirán normalmente para la separación de las bandas desiguales. Fig 3. Separación de dos bandas como una función de la resolución (Rs) y el tamaño relativo de banda. La resolución depende de la retención y selectividad de manera proporcional. A mayor selectividad y retención k´, mayor resolución. Para tener una buena separación se recomienda utilizar k´ > 2, y generalmente se trabaja con α ≈1. Para el análisis cuantitativo es deseable (pero no siempre práctico) alcanzar la resolución de al menos Rs = 1.5, desde que esto corresponde a la separación a la línea base de bandas de tamaño similar. La separación de la línea base hace que los sistemas de datos midan el tamaño de cada banda apropiadamente y esto significa una cuantificación fiable. Una resolución pobre puede deberse a que el método utilizado no es apropiado, pues no discrimina entre los solutos, o que hay mucha muestra en proporción a la columna cromatográfica. Capítulo 2 21 Parámetros cromatográficos 2.8 Cuantificación en cromatografía Para llevar a cabo un análisis cuantitativo, es necesario que los datos obtenidos sean confiables, tanto para la construcción de la curva de calibración como para el tratamiento del analito mismo. Primero es necesario llevar a cabo un análisis cualitativo, en el cual se encuentren las condiciones óptimas. Para evaluar esto, se calculan los parámetros cromatográficos de cada ensayo que sirven como referencia. De ésta manera se eliminan fuentes de error, pues se trabaja en las mejores condiciones posibles, acercándose a resultados reproducibles y por tanto precisos, y lo más exactos que ésta técnica nos permite. Si no se obtiene una buena resolución, primero debe repetirse el ensayo con menor cantidad de muestre, sino puede probarse utilizando una fase móvil distinta, y si esto no ayuda, puede utilizarse otra fase estacionaria. En caso de que la selectividad no sea buena, debe revisarse la temperatura de trabajo, y analizar la naturaleza química de las distintas fases así como solutos, para probar con otra fase sea estacionaria o móvil. No debe perderse de vista, que a menor tiempo de operación mejor, y que una vez encontradas las mejores condiciones, no deben alterarse. Para realizar la curva de calibración puede usarse el método de estándar externo o interno, siendo el segundo apropiado para eliminar posibles errores de operación. 2.9 Resumen La cromatografía es una técnica de separación que se basa en la diferencia de distribución que existe entre dos componentes de una mezcla en las fases estacionaria y móvil. Así, cada soluto tendrá un tiempo de retención distinto y podrán analizarse por separado. Los parámetros cromatográficos son claves para el diseño de un análisis, pues son herramientas que ayudan a evaluar las condiciones en las que se está llevando a cabo. Cada soluto tendrá asociado un tiempo de retención distinto que se puede expresar como el factor de capacidad, y la relación de los tiempos de retención o factores de capacidad de los solutos nos indicará si la selectividad del sistema es buena. Al calcular la resolución se podrá evaluar si la separación entre los tiempos de retención se los solutos es suficiente o excesiva. El cálculo de los platos teóricos nos indicará la eficiencia del sistema. Una vez evaluados los parámetros cromatográficos, se podrá determinar si el cromatograma obtenido es aceptable, o si es necesario modificar alguna condición para optimizarlo. Capítulo 2 22 Parámetros cromatográficos 2.10 Bibliografía Heftmann, E. Chromatography: Fundamentals and Applications of chromatography and related differential migration methods, Elsevier Science Publishers B.V,5a. edición, EUA, 1992, pp. 2-14 Rojo Callejas F., Manual de Química Analítica Instrumental II, Anexo III, Facultad de Química UNAM, México 2002 http://www.textoscientificos.com/quimica/cromatografia http://www.uam.es/personal_pdi/ciencias/manchi/alim/Trabajo5.pdf http://mazinger.sisib.uchile.cl/repositorio/ap/ciencias_quimicas_y_farmaceuticas/apquim-an-instr14/skoog/26d.html Capítulo 2 23 Cromatografía en fase líquida a columna abierta Capítulo 3 Cromatografía en fase líquida a columna abierta 3.1 Procedimientos de empaquetamiento En cromatografía de fase líquida, la fase móvil es un líquido y éste desciende a través de la columna. La fase estacionaria es frecuentemente un líquido que recubre el interior de un tubo capilar o la superficie de partículas sólidas empaquetadas dentro de la columna. Alternativamente, las mismas partículas sólidas pueden ser la fase estacionaria (como se muestra en la figura 1, en cualquier caso, el reparto de solutos entre las fases móvil y estacionaria dan lugar a la separación. Fig 1. Representación esquemática de una separación cromatográfica. El soluto A, que tiene una mayor afinidad que el soluto B para la fase estacionaria, permanece en la columna más tiempo. Las columnas pueden ser empaquetadas o de tubo abierto. Una columna empaquetada se llena con partículas que contienen la fase estacionaria y los materiales más usados para los tubos de las columnas son de acero inoxidable y de vidrio, siendo el primero preferido por la manipulación más fácil. Para el caso de la columna de tubo abierto, se trata de un capilar hueco estrecho con la fase estacionaria cubriendo las paredes interiores. En las columnas empaquetadas, el soporte debe ser un sólido poroso con gran área superficial, inerte y con una buena resistencia mecánica. Los compuestos empleados para empaquetamiento varían, entre los cuales podemos encontrar: tierra de diatomeas (más empleado en CG), alúminas, resinas de intercambio iónico o compuestos de sílice como SiO2 amorfo, sin embargo, éste debe de someterse a un tratamiento para hacerlo aún más inerte y que pueda ser empleado sin problemas. Es característico que Capítulo 3 Página 24 Cromatografía en fase líquida a columna abierta el tamaño de las partículas y de los poros deben ser lo más uniforme posible. El procedimiento del empaquetado es muy sencillo y se resume en los dos siguientes pasos: 1. Colocación de la fase estacionaria y/o compuestos de empaquetamiento de manera uniforme y compacta en la columna (longitud y diámetro apropiados). 2. Verificación de ausencia de espacios vacíos en la columna (para evitar que los picos del cromatograma sean más anchos y de menor eficiencia. 2.2 Aplicación de la muestra Una vez que la columna se encuentra correctamente empaquetada, se procede a la aplicación de la disolución de la muestra. Cuando se introduce en la columna cierta cantidad de muestra, ésta se queda en una zona de cierta altura en la columna. Si entonces se introduce el disolvente, comienza el desplazamiento de la zona a lo largo de la columna. La parte superior de la zona se disuelve poco a poco y, empujado por la disolución que se forma, el frente de la zona avanza hacia la base de la columna. Si el disolvente utilizado para la elusión es el mismo que la disolución problema, los platos teóricos comprendidos en la parte central de la zona, reciben una fase móvil en equilibrio con ellos y las concentraciones permanecen estacionarias. El movimiento de la zona sólo es aparente en sus extremos por lo que su estudio se puede hacer sobre una zona que se supone no tiene más espesor de un plato teórico. 2.3 Procedimientos de elución La diferente capacidad de los distintos disolventes para eluir un determinado soluto es prácticamente independiente de la naturaleza del soluto. Se puede describir la elución como el desplazamiento de un soluto de la fase estacionaria por un disolvente. Para el caso específico de cromatografía de adsorción, existe una ordenación de los disolventes de acuerdo a su capacidad relativa para desplazar solutos de un determinado adsorbente, llamada serie eluotrópica. La fuerza eluyente es una medida de energía de adsorción del disolvente, supuesto valor cero para el sistema pentano/sílice pura. Cuanto más polar es el disolvente, mayor es su fuerza eluyente respecto a la sílice en cromatografía de adsorción. Cuanto mayor es la fuerza eluyente del disolvente, tanto más rápidamente se eluirán los solutos de la columna. Capítulo 3 Página 25 Cromatografía en fase líquida a columna abierta La cromatografía de adsorción sobre sílice pura es un ejemplo de cromatografía de fase normal, que se caracteriza por usar una fase estacionaria polar y un eluyente menos polar. Un disolvente más polar tiene fuerza eluyente mayor. La cromatografía de fase inversa, que es la más utilizada, se caracteriza porque la fase estacionaria es no polar o débilmente polar y el disolvente es más polar. Un disolvente menos polar tiene mayor fuerza eluyente. La cromatografía de fase inversa elimina las colas de los picos porque la fase estacionaria tiene pocos puntos que adsorban con fuerza a ningún soluto originando colas. La cromatografía de fase inversa también es menos sensible a impurezas polares (como el agua), que pueda haber en el eluyente. 2.4 Elución isocrática y gradiente La elución isocrática se lleva a cabo con un único disolvente (o una mezcla de disolventes fija). Si un disolvente no permite una elución suficientemente rápida de todos los componentes, se puede usar una elución gradiente. En este caso, se van añadiendo cantidades crecientes del disolvente B al disolvente A, produciendo un gradiente continúo. Fig 1. Las moléculas del disolvente y las moléculas del soluto compiten entre sí en su reacción con los puntos activos de la fase estacionaria. Cuanto mayor es la fuerza del eluyente, más fácilmente se desplaza el soluto. 2.5 Cromatografía de absorción y de partición de columna Estas son dos técnicas de análisis cromatográfico, la diferencia entre ellas es que la retención del analito en la columna depende de la naturaleza de su interacción con la fase estacionaria. En el dibujo de abajo, observamos las diferentes formas en que interacciona el soluto, nuestro analito, con la fase estacionaria, en la de absorción, vemos que de pega a la superficie, mientras que en la de partición de columna, el analito se disuelve dentro de la fase estacionaria. Capítulo 3 Página 26 Cromatografía en fase líquida a columna abierta 2.6 Cromatografía de adsorción La absorción es un fenómeno de superficie, por lo tanto físico, en el cual diversas sustancias son atrapadas en la superficie de un material, es decir, se acumulan en una determinada superficie interfacial, formándose una película del material retenido en la superficie del cuerpo adsorbente, sea sólido o líquido, aunque este proceso implica una interacción de diferentes fuerzas, sean estas físicas o químicas, por ello se habla de fisisorción, cuando la naturaleza de las fuerzas que retienen al material es solo física, y quimisorción, cuando la retención implica enlaces con el adsorbente. Así pues, en la cromatografía de adsorción, se aprovecha esta propiedad del analito que queremos separar, dado que si es en una mezcla, cada componente de la mezcla se adsorbe con distinta fuerza, lo cual origina que el analito se retenga en la fase estacionaria, es decir, se adsorba, y los demás componentes se eliminen. Durante la separación, se filtra una solución a través de una columna de adsorbente, que es la fase móvil, esta es adsorbida en la superficie de la fase estacionaria sólida, formando una “cama” de partículas que se dividen de modo que el área de absorción sea máxima, o sea, se distribuyen a lo largo del sólido para atrapar la mayor cantidad posible del analito, estos se llaman sitios de absorción. El equilibrio entre la fase estacionaria y móvil permite la separación del analito. Si vemos la ilustración de abajo, los puntos negros representan el analito, y la mancha gris con protuberancias es la fase sólida, los huecos en ella son los sitios de absorción, donde el analito interacciona con la fase estacionaria y se retiene. Como fase estacionaria se han usado muchos materiales, aunque generalmente encontramos la Hidroxiapatita (Ca3(PO4)2.Ca(OH)2), alumina (Al2O3) y carbonato de magnesio. Capítulo 3 Página 27 Cromatografía en fase líquida a columna abierta Aplicaciones. Esta es una de las técnicas cromatográficas mas antiguas, su uso depende del tipo de compuesto que estemos separando, o sea de su naturaleza química, en particular se usa para la separación de compuestos isoméricos, especies no polares, hidrocarburos alifáticos, y para compuestos con grupos funcionales capaces de formar puentes de hidrógeno fuertes, por ello es utilizada para separar azúcares, especialmente azucares aminados y glicoproteinas así como oligosacáridos, y también se ha usado para obtener proteínas biliares. 2.7 Cromatografía de partición de columna Esta técnica se basa en la retención del analito en una columna sólida como granos de fibra o cualquier otro material inerte químicamente, sobre la cual hay distribuida una capa fina de un disolvente afín al analito; y una fase móvil liquida o gaseosa; cuando la fase móvil pasa a través de la columna, el equilibrio entre esta y la estacionaria esta mediado por el analito, que se reparte entre ambas y queda al final disuelto en la fase estacionaria. En el dibujo de abajo, vemos las flechas, que son el flujo de la fase móvil y que llevan el analito, pasan por la fase estacionaria, los círculos, alrededor de los cuales hay una capa donde se quedan retenidos los analitos, esta capa es de un material que interacciona con el analito, donde este se disuelve. Las moléculas del analito se equilibran entre la fase estacionaria y la móvil, a esto se le llama partición puesto que se distribuyen, o sea se reparten dependiendo de la fisicoquímica del sistema, entre las dos fases. La retención depende de la solubilidad del analito con la capa de sorbente adherida a la matriz sólida, y de que tanto la fase móvil pueda arrastrarlo al fluir, en el dibujo de abajo, vemos como el analito, los puntos negros, se quedan disueltos en la superficie de la fase estacionaria, dentro de la capa Capítulo 3 Página 28 Cromatografía en fase líquida a columna abierta de solvente adherida a la matriz sólida, y como vemos también, el analito se distribuye entre la capa de solvente de la fase estacionaria y otra entre la fase estacionaria, a esto se le llama reparto, y como esta sobre una columna, por ello se le llaman partición de columna. La solubilidad en ambas fases esta dada por el coeficiente de partición, KD: 𝐾𝐷 = 𝑎𝑛𝑎𝑙𝑖𝑡𝑜 𝑒𝑛 𝑙𝑎 𝑓𝑎𝑠𝑒 𝑚ó𝑣𝑖𝑙 𝑎𝑛𝑎𝑙𝑖𝑡𝑜 𝑒𝑛 𝑙𝑎 𝑓𝑎𝑠𝑒 𝑒𝑠𝑡𝑎𝑐𝑖𝑜𝑛𝑎𝑟𝑖𝑎 Eso se debe a que el analito tiene diferentes afinidades para las dos diferentes fases, o sea tiene diferentes preferencias por estar en una u otra fase, de esta forma, la separación del analito de la mezcla se logra por migración diferencial de este, y dada la afinidad que tiene el analito pro el disolvente de la columna, el reparto en esta será mayor que en la fase móvil. Aplicaciones. Esta técnica se usa para separa sustancias en la misma fase, o líquidos inmiscibles. 2.8 Cromatografía en gel. La separación basada en el tamizado molecular se puede llevar a cabo en sustancias sin carga durante una migración osmótica a través de geles. La filtración en gel es el método de separación que consiste en la separación de moléculas basado en el tamaño relativo de las mismas. En la cromatografía en gel, la fase estacionaria es una matriz porosa polimérica cuyos poros se llenan con el solvente que se usará en la fase móvil. El flujo de la fase móvil provocará que moléculas mayores pasen a través de la columna libremente, sin penetrar la matriz del gel, mientras que partículas más pequeñas tardarán más tiempo en salir dependiendo de la penetración que tengan sobre el gel. Los componentes de una mezcla emergen de la columna de acuerdo a masa molecular relativa. Capítulo 3 Página 29 Cromatografía en fase líquida a columna abierta El volumen total de la columna, Vt es la suma del volumen del líquido afuera de la matriz de gel (V0, volumen de la fase móvil que eludirá a una molécula completamente excluida ), el volumen del líquido dentro de la matriz (Vi) y el volumen de la matriz de gel (Vm). El volumen de elusión (Ve) se puede relacionar con V0 y Vi: Ve V0 K d Vi Kd es el coeficiente de distribución volumétrica: K d (Ve V 0) / Vi Kd caracteriza el comportamiento de retención del soluto. Representa la fracción del volumen del gel que es accesible a la molécula en cuestión. Si Kd se grafica contra el logaritmo del peso molecular se una serie de solutos similares en forma y densidad molecular, se obtiene: En un rango pequeño de Kd, el rango de fraccionamiento, la curva se aproxima a una línea recta: K d A B log M La separación por cromatografía en gel está influenciada por las propiedades de los poros de la red tridimensional, y la naturaleza del material que forma el poro. Se distinguen dos tipos de material para el gel: xerogeles y aerogeles. Los xerogeles son geles simples; consisten en polímeros entrecruzados que se engrosan en contacto con el solvente para formar un medio porosos relativamente suave. Los aerogeles son sólidos porosos que son penetrados por el disolvente; algunos ejemplos es el vidrio y silica porosos. El nombre de los geles más comúnmente utilizados: Sephadex, Strygel, Biogel Capítulo 3 Página 30 Cromatografía en fase líquida a columna abierta Aplicaciones. Desalinización: Largas moléculas de origen biológico son separadas de especies ionizables o inorgánicas. Un ejemplo en la separación de la hemoglobina y NaCl en un gel Sephadex. La cromatografía en gel se utiliza para separar proteínas, péptidos, ácidos nucléicos, polisacáridos, enzimas, hormonas y los químicos en polímeros, para determinar distribuciones de pesos moleculares en mezclas de polímeros. 2.9 Cromatografía de intercambio iónico. El intercambio iónico es un proceso donde una solución de un electrolito se pone en contacto con una resina de intercambio iónico y los iones activos de la resina son remplazados por las especies iónicas de carga similar del analito. Ocurre un intercambio de iones de signo igual entre una solución y un sólido esencialmente insoluble en contacto con la solución. Las resinas cambiadoras de iones están constituidas por una red tridimensional de cadenas polímeras entrelazadas con cadenas cortas que tienen grupos funcionales ionizables. Se tiene una fase insoluble con sitios iónicos fijos de una sola carga, muestra que las especies de carga contraria se mueven libremente a su alrededor se pueden sustituir por otros iones de la misma carga, a condición de que se mantenga la electroneutralidad. Una resina típica se prepara por polimerización de estireno y divinilbenceno. Cambiadores catiónicos. Los grupos funcionales ácidos se pueden introducir fácilmente, tienen el protón disociado, pero no libres para abandonar la resina, excepto cuando se sustituyen por otros iones positivos. Cambiadores aniónicos. Si se introducen grupos funcionales básicos, la resina intercambia aniones. Los cambiadores aniónicos se preparan con aminas, obteniéndose in grupo amonio, y por tanto, un medio básico. Las propiedades más importantes que determinan el funcionamiento de una resina: o Tamaño de las partículas – velocidad de intercambio y permeabilidad de la columna. o Naturaleza de los grupos funcionales – tipo de los iones intercambiables. o Fuerza de los grupos funcionales – coeficiente de distribución. o Número de grupos funcionales – capacidad de la resina. Capítulo 3 Página 31 Cromatografía en fase líquida a columna abierta A partir de la sustitución de cantidades equimolares de iones de cargas iguales, se obtienen equilibrios de intercambio. Se aplica la ley de acción de masa, donde K(coeficiente de selectividad), es la constante de equilibrio del sistema. Y donde el coeficiente de distribución KD: KD K [ HR] [H ] Efecto del pH del eluyente. La carga eléctrica de una especie se puede aumentar, disminuir o invertir por cambio del pH. Disponemos de un medio para influir sobre la relación de distribución o evitar un intercambio total. El efecto del pH sobre la elusión: Efecto de agentes complejantes. Los aniones complejan muchos iones metálicos dando iones complejos de carga negativa. En esta forma, los cationes metálicos que se separen mal en cambiadores catiónicos, se pueden complejar y separar en cambiadores aniónicos. Aplicaciones. Eliminación de iones. Un agua completamente desionizada se obtiene pasándola a través de un cambiador de cationes, y después a través de un cambiador de aniones. Separación de aminoácidos. La naturaleza anfótera de este grupo permite cambiar el signo de su carga o eliminar la carga neta, de modo que un ácido determinado se puede intercambiar en una resina aniónica, en una resina catiónica o en ninguna de las dos, mediante control de pH de la solución. 2.10 Cromatografía covalente Es un sistema especial de cromatografía, el cual es altamente selectiva la fase estacionaria, en el que se forma un enlace covalente reversible entre la fase estacionaria y la biomolécula a separar, las separaciones se basan en el acoplamiento “llave-cerradura” típico de la biología molecular; por lo cual es muy utilizado en esta área, además de tener un pequeño inconveniente de que al ser tan selectiva, solo se puede separar un solo analito. Capítulo 3 Página 32 Cromatografía en fase líquida a columna abierta 2.11 Conformación de la cromatografía covalente a) Ligandos: Se trata de biomoléculas que se encuentran en una base sólida, siendo estas las responsables de la adsorción específica de los solutos-analitos. Los ligandos se pueden clasificar de 2 maneras: Ligandos específicos, como los anticuerpos, que se enlazan reversiblemente a un solo soluto, y los ligandos generales enlazados con un determinado grupo de compuestos bioquímicos, como las lectinas y nucleótidos. b) Soportes: Es la superficie en donde se establece el enlace covalente con el ligando de afinidad Debe poseer propiedades como tener una gran superficie, porosidad controlable, carácter suficientemente hidrofílico para evitar adsorciones no específicas de proteínas u otras moléculas, estabilidad mecánica, en especial para trabajar a alta presión, estos soportes generalmente geles orgánicos derivados de los polisacáridos como la agarosa (sepharosa), polímeros de acrilamida, fractogel TSK y sílices CPG. 2.12 Fundamento de la cromatografía de afinidad Un volumen no excesivamente grande de muestra de naturaleza biológica se introduce en la columna que contienen un soporte polimérico inerte que retienen a la sustancia activa enlazado covalentemente y que se denomina ligando de afinidad. Sólo existe interacción específica entre este ligando y un soluto-analito (proteína) de la muestra insertada que queda retenido. Se procede a la elusión de los demás componentes de la muestra mediante una primera fase móvil que no influye en el acoplamiento. A continuación se introduce una nueva fase móvil que desactiva el acoplamiento por alteración reversible de los sitios activos del inhibidor-ligando, del soluto (proteína) o de ambos; generalmente se utiliza un cambio de pH que modifica las características de los sitios activos de esta manera se eluye el soluto de interés. Una vez finalizada la separación, se procede a la regeneración de la columna, lo que generalmente se hace mediante el empleo de la primera fase móvil siendo una etapa rápida. Capítulo 3 Página 33 Cromatografía en fase líquida a columna abierta 2.13 Cromatografía Flash Es un método cromatográfico, el cual nos permite separar de una manera rápida, fiable y económica; principalmente utilizada para la separación y purificación de proteínas. La Cromatografía Flash consiste en una bomba y una columna que permita resistir alta presión para la separación puede llevarse a cabo con relativa rapidez. El sistema incluye dos bombas de flujo continuo en la depuración de las columnas (variables por el usuario), una bomba peristáltica puede utilizarse para deposito en la columna, seguido de la radiación ultravioleta para detectar las biomoléculas en la columna, un mezclador para producir un gradiente de elusión, las válvulas de motor puede ser utilizados para ayudar en la inyección de la muestra, la selección de la columna o corriente de inversión, y un controlador con una función de integración computadorizada, en el cual se muestran los resultados de el análisis. Aplicaciones. En farmacia la cromatografía líquida rápida de proteínas (FPLC) se utiliza para la depuración de grandes cantidades de diferentes biomoléculas (es decir, las proteínas y el ADN). 2.14 Resumen Las columnas pueden ser empaquetadas o de tubo abierto. Una columna empaquetada se llena con partículas que contienen la fase estacionaria y la columna de tubo abierto, es un capilar hueco estrecho con la fase estacionaria cubriendo las paredes interiores. El procedimiento del empaquetado consta de dos sencillos pasos: a) colocación de fase estacionaria y/o compuestos de empaquetamiento, y b) Verificación de compactación (no espacios vacíos). El desplazamiento de la muestra a través de la columna, depende de la afinidad por ésta y del disolvente empleado para el proceso. Capítulo 3 Página 34 Cromatografía en fase líquida a columna abierta La elución es el desplazamiento de un soluto de la fase estacionaria por un disolvente. Existen dos tipos de elusiones: Elusión por gradiente (cambio continuo de la composición del eluyente en sentido de aumento de fuerza eluyente) y elusión isocrática (un solo disolvente). Cromatografía de adsorción: esta técnica aprovecha el fenómeno de la absorción de sustancias en una superficie para separar analitos de mezclas al hacerlos pasar por una columna con un material adsorbente, que retiene el analito cuando este pasa a través de su superficie. Cromatografía de partición de columna: sta técnica permite separar analitos al hacerlos pasar por una columna sobre la cual hay una capa fina de un disolvente químicamente similar al analito, en el que este queda disuelto y retenido cuando pasa la fase móvil, se separa cuando se da un reparto entre las fases, en el que se espera que la mayor parte se quede en el disolvente de la columna. Cromatografía en gel: consiste en la filtración en gel que consiste en la separación de moléculas basado en el tamaño relativo de las mismas. El flujo de la fase móvil del sistema provocará que moléculas mayores pasen a través de la columna libremente, sin penetrar la matriz del gel, mientras que partículas más pequeñas tardarán más tiempo en salir dependiendo de la penetración que tengan sobre el mismo. Los componentes de una mezcla emergen de la columna de acuerdo a masa molecular relativa. Cromatografía de intercambio iónico: el intercambio iónico es un proceso donde una solución de un electrolito se pone en contacto con una resina de intercambio iónico y los iones activos de la resina son remplazados por las especies iónicas de carga similar del analito. Se tiene una fase insoluble con sitios iónicos fijos de una sola carga, muestra que las especies de carga contraria se mueven libremente a su alrededor se pueden sustituir por otros iones de la misma carga. Cromatografía covalente: las separaciones se basan en el acoplamiento “llave-cerradura” típico de la biología molecular; por lo cual es muy utilizado en esta área, además de tener un pequeño inconveniente de que al ser tan selectiva, solo se puede separar un solo analito. Permite la separación de anticuerpos y enzimas. Cromatografía Flash: consiste en una bomba y una columna que permita resistir alta presión para la separación puede llevarse a cabo con relativa rapidez. Es una técnica muy utilizada en el área de bioquímica ya que nos permite separar biomoléculas y proteínas. Capítulo 3 Página 35 Cromatografía en fase líquida a columna abierta 2.15 Bibliografía BERMEJO, F. - Química Analítica General, Cuantitativa e Instrumental - Vol 1. - Ed. Paraninfo 7ma edición, Madrid 1991. HARRIS, D. - Análisis Químico Cuantitativo - Ed. Reverté - 2ª edición - España, 2001. SKOOG, WEST, HOLLER, CROUCH - Química Analítica - 7ª ed - McGraw Hill – México, 2001. ROUESSAC - Métodos y técnicas instrumentales modernas. Análisis Químico - McGraw Hill – USA, 2003. SHARON, N. - Complex Carbohydrates: their chemistry, Biosíntesis and Functions - Addison-Wesley - New York, 1975. PECSOK, Robert – Métodos Modernos de Análisis Químico – Editorial Limusa – México, 1981. BRAITHWAITE, SMITH – Chromatografic Methods – 4° edición - Chapman & Hall – UK, 1994. http://www.srigc.com/2003catalog/cat-86.htm http://holivo.pharmacy.uiowa.edu/separation/chromatography.pdf http://www.che.utexas.edu/cache/newsletters/spring2001_chemmicro.pdf http://www.resonancepub.com/chromtutorial.htm www.javeriana.edu.co/Facultades/Ciencias/ www.um.es/bbmbi/AyudasDocentes www.um.es/bbmbi/AyudasDocentes www.gmi-inc.com/Products/PharmaciaFPLC.htm Capítulo 3 Página 36 Cromatografía de gases Capítulo 4 Cromatografía de gases Esta técnica permite el análisis rápido y exacto de gases, vapores líquidos; permite identificar los componentes individuales de las mezclas gaseosas. Iniciada a finales de 1952, se ha desarrollado notablemente en separación, identificación y determinación de compuestos volátiles (gases y líquidos) de puntos de ebullición de hasta 350-400°C. El método tiene las ventajas de ser sensible, rápido y sencillo, y si se maneja con suficiente cuidado, suministra información cuantitativa exacta con cantidades muy pequeñas de muestra. En cromatografía de gases (GC) la muestra se volatiliza y se inyecta en la cabeza de una columna cromatográfica. La elución se produce por el flujo de un fase móvil de un gas inerte a diferencia de los otros tipo de cromatografía, la fase móvil no interaccionan con la moléculas del analito; su única función es la de transportar el analito a través de la columna.Existen dos tipos de cromatografía de gases:la cromatografía Gas-Sólido (GCS) y la cromatografía gas liquido (GLC).La cromatografía gas liquido tiene gran aplicación en todos los campos de la ciencia y su denominación se abrevia normalmente como cromatografía de gases (GC). En la cromatografía gas sólido se produce la retención de los analitos de una fase estacionaria como consecuencia de la absorción física. La cromatografía gas sólido ha tenido una aplicación limitada debido a la retención semipermanente de las moléculas activas o polares y a la obtención de picos de elusión con colas muy significativas (como consecuencia del carácter no lineal del proceso de absorción), de modo que esta técnica no ha encontrado una gran aplicación excepto para la separación de ciertas especies gaseosas de bajo peso molecular. La cromatografía gas líquido se basa en la distribución del analito entre una fase móvil gaseosa y una fase líquido inmovilizada sobre la superficie de un sólido inerte. 4.1 Instrumentación Los componentes básicos de un instrumento para la cromatografía de gases se muestran a continuación, el caudal de gas se divide antes de entrar en la columna, este tipo de disposición se utiliza cuando el detector empleado mide un cambio en las propiedades de la corriente de gas por la presencia de las moléculas de analito. Capítulo 4 37 Cromatografía de gases Gas portador. Entre los gases portadores deben ser químicamente inertes, se encuentran el helio, nitrógeno y el hidrogeno, con el suministro de gas se encuentran asociados los reguladores de presión, manómetros y medidores de caudal. Además el sistema de gas portador contiene a menudo un tamiz molecular para eliminar el agua u otras impurezas. Los caudales se controlan normalmente mediante un regulador de presión de dos niveles colocando en el cilindro de gas, y algún tipo de regulador de presión o regulador de flujo instalado en el cromatógrafo. Sistema de inyección de muestra. La eficacia de la columna requiere que la muestra sea de un tamaño adecuado y que sea introducida como un <tapón> de vapor, la inyección lenta de la muestras demasiado grandes provoca un ensanchamiento de la bandas y una pobre resolución .El método mas común de inyección de muestra implica el uso de una micro jeringa para inyectar una muestra liquida o gaseosa a través de un diafragma o <septum> de goma de silicona en una cámara de vaporización instantánea situada en la cabeza de la columna (la cámara demuestra normalmente esta unos 50°C por encima del punto de ebullición del componente menos volátil de la muestra. Configuración de la columna y del horno para la columna. En cromatografía de gases se usan dos tipos generales de columnas, las rellenas, y las abiertas o capilares. Hasta la fecha la mayor parte de las cromatografía de gases se ha realizado con columnas rellenas. Sin embargo, en la actualidad esta situación está cambiando rápidamente y parece probable que en un futuro próximo, Capítulo 4 38 Cromatografía de gases excepto para ciertas aplicaciones especiales, las columnas de relleno sean sustituidas por columnas abiertas más eficaces y rápidas. Las columnas cromatográficas varían desde menos de 2 hasta 50m de longitud o más. Están construidas con acero inoxidable, vidrio, sílice fundida o teflón. La temperatura de la columna es una variable importante para que un trabajo preciso ha de regularse a las décimas de grado, por ello la columna normalmente se introduce dentro de un horno de temperatura controlada , la temperatura optima de la columna depende del punto de ebullición de la muestra y del grado de separación requerida. Sistemas de detección. El detector ideal para cromatografía de gases tiene las siguientes características. 1.- adecuada sensibilidad 2.- buena estabilidad y reproducibilidad 3.-respuesta lineal para los solutos que se extienda a varios órdenes de magnitud 4.-intervalo de temperaturas de trabajo comprendido desde la temperatura ambiente hasta al menos 400°C 5.-tiempos de respuesta cortos que sean independientes del caudal 6.-alta fiabilidad y manejo sencillo 7.-respuesta semejante para todos los soluto s o por el contrario una respuesta selectiva y altamente predecible para uno o más tipos de soluto 8.-no destructivo de la muestra. Resolución (Rs). Existe una magnitud que expresa el poder de resolución de la columna y que se denomina resolución de la columna y se define: 𝑅𝑠 = 2 𝑡𝑟2 − 𝑡𝑟1 𝑊𝑏1 + 𝑊𝑏2 Si se toma W como el ancho medio de los picos, la ecuación queda: 𝑅𝑠 = 2 𝑡𝑟2 − 𝑡𝑟1 𝑊 Para los picos que están próximos entre sí Wb1 y Wb2 serán lo suficientemente parecidos como para que baste medir sólo uno. Capítulo 4 39 Cromatografía de gases Como lo muestra la figura, la resolución 1.5 nos da una separación prácticamente completa de los solutos, mientras que una resolución de 0.75 no lo hace. A una resolución de 1, la zona X contiene casi 4% de Y y viceversa, a una resolución de 1.5 la superposición es de aproximadamente 0.3%. Para el mismo tipo de empaque, la resolución puede mejorarse alargando la columna y por lo tanto aumentando el número de platos teóricos. Tipos de Columnas. Existen dos tipos de columnas, principalmente, de relleno y capilares o semicapilares. Contiene la fase estacionaria y en ella tiene lugar la separación cromatográfica Las columnas de relleno suelen ser de cobre, acero inoxidable, aluminio, vidrio y teflón; con un diámetro interior entre 2 y 4 mm y una longitud entre 2 y 3 m. La eficacia está en torno a 1.000-2.000 (platos teóricos/m). Suelen emplearse para muestras poco complejas, máximo 10 componentes. La columna está rellena de un material sólido (soporte), finamente dividido y homogéneo; recubierto, por una capa de 0,05-1 μm de espesor, de fase estacionaria líquida. Está configurada en forma helicoidal, con un diámetro de unos 15 cm, para poder ser instalada en el horno termostatizado Fase estacionaria. Elección de la fase estacionaria de una columna cromatográfica ha de reunir una serie de requisitos como: o Baja volatilidad, su punto de ebullición debe de ser por lo menos 100 C superior a la temperatura máxima de operación de la columna. Estabilidad térmica. Capítulo 4 40 Cromatografía de gases Inercia química. Los valores del factor de capacidad (k´) y del factor de selectividad (α) de los analitos deben estar dentro de los intervalos aconsejados. La separación en cromatografía gas-líquido se debe a los diferentes coeficientes de reparto del analito entre la fase móvil y la fase estacionaria y tiene que haber un cierto grado de solubilidad de los compuestos con la fase estacionaria. Por ello, una característica muy importante de la fase estacionaria es la polaridad. Siguiendo el principio de "semejantes disuelven a semejantes" los solutos se retienen más en las fases líquidas de polaridad parecida, lo que permiten obtener mejores separaciones. Las fases estacionarias compuestas de hidrocarburos y de dialquilsiloxanos son poco polares, las compuestas por poliéster son muy polares. En cuanto a posibles compuestos objeto de separación, los alcoholes, ácidos y aminas son polares; los ésteres cetonas y aldehidos tienen polaridas intermedia; y son de baja polaridad los hidrocarburos saturados. Otro factor a tener en cuenta son los límites de temperatura que puede soportar la fase estacionaria, el límite inferior será el punto de fusión o temperatura a la que la viscosidad de la fase líquida es muy elevada, lo que haría disminuir la eficacia. El límite superior es la temperatura a la que la o presión de vapor de esta fase sea 0,1 mm Hg (250 C inferior al punto de ebullición) por encima de esta temperatura se produce arrastre de la fase líquida, lo que lleva consigo efectos en el detector (ruido, suciedad), interferencias con los solutos y en suma, deterioro de la columna. En una fase líquida cualquiera, una serie homóloga se eluye según orden creciente de número de átomos de carbono. Si la fase es no polar, los solutos no polares eluyen según orden creciente de punto de ebullición. En una fase polar se retendrán más los solutos polares que los no polares a igualdad de puntos de ebullición. Otro aspecto a desarrollar, relacionado con la fase estacionaria, es el soporte sólido empleado en las columnas de relleno; el objetivo es el de proporcionar una superficie elevada donde depositar la fase líquida en forma de película muy fina para proporcionar una mayor superficie de contacto entre fase móvil y fase estacionaria. 2 Las características de los soportes sólidos son, una elevada superficie específica (1m /g); una superficie homogénea; estabilidad térmica; geometría adecuada; baja dispersión de tamaño de las partículas, entre 150-250 μm; dureza mecánica; inercia química y naturaleza porosa. Capítulo 4 41 Cromatografía de gases Los soporte, más generalizados, son los de silíceo como las tierras de diatomeas o sintéticos; de vidrio y de polifluorocarbonados (teflón). Los soportes de diatomeas están constituidos por residuos de algas unicelulares diatomáceas que se unen formando filamentos. Hay unas 10.000 especies de diatomeas con esqueletos diferentes, pero parecida estructura. Tienen 2 una superficie específica de 1 m /g. El constituyente mayoritario es de anhídrido silícico SiO (90%), 2 o tratado con un fundente alcalino a 1.600 C se obtiene el producto comercial conocido como Chromosorb W de superficie poco adsorbente por lo que es muy útil para separar compuestos polares. Otro soporte es el de polvo de ladrillo refractario conocido como Chromosorb P, C-22 y Sterchomel, tiene mayor resistencia mecánica y superficie específica que el anterior, pero es muy activo y no se puede utilizar con compuestos polares. Los soportes de silicatos en columnas de relleno y en las paredes interiores de sílice fundida, en las columnas capilares, provocan adsorciones no deseadas de analitos polares, lo que da como resultado picos cromatográficos distorsionados. Se ha demostrado que este fenómeno se debe a los grupos silanol (-SiOH) que se forman en la superficie de los silicatos debido a la humedad. El silanol tiene gran afinidas por las moléculas orgánicas polares. Este proceso puede desactivarse por sililación con dimetilclorosilano , Cl Si(CH ) , que elimina el 2 3 2 H del silanol formando ClH. Columnas capilares. Las columnas capilares son de dos tipos básicos: WCOT, de pared recubierta y SCOT, de soporte recubierto. Las WCOT son simplemente tubos capilares donde la pared interna se ha recubierto con una finísima capa de fase estacionaria. Las columnas SCOT tienen en su parte interna una fina capa de material adsorbente como el empleado en las columnas de relleno donde se ha adherido la fase estacionaria. Las ventajas de las SCOT frente a las WCOT es la mayor capacidad de carga de esta última, ya que en su fabricación se emplean mayores cantidades de fase estacionaria, al ser la superficie de intercambio mayor. Por orden de eficacia, en primer lugar están las WCOT, luego las SCOT y por último las columnas de relleno. Las columnas WCOT se fabrican a partir de sílice fundida, conocidas como columnas tubulares abiertas de sílice fundida o FSOT. Estas columnas se fabrican a partir de sílice especialmente pura, sin Capítulo 4 42 Cromatografía de gases apenas contenido de óxidos metálicos. Debido a la fragilidad inherente a este material, en el mismo proceso de obtención del tubo se recubre con una capa de poliamida, de esta forma la columna puede enrollarse con un diámetro de unos pocos centímetros, ya que la longitud de estas columnas es varia de 2 a 50 metros, por lo que se tiene la necesidad de enrollarlas en una forma helicoidal con diámetros de 10 a 30 cm, dependiendo del tamaño del horno.. Estas columnas, con propiedades como baja reactividad, resistencia física y flexibilidad, han sustituido a las WCOT clásicas. Las columnas FSOT tienen diámetros internos variables, entre 250 y 320 μm y 150-200 μm para columnas de alta resolución. Estas últimas requieren menor cantidad de analito y un detector más sensible, al eluir menor cantidad de gas. Existen asimismo columnas macrocapilares con diámetros de hasta 530 μm, que admiten cantidades de analito comparables a las de relleno pero con mejores prestaciones. En estas columnas existe un problema debido a la adsorción del analito sobre la superficie de la sílice fundida, adsorción debida a la presencia de grupos silanol, los cuales interaccionan fuertemente con moléculas polares orgánicas. Este inconveniente se suele solventar inactivando la superficie por sililación con DMCS. La adsorción debida a los óxidos metálicos se ve paliada en gran parte por la elevada pureza de la sílice empleada. 4.2 Cromatografía gas-sólido En la cromatografía gas-sólido se produce la retención de los analitos en una fase estacionaria sólida como consecuencia de la absorción física. La cromatografía gas-sólido ha tenido una aplicación limitada debido a la retención semipermanente de las moléculas activas o polares y a la obtención de picos de elusión con colas muy significativas (como consecuencia del carácter no lineal del proceso de adsorcion), de modo que esta técnica no ha encontrado una gran aplicación excepto para la separación de ciertas especies gaseosas de bajo peso molecular. La cromatografía gas-sólido se basa en la adsorcion de sustancias gaseosas sobre superficies sólidas. Los coeficientes de distribución son generalmente mucho mayores que en el caso de la cromatografía gas-liquido. En consecuencia la cromatografía gas-sólido es útil para la separación de especies que no se retienen en columnas de gas-liquido, tales como los componentes del aire, sulfuro de hidrogeno, disulfuro de carbono, óxidos de nitrógeno, monóxido de carbono, dióxido de carbono y gases nobles. Capítulo 4 43 Cromatografía de gases La cromatografía gas-sólido se lleva a cabo tanto en columnas de relleno como en columnas abiertas. En estas últimas, se fija en las paredes del capilar una delgada capa del adsorbente; estas columnas a veces se denominan columnas abiertas de pared porosa, o columnas PLOT. Se encuentras dos tipos de adsorbente: los tamices moleculares y los polímeros porosos. 4.3 Tamices moleculares. Los tamices moleculares son intercambiadores de iones de silicato de aluminio, cuyo tamaño de poro depende del tipo de catión presente. Los preparados comerciales de esos materiales están disponibles en tamaños de partícula de 40/60 mesh a 100/120 mesh. Los tamices se clasifican según el diámetro máximo de las moléculas que pueden penetrar en los poros. Los tamices moleculares comerciales ser encuentran con tamaños de poro de 4, 5, 10 y 13 amstrongs. Las moléculas más pequeñas que las dimensiones anteriores penetran en el interior de las partículas donde tiene lugar la adsorcion, para estas moléculas el área de la superficie disponible es enorme cuando se compara con el área disponible para las moléculas más grandes. Por ello, los tamices moleculares se pueden utilizar para separar las moléculas pequeñas de las grandes. Por ejemplo, un relleno de 5 amstrongs y 183 cm de largo, a temperatura ambiente, separar fácilmente una mezcla de helio, oxigeno, nitrógeno, metano y monóxido de carbono en ese orden. En la primer figura de la siguiente hoja se muestra un típico cromatograma obtenido con tamices moleculares. En esta aplicación se utilizan dos columnas de relleno: una es una columna de gas-liquido ordinaria y la otra es una columna de tamices moleculares. La primera retiene solamente el dióxido de carbono y deja pasar los restantes gases a una velocidad que corresponde a la velocidad del portador. Cuando el dióxido de carbono eluye de la primera columna, un conmutador desvía el flujo de la segunda columna para evitar la adsorcion permanente del dióxido de carbono en los tamices moleculares. Una vez que la señal de dióxido de carbono ha vuelto a cero el flujo se reconduce a traves de la segunda columna, y de este modo se produce la separación y elusión del resto de los componentes de la muestra. 4.4 Polímeros porosos: Las bolitas de los polímeros porosos de tamaño uniforme se fabrican a partir de estireno polimerizado con divinilbenceno. El tamaño de poro en estas bolitas es uniforme y se controla por el grado de polimerización. Los polímeros porosos han encontrado una gran aplicación en la separación de especies polares gaseosas tales como sulfuro de hidrogeno, óxidos de nitrógeno, agua, dióxido de carbono, metanol y cloruro de vinilo. Capítulo 4 44 Cromatografía de gases En la segunda figura de la siguiente hoja se muestra una aplicación característica de una columna abierta revestida con un polímero poroso (columna PLOT). 4.4 Aplicaciones. Se sabe que los ácidos grasos trans incrementan el colesterol LDL y disminuyen el colesterol HDL en sangre. Consecuentemente una ingesta excesiva de ácidos grasos trans aumenta el riesgo de enfermedad cardiovascular. La aumentada preocupación por los ácidos grasos trans requiere de métodos precisos y convenientes para el análisis de productos comerciales. Los métodos analíticos disponibles actualmente incluyen: cromatografía de gases, espectroscopia de infrarrojo, y cromatografía de gases de capa fina (AgNO3- TLC- GC, por sus siglas en inglés). Dentro de estos métodos la cromatografía de gases se ha usado preferencialmente dado su conveniencia y su sensibilidad. Bis cianopropil polisiloxano o bis cianopropilsiloxano polisilfenileno son las fases estacionarias comúnmente usadas en el análisis de ácidos grasos trans en correspondencia a la necesidad por una alta resolución entre los picos de los metil- esteres de los ácidos grasos (FAME). Existen varios métodos analíticos oficiales utilizando estas fases estacionarias tales como los métodos del AOAC (Asociación Oficial de Químicos Agricultores) y AOCS (Asociación Americana de Químicos y Aceites) (por sus siglas en inglés). A pesar de que el método de AOCS es muy preciso tiene varias desventajas, tales como: un tiempo de análisis mayor debido a la longitud de la columna (100 m), la dificultad en la identificación de los picos debido a su fase estacionaria de bis cianopropil polisiloxano y los grandes costos económicos debidos a el uso de un estándar caro. Por lo tanto debe buscarse un método más conveniente de GC, particularmente para el propósito de control de calidad. Capítulo 4 45 Cromatografía de gases Método 1 TM Método 2 1 TM Columna TC- 70 Fase estacionaria bis cianopropilsiloxano polisilfenileno bis cianopropil polisiloxano Longitud 30m 100m Diámetro interno 0.25mm 0.25mm Grosor de película 0.25μm 0.20μm Temperatura 190ºC 180ºC Inj/Det 250ºC/260ºC 250ºC/250ºC Gas acarreador 1 ml/min Helio 1 ml/min Helio Split 100:1 100:1 1μl 1μl Volumen inyectado 3 Tiempo de corrida (GL- Science) Método 3 60m 18min Tabla 1. Ejemplo del trabajo hecho por determinación de ácidos grasos trans 40 min SP-2560 (Supelco Inc.) 2 74min Shirasawa y colaboradores para encontrar un mejor método de 1) Prueba control: AOCS celh-05 2) 180ºC (60 min)- (10ºC/min)- 220ºC (10 min) 3) 1% del aceite en hexano. Por otro lado, derivados de las anfetaminas tales como: 3,4-metilendioxi(MDA), 3,4 metilendioxietilanfetamina (MDEA) y 3,4- metilendioximetilanfetamina (MDMA) constituyen un grupo de estimulantes del sistema nervioso que producen efectos cardiovasculares y del comportamiento bien identificados. El abuso de estas drogas es especialmente problemático entre la población joven y a pesar de que las intoxicaciones agudas han sido ampliamente documentadas el potencial de toxicidad por lapsos mayores de tiempo para el sistema nervioso es el tema de muchas controversias. MDMA se excreta en la orina generalmente sin cambio alguno junto con otros derivados de las anfetaminas como HHMA, HHA, HMMA y HMA. La cromatografía de gases acoplada a espectrometría de masas es la técnica instrumental más comúnmente empleada en el análisis de anfetaminas y sus derivados. La derivatización es requerida para mejorar la cromatografía, en cuanto a la sensibilidad y reproducibilidad de aminas primarias y secundarias. Capítulo 4 46 Cromatografía de gases Tabla 2. Ejemplo del trabajo de Pirnay y colaboradores en la cuantificación de derivados de anfetaminas en orina por GC-MS 4.5 Resumen La cromatografía de gases constituye un poderoso instrumento en la determinación de los componentes de una muestra, al permitir tanto la separación de éstos como su detección individual. Una gran ventaja de este método es la rapidez y su límite de detección, un requisito indispensable para el análisis es que la muestra debe ser volátil. Dependiendo de la fase estacionaria utilizada la cromatografía de gas se subdivide en: gaslíquido y gas sólido. En el caso de la cromatografía de líquidos, los parámetros dependen del poder del eluyente (la polaridad de este); para la cromatografía de gases, los parámetros dependen de la temperatura a la cual se esté trabajando y del flujo del gas de arrastre. Las partes esenciales de un equipo de cromatografía son: fuente de gas portador, sistema de regulación de caudales, bloque termostatado de inyección de las muestras, columna termostatada, detector termostatado, con amplificador de señal y registro gráfico y caudalímetro de precisión. 4.6 Bibliografía http://www.upct.es/~sait/_sit/html/recursos_masas_y_gases.htm http://instrumental.uprh.edu BERMEJO, Francisco. BERMEJO, Pilar. BERMEJO, Adela. Química Analítica generla, cuantitativa e instrumental. Vol. 2. 6ta. Edición. Ed. Paraninfo. Madrid 1991. SKOOG, Douglas A. y Leary, James J. (1994), Análisis Instrumental,Madrid: McGraw-Hill. Pirnay et al. Sensitive Gas Chromatography – Mass Spectrometry Method for Simultaneous Measurement of MDEA, MDMA, and metabolites HMA, MDA, and HMMA in human urine. Drug Monitoring and Toxicology. Clinical Chemistry 52:9. 1728-1734. (2006). Shirasawa et al. A Rapid Method for Trans- Fatty Acid Determination Using a Single Capillary GC .Journal of Oleo Science. J. Oleo Sci. 56 (2) 53- 58. (2007) Rubinson, Kenneth. Analisis Instrumental. Editorial Pearson Education, Madrid 2001. pps 680700. Capítulo 4 47 Cromatografía de líquidos de alta resolución Capítulo 5 Cromatografía líquida de alta resolución (HPLC) La cromatografía es un método físico de separación, basado en la distribución de los componentes de una mezcla entre dos fases inmiscibles, una estacionaria y otra móvil. En cromatografía líquida, la fase móvil es un líquido que fluye a través de una columna que contiene a la fase estacionaria. La cromatografía líquida se lleva a cabo en una columna de vidrio. Después se coloca la muestra por la parte superior y se hace fluir la fase móvil a través de la columna por efecto de la gravedad. Para aumentar la eficiencia en las separaciones, en la cromatografía de alta resolución; el tamaño de las partículas de fase fija se disminuye hasta los micrones, usando altas presiones para lograr que la fase móvil pueda fluir. 5.1 Tipos de cromatografía en HPLC y aplicaciones La Cromatografía de líquidos, es una técnica de análisis químico ampliamente utilizada, la cual permite separar físicamente y cuantitativamente los distintos componentes de una solución por la absorción selectiva de los constituyentes de una mezcla, consta de dos fases, una fija que suele llamarse fase estacionaria, y una móvil (fase móvil) que fluye permanente durante el análisis, y que en este caso es un líquido. La tabla 1 nos muestra los tipos de cromatografía líquida que existen así como su separación cromatográfica. Nombre Tipo de fase móvil Tipo de fase estacionaria Método de fijación de la fase estacionaria Partición Líquido Líquido Adsorbida en un sólido poroso sostenido en una columna tubular Adsorción Líquido Sólido Sostenida en una columna tubular Papel Líquido Líquido Sostenida en los poros de un papel grueso Capa delgada Líquido Líquido o sólido Sólido finamente dividido sostenido sobre una placa de vidrio: el líquido puede absorberse sobre las partículas Gel Líquido Líquido Sostenido en los intersticios de un polímero sólido Intercambio iónico Líquido Sólido Resina de intercambio iónico finamente dividida en una columna tubular Tabla1.- Clasificación de las separaciones cromatográficas Capítulo 5 48 Cromatografía de líquidos de alta resolución 5.2 Cromatografía de adsorción Es la técnica más usada para la separación de compuestos orgánicos neutros, es adecuada para compuestos no polares probablemente con masas moleculares inferiores a 5 000. Se basa en la afinidad de adsorción, su fase estacionaria es la superficie de un sólido finamente dividido. El soluto compite por los sitios sobre la superficie del sólido con el disolvente utilizado como eluyente; la retención es el resultado de la adsorción. Las únicas fases que se utilizan en este tipo de cromatografía son la sílice y la alúmina, siendo la primera la preferida. La elección de la fase móvil es fundamental para el éxito de una cromatografía líquido-sólido; variando el disolvente, si este presenta una alta fuerza de elución eluirá las especies absorbidas más rápidamente que uno que tenga un valor más bajo. La cromatografía de capa fina (TLC) es un ejemplo especial de cromatografía por adsorción en la cual la fase estacionaria es un plano, en la forma de un soporte sólido en un plato inerte. Aplicaciones: se utiliza para compuestos no polares con masas <5000, muestras solubles en disolventes no polares y es capaz de diferenciar entre compuestos isómeros. Es particularmente adecuada para el análisis de moléculas no ionizantes, insolubles en agua y relativamente simples, que frecuentemente son isómeros o compuestos muy relacionados. Se utiliza en la separación de la vitamina D3 y sus metabolitos las vitaminas A, D y E (y compuestos muy relacionados a estas vitaminas), muchas drogas de abuso (LSD, por ejemplo), antidepresivos tricíclicos, bloqueadores beta y los PTHaminoácidos. Los aceites naturales y los extractos de esencias se analizan fácilmente y lospigmentos menos polares de las plantas, tales como los carotenoides y las porfirinas, La tendencia a ser adsorbido disminuye en el siguiente orden: ácido>alcohol>carbonilo>éster>hidrocarburo. A continuación se muestra una cromatografía de absorción. Figura 1.- Cromatografía de Adsorción Capítulo 5 49 Cromatografía de líquidos de alta resolución 5.3 Cromatografía de partición Cromatografía líquido-líquido La separación de las sustancias se logra por migración diferencial de los solutos. La fase estacionaria de la cromatografía de partición es un líquido soportado en un sólido inerte, el más usado es el ácido silícico o gel de sílice. La fase móvil puede ser un disolvente puro o una mezcla de disolventes, la polaridad puede ser notablemente diferente al la del líquido estacionario. Esta consiste en aplicar una gota de la solución con la mezcla de substancias a separar en la parte inferior de una tira de papel o en una delgada capa de un material inerte, como silicagel o celulosa, el papel o material inerte (los denominaremos soportes) se colocan en un recipiente con un solvente, de manera que, este los vaya impregnando; el disolvente se elige de manera tal que uno de ellos se adsorba más al soporte que el otro; a medida que el disolvente avance a lo largo del soporte los líquidos suben espontáneamente por capilaridad, aquellos componentes de la muestra que sean más solubles en el líquido que queda adsorbido serán retenidos, y los que tengan mayor afinidad por el que no se adsorbe serán arrastrados por este. Aplicaciones: es un poderoso instrumento para la separación de sustancias estrechamente relacionadas. Ejemplos típicos son la resolución de los numerosos aminoácidos formados en la hidrólisis de una proteína, la separación y análisis de alcoholes alifáticos y la separación de derivados de azucares. Fig. 2 Separación de moléculas pequeñas por cromatografía de partición. 5.4 Cromatografía de intercambio iónico El disolvente por lo general es agua y las especies a separar son iones, es muy utilizada en la química inorgánica, también se usa para la separación de aminoácidos y otros ácidos y bases orgánicas. Está basada en la atracción entre iones del soluto y puntos cargados que existen en la fase estacionaria. El intercambio iónico es un proceso en el cual ocurre un intercambio de iones de signo igual entre una solución y un sólido esencialmente insoluble en contacto con la solución. La fase estacionaria es una resina de intercambio iónico que contiene grupos cargados, teniendo la propiedad de separar especies ionizadas (Cationes o Aniones); la Fase Móvil es generalmente una solución amortiguadora de pH. Capítulo 5 50 Cromatografía de líquidos de alta resolución Dentro de los intercambiadores encontramos: arcillas y las ceolitas, así como resinas sintéticas. Estas últimas pueden ser usadas para intercambio de cationes se usan: resinas ácidas fuertes las cuales contienen grupos de ácidos sulfónico y resinas ácidas débiles con grupos de ácido carboxílico. En el caso de intercambiadores aniónicos, contienen grupos funcionales básicos adheridos a la molécula de polímero generalmente son aminas. La Fase Estacionaria es una resina de intercambio iónico que contiene grupos cargados, teniendo la propiedad de separar especies ionizadas (Cationes o Aniones); la Fase Móvil es generalmente una solución amortiguador de pH. Aplicaciones: encuentra mucho uso en los analizadores para aminoácidos, se ha aplicado a una gran variedad de sistemas orgánicos y bioquímicos incluyendo fármacos y sus metabolitos, sueros, conservantes de alimentos, mezclas de vitaminas, azúcares y preparaciones farmacéuticas. Figura 3.-Cromatografía Iónica (a) Separación de aniones en una columna de intercambio aniónico. (b) separación de iones alcalinotérreos en una columna de intercambio catiónico . 5.5 Cromatografía en gel Es una técnica de caracterización de polímeros que proporciona la distribución completa de pesos moleculares de una muestra y sus distintos promedios. El fraccionamiento se basa, por lo menos en parte, en el tamaño y la forma molecular de las especies de la muestra, también se le conoce como cromatografía de permeación en gel, cromatografía de exclusión y cromatografía de tamizado molecular. Se efectúa en una columna por el método de elusión. La fase fija está formada por partículas poliméricas o de sílice que contienen una red uniforme de poros por los que pueden penetrar las moléculas de pequeño tamaño. Las moléculas de tamaño grande se excluyen totalmente y son eluidas en primer lugar, mientras que las de pequeño tamaño tienen acceso a todo el volúmen poroso y son las últimas que se eluyen, los factores que determinan la separación de las moléculas son el tamaño del poro, el tamaño de la partícula y el flujo de elución. Los diferentes tipos de partículas usadas deben ser estables, mecánica y químicamente, tener bajo contenido en grupos iónicos, uniformidad de poro y Capítulo 5 51 Cromatografía de líquidos de alta resolución tamaño. Los compuestos pueden ser derivados de dextranos (Sephadex), derivados de agarosa (Sepharosa), derivados de acrilamidas (Biogel P) y esferas de vidrio. Hay diferentes tamaños de partícula para un gel: a menor tamaño mayor resolución y menor gasto en la columna. Aplicaciones: Este técnica se emplea en la separación de proteínas de alimentos, determinación de glucosa y fructosa en zumos de fruta, así como para la separación de compuestos de alto peso molecular- proteínas y polímeros, ácidos grasos, azucares. Cambios de buffers, después de cromatografías de intercambio iónico, afinidad o de interacción hidrofóbica; desalado, proteínas, polisacáridos, polipéptidos etc. pueden ser desalados antes de ser concentrados o liofilizados; eliminación de fenol de las preparaciones de ácidos nucléicos; eliminación de compuestos de bajo peso molecular marcados. I125, FITC de las soluciones de marcaje de proteínas; para reacciones entre macromoléculas y reactivos de bajo peso molecular; eliminación de productos, cofactores, inhibidores, etc. de las enzimas; Purificación de macromoléculas; determinación del peso molecular de las proteínas. Figura 4.- Cromatograma Sephadex G-200 superfine. Peaks: 1. catalase; 2. aldolase; 3. Bovine serumalbumin; 4. ovoalbumin; 5 chymotrypsinogen A; 6 ribonuclease A. 5.6 Cromatografía plana La separación se produce sobre una capa de un sólido finamente dividido que se ha fijado sobre una superficie plana, es un método notablemente simple y de bajo costo. Técnica de separación en la que la fase estacionaria está en forma de plano o sobre un plano, éste puede ser un papel, que esté impregnado con una sustancia que actúe de fase estacionaria o una capa de partículas sólidas extendida sobre un soporte, tal como una placa de vidrio. A veces a la se la llama Cromatografía de Lecho Abierto. La cromatografía plana tiene como fase móvil un líquido y como fase estacionaria un líquido o un sólido dispuestos sobre una superficie plana; existen tres tipos, cromatografía en papel, donde una hoja o tira de papel de filtro sirve como fase estacionaria y medio de separación; cromatografía en capa fina, en la que la separación se produce sobre una capa de sólido finamente dividido que se ha fijado Capítulo 5 52 Cromatografía de líquidos de alta resolución sobre una superficie plana; y la electroforésis. La fase móvil se mueve a través de la fase estacionaria por capilaridad, a veces es ayudado por gravedad o por potencial eléctrico. Aplicaciones: se utiliza para separar e identificar los componentes de pequeñas muestras de substancias inorgánicas, orgánicas y bioquímicas, se usa en determinadas aplicaciones con una finalidad didáctica, también para la separación de muestras clínicas y bioquímicas así como en otras aplicaciones analíticas generales por su gran sencillez y bajo costo. 5.7 Cromatografía en papel Se utilizan papeles especiales de elevada pureza, Es una técnica muy sencilla, se utiliza una tira de papel de filtro (Whatman nº 1 por ejemplo) como soporte para la separación. El mecanismo que interviene en la separación fundamentalmente de reparto. La fase estacionaria esta constituida por el agua absorbida sobre las moléculas de celulosa del papel. El papel utilizado puede ser papel de filtro estándar o papel, La fase móvil se selecciona empíricamente, se emplean mezclas consistentes en compuestos orgánicos, agua y otras especies (ácidos, bases, agentes complejantes) que modifiquen la solubilidad de los compuestos de la muestra. 5.7 Cromatografía en capa fina Se realiza en placas de vidrio o plástico recubiertos con una capa delgada de partículas finamente divididas contenidas en la fase estacionaria. El mecanismo predominante es la adsorción. Los materiales más usados han sido: gel de sílice, alúmina, celita, poliamidas. La fase móvil el disolvente a utilizar debe reunir una serie de características: inerte frente a los componentes de la muestra y de la fase estacionaria, elevada pureza, adecuada viscosidad y tensión superficial, bajo punto de ebullición para facilitar el secado de las placas, económico y baja inflamabilidad y toxicidad. Figura 5.- Cromatograma en capa fina bidimensional (gel de sílice) de algunos aminoácidos. Disolvente A: tolueno/2- cloroetanol/piridina. Disolvente B: cloroformo/alcohol bencilico/ácido acético. Aminoácidos: (1) ácido aspártico, (2) ácido glutámico, (3) serina, (4) f3-alanina, (5) glicina, (6) alanina, (7) metionina, (8) valina, (9) isoleucina y (10) cisteína. Capítulo 5 53 Cromatografía de líquidos de alta resolución El HPLC se utiliza para: detección y cuantificación de sacarina benzoatos, cafeína y aspartame en refrescos; detección de ciclomato en jugos de fruta; separación de esteres de ácidos grasos por fase inversa y con argentación de columnas( Silicato de Al con Ag); separación de triglicéridos por la longitud de la cadena y el grado de instauración por fase inversa y detector de dispersión luminosa, para el estudio de aceites y grasas adulteradas; determinación del contenido de Vit A y sus precursores ( α y β caroteno) en margarina y aceites de hígado por fase inversa; determinación del contenido en Vit D en leche en polvo y cereales por fase normal. 5.8 Instrumentación. Un equipo para HPLC puede ser representado por la siguiente figura 6 Figura 6.-Esquema de los componentes de un HPLC La fase móvil puede ser un disolvente puro o una mezcla de disolventes; algo importantes es que deben ser grado HPLC, esto implica un 99% o mas de pureza para evitar contaminantes que puedan interferir en la elución de la muestra o bien que contengan algunas pequeñas partículas que puedan tapar la columna; por lo que es necesario filtrarlos antes de que entren a la columna. Dependiendo del equipo, cuando se trata de una mezcla de disolventes; se puede o no programar la bomba para que tome las cantidades adecuadas de cada disolvente o bien, algunas otras bombas (las más antiguas) no tienen la capacidad de realizar esta mezcla y por lo tanto esta se tiene que preparar por nosotros. Cuando durante toda la separación se usa el mismo disolvente, se denomina isocrática. La bomba envía el disolvente hacia la válvula inyectora que es una válvula de seis vías que permite introducir al disolvente, la muestra contenida en el loop de volumen calibrado. Luego de que se produzca la separación en la columna, los componentes de la mezcla pasan al detector. El cual da una señal eléctrica proporcional a la cantidad de materia; esta señal es enviada al registrador que a su vez da un cromatograma de intensidad en función del tiempo (figura 7); en el cual, lo ideal es obtener picos gaussianos los cuales corresponden cada uno a un componente diferente de la muestra. Capítulo 5 54 Cromatografía de líquidos de alta resolución Figura 7.- Cromatograma típico obtenido por un HPLC En HPLC existen dos tipos básicos de detectores: Los basados en una propiedad de la disolución. Los basados en una propiedad del soluto Algunos de los detectores más usados son: detectores de absorbancia, detectores de fluorescencia, detectores de Índice de refracción, detector de dispersión de luz, detectores electroquímicos, detectores por espectrometría de masas. El integrador calcula el área de cada pico, la cual se puede relacionar con la concentración del componente si se tiene una curva patrón; si no se cuenta con ella, sólo sería cualitativa. Debido a que los detectores que se usan en estos equipos no son destructivos, es posible recuperar los productos que salen de él, y de esta manera realizar otro tipo de separaciones (por ejemplo) analíticas (también depende del tamaño del loop, de la columna y del tipo de bomba). 5.9 Volumen y tiempo de retención El volumen de fase móvil (o tiempo para Tr) necesario para transportar la banda de soluto desde el punto de inyección a través de la columna, hasta el detector, en el punto máximo del pico del soluto se define como volumen de retención (Vr). El volumen muerto (V0) representa lo que se conoce como espacio muerto o volumen de retraso de la columna, incluye las contribuciones efectivas del volumen del inyector, tubería, conexión, columna y detector. Capítulo 5 55 Cromatografía de líquidos de alta resolución 5.10 Factor de capacidad (K) Actualmente, se conoce como factor de retención (k). El factor de retención es un parámetro experimental importante que se utiliza para describir las velocidades de migración de los solutos en columnas. Para el soluto A, el factor de retención, kA se define como: 𝐾𝐴 = 𝐾𝐴 ∗ 𝑉𝑆 𝑉𝑀 donde KA es la constante de distribución del soluto A, Vs es el volumen del soluto en la fase estacionaria y Vm el volumen del soluto en la fase móvil (Skoog y cols., 2001). 5.11 Selectividad El factor de selectividad α de una columna para dos solutos, A y B, se define como la relación de la constante de distribución del soluto retenido con más fuerza, B, y la constante de distribución del soluto retenido con menos fuerza, A: 𝛼= 𝐾𝐵 𝐾𝐴 donde KB es la constante de distribución de la especie retenida con más fuerza, especie B, y KA es la constante de la especie retenida con menos fuerza, es decir, la especie A, que eluye más rápido. De acuerdo con esta definición, α siempre es mayor que la unidad (Skoog y cols., 2001). 5.12 Eficiencia La eficiencia de una columna cromatográfica depende del ensanchamiento de banda que ocurre cuando un compuesto pasa a través de la columna. Para las mediciones cuantitativas de la eficiencia de las columnas cromatográficas se emplean dos términos: (1) altura del plato H y (2) cantidad de platos o número de platos teóricos N. Los dos están relacionados por la ecuación: 𝑁= 𝐿 𝐻 donde L es la longitud del empaque de la columna (en cm). La eficiencia de las columnas cromatográficas aumenta a medida que es mayor el número de platos N y la altura H es menor. Se observan grandes diferencias en la eficiencia de las columnas como resultado de las diferencias en el tipo de columna y de las fases móvil y estacionaria. En términos de número de platos teóricos, la eficiencia puede variar desde unos centímetros hasta varios cientos de miles; la altura de los platos varía desde unas décimas hasta milésimas de centímetro y son comunes incluso mas pequeñas (Skoog y cols., 2001). Capítulo 5 56 Cromatografía de líquidos de alta resolución 5.13 Resolución cromatográfica Es una medida cuantitativa de su capacidad para separar dos analitos A y B. la resolución de cada columna queda definida como: 𝑅𝑆 = 2Δ𝑍 2 𝑡𝑟𝐴 − 𝑡𝑟𝐵 = 𝑊𝐴 + 𝑊𝐵 𝑊𝐴 + 𝑊𝐵 Se puede mejorar la resolución para una fase estacionaria determinada alargando la columna, lo que incrementa el número de platos. Sin embargo, una consecuencia adversa de añadir platos es un incremento en el tiempo necesario para la separación de los componentes (Skoog y cols., 2001). 5.14 Número de platos teóricos Expresada como una cantidad adimensional, refleja el número de veces que el soluto se reparte entre las dos fases durante su paso a través de la columna. 𝑁= 𝐿 𝐻 5.15 Asimetría (AF) El factor de asimetría del pico (AF, de asymetry factor) se define como la razón de las mitades del ancho del pico a una altura dada. Conforme se mida más abajo la asimetría del pico AF es mayor, debido al ruido del detector, un compromiso aceptable es medir AF en 10% de la altura del pico. SF=A/B A B Representación esquemática del factor de asimetría. Skoog, 2001 Capítulo 5 57 Cromatografía de líquidos de alta resolución 5.16 Instrumentación Los componentes básicos de un sistema para HPLC son: A) Depósitos para la fase móvil (disolventes) B) Sistema de bombeo para proporcionar presión a la fase móvil C) Sistema de inyección de muestras D) Columna cromatográfica E) Termostatos para las columnas F) Detectores G) Sistema para el tratamiento de datos y registrador Componentes básicos de un sistema para HPLC. Hernández, 2002. Como algunas de las fases móviles usadas en HPLC pueden ser químicamente activas como ácidos, bases o líquidos corrosivos, es esencial que los componentes del sistema estén fabricados con materiales resistentes, por lo que la mayoría de las partes en contacto con la fase móvil suelen estar fabricadas con acero inoxidable (Hernández L. 2002). Los disolventes más usados en HPLC son agua, disoluciones tampón acuosas y disolventes orgánicos como el metanol. Deben ser espectroscópicamente puros, exentos de partículas sólidas y degasificados, esto se lleva a cabo con un gas inerte muy poco soluble como el helio. Como fase estacionaria lo más común es usar partículas microporosas esféricas de sílice muy puro, que son permeables al disolvente (Harris, D. 2001). Capítulo 5 58 Cromatografía de líquidos de alta resolución A) Los recipientes que se utilicen para almacenar la fase móvil tienen que ser inertes, es decir, el disolvente no deberá extraer especie alguna del material con el que estén construidos. Suelen ser botellas de vidrio y tubos de teflón. Están provistos de unos filtros, indispensables para eliminar los gases disueltos y partículas que pueda contener la fase móvil (Harris, . 2001). B) Debido a las elevadas presiones de trabajo y al pequeño tamaño de las partículas de la fase estacionaria, se utiliza una bomba que es la encargada de introducir la fase móvil o disolvente a través de la columna. Los sistemas de bombeo deberán reunir las siguientes características: (Hernández, L. 2002). Generar presiones superiores a 6000 psi. Capaces de cubrir un amplio rango de flujo entre 0,1 y 10 ml/min con una precisión del 0,5 % y que esté libre de pulsaciones. Construidos con materiales inertes respecto a los disolventes empleados. Las bombas empleadas en HPLC son de tres tipos: Bombas recíprocas o de vaivén, son las más utilizadas. Están formadas por una pequeña cámara cilíndrica que se llena y luego se vacía por oscilación de un pistón de zafiro. El bombeo produce un flujo pulsado que después debe amortiguarse. Sus principales ventajas son que se consiguen presiones elevadas y se suministra un caudal constante, pudiéndose adaptar a la técnica de elución con gradiente, debido a su pequeño volumen interno (Skoog, D. A. et al. 2001). Bombas neumáticas o de presión constante, hacen uso de la presión de un gas aplicado al recipiente conteniendo la fase móvil. Son sencillas, no provocan pulsaciones pero están limitadas a presiones relativamente bajas. (Hernández, L. 2002). Bombas de desplazamiento o tipo jeringa, consisten en una cámara equipada con un mecanismo de tornillo. Suministran un flujo libre de pulsaciones pero con una capacidad limitada a unos 250 ml. (Harris,D.C. 2001) C) Los volúmenes que se inyectan de muestra deberán ser pequeños para evitar la sobrecarga de la columna. Hay varios tipos: El método más simple es la utilización de una jeringa de alta presión con un diafragma (“septum”) a la entrada de la columna. Está limitado a una presión máxima de operación de 1500 psi. Capítulo 5 59 Cromatografía de líquidos de alta resolución Las válvulas de inyección con bucles de volumen conocido, es el método más utilizado (Harris, D. C. 2001). D) En las columnas cromatográficas es donde se produce la velocidad diferencial de los solutos que permite su separación (Harris, D. C. 2001). El material de las columnas cromatográficas suele ser de acero inoxidable cuya longitud varía de 5 a 30 cm y un diámetro de 1 a 5 mm. La eficacia de las columnas aumenta al disminuir el tamaño de las partículas de la fase estacionaria. El tamaño típico de las partículas es de 3-10 um (Harris, D. 2001). Columna para HPLC. Fuente: P.V.G. Lab. 3-D, Facultad de Química, 2007 Las columnas son caras y se degradan con facilidad, por eso, se protege la entrada de la columna con otra más corta, la precolumna, que retiene por adsorción las impurezas de forma irreversible (Harris, D. 2001). E) No es necesario el control estricto de la temperatura de la columna, pero las separaciones resultan más reproducibles cuando la temperatura se mantiene constante. Los instrumentos comerciales modernos están equipados con calentadores que regulan la temperatura de la columna. F) El papel del detector es indicar los momentos de aparición de los componentes, y proporcionar indicación cuantitativa y cualitativa de los mismos. El detector utilizado depende de la naturaleza de la muestra y deberá reunir una serie de características como son, tener una sensibilidad elevada, buena estabilidad y reproducibilidad. Amplio margen de respuesta lineal, insensible a cambios en la presión y la temperatura. Se pueden clasificar de la forma siguiente: (Hernández, L. 2002) El detector se coloca al final de las columnas, responde a la concentración del soluto y se registra en función del tiempo y obteniéndose una serie de picos, generándose un grafico que se denomina cromatograma. La posición de los picos en el eje del tiempo puede servir para identificar los componentes de la muestra. Capítulo 5 60 Cromatografía de líquidos de alta resolución Los detectores basados en una propiedad del soluto que no la suele presentar la fase móvil. Suelen ser muy selectivos y sensibles: Detectores de absorbancia ultravioleta, son los más utilizados. Su fundamento es la espectrofotometría de absorción de luz visible y ultravioleta de un componente a una determinada longitud de onda. Los más potentes son los que utilizan un montaje de fotodiodos para registrar el espectro completo de cada soluto que pasa por el detector. Los datos de absorbancia se representan en función de la longitud de onda y del tiempo. Detectores de fluorescencia, son muy sensibles y selectivos, el principio de operación se basa en la irradiación con la luz UV al componente de interés y la posterior medida de la luz fluorescente emitida por éste.( Harris, D. C. 2001) Detectores electroquímicos, ofrece ventajas debido a su especificidad, sensibilidad y amplia aplicabilidad, especialmente para compuestos orgánicos. Responde a analitos que puedan oxidarse o reducirse. Es el ejemplo de fenoles, aminas, peróxidos (pueden detectarse por oxidación) y cetonas, aldehídos (detectados por reducción). Las técnicas electroquímicas más utilizadas con esta finalidad son la amperometría, voltamperometría y culombimetría. Los detectores basados en una propiedad de la disolución, responden a un conjunto amplio de solutos, pero suelen ser poco sensibles: Detectores de índice de refracción, está formado por una celda con dos compartimentos, en uno se introduce el disolvente puro y en el otro la muestra, se hace pasar luz visible paralela. Cuando entra en la celda soluto de distinto índice de refracción al disolvente el has se desvía y varía la señal dada por la fotocélula (Harris, D. C. 2001). El principal inconveniente es que son muy sensibles a los cambios de temperatura y no resultan apropiados para trabajar con la modalidad de elusión con gradiente. Detectores de conductividad, son los más utilizados cuando los solutos eluidos son iónicos, como ácidos y bases, así como cationes y aniones inorgánicos después de su separación por cromatografía de cambio iónico. Tienen elevada sensibilidad, baratos y de larga duración. Características de los detectores: Sensibilidad. Medida de la efectividad de un detector para convertir la muestra en una señal eléctrica medible. Linealidad. Rango de masa ó concentración de muestra sobre el cual el detector mantiene una sensibilidad constante sin una desviación arbitraria. Capítulo 5 61 Cromatografía de líquidos de alta resolución El significado práctico de la linealidad del detector es el que le indica al analista la concentración para la cual el detector es confiable. Hay dos límites en la curva de linealidad: El límite de concentración inferior, que es dado por el límite de detección. El límite Superior, definido por un porcentaje de desviación arbitrario de la curva de linealidad. Rango Dinámico Lineal. Rango sobre el cual la sensibilidad del detector es constante. Ruido. Es cuantificado por el promedio de la amplitud pico-pico de la señal. El significado de conocer el nivel de ruido de un detector es un factor determinante en la determinación de la cantidad mínima detectable y el límite inferior del rango lineal. Límite de Detección. Está definido como la mínima cantidad de sustancia que puede producir una señal que sea el doble del nivel de ruido. Corriente de Fondo. Señal constante de salida generada por el proceso en el que un detector está operativo sin que alguna sustancia pase a través de él. Esta señal es muy importante, ya que permite diagnosticar el buen o mal funcionamiento del detector. La separación efectuada se conserva en un registro individual llamado cromatograma. Un cromatograma es una imagen que traduce visualmente en una pantalla o en un papel la evolución, en función del tiempo, de un parámetro que depende de la concentración instantánea del soluto a la salida de la columna. Este gráfico se obtiene gracias a un detector situado a la salida de la columna (Rouessac y Rouessac, 2003). Montaje cromatográfico y cromatograma (Skoog, A. D. Et. Al, 2001). Capítulo 5 62 Cromatografía de líquidos de alta resolución 5.17 Bibliografía BERMEJO, M. F. – “Química analítica general, cuantitativa e instrumental “. Vol. 2. Ed. Paraninfo, S. A. Madrid. 1991. HARRIS, D. C. – “Análisis químico cuantitativo”. Ed. Reverte, S. A. Barcelona. 2001. HERNÁNDEZ, L. y GONZÁLEZ, C. – “Introducción al análisis instrumental”. Ed. Arial Ciencia. 2002. KIRK, R. S., SAWYER, R., EGAN, H. – “Compuestos y análisis de alimentos de Pearson” Ed. Continental, S. A. México. 1996. LORO, J. F. – “Manual de cromatografía”. Colección Textos Universitarios. 2001 ROUESSAC, F. y ROUESSAC, A. – “Análisis químico: Métodos y técnicas instrumentales modernas”. Ed. Mc Graw Hill. 2003. SKOOG, D. A., WEST, D. M., HOLLER, F. J. – “Química analítica”. Ed. Mc Graw Hill. 7ª edición. 2001. SKOOG, A y LEARY – “Análisis instrumental”. Ed. Mc Graw Hill. 1998 Capítulo 5 63 Cromatografía de líquidos de alta resolución Cromatografía líquida de alta resolución II 5.18 Cromatografía en fase reversa En esta técnica una mezcla de agua/solvente orgánico es comúnmente usada como fase móvil, y un sólido de área de superficie altamente no polar es empleado como fase estacionaria. La última es usualmente un empaque de alcanos adheridas a sílice, por ejemplo, con grupos alquilo de 8 o 18 carbonos cubriendo la superficie de sílice. RPC (reverse phase chromatography) es actualmente el método más popular de cromatografía en líquidos; mas del 70 % de todas las separaciones por HPLC son llevadas a cabo por este método. La base de la retención de soluto en RPC es aun algo controversial; algunos trabajadores prefieren un proceso de adsorción, mientras que otros creen en la partición del soluto dentro de la fase estacionaria no polar. Probablemente ambos procesos son importantes para algunas muestras. La retención de un soluto, X, por la adsorción competitiva en la superficie de la fase estacionaria puede ser representado por: Xm + zMs ↔ Xz + zMm Donde M es una molécula de fase móvil. Los subíndices m y s refirieren a las moléculas en la fase móvil o estacionaria respectivamente. La ecuación 1.1 asume que una competencia entre el soluto y las moléculas de la fase móvil por un lugar en la superficie de la fase estacionaria existe. Esto es, una molécula adsorbida, X, desplazara un numero z de moléculas M previamente adsorbidas. Las pruebas que favorecen un proceso de particiones en RPC son de varias clases: Primero, la retención de una serie de alcanos sustituidos del tipo Cn-substituidos en el empaque de una columna, muestra una discontinuidad diferente cuando el soluto alquilo sustituido es igual en tamaño con el grupo Cn de la fase estacionaria. Esto indica que los grupos n-alquilo en un soluto la molécula puede penetrar (partición) dentro de la fase estacionaria, mientras no sean demasiado largos [1] 5.19 Cromatografía quiral Los compuestos ópticamente activos han atraído una gran atención porque los sistemas de la vida son quirales. Proteínas, ácidos nucleicos y polisacáridos poseen características quirales las cuales poseen una estrecha relación con sus funciones. Capítulo 5 64 Cromatografía de líquidos de alta resolución Los estereoisomeros son isómeros con una constitución idéntica pero un arreglo espacial diferente. Los estereoisomeros se clasifican de acuerdo a si factor de de simetría en un carbono denominado el carbono quiral. Los enantiomeros tienen propiedades físicas idénticas excepto por el signo de la rotación óptica. Generalmente los enantiomeros se obtienen en una mezcla racemica, mezcla en la cual la cantidad de los enantiomeros es igual. Modos de separación. La separación cromatografica de enantiomeros se puede conseguir por varios métodos; sin embargo, es siempre necesario el uso de un discriminador de enantiomeros o selector. Dos diferentes tipos de selectores pueden distinguirse: un aditivo quiral en la fase móvil o una fase estacionaria quiral. El mecanismo de separación es dependiente del modo de separación usado. 5.20 Cromatografía de derivados diastoméricos. Este el más antiguo y ampliamente usado método cromatográfico para distinguir enantiomeros. La columna de derivación de un soluto ópticamente activo con otra molécula ópticamente activa depende de la habilidad de separar a la molécula buscada. Un número importante de de grupos que desvían la actividad de los compuesto a separar se han estudiado entre ellos están los grupos amino, (separados con amidas, carbamatos, ureas, tioureas, y sulfoaminas) grupos hidroxilo, (separados con esteres, carbonatos, y carbamatos) grupos carboxilo (esteres y amidas), epóxidos (isotiocianatos), olefinas (con complejos quirales de platino), y tioles (con tioeteres). Las ventajas de esta técnica son: 1. La metodología ha sido ampliamente estudiada, haciendo que la aplicación sea relativamente fácil y accesible. 2. La detección de los productos puede ser llevada a cabo mediante la selección adecuada del agente derivante (separante) con un fuerte cromoforo o fluoroforo. Las principales limitantes: 1. La síntesis de derivados diastomericos requiere del aislamiento de los compuestos necesarios para la separación. 2. Las muestras se contaminan con el derivado diastomérico. 3. Los productos de interés pueden tener reacciones con los aditivos de la columna. Capítulo 5 65 Cromatografía de líquidos de alta resolución 5.21 Separación usando aditivos quirales a la fase móvil. La separación de compuestos enantiomericos se ha logrado a través de la formación de complejos diastoisomericos con una molécula quiral añadida a la fase móvil. La separación quiral es posible dada la diferencia de los complejos diastoisomericos formados, la solvatación en la fase móvil o la unión a la fase solida. Una visión general del fundamento de este método ha sido publicada por Lindner y Pettersson [2]. Existen tres principales tipos de aditivos para la formación de los complejos: A) Complejos de metales de transición (intercambiadores de ligandos) B) Apareamiento de iones C) Inclusión molecular 5.22 Separación usando aditivos quirales en la fase estacionaria. A diferencia del método anterior en este método los complejos se forman en la fase estacionaria, donde los aditivos se encuentran ya fijados. El enlace que se forma con uno de los enantiomeros hace posible la separación de estos mismos. Existen 5 tipos principales de sistemas usados en esta técnica. 1) Es los complejos soluto fase estacionaria son formados por interacciones atractivas, puentes de hidrogeno, interacciones pi, dipolos instantáneos, etc., entre el soluto y la fase estacionaria. 2) El mecanismo para la formación de complejos soluto-fase estacionaria es a través de interacciones atractivas en donde la inclusión de complejos juega un papel importante. 3) El soluto entra dentro de cavidades quirales en la fase estacionaria para formar los complejos incrustados. 4) El soluto es parte de un complejo metálico diastomérico. 5) La fase estacionaria es una proteína y el complejo soluto-fase estacionaria se basa en combinaciones e interacciones hidrofóbicas y polares. Aplicaciones. Las principales aplicaciones e encuentran en la industria farmacéutica donde los compuestos quirales tienen diferentes actividades biológicas en un organismo, la adecuada separación de estos compuestos es de vital importancia en esta industria [3]. Capítulo 5 66 Cromatografía de líquidos de alta resolución 5.23 Cromatografía de intercambio iónico Se utiliza principalmente para la separación de iones o de una sustancia fácilmente ionizable, en donde una de los principales factores que facilitan la retención, es la atracción electrostática entre los iones de la fase móvil y aquellos situados en la fase inerte estacionaria. En este tipo de cromatografía los iones del analito se separan basándose en las diferencias entre sus afinidades relativas por la fase estacionaria (iones de la fase inerte), contra aquellos que se encuentran en la fase móvil, de forma tal, que se establece un continuo intercambio de cargas en donde los iones de la matriz son reemplazados por los de la muestra a su paso por la columna. Los iones que se intercambian más frecuentemente son cationes como NH4+, metales grupo I y II, NO2-, NO3-, PO42-, haluros, SO42-. Fundamento Es un proceso en el que una solución de un electrolito es puesta en contacto con una resina intercambiadora, en donde los iones activos de la resina son reemplazados por aquellos iones de la misma carga presentes en el analito. 5.24 Intercambiadores catiónicos y aniónicos Un intercambiador catiónico es aquel en el que los iones activos sobre la matriz son cationes y el proceso de intercambio involucra cationes, entre los más comunes se encuentran: -SO3H, -CO2H, -OH, -PO4H3 Los grupos más comunes en una resina aniónica son grupos amino terciarios y cuaternarios y se intercambian de manera análoga a una resina catiónica. Resinas. Inicialmente la fase estacionaria constaba de resinas preparadas por policondensación, de fenoles ya minas aromáticas con formaldehído. Algunos grupos inorgánicos eran introducidos por condensación de formaldehído con sulfo o carboxil derivados. Actualmente estas se han reemplazado por materiales basados en estireno (poliacrilatos y divinilbencen derivados) los cuales pueden ser modificados para adaptar el tamaño de partícula, así como para mejorar la selectividad y la capacidad de retención de la columna. Capítulo 5 67 Cromatografía de líquidos de alta resolución Las resinas se preparan por copolimerización de estireno con divinilbenceno, de la cual se origina una maya tridimensional que puede ser modificada con gradientes de concentración para obtener diferencias en el tamaño de poro y así aumentar la selectividad. Para las resinas aniónicas, se realiza una clorometilación de la matriz polimérica seguida de un tratamiento con la correspondiente amina. Resinas catiónicas de ácido fuerte. Intercambian iones positivos (cationes). Funcionan a cualquier pH. Es la destinada a aplicaciones de suavizado de agua, como primera columna de desionización en los desmineralizadores o para lechos mixtos. Elimina los cationes del agua y necesitan una gran cantidad de regenerante, normalmente ácido clorhídrico (HCl). Resinas catiónicas de ácido débil. Tienen menor capacidad de intercambio. No son funcionales a pH bajos. Elevado hinchamiento y contracción lo que hace aumentar las pérdidas de carga o provocar roturas en las botellas cuando no cuentan con suficiente espacio en su interior. Se trata de una resina muy eficiente, requiere menos ácido para su regeneración, aunque trabajan a flujos menores que las de ácido fuerte. Es habitual regenerarlas con el ácido de desecho procedente de las de ácido fuerte. Resinas aniónicas de base fuerte. Intercambian iones negativos (aniones). Es la destinada a aplicaciones de suavizado de agua, como segunda columna de desionización en los desmineralizadores o para lechos mixtos. Elimina los aniones del agua y necesitan una gran cantidad de regenerante, normalmente sosa (hidróxido sódico - NaOH). Resinas aniónicas de base débil. Se trata de una resina muy eficiente, requiere menos sosa para su regeneración. No se puede utilizar a pH altos. Pueden sufrir problemas de oxidación o ensuciamiento. Propiedades de las resinas: 1. Poseer grupos intercambiadores monofuncionales 2. Un tamaño de partícula pequeño 3. Factor de retención elevado Tamaño de la partícula Estos se basan en los requerimientos estándares (50 mesh) Capítulo 5 68 Cromatografía de líquidos de alta resolución 100-200 mesh: separaciones a macroescala; cuantitativa 50-100 mesh: preparativas 14-50 mesh: separaciones a escala industrial Capacidad intercambiadora. Es la medida de la habilidad de la resina para intercambia un número determinado de iones o de carga por gramo de resina. Está determinada por la accesibilidad del grupo que se desea intercambiar, la concentración del eluyente, fuerza iónica, pH, tamaño de partícula y la fuerza de la unión resina-analito Para una resina catiónica, esta cantidad se define como la capacidad de intercambiar H+, mientras que para una resina aniónica esta capacidad esta e función de la cantidad de iones Cl- que pueda intercambiar. Selectividad de la Resina. La afinidad entre la resina y la partícula a intercambiar, es función de la resina y de los iones. Esto se puede expresar como un equilibrio de la siguiente forma: n(R- H+ ) + Mn+1 ( R- )n Mn+ + n H+ Donde R representa la matriz y puede establecerse la constante de equilibrio como se muestra: Kd = [Mn+]r [H+]n / [M m+] [H+]n r Donde el último término representa las concentraciones de los iones. Entre mayor sea el valor se Kd mayor será la afinidad de la partícula por la resina intercambiada y en consecuencia aumentará la capacidad de intercambio. Naturaleza de la Resina. El coeficiente de selectividad Kd está determinado por algunos de los factores de capacidad de la resina; la selectividad se afecta de acuerdo al grado de fuerza relativa en el enlace ion-resina. Los pequeños tamaños de partícula excluyen en su totalidad a aquellas de mayor tamaño, aumentando la selectividad de la resina; así como la temperatura. La afinidad de los cationes de las resinas e solución acuosa se incrementa caudno la carga se incrementa; para cationes de la misma carga, la afinidad es inversamente proporcional al radio del ion hidratado. Algunas secuencias de afinidad se muestran a continuación: 1. 2. 3. Capítulo 5 Na < Ca < Al < Th Li < Na < NH < K < Ag Mg < Cu < Sr < Ba 69 Cromatografía de líquidos de alta resolución Para las resinas aniónicas el intercambio depende del grado de polarizabilidad del anión por el intercambiador; en la medida en la que la polarización sea mayor, mayor será la fuerza con la cual se desplaza el ion de la matriz por el ion de la muestra. En general los iones polivalentes tienen una mayor afinidad que los monovalentes; y para los aniones de la misma carga el ion de mayor tamaño tendrá una afinidad: F- < HCO3- < Cl- < HSO3- < CN- < Br- < NO3- < I- << SO4 2En general: Parámetro Fuerza iónica pH Temperatura Buffer Observaciones La fuerza del solvente incrementa la fuerza iónica y en consecuencia la selectividad se ve afectada La retención disminuye en la resina catiónica y se incrementa en la resina aniónica cuando se modifica el pH a valores a mayores Elevando la temperatura se aumenta el grado de intercambio entre la fase móvil y la fase estacionaria Pude aumentar el valor de Kd cuando se modifica la fase [4] estacionaria . 5.25 Aplicaciones Esta técnica tiene una de sus aplicaciones primordiales en el campo del análisis clínico, en donde se utiliza comúnmente para la determinación de la presencia de desórdenes metabólicos cuando se separan aminoácidos y otras aminas de importancia fisiológica de las muestra de las pacientes. En condiciones normales, los aminoácidos se separan en su forma protonada como ácidos, utilizando diferentes combinaciones de buffers de citratos y boratos. Posteriormente estos aminoácidos pueden ser caracterizados por medio de técnicas como espectroscopia UV y fluorescencia. En la separación de carbohidratos, en las cuales se utilizan ligantes sobre un resina polimérica, con algunos metales como Ca2+ (recomendado para alcoholes) y Ag+ (para estructuras oligomericas), utilizando agua destilada o una mezcla de solventes orgánicos como fase móvil. La retención en este caso se ve determinada por la atracción electrostática entre grupos electronegativos (OH-) y electropositivos (Ca2+) y además por el impedimento estérico entre grupos [5]. Capítulo 5 70 Cromatografía de líquidos de alta resolución 5.26 Cromatografía de apareamiento iónico La cromatografía iónica, que es una versión de cromatografía de intercambio iónico de alta eficacia, se ha convertido en el mejor método de análisis de iones. Por ejemplo se usa en la industria de semiconductores para controlar niveles de 0.1 ppm de aniones y cationes en agua desionizada. La cromatografía de pares iónicos utiliza una columna HPLC de fase inversa, en lugar de una columna de intercambio iónico. Para separar una mezcla de cationes se añade a la fase móvil un surfactante aniónico, como n-C8H17-SO3. El surfactante se aloja en la fase estacionaria convirtiéndola eficazmente en un intercambiador iónico. Cuando los cationes del analito pasan a través de la columna se pueden unir a la fase estacionaria por atracción electrostática con los aniones del surfactante. El mecanismo de retención es una mezcla de interacciones con fase inversa y de intercambio iónico. Para separar los analitos aniónicos se pueden añadir a la fase móvil sales de tetrabutilamonio, como reactivo de par iónico. La cromatografía de pares iónicos es más compleja que la cromatografía de fase inversa, por que el equilibrio del surfactante con la fase estacionaria es lento, la separación es más sensible a variaciones de temperatura y pH, y la concentración del surfactante no afecta la separación. El disolvente a elegir es el metanol debido a que los surfactantes iónicos son mas solubles en mezclas metanol/agua que en mezclas acetonitrilo/agua. Las estrategias para el desarrollo de un método dependen de las variaciones en el pH y la concentración de surfactante, para una concentración de metanol y temperatura fija. Dada la lentitud del equilibrio entre el surfactante y la fase estacionaria, no se recomienda una elución gradiente en cromatografía de pares iónicos. Figura 1. Fundamento de la cromatografía de pares iónicos. EL surfactante octamonosulfato sódico añadido a la fase móvil se une a la fase estacionaria no polar. Los grupos sulfonato negativos, que sobresalen de la fase estacionaria, actúan como puntos activos de intercambio iónico frente analitos catiónicos, como bases orgánicas protonadas, BH+ [6] Capítulo 5 71 Cromatografía de líquidos de alta resolución 5.26 Cromatografía de exclusión de tamaño La cromatografía de exclusión, también llamada cromatografía de permeación en gel es un modo de separación no interactivo. Las partículas del empaque de la columna tienen varios tamaños y estructuras de poro, de forma que las moléculas son retenidas o excluidas con base en su volumen molecular hidrodinámico; estos es, su tamaño y forma. Estrictamente, la separación en cromatografía de exclusión no se basa en el peso molecular. Conforme la muestra pasa por la columna las moléculas del soluto se ordenan. Las moléculas muy grandes no pueden entrar en muchos de los poros e incluso penetran menos en las regiones comparativamente abiertas del empaque. Las moléculas muy pequeñas difunden hacia dentro de todos o muchos de los poros accesibles a ellas. Con un mayor volumen de la columna a su disposición, las moléculas pequeñas tardan más en salir de la columna. Empaques de la columna. Los empaques para la columna pueden ser semirrígidos (polímeros macromoleculares entrecruzados) o rígidos (vidrios o sílices de tamaño de poro controlado). Los primeros están limitados para una presión máxima de 300 psi. Las perlas de poliestireno parcialmente sulfonadas son compatibles con los sistemas acuosos y las no sulfonadas con los no acuosos. Los vidrios y sílices porosos cumplen una amplia gama de tamaños de poro. A continuación se presentan unos ejemplos y los correspondientes intervalos de operación. Diámetro del poro (nm) Intervalo de operación (daltons) 4 1 000 – 8 000 10 1 000 – 30 000 25 2 500 – 125 000 55 11 000 – 350 000 150 100 000 – 1 000 000 250 200 000 – 1 500 000 Estos empaques son químicamente resistentes a valores de pH menores de 10 y pueden usarse con disolventes acuosos y orgánicos polares. Capítulo 5 72 Cromatografía de líquidos de alta resolución Disolventes. Se requiere de un solo disolvente en el que se disuelve y cromatografía la muestra. Si la diferencia en la viscosidad entra una muestra inyectada y la fase móvil es muy grande, puede producirse la distorsión del pico y anomalía en los tiempos de elusión. Detectores. Los detectores más utilizados son el refractómetro diferencial y los espectrofotométricos que operan en las regiones ultravioleta e infrarroja. Un detector de dispersión de luz laser de ángulo bajo (LALLS) hace posible la determinación directa de los pesos moleculares ya que responde al peso molecular del analito, no únicamente a la concentración. Un detector de infrarrojo proporciona información sobre la composición del copolímero, ramificación y tacticidad, esta se refiere a la estereorregularidad que se encuentra en ciertos tipos de polímeros. Comportamiento de la retención. El comportamiento esencial de un soluto y la característica de los empaques de las columnas se puede describir en términos muy simples. Si se supone que el tiempo que toma un soluto para difundirse dentro de un poro es pequeño con respecto al tiempo que la molécula está en la vecindad del poro, entonces el proceso de separación es totalmente independiente del proceso de difusión, esto se representa de la siguiente forma (coeficiente de distribución: K VR VM VS Se puede enunciar como la fracción interna del volumen de poro que es accesible al soluto. Donde VR (volumen de retención): es el volumen de eluyente que fluye de una columna entre la inyección de la muestra y su salida en el eluyente; VM: es el volumen ocupado por la fase móvil (esto es, en los intersticios entre las partículas porosas llenas de disolvente), que s e estima con la elución de un soluto totalmente excluido; VS: es el volumen interno acumulado dentro de los poros de las partículas y disponible para un soluto totalmente incluido o para las moléculas del disolvente. Las moléculas totalmente excluida eluyen en un volumen vacío, esto es, VR=VM y por lo tanto K=0. Para las moléculas pequeña que pueden entrar en todos los poros del empaque, VR=VM+VS y, por lo tanto, K=1. Las moléculas de tamaño intermedio eluyen entre estos dos límites y K se encuentra en el intervalo de 0 a 1. Capítulo 5 73 Cromatografía de líquidos de alta resolución 5.27 Aplicaciones Mucho del trabajo en la industria de los alimentos y las bebidas o en muestras fisiológicas en las que la mayoría de los ácidos del ciclo de Krebs están presentes, se realiza con facilidad con columnas de exclusión de aniones. Vino, cerveza, jugo de fruta y muchos productos lácteos se analizan rápidamente con una preparación mínima de la muestra (generalmente sólo filtración o centrifugación). Otra aplicación es la separación de oligosacáridos y azúcares de alcohol con una columna de exclusión de aniones en la forma iónica de calcio con agua como eluyente y una temperatura de 90°C. La maltotriosa y otros oligosacáridos superiores se separan del mono y los disacáridos por efectos de exclusión estérica. En bioquímica la separación de péptidos, proteínas y demás moléculas biológicas es importante y se puede separar efectivamente por este método [7]. 5.28 Cromatografía líquido-líquido Los empaques de columna más utilizados más ampliamente para cromatografía de reparto líquido-líquido, son aquellos con fases estacionarias orgánicas enlazadas. Reemplazan a los empaques clásicos en los que el líquido estacionario recubre un material de soporte. El reparto ocurre entre la fase enlazada y una fase móvil líquida. Preparación de soportes con fase enlazada Los soportes con fase enlazada se preparan uniendo covalentemente a la superficie de la sílice una especie orgánica de hidrocarburo. Los soportes incluyen geles de sílice de poro grandes, cuentas con lechos porosos y micropartículas. El siloxano se ha convertido en el estándar de las fases enlazadas comerciales. Es poco probable que la porción hidrocarbonada de octilo u octadecilo se extienda totalmente dentro de la fase móvil. Las fases monoméricas responden rápidamente a los cambios en la composición de la fase móvil, cuando son mojadas por ellas. La falta de mojado causa una eficiencia pobre, presentándose adsorción en la interfase o entrecara adsorbente-disolvente en adición al equilibrio de reparto líquido – líquido esperado. Una fase enlazada popular es un hidrocarburo de cadena lineal. El grupo alquilo puede ser de diversas longitudes, normalmente es un grupo etilo (C-2), octilo (C-8) u octadecilo (C-18). Este último puede utilizarse para las aplicaciones donde se requiere un máximo de retención. Capítulo 5 74 Cromatografía de líquidos de alta resolución 5.29 Cromatografía líquido-liquido de fase normal Utiliza una fase estacionaria polar (frecuentemente hidrofílica) y una fase móvil menos polar. Para seleccionar una fase óptima, es mejor empezar con una fase móvil de un hidrocarburo puro como el heptano. Si la muestra se retiene fuertemente, la polaridad de la fase móvil debe aumentarse, quizá añadiendo pequeñas cantidades de metanol o de dioxano. 5.30 Cromatografía de fase inversa Utiliza un empaque enlazado hidrofóbico, usualmente con un grupo funcional octadecilo (C-18) u octilo(C-8) y una fase móvil polar, frecuentemente una fase móvil parcial o totalmente acuosa. Conforme aumenta el carácter hidrofóbico de los solutos, la retención aumenta. El agua es el eluyente más débil. El metanol y el acetonitrilo son disolventes populares porque tienen baja viscosidad y son fáciles de conseguir con excelente pureza. En cromatografía de fase inversa la fuerza de la retención no es la interacción favorable del soluto con la fase estacionaria, sino el efecto del disolvente de la fase móvil para forzar al soluto hacia dentro de la capa hidrocarbonada enlazada. Aplicaciones de cromatografía de fase inversa La técnica de fase inversa en sus varias formas es el modo más ampliamente utilizado en HPLC e incluye cerca de la mitad de los métodos de cromatografía líquida. Esta técnica es la que probablemente proporcionará retención y selectividad óptimas cuando los compuestos no tienen grupos para enlaces de hidrógeno o no tienen un carácter predominantemente alifático o aromático. El método es muy apropiado para separar solutos con base en el tamaño y estructura de los grupos alquilo. En química clínica cada vez se realiza con más frecuencia el análisis cuantitativo de las drogas de abuso por medio de esta técnica. Los productos farmacéuticos que se analizan en forma rutinaria incluyen a los barbitúricos, drogas antiepilépticas, analgésicos y sedantes. Aplicaciones adicionales de los métodos de fase inversa son los preservativos alimentarios, herbicidas y azúcares. En el campo farmacéutico ha venido en aumento el uso de esta técnica a costa de la cromatografía de adsorción. Un amplio espectro de biomoléculas, lipofílicas o iónicas, pequeñas o grandes, pueden ser cromatografiazas debido a que el contenido de agua en la fase móvil puede variar desde el 100% hasta porcentajes muy bajos o ninguno en absoluto. Los compuestos lipofílicos, tal como los triglicéridos, que tienen una solubilidad muy pobre en los disolventes acuosos de la fase inversa, con Capítulo 5 75 Cromatografía de líquidos de alta resolución frecuencia pueden separarse con una fase inversa no acuosa utilizando una columna empacada con octadecilo y disolventes orgánicos polares como el acetonitrilo o el tetrahidrofurano[7]. Bibliografía [1] L. R. Snyder. (1992). Chromatography, Part A: fundamentals and techniques. 5th edition. Editado por E.Haftman Del Journal of Chromatography Library, p. A25-A26. [2] J.Gal, LC-GC, 5, 106 (1987) [3] Satinder Ahuja, (1990). Chiral Separations by Liquid Chromatography. 1st Edition. Maple pres: New York. [4] Baithwaite. Chromatographic methods. Chapman & Hall U.K., 1977 [5] Poole. Chromatography Today. Elsevier U.K., 1991 [6] Harris Daniel C., (2001), Análisis Químico Cuantitativo, 2da Edición, Editorial Reverte, España, pp.740-743. [7] Willard. (1991) “Métodos instrumentales de análisis” Edit. Iberoamérica S.A. de C.V. México. Capítulo 5 76 Técnicas acopladas Capítulo 6 Cromatografía y técnicas espectroscópicas acopladas Los métodos de acoplamiento combinan las capacidades de separación de la cromatografía con las de detección cuantitativa y cualitativa de los métodos espectrales tales como infrarrojo, resonancia magnética nuclear y masas, entre otros. A estas técnicas se les denomina en ocasiones técnicas hifenadas. En los primeros métodos de esta categoría los eluyente de la columna cromatográfica se recogían como fracciones separadas en una trampa fría, después de lo cual se usaba un detector no selectivo ni destructivo para la identificación de su presencia. Posteriormente, se investigaba la composición de cada fracción por espectroscopia de resonancia magnética nuclear, infrarroja, masas o con medidas electroanalíticas. Una importante limitación de esta aproximación era la pequeña cantidad (usualmente de micromoles) de soluto contenida en las fracciones. Sin embargo, hoy día muchos métodos hifenados monitorizan el efluente de la columna cromatográfica de manera continua, con métodos espectroscópicos dando como resultado una combinación de dos técnicas, que basadas en principios distintos, permiten lograr una enorme selectividad. Características y requerimientos de las técnicas acopladas 6.1 Cromatografía HPLC acoplada a Espectroscopia de Masas (MS) Aunque la espectroscopia de masas es una poderosa herramienta para la identificación de compuestos puros, la complejidad de los espectros de masas dificulta enormemente el análisis de mezclas, incluso simples. Por ello para el análisis de muestras complejas se emplean acoplamientos de esta técnica con técnicas potentes de separación, como es la cromatografía líquida. Así el acoplamiento Cromatografía Líquida-Espectroscopia de Masas (GL-MS), es una herramienta más poderosa al alcance de los químicos para el análisis de muestras complejas. En este caso se efectúan los espectros de masas de los compuestos que salen de la columna de cromatográfica. Es necesaria una interfase entre ambos instrumentos que tiene como misión fundamental eliminar el eluyente (fase móvil) antes de la introducción de los analitos en el espectrómetro. Capítulo 6 81 Técnicas acopladas Estos acoplamientos permiten reunir las ventajas de una técnica poderosa de separación con una herramienta poderosa de identificación. Pueden registrarse espectros de masas a lo largo de toda la separación cromatográfica. Así pueden registrarse inequívocamente los picos y comprobar incluso su pureza, es decir si la separación ha sido completa o se ha producido en algún momento la co-elución de dos compuestos. Espectroscopia de Masas acopla a Cromatografía Líquida de Alta Resolución Tal vez una de las desventajas de esta técnica es que los espectros de masas de los compuestos orgánicos analizados por esta técnica, dependen de las condiciones de análisis, principalmente del tipo de fase móvil y del potencial de ionización aplicado; ya que comúnmente a partir del espectro de masas se obtiene normalmente el ión molecular y fragmentos que proporcionan a su vez el peso molecular del compuesto. En tanto que en la técnica del HPLC-MS, el ión molecular forma fragmentos con el disolvente empleado en la fase móvil y por tanto, no corresponden directamente con el peso molecular del compuesto analizado. Razón por la cual, se hace necesario inyectar disoluciones patrón que contengan los compuestos objetos del análisis, en las mismas condiciones experimentales (eluyente potencial y ionización), para conocer el espectro de masas característico del compuesto en cada caso. Sin embargo pese a ello esta técnica resulta extremadamente útil en el análisis de ultratrazas (ng/kg) de compuestos orgánicos. Donde un análisis de tan bajos niveles de concentración (ultratrazas) puede resultar necesario cuando se trata de compuestos de muy altos niveles de toxicidad y que se cumula en los organismos vivos. Es el caso de las dibenzoparadioxinas policloradas (PCDDs), los dibenzofuranos policlorados (PCDFs), o los bifenilos policlorados. Se analizan en los suelos y organismos vivos principalmente ya que se acumulan en la materia orgánica del suelo. Capítulo 6 82 Técnicas acopladas Otra de las aplicaciones de esta técnica, es la identificación de analitos desconocidos, ya sea como productos secundarios de síntesis, análisis de herbicidas y pesticidas, impurezas de drogas comerciales, ó la identificación inequívoca de sustancias de estructura similar, para las que un simple espectro masas es insuficiente; cabe mencionar que el acoplamiento HPLC-MS es una herramienta muy selectiva que se emplea hoy día permite la identificación de algunos péptidos y proteínas. Equipo de HPLC acoplado a MS 6.2 Cromatografía HPLC acoplada a Ultravioleta (UV) El Detector de Ultravioleta Visible es el más común acoplado a HPLC, se usa cuando los componentes absorben radiación UV- visible, como los compuestos aromáticos, alquenos, moléculas con enlaces C-O, C-N, C-S. Pueden ser de longitud de onda fija o variable (arreglo de diodos). La detección UV-visible no es destructiva, es estable en el tiempo y se afecta poco por la temperatura, por lo que se puede colectar los componentes por separado. Se considera a la detección UV como selectiva ya que se elige la longitud de onda de mayor absorbancia de acuerdo con la sustancia que se desea conocer, pero debe tenerse en cuenta que en el rango del UV lejano (190-220nm) la mayoría de los compuestos está en el orden de los nanogramos (ng), lo que posibilita el análisis de cantidades trazas. Capítulo 6 83 Técnicas acopladas El detector de UV-visible es de longitud de onda variable o espectrofotométrico es el más usado ya que ofrece como ventaja la libre selección de longitud de onda de trabajo sin necesidad de cambiar filtros o lámparas. Permite trabajar desde 190 hasta 700 u 800nm debido a que utiliza una red de difracción consistente en un prisma que posibilita, mediante su rotación, seleccionar la longitud de onda de trabajo de forma continua y no discreta como los detectores fotométricos. Estos detectores utilizan también como fuente de luz una lámpara de deuterio o halógeno, que en algunos modelos puede ser intercambiada por una de tungsteno para la detección en el rango visible. Al ser constructivamente más complejos que los detectores fotométricos son algo menos sensibles y más costosos. La espectroscopia de ultravioleta (UV) es de mucho valor, especialmente en HPLC. El espectro UV es muy utilizado para corroborar la identidad, ya que la identificación basada en una sola longitud de onda del rango UV puede presentarse a interferencias. En los detectores de arreglo de diodos (DAD) el espectro UV puede ser registrado en computadora y comparado con el del compuesto sospechosos para corroborar su variedad Así mismo en la industria láctea el HPLC acoplado a UV se emplea para la determinación de vitamina D3 en determinación de productos derivados de la leche y en premezclas vitamínicas. Esto debido a que la vitamina D3 es una vitamina liposoluble normalmente presente en la leche y sus derivados en cantidades muy bajas. Por esta razón, industrialmente se adiciona la misma a algunos productos lácteos para mejorar sus propiedades nutritivas. Por ello la determinación del contenido de vitamina D3 por HPLC acoplado a UV a 264nm constituye parte del control de calidad del producto lácteo terminado. Otra de las aplicaciones de esta técnica es la determinación de sulfonamidas, nitrofuranos y cloranfenicol en leche. Esto debido a que los residuos de antibióticos y otros quimioterapéuticos inhibidores del crecimiento bacteriano en la leche traen aparejadas serias dificultades al procesamiento industrial de la misma, además de ser nocivos para la salud humana, entre otras causas, por la aparición de cepas de microorganismos patógenos resistentes a los mismos. El control de estas sustancias se basa en la extracción selectiva de los residuos de sulfonamidas, nitrofuranos y cloranfenicol presentes en la leche con una mezcla cloroformo-acetona. El extracto se evapora hasta sequedad a presión reducida y se redisuelve en buffer fosfato de potasio 0.1M desgrasándose por partición con hexano. La fase acuosa (buffer) contendrá los residuos de sulfonamida, Capítulo 6 84 Técnicas acopladas nitrofuranos y cloranfenicol. El extracto se filtra por membrana e inyecta al cromatógrafo de líquidos de alta resolución con detección de UV-visible y columna de fase reversa (C18). Así mismo en la industria láctea el HPLC acoplado a UV se emplea para la determinación de vitamina D3 en determinación de productos derivados de la leche y en premezclas vitamínicas. Esto debido a que la vitamina D3 es una vitamina liposoluble normalmente presente en la leche y sus derivados en cantidades muy bajas. Por esta razón, industrialmente se adiciona la misma a algunos productos lácteos para mejorar sus propiedades nutritivas. Por ello la determinación del contenido de vitamina D3 por HPLC acoplado a UV a 264nm constituye parte del control de calidad del producto lácteo terminado. Otra de las aplicaciones de esta técnica es la determinación de sulfonamidas, nitrofuranos y cloranfenicol en leche. Esto debido a que los residuos de antibióticos y otros quimioterapéuticos inhibidores del crecimiento bacteriano en la leche traen aparejadas serias dificultades al procesamiento industrial de la misma, además de ser nocivos para la salud humana, entre otras causas, por la aparición de cepas de microorganismos patógenos resistentes a los mismos. El control de estas sustancias se basa en la extracción selectiva de los residuos de sulfonamidas, nitrofuranos y cloranfenicol presentes en la leche con una mezcla cloroformo-acetona. El extracto se evapora hasta sequedad a presión reducida y se redisuelve en buffer fosfato de potasio 0.1M desgrasándose por partición con hexano. La fase acuosa (buffer) contendrá los residuos de sulfonamida, nitrofuranos y cloranfenicol. El extracto se filtra por membrana e inyecta al cromatógrafo de líquidos de alta resolución con detección de UV-visible y columna de fase reversa (C18). 6.3 Cromatografía Líquida de Alta Resolución (HPLC) acoplada a RMN El acoplamiento de HPLC y RMN requiere el ajuste de los sistemas de análisis. El flujo de la fase móvil lleva a un período de exposición limitada (m) para los núcleos de células de la corriente. El tiempo (m) es definido como la proporción del volumen de detección de la velocidad de flujo. Por otra parte, el estado de equilibrio se alcance en un tiempo más corto permitiendo un rápido tiempo de la tasa de repetición de la exposición de un espectro y, por tanto, un aumento en la sensibilidad. En la técnica de HPLC la mayoría de las separaciones se realizan con materiales de fase invertido usando mezclas binarias de disolventes como acetonitrilo / agua, acetona / agua o metanol / agua como móvil fases. La elección de la fase móvil debe ser adaptada a la espectroscopía de RMN. Una ventaja evidente es la de obtener un número pequeño de señales del disolvente en el espectro de RMN, ya que el disolvente puede ocultar las señales de la muestra espectros. En general, las condiciones en Capítulo 6 85 Técnicas acopladas cromatografía HPLC-RMN los experimentos son los mismos que en la cromatografía convencional, pero el agua se sustituye por agua deuterada. El uso de disolventes deuterados orgánicos en general es demasiado caro. Para un ajuste correcto del receptor de la ganancia RMN instrumento, la intensidad de las señales de disolvente debe reducirse a la altura de la muestra mediante la aplicación de una técnica de represión del disolvente. La combinación de técnicas de separación cromatográfica con espectroscopía de RMN es uno de los más poderosos y los métodos de ahorro de tiempo para la separación y la determinación estructural de compuestos desconocidos y mezclas. Especialmente para la elucidación de la estructura de sustancias sensibles a la luz y el oxígeno, por ejemplo ácidos de lúpulo amargo y estereoisómeros de carotenoídes. 6.4 Cromatografía HPLC acoplada a espectroscopía de IR En esta técnica de acoplamiento se presenta un problema importante que la diferencia de la CGIR: la mayoría de los disolventes (y solutos) empleados como fase móvil en HPLC presenta una considerable absorción en la zona infrarroja del espectro por lo que la caracterización de los analitos se hace más problemática. Existen dos tipos de acoplamiento: 1.- Acoplamiento directo: parecido al de CG-IR, se emplea un tubo lumínico que actúa de célula de flujo. Los interferogramas obtenidos durante todo el proceso cromatográficos son almacenados y posteriormente se ofrecen los espectros IR convencionales de los analitos cuando el ordenador ha sustraído en los mismos las bandas de absorción correspondientes a la fase móvil. Para evitar un bloqueo excesivo del espectro por parte del disolvente se reducen las dimensiones de la célula (el paso de la luz de 0.1 a 0.2 mm y el volumen entre 3 y 12 µL) y aun así existen inconvenientes: no se puede usar gradiente de elución no se puede obtener información segura de la zona del espectro en el que el disolvente absorbe fuertemente algunos disolventes ofrecen dificultades especiales para la sustracción la sensibilidad es veinte veces menor a la de CG-IR, comparable a la que se obtiene con un detector de índice de refracción convencional. 2.- Acoplamiento con eliminación del disolvente (previa a la identificación IR): debe cumplirse la condición de que los analitos sean mucho menos volátiles que los componentes de la fase móvil para lograr una volatilización selectiva. Se usa un muestreador con varios pocillos que contienen mg de KCl Capítulo 6 86 Técnicas acopladas soportados sobre una placa metálica. El efluyente cromatográfico es rociado en un tubo concentrador, usando nitrógeno que evaporara un 90 % del disolvente; al entrar un pico a dicho tubo se abre una válvula y se vierte el contenido en los pocillos del muestreador (se vierte de pozo en pozo al cerrarse y abrirse la válvula). La detección se realiza por reflectancia difusa. 6.5 Cromatografía de gases acoplada a Espectrometría de Masas (CG-MS) El espectrómetro GC/MS es un equipo de gran importancia en los laboratorios analíticos debido a su gran sensibilidad (límite de detección de picomoles), el compuestos al que se realice el análisis por GC/MS, debe ser térmicamente estable (no descomponerse) a las temperaturas de la columna, y debe ser lo suficientemente volátil como para estar en fase gaseosa en el proceso de separación cromatográfica (punto de ebullición < 250ºC). En la técnica de GC/MS se utiliza la espectrometría de masas (que no se debe confundir con espectroscopia, ya que no hay absorción o emisión de radiación) como método de detección para identificar los analitos separados en la columna cromatográfica. El espectrómetro de masas está acoplado de modo hermética y directamente a la salida de la columna cromatográfica a través de un capilar. El cromatograma, que registra los picos de elución de los analitos a su salida de la columna y sus tiempos de retención. Esta información es de especial interés en series homólogas (compuestos de la misma familia que se distinguen entre sí sólo por la longitud de la cadena alifática, por ejemplo, la serie homóloga de los alcoholes ya que permite hacerse una idea sobre el peso molecular de la especie. El espectro de masas correspondiente a cada pico de elución, que permite identificar el pico con un compuesto determinado. La espectrometría MS consiste en la ionización de moléculas en fase gaseosa y la separación de los iones resultantes de acuerdo a su relación masa/carga (m/z). Un haz de electrones colisiona con las moléculas que entran en la cámara de ionización del espectrómetro de masas y, paradójicamente, les arranca un electrón, dando lugar a diversos fragmentos cargados positivamente. Estos fragmentos son acelerados en un campo electromagnético y llegan al detector. Capítulo 6 87 Técnicas acopladas Los instrumentos de CG/MS se han utilizado para la identificación y caracterización de sabores, olores en los alimentos, identificación de contaminantes en el agua, llevar a cabo diagnostico medico basado en componentes del aliento y estudio sobre los metabolitos de drogas. 6.6 Cromatografía de gases acoplada Infrarrojo (CG-IR) Una ventaja sustancial de esta hibridación es que la fase móvil cromatográfica no absorbe en la zona de IR, por lo que no es precisa su separación previa de los analitos., además del carácter no destructivo del detector acoplado. Este acoplamiento puede llevarse a cabo mediante dos planteamientos técnicos diferentes: Discontinuo , cuando no se dispone de un instrumento IR-TF(IR-Transformada de Fourier) Continuo , mediante una interfase ,lo que exige la mayor sensibilidad y alta velocidad de barrido del instrumento (IR-TF) a) En la figura se muestran un diagrama de bloque de combinación CG-IR. La muestra se inyecta en el cromatógrafo de gases y los analitos se separan en la columna capilar .El efluyente cromatográfico se dirige a una cedula de flujo especial denominada “tubo lumínico” que constituye la interfase de la hibridación. Después el fluido regresa al cromatógrafo donde realiza la detección continua convencional b) Tubo lumínico, está cubierto de oro en su interior para reflejar la luz, y calentado eléctricamente. Se sitúa alineado axialmente a la radiación de IR modulada incidente. Sus extremos so de material transparente a esta radiación, generalmente KCl. El flujo gaseoso entra y sale mediante dos orificios. Para obtener la información del acoplamiento: 1. Se obtiene el cromatográma ordinario a través del detector de CG puede o no ser destructivo 2. El espectrómetro IR pude realizar varias misiones una vez aplicado el algoritmo y obteniendo el espectro de IR convencional: (a) proporcionar espectros IR de cada pico (b) proporcionar un Capítulo 6 88 Técnicas acopladas cromatográma (c) comprobar la pureza de espectral. (d) Comparar los espectros obtenidos con los almacenados para identificar a los analitos. Muestra representativa de una determinación de herbicidas en los sedimentos de una plata industrial 6.7 Cromatografía de Gases acoplada a Espectroscopia Atómica (CG-EAA) Se trata de las combinaciones más simples, ya que las correspondientes interfases son atomizadores comerciales, con escasas variaciones. La facilidad y sencillez de los montajes aumenta en el siguiente sentido Hornos de grafito< llamas < plasmas. Los plasmas ICP, DCP, MIP y los atomizadores de llama pueden acoplarse directamente con el efluyente de un cromatógrafo de gases debido a que el caudal cromatográfico es generalmente compatible con el de introducción a estos atomizadores. En el caso de los plasmas, el argón o helio deben ser los gases portadores usados en el cromatógrafo. Las ventajas del uso de emisión atómica en plasma son: Mayor sensibilidad, posibilidad de multidetección atómica con instrumentos comerciales, mayor campo de aplicación. El acoplamiento de un cromatógrafo de gases con la espectroscopia de absorción atómica con una llama como interfase puede ser directo, aun que la mejor alternativa es utilizar un tubo de cuarzo o de cerámica calentando en la llama a través del cual circula el efluyente gaseoso. En general los diferentes diseños descritos tienen por objetivo aumentar el tiempo de los átomos en la zona de absorción lumínica. Los analitos más frecuentemente determinados mediante estas hibridaciones instruméntales son compuestos órgano metálicos procedentes de su introducción como contaminantes a partir de productos comerciales (estabilizadores de plásticos aditivos de gasolina, diacidas o de alquilación en Capítulo 6 89 Técnicas acopladas procesos naturales). Pese a que estas técnicas presentan una gran sensibilidad (generalmente el límite de detección es inferior al ng), siempre son necesarias técnicas de preconcentración debido a que se encuentran en niveles sumamente bajos en la naturaleza. Lo limites de detección alcanzados para compuestos organometálicos de plomo son 250pg Pb/m3 para tretrametilplomo y 375pg Pb/m3 para tetraetilplomo. Figura: Una de dichas técnicas presente dos etapas: En la primera tiene lugar la retención de los analitos en un tubo de absorción de polímetro poroso. La segunda etapa la desorcion-determinación continúa con la hibridación CGEAA. Se utiliza una corriente adicional de hidrogeno y un tubo de cuarzo sobre la llama. 6.8 Cromatografía (HPLC) acoplada a Espectroscopia Atómica (CG-EAA) El caudal de aspiración de las muestras en los plasmas es de 1y 2 mL/min. Lo que los hace compatibles con los caudales de los efluyentes cromatográficos líquidos. Cuando el disolvente de la fase móvil es fundamentalmente acuoso (en cromatografía de líquidos en fase invertida, cromatografía de intercambio iónico) un nebulizador convencional es la interfase CG-EAE. Cuando los disolventes son hidrocarburos o compuestos orgánicos halogenados, debe unirse un nebulizador especial de impacto. Los plasmas de microondas (MIP) no son recomendables para ser hibridados en HPLC. Los métodos atómicos de llama tienen un caudal entre 3 y 6 mL/min, superior el de los caudales usuales de salida en HPLC por lo cual puede usarse un flujo adicional un que los analitos sufres una dilución excesiva, una solución interesante es la propuesta por Slavin que se basa en la formación de una gota a la salida del efluyente. Cuando esta alcanza un determinado tamaño (≈100l) se desprende y cae sobre un micro embudo de teflón conectado directamente al nebulizador. Las interfases más complejas son las que se necesitan para acoplar un HPLC con un espectrofotómetro de absorción atómica con vaporización electrotérmica. Esto se debe a la Capítulo 6 90 Técnicas acopladas discontinuidad implícita de la detección por la necesidad de establecer ciclos precisos de incrementos de temperatura para cada medición por eso todo acoplamiento de este tipo debe ser de modo discontinuo Otra posibilidad es HPLC-EAA (cámara de grafito); en general constan de dos válvulas de apertura/cierre, una múltiple de desvió y otra de inyección cuyo funcionamiento controlado por un secuenciador es clave. Este secuenciador controla además el funcionamiento de la bomba a alta presión, los ciclos de temperatura en la atomización y el funcionamiento del EAA .La válvula de inyección introduce alícuotas a través de u capilar de tántalo. Las dos válvulas de apertura/cierre permanecen cerradas durante el periodo de calentamiento programado del tubo de grafito y posteriormente son abiertas cuando se realiza la inyección de la siguiente muestra, Estas válvulas también pueden permanecer abiertas y así la mayor parte del efluyente no es enviado al instrumento de de medida, inyectando alícuotas después de cada ciclo de calentamiento. Una posibilidad mas es de conectar HPLC con EAA (cámara de grafito) se utiliza un muestreador comercial adaptado a la absorción atómica con cámara de grafito. El eluyente cromatográfico puede ser recogido en su totalidad en los pocillos a intervalos regulares de tiempo o bien puede programarse de tal forma que el eluyente valla alternativamente a los pocillos receptores y al desecho según lo programado. Estas interfases son más complejas respecto a las que origina un atomizador de llama, pero la vaporización electrotérmica origina sensibilidades superiores, de ahí su atractivo. 6.9 Resumen El acoplamiento instrumental se define como la combinación a través de una interfase adecuada de dos técnicas analíticas independientes, que genera información única e integral de la composición de la muestra, la cual se caracteriza por ser más completa que la información alcanzada independientemente por cada técnica. La razón es que se combinan el elevado poder de separación de la cromatografía para una amplia mezcla de analitos y el elevado poder de discriminación e identificación que poseen estas técnicas determinativas. En general todas las técnicas de acoplamiento constan de una interfase entre los dos instrumentos que constituyen la conexión que produce el fluido que emerge de la columna cromatográfica y el sistema de detección. Es imprescindible el control coordinado del funcionamiento de ambos instrumentos (separativo y determinativo). Capítulo 6 91 Técnicas acopladas La gran cantidad de datos generada exige un sistema de almacenamiento, tratamiento, interpretación y presentación de resultados. Hoy las técnicas acopladas son una gran herramienta para los químicos y la ciencia en general y son por este orden las más usadas: Espectrometría de masas (EM), Espectroscopia de Absorción Infrarroja con transformada de Fourier (IR-TF), Técnicas Espectroscópicas Atómicas de Emisión (ICP) ó Absorción Atómica (EAA) y Resonancia Magnética Nuclear (RMN). 6.10 Bibliografía Revisiones de los Métodos combinados en C.L. Wilkins, Science. 1983.222, 251; Anal. Chem., 1989, 59, 517A. Thomson, Skoog, West, Holler y Crouch. Fundamentos de Química Analítica. Intenational Thomson Editores S. A. de C.V., 8 ª Edición, México, p.p. 970, 2005. http://www.analytical-tech.com/productos/HPLC_MS.html Hirschmugl, C.J. Frontiers in infrared spectroscopy at surfaces and interfaces. Surf. Sci. 500, 577-604 (2002 http://www.quimica.izt.uam.mx/Docencia/DocenciaQA/CursoCromatografia2005.pdf Noa P. M., Pérez Fl. N., Díaz G. G., Vega L. S. Cromatografía de gases y de líquidos de alta resolución. División de Ciencias Biológicas y de la salud. Depto. de producción agrícola y animal. UAM, unidad Xochimilco. 1ª edición. México D.F. pp.165-169, 175, 299,.2005. Varcárcel M., Goméz H. Técnicas Analíticas de Separación. Editorial Reverte S.A. Capitulo22. 1994. Duglas A., Leary J., Skoog. Análisis Instrumental. Editorial McGraw Hill, 4 ta. Edición. p.p.722-727.1992. Capítulo 6 92 Detectores para cromatografía de gases y líquidos Capítulo 7 Detectores para cromatografía de gases y líquidos Una parte importante de la cromatografía de gases y líquidos son los detectores, Un detector es un dispositivo para revelar la presencia de las sustancias eluídas a la salida de la columna cromatográfica. El Detector es un dispositivo capaz de convertir una propiedad física, no medible directamente, en una señal elaborable y ofrecernos información sobre la naturaleza y magnitud de la propiedad física. En cromatografía un detector funciona comparando una propiedad física entre el eluyente puro y el mismo eluyente llevando cada uno de los componentes que previamente se han separado en la columna, esta acción se traduce en una señal tipo eléctrica, que posteriormente se amplificará mediante un registrador gráfico ó integrador permitiendo indicar el momento que salen de la columna los componentes. 7.1 Clasificación de los detectores Detectores según su Grado de Selectividad. Universales. Responde a la mayoría de los solutos que pasan por él. Específicos ó Selectivos. Exhibe una gran respuesta a un grupo particular de substancias con un mínimo de respuesta a otras. Detectores Destructivos y No destructivos. Esta clasificación, obviamente, es en referencia a si la muestra es destruida o no. Detectores según su Modo de Respuesta: Dependientes del Flujo Másico. Producen una señal que es proporcional a la cantidad de soluto que pasa a través de él en la unidad de tiempo pero es independiente del volumen de gas portador requerido para la elución. Dependiente de la Concentración. Dan una señal proporcional a la cantidad de soluto por unidad de volumen de gas portador que pasa a través de él. Detectores según el proceso de detección Ionización, Óptico-espectroscópico, Electroquímico, etc. 7.2 Características de los Detectores Sensibilidad. Medida de la efectividad de un detector para convertir la muestra en una señal eléctrica medible. Capítulo 7 93 Detectores para cromatografía de gases y líquidos Linealidad. Rango de masa ó concentración de muestra sobre el cual el detector mantiene una sensibilidad constante sin una desviación arbitraria. El significado práctico de la linealidad del detector es el que le indica al analista la concentración para la cual el detector es confiable. Hay dos límites en la curva de linealidad: Límite de concentración inferior, que es dado por el límite de detección y, límite Superior, definido por un porcentaje de desviación arbitrario de la curva de linealidad, normalmente se toma un 5% de desviación. Rango Dinámico Lineal. Rango sobre el cual la sensibilidad del detector es constante. Ruido. Es cuantificado por el promedio de la amplitud pico-pico de la señal. El significado de conocer el nivel de ruido de un detector es un factor determinante en la determinación de la cantidad mínima detectable y el límite inferior del rango lineal. Límite de Detección. Está definido como la mínima cantidad de sustancia que puede producir una señal que sea el doble del nivel de ruido. Corriente de Fondo. Señal constante de salida generada por el proceso en el que un detector está operativo sin que alguna sustancia pasa a través de él. Esta señal es muy importante, ya que permite diagnosticar el buen o mal funcionamiento del detector. 7.3 Detectores en Cromatografía de Gases Detector de Conductividad Térmica (DCT). Mide la conductividad térmica del gas portador, ocasionada por la presencia de substancias eluídas. El funcionamiento del DCT esta basado en el hecho que la velocidad de pérdida de calor de un cuerpo caliente para un cuerpo más frío es proporcional, entre otros factores, a la conductividad térmica del gás que separa estos cuerpos. Un filamento metálico muy delgado (de W, Au o aleación W-Re) es calentado por el pasaje de una corriente eléctrica constante. Este filamento está colocado dentro de un orificio en un bloque metálico (celda), calentado a una temperatura más baja que aquella del filamento, por donde el gas de arrastre proveniente de la columna pasa continuamente (Fig.1). Mientras pasa gas de arrastre puro por la celda, la proporción de pérdida de calor del filamento para el bloque es constante y la temperatura del filamento no varía. Cuando un componente es eluido de la columna, este sale mezclado con el gas de arrastre y pasa por el detector. Si la conductividad de esta mezcla es diferente de aquella del gas de arrastre puro, el filamento pasa a perder calor para el bloque en una proporción diferente de aquella del equilibrio. Por ejemplo, si la proporción de pérdida de calor disminuye, el filamento se calienta cuando la muestra es eluida. El calentamiento del filamento causa una variación en Capítulo 7 94 Detectores para cromatografía de gases y líquidos su resistencia eléctrica y la resistividad de un metal aumenta con la temperatura. El filamento es montado en un circuito puente de Wheatstone, que transforma la variación en resistencia eléctrica del filamento en una variación de voltaje, que es colectada en un registrador generando el cromatograma. Figura 1. Celda de un detector de conductividad térmica. El DCT es un detector universal, sensible a la concentración del soluto en el gas de arrastre. Generalmente, cuando se usa DCT, el gas de arrastre es He o H2. Por el hecho de que estos gases tienen conductividades térmicas altas, las mezclas gas de arrastre más soluto siempre tendrán conductividades térmicas menores que la del gas de arrastre puro, lo que impede señales negativas, además de obtenerse factores de respuesta más grandes. Sin embargo, es considerado un detector poco sensible. La CMD de un modelo moderno, para propano, es de 400 pg/ml de gas de arrastre, con un rango lineal de 106. A pesar de eso, el hecho de ser universal, barato y de funcionamiento simple, lo hace extremamente útil para análisis que no necesitan de alta sensibilidad. Detector de Ionización a la Llama. Basado en la medida de las variaciones de la corriente de ionización en una llama oxígeno-hidrógeno debido a la presencia de substancias eluídas. Básicamente es un quemador de hidrógeno/oxígeno, donde se mezcla el efluente de la columna (gas portador y analito) con hidrógeno. Inmediatamente, este gas mezclado se enciende mediante una chispa eléctrica, produciéndose una llama de alta temperatura. La mayoría de compuestos orgánicos al someterse a altas temperaturas pirolizan y se producen iones y electrones, que son conductores eléctricos. Este hecho se aprovecha estableciendo una diferencia de potencial de unos centenares de voltios entre la parte inferior del quemador y un electrodo colector situado por encima de la llama. La corriente generada es baja (del orden de los 10-12 A), por lo tanto debe ser amplificada mediante un amplificador de alta impedancia (Fig. 2). El proceso de ionización que se da en la llama es complejo, pero se puede aproximar el número de iones producidos al número de átomos de carbono transformados en la llama. Esto produce que sea Capítulo 7 95 Detectores para cromatografía de gases y líquidos un detector sensible a la masa (al número de átomos de carbono que salen de la columna) más que a la concentración, por lo tanto no le afectan demasiado los cambios en el flujo de salida. Existen algunos grupos funcionales que no dan respuesta en este detector, como el carbonilo, alcohol, halógeno o amina, y tampoco responden gases no inflamables como el CO2, SO2, agua y óxidos de nitrógeno. Este hecho, más que limitar el ámbito de aplicación de este detector, permite el análisis de muestras contaminadas con alguno de los compuestos mencionados. Fig. 2. Detector de ionización de llama. Ventajas: ♠ Alta sensibilidad, del orden de 10 -13 g/s. 7 ♠ Amplio intervalo lineal de respuesta, 10 unidades. ♠ Bajo ruido de fondo (elevada relación señal/ruido). ♠ Bajo mantenimiento, fácil de fabricar. Desventajas: ♠ Destruye la muestra (la piroliza). Detector de Captura Electrónica. Basado en la electronegatividad de las substancias eluídas, y su habilidad para formar iones negativos por captura de electrones. El detector de captura electrónica es sensible en particular a las moléculas que contienen halógeno, carbonilos conjugados, nitrilos, nitrocompuestos y compuestos organometálicos, pero es relativamente insensible a los hidrocarburos, alcoholes y cetonas. El gas portador o el complementario tiene que ser o N2 o Ar con un 5% de metano. La humedad disminuye la sensibilidad. El gas que entra en Capítulo 7 96 Detectores para cromatografía de gases y líquidos el detector se ioniza por electrones de gran energía ("rayos beta") emitidos por una lámina que contiene 63 Ni radiactivo. Los electrones así formados son atrapados por un ánodo, produciendo una pequeña corriente continua. Cuando llegan moléculas de analito de gran electroafinidad captan algunos electrones. El detector responde modificando la frecuencia de los impulsos de voltaje entre el ánodo y el cátodo para mantener constante la corriente. Cuando estos compuestos se difunden a la estratosfera, catalizan la descomposición de las moléculas de ozono Detector de Fotometría a la Llama. Basada en la medida de la intensidad de la emisión molecular de la fluorescencia de heteroátomos en las moléculas orgánicas. La fotometría de llama se emplea para determinar el sodio y el calcio en una muestra biológica, debemos conseguir que ese sodio, en la forma que este en la muestra, pase a estar en forma de átomo de sodio libre en fase gaseosa. Debe producirse la activación de ese átomo pasando el electrón de valencia del nivel fundamental a niveles excitados, al volver de ese nivel al fundamental emite energía, se trata de cuantificar la intensidad de la energía emitida por los electrones al volver a su nivel fundamental. Necesitamos: Fuente de radiación, monocromador, detector. La fuente de radiación que provoca la activación de los átomos es una llama. El monocromador será en aparatos complejos filtros interferenciales y en aparatos sencillos redes de bajo poder de resolución. Los detectores podrán ser células fotovoltaicas o fototubos, pueden medir intensidades relativamente altas. Detector de Ionización de Llama Alcalina. El detector de nitrógeno fósforo, también llamado detector de llama alcalina, es un detector de ionización de llama modificado, que es especialmente sensible a compuestos que contienen nitrógeno y fósforo (pero que, en general, responde también a hidrocarburos.) En particular, tiene interés en análisis de medicamentos, pesticidas y herbicidas. Cuando estos elementos se ponen en contacto con una bola de vidrio, que contienen Rb2SO4 y que está en la punta de un mechero, producen iones que crean una corriente que se puede medir. Desde luego, no se puede usar N2 como gas portador, si se analizan muestras que contienen nitrógeno. Otros detectores de cromatografía de gases: Fotómetro de llama: ciertos elementos muy concretos, como P, S, Sn, Pb. De fotoionización: compuestos aromáticos e insaturados. De quimiluminiscencia de azufre: S Capítulo 7 97 Detectores para cromatografía de gases y líquidos De quimiluminiscencia De nitrógeno: N De emisión atómica: la mayoría de los elementos (seleccionados individualmente). Espectrómetro de masas: la mayoría de los analitos Espectrómetro de infrarrojos: la mayoría de los analitos Un detector fotométrico de llama mide la emisión óptica procedente del fósforo, azufre, plomo, estaño, y algún otro elemento concreto. Cuando el eluato pasa por una llama de H2-aire, análoga a la de un detector de ionización de llama, los átomos excitados emiten luz característica. Las emisiones del fósforo a 536 nm y del azufre a 394 nm se pueden aislar con un filtro interferencial de banda estrecha y detectar con un tubo fotomultiplicador. El detector de fotoionización utiliza una fuente ultravioleta de vacío para ionizar compuestos aromáticos y no saturados, pero apenas responde a hidrocarburos saturados. Recoge y mide los electrones producidos por ionización de estos compuestos. Un detector de quimiluminiscencia de azufre recoge los productos que salen de un detector de ionización de llama, entre los que se puede encontrar SO producido por oxidación de azufre, y lo mezcla con ozono, formándose SO2 en estado excitado, que emite luz azul y radiación ultravioleta. La intensidad de emisión es proporcional a la cantidad de azufre eluido, independientemente del compuesto de que procede, con una sensibilidad 107 veces mayor que la que tiene frente al carbono. Un detector de quimiluminiscencia de nitrógeno funciona de forma semejante. El eluato se quema a 1800 ºC, para transformar el nitrógeno en NO, que al reaccionar con el O3, produce un producto quimiluminiscente. Así mismo, la sensibilidad frente al nitrógeno es 107 veces mayor que frente al carbono. Un detector de emisión atómica conduce el eluato a un plasma de helio generado en una cavidad de microondas. Todos los elementos de la Tabla Periódica producen emisión atómica característica que se puede detectar con un policromador de fila de fotodiodos. Se puede sintonizar el detector para observar casi cualquier elemento presente en el analito que emerge de la columna. Por ejemplo, observando la emisión del selenio se pueden identificar trazas de compuestos de selenio, que dan el característico olor a ajos. El contenido total de selenio de ajos naturales es sólo de 0,28 μg de selenio por gramo de ajos. Capítulo 7 98 Detectores para cromatografía de gases y líquidos 7.4 Detectores en Cromatografía de Líquidos Los tipos de detectores en cromatografía de líquidos se clasifican en: Detectores basados en una propiedad de la fase móvil. Detectores basados en una propiedad de la sustancia a separar. Los detectores más utilizados en cromatografía de líquidos son: Detector UV. Hay básicamente tres tipos: Detector de Longitud de Onda Fija Detector de Longitud de Onda Variable Detector de Arreglo de Diodos El detector más común en HPLC es el detector de ultravioleta, que utiliza una celda de flujo como la que se muestra en la figura 3, porque muchos solutos absorben luz ultravioleta. Los sistemas más simples utilizan la intensa raya de emisión a 254 nm de una lámpara de mercurio. Los instrumentos más versátiles tienen lámparas de deuterio, xenón o volframio, y un monocromador, con el que se puede elegir la longitud de onda óptima, de ultravioleta o visible, para detectar los analitos estudiados. El sistema de la figura 4 utiliza una fila de fotodiodos, para registrar todo el espectro de cualquier soluto que pasa por el detector. Los detectores de gran calidad tienen intervalos de escala completa desde 0,0005 a 3 unidades de absorbancia. En la escala más sensible, una absorbancia de 0,0005, daría una señal del 100%, con un nivel de ruido del 1 %, de fondo de escala. El intervalo lineal cubre más de cinco órdenes de magnitud de concentración de soluto (que es otra manera de decir el intervalo en que se cumple la ley de Beer). Los detectores de ultravioleta están indicados para elución gradiente, y para disolventes que no absorben a la longitud de onda de trabajo. Detector de Índice de Refracción. Existen muchos diseños de estos detectores, pero solamente existen ahora dos tipos: Tipo Deflexión Tipo Fresnel Capítulo 7 99 Detectores para cromatografía de gases y líquidos Fig. 3. Camino óptico de una microcelda de un detector espectrofotométrico. Una celda ordinaria contiene un camino óptico de 0,5 cm y contiene sólo 8μl de líquido. Fig. 4. Detector ultravioleta de fila de diodos para HPLC. Un detector de índice de refracción responde a casi cualquier soluto, pero su límite de detección es aproximadamente 1000 veces peor que el de un detector de ultravioleta. El detector del tipo de deflexión, que se muestra en la figura 5, tiene dos compartimientos triangulares de 5 a 10 μl: a través de uno pasa disolvente puro, y a través del otro eluato. Para eliminar la radiación infrarroja (que calentaría la muestra), se hace pasar luz visible colimada (paralela) a través de la celda, con disolvente puro en los dos compartimientos, y se dirige a la fotocélula mediante la placa de deflexión. Cuando entra en la celda soluto de diferente índice de refracción, el haz se desvía, y varía la señal dada por la fotocélula. Los detectores de índice de refracción no sirven en elución gradiente, porque es imposible ajustar exactamente la muestra y la referencia mientras varía la composición del disolvente. Los detectores de índice de refracción son sensibles a las variaciones de presión y temperatura (~0,01 °C). Debido a su baja sensibilidad, los detectores de índice de refracción no sirven en análisis de trazas. Tienen intervalos Capítulo 7 100 Detectores para cromatografía de gases y líquidos pequeños de linealidad, que se extiende sólo en un factor de 500 de concentración de soluto. La principal ventaja de este detector es que es casi universal, respondiendo a todos los solutos, incluso a aquellos que no absorben en el ultravioleta. Fig. 5. detector de índice de refracción de tipo de deflexión. Detector de dispersión de luz. Responde a todos los solutos que son claramente menos volátiles que la fase móvil. Como se ve en la figura 6, el eluato entra en este detector por la parte superior. En el nebulizador, el eluato se mezcla con nitrógeno, y se le fuerza a pasar por un capilar, a cuya salida forma una fina dispersión de gotitas. El disolvente se evapora en un tubo caliente que se le hace recorrer, dejando una nube de finas partículas sólidas, que entran en la zona de detección por la parte inferior. Las partículas se detectan por la luz que procede de un diodo láser y llega al fotodiodo por dispersión. El detector de dispersión de luz responde a la masa del analito, no a su estructura o peso molecular. Si se observa un pico grande y uno pequeño, se puede estar seguro de que el pico pequeño corresponde a menos cantidad que el pico grande. Con un detector de ultravioleta, una pequeña cantidad de un analito que absorbe mucho da una señal más intensa que una cantidad grande de un analito que absorbe poco. La respuesta de un detector de dispersión de luz no es lineal, de modo que frecuentemente se recurre a polinomios para construir la curva de calibrado. El detector de dispersión de luz es compatible con una elución gradiente. Además, no hay picos asociados con el frente del disolvente, y así no se dan interferencias con los picos que se eluyen al principio. Capítulo 7 101 Detectores para cromatografía de gases y líquidos Si se usa un tampón en el eluyente, debe ser volátil, o de lo contrario se evaporara formando partículas sólidas, que dispersaran la luz y no dejaran discernir la señal del analito. Se pueden usar tampones de baja concentración a partir de ácido acético, fórmico o trifluoroacético, acetato de amonio, fosfato de diamonio, amoníaco o trietilamina. Fig. 6 Funcionamiento de un detector de dispersión de luz . Detector de Fluorescencia. Este detector solamente puede detectar compuestos que tengan fluorescencia nativa o inducida. La fluorescencia se detecta por medio de un detector fotoeléctrico colocado perpendicularmente respecto al haz de excitación. Semejantes a los detectores de absorbancia. Son altamente sensitivos Detectores Electroquímicos. Pueden ser clasificados en tres tipos: Detector Amperométrico, Detector Conductimétrico y Detector Potenciométrico Se basan en métodos electroquímicos (amperometría, voltamperometría, conductimetría y coulombimetría). Elevada sensitividad. El analito debe contener grupos susceptibles de sufrir procesos redox. Se deben tener algunas precauciones con los detectores electroquímicos para asegurarse análisis reproducibles: Checar que estén conectados adecuadamente a tierra la bomba, el detector y registradorintegrador. Usar bombas reciprocantes de doble pistón Mantener en todo momento el flujo de la fase móvil en el detector. Operar con el voltaje adecuado Capítulo 7 102 Detectores para cromatografía de gases y líquidos Monitorear la altura de los picos para observar cambios en la eficiencia que nos indique la necesidad de reacondicionar los electrodos. Tener electrodos de referencias extras en solución 3M de NaOH y reemplazar el electrodo de referencia en la celda 1 ó 2 veces a la semana. Desconectar el detector electroquímico cuando este limpiando las columnas. Utilizar agua, buffer y solventes orgánicos de alta pureza. Comparación de detectores comerciales usados en HPLC: a Detector límite de detección aproximado (ng) ¿útil en elución gradiente? De ultravioleta 0,1-1 Sí De índice de refracción 100-1000 No De dispersión de luz 0,1-1 Sí Electroquímico 0,01-1 No De fluorescencia 0,001-0,01 Sí De conductividad 0,5-1 No De espectrometría de masas 0,1-I Sí De infrarrojos con transformada de Fourier 1000 Sí 7.5 Bibliografía http://www.relaq.mx/RLQ/tutoriales/cromatografia/Gas.htm#detectores http://www.relaq.mx/RLQ/tutoriales/cromatografia/hplc.htm http://www.chemkeys.com/esp/md/mds_7/cgced_1/edpct(_4/edpct(_4.htm http://es.wikipedia.org/wiki/Detector_de_ionizaci%C3%B3n_de_llama http://mazinger.sisib.uchile.cl/repositorio/ap/ciencias_quimicas_y_farmaceuticas/apquim-aninstr14/harris/c24b.html http://www.elergonomista.com/tecnicas/fotometria.htm http://webservmida.mida.gob.pa/CYTED/CYTED%20PDF/tallerhomonologacionpma/anexos/CROMATOG RAFIA%20LIQUIDA%20HPLC.pdf http://mazinger.sisib.uchile.cl/repositorio/ap/ciencias_quimicas_y_farmaceuticas/apquim-an-instr14/harris/c25b.html http://www.pucpr.edu/titulov/Q420/examen%203/Cap%2028.pdf Capítulo 7 103 Procesamiento de datos cromatográficos Capítulo 8 Procesamiento de datos cromatográficos Cromatogramas Es la Representación de la respuesta o señal del sistema de detección continuo (CLAR o CG) o discontinuo en función del tiempo, volumen del eluyente o distancia en el lecho Cromatográfico. El cromatograma puede tomar varias diversas formas: un registro en la carta de un registrador o plotter, una mancha en cromatografía plana, o un número determinado de datos tomados por un microcomputador después de la correspondiente transformación. Según el tipo de detector continuo, el cromatograma puede ser diferencial, cuando la señal solo se origina al pasar analito por el mismo, e integral, cuando la señal se acumula (Figura 1). Figura 1. Tipos de Cromatogramas: (1) Diferencial, (2) Integral. El cromatograma contiene la información analítica relativa a la muestra (complejidad: número de picos, detección cualitativa y/o cuantitativa de uno o varios componentes) o del funcionamiento del sistema cromatografico. Los parámetros que definen el comportamiento cromatográfico de un soluto en un sistema cromatográfico de elución, y que sirve de base para los cálculos analíticos, que a continuación se comentaran. Para ello se usará el esquema de la Figura 2. Capítulo 8 104 Procesamiento de datos cromatográficos Figura 2. Parámetros que definen un cromatograma . Los aspectos termodinámicos y cinéticos de la separación cromatográfica quedan reflejados en el cromatograma, es decir, en la situación y forma del pico. Ajuste de datos El software realiza en primer lugar un ajuste de datos, con la finalidad de eliminar el ruido y permitir el correcto funcionamiento del algoritmo de integración. Con tal finalidad, se probaron distintos métodos optándose finalmente por splines de segundo orden con derivada primera continua. Este método permitió una mejor identificación del comienzo de pico y de los máximos, aún en condiciones de alto ruido. En la Figura 4 se muestra en detalle un trozo de cromatograma, en el que puede observarse la curva continua del ajuste y los datos con el ruido de la señal. Integración de los picos El algoritmo de integración del cromatograma es capaz de distinguir diferentes casos de picos cromatográficos en los cuales la línea de base se fija de distinta manera. Para ello se tiene en cuenta el valor de la señal, su derivada (D1) y su derivada segunda (D2). Picos simples: Se considera detectado el comienzo del pico cuando la derivada primera de la señal supera un valor prefijado (D1 > Ls) punto 2 de la Figura 5. Cuando la derivada pasa por cero siendo la derivada segunda negativa, se detecta el máximo (D1=0, D2<0) Punto 3 de la Figura 5. El final de pico se detecta cuando la derivada segunda es positiva y la derivada primera supera el valor prefijado (D1>Li) Punto 4 de la Figura 5. Los valores de Ls y Li pueden ser variables a lo largo del cromatograma, lo cual permite compensar el efecto de ensanchamiento de los picos. El tiempo de retención que identifica al componente es aquel en el cual ocurre el máximo, punto 3 de la Figura 5. Capítulo 8 105 Procesamiento de datos cromatográficos La línea de base es una recta que une los puntos 2 y 4, Figura 5. Para calcular el área se resta el área bajo la línea de base del área bajo la curva de la señal. Figura 5 Principio para la detección de los picos. Picos superpuestos: En el caso de dos o más picos fusionados o superpuestos se traza una línea de base entre el comienzo del primero y el fin del último. El criterio usual es trazar rectas verticales desde los valles hasta la línea de base común y en base a esta división se asignan las áreas. Figura 6 Detección de picos superpuestos Picos tangentes: Los picos pequeños montados sobre uno mucho mayor se denominan tangentes y su línea de base se traza de manera especial. Para detectarlo un criterio común es comparar alturas con la misma diferencia medida en el pico anterior, si este valor cae por debajo de un límite prefijado, el pico será considerado tangente. Figura 7 Ejemplo de pico tangente Colección y procesamiento de datos Capítulo 8 106 Procesamiento de datos cromatográficos El procesamiento de datos en cromatografía incluye tres objetivos: colectar y procesar la señal proveniente del detector de modo de producir un cromatograma y la información correspondiente como área de los picos, tiempos de retención y ancho de picos colectar y analizar los datos de modo de obtener información cualitativa y cuantitativa y generar los reportes correspondientes optimizar los parámetros cromatográficos. Detección El Detector es un dispositivo capaz de convertir una propiedad física, no medible directamente, en una señal elaborable y ofrece información sobre la naturaleza y magnitud de la propiedad física. Es la parte del cromatógrafo que se encarga de determinar cuándo ha salido el analito por el final de la columna. En cromatografía un detector funciona comparando una propiedad física entre la muestra a analizar y la sustancia portadora llevando cada uno de los componentes que previamente se han separado en la columna, esta acción se traduce en una señal tipo eléctrica, que posteriormente se amplificará mediante un registrador gráfico ó integrador permitiendo indicar el momento que salen de la columna los componentes. *Detectores según su Grado de Selectividad : *Detectores según el proceso de detección Ionización, Óptico-espectroscópico, Electroquímico, etc. *Detectores según su Modo de Respuesta: Características de los Detectores Sensibilidad. Medida de la efectividad de un detector para convertir la muestra en una señal eléctrica medible. o Linealidad. Rango de masa ó concentración de muestra sobre el cual el detector mantiene una sensibilidad constante sin una desviación arbitraria. El significado práctico de la linealidad del detector es el que le indica al analista la concentración para la cual el detector es confiable. Rango Dinámico Lineal: Rango sobre el cual la sensibilidad del detector es constante. Ruido. Es cuantificado por el promedio de la amplitud pico-pico de la señal. El significado de conocer el nivel de ruido de un detector es un factor determinante en la determinación de la cantidad mínima detectable y el límite inferior del rango lineal. Capítulo 8 107 Procesamiento de datos cromatográficos Límite de Detección. Está definido como la mínima cantidad de sustancia que puede producir una señal que sea el doble del nivel de ruido. Corriente de Fondo. Señal constante de salida generada por el proceso en el que un detector está operativo sin que alguna sustancia pasa a través de él. Esta señal es muy importante, ya que permite diagnosticar el buen o mal funcionamiento del detector. El detector debe ser sensible a los efluentes de la columna y capaz de suministrar un registro de la cromatografía en la forma de un cromatograma. La señal del detector debe ser proporcional a la cantidad de cada soluto (analito). Con lo cual debe ser posible realizar un análisis cuantitativo. Registro e integración La señal proveniente del detector, se transmite a un sistema de registro e integración, el cual genera un cromatograma que representa un registro del análisis. Los datos obtenidos son procesados mediante un registrador e integrador sin ninguna o muy poca capacidad de procesamiento, o a través de sistemas computarizados que incluyen un procesamiento posterior (post-run analysis) mas sofisticado y completo mediante el empleo de un software apropiado. En la mayor parte de los casos, el sistema integra automáticamente el área de cada pico, realiza los cálculos e imprime un reporte con los resultados cuantitativos y los tiempos de retención. Cálculo de área del pico (análisis cuantitativo) La cromatografía tuvo un gran crecimiento durante las anteriores cuatro décadas, en parte a que se trata de una técnica rápida, sencilla, de bajo costo y esencialmente a su gran aplicación como herramienta de separación. No obstante su gran éxito se debe sin duda a que también proporciona información cualitativa acerca de las especies separadas. La cromatografía en columna cuantitativa tiene como principio la comparación de las alturas, o de las áreas del pico del analito con la de uno o más patrones, en la cromatografía en plano el área ocupada por las especies separadas sirve como parámetro analítico, si las condiciones son controladas adecuadamente esos parámetros varían linealmente con la concentración, es decir, que el área del pico cromatografico es directamente proporcional a la concentración del analito, así es fundamental para la confiabilidad del análisis que el área de los picos sea medida lo más exacto y reproduciblemente posible. Los instrumentos cromatográficos modernos estas equipados con integradores electrónicos digitales, los cuales permiten una precisa estimación de las áreas de los picos, si no se dispone de tales equipos tienen que hacerse una estimación manual. Existen varias maneras de medir el área de los picos cromatográficos. Una de Capítulo 8 108 Procesamiento de datos cromatográficos esta técnicas consiste en suponer que el pico se asemeja a un triangulo isósceles, para lo cual se mide la altura del pico (h) y el ancho de la base del pico (Wb) o ya sea la mitad de la altura (Wh) y se calcula el área del pico empleando la formula usada para el calculo del área de un triangulo. 𝑨= 𝒃×𝒉 𝒉 × 𝑾𝒃 ∴ 𝑨= ó 𝑨 = 𝒉 × 𝑾𝒉 𝟐 𝟐 Las limitantes de esta técnica es que su utilización depende de la simetría y del ancho del pico. Otra técnica utilizada para determinar el área del pico es la llamada corte y pesada esta técnica consiste en recortar el pico del cromatograma, luego se pesa en una balanza analítica y se determina su peso relativo al peso de un área conocida de papel de registro. Dicho recorte y pesada depende mucho de la habilidad del operador, pueden introducirse errores por cambios en la humedad del papel, homogeneidad del papel, etc. Para la utilización de esta técnica se recomienda utilizar una fotocopia del cromatograma para no destruir el original. En general las técnicas de integración manual proporcionan áreas que son reproducibles a un nivel del 2 al 5 por 100; por el contrario los integradores digitales son al menos un orden de magnitud más precisos. Como ya se menciona anteriormente los cromatógrafos modernos contienen integradores electrónicos los cuales son dispositivos que se basan en microprocesadores que colectan la señal cromatografica, digitalizándola, es decir, que transforma una señal eléctrica en números, detectan la presencia del pico y calcula su área, los integradores son más precisos y rápidos que cualquier método manual para determinar el área del pico cromatografico, aunque son dispositivos caros, cuando un análisis requiere de rapidez en la producción de resultados su uso es casi indispensable. Otra manera de obtener el área del pico es sustituir el integrador por un computador el cual es un dispositivo que convierte la señal eléctrica en números que puedan guardarse en una memora (conversor analogo-digital) y que disponga de un programa para la realización del análisis del cromatograma digitalizado, Capítulo 8 109 Procesamiento de datos cromatográficos el costo del computador con los accesorio necesarios para hacer el análisis es por lo general inferior al de un buen integrador, además con el computador se obtiene resultados más confiables. Desarrollo y optimización de métodos Cromatografía de gases (rampas de temperatura) El control de la temperatura de la columna es una de las formas más sencillas y más efectivas de influenciar la separación de los componentes. La columna se fija entre un inyector mantenido a una temperatura de inyección, y un detector mantenido a una temperatura también predeterminada. Por lo tanto, se debe definir la temperatura a la que cada uno de los componentes opera. En general, caben dos posibilidades de modificación de las condiciones de trabajo cromatográficas. En cromatografía gaseosa, la temperatura debe ser mantenida por encima del “punto de rocío” de la muestra, pero no por encima de su punto de ebullición. (a) Modalidad Isocrática, en la que las condiciones experimentales permanecen constantes durante el proceso cromatográfico. (b) Modalidad no Isocrática, en la que durante el proceso de separación cromatográfica se varia de manera gradual y estrictamente controlada el parámetro de temperatura. La temperatura es una variable importante, ya que de ella va a depender el grado de separación de los diferentes analitos. Para ello, debe ajustarse con una precisión de décimas de grado. Dicha temperatura depende del punto de ebullición del analito o analitos, y por lo general se ajusta a un valor igual o ligeramente superior a él. Si tenemos varios componentes con diferentes puntos de ebullición, se ajusta la llamada rampa de temperatura con lo cual ésta va aumentando ya sea de forma continua o por etapas. En muchas ocasiones, el ajustar correctamente la rampa puede significar separar bien o no los diferentes analitos. Es recomendable utilizar temperaturas bajas para la elución, pero conforme la temperatura es mayor la elución es más rápida, pero corriendo el riesgo de descomponer el analito. Por ejemplo: Si al meter a eluir una muestra nos salen cuatro analitos con tiempos muy diferentes de elución la temperatura adecuada para que eluyan todos se calcula con la rampa de temperatura: Capítulo 8 110 Procesamiento de datos cromatográficos Se toman los primeros 5 minutos como 60 °C y a partir de estos 5 minutos cada minuto adicional se le aumenta 10°C, por ello observamos en el primer pico 80°C, ya que los primeros 5 minutos se toman como 60° más 2 minutos se le suman 10°C esto es precisamente 80°C y así, se trabaja con los siguientes picos, tomando en cuenta la escala de temperatura. Esto es muy importante ya que si en nuestra muestra solo se vieran dos picos uno en 11 minutos y otro en 20 la temperatura e elución correcta seria una que estuviera entre estos dos puntos, con lo que se correría muy bien la muestra y no tendríamos que esperar mucho entre a y otra. HPLC (Cambios en la fase móvil.) El proceso de elución en cromatografía de adsorción, el disolvente compite con las moléculas de soluto por ocupar los sitios activos de la fase estacionaria. La diferente capacidad de los distintos disolventes para eluir un determinado soluto del adsorbente es independiente de la naturaleza del soluto. Se puede describir la elución como el desplazamiento de un soluto de la fase estacionaria por un disolvente La fuerza eluyente (ε0 ) es una medida de la energía de adsorción del disolvente, supuesto el valor de cero para el sistema pentano/ sílice pura. Cuanto más polar es el disolvente, mayor es su fuerza eluyente Capítulo 8 111 Procesamiento de datos cromatográficos respecto a la sílice en cromatografía de adsorción. Cuanto mayor es la fuerza eluyente del disolvente , tanto más rápidamente se eluirán los solutos de la columna. La cromatografía de adsorción sobre sílice pura es un ejemplo de cromatografía de fase normal, que se caracteriza por usar una fase estacionaria polar y un eluyente menos polar. Un disolvente más polar tiene fuerza eluyente mayor. La cromatografía de fase inversa, que es la más utilizada, se caracteriza por que la fase estacionaria es no polar o débilmente polar y el disolvente es más polar. Un disolvente menos polar tiene mayor fuerza eluyente. La elución isocrática se lleva a cabo con un único disolvente (o una mezcla de disolventes fija). Si un disolvente no permite una elución suficientemente rápida de todos los componentes, se puede usar una elución en gradiente; en cual, hay un cambio continuo de la composición del eluyente en sentido de aumento de fuerza eluyente. Para eluir solutos fuertemente retenidos hay que aumentar la fuerza eluyente del disolvente. En este caso, se van añadiendo cantidades crecientes del disolvente B al disolvente A, produciendo así un gradiente continuo el análisis utilizando el método de elución por gradiente permite mantener una resolución deseada y al mismo tiempo disminuir la duración del análisis. Por ejemplo en la figura 15 muestra el efecto de aumentar la fuerza eluyente de la elución isocrática de ocho componentes en una columna de fase inversa. En una separación con fase inversa , la fuerza eluyente disminuye a medida que el disolvente se hace más polar . El primer cromatograma (superior izquierda) se obtuvo con un disolvente que tenía 90% de acetonitrilo y 10% de tampón acuoso. El acetonitrilo tiene una gran fuerza eluyente, y eluye todos los compuestos rápidamente. Se observan sólo 4 picos por que los demás están solapados. Es habitual llamar disolvente A al componente acuoso, y disolvente B al orgánico. El primer cromatograma se obtuvo con un 90% de B. Cuando se redujo la fuerza eluyente utilizando un disolvente con 80% de B, se consigue una separación un poco mejor, observándose 5 picos. Con 60% de B, se empiezan a ver 6 picos. Con 40% de B se ven claramente 8 picos. Con 30% de B, todos los picos quedan bien resueltos, pero la separación tarda demasiado (cerca de 2 horas). A partir de los datos obtenidos con las elusiones isocráticas de figura 13, se eligió el gradiente que se presenta en la figura 25.11, con el que se consiguió resolver todos los picos en 38min. Primero se trabajo con 30% de B (B=acetonitrilo) durante 8 minutos, para separar los componentes 1,2,3. A continuación se fue aumentando de forma continua la fuerza eluyente durante 5 minutos hasta alcanzar un 45% de B, y se mantuvo durante 15 minutos, para eluir los picos 4 y 5. Finalmente se modifico el disolvente hasta alcanzar un 80% de B en 2 minutos, manteniéndose esa composición para eluir los últimos picos. Capítulo 8 112 Procesamiento de datos cromatográficos Figura 15. Separación isocrática en HPLC de una mezcla de compuestos aromáticos en una columna Hypersil ODS (C18 sobre sílice de 5μm), de o.46 x 25cm, caudal de 1.0ml/min,temperatura ambiente ( ˜22°C). (1) alcohol bencílico, (2) fenol, (3)3¨,4¨-dimetoxiacetofenona, (4) benzoína, (5) benzoato de etilo, (6) tolueno, (7)2,6-dimetoxitolueno, (8) ometixibifenilo.El eluyente estaba formado por un tampón acuoso (designado por A ) y acetonitrilo (designado por B). El tampón contenía KH2PO4 25mM y 0.1 g/L de azida de sodio con el pH ajustado a 3.2 con HCl Aplicaciones La cromatografía es utilizada para análisis del principio activo de un producto farmacéutico, por ejemplo la determinación de cafeína y ácido acetilsalicílico en cafiaspirina , los siguientes datos fueron obtenidos para determinar la cantidad de cafeína y ácido acetilsalicílico contenidos una tableta de cafiaspirina. En este caso se realizo el análisis en HPLC Para tal análisis se tomo como estándar las siguientes concentraciones de cafeína y ácido acetilsalicílico: Ccafeína = 0.03mg/mL CAAS = 0.53mg/mL Se realizo el análisis y se obtuvieron los siguientes resultados. Cromatograma de la inyección número 1 del estándar Capítulo 8 Cromatograma de la inyección número 3 del estándar 113 Procesamiento de datos cromatográficos Cromatograma de la inyección número 4 del estándar Cromatograma de la inyección número 5 del estándar Cafeína Ácido Acetilsalicílico Núm. Inyección Área Núm. Inyección Área 1 149019 1 615352 2 187639 2 784811 3 162884 3 684670 4 201409 4 874595 5 184141 5 797979 Ã=177018.4 Ã=751481.4 A partir de estos datos se obtuvo el factor de respuesta para cada componente en estudio con la siguiente ecuación A=FrC. Fr cafeína = 177018.4 / 0.03 = 5900613.33 Fr AAS = 751481.4 / 0.53 = 1417889.43 Análisis de la tableta de CAFIASPIRINA Posteriormente se analizo la tableta de Cafiaspirina (lote 7605H2, fecha de caducidad Julio 2009), obteniendo los siguientes datos. Capítulo 8 114 Procesamiento de datos cromatográficos Cromatograma de la inyección #1 de la muestra problema Cromatograma de la inyección #2 de la muestra problema Cromatograma de la inyección # 3 de la muestra problema Cafeína Ácido Acetilsalicílico Núm. Inyección Área Núm. Inyección Área 1 177343 1 678496 2 178856 2 717425 3 182458 3 723643 Ã= 179552.3 Ã= 706521.3 Con los datos de las áreas y el factor de respuesta se obtuvieron la cantidad presente del de cafeína y ácido acetilsalicílico en cada tableta. Capítulo 8 115 Procesamiento de datos cromatográficos C cafeína = 179552.3 / 5900613.33 = 0.0304 0.0304 * 10/ 1 * 100 = 30.4 mg Cada tableta de cafiaspirina contiene 30.4 mg de cafeína CAAS = 706521.3 / 1417889.43 = 0.4983 0.4983 * 10 / 1 * 100 = 498.3 mg Cada tableta de cafiaspirina contiene 498.3 mg de ácido acetilsalicílico Determinación del grado alcohólico de una bebida Conocer el contenido de etanol en muestras analizadas a partir del método del estándar interno. Se analiza el tequila comercial “Herradura”, EtOH 40% Se trabajo a 70°C y se realizaron corridas a diferentes concentraciones de EtOH (2, 4, 6, 8, 10%v/v) y una concentración constante de Acetona (6%v/v; estándar interno. Para la curva patrón, las diferentes disoluciones se realizaron a partir de una solución Stock de EtOH al 20%. En los cromatogramas aparecen 2 picos con 2 áreas diferentes. El primer pico y área corresponde a la acetona, ya que es el compuesto menos polar. El segundo correspondo al del EtOH. Los valores de área para la curva patrón: Área [Acet] [Acetona](%) Área [EtOH] [EtOH](%) 752578 6 225296 2 394769 6 125961 2 175540 6 57133 2 Promedio 440962.3333 Promedio Capítulo 8 136130 270166 6 198196 4 251991 6 183560 4 168917 6 142527 4 230358 174761 183014 6 207592 6 177426 6 219475 6 116 Procesamiento de datos cromatográficos 234651 Promedio 6 282560 198363.6667 Promedio 236542.3333 276769 6 382117 8 145605 6 228924 8 211187 Promedio 6 305520.5 178488 6 341568 10 168642 6 350407 10 266046 6 486201 10 204392 392725.3333 Curva Patrón - Estándar Interno 2.5 Ae/Aei 2 1.5 1 0.5 0 0 0.5 1 1.5 Ce/Cei 2 y = 1.2536x - 0.075 R2 = 0.9868 Frr=1.2536 Ae/Aei=Frr*(Ce/Ci) Tequila "Herradura" (EtOH 40%) E.I. Promedio Capítulo 8 Área [Acet] [Acet] (%) Área [EtOH] R=[EtOH] (%) 250005 6 300125 5.789146394 213666 6 288804 6.518217346 294464.5 6.125112357 231835.5 117 Procesamiento de datos cromatográficos Los resultados obtenidos en la tabla se obtuvieron con cálculos de regresión lineal, de la misma manera que se realizaron para la muestra del destilado. Tomando en cuenta el factor de dilución utilizado se encontrará la concentración (%v/v) del etanol en la muestra de Tequila. Para hacer la dilución de la muestra se tomaron 1.5ml del tequila, y se aforaron a 10ml para llevarlo a una concentración del 6% (equivalente a la de la acetona, el estándar interno). E.I.: 6.12511 10ml 30.6255% 2ml Según la información del producto, este indicaba que el porcentaje de etanol que contenía es del 40%. El estudio cromatográfico, encontró una concentración del 30%. El estudio de la muestra a través del estándar interno, disminuye el error caudado por la inyección es por ello que el resultado es confiable. La cromatografía tiene una gran aplicación una de ellos es la determinación del grado alcohólico de una muestra para comparar con el porcentaje reportado por el fabricante. Capítulo 8 118 Procesamiento de datos cromatográficos RESUMEN Los cromatogramas. son la representación de la respuesta o señal del sistema de detección continuo (CLAR o CG) o discontinuo en función del tiempo, volumen del eluyente o distancia en el lecho cromatográfico. Contiene la información analítica relativa a la muestra (complejidad: número de picos, detección cualitativa y/o cuantitativa de uno o varios componentes) o del funcionamiento del sistema cromatográfico. Los aspectos termodinámicos y cinéticos de la separación cromatográfica quedan reflejados en el cromatograma, es decir, en la situación y forma del pico. La colección y procesamiento de datos así como su análisis se realiza con el fin de obtener información cualitativa y cuantitativa y generar los reportes correspondientes, obteniéndose los datos a partir de un detector; éste es un dispositivo capaz de convertir una propiedad física, no medible directamente, en una señal elaborable que ofrece información sobre la naturaleza y magnitud de la propiedad física. La señal proveniente del detector se transmite a un sistema de registro e integración, el cual genera un cromatograma. Los datos obtenidos son procesados mediante un registrador e integrador. La cromatografía en columna cuantitativa tiene como principio la comparación de las alturas, o de las áreas del pico del analito con la de uno o más patrones, en la cromatografía en plano el área ocupada por las especies separadas sirve como parámetro analítico, el área del pico cromatografico es directamente proporcional a la concentración del analito, por lo cual es fundamental para la confiabilidad del análisis que el área de los picos sea medida lo más exacto y reproduciblemente posible. Existiendo así varias maneras manuales de medir el área de los picos cromatográficos; sin embargo, son los instrumentos cromatográficos modernos equipados con integradores electrónicos digitales son los más precisos y rápidos que cualquier método manual para determinar el área del pico cromatografico. Se ha logrado un desarrollo y optimización de métodos a través de ciertas modificaciones en las condiciones de trabajo tales como las rampas de temperatura utilizadas en cromatografía de gases en donde el control de la temperatura de la columna es una de las formas más sencillas y más efectivas de influenciar la separación de los componentes. En general, caben dos posibilidades de modificación de las condiciones de trabajo cromatográficas. La modalidad Isocrática, en la que las condiciones experimentales permanecen constantes durante el proceso cromatográfico y la modalidad no Isocrática, en la que durante el proceso de separación cromatográfica se varia de manera gradual y estrictamente controlada el parámetro de temperatura. Capítulo 8 119 Procesamiento de datos cromatográficos Otra modificación en el caso de cromatografía de líquidos de alta eficiencia es cambios de composición de la fase móvil. En donde sí un disolvente no permite una elución suficientemente rápida de todos los componentes, se usa una elución en gradiente; en cual, hay un cambio continuo de la composición del eluyente en sentido de aumento de fuerza eluyente. Para eluir solutos fuertemente retenidos hay que aumentar la fuerza eluyente del disolvente con el fin de producir análisis manteniendo una resolución deseada y al mismo tiempo disminuir la duración de éste. La cromatografía es utilizada para análisis del principio activo de un producto farmacéutico, Otra de las aplicaciones de la cromatografía en este caso la de líquidos, es la determinación del grado alcohólico de una bebida. Entre muchas otras más. BIBLIOGRAFÍA M. Valcárcel Cases, A. Gómez Hens, Técnicas analíticas de separación, Editorial Reverte, España, 1990, pp778 Lucas Hernández Hernández, Claudio González Pérez, Introducción al análisis instrumental,1ª Edición, Editorial Ariel S.A, Barcelona, 2002. pp 456 Douglas A. Skoog, F. James Holler, Principios de Análisis Instrumental, Quinta Edición, Editorial Mc. Graw Hill, España 2001, pp 753 Daniel C. Harris, Análisis Químico Cuantitativo, Segunda Edición en Español, Editorial Reverte, España 2001. http://www.relaq.mx/RLQ/tutoriales/cromatografia/Gas.htm Capítulo 8 120 Procesamiento de datos cromatográficos Determinación de Azúcares por CGL a través del método de derivados TMS Este método se recomienda para la identificación y cuantificación de los monosacáridos y disacáridos presentes en diversos alimentos frescos o industrializados, después de la correspondiente derivatización de éstos para su análisis por CGL con detector FID. Fundamento del Método. Se basa en la obtención de derivados de trimetilsililados (TMS) de los carbohidratos presentes en el alimento para su posterior análisis en CGL. La derivatización se realiza en 2 pasos: formación de la oxima del azúcar correspondiente con hidroxilamina clorhidrato en piridina y silanización con hexamil disilazano (HMDS) y ácido trifluoroacético (TFA). La identificación se efectúa por los tiempos de retención y la cuantificación por el método del estándar interno. Procedimiento. Para productos lácteos (yogur o leche) se debe liofilizar una alícuota de aproximadamente 100mg. Para vegetales frecos se extrae en baño de agua a 50ºC durante 1 hora con etanol 70%. El extracto se rotovapora a sequedad en evaporador rotatorio de vacío a 50ºC como máximo. Añadir a cada frasco de muestra 1 mL de solución de estándar interno (2mg/mL de m-inositol o 1mg de xilosa) antes de liofilizar. Preparación Estándar. Pipetear 1mL de cada solución de estándares y estándar interno en etanol en un matraz de fondo cónico y boca esmerilada. Llevar a sequedad en el evaporador rotatorio a una temperatura no mayor de 40ºC. Derivatización. Se agrega a cada frasco de muestra liofilizada y a la mezcla de estándares 1mL de solución de hidroxilamina clorhidrato lavando cuidadosamente el recipiente. Se trasvasa hacia un tubo de tapa rosca y se coloca durante media hora en baño de agua a 75-85ºC con agitación ocasional. Se toman 100 µL de la solución de piridina de la muestra y se pasa a un vial ambar se añaden 150 µL de HMDS y 1 gota de TFA, se deja reposar hasta la aparición de precipitado blanco. Análisis por CGL Condiciones operacionales Flujo de gas acarreador (nitrógeno): 35mL /min. Temperatura: Detector: 300ºC, Inyector: 250ºC, Horno (programado): 180ºC (2min) a 8ºC/min, hasta 250ºC. Se inyectará 1 µL de la mezcla de estándares hasta obtener reproducibilidad en la respuesta del equipo. Es decir que el factor de respuesta (km) debe de ser mínima la variación. Ajustar las demás Capítulo 8 121 Procesamiento de datos cromatográficos condiciones de operaciones de tal forma que el estándar de glucosa brinde una señal aproximadamente igual a la mitad de la escala del registrador o el dispositivo de salida del cromatógrafo de gases. Los azúcares se identifican por comparación con los tiempos de retención de cada estándar correspondiente. La concentración se determina por el método de estándar interno utilizando las fórmulas siguientes. Para la inyección de la mezcla de estándares puros: (1) Donde: Km = Factor de respuesta para el azúcar (ej glucosa) AM = Área del pico del estándar correspondiente (ej. Glucosa) Mi = Masa tomada del estándar interno utilizado (2mg para inositol o 1 mg para xilosa) Ai = Área del pico estándar interno MM = Masa tomada del estándar correspondiente (1mg de glucosa) Se calculan tantos valores de km como azúcares se cuantifiquen. Para la muestra el peso de cada azúcar se calcula por la siguiente fórmula: (2) Donde: MM = peso del azúcar correspondiente en la muestra AM = área del pico del azúcar en la muestra (mm) Mi = masa del estándar interno añadido a las muestras (mg) Ai = área del pico estándar interno añadido a las muestras (mm) Km = valor promedio obtenidos mediante la fórmula (1) para el azúcar correspondiente. Un cromatograma típico de la separación de algunos TMS derivados de azúcar se muestra en la figura1. Capítulo 8 122 Procesamiento de datos cromatográficos 2. Se desea determinar el contenido de etanol en una bebida alcohólica. Para ello se realizó un análisis cromatográfico utilizando una curva de calibración relativa. Preparación de soluciones Solución de estándar interno (EI). Solución de acetona 10% Solución patrón (SP). Solución de etanol al 10% Empleando estas soluciones se preparó la siguiente curva de calibración Tabla 2. SP (mL) EI (mL) Volumen final (mL) 0.1 1 10 0.5 1 10 1.0 1 10 1.5 1 10 2.0 1 10 De cada concentración de la curva se inyectó 1µL en el cromatógrafo. Preparación de la muestra Se tomaron 5 mL de licor, se adicionó 1mL de acetona (EI), se aforó a 10ml con agua. La muestra se inyectó en el cromatógrafo 5 veces (1µL). Tr acetona = 0.79min y tr etanol = 1.38min Capítulo 8 123 Procesamiento de datos cromatográficos Condiciones cromatagráficas Cromatógrafo de gases integrado con inyector split/splitless, detector de ionzación de flama y una columna de sílice fundida SP 1000, fase polar (30m x0.32mm x 0.251µm). Temperatura del inyector y detector 200ºC. Programa de temperatura: temperatura inicial 80ºC / 1.3 min, incrementandose a 20ºC/min hasta 130ºC y mateniendose a esa temperatura durante 5 min. Resultados Curva estándar (n=3) Tabla 3. Área EtOH Àrea EI Promedio 1 2 3 1 2 3 Promedio 29054.33 30556 25864 30743 290080 249370 287220 275556.6 123820 109170 123050 139240 214770 246810 266110 242563.3 243853.3 228380 247660 255520 241720 263180 261540 255480 360023.3 321700 371000 387370 220850 254990 265390 247076.6 468270 489390 481950 433470 261410 261700 247950 257020 Con respecto a estos resultados obtenidos se calculo el promedio de cada repetición. Muestra (n=5) Tabla 4. Capítulo 8 Inyección Área de etanol Área acetona 1 296800 187150 2 290840 185900 3 287960 185880 4 285800 183680 5 291010 187180 Promedio 29.0482 186078 124 Procesamiento de datos cromatográficos Tomando en cuenta los promedios de las áreas tanto de Etanol como del Estándar Interno (tabla 3.) así como la relación entre éstas y de las concentraciones tanto de la solución patrón como la interna que se muestran en la tabla 2. Se grafica y se obtiene la pendiente que en este caso es el Frr (factor de respuesta relativo); en base a éste dato se puede determinar la concentración de etanol en la muestra. Área de Etanol Área EI AEtOH / AEI [sp] /[EI] 29054.33 275556.6 0.1054 0.1 123820 242563.3 0.5105 0.5 243853.3 255480 0.9545 1 360023.3 247076.6 1.4571 1.5 468270 257020 1.822 2.0 Curva Patrón y = 0.9114x + 0.0403 R2 = 0.9974 AEtOH / AEI 2 1.5 1 0.5 0 0 0.5 1 1.5 2 2.5 [sp] /[EI] Por lo tanto tenemos que: m = Frr =0.9114 Y sabemos que: Frr AEtOH EI AEI EtOH Por lo tanto despejando y tomando en cuenta los promedios de la tabla 4. con las respectivas áreas de la muestra tenemos que: EtOH 290,4820.1 0.1713 0.911 186078 [EtOH] v/v = 0.1713 x 100 = 17.13 % de Etanol por cada 100ml de bebida alcohólica en este caso licor. Capítulo 8 125 Procesamiento de datos cromatográficos Estudio sobre los ácidos grasos libres en queso blanco Fundamento del método. Los ácidos grasos libres juegan un papel importante en el sabor de muchos quesos y se producen por la hidrólisis de los triglicéridos de la grasa por las lipasas nativas de la leche, las lipasas microbianas y las lipasas de las células somáticas. También se liberan durante el metabolismo de los carbohidratos y aminoácidos por las bacterias. Los ácidos grasos de bajo peso molecular son consecuencia de la fermentación bacteriana, mientras que los ácidos grasos restantes son el resultado de la acción de la lipasa [1]. Se ha demostrado que los ácidos grasos libres deben estar presentes dentro de un rango específico y a un nivel óptimo de concentración para un sabor deseable. Cantidades excesivas de ácidos grasos libres se asocian con rancidez hidrolítica y causan sabores desagradables. Procedimiento. Los ácidos grasos libres (AGL) analizados fueron: butírico (C4), caproico (C6), caprílico (C8), caprico (C10), laurico (C12), miristico (C14), palmitico (C16), estearico (C18), oleico (C18:1) y linoleico (C18:2). Extracción de los lípidos del queso. Se mezclaron 2 g de queso rayado con 6 g de sulfato de sodio hidratado para absorber la humedad y 0,6 ml de ácido sulfúrico 2,5 mol/l. Se procedió a la extracción de los lípidos con 6 ml de eter/heptano (1:1 v/v) en un tubo de centrífuga de 75 ml mediante centrifugación a 2.500 rpm/min por 2 min. Esta extracción se repitió dos veces añadiendo la misma cantidad de eter/heptano (1:1 v/v) al residuo. Los tres extractos se mezclaron. Separación de los ácidos grasos libres. Se utilizó como estandar interno el ácido pelargónico (C9) (0,043 mg) el cual se adicionó al extracto lipídico antes de proceder al aislamiento de los ácidos grasos libres. Para tal fin se utilizaron columnas aminopropílicas de 3 ml (Supelclean LC-NH2 de Supelco, Inc) acondicionadas con 2 ml de heptano. El volumen total del extracto se aplicó a la columna con presión positiva. Los lípidos neutros fueron eluídos con 3 ml de cloroformo/2-propanol 2:1 v/v. A continuación los ácidos grasos libres adsorbidos en la columna fueron eluídos con 3 ml de dietil eter con 2% de ácido fórmico. El primer ml se descartó por no contener ácidos grasos libres. De los siguientes 2 ml eluídos, con la totalidad de los AGL, se inyectó 1 ml al cromatógrafo para su determinación. Se realizaron 2 extracciones de cada muestra de queso y cada extracción se inyectó por duplicado al cromatógrafo [2,3]. Las condiciones de operación en el cromatógrafo fueron: Temperatura inicial 90ºC Variación de temperatura 20ºC/min Tiempo Inicial 1 min Temperatura final 220ºC Capítulo 8 126 Procesamiento de datos cromatográficos Tiempo final 10 min Flujo de hidrógeno 30 ml/min 500 ml/min Temperatura del detector 250ºC Flujo de aire Temperatura del inyector 250ºC Purga Flujo de helio 20 ml/min 1 min Cuantificación de los AGL. Se utilizó un cromatógrafo de gas Hewlett Packard 5890 equipado con un detector de ionización de llama y una columna capilar de sílica de 15 m x 0,53 mm (Nukol de Supelco, Inc.) calibrado con soluciones de referencia de los ácidos grasos a analizar (Sigma) [6]. Se adaptó inyección directa debido a las bajas concentraciones de los ácidos grasos. La cuantificación se realizó relacionando las áreas de los picos con el área del estándar interno (C9) mediante un integrador Hewlett Packard 3393A. Se obtuvo la media y la desviación estandar de todos los AGL. Resultados La Tabla 1 y las Figuras 1 y 2 muestran la composición y los cromatogramas de los ácidos grasos libres de los quesos Palmita y Semiduro. Se ha podido comprobar la presencia en los quesos de los AGL saturados del butírico (C4) al palmítico (C18), así como de los mono-insaturados y poliinsaturados, el ácido oleico (C18:1) y linoleico (C18:2) en concentraciones muy diversas entre 2.3 g/g (C6) en el queso Palmita y 185.1 g/g (C18:1) en el queso Semiduro, con el predominio de los de mayor peso molecular, palmítico (C16) y oleico (C18:1), sobre los de menor peso molecular, entre los cuales resalta el butírico (C4). Capítulo 8 120 Procesamiento de datos cromatográficos Se observan amplias variaciones entre las muestras, debido posiblemente a la influencia de las diferentes condiciones del proceso de elaboración en la composición de los quesos, así como la manipulación y almacenamiento durante el período de comercialización. La columna utilizada proporcionó excelentes resultados sin la necesidad de derivatizar para obtener los ésteres metílicos previo a su cuantificación. Al disminuir una etapa del análisis se evita una posible fuente de error y se consigue un ahorro de tiempo. Estudio de validación de la Metodología para la determinación de Vitamina A en Alimentos infantiles instantáneos por HPLC Fundamento. Los alimentos infantiles instantáneos, en su gran mayoría, son fortificados normalmente utilizando formulaciones especiales que mejoran su estabilidad y valor nutritivo, siendo la ingesta diaria recomendada de 400-700 g devitamina A, para niños de 1-10 años de edad 3. En ese sentido, y buscando una óptima metodología de análisis, en el presente estudio se realizó la validación de la metodología por HPLC para la determinación de vitamina A contenida en alimentos infantiles instantáneos. Procedimiento. Se utilizó un equipo de cromatografía líquida de alto rendimiento (HPLC), marca Shimadzu, modelo LC10A, con inyector automático, bomba con sistema de degasificación, con integrador o sistema de registro, software para el procesado de datos cromatográficos, detector de arreglo de diodos (longitud de onda variable). La columna cromatográfica fue de acero inoxidable LC-18 para fase reversa, de 25 cm x 4,6 mm de diámetro interno, 5 m de diámetro de partícula, y guarda columna con cartucho C18, Supelco. Las condiciones típicas de operación fueron: sistema isocrático, flujo 1,2 mL/min, volumen de inyección 20 l, detección UV 242 nm, temperatura de horno, ambiente, y tiempo de corrida 7 min. La fase móvil fue metanol al 100% grado HPLC. Se pesaron 20 g de la muestra de alimento infantil en un balón de base plana, se colocó un magneto y se adicionó 70 mL de etanol absoluto. Se colocaron los tubos de reflujo y se agitó la mezcla hasta su ebullición con corriente de nitrógeno. Luego, se adicionó 20 mL de solución de KOH al 50% y se saponificó la mezcla por 30 minutos con agitación moderada. Antes de finalizar esta etapa se enjuagó el contenido con 50 mL de agua en 3 porciones. La solución se enfrió a temperatura ambiente y luego se filtró al vacío. Se colocó inmediatamente el contenido en una pera de separación de 250 mL y se adicionó 50 mL de hexano p.a. para proceder a la extracción. Se agitó la mezcla por 20 segundos y al separarse las fases se colocó la capa superior (fase orgánica) en un balón de 250 mL de base redonda Capítulo 8 120 Procesamiento de datos cromatográficos que contenía aproximadamente 0,5 g de BHT ó ácido ascórbico. Se efectuó dos veces más la extracción y se juntaron los extractos en el balón de 250 mL. Se evaporó a sequedad el solvente, haciendo uso de un rotavapor con baño de agua a 40°C, se diluyó inmediatamente con metanol grado HPLC el residuo, y se llevó a volumen en un matraz volumétrico de 10 mL. Finalmente, se pasó la solución final por un filtro de 0,2 m, llenándolos en viales ámbar de 2 mL para colocarlos en el inyector del cromatógrafo. Cálculos: Donde: Am (Área del pico de vitamina A en la muestra), As (Área del pico de vitamina A en el estándar), Cs (Concentración de vitamina A en el estándar, mg/ml), D (Factor de dilución), Wm (Peso de la muestra. Se analizaron muestras de papilla para cuantificar la vitamina A expresada en g/g. El analito se adicionó a la matriz, disuelto en el mismo solvente usado para la extracción, y se dejó en contacto con ésta por varias horas antes de la extracción, para permitir su interacción. Se tomaron 0,76 mL, 1,27 mL y 1,78 mL de una solución estándar de 100 ppm de vitamina A y se adicionaron a 6 muestras de papilla por nivel. Se dejó en contacto la solución estándar con las muestras durante toda la noche bajo refrigeración y, al día siguiente, se realizó el tratamiento y se hicieron las lecturas de los resultados de las 18 muestras. Resultados En la Figura 1 se muestran los cromatogramas representativos de dos concentraciones (mayor y menor) de vitamina A para la curva de calibración. El tiempo de retención de la vitamina A fue 3,91 ± 0,5 min, siendo el tiempo total de análisis para una muestra de 7 min. La respuesta de detección fue lineal en el rango de concentraciones de 5- 15 g/g Figura ( 2), obteniéndose un coeficiente de correlación r=0,99. El límite de detección del método en base a la señal / ruido (S/N) fue de 0,06 g/g y el límite de cuantificación de 0,58 g/g Capítulo 8 121 Procesamiento de datos cromatográficos La recuperación obtenida fue de 97,98%. Determinación de óxido de etileno en aire. Método de muestreadores pasivos por difusión / Cromatografía de gases El oxido de etileno es una sustancia utilizada en la industria farmacéutica para la esterilización de embases, viales, ampolletas, tapones etc. Utilizados en la fabricación de inyectables. Toma de muestras. Para el muestreo personal colocar el muestreador pasivo 3M-3551 en la zona de respiración. Una vez terminado el período de captación desmontar la membrana y el aro de sujeción colocando en su lugar la tapa para la desorción, asegurando la hermeticidad de ésta y los tapones de la misma. Preparación de la muestra. El muestreador se desorbe con 1,5 ml de la disolución de 10% de cloruro de metileno en metanol por 30 min. Una vez realizada la desorción se diluyen las muestras diez veces con la disolución de 50% de acetonitrilo en tolueno. Calibración. La disolución patrón se prepara por triplicado, añadiendo una cantidad determinada de 2bromoetano a un volumen de disolución desorbente de 10% de cloruro de metileno en metanol a fin de obtener una disolución patrón de concentración similar a la muestra a analizar. Dicha concentración se debe expresar en mg/ml de disolución desorbente. Desarrollo de una curva de calibrado. Para el desarrollo de la misma, se preparan cinco disoluciones de calibración de los analitos de interés que cubran el intervalo de concentraciones de aplicación del método. Analizar los patrones en las mismas condiciones que las muestras. Se recomienda un mínimo de seis inyecciones. Calcular para cada concentración y analito el promedio de las respuestas obtenidas y la desviación típica correspondiente. Las curvas de calibración se construyen representando los intervalos Capítulo 8 122 Procesamiento de datos cromatográficos de los valores promedios ±2 desviaciones típicas, frente a las concentraciones en mg/ml de cada analito. Comprobar diariamente la curva de calibrado mediante el análisis de uno de los patrones de calibración. El valor obtenido para cada analito debe encontrarse dentro del intervalo asociado a ese patrón. Las condiciones de trabajo para el cromatógrafo de gases equipado son las siguientes: Temperatura del inyector 200 ºC Gas portador nitrógeno 25 ml/min Temperatura del horno 120 ºC Gas auxiliar nitrógeno 50 ml/min Temperatura del detector 250 ºC Cálculos Se calcula el factor de respuesta del analito con los datos obtenidos mediante la expresión: Fz = m/A donde: m= es la cantidad de analito en las disoluciones patrón A es el área promedio correspondiente al analito en las disoluciones patrón. La concentración en miligramos por mililitro de analito en las disoluciones de desorción de cada muestra, se determina según la expresión: co = Ao x FR donde: co= es la concentración de analito en mg/ml de disolución. Ao= es el área correspondiente al pico de analito en la muestra. FR= es el factor de respuesta. Calibración multinivel. Leer la concentración en miligramos por mililitro en la curva de calibración realizada. Capítulo 8 123 Procesamiento de datos cromatográficos Conversión de la cantidad de 2-bromoetanol a su equivalente en óxido de etileno Se realiza mediante la siguiente expresión: Ci = Co * 44.05/124.98 donde: ci= es la concentración de óxido de etileno en mg/ml de disolución. 44,05 es el peso molecular de óxido de etileno. 124,98 es el peso molecular de 2-bromoetanol. Determinación de la cantidad de óxido de etileno presente en la muestra Una vez determinada la cantidad de óxido de etileno en la disolución de desorción, se calcula la cantidad en mg de compuesto mediante la siguiente expresión: ms = ci x Vd ms= es la masa de óxido de etileno captada por el muestreador. Vd= es el volumen de desorción (1,5 ml). Determinación de la concentración ambiental de óxido de etileno Se calcula la concentración de óxido de etileno en aire muestreado, en miligramos por metro cúbico, por medio de la siguiente ecuación: 𝐶𝑖 = 𝑚𝑠 × 106 𝑆𝑅 × 𝐶𝑅 × 𝑡 donde: Ci= es la concentración ambiental del contaminante (mg/m3). ms= es la masa de contaminante captada (mg). SR= es el caudal de muestreo específico para el óxido de etileno (49,3 ml/min 760 mm Hg, 25 % Humedad relativa) . CR= es el coeficiente de recuperación específico para óxido de etileno en tanto por uno (0,92). t es el tiempo de exposición en minutos. La concentración de óxido de etileno en aire, expresada en mililitro por metro cúbico (ppm), se calcula por medio de la siguiente expresión: Cppm = Ci (24.0/44.05)*(101.3/P)*(T+273.15/293.15) donde: P= es la presión del aire muestrado en kPa (103 N/m3). T= es la temperatura del aire muestreado en ºC. 44,05 = es el peso molecular de óxido de etileno en g/mol. Capítulo 8 120 Procesamiento de datos cromatográficos Análisis cuantitativos de los principales constituyentes químicos de raíces de Echinacea purpurea y E. angustifolia producidas. Fundamento. Se cuantificó los fenilpropanoides libres: ácido clorogénico y ácido cichórico, glicosídicos así como las alcamidas presentes en extractos de raíces de las plantas medicinales Echinacea purpurea y E. angustifolia. Procedimiento. Se recolectaron 300 g de material vegetal, fue macerada con una mezcla 80:20 de etanol al 95%: agua. Después de realizar 3 maceraciones, el extracto hidroalcohólico fue concentrado en un evaporador rotativo a presión reducida y 45[grados]C, obteniéndose 1,5 litros de extracto concentrado. Análisis por cromatografía líquida de alta eficiencia (HPLC) La detección y cuantificación de los fenilpropanoides libres y del fenilpropanoide glicosilado, se realizó por medio de un cromatógrafo líquido de alta eficiencia (HP 1090 A). Los fenilpropanoides fueron separados utilizando un gradiente lineal de fase móvil (20 min) de un 5% de acetonitrilo [agua.sup.-1] hasta un 25% de acetonitrilo. El flujo fue de 1,0 ml [min.sup-1]. Además, la fase móvil contenía un 1% (v/v) de ácido fosfórico 0,1 N., para lograr una mejor resolución de los picos presentes en el cromatograma. La separación fue monitoreada a 330 nm. Las alcamidas fueron identificadas según los tiempos de retención y los espectros de masas de los picos obtenidos en los cromatogramas, comparados con los obtenidos con las disoluciones de sustancias puras, previamente analizados y almacenados en una base de datos. La cuantificación se realizó con el método de estándar externo empleando patrones en rango de concentración de 0-1,5% en peso. Resultados La mayor concentración de extractos de E. purpurea fue de 5,16% de la region de Santos y contiene 1,12% más ácido clorogénico y cichórico que la muestra reportada de EE.UU. El análisis por HPLC se obtuvo que los contenidos de metabolitos secundarios producidos tanto en Echinacea purpurea como en E. angustifolia, en condiciones de Costa Rica, son mayores que los reportados en los EE.UU., de donde es originaria esta planta. Se encontró que E. angustifolia, además produce un equinacósido que no sintetiza E. purpurea y que es de suma importancia por ser uno de los principales compuestos para mejorar el sistema inmunológico de los seres humanos. Capítulo 8 121 Procesamiento de datos cromatográficos Anfotericina b: determinación en diversos fluidos biologicos por cromatografía líquida. Aplicación a estudios farmacocinéticos y de estabilidad química. La anfotericina B (AnB), comercializada en el año 1956, continúa siendo el fármaco de elección para el tratamiento de las micosis sistémicas, aunque su utilización no está exenta de toxicidad. Procedimiento. Se determinó analizando 10 veces dentro de la misma serie analítica, dos muestras, una de concentración de AnB de 0.5 -g/mL y otra de 2.5 -glmL, preparadas a partir de un "pool'Lde sueros al cual le fue adicionado el fármaco. En este estudio se presenta un método de determinación de AnB en suero humano por cromatografía líquida de alta resolución (HPLC) en fase reversa y columna corta (30 mm). El desarrollo y puesta a punto de la técnica se realizó en base a los métodos de determinación de AnB por HPLC previamente descritos y resumidos en la siguiente tabla. 405 (1) Fase móvil EI Procedimiento de extracción Tampón Na2HPO4-KH2-PO4 1-Amino-4-Nitro Precipitación 1.6 mM, acetonitrilo naftaleno metanol (lleva incorporado el estándar proteínas sétricas con (63/37, v7v) pH=7 interno). Centrifugación inyección directa Flujo 1.5 mL/min sobrenadante. 382 Na2EDTA 2.5 mM, (2) Acetonitrilo con (55/45, v/v) (60/40/,v/v) Flujo 1 mL/min Inyección del eluido 405 (3) Tr NO Acetonitrilo, 25mM NO Extracción proteínicas séricas con metanol acetronilo Precipiatación Na2EDTA proteínas 2.5 mM séricas con EDTA, metanol metanol. Centrifugación inyección directa (30:20:50 v/v/v) del sobre nadante 5.5 min 6-7 min 4 min Flujo 1.6 mL/min 405 (4) Metanolm tampón fosfato 1-Amino-4 Precipitación proteninas séricas con (10 mM KH2PO4, 5mM Nitronaftaleno metanol (lleva incorporado el estándar EDTANa2 pH=4) interno). Centrifugación Inyección directa (80:20, v/v) del sobresedante. Acetronitirilo, tampón fosfato (10 mM KH2PO4, 5 6.8 +- 0.5 min 8.4 +- 0.5 min mM EDTANa2 pH=4) (40:60, v/v) Capítulo 8 122 Procesamiento de datos cromatográficos 382(5) EDTA 0.01 M, acetronitrilo 1-Amino-4 Si bilirrubinas < + mg/dL: (60/40 v/v) pH=4.2 Flujo 1.5 Nitronaftaleno Precipitación mL/min proteínas 4.9 séricas con +- 0.8 min metanol. Centrifugación. Inyección directa N- del sobrenadante. acetilanfotericina B Si bilirrubinas > 3 mg/dL: Precipitación preteínas séricas con metanol. Extracción en fásesólida con columna SuperClean C18 (vacmetanol e inyección del eluído) Resultados Se realizó una curva de calibración, misma que se calculó por regresión lineal a partir de las áreas de los picos correspondientes a los estándares, procesados en cada serie analitica. La concentración de las muestras se obtuvo por interpelación de su área en la recta de calibración. Se resenta un método de determinación de AnB en suero humano por cromatografía líquida de alta resolución (HPLC) en fase reversa y columna corta (30 mm). Para ello. se escogib una columna de octadecilsilano (C18), más corta (30 mm x 4.6 mm ID) que en los métodos de referencia (125 a 300 mm) (108,119,120,129,131), con el fin de obtener tiempos de retención más pequeños y se utilizó la fase móvil del método descrito por Wang et al (131) (acetonitrilo y tampón EDTA ). Bibliografia: Sanchez Ma. Dolores. Venezuela. 2004. Estudio sobre los ácidos grasos libres en queso blanco, Universidad de Los Andes. 46:2. Perez C. Ruth. Estudio de validación de la Metodología para la determinación de Vitamina A en Alimentos infantiles instantáneos por Cromatografía Líquida de alto rendimiento (HPLC). Rev. perú. med. exp. salud publica, 2000, vol.17, no.1-4, p.26-29. ISSN 1726-4634. Serie Académicos CBS. Cromatografía de gases y de líquidos de alta resolución México 2000. pag. 215-231 255-305. Khoo SH, Bond J, Denning DW. Administering arnphotericin B-a practica1 approach. J Antimicrob Chemother 1994;33:203-13. Capítulo 8 123