Las tres leyes de la termodinámica. Los potenciales termodinámicos
Anuncio

Repaso de Termodinámica
Las tres leyes de la termodinámica.
Los potenciales termodinámicos y las relaciones de Maxwell.
1
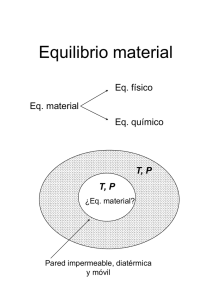
Descripción macroscópica de sistemas termodinámicos
Un sistema termodinámico es cualquier cantidad de materia o radiación lo
suficientemente grande como para ser descrito por parámetros macroscópicos, sin
ninguna referencia a sus componentes individuales (microscópicos).
Para una descripción completa del sistema también se necesita un descripción del
contorno (los límites), y de las interacciones que este permite con el entorno. Los
contornos pueden permitir el paso de materia y energía.
Sistema aislado: no intercambia energía ni masa con su entorno.
Sistema cerrado: sólo puede intercambiar energía.
Sistema abierto: puede intercambiar materia y energía.
Sistema móvil / rígido: las paredes permiten (o no) transferir energía en forma de
trabajo mecánico.
Sistema diatérmico: transferencia de calor sin trabajo.
Sistema adiabático: no hay transferencia de calor por las paredes.
Sistemas en contacto térmico, permeables, en contacto difusivo, etc
Parámetros termodinámicos: variables termodinámicas que describen el
macroestado del sistema.
Los macroestados se pueden describir en términos de un pequeño número de
variables de estado. (Ej: macroestado de un gas: masa, presión y volumen lo
describen totalmente)
2
Variables intensivas: independientes de la masa (ej: temperatura)
Variables extensivas: proporcionales a la masa (ej: energía interna)
Cantidades específicas: expresadas por unidad de masa.
Cantidades molares: expresadas por mol.
(EJ: Capacidad calorífica específica y molar)
Un sistema está en equilibrio termodinámico cuando sus variables de estado son
constantes a escala macroscópica.
No se requiere que los parámetros termodinámicos sean estrictamente
independientes del tiempo.
Los parámetros termodinámicos son promedios macroscópicos del movimiento
microscópico, por tanto habrá fluctuaciones. El valor relativo de estas fluctuaciones
es casi despreciable en sistemas macroscópicos, excepto cerca de las transiciones
de fase.
Sistema homogéneo: los parámetros intensivos son los mismos en todo el sistema.
Sistema inhomogéneo: uno o mas de los parámetros intensivos presenta variaciones
espaciales.
Un sistema inhomogéneo puede estar formado por distintas fases, separadas por
contornos de fase, de forma que cada fase sea homogénea. (EJ: En el punto triple del
agua coexisten hielo, agua y vapor de agua.)
Los contornos pueden ser arbitrarios, pero la termodinámica sigue siendo válida.
3
Ecuación de estado: es una relación funcional entre los parámetros de un sistema en
equilibrio.
Un estado de un sistema descrito por los parámetros p,V y T tendrá una ecuación de
estado f[p,V,T]=0, y por tanto reduce en uno el número de variables independientes.
Esta ecuación describe una superficie, la superficie de equilibrio.
Un estado de equilibrio puede ser representado por un punto en esta superficie. Un punto
fuera de ella es un estado de no-equilibrio.
Diagrama de estado: una proyección de una curva en la superficie de estado. (ej:
diagrama P-V)
Superficie de equilibrio para un
gas ideal con número fijo de
partículas.
U
3
2
pV
4
Leyes de la Termodinámica
Ley Cero (o principio cero) de la Termodinámica
Si dos sistemas están por separado en equilibrio con un tercero, entonces también deben
estar en equilibrio entre ellos.
Si tres o mas sistemas están en contacto térmico y todos juntos en equilibrio, entonces
cualquier par está en equilibrio por separado.
El concepto de temperatura se basa en este principio cero.
5
Primera Ley de la Termodinámica
Es una adapación para la termodinámica de la ley de conservación de la energía.
Se define la energía interna del sistema, E, como su energía respecto del SR del centro de masa.
El trabajo necesario para cambiar el estado de un sistema aislado depende unicamente de los estados
inicial y final, y es independiente del método usado para realizar el cambio.
Por tanto, existe una función de estado que identificamos como la energía interna. El trabajo
realizado sobre el sistema es W. Por tanto, el cambio de la energía interna durante una
transformación adiabática es ∆ E = W.
El sistema también puede variar su energía sin realizar trabajo mecánico, se transfiere de otra forma,
como calor.
Definición de calor: La cantidad de calor Q absorbido por un sistema es el cambio en su energía interna
que no se debe al trabajo.
La conservación de energía será:
∆ E = Q + W.
Si realizamos variaciones cuasiestáticas (p.ej., de volumen) escribiremos: δ W = - p dV.
Si movemos el pistón muy rápido el gas no hará trabajo sobre el pistón y δ W → 0 aunque varíe el
volumen.
Usamos d para una diferencial (propia) que depende sólo del cambio de estado. Usamos δ para indicar una diferencial
(impropia) que también depende del proceso usado para cambiar el estado.
Por tanto se escribe:
d E = δ Q + δ W.
6
Segunda Ley de la Termodinámica
La base de esta ley es el hecho de que si mezclamos partes iguales de dos gases nunca los
encontraremos separados de forma espontánea en un instante posterior.
Enunciado de Clausius: No hay ninguna transformación termodinámica cuyo único efecto sea transferir
calor de un foco frío a otro caliente.
Enunciado de Kelvin: No hay ninguna transformación termodinámica cuyo único efecto sea extraer calor
de un foco y convertirlo totalmente en trabajo.
La segunda ley proporciona la base para el concepto termodinámico de entropía.
Principio de máxima entropía: Existe una función de estado de los parámetros extensivos de cualquier
sistema termodinámico, llamada entropía S, con las siguientes propiedades:
1. los valores que toman las variables extensivas son los que maximizan S consistentes con
los parámetros externos,
2. la entropía de un sistema compuesto es la suma de las entropías de sus subsistemas.
(2º y 3º postulados de Callen)
Introducción de la definición de temperatura. Sistema hidrostático: E=E(S,V,ni)
dE =
∂E
∂E
dS +
dV +
∂S
∂V
∂S
>0
∂E
i
∂E
dni = T dS − pdV +
∂ni
1
>0
T
µ i dni
i
T >0
7
Teorema de Clausius:
Para cualquier ciclo cerrado
Q
T
0
donde δ Q es el calor absorbido por el sistema de un foco a temperatura T.
Si el ciclo es reversible
Q
0
T
Qrev
T S
es el calor absorbido durante un proceso reversible el que
la temperatura cambia en δ T
Podemos definir la capacidad calorífica:
Cx
Qrev
T
x
CX = T
Relacionamos capacidad calorífica y entropía:
∂S
∂T
dE = δQ + dW = T dS − pdV
Capacidad calorífica a volumen constante: dV = 0
dE = TdS
X
CV = T
Capacidad calorífica a presión constante:
(entalpía)
H = E + pV
dp = 0
∂S
∂T
=
V
∂E
∂T
V
dH = TdS + Vdp
dH = TdS
CP = T
∂S
∂T
=
P
∂H
∂T
P
8
Tercera Ley de la Termodinámica
Terorema de Nerst: Una reacción química entre fases puras cristalinas que ocurre en el cero absoluto
no produce ningún cambio de entropía.
Enunciado de Nerst-Simon: El cambio de entropía que resulta de cualquier transformación isoterma
reversible de un sistema tiende a cero según la temperatura se aproxima a cero.
lim
T 0
ST
0
Enunciado de Planck: Para T→0, la entropía de cualquier sistema en equilibrio se aproxima a una
constante que es independiente de las demás variables termodinámicas.
Teorema de la inaccesibilidad del cero absoluto: No existe ningún proceso capaz de reducir la
temperatura de un sistema al cero absluto en un número finito de pasos.
4º Postulado de Callen: La entropía de cualquier sistema se anula en el estado para el cual
∂E
∂S
=0
V , ni
9
Potenciales termodinámicos
Necesitamos relaciones entre las funciones termodinámicas, definir diferentes
potenciales termodinámicos dependiendo de qué se mantiene constante en el
sistema.
Partimos de:
µ i dni ,
dE = T dS − pdV +
E = E ( S , V , ni )
i
µ i dni )
Definimos los potenciales termodinámicos: (todos con +
i
Energía interna:
E = E (S ,V )
Entalpía:
H = E − PV
Energía libre de Helmholtz:
dE = T dS − pdV
H (S , P)
F = E − TS
Energía libre de Gibbs (entalpía libre):
dH = T dS + Vdp
F (T , V )
G = H − TS
dF = − SdT − pdV
G (T , p )
dG = − SdT + Vdp
Esto se obtiene usando las transformaciones de Legendre:
Sea:
f ( x, y ) → df = u dx + v dy
Si definimos:
g ≡ f −ux
Entonces:
g (u , y ) → dg = − x du + v dy
10
Principio de energía mínima: (es un corolario del de S máxima):
El valor de equilibrio de cualquier parámetro interno sin ligadura es tal que hace mínima la
energía interna para el valor dado de la entropía.
Principios extremales:
Los valores de equilibrio de los parámetros internos sin ligadura minimizan los potenciales
termodinámicos correspondientes:
F: sistema en contacto con foco térmico, T cte.
H: sistema en contacto con fuente de presión, P cte.
G: sistema en contacto con ambps focos, T y P ctes.
11
Relaciones de Maxwell
Se obtienen usando las transformaciones de Legendre, más la diferencial exacta:
f ( x, y ) → df = u dx + v dy
∂2 f
∂2 f
=
⇔
∂x∂y ∂y∂x
E = E (S ,V )
H = E − PV
F = E − TS
G = H − TS
∂u
∂y
=
x
∂v
∂x
y
dE = T dS − pdV →
dH = T dS + Vdp →
dF = − SdT − pdV →
dG = − SdT + Vdp →
∂T
∂V
∂T
∂P
∂S
∂V
∂S
∂P
=−
S
=
S
=
T
=
T
∂P
∂S
∂V
∂S
∂P
∂T
∂V
∂T
V
P
V
P
12
Aplicaciones:
Relaciones entre capacidades caloríficas, y coeficientes de dilatación y de compresibilidad.
dS =
dS =
∂S
∂S
∂P
dV → TdS = CV dT + T
dT +
∂T
∂V
∂T
∂V
∂S
∂S
dP → TdS = C P dT − T
dT +
∂T
∂P
∂T
∂V
C P − CV = T
∂T
α≡
1 ∂V
V ∂T
P
∂P
∂T
κT oS ≡
P
α2
,
C P − CV = T V
κT
V
∂P
= −T
∂V
1 ∂V
V ∂P
T
∂V
∂T
dV
V
dP
P
2
P
T oS
Constante adiabática : γ =
CP κT
=
CV κ S
13
Ecuación de Gibbs-Duhem
Si comparamos la expresión diferencial para E con la relación fundamental:
µ i dni ,
dE = T dS − pdV +
i
µ i dni +
dE = T dS + S dT − Vdp − pdV +
i
n i dµ i ,
i
... vemos que los parámetros intensivos no son independientes, sino que cumplen la
relación de Gibss-Duhem:
ni dµ i = 0
S dT − Vdp +
i
Y para la energía libre de Gibbs tenemos:
E = TS − pV + µN ,
G = E − TS + pV = µN
Es decir, la energía libre de Gibbs molar es igual al potencial químico.
14
Estabilidad termodinámica.
Principio de Le Chatelier: Si un sistema está en eauilibrio estable, cualquier perturbación
produce procesos que tienden a devolver al sistema a su estado original de equilibrio.
Cómo responde el sistema a fluctuaciones locales de los distintos parámetros.
El principio de máxima entropía requiere:
S 0
2
La estabilidad térmica requiere: Cp
S
CV
0
Partiendo de fluctuaciones de presión:
Ti
Si
0
0
Vi
Vi
Vi
T
Vi
S
T,i
S,i
0
pi
pi
0
2
S
2
S
1
Vi
2
1
Ti
Vi S,i
2
i
T,i
Ti
i
S
pi
2
pi
2
1
V
V
p
T
S
0
0
S
15
TRANSICIONES DE FASE Y FENÓMENOS CRÍTICOS
Transiciones de fase de primer orden.
Transiciones de fase de orden superior y fenómenos críticos.
Teoría de Landau y parámetro de orden.
Exponentes críticos y leyes de escala.
[CAL-9,10; HUA-16,17; YEO-1,2,4]
16
Transiciones de fase de primer orden.
Transiciones de fase de orden superior y
fenómenos críticos.
17
TRANSICIONES DE FASE
Si no se satisfacen los criterios de estabilidad por la ecuación fundamental del sistema,
este se separa en dos o más fases.
Fase: sistema o subsistema con composición química y estructura física
homogénea, limitado por una superficie a través de la cual dichas propiedades
cambian bruscamente.
En una fase los parámetros intensivos son uniformes.
18
Ejemplo: Gas real
Ec. van der Waals
P/Pc
Criterio de estabilidad:
∂p
∂v
1
0
1
2
3
V/Vc
4
a
(v − b ) = R T
2
v
~
t
3
8
~
p=
− 2
~
3v − 1 v~
T/Tc = 1
0.9
0.85
0.84
1.1
1.2
1.3
2
P+
5
≤0
T
Hay zonas de las curvas
donde no se cumple, habrá
un cambio de fase
19
El estado estable será el de menor G (o µ).
∂µ
∂T
= −s
p
∂µ
∂p
=v
T
dg = dµ = − sdT + vdp
ssólido < slíquido < s gas vsólido ≈ vlíquido <<< v gas
Diagrama de fases
20
Diagrama presión-volumen
•
•
•
•
•
En este diagrama se ve la influencia del
cambio de fase en el volumen.
Las isotermas se hacen horizontales
durante el cambio de fase.
El inverso de la pendiente de las
isotermas es proporcional al coeficiente
de compresibilidad de la fase.
La curva binodal une los puntos de líquido
saturado, como a, el punto crítico y los
puntos de vapor seco, como b.
Si ya e yb son las fracciones molares del
líquido y del vapor:
ya
v −v
bc
=
= b
yb
ac
v − va
ya + yb = 1
Estabilidad de las fases
To es la temperatura de cambio de fase.
Los mínimos absolutos son los estados estables.
Los mínimos relativos estados “metaestables”.
21
Ecuación de Clapeyron
En el equilibrio de las fases 1 y 2 de un cuerpo puro se cumple d µ 1 = d µ 2 y a lo largo de
su línea de coexistencia: µ 1 = µ 2
(Ec. Gibbs-Duhem)
Sustituyendo los potenciales químicos:
− s 1 dT + v 1 dp = − s 2 dT + v 2 dp
donde
∆s = s2 − s1 = ( h2 − h1 ) / T = ∆h / T
y
∆v = v2 − v1
discontinuidades en S y V: transición de fase de 1er orden
dp
L
∆s
∆h
∆h
=
=
=
= − ρ1ρ 2
dT
∆v
T∆v
T∆v
T∆ρ
Calor latente: L = T ∆S
22
Aproximación de Clausius
•
•
•
Aplicable sólo cuando una fase es vapor.
El volumen específico de la fase condensada se desprecia frente al del vapor.
La fase vapor se considera un gas ideal:
vsólido ≈ vlíquido <<<< vvapor
pvvapor ≈ RT
∆h
∆h
∆h
dp
=
≈
= p
RT 2
dT T ∆ v Tv vapor
R = kB / N A
d ln p
∆h
=
dT
RT 2
Es la ecuación de Clausius-Clapeyron:
23
Clasificación de Ehrenfest : orden de la transición
C p ≥ Cv ≥ 0
Criterios de estabilidad:
cp > 0
κT > 0
dG =
∂G
∂T
= −S
P
∂G
∂T
T
∂S
∂T
>0
P
1 ∂V
−
V ∂P
T
∂G
∂P
dT +
P
∂G
∂P
>0
=V
T
κT ≥ κ S ≥ 0
∂S
∂T
∂V
∂P
>0
P
<0
T
dP = − SdT + VdP
T
Las discontinuidades en S y V de las
transiciones de fase de primer orden
son discontinuidades en las derivadas
primeras de G.
Hacemos derivadas segundas de G:
24
Las discontinuidades en las derivadas primeras de G
implican divergencias en las derivadas segundas
cp = T
1
κT = −
V
1
α =
V
∂S
∂T
= −T
P
∂V
∂P
∂V
∂T
T
P
∂ 2G
∂T 2
1
=−
V
1
=
V
P
∂ 2G
∂P 2
∂ 2G
∂T ∂P
T
P ,T
25
Si no hay discontinuidades en las derivadas primeras: Las discontinuidades en las
derivadas segundas de G indican transiciones de fase de segundo orden (etc)
26
Ecuaciones de Ehrenfest
(son análogas a la ecuación de Clausius-Clapeyron)
Si V y S son continuas en la transición:
dv 1 = dv
dv =
∂v
∂T
dT +
P
TdS = C p dT − T
∂v
∂P
2
; dS
1
= dS
2
dP = α v dT − κ v dP
T
∂V
∂T
Tds = C p dT − T α v dP
dP
P
∂P α 2 − α 1
=
κ 2 − κ1
∂T
C P , 2 − C P ,1
∂P
=
∂ T vT (α 2 − α 1 )
v T (∆α ) 2
=1
∆C P ∆κ T
27
28
Muchos comportamientos son
similares en el punto crítico.
Para ello, se usan las variables
reducidas:
p
v
T
ρ
π= ; φ= ; θ = ; y d=
ρc
pc
vc
Tc
29
Diagrama
presión-volumen-temperatura
30
Teoría de Landau y parámetro de orden.
Exponentes críticos y leyes de escala.
31
Los fenómenos críticos
•
•
•
•
•
•
Desde 1905, las técnicas de medida permitieron detectar
saltos abruptos en el calor específico de ciertos cuerpos.
Por ejemplo, el helio.
Ese mismo comportamiento aparecía en otros coeficientes
termodinámicos.
Las primeras fueron “transformaciones”, porque se creyeron
transiciones de fase.
Así, la “transformación lambda” se le dio por la forma de la
curva.
Después, Ehrenfest creyó que su origen era una
discontinuidad en la segunda derivada del potencial entalpía
libre, de ahí el nombre de “transición de segundo orden”.
•
•
La denominación actual es la de “fenómenos críticos”.
Este nombre deriva de las similitudes que se han encontrado entre el punto crítico de los
gases y ciertos puntos característicos de los líquidos y los sólidos, como el paso de helio
normal a superfluido y el punto de Curie de los materiales ferromagnéticos.
•
•
Los resultados experimentales ha inducido a creer que este comportamiento es universal.
El fenómeno crítico es una característica general de la naturaleza, que se refleja con
diversos parámetros en distintos cuerpos.
Los sistemas mejor conocidos son los expansivos y los magnéticos, aunque hay muchos
32
otros ejemplos.
•
Características comunes
•
•
La “temperatura crítica”, Tc , es aquella en la que se produce el máximo del coeficiente.
Los fenómenos críticos se producen en un intervalo de temperatura pequeño, ∆T < 5 K. La
“desviación relativa de temperatura” es:
t=
(T − Tc )
Tc
La temperatura crítica separa la “forma ordenada” y la “forma desordenada”.
La forma ordenada se conoce porque posee porciones internas distinguibles,
no fases. Siempre se presenta a las temperaturas inferiores a la crítica:
T < Tc , t < 0
La forma desordenada es homogénea y carece de porciones internas
distinguibles. Siempre se presenta a las temperaturas mayores que la crítica:
T > Tc , t > 0
Parámetro de orden
•
•
•
•
Con el fin de describir lo dicho antes, se introduce el “parámetro de orden”, φ, con las
siguientes propiedades:
Es una característica interna del sistema que no puede imponerse desde el exterior.
Posee valor en la fase ordenada y se anula en la desordenada.
Debe definirse en cada problema.
33
Ejemplos de parámetro de orden
•
Alrededor del punto crítico de
una gas hay dos fases que
llegan a confundirse.
El parámetro de orden puede
ser la diferencia de
densidades de las fases:
•
φ = ρlíquido − ρvapor
•
•
Un material ferromagnético
pueden tener dos
imanaciones remanentes
opuestas.
Tras el punto de Curie, se
hace paramagnético. El
parámetro de orden es:
φ =M
34
Exponentes críticos
•
Alrededor del punto crítico las propiedades tienden a depender de t exponencialmente.
Por ejemplo, el parámetro de orden:
φ = B (− t )β {1 + C (− t ) x + ...}
donde x tiende a cero cuando t lo hace, y β se conoce como “exponente crítico”:
β = lím t → 0
ln φ
ln (− t )
•Obsérvese que φ sólo existe para t < 0.
35
Exponentes críticos: fluidos
Las discontinuidades en las derivadas primeras de G
implican divergencias en las derivadas segundas
cp = T
1
κT = −
V
∂S
∂T
= −T
P
∂V
∂P
T
∂ 2G
∂T 2
1
=−
V
1
α =
V
P
∂ 2G
∂P 2
∂V
∂T
P
1
=
V
∂ 2G
∂T ∂P
P ,T
T
Divergencias del calor
específico en la transición:
y de la compresibilidad:
C P ,V ∝ T − TC
κ T ∝ T − TC
−α
−γ
36
Exponentes críticos: fluidos
La forma de la isoterma crítica cerca del punto crítico es:
P − PC ∝ ρ − ρ C
δ
signo ( ρ − ρ C )
δ >0
La forma de la curva de coexistencia en el plano ρ-T cerca del punto crítico, para T<Tc es:
ρ L − ρ G ∝ ( T − TC ) β
Diagrama P-V
Diagrama P- ρ
Estos son los exponentes críticos primarios
que nos indican como divergen las distintas
magnitudes en la transición de fase.
α, β, δ, γ
37
Para un sistema ferromagnético en presencia de campo magnético:
38
Teoría de Landau
En 1937, Lev Landau propuso una teoría en la que el potencial termodinámico del
sistema se hacía función continua del parámetro de orden.
Para un sistema expansivo se cumple:
G = G (p,T ,φ )
y en el equilibrio:
∂G
∂φ
=0
p ,T
En el entorno del punto crítico, Landau aceptó como válido el desarrollo (φ es pequeño):
G(T, p,φ ) = Go (T , p) +αφ + Aφ 2 + Bφ3 + Cφ 4 + ...
donde los coeficientes α, A, ... son funciones de la temperatura y de la presión.
Como la entalpía libre es un mínimo, debe ser una función par de φ, es decir:
G (φ
por lo que:
)=
G (− φ
)
G(T , p, φ ) = Go (T , p ) + Aφ 2 + Cφ 4 + ...
39
Aceptando la cuarta potencia como una aproximación
suficiente, en el equilibrio:
φ =0
que tiene dos tipos de soluciones:
∂2G
∂φ 2
= 2 Aφ + 4 C φ 3 = 0
T,p
T ≥ Tc
φ =± −
Aplicando la condición de estabilidad:
∂G
∂φ
A
2C
T < Tc
= 2 A +12 C φ 2 ≥ 0
T,p
Llevando las dos soluciones anteriores:
φ =0
φ =± −
A
2C
T ≥ Tc
A≥0
T < Tc
A≤0
40
A≥0
Se debe cumplir:
T ≥ Tc
A≤0
y
T < Tc
A( p, T ) = 0
Además A debe ser una función continua. Por tanto, en Tc :
Así que Landau hizo la siguiente aproximación:
A = a(T − Tc ) = Bt
B = const. C = const.
Go (T, p)
G(T, p,φ ) = Go (T, p) + Btφ 2 + Cφ 4 =
Representación gráfica:
t ≥0
B2t 2
t <0
Go (T, p) −
4C
Energía Libre v s Magnetización
Energía Libre v s Magnetización
Energía Libre v s Magnetización
80
250
60
200
1400
Energía Libre
Energía Libre
1000
800
600
400
200
40
Energía Libre
1200
20
0
150
100
50
-20
0
0
-40
-200
-6
-4
-2
0
Magnetización
2
4
6
-6
-4
-2
0
Magnetización
Si T > Tc hay una forma estable, si T < Tc
las que el sistema fluctúa.
2
4
6
-6
-4
-2
0
2
4
6
Magnetización
hay dos y si T = Tc existen infinitas entre
41
Exponente β del parámetro de orden:
φ= −
A
Bt
B
= −
=
(−t)1/ 2
2C
2C
2C
β=
1
2
Exponentes α y α’ de las capacidades caloríficas:
∂2G
Cφ = −T
∂T 2
∂2Go
≈ −Tc
2
∂
T
η
∂2 B2t 2
+ Tc
2
∂
2C
T
φ
= Cφo +
φ
B
CTc
α = α '= 0
Exponentes γ y γ’ del coeficiente de compresibilidad, si φ = ∆ρ , y de la susceptibilidad
magnética, si φ = M :
∂2G
χT =
∂φ 2
∂2
= − 2 Btφ 2 = 2 B(−t )
∂φ
(
T
)
γ '=1
Los exponentes experimentales se aproximan a estos valores, pero en ningún caso se
hacen iguales a ellos.
42
Se cumplen las siguientes relaciones:
Relación de Rushbrooke:
α '+2 β + γ ' = 2
Relación de Griffiths:
α '+ β (δ + 1) ≥ 2
Relación de Widom:
β (δ − 1) ≤ γ '
Relación de Fisher:
γ = υ (2 − η )
Relación de Josephson:
υ n = 2 −α
Landau
α
β
γ
δ
υ
η
Longitud de correlación y
Fluctuaciones del
parámetro de orden
Experimental
0
1/ 2
− 0 . 2 ... 0 . 2
0 . 3 ... 0 . 4
1
3
1/ 2
0
1 ... 1 . 4
4 ... 5
0 . 6 ... 0 . 7
≈ 0 . 05
43
Fluctuaciones y funciones de correlación
χT
y κT
divergen justo por encima de Tc, por tanto habrá grandes fluctuaciones
en el parámetro de orden (densidad o magnetización)
Gas cerca del punto crítico: muy denso, índice de refracción muy alto, fluctuaciones
grandes en la escala de la luz visible (500nm), opalescencia crítica.
Longitud de correlación: tamaño de los ‘bloques’ del sistema en los que fluctúa el
parámetro de orden.
Por ejemplo, para un sistema de momentos magnéticos:
Se define la función de correlación:
Se asume que cerca del punto crítico varía de la forma:
Donde
es la longitud de correlación
44
Experimentalmente, se encuentra que la longitud de correlación diverge en el punto crítico:
Esto implica que puntos lejanos están correlacionados, es decir, dominan las
fluctuaciones con longitud de onda grande. Por tanto, el sistema cerca de una transición
de fase de segundo orden pierde memoria de su estructura microscópica y comienza a
presentar nuevas correlaciones macroscópicas de largo alcance.
Justo en el punto crítico la función de correlación sigue una ley de potencias:
Donde d es la dimensionalidad espacial del sistema.
Estos exponentes críticos, ν y η no se pueden obtener a partir de la teoría de Landau,
pues es una teoría de campo medio y no considera las correlaciones entre los valores
microscópicos.
Esta pérdida del detalle microscópico en la transición hace que sistemas
distintos tenga el mismo tipo de comportamiento: Universalidad
45