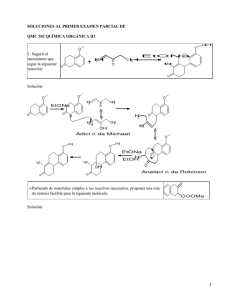
SÍNTESIS ORGÁNICA
Anuncio

CURSO SÍNTESIS ORGÁNICA BIBLIOGRAFÍA Organic Synthesis: Strategy and Control By Paul Wyatt, Stuart Warren ISBN-13: 9781118681442 Wiley, John & Sons, Incorporated, 2013 Organic Synthesis: The Disconnection Approach By Stuart Warren, Paul Wyatt ISBN-13: 9781119965534 Wiley, 2011 Workbook for Organic Synthesis: The Disconnection Approach By Stuart Warren, Paul Wyatt ISBN-13: 9781119965558 Wiley, 2011 Organic Synthesis: Concepts and Methods / Edition 3 By Jurgen-Hinrich Fuhrhop, Guangtao Li, E,J, Corey, Gustav Penzler ISBN-13: 9783527302734 Wiley, 2003 Classics in total synthesis : targets, strategies, methods. Nicolaou, K. C.; Sorensen, E. J. (1996). VCH. ISBN 3-527-29284-5. The Logic of Chemical Synthesis / Edition 1 By E. J. Corey, Xue-Min Cheng. ISBN:0471115940, Wiley, 1995 Modern Methods of Organic Synthesis / Edition 4 By W. Carruthers, Iain Coldham ISBN-13; 9780521778305, 2010. Ed. Cambridge University Press EVALUACIÓN 3 EXÁMENES PARCIALES Examen parcial + 1.5 puntos por tareas entregadas LAS TAREAS SE ENTREGAN A LA SIGUIENTE CLASE NO SE ACEPTAN TAREAS YA CALIFICADAS Y ENTREGADAS AL GRUPO 4ª CALIFICACIÓN ANÁLISIS RETROSINTÉTICO DE UNA MOLÉCULA OBJETIVO SÍNTESIS ORGÁNICA INTRODUCCIÓN La síntesis orgánica es la construcción planificada de moléculas orgánicas mediante reacciones químicas. La síntesis de compuestos orgánicos se ha convertido en uno de los ámbitos más importantes de la química orgánica. Flutriazol Fungicida Periplanona-B Feromona de la cucaracha Vitamina B12 Deltametrina piretroide insecticida y acaricida De gran uso en el mercado de los insecticidas (S)-L-(-)-DOPA Se utiliza en el tratamiento de la enfermedad de Parkinson Se hace uso de una hidrogenación catalítica durante su síntesis comercial Hay dos campos de investigación principales dentro del campo de la síntesis orgánica: • la síntesis total • la síntesis parcial Se diferencian por el origen y complejidad de los precursores químicos utilizados. SÍNTESIS TOTAL Las materias primas son, en general, compuestos derivados del petróleo, de estructura simple SÍNTESIS PARCIAL Las materias primas son productos naturales de estructura más compleja. Objetivos 1) Llevar a cabo la síntesis de productos naturales que presentan un especial interés. Taxol Un antitumoral Objetivos 2) Llevar a cabo la síntesis de compuestos para su aprovechamiento en diferentes campos como el farmacéutico, la industria alimentaria, colorantes, plaguicidas, etc. Fluoxetina (Prozac) Un antidepresivo. Objetivos 3) La síntesis de compuestos para el estudio de propiedades físicas o químicas (investigación básica) Síntesis de moléculas interesantes Las moléculas objetivo pueden ser compuestos con propiedades artísticas o antropomorficas … Nanoatleta Nanoguasón Nanopilgrim Nanomonarca Nanogreenbeteret (boina verde) Nanotexano NanoPutianos Nanoescolar Nanobaker Nanochef TIPOS DE SÍNTESIS ORGÁNICA SÍNTESIS ORGÁNICA SÍNTESIS ORIENTADAS A LA DIVERSIDAD (QUÍMICA COMBINATORIA) SÍNTESIS ORIENTADAS A MOLÉCULAS OBJETIVO (SÍNTESIS TOTALES) MÉTODOS ORIENTADOS A SÍNTESIS (METODOLOGÍA SINTÉTICA) LIBRERÍAS DE FARMOQUÍMICOS PRODUCTOS NATURALES REACTIVOS DISEÑO DE MOLÉCULAS ESTRATEGIAS LIBRERÍAS DE CATALIZADORES MOLECULAS PARA MATERIALES MOLECULAS INTERESANTES CANDIDATOS PARA FARMOQUÍMICOS CATALIZADORES TÁCTICAS Síntesis orgánica actual Materias primas Comercialmente accesibles Síntesis ideal Medio ambientalmente aceptable Evitar la problemática de los residuos (ruta sostenible). Evitar la generación de intermediarios tóxicos (la ruta menos tóxica) Económicamente aceptable Costo de materiales (la ruta más barata) Novedosa (patentable) Síntesis ideal Eficiencia alta Simple, con alto rendimiento, convergente pocos papsos) QUÍMICA SOSTENIBLE Segura Evitar los procedimientos de riesgo (ruta más segura) Robusta Que se pueda escalar con facilidad, procedimientos reproducibles LA QUÍMICA ORGÁNICA SE DIRIGE E INTERACTUA CON OTRAS DISCIPLINAS MEDICINA BIOLOGÍA CIENCIA DE MATERIALES NUEVAS MOLÉCULAS ORGÁNICAS FÍSICA NANOTECNOLOGÍA Tipos de síntesis Síntesis total Una síntesis total es la síntesis química de moléculas orgánicas complejas partiendo de moléculas simples comercialmente asequibles, habitualmente derivadas del petróleo. En una síntesis lineal existen una serie de pasos que se llevan a cabo uno tras otro hasta que se obtiene la molécula objetivo. Esto es a menudo adecuado para una estructura simple. A los compuestos químicos producidos en cada paso se les denomina intermedios sintéticos. APLICACIONES INDUSTRIALES Fungicida FLUTRIAFOL AstraZeneca molécula objetivo Flutriazol Fungicida intermedios sintéticos SÍNTESIS LINEAL Para moléculas más complejas una síntesis convergente es con frecuencia preferible. Esto es así cuando varias "piezas" (intermedios clave) del producto final son sintetizadas separadamente y a continuación unidas, a menudo cerca del final del proceso de síntesis. Rendimiento 78% Rendimiento 83% síntesis convergente Rendimiento 76% Flexibileno Rendimiento 52% Tipos de síntesis Síntesis parcial o semisíntesis Síntesis donde se parte de un producto natural, que no ha sido previamente sintetizado, sino extraído y purificado de organismos por métodos de separación de mezclas, que sí es fácilmente accesible. Se usa cuando es una alternativa mejor a una síntesis total. Un ejemplo sería la síntesis del LSD (TAREA). Aproximación directa asociativa El químico reconoce directamente dentro de la estructura de la molécula objetivo un número de subunidades estructuras fácilmente accesibles, las cuales se unen en forma apropiada usando reacciones estándar En la síntesis de péptidos, se pueden reconocer con facilidad en forma inmediata. Sin embargo, el llevar a cabo dicha síntesis en el laboratorio puede llegar a ser uno de los temas más arduos con las cuales se enfrenta un químico orgánico Historia de la síntesis orgánica en un vistaso Era pre Segunda Guerra Mundial Terpineol (Perkin, 1904) Tropinona (Robinson, 1917) Premio Nobel de Química (1947) Haemina (Fischer, 1929) Premio Nobel de Química (1929) Clorhidrato de Piridoxina (Folkers, 1939) Las síntesis antiguas, se basaban en la disponibilidad de las materias primas que contenían una porción importante de la estructura atómica final. Estas síntesis del Siglo XX dependían del conocimiento de reacciones adecuado para formar moléculas policíclicas y en la planificación detallada de encontrar una manera aplicar estos métodos La Era Corey (1960–1990) Longifoleno ( 1961) Eritronólido B ( 1975) (+)-Biotina ( 1988) Prostaglandina F2a ( 1969) El logro del método de Corey en la síntesis total, fue marcado por dos elementos distintivos: el análisis retrosintético y el desarrollo de nuevos métodos de síntesis como parte integral de la aproximación, a pesar de que Woodward (consciente o inconscientemente) debió hacer uso de tales prácticas Diseño de la síntesis El diseño de una síntesis se basa en el análisis retrosintético, que es un enfoque del diseño de síntesis aportado por el químico estadounidense Elías James Corey. Análisis retrosintético Desconexiones Fostriecina (Cl-920) Con esta técnica el diseño de la síntesis se planifica hacia atrás partiendo desde el producto final hasta llegar a unos compuestos de partida asequibles, mediante una secuencia de pasos lógicos donde cada vez las estructuras precursoras son más sencillas. ANÁLISIS RETRO SINTÉTICO Un sintón se define como una unidad estructural, sin ser una molécula pero que está íntimamente relacionada con una reacción sintética. Elias James Corey 1928 – Premio Nobel de Química 1990 Propuso en 1967 que la palabra sintón fuese usada para denominar un bloque de construcción en síntesis de una manera más sencilla que nombrarles "estructuras de fragmentación retrosintética". ". . . the grand thing is to be able to reason backwards. That is a very useful accomplishment, and a very easy one, but people do not practice it much." Sherlock Holmes, in "A Study in Scarlet" "The end is where we start from...." T. S. Eliot, in "The Four Quartets" Una gran cosa es ser capaz de razonar hacia atrás . Eso es un logro muy útil, y uno muy fácil, pero la gente no lo practica mucho El fin es de donde partimos .. "...even in the earliest stages of the process of simplification of a synthetic problem, the chemist must make use of a particular form of analysis which depends on the interplay between structural features that exist in the target molecule and the types of reactions or synthetic operations available from organic chemistry for the modification or assemblage of structural units. The synthetic chemist has learned by experience to recognize within a target molecule certain units which can be synthesized, modified, or joined by known or conceivable synthetic operations...it is convenient to have a term for such units; the term "synthon" is suggested. These are defined as structural units within a molecule which are related to possible synthetic operations... a synthon may be almost as large as the molecule or as small as a single hydrogen; the same atoms within a molecule may be constituents of several overlapping synthons..." from "General Methods for the Construction of Complex Molecules" E. J. Corey, Pure Appl. Chem. 1969, 14, 19 "I for one will not conceal my hope, contrary though it may be to the often too narrowly utilitarian spirit of the day, that synthesis for its own sake will continue. There is excitement, adventure, and challenge, and there can be great art, in organic synthesis. These alone should be enough, and organic chemistry will be sadder when none of its practitioners are responsive to these stimuli." R.B. Woodward in "Perspectives in Organic Chemistry", 1956 " Por mi parte, no voy a ocultar mi esperanza, al contrario de lo que podría pensarse aunque puede ser el espíritu práctico del día, que la síntesis por su propio bien continuará. En la síntesis orgánica hay emoción, aventura, desafío y puede haber gran arte. Esto por si mismo debería ser suficiente. La química orgánica será más triste cuando ninguno de sus practicantes respondan a estos estímulos " . ¿Hay alguna estrategia estándar para analizar cualquier molécula objetivo? ¿Hay alguna manera preferencial a proceder? No exactamente, la libertad, la imaginación y el riesgo son palabras comunes en síntesis orgánica, es una actividad heurística (hallar, inventar, el arte o la ciencia del descubrimiento) y de alguna manera artística, en el que conceptos como la belleza o elegancia a menudo aparece CEREBRO PARTE IZQUIERDA CEREBRO PARTE DERECHA Lógica Intuición Análisis Emoción Organización Espiritualidad Conocimiento / hechos Creencia Detalle Arte / música Panorama Matemáticas & ciencia Táctica Estrategia El objetivo final de Síntesis Orgánica es ensamblar un compuesto orgánico (molécula objetivo) a partir de materiales de partida fácilmente disponibles y reactivos de la manera más eficiente. Este proceso suele comenzar con el diseño de un plan sintético (Estrategia) Definiciones Molécula objetivo (TGT, de target) El compuesto final deseado. Flutriazol Fungicida Periplanona-B Feromona de la cucaracha Vitamina B12 Definiciones Desconexión Es un proceso mental, imaginario donde se rompen enlaces de una forma lógica dando lugar a fragmentos o sintones. Una desconexión se puede considerar lógica si: • Existe un mecanismo de "reconexión" razonable. • Conduce a fragmentos relativamente estables. • Representa la mayor simplificación posible. Igualmente nada impide recurrir durante el análisis a desconexiones aparentemente ilógicas si se consideran útiles. Definiciones Transformada Exactamente lo contrario de una reacción. Es una operación retrosintética imaginaria a través de la cual se transforma la molécula objetivo en una molécula precursora de una manera tal que los enlaces se reformen (o rompan) por reacciones sintéticas razonables Reacción: A B Transformada: B La flecha A equivaldría a la expresión: “proviene de". Algoritmo de Estrategia Las instrucciones, paso a paso, para realizar una operación retrosintética Objetivo de este curso Retrón Elemento estructural necesario para poder llevar a cabo una cierta transformada (o desconexión). " Retron: es el elemento subestructural mínimo en una estructura objetivo que es clave para la aplicación directa de una transformada para generar un precursor sintético " from E. J. Corey and X.-M. Cheng, "The Logic of Chemical Synthesis", 1989 Transformada: operación inversa en el sentido retrosintético Retrón Flechas de retrosíntesis Sintón Fragmento, idealizado, de la molécula. El compuesto orgánico (o reactivo) equivalente al sintón sería su equivalente sintético. sintón equivalente sintético Corey definió al sintón en 1967 como: unidades estructurales dentro de una molécula que se relacionan con posibles operaciones sintéticas o unidades que se pueden formar y/o ensamblar por medio de operaciones sintéticas concebibles Corey, E. J. Pure & Appl. Chem. 1967, 14, 19. .. Pero después, el mismo evita usar este término y utiliza precursor sintético en lugar del anterior. Corey, E. J. The Logic ...; Angew. Chem. Int. Ed. Eng. 1990, 1320 Sin embargo, el concepto sintón fue fácilmente arraigado en el lenguaje sintético y hoy en día es común su uso. Sintones polares se han clasificado Teniendo en cuenta que las reacciones sintéticas más comunes son del tipo polar, las que pueden ser vistas como la combinación de: 1) Un sintón que posea un átomo de carbono (electronegativo) polarizado negativamente, o donador de electrones, d. 2) Con otro sintón que posea un átomo de carbono (electropositivo) polarizado positivamente, o aceptor electrones, a. Los sintones están numerados (d0 , d1 , d2 , ... o a0 , a1, a2 , .... ) con respecto a las posiciones relativas de un grupo funcional (FG) y el sitio de reacción 0 0 1 Sintones donadores 0 2 1 3 0 2 1 Sintones " d " Tipo Ejemplo Equiv. Sint. Grupo funcional Sintones " a " Tipo Ejemplo Equiv. Sint. Grupo funcional Sintones donadores Sintón Equivalente (s) sintético (s) Sintón Equivalente (s) sintético (s) (C=O enmascarado) Reformasky (C=O enmascarado) Sintones aceptores Sintón Equivalente (s) sintético (s) Sintón Equivalente (s) sintético (s) (C=O enmascarado) Sintones a1 Especies equivalentes Las iminas están muy relacionadas con aldehídos, pero muestran una baja reactividad X = Cl, Oac, SR’, OR’ Las sales de iminio se prepararn con facilidad (i.e. Mannich) y son muy reactivas Friedel-Crafts CO2 Vilsmeier-Haack Equivalente sintético Sintones naturales NaBr NaN3 RONa a partir de ROH RSNa a partir de RSH Alquilo, alílico o bencílico Alquilo, alílico o bencílico Vinilo o arilo Alquilo, alílico o bencílico O Cuprato para adición de Michael Alquilo, alílico o bencílico O Cuprato para adición de Michael Vinilo o arilo o o o o Es frecuente el uso de acetoacetato de metilo o malonato de dimetilo como materias primas Sintones no naturales O2+ o HO+ Equivalente sintético Br2 SeO2 o dimetildioxirano RSCl o RSSR Anión acilo Después reducción (LiAlH4 o H2/Pd-C) Homoenolato Homoenolato (C=O enmascarado) o "Retron", termino propuesto por EJ Corey, se puede utilizar para referirse a una porción particular de una molécula que indica lo que la transformada retrosintética ( lo contrario de una reacción sintética en sentido directo ) se puede utilizar para reducir la complejidad estructural. Así, por ejemplo, acetoacetato de etilo presenta el Retron (es decir, el 1,3 -dicarbonilo) para una transformada de una condensación de Claisen. "Sinton" se utiliza en el "método de desconexión " ó retrosíntesis , y se refiere a los fragmentos imaginarios que resultan de romper un enlace heterolíticamente . Los sintones que resultan de desconectar acetoacetato de etilo en el enlace entre el carbono 2 y 3 y dando ambos electrones carbono 2 son un catión de acilio y un anión enolato . Estos sintones se sustituyen por "equivalentes sintéticos " o "precursores" en la síntesis hacia adelante , es decir, dos moléculas de acetato de etilo Molécula objetivo Retrón Transformada Materias primas Molécula objetivo (TGT) Retrón Transformada Adición ionica a C=O Reacción aldol Reacción Michael Anillación de Robinson Rearreglo de Claisen Precursores (materias primas) Molécula objetivo (TGT) Retrón Transformada Oxidación alílica O-Metalación y carboxilación cis-hidroxilación y dihidroxilación de Sharpless Epoxidación de Sharpless Precursores (materias primas) Molécula objetivo Retrón Transformada Anillación de Robinson Aldol Precursores sintéticos TAREA ¿CUÁL SERÍA EL RETRÓN PARA LAS SIGUIENTES MOLÉCULAS? Hidrogenación catalítica Reacción de Simmons-Smith Árbol retrosintético Representación gráfica en forma de árbol de varias de las posibles rutas retrosintéticas. Sintón Sintón O Equivalente sintético Árbol retrosintético Árbol TGT = extensión infinita de ramificaciones El primer principio de la planificación retrosintética: estrategias convergentes son las estrategias más eficientes para el ensamblado (unión) de moléculas complejas El poder de la síntesis convergente Considere una secuencia de tres pasos: Si el rendimiento promedio de cada uno es del 90 %, ¿Cuál será el rendimiento global? Rendimiento global: 73 % Se pierde ¼ de material Si consideramos 70 % (más realista) en cada paso, Se obtendrán al final un rendimiento global de Rendimiento global: 34 % Se pierde 2/3 de material Si la secuencia constara de 15 pasos y si consideramos 70 % de rendimiento para cada paso: Rendimiento global: 47 % Se pierde 999/100 de material Esto significaría que se necesita empezar con 1000 veces del material que se necesita SELECTIVIDAD Regioselectividad (en cual sitio de la molécula) Favorecido Regioisómeros SELECTIVIDAD Quimioselectividad (en cual grupo de la molécula) TIENE 2 ENLACES p Favorecido SELECTIVIDAD Diastereoselectividad (cual diastereoisómero) Favorecido Diasterómeros SELECTIVIDAD Enantioselectividad (cual enantiómero) Enantiómeros ¿Qué esta descrito en la literatura? ¿Qué tan cercana es la analogía a la reacción que se quiere hacer? 1 hora en la biblioteca = 1 mes en el laboratorio Exacto vs. Cercano Si se acerca, considere: • FG • tamaño del anillo • Sustituyentes • estereoquímica Todo puede afectar el resultado • • • • • • • • Residuo generados Costo Toxicidad Contaminantes Seguridad Escala Materiales de partida disponibles Cuestiones de propiedad intelectual Estrategia para el ensamblaje de compuestos cíclicos Ciclización Anillación Cicloadiciones concertadas Anillación no concertada «un sola operación» Estrategias de anillación en varios pasos LA REACCIÓN DE DIELS-ALDER Our results will play a role not only in the discussion of theoretically interesting questions . . . . but probably also will yield greater significance in a practical sense. Thus it appears to us that the possibility of synthesis of complex compounds related to or identical with natural products such as terpenes, sesquiterpenes, perhaps also alkaloids, has been moved to the near prospect. . . . . . We explicitly reserve for ourselves the application of the reaction discovered by us to the solution of such problems. Otto Diels and Kurt Alder Justus Liebigs Annalen der Chemie, 1928, 460, 98 Nuestros resultados tendrán un papel no sólo en la discusión de cuestiones teóricamente interesantes, pero probablemente también tendrá una mayor importancia en el sentido práctico. Así nos parece que la posibilidad de una síntesis de compuestos complejos relacionados con o bien idénticos a productos naturales tales como terpenos, sesquiterpenos, quizás también alcaloides , se ha movido hacia esta perspectiva. Nos reservamos explícitamente la aplicación de la reacción descubierta por nosotros para la solución de tales problemas . EL DESCUBRIMIENTO DE LA REACCIÓN DE DIELS-ALDER Benceno, 10 oC Dienófilo Doble enlace-cis enlaces sin Aquí la reacción de Diels-Alder es apropiada debido a que es una reacción estereoespecífica Reacción antitética IGF (FGI) DESCONEXIÓN IGF (FGI) 1-fenil1,3-butadieno Anhídrido maleíco Otras directrices para retrosíntesis se dan a continuación: 1. Es mejor utilizar el enfoque convergente en lugar del divergente para muchas moléculas complejas. 2. De preferencia utilice solamente desconexiones que correspondan a desconectar enlaces C-C y C-X siempre que sea posible. 3. Desconectar para generar sintones fácilmente identificables mediante el uso de reacciones conocidas (transformar). 4. La síntesis debe ser corta. 5. Es mejor utilizar reacciones que no forman mezclas. 6. La atención se centra en la eliminación de estereocentros bajo estereocontrol. Se puede lograr el estereocontrol a través de un control mecanistico o bien por el control de la estereoquímica del sustrato. ¿Qué desconectar y que mantener? DESCONECTAR 1) Para obtener fragmentos simétricos 2) Enlaces C–X (C–heteroátomo, ésteres, amidas, etc), Enlaces dobles (E o Z), Enlaces (de 1 a 3) alejados del grupo funcional 3) Enlaces que unan anillos con cadenas (así se produce un fragmento largo MANTENER 1) Grupos que forman parte de la estructura carboxíclica (alquilo , arilo) 2) Estereocentros alejados (más de 3 C es alejado) 3) Enlaces de la cadena próximos a estereocentros ¿Dónde debería elegir para desconectar? Las desconexiones muy a menudo tienen lugar en la posición inmediata adyacente (o muy cerca) a grupos funcionales presentes en la molécula objetivo desconexión cercana al grupo -OH mala buena desconexión bastante alejada del grupo -OH ¿Cómo puedo reconocer una buena desconexión? Una buena desconexión simplifica visiblemente la molécula objetivo. De lo contrario, el reto de la síntesis no será nada fácil !. mala Nivel similar de complejidad a que la molécula objetivo buena Los dos sintones son considerablemente más simples que la molécula objetivo ¿Cómo decido que síntón lleva que carga? Un buen truco aquí es considerar si usted puede dibujar una forma de resonancia del sintón, la cual se parezca más a un verdadero intermedio ... Si usted es capaz de hacerlo, es claro que ha hecho una buena elección de polaridad Desconexión Reacción antitética Reacción antitética: la opuesta a la reacción sintética Sintón Sintón Equivalente sintético Equivalente Este equivalente sintético sintético esta muy relacionado con la forma de resonancia del sintón ¿Cómo se podría obtener el siguiente compuesto? Nomenclatura de Seebach para sintones: d2, a3, etc. Cualquier átomo de carbono funcionalizado en la molécula objetivo (es más común un grupo carbonilo) se numera como 1 y a partir de el se numeran el resto de los carbonos de la cadena 3 2 1 Sinton: a3 Reactivo = enona 2 1 Sinton: d2 Reactivo = enolato Sintón d2: Representado por un enolato o su equivalente 1 2 Sintón a3: Representado por un compuesto cabonílico a,b-insaturado 3 2 1 En general tienen polaridad natural los: Sintones donadores con numeración par (d2, d4, etc.) y Sintones aceptores con numeración impar (a1, a3, etc.) En general tienen polaridad no natural ó umpolong (invertida) los: Sintones donadores con numeración impar (d1, d3, etc.) y Sintones aceptores con numeración par (a2, a4, etc.) Estrategias para la desconexión de enonas Desconexión aldol: sintones a1 y d2: sinton a1 sinton d2 enolato Acilación de un anión vinílico: sintón a1 Vinil metálicos Aproximadamente a la mitad de la molécula sinton a1 Agente acilante (X = Cl ó –OR) Desconexiones que involucran alquilación de enolatos: sintones d2 y d4 sinton a1 sinton d2 sinton d2 sinton a1 sinton d4 sinton a1 Desconexiones que involucran la acilación de compuestos organometálicos: sintones a1 y d1 sinton a1 sinton d1 sinton d1 Desconexiones que involucran adición de Michael sin perdida del alqueno: sintones a3 insaturados sinton d1 sinton a3 insaturado sinton a1 X= grupo saliente ANÁLISIS RETROSINTÉTICO 1) Maximice la convergencia 2) Minimice el número de pasos a)Busque multiples ruta b)Evite la interconversión de grupos funcionales (FGI) y grupos protectores, tanto como sea posible 3) Adiciones FG si estos pueden ayudar 4) Los enlaces C-X & C-CX es usual que sean buenas desconexiones 5) Desconecte estereocentros cuando sea posible (eliminelos) 6) Minimice los anillos de tamaño medio y grandes ( o tenga a la mano un buen plan) 6) Minimice los anillos de tamaño medio y grandes ( o tenga a la mano un buen plan) Cetona de Wieland-Miescher H2 ó H:- Desconexión pobre: anillo de 10 miembros Adición de grupo funcional (FGA) Anillación de Robinson 6) Minimice los anillos de tamaño medio y grandes ( o tenga a la mano un buen plan) 7) Desconecte los grupos inestables (lábiles) al principio Indolizomicina inestable a pH 7, 25 oC Nota: la molécula requiere de una desconexión de un anillo medio. Es necesario tener un buen plan de síntesis N-TEOC = 2-trimetilsililetoxicarbonil 8) Reconozca la simetría interna (±)carpanona 2 pasos 9) Identifique moléculas complejas dentro de la estructura Ambiguina (S)-carvona LA (S)-carvona proviene de las semillas de alcaravea. La obtención de los centros quirales ya existentes en materias primas fácilmente disponibles como en los productos naturales a menudo se llama estrategia de "pool quiral" 10) Use la topología (forma) de la molécula para guiar desconexiones (-)-morfina reacción tandem Pd-p-alilo Haga el modelo de la molécula Directrices básicas: 1. Utilice desconexiones que correspondan a reacciones confiables conocidas. Elija la desconexión correspondiente al mayor rendimiento en la reacción (convergencia). Sal de diazonio y reactivo de Grignard (propargílico) Grignard (fenilo) y halogenuro propargílico sintones Equivalentes sintéticos Halogenuro de bencilo y Grignard (propino) Grignard (bencilo) y halogenuro (haluro de propino) 2. Desconecte el enlace C-C de acuerdo con los FG (grupos funcionales) presentes en la molécula: Enlace C-C con un sustituyente O Enlace C-C alílico Enlace C-C con dos sustituyentes O en posiciones-1,3 3. Que su objetivo sea la simplificación : desconectar en el centro de la molécula Esta pequeña hormiga ( largo 2mm ) vive en todo el mundo , pero sólo puede sobrevivir en el Reino Unido en lugares cálidos , como los edificios con calefacción . Es un insecto difícil de controlar , en parte debido a su tamaño y también en la forma en que se vive . Una colonia se dividirá en varias colonias más pequeñas y sobrevivir a los intentos de controlarla . La hormiga faraón puede ser un problema importante en los hospitales , ya que puede entrar en máquinas e instrumentos estériles etc. Faranal Feromona de rastro de la hormiga faraón (E,Z)-3,4,7,11-Tetramethyl-6,10-tridecadiena Faranal, Feromona de rastro de la hormiga faraón Es necesario proteger 3. Objetivo de la simplificación: • Desconexión en un punto de ramificación • El uso de la simetría • El uso de la simetría Tropinona • El uso de la simetría Nonactina • El uso de los rearreglos (transposiciones) Claisen Ciclización aniónica Timo V. Ovaska,* Sarah E. Reisman, and Meghan A. Flynn Oxi-Cope Oxi-Cope Arthur Clay Cope 1909 - 1966 Oxi-Cope CALOR A Synthesis of Ketones by the Thermal Isomerization of 3-Hydroxy-1,5-hexadienes. The Oxy-Cope Rearrangement Jerome A. Berson, Maitland Jones, , Jr. J. Am. Chem. Soc. 1964; 86(22); 5019– 5020. doi:10.1021/ja01076a067 Stepwise Mechanisms in the Oxy-Cope Rearrangement Jerome A. Berson and Maitland Jones pp 5017 – 5018; J. Am. Chem. Soc. 1964; doi:10.1021/ja01076a066 Evans, D.A.; Golob, A.M. J. Am. Chem. Soc. 1975, 97, 4765–4766. doi:10.1021/ja00849a054 Dos estados de transición son posibles y el resultado de la reacción se puede predecir sobre la base de la superposición más favorable de los orbitales del doble enlace, la cuales influenciada por factores estereoelectrónicos : Paquette, L.A.;* Gao, Z.; Ni, Z.; Smith, G.F. J. Am. Chem. Soc., 1998, 120, 2543-2552. Aza Cope trans:cis 1:1 AZA-COPE MANNICH METATESIS TRANSPOSICIÓN DE CLAISEN ALQUILACIÓN OXI-COPE REACCIÓN DE METATESIS DE GRUBS C6H6, 0 OC, 1 h C6H6, 0 OC, 1 h Metatesis con cierre de anillo (RCM) Metatesis cruzada (CM) Metatesis con apertura de anillo (ROM) Metatesis de enino (intramolecular) Polimerización por metatesis con apertura de anillo (ROMP) Polimerización por metatesis Dieno acíclica (ADMET) Catalizador de Schrock 1990 muy sensible Catalizador de Schrock Más reactivo que el catalizador de Grubs Catalizador de Grubs 1996 muy usado, comercial 1ªestable generación Más y más Tolerante frente a otros grupos funcionales Menos reactivo Catalizador de Grubs 1999 muy usado, comercial 2ª generación Uno de los más eficientes Cy = ciclohexilo Mes = 2,4,6-trimetilfenilo Total Synthesis of (+)-Lysergic Acid Qiang Liu, Yu-An Zhang, Ping Xu, and Yanxing Jia* METATESIS OLEFÍNICA 1. 2. 3. 4. 5. 6. 7. Es la reacción perfecta El proceso es catalítico (1 5 % mol) Altos rendimientos bajo condiciones suaves Altos niveles de quimio, regio y estereoselectividad La reacción es reversible Materias primas fácilmente accesibles Los productos olefínicos son apropiados para una posterior elaboración en su estructura Hay 3 variaciones principales en el tema de metatesis: a) Metatesis cruzada b) Metatesis con cierre de anillo o por apertura de anilllo (RCM & ROM) c) Metatesis enino (RCM = RING FORMING METATHESIS) Proceso catalítico Procesos Inter o Intramoleculares Reversible Se forman 4 nuevos estereocentros Son posibles reacciones Carbono y hetero-Diels-Alder Proceso catalítico Procesos Intramoleculares Reversible No se forman estereocentros Son posibles reacciones Carbonó y hetero-RCM Metatesis olefínica El potencial de la RCM: Síntesis de Laulimalida por Ghosh y Mulzer Transformación catalítica pionera: Síntesis de Sch38516 por Hoveyda Dobles enlaces Ciclización Domino mediada por metatesis: Grubbs Desconexión en un enlace C-X (R-C(=O)-X) Cuadrona sesquiterpeno Desconecte anillos de cadenas 4. Anillos carbocíclicos Si uno o más unidades carbocíclicas de 6 miembros están presentes en la molécula objetivo, considerar un conjunto de desconexiones disponibles para la construcción de anillos de 6 miembros: Reacción de 1. 2. 3. 4. 5. 6. Diels -Alder, Anillación de Robinson Aldolica intramolecular Dieckmann (Claisen intramolecular) SN2 interna Reducción de Birch, etc. Algunos tipos de desconexiones de Diels -Alder : 5. Ejemplos de ruptura del enlace C-C como una reconexión sintética Rearrangements- Shapiro reaction Eschenmoser fragmentation Desconexión de moléculas de acuerdo con los GF presentes en la molécula: El gran potencial de la funcionalidad carbonilo Polaridad invertida (umpolong): Polaridad normal: (Adición-1,4 a enonas) (Adición-1,2) (via el enolato) Polaridad latente es el patrón imaginario de alternancia de cargas positivas y negativas utilizadas para ayudar en la elección de las desconexiones y sintones. Considerar la polaridad latente suele dar la mejor opción de sintones. Adición de un nucleófilo o Así: Sintones naturales Desprotonación para dar un enolato o Así: Sintones naturales Aplicando esto a diferentes grupos funcionales Polaridad latente de un grupo carbonilo Obtenga estos dos a partir de un grupo carbonilo POLARIDADES LATENTES La presencia de un heteroátomo en una molécula imparte un patrón de electrofilia y nucleofilia al átomo de la molécula. El concepto de polaridades alternas o polaridades latentes (cargas imaginarias) a menudo permite identificar las mejores posiciones para hacer una desconexión dentro de una molécula compleja Los grupos funcionales se pueden clasificar de la siguiente manera . Clase E: Grupos que confieren carácter electrofílico al carbono unido (+): -NH2 , -OH , -OR, = O, = NR , -X ( halógenos) Clase G: Grupos que confieren carácter nucleófilo al carbono unido (-): -Li , -MgX , -AlR2 , -SiR3 Clase A: Los grupos funcionales que presentan carácter ambivalente (+ o -) : -Br2 , C = CR2 , CCR3 , -NO2 , N , -SR , -S (O ) R, -SO2R Patrón Consonante : La carga positiva se colocan en el átomo de carbono unido a los grupos de la clase E . Patrón Disonante : Una clase E está unido a un carbono con una carga positiva, mientras que el otro grupo de clase E reside en un carbono con una carga negativa. Síntesis simple De acuerdo con estas ideas, es posible identificar las relaciones difuncionales (consonante o disonante) entre los grupos funcionales en una TGT Relación disonante 1,2-difuncional Relación consonante 1,3-difuncional Relación consonante 1,4-difuncional Relación consonante 1,5-difuncional En general las relaciones consonantes permiten elaborar desconexiones fáciles. Sin embargo, las relaciones disonante a menudo requieren introducir tácticas umpolung, reacciones por radicales o pericíclicas Relación consonante (concuerdan) Arreglo polar por X Arreglo polar por Y Relación disonante (no concuerdan) Arreglo polar por X Arreglo polar por Y EJEMPLOS DE DESCONEXIONES DISONANTES Los compuestos difuncionalizados-1,2 no pueden ser preparados por este tipo de desconexión, la cual si se usa para compuestos difuncionalizados-1,3 y 1,5 Acilanión enmascarado: umpolung Disponible comercialmente Utilice: RCH=O Sintón inusual (umpolung) Problema: la reactividad poco usual requiriría un sintón nucleofílico umpolung Solución: diseñe un reactivo para el sinton que se requiere o bien, evite el problema empleando una estrategia diferente Usar: RCH=O Usar el anión acilo apropiado Aniones acilo A partir de aniones acetiluros Oximercuración de acetileno A partir de tioacetales Hidrólisis tioacetal Equivalentes de aniones acilo Compuestos Nitro Ion cianuro Usar: RCH=O SÍNTESIS EJEMPLOS DE DESCONEXIONES CONSONANTES COMPÚESTOS 1,3-DIFUNCIONALIZADOS COMPUESTOS 1,5-DIFUNCIONALIZADOS + - + - + Reactivo de Reformatsky (enolato de zinc nucleofílico) Reacción de Michael b-hidroxicetonas Sintones Reacción antitética Reacción sintética Usar Compuestos carbonílicos a,b-insaturados Ácido o base eliminación Por lo tanto Epóxidos Sintones Usar epóxido Usar Reacción sintética calor El enolato ataca al epóxido por al cara opuesta al átomo de O (SN2) Sintones nucleofílicos no naturales En las dos aproximaciones anteriores se necesita un sinton invertido (umpolung) que se adicione-1.4 (Michael) Si se necesita: Se puede usar: ó Si se necesita: Se puede usar: Carbanión estabilizado por resonancia pKa ≈ 10 Análisis Reacción sintética Reacción de Nef La reacción de ciclización da la enona más estable COMPÚESTOS 1,4-DIFUNCIONALIZADOS DESCONEXIÓN A LA MITAD + - + REACTIVIDAD USUAL ENOLATO SINTON CON INVERSIÓN (UMPOLUNG) ALGUNOS ACEPTORES UMPOLUNG a-HALOCARBONILOS C=O ACIDO DURO C-Br ÁCIDO BLANDO Reacción sintética ENAMIMA NUCLEOFILO BLANDO SE NECESITA UN EQUIVALENTE DE ENOLATO QUE SEA BLANDO COMPÚESTOS 1,5-DIFUNCIONALIZADOS Con frecuencia se genera in situ • Se requiere enolato suave • Se usa un grupo activante para asegurar la enolización y la adición de Michael (éster) Desconexión en el punto de ramificación Es necesario un grupo activante (hidrógeno a más ácido) Usar Malonato de dietilo COMPÚESTOS 1,5-DIFUNCIONALIZADOS Reacción sintética Hidrólisis del éster y descarboxilación COMPUESTOS 1,5-DIFUNCIONALIZADOS Desconexión en el punto de ramificación El enolato del aldehído es demasiado reactivo. Posible autocondensación Usar: enamina Usar: Reacción sintética COMPUESTOS 1,6-DIFUNCIONALIZADOS Sinton difícil si nos basamos en la química del grupo carbonilo Usar Estrategia alterna: Una reconexión de grupo funcional, más que una desconexión: MOLOZÓNIDO OZÓNIDO PRIMARIO OZÓNIDO OZÓNIDO SECUNDARIO GEM-DIOL IGF RECONECTAR IGF Reacción sintética ESTRATEGIAS BASADAS EN GRUPOS FUNCIONALES Corey clasifica a los grupos funcionales (FG) en: 1er. nivel: los FG más importantes alquenos arenos Aldehídos, R = H Cetonas, R ≠ H alquinos Ácidos, X = OH Ésteres, X = OR’ Amidas, X = NR2 alcoholes nitro aminas ciano 2º. nivel: los FG menos importantes diazo disulfuro fosfina 3er. nivel: periféricos, los cuales están asociados con reactivos útiles que proporcionan activación o control en procesos químicos, o una combinación de más grupos fundamentales haluros fosfonio sulfonas enamina boranos enona Pueden estar asociados en grandes familias, dependiendo de su comportamiento electrónico EWG (Electroatractores: CO, CN, -SOR. NO2) o EDG (Electrodonadores: OR, NR2) ESTRATEGIAS BASADAS EN GRUPOS FUNCIONALES Muchos retrones contienen un solo FG, mientras que otros consisten de un par de FG, separados por una cadena de carbonos o conexiones 1) Transformadas que involucran a un solo grupo funcional Eliminación de un grupo FGI interconversión de grupo funcional Rearreglo o transposición 2) Transformadas que requieren dos FG A través de una desconexión C-C Formación de una nueva estructura Modificación de funcionalidad sin alterar la cadena Si se toma en cuenta que la mayoría de las reacciones sintéticas son polares, el proceso de formación de un enlace (y su correspondiente transformada) se pueden visualizar como una combinación de sintones donadores, d y aceptores a Se pueden aplicar reglas obvias para ordenar la funcionalidad en el producto. Para una molécula conteniendo n FG hay: Alquilo a + Alquilo d Alquilo a + d1 ó Producto no funcional Alquilo d + a1 Producto monofuncional a1 + d1 a1 + d2 a1 + d3 ó ó a2 + Producto 1,2-difuncional a2 + d2 d1 ó Relación consonante a3 + Producto 1,3-difuncional d1 Producto 1,4-difuncional Combinación de sintones No funcional Alquilo a Monofuncional 1,2-Difuncional Alquilo d 1,3-Difuncional 1,4-Difuncional a1 d3 Sistemas 1,2-difuncionales: Combinación a1 + d1 Síntesis de Strecker Ácido ciclopentilaspártico Sistemas 1,2-difuncionales: Combinación a1 + d1 Sistemas 1,3-difuncionales: Combinación a1 + d2 Reacción aldólica Reacción Wittig Reacción retro-Claisen Sintones d2: enol, enolato y equivalentes sintéticos Sintones a1: aldehídos, cetonas y ésteres Punto de referencia: Síntesis de helmintosporal por Corey Helmintosporal, toxina del hongo helminthosporium savitum Condiciones experimentales y resultados obtenidos: Este compuesto 1,5-difuncional puede reaccionar de dos maneras diferentes: Catalisis ácida Catalisis básica Síntesis de helmintosporal Protocolo sintético Una molécula objetivo polifuncional: núcleo 18-epi-tricíclico de Garsubelina A sintetizada por Shibasaki Garsubelina A Org. Lett. 2002, 859 Aplicando la relación Modelo de la Garsubelina A Org. Lett. 2002, 859 6 FG: 15 posibles pares de FG Las relaciones 1,3-difuncionales juegan un papel importante (crucial) El análisis retrosintético se basa en las siguientes desconexiones a1 a3 d2 Y todavía una tercera posición d2 Posición mas ácida Posición menos ácida Acoplamiento de Stille d 2 + a3 d 2 + a1 d 2 + a1 d 1 + a1 d 2 + a2 d 0 + a3 La estrategia marca el camino, pero la táctica explica el éxito: regiocontrol en la formación de enolato Control cinético del enolato resultante, evita los problemas de regioselectividad Región más accesible para la desprotonación con el hexametildisilamiduro de potasio (KHDMS) base voluminosa más estable pero no se forma por impedimento estérico Pasos finales El enlace Si-Si bond en hexametildisilano es rompe tanto por nucleófilos fuertes como por electrófilos. <los compuestos de alquil-lito reaccionan de la siguiente manera: Si2Me6 + RLi → RSiMe3 + LiSiMe3 Con yodo se obtiene el yoduro de trimetilsilicio Me3Si−SiMe3 + I2 → 2 SiMe3I Fleming-Tamao Oxidation Tamao-Kumada Oxidation periodinano Dess–Martin J. Org. Chem., 1992, 57, 3994. Proceso Wacker Intramolecular Cerium (IV) ammonium nitrate ((NH4)2Ce(NO3)6) is a oneelectron oxidizing agent that is used for oxidative addition reactions of electrophilic radicals to alkenes Proceso Wacker Intramolecular Adición nucleofílica tautomería Eliminación de hidruro b (Eliminación de hidruro de paladio) Procesos catalizados con metales de transición Reacciones de entrecruzamiento Metatesis: meta (cambio) & tesis (posición) Reacciones catalizadas con Pd (0) (X = OTf ó Hal. Ar = Arilo. Y = O ó NH. L-Grupo saliente) Heck Fuente de Pd (0) + Base Suzuki Fuente de Pd (0) + Base Stille Fuente de Pd (0) Hiyama Fuente de Pd (0) + Bu4NF Sonogashira Fuente de Pd (0) + Fuente de Cu (I) + Base (alquiniluro de cobre) Buchwald-Hartwig Fuente de Pd (0) + Base Reacciones catalizadas con Pd (0) (X = OTf ó Hal. Ar = Arilo. Y = O ó NH. L-Grupo saliente) Tsuji-Trost sustitución p-alilica Fuente de Pd (0) + Nu-H LG grupo saliente Wacker Fuente de Pd (0) + H 2O Metaloide-carbenos Metatesis de olefinas LnM=CH-R Grubs catalizador (Tipo 1, 2) Ru: Grubs-Hoveyda Ru; Schrock Mo Tebbe Cp2Ti (CH2)ClAlMe2 Reacciones Co-catalizadas Pauson-Khand Co(CO)8 + CO Estas reacciones tienen los mismos pasos clave: 1) Adición oxidativa (O). 4) Eliminación reductiva (RE) 2) Inserción (I) 5) Transmetalación (T) 3) b-eliminación (BE) Heck Fuente de Pd (0) + Base Suzuki Fuente de Pd (0) + Base Stille Fuente de Pd (0) Hiyama Orden mecanístico: 1) O; 2) I; 3) Rotación Interna ; 4) sin-BE; 5) RE Orden mecanístico: 1) O; 2) T; 3) Isomería trans-cis; 4) cis-RE Orden mecanístico: 1) O; 2) T; 3) Isomería trans-cis; 4) cis-RE Fuente de Pd (0) + Bu4NF Orden mecanístico: 1) O; 2) T (de Si «ato»); 3) Isomería trans-cis; 4) cis-RE Sonogashira Fuente de Pd (0) + Fuente de Cu (I) + Base Orden mecanístico: (alquiniluro de cobre) Buchwald-Hartwig 1) O; 2) T (con alquino-Cu); 3) Isomería trans-cis; 4) cis-RE Fuente de Pd (0) + Base Orden mecanístico: 1) O; 2) Intercambio de ligando (cf I) 3) RE Tsuji-Trost sustitución p-alilica Fuente de Pd (0) + Nu-H LG grupo saliente Wacker Fuente de Pd (0) Orden mecanístico: Doble desplazamiento a través de un intermediario h3-Pd + H 2O Orden mecanístico: 1) I con ataque nucleofílico concertado por H2O; 2) BE: 3) tautomería ceto-enólica Metaloide-carbenos Metatesis de olefinas LnM=CH-R catalizador Grubs (Tipo 1, 2) Ru: Grubs-Hoveyda Ru; Schrock Mo Orden mecanístico: 1) [2 + 2]; 2) retro [2 + 2] 3) [2 + 2]; 4) retro [2 + 2] Tebbe Cp2Ti (CH2)ClAlMe2 Orden mecanístico: 1) Retro [2 + 2]; 2) [2 + 2] 3) retro [2 + 2] Reacciones Co-catalizadas Pauson-Khand Co(CO)8 + CO Orden mecanístico: 1) Intercambio de ligando x 2 (coordinación alquino) ; 2) O 3) I (alqueno) ; 4) I (CO); 5) migración; 6) RE Eliminación reductiva Adición oxidativa Reactivo borónico u otro reactivo organometálico Fuhrhop y Li identifican al siguiente intermediario como un sintón a1: i.e. Reacción de Heck O CCH3 + Br O CCH3 Pd(PPh3)4 CH2 CH2 (CH3CH2)3N Pd(PPh3)4 (CH3CH2)3N El haluro debe ser arilico o vinilico CH CH2 CH3O Reacción de Stille Br Pd(Ph3)4 + H2C CHSn(CH 2CH2CH2CH3)3 OTf + Sn(CH2CH2CH2CH3)4 CH CH2 THF Pd(Ph3)4 CH2CH2CH2CH3 THF El haluro debe ser arilico, vinílico o bencílico 228 Acoplamiento de Suzuki CH2 Br H3C + CH3CH2CH2 Br H O + CH3CH C B O O B O Pd(PPh3)4 CH2 CH2CH2CH3 NaOH Pd(PPh3)4 H3C CH CHCH3 NaOH El haluro debe ser arílico, vinílico o bencílico 229 Acoplamiento de Stille Garsubelina A [1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) METATESIS DE OLEFINAS Garsubelina A El poder de las reaccion es de metatesis: síntesis de laulimalida realizada por Ghosh y Mulzer ¿Cuál debe ser el análisis en el caso de las relaciones disonantes? Recuerde considerar la posibilidad de: Desconexiones por radicales Procesos basados en reacciones redox Reconectar anillos Alternar la reactividad: umpolung o inversión de la reactividad reversible Desde el punto de vista retrosintético, si una desconexión es considerada como estratégica, pero no es permitida por el grupo funcional presente, la sustitución de dicho grupo por uno equivalente que si permita dicha desconexión, se convierte en una submolecula objetivo. Obviamente, esta operación requiere una etapa de síntesis (FGI) que permite invertir (umpolung) la reactividad: pasar de un aceptor a un donador o de un donador a un aceptor UMPOLUNG CARBONILO: ACILANIÓN ENOLATO UMPOLUNG: CATIÓN a-CARBONILO Sintón formilmetilo Con frecuencia reacciones no deseadas Sintón formiletilo Las espongistatinas: Productos Naturales arquitectónicamente complejos. Análisis por concepto umpolung Wittig Macrolactonización Alquilación Espongistatina 1 Angew. Chem. Int. Ed. 2001, 40, No. 1, 191-195 Organic Letters, 2002, 783 Fragmento A-B Relaciones 1,3 -consonante: ¿La reacción Aldol podría ser la respuesta? Si podría ser, pero se puede imaginar otra desconexión Fragmento C-D Fragmento A-B 1HMPA: hexametilfosforotriamida, se usa como un agente de coordinación de litio y otras sales metálicas. Se utiliza para disociar reactivos organometálicos de litio, mejorando la nucleofilicidad y basicidad del anión. COMPUESTOS ORGANOMETÁLICOS Los compuestos organometálicos tienen al menos un enlace carbono a metal, de acuerdo a la mayoría definiciones. Este enlace puede ser 1) Un carbono unido directamente al metal (enlace σ) 2) A través de un complejo metálico (enlace π ). Se incluyen en este tipo de compuestos a los que contienen un enlace metal con hidrógeno, así como algunos compuestos que contienen elementos no metálicos (metaloide) unidos a carbono. COMPUESTOS ORGANOMETÁLICOS 1760 Louis Claude Cadet Sintetizó metil arsénico, relacionado con cacodilo 1827 COMPUESTOS ORGANOMETÁLICOS El primer complejo metalico identificado como un compuesto organometálico fue una sal, K(C2H4)PtCl3, obtenida por William Zeise en 1825 por medio de la reacción de etileno con cloruro de platino (II). William Christopher Zeise (1789 – 1847) Sal de Zeise: tricloro(eteno) platinato (II) de potasio No fue hasta mucho más tarde (1951-1952) que la estructura correcta del compuesto de Zeise fue reportada en conexión con la estructura del metaloceno conocido como ferroceno 1848 COMPUESTOS ORGANOMETÁLICOS Edward Frankland descubrió el dietil zinc Edward Frankland (1825 – 1899) 1863 Prepararon organoclorosilanos SiCl4 + (k/2)ZnR2 → RkSiCl4-k + (k/2)ZnCl2 Charles Friedel (1832 – 1899) James Crafts (1839-1917) 1890 Ludwig Mond descubrió el Ni(CO)4, Ludwig Mond (1839 – 1909) 1899 Introducción del reactivo de Grignard (compuestos de organomagnesio) François Auguste Victor Grignard (1871 -1935) 1900 CO2 + 4 H2 → CH4 + 2 H2O + energy ∆H = −165.0 kJ/mol Catalizador de Ni Paul Sabatier (1854 – 1941) 1909 Paul Ehrlich introduce Salvarsan en el tratamiento de la Sifilis 1912 Premio Nobel de Química a Victor Grignard y Paul Sabatier 1930 Henry Gilman trabajó en química de cuprato de litio 1951 Walter Hieber recibió el premio Alfred Stock por su trabajo en química de metal carbonilo 1951 Descubrimiento del Ferroceno Se preparó primero involuntariamente . En 1951 , Pauson y Kealy de la Universidad de Duquesne informaron de la reacción de bromuro de magnesio ciclopentadienilo y cloruro férrico con el objetivo de acoplar oxidativamente el dieno para preparar fulvaleno 1963 Premio Nobel de Química a Karl Ziegler y Giulio Natta por el catalizador Ziegler-Natta TiCl4 como ingredient e activo y MgCl2 como soporte 1968 Reacción de Heck Richard Heck ( 1931 ) 1973 Premio Nobel de Química a Geoffrey Wilkinson y Ernst Otto Fischer por los compuestos sandwich Geoffrey Wilkinson (1921 - 1996) Ernst Otto Fischer 1918 – 2007) 2005 Premio Nobel de Química a Richard R. Schrock (1945,) Yves Chauvin (1930 – 2015) Robert H. Grubbs (1942, Metatesis de alquenos catalizada con metales 2010 Premio Nobel de Química Richard Fred Heck (1931,) Eiichi Negishi (1935,) Akira Suzuki (1930,) A partir de la década de 1920 se patenta su uso mezclándolo con la gasolina como un refuerzo octanaje, gracias a lo cual la compresión del motor se eleva sustancialmente, lo que a su vez aumenta el rendimiento del vehículo o el ahorro de combustible Algunas propiedades comunes de los compuestos organometálicos son puntos de fusión relativamente bajos, insolubilidad en agua, solubilidad en éter y en otros disolventes relacionados, toxicidad, oxidables y una alta reactividad. 323.44 g·mol −1 Masa Molar Apariencia Líquido incoloro Densidad 1.653 g cm Punto de fusión −136 °C (−213 °F; 137 K) Punto de ebullición 84 °C (183 °F; 357 K) 15 mmHg Índice de Refracción 1.5198 −3 Actualmente esta prohibido su uso en los Estados Unidos Nomenclatura Los compuestos organometálicos normalmente se nombran como metales sustituidos, por ejemplo, de alquilo metal o haluros de alquilo metálicos. Compuestos de organomagnesio se denominan generalmente como reactivos de Grignard . Ejemplos : CH3Li = metil-litio , CH3MgBr = bromuro de metil magnesio. Propiedades Físicas: los compuestos organometálicos generalmente se mantienen en disolución en disolventes orgánicos debido a su alta reactividad (especialmente con H2O , O2 etc.) Estructura: organosodio y compuestos organopotasio son esencialmente compuestos iónicos. Organolitiados y organomagnesio tienen como enlace entre un átomo de C y el metal: C-M Estos son enlaces covalentes polares debido al carácter electropositivo de los metales. Observe las electronegatividades de los metales Li , Na, K y Mg comparado con C y los otros átomos ( por ejemplo, N , O, F , Cl , etc ). Vea cómo el átomo de C es más electronegativo que el metal Tabla periódica de parcial con Electronegatividades de Pauling Cloruro de metilo Menor densidad electrónica Mayor densidad electrónica Metil litio Mayor densidad electrónica Bromuro de metil magnesio Menor densidad electrónica Menor densidad electrónica Mayor densidad electrónica En el haluro de alquilo , el grupo metilo tiene una densidad de electrones inferior ( azul), y es un electrófilo Cloruro de metilo En el metil-litio, el grupo metilo tiene mayor densidad de electrones (rojo) y es un nucleófilo Metil litio En el bromuro de metil magnesio, el grupo metilo es menos rico en electrones que metil-litio Bromuro de metil magnesio Por lo tanto, los compuestos organometálicos de carbono reaccionan como ricos en electrones, es decir, como carbaniones: funcionarán ya sea como bases o nucleófilos . Es razonable pensar que estos compuestos organometálicos como R:- M + BASICIDAD La siguiente escuación representa la pérdida de protón de un hidrocarburo para formar un carbanión: Hidrocarburo Carbanión Los compuestos organolitiados y de organomagnesio son bases fuertes ya que la carga negativa se encuentra sobre el carbono. Los carbaniones simples son bases fuertes, (ver pKa a continuación) ya que el C no es muy electronegativo (en comparación con N o O) Compuesto Estructura pKa 2-metilpropano 71 Etano 62 Metano 60 Eteno 45 Benzeno 43 Amoniaco 36 Etino 25 Etanol 16 Agua 15.7 La tabla muestra los pKa de una selección de representante sistemas. Tenga en cuenta que los hidrocarburos son ácidos muy débiles, lo que implica que los carbaniones serán bases fuertes. • Los compuestos organolitiados se forman por la reacción de haluros de alquilo con el metal de litio. • Los disolventes típicos son éter dietílico anhidro pero normalmente pentano o hexano también se pueden utilizar. • El grupo alquilo puede ser primario , secundario o terciario. • Reactividad del haluro : I> Br > Cl • R puede ser alquilo , vinilo o arilo • Se pueden utilizar otros metales del Grupo I (Na, K) en lugar de Li • Los compuestos organomagnesio se forman por la reacción de haluros de alquilo con magnesio metálico. • Los disolventes típicos son normalmente éter dietílico anhidro o tetrahidrofurano. • El grupo alquilo puede ser primario, secundario o terciario. Reactividad de haluro : I> Br > Cl • R puede ser alquilo, vinilo o arilo. En presencia de ácidos débiles (como los alcoholes), RLi y RMgX se protonan dando el hidrocarburo .correspondiente (ácido conjugado) Preparación de Reactivos de organocobre • Los reactivos de organocobre más útiles son los dialquilcupratos de litio, R2CuLi • Los dialquilcupratos de litio se forman por la reacción de 2 equivalentes de un organolitio con un haluro de cobre (I). • Los disolventes típicos son normalmente éter dietílico anhidro o tetrahidrofurano. • El grupo alquilo es generalmente primario. Con los grupos Secundarios y terciarios son propensos a la descomposición. • Reactividad de haluro: I> Br > Cl • R puede ser alquilo, vinilo o arilo. 1936 Henry Gilman preparó metilcobre 1941 Kharash descubrió la reacción de un reactivo de Grignard con ciclohexenona en presencia de Cu(I) (adición-1,4 en lugar de una adición-1,2 1952 Gilman investigó por primera vez los dialquilcupratos Preparación de Reactivos de organozinc • Reactivos organometálicos de cinc, RZnX, se preparan de una manera análoga a la de los reactivos de organomagnesio RMgX. • Son mucho menos reactivos que RLi o RMgX a aldehídos y cetonas. • La aplicación más común de reactivos organometálicos de cinc está en la reacción de Simmons -Smith • Reactivos de Grignard, haluro de acetilén magnesio (RC≡CMgX) • Son preparados por una reacción ácido-base del acetileno terminal con un segundo reactivo de Grignard. • Los reactivos de haluro de acetilén magnesio reaccionan de una manera similar a otros reactivos de Grignard. 1. Sustitución nucleofílica Aplicación organometálica R2CuLi con haluros de alquilo o tosilatos para dar alcanos 2. Adición nucleofílica RLi o RMgX con aldehídos o cetomas para dar alcoholes 2os. ó 3os. 3. Sustitución nucleofílica sobre el grupo acilo RLi o RMgX con ésteres para dar a través de una SNAC y una adición alcoholes 3os Limitaciones: 1) Compuestos de organolitio, RLi , y de organomagnesio, RMgX a) Sustitución nucleofílica alifática Los reactivos son típicamente demasiado básicos para ser utilizado en reacciones de sustitución nucleófila con haluros de alquilo o tosilatos donde tienden a causar reacciones de eliminación u otras reacciones secundarias. b) Reacciones de adición con compuestos carbonílicos, epóxidos. b) Sustitución nucleofílica de acilo se observa con mayor frecuencia con ésteres. 2) Organocupratos , R2CuLi, a) Sustitución nucleofílica alifática Los R2CuLi, reaccionan con haluros de alquilo o tosilatos para dar alcanos sin eliminación. b) Reacciones de adición. Los R2CuLi, son menos reactivos y no reaccionan con aldehídos, c) Sustitución nucleofílica sobre el grupo acilo. Los R2CuLi no la presentan, aún con ésteres Revisión reactivo de Grignard R’ = alquilo, vinilo, arilo Mg / éter ó medio ácido Procesamiento final típico para estas reacciones: 1) Diluir con ácido acuoso o 2) Cloruro de amonio acuoso Reacciones de RLi y RMgX con Aldehídos y cetonas Es usual que estas reacciones se lleven a cabo en éter o THF, seguido de un tratamiento final con H3O+ • Los compuestos de organolitio o de Grignard reaccionan con el grupo carbonilo, C = O, de aldehídos o cetonas para dar alcoholes. • Los sustituyentes en el carbonilo determinan la naturaleza del producto alcohol. • Solo el formaldehído da alcoholes primarios. Cualquier otro aldehído da alcoholes secundarios. • Las cetonas dan alcoholes terciarios. • El tratamiento ácido convierte una sal de alcóxido de metal, en el alcohol deseado mediante una reacción ácido-base simple. Es usual que estas reacciones se lleven a cabo en éter , seguido de un tratamiento final con H3O+ • Los reactivos acetiluro con el grupo carbonilo, C=O, de aldehídos o cetonas para dar alcoholes. • Los sustituyentes en el carbonilo dictan la naturaleza del alcohol producto. Solo el formaldehído da alcoholes primarios. Los otros aldehídos dan alcoholes secundarios. Las cetonas dan alcoholes terciarios. • El tratamiento ácido convierte una sal de alcóxido de metal en el alcohol deseado mediante una reacción ácido-base simple. Aplicaciones Sintéticas nucleofilo Sólo la reacción con el anión acetiluro ofrece los medios para hacer un nuevo enlace CC y una molécula más grande . Se necesita un alquino terminal. Es usual que estas reacciones se lleven a cabo en éter , seguido de un tratamiento final con H3O+ • Ésteres carboxílicos, R'CO2R’' , reaccionan con 2 equivalentes de organolitio o reactivos de Grignard para dar alcoholes terciarios. • El alcohol terciario contiene 2 grupos alquilo iguales (R) • . • La reacción transcurre a través de una cetona intermedia que luego reacciona con el segundo equivalente del reactivo organometálico. • Dado que la cetona es más reactiva que el éster, la reacción no se puede utilizar para preparar cetonas. Un reactivo de Grignard tiene un carbanión reactivo. Con el anillo de oxirano Bromuro de n-butilmagnesio Resultado netos 1-hexanol Alcóxido de magnesio Nuevo enlace formado El tamaño del grupo alquilo se incrementa por 3 carbonos R-OH R-X R-Mg-X R-CH2-CH2-OH. El grupo funcional (OH) permanece en el extremo de la cadena. Se podría repetir la secuencia El ataque es bajo condiciones SN 2 Con un oxirano sustituído Nuevo enlace formado Ejemplo de síntesis Análisis Retrosintético OH OH O CH2CH=CH2 CH2=CH - CH2MgBr La reacción de un nucleófilo con un oxirano da un patrón: HO-C-C-Nu. El ataque bajo condiciones SN2 da una apertura anti. La geometría Trans sugiere probar con un oxirano. ¿Cual podría ser el nucleófilo? El grupo alilo deberá ser el nucleófilo (usar un Grignard (o Gilman). • Los Organo cupratos de litio, R2CuLi , reaccionan con haluros de alquilo para formar un nuevo C-C , dando alcanos • Los Yoduros de alquilo primarios son los mejores sustratos, de otro modo la eliminación puede ser un problema. • El grupo R del cuprato puede ser arilo o vinilo. • El grupo R ' en el haluro también puede ser arilo o vinilo. • No obstante que el mecanismo se ve como un SN2 , es más compleja y actualmente no se entiende bien. Reacciones del Reactivo de Gilman Reacción de acoplamiento: se usa para formar nuevos enlaces C – C. Resultado global: R-X + R’-X R – R’ Detalles necesarios Li Formación del reactivo de Gilman CuI R-Li R-X R2CuLi electrófilo R'-X Siguiente paso: R2CuLi R - R' Restricciones en el proceso El grupo R del reactivo de Gilman puede ser CH3, 1o (de preferencia no 2o o 3o), alílico, vinílico (poco usual), arilo Alquilo (no 3o), vinílico nucleófilo Reactivo de Gilman: particularmente útil en la reacción con haluros de vinilo para formar alquenos Dietil éter O THF trans Se retiene la estereoquímica del alqueno REACTIVOS DE GILMAN En general son: • Solubles • Térmicamente inestables • Es usual generarlos in situ • Con frecuencia la "receta " que se utiliza para hacer el reactivo y / o llevar a cabo la reacción con el sustrato es crítica para el buen éxito • Se han descubierto su reactividad en forma empírica • Se pueden utilizar y para transferir prácticamente cualquier carbono con hibridación sp2 o sp3 Debido a su baja basicidad, los diorganocupratos pueden llevar a cabo reacciones de alquilación con una gran variedad de electrófilos; generalmente con altos niveles de inversión y poca eliminación Reaccionan normalmente reaccionan por un mecanismo SN2 ', si es posible ORDEN DE REACTIVIDAD primario > secundario > > terciario yoduro > bromuro > cloruro haluros de alquenilo y triflatos funcionan tan bien, con retención de la configuración ( cis , trans) RCOCl > aldehídos > tosilatos ~ epóxidos > yoduros > cetonas > ésteres > nitrilos Algunos ejemplos REACTIVOS DE GILMAN Presentan reacciones de adición electrófilos conjugados α,β-insaturados; el enolato intermedio puede ser atrapado con una gran variedad de electrófilos • Cetonas – son las más reactivas, la rapidez se puede ver ligeramente disminuída con sustitución en la posición α o β. • Aldehídos – hay competencia con adición-1,2 • Ésteres – son menos reactivos que las cetonas, la rapidez se ve disminuída en forma dramática con sustitución en las posiciones α o β • Sulfonas - son sustratos competentes • Ácidos carboxílicos - no reaccionan • Derivados de ácidos carboxílicos - Amidas y anhídridos tienen un uso limitado • Ésteres – son menos reactivos que las cetonas, la rapidez disminuye en forma dramática con sustitución en posiciones α o β • La adición de ligandos de fosfina con frecuencia acelera a las reacciones que presentan problemas de reactividad Algunos ejemplos Gilman y oxiranos 1. R2CuLi HO O 2. H2O, HCl R R es el reactivo de Gilman y es el nucleofilo (organometálico). Debido al medio básico (el ácido destruye al reactivo de Gilman) el oxirano no puede ser protonado. El ataque ocurre bajo condiciones SN2 Análisis Sintético Similar al análisis del reactivo de Grignard 1. R2CuLi HO O 2. H2O, HCl R Enlace recién formado. Es importante su posición relativa a la OH Ejemplo de análisis retrosintético Diseño de una síntesis usando oxiranos El anillo de oxirano puede estar en cualquier lado del OH. Analizar las dos posibilidades El Nucleofilo puede venir en una sola posición del anillo del oxirano, en la que el C no debe estar unido al OH OH Ph OH OH o Ph A la izquierda, localizado aquí. Apertura del oxirano por aquí El Nucleofilo forma este enlace Ph A la derecha, localizado aquí La apertura del oxirano por este lado. O (PhCH2)2CuLi El Nucleofilo forma este enlace 2 rutas sintéticas posibles O Ph LiCu(CH2CH3)2 M = Li, MgBr o Cu Rendimiento altos, adición-1,4 con >99 % Tiempos de reacción cortos (< 1h) Ejemplo de Síntesis Proponga como llevar a cabo la siguiente transformación, empleando tantos pasos como sean necesarios Br O OCH3 O TGT ó MO OH Br O OCH3 La oxidación de un alcohol 2o forma una cetona OCH3 La relación de un nucleofilo (OCH3) luego C-C y al final OH. Usar un oxirano Los oxiranos provienen de alquenos con perácidos Alquenos: a partir de haluros a través de una E2. • Esta es la reacción más importante que implica un reactivo orgánico de zinc. • Se conoce como la reacción de Simmons–Smith. • El yoduro de yodometil zinc se prepara generalmente con Zn activado y Cu. • El yoduro de yodometil zinc reacciona con un alqueno para dar un ciclopropano. • La reacción es estereoespecífica con respecto a al alqueno (mecanismo concertado) Los sustituyentes que se encuentran trans en el alqueno se encuentran trans en el ciclopropano (y lo mismo para el alqueno cis) Carbenos, :CH2 Preparación de carbenos simples 1. carbeno 2. diclorocarbeno Mecanismo de la a-eliminación diclorocarbeno Reacciones de Carbenos, :CH2 (no para síntesis) Adición a dobles enlaces Inserción en un enlace C-H Formación del iluro liquid Reacción de Simmons Smith (para síntesis, adición a alquenos para formar ciclopropanos) CH2I2 + Zn(Cu) ICH2ZnI Carbenoide, propiedades similares a los carbenos Dietil éter Metilénciclopentano Espiro[4.2]heptano Plantilla para reacciones OH OH H H CH2I2 Zn(Cu) ¿Por que razón es estereoespecífica, al estar el anillo del mismo lado que el grupo OH? H H estereoespecífica, stereospecific as shown ICH2ZnI H OH H Interacción con el metal mantiene al carbenoide en la parte superior Estructura Electrónica carbeno Orbital p vacío: semejante a un carbocatión Estructura del orbital de CH2 (metileno) Par de electrones libre en un orbital sp2, semejante a un carbocatión Electrones apareados, singulete Metileno Triplete y singulete Forma dominante en disolución Forma dominante en fase gaseosa CH2N2 Carbeno singlet carbene singulete Carbeno triplet carbene triplete La rotación puede ocurrir alrededor de este enlace pi electrons Electrones p CH2 + stereospecific addition Adición estereoespecífica diradical diradical non-stereospecific Adición no estereoespecífica • En general, la transformación C=C a H-C-C-OH • Este es un método alternativo para la hidratación de alquenos para dar alcoholes • Los reactivos típicos son el acetato de mercurio, Hg(OAc)2 en THF acuoso • Problema: la toxicidad tan alta de los compuestos de mercurio • Regioselectividad de acuerdo a la regla de Markovnikov (el producto es el alcohol más sustituido) . • La reacción no es estereoselectiva La reacción procede a traves de la formacion del ion mercurinio Azúcares como materiales de partida quirales: algunos ejemplos de enfoque retrosíntesis TROMBOXANO B2 SWAINSONINA D-glucosa D-manosa Elaboración de la glucosa en la síntesis de tromboxano B2 Elongación de la cadena Formación enlace C-C inversión desoxigenación Tromboxano B2 Elongación de la cadena D-glucosa Síntesis de tromboxano B2 Retrosíntesis de cis-3-oxabiciclo[3.3.0]octan-7-ona ozonólisis Retrosíntesis de un intermediario para la carbaprostaciclina Retrosíntesis de Minaprina (antidepresivo) Swainsonina, síntesis descrita por Fleet Swainsonina Se requieren dos inversiones D-manosa La swainsonina es un derivado de la indolizidina que actúa como alcaloide Se puede encontrar en la naturaleza en varias plantas llamadas locoweed en Estados Unidos (+)-Meroquinona síntesis descrita por Hanessian Tetrahedron, 1990, 231 D-glucosa (+)-Meroquinona Es evidente que todos los grupos hidroxilo en la D-glucosa deben ser destruidos en ruta hacia la construcción del esqueleto de carbono de la (+)-Meroquinona, que puede ser considerado como un procedimiento estereoquímicamente de desperdicio. Sin embargo, la estructura de la D-glucosa es utilizado en forma eficiente para instalar los dos sustituyentes vecinales por una secuencia estereocontrolada de un solo paso de adición conjugada y atrapando el enolato, en un protocolo en un enona fácilmente disponible Pure & Appl. Chem. 1993, 1189 Otros materiales quirales de partida: aminoácidos, hidroxiácidos, terpenos Estructura hidrocarbonada Amino ácidos Acíclica (excepto prolina) 3 a 6 átomos de carbono Hidroxiácidos Acíclica 3 a 4 átomos de carbono Terpenos Acíclica Cíclica Centros asimétricos Sentido de la quiralidad Funcionalidad secuencial 1ó2 En general L a-aminoácido Ácido a-amino o bsustituído 1ó2 En general 1ó2 Combinaciones RóS a-hidroxi ácido a,b-dihidroxiácido RóS Enona cetona a-sustituída Algunos ejemplos de retrosíntesis con casos individuales de aminoácidos, terpenos y los hidroxiácidos AMINOÁCIDOS CEFALOSPIRINA Cisteína HIDROXIÁCIDOS Leucina TETRAHIDROLIPSTATINA Ácido L-málico TERPENOS d,l-sirenina Geraniol Obtención de Cefalosporina C, sintetizada por Woodward CEFALOSPIRINA Cisteína Esta posición requiere de una posterior funcionalización