Síntesis y Caracterización Estructural de los Materiales
Anuncio

Síntesis y Caracterización
Estructural de los Materiales
Ángel Carmelo Prieto Colorado
Física de la Materia Condensada, Cristalografía y Mineralogía.
Facultad de Ciencias.
Universidad de Valladolid.
Estructura Estática y Dinámica de la Materia
Tema 5
Estructura dinámica
Vibraciones atómico – moleculares
Coordenadas de movimiento
Modos Normales de Vibración (MNV): frecuencias
fundamentales
©A. Carmelo Prieto Colorado
Estructura dinámica
En una red cristalina, a efectos de su estructura dinámica, podemos
distinguir dos tipos de casos a estudiar:
Densidades electrónicas, asociadas a las zonas nucleares, es decir
algo que se repite periódicamente en la red y por tanto se puede
estudiar y representar mediante una función periódica del tipo
donde
©A. Carmelo Prieto Colorado
Si consideramos que los motivos no están fijos, si no que se
trasladan, vibran y rotan respecto de la posición de equilibrio, o sea,
existen excitaciones en la red, ya no se pueden representar las
densidades electrónicas por una función periódica, puesto que, las
excitaciones en la red fluctúan temporalmente, y se precisan de
funciones de onda o de “estado”, para un tiempo t.
Así pues, el estado del cristal se puede definir como una parte temporal
o fase y otra posicional o espacial, (Teorema de Bloch) donde:
La descripción de las vibraciones se puede hacer a través de su
función de estado Ψκ(r), donde k representa el número de onda de la
vibración.
©A. Carmelo Prieto Colorado
Para resolver la estructura dinámica es relevante la determinación de las
vibraciones.
En consecuencia, la descripción de todos los valores del vector de onda
de los espectros de vibración se pueden hacer en el marco de la red
recíproca asociada a cada estructura atómica o molecular particular.
Así, los átomos en un cristal están vibrando a cualquier temperatura,
incluso en las proximidades del cero absoluto, y vibran de un modo
peculiar, dado que los átomos no reciben impulso externo alguno, si no
que son debido al aumento de su energía cinética y aumentan con la
temperatura. Luego las posiciones atómicas son solo posiciones medias
o de equilibrio de un conjunto de osciladores acoplados.
Por otra parte los elementos de simetría del cristal imponen los tipos de
movimientos que los átomos pueden tener según las posiciones
cristalográficas que ocupen en equilibrio. Como resultado, el formalismo
de las propiedades estructurales de simetría (es decir, la teoría de los
grupos cristalográficos) es una herramienta fundamental para el estudio
de las vibraciones atómico-moleculares y la estructura dinámico
vibracional de redes cristalinas.
©A. Carmelo Prieto Colorado
Vibraciones atómico – moleculares
La única razón que hace que los átomos formen estructuras cristalinas
es que sus energías cinéticas sean menores que las fuerzas de enlace,
además como los osciladores ligados a un enlace químico no son
armónicos, el cristal deja de ser simétrico y regular en cada instante t.
No obstante la periodicidad no se ve “muy” alterada por estas
vibraciones atómicas, dado que son de amplitud muy baja. Al ser la
fuerza de enlace, la energía dominante, las vibraciones de un átomo se
propagan a los vecinos y consecuentemente su resolución es una serie
compleja de ondas transversales (TO) y longitudinales (LO), con
frecuencias y longitudes de onda que varían según sea el tipo de
estructura, la composición química del material y la temperatura.
Por utilidad práctica se resuelve la función de estado dentro de un
tratamiento clásico de mecánica Newtoniana, donde se efectúa una
aproximación de la energía potencial a la de un oscilador armónico que
permite una resolución no compleja, sin perder sentido físico cualitativo.
©A. Carmelo Prieto Colorado
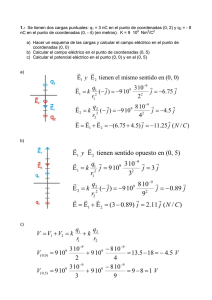
La imagen de la izquierda muestra un oscilador armónico, donde la
rayas horizontales indican las energías potenciales permitidas. A la
derecha se observa una imagen mas realista del enlace químico, donde
el oscilador inharmónico no tiene espaciados regularmente sus niveles
vibracionales y el potencial V(r) aumenta más, cuando disminuimos la
distancia de enlace (r), por debajo del equilibrio, que cuando la
aumentamos.
©A. Carmelo Prieto Colorado
Así, cada átomo está vinculado a su posición de equilibrio por una
fuerza F, que se describe en la aproximación armónica mecánica como
F = -f∆r. La aplicación directa de la segunda ley de Newton, F-ma = 0,
implica que -f∆r-ma = 0 ma = -f∆r.
Una molécula con N núcleos dispone de 3N grados de libertad en su
movimientos nucleares: 3 de traslación (Tx, Ty, Tz) 3 de rotación (Rx, Ry,
Rz) (2 en moléculas lineales), y el resto 3N-6 (o 3N-5 en moléculas
lineales) de vibración.
Para una configuración nuclear con posiciones cercanas al equilibrio los
N núcleos tendrán coordenadas de posición, o de desplazamiento
velocidades y energía cinética tales como:
©A. Carmelo Prieto Colorado
Si consideramos las coordenadas de desplazamiento ponderadas a
cada masa nuclear de modo que:
por tanto la velocidad será:
y la energías cinética y potencial serán:
©A. Carmelo Prieto Colorado
El desarrollo de la energía potencial en series de Taylor, en torno a q0:
es
donde, al ser la configuración de equilibrio un
mínimo de la energía potencial, su derivada
es nula y tomo el valor Vo como referencial
por tanto:
©A. Carmelo Prieto Colorado
Si consideramos la matriz Hessiana (matriz cuyos elementos son las
segundas derivadas parciales de la función potencial) del tipo fij = fji,
son las constantes de fuerza “cartesianas” de interación entre
partículas, que determinan como vibra una molécula:
Podemos determinar las trayectorias de los núcleos resolviendo las
ecuaciones del movimiento mecánico clásicas, aplicando de nuevo la
segunda ley de Newton (ma = -f∆r):
usando las coordenadas de desplazamiento ponderadas {qk}:
©A. Carmelo Prieto Colorado
de modo que las ecuaciones del movimiento quedarán como:
de modo mas compacto, quedar como:
Las soluciones son del tipo:
por tanto tendremos que :
©A. Carmelo Prieto Colorado
Es decir la ecuación correspondiente a las coordenadas ponderales qk,
contiene a todas las 3N coordenadas de desplazamiento ponderadas y
el sistema será mas fácil de resolver si desacoplamos las ecuaciones.
Las incógnitas son las amplitudes Aj, y el sistema tiene soluciones no
triviales para los valores que anulan el determinante secular de los
coeficientes Aj.
donde λ son los valores propios de la matriz [fij] y sus correspondientes
vectores propios, los cuales se denominan modos normales de
vibración. Los valores propios λ se obtienen de la ecuación secular
junto con las condiciones de ortogonalidad.
Hay 3N valores de λ que son
soluciones del sistema y seis
valores serán nulos (λ=0),
correspondiendo con los tres
modos globales de traslación y
tres de rotación molecular.
©A. Carmelo Prieto Colorado
Es una ecuación algebraica de grado 3Nx3N. Los modos de traslación y
rotación tendrán un valor propio nulo. Las verdaderas vibraciones
deben tener λκ > 0. La simetría molecular puede dar lugar a la
degeneración de uno o más modos de vibración.
La obtención de los autovalores λκ y los coeficientes de los autovectores
asociados a las amplitudes Ai, nos permite describir los movimientos de
los N átomos de la molécula y referenciarlos a sus posiciones de
coordenadas cartesianas. Pero esto solo puede hacerse de modo
relativo entre amplitudes o referencial, dado que el sistema no permite
una solución absoluta de las amplitudes.
Para paliar esta incertidumbre se hace una suposición muy interesante
que es considerar a la molécula como si fuera como un todo uno,
moviendose mediante una combinación lineal de λκ en la cual para
cada valor todos los átomos se mueven sincrónicamente pero con
diferentes amplitudes. Estas vibraciones de conjunto, donde cada
átomo pasa por la posición de equilibrio al mismo tiempo, se las
conoce como vibraciones fundamentales o modos normales de
vibración (MNV).
©A. Carmelo Prieto Colorado
Coordenadas de movimiento
Las coordenadas que describen los modos normales de vibración
(MNV) se denominan Coordenadas Normales (Q), y se relacionan con
las coordenadas ponderales, del siguiente modo:
Las coordenadas normales Qk nos permiten simplificar las expresiones
de las energías cinética y potencial al pasarlas a forma matricial,
mediante una transformación ortogonal L, que permite diagonalizar
simultáneamente las energías cinética y potencial T y V.
©A. Carmelo Prieto Colorado
Modos Normales de Vibración (MNV): frecuencias fundamentales
Con el cambio de coordenadas de posición y ponderadas a
coordenadas de movimiento, las ecuaciones del movimiento se
simplifican de modo patente, dado que:
La energía total del sistema será:
La energía cinética es invariante a la introducción de las coordenadas
normales, mientras que la energía potencial pasa de decribirse como la
suma de pares de coordenadas a describirse como un sumatorio simple
sobre modos normales de vibración λκ.
Las ecuaciones del movimiento en coordenadas normales son:
©A. Carmelo Prieto Colorado
El sistema es de fácil solución, cuando se definen “bien” las
coordenadas normales Qk, y para ello se utiliza la transformada inversa
Se precisa conocer lik y lik-1, los cuales se obtienen al resolver el
problema dinámico, en términos de coordenadas ponderadas qi.
Así pues, las 3N ecuaciones de movimiento son ahora independientes,
y cada modo normal de vibración da lugar a su propia ecuación
diferencial. De hecho, las ecuaciones son equivalentes a las de un
movimiento armónico simple en el que los valores propios λκ son las
constantes recuperadoras elásticas.
Por consiguiente para resolver directamente un problema de dinámica
molecular, solo hay que resolver su ecuación secular. Conocida la
estructura estática promedio (disposición geométrica), composición
química elemental y fuerzas interatómicas de enlace, la solución a la
estructura dinámica consiste en obtener las coordenadas normales Qk.
©A. Carmelo Prieto Colorado
En el caso de un oscilador armónico ligado a los movimientos
moleculares inter-atómicos, la frecuencia del oscilador es proporcional a
la fuerza de enlace fij, e inversamente proporcional a las masas
reducidas (μ=∑mi / ∏mi), de los átomos implicados en el enlace.
En resumen, para la determinación de los modos fundamentales de
vibración, el proceso implica conocer:
3N coordenadas cartesianas de desplazamiento (xi,yi,xi)
3N coordenadas ponderables (qi = √mi xi)
3N-6 coordenadas normales de vibración (Qi = [L]-1 qi)
Aquí seria el resultado final, pero paradójicamente el proceso suele ser
inverso, dado que se asume una geometría molecular, una composición
química y un conjunto experimental frecuencias, obtenidas mediante
espectroscopía y se tratará de estimar las fuerzas intermoleculares fij.
©A. Carmelo Prieto Colorado
Para la asignación correcta de las frecuencias experimentales a modos
normales de vibración se representa en términos de un conjunto de
coordenadas que tienen una geométrica precisa y un significado físico
derivado directamente de la estructura molecular.
Las dos representaciones más usados se basan en las coordenadas
internas y las de simetría.
Coordenadas internas
Se asocian a las variaciones de las distancias y relaciones
angulares del enlace entre átomos.
1. Tensiones: variaciones de la longitud del enlace
2. Deformaciones angulares: variaciones del ángulo entre enlaces
adyacentes con un átomo común y fijo.
3. Deformaciones fuera del plano: variaciones del ángulo entre un
enlace y el plano soporte de la estructura molecular con un
átomo en común.
4. Torsión: variación de la orientación de un enlace por giro
impedido de un enlace respecto de los enlaces adyacentes.
©A. Carmelo Prieto Colorado
Coordenadas de simetría
Si las moléculas de un material presentan una configuración
geométrica asociada a un grupo de simetría, los MFV se asocian al
grupo de simetría. Para utilizarlas es necesario:
1. Conocer el GSP (Grupo de Simetria Puntual) de la molécula.
2. Conocer la representaciones irreducibles RI, del grupo
(tabuladas para los GSP)
3. Clasificar los MFV en las especies de Simetria del GSP.
4. Calcular el nº de veces que cada RI, interviene en la
representación global.
©A. Carmelo Prieto Colorado
Tensión simétrica
Tensión antisimétrica
Deformación simétrica
Wagging (x)
Roking (y)
Twisting (z)
©A. Carmelo Prieto Colorado
©A. Carmelo Prieto Colorado
Ángel Carmelo Prieto Colorado
Física de la Materia Condensada, Cristalografía y Mineralogía.
Facultad de Ciencias.
Universidad de Valladolid.