panhipopituitarismos - Elsevier Instituciones
Anuncio

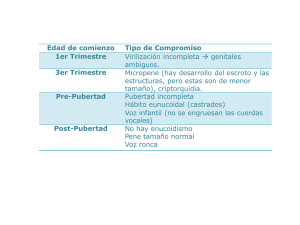
PANHIPOPITUITARISMOS J.L. Herrera Pombo Servicio de Endocrinología. Fundación Jiménez Díaz. Universidad Autónoma. Madrid. Introducción El hipopituitarismo es una insuficiencia del lóbulo anterior de la hipófisis de etiología múltiple que puede afectar a una o varias de estas hormonas o incluso a todas ellas (panhipopituitarismo). En la destrucción crónica de la hipófisis anterior los defectos hormonales se producen generalmente por este orden: hormona del crecimiento (GH), gonadotropinas, hormona estimulante del tiroides (TSH), hormona hipofisaria corticotropina (ACTH) y prolactina, mientras que en la destrucción hipofisaria aguda (apoplejía hipofisaria) lo primero en aparecer es el fallo suprarrenal. Los defectos aislados de GH o de gonadotropinas se ven con más frecuencia que los de ACTH o TSH, y los de prolactina son infrecuentes excepto en el infarto hipofisario. El tratamiento prolongado con corticoides puede inducir un déficit temporal de ACTH. Etiopatogenia del hipopituitarismo El hipopituitarismo se puede producir por una lesión en el lóbulo anterior de la hipófisis o por alteraciones en el área hipotalámica, en relación con causas congénitas, con aplasia o hipoplasia hipotalámica o hipofisaria que coinciden con anomalías del sistema nervioso central (SNC) incompatibles con la vida; en este grupo se incluye el síndrome del encefalocele y la displasia septo-óptica. Diversas lesiones del área hipotalámica o del tallo hipofisario pueden conducir a hipopituitarismo (tabla 1). Los tumores hipotalámicos como el craneofaringioma, meningioma, quistes del tercer ventrículo, glioma del nervio óptico, cordoma, y germinoma pueden inducir hipofunción hipofisaria y ésta también puede suceder en relación con metástasis hipotalámicas o hipofisarias (mama y pulmón), que pueMedicine 2000; 8 (16): 855-859 den inicialmente afectar a la neurohipófisis produciendo diabetes insípida. Los adenomas hipofisarios tanto secretores como no secretores y el craneofaringioma intraselar pueden cursar con hipopituitarismo. Las enfermedades infiltrativas que afectan al hipotálamo y a la hipófisis como las granulomatosis, sarcoidosis, granuloma eosinófilo (histiocitosis X), granuloma de células gigantes, y granulomatosis de Wegener pueden inducir hipofunción hipofisaria. Esto mismo puede ocurrir por infiltración de hierro en la hipófisis en la hemocromatosis y con diversas infecciones como la tuberculosis, micosis, toxoplasmosis y sífilis. La hipofisitis linfocítica es una causa rara de hipopituitarismo, es de etiología autoinmune y ha sido descrita sobre todo en mujeres en relación próxima con el TABLA 1 Causas de hipopituitarismo Anomalías congénitas Tumores hipotalámicos (craneofaringiomas, meningiomas, quistes del tercer ventrículo, gliomas del nervio óptico, cordomas, germinomas) Metástasis de otros tumores (mama, pulmón, colon, próstata) Tumores hipofisarios: adenomas secretores (prolactina, GH, ACTH, TSH, gonadotropinas, subunidad alfa de las hormonas glucoproteicas), adenomas no secretores, craneofaringiomas, quiste de la bolsa de Rathke Enfermedadades infiltrativas (hipotalámicas o hipofisarias): granulomatosis (sarcoidosis, granuloma eosinófilo, granuloma de células gigantes, granulomatosis de Wegener), hemocromatosis, hipofisitis linfocítica (autoinmune), infecciones (tuberculosis, micosis, toxoplasmosis, sífilis) Apoplejía hipofisaria Postcirugía hipofisaria o sección del tallo hipofisario Postcirugía (hipófisis, craneal, nasofaringe) Vascular: necrosis hipofisaria postpartum (síndrome de Sheehan), aneurisma de la carótida interna Otras causas: silla turca vacía, traumatismos craneales Idiopática GH: hormona del crecimiento; ACTH: hormona hipofisaria corticotropina; TSH: hormona estimulante del tiroides. embarazo, aunque hay casos fuera del embarazo y también en hombres. Puede presentarse con posibles síntomas compresivos en relación con el aumento de la masa glandular y con diversos grados de hipopituitarismo, y es rara la afectación neurohipofisaria, pero existe algún caso asociado con la diabetes insípida. Con la hipofisitis linfocítica se pueden asociar otros procesos autoinmunes como la tiroiditis de Hashimoto, y en algún caso se han referido anticuerpos antiprolactina. En algunos hipopituitarismos citados, como los idiopáticos, debería tenerse en cuenta esta posibilidad etiológica. La apoplejía hipofisaria es una destrucción aguda de la hipófisis por un infarto o hemorragia intrahipofisaria que puede ocurrir en un adenoma. La clínica es de cefalea intensa, pérdida de visión, parálisis de pares craneales (motores oculares) y afectación sensorial, a veces puede cursar de forma asintomática y es importante estar atento a una posible insuficiencia aguda del eje hipófiso-suprarrenal, que debe ser tratada desde el principio, ya que puede conducir a un estado de shock. La diabetes mellitus, radioterapia, coagulopatías, traumas craneales, y hemorragia postpartum (síndrome de Sheehan) pueden precipitar una apoplejía hipofisaria. En la fase aguda las determinaciones hormonales pueden ser normales, en días sucesivos disminuye el cortisol y los esteroides sexuales, y más lentamente las hormonas tiroideas, por todo lo cual deben ser tratados desde el primer momento con hidrocortisona. Muchos casos pueden precisar una descompresión aguda de la hipófisis mediante cirugía transesfenoidal (CTE). La cirugía hipofisaria puede provocar déficit hormonal posterior dependiendo del tamaño del tumor, de la cantidad del tejido peritumoral que permanezca indemne y de la posibilidad de que se puede resecar todo el tumor dejando el resto hipofisario normal. La radioterapia puede producir daños a nivel hipotalámico o hipofisario con la aparición posterior de hipopituitarismo parcial o total, que puede llevar un tiempo de evolución muy variable, a veces años, por lo que es preciso un seguimiento y evaluación periódica. En casos de neoplasias nasofaríngeas o de la cabeza y cuello que se tratan con radioterapia se pueden desarrollar también alteraciones hormonales por daño en hipotálamo, hipófisis y tiroides. 855 © DOYMA 2001 ENFERMEDADES ENDOCRINOLÓGICAS (II) El síndrome de Sheehan se produce por una necrosis isquémica de la hipófisis, en una situación de shock hipovolémico por importante hemorragia postpartum. La hipófisis, hipertrófica durante el embarazo, sufre una trombosis de los vasos hipofisarios en el seno de una coagulación intravascular generalizada con isquemia y destrucción que conduce a hipopituitarismo. El síntoma clínico inicial es la agalactia (incapacidad para lactar) por déficit de prolactina, el cuadro clínico se completa con amenorrea, hipotiroidismo, e insuficiencia suprarrenal y algunas veces puede acompañarse de diabetes insípida. El síndrome de Sheehan puede evolucionar de una forma muy lenta y a veces el diagnóstico definitivo se hizo al cabo de mucho tiempo cuando se manifestó la insuficiencia tiroidea y suprarrenal. Un aneurisma de la arteria carótida interna puede producir ocasionalmente hipopituitarismo por rotura de la dilatación aneurismática y destrucción hemorrágica de la hipófisis. Una angiografía carotídea puede confirmar el diagnóstico y decidir el tratamiento. En la tomografía axial computarizada (TAC) se aprecia a veces una masa hipofisaria uniforme que puede confundirse con un adenoma hipofisario. Un traumatismo craneal puede desarrollar hipopituitarismo por lesión hipofisaria o hipotalámica. La clínica de la insuficiencia hipofisaria se instala a veces de forma inmediata, pero no es infrecuente que se presente bastante tiempo después (incluso años), y los defectos hormonales iniciales son casi siempre de GH y gonadotropinas, aunque puede desarrollarse posteriormente panhipopituitarismo. En bastantes casos existe hiperprolactinemia que sugiere desconexión hipotálamohipofisaria y algunos desarrollan diabetes insípida transitoria o permanente. En el síndrome de la silla turca vacía (STV) puede aparecer hipopituitarismo, pero en muchos casos de STV primaria la función hipofisaria es normal. En la STV existe un diafragma selar incompleto que permite la entrada de la aracnoides y del líquido cefalorraquídeo en la misma y puede comprimir la hipófisis e incluso dejarla reducida a un resto de tejido. Se habla de STV primaria cuando el defecto del diafragma selar es congénito mientras que la STV secundaria es la que se produce postcirugía, radioterapia, o necrosis de un adenoma hipofisario. En algunos casos puede haber hiperprolactinemia en probable relación con la compresión y desviación lateral del tallo hipofisario, aunque cabe la posibilidad de un microprolactinoma. Los pacientes que desarrollan hipofunción hipofisaria suelen tener mayor volumen selar y esto es más aparente en el síndrome de silla turca parcialmente vacía por un adenoma hipofisario que se necrosó espontáneamente o como hemos visto en más de una ocasión en los prolactinomas secundarios al tratamiento con bromocriptina. A veces se ha observado STV primaria en mujeres obesas multíparas, con hipertensión arterial y cefaleas, en la mayoría sin apreciarse alteraciones endocrinas, excepto hiperprolactinemia en algunos casos y confirmándose el diagnóstico en un estudio de imagen. Es imprescindible una TAC o resonancia magnética (RM) para confirmar la existencia de una STV comprobando la presencia del líquido cefalorraquídeo intraselar. Cuadro clínico del hipopituitarismo La clínica del hipopituitarismo puede ser muy variable y depende de las hormonas hipofisarias comprometidas, si el déficit hormonal es total o parcial, de la edad del paciente y de la causa (hipotalámica o hipofisaria) del trastorno. En el hipopituitarismo los síntomas y signos clínicos son en general semejantes a los que se ven en la insuficiencia primaria de las glándulas dianas, y aunque existen matices que comentaremos, algunos pacientes sólo presentan un déficit hormonal aislado y su cuadro clínico correspondiente, pero pueden existir defectos parciales que será necesario descubrir mediante las pruebas oportunas. El déficit de GH produce en el niño enanismo hipofisario y la falta de GH en el adulto está siendo ahora reconocida y le dedicaremos algunos comentarios en la segunda parte. El déficit de gonadotropinas es el que con más frecuencia puede verse aislado y condiciona un hipogonadismo hipogonadotrópico. El cuadro mostrará algunas diferencias según haya comenzado antes o después de la pubertad. Si se asocia con otros defectos de hormonas tróficas hipofisarias su talla será baja, y si el déficit gonadotrópico es exclusivo su altura será 856 © DOYMA 2001 excesiva con hábito eunucoide y desproporción entre el desarrollo del tronco y las extremidades. Completaremos su descripción posteriormente al hablar de las deficiencias hipofisarias aisladas. En el síndrome de Sheehan (necrosis hipofisaria postpartum), la mujer experimenta inmediatamente después del parto involución de las mamas y fallo de la lactación, el ciclo menstrual no se reinstaura y progresivamente van apareciendo astenia, adinamia, somnolencia y demás sintomatología de hipofunción adrenal y tiroidea que caracteriza al hipopituitarismo, a veces puede evolucionar a muy largo plazo o en otras ocasiones se manifiesta de forma más inmediata. El cuadro clínico del hipotiroidismo secundario (hipofisario) o terciario (hipotalámico) es indistinguible del hipotiroidismo primario. El déficit de ACTH condiciona un cuadro de insuficiencia suprarrenal con astenia, náuseas, vómitos, anorexia, pérdida de peso, hipotensión, a veces tienen la presión arterial normal y sin embargo refieren hipotensión ortostática, pero una insuficiencia suprarrenal grave puede conducir a una crisis adrenal, con hipotensión y shock. En muchos casos existe hiperprolactinemia por un tumor secretor de prolactina, o bien porque está comprometido el paso de dopamina desde el hipotálamo a la hipófisis y por tanto desaparece el efecto tónico inhibitorio sobre la secreción de prolactina; en este último caso el nivel de la hormona no suele ser muy elevado y por eso puede faltar amenorrea-galactorrea. El hipopituitarismo puede asociarse con diabetes insípida, lo cual induce a pensar que el trastorno está en el hipotálamo o en la porción alta del tallo hipofisario, y entonces se combinan hipofunción hipofisaria, hiperprolactinemia no muy intensa y diabetes insípida. Diagnóstico del hipopituitarismo El cuadro clínico sugiere la existencia de una insuficiencia gonadal, tiroidea, o adrenal, pero es preciso disponer de determinaciones hormonales y pruebas dinámicas del eje hipotálamo-hipofisario así como de estudios de imagen. La valoración del eje hipófiso-suprarrenal es muy importante. Una cifra de cortisol PANHIPOPITUITARISMOS plasmático muy baja o indetectable, si el paciente no toma esteroides, indica que existe insuficiencia suprarrenal, pero una cifra normal no descarta totalmente la existencia de una insuficiencia parcial. La existencia de un cortisol plasmático bajo (8 h), con ACTH plasmática normal o baja corresponde a un déficit de ACTH. El déficit de TSH ocasiona un hipotiroidismo de origen hipotalámico o hipofisario y cursa con niveles bajos de T4-libre y TSH normal o baja pero en todo caso inadecuada para la T4. En la valoración del eje hipófiso-gonadal es importante el cuadro clínico (historia menstrual en la mujer). Si un hombre tiene disminución de la testosterona o una mujer deficiencia estrogénica y no coexiste con aumento de las gonadotropinas hipofisarias, verosímilmente hay un fracaso hipofisario en la producción de hormona foliculoestimulante (FSH) y hormona luteinizante (LH). Diversas pruebas (hipoglucemia insulínica, glucagón) pueden emplearse para valorar un posible déficit de GH. Las pruebas para investigar el eje hipotálamo-hipófisis y los estudios de imagen son tratados en otro capítulo de esta monografía. Tratamiento El tratamiento hormonal sustitutivo en el hipopituitarismo es absolutamente necesario, ya que hay que tener en cuenta que cortisol y tiroxina son imprescindibles para la vida. El déficit del eje gonadotrópico produce hipogonadismo tanto en la mujer como en el hombre y puede conducir a osteopenia. Siempre que exista insuficiencia del eje hipófiso-suprarrenal debe iniciarse el tratamiento con hidrocortisona, ya que tratar con hormona tiroidea a un paciente que tenga déficit de ACTH-cortisol no sustituido puede precipitar una crisis adrenal aguda. Para la sustitución terapéutica se puede administrar hidrocortisona en dosis de 20 mg antes del desayuno y 10 mg a media tarde, como alternativa puede emplearse prednisona 5 mg por la mañana y 2,5 mg por la tarde. Con estas dosis debe valorarse si la sustitución es adecuada porque a veces resulta excesiva para algunos pacientes y otros sin embargo tienen astenia, malestar general, cefaleas y puede ser preciso en ellos fraccionar la dosis total en tres tomas diarias. Es difícil valorar la dosis precisa de hidrocortisona, ya que el cortisol plasmático y la cortisoluria no dan una idea adecuada de esto, aunque sí pueden servirnos cuando estamos administrando dosis excesivas. Estos pacientes pueden requerir otras pautas terapéuticas en situaciones de estrés, infecciones, etc., como se describe en los capítulos que tratan de la insuficiencia suprarrenal. El déficit hipófiso-tiroideo se trata con L-tiroxina en una sola dosis diaria, comenzando por 25-50 µg y ajustando posteriormente la dosis, midiendo los niveles de T4-libre sanguínea, ya que al contrario de lo que ocurre en el hipotiroidismo primario la TSH lógicamente no es el parámetro adecuado en estos casos. Si existe déficit gonadotrópico, las mujeres en edad premenopáusica deben ser tratadas con la combinación de estrógeno y progesterona de forma cíclica, para reproducir un ciclo normal. En el hombre se utiliza sobre todo enantato de testosterona (depot) en dosis de 250 mg cada tres o cuatro semanas. No disponemos todavía en nuestro país de testosterona por vía dérmica (parches) que pueden conseguir unos niveles más adecuados de testosterona circulante. En algunos pacientes con déficit gonadotrópico que quieran conseguir fertilidad se puede hacer un tratamiento combinado con gonadotropinas, coriónica humana (HCG) y menopáusica humana (HMG). En caso de déficit hipotalámico se ha intentado el tratamiento de la infertilidad con análogos de la hormona hipotalámica estimuladora de las gonadotropinas (GnRH). Actualmente, en España el déficit de hormona del crecimiento en adultos puede ser también sustituido siempre que coexista al menos con otro déficit hormonal hipofisario, que no sea prolactina, y esté adecuadamente sustituido antes de iniciar el tratamiento con GH. La dosis inicial recomendada de GH en las primeras cuatro semanas es de 0,03-0,1 UI/kg/semana, dividida en siete inyecciones/semana sin que la dosis total diaria sea superior a 1,5 UI (0,5 mg). La dosis debe ajustarse mensualmente durante al menos los tres primeros meses y dependiendo de los niveles de factor de crecimiento similar a la insulina-1 (IGF-I). En caso de desarrollar efectos secundarios debe reducirse la dosis a la mitad o suspenderla recomendando revisión a los 30 días. La dosis de mantenimiento de GH recomendada es 0,250 UI/kg/semana, sin que la dosis total diaria sea superior a 3 UI (1 mg/día). Los pacientes hipopituitarios que tienen diabetes insípida asociada deberán recibir el tratamiento adecuado con desmopresina. Deficiencias hipofisarias aisladas Déficit aislados de GH o de gonadotropinas se pueden ver con más frecuencia que el de ACTH o TSH, aunque ocasionalmente algunos pacientes pueden tener deficiencia de ACTH o de TSH o de ambas como cuadro inicial de su hipopituitarismo. Deficiencia aislada de gonadotropinas El déficit de gonadotropinas puede producirse como consecuencia de un fallo hipofisario o bien por un déficit hipotalámico de LHRH. Existen alteraciones funcionales que afectan a LHRH y que representan formas transitorias de hipogonadismo hipogonadotrópico, esto se da con más frecuencia en mujeres con amenorrea hipotalámica en relación con factores como pérdida de peso, ejercicio excesivo, estrés, y si se corrigen los factores desencadenantes puede normalizarse la secreción de LHRH y el ciclo menstrual. Un síndrome clínico funcional parecido a éste no ha podido ser confirmado en el hombre; sin embargo, en los atletas el empleo de anabolizantes puede conducir a hipogonadismo hipogonadotrópico funcional con disminución de la testosterona y alteración de la espermatogénesis, cuadro que suele ser reversible pero nunca antes de unos cuatro meses después de retirar los anabolizantes. Se incluyen como idiopáticos una serie de hipogonadismos hipogonadotrópicos, algunos con un perfil ya más definido como el síndrome de Kallmann. El síndrome de Kallmann consiste en un defecto aislado en la secreción de GnRH que se asocia con anosmia por agenesia de los bulbos olfatorios. Aunque hay formas de hipogonadismo hipogonadotrópico idiopático que cursan sin anosmia, el hecho de que ambas formas, con y sin anosmia, 857 © DOYMA 2001 ENFERMEDADES ENDOCRINOLÓGICAS (II) se hayan comprobado dentro de una misma familia ha hecho pensar que se trata de variantes de un mismo rasgo. Además de anosmia pueden estar presentes otras anomalías como sordera neurológica, labio leporino, criptorquidia, ceguera para los colores, ataxia cerebelosa, agenesia renal. Dentro de las rarezas del cuadro existen formas familiares y esporádicas siendo más frecuentes en hombres. En el síndrome de Kallmann se han descrito dos patrones de herencia, autosómica y ligada al cromosoma X y en esta forma se han encontrado pacientes que tenían deleciones en un gen KAL que codificaba una proteína llamada “anosmina” relacionada con la migración de las neuronas de GnRH desde la región olfatoria a la preóptica del hipotálamo. Hay algunos casos de hipogonadismo hipogonadotrópico en los que se han encontrado mutaciones del gen del receptor de GnRH. Se ha descrito, además de las formas congénitas, un hipogonadismo hipogonadotropo idiopático de comienzo adulto, pacientes que tienen una pubertad normal y posteriormente disminución de la libido y de la fertilidad, con testes que suelen conservar su volumen normal. Desde el punto de vista bioquímico no muestran diferencias con las formas congénitas, y presentan ausencias del pulso secretorio de LH y testosterona disminuida y buena respuesta al tratamiento con LHRH. No se conoce con certeza la base etiopatogénica de esta forma adquirida (?) de comienzo adulto, aunque podrían constituir un grupo de formas incompletas de déficit de GnRH, con hipogonadismo hipogonadotrópico ocasionalmente reversible con la administración exógena de LHRH. Existe una forma incompleta de déficit de GnRH que es el llamado síndrome del eunuco fértil en el que existe suficiente cantidad de GnRH para conseguir adecuada testosterona intratesticular para conservar la espermatogénesis pero no para conseguir los niveles de testosterona sistémicos necesarios para una virilización normal. Algunos autores han sugerido que la pubertad diferida que luego se sigue de un desarrollo sexual normal, puede ser una forma leve evolutiva del síndrome de deficiencia de GnRH. Existe una gran heterogeneidad clínica y bioquímica en el síndrome de déficit aislado de GnRH y gran variedad de patrones de pulso de secreción anormal de LHRH. Puede observarse tanto en hom- bres como en mujeres y también con carácter familiar, todo lo cual hace pensar en la existencia de diversos determinantes genéticos que controlan la expresión de la secreción de GnRH. Es evidente la heterogeneidad genética en este síndrome que puede heredarse de forma autosómica dominante o recesiva o ligada al cromosoma X, con variable expresividad como se deduce de los estudios realizados en diversos miembros de familias con síndrome de Kallmann. Además del defecto del gen KAL (del síndrome de Kallmann) se ha demostrado otro gen en el cromosoma X que produce hipogonadismo hipogonadrotópico en el seno de un síndrome de hipoplasia adrenal congénita, este gen se ha denominado DAX-1; estos pacientes tienen insuficiencia suprarrenal en la infancia y con el tratamiento adecuado han llegado a la edad adulta demostrándose entonces en ellos hipogonadismo hipogonadotrópico. Para concluir sobre los interesantes aspectos genéticos del síndrome de deficiencia de GnRH podríamos decir que un 66% son formas esporádicas y que de éstos un 5% tienen una mutación en el gen KAL; un 34% tienen carácter familiar y de este grupo 64% tienen herencia autosómica dominante, 25% autosómica recesiva y un 11% ligado al cromosoma X. Existen otros síndromes genéticos donde puede haber hipogonadismo hipogonadotrópico así como por ejemplo el síndrome de Prader-Willi, que se asocia además con obesidad, retraso intelectual, hipotonia, que se pone en relación con deleciones de la porción proximal (derivada vía paterna) del cromosoma 15 q, pero los genes responsables del síndrome no han sido identificados. En el síndrome de Laurence-Moon-Biedl, la retinitis pigmentaria, obesidad, polisindactilia entre otros datos se asocian con hipogonadismo hipogonadotrópico cuya fisiopatología no es bien conocida. El cuadro clínico del déficit gonadotrópico va a depender de la edad de comienzo, si es congénito o adquirido, la intensidad del mismo, es decir, si es una forma completa o parcial y por último la duración del proceso si es funcional o permanente. Un déficit aislado de GnRH podría tal vez diagnosticarse en período neonatal (criptorquidia y micropene). En los casos de hipogonadotropinismo prepúber ya dijimos en el capítulo anterior 858 © DOYMA 2001 que existe un fallo para el desarrollo puberal, con ausencia de características sexuales secundarias, hábito eunucoide, testículos que permanecen prepuberales y en la mujer existirá amenorrea primaria, ausencia de telarquía, y en ambos sexos la edad ósea está retrasada. En los hipogonadismos hipogonadotrópicos postpuberales se producirá una atrofia gonadal importante, la mujer mostrará alteraciones de la función menstrual, atrofia de mamas, útero y vagina y disminución de la libido, pérdida de vello axilar y del vello púbico si existe al mismo tiempo insuficiencia hipófiso-suprarrenal. En el hombre existe atrofia testicular progresiva, con testículos más blandos, disminución del vello corporal y de la barba, arrugas en la cara, eyaculado más escaso, oligoespermia, disminución de la libido e impotencia, además de ginecomastia; todos los datos estarán presentes en un hipogonadismo de más larga evolución, a veces durante años. En la valoración del eje hipófiso-gonadal en la mujer es importante su historia menstrual, ya que la presencia de reglas normales y regulares indica la existencia de gonadotropinas normales. Como las gonadotropinas se segregan por pulsos éstas pueden ser normales en mujeres con amenorrea. Las mujeres postmenopáusicas tienen gonadotropinas elevadas, y si sus cifras son normales es que tienen déficit gonadotrópico. En el hombre que tiene testosterona plasmática y espermatogénesis normales, las gonadotropinas son normales. Si un hombre tiene testosterona disminuida o una mujer los estrógenos deficientes y no hay aumento de las gonadotropinas hipofisarias, verosímilmente tienen un fracaso hipofisario en la producción de FSH y LH. En el diagnóstico diferencial es importante tener en cuenta las distintas causas que hemos comentado, causantes de hipogonadismo hipogonadotrópico y en todo caso será necesario completar con las determinaciones hormonales y pruebas pertinentes (véase capítulo correspondiente). Tratamiento Con relación al hipopituitarismo se han comentado las bases terapéuticas del hipogonadismo hipogonadotrópico que completamos a continuación. En pacientes prepúber se deben iniciar dosis de enantato de testosterona de 50 mg PANHIPOPITUITARISMOS mensualmente y aumentar la dosis en 50 mg cada tres a seis meses hasta que se consiga la maduración sexual completa. Algunos utilizan HCG 200-500 UI a días alternos por vía intramuscular subcutánea. En las mujeres se puede iniciar con 5 µg de etinilestradiol ó 0,3 mg de estrógenos conjugados diariamente durante seis meses, para conseguir un adecuado desarrollo mamario y después debe añadirse de forma cíclica un progestágeno. En los adultos ya indicamos la pauta a seguir y en caso de que se intente conseguir fertilidad hay que utilizar FSH recombinante que se ha mostrado muy efectiva (70-150 UI a días alternos) añadiendo a la HCG (1.000 UI a días alternos). En lugar del tratamiento con gonadotropina se puede emplear administración pulsátil de LHRH que se puede incluso administrar con una bomba de infusión portátil programable, aunque no debemos olvidar que en pacientes con lesión hipofisaria importante es de presumir baja respuesta a la LHRH y sería preferible emplear gonadotropinas. Deficiencia de hormona del crecimiento Hasta hace no mucho tiempo se había prestado poca atención al posible déficit de GH en los cuadros de hipofunción hipofisaria, ahora sabemos que la mayoría de los pacientes que por diversas causas tienen bajos niveles de otras hormonas hipofisarias también lo tienen de GH y que este defecto aparece precozmente en el hipopituitarismo. Los síntomas y signos clínicos que se han relacionado con el defecto de GH son: obesidad troncular, piel seca y delgada, anomalía en la composición corporal con aumento de la grasa y reducción del agua extracelular, sequedad de la piel, alteraciones psicológicas, reducción de la fuer- za muscular y de resistencia al ejercicio, anomalías en la función cardíaca, presencia de factores de riesgo cardiovascular, como hiperlipidemia y disminución de la fibrinolisis, disminución de la densidad ósea, y alteración en la función renal (disminución del filtrado glomerular). Además de estos datos clínicos debe confirmarse el déficit de GH mediante las pruebas oportunas que se describen en otro capítulo. Respecto al tratamiento con GH hay que indicar que en nuestro país no está autorizado en el caso de déficit aislado de esta hormona. Deficiencia de TSH Es muy rara de forma aislada, y acompaña generalmente a otros déficits hipofisarios pero cuando se encuentren datos como T4-libre baja y TSH normal o baja deben descartarse otras posibilidades como el síndrome de deficiencia de la tiroxina ligada a la globulina (TBG), que cursa con T4-libre baja y TSH normal o el síndrome del enfermo eutiroideo con patrón bioquímico parecido. El cuadro clínico del hipotiroidismo secundario (hipofisario) o terciario (hipotalámico) es indistinguible del hipotiroidismo primario, de tal forma que los pacientes muestran bradipsiquia, fatigabilidad, intolerancia al frío, disfonía, estreñimiento, calambres musculares, parestesias y sequedad de la piel que da una apariencia típica de la cara que muestra un aspecto de vejez prematura con arrugas alrededor de los ojos y de la boca. Los aspectos terapéuticos ya han sido tratados previamente. Deficiencia aislada de ACTH Es muy rara y ya hemos comentado en el capítulo anterior algunos de los aspectos clínicos y terapéuticos. Un hecho distintivo importante en el diagnóstico diferencial de la insuficiencia suprarrenal primaria (enfermedad de Addison) es la hiperpigmentación que muestran estos pacientes frente a los casos de insuficiencia adrenal secundaria (hipotálamo-hipofisaria), los cuales incluso suelen tener la piel muy pálida. En la insuficiencia suprarrenal de larga evolución las mujeres pueden tener pérdida del vello axilar y púbico. En el déficit de ACTH no se afecta de forma fundamental la aldosterona que primariamente es regulada por el sistema reninaangiotensina, por lo cual estos pacientes tienen un potasio sérico normal y si existe déficit de cortisol importante puede estar presente hipoglucemia e hiponatremia. Debe tenerse precaución porque el cuadro clínico puede empeorarse de forma gradual y cualquier enfermedad concomitante puede precipitar una insuficiencia suprarrenal si no se han tomado las debidas precauciones. Las pruebas diagnósticas y el tratamiento se tratan en los capítulos correspondientes. BIBLIOGRAFÍA RECOMENDADA Hayes FJ, Seminara StB, Crowley WF. Hypogonadotropic hypogonadism. Endocrinol Metab Clin North Am 1998; 27: 739-763. Herrera Pombo JL. Enfermedades del hipotálamo y la hipófisis anterior. En: Rodés J, Guardia J, eds. Medicina Interna. Barcelona: Edit. Masson, 1997; Tomo II: 2.494-2.514. Lamberts SWJ, de Herder W W, van der Lely JA. Pituitary insufficiency. Lancet 1998; 352: 127-134. Oelkers WHK. Hypopituitarism. En: Bardin CW, ed. Current Therapy in Endocrinology and Metabolism (6th ed.). St Louis: Mosbi, 1997; 27-31. Schmidt DN, Wallace K. How to diagnose hypopituitarism. Learning the features of secondary hormonal deficiencies. Postgrad Med 1998; 104(1): 81-88. Thorner MO, Vance ML, Laws ER, Horvath E, Kovacs K. The anterior pituitary. En: Wilson JD, Foster DE, Kronenberg HM, Larsen PR, eds. Williams Textbook of Endocrinology (9th Ed). Filadelfia: SW Saunders Co, 1998; 249-340. 859 © DOYMA 2001