Universidad Autónoma de Querétaro Facultad de Ciencias Naturales Licenciatura en Nutrición Trastornos de las glándulas tiroides, paratiroides, páncreas, suprarrenales y sus consecuencias en el metabolismo de macronutrimentos. Trabajo de Investigación Que como parte de los requisitos para obtener el grado de Licenciado en Nutrición Presenta: Montserrat Hernández Barredo Dirigido por: Dra. Margarita Teresa de Jesús García Gasca SINODALES Dra. Margarita Teresa de Jesús García Gasca ______________ M. en C. Diana Beatriz Rangel Peniche _______________ Dr. Pablo García Solís _______________ Dra. Ludivina Robles Osorio _______________ M. en C. María del Rocío Arellano Jiménez _______________ Centro Universitario Querétaro, Qro. México Mayo, 2011 i México Trastornos de las Glándulas Tiroides, Paratiroides, Páncreas, Suprarrenales, Consecuencias en el Metabolismo de Macronutrimentos y Manejo dietético Trabajo de Investigación ii ÍNDICE Página Índice.......................................................................................................................iii Índice de cuadros.....................................................................................................v Índice de figuras......................................................................................................vi Agradecimientos……………………………………………………………....................1 Resumen……………………………………………………………………………..........2 Introducción..............................................................................................................3 Capítulo 1: Tiroides 1.1 Definición de la glándula Tiroides......................................................................5 1.2 Fisiología y Mecanismo de acción de hormonas Triyodotironina (T3) y Tetrayodotironina (T4).............................................................................................6 1.3 Efectos Metabólicos de las Hormonas Tiroideas...............................................8 1.3.1 Metabolismo de Hidratos de Carbono...............................................11 1.3.2 Metabolismo de Lípidos.....................................................................11 1.3.3 Metabolismo de Vitaminas.................................................................12 1.4 Pruebas de funcionamiento tiroideo y valores séricos normales………………12 1.5 Patologías más frecuentes de la glándula tiroides y sus alteraciones en el metabolismo............................................................................................15 1.5.1 Hipertiroidismo...................................................................................16 1.5.2Hipotiroidismo.....................................................................................22 1.6 Recomendaciones generales adicionales para pacientes con hipertiroidismo e hipotiroidismo........................................................................29 Capítulo 2: Paratiroides 2.1 Definición de la glándula paratiroides..............................................................30 2.2 Fisiología de la hormona paratiroidea..............................................................30 2.3 Relación del metabolismo de calcio, fósforo, calcitonina y vitamina D con la glándula paratiroides...........................................................33 2.3.1 Metabolismo de calcio y fósforo........................................................33 2.3.2 Efecto de la calcitonina en la regulación de calcio en sangre...........34 2.3.3 Regulación del metabolismo de la vitamina D..................................36 2.4 Pruebas de funcionamiento paratiroideo y valores séricos normales.............37 2.5 Patologías más frecuentes de la glándula paratiroides...................................38 2.5.1 Hiperparatiroidismo...........................................................................39 2.5.2 Hipoparatiroidismo............................................................................42 2.5.3 Raquitismo y osteomalacia...............................................................47 2.5.4 Osteoporosis.....................................................................................49 2.6 Alteraciones óseas debidas a deficiencias y excesos nutricionales diversos............................................................................................................50 2.7 Valores nutrimentales de Referencia de calcio y fósforo.......................51 Capitulo 3: Páncreas 3.1 Definición de la Glándula Pancreática.............................................................53 3.2 Fisiología de la insulina....................................................................................55 3.3 Efecto de la insulina sobre el flujo global de sustratos energéticos………......60 iii 3.3.1 Efecto de la insulina sobre el metabolismo de los hidratos de carbono……………………………………………...60 3.3.2 Efecto de la Insulina y sobre el metabolismo de los Lípidos....................................................................................63 3.3.3 Efecto de la insulina sobre el metabolismo de las proteínas................................................................................65 3.4 Glucagon.........................................................................................................66 3.5 Regulación de la secreción de insulina y glucagon.........................................67 3.6 Pruebas de Laboratorio para determinar el estudio de la patología del páncreas....................................................................................67 3.7 Niveles de glucosa en sangre.........................................................................69 3.8 Patologías más frecuentes en el páncreas 3.8.1 Diabetes mellitus..............................................................................70 3.8.1.1 Tratamiento nutricio para pacientes con diabetes mellitus..............................................................................72 3.8.2Pancreatitis aguda.............................................................................76 3.8.3 Pancreatitis crónica..........................................................................80 Capitulo 4: Suprarrenales 4.1 Definición de las glándulas suprarrenales.......................................................83 4.2 Efectos relacionados con nutrición de la aldosterona y de cortisol.................85 4.2.1 Aldosterona.......................................................................................85 4.2.2 Cortisol..............................................................................................87 4.3 Efecto del cortisol en el metabolismo de hidratos de carbono………………...88 4.4 Efecto del cortisol en el metabolismo de Lípidos.............................................89 4.5 Efecto del cortisol en el metabolismo de Proteínas.........................................90 4.6 Datos de laboratorio para diagnosticar la presencia de enfermedad de la glándula suprarrenal...............................................................................90 4.7 Patologías más frecuentes de la corteza suprarrenal.....................................91 4.7.1 Enfermedad de Addison......................................................................91 4.7.2 Sindrome de Cushing..........................................................................95 Bibliografías...........................................................................................................99 Anexos.................................................................................................................109 Anexo 1. Fuentes de vitaminas y nutrimentos inorgánicos…………………........109 Anexo 2. Recomendaciones para disminuir los niveles de colesterol y triglicéridos………………………………………………………………..111 Anexo 3. Recomendaciones para dieta hiposódica e hipoprotéica….................112 Anexo 4. Raciones ó porciones diarias……………………………………………..114 Anexo 5. Metas básicas del tratamiento y criterios para evaluar el grado de control del paciente…………………………………………………….115 Anexo 6. Recomendaciones para pacientes diabéticos………………………….116 Anexo 7.Recomendaciones para dieta blanda…………………………………….117 iv INDÍCE DE CUADROS Cuadro Página 1. Efectos metabólicos de las hormonas Tiroideas..................................................11 2. Parámetros cinéticos de las hormonas tiroideas..................................................13 3. Interpretación del perfil tiroideo............................................................................15 4. Comparación de las características que se presentan en las patologías de hipotiroidismo e hipertiroidismo.......................................................................16 5. Recomendaciones para la ingestión de Yodo......................................................24 6. Contenido de yodo en alimentos de consumo común..........................................28 7. Ingestión diaria sugerida (IDS) de Calcio por grupo de edad...............................52 8. Requerimiento nutrimental promedio (RNP), ingestión diaria recomendada (IDR) e ingestión diaria sugerida (IDS) de fósforo por grupos de edad………….52 9. Factores y estados que aumentan ó disminuyen la secreción de insulina………59 10. Principales acciones de la insulina.....................................................................60 11. Efectos de la insulina sobre enzimas del metabolismo de hidratos de carbono y de lípidos............................................................................................63 12. Efectos del metabolismo de hidratos de carbono, grasas y proteínas………….65 13. Comparación entre diabetes miellitus dependiente de insulina y la no dependiente de insulina...........................................................................71 14. Distribución de la energía ( % ) de acuerdo al tipo de insulina...........................75 15. Criterios de gravedad de Ranson........................................................................77 16. Clasificación de los fármacos potencialmente útiles en el tratamiento médico del Síndrome de Cushing en función de su mecanismo de acción……………....97 v INDÍCE DE FIGURAS Figura Página 1. Localización de la glándula tiroides.........................................................................5 2. Producción y almacenamiento de las hormonas tiroideas......................................6 3. Hormonas Metabólicas Tiroideas............................................................................9 4. Relación aproximada entre la secreción diaria de hormona tiroidea (T4 y T3) y el metabolismo basal…………………………………………………………………10 5. Hipótesis compuesta para los sitios posibles de acción de las hormonas Tiroideas…………………………………………………………………………………20 6. Mecanismo a través del cual la deficiencia de yodo causa bocio.........................26 7. Localización de las glándulas paratiroides............................................................31 8. Acciones de la hormona paratiroidea y control de su secreción...........................32 9. Metabolismo del calcio..........................................................................................34 10. Control por retroinhibición de la secreción de calcitonina...................................35 11. Vías de 1,25-(OH)2D3, Hormona paratiroidea (PTH), y calcitonina (CT)..............36 12. Vías de la producción de Vitamina D..................................................................37 13. Manifestaciones bioquímicas en hiperparatiroidismo e hipoparatiroidismo…....38 14. El páncreas y sus islotes (islotes de Langerhans)..............................................53 15. Estructura de la insulina Humana.......................................................................55 16. Principal mecanismo de liberación de la insulina...............................................56 17. Efecto de la insulina sobre el flujo global de sustratos energéticos....................61 18. Efecto de la toma de alimento y del ayuno sobre el metabolismo......................62 19. Regulación de la secreción de insulina y glucagon............................................68 20. Producción de insulina en la diabetes mellitus...................................................70 21. Tratamiento dietético para pacientes diabéticos obesos…………………………74 22. Tratamiento dietético para pacientes diabéticos no obesos...............................75 23. Algoritmo de decisión para el tratamiento de pancreatitis aguda.......................78 24. Algoritmo de la terapia nutricional en la maldigestión debida a insuficiencia pancreática.....................................................................................82 25. Localización de las glándulas suprarrenales......................................................83 26. Estructura de la glándula suprarrenal.................................................................84 27. Estructura química de la aldosterona.................................................................85 28. Principales Acciones Fisiológicas del cortisol.....................................................88 29. Sistema renina-angiotesina-aldosterona............................................................92 30. Producción insuficiente ó excesiva de las hormonas adrenales.........................93 31. Manifestaciones clínicas del síndrome de Cushing............................................96 vi Agradecimientos Quiero expresar mi agradecimiento En primer lugar a Dios por estar siempre presente en mi vida, por ser mi brújula, y guiar mis pasos hacia este maravilloso momento que me llena de felicidad y satisfacción. Por haber puesto en mí camino aquellas personas que han sido mi soporte y compañía durante todo el periodo de estudio. A mi Universidad por ser testigo del camino que he recorrido durante estos años y contribuir en mi desarrollo personal y académico, dejándome la inquietud de continuar mi crecimiento, llevando conmigo los valores de la salud y la ética. A mi Directora de Tesis, Dra. Margarita Teresa de Jesús García Gasca por su generosidad al brindarme la oportunidad de recurrir a su capacidad y experiencia científica en un marco de confianza, afecto y amistad, fundamentales para la realización de este trabajo. A mis Asesores M en C. Diana Beatriz Rangel Peniche, M. en C. Rocío Arellano Jiménez, Dra. Ludivina Robles Osorsio y Dr. Pablo García Solís por su paciencia y dedicación, por honrarme al aceptar contribuir en el desarrollo del presente trabajo. A todos mis maestros, por ser mi ejemplo a seguir, por hacerme portadora de conocimientos y herramientas que me permitirán desarrollarme como Licenciada en Nutrición. A mi familia, por que el cariño y apoyo que me brindan me dan la fortaleza para seguir adelante, por ser la motivación más grande que tengo en mi vida En general quisiera agradecer a todas y cada una de las personas que han vivido conmigo mi trayecto para ser Licenciada en Nutrición, desde los más profundo de mi corazón les agradezco el haberme brindado todo el apoyo, colaboración, ánimo y sobre todo cariño y amistad. 1 Resumen El sistema endocrino y sus secreciones, las hormonas, regulan el crecimiento, el desarrollo, el metabolismo energético e hidroelectrolitico, así como las funciones sexuales y la reproducción de los seres humanos. En el presente trabajo se abordaron algunas patologías endocrinas y su manejo nutricional, en especial enfermedades de las glándulas tiroides, paratiroides, páncreas y suprarrenales. Se abordan también algunos aspectos fisiopatológicos de cada glándula para brindar un contexto global. Con lo anterior, se pretende que el este trabajo sirva como apoyo para nutriólogos que se enfrentan al tratamiento de pacientes con disfunciones endocrinas. Palabras clave: sistema endócrino, tiroides, paratiroides, páncreas, glándulas suprarrenales, nutrición, dietoterapia, hormonas. 2 INTRODUCCIÓN La endocrinología es la rama de la medicina que estudia la fisiología y patología de las glándulas de secreción interna (Ríos, 2008). Las hormonas son sustancias secretadas por células especializadas, localizadas en glándulas de secreción interna o glándulas endocrinas (carentes de conductos), o también por células epiteliales e intersticiales con el fin de afectar la función de otras células (Lisci, 2008). Los primeros estudios de las glándulas endócrinas se iniciaron con Berthold en 1849 castrando animales de experimentación, detectando cambios en el desarrollo morfológico y en el comportamiento. Muchos de los órganos endocrinos fueron identificados por anatomistas desde el siglo XIX, su mecanismo de acción se descubrió hasta los inicios del siglo XX. Haciendo referencia a las primeras comunicaciones, en 1902 se publicaron las primeras experiencias sobre la secreción del páncreas otorgándole el nombre de secretina al primer producto identificado (Ríos, 2008). Ya que la endocrinología es una especialidad relativamente reciente en el amplio horizonte de las ciencias médicas, resulta impresionante conocer los pasos agigantados con los que avanza dentro de la modernidad científica. La integración de la unidad hipotálamo-hipofisaria con las glándulas regidas por ella, la interrelación que tienen los ejes con el medio ambiente es por demás apasionante, como también la patología derivada de cualquier falla en esta organización prácticamente perfecta (Lisci, 2008). La regulación del anabolismo (síntesis) y catabolismo (degradación) está orientada a regular la energía necesaria para las actividades celulares, el crecimiento y el desarrollo. En situaciones adversas como por ejemplo, la desnutrición, el organismo deja de crecer y desarrollarse para mantener las actividades celulares (Jameson, 2006). La concentración de hormonas secretadas por las glándulas tiroides, paratiroides, suprarrenales y páncreas intervienen directamente en el control endócrino del metabolismo de los macronutrimentos (hidratos de carbono, lípidos y proteínas). Sin embargo, 3 cuando se presenta una secreción insuficiente, resistencia hormonal o una secreción excesiva por dichas glándulas, la concentración secretada de hormona altera el control endócrino del metabolismo desencadenando patologías tales como: resistencia a la insulina asociada a obesidad abdominal, hipotiroidismo, hipertiroidismo, enfermedad de Addison, síndrome de Cushing, pancreatitis, hiperparatiroidismo e hipoparatiroidismo, entre otras, las cuales han tomando mayor relevancia debido a los problemas de salud que se presentan en la actualidad. Su tratamiento requiere un plan de alimentación específico para regular el metabolismo y en consecuencia mejorar la salud (Fox, 2003). Contar con información sobre cómo se desarrollan dichas patologías y la alteración que se produce en el metabolismo es necesario para implementar un tratamiento dietético para la población afectada. En este sentido, se requiere que el nutriólogo se involucre en el tratamiento de las enfermedades del sistema endócrino ya que la alimentación es una herramienta indispensable para el control, tratamiento y prevención de estas alteraciones (Ríos, 2008). Por lo anterior, el objetivo del presente trabajo es integrar el conocimiento sobre la disfunción de las glándulas del sistema endocrino dirigido a los nutriólogos con enfoque hacia la alimentación y la salud. 4 Capítulo 1: Tiroides 1.1 La glándula tiroides La glándula tiroides es la más grande de las glándulas endócrinas puras con un peso de 20 a 25 g, se encuentra inmediatamente por detrás de la laringe, sus dos lóbulos están colocados a cada lado de la tráquea y unidos en la parte anterior por una masa medial de tejido tiroideo denominada istmo (Figura 1) (Fox, 2003). Figura 1. Localización de la glándula tiroides (Adaptado de Fox, 2003). La función tiroidea es regulada por la hormona estimulante de la tiroides (TSH, tirotropina) de la hipófisis anterior. A su vez, la secreción de la TSH es regulada principalmente por la hormona liberadora de tirotropina (TRH) del hipotálamo. Por otra parte las hormonas tiroideas regulan por retroalimentación negativa las secreciones de la TSH y TRH en la hipófisis anterior y el hipotálamo, respectivamente. (Gangog, 2004). Existen tres hormonas importantes que son secretadas por la tiroides: la tiroxina o tetrayodotironina (T4), la triyodotirionina (T3), y la triyodotirionina reversa (T3r). La T4 y T3 inducen un notable aumento del metabolismo del organismo. La ausencia de hormonas tiroideas provoca con frecuencia descensos metabólicos de hasta un 50% inferiores del valor normal, mientras que la secreción excesiva incrementa el metabolismo entre un 60% hasta un 100% por encima de lo normal. Por otra parte la T3r es biológicamente inactiva (Leibel y Pat, 2007). La glándula tiroides secreta además, calcitonina, una hormona importante para el metabolismo del calcio (Guyton y Hall, 2006). 5 1.2 Fisiología y mecanismo de acción de hormonas triyodotironina (T3) y tetrayodotironina (T4) Desde el punto de vista microscópico, la tiroides está constituida por numerosas estructuras esféricas y huecas denominadas folículos tiroideos (Figura 2). Estos folículos están revestidos por un epitelio cuboideo simple constituido por células foliculares que sintetizan y secretan a las hormonas tiroideas. Dentro de los folículos se encuentra el coloide, un líquido con abundantes proteínas. Además de las células foliculares, la tiroides también contiene células parafoliculares que secretan a la calcitonina (Marino y McCluskey, 2000; Fox, 2003). Figura 2. Producción y almacenamiento de las hormonas tiroideas. El yoduro se transporta de manera activa a las células foliculares. En el coloide, se convierte en yodo y se une a aminoácidos tirosina en la proteína trioglobulina. La MIT (monoyodotirosina) y la DIT (Diyodotirosina) se utilizan para producir T3 y T4 en el coloide. Tras su estimulación por la TSH, las hormonas tiroideas unidas a la trioglobulina son captadas por las células libres foliculares mediante pinocitosis. Las reacciónes de hidrólisis dentro de las células foliculares dan lugar a la liberación de T4 y T3 que secretan (Tomado de Guyton y Hall, 2006. Esquema modificado de Fox, 2003). 6 Los folículos tiroideos acumulan de manera activa yoduro (I-) a partir de la sangre y lo secretan al coloide. Una vez que el yoduro ha entrado en el coloide, se oxida a yodo y se une a residuos de tirosina contenidos en la trioglobulina. La unión de un átomo de yodo a una tirosina da lugar a la monoyodotirosina (MIT), la unión de dos yodos da lugar a la diyodotirosina (DIT). En el interior del coloide, las enzimas modifican la estructura de la MIT y la DIT y dan lugar a su acoplamiento. Cuando se unen dos moléculas de DIT, se forma la molécula de T4. La combinación de una molécula de MIT con otra de DIT da lugar a la formación de T3. En este momento, la T4 y la T3 todavía están unidas a la tirioglobulina. Tras su estimulación por la TSH, las células del folículo captan un pequeño volumen del coloide mediante pinocitosis, hidrolizan a la T3 y a la T4 de la tirioglobulina y secretan las hormonas libres a la sangre (Marino y McCluskey, 2000; Fox, 2003). Si el ritmo de producción y secreción hormonal es normal, diariamente entran alrededor de 120 µg de yodo a la tiroides. La tiroides secreta 80 µg de yodo cada día unido a las hormonas tiroideas y diariamente difunden 40 µg de yodo al líquido extracelular. Las hormonas tiroideas secretadas se metabolizan en el hígado y otros tejidos, con lo cual se liberan diariamente 60 µg de yodo al líquido extracelular. Algunos derivados se excretan en la bilis y parte del yodo que contiene es reabsorbido por la circulación entero hepática, pero existe una pérdida neta de yodo en las heces que se aproxima a 20 µg/día. Por tanto, la cantidad total de yodo que entra al líquido extra celular es de 600 µg/día; 20% de esta cantidad entra a la tiroides, mientras que el restante 80% se excreta en la orina (Gangog, 2004). Por otra parte, aproximadamente el 99.96% de la T4 se transporta por la sangre unida a proteínas transportadoras (principalmente a tiroglobulina o TBG) de esta forma dichas proteínas forman un reservorio de la hormona. La glándula tiroides también secreta pequeñas cantidades de T3. Sin embargo, las proteínas transportadoras presentan mayor afinidad por la T4 que por la T3 y, debido a ello, la cantidad de T3 que permanece en el plasma sin unir a proteínas (libre) es aproximadamente 10 veces mayor que la cantidad de T4 libre. Sólo la T4 libre y la T3 pueden introducirse en las células diana. Una vez 7 que la T4 libre se introduce en el citoplasma de la célula diana se convierte enzimáticamente en T3, la forma activa (Fox, 2003). Los receptores de las hormonas tiroideas se localizan en el núcleo de las células y se definen como factores de transcripción dependientes de ligando y forman parte de la super familia de receptores de nucleares que se unen al ADN mediante un dominio denominado dominio de la unión al ADN también está el dominio de unión al ligando (hormona) y dominios hipervariables con sitios de dimerización y transactivación. Usualmente los receptores a hormonas tiroideas funcionan como heterodimeros con el receptor del retinoide x (9-cis ácido retinoico) y en algunas ocasiones funcionan como homodímeros, mientras no se una la hormona no tiene ningún efecto sobre la transcripción génica. Una vez que se unen los respectivos ligandos al homo ó heterodímero se regula la transcripción de los genes blancos. Dicha regulación positiva ó negativa (Figura 3) (Fox, 2003). Entre las funciones más importantes de los receptores de hormona tiroidea se encuentran la regulación del metabolismo y de la frecuencia cardiaca (Yen, 2001).Se han descrito dos isoformas del receptor de hormona tiroidea (TR) codificados por genes distintos denominadas alfa y beta, que son capaces de unir dicha hormona. Además, se obtienen dos variantes del TR-alfa mediante splicing alternativo del gen THRA, y otras dos variantes del TR-beta mediante splicing alternativo del gen THRB (Flamant y col., 2006). TR-α1: Expresado en diversos tejidos, con elevados niveles en músculo esquelético y el músculo cardiaco. TR-α2: homólogo del oncogén viral c-erb-A, también expresado en diversos tejidos pero incapaz de unir la hormona.TR-β1: expresado de forma predominante en cerebro, hígado y riñón.TR-β2: expresión limitada al hipotálamo y a la pituitaria (Flamant y col., 2006). 1.3 Efectos metabólicos de las hormonas tiroideas El gasto metabólico en condiciones de reposo se conoce como metabolismo basal (MB), el cual posee dos componentes, uno regulado por la T4 y otro independiente de la misma. La T4 fija el punto de equilibrio del metabolismo basal de tal manera que si se encuentra en exceso, tiende a elevarlo hasta en 8 un 60% a 100%. Por el contrario, cuando no se produce hormona tiroidea, el metabolismo basal disminuye hasta la mitad de lo normal (Figura 4) (Guyton y Hall, 2006). Figura 3. Hormonas Metabólicas Tiroideas. La tiroxina (T4) y la triyodotironina (T3) difunden fácilmente a través de la membrana celular. LaT4 se desyoda enzimáticamente y se forma T3, que se une a su receptor. Usualmente el receptor a T3 forma heterodímeros con el receptor del retinoide X (9-cis ácido retinoico). La unión de la hormona con su receptor promueve la transcripción de ciertos genes que tienen en su región promotora secuencias específicas denominadas elementos de respuesta a hormonas (Tomado de Guyton y Hall, 2006). 9 La dieta también tiene un efecto definitivo en la conversión de T4 en T3. En los sujetos que permanecen en ayuno, la T3 plasmática disminuye de 10 a 20% en 24 h y en casi 50% después de 3 a 7 días en ayuno. Figura 4. Relación aproximada entre la secreción diaria de hormona tiroidea (T4 y T3) y el metabolismo basal (Tomado de Guyton y Hall, 2006). La T4 (a través de su conversión en T3) estimula la velocidad de respiración celular en casi todas las células del cuerpo, un efecto que se atribuye a un descenso de las concentraciones celulares de ATP. Este efecto se produce por: 1) La síntesis de proteínas desacopladoras, 2) Estimulación de las bombas de sodio/potasio de transporte activo, que constituyen un sumidero de energía que disminuye todavía más las concentraciones de ATP, el ATP ejerce una inhibición como producto final de la respiración celular, de forma que cuando disminuyen las concentraciones de ATP, aumenta la velocidad de respiración celular. Dado que la T4 estimula la producción de proteínas de desacoplamiento y la velocidad de la respiración celular, incrementa la producción del calor metabólico (Cuadro 1) (Fox, 2003). En enfermedades sistémicas o en neonatos, los niveles de T3 son bajos y los de T3 reversa son elevados. La T3 reversa es, por tanto útil como ayuda al diagnóstico en el tratamiento del hipotiroidismo fetal e infantil (Fox, 2003). 10 Cuadro 1. Efectos metabólicos de las hormonas tiroideas (Tomado de Mendoza, 2008). 1.3.1 Metabolismo de hidratos de carbono Las hormonas tiroideas estimulan casi todas las fases del metabolismo de hidratos de carbono, entre ellos, aumentan la captación de glucosa por las células intestinales y promueven la secreción de insulina, aumentan la glucólisis, incrementan de la síntesis de glucógeno. Toda esta actividad obedece, probablemente, a la expresión general de las enzimas metabólicas inducidas por la hormona tiroidea (Guyton y Hall, 2006). 1.3.2 Metabolismo de lípidos Las hormonas tiroideas regulan el metabolismo de los lípidos se ha observado que cuando hay un incremento de hormonas tiroideas hay un descenso de la concentración plasmática de colesterol, fosfolípidos y triglicéridos, pero eleva los ácidos grasos libres. Por el contrario, la disminución de la secreción tiroidea eleva en gran medida la secreción plasmática del colesterol, fosfolípidos y triglicéridos y casi siempre origina un depósito excesivo en el hígado, y disminuye la expresión del receptor del colesterol de baja densidad (LDL) en el 11 hígado, el caul tiene la función de captar el colesterol LDL de la circulación, por lo que al disminuir su expresión se manifiesta con hipercolesterolemia. El gran aumento del colesterol plasmático observado en el hipotiroidismo prolongado se asocia a menudo una arterosclerosis grave (Guyton y Hall, 2006). Uno de los mecanismos mediante los cuales la hormona tiroidea reduce la concentración plasmática de colesterol consiste en el notable aumento de secreción de colesterol hacia la bilis y su pérdida consiguiente por las heces, así como un aumento de la depuración del hígado (Guyton y Hall, 2006). 1.3.3 Metabolismo de vitaminas Dado a que las hormonas tiroideas incrementan la cantidad de numerosas enzimas y que las vitaminas suponen una parte esencial de algunas enzimas o coenzimas, las hormonas tiroideas aumentan las necesidades de vitaminas. Por consiguiente, a veces aparece un déficit vitamínico cuando se secreta una cantidad excesiva de hormona tiroidea, salvo que el organismo disponga al mismo tiempo de mayor cantidad de vitaminas (Guyton y Hall, 2006). 1.4 Pruebas de funcionamiento tiroideo y valores séricos normales El perfil tiroideo sirve para medir el desequilibrio en las hormonas tiroideas. Los valores séricos normales de algunos parámetros de la función tiroidea se muestran en el Cuadro 2. Entre las pruebas clínicas para determinar la función tiroidea están (González y col., 1988): I. Pruebas que miden la concentración y unión de las hormonas tiroideas en sangre. a) Determinación de hormonas unidas a proteínas transportadoras 1. Yodo proteínico 2. Tiroxina total 3. Triyodotiroxina total 4. Triyodotrionina reversa 12 Cuadro 2. Parámetros cinéticos de las hormonas tiroideas (Tomado de Mendoza, 2008). T3 T4 Producción diaria 25µg 75µg Reserva 54µg 800µg Concentraciones séricas totales 95-190 (ng/dL) 5-11 (µg/dL) Fracción libre Equivalentes a 1.5-2.9 nmol/L 0.2-0.52 ng/dL Equivalentes a 64-132 nmol/L 0.7-1.86 ng/dL Unión a proteínas (%) Equivalentes a 3-8 pmol/L 99.99 Equivalentes a 9-24 pmol/L 99.96 TBG 60 50 Albúmina 40 35 TBPA Absorción oral (%) -95 15 80 Volumen de distribución 40L 10L Recambio por día (%) 60 10 Depuración por día 24L 1.1 L Tiempo de vida media <2 días 6-7 días Potencia biológica relativa 4 1 b) Determinación de la capacidad de saturación de proteínas transportadoras. 1. Captación de T3 por resinas c) Determinación de la hormona tiroidea libre 1. Tiroxina libre 2. Triyodotrionina libre 3. Índice de T4 libre (calculado por fórmula, se ha dejado de utilizar debido a la disponibilidad de la medición de tiroxina libre). d) Determinación de las proteínas transportadoras de las hormonas tiroideas 1. Globulina fijadora de tiroxina (TBG) 13 II. Pruebas que miden la acumulación del yodo por la tiroides a) Captación de I131 a las 24h b) Captación de pertecnetato por la tiroides c) Índice de depuración de yodo por la tiroides d) Excreción urinaria del yodo e) Prueba de descarga por perclorato ó tiocianato III. Pruebas que informan sobre la morfología del tiroides a) Gammagrafía b) Ecografía c) Tomografía axial computarizada d) Linfangiografía del tiroides e) Placa simple de cuello f) Gammagrama fluorescente g) Resonancia magnética de la tiroides IV. Pruebas que informan sobre la relación hipófisis-tiroides a) Tirotropina en sangre b) Prueba de la estimulación con TRH c) Prueba de la inhibición con T3 d) Prueba de la estimulación con TSH. En el Cuadro 3 se muestra la interpretación del perfil tiroideo (Kronenberg y col., 2009). Los valores normales de la TSH son de 0.4 a 4.0 ml U/L (miliunidades internacionales por litro). Un valor normal de la hormona T4 es de 4.5 a 11.2 14 microgramos por decilitro (mcg/dL). El nivel normal de la hormona T4 libre es de 0.8 - 1.9 ng/dl. El rango para los valores normales es de 100 a 200 ng/dL (nanogramos por decilitro). El rango para los valores normales de T3 es de 100 a 200 ng/dL (nanogramos por decilitro).La T3 tiene una menor afinidad por las proteínas transportadoras, por ese motivo la concentración de T3 libre es mayor que la T4 libre. Los niveles normales de T3 libre oscilan entre 2,6-5,4 pg/dl (4-8,3 pmol/L) (Kronenberg col., 2009). Cuadro 3. Interpretación del perfil tiroideo (Modificado de Kronenberg col., 2009). N: normal, alto, bajo. 1.5 Patologías más frecuentes de la glándula tiroides y sus alteraciones en el metabolismo Las enfermedades tiroideas se encuentran entre las alteraciones endocrinológicas más frecuentes y presentan una sintomatología poco específica que es común a muchas situaciones clínicas. En su diagnóstico tienen un papel importante las determinaciones bioquímicas (Velazco y Rodriguez, 1998). Las manifestaciones metabólicas de las enfermedades tiroideas se relacionan con una producción excesiva de hormonas tiroideas (hipertiroidismo) o déficit de secreción de las mismas (hipotiroidismo), las cuales afectan a la población, en especial a las mujeres, el grupo más 15 vulnerable, en que la presencia de una disfunción tiroidea alcanza cifras cercanas al 10%, lo que equivale a tres veces más que en los hombres (Rodríguez y col., 2007). La patología de la glándula tiroides afecta a casi todas las funciones del organismo, destacando su repercusión sobre el desarrollo del sistema nervioso y del crecimiento (Rodríguez y col., 2007). En un estudio se investigó la asociación entre TSH, T4, T4 libre, T3 y T3 reversa y se demostró que la TSH y/o los niveles de T4 son pobres indicadores de los niveles de tejido de la tiroides y, por lo tanto, no se pueden utilizar para saber si la persona es eutiroidea (niveles normales de la tiroides). La relación T3/T3r es actualmente el mejor indicador de los niveles del tejido de la tiroides (Vandenbeld y col., 2005). En el Cuadro 4 se muestran efectos relacionados con hipotiroidismo e hipertiroidismo. Cuadro 4. Comparación de las características que se presentan en las patologías de hipotiroidismo e hipertiroidismo (Tomado de Fox, 2003). Característica Crecimiento y Desarrollo Hipotiroidismo Crecimiento disminuido Actividad y Sueño Letargo; aumento de sueño Tolerancia a la temperatura Características de la piel Sudoración Pulso Hipertiroidismo Crecimiento acelerado Aumento de la actividad; disminución del sueño Intolerancia al frío Intolerancia al calor Piel oscura y seca Ausente Lento Síntomas Gastrointestinales Estreñimiento, disminución del apetito, aumento del peso corporal Reflejos Aspectos psicológicos Concentraciones plasmáticas de T4 Lentos Depresión y apatía Piel normal Excesiva Rápido Movimientos intestinales frecuentes; aumento del apetito; disminución del peso corporal. Rápidos Nerviosismo Disminuidas Aumentadas 1.5.1 Hipertiroidismo El síndrome clínico se debe a un exceso de hormonas tiroideas en la sangre, y los signos y síntomas clínicos que se presentan se deben a una exageración de las acciones fisiológicas de las hormonas tiroideas, produciéndose una acción o elevación general del metabolismo. Algunas características clínicas del hipertiroidismo son temblor, taquicardia (a veces con arritmia), pérdida de masa corporal a pesar de un aumento de apetito, fatiga y debilidad muscular, 16 hiperhidrosis con intolerancia al calor, piel fina, húmeda y caliente, trastornos menstruales, aumento de la motilidad intestinal con diarrea y en algunas ocasiones ansiedad grave y cambios emocionales. En enfermos de edad avanzada, el hipertiroidismo puede manifestarse por un agravamiento de trastornos ya existentes (Velazco y Rodríguez, 1998). Las causas principales de hipertiroidismo no mediado por mecanismos inmunitarios son los adenomas, ya sean solitarios (adenoma hiperfuncionante) ó múltiples (bocio multinodular). El bocio multinodular es frecuente en ancianos y suele ser poco significativo clínicamente. Ocasionalmente, la producción de hormona tiroidea por parte de un adenoma sobrepasa las necesidades diarias y esto lleva a un hipertiroidismo. Otras causas comunes son la ingestión de hormonas tiroideas, bocio tóxico difuso (enfermedad de Graves), adenoma hiperfuncionante (nódulo tóxico), bocio multinodular tóxico, tiroiditis indolora, tiroiditis subaguda, adenoma secretor de hormona estimulante del tiroides y tumores secretores de gonadotropina coriónica humana (Page y col., 1998). En el hipertiroidismo suele alterarse tanto la concentración de T4 en plasma como la de T3, aunque la glándula tiroidea aumenta más en proporción a la producción de T3, por lo que el incremento de su concentración en plasma es mayor y más precoz que la de T4. Las concentraciones anormalmente elevadas de T4 en plasma inhiben la secreción de tirotropina la cual, además, no responde a la estimulación con hormona liberadora de tirotropina (TRH) (Velazco y Rodríguez, 1998). La enfermedad de Graves es la causa más frecuente del hipertiroidismo. Es una enfermedad autoinmune que se caracteriza por la presencia en plasma de anticuerpo estimulante de tiroides que se une a los receptores de tirotropina de las células tiroideas, estimulando así la producción de hormonas tiroideas. Se presenta con mayor frecuencia en jóvenes y puede actuar como factor precipitante los estados de tensión. El tratamiento de la enfermedad de Graves puede realizarse mediante fármacos antitiroideos, yodo radioactivo o cirugía (tiroidectomía subtotal) (Herranz, 2010). El seguimiento a largo plazo de los pacientes ya tratados es muy importante debido a que con relativa frecuencia se producen recidivas de la enfermedad en aquellos pacientes que han sido 17 tratados con fármacos antitiroideos, en los cuales puede presentarse incluso después de varios años de permanecer asintomáticos. Además, el tratamiento, especialmente si se ha realizado con yodo radioactivo o cirugía, puede causar la aparición de hipotiroidismo, que puede ser temporal o persistir durante años. Por último, puede encontrarse en estos pacientes concentraciones de T4 en plasma dentro del intervalo de referencia junto a una ligera elevación de tirotropina que puede persistir a pesar de que el paciente permanezca eutiroideo clínicamente (Velazco y Rodríguez, 1998). Tras la tiroidectomía subtotal, la concentración de T4 en el plasma desciende inmediatamente y la de tirotropina permanece inicialmente baja, pero luego se puede elevar en respuesta a las concentraciones disminuidas de T4 y estimular las células tiroideas. Con esto se consigue mantener la concentración de T4 en plasma dentro del intervalo de referencia con el tejido tiroideo restante gracias a una mayor producción de tirotropina (Page y col., 1998). Otra causa de hipertiroidismo, aunque menos frecuente que la enfermedad de Graves, son los bocios nodulares tóxicos, que se pueden clasificar en adenomas tiroideos y bocio multinodular tóxico. En ambos casos puede producirse una secreción autónoma de hormonas tiroideas que causará el hipertiroidismo. También es más frecuente esta enfermedad en mujeres, pero se presenta en edades más avanzadas que la enfermedad de Graves. La secreción autónoma de hormonas tiroideas que se produce en estos casos inhibe la secreción de tirotropina, con lo que ésta no estimula el tejido tiroideo sano que suele volverse hipofuncionante (Herranz, 2010). Aún es difícil separar las acciones normales de las hormonas de la tiroides de los que ocurren durante la tirotoxicosis. En este último caso, los efectos tóxicos pueden anular la función de control normal por ejemplo, incrementa el transporte de glucosa a las células (Danoff y Laster, 1962), la movilidad intestinal se afecta (Johansson, 1966), se presenta mayor transporte de sodio (Green y Matty, 1963), se alteran el fosfato y el manejo del agua renal (Parsons y Anderson, 1964) y hay un cambio en la respuesta celular y en la fijación de digoxina (Doherty y Perkins, 1966). Éstos y muchos otros cambios metabólicos se relacionan con la alteración de síntesis de proteínas, inflamación 18 mitocondrial y desacoplamiento de la fosforilación oxidativa (Figura 5) (Parsons y Ramsay, 1968). También existe un incremento del recambio óseo cuando existe un aumento de los niveles de hormonas tiroideas (Mataix y Vázquez, 2009). El exceso de hormonas tiroideas condiciona alteraciones mentales importantes en quienes lo padecen. La ansiedad y la inquietud suelen ser de los primeros síntomas, llegando a catalogarse como solo un trastorno de ansiedad. En las personas de la tercera edad, el cuadro tiene características propias entre las que prevalecen la indiferencia afectiva y el desorden mental. En casos graves puede llegar hasta el estupor y el coma. En determinados pacientes existe deterioro de la memoria, de la orientación y del juicio. En situaciones más graves todavía, incluso hay síntomas maniacos y esquizofreniformes, matizados de ideas delirantes y alucinaciones visuales y auditivas. La evolución de los trastornos mentales mejora cuando el enfermo llega al eutiroidismo. Los trastornos del estado de ánimo y los psicóticos pueden requerir tratamiento adicional para su resolución (Valdivieso y col., 2006). El tratamiento para pacientes con hipertiroidismo es con base en fármacos y en algunos casos quirúrgico sin embargo, deben tenerse en cuenta algunas normas dietéticas. Así, la pérdida de peso severa que presentan algunos pacientes, que suele ir acompañada de una desnutrición, obliga a la recuperación nutricional. Esta repercusión debe cuidarse especialmente en los que van a sufrir tiroidectomía, prestando una especial atención no solo al aporte energético y proteínico sino a todos los micronutrimentos en general. Por otra parte, una vez tratado el paciente y en situación de normalidad metabólica, puede presentar un apetito aumentado, respecto a los menores requerimientos energéticos que presenta en esa nueva condición, lo que habría que tener en cuenta por el riesgo de obesidad que conlleva (Mataix y Vázquez, 2009). 19 Figura 5. Hipótesis compuesta para los sitios posibles de acción de las hormonas tiroideas. A) Transporte de iones a través de aceleración de ATPasa. B) Acciones de la permeabilidad mitocondrial. C) Desacoplamiento de la fosforilación oxidativa. D) Aumento de enzimas lipolíticas. E) y F) Alteraciones en la síntesis de proteínas a través de cambios en la síntesis de ARN y estructura ribosomal. I) Sitios de la acción inhibitoria de citrato y ácidos grasos libres en la vía de Embden Meyerhof (Tomado de Parsons y Ramsay, 1968). Los objetivos del tratamiento nutricio para pacientes con hipertiroidismo son (Landa y col., 2006): 1. Prevenir o tratar las complicaciones que incluyen desmineralización ósea. 2. Corregir el balance negativo de nitrógeno. 20 3. Reemplazar las pérdidas de líquidos por diarrea, diaforesis y aumento de la respiración. 4. El exoftalmos, causado por un aumento de líquido extracelular en los ojos puede requerir restricción de líquidos y sal. 5. Vigilar la intolerancia de las grasas y esteatorrea. Las recomendaciones dietéticas para pacientes con hipertiroidismo son: 1. Aumentar el consumo energético (40 Kcal/Kg/día), mientras se normalizan los niveles hormonales con el tratamiento administrado. Las necesidades energéticas del paciente pueden estar aumentadas en un 50-60% (10-30% casos leves) por el aumento de la tasa del metabolismo basal; si hay un aporte adecuado de energía será más fácil mantener un peso saludable (Mahan y Arlin, 1995). 2. Ingestion elevada de hidratos de carbono y fibra. La fibra está presente en alimentos integrales de grano entero y en la cáscara de frutas y verduras, y ayuda a satisfacer el exceso de apetito que presentan estos pacientes (Landa y col., 2006) (Anexo 1). 3. Consumir 1-1.75 g/Kg/ día de proteína. El metabolismo de las proteínas se encuentra muy aumentado en esta enfermedad, sobre todo su degradación; lo que se traduce en debilidad muscular y pérdida de peso (Mahan y Arlin, 1995). 4. Tomar de 2-3 litros de agua al día, ya que es importante mantenerse bien hidratado, para contrarrestar la fatiga presente en el hipertiroidismo. Es común que las personas que padecen esta enfermedad presenten diarreas ocasionales y sudoración excesiva lo que hace necesario aumentar el consumo de líquidos. Sólo se contraindicaría en caso de padecer problemas cardiacos o renales (Mahan y Arlin, 1995). 5. Evitar alimentos procesados y refinados. Este tipo de alimentos que incluyen harina blanca y azúcar tienen una alta densidad energética y poco contenido de nutrimentos por lo que hay que limitar este tipo de productos (Mahan y Arlin, 1995). 21 6. Incluir variedad de frutas y verduras (Anexo 1). Las frutas y verduras, aparte de ofrecer fibra, contienen vitaminas y nutrimentos inorgánicos que son necesarios para las correctas funciones fisiológicas del organismo. Son parte esencial de la dieta (Degroot y col., 1996). 7. Preferir las carnes magras (Anexo 2). Consumir pescado, atún en agua o pollo sin piel con más frecuencia porque tienen menos grasa saturada y colesterol. Evitar las carnes rojas, embutidos, cerdo y quesos grasosos. Limitar el consumo de alimentos ricos en yodo. Alimentos como ajo, mariscos, yemas de huevo, perejil, albahaca, algas y sal yodada. Es importante no sobre consumir yodo, ya que tiene un rango relativamente estrecho de ingestión que apoya el funcionamiento correcto de la tiroides (100 a 300 microgramos al día) (Degroot y col., 1996). 8. Consumir alimentos ricos en hierro (Anexo 1). Se recomienda la avena, pan integral, lentejas, espinacas, acelgas, pistaches, nueces, almendras, pasas, ciruelas, garbanzos, dátiles e hígado. Es común que los pacientes con hipertiroidismo presenten anemia (Degroot y col., 1996). 9. Aumentar el consumo de alimentos ricos en calcio (lácteos) (Anexo 1). El exceso de hormonas tiroideas parece actuar directamente sobre el hueso favoreciendo su reabsorción, por lo que puede producirse una desmineralización del hueso. Se recomienda tomar 1 litro de leche que además del calcio provee una adecuada cantidad de fósforo y vitamina D (Degroot y col., 1996). Dada las tendencias actuales de nutrición y en línea acorde con los otros lineamientos la leche deberá consumirse descremada ó semidescremada. 10. Se recomienda suplementarla dieta con vitaminas A y C, también de complejo B especialmente tiamina, riboflavina, B6 y B12 (Landa y col., 2006) (Anexo 1). 1.5.2Hipotiroidismo El hipotiroidismo es una situación clínica caracterizada por una secreción deficiente de hormonas tiroideas debida a una alteración orgánica o funcional 22 de la propia glándula, o por un déficit de estimulación de la tirotropina (Ferri, 2006).Se puede presentar a cualquier edad, pero su frecuencia es mayor en edades avanzadas de la vida. Si se produce durante la vida intrauterina puede dar lugar, si no se trata precozmente, a una alteración neurológica importante e irreversible. Si el hipotiroidismo se desarrolla después del nacimiento, en la infancia o en la edad adulta, la característica principal será un enlentecimiento general del metabolismo (Velazco y Rodríguez, 1998). Cuando el hipotiroidismo se origina por alteraciones tiroideas se denomina primario mientras que el que depende de una secreción de tirotropina insuficiente puede ser secundario si el fallo es hipofisario o terciario si es hipotalámico. El hipotiroidismo subclínico incluye una serie de situaciones asintomáticas en las que la reducción de la función tiroidea queda compensada por un aumento en la secreción de tirotropina (Swearingen, 2008). Las alteraciones en la síntesis de las hormonas tiroideas pueden producirse por defectos congénitos de cualquiera de las enzimas que intervienen en la síntesis de la T4; en la mayor parte de los casos la estimulación prolongada que ejerce la tirotropina. Debido a la disminución de concentración de hormonas tiroideas se genera un agrandamiento de la glándula tiroidea. Dentro de este grupo, la forma más frecuente es la debida a un fallo en la incorporación de yodo a tirosina. La deficiencia de yodo da lugar a bocio endémico y la ingesta de bociógenos exógenos también puede causar hipotiroidismo por alteración de las hormonas tiroideas (Velazco y Rodíguez, 1998). En el Cuadro 5 se muestran las recomendaciones para la ingestión de yodo en los diferentes grupos de edad. La tiroiditis de Hashimoto es una de las causas más frecuentes de bocio esporádico. Tiene una base autoinmune; en el plasma de estos pacientes se encuentran concentraciones elevadas de anticuerpos contra la tirioglobulina y contra la peroxidasa microsomal tiroidea y en algunas ocasiones contra el segundo antígeno coloide. En una primera fase puede acompañarse de hiperfunción tiroidea (Ferri, 2006).La tiroiditis de De Quervain o sub aguda al igual que la anterior, puede presentar hiperfunción tiroidea, debido a la liberación de hormonas por la destrucción del tejido tiroideo, posteriormente 23 pasan por una fase de hipotiroidismo por agotamiento de las hormonas que se encontraban almacenadas en la tiroides durante la primera fase de inflamación y tiroiditis, posteriormente se recuperan llegando a tener niveles normales de hormonas tiroideas (eutiroidismo) y sólo en raras ocasiones el hipotiroidismo será permanente. La secuencia de la tiroiditis posparto es similar (Velazco y Rodríguez, 1998). Cuadro 5. Recomendaciones para la ingestión de yodo (Tomado de Martínez y col., 2005). Grupos de edad Lactantes 0-6 meses 7-12 meses Niños menores 1-3 años 4-8 años Niños mayores y adolescentes 9-13 años 9-13 años 14-18 años 14-18 años Adultos 19-50 años 51-70 años Más de 70 años Menos de 18 años 19-30 años 31-50 años Lactancia M: masculino, F: femenino. Sexo M+F M+F M+F M+F M F M F M+F M+F M+F IDS (µg/d) IDR (µg/d) LSC (µ/d) 110 130 // // // // // // // // // // // // // // // // // // // // 65 65 200 300 73 73 95 95 600 600 900 900 150 150 150 220 220 220 290 1 100 1 100 1 100 900 1 100 1 100 1 100 IDR : Ingesta diaria recomendada IDS: Ingesta diaria sugerida LSC: Limite superior de consumo El hipotiroidismo idiopático es la forma más común de presentación en el adulto de hipofunción tiroidea no secundaria a destrucción iatrogénica del tiroides. 24 Puede representar el estadio final de una tiroiditis autoinmune atrófica, la cual da lugar a la destrucción progresiva de la glándula. Otras formas bastante frecuentes de hipotiroidismo en el adulto son las secundarias al tratamiento del hipertiroidismo mediante cirugía o con yodo radioactivo, que en algunos casos puede conducir a hipofunción tiroidea definitiva (Swearingen, 2008). El hipotiroidismo congénito suele deberse a defectos del desarrollo de glándula tiroidea (agenesia, hipoplasia, o, más frecuentemente, ectopia tiroidea) o alteraciones en la síntesis de las hormonas tiroideas (dishormonogénesis). También puede producir hipotiroidismo congénito un aporte deficiente de yodo durante la gestación, como puede ocurrir en zonas de endemia bociosa, que da lugar al llamado cretinismo endémico que se manifiesta por un déficit intelectual grave y signos clínicos y alteraciones bioquímicas relacionados con trastornos del sistema nervioso central y de la función tiroidea (Figura 6). El hipotiroidismo congénito tiene una elevada incidencia y una morbilidad grave además, la sintomatología puede pasar desapercibida en la mayoría de los casos y existe un tratamiento eficaz mediante la administración de hormonas (Netter y col., 2005) Los cambios bioquímicos más importantes que se producen en el hipotiroidismo primario son un descenso de la concentración de T4 en plasma que por retroalimentación negativa estimulará la secreción de tirotropina desde la hipófisis. No obstante, la T4 puede permanecer inicialmente dentro de los intervalos de referencia, por lo que la medida de utilidad para el diagnóstico de enfermedad en sus fases iniciales es la concentración de tirotropina en el plasma (Fox, 2003). En el hipotiroidismo secundario a lesión hipofisaria, a diferencia del primario, suele encontrarse un descenso de concentración de tirotropina en plasma, aunque no es infrecuente el hallazgo de concentraciones dentro de los límites de referencia, o incluso, ligeramente elevados. Lo anterior se debe a la secreción de formas moleculares anormalmente glicadas, desprovistas de potencia biológica pero que conservan su actividad inmunoreactiva (Velazco y Rodríguez, 1998). 25 Figura 6. Mecanismo a través del cual la deficiencia de yodo causa bocio. La cantidad insuficiente de yodo en la alimentación interfiere con el control de retroacción negativa de la secreción de TSH, lo que da lugar a la aparición de un bocio endémico (Tomado de Fox, 2003). Los síntomas psiquiátricos pueden ser la manifestación inicial de la enfermedad. En los primeros años de la existencia, el déficit de hormonas tiroideas condicionan retraso del desarrollo mental irreversible conocido como cretinismo. En los adultos, el cuadro clínico psiquiátrico se manifiesta como incapacidad psicomotora difusa con menoscabo de las funciones cognoscitivas, trastornos afectivos, cambios en el humor tales como apatía y desinterés en el ambiente. Se habla de severidad cuando aparece la psicosis, ya sea aguda o subaguda y con predominio de actitud paranoide. Si la evolución de la deficiencia hormonal es prolongada, pueden presentarse daño del intelecto y de la memoria. Se debe ser cauto con la administración de los psicofármacos. Es posible utilizar antidepresivos y antipsicóticos aunque las fenotiacinas están formalmente contraindicadas por el riesgo potencial de llevar al paciente a un estado de coma (Valdivieso y col., 2006). 26 Los objetivos del tratamiento nutricio para pacientes con hipotiroidismo son (Landa y col., 2006): 1. Controlar el aumento de peso que resulta por una reducción del gato energético basal del 15 a 40 %, sobre todo en los pacientes no tratados. El paciente se debe pesar frecuentemente para detectar pérdidas o retención de líquidos. 2. Corregir las razones para el desequilibrio, el cual puede deberse a la ingestión inadecuada de yodo, aumentando la ingestión de bociógenos o desequilibrio congénito. La Norma Oficial Mexicana (NOM-040-SSA1-1993) tiene como propósito establecer los límites de ion yodo que debe contener el producto denominado sal yodada, con el fin de prevenir las enfermedades provocadas a la población por deficiencia de dichos elementos. Un cuarto de cucharadita de sal yodada proporciona 95 microgramos de yodo. Una porción de 6 onzas (170 g) de pescado de mar suministra 650 microgramos de yodo. La mayoría de las personas pueden satisfacer las recomendaciones diarias consumiendo mariscos, sal yodada y plantas cultivadas en suelos ricos en yodo (Mason, 2007). 3. Revertir las anemias por deficiencia de vitamina B12 o hierro, si están presentes. 4. Mejorar los niveles de energía, reducir la fatiga. Las recomendaciones dietéticas para el paciente con hipotiroidismo son: 1. Dieta hipoenergética equilibrada. Consumir la energía necesaria deacuerdo a la edad, estatura, peso y actividad física del paciente (Landa y col., 2006). 2. Suplementar Zn y Cu. Para un adecuado funcionamiento de la tiroides (Mahan y Arlin, 1995). 3. Consumir un aporte adecuado de fibra 25 a 35g al día (Mahan y Arlin, 1995). (Anexo 1). 27 4. Aumentar el consumo de alimentos con alto contenido en yodo (Cuadro 6) (Mahan y Arlin, 1995). 5. Consumir alimentos que estimulen la absorción de yodo por la tiroides (Mahan y Arlin, 1995). Pescados y productos del mar. La Norma Oficial Mexicana (NOM-040-SSA1-1993) tiene como propósito establecer los límites de ion yodo que debe contener el producto denominado sal yodada, con el fin de prevenir las enfermedades provocadas a la población por deficiencia de dichos elementos. Un cuarto de cucharadita de sal yodada proporciona 95 microgramos de yodo. Una porción de 6 onzas (170 g) de pescado de mar suministra 650 microgramos de yodo. La mayoría de las personas pueden satisfacer las recomendaciones diarias consumiendo mariscos, sal yodada y plantas cultivadas en suelos ricos en yodo (Mason, 2007). Cuadro 6. Contenido de yodo en alimentos de consumo común (Tomado de Martínez y col., 2005). Alimento Yodo (µg/100) Alimento Yodo (µg/100) Leche de vaca 3.3 Arroz 2.0 Queso procesado 5.1 Harina de Trigo 5.8 Huevo de gallina 9.5 Zanahoria 1.6 Carne de res 5.4 Manzana 820 Carne de puerco 4.5 Naranja 851 Atún en aceite 149 Plátano 2.0 Maíz 2.6 Sandía 10 6. Moderar el consumo de alimentos bociógenos ya que pueden bloquear la captación de yodo por las células corporales. Si se consumen, se recomienda que sean cocidos, ya que se inactivan por el calor (Degroot y col., 1996). Col, nabos, cacahuates, coliflor, brócoli, camote, la semilla de colza (que se usa para hacer aceite de canola) (Degroot y col., 1996). 28 7. Disminuir el consumo de los alimentos que aumentan la excreción fecal de T4. Harina de soya, nueces y sus aceites, aceite de girasol, cacahuate y sus aceites (Degroot y col., 1996). 8. Someterse al tratamiento que el médico le recete al paciente será suficiente para normalizar el ritmo metabólico de la persona. Los cambios en la dieta del paciente con hipotiroidismo son sólo un complemento del tratamiento y no pueden sustituir a los medicamentos (Landa y col., 2006). 1.6 Recomendaciones generales adicionales para pacientes con hipertiroidismo e hipotiroidismo (Mataix y Vázquez, 2009): Motivar al paciente a que tome comidas a su debido tiempo de una manera tranquila y placentera. Evitar el uso de alcohol el cual puede causar hipoglucemia. Para evitar la obesidad ajustar la dieta del paciente según sus condiciones personales. Estar alerta de hiperglucemia después de las comidas ricas en hidratos de carbono. Comida en pequeñas cantidades (frutas, ensaladas, galletas) podrían ser necesarias. Excluir estimulantes (cafeína) 29 Capitulo 2: Paratiroides 2.1 Definición de la glándula paratiroides El ser humano posee cuatro glándulas paratiroides situadas inmediatamente por detrás de la glándula tiroides, una detrás de cada uno de los polos superiores e inferiores del órgano. Cada glándula mide unos seis milímetros de longitud, tres milímetros de ancho y dos milímetros de espesor, tiene un aspecto macroscópico de grasa parda oscura. Las glándulas paratiroides son difíciles de localizar durante las intervenciones de tiroides debido a que con frecuencia parecen sólo un lobulillo más de la tiroides. La extirpación de la mitad del tejido paratiroideo suele causar pocas alteraciones fisiológicas sin embargo, si se extirpan tres de las cuatro glándulas paratiroideas normales se producirá un hipoparatiroidismo transitorio. No obstante, basta conservar una pequeña cantidad de tejido paratiroideo, porque normalmente es capaz de hipertrofiarse de forma satisfactoria para realizar todas las funciones de las glándulas. Las glándulas paratiroides secretan la hormona paratiroidea (PTH) y es la hormona más importante que regula la concentración de calcio en la sangre ya que induce su incremento debido a sus efectos sobre el hueso, el riñón y el intestino (Guyton y Hall, 2006). 2.2 Fisiología de la hormona paratiroidea La glándula paratiroides del ser humano adulto (Figura 7) está compuesta sobre todo por células principales y contiene un moderado número de células oxífilas que tienen citoplasma granular y acidófilo (Dorantes y col., 2008).Sin embargo, no existen en muchos animales y en los seres humanos jóvenes. Se cree que las células principales secretan la mayoría, si no toda, la PTH. No está clara la función de las células oxífilas; se cree que son células principales modificadas o vacías, que ya no secretan la hormona (Dorantes y col., 2008). La función principal de la PTH es mantener la concentración de calcio en el líquido extracelular dentro del estrecho margen normal. La hormona actúa directamente sobre el hueso y el riñón e indirectamente sobre el intestino a través de sus efectos en la síntesis de 1,25-dihidroxivitamina D para elevar las 30 concentraciones de calcio en el suero y a su vez, la producción de PTH está regulada estrictamente por la concentración sérica del calcio ionizado. Este sistema de retroalimentación es el mecanismo homeostático esencial que mantiene el calcio del líquido extracelular. Toda tendencia hacia la hipocalcemia, como la que podrían inducir las dietas pobres en calcio, queda contrarrestada por una mayor secreción de PTH. Esta a su vez: 1) aumenta la disolución del mineral óseo, lo que va seguido de mayor flujo de calcio desde el hueso a la sangre; 2) disminuye la eliminación renal del calcio, con lo que vuelve al líquido extracelular mayor proporción del calcio filtrado en el glomérulo, y 3) aumenta la eficiencia de la absorción del calcio en el intestino al estimular la producción de 1,25-dihidroxivitamina D (Jameson y Weetman, 2006). Figura 7. Las cuatro glándulas paratiroides se localizan por detrás de la glándula tiroides. Casi toda la PTH se sintetiza y se secreta en las células principales. La función de las células oxífilas se desconoce, pero pueden ser células principales modificadas o vacías que han dejado de secretar PTH (Tomado de Guyton y Hall, 2006). La regulación inmediata de la calcemia se debe a los efectos de la hormona sobre el hueso y, en menor grado, sobre la eliminación renal (Figura 8). Por otro lado, probablemente el calcio mantenga un estado de equilibrio constante gracias a los efectos de 1,25-dihidroxivitamina D sobre la absorción intestinal. Las acciones renales de la hormona se ejercen a múltiples niveles, como la 31 inhibición del transporte de fosfato (túbulo proximal), el incremento de la reabsorción de calcio (túbulo distal) y la estimulación de la 25(OH) D-1 hidroxilasa. Cada día se intercambian hasta 12 mmol (500 mg) de calcio entre el líquido extracelular y el hueso (una gran cantidad en relación con la reserva de calcio en el líquido extracelular) y el principal efecto de la PTH incide en este intercambio (Jameson y Weetman, 2006). Figura 8. Acciones de la hormona paratiroidea y control de su secreción. El incremento de la concentración de hormona paratiroidea da lugar a la liberación de calcio por parte de los huesos y a la retención de calcio por parte de los riñones, con disminución de su concentración en orina. El aumento de calcio en la sangre puede ejercer una inhibición por retroacción negativa sobre la secreción de la hormona paratiroidea ( Tomado de Fox, 2003). La hormona paratiroidea parece tener dos efectos en hueso, ambos destinados a favorecer la resorción del calcio y fosfato. Uno es una fase rápida que se inicia en minutos y aumenta progresivamente durante varias horas. Esta fase es el resultado de activación de las células óseas ya existentes (sobre todo de los osteocitos) para provocar la resorción de calcio y de fosfato. La segunda fase es más lenta y requiere para su desarrollo pleno varios días ó incluso semanas; es el resultado de la proliferación de los osteoclastos seguida de un 32 gran incremento de la resorción osteoclásica del propio hueso y no sólo de las sales fosfato cálcico que contiene (Fox, 2003). 2.3 Relación del metabolismo de calcio, fósforo, calcitonina y vitamina D con la glándula paratiroides. 2.3.1 Metabolismo de calcio y fósforo El metabolismo del calcio está regulado por tres hormonas principales. El 1,25dihidroxicolecalciferol, es una hormona esteroidea formada a partir de la vitamina D por hidroxilaciones sucesivas en el hígado y riñones. Su principal acción es aumentar la absorción del calcio en el intestino. La PTH se produce en las glándulas paratiroides, su acción principal es movilizar el calcio del hueso y aumentar la excreción urinaria del fosfato. La calcitonina se produce principalmente en las células de la glándula tiroides, inhibe la reabsorción del hueso. Es probable que las tres hormonas operen en conjunto para mantener la constancia de la concentración del calcio en los líquidos corporales. Una cuarta hormona local, la proteína relacionada con la PTH (PTHrP), actúa sobre uno de los receptores para PTH y es importante en el desarrollo intrauterino del esqueleto. Los glucocorticoides, la hormona del crecimiento, los estrógenos y diversos factores de crecimiento también influyen en el metabolismo del calcio (Figura 9) (Gangog, 2004). La mayor parte del fósforo del organismo (unos 600 g) se encuentra como fosfato inorgánico. El 70% del fosfato en plasma y la mayoría del fosfato intracelular se encuentra como fosfato orgánico. Constituye, junto con el calcio, la fase mineral del hueso, representando el 85% del total del fósforo del organismo. Un 10% del fosfato en plasma circula unido a proteínas siendo, por tanto, la mayoría ultrafiltrable. La diferencia de concentración entre el fosfato intracelular y extracelular es de unas dos veces, por ello no es necesario un mecanismo de regulación tan fino como en el caso del calcio (Gómez y col., 2005). 33 Figura 9. Metabolismo del calcio (Tomado de Marx, 2000). 2.3.2 Efecto de la calcitonina en la regulación de calcio en sangre La calcitonina es una hormona peptídica secretada por la glándula tiroides que tiende a reducir las concentraciones plasmáticas de calcio y, en general, sus efectos se oponen a los de la PTH. No obstante, desde el punto de vista cuantitativo, el papel que desempeña la calcitonina es mucho menor que el de la PTH en lo relativo a la regulación de la concentración de los iones de calcio. La síntesis y la secreción de calcitonina tiene lugar en las células parafoliculares o células C, situadas en el líquido intersticial entre los folículos de la glándula tiroides. Estas células constituyen sólo alrededor de 0.1% de la glándula tiroides humana. La calcitonina es un polipéptido grande, de un peso molecular aproximado de 3400 y una cadena de 32 aminoácidos (Guyton y Hall, 2006). El estímulo principal para la secreción de calcitonina es el incremento de la concentración plasmática de calcio iónico. Este efecto es contrario al que 34 afecta la secreción de la PTH, que aumenta cuando la concentración del calcio disminuye (Guyton y Hall, 2006). La calcitonina reduce la concentración plasmática de calcio: inhibiendo la actividad de los osteoclastos y, de este modo, reduciendo la resorción ósea y estimulando la excreción urinaria de calcio y fosfato a través de la inhibición de su reabsorción en los riñones (Figura 10) (Fox, 2003). La función clásica de la hormona de vitamina D es la de elevar los niveles de calcio y fósforo en sangre que se requieren para la mineralización del hueso y además para prevenir el raquitismo y la osteomalacia. Esta hormona lleva a cabo su acción en el intestino, huesos y riñón. Cuando el calcio y el fosfato son muy bajos en la dieta, el efecto de la vitamina D en el riñón y en el hueso es retenerlos para realizar la mineralización del hueso. Si el calcio y el fosfato son adecuados en la dieta, el papel de la vitamina D en el intestino domina ya que se suspende la secreción de la PTH. Esta supresión se da gracias a los niveles de calcio en sangre normales que actúan en el receptor de calcio en la glándula paratiroidea. La ausencia de PTH apaga la resorción del hueso y el calcio del hueso se mantiene. En este caso, el calcio se utiliza para las funciones neuromusculares y de mineralización (Plum y De Luca, 2009). Figura 10. Control por retroinhibición de la secreción de calcitonina. La acción de calcitonina antagoniza a la hormona paratiroidea (Tomado de Fox, 2003). 35 2.3.3 Regulación del metabolismo de la vitamina D Los mecanismos por los cuales la vitamina D actúa para conservar concentraciones plasmáticas normales de Calcio y fosfato constan de: facilitación de su absorción por el intestino delgado, interacción con la hormona paratiroidea para aumentar su movilización desde los huesos, y decremento de su excreción por los riñones (Plum y De Luca, 2009). El propósito de 1,25-(OH)2D3 y PTH, cuando existe una dieta baja en calcio y/o hipocalemia, es la de proveer calcio a la sangre para la función neuromuscular (Figura 11) (Plum y De Luca, 2009). Figura 11. Vías de 1,25-(OH)2D3, Hormona paratiroidea (PTH) y calcitonina (CT) mediante las que se alteran los niveles de calcio en sangre (Modificado de Plum y De Luca, 2009). La síntesis de vitamina D en la piel y su metabolismo en 1,25-(OH)2D3 están cuidadosamente controlados en el cuerpo. La luz del sol regula la producción total de de pre-vitamina D3 y vitamina D3 en la piel. Una vez que la vitamina D entra en la circulación puede almacenarse en la grasa para el uso posterior o puede metabolizarse en el hígado en 1,25-dihidroxivitamina D. La etapa de hidroxilación se regula mediante retroalimentación. El paso fundamental en el metabolismo de vitamina D es la producción de 1,25-dihidroxivitamina D por el riñón (Figura 12). Durante los períodos de privación del calcio, declinan las concentraciones de calcio ionizado circulante. Las glándulas paratiroideas detectan de inmediato esta disminución e incrementan la producción y 36 secreción de PTH que aumenta la reabsorción tubular de calcio y la producción renal de 1,25-dihidroxivitamina D.la cual se desplaza al intestino delgado donde aumenta la eficiencia de la absorción del calcio intestinal. 1,25-dihidroxivitamina D junto con la PTH, actúan de manera sinérgica sobre los osteoclastos, que a su vez movilizan las reservas de calcio en el hueso (Holick, 2002). Figura 12. Vías de la producción de Vitamina D (Tomado de Holick, 2002). 2.4 Pruebas de funcionamiento paratiroideo y valores séricos normales La liberación de la PTH es controlada por el nivel de calcio en la sangre. Los niveles bajos de calcio en la sangre provocan un aumento en la liberación de esta hormona, mientras que los niveles altos de calcio en la sangre inhiben su liberación. Se puede hacer un examen de laboratorio para medir la cantidad de PTH en la sangre. Los valores normales son de 10 a 55 pg por mililitro. Los rangos de los valores normales pueden variar ligeramente entre diferentes 37 laboratorios. En cuanto a los valores de referencia para el calcio sérico, el límite superior se considera 10,5 mg/dL (2.6 mmol/L) (Bilezikian y col., 2002). 2.5 Patologías más frecuentes de la glándula paratiroides Si las glándulas paratiroideas producen cantidades excesivas o muy bajas de hormona, alteran el equilibrio. Si segregan demasiada PTH, el cuadro se denomina hiperparatiroidismo y el nivel de calcio en la sangre aumenta. Si no se produce suficiente cantidad de PTH, el cuadro se denomina hipoparatiroidismo, la sangre tendrá muy poco calcio y una excesiva cantidad de fósforo (Figura 13). Hipoparatiroidismo 1° Seudo Valores fisiológicos Hiperparatiroidismo 1° y 3° 2° Calcemia Calciuria Fosfatemia Fosfaturia Figura 13. Manifestaciones bioquímicas en hiperparatiroidismo e hipoparatiroidismo (Mataix y Riobó, 2009). 38 2.5.1 Hiperparatiroidismo El hiperparatiroidismo es la elevación de la PTH, el cual se puede clasificar en primario, secundario y terciario, basándose en la causa de elevación que causa la hipercalcemia (Morales y Bahena, 2003): a) Hiperparatiroidismo primario. Esta entidad es definida como la presencia de hipercalcemia con la elevación de la PTH. b) Hiperparatiroidismo secundario. Es la estimulación crónica de las glándulas paratiroideas causadas por calcio sérico bajo. La condición más frecuente es la insuficiencia renal crónica, otras causas son acidosis tubular renal, síndrome de malabsorción, deficiencia de vitamina D y calcio, resistencia del tejido a vitamina D e hipomagnesemia grave. Hay un aumento adaptativo del tamaño de la glándula. c) Hiperparatiroidismo terciario. A pesar de corregir el estímulo subyacente las glándulas paratiroides continúan secretando niveles altos de la PTH; esto resulta de hiperparatiroidismo secundario a largo plazo o durante largo tiempo por falla renal. Esto es, un hiperparatiroidismo autónomo en pacientes con hiperparatiroidismo secundario. Manifestaciones clínicas: Las manifestaciones clínicas del hiperparatiroidismo se deben generalmente a hipercalcemia crónica, siendo raras las crisis de hipercalcemia aguda. Debe recalcarse que con frecuencia el hiperparatiroidismo primario se presenta en forma silenciosa y que solo se puede detectar por programas de tamiz selectivo mediante la presencia de hipercalcemia (Netter y col., 2005). Entre las manifestaciones se encuentran (Dorantes, 2008): 1. Manifestaciones inespecíficas: los síntomas más frecuentes son poliuria, polidipsia, astenia, anorexia y pérdida de peso. El dolor abdominal con náuseas y vómitos frecuentes a veces es el síntoma principal. También puede presentarse retardo mental, convulsiones y ceguera, otras manifestaciones son labilidad emocional, disminución del rendimiento intelectual y depresión. 39 2. Manifestaciones esqueléticas: debidas a la actividad osteolítica de la hormona paratiroidea sobre el hueso; se refieren en ocasiones al dolor de espalda o extremidades (o articulaciones), trastornos de la marcha, genu valgo, facturas y tumores. La estatura puede estar disminuida por compresión vertebral. 3. Manifestaciones renales: a nivel renal la hipercalcemia prolongada puede originar nefrocalcinosis con disminución progresiva de la función renal y litiasis renal con cólicos nefríticos y hematuria (50 a 70% de los pacientes). Un porcentaje importante de litiasis en niños son consecuencia del hiperparatiroidismo, las manifestaciones esqueléticas junto con las renales son las determinantes de la gravedad del proceso. 4. Manifestaciones digestivas: a veces hay dolor abdominal importante debido si se padece pancreatitis aguda. 5. Manifestaciones neuropsíquicas: bradipsiquia, tendencia a la depresión, labilidad emocional, normalmente benignas, poco frecuentes en niños. Se han relacionado con la alteración de metabolismo de monoaminas en el sistema nervioso central por la hipercalemia y desaparecen al corregirse ésta. 6. Hipertensión arterial: se debe quizás a la acción calcio sobre las paredes vasculares y es proporcional a la intensidad de hipercalcemia, remitiendo, si es moderada, al hacerlo ésta. Se presenta en 75% de los adultos, y en niños y adultos es menos frecuente. 7. Crisis hiperparatiroidea: Consiste en grandes elevaciones de la calcemia (más de 15 mg/dL), que se acompañan de oliguria, elevación de la creatinina e importante afectación neurológica central (estupor y coma). Manifestaciones Bioquímicas (Mawer y Davies, 2001): 1. Hipercalcemia, por aumento de la resorción ósea y reabsorción tubular renal, así como indirectamente a por aumento de absorción un mayor nivel de la intestinal hormona D3. debido En el hiperparatiroidismo secundario la calcemia puede ser normal o baja, pues la hiperfunción glandular sólo intenta compensar una calcemia normalmente baja. 40 2. Hipercalciuria, consecuencia de la gran hipercalcemia, no compensada por la mayor reabsorción renal del calcio. 3. Hipofosfatemia, por el efecto inhibidor de la PTH sobre la reabsorción renal del fosfato. A veces el fosfato sanguíneo aumenta por la incapacidad renal de excretar con suficiente rapidez todo el fosfato que se encuentra en el plasma. Esto explica la formación de fosfato monocálcico, que se deposita en diversos tejidos blandos, en túbulos renales, alvéolos pulmonares, glándula tiroides, paredes arteriales entre otros, que puede conducir finalmente hasta la muerte. 4. Hiperfosfaturia, debido a que el efecto renal sobre el fosfato es muy fuerte. 5. PTH plasmática aumentada. Manifestaciones Psiquiátricas (Valdivieso y col., 2006). El cuadro clínico mental dependerá del grado de hipercalemia. Presente en más de las dos terceras partes de los casos es, en muchas ocasiones el único síntoma, por lo que el diagnóstico del hiperparatiroidismo suele escaparse con frecuencia. Estos pacientes cursan con depresión, debilidad, irritabilidad y gran inestabilidad emocional. Durante las crisis paratiroideas pueden producirse reacciones psicóticas agudas, con alucinaciones visuales y auditivas, paranoia, agresividad y delirio. Se recomienda investigar la existencia de un hiperparatiroidismo en los casos de trastorno afectivo crónico, deterioro mental progresivo aunado a cambios de la personalidad inespecíficos, sobretodo en personas que carecen de antecedentes personales o familiares de estos tipos de alteraciones del humor. Con la normalización de los valores del calcio se obtiene por lo general la remisión completa de esta psicopatología La paratiroidectomía es la solución quirúrgica a la hiperfuncionalidad paratiroidea más usual, la cual debe ser completada con las medidas médicas relacionadas con la etiología de la enfermedad (Mataix y Riobó, 2009). 41 Tratamiento nutrício: En cuanto al tratamiento nutricional lo esencial es la dietoterapia con calcio vía oral o intravenosa con el fin de intentar mejorar la mineralización ósea (Mataix y Riobó, 2009). Algunos estudios han comprobado la eficacia de la dieta hipoproteínica (Anexo 3) complementada con cetoácidos para reducir los síntomas urémicos y mantener parámetros nutricionales y un balance nitrogenado adecuado. Además, mejora el hiperparatiroidismo al aumentar los niveles séricos de calcio y de 1,25- dihidroxicolecalciferol y al reducir los niveles séricos de fósforo y fosfatasa alcalina. Asimismo, las dietas hipoproteínicas mejoran la acidosis metabólica pues generan menos iones H+. Al corregir la acidosis metabólica se produce una disminución del catabolismo proteinico además de que mejora el balance nitrogenado. Además, las dietas complementadas con cetoácidos parecen aumentar la tolerancia a la glucosa. Es importante mencionar que el suplemento de aminoácidos esenciales o cetoácidos sólo da resultado si el paciente cumple con la dieta de restricción protéica (0.3g/Kg/día). En caso de una ingesta protéica mayor, los suplementos simplemente son oxidados (Riella y Martinis, 2004) 2.5.2 Hipoparatiroidismo El hipoparatiroidismo se refiere a un espectro de trastornos en los que hay disminución de la acción de la hormona paratiroidea, ya sea por la alteración de la síntesis de paratohormona o de su acción periférica, dando lugar a hipocalcemia e hiperfosfatemia (Dorantes, 2008). Se pueden distinguir dos formas fisiopatológicas, aunque una de ellas es un pseudohipoparatiroidismo (Mataix y Riobó, 2009). a) Hipoparatiroidismo primario. Se produce por una extirpación involuntaria de la glándula paratiroides en tiroidectomía, paratiroidectomía parcial o en determinadas manipulaciones quirúrgicas en el cuello. La falta de la hormona PTH se manifiesta en los tres órganos sobre los que actúa, hueso, riñón e indirectamente sobre el intestino. b) Pseudohipoparatiroidismo. No existe una alteración paratiroidea, sino una falta de respuesta efectora de los órganos “diana” de la PTH, hueso y riñón. 42 Sin embargo, sí se produce el efecto indirecto sobre el intestino, a través de la hormona D3, favoreciendo la absorción del calcio. Al contrario que en el caso anterior, existe una elevación de la secreción de la hormona, debido a la hipocalcemia existente (Mataix y Riobó, 2009). El hipoparatiroidismo causa hipocalcemia debido a que la secreción de PTH es inadecuada para movilizar el calcio del hueso, reabsorber el calcio en el nefrón distal y estimular la actividad renal de la 1a-hidroxilasa. Como resultado, se produce una deficiencia de la 1,25-dihidroxivitamina D (1,25[OH]2 vitamina D) generada por la eficiente absorción intestinal de calcio (Mawer y Davies, 2001). Según Shobak (2008) el hipoparatiroidismo adquirido es el resultado de la extirpación inadvertida de las glándulas paratiroides o el daño irreversible de las mismas. Lo más común es la interrupción del aporte sanguíneo durante la tiroidectomía, la paratiroidectomía o la disección radical del cuello. Las definiciones de hipoparatiroidismo posquirúrgico permanente varían, pero la más aceptada es la insuficiencia de PTH para mantener la normocalcemia durante los 6 meses posteriores a la cirugía. Se calcula que el hipoparatiroidismo ocurre en el 0.5-6.6% de las tiroidectomías, siendo esta tasa aún más elevada en algunos casos pero en algunos centros especializados no supera el 1.6%. Depende de la experiencia del cirujano, la extensión de la resección tiroidea y de los ganglios en el cáncer, pero la enfermedad tiroidea subyacente, con bocio subesternal, cáncer o enfermedad de Graves, aumenta el riesgo. El riesgo también aumenta si una o más glándulas paratiroides no son identificadas durante la operación y si el procedimiento quirúrgico es una re-operación. El hipoparatiroidismo también puede estar causado por la acumulación de hierro en las glándulas paratiroides (hemocromatosis o talasemia tratada con transfusiones) o cobre (enfermedad de Wilson) o, en casos raros, por el tratamiento con I131 por una enfermedad tiroidea o la infiltración metastásica tumoral de las glándulas paratiroideas (Shobak, 2008). El magnesio es esencial para la secreción de PTH y la activación del receptor de PTH. En la hipomagnesemia, los niveles de PTH son inapropiadamente 43 bajos o están en el límite inferior normal, en presencia de hipocalcemia leve, porque la paratiroides es incapaz de secretar hormona suficiente y las respuestas del riñón y del esqueleto a la PTH están atenuadas. En casos raros, cuando se administra magnesio por vía parenteral (por ej., el tratamiento tocolítico) o se acumula en presencia de insuficiencia renal, aumentando sus niveles, la secreción de PTH es inhibida. El magnesio, como el calcio, puede activar los receptores extracelulares de calcio y suprimir la liberación de PTH. Una vez que se corrige la magnesemia se reanuda la capacidad de secreción de la PTH y la respuesta correspondiente (Mawer y Davies, 2001). Los trastornos genéticos también deben ser considerados como causas posibles de hipocalcemia: síndrome de DiGeorge o velocardiofacial; el hipoparatiroidismo familiar por disgenesia de las glándulas paratiroides; el síndrome hipoparatiroidismo, retardo y dimorfismo, trastornos por defectos genéticos mitocondriales (Shobak, 2008). A veces se emplea PTH para tratar el hipoparatiroidismo. Sin embargo, debido al costo de esta hormona y a que su efecto dura sólo cuando mucho unas pocas horas y a que la tendencia del organismo a desarrollar anticuerpos contra ella reduce poco a poco su eficacia, el tratamiento del hipoparatiroidismo con PTH es raro en la actualidad. En la mayoría de los pacientes con hipoparatiroidismo, la administración de vitamina D, de hasta 100,000 unidades diarias, junto con la ingestión de 1 a 2 gramos de calcio, bastará para mantener la concentración de calcio iónico en los límites normales. A veces puede ser necesario administrar 1,25-dihidroxicolecalciferol en vez de la forma no activada de la vitamina D debido a que su potencia y rapidez de acción son mucho mayores. Esto puede causar también efectos adversos porque a veces es difícil evitar la hiperactividad de la forma activa de vitamina D (Guyton y Hall, 2006). Manifestaciones clínicas (Dorantes, 2008): 1. Hiperparatiroidismo secundario. 2. Aumento de la excitabilidad neuromuscular, al disminuir el potencial umbral que se aproxima al potencial de reposo, pudiendo provocar 44 tetania en donde hay hiperexcitabilidad sensitiva y motora y asimismo existe la hiperexcitabilidad vegetativa. 3. Manifestaciones sensitivas. Parestesias (hormigueo) en boca, manos y pies. 4. Manifestaciones motoras. Pueden presentarse crisis epilépticas. La tetania puede afectar los músculos de la laringe y es precisamente el espasmo tetánico lo que puede conducir a la muerte. 5. Manifestaciones vegetativas. Pueden ser disnea asmática por espasmo bronquial, dolor intestinal por contracción violenta de la fibra muscular lisa. 6. Afectación cardiaca. 7. Calcificaciones. La alteración de la relación Ca/P puede explicar ciertas calcificaciones como ocurre en núcleos encefálicos, entre otros. Manifestaciones Bioquímicas (Mawer y Davies, 2001): 1. Hipocalcemia, al disminuir la resorción ósea, la reabsorción tubular y la absorción intestinal, esto último indirectamente por la deficiencia de la hormona D3. 2. Hipocalciuria, por hipocalemia no compensada por la disminución de absorción tubular renal (que facilitaría la excreción del calcio). 3. Hiperfosfatemia al aumentar la reabsorción tubular renal del fósforo, lo que conduce a una fosfaturia baja. 4. PTH plasmática disminuida, y al contrario en pseudohipoparatiroidismo, en que está aumentada). Manifestaciones Psiquiátricas: El cuadro clínico está en relación directa con la velocidad de instalación de la hipocalemia. En los casos agudos, los síntomas psíquicos más comunes son el delirio y la psicosis. En la hipocalcemia crónica pueden verse con mayor frecuencia ansiedad, irritabilidad, labilidad emocional, depresión, alteración de la memoria y de la concentración, deterioro cognitivo y en ocasiones, retraso mental. El compromiso intelectual está presente en la mitad de los casos de 45 hipoparatiroidismo. La normalización de la calcemia mejora con rapidez la sintomatología psiquiátrica (Valdivieso y col., 2006). Manifestaciones óseas: El hiperparatiroidismo se asocia de forma estadísticamente significativa a una serie de enfermedades cuya relación causa-efecto con el exceso de PTH o la hipercalemia no está aún bien aclarada. Entre ellas caben destacar: las enfermedades óseas. Aunque en el hiperparatitroidismo leve se puede depositar hueso nuevo con la suficiente rapidez como para compensar el aumento de la resorción osteoclástica del hueso, en el hiperparatiroidismo grave la actividad osteoclástica supera pronto al depósito osteoblástico y el hueso puede ser devorado casi por completo. De hecho, la razón por la que los pacientes con hiperparatiroidismo consultan al médico, es con frecuencia, una fractura ósea; las radiografías óseas muestran una descalcificación extensa, los huesos debilitados pueden sufrir múltiples fracturas (Stiges-Serra, 2010). La actividad osteoblástica de los huesos también aumenta mucho en un intento vano de formar suficiente hueso nuevo para compensar la destrucción del hueso viejo asociada a la actividad osteoclástica. Cuando los osteoblastos se activan, secretan grandes cantidades de fosfatasa alcalina, por tanto, uno de los hallazgos diagnósticos importantes del hiperparatiroidismo es una concentración elevada de fosfatasa alcalina (Guyton y Hall, 2006). Tratamiento nutrício: El objetivo del tratamiento es restaurar el equilibrio del calcio y otros nutrimentos inorgánicos en el organismo. Usualmente, el suministro de suplementos de carbonato de calcio oral y vitamina D es una terapia de por vida y se miden regularmente los niveles en la sangre para asegurarse de que la dosis sea correcta. Se recomienda una dieta alta en calcio (Anexo 1) y baja en fósforo (Mataix y Riobó, 2009). El tratamiento crónico del hipoparatiroidismo pretende mantener las cifras de calcio sérico en el rango bajo de la normalidad (entre 8,5 y 9 mg/dL) ya que calcemias más altas en ausencia de PTH pueden provocar hipercalciuria 46 debido a la baja reabsorción tubular del calcio. Para lograr este objetivo se administra calcio oral 1-2 g/día y vitamina D en la dosis eficaz más baja (siempre se corre el riesgo de provocar una intoxicación). Conviene evitar las sales de fosfato cálcico, la leche y sus derivados, ya que por su alto contenido en fosfato empeoran la hiperfosfatemia. A medida que se normalizan las cifras de calcio, el túbulo renal es capaz de excretar más fosfato. La hipocalcemia puede ocasionalmente producir malabsorción de grasa o déficit de vitamina B12. Durante la hipocalemia se puede ver la elevación marcada de enzimas séricas, transaminasas y LDH, que desaparecen al corregir la hipocalcemia (Freijanes y Amado, 1995). 2.5.3 Raquitismo y osteomalacia El raquitismo en niños y la osteomalacia en adultos son dos enfermedades óseas caracterizadas por una descalcificación del hueso que conduce a distintas anormalidades del mismo (Anderson, 2001). El raquitismo es un desorden infantil caracterizado por un fallo del crecimiento del cartílago que no permite una adecuada maduración y mineralización. El crecimiento se detiene y ocurren diferentes deformidades en las placas de crecimiento. La osteomalacia es el desorden correspondiente en adultos en la cual fallos en la matriz ósea depositada impiden la adecuada mineralización (Favus, 1996). En ambas condiciones, la formación de nueva matriz ósea se enlentece y aún es más acusado este enlentecimiento en el proceso de mineralización. De esta manera, la relación mineral/matriz disminuye, llegando en ocasiones a un valor tan bajo que los huesos pierden su dureza y se deforman ostensiblemente (Díaz y Riobó, 2006). Las causas del raquitismo y la osteomalacia pueden ser primarias o secundarias. Dentro de las primarias se encuentra una deficiencia en la ingestión de vitamina D y/o una mala absorción de la misma (Anderson, 2001). La consecuencia de los bajos niveles de vitamina D y en términos más generales de la hormona D, conduce a baja concentración plasmática del calcio, esto provoca el aumento de PTH. Dado que uno de los efectos de esta hormona es aumentar la depuración renal del fosfato, los niveles del mismo, ya 47 de por si disminuidos, aún se reducen más, lo que conduce a una deficiencia severa de fosfato, en principio localizada en la vecindad de osteoblastos y condrocitos y después en otros tejidos, produciendo por ejemplo debilidad muscular, sensibilidad excesiva y dolor (Díaz y Riobó, 2006). Para prevenir el raquitismo y la deficiencia subclínica de vitamina D, el Comité de Nutrición de la AAP (Academia Americana de Pediatría) apoya estas recomendaciones y aconseja la suplementación con 200 UI de vitamina D/día en los siguientes casos (Mataix y Monereo, 2009): 1. Lactantes alimentados con lactancia materna exclusiva. 2. Lactantes que ingieren menos de 500 mL de fórmula fortificada con vitamina D. 3. Niños y adolescentes que no tienen una exposición solar regular, no toman al menos 500 mL de leche fortificada o un suplemento vitamínico que contenga al menos 200 UI de vitamina D. Deficiencia primaria. El raquitismo de origen nutricional responde rápidamente a la administración de 2.000-4.000 UI de vitamina D al día, siendo conveniente un suplemento de calcio, para que haya el adecuado aporte que requiere una rápida mineralización (Riobó y Rapado, 2002). Deficiencia secundaria. Los individuos con osteomalacia secundaria a malabsorción requieren dosis elevadas de vitamina D bien vía oral ó parenteral, según la capacidad digestiva de absorción (Riobó y Rapado, 2002). Cuando el fallo es la biosíntesis renal de la vitamina D3, se debe administrar esta hormona (y no el precursor vitamínico o análogo) .El uso de protectores solares muy fuertes, la exposición limitada del cuerpo a la luz solar, los días de luz solar cortos y la contaminación son factores que disminuyen la formación de la vitamina D dentro del cuerpo. Las personas de edad avanzada y quienes evitan tomar leche tienen un mayor riesgo de padecer osteomalacia (Mawer y Davies, 2001). 48 2.5.4 Osteoporosis La osteoporosis (literalmente, hueso poroso) se define como una condición de fragilidad esquelética, debido a una disminuida masa ósea y un deterioro de la micro arquitectura con el consiguiente riesgo de fractura. El calcio y el fósforo son dos nutrimentos inorgánicos esenciales para la formación normal del hueso. A lo largo de la juventud, el cuerpo utiliza estos nutrimentos inorgánicos para producir huesos. Si uno no obtiene suficiente calcio o si el cuerpo no absorbe suficiente calcio de la dieta, se puede afectar la formación del hueso y los tejidos óseos (Sitges-Serra, 2010). Los objetivos de tratamiento nutrício son (Mataix y Monereo, 2009): 1. Reducir el riesgo de fractura 2. Mejorar la masa ósea y favorecer su retención 3. Aliviar los síntomas de fracturas y deformidades óseas en el caso de estar presentes. 4. Maximizar la funcionalidad física en aquellos casos en que se halle comprometida. Aspectos a considerar en la dieta (Mataix y Monereo, 2009): 1. Dieta normoenergética o hipoenergética en caso de obesidad o sobrepeso (Anexo 4). 2. Moderado contenido proteínico: 0,8 g a 1 g de proteína por kg de peso. 3. Contenido de calcio entre 1200 y 1500 mg/día: consumo diario de 3 a 4 raciones de lácteos (Anexo 1). 4. Aporte de vitamina D en meses de invierno y asegurar una exposición solar. 5. Correcto aporte de potasio, limitar a una ración diaria de alimentos proteínicos: Carnes (100 g), pescados (100-200 g) y huevos (dos unidades). 6. Moderado contenido de sal: Evitar la sal añadida en la mesa y el consumo habitual de sazonadores, embutidos, conservas, salsas comerciales y precocinados. 49 2.6 Alteraciones óseas debidas a deficiencias y excesos nutricionales diversos. Deficiencias (Anderson, 2001): a) Vitamina C. La vitamina C es esencial en la hidroxilación de residuos de prolina dando hidroxiprolina, que es fundamental para la formación del colágeno, Cuando hay deficiencia, la organización del colágeno en fibras no se produce y son evidentes las alteraciones óseas, como las que caracterizan el escorbuto en niños. En el caso de los adultos, la deficiencia vitamínica C no parece afectar al hueso. b) Vitamina B6. La Vitamina B6 o piiridoxal es la coenzima de la cistationasa, Su déficit conduce a homocisteinuria, en dónde se pueden observar alteraciones óseas como huesos osteoporóticos y otras anormalidades. c) Cobre. El cobre es cofactor de la lisil oxidasa que cataliza la formación de aldehídos de lisina, los cuales son importantes en la configuración de enlaces cruzados en la estructura del colágeno. Su deficiencia conduce también a enfermedad ósea. Esta situación suele presentarse en niños de bajo peso, con una ingestión insuficiente de cobre, bien a través de preparados lácteos o de la nutrición parenteral. d) Fosfato. Ya se ha comentado que su déficit se asocia a un aumento de la formación de hormona D3. e) Magnesio. La deficiencia de este mineral se produce sobretodo por malabsorción intestinal severa como enfermedad celiaca, fístulas o resección ileal, entre otros, especialmente en dietas altas en grasa o por pérdidas urinarias debido a alteraciones tubulares renales. El magnesio inicialmente impide la respuesta ósea a la PTH, lo que conduce a hipocalemia a pesar de los niveles elevados de la hormona, y posteriormente, a medida que la deficiencia progresa, la secreción de PTH se reduce. Excesos (Anderson, 2001): También se encuentran afectaciones óseas como consecuencia de excesivas ingestas de determinados nutrimentos. 50 a) Vitamina A. El exceso de vitamina A afecta a la función osteoblástica dando lugar a hiperostosis cervical en niños y osificación ectópica de ligamentos en adultos. Así, el uso continuado de derivados del ácido retinóico en enfermedades cutáneas como psoriasis e ictiosis conduce a calcificación de ligamentos, especialmente alrededor de la columna, donde existe neoformación ósea, provocando rigidez y movilidad disminuida. b) Aluminio. El aluminio en exceso conduce a osteodistrofia y así ocurre en diálisis el cual pasa al medio interno desde el líquido de diálisis. También puede ocurrir en individuos sometidos a nutrición parenteral prolongada. Por otra parte, es un componente importante en antiácidos (que a veces se usan además en enfermos renales para bloquear absorción de fosfato) y es ampliamente usado como utensilio. El aluminio se acumula en los lugares de mineralización en el proceso de remodelación ósea provocando así la correspondiente alteración ósea. c) Floruro. Un exceso de ingestión de floruro puede ocurrir en la fluorosis endémica o cuando se administra en el tratamiento de la osteoporosis. d) Cadmio. Un exceso de cadmio, como puede ocurrir en determinados casos de contaminación es también causa de alteraciones óseas. La razón de esta alteración es el daño renal a nivel del túbulo que produce el cadmio. 2.7 Valores nutrimentales de referencia de calcio y fósforo. Las recomendaciones de calcio y fosforo en diversas partes del mundo han ido aumentando desde lo establecido por la FAO en 1961 a la fecha (Bourges, 1976; Dietary Reference Intakes, 2002). Las recomendaciones para el calcio y el fósforo parten por un lado de observaciones realizadas en México para otros fines y por estudios de otras poblaciones y constituyen ingestiones diarias sugeridas (Cuadros 7 y 8) (Torún y col., 1994). 51 Cuadro 7. Ingestión diaria sugerida (IDS) de calcio por grupo de edad (Tomado de De Santiago y col., 2005). Grupos de Edad Basado en 0-6 meses Contenido de calcio en leche humana IDS (mg/día) 210 7-12 meses Leche humana + comida sólida 270 1-3 años Extrapolación de la retención máxima 500 de calcio edades de 4-8 años 4-8 años Retención máxima de calcio 800 9-13 años Retención máxima de calcio 1 300 14-18 años Retención máxima de calcio 1 300 19-30 años Retención máxima de calcio 1000 31-50 años Balance de calcio 1000 51-70 años Retención máxima de calcio 1 200 Más de 70 años Extrapolación de la retención máxima 1 200 de calcio en edades de 51-70 años Embarazadas y lactantes <= 18 años Masa mineral del hueso 1 300 19-50 años Masa mineral del hueso 1 300 Cuadro 8. Requerimiento nutrimental promedio (RNP), ingestión diaria recomendada (IDR) e ingestión diaria sugerida (IDS) de fosforo por grupos de edad (Tomado de De Santiago y col., 2005). Grupos de Edad 0-6 meses Indicador Contenido de fósforo en la leche humana 7-12 meses Leche humana + comida sólida RNP (mg/día) // IDR (mg/día) // IDS (mg/día) 100 // // 275 1-3 años Aproximación factorial 380 460 // 4-8 años Aproximación factorial 405 500 // 9-13 años Aproximación factorial 1055 1250 // 14-18 años Aproximación factorial 1055 1250 // 19-30 años Concentración de Fósforo inorgánico 580 700 // 31-50 años Concentración de Fósforo inorgánico 580 700 // 51-70 años 580 700 // Más de 70 años <= 18 años Extrapolación de concentración de Fósforo inorgánico en edades de 19 a 50 años Extrapolación de concentración de Fósforo inorgánico en edades de 19 a 50 años Aproximación factorial 580 700 // 1055 1250 // 19-50 años Concentración de Fósforo inorgánico 580 700 // 52 Capitulo 3: Páncreas 3.1 Definición de la glándula pancreática El páncreas es una glándula que tiene un componente endocrino y otro exocrino. La porción endocrina del páncreas está constituida por grupos celulares diseminados denominados islotes pancreáticos o islotes de Langerhans (Figura 14) (Fox, 2003). El páncreas humano pesa menos de 100 g y diariamente se secreta 1 litro (10 veces su masa) de jugo pancreático. Las secreciones exocrinas del páncreas son importantes en la digestión. El jugo pancreático consta de un componente acuoso, rico en bicarbonato, que ayuda a neutralizar el contenido duodenal y un componente enzimático, que contiene enzimas para la digestión de hidratos de carbono, proteínas y grasas. La secreción exocrina del páncreas está controlada por señales nerviosas y hormonales originadas sobre todo por la presencia de ácido y productos de digestión en el duodeno. (Steinberg, 1994). Figura 14. El páncreas y sus islotes (islotes de Langerhans). Las células alfa secretan glucagon y las células beta secretan insulina. El páncreas también es una glándula exocrina que produce jugo pancreático que alcanza el duodeno del intestino delgado a través del conducto pancreático (Modificado de Steinberg, 1994). 53 Los islotes de Langerhans del páncreas secretan cinco polipéptidos con actividad hormonal. Dos de estas hormonas, la insulina y el glucagon, tienen funciones importantes en la regulación del metabolismo energético. La tercera hormona, somatostatina, participa en la regulación de la secreción de las células de los islotes y la cuarta y la quinta son el polipéptido pancreático y la amilina, cuya función no se conoce. Es posible que las células de la mucosa del tubo digestivo también secreten glucagon, somatostatina y el polipéptido pancreático (Gangog, 2004). Los islotes de Langerhans son agrupaciones ovoideas de 76x175 µm de células dispersas en todo el páncreas, aunque son más abundantes en la cola que en el cuerpo y en la cabeza de éste. Constituyen alrededor de 2% del volumen granular, la porción exocrina ocupa el 80% y los conductos y vasos sanguíneos representan el resto. En los humanos hay 1 ó 2 millones de islotes, cada uno tiene una irrigación abundante y la sangre que sale a los islotes drena a la vena porta. (Gangog, 2004). Los islotes de Langerhans contienen tres tipos de células fundamentales, alfa, beta y delta que se diferencian entre sí por sus características morfológicas y de tinción (Guyton y Hall, 2006). Las células beta representan casi el 60% de la totalidad de las células de los islotes y se encuentran sobre todo en el centro de cada uno y secretan insulina y amilina, hormona que suele liberarse en paralelo con la insulina. Las células alfa que componen casi el 25 % del total secretan glucagon y las células delta, que representan el 10%, somatostatina. Además, existe por lo menos otro tipo de célula, la célula PP que secreta al polipéptido pancreático. Las relaciones íntimas entre los tipos celulares de los islotes de Langerhans facilitan la comunicación intercelular y el control directo de la secreción de algunas de las hormonas. Por ejemplo, la insulina inhibe la secreción del glucagon; la amilina inhibe la secreción de insulina y la somatostatina, la de la insulina y el glucagon (Guyton y Hall, 2006). 54 3.2 Fisiología de la insulina La insulina es una hormona polipeptídica producida por las células beta de los islotes de Langerhans. Es la hormona más importante que regula el uso de combustibles en los tejidos. Sus efectos metabólicos son anabólicos, favorecen la síntesis de glucógeno, triglicéridos y proteínas. La insulina tiene un peso molecular de 5,808 Da y se compone de 51 aminoácidos dispuestos en dos cadenas polipeptídicas, designadas A y B, que están unidas por dos puentes disulfuro y adicionalmente la cadena A contiene un puente disulfuro intramolecular (Figura 15) (Champe y col., 2005). Figura 15. Estructura de la insulina Humana (Modificado de Champe y col., 2005). Se sintetiza en las células beta con la maquinaria celular habitual para la síntesis de proteínas acopladas al retículo endoplásmico y se forma una preprohormona insulínica. Esta preprohormona inicial tiene un peso molecular aproximado de 11,500 Da, pero luego se desdobla en el retículo endoplásmico para formar proinsulina, con un peso molecular de 9,000. Casi toda la proinsulina sigue escindiéndose en el aparato de Golgi a insulina y fragmentos peptídicos antes de empacarse en los gránulos secretores. No obstante, casi una sexta parte del producto final secretado persiste en forma de proinsulina con poca actividad biológica (Champe y col., 2005). Las células beta de los islotes de Langerhans liberan la insulina en dos fases. La primera fase de la liberación de insulina se desencadena rápidamente en respuesta al aumento 55 de los niveles de glucosa en la sangre. La segunda fase produce una liberación sostenida y lenta de las recién formadas vesículas que se activan independientemente de la cantidad de azúcar en la sangre (Figura 16). En la primera fase, la liberación de la insulina ocurre de manera inmediata (Maitra y Abbas, 2005): Figura 16. Principal mecanismo de liberación de la insulina (Tomado de Maitra y Abbas, 2005). 1. La glucosa entra en las células beta a través del transportador de glucosa GLUT2. 2. La glucosa pasa a la glucólisis y el ciclo respiratorio, donde se producen por oxidación varias moléculas de ATP. 3. Los canales de potasio (K+) dependientes de los niveles de ATP y, por tanto, de los niveles de glucosa en sangre, se cierran y la membrana celular se despolariza. 4. Con la despolarización de la membrana los canales de calcio (Ca2+) dependientes de voltaje se abren y el calcio entra la célula. 56 5. Un aumento en el nivel de calcio intracelular produce la activación de fosfolipasa C, que desdobla fosfatidil inositol 4,5-bifosfato en inositol 1,4,5-trifosfato y diacilglicerol. 6. El inositol 1,4,5-trifosfato (IP3) se une a los receptores proteínicos sobre la membrana del retículo endoplásmico (RE). Esto permite la liberación de Ca2+ del RE a través de los canales IP3 aumentando más aún la concentración intracelular de calcio. 7. Estas cantidades significativamente mayores de calcio dentro de las células provoca la liberación de insulina previamente sintetizada y almacenada en las vesículas secretoras. Cierta liberación de insulina ocurre además con la ingestión de alimentos, no sólo de glucosa o hidratos de carbono y las células beta son también en cierta medida influenciadas por el sistema nervioso autónomo. Los mecanismos de señalización que controlan estos vínculos no son del todo comprendidos. Otras sustancias que pueden estimular la liberación de insulina incluyen los aminoácidos de las proteínas ingeridas, la acetilcolina liberada de las terminaciones del nervio vago (sistema nervioso parasimpático), la colecistocinina secretada por células enteroendocrinas de la mucosa intestinal y el péptido insulinotrópico dependiente de glucosa (GIP). Tres aminoácidos (alanina, glicina y arginina) actúan de manera similar a la glucosa alterando el potencial de membrana de la célula beta. La acetilcolina desencadena la liberación de insulina a través de la fosfolipasa C, mientras que la colecistocinina actúa a través del mecanismo de adenilato ciclasa (Cuadro 9) (Maitra y Abbas, 2005). 57 Cuadro 9. Factores y estados que aumentan ó disminuyen la secreción de insulina ( Tomado de Guyton y Hall, 2006). Aumento de la secreción de insulina Aumento de la glucemia Aumento de los ácidos grasos libres en sangre Hormonas gastrointestinales (gastrina, colecistocinina, secretina, péptido inhibidor gástrico) Glucagon, hormona del crecimiento, cortisol Estimulación parasimpática; acetilcolina Estimulación β-adrenérgica Resistencia a la insulina; obesidad Sulfonilureas (gliburida, tolbutamida) Disminución de la secreción de insulina Disminución de la glucemia Ayuno Somatostatina Actividad α- adrenérgica Leptina El sistema nervioso simpático, a través de la estimulación de receptores adrenérgicos alfa 2, como lo demuestran los agonistas de la clonidina o la metildopa, inhiben la liberación de insulina. Sin embargo, cabe señalar que la adrenalina circulante activará los receptores beta 2 en las células beta de los islotes pancreáticos para promover la liberación de insulina. Esto es importante ya que los músculos no pueden beneficiarse de los incrementos de glucosa en la sangre como consecuencia de la estimulación adrenérgica (aumento de la gluconeogénesis y glucogenólisis con los niveles bajos de la insulina en sangre por el glucagon) a menos que la insulina esté presente para permitir la translocación de GLUT-4 a nivel de los tejidos. Por lo tanto, comenzando con la inervación directa, la noradrenalina inhibe la liberación de insulina a través de los receptores alfa 2 y, subsecuentemente, la adrenalina circulante proveniente de la médula suprarrenal estimulará los receptores beta 2 promoviendo así la liberación de insulina (Alemzaden y Wyatt, 2004). Cuando el nivel de glucosa se reduce al valor fisiológico normal, la liberación de insulina de las células beta frena o se detiene. Si los niveles de glucosa en sangre se vuelven inferiores a ese nivel, especialmente a niveles peligrosamente bajos, la liberación de hormonas hiperglicémicas, la más prominente de las cuales es el glucagon de los mismos islotes de Langerhans pero de células alfa, obligan a la liberación de glucosa en la sangre a partir de los almacenes celulares, principalmente el almacenamiento de glucógeno en 58 las células del hígado. Mediante el aumento de glucosa en la sangre, las hormonas hiperglucémicas previenen o corrigen la hipoglucemia que pone en peligro la vida del individuo. La liberación de insulina está fuertemente inhibida por la hormona del estrés noradrenalina, lo que conduce a un aumento de los niveles de glucosa en sangre durante momentos de estrés. Pruebas de alteración de la primera fase de liberación de insulina se pueden detectar en la prueba de tolerancia a la glucosa, demostrado por una sustancial elevación de nivel de glucosa en sangre en los primeros 30 minutos, un marcado descenso durante los siguientes 60 minutos y un constante ascenso de nuevo a los niveles de referencia en las siguientes horas (Alemzaden y Wyatt, 2004). La mayor parte de la insulina liberada hacia la sangre circula de forma no ligada: su vida media plasmática es de unos 6 minutos y desaparece de la circulación en unos 10 a 15 minutos. Con excepción de la parte de insulina que se une a los receptores de las células efectoras, el resto se degrada por efecto de la enzima insulinasa sobre todo en el hígado y, en menor medida, en los riñones y en los músculos, y menos aún en casi todos los demás tejidos. Su desaparición inmediata del plasma tiene interés porque es tan importante desactivar con rapidez el efecto de la insulina como activar sus funciones reguladoras (Gómez y col., 1997). Los efectos fisiológicos de la insulina son muy amplios y complejos. Por conveniencia, se dividen en acciones rápidas, intermedias y tardías (Cuadro 10). El que se conoce en mayor medida es el efecto hipoglucémico, pero tiene más efectos en el transporte de aminoácidos y electrolitos, en muchas enzimas y en el crecimiento. El efecto neto de la hormona es el almacenamiento de hidratos de carbono, proteína y grasa. Por tanto, es conveniente llamar a la insulina “la hormona de la abundancia” (Gangog, 2004). 59 Cuadro 10. Principales acciones de la insulina (Tomado de Gangog, 2004). Principales acciones de la insulina Rápida (segundos) + Aumenta el transporte de glucosa, aminoácidos y K al interior de las células sensibles a la insulina. Intermedia (minutos) Estimula la síntesis de proteína Inhibe la degradación de proteínas Activa enzimas glucolíticas y sintetasa del glucógeno Inhibe la fosforilasa y las enzimas gluconeogénicas Tardías (horas) Aumenta mRNA para enzimas lipogénicas y otras Tejido adiposo Aumenta la entrada de glucosa Incrementa la síntesis de ácidos grasos Incrementa la síntesis de fosfato de glicerol Intensifica el depósito de triglicéridos Activa la lipasa de lipoproteínas Inhibe la lipasa sensible a hormonas + Aumenta la captación de K Hígado Disminuye la cetogénesis Aumenta la síntesis de proteínas Incrementa la síntesis de lípidos Aminora el gasto de glucosa por la disminución en la gluconeogénesis, intensificación de la síntesis del glucógeno e incremento de la glucólisis. General Aumenta el crecimiento celular 3.3 Efecto de la insulina sobre el flujo global de sustratos energéticos La captación tisular de glucosa y aminoácidos es estimulada por la insulina; la liberación tisular de glucosa, aminoácidos y ácidos grasos libres resulta inhibida por la hormona, al igual que la cetogénesis (Figura 17). El resultado neto es un descenso de la concentración plasmática de estos sustratos (Genuth, 2006). 3.3.1 Efecto de la insulina sobre el metabolismo de los hidratos de carbono Inmediatamente después de consumir una comida rica en hidratos de carbono, la glucosa absorbida hacia la sangre induce una secreción rápida de insulina. A su vez, la insulina provoca la captación rápida, el almacenamiento y el aprovechamiento de la glucosa por casi todos los tejidos del organismo, pero sobre todo por el hígado, músculo y el tejido adiposo (Figura 18) (Guyton y Hall, 2006). 60 Figura 17. Efecto de la insulina sobre el flujo global de sustratos energéticos (Tomado de Genuth, 2006). La insulina influye en el metabolismo de la glucosa, especialmente en el hígado, donde inhibe la glucogenólisis (degradación de glucógeno) y la gluconeogenesis (síntesis de glucosa a partir de precursores no glucídicos) y aumenta la utilización de glucosa (glucólisis), aunque la consecuencia final es el incremento de los depósitos hepáticos del glucógeno. En el músculo, a diferencia de lo que sucede en el hígado, la captación de glucosa es lenta y constituye el paso limitante del metabolismo de los hidratos de carbono. El principal efecto de la insulina consiste en aumentar el transporte facilitado de la glucosa por un transportador denominado GLUT-4 y en estimular la síntesis de glucógeno y de glucólisis. La insulina incrementa la captación de glucosa por GLUT-4 en el tejido adiposo y el músculo, favoreciendo así el metabolismo de la glucosa. Uno de los productos finales del metabolismo de la glucosa en el tejido adiposo es el glicerol, que se esterifica con ácidos grasos para formar triglicéridos, influyendo de este modo en el metabolismo de las grasas (Rang y col., 2008). 61 Figura 18. Efecto de la toma de alimento y del ayuno sobre el metabolismo. El consumo de alimentos inicia el equilibrio metabólico hacia el anabolismo mientras que el ayuno lo hace hacia el catabolismo. Esto sucede debido a una relación inversa entre la secreción de insulina y la del glucagon. La secreción de insulina se eleva y la del glucagon disminuye durante la absorción de los alimentos, mientras que durante el ayuno sucede lo contrario (Tomado de Fox, 2003). La insulina estimula el metabolismo oxidativo (glucólisis, vía de las pentosas fosfato, entrada del piruvato al ciclo de krebs) y no oxidativo (síntesis de glucógeno) de la glucosa por medio de la desfosforilación de enzimas clave y la inducción enzimática (Dvorkin y Cardinali, 2009). Favorece el transporte de glucosa y su utilización por casi todas las demás células del organismo de la misma manera que modifica el transporte y el uso de glucosa por las células musculares. El transporte de glucosa a las células adiposas aporta sobretodo la fracción glicerol de la molécula grasa fomentando el depósito de grasa en estas células (Cuadro 11) (Guyton y Hall, 2006). 62 Cuadro 11. Efectos de la insulina sobre enzimas del metabolismo de hidratos de carbono y lípidos ( Tomado de Dvorkin y Cardinali, 2009). Enzima Tejido Efecto Glucógeno sintetasa Adiposo, hígado y músculo esquelético Aumento de la síntesis de glucógeno Glucógeno fosforilasa Adiposo, hígado y músculo esquelético Disminución glucogenólisis Piruvato cinasa Hígado Aumento glucólisis Piruvato deshidrogenasa Adiposo e hígado Formación de Acetil-CoA a partir del piruvato Triacilglicerol lipasa Adiposo Inhibición de la lipólisis (estimulada por AMPc) HMG- CoA Hígado Aumento de la síntesis de colesterol de de la tasa 3.3.2 Efecto de la Insulina y sobre el metabolismo de los lípidos Pese a no resultar tan evidentes como sus efectos agudos sobre el metabolismo de los hidratos de carbono, las acciones de la insulina sobre el metabolismo lipídico resultan igualmente importantes a largo plazo. En particular, destaca el efecto a largo plazo de la falta de insulina, que produce una aterosclerosis marcada, a menudo con infartos de miocardio, ictus cerebrales y otros antecedentes vasculares (Guyton y Hall, 2006). La insulina promueve la síntesis de lípidos. En el tejido adiposo, la insulina facilita la transferencia de lípidos circulantes a los adipocitos al inducir la acción de la enzima lipoproteinlipasa. Por tanto se liberan más ácidos grasos libres desde las lipoproteínas circulantes y se captan rápidamente por los adipocitos, donde se reesterifican. Así, los lípidos de la dieta no utilizados para la generación inmediata de energía se almacenan. De igual o mayor importancia es el hecho de que la insulina inhibe profundamente la reacción inversa (es decir, la lipólisis de los triglicéridos almacenados) al actuar sobre la enzima necesaria para el proceso, la lipasa sensible a hormonas del tejido adiposo. De este modo se suprimen en gran medida la liberación de ácidos grasos libres y su aporte a otros tejidos. En el hígado, la insulina favorece que los ácidos grasos libres que le llegan se desvíen de la beta-oxidación al inhibir su 63 la de transferencia hacia las mitocondrias y dirigirlos hacia la esterificación al aumentar la producción de glicerol fosfato. Al disminuir la beta-oxidación se produce menos cantidad de beta-hidroxibutirato y acetoacetato. Por tanto, la insulina tiene una potente acción anticetógena (Genuth, 2006). La insulina también estimula la síntesis de novo de ácidos grasos libres a partir de glucosa. La acetil coenzima A (acetil-CoA) citoplásmica derivada del piruvato se desvía hacia la formación de ácidos grasos por que la insulina aumenta las enzimas clave acetil-CoA carboxilasa y sintasa de ácidos grasos. Además, estimula la síntesis de colesterol a partir de acetil-CoA al activar la enzima clave, hidroximetilglutaril-CoA reductasa. El efecto neto de la insulina es, por tanto, incrementar el contenido de lípidos en el hígado y, en algunos casos, una mayor liberación de lipoproteínas de muy baja densidad por ese órgano (Genuth, 2006). La insulina aumenta 20-30 veces la velocidad de transporte de glucosa a través de la membrana de los adipocitos. Estos efectos son agudos y se producen en un tiempo medio de dos o tres minutos. El mecanismo es la traslocación, desde el “pool” intracelular a la superficie de la membrana de los transportadores para la glucosa. La insulina activa al GLUT-4 preexistente, al tiempo que aumenta la expresión del gen que lo codifica. La señal molecular inicial implica la activación del receptor insulina (tirosincinasa). Este paso culmina con la fosforilación de sustratos del receptor de IRS en múltiples residuos de tirosina que actúan como sitios de fijación para ciertas proteínas, incluyendo la subunidad regulatoria p85 y la subunidad P13K. Si bien la activación de la P13K es necesaria para la completa estimulación del transporte de glucosa inducido por insulina, la evidencia reciente apunta a que también se requiere otra vía metabólica. En condiciones basales, el GLUT-1 es el principal responsable del ingreso de la glucosa en las células. La insulina duplica la acción del GLUT-1 y multiplica por diez la actividad del GLUT-4. Por esa razón en condiciones posprandiales la glucosa es desviada hacia el tejido adiposo y el músculo. Debido a que en el adipocito del 75% a 90% de los GLUT son del tipo 4, la insulina se convierte en el principal estímulo para el ingreso de glucosa al tejido adiposo. En los estados insulino-resistencia como la diabetes, 64 la obesidad y el ayuno, disminuye el transporte de la glucosa en respuesta a la insulina, aunque con diferentes niveles de sensibilidad en los tejidos adiposo y muscular (Dvorkin y Cardinali, 2009). 3.3.3 Efecto de la insulina sobre el metabolismo de las proteínas La insulina estimula la captación de aminoácidos por el músculo y aumenta la síntesis proteica. También disminuye el catabolismo proteico e inhibe la oxidación hepática de aminoácidos (Rang y col., 2008). El aumento de la síntesis de proteínas y el bloqueo de su degradación es facilitado por la TORcinasa. En el Cuadro 12 se muestras los efectos de la insulina en cada tejido respecto a los macronutrimentos (García y col., 2008). Cuadro12. Efectos del metabolismo de hidratos de carbono, grasas y proteínas (Tomado de Rang y col., 2008). Tipo de metabolismo Células hepáticas Célula adiposa Músculo Gluconeogenia Captación de Captación de Metabolismo de Glucogenólisis glucosa glucosa hidratos de carbono Glucólisis Síntesis de glicerol Glucólisis Metabolismo de Glucogenia Lipogenia Síntesis de grasas Lipólisis triglicéridos Glucogenia Síntesis de ácidos grasos ---- Lipólisis Metabolismo de Degradación de Captación de proteínas aminoácidos proteínas ---- Síntesis de proteínas Además de facilitar el transporte de iones (K+; Ca2+) al interior de la célula, la insulina desempeña un papel muy importante en la estimulación, la proliferación celular, el crecimiento y el desarrollo. Estas complejas funciones están mediadas por la vía Ras. Su activación da lugar a una cascada de serin65 cinasas, que culmina con la activación de ERK (extracelular signal-regulated kinase MAP-kinase, por sus siglas en inglés). Ésta, una vez translocada al interior del núcleo, cataliza la fosforilación de factores de transcripción iniciando un programa que conduce a la proliferación y diferenciación celulares (García y col., 2008). 3.4 Glucagon El glucagon es un péptido monocatenario de 29 aminoácidos, con un peso molecular de unos 3.5 kDa. Es sintetizado por las células alfa del islote de Langerhans en forma de prohormona de 9 kDa, el preproglucagon, que a diferencia de lo que ocurre con la preproinsulina, da origen a numerosos péptidos con distinta actividad biológica (Dvorkin y Cardinali, 2009). El glucagon junto con la adrenalina, cortisol y la hormona del crecimiento (las “hormonas contrareguladoras”) se oponen a muchas de las acciones de la insulina. Lo más importante es que el glucagon actúe para mantener los niveles sanguíneos de glucosa mediante la activación de la glucogenólisis y gluconeogénesis hepáticas (Champe y col., 2005). El efecto más espectacular del glucagon consiste en estimular la glucogenólisis hepática que, a su vez, aumenta la glucemia en unos minutos. Esta secuencia sigue una cascada compleja de acontecimientos (Guyton y Hall, 2006): 1. El glucagon activa a la adenilato ciclasa de la membrana de los hepatocitos, lo que determina la síntesis del AMP cíclico 2. Que activa a la proteína reguladora de la proteincinasa 3. Que activa la fosforilasa B cinasa 4. Que transforma la fosforilasa B en fosforilasa A 5. Lo que estimula la degradación de glucógeno a glucosa-1 fosfato, 6. Que por último se desfosforila para que el hepatocito libere glucosa. Esta secuencia de acontecimientos pone de relieve un sistema de cascada en el que cada producto sucesivo se fabrica en cantidad superior a la de su precursor (amplificación metabólica). Así se explica porque basta con unos microgramos de glucagon para que la glucemia se duplique o aumente incluso más a los pocos minutos. La infusión de glucagon durante unas 4 horas puede 66 causar tal glucogenólisis hepática que agote todos los depósitos de glucógeno en el hígado (Guyton y Hall, 2006). 3.5 Regulación de la secreción de insulina y glucagon La secreción de insulina y glucagon está regulada por la concentración plasmática de glucosa y, en menor medida, por los aminoácidos. Por lo tanto las células alfa y beta actúan como sensores y como efectores de este sistema de control. Dado que las concentraciones plasmáticas de glucosa y aminoácidos se elevan durante la absorción de una comida y caen durante el ayuno, la secreción de insulina y glucagon fluctúan según el estado absortivo y postabsortivo. Las variaciones de la secreción de insulina y glucagon provocan cambios de las concentraciones de glucosa y aminoácidos plasmáticos y ayudan a mantener la homeostasis a través de bucles de retro inhibición (Figura 19). Los mecanismos que regulan la secreción de insulina y de glucagon y las acciones de estas hormonas evitan en condiciones normales que la concentración plasmática de glucosa supere 170 mg por 100 mL después de una comida. Esta regulación es importante debido a que una glucemia anormalmente elevada puede lesionar determinados tejidos y una glucemia anormalmente baja puede provocar daño cerebral (Fox, 2003). 3.6 Pruebas de laboratorio para determinar el estudio de la patología del páncreas. Las pruebas de laboratorio más útiles para el estudio de la patología del páncreas exocrino son (Laso, 2010): 1. Determinación de enzimas pancreáticas en el suero: se valora fundamentalmente la amilasa y la lipasa. 2. Pruebas de función pancreática exocrina: puede realizarse el análisis directo del jugo pancreático (agua, bicarbonato, enzimas) obteniendo por sondaje duodenal en condiciones basales y tras la estimulación de la secreción enzimática del páncreas con colecistocinina y de la de agua y electrolitos con secretina (test de secretina- colecistocinina). También es posible valorar indirectamente la secreción pancreática exocrina 67 administrando por vía oral un sustrato de las enzimas secretadas por el páncreas y determinando posteriormente los productos resultantes de la acción enzimática. Figura 19. Regulación de la secreción de insulina y glucagon. La secreción de las células beta y alfa de los islotes pancreáticos está regulada en gran medida por la concentración sanguínea de glucosa. (A) una concentración sanguínea elevada de glucosa estimula la secreción de insulina e inhibe la del glucagon. (B) una concentración baja de glucosa en sangre estimula la secreción del glucagon e inhibe la de insulina (Tomado de Fox, 2003). Entre los estudios morfológicos del páncreas, los que tienen más interés son (Laso, 2010): 1. Técnicas de computarizada imagen: y ultrasonografía colangiopancreatografía abdominal, retrógada tomografía endoscópica incluyendo un producto de contraste en el conducto Wirsung. 2. Punción-aspiración del páncreas con una aguja fina: se realiza identificando al mismo tiempo la víscera mediante ultrasonografía o tomografía computarizada 68 3.7 Niveles de glucosa en sangre Los niveles varían de acuerdo con el laboratorio pero en general hasta 100 mg/dL se consideran normales para un examen de glucemia en ayunas. Las personas con niveles entre 100 y 125 mg/dL tienen una alteración de la glucosa en ayunas o prediabetes. Se considera que estos niveles son factores de riesgo para desarrollar diabetes tipo 2 y sus complicaciones. La diabetes se diagnostica en personas con niveles de glucemia en ayunas que sean de 126 mg/dL o mayores. Los niveles de glucosa en la sangre superiores a los normales (hiperglucemia) pueden ser un signo de diabetes. En alguien que tenga diabetes, puede significar que la enfermedad no está bien controlada. El aumento en los niveles también pueden deberse a (Thompson y col., 2008): Acromegalia (muy poco común) Síndrome de Cushing (poco común) Glucagonoma Diabetes mellitus Alteración de la glucosa en ayunas (también llamada "prediabetes") Hipertiroidismo Cáncer pancreático Pancreatitis Feocromocitoma (muy poco común) Los valores inferiores al nivel normal (hipoglucemia) pueden ser indicio de (Thompson y col., 2008): Hipopituitarismo Hipotiroidismo Insulinoma (muy poco común) Muy poco alimento Demasiada insulina u otros medicamentos para la diabetes 69 3.8 Patologías más frecuentes en el páncreas 3.8.1 Diabetes mellitus La diabetes mellitus (DM) es un conjunto de trastornos metabólicos, que afecta a diferentes órganos y tejidos, dura toda la vida y se caracteriza por un aumento de los niveles de glucosa en la sangre: hiperglucemia. Es causada por varios trastornos, incluyendo la baja producción de insulina o por su inadecuado uso por parte del cuerpo, lo que repercutirá en el metabolismo de los hidratos de carbono, lípidos y proteínas (Figura 20) (Tébar y Ferrer, 2009). Los síntomas principales de la diabetes mellitus son emisión excesiva de orina (poliuria), aumento anormal de la necesidad de comer (polifagia), incremento de la sed (polidipsia), y pérdida de peso sin razón aparente. La Organización Mundial de la Salud reconoce tres formas de diabetes mellitus: tipo 1, tipo 2 y diabetes gestacional, cada una con diferentes causas (Cuadro 13) y con distinta incidencia. Varios procesos patológicos están involucrados en el desarrollo de la diabetes como el carácter autoinmune característico de la DM tipo 1 o hereditario y resistencia del cuerpo a la acción de la insulina como ocurre en la DM tipo 2 (Morgan y Weinsier, 2000). Figura 20. Producción de insulina en la diabetes mellitus. En la diabetes mellitus el páncreas no produce o produce muy poca insulina (DM Tipo I) o las células del cuerpo no responden normalmente a la insulina que se produce (DM Tipo II). Esto evita o dificulta la entrada de glucosa en la célula, aumentando sus niveles en la sangre hiperglucemia (Modificado de Morgan y Weinsier, 2000). 70 Este padecimiento causa diversas complicaciones, dañando frecuentemente a ojos, riñones, nervios y vasos sanguíneos. Sus complicaciones agudas (hipoglucemia, cetoacidosis, coma hiperosmolar no cetósico) son consecuencia de un control inadecuado de la enfermedad mientras sus complicaciones crónicas (cardiovasculares, nefropatías, retinopatías, neuropatías y daños microvasculares) son consecuencia del progreso de la enfermedad (Tébar y Ferrer, 2009). Cuadro13. Comparación entre diabetes miellitus dependiente de insulina y la no dependiente de insulina (Tomado de Fox, 2003). Característica Dependiente de insulina No dependiente de insulina (tipo1) (tipo2) Edad habitual de comienzo Menores de 20 años Mayores de 40 años Aparición de síntomas Rápida Lenta Porcentaje de la población Aproximadamente 10% Aproximadamente 90% Aparición de cetoacidosis Frecuente Raro Asociación a obesidad Rara Frecuente Células beta de los islotes Destruidas No destruidas Secreción de insulina Disminuida Normal ó aumentada Autoanticuerpos contra las Presentes Ausentes si No está clara Inyecciónes de insulina Dieta y ejercicio; estimulantes diabética (al comienzo de la enfermedad células de los islotes Asociación a determinados antígenos MHC Tratamiento orales de la secreción de insulina Se establece el diagnóstico de diabetes si cumple cualquiera de los siguientes criterios (Modificación NOM-015-SSA2-1994): 1. Presencia de síntomas clásicos y una glucemia plasmática casual >200 mg/dL (11.1 mmol/L); glucemia plasmática en ayuno >126 mg/dL (7 mmol/L); o bien glucemia >200 mg/dL (11,1 mmol/L) a las dos horas después de carga oral de 75 g de glucosa disuelta en agua. En ausencia de 71 hiperglucemia inequívoca, con descompensación metabólica aguda, el diagnóstico debe confirmarse repitiendo la prueba otro día. 2. Se establece el diagnóstico de glucosa anormal en ayuno, cuando la glucosa plasmática o en suero es >110 mg/dL (6,1 mmol/L) y <126 mg/dL (6,9 mmol/L). 3. Se establece el diagnóstico de intolerancia a la glucosa, cuando la glucosa plasmática, a las dos horas poscarga, es >140 mg/dL (7,8 mmol/L) y <200 mg/dL (11,1 mmol/L). 3.8.1.1 Tratamiento nutricio para pacientes con diabetes melltus Los objetivos del tratamiento nutricio son (Araiza y Nazor, 2001): 1. Mantener cifras de glucemia normales o cercanas a lo normal (Anexo5). 2. Proporcionar energía necesaria para mantener un peso adecuado, saludable en adultos, mujeres embarazadas, ancianos, niños y adolescentes. 3. Lograr y mantener cifras óptimas del colesterol total, del colesterol de las lipoproteínas de baja densidad, de las lipoproteínas de alta densidad y de las lipoproteínas de muy baja densidad, así como de los triglicéridos (Anexo5). 4. Si existe hipertensión arterial, favorecer su control. 5. Modificar los hábitos alimentarios dañinos. 6. Respetar y promover los hábitos alimentarios correctos. 7. Inducir a la costumbre de realizar actividad física. 8. Promover la salud y la buena calidad de vida. No existe una dieta ideal única para la DM, tanto en lo que respecta a los nutrimentos como a las diferentes formas clínicas y circunstancias de cada paciente. Un exceso de hidratos de carbono puede empeorar la glucemia; un alto aporte de grasa puede aumentar el riesgo de enfermedad cardiovascular, un gran aporte de proteína en la dieta puede empeorar la nefropatía diabética (Valero y Leon, 2010). El tratamiento nutricional debe coincidir con los otros aspectos de la terapéutica de la DM, actividad física y medicación (Valero y Leon, 2010). 72 La recomendación de la cantidad y tipo de hidratos de carbono de la dieta se rige con base en la respuesta glucémica y pueden ser de 50 a 60 % de la energía total, los cuales deben ser complejos ( cereales integrales) ya que tienen un bajo indice glucémico y son ricos en fibra (Araiza y Nazor, 2001). Se debe evitar en lo posible la ingestión de hidratos de carbono simples, ya que su absorción es muy rápida y pueden generar hiperglucemia posprandial temprana y grave. La fibra dietética soluble que se encuentran en frutas, verduras, cebada y en especial avena y leguminosas, mejora el control de la glucemia porque hace más lento el vaciamiento gástrico, aumenta el tránsito intestinal y retrasa la absorción de la glucosa (Mahan y Arlin, 1995). La cantidad de fibra que se recomienda en la dieta del paciente diabético es de 40g/d o 25 g/1000 kcal, con cantidades iguales de fibra soluble e insoluble (Mahan y Arlin, 1995). Cuando los pacientes inician una dieta alta en fibra debe vigilarse muy de cerca las necesidades de insulina y la glucemia y reducirse la dosis de la hormona según se requiera. Los estudios hechos sobre la ingestión alta en fibra a largo plazo demostraron que en esas cantidades no afectan la absorción de nutrimentos inorgánicos. Ante cualquier incremento de fibra de la dieta también debe hacerse un incremento de agua (Araiza y Nazor, 2001). Las necesidades de proteínas son las mismas para personas diabéticas que sanas a la edad pueden variar de 0.8 a 2 g/kg de peso corporal ideal. Esto significa una ingestión de proteínas de 10 a 20 % de la energía total. Las mujeres embarazadas y lactantes tienen necesidades adicionales al igual que las personas con estrés catabólico. En niños equivaldría a un 12 a 15 % del total de la energía (Morgan y Weinsier, 2000). Los pacientes diabéticos tienen un riesgo elevado de desarrollar ateroesclerosis y enfermedad coronaria, para disminuir este riesgo se recomienda restringir la ingestión total de lípidos a 30 % o menos del valor energético y la ingestión total de colesterol a menos de 300 mg/dL, limitar el consumo de ácidos grasos saturados de 6 a 8 %, 10% de poliinsaturados y entre 10 y 15% ácidos grasos monoinsaturados (Mahan y Arlin, 1995). Cuando la dieta es adecuada y se controla la glucosuria no es necesario administrar suplementos de vitaminas y nutrimentos inorgánicos (Anexo 6) 73 (Tébar y Ferrer, 2009). El nutriólogo responsable del tratamiento indicará la dieta apropiada para cada paciente, conforme a las kcla/kg recomendadas al día y según las condiciones clínicas. En las Figuras 21 y 22 se muestran recomendaciones para el plan alimentario según condiciones específicas de los pacientes (Modificación NOM-015-SSA2-1994). Figura 21. Tratamiento dietético para pacientes diabéticos obesos (Tomado de Modificación NOM-015-SSA2-1994) Para el paciente con DM tipo 1 es importante considerar el horario de las comidas, la composición y contenido energético de la dieta así como su actividad física. En tanto sea posible, debe ajustarse la insulina a un plan de alimentación preferido en lugar de ajustar la dieta a la dosis (Cuadro14). Seguir un plan de alimentación es importante para evitar la hiperglucemia, prevenir la hipoglucemia y mantener un balance metabólico (Mahan y Arlin, 1995). 74 Figura 22. Tratamiento dietético para pacientes diabéticos no obesos (Tomado de Modificación NOM-015-SSA2-1994). Cuadro 14. Distribución de la energía en porcentaje de acuerdo al tipo de insulina (Tomado de Mahan y Arlin, 1995). Insulina Desayuno Colación Comida matutina 10 Cena vespertina Rápida 25 Intermedia 20 30 Lenta 20 25 Intermedia 20 10 Colación 30 30 10 10 Colación nocturna 25 10 30 10 35 20 20 10 y rápida 3.8.2 Pancreatitis aguda Las pancreatitis aguda, es una inflamación no bacteriana del páncreas caracterizada por la activación intraglandular de las pro-enzimaspancreáticas y autodigestión concomitante de los acinos. Habitualmente va seguida de una total restauración estructural y funcional de la glándula. Anatómicamente se describen dos tipos con marcada diferencia evolutiva (Maldonado y Mataix, 2009): 75 a) Forma intersticial o edematosa. Predominan los fenómenos de edema, necrosis grasa peripancreática, inflamación y exudado y sin signos de necrosis pancreática Es la forma más frecuente y generalmente tiene un curso benigno y de evolución breve. b) Forma necrohemorrágica. Predominan la necrosis grasa peri e intrapancreática; necrosis del parénquima y hemorragia pancreática con desaparición prácticamente total de las estructuras anatómicas pancreáticas, de evolución mucho más prolongada y de pronóstico grave. Puede conducir a un desenlace fatal si da lugar a choque o insuficiencia renal o respiratoria. La pancreatitis intersticial leve aguda se caracteriza por dolor abdominal, náuseas, vómito, anorexia y una mortalidad baja o nula. En el curso de la enfermedad pueden presentarse algunas complicaciones locales, son poco frecuentes y la resolución de los síntomas ocurre en algunos días. En estos pacientes basta con un tratamiento médico simple de sostén y la nutrición parenteral se requiere en muy pocos casos. Sin embargo, en la pancreatitis necrótica grave, que se caracteriza por insuficiencia del órgano, la mortalidad puede ser hasta de 30% si el tejido necrosado se infecta. Estos individuos ameritan nutrición parenteral total (NPT). Tan pronto como se reconoce la pancreatitis grave porque es muy posible que el curso clínico se complique y prolongue (Raimondo y Dimagno, 2002). La gravedad de la pancreatitis aguda es el principal factor predictivo de la evolución de un ataque de pancreatitis e indica el tipo de tratamiento que los pacientes deben recibir. Por eso resulta indispensable predecir la gravedad de la enfermedad al momento de la presentación. Se proponen diversos datos que permiten anticipar la gravedad y muchos de ellos llevan el nombre del investigador que los definió. Estos datos clínicos incluyen los criterios de Ranson (Cuadro 15), el primer grupo de criterios que se estableció. Además se cuenta con otros criterios propuestos por Bank, Agarwal y Pitchumoni, e Imrie. Todos tienen la desventaja de requerir 48 horas para medir cuando menos algunos de sus componentes. Dos signos o menos predicen una mortalidad de cero; tres a cinco criterios de 10 a 20% y seis o más una mortalidad que 76 excede del 50%. Los pacientes con tres signos o más tienen un riesgo alto de sufrir necrosis del páncreas y complicaciones sistémicas (Raimondo y Dimagno, 2002). Cuadro 15. Criterios de gravedad de Ranson Al ingreso: Edad > 55 años Cuenta leucocitaria > 16000/mm 3 Glucosa > 200 mg/100 mL LDH > 350 UI/L AST > 250 U/L Durante las primeras 48 horas Disminución del Hto > 10 Aumento del BUN > 5mg/100mL 2+ Ca < 8mg/ 100mL PaO2 < 60 mmHg Deficiencia de bases > 4 meq/L Secuestro de líquidos > 6L En la pancreatitis edematosa generalmente se puede reinstaurar la ingestión de alimentos a los 3-7 días tras el inicio del proceso, una vez que haya desaparecido el dolor, exista peristaltismo y se compruebe que las cifras de las enzimas pancreáticas se han normalizado. La enfermedad tiene muy poco impacto sobre el estado nutricional y sobre el metabolismo de los sustratos energéticos. No hay evidencias de que el apoyo nutricio tenga un efecto benéfico en la evolución clínica y solamente se considera la nutrición enteral o parenteral en aquellos casos en que la alimentación por vía oral deba posponerse más de siete días, evitando de este modo los efectos de la desnutrición (Figura 23) (Maldonado y Mataix, 2009). Es importante revisar en forma periódica al enfermo en busca de síntomas como dolor, náuseas o vómito, la dieta debe evolucionar, según la tolere hasta alimentos de fácil digestión con poca grasa y a partir de ese momento avanzar 77 hasta dónde sea factible. Los alimentos pueden ser tolerados mejor si se dividen en seis raciones pequeñas (Anexo 7) (Aiello y Nickleach, 1998). Figura 23. Algoritmo de decisión para el tratamiento de pancreatitis aguda (Tomado de Maldonado y Mataix, 2009). La dieta será rica en hidratos de carbono y proteínas y baja en grasas, con aporte importante de verduras. Los alimentos se introducirán en forma progresiva, cada 1-2 días, según lo aconseje la tolerancia del paciente. En una primera fase se iniciará la administración de líquidos totalmente exentos de grasa (infusiones sin azúcar, agua, caldos vegetales desgrasados). Posteriormente se introducirán alimentos ricos en hidratos de carbono, como puré de papa, pastas, arroz, frutas (no cítricas) y pan tostado. Si esta fase se tolera bien, se introducen progresivamente las proteínas animales, iniciando por pequeñas cantidades (50 g en la comida y en la cena), comenzando por el pescado. En esta fase puede existir un ligero aumento de la amilasa en sangre y orina, que si se mantiene o aparecen otros síntomas obliga a volver a la fase anterior y reiniciar el ciclo. En las fases finales se aumenta el aporte de proteínas unos 100 g por tiempo de comida y se introducen verduras cocidas y carne cocida o asada, primero de pollo y posteriormente las blancas y las rojas. Las preparaciones deben excluir las formas grasas, fritos y guisados (Maldonado y Mataix, 2009). 78 En casos de pancreatitis aguda a menudo se identifica hipocalemia y entre sus causas posibles están hipoalbuminemia, con paso ulterior del líquido al tercer espacio, el calcio que está ligado a la albúmina sale de la circulación. Otra explicación posible sería la saponificación, en la que intervienen el calcio y los ácidos grasos producidos por la necrosis grasa. Un método para cuantificar el calcio disponible es medir el nivel del mineral ionizado (Havala y col., 1989). En las pancreatitis graves se recurrirá a la nutrición parenteral o enteral por sonda nasoyeyunal para evitar el deterioro del estado nutricio debido al estrés metabólico y al ayuno prolongado. El 80% de todos los pacientes presentan un estado catabólico, con aumento de las necesidades energéticas y catabolismo proteínico. El balance negativo de nitrógeno puede ser hasta de 40 g/día, lo que puede tener efectos deletéreos sobre el estado nutricio y la evolución de la enfermedad (Havala y col., 1989). La nutrición enteral tiene ventajas sobre la parenteral, por ello se recomienda en todos los pacientes que la toleren y se debe iniciar tan pronto como sea posible. El paso a la alimentación oral será gradual y de acuerdo a la situación clínica del paciente y a la evolución de la enfermedad (Maldonado y Mataix, 2009). Los requerimientos energéticos oscilan entre 25 y 35 Kcal/Kg/día y normalmente es suficiente aportar entre 3 y 6 g/Kg/día de hidratos de carbono. El aporte proteínico óptimo se estima entre 1.2 y 1.5 g/Kg/día, se aumentará si existe un balance de nitrógeno muy negativo y a veces es necesario disminuirlo en pacientes con falla renal o hepática. Puede ser suficiente un aporte de grasa de 2g/Kg/día, tolerándose mejor los triglicéridos de cadena media, pero se deben monitorizar estrictamente las concentraciones séricas de triglicéridos por el peligro de que se presente hipertrigliceridemia, que ya de por si suele estar presente en la pancreatitis aguda (Maldonado y Mataix, 2009). El uso de la nutrición parenteral quedaría solo para aquellos casos en que no sea viable la utilización del tubo digestivo o no se pueda colocar la sonda nasoyeyunal, si se reagudiza la pancreatitis al inicio de la nutrición enteral, o cuando sea necesario un apoyo nutricional mixto al no poderse alcanzar los objetivos nutricionales únicamente con la nutrición enteral (Maldonado y Mataix, 2009). 79 3.8.3Pancreatitis crónica La pancreatitis crónica es una enfermedad en la que se produce una alteración progresiva e irreversible de la glándula pancreática, que da lugar a un cuadro clínico caracterizado por la presencia de dolor abdominal y signos de insuficiencia glandular, tanto exocrina como endocrina. En general, la enfermedad sigue un curso progresivo en el que ocurren varios episodios de pancreatitis aguda poco sintomáticos o silentes. La pancreatitis crónica cursa clínicamente con dolor abdominal, esteatorrea e intolerancia a los hidratos de carbono, aunque en ocasiones los pacientes pueden permanecer asintomáticos por largo tiempo. El tratamiento de la pancreatitis crónica se apoya en los siguientes pilares (García y col., 2010): 1. Tratamiento del dolor. 2. Sustitución con enzimas pancreáticas. 3. Evaluación y tratamiento nutricional. 4. Diagnóstico y tratamiento de posibles complicaciones. Las recomendaciones dietéticas se basan en el mantenimiento de un aporte energético adecuado, comidas frecuentes y poco copiosas y la ingestión abundante de hidratos de carbono. El consumo de hidratos de carbono sólo suele restringirse cuando hay presencia de diabetes mellitus (Casellas, 2008). Las enzimas pancreáticas suplementarias pueden aliviar el dolor al disminuir la secreción exocrina de la glándula. Cuando la función en el órgano ha disminuido aproximadamente a un 90%, la producción y la secreción de enzimas no son suficientes. En estos casos la digestión deficiente y malabsorción de proteínas y grasas se tornan problemáticas (Holt, 1993). La ingestión de grasas, especialmente de grasas de origen vegetal, puede permitirse para conseguir un correcto aporte energético siempre que sean toleradas. Si no se controla la esteatorrea y hay disminución del peso corporal pueden sustituirse las grasas de la dieta por triglicéridos de cadena media (TCM). Los TCM se absorben a través de la mucosa intestinal sin necesidad de lipasa pancreática, pasando directamente a la circulación general por vía portal. Esta sustitución de grasa también tiene sus limitaciones porque para conseguir 80 una buena absorción de los TCM también se necesita administrar suplementos de enzimas pancreáticas que pueden provocar intolerancia en forma de diarrea (Casellas, 2008). En sujetos con esteatorrea notable se observa a veces malabsorción de vitaminas liposolubles; también la deficiencia de la proteasa pancreática, necesaria para separar la vitamina B12 de su proteína transportadora, puede ocasionar deficiencia de vitamina B12. (Hacker, 1988). Con frecuencia debe recurrirse a la administración de suplementos exógenos de enzimas pancreáticas. La finalidad de estos es permitir un correcto mantenimiento del peso corporal y reducir la esteatorrea, la dosis de suplementos debe ser establecida individualmente. La actividad de la lipasa intraduodenal conseguida con la sustitución de enzimas pancreáticas debe ser superior al 10% de la actividad fisiológica. El control del cumplimiento y la respuesta al tratamiento sustitutivo enzimático en la insuficiencia pancreática es clínico, basándose en la recuperación del peso corporal, la normalización del aspecto de las deposiciones y la desaparición de los síntomas. Sin embargo, es aconsejable complementar las medidas clínicas con otras medidas objetivas como la desaparición de esteatorrea o la normalización del test de aliento con triglicéridos (Casellas, 2008). La secreción de bicarbonato por el páncreas puede ser deficiente, razón por la cual la asistencia médica incluye también conservación del pH intestinal óptimo para facilitar la activación de enzimas. Para lograr este efecto cabe utilizar los antiácidos u otros agentes que aminoren la secreción de ácido estomacal (Aiello y Nickleach, 1998). Diversas situaciones pueden conducir al fracaso terapéutico, como son el desarrollo de un sobre-crecimiento bacteriano o la precipitación de ácidos biliares en un medio ácido, que actúan como factor favorecedor de la malabsorción no relacionada con la actividad de la lipasa. Por este motivo se recomienda el tratamiento anti-secretor con inhibidores de la bomba de protones y, eventualmente, el tratamiento cíclico con antibióticos (Figura 24) (Casellas, 2008). 81 En casos crónicos con destrucción extensa del páncreas, disminuye la capacidad insulinógena de la víscera y surge intolerancia a la glucosa. En estos casos hay que recurrir a la insulina y la atención nutricional semejante a la que se usa en el diabético. Estos pacientes también pueden mostrar hipoglucemia refractaria por deficiencia de glucagon. La asistencia y el tratamiento son delicados y deben orientarse al control de los síntomas y no a la normoglucemia como objetivo (Holt, 1993). Figura 24. Algoritmo de la terapia nutricional en la malabsorción debida a insuficiencia pancreática (Tomado de Casellas, 2008). Capitulo 4: Suprarrenales 4.1 Definición de las glándulas suprarrenales Las glándulas suprarrenales (adrenales) son glándulas endócrinas, bilaterales, situadas en la parte posterior del abdomen, debajo y delante del diafragma, arriba los riñones, sobre la cara anterolateral de la parte superior de la columna lumbar. Cada glándula suprarrenal está formada por dos partes, de orígen 82 embriológico diferente: la corteza suprarrenal, mesodérmica, y la médula suprarrenal, ectodérmica, ligada al desarrollo del sistema nervioso simpático (Figura 25). Cada glándula mide en promedio 30 mm de alto por 25 mm de ancho y de 7 a 8 mm de espesor. Su peso oscila alrededor de los 12 g, su coloración es castaño amarillento (Latarjet y Ruiz, 2008). Figura 25. Localización de las glándulas suprarrenales. Existen dos suprarrenales, una derecha y otra izquierda, cuya forma es algo diferente: la glándula derecha es regularmente triangular, aplanada de adelante hacia atrás, la glándula izquierda es más gruesa en el sentido medial. Cada glándula tiene una superficie cóncava en la base, que se aplica sobre la extremidad superior del riñón (Modificado de Latarjet y Ruiz, 2008). La porción externa, o corteza, de la glándula suprarrenal rodea completamente a la porción interna, o médula, y constituye la mayor parte de la masa glandular. La médula suprarrenal secreta catecolaminas: adrenalina y noradrenalina, mientras que la corteza secreta un grupo de hormonas denominadas corticosteroides. En el hombre adulto, la corteza suprarrenal está constituida por tres zonas histológicamente distintas; la zona más externa que se encuentra inmediatamente debajo de la cápsula, se denomina zona glomerular, responsable de la producción de mineralocorticoides. La capa media y más gruesa de la corteza es la zona fascicular, que sintetiza 83 glucocorticoides. Y por último la capa interna de la corteza, la zona reticular que segrega andrógenos suprarrenales (Figura 26) (Modificado de Gal y col., 2001). Figura 26. Estructura de la glándula suprarrenal. La zona glomerular secreta los mineralocorticoides (incluyendo la aldosterona), mientras que las otras zonas segregan glucocorticoides (incluyendo el cortisol) (Adaptado de Gal y col., 2001). Los mineralocorticoides reciben este nombre por que controlan el metabolismo hidroelectrolítico, siendo la principal hormona esteroide mineralocorticoide la aldosterona. Los glucocorticoides se denominan así por su capacidad de incrementar los niveles plasmáticos de glucosa. El cortisol es el principal glucocorticoide secretado por las glándulas suprarrenales. Las glándulas suprarrenales son esenciales para la supervivencia, sin ellas las alteraciones del metabolismo electrolítico y de hidratos de carbono producen la muerte por colapso circulatorio y coma hipoglucémico. También permiten que el organismo sea capaz de soportar los traumatismos físicos y psíquicos y el estrés (Gal y col., 2001). 84 4.2 Efectos relacionados con nutrición de la aldosterona y del cortisol 4.2.1 Aldosterona La aldosterona es una hormona esteroidea de la familia de los mineralocorticoides, producida por la sección externa de la zona glomerular de la corteza adrenal en la glándula suprarrenal. Actúa en la conservación del sodio, secretando potasio e incrementando la presión sanguínea (Figura 27) (Ochoa y García, 2008). Figura 27. Estructura química de la aldosterona (Tomado de Ochoa y García, 2008). La función más importante de la aldosterona es el transporte de sodio y potasio a través de las paredes de los túbulos renales. La aldosterona induce la reabsorción de sodio y la secreción simultanea de potasio por las células epiteliales tubulares en el túbulo colector, túbulo distal y conducto colector (conserva el sodio en el liquido extracelular y secreta potasio a la orina). Junto con la reabsorción de sodio a nivel tubular se reabsorbe, simultáneamente, y por mecanismos osmóticos, agua (el volumen de liquido extracelular aumenta en proporción directa al sodio retenido). El aumento del volumen extracelular produce un aumento de la presión arterial y aumento de la eliminación renal de sodio y agua (diuresis de presión). Cuando disminuye la secreción de aldosterona, se pierden grandes cantidades de sal en la orina y se produce una deshidratación celular interna (shock circulatorio). La aldosterona también induce secreción tubular de iones hidrógeno, igualmente intercambiados por sodio (disminuye la concentración de hidrógeno intracelular, lo que produce una alcalosis metabólica leve) (Bruce y Bruce, 2006). 85 El exceso de aldosterona produce hipopotasemia. La pérdida excesiva de iones K desde el liquido extracelular hacia la orina disminuye su concentración plasmática (valor normal 4,5 mEq/L). Suele producirse una debilidad muscular, como consecuencia de la hiperpolarización de las membranas de las fibras musculares y nerviosas, que impide la transmisión de los potenciales de acción (Bruce y Bruce, 2006). La hiperpotasemia, por su parte, se produce por falta de aldosterona. Puede aparecer una grave toxicidad cardiaca, incluyendo debilidad en la contracción y arritmias (Bruce y Bruce, 2006). Son tres los mecanismos primarios encargados del control de la secreción suprarrenal de aldosterona: sistema de renina y angiotensina, potasio y ACTH (hormona adrenocorticotropica). No está claro si estos son también los mecanismos reguladores primarios que modifican la producción no suprarrenal. El sistema de renina y angiotensina controla el volumen del líquido extracelular mediante regulación de la secreción de aldosterona. El sistema de renina y angiotensina conserva constante el volumen de la sangre circulante al desencadenar la retención de sodio inducida por aldosterona durante la deficiencia de volumen y al disminuir la retención de sodio dependiente de la aldosterona cuando el volumen es amplio. Se cuenta con numerosas pruebas que indican que algunos tejidos, además del renal, producen angiotensina II y podrían participar en la regulación de la secreción de aldosterona por vías suprarrenales o extrasuprarrenales. Es intrigante que la propia corteza suprarrenal es capaz de sintetizar angiotensina II. Prácticamente no se ha identificado función alguna de la producción extrarenal de angiotensina II en la fisiología normal. Sin embargo, el sistema hístico de renina y angiotensina se activa dentro del útero como reacción al crecimiento y al desarrollo, y durante la vida extrauterina como reacción a las lesiones (Williams y Dluhy, 2006). 4.2.2 Cortisol El cortisol es producido por la zona fasciculada de la corteza suprarrenal, una de las dos partes de la glándula suprarrenal. Esta liberación está controlada por el hipotálamo en respuesta al estrés o a un nivel bajo de glucocorticoides en la sangre. La hormona suprarrenal corticotropina (ACTH) es transportada por la 86 sangre hasta la corteza suprarrenal, en la cual desencadena la secreción de glucocorticoides (Pocock y Richards, 2005). El cortisol es esencial para la vida. Junto con otros glucocorticoides produce numerosas acciones en todo el organismo (Figura 28). Sus efectos fisiológicos más importantes son los que se producen sobre el metabolismo de los hidratos de carbono, las proteínas y, en menor grado, las grasas. Los glucocorticoides también desempeñan un papel decisivo en las respuestas del organismo a diversos estímulos estresantes. Son imunosupresores, antiinflamatorios y antialérgicos y poseen una débil actividad mineralcorticoide. Esta actividad es importante en extremo por que los niveles plasmáticos de cortisol son mucho mayores que los de la aldosterona. Hay datos recientes que indican que las enzimas de los tejidos diana son muy importantes para inactivar los distintos esteroides como para activarlos (Pocock y Richards, 2005). Sin embargo, uno de los efectos secundarios del cortisol es que disminuye la formación ósea. El cortisol (hidrocortisona) se usa para tratar varias dolencias y enfermedades como la enfermedad de Addison, enfermedades inflamatorias, reumáticas y alergias. La hidrocortisona de baja potencia, disponible sin receta en algunos países, se utiliza para tratar problemas de la piel tales como erupciones cutáneas y eczemas, entre otros (Guyton y Hall, 2006). La hidrocortisona previene la liberación en el cuerpo de sustancias que causan inflamación. Estimula la gluconeogénesis, la descomposición de las proteínas y las grasas para proporcionar metabolitos que pueden ser convertidos en glucosa en el hígado; también activa las vías antiestrés y antiinflamatorias (Guyton y Hall, 2006). 87 Figura 28. Principales Acciones Fisiológicas del cortisol (Tomado de Pocock y Richards, 2005). El cortisol, a diferencia de los otros esteroides suprarrenales, ejerce un control por realimentación negativa sobre la síntesis de ACTH al suprimir la transcripción del gen de la ACTH en la hipófisis y suprime la formación de la hormona liberadora de hormona adrenocorticotropica (Guyton y Hall, 2006). 4.3 Efecto del cortisol en el metabolismo de hidratos de carbono Los glucocorticoides aumentan las concentraciones de glucosa en sangre por medio de su acción sobre el metabolismo del glucógeno, proteínas y lípidos. En términos generales, puede afirmarse que los efectos del cortisol son contrarios a los de la insulina. Estos efectos varían en cada tejido diana, pero siempre conducen a un aumento de la concentración plasmática de glucosa (Pocock y Richards, 2005). 88 En el hígado, el cortisol estimula el depósito de glucógeno al aumentar la glucógeno sintasa e inhibir la glucógeno fosforilasa, enzima movilizadora del glucógeno. Aumenta la producción hepática de glucosa por medio de la activación de las enzimas clave relacionadas con la gluconeogénesis, principalmente la glucosa-6-fosfatasa y la fosfoenolpiruvato cinasa (PEPCK) (Stewart, 2009). El cortisol también reduce, aunque en grado moderado, el ritmo de utilización de glucosa por la mayoría de las células del cuerpo. La base del mecanismo propuesto se encuentra en la observación de que los glucocorticoides disminuyen la oxidación del dinucleótido de nicotinamida y adenina (NADH) para formar NAD. Como el NAD debe oxidarse para permitir la glucólisis, este efecto quizá explique la menor utilización celular de la glucosa (Guyton y Hall, 2006). Los glucocorticoides tienen también un efecto permisivo sobre otras hormonas, como las catecolaminas y el glucagon. El efecto resultante es una resistencia a la insulina y un aumento de las concentraciones de glucosa en sangre a expensas del catabolismo de las proteínas y de los lípidos (Stewart, 2009). El incremento de glucemia se debe tanto al incremento de la gluconeogenia como a la reducción moderada de la utilización celular de la glucosa. A su vez, el aumento de la concentración de glucosa estimula la secreción de insulina. Los valores elevados de glucocorticoides reducen la sensibilidad de muchos tejidos, en particular del músculo esquelético y del tejido adiposo, a los efectos favorecedores de la captación y utilización de la glucosa característicos de la insulina. El incremento de la glucemia alcanza a veces tal proporción (50% o más sobre el límite normal) que se llega a un estado conocido como diabetes suprarrenal. En ésta, la administración de insulina reduce la glucemia sólo de manera moderada (no tanto como en la diabetes pancreática), por que los tejidos adquieren resistencia a los efectos de la hormona (Guyton y Hall, 2006). 4.4 Efecto del cortisol en el metabolismo de lípidos En el tejido adiposo se activa la lipólisis, lo que da lugar a la liberación de ácidos grasos libres a la circulación. Se observa un aumento de colesterol y en los triglicéridos circulantes totales, pero disminuye la concentración de colesterol HDL. Los glucocorticoides estimulan la diferenciación de los 89 adipocitos, promoviendo la adipogénesis por medio de una activación transcripcional de genes clave de diferenciación, como la lipoproteinlipasa, el glicerol-3-fosfato deshidrogenasa y la leptina. A largo plazo los efectos de un exceso de glucocorticoides sobre el tejido adiposo son más complejos, por lo menos en los humanos, en los que el depósito de tejido adiposo visceral o central se halla estimulado, lo que aporta un signo discriminatorio útil para el diagnóstico del síndrome de Cushing (Stewart, 2009). Si bien el cortisol puede provocar una movilización moderada de los ácidos grasos en el tejido adiposo, las personas que presentan una secreción excesiva de cortisol desarrollan una obesidad peculiar: la grasa sobrante se deposita en el tórax y en la cabeza y produce el cuello de búfalo y la cara redonda de luna llena (Guyton y Hall, 2006). 4.5 Efecto del cortisol en el metabolismo de proteínas El cortisol disminuye la reserva proteínica de casi todas las células del cuerpo, exceptuando las del hígado, por una disminución en la síntesis y un aumento del catabolismo de las proteínas celulares. Estimula el transporte de aminoácidos hacia las células hepáticas y el aumento de enzimas para el anabolismo proteínico (las proteínas del plasma que se sintetizan en el hígado y luego pasan a la sangre, aumentan al igual que las proteínas hepáticas), disminuye el transporte de aminoácidos hacia las células musculares y otras células extrahepáticas (disminuye la concentración de aminoácidos y la síntesis de proteínas) (Guyton y Hall, 2006). 4.6 Datos de laboratorio para diagnosticar la presencia de enfermedad de la glándula suprarrenal (González y col., 2001). 1. Hiponatremia: Los niveles séricos bajos de sodio se deben a su pérdida por la orina por déficit de aldosterona y al desplazamiento del sodio hacia el compartimento intracelular. Esta pérdida de sodio extravascular reduce el volumen plasmático y acentúa la hipotensión. 90 2. Hipercalemia: Aumento de los niveles séricos de potasio. Se debe a los efectos combinados del déficit de aldosterona, la reducción del filtrado glomerular y la acidosis. 3. Hipocortisolemia: Los niveles de cortisol y aldosterona son bajos y no aumentan con la administración de ACTH.. 4. Hipercalcemia: Aumento de los niveles séricos de calcio. Ocurre en un 10-20% de los pacientes de causa desconocida. 5. Cambios electrocardiográficos: Suelen ser inespecíficos, aunque con lentificación generalizada del trazado. 6. Hemograma: Puede haber anemia normocítica, linfocitosis relativa y eosinofilia moderada. 7. Prueba de estimulación de ACTH: Prueba principal que confirma el diagnóstico de insuficiencia suprarrenal, al evaluar la capacidad de las suprarrenales para producir esteroides, que suelen estar ausentes o disminuidos tanto en sangre como en orina tras la estimulación de ACTH. 8. Determinación de la ACTH: En la insuficiencia suprarrenal primaria o Enfermedad de Addison, la ACTH y sus péptidos afines están elevados en plasma ante la pérdida del mecanismo de retroalimentación del eje hipotálamo-hipofisario-suprarrenal. 9. Hipertermia: La hormona del hipotálamo no controla la homeostasis. 4.7 Patologías más frecuentes de la corteza suprarrenal Las enfermedades de la corteza suprarrenal son relativamente infrecuentes, pero su importancia radica en la morbilidad y mortalidad en caso de que no se traten, junto con la facilidad relativa del diagnóstico y la existencia de un tratamiento efectivo. Lo más fácil es clasificarlas atendiendo a la existencia de exceso o déficit hormonal (Stewart, 2009). 4.7.1 Enfermedad de Addison La enfermedad de Addison es el nombre que recibe una insuficiencia primaria de las glándulas suprarrenales con ausencia de secreción de cortisona y aldosterona. En consecuencia se produce una mayor cantidad de hormona 91 adrenocorticotropica y se activa el sistema renina-angiotensina (Figura 29). La enfermedad tiene una prevalencia de aproximadamente 9/100.000 y afecta predominantemente a las personas de 30 a 50 años. Además de la forma primaria existen una forma secundaria (reducción de la secreción de ACTH en la hipófisis) y la forma terciaria (causas hipotalámicas) (Netter y col., 2005). Figura 29. Cuando disminuye la presión arterial se libera renina (una enzima renal). La renina a su vez activa la angiotensina, una hormona que contrae las paredes musculares de las arterias pequeñas (arteriolas) y, en consecuencia, aumenta la presión arterial. La angiotensina también estimula la secreción de la hormona aldosterona de la glándula suprarrenal, provoca la retención de sal (sodio) en los riñones y la eliminación de potasio. Como el sodio retiene agua, se expande el volumen de sangre y aumenta la presión arterial) (Tomado de Netter y col., 2005). La enfermedad tiene casi siempre un avance lento y puede descompensarse a causa de enfermedades complementarias o estrés (las denominadas crisis addisionianas). Los síntomas de la insuficiencia crónica son debilidad, cansancio, reducción del rendimiento, trastornos gastrointestinales, pérdida de peso, deshidratación, hiperpigmentación de piel y mucosas (sobre todo cicatrices y pliegues de las palmas de las manos; solamente en la forma primaria), hipotensión, hipovolemia, hipoglucemia, acidosis y hambre de sal) (Netter y col., 2005). Las crisis addisionianas se producen a causa de un fallo total de las glándulas suprarrenales (a consecuencia del estrés o por una enfermedad crónica 92 desconocida) (Figura 30). Constituyen una situación de riesgo de muerte que se manifiesta mediante deshidratación, descenso de la tensión sanguínea, shock, dolor muscular y articular, dolor abdominal, fiebre y trastornos de la conciencia, llegando hasta somnolencia y coma (Netter y col., 2005). Los factores de riesgo para la enfermedad de Addison de tipo autoinmunitario incluyen otras enfermedades autoinmunitarias: tiroiditis crónica, dermatitis herpetiforme, enfermedad de graves, hipoparatiroidismo, hipopituitarismo, miastenia grave, anemia perniciosa, disfunción testicular, diabetes tipo 1, vitiligo (Stewart, 2009). Figura 30. Disfunción de las glándulas suprarrenales. Las glándulas suprarrenales pueden dejar de funcionar cuando uno de los dos, bien hipófisis o hipotálamo no producen una suficiente cantidad de hormonas adecuadas. La producción insuficiente o excesiva de las hormonas adrenales puede provocar enfermedades graves como la enfermedad de Addison (Tomado de Netter y col., 2005). Algunas veces se secretan enormes cantidades de glucocorticoides como respuesta a distintos tipos de estrés físico o mental. En las personas con enfermedad de Addison, la producción de glucocorticoides no aumenta durante 93 el estrés. Sin embargo, ante un traumatismo, enfermedad u otra clase de estrés como una intervención quirúrgica, es muy probable que se necesiten mayores cantidades de glucocorticoides. En general, para evitar su muerte, se requiere una dosis de glucocorticoides al menos 10 veces superior a la normal. Esta necesidad crítica de glucocorticoides suplementarios y la debilidad asociada a los períodos de estrés se conoce como crisis addisoniana (Guyton y Hall, 2006). El tratamiento de la enfermedad de Addison consiste en la sustitución hormonal de los glucocorticoides y mineralocorticoides que no se sintetizan. No debe olvidarse aumentar las dosis de glucocorticoides de manera profiláctica ante situaciones de estrés, como son infecciones, partos o cirugías para evitar las crisis addisonianas. El tratamiento se realiza en pacientes sintomáticos, no en aquéllos en los que tan sólo se aprecian anormalidades hormonales (Candel y col., 2001). El fármaco de elección para el tratamiento sustitutivo del déficit hormonal es la hidrocortisona o la cortisona, que presentan un efecto dual, tanto glucocorticoide como mineralocorticoide. La dosis de mantenimiento es de 2025 mg/día de hidrocortisona y 25-35 mg/día de cortisona, no olvidando la forma de administración fraccionada: 2/3 por la mañana y 1/3 por la tarde, emulando su ciclo circadiano. Pero la dosis se debe reducir paulatinamente a la mínima capaz de mantener al paciente asintomático (sin reducción de la fuerza física, entre otros) con objeto de evitar los efectos adversos originados por el uso de corticoides. La alternativa terapéutica similar en resultados es la asociación de dexametasona (2.5-7.5 mg/día) o de prednisona (0.25-0.75 mg/día) con un mineralocorticoide, por ejemplo la fludrocortisona a dosis de 100 mg/día, pero que puede oscilar entre 50-200 mg/día (0.05-0.1 mg/día). En los cuadros clínicos en los que no se pueda garantizar la absorción del 100% del fármaco, como en diarreas, la dosis podría administrarse por vía intramuscular 50 mg cada 12 horas hasta su total recuperación (Candel y col., 2001). 94 4.7.2 Síndrome de Cushing Cushing describió un síndrome constituido por obesidad del tronco, hipertensión, fatiga fácil y debilidad, amenorrea, hirsutismo, estrías abdominales purpúreas, edema, glucosuria, osteoporosis y un tumor basófilo de la hipófisis (Figura 31). A medida que se ha ido conociendo mejor, el diagnóstico del síndrome de Cushing se ha ampliado (Gangog, 2004). Independientemente de la etiología, todos los casos de síndrome de Cushing endógeno se deben al aumento de la producción de cortisol por la suprarrenal. La mayor parte de las veces la causa es una hiperplasia suprarrenal bilateral debida a hipersecreción de ACTH por la hipófisis o a la producción ectópica de ACTH por un tumor no hipofisario. La incidencia de hiperplasia suprarrenal dependiente de la hipófisis es tres veces mayor en las mujeres que en los varones y la edad de comienzo más frecuente es el tercero o cuarto decenio de la vida. La mayor parte de los datos indican que el defecto primario depende del desarrollo de novo de un adenoma hipofisario, pues en más de 90% de los pacientes con hiperplasia suprarrenal dependiente de la hipófisis se encuentran tumores (Williams y Dluhy, 2006). Tradicionalmente, sólo se define como caso de enfermedad de Cushing al individuo que tiene un tumor hipofisario productor de ACTH, en tanto que el término síndrome de Cushing se refiere a todas las causas de exceso de cortisol: tumor productor de ACTH exógena, tumor suprarrenal, tumor hipofisario secretor de ACTH o tratamiento con exceso de glucocorticoides (Williams y Dluhy, 2006). Los pacientes con síndrome de Cushing tienen deficiencia de proteínas a causa del catabolismo proteínco excesivo. Por tanto la piel y los tejidos subcutáneos son delgados y los músculos están poco desarrollados. Muchos de los aminoácidos liberados de las proteínas degradadas se convierten en glucosa en el hígado y la hiperglucemia resultante con disminución de la utilización periférica de glucosa pueden ser suficientes para desencadenar diabetes resistente a la insulina, sobretodo en pacientes con predisposición genética a la diabetes, pero por lo general la acidosis no es grave. El exceso de glucocorticoides induce la disolución ósea por descenso en la formación del 95 hueso y aumento en la resorción del mismo, lo que origina osteoporosis (Gangog, 2004). Figura 31. Manifestaciones clínicas del síndrome de Cushing (Tomado de O´Sullivan, 2000). La terapia farmacológica actualmente en uso clínico en los pacientes con síndrome de Cushing se reparte entre los fármacos de moduladores de la secreción de ACTH, los inhibidores de la biosíntesis de cortisol y los bloqueadores de receptores de glucocorticoides (Cuadro 16). Los inhibidores de la esteroidogénesis han demostrado una eficacia clínica superior y son los preferidos por la mayoría de los autores. La variedad de tratamientos médicos disponibles no son más que un reflejo de las imperfecciones de la terapia de un síndrome tan complejo y con un comportamiento biológico tan variable como el síndrome de Cushing. Los agentes farmacológicos en ningún caso constituyen el tratamiento definitivo del hipercortisolismo (Díez, 1999). La curación completa sólo es esperable tras una extripación quirúrgica completa del tumor, 96 ya sea adenoma hipofisario, tumor ectópico secretor de ACTH o tumor suprarrenal productor de cortisol, por lo que este procedimiento representa el tratamiento de elección en la mayoría de los pacientes. La radioterapia hipofisaria es un método que también ha demostrado a largo plazo una remisión completa del hipercortisolismo (Estrada y col., 1997). El papel del tratamiento farmacológico queda, por tanto, restringido principalmente a los pacientes que se encuentran en espera de una intervención quirúrgica o en espera del efecto de la radioterapia, o aquellos que presentan persistencia de la enfermedad tras cirugía, recidiva tras un período de remisión o contraindicación para la cirugía (Howlett y col., 1985). Cuadro 16. Clasificación de los fármacos potencialmente útiles en el tratamiento médico del Síndrome de Cushing en función de su mecanismo de acción (Tomado de Díez, 1999). Inhibición de la secreción de ACTH Inhibición de ACTH hipofisiaria Agonistas dopaminérgicos Bromocriptina Antagonistas Serotononérgicos Ciproheptadina Metergolina Ritanserina Agonistas GABA-érgicos Valproato sódico Inhibidores del almacenamiento de catecolaminas Reserpina Inhibición de ACTH ectópica Análogos de somatostatina Octreótida Lanreótida Vapreótida Inhibición de la secreción de cortisol suprarrenal Adrenocorticóliticos Mitotane Inhibidores de la P450scc Aminoglutetimida Inhibidores de la P450c11 Metopirona Inhibidores Múltiples Ketonazol Etomidato Bloqueadores de receptores de cortisol Mifepristone (RU 486) 97 Los cuidados preoperatorios son el conjunto de acciones realizadas al paciente previo al acto quirúrgico, destinadas a identificar condiciones físicas y psíquicas que puedan alterar la capacidad del paciente para tolerar el stress quirúrgico y prevenir complicaciones postoperatorias (O´Sullivan, 2000). Los cuidados pre-operatorios para los pacientes que padecen Enfermedad de Addison y Síndrome de Cushing son (O´Sullivan, 2000) (Anexo 7): 1. Control de hipertensión e hiperglucemia 2. Corrección de hipopotasemia con dieta y suplementos de potasio. 3. Dieta rica en proteínas (huevos, aves, pescados, carnes, productos lácteos, soya, frutos secos, champiñones, leguminosas y cereales). 4. Restringir alimentos ricos en sodio y en hidratos de carbono 5. Aumentar alimentos ricos en potasio (manzana, plátano, zanahoria, papa, brócoli, naranja, leche, yogurt). 6. Consumir alimentos ricos en calcio y vitamina D (lácteos, cereales, verduras de hoja verde, semillas de ajonjolí, almendras). 98 Referencias Bibliográficas. Aiello B, Nickleach J. 1998. Atención nutricional en enfermedades del hígado, vías biliares y porción exócrina del páncreas. En: Mahan K, EscottStump S. Nutrición y dietoterapia de Krause. McGraw-Hill Interamericana. 10ª edición México. pp 674-675. Alemzaden R, Wyatt D. 2004. Diabetes mellitus infantil. En: Behrman R, Kliegman R, Jenson H. Nelson Tratado de Pediatría. Elsevier. 17ª edición. España. pp 1955-1970. Anderson J. 2001. Nutrición para la salud ósea. En: Kathleen L y Escote-Stump S. Nutrición y dietoterapia de Krause. Interamericana McGraw-Hil. 10ª edición. México D.F. pp 663-88. Araiza R, Nazor N. 2001. Diabetes mellitus y nutrición. En: Casanueva E, Kaufer M, Pérez A. Nutriología Médica. Editoriam Médica Panamericana. 3° edición. México, D.F. pp 371-373 Bilezikian J, Potts J, Fuleihan G. 2002. Summary statement from a workshop on asymptomatic primary hyperparathyroidism: a perspective for the 21st century.J Bone Miner Res. 17(Suppl2):N2-N11. Bourges H. 1976. Recomendaciones de nutrimentos para la población mexicana. Cuadernos de nutrición. 1:273-94. Bruce M, Bruce S. 2006. Transporte de agua y solutos a lo largo de la nefrona: función tubular. En: Berne y Levy Fisiología. 4° edición. España. pp 578628. Candel F, Matesanz M, Candel I. 2001. Insuficiencia corticosuprarrenal primaria. Enfermedad de Addison. An. Med. Interna (Madrid) 18:9. Casellas J. 2008. Modificaciones dietéticas ante la diarrea. En: Salas J, Bonada A, Saló M, Burgos R. Nutrición y Dietética Clínica. Elsevier. 2 a edición. España. pp 516-517. 99 Champe P, Harvey R, Ferrier D. 2005. Bioquímica. McGraw-Hill. 4° edición México. pp 97-99, 349-360. Chávez M, Ledesma J. 2002. Tablas de valor nutritivo de los alimentos de mayor consumo en Latinoamérica. Mc GrawHill. 8ava edición. México. Pp 1-313. Danoff D, Laster L.1962. In vitro endocrine stimulation of active transport n the small intestine of hamster. J ClinInvest. 41:1376. Degroot L, Larsen P, Hennemann G. 1996. The thyroid and its diseases. New York: Churchill Livingtone. pp 823. De Santiago S, Halhali A, Frenk S, Bourges H. 2005. Calcio y Fosfato. En: Bourges H, Casanueva E, Rosado JL. Recomendaciones de la ingestión de nutrimentos para la población mexicana. Panamericana. Tomo 1. México. pp 217-229. Díaz C, Riobó P. 2006. Nutrición en las enfermedades del tejido conjuntivo y sistema óseo en el adulto. En: Gil A. ed. Tratado de Nutrición. Editorial Médica Panamericana. 2° edición. España. Tomo IV. Capítulo 4.34. pp 1019-36. Díez, J. 1999. Tratamiento farmacológico del síndrome de Cushing. Rev Cubana Med. 38(1):35-66. Dietary Reference intakes: Calcium and phosphorus. Institute of medicine. National Academy Press. 2° edición. Washington, DC. pp 30. Doherty JE, Perkins WH. 1966. Didoxin metabolism in hypo and hyperthyroidism. Studies with tritiated digoxin in thyroid disease. Ann Intern Med. 64:489. Dorantes LM. 2008. Metabolismo de calcio y sus alteraciones. En: Dorantes AY, Martínez C, Guzmán A. Endocrinología Clínica. El Manual Moderno. 2°edición. México. pp 179-191. 100 Dvorkin D, Cardinali C. 2009. Regulación del metabolismo. Diabetes Mellitus. En: Best y Taylor. Bases Fisiológicas de la Práctica Médica. Editorial Médica Panamericana. 13°edición. España. pp 761-778. Estrada J, Boronat M , Mielgo M, Magallón R, Millán I, Díez S.1997. The longterm outcome of pituitary irradiation after unsuccessful transsphenoidal surgery in Cushing’s disease. N Engl J Med. 336:172-177. Favus M. 1996. Primer on the metabolic bone diseases and disorders from mineral metabolism. Lippincott- Reaven 3ª edición. Philadelphia, EU pp 120-123. Ferri F. 2006. Hipotiroidismo En: Consultor Clínico, claves diagnósticas y tratamiento. Elsevier. 3° edición. Madrid, España. p 452. Flamant F, Baxter JD, Forrest D, Refetoff S, Samuels H, Scanlan TS, Vennstrom B, Samarut J. 2006. The pharmacology and classification of the nuclear receptor superfamily: thyroid hormone receptors. Pharmacol Rev 58 (4):705–11. Fox S. 2003. Fisiología humana. McGraw-Hill. 10° edición. España. pp 292327, 624-652. Freijanes J, Amado J. 1995. Alteraciones en el metabolismo mineral y el metabolismo del hueso. En: Gaztambide. Endocrinología clínica. Díaz de Santos. 1° edición. España. pp 119-130. Gal B, López M, Martín A, Prieto J. 2001. Glándulas suprarrenales. En: Bases de la fisiología. Tébar. 2° edición. Madrid. pp 407-428. Gangog W. 2004. Fisiología Médica. El Manual Moderno. 18°edición. México, D.F. pp 349-363, 365-387, 389-415, 417-430. García M, Iglesias M, Moratinas J. 2008. Fármacos antidiabéticos, insulinas y antidiabéticos orales. En: Lorenzo P, Moreno A, Lizasoain I, Leza JC, Moro MA, Portoles A. Velazquez Farmacología Básica y Clínica. Editorial Médica Panamericana. 18° edición. Madrid, España p 625. 101 García P, Olveira G, Bretón I. 2010. Nutrición en las enfermedades del páncreas exócrino. En: Gil A. Tratado de Nutrición. Nutrición clínica. Editorial Médica Panamericana. 2° edición. España. pp 771-789. Genuth S. 2006. Hormonas de los islotes pancreáticos. En: Levy M, Bruce K, Bruce S. Berne y Levy Fisiología. Elsevier. 4° edición. España. pp 617627. Gómez C, Rodríguez M, Cannata J. 2005. Metabolismo de calcio, fósforo y magnesio. En: Manual práctico de osteoporosis y enfermedades del metabolismo mineral. Editorial Médica Panamericana. 2° edición. España. pp 10-11. Gómez F, Del Rincón J, Morales H.1997. Biosíntesis y secreción de la insulina. En: Gómez-Pérez y Rull. Tratado de diabetología. Instituto Nacional de la Nutrición Salvador Zubirán. Editorial Médica Panamericana. 1° edición. México. pp 15-34. González C, Matesanz D, Monserrate C. 2001. Insuficiencia corticosuprarrenal primaria: Enfermedad de Addison. Med. Interna. Madrid, 18(9):48-54. González M, Pérez A, Gómez E. 1988. Tiroides. En: Flores F, Cabeza A. Endocrinología. Mc Editor. 2°edición. México, D.F. pp 420-424. Green K, Matty AJ. 1963. Action of thyroxine on active sodium transport in insolated membranes. Gen Comp Endocr. 3:244. Guyton, A., y E. Hall. 2006. Tratado de fisiología médica. Elsevier. 11° edición. Madrid, España. pp 955-967, 968-975, 985-996, 1002-1016 . Havala T, Shronts E, Cerra F. 1989. Nutritional support in acute pancreatitis. Gastro Clin North Amer 18:3 pp 525-542. Hacker J. 1988. Chronic pancreatitis. In: Chobanian SJ and Van Ness MM (eds): Manual in clinical problems in gastroenterology. Mc Graw Hill. Num edición 9. Boston. p. 103. 102 Herranz L. 2010. Hipertiroidismo En: Pallardo L, Morante T, Marazuela M, Rovira A. Endocrinología clínica. Díaz de Santos. 10° edición. España. pp 53-62. Holick W. 2002. Vitamina D. En: Shils M, Olson J, Shike M, Ross A. Nutrición en salud y enfermedad. McGrawHill. 9° edición. México. pp 390. Holt S. 1993. Chronic pancreatitis. South Med J. 86:201 Howlett TA, Rees LH, Besser GM. 1985. Cushing´s syndrome. Clin Endocrinol Metab. 14:911-945. Jameson L. 2006. Principios de endocrinología. En: Kasper DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL, Isselbacher KJ. Harrison Principios de Medicina Interna. McGrawHill. 16° edición. México. p 11011. Jameson L, Weetman A. 2006. Trastornos de la glándula tiroides. En: Kasper, DL, Braunwald E, Fauci AS, Hauser SL, Longo DL, Jameson JL, Isselbacher KJ. Harrison Principios de Medicina Interna. McGrawHill. 16° edición. México. pp 11167-11186. Johansson H. 1966. Gastrointestinal motility functions related to rhyroid activity. Acta Chir Scand. Suppl.359:1. Kronenberg H, Melmed S, Polonsky K, Larsen R. 2009. Fisiología de la tiroides y evaluación diagnóstica de los pacientes con trastornos tiroideos. En: Williams Tratado de Endocrinología. Editorial Elsevier. 11° edición. España. pp 326-382. Landa L, Rosas I, Serrano M. 2006. Recomendaciones dietéticas en Hipertiroidismo. http://www.infarmate.org.mx/PDF2/INDICE.pdf Laso F. 2010. Patología del páncreas. En: Laso F. Introducción a la medicina clínica fisiopatología y semiología. Elsevier. 2° edición. España. pp 135137. 103 Latarjet M, Ruiz A. 2008. En: Anatomía Humana. Editorial Médica Panamericana. 4° edición. China. pp 1693-1699. Leibel N, Pat M. 2007. Disorders of the thyroid hormone. En: Zaoutis LB, Chiang VW. Comprehensive pediatric hospital medicine. Elsevier. Num edición. Filadelfia, Pennsylvania, EU. pp 584. Lisci A. 2008. Historia de la endocrinología. En: Dorantes AY, Martínez C, Guzmán A. Endocrinología Clínica. El Manual Moderno. 1° edición. México. pp 3-7. Mahan K, Arlin M .1995. Cuidado nutricional en diabetes melitus e hipoglicemia reactiva En: Kathleen L. y Escote-Stump S. Nutrición y Dietoterapia de Krause. Interamericana McGraw-Hill. 10° edición. México D.F. pp 535564. Maitra A, Abbas AK. 2005. El sistema endocrino. En: Kumar V, Abbas A, Fausto N. Patología Humana. Elsevier. 1° edición. España. p 1195. Maldonado J, Mataix J. 2009. Páncreas exócrino. En: Mataix J. Tratado de Alimentación y Nutrición. Situaciones fisiológicas y patológicas. Ergón. 2° edición Barcelona, España. pp 1344-1354. Marino M, McCluskey R. 2000. Role of thyroglobulin endocytic pathways in the control of thyroid hormone release. Am J Physiol Cell Physiol. 279:12951306. Martínez H, Castañeda R, González M, Ramos R, Velásquez L. 2005. Iodo. En: Bourges, H, Casanueva E, Rosado JL. Recomendaciones de la ingestión de nutrimentos para la población mexicana. Panamericana. Tomo1. México. pp 303-313. Marx SJ. 2000. Hyperparathyroid and hypoparathyroid disorders. N Engl J Med 343:1863-1875. 104 Mason, MB. 2007. Vitamins, trace minerals, and other micronutrients. En: Goldman L, Ausiello D. Cecil Medicine. Elsevier. 23 edición. Philadelphia. pp 237. Mataix J, S Monereo. 2009. Enfermedades óseas: osteoporosis, raquitismo y osteomalacia. En: Mataix J. Tratado de Alimentación y nutrición, Situaciones fisiológicas y patológicas. Ergón. 2° edición. Barcelona, España. pp 1595-1624. Mataix J, Riobó P. 2009. Paratiroides. En: Mataix J. Tratado de Alimentación y nutrición, Situaciones fisiológicas y patológicas. Ergón. 2° edición. Barcelona, España. pp 1640-1643. Mataix J, Vázquez C. 2009. Tiroides. En: Mataix J. Tratado de Alimentación y nutrición, Situaciones fisiológicas y patológicas. Ergón. 2° edición. Barcelona, España. pp 1640-1643. Mawer B, Davies M. 2001. Vitamin nutrition and bone disease in adults. Rev Endocr Metab Disord. 2:153-164. Mendoza N. 2008. Farmacología Médica. Editorial Médica Panamericana. 3° edición. México. pp 452-465. Morales G, Bahena R. 2003. Hiperparatiroidismo primario: Características clínicas e histopatológicas. Revisión basada en evidencias. Médica Sur. 10:4-17. Morgan L, Weinsier R. 2000. Nutrición clínica. Editorial Harcourt. 2° edición. Madrid, España. pp 25-31. Netter F, Bottcher T, Engelhardt S, Kortenhaus M. 2005. Medicina Interna. Masson. Barcelona, España. p 62-65. Norma oficial mexicana NOM-040-SSA1-1993, Bienes y servicios. sal yodada y sal yodada fluorurada. especificaciones sanitarias. Consultado el: 10 de mayo de 2011 http://www.salud.gob.mx/unidades/cdi/nom/040ssa13.html. 105 en: Norma Oficial Mexicana NOM-015-SSA2-1994, Para la prevención, tratamiento y control de la diabetes. Consultado el: 13 de enero de 2011 en: http://www.salud.gob.mx/unidades/cdi/nom/m015ssa24.html. Norma oficial mexicana Nom-043-ssa2-2005, servicios básicos de salud. Promoción y Educación para la salud en materia alimentaria. Criterios para brindar Orientación. Consultado el: 13 de enero de 2011 en: http://www.nutrinfo.com/pagina/info/nom.pdf Ochoa C, García J. 2008. Alteraciones de la secreción de mineralocorticoides. En: Dorantes, A.Y., C. Martínez, A. Guzmán. Endocrinología clínica. El Manual Moderno. 3° edición. México. pp 221-237. O´Sullivan S. 2000. Cuidados de enfermería de los adultos con trastornos del hipotálamo, de la hipófisis ó de las suprarrenales. En: Beare y Myers: Enfermería médico quirúrgica. Tébar. 3ª edición Madrid. pp 1370-1375. Page C, Curtis M, Sutter M, Walker M, Hoffman B. 1998. Fármacos y sistemas del sistema endocrino y metabólico. En: Farmacología Integrada. Harcourt. 1° edición. España. pp 253-262. Parsons V, Anderson J. 1964. The maximal tubular resorptive rate for inorganic phosphate in thyrotoxicosis. Clin Sci. 27:314. Parsons V, Ramsay I. 1968. Thyroid and adrenal relationships. Postgrad Med J. 44:377-384. Pérez A, Palacios B, Castro A. 2008. Sistema Mexicano de Alimentos Equivalentes. Ogali. México, D.F. pp 14-89. Plum L, De Luca H. 2009. The functional metabolism and molecular biology of vitamin D action. Clin Rev Bone Miner Metab. 7:20-41. Pocock G, Richards C. 2005. Fisiología Humana, la Base de la Medicina. Masson. 2° edición. Barcelona, España. pp 235-242. 106 Raimondo M, Dimagno E. 2002. Nutrición en enfermedades pancreáticas. En: Shils M, Olson J, Shike M, Ross A. Nutrición en Salud y Enfermedad. McGraw-Hill. 9° edición México. pp 1351-1358. Rang H, Dale M, Ritter J, Flower R. 2008. Páncreas endocrino y control de la glucemia. En: Rang y Dale. Farmacología. Elsevier. 6° edición. España. pp 399-341. Riella M, Martinis C. 2004. Nutrición en la progresión de la insuficiencia renal crónica. En: Nutrición y Riñón. Editorial Médica Panamericana. 1° edición. Argentina. pp 102-103. Riobó P, Rapado A. 2002. El cuerpo humano: sistema óseo. En: Torre A. Técnicas y métodos de investigación en nutrición humana. Glosa. 1° edición. Barcelona, España. pp 219-30. Ríos J. 2008. Principios de endocrinología. En: Dorantes AY, Martínez C, Guzmán A. Endocrinología Clínica. El Manual Moderno. 2° edición. México. pp 9-14 Rodríguez A, Roldán B, Rodríguez D. 2007. Patología tiroidea. Pediatr Integral. 7:581-592. Shobak D. 2008. Hypoparathiroidism. New Engl J Med. 359:391-403. Steinberg T. 1994. Acute Pancreatitis. New Engl J Med. 30:1. Abrev revista Stewart P. 2009. La corteza suprarrenal. En: Kronenberg H, Melmed S, Polonsky K, Larsen R. Williams Tratado de Endocrinología. Elsevier. 11°edición. España. pp 453-503. Stiges-Serra A. 2010. Hiperparatiroidismo primario. En: Parrilla P, Landa J. Cirugía AEC. Editorial Médica Panamericana. 2° edición. España. pp 845-850. Swearingen P. 2008. Hipotiroidismo. En: Manual de enfermería médico quirúrgica. Elsevier. 6° edición. España. pp 537-542. 107 Tébar F, Ferrer M. 2009. Concepto, clasificación y diagnostico de la diabetes mellitus. En: Tébar F, Escobar F. La diabetes mellitus en la práctica clínica. Editorial Médica Panamericana. 1°edición. Buenos Aires, Argentina. pp 1-20. Thompson M, Nussbaum R, Thompson J, Mclnnes R, Williard F. 2008. Diabetes mellitus no insulinodependiente. En: Thompson y Thompson Genética en medicina. Masson. 5°edición. España. pp 292-298. Torún B, Menchú M, Elías L.1994. Recomendaciones dietéticas diarias del INCAP. Instituto de Nutrición de Centro América y Panamá (INCAP). Organización Panamericana de la Salud (OPS). Nutr Res. 20:215-224 Vandenbeld AW, Visser TJ, Feelders RA, Grobbe EG, Lamberts WJ. 2005. Thyroid hormone concentrations, disease physical function, and mortality in elderly men. J Clin Endocrinol Metab 90:6405-6409. Valdivieso S, Kripper C, Ivelic J, Fardella C, Gloger S, Quiroz D. 2006. Alta prevalencia de disfunción tiroidea en pacientes psiquiátricos hospitalizados. Rev Med Chile. 134:623-628. Valero M, Leon M. 2010. Nutrición en la diabetes mellitus. En: Gil A. Tratado de Nutrición. Nutrición clínica. Médica Panamericana. 2°edición. Madrid, España. pp473-450. Velazco J, Rodriguez J.1998. Bioquímica Clínica y Patología Molecular. Reverté. 2° edición. Barcelona, España. pp 915-928. Williams G, Dluhy R. 2006. Hiperfunción de la corteza suprarrenal. En: Kasper D, Braunwald E, Fauci A, Hauser S, Longo D, Jameson J y Isselbacher K. Harrison principios de medicina interna. McGrawHill. 16° edición. México. pp 11278-11295. Yen PM. 2001. Physiological and molecular basis of thyroid hormone action. Physiol Rev 81 (3): pp. 1097–142. 108 Anexo 1. Fuentes de Vitaminas y Nutrimentos inorgánicos (Nom-043SSA2-2005) Fuentes de Vitaminas y Nutrimentos inorgánicos Fuentes de Hierro: Alimentos de origen animal.- hígado, moronga, carne de res seca, carnes rojas, huevo, mariscos. Leguminosas.- frijol, lenteja, habas, garbanzos secos, soya. Verduras.- chiles secos, calabacita, acelgas, espinacas, verdolagas, quelites, hojas de chaya, tomatillo, chile poblano, hongos, col. Cereales.- productos elaborados con harinas adicionadas. Otros.- frutas secas, cacahuates y nueces. Fuentes de Zinc: Alimentos de origen animal.- leche y derivados, carnes, huevo, mariscos (ostras). Leguminosas.- alubias Verduras.- germen de trigo Otros.- levadura de cerveza, cacahuate, semillas de girasol, semillas de calabaza, nuez, almendras. Calcio Cereales: tortillas y productos elaborados con maíz nixtamalizado. Alimentos de origen animal: queso, leche, yogurt, acociles, sardinas, charales, boquerones. Fósforo Cereales: salvado de trigo, germen de trigo. Alimentos de origen animal: carne, pescado, lácteos. Oleaginosas: semillas de ajonjolí, semillas de girasol. Fibra dietética: Cereales: tortillas y otros productos elaborados con maíz nixtamalizado, cebada, salvado, harinas integrales, avena, pan y cereales integrales. Verduras (de preferencia crudas y con cáscara): brócoli, col, zanahoria, coliflor, elote, 109 chícharos, espinacas, nopales, acelgas, verdolagas y berros. Frutas (de preferencia crudas y con cáscara): chabacano, plátano, moras, dátiles, higos, guayaba, naranja y toronja en gajos, pera, manzana, mango y tamarindo. Leguminosas: frijol, lentejas, cacahuate, habas, garbanzos, soya. Otros: ciruela pasa, pasas. Vitamina C: Verduras (principalmente crudas): chile poblano, hojas de chaya, chile, col, pimiento rojo, coliflor, brócoli, miltomate (tomate verde o tomatillo), chile seco, habas verdes, tomatillo. Frutas: guayaba, marañón, kiwi, zapote negro, mango, limón, mandarina, papaya, fresa, toronja, naranja, tejocote, melón. Vitamina B12 Alimentos de origen animal.- Hígado, corazón, riñón, carne, aves, pescado, leche, queso, huevo, cereales fortificados. Acido fólico: Alimentos de origen animal.- hígado y otras vísceras. Verduras.- berro, espinaca, lechuga, espárrago, betabel, acelga, alcachofas, brócoli, coliflor, chícharo, aguacate, col, elote. Frutas.- naranja, plátano. Cereales.- productos elaborados con harinas adicionadas. 110 Anexo 2. Recomendaciones para disminuir los niveles de colesterol y triglicéridos (Chávez y Ledesma, 2002; Pérez y col., 2008). Alimentos Permitidos: - Cortes magros de carne: pescado, bistec de res, atún en agua, sardinas. Pollo y pavo sin piel. Claras de huevo. Leche y yogurt descremados (light). Quesos: panela, requesón y cottage. Grasas: aceite de cártamo, maíz, olivo y aguacate. Cereales: arroz, pastas, tortilla de maíz, pan integral, papas, bolillo, galletas marías, galletas habaneras, avena, cereales con alto contenido en fibra. Frutas y verduras: todas. Evitar los siguientes Alimentos por su alto contenido de grasa: - Carne de res, cerdo y cordero grasos. Vísceras: hígado, riñón, tripas, corazón, pancita. Pollo y pavo con piel. Carnes frías y embutidos: mortadela, jamón de cerdo, salami, tocino, salchichón, chistorra, chorizo, queso de puerco, pastel de pollo. Yema de huevo. Leche y Yogurt entero. Quesos grasosos: Oaxaca, chihuahua, manchego, amarillo. Grasas: manteca, mantequilla, margarina, mayonesa, crema, aderezos comerciales, aceitunas, sopas de crema. Alimentos con alto contenido de azúcares simples: helados, chocolate, pan dulce, galletas dulces, postres (flan, pay, pastel, gelatina…), mermelada, miel, cajeta, leche condensada, refresco, jugo natural y artificial. Recomendaciones generales: - Fija el horario de tus comidas. Consume fruta 3 veces al día y 2-3 veces al día verdura. Aumenta el consumo de fibra (avena, cereales, panes integrales, frutas y verduras con cáscara). Consume los alimentos cocidos ó asados. Deja de fumar ó tomar bebidas alcohólicas progresivamente. Practica un deporte ó actividad física mínimo 30 minutos diarios. Toma mínimo 8 vasos de agua al día. Recuerda tomar tus medicamentos según las indicaciones de tu Doctor. 111 Anexo 3. Recomendaciones para paciente con hiperparatiroidismo dieta hiposódica e hipoproteínica (Chávez y Ledesma, 2002; Pérez y col., 2008). Alimentos permitidos: • Cereales: pan blanco, pan integral, harina de trigo integral, arroz, arroz integral. • Verduras: pepino, cebolla, pimiento, rábano, tomate, berenjena, aguacate, calabaza, lechuga, champiñones, setas, espárragos, papa, habas verdes, alcachofas, col ó Si la dieta es estricta en sal, se deben restringir: zanahorias, espinacas, col y acelgas. • Frutas: manzana, pera, sandía, limón, níspero, mango, piña, ciruelas, melón, pomelo, uvas, fresa, cereza, mandarina, naranja, frambuesa, fruta en almíbar, kiwi, higos, plátano, uva, pasa, ciruela seca, higos secos. • Alimentos de Origen Animal: - Eliminando todos aquellos elaborados con sal: ahumados, secados, embutidos, enlatados… • Lácteos y derivados: leche de cabra, vaca, flan, natillas, arroz con leche, helados, yogur de frutas y natural. • Grasas y aceites: aceites, margarina, mantequilla, mayonesa sin sal. • Azúcares y dulces: mermelada, miel. • Bebidas: agua de frutas, te. Evitar los siguientes alimentos (por su alto contenido proteínico): • Cereales y derivados: habas secas, garbanzos, soya en grano. • Oleaginosas: nueces, almendras con cáscara, cacahuetes sin cáscara. • Alimentos de origen animal: embutidos (chorizo, salami, peperoni…), carnes frías (tocino, pastel de pollo, queso de puerco), alimentos fritos, capeados, empanizados o dorados. 112 • Lácteos: leche en polvo, leche condensada azucarada, cuajada, queso parmesano, roquefort, de bola, gruyere, manchego. • Grasas y aceites: manteca de cerdo y crema. • Azúcares: cajeta, dulces, bebidas carbonatadas e industrializadas. • * Si está indicada una dieta hiposódica estricta estará prohibido todo el marisco. Para hacer más sabrosa la cocina "sin sal" es recomendable potenciar el sabor natural de los alimentos: • Cociéndolos al vapor, la cocción simple con agua diluye el sabor de los alimentos. • También se puede envolver el alimento en papel de horno, estofados y guisados o a la plancha. • Utilizando potenciadores de sabor como: ácidos: vinagre, limón (los asados de carne roja o blanca con limón se dará un mejor sabor de las mismas e incluso un filete a la plancha gana en sabor), condimentos: ajo, cebolla, especias: pimienta, curry, azafrán, canela, mostaza sin sal, Hierbas aromáticas: albahaca, comino, laurel, menta, perejil, romero, tomillo… • Utilizando aceite con sabor, como el de oliva. El vinagre y el aceite pueden ser aromatizados con finas hierbas. Recomendaciones generales: - Fija el horario de tus comidas. Consume fruta 3 veces al día y 2-3 veces al día verdura. Aumenta el consumo de fibra (avena, cereales, panes integrales, frutas y verduras con cáscara). Consume los alimentos cocidos ó asados. Deja de fumar ó tomar bebidas alcohólicas progresivamente. Practica un deporte ó actividad física mínimo 30 minutos diarios. Toma mínimo 8 vasos de agua al día. Recuerda tomar tus medicamentos según las indicaciones de tu Doctor. Elimina los alimentos prohibidos de la alacena. 113 Anexo 4. Raciones o porciones diarias para un adulto sano (Nom043-ssa2-2005) Grupo de Alimento Cereales Porciones 6-8 Características Principal fuente de energía y fibra. Leguminosas 1-2 Energía, proteínas, magnesio, potasio y fibra. Verduras 4-5 Ricos en potasio, fibra y antioxidantes. Frutas Alimentos de origen animal 5-6 2-4 Ricos en potasio, fibra y antioxidantes. Deberá promoverse el consumo de carnes y quesos con bajo contenido de grasas saturadas (aves sin piel, pescado, queso cottage y tipo panela). Moderar el consumo de vísceras. Leche (descremada) Grasas y oleaginosas 1-2 3-4 Consumo de yema de huevo no deberá exceder dos piezas a la semana. bajo contenido en grasas Las grasas saturadas (origen animal) no deberán representar más del 10% de total de las calorías. Se recomienda el uso de grasas poliinsaturadas. 114 Anexo 5. Metas básicas del tratamiento y criterios para evaluar el grado de control del paciente diabético (Nom-043-SSA2-2005) Metas del tratamiento Glucemia en ayunas (mg/dl) Glucemia postprandial de 2 h. (mg/dl) Colesterol total (mg/dl) Triglicéridos en ayuno (mg/dl) Colesterol HDL (mg/dl) P.A. (mm de Hg) IMC HbA1c* Bueno Regular Malo <110 <140 110-140 <200 >140 >240 <200.0 <150 >40 <120/80 <25 <6.5%mg/dl 200-239 150-200 35-40 121-129/81-84 25-27 6.5-8%mg/dl >240 >200 <35 >130/85** >27 >8%mg/dl 115 Anexo 6.Recomendaciones para pacientes diabéticos (Chávez y Ledesma, 2002; Pérez y col., 2008). Alimentos Permitidos: - Cortes magros de carne: pescado, bistec de res, atún en agua, sardinas, pollo y pavo sin piel. Huevo: solo 2 piezas enteras a la semana. Leche y yogurt descremados (light). Quesos: panela, requesón, cottage. Grasas: aceite de cártamo, maíz, olivo y aguacate. Cereales: arroz, pastas, tortilla de maíz, pan integral, papas, bolillo, galletas marías, galletas habaneras, avena, cereales con alto contenido de fibra. Leguminosas: frijoles, habas, lentejas, garbanzos, soya, alubias, germen de trigo. Frutas y verduras. Alientos de consumo Moderado (1-2 veces por semana): - ½ taza de betabel o de zanahoria, ½ pieza de mango o de plátano, 10 piezas de uva, 2 piezas de higo, 1-2 piezas de huevo entero. Si se consume alguno de éstos alimentos hacerlo una sola vez al día en la cantidad indicada 1-2 veces por semana. Evitar los siguientes alimentos por su alto contenido de azúcar y grasa: - - Alimentos con alto contenido de azúcares simples: azúcar, miel, cajeta, leche condensada, mermelada, jalea, chocolate, pastel, galletas, pay, gelatina, flan, nieve, helado, fruta en almíbar, pan dulce, dulces, yogurt de frutas, jugos artificiales y naturales, refrescos, bebidas alcohólicas. Alimentos con alto contenido en grasas: Quesos grasos (oaxaca, chihuahua, manchego, gouda, queso amarillo), carnes grasosas (barbacoa, menudo, carnitas, sesos, cordero, arrachera etc.) tocino, longaniza, chorizo, queso de puerco, chistorra, leche y yogurt entero, pollo con piel, alimentos fritos, capeados, empanizados o dorados. Recomendaciones: - Aumenta el consumo de fibra (avena, cereales, panes integrales, frutas y verduras con cáscara). Consume los alimentos cocidos ó asados. Deja de fumar o tomar bebidas alcohólicas progresivamente. Practica un deporte ó actividad física mínimo 30 minutos diarios. Toma mínimo 8 vasos de agua al día. Recuerda tomar tus medicamentos según las indicaciones de tu Doctor. Elimina los alimentos prohibidos de la alacena. 116 Anexo 7.Recomendaciones para dieta blanda (Chávez y Ledesma, 2002; Pérez y col., 2008). Alimentos Permitidos: - - - Carnes: Pescado, pollo, carne de res ( sin freír) Quesos bajos en grasa: Panela, requesón ó cottage. Leche ó yogurt descremados Cereales: Tortilla de maíz, pasta, arroz, pan blanco, pan tostado, atole, amaranto, bolillo, avena, galletas maría ó de animalito, pan integral, pasta sin freír, cereal integral, papa y camote según tolerancia. Frutas de preferencia cocidas sin cáscara: Manzana, pera, papaya, ciruela, durazno, plátano. Verduras de preferencia cocidas y sin cáscara: Acelgas, jitomate, tomate, zanahoria, chayote, flor de calabaza, hongos, Preferentemente consuma verduras cocidas. Grasas: Aceite, aguacate, almendras, nueces. Otros: te claro. Alimentos de consumo Moderado (1-2 veces por semana): - Consumir 3-4 piezas de huevo (revuelto, sin freír, tibio, duro) como máximo en una semana. Moderar el consumo (máximo 2 veces a la semana) de productos industrializados (enlatados, refinados, ahumados, embutidos bajos en grasa). Evitar los siguientes Alimentos: - Alimentos de Origen Animal: carnes grasosas como barbacoa, menudo, arrachera, chorizo, queso de puerco, tocino, carnitas, moronga. Leche ó yogurt entero. Quesos: Grasos como el chihuahua, manchego, oaxaca, queso amarillo. Cereales: Tamal, pastel. Frutas: Tuna, guayaba, piña, fresa, melón, sandía, kiwi, uva y cítricos según tolerancia. Verduras: calabaza, cebolla, betabel, chícharos, pimiento, pepino, rábano, brócoli, col, coliflor, jícama, lechuga, calabaza, ejote, elote, nopal Leguminosas: Todas Grasas: Manteca, mantequilla, crema, mayonesa. Oleaginosas: a tolerancia. Azúcares: Refrescos, nieve, chocolate, cocoa, jugos embotellados, postres. Otros: Tabaco, irritantes (Condimentos, mole, frituras, empanizados, capeados, chile ó salsa, vinagre). Bebidas: Té negro, café, alcohol. 117 Recomendaciones: - Fija el horario de tus comidas. Consume fruta 3 veces al día y 2-3 veces al día verdura. Aumenta el consumo de fibra (avena, cereales, panes integrales, frutas y verduras con cáscara). Consume los alimentos cocidos ó asados. Deja de fumar ó tomar bebidas alcohólicas progresivamente. Practica un deporte ó actividad física mínimo 30 minutos diarios. Toma mínimo 8 vasos de agua al día. Recuerda tomar tus medicamentos según las indicaciones de tu Doctor. Elimina los alimentos prohibidos de la alacena. 118
 0
0
Anuncio
Documentos relacionados
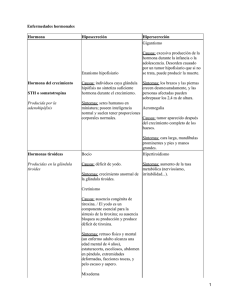
Descargar
Anuncio
Añadir este documento a la recogida (s)
Puede agregar este documento a su colección de estudio (s)
Iniciar sesión Disponible sólo para usuarios autorizadosAñadir a este documento guardado
Puede agregar este documento a su lista guardada
Iniciar sesión Disponible sólo para usuarios autorizados