Manual eTO
de Medicina y Cirugía
,
INMUNOLOGIA
Grupo CTO
Editorial
Manual eTO
de Medicina y Cirugía
@a
edición
,
INMUNOlOGIA
Autora
Sara CaLLeja Antolín
Director de la obra
Juan José Ríos Blanco
ERRNVPHGLFRVRUJ
Grupo CTO
Editorial
NOTA
La medicina es una ciencia sometida a un cambio constante. A med ida que la investigación y la experiencia
cl ínica amplían nuestros conocimientos, son necesarios cambios en los tratamientos y la farmacoterapia.
Los editores de esta obra han contrastado sus resultados con fuentes consideradas de confianza,
en un esfuel70 por proporcionar información completa y general, de acuerdo con los criterios aceptados
en el momento de la publicación. Sin embargo, debido a la posibil idad de que existan errores humanos
o se produzcan cambios en las ciencias médicas, ni los editores ni cualqu ier otra fuente implicada
en la preparación o la publicación de esta obra garantizan que la información contenida en la m isma sea
exacta y completa en todos los aspectos, ni son responsables de los errores u om isiones ni de los resultados
derivados del empleo de dicha información. Por ello, se recomienda a los lectores que contrasten dicha
información con otras fuentes. Por ejemplo, y en particular, se aconseja revisar el prospecto informativo
que acompaña a cada med icamento que deseen admin istrar, para asegurarse de que la información
contenida en este libro es correcta y de que no se han producido modificaciones en la dosis recomendada
o en las contraindicaciones para la administración. Esta recomendación resulta de particular importancia
en relación con fármacos nuevos o de uso poco frecuente. Los lectores también deben consu ltar
a su propio laboratorio para conocer los va lores normales.
No está permitida la reproducción total o parcial de este libro, su tratamiento informático, la transmisión
de ningún otro formato o por cualquier med io, ya sea electrónico, mecán ico, por fot ocopia, por registro
y otros medios, sin el permiso previo de [os titulares del copyright.
e CTO ED[TOR[AL, S.L. 2018
Diseño y maquetación: CTO Editoria [
C! A[barradn, 34; 28037 Madrid
Tfno.: (0034) 91 7824330 - Fax: (0034) 91 782 4343
E-mai[: ctoeditoria [@ctomedicina.com
Página Web: www.grupocto.es
[SBN Obra completa: 978-84-17095-00-0
[SBN Inmunología: 978-84-17095- 16-1
[SBN Genética: 978-84- 17095-17-8
Depósito legal: M-1939B-2017
Manual eTO
de Medicina y Cirugía
@a
edición
Grupo CTO
Editorial
,
•
Ice
01. Estructura del sistema inmunitario
05. Respuesta inmunitaria .
1
1.1.
Introducción. Inmunidad __
1
1.2.
Órganos del sistema inmunitario.
1
5.1.
5.2.
5.3.
02. lnmunoglobulinas
4
2.1.
2.2.
Estructura y función de las inmunoglobulinas_
Clases de inmunoglobulinas __
2.3.
Antígeno, inmunógeno, epítopo,
idiotipo, hapteno e isotipo
Unión antígeno-anticuerpo: afinidad yavidez.
Cambio de clase de inmunoglobulina_
2.4.
2.5.
03. Células del sistema inmunitario
5.4.
5.5.
5.6.
4
s
Respuesta inmunitaria __
16
Respuesta de anticuerpos primaria
y secundaria
16
Respuestas de las células T. Cooperación
y citotoxicidad.
17
Alorreactividad _
18
Tolerancia.
18
Envejecimiento e inmunidad.
19
6
7
06. Complemento .
...
....
....
....
....
20
7
6.1.
6.2.
6.3.
6.4.
6.5.
6.6.
6.7.
8
3.1.
3.2.
Linfocitos T.
Linfocitos B.
8
11
3.3.
3.4.
Linfocitos granulares grandes. Células NK.
Células presentadoras de antígeno.
11
11
Funciones del complemento ..
20
Vías de activación del complemento
20
Vía común.
20
Regulación del complemento.
20
Receptores para el complemento_
21
Complemento e inflamación ..
21
Cascada de las cininas.
21
07 . Inmunología clínica . . . .
04. Complejo principal
de histocompatibilidad .
16
7.1.
7.2.
7.3.
7.4.
13
4.1.
Introducción_
13
4.2.
4.3.
Moléculas HLA de clase I y de clase 11 _
13
Genética del sistema HLA y nomenclatura ..
14
4.4.
HLA Y enfermedad __
14
VI
....
...
....
22
Trasplante de órganos ..
22
Reacciones de hipersensibilidad .
23
Alergia
24
Inmunidad tumoral
2S
ERRNVPHGLFRVRUJ
,
Indice
INMUNOLOGíA
08. lnmunodeficiencias
8.1.
8.2.
8.3.
8.4.
8.5.
8.6.
8.7.
8.8.
8.9.
09. lnmunoterapia
28
Concepto de inmunodeficiencia
28
Clínica de los defectos inmunitarios
28
Inmunodeficiencias secundarias.
Inmunodeficiencias primarias __
29
Inmunodeficiencias primarias humorales
30
Inmunodeficiencias primarias combinadas.
32
Defectos primarios de la función fagocítica
Defectos primarios
por disregulación inmunológica
33
9.1.
9.2.
9.3.
9.4.
29
Síndromes bien definidos que cursan
con inmunodeficiencia (primaria)_
Inmunosupresores clásicos __
37
Anticuerpos monoclonales y proteínas
de fusión
37
Gammaglobulinas
Otros fármacos _
Bibliografía .
33
33
8.10. Deficiencias primarias del complemento
8.11. Síndromes autoinflamatorios_
8.12. Evaluación de la inmunidad.
34
34
3S
VII
37
ERRNVPHGLFRVRUJ
38
39
40
ERRNVPHGLFRVRUJ
Estructura
del sistema inmunitario
En este tema se trilla la estructura general del >istema i nmun ~ario, Se debe
prestar iIIffición a las células y molk ulas JIeI1enecientes a la inmunidad inn"la
ya lo adaptativa. En los últimos años han (Obrado importancia en elexamen
cuestiones relationad.Js con los rffeptores presentes en las (BuloS
de la inm unidad innata.
La inmun idad innata o natural está constitu ida, entre otros, por los sigu ien-
tes componentes:
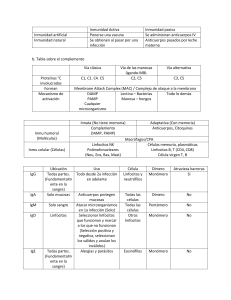
Introducción. Inmunidad
•
Las barreras epiteliales.
•
Inmunidad innata celular: fagocitos (monocit os-macrófagos y leucocitos
polimorfonucleares [PMN)) y células agresoras natura les (células natu-
La inmunología es la ciencia que estudia el sistema inmunitario (SI) y las
rol killer) V cé lulas déntricas.
patologías con él re lacionadas. El sistema inmunitario es el encargado de
•
proteger al individuo de las agresiones procedentes tanto del medio externo
Inmunidad innata humoral: lisozima, complemento e interferones (MIR
13· 14, 61).
como del medio interno, así como de ser capaz de aprender a to lerar los
RECUERDA
agentes no patogén icos.
Los linfocitos T no son componentes de la inmun idad natural.
Los diversos componentes que lo forman (células y moléculas solubles), se
d istribuyen por todos los sistemas del organismo siendo el aparato digestivo
el de mayor concentración.
Inmunidad adaptativa o específica
Esta disposición ubicua hace que sea imprescindible la existencia de complejos y precisos mecanismos de intercomunicación y coordinación, así como de
"seña les" de recirculac ión que permitan la movilidad de estas células. Clási-
Se caracteriza por la especificidad de sus compon entes por el antígeno y por
camente, se pueden diferenciar dos mecanismos de inmunidad, la innata o
taria, cada vez más potente y rápida, frente al antígeno en cuestión).
poseer memoria (exposicion es posteriores producen una respu esta inmun i-
tamb ién llamada inespecífica, y la adaptativa o específica.
Tras la entrada, por primera vez, de un germen en el organismo se desarrolla
Inmunidad innata o inespecífica
una respu est a inm unita ria primaria. Dicha respuesta se puede estructurar
en tres etapas:
Sus componentes se encuentran siempre presentes y dispuestos para act uar
•
Reconocim iento del antígeno.
inmediatamente sin requer ir tiempo de latencia para el desencadenam iento
•
Periodo de latencia, que dura varios días, en los que los li nfocit os espe-
de las acciones defensivas. La inmunidad innata no es específica de antígeno
cíficos amplifican su número (expa nsión clonal), a la vez que se d iferen-
ycarece de memoria. Es decir, sus respuestas no registran un aumento de su
cian en células efectoras.
eficacia en sucesivas exposiciones al mismo (MIR 10-11, 209).
•
Respuesta efectora, que consiste en:
Secreción de anticuerpos específicos.
Aunque no es específica de antígeno, sí que es capaz de diferenciar patro-
Desarrol lo de actividad citolítica específica.
nes de estructuras microbianas conservadas o pertenecientes a grandes
Liberación de factores que activan las células fagocíticas.
grupos de m icroorganismos, denominados PAMP (pathogen -associated
Ad qu isición de memoria inmunit aria.
molecular patterns) (MIR 11· 12, 215) (LPS, secuencias de ADN viral, ADN
bacteriano ... ), activando así d iferentes mecanismos de activación intra ce lula r, que van a condicionar u orientar la respu esta adaptativa que van
a reclutar (MIR 09-10, 214). También tiene la capacidad de reconocer
Órganos del sistema inmunitario
señales endógenas de daño celu lar (como, por ejemplo, el ácido úrico),
llama dos en su conjunto DAMP (damage-associated molecular patterns).
Los PAMP y los DAMP son reconocidos por receptores que las células de
la inmunidad innata poseen de forma mayorita ria, aunque no exclusiva -
Los linfocitos son las principales células re sponsables de la respue st a inmu-
mente, en su membrana plasmática y que de forma genérica reciben el
nombre de PRR (pattern recognitlan receptors), siendo los princi pa les los
nitaria adaptativa. Est án distribu idos por todo el organismo en órganos bien
delimitados, o en form a de acumulaciones d ifusas; al conjunto de estas
receptores TLR (ta/l like receptor) y los NLR (nod like receptor). Muestran
estructuras se le denom ina sistema linfático y están en intercomun icación
especial interés el TLR2 y el TLR4 por su implicación en el reconocimiento
continua gracias al tránsito, desde unas a otras, de los linfocitos a través de
de po lisacáridos (M IR 09· 10, 217). Algunos polimorfismos genéticos en la
las circulaciones sanguínea y linfática. Los órganos linfoides se dividen en
secuencia del gen NOD2 se han asociado con susceptibilidad a enfermedad
dos grandes categorías: órganos linfoides primarios (centrales) y secundarios
inflamatoria intestinal.
(periféricos).
1
ERRNVPHGLFRVRUJ
INMUNOLOGÍA
órganos linfoides primarios (centrales)
Ganglios linfáticos
Se consideran órganos linfoides primarios aquéllos en los que se originan y
A través de la linfa, los antígenos procedentes del medio extracelu lar de
maduran, hast a alcanzar su competencia funcion al, las células del sistema
los tejidos son conducidos hacia los ganglios linfáticos, bien directamente o
inmun itario.
mediante células presentadoras de antígenos procedentes de esos tejidos.
Médula ósea
La localización anatómica de los ganglios linfáticos se sitúa en zonas de confluenci a de varios vasos linfáticos (Figura 1).
Los linfocitos proceden de las células madre hematopoyéticas, ést as son de
origen mesodérmico y aparecen inici almente en el saco vitelina del embrión
para luego trasladarse al hígado (en la sexta semana) y más tarde (a partir de l
quinto mes) a la médula ósea, que es el órgano hematopoyético fundame n-
ta l pa ra el resto de la vida .
Los linfocitos que maduran (se diferencian) en la médula ósea se denominan
linfocitos B (del inglés bone marrow) y están especi alizados en la producción de
Sazo
Pu lmón
anticuerpos y, por tanto, son los principales actores de la inmunidad humoral.
Piel
El microambiente de la médula ósea que determina la maduración de los
Placa~
I¡nfático~
de Peyer
linfocitos B no se conoce con precisión. Se cree que im plica la liberación de
factores solubles (como la IL·7) y estimulaciones yuxtacrin as (entre células
periféricos
Linfát ico~ eferentes
adyacentes) que llevan a cabo las células del estroma medular. También es
Esquema de La circulación Linfática
muy importante la interacción de las células inmaduras con proteínas de la
matriz extracelular.
Tienen una forma sim ilar a la del riñón, con una longitud y grosor, respectiva-
Timo
mente, infe rio res a 1 y 0,5 cm, en condiciones fisiológicas.
Es un órgano linfoepitel ial, de forma bi lobulada, que resu lta imprescindible
Cuando se desencadena una respuesta, su tamaño aument a. Histológica-
para la adquisición de la inmunocompetencia de los linfocitos T durante los
mente, se distinguen tres zonas (Figura 2).
primeros años de la v ida. Aunque es en el timo donde los linfocitos T adquiere n su diferenciación y madurez, no se debe olvidar que sus precursores se
originan, al igual que ocurre con los linfoc itos B, en la médula ósea desde la
Cápsula - ,
que migran hacia el timo. El periodo clave de este proceso lo constituirían el
,
desarrollo ontogén ico y la infancia, ya que la extirpación del timo a un adu lto
Corteu¡ -
(o al final de la adolescencia, con el desarrollo completo del sistema inmuni-
,
~_ Cordones
/
Linfático
aferente ---,
tario), no implica un déficit inmunitario.
- Médula
El órgano deriva de un esbozo epitelial formado a partir de la tercera y cuarta
medu la res
, __ Linfático
;:::
eferente
bolsas faríngeas, y es el primer órgano linfoide que aparece. El ta maño del
timo aumenta a lo largo de la vida feta l y posnata l hasta alrededor de la
pubertad, momento a partir de l que empieza a involucionar. En el adulto,
' --
la producción y maduración de los linfocitos T tiene lugar en la médula. Es
importante conocer que el desarrollo de estos linfocitos en el timo sigue una
d istribución corticomedular, situándose en la médula, de forma mayoritaria,
los linfocit os T con mayor grado de m adu rez, desde donde ci rcu larán a los
Folículo _ J
órganos linfoides secundarios.
primario
,;;;:::~~;;;:''I
FolicuJo
L_ se<undario
órganos linfoides secundarios (periféricos)
B
Son los órganos donde los linfocit os ya maduros, e inmunológicamente
competentes, toman contacto con los antígenos y donde se producen las
respuestas inmun itarias frente a los estímulos antigén icos. Básicamente,
existen tres tipos de órganos lin foides secundarios: los ganglios linfáticos,
el bazo y el tejido linfoide asociado a mucosas (MALT) (MIR 08-09, 242). El
funcionamiento de los tres es simi lar, distinguiéndose básicamente por la
procedencia de los antígenos que penetran en ellos y que provienen, respectivamente, de:
1.
Linfa (medio extracelular de los tejidos), en el caso de los gangl ios linfáticos.
2.
Sangre, en el caso del bazo.
3.
Luz intestinal, en el caso de las pl acas de Peyer (tej ido MALT del intestino).
Áreas funcionaLes del ganglio linfático
2
ERRNVPHGLFRVRUJ
Hilio
al . Estructura del sistema Inmunitario
Manual CTO de Medicina y Cirugía, 10. 3 edición
•
Corteza. Donde se localizan los linfocitos S, formando los fo lículos lin-
secundario donde los linfocitos T y S vírgenes entran en contacto con los
foides primarios y secundarios, en los que se sitúa el centro germ ina l.
antígenos circulantes en la sangre, para poner en marcha la respuesta
Esta estructura (el centro germ inal) es la zona en la que se genera el
inmunitaria adaptativa; hay que recorda r que e l bazo carece de circu la-
microambiente adecuado para la presentación antigénica entre los lin-
ción linfática.
focitos S y los linfocitos T, así como para el desarrollo, a partir de esos
linfocitos S, de células plasmáticas y linfocitos S memoria.
La esplenectomía aumenta el riesgo de padecer infecciones por bacterias
•
Paracorteza. Poblada por linfocitos T dispuestos de manera difusa.
encapsuladas, ya que es el órgano en el que mayoritariamente se produce
•
Médula. Contiene linfocitos S y T. Los cordones medulares, que parten
su el iminación mediante la fagoc itosis de estas bacterias, una vez han sido
de la paracorteza como prolongaciones de tejido linfoide en la médula,
opsonizadas (rodeadas por inmunoglobu linas).
contienen la m ayor parte de las células plasmáticas que existen en el
gangl io.
Los linfocitos T son la población linfocitaria mayoritaria en el ganglio,
considerado en conjunto.
Tejido linloide asociado a las mucosas (MALT)
En la submucosa de los principa les puntos de posible entrada de microorganismos, se sitúan agregados de tej ido linfoide, difusos en la lámina propia
y/o en nódulos como las amígdalas y las adenoides (en la nasofaringe), o las
placas de Peyer (en el intestino).
También existen linfocitos intraepitel iales situados entre las células del epitelio, por encima de la membrana basal.
RECUERDA
En el MALT, la población linfocitaria mayoritaria son los linfocitos T.
El MALT desempeña un papel importante en la respuesta inmunit aria local
Células del sistema inmunitario asociadas a los bronquiolos terminales
y alvéolo
de la superficie de las mucosas (Figura 3).
Bazo
En el bazo se eliminan los hematíes envejecidos (pu lpa roja), pero además es un órgano linfoide secundario (pu lpa blanca) y en situaciones
PREGUNTAS ·
MIR
extremas puede producir hematopoyes is extramedular, al igua l que e l
hígado. El tejido linfo ide se organiza alrededor de las arteriolas a modo
de manguitos (tejido linfoide periarteriolar) y contiene áreas de linfocitos
,/ MIR 13·14, 61
,/ MIR 11·12, 215
,/ MIR 10·11, 209
,/ MIR 09·10, 214, 217
,/ MIR 08-(19, 242
Ty B, siendo los linfocitos S los mayoritarios. El bazo es e l órgano linfo ide
Ideasel ve
,/ Es posible clasificar los componentes del sistema inmunitario en los pertenecientes a la inmunidad innata y los pertenecientes a la inmun idad
adaptativa.
culares de diferentes grupos de patógenos y para moléculas que seña lizan daño celular.
,/ La inmunidad adaptativa posee receptores específicos de antígenos
y memoria inmunológica que optim iza la respuesta en sucesivos con tactos.
,/ La inmun idad innata no posee receptores específicos de antígeno y ca rece de memoria; sin embargo, posee receptores para patrones mole-
3
ERRNVPHGLFRVRUJ
Inmu
Es i m pr5{ind ib l ~ tener claros los conceptos que se desoibenerl este capítulo
¡x¡rque rIO sólo son fundamentl les y plJl'derl ser objeto depreg untls, sino
¡x¡rque van aser nt'{!'S.l rios pilrapoder aOOrdar cuestiorll'S relacionadas
con la inmunología que puedenaflilltCer en otfilS asignaturas como Infecciosas,
Pediillría, entre otfil~.
Estructura
y función de las inmunoglobulinas
Los anticuerpos son glucoproteínas sintetizadas por los linfocitos B (en los
que se pu ede encontrar en forma de receptores de membrana) V células
C
plasmáticas (que los secreta n como proteínas solubles) en respuesta al estímu lo antigénico.
RECUERDA
Reg la mnemotécnica de las cinco clases básicas de Ig: GAMDE:
IgG, IgA, IgM, IgO e IgE.
C2
Su caracte rística fundamental es que tienen la propiedad de unirse especificamente al antígeno que indujo a su formación, son por ello uno de los elemen tos f undamentales de la respuesta inmunitaria específica. Se las denomina
inmunoglobulinas (Ig) porque son proteínas formadas por grupos globulares
V son capaces de transferir pasivamente la inmunidad al administrarse a otro
individuo. Clásicamente, reciben también el nombre de gammaglobu li nas por
su migración electroforética en la fracción gamma (y) en un proteinograma.
Dominio de las inmunoglobulinas
Existen cinco clases básicas o isotipos de Ig que, agrupadas de mayor a menor
concentración en el suero de un adu lto normal, son: IgG, IgA, IgM, IgO e IgE.
Ún icam ente existen dos t ipos de cadenas ligeras: kappa (K) y lambda (A). las
la frecuencia de una determinada clase de Ig en los mielomas es directa-
19 con caden as ligeras kappa (K) predom inan sobre las de tipo lambda (A),
mente proporcional a la concentración de dicha Ig en suero (G, A, M, O Y E).
en una proporción aproximada de 2:1. En una molécula de inmunoglobu lina
determinada, las dos cadenas ligeras son siempre idénticas, independiente-
Estructura de las inmunoglobulinas
mente de las cadenas pesadas a las que estén un idas; es decir, que pueden
existir molécu las de inmunoglobu lina de clase G (\1) con caden as ligeras K y
Primeramente hay que referirse, como modelo básico, a la molécula de IgG,
moléculas de inmunoglobulina de clase G (y) con cadenas ligeras A, y así para
y posteriormente se analizarán las diferencias de ésta con las otras cl ases.
cada clase de inmunoglobul ina.
Se trata de un tetrámero (est á formado por cu atro caden as peptídicas), for-
Cad a cadena ligera está unida a una de las pesadas med iante enlaces disu l-
mado por dos cad enas pesadas H idénticas entre sí en una misma molécu la
furo, y las pesadas tamb ién están unidas entre sí por puentes disulf uro.
de inmunoglobul ina (de heavy: 'pesado', en inglés) y dos cadenas ligeras l
Estas un iones son enlaces cova lentes que constituyen las #regiones bisagra"
(de Iight: 'l igero'), t ambién idénticas, que se ens amblan adoptando una con-
de las inmunoglobulinas, siendo éstas las zonas m ás sensibles a la degra-
figurac ión espacial en forma de #Y# (Figura 4).
dación enzimática. las caden as de las Ig, tanto pesadas como ligeras, presentan una parte o región variable (V) en el extremo aminoterminal, y otra
la secuencia de aminoácidos de la cadena pesada es la que determ ina la clase
constante (C) en la porción ca rboxiterm inal. Se nombran como Vl y Cl para
y la subcl ase de la Ig (es decir, IgG4, IgO... ). Existen cinco clases básicas de cade-
las cadenas ligeras y VH y CH para las cadenas pesadas. Est a región variable
nas pesadas, que se designan con la letra minúscula griega homóloga de la
es la que determ ina la especificidad de la inmunoglobul ina por el antígeno.
latin a con la que se nombra la molécu la de Ig completa: gamma (\1) (lgG), alfa
El conjunto de inmunoglobulinas de un ind ividuo es capaz de reconocer
(a) (lgA), mu (11) (lgM), delta (6) (lgO) yépsilon (E) (lgE). Asu vez, existen cuatro
millones de antígenos diferentes, pero cada molécu la es específica para un
subcl ases de cadena gamma (y) y dos de alfa (a) (MIR 12· 13, 212).
ún ico antígeno.
4
ERRNVPHGLFRVRUJ
Manual CTO de Medicina y Cirugía, 10. 3 edición
02 . Inmunoglobullnas
RECUERDA
Digestión enzimática de las inmunoglobulinas
La región Fe es la zona de la Ig a la que se fija el comple mento.
Si se rea li za con papaína se obtienen tres f ragmentos (Figura S):
•
Dos idént icos llam ados Fab; cada fragmento Fab cont iene la zona de
la mo lécu la responsabl e de la unión al antígeno (fracción antigen
binding)
Un Fab está constituido por la mitad aminoterminal de una cadena
pesada unida a la cadena ligera (contiene los dominios variab les y un
Clases de inmunoglobulinas
domin io constante de la cadena pesada y de la ligera).
•
Un fragmento Fc (f racción crista lizable), formado por las dos mitades
carboxiterm ina les de las cadenas pesadas (sólo contiene dominios
constantes). Ejerce las funciones efectoras de las inmunoglobulinas
La inmunoglobulina predominante en el suero y en el espacio extravascular
(activación del complemento, unión a re ceptores de Fe presentes en las
es la IgG; se dif unde muy bien a través de las membranas y es también la que
membranas de algunas células) (MIR 08-09, 239).
predomina en las secreciones internas. Es la única Ig que atraviesa la placenta:
la IgG proceden te de la mad re es la principa l inmunoglobu lina del feto y del
rec ién nacido, y persiste en la circulac ión del niño durante los primeros 6-8
F,b
meses de vida (MIR 15-16, 46).
Existen cuatro subclases, determinadas por pequeños camb ios de aa (aminoácidos) en sus cadenas pesadas, denom inadas IgG1, IgG2, IgG3 e IgG4,
cuya pro porc ión re specto del tota l de IgG séric a es 70, 20, 6 Y 4%, respectivamente, es decir, son tanto m ás abundantes cuanto menor es el número de
su subtipo. Es importante re cordar que la subclase IgG4 es la única IgG que
~
~
no fija complemento por la vía clásica (Tabla 1).
--
0
0
~c
---- ~
pFc l -----¡::o¡¡
Tabla 1
Digestión enzimática de inmunoglobuLina G
Con pepsina se consigue un fragmento bivalente (que reconoce dos antígenos), llamado F(ab)2 (fracción Fab doble) y dos péptidos grandes llamados
pFc', así como pequeños fragmentos peptídicos que derivan de la zona de la
IgGl
IgG2
IgGl
(%) de la IgG en 1'1 suero
70
lO
6
4
Paso de la placenta
+++
+
+++
+++
Fijación de complemento
+++
+
+++
Unión a Fede células
+++
+
+++
+
Vida media (días)
2l
2l
7
2l
Tratamiento quirúrgico de la pancreatitis aguda grave
molécula situada entre F(ab)2 y pFc'.
RECUERDA
Funciones de las inmunoglobulinas
La IgG es la inmunoglobulina de m ayo r vida med ia.
Las inmunoglobul inas func ionan como "enlaceN entre el antígeno que reconocen mediante el Fab y la respuesta inmunitaria que desencadenan a través
del Fe, que puede interaccionar con diversos componentes solubles (com-
Características de las otras clases
de inmunoglobulinas
plemento) y ce lulares (macrófagos, célul as NK) a los que activa.
•
Unión específica con el antígeno. Reside en el f ragmento Fab, en una
hend idura que se forma en la conjunción de las regiones VH y VL, es
•
decir, los dominios variables de las cadenas ligera y pesa da. El grado de
•
IgM. La forma secretada es un pentámero de cinco moléculas de IgM. Tam-
complementariedad para el antígeno (Ag) que presenta esta hend idura
bién existe en su forma monomérica, como proteína de membrana en la
es lo que determ ina la especific idad del antícuerpo. Dentro de las regio-
superficie de los linfocitos 8. Cada uno de los cinco monómeros de la fo rma
nes VH y VL existen tres regiones hipervariables (HR 1, 2 Y 3), que son
secretada se mantiene unido gracias a puentes disulfuro intermonóme-
las que fo rman las paredes del sitio de comb inación con el antígeno y
ros, situados en el dominio CH3. La polimerización está determinada por
determinan su complementariedad para éste.
Funciones efectoras. Mediadas por los dominios constantes de las cade-
la cadena J (proviene del inglésjunction), que es sintetizada por las propias
células secretoras de anticuerpos (células plasmáticas) y que se une cova-
nas pesadas, concreta mente CH2 y CH3 (que pertenecen a la región Fe).
lentemente a través de un puente disu lfuro a la cadena pesada).1.
Las más importantes son:
El carácter pentamérico confiere a los anticuerpos de clase IgM una gran
Activac ión del comp lemento (vía clásica).
eficiencia para activar el complemento y para aglutinar antígenos par-
Unión a los receptores para el Fe de las células fagocíticas, con lo
ticu lados, ya que, al contener cinco regiones Fc, lógicamente son cinco
que faci li ta la fagoc itosis.
veces más potentes que una fo rm a monomérica. Como desventaja, por
Unión a los receptores para el Fe de los mastocitos, basófi los y eosi-
su gran peso molecular, la IgM no d ifunde fuera de los vasos, siendo
nófi los, induciendo así su degra nu lación.
por ello exclusivamente intravascular y no cruzando tampoco la barrera
Unión a los receptores para el Fe de la membrana de las células NK.
fetop lacentaria.
•
Capacidad de atravesar membrana s del organismo, como la pl acenta (sólo la IgG).
IgA. Está presente en suero y secreciones. Es la Ig predominante en las
mucosas y secreciones externas: tubo digestivo, árbol traqueobron -
s
ERRNVPHGLFRVRUJ
INMUNOLOGÍA
Tabla 2
quía l, nasofaringe, leche y calostro, saliva, lágrimas, bi lis y flujo vaginal,
donde actúa localmente neutralizando posibles patógenos.
RECUERDA
La IgM es la inmunoglobulina más efi caz para fijar comp le-
mento, al ser un pentámero.
I I,G
I,A
I I,M
Concentración en suero (mg/dl)
1.200
200
120
l
0,05
Vida media en suero (días)
II
6
5
l
2
Paso por placenta
+
,.,.
,.,.
+++
,.,.
Actividad reagín ica
Existen dos subcl ases de IgA: IgAl e IgAl (en función de cambios de
Actividad antibacte riana
aminoácidos en su cadena pesada a). La IgA2 constituye sólo el 10% de
Actividad antivíri ca
la IgA sérica, mientras que en las secreciones e s algo superior al 50%.
Zona bisagra sensible a enzimas
proteolíticas
La IgA sérica es, en su mayor parte, monomérica (más del 80%); no obstante, existe t ambién una IgA d imérica, que es la forma mayoritaria en
+
+
+
+++
+++
+++
+++
+
,..,
,..,
+++
Clases de inmunogLobuLinas G
las secreciones, que está constituida por dos moléculas de IgA unidas
por una cadena J. Esta IgA dimérica predominante en las secreciones y
mucosas contiene, además, un pol ipéptido denominado componente
secretor (CS) (Figura 6), que no es sino un f ragmento que provien e del
receptor de la membrana bas al de la cé lula epitelial de las mucosas a
Antígeno, inmunógeno, epítopo, idiotipo,
través de la que ésta capt a de forma selectiva a la IgA dimérica para ser
secretada. La unión del CS a la IgA confiere adem ás una mayor resisten -
hapteno e isotipo
cia al ataque de enzimas proteolíticas presentes en el med io extracelular. Al cubrir zonas sensibles a dicho ataque, como la "región bisagra",
•
lo que permite es que los anticuerpos de clase IgA puedan actuar en
las secreciones y proteger las mucosas, impidiendo o bloqueando la
Antígeno. Supone cualquier molécula que pueda ser reconocida por
una inmunoglobu lina o por el receptor de la célu la T (RCT) (Figura 7).
adhesión de los microorganismos. Algunos autores sostienen que la IgA
también puede actua r como una ba rrera contra alérgenos alimentarios
(M IR 16·17, 50).
•
IgD. Su concentración sérica es muy baja en los sujetos sanos. los linfocitos B vírgenes, cuando alcanzan el estad io de plena madurez inmunológica, coexpre san IgD de membrana junto con IgM; se sugiere que el
papel fisiológico de la IgD reside, sobre todo, en actuar como receptor
•
de los linfocitos B para el antígeno.
IgE. la concentración sérica de IgE es muy pequeña en sujetos sanos.
Int erviene f undamentalmente en la defensa frente a helmintos, gracias a su unión a receptores de membrana específicos para la Fc de la
Idiotipo
IgE (RFclgE) presentes en los eosinófi los, y también genera las reacciones alérgicas, por su capacidad para un irse a los basófilos y mast ocitos,
mediante receptores de gran afinidad que estas cé lulas poseen para su
extremo Fe. La activación de los eosinófilos por medio de estos receptores
produce la liberación de la proteína catiónica del eosinófilo (PCE), mientras
que los basófilos y mastocitos liberan múltiples moléculas vasoactivas e
Antígeno y epítopos
inflamatorias, destacando la histamina (Tabla 2).
•
Inmunógeno. Son aquellos antígenos capaces
de desencadena r una respuesta inmunitaria,
de m anera más concreta se suele aplicar a
aquellos antígenos capaces de inducir la acti vación del clan de linfocitos B, que lo ha reco-
Captadón
yendocltosis
de la IgA
nocido de manera específica. No todos los
antígenos son inmunógenos.
El complejo IgA-receptor pa>a
al otro polo de la céh, la epitelial
Receptor poli Ig
o O O
•
geno a la que se une el anticuerpo (entre 15 y 20
aminoácidos). Un antígeno puede tener var ios
0000
Célul.JIS epltel1ale
Epítopo. Representa la región concreta del antí-
epítopos distintos, que serán reconocidos por
distintos anticuerpos. A los epítopos también se
les llama determinantes antigénicos.
Luz del bronquio. intestino ..•
Liberadón a la luz de la IgA con parte
del receptor:el componente secretor
~
•
bles de las cadenas pesadas y ligeras).
•
Proceso de secreción de la IgA y componente secretor
6
ERRNVPHGLFRVRUJ
Idiotipo. Es la zona del anticuerpo que se une
al epítopo (se loca liza en los dominios var iaHaptenos. Son sustancias no proteicas de
poco peso molecular; que por sí solas no son
Manual CTO de Medicina y Cirugía, 10. 3 edición
•
02 . Inmunoglob ullna s
inmunógenas, pero que pueden comportarse como tales si se unen covalente-
sufren un proceso de activación, proliferación e interacción con los linfo-
mente a otra molécula más grande (a la que se denomina portador o camer).
citos T.
Isotipo. Es sinónimo de clase de inmunoglobu lina y viene definido por el
tipo de caden a pesada que lleve (G, A, M, D, Ej.
Durante este proceso dejan de expresar IgD y las células plasmáticas pasan a
sintetizar la misma IgM que antes se expresaba en la membrana, pero ahora
en forma de molécula de secreción.
Algunos de los miembros del clon experimentan el cambio de clase de la Ig,
Unión antígeno-anticuerpo:
pasando a secretar IgG o IgA en lugar de IgM, pero conservando la misma
región VH -VL propia de dicho clon, es decir, la misma especificidad de reco-
afinidad y avidez
nocimiento del antígeno. Este cambio de cl ase es inducido en el linfocito B
por la interacción en la sinapsis inmunológica con el linfocito T de los receptores de membrana CD40, del linfocito B con CD40L (CD154) del linfocito T.
La unión antígeno-anticuerpo se produce por en laces débiles o no covalentes, siendo por t anto reversible.
El mecanismo genético de base es una re ordenación en la que intervienen
•
Afinidad. Fuerza de unión entre el epítopo y el anticuerpo. Pueden exis-
las regiones 5 (conmutador, del inglés switch) que existen delante de cada
tir anticuerpos de igual especificidad, pero diferente afin idad. La afinidad
gen C.
aumenta en las sucesivas reexposiciones al antígeno (ésta es precisa-
•
•
Exclusión iso típica. Una misma cé lula B y su c lan (células deriva-
mente una de las características de la respuesta inmunitaria secundari a).
das de una m isma célula progenitora por división celu lar) solamente
Avidez. Es la fuerz a de unión global del anticuerpo por el antígeno. Un
expresan cadenas ligeras K o "J., V jamás ambos tipos simultánea-
conjunto de anticuerpos de baja afinidad (por ejemplo, IgM pentamé-
mente .
Exclusión al é lica. Una cé lula B só lo expresa los genes de las cadenas
rica) pueden dar lugar al reconocimiento de un antígeno con avidez alta.
•
pesadas y ligeras de uno de los alelos de los cromosomas homó logos (e l materno o el paterno). El otro jamás será expresado por esa
célula.
Cambio de clase de inmunoglobulina
,/ MIR 16-17, 50
,/ MIR 15-16, 46
,/ MIR 12-13, 212
,/ MIR 08-09, 239
Los linfocit os B maduros pero vírgenes, que presentan como receptores
de membrana IgM e IgD, tras el reconoc imiento específico de l antígeno,
Ideasclave
,/ Existen cinco clases de inmunoglobulinas que, ordenadas de mayor a
menor abundancia en el suero, son: IgG, IgA, IgM, IgD V IgE (palabra
mnemotécnica GAMDE).
,/ La primera inmunoglobulina que se fabrica en respuesta a un antígeno
es IgM; las otras inmunoglobu linas se secretan fundamentalm ente en
la respuesta secundaria.
,/ La inmunoglobulina prototipo está formada por dos cad enas pesad as
(H) y dos cadenas ligeras (L). Existen cinco tipos de cadenas pesad as
,/ IgA es la inmunoglobu lina de las secreciones externas (mucosas, leche
m aterna ... ). En las secreciones se presenta como d ímeros, mientras que
en el suero predomin a la forma monomérica.
(y, a, li, 05 VE).
,/ La clase de inmunoglobu lina viene determ inada por la cadena pesada
que tiene.
,/ Las únicas Ig capaces de activar el complemento por la vía clásica son la
IgG (excepto IgG4) y la IgM.
,/ La zona de unión al antígeno se forma en el extremo term inal de las
cadenas ligera y pesada.
,/ IgG predomina en el medio interno: su ero, medio extracelular y flu idos
corporales (LCR, líqu ido pleuraL .. ).
7
ERRNVPHGLFRVRUJ
Células
Este es untema que se debe estudiar en profundidad, ya que, adem~s
de caer prelJuntas dirfftas sorne B, es totalmente nffes.J riodominarlo
para podereflleooer la respuesta inmunitaria que se trata en elÚlpítu/a 5
y los ITle(anismos de algunos tifXJS de ItChazos (Capítulo 7) yde algunos
fármacos inmunosupresores (Capítulo 9).
Los linfocitos son las cé lulas leucocit arias de estirpe linfoide. En reposo, son
dad: una gran proporción de los linfocitos T de un individuo son capaces
células pequeñas, redondas, de muy escaso citoplasma. Se han identificado
de reconocer como extrañas las moléculas del CPH de otro individuo de
tres clases principales de lin foc itos: B, T Y NK.
su misma especie (antigénicamente distintas de las suyas) sin que med ie
inmunización previa. El linfocit o percibe la diferencia con las moléculas
La tasa de renovación linfocitaria es muy elevada; se ca lcu la que cada día se
CPH propias, e interpreta que se trata de su propio CPH, pero que lleva
producen 10" linfocitos en los órganos linfoides primarios y que diariamente
incorporado un péptido antigénico. Este fenómeno es la base del rechazo
se renueva el 2% de los linfocitos. En un organ ismo humano sano existen
agudo del trasplante alogén ico, como se expondrá más adelante.
alrededor de 10" célu las linfoides.
Los li nfocitos implicados en la respuesta inmunitaria adaptativa son los B y T.
~stos reconocen antígenos específicos, y tras el estímulo antigén ico, desarrollan una serie de tra nsformaciones (proceso que se conoce como activación),
que consiste en un proceso de proli ferac ión (expansión clonal) y diferenciación
a célu las efectoras.
Ellinfograma norma l presenta 75-85% de linfocitos T (aproximadamente dos
terc ios son C04+ y un tercio C08+), 5-15% de linfocitos By 5-15% de células
NK. Estos valores son dinámicos y presentan diferencias según la edad del
' CT
individuo (MIR 12-13, 2IS).
Cllopl.llJINI ftl l.nfodlOT
Linfocitos T
Restricción histocompatible
Se pueden distingu ir cuatro rasgos generales que diferencian la biología de
Receptor de la célula T (RCT)
los linfocitos T (LT) respecto de los otros linfocitos.
•
•
•
•
Se desarrol lan en el timo, a partir de los progenitores linfoides derivados
de la CHP (célula hematopoyética pluripotencia l). Se les denom ina T por
El RCT (TCR en terminología anglosajona) es bastante similar, bioquímica,
originarse en el timo (los B lo hacen en la médu la ósea).
funcional y genéticamente, a las inmunoglobu linas. Son moléculas que varían
Poseen el receptor de la célu la T (RCT). Es una molécula de reconoci-
en su compos ición quím ica para adaptarse a antígenos concretos, uniéndose
m iento específica para cada antígeno, como las inmunoglobulinas, pero
de modo específico. No obstante, las inmunoglobulinas y el RCT son molé-
únicamente está presente en la membrana y no es liberado al med io
cu las distintas, codificadas por genes diferentes. El RCT es un heterodímero
extracelular en fo rma soluble en respuest a al antígeno. Todo esto se
compuesto por dos cadenas polipeptid icas d istintas unidas por un enlace
expondrá con más detal le en otros apartados del capítu lo.
d isulfuro; siem pre se presenta como una molécula integral de la membrana
Present an diversidad de funciones, de esta forma, existen linfocitos T
plasmática del linfocito T (no existen formas solubles); es decir, tiene una
reguladores, colaboradores y citotóx icos.
porción extracelular, otra transmembrana y una co la intracitop lásmica.
El RCT sólo reconoce al antígeno cuando éste es Npresentado", form ando
un complejo con las moléculas del CPH (compleja principa l de histo-
El RCT está compuesto por dos cadenas que pueden ser a y I}, o, y y 8. E195%
compatibil idad) (MIR 11-12, 216), bien de clase I o de clase 11, propias
de los linfocitos T de sangre periférica tienen el RCT tipo 2 (RCT- 2), form ado
del individuo, en cuyo timo se desarrollan. Este cond icionamiento del
por una cadena a y otra j3 (linfocitos T-aJ3). Menos del 5% de linfocitos T
reco nocimiento del antígeno a su asociación con las moléculas del CPH
expresan el RCT-l, fo rm ado por cadenas y y 6, y se les denomina linfocitos
(moléculas HLA) se conoce como rest ricción hist ocompatible (Figura 8)
T-y6. Los linfocitos T-y6, de forma mayoritaria, no expresan en su membrana
o restricc ión por el CPH.
ni C04, ni CD8, por lo que también se les denomina cé lulas Ndobles negati-
La excepción a la restr icción histocompatible son los superantígenos. Una
vas". No se sabe con exactitud cuá l es su cometido ni cómo funciona el pro-
característica del fenómeno de la restricción por el CPH es la alorreactivi-
pio receptor yo, pero parecen estar implicados en fenómenos de toleranc ia.
8
ERRNVPHGLFRVRUJ
Manual CTO de Medicina y Cirugía, 10. 3 edición
03 . Células del sistema Inmunltarlo
RECUERDA
El porcentaje de linfocitos T-yo es superior en los linfocitos intraepite liales
en mucosas que en sangre periférica, por lo que se supone que juegan un
Las cé lulas presentadoras de antígenos e)(presan 87 en su
superficie cuando ingieren antígenos e)(traños.
papel importante en la defensa de éstas. El aumento de la población yo en
sangre periférica aparece en diversas patologías, como son los síndromes
linfoproliferativos neoplásicos. En la mucosa duodenal, se ha observado que
Hasta tal punto es totalmente necesaria esta interacción para la activación
los pacientes con enfermedad celíaca presentan un mayor porcentaje de linfocitos T-y6 intraepitel iales que los individuos no celíacos.
del linfocito T que, si no sucede, se produce el fenómeno de anergia, en el
que el LT no es capaz de transformarse en una cé lula efectora. Esta anergia
La estruct ura molecular, la organización y el reordenamiento de los genes
que codifican las cadenas del RCT son bastante sim ilares a los de las inmuno-
clona l es uno de los mecan ismos de adqu isición de tolerancia inmunológica
a nivel periférico (en órganos linfoides secundarios).
globu linas. Las cadenas ( l y y son muy parecidas genéticamente a las cadenas
ligeras, únicamente tienen genes Vy J (lo que supone una sim ilit ud con las
En algunos linfocitos T la anergia induce su apoptosis med iada por la vía FAS/
cadenas ligeras de las inmunoglobinas), mientras que las 13 y o poseen genes
FASL (C095/CD95L). Por tanto, sólo podrá activarse una célula T si ambas
V, D Y J, como las cadenas pesadas.
señales (TCR -C03 y CD28) están presentes.
El RCT reconoce péptidos unidos a las moléculas del CPH de antígenos que
previamente han sido procesados por otra célula. La e)(cepción es la capa-
El RCT presenta una gran especificidad pero baja afinidad por el antígeno,
cidad de ciertos antígenos (superantígenos). Asociado al dímero RCT se
se necesita a las denominadas moléculas accesorias para estabilizarla. Así,
encuentra un complejo de moléculas encabezado por C03, que está invo-
las moléculas C04 y CD8 son capaces de reconocer y un irse a la molécula del
lucrado en la transmis ión de la seña l de activación a través de la membrana
CPH en la que está siendo presentado el antígeno (CD4 se une a CPH de clase
plasmática (transducción) y es un marcador característico del linfocito T.
11 y C08 a CPH de clase 1).
Sinapsis inmunológica
En fases inicia les de la activación, aparece una nueva molécu la en la mem-
por lo que en la un ión entre la CPA (célula presentadora de antígeno) y el LT
brana, C0152 (CTLA4) que interacciona con 87 (CD80/CD86) de forma muy
Se denomina sinapsis inmunológica al conjunto de interacciones que se pro-
similar a C028, compitiendo con ella. La principal diferencia entre ambas molé-
ducen entre el linfocito T y la célula presentadora de antígeno (CPA), con
culas es que CD1S2 codifica una seña l negativa que desactiva el linfocito T.
la finalidad de producir la activación del linfocito T y poner, así, en marcha
la respuesta inmunitaria. Est as interacciones consisten, principalmente, en
Se trata de una seña l reguladora fis iológica que sirve para inhibir la respuesta
señales recibidas por receptores de membrana.
inmunitaria, una vez vencida la infección.
Secuencia de la sinapsis inmunológica
Sinapsis inmunológica línfocito T activo (Figura 9):
CD69, CD2S, CPH 11 (DR)
En primer término, tiene lugar el reconocimiento específico por el RCT del
antígeno presentado por moléculas del CPH. Tras este reconocimiento anti-
Cambio isotipo Ig:
CD40-CD4OL
génico, se produce la transducción de la primera seña l de activación mediada
Primera señal:
por C03. Sin embargo, para la activación completa del linfocito T es totalmente
HLA-~-TCR-CD3
necesario que se produzca una segunda señalo señal coestimuladora.
Segunda señal:
C028--- 87 (coestimu lación, señales antiapoptóticas)
La segunda señal (coestimulación antigénica) se produce tras la interacción
CTLA4--- 87 (inh ibición)
entre el receptor CD28 (presente en la superficie del linfocito T) y el 87 (C080
Si no hay segunda señal:
o C086 de la célula presentadora de antígeno).
Anergia (tolerancia/apoptosis)
Figura 9
Célulil T
Sel\all
s.ñil12
Setailll
TCR
B7
Célu la
dendrítica
Cé lula
d endrítica
Segundas señales de activación (CD28) e inhibición posterior a La resoluc ión de la infección (CD152)
9
ERRNVPHGLFRVRUJ
INMUNOLOGÍA
Activación linfocitaria
•
Selección negativa. Los timocitos cuyo RCT tiene una muy alta afinidad
por las moléculas del CPH propias son eliminados porque, si saliesen del
timo, se comportarían como linfocitos autoinmunitarios. Los timocitos
Los linfocitos T se clasifican según el grado de activac ión que posean.
•
•
N
linfocitos T quiescentes. También llamados #vírgenes o en reposo. Son
los que no han tomado contacto todav ía con su antígeno.
linfocitos T activados (también llamado efectores) . Son aqué llos a los
que les ha sido presentado su antígeno específico y han rec ibido, además,
las seña les de coestimulación de la célula presentadora de antígeno. Tras
activarse, este tipo de linfocitos expresan:
capaces de interaccionar con las moléculas CPH de clase II se convierten en
linfocitos T CD4 y los que lo hacen con CPH de cl ase 1, en linfocitos T COs.
La principal diferencia entre los linfocit os T C04+ y los C08+ es la clase de
CPH que son capaces de reconocer. Existen linfocitos, tanto T CD4+ como T
C08+, colaboradores y citotóxicos.
Receptor de alta afinidad para IL-2 ((D25), que a su vez es una
Fenotipo de los linfocitos T adultos
interleucina estimuladora de la actividad de estas células.
CPH de clase 11. Todos los linfocitos T tienen CPH de clase 1, pero
únicamente los activados tienen también CPH de clase 1I (marcador
Los linfocitos T maduros presentes en la periferia se caracterizan fenotípica-
tardío de activación).
mente por expresar las siguientes molécu las de superficie: RCT, CD2, CD3,
CD69 (marcador precoz de activación).
receptor para las lectinas, fitohemaglutin ina y concanava lina A (m itógenos)
y, además, uno de los siguientes (pero no los dos):
Diferenciación de los linfocitos T
•
CD4. Los linfocit os T C04+ son los que reconoce n antígenos presentados
La maduración de los linfocitos T, tanto en el niño como en el adolescente,
junto con el CPH de clase 11. Predominan sobre los CD8 en una rel a-
se produce en el timo, a partir de precursores provenientes de la médu la
ción 2:1. Esta relación se invierte en la infección por VIH (por linfopen ia
ósea. En el adulto, dicho órgano se va atrofiando y se acepta que en parte
selectiva C04) y, de forma transitoria, en otras infecciones virales (por
los linfocitos T pueden madurar en la médula ósea, con un papel no claro de l
expansión de la población C08). La mayor parte de los CD4+ desarrollan
intestino. Los linfocitos T inmaduros reciben el nombre de timocitos y, según
funciones co laboradoras (he/per), tanto para la respuesta de anticuer-
el estadio de diferenciación, se pueden subdividir en tres grandes subpobla-
pos como de inmunidad celular, aunque también existen T C04+ con
ciones cuyo estudio es de gran interés para comprender las leucem ias de
actividad citotóxica (el 10%) que participan en reacc iones de hipersen-
células T:
sibilidad retardada.
•
•
•
Pretimocitos. Son los progenitores li nfoides derivados de la CHP (célu la
•
CDS. Los linfocitos T C08+ reconocen antígenos present ados junto
hematopoyética pluripotencial) de la médula ósea. No expresan CD4 ni
con el CPH de clase 1. La mayoría son citotóxicos, pero también exis-
CD8 ("dobles negativos N ). Viajan de la médula ósea al timo para seguir
ten co laboradores. Los linfocitos T C08+ he/per 2 colaboran en la res-
madurando ahí.
puesta de anticuerpos, igual que los C04 he/per 2. En los pacientes
Timocitos comunes. Se caracterizan por la expresión de CD4 y CD8
("dobles positivos").
con SIDA existe una cantidad de inmunoglobulinas séricas superior a
Timocitos tardíos. Caracterizados por expresar RCT con abundancia y
además una u otra de las moléculas CD4 o CD8 (nunca "dobles positivos
elaboración de anticue rpo s en los pacientes con SIDA son fundamen -
la de los sujetos sanos. Los linfocitos colaborado res que coordinan la
talmente CD8+.
o negativos N ). Sus características funcionales y los marcadores de super-
•
ficie son indistinguibles de los linfocitos T maduros de la periferia.
Linfocitos de memoria. Son los que se activaron durante una respuesta primaria y que, una vez pasada ésta, permanecen en reposo
durante mucho tiempo (incluso toda la vida). Están preparados para,
RECUERDA
cuando se vuelven a encontrar con el antígeno (respuest a secundaria),
responder de un modo más rápido, se lectivo e intenso. Son difíciles
La suma del CD4 y CD8 no es el total de CD3, ya que hay que
tener en cuenta a los linfocitos y5 dobles negativos.
de distingu ir de los activados T y ambos circulan por la sangre y el
sistema linfático. Una característica distintiva es que los de memoria
expresan C045 Ro (los linfocitos vírgenes expresan C045 Ra) y carecen
Procesos de tolerancia central del linfocito T
de C062 L.
Selección de los linfocitos T
Activación linfocitaria por superantígenos
Durante la maduración de los linfocitos T en el timo, tiene lugar una serie de
La inmensa mayoría de los antígenos se sitúan en el surco creado entre los
procesos encam inados a lograr la tolerancia de los mismos, es decir; impedir
extremos de las cadenas a y ~ del CPH de clase II y son reconocidos, asi-
que existan linfocitos T autorreactivos (capaces de reconocer antígenos pro-
mismo, por los extremos de las cadenas a y ~ del receptor de la célu la T. Se
pios). Los procesos de to lerancia que tienen lugar en el timo se denominan
trata, pues, de una interacción similar a la del antígeno con el idiotipo de las
procesos de tolerancia "centra les N , entre los que destacan los procesos de
inmunoglobul inas.
selección. La selección está determinada por la interacción entre el RCT que
adquieren los timocitos en desarrollo y las moléculas del complejo principa l
Los superantígenos, a d iferencia de los antígenos convenciona les, se
de histocompatibilidad (CPH) expresad as por las células del estroma del timo.
unen directamente a una zona latera l de la cadena ~ del RCT que es muy
•
Selección positiva. Los timocit os con un RCT que reconozcan las molé-
poco polimórfica, sin tomar contacto con la zona polimórfica (donde se
culas del CPH son seleccionados. El resto son eliminados (apoptosis,
sitúa la especificidad del RCT por e l ant ígeno). Al no ser capaces de d is-
muerte celular program ada, vía FAS;FASL [C095;C095L]). Los timocitos
criminar selectivamente los RCT específicos, los sup erantígenos pueden
que no reconocen el CPH tampoco serían capaces de reconocer el sistema
estimular de modo totalmente inespecíf ico, hasta el 20% de la totalidad
HLA-péptido antigénico, por lo que jamás podrían llegar a activarse, es
de los linfocitos T periféricos que, al activarse, secretarán citoc inas e
decir, son elim inados porque nunca van a ser úti les al organismo (MIR
interleucinas masivamente. La enorme cantidad de ci t ocinas actuando
10-11, 222).
sobre sus correspondientes receptores es la responsable del cuadro clí-
10
ERRNVPHGLFRVRUJ
Manual CTO de Medicina y Cirugía, 10. 3 edición
03 . Células del sistema Inmunltarlo
nico. Un ejemplo de enfermedad inducida por superant ígenos es e l shock
tóxico estafilocócico.
Linfocitos granulares grandes.
Células NK
Linfocitos B
Los térm inos LGL (Jorge granular Iymphocyte) y linfocito NK (células agresoras naturales o natural killer) son prácticamente sinón imos y constituyen
Los linfocitos B son célu las especializadas en la producción de anticuerpos.
el 5-15% de las célu las mononucleadas de la sangre periférica en personas
Se desarrollan a partir de la CHP y, una vez maduros, expresan el receptor
sanas, tienen un tamaño algo superior al de los típicos linfocitos pequeños
de la célula B, que consiste en inmunoglobulinas de membrana asociadas
y una granulación azurófila en su citoplasma. Los LGL son muy importantes
a otras moléculas. También tienen receptores para las lectinas pokeweed
en los primeros momentos de una infección vírica, cuando el virus se está
(sólo presentes en los linfocitos B) y fit ohemaglutin ina que, se debe recor-
mu ltiplicando y todavía no se ha desarrol lado la respuesta de linfocitos T. Su
dar, también tienen los linfocitos T. Su denominación como linfocitos B se
misión, cons iderada como perteneciente al sistema de inmunidad natura l
debe a su origen en la médula ósea (en inglés, bone morrow). Los linfocitos
(innata), es destruir células anormales (neoplásicas o infectadas) y contener
B maduros circulan por la sangre y el sistema linfático y, cuando encuentran el
la infección hasta que el sistema de linfocitos T se encuentre plenamente
antígeno (Ag) para el que son específicas sus inmunoglobulinas de membrana,
experimentan una serie de cambios madurativos caracterizados por prolifera-
operativo.
ción y d iferenciac ión hacia célu la secretora de anticuerpos (célula plasmática),
Una de las principales f unciones biológicas de las células NK es la de destruir
que secreta grandes cantidades de inmunoglobu li na con las mismas regiones
células que carecen de CPH clase 1. Dado que el bloqueo de la expresión del
variables (misma especificidad) que las que expresaban en la membrana antes
CPH en la célula infectada es una estrategia viral para burlar al sistema inmu-
de ser estimulados por el antígeno. Los linfocitos B, tanto en reposo como acti-
nitario, eso les convierte en un mecanismo alternativo de defensa antiviral y,
vados, expresan CPH de clase I y también CPH de clase II (pueden actuar como
en determinadas ocasiones, de defensa antitumoral, ya que algunas células
célu las presentadoras de antígeno).
tumorales también pierden la expresión de CPH clase I y se convierten así en
d ianas de los NK. Asimismo, los linfocitos NK poseen recepto res activadores,
Receptor de la célula B
KAR (killer act1vat1on receptar), que reconocen diversos antígenos m icrobianos. El linfocito NK posee, además, la capacidad de ampl ificar la respuesta
El receptor característico del linfocito S y el que le proporciona la especifi-
de inmunidad, específica o adaptativa, de anticuerpos; esta capacidad viene
cidad para el antígeno es la inmunoglobu lina de superficie (de membrana).
dada por la existencia de receptores para Fe de la IgG en su membrana
Asociadas a la inmunoglobulina de superficie, existen una serie de moléculas
(CD16). Esta aptitud para reconocer anticuerpos constituye el nexo de la
cuyo conjunto constituye el receptor de la cé lula B (RCB). La misión de éste
célula NK con la inmunidad adaptativa. Fenotípicamente, las moléculas que
es activar la célula cuando se fije en él el antígeno.
definen a los linfocitos NK son C094, C056 y C016 (Tabla 3).
RECUERDA
Tabla 3
Tipo cl!lular
Los linfocitos S no presentan restricción histocompatible.
Marcador caractl!rfrtlco
Linrocito 8
Ig de superficie, C019, C020, C021
LinrocitoT
C02, COl, COS, C07
Las principa les moléculas que forman parte del receptor son las siguientes:
NK
C016, C056
•
Mil'loide
C014
Leucocilos
C045
Inmunoglobulina. Generalmente es IgM, pero tamb ién puede ser IgO
(linfocito B maduro pero v irgen, que expresa IgM e IgO).
•
CD 19. Forma un complejo con el C021 que contiene una tirosincinasa.
•
CD 21. Receptor para el fragmento C3d del complemento y virus
Marcadores celulares (MIR 14-15, 217)
Epstein-Barr.
•
CD 20. Interviene en la formación de un cana l de ca lcio. Es la diana tera -
Los receptores KIR (killer cell immunoglobulin -Iike receptor), como el Ly-49, al
péutica del antícuerpo monoclonal rituximab (véase Capitulo 9, Inmu-
unirse al CPH de las hipotéticas cé lulas diana, apaciguan a las células NK cito-
noterapia) (MIR 14-15, 217).
tóxicas. Si la célula carece de CPH, el receptor KIR dejará de transmitir la señal
inhibitoria y la cé lula NK desencadenará el mecanismo efector cit olítico sobre
En el proceso de activación del linfocito S, del mismo modo que en el del T,
la célula diana. Los genes de los receptores KIR son muy polimórficos, con gran
es necesaria la interacción de otras moléculas de membrana, además del
d iferenc ia interindividual; d iferentes polimorfismos se han asociado con diver-
propio receptor antigénico.
sas presentaciones clínicas de la infección por virus de la familia de los herpes.
linfocitos B CD5+. Una subpob lación de los linfocitos S maduros expresa
la molécula CDS, que paradójicamente es característica de las cé lulas T, y
se les denomina linfocitos B- 1. La población mayoritaria de linfocitos B (lin focitos B-2) no expresan en su membrana la molécu la C05. Estos linfocitos
Células presentadoras de antígeno
S C05+ secretan abundante IgM y algo de IgG e IgA. Est a subpoblación
de linfocitos B tienen un importante papel en la respuesta de anticuerpos
timoindependientes, como por ejemplo, la producida frente a bacterias
Se denomina cé lula presentadora de antígeno (CPA) a aquélla que es capaz
encapsuladas.
de presentar antígenos de origen externo a través de moléculas CPH de clase
11
ERRNVPHGLFRVRUJ
INMUNOLOGÍA
11. Estas células son capaces de internal izar el m icroorgan ismo, digerirlo y
los órganos (piel, corazón, pulmón, hígado, intestino ... ). Cuando toman
procesarlo. Se considera que pertenecen a esta clase las células de estirpe
monocitomacrofágica, las células dendr íticas y los linfocitos B. Se debe reco rdar que estas células, al igual que todas las células nucleadas del organismo,
contacto con un Ag, migran a través de los vasos linfáticos hacia la para corteza de los ganglios linfáticos regionales; al lí se transforman en célu-
tamb ién expresan CPH de clase l.
linfocitos T he/pero El prototipo de célula dendrítica interdigitante es la
Los monocitos-macrófagos, al igual que los linfocitos NK, poseen (D16, e l
célu la de Langerhans (célul as dendríticas de la piel).
Células dendríticas foliculares. Se localizan en los órganos linfoides
las dendríticas interd igitantes encargadas de presentar antígenos a los
•
receptor para la región Fe de las inmunoglobulinas. Es importante reco r-
secundarios (sobre todo, el bazo y los gangl ios), en áreas ricas en linfo-
dar que se consideran manocitas a las células de esta estirpe que están
citos 6, como los folículos (a lo que deben su denominación). No tienen
circulando por el torrente sanguín eo, mientras que cuando se local izan en
CPH de clase 11, pero sí receptores para complemento e inmunoglobuli-
tejidos, se les llama macrófagos. En algunos casos, estos macrófagos reci-
nas, y están re lacionadas con el aclaramiento de inmunocomplejos y el
ben nombre propio, en función del tejido en el que se ub iquen (célu las de
Kupffer, en el hígado; osteoclastos, en el hueso; microglía o cé lulas Nde l
desarrollo de los linfocitos 6 de memoria.
Río Hortega N , en el sistema nervioso). Su función principal es localizar a los
Las cé lulas dendríticas foliculares no func ionan como CPA de los linfocitos T; se
invasores e iniciar las respuest as destinadas a restaurar el daño producido
cree que son fundamentales para presentar el antígeno a los linfocitos 6 del
por los mismos; es decir, comenzar la lu cha frente a los m ismos y los meca-
folícu lo y para generar las respuestas secundarias de anticuerpos. Los mono-
nismos de reparación de los tej idos dañados, para lo que secretan diversos
citos producen IL-l y otras citoc inas importantes para que los li nfocitos T pue-
tipos de citocinas e interleucinas (I L-1, TNF, IL-6, POGF, VEGF, interferones,
dan activarse. Los linfocitos 6 activados también pueden producir IL-1, pero no
q u imioci nas ... ).
está claro que lo hagan las cé lulas dendríticas. El tipo de respu esta inflamatoria
que ponga en marcha la CPA condiciona y polariza el tipo de respuesta inmuni-
Células dendrítícas
taria adaptativa que va a tener lugar.
Son células presentadoras de antígeno que tienen unas prolongaciones alargadas en su membrana con la finalidad de obtener una mayor superficie de
PREGUNTAS ·
contacto. Existen dos clases distintas:
MIR
•
Células dendríticas interdigitantes. Expresan en su membrana una gran
,/ MIR 14·15, 217
,/ MIR 12-13, 215
,/ MIR 11·12, 216
,/ MIR 10-11, 222
cantidad de CPH de clase II y se loca lizan intersticial mente en casi todos
Ideasclave
,/ Los linfocitos T se caracter izan por expresar en su membrana una molécula para reconocer antígenos: el RCT (receptor antigénico de la célula
T). Asociado a esta molécula se encuentra C03, por lo que se puede
afirmar que todos los linfocitos T son CD3 positivos.
mismo la expresión de: 1) C02S (receptor de alta afinidad para IL-2), 2)
HLA de clase 11, y 3) CD69.
,/ Los lin focitos T colaboradores se subdividen tomando como base prin cipalmente las citocinas que producen y su func ión. Destacan TH1, TH2,
T reguladores y TH17.
,/ Los linfocit os T se pueden d ividir en dos grupos básicos: los C04+ (la
mayoría son colaboradores) y los C08+ (la mayoría son citotóxicos).
,/ Los linfocitos 6 se caracterizan por expresar en su membrana inmunoglobulina de superficie (su molécu la para reconocer antígenos). La Ig
de superficie se asocia a la molécu la C019, por lo que se puede afirmar
que los linfocitos 6 son C019+.
,/ Los lin focitos T sólo pueden reconocer antígenos si éstos les son presen t ados en el interior de moléculas del complejo principal de histocompa tibilidad (HLA). Los CD4+ reconocen antígenos presentados en el HLA de
clase II y los C08+ reconocen en el HLA de clase 1.
,/ Los linfocitos NK son células cit otóxicas que identifican y eliminan células infectadas por virus o con muta ciones. Se caracterizan por expresar
CD16, CD56 Y CD94.
,/ El acto de presentación supone la formación de una sinapsis inmunitaria entre la célula que presenta el antígeno (HLA) y el li nfocito T. En este
proceso se intercambia información, en forma de interacciones moleculares, entre ambas cé lulas.
,/ Las células present adoras de antígenos (CPA) son las que pueden pre sentar antígenos a todos los linfocitos T (tanto C04 como C08), porque
expresan tanto HLA de clase I (como todas las células nucleadas) como
de clase 11.
,/ Las tres señales fundamentales de la sinapsis inmun itaria son: 1)
presentación del antígeno, 2) señal de coestimulación (67;C028) y
3) c itocinas que modulan la respuesta (IL-4, IL- 12 ... ).
,/ Los superantígenos son moléculas capaces de activar hasta un 20% de
linfocitos T de sangre periférica de forma inespecífica.
,/ Tras formarse la sinapsis inmunitaria y reconocerse el antígeno, los linfocitos T se activan y present an un fenotipo distinto, destacando en el
12
ERRNVPHGLFRVRUJ
Complejo principal
de histocompatibilidad
La m.Jteria tratl daen este c.lpítuloes (om!liemffitari.l de laquese estudió
en el Úlpítulo J Palil resolver una preg untl MIRrelaciofl3da co n la activación
linfocitl riao latoleran(ia, es esendal manejilr los COfKeptos expuestos
en los tres temas (3, 4YS).También se trata ráffi los meGInismos básicos
de rechazo de Ófl¡anos, ¡¡or loquese estl ante un tem.J de m.lximaim¡¡ortancja
queno se puede dejar de estudiar.
clase I clásicas son HLA-A, HLA-B Y HLA-C; se trata de molécu las de
expresión ubicua. De entre las no clás icas destaca HLA-G; su expresión
Introducción
queda restringida a tejidos fetales y hepáticos, por lo que se intuyó su
implicación en los fenómenos de tolerancia entre tej idos mediante la
inhibición de las cé lulas N K (linfocitos natural killer). Se ha demostrado
La d iscriminación entre lo propio y lo extraño es esencial para que el sistema
la re lación entre el desarrollo de preeclampsia y la baja expres ión de
inmun itario pueda destruir cualquier agente invasor, una vez reconocido
HLA-G en tejidos feta les (Figura 10).
como ajeno o dañino al organismo. Los linfocit os T no son capaces de reconocer directamente a los antígenos, sino que tienen que ser mostrados junto
HLA d<lU I
con moléculas del comp lejo principa l de histocompatibilidad (CPH).
RECUERDA
La función del HLA es presentar antígenos peptídicos a los
linfocitos T.
Este comp lejo CPH -péptido antigénico sí puede ser identificado por los linfoc itos T por medio de su receptor específico (RCT) y, una vez rea lizado el
reconocimiento del antígeno, se desencadena la respuesta inmun itaria. Los
antígenos de histocompatibilidad deben su nombre a que se descubrieron
por su participación en los mecanismos de rechazo de órganos trasp lantados
entre ind ividuos genéticamente distintos. Se ha descrit o CPH en todos los
vertebrados estudiados, su equivalencia en lengua inglesa es MHC (compleja
mayor de histocompatibi lidad). El CPH humano y de grandes simios recibe
el nombre de HLA, por human leukocyte antigen (antigenos leucocitarios
humanos). A partir de ahora, se emplea rán de manera indistinta ambas terHlA clase 1 c 'c' __________________________________________
m inologías : CPH y HLA.
•
HLA de clase 11 (HLA· II). Compuestas por dos cadenas, una cadena
llam ada a y la otra f), conteniendo regio nes pol imórficas. Presenta
péptidos de origen exógeno, es decir, de antígenos que han sido capta -
Moléculas HLA de clase I y de clase 11
dos del exterior por las cé lulas que los presentan. Sólo tendrán HLA-II
aq uellas célu las con capac idad endocítica vIo fagocítica, las denominadas CPA: macrófagos -monocitos, células dendríticas y linfocitos B
Las moléculas HLA son glucoproteínas de membrana. Se d istingu en dos cIa-
(MIR 09-10, 215).
ses de HLA:
•
HLA de clase I (HLA· I). Están compuestas por una cadena a que con -
Como excepción cabe reco rda r que los linfocitos 1; sólo cuando están
tiene zonas polimórficas V una cadena f) constante, la 13, microglobu -
trans itoria. Las tres molécu las HLA-I I principales son HLA-DR, HLA-DP Y
Ii na.
HLA-DQ (MIR 13-14, 57).
activados (pero no de manera constitutiva), expresan HLA-II de forma
Es importante aclarar que, en la molécula de HLA-I, únicamente la
cadena a es cod ificada por los genes HLA. Se encarga de la presen -
Las moléculas HLA forman parte estructura lmente de la superfamilia de las
tac ión de péptidos endógenos, provenientes de la síntesis prote ica
de la m isma célu la que los presenta. Es como un "control de cal i dad~
inmunoglobu li nas, al igual que el RCT (curiosamente son tres moléculas capa ces de reconocer o interactuar con antígenos). Sus aminoácidos se d isponen
intrace lu lar. Se encuentran en la membrana de prácticamente todas
formando dominios globulares similares a los que forman las Ig (Tabla 4).
las cé lulas nucleadas y plaquetas. No expresan CPH de clase I hematíes, sincitiotrofob lasto y algunos timoc itos. Se distinguen dos tipos de
El hecho de que los linfocitos T no reconozcan e l antígeno m ás que en
molécu las HLA-I, las clásicas y las no clásicas. Las molécu las HLA de
combinac ión con moléculas HLA, añade a la fase de reconocimiento
13
ERRNVPHGLFRVRUJ
INMUNOLOGÍA
inmunita rio un grado adicional de complejidad que puede tener repercu-
Los genes HLA siguen un mecanismo de herencia autosómica codomi·
siones funcionales. Las moléculas CPH deben poseer la cualidad de poder
nante, es decir, no sólo existen dos alelas para cada gen (ya que somos
combinarse con cualquier péptido, aunque la afinidad de esta combina-
diploides), una copia de origen materno y otra de origen paterno, sino
ción dependa de la est ructura del péptido V de la molécula CPH corres-
que cada uno de esos alelas se expresará dando luga r a una proteína. Por
pondient e .
ejemplo, en la membrana de un macrófago de un individuo existirán dos
variantes de la mo lécula HLA-A (la correspondiente al alelo HLA-A here-
"U-I
lo expresan
Todas las células no GR
dado de l padre y la del alelo HLA-A heredado de la madre), dos var iantes
I
de la molécula HLA-B, dos de HLA-C, dos de HLA-DR, y así sucesivamente.
"U-U
El tipaje HLA de un ind ividuo viene definido, entonces, por las dos var iantes
CPA (linfocitos 8,
de cada gen HLA.
monocitos-macr6fagos
ycélulas dendríticas)
ylinfocitos Tactivados
Composición
Cadena ~sada a + 13 1
Los genes HLA se heredan en haplotipo (término que define a un conjunto
de genes que se heredan juntos) y los fenómenos de recomb inación genética
Cadena a + Cadena 13
en estos genes son muy poco f recuentes.
HLA DR, DP. DQ
El sistema de genes HLA es muy polimórfico. La definición clásica de poli·
miaoglobulina
Tipos
HLA A, 8, (
Origen del AG
Intracelular RER
Extracelular
Procesado delAG en
Citoplasma
Fagolisoma
morfismo genético es la de una variante que aparece en más de l 1% de la
población sana, es decir, una variante de la normalidad. Los genes HLA admi ten variaciones en su secuencia de nucleótidos sin alterar su func ional idad.
Las posiciones polimórficas de estos genes se concentran en las regiones que
AG: antígeno; RER: retículo endopl~smko rugoso
van a codificar las zonas de la molécu la HLA donde se presenta el antígeno.
Características de Los distintos tipos de molécula HLA
Estas variantes para cad a locus (gen) HLA hace necesaria una nomenclatura
que nombre cada una de ellas, así, por ejemplo, en el gen HLA-B se puede
El hecho de que cada individuo posea var ias molécu las de clase I y de clase 11
encontrar diferentes variantes como HLA-B27, HLA-B5, entre otras.
puede constit uir una ventaja, pues perm itirá combinar más eficazmente un
mayor número de péptidos. La co lección de molécu las CPH que cada indivi-
La casi imposibilidad de encontrar dos individuos no emparentados, absolu·
duo posee le confiere un carácter específico de individua lidad para organizar
tamente idénticos, permite la aplicación del sistema en los estud ios de pater-
la respuesta inmun itaria.
nidad dudosa y en la identificación de ind ividuos a partir de restos humanos,
pero también es la base de los fenómenos de rechazo agudo de trasp lantes
en parejas donante-receptor no HLA idénticas.
Genética del sistema HLA y nomenclatura
HLA Y enfermedad
Los genes HLA se localizan en el brazo corto del cromosoma 6 (Cr6p)
humano. Los genes de clase 11 (DR, DQ Y DP) se sitúan más centroméricos
y los de clase I más teloméricos. Entre los genes de clase II y los de clase I
se encuentra un fragmento de l Cr6p al que se denominó #región genética
afect ados de ciertas enfermedades, f undamenta lmente autoinmun itarias e
del HLA de clase 11 1". Ese nombre es meramente descriptivo, pues la región
inflamatorias. Por ejemplo, el 95% de los individuos con espondilitis anqu ilo-
del HLA de clase 11 1 no contiene genes que den lugar a proteínas HLA, es
poyética son HLA-B27 positivos, mientras que la f recuencia de este antígeno
decir, como proteína no existe el HLA-II I (Figura 11).
en la población genera l es inferior al 10%.
Algunos alelas HLA se encuentran con gran frecuencia entre los pacientes
Clase 11
Centrómero
Clase 111
Clase I
DP
C4
Bf
C2
TNFo:
Genética deL HLA
14
ERRNVPHGLFRVRUJ
TNF~
• e
A
Manual CTO de Medicina y Cirugía, 10. 3 edición
04 . Complejo principal de hlstocom patlbllldad
La susceptibilidad que ciertos antígenos parecen conferir ante algunas enfer-
nes se han heredado en desequilibrio de ligamiento de forma ancestral (por
medades puede cuantificarse med iante el cálculo de l riesgo relativo (RR). No
ejemplo, en mutaciones del gen HFE, causantes de hemocromatosis heredi-
se ha encontrado ninguna asociación absolut a entre una molécula del CPH y
t aria y algunas deficiencias del complemento como C2 y C4).
ninguna enfermedad, es decir, nunca se ha encontrado un antígeno presente
en exclusividad en los enfermos y ausente en la población libre de la enfer-
En las enfermedades inmunomed iadas, como la artritis reumato ide, se han
medad, por tanto, la presencia del alelo HLA asociado sería un factor más
encontrado condicionantes genéticos, de los que el m ás importante es el
de predisposición a la enfermedad en cuestión. No obstante, la asociación
HLA. No obstante, el mecanismo patogénico es complejo, por lo que pue -
clásicamente considerada más fuerte de un HLA con una enfermedad es la
den considerarse como enfermedades poligénicas mod ificadas con factores
del DR1S (DR2) y HLA-DQB l ·06:02 con la narcolepsia, la enfermedad ce líaca
ambientales. Para entender el complejo papel del HLA en estos procesos
presenta una fuerte asociación con DQ2 y DQ8 con un alto VPN (va lor pre-
autoinmunit arios se debe considerar el papel fi siológico de las molécu-
d ictivo negativo, en este caso no se r DQ2 y/o DQ8 excluye, con una altísima
las CPH en la respuesta inmunit ari a: un combinado HLA-péptido particu lar
probabilidad, la ce liaquía).
puede semejarse espacialmente y, por tanto, parecer idéntico a la combi nación formada por otra molécula CPH del mismo ind ividuo y un antígeno
propio, lo que explicaría ciertas reacc iones autoinmunitarias.
Otras enfermedades con importante asociación con HLA son la coriorretinopatía en perdigonada o enfermedad de Birdshot (HLA-A29), artritis reuma toide (HLA-DR4 y DR1), enfermedad de Beho;:et (HLA-BS l ), la forma cl ínica de
PREGUNTAS
psoriasis guttata (HLA-Cw6) y la hipersensibilidad al antirretrovira l abacavir
MIR
(HLA-B· S7:01). En algunas enfermedades, la asociación con unas variantes
HLA determinadas se produce debido a que son enfermedades genéticas
,/ MIR 13· 14, 57
,/ MIR 09-10, 215
cuyos genes responsables se loca lizan cercanos a los del HLA y las mutacio-
tema retículo endotel ial, linfocitos B y linfocitos T activados (los T en
reposo no lo expresan).
,/ El complejo principal de histocompatibil idad (CPH) está compuesto
por un grupo de mo léculas que, por su estructura y función, se clasifican en dos tipos: CPH de clase I y CPH de clase 11. A estas moléculas
también se les denomina con la nomenclatura HLA (Human Leukocyte
Antigen).
,/ Se denominan células presentadoras de antígenos profesionales a
aquéllas que expresan CPH de clase 11.
,/ Las moléculas HLA de clase I presentan antígenos sintetizados en la
propia célula que los expres a. Los de clase II p resentan antígenos exógenos, que han sido capturados y fagocitados por las células que los
exp resan.
,/ El sistema genético que cod ifica el CPH es uno de los m ás polimórficos
que se conocen, está en el cromosoma 6 (brazo corto) y se hereda de
modo autosómico codominante.
,/ El conj unto de genes CPH de un cromosoma 6 se hereda en bloqu e,
como si fuese uno solo (haplotipo).
,/ Todas las cé lulas del cuerpo, menos los hematíes, expresan en su membrana moléculas CPH de cl ase l. Los principales son los HLA-A, HLA-By HLA-C.
,/ La herencia del HLA tiene pocas posibilidades de recombinación, por lo
que la probabi lidad de tener un hermano HLA idéntico es aproximadamente del 2S%.
,/ Los CPH de clase 11 (HLA-DR, DP Y DQ) sólo los expresan conjuntos concretos de células: monocitos, macrófagos, células dendríticas y del sis-
15
ERRNVPHGLFRVRUJ
Respuesta inmunitaria
Tema pri oritario. Se debe enlazar con los (apítulos 3y 4. Se debe prestar
!'Spt'(ialatención a los gruflOS de linfocitos Tdesde el puntode vista funciúnal,
ya las novedades ro la wacteriz.Jción y fu nción de lús Tcolaboradores
mm yTreg uladores).
•
La respuesta secundaria tiene lugar cuando el sistema inmun itario
encuentra un antígeno por segunda vez o en subsiguientes ocasiones.
Respuesta inmunitaria
Se distingue de la primaria por:
Mayor rapidez en instaurarse, es decir, presenta una fase de latencia más corta.
La respuesta inmun itaria abarca el conjunto de procesos que desarrollan las
Los anticuerpos duran más tiempo en el suero (fase de meseta más
células del sistema inmunitario cuando penetra una sustancia inmunogén ica
prolongada).
en el organismo. En la elaboración de est a respuesta hay una serie de fases:
El título de anticuerpos alcanza un va lor mucho más alto (mayor
•
Reconocim iento del antígeno.
potencia).
•
Identificación, activación y expansión de los escasos linfocit os específicos para dicho antígeno, formando clones.
Camb io de clase: los anticuerpos, en vez de IgM son IgG, IgA o
IgE (revisar cambio de clase o isot ipo del linfocito B en el Capi-
•
Diferenciación efectora de las células del sistema inmunitario.
tulo 3, apartado Secuencia de la sinapsis inmunológica) (MIR
•
Desarrol lo de la respuesta: acción de las células, o sus productos (anticuerpos), sobre el antígeno.
09· 10,216).
La afinidad de los anticuerpos por el antígeno es mayor.
RECUERDA
Clásicamente, se distinguen dos grandes tipos de respuesta efectora:
•
Respuesta humoral, desarrol lada por los linfocitos B y coordinada por
La presencia de IgM específica para el microorganismo
sospechado permite detectar primoinfecciones agudas.
los TH2.
•
Respuesta celular, desarrollada, fundamentalmente, por los linfocitos T
citotóxicos y coord inada por los TH1; ésta puede ser muy heterogénea.
Las características de mayor potencia y rapidez de la respuesta secundaria
se deben a:
•
Un mayor número de linfocit os 8 yT, seleccionados para el Ag, que en la
respuesta primaria (células de memoria). Las estrategias de vacunac ión
Respuesta de anticuerpos primaria
se basan en generar linfocitos de memoria por exposición a antígenos
atemperados respecto a su patogenicidad, de modo que, en caso de
infección por el patógeno, se pueda establecer rápidamente una res~
y secundaria
puesta secundaria.
•
Las células B de memoria generadas han experimentado hipermutacio-
La respuesta de anticuerpos (AC) juega un gran papel en la defensa f rente
nes somáticas puntua les en la zona de unión al antígeno que les confie-
a bacterias, antígenos solubles (toxinas), virus, protozoos y otros parásitos
ren mayor afinidad por éste (MIR 15·16, 49).
extracelulares.
RECUERDA
Puede ser de dos tipos: primaria y secundaria.
•
La respuesta primaria está mediada por células vírgenes y la
secundaria por células de memoria.
La respuesta primaria se produce la primera vez que el sistema inmunitario entra en contacto con el antígeno en cuestión. Se caracteriza porque,
después de la exposición al antígeno, se originan las siguientes fases:
Antígenos T-dependientes
Fase de latencia (5· 7 días). En esta fase todavía no aparecen anti-
cuerpos.
Fase de incremento. La concentración de los anticuerpos séricos
La mayoría de los linfocitos B específicos necesitan la ayuda de linfocitos T
aumenta en progresión geométrica hasta alcanzar la siguiente:
Fase de meseta. La secreción se mantiene durante unos días (3 -5)
co laboradores para activarse, proliferar y diferenciarse hacia células secre-
y, a continuación, desciende lenta, pero progresivamente, en los
toras de anticuerpos. Estos linfocitos B productores de la respuesta de
anticuerpos T-depend iente (timo-dependiente) se loca lizan en los folículos
siguientes 10-15 días.
linfoides de los ganglios y en la médula ósea.
En la respuest a primaria, los anticuerpos son siempre de la clase IgM y
La cooperación T-8 se establece gracias al papel de los linfocitos 8 como célu-
con baja afin idad por el antígeno.
las presentadoras de Ag (ePA).
16
ERRNVPHGLFRVRUJ
Manual CTO de Medicina y Cirugía, 10. 3 edición
05 . Respuesta Inmunitaria
Los linfocitos B, específicos para un epítopo, tras reconocer el Ag con su Ig de
mecanismos efectores. Se puede clasificar al linfocito T desde el punto de
superficie, endocit an todo el antígeno, lo procesan (degradación y desnatu-
v ista func iona l en T citotóxico y en T colaborador (cooperador o he/perlo
ralización) y pasan a expresar péptidos del antígeno en su membrana, un idos
a las moléculas CPH de clase 11.
Linfocitos T citotóxicos
Los linfocitos T colaboradores (he/per) 2, con un RCT capaz de reconocer el
Este tipo de respuestas son esenciales en la defensa contra virus y en la eli-
antígeno unido al CPH de clase 11, se unen a él y se activan, transm itiendo a
minación de otros microorganismos intracelulares: Condjda, PneumocysHs,
su vez señales de activación al linfocito S:
Toxop/asma y mycobacterjas, entre otros.
•
IL-4 promueve la proliferación de los linfocitos S activados, así como la
d iferenciación de los linfocitos S que están proliferando.
En la respuesta de citotoxicidad específica, los linfocitos T he/per juegan un
•
IL-6 actúa promoviendo la d iferenciación.
papel fundamental como cé lulas colaboradoras (TH1). La función cooperadora
•
Interacción C040 (célula S) con CD40L (CD154) de la célula T colabora-
depende, en su mayor parte, de la acción de las interleucinas (IL- 2, INF-y ... ) que
dora (he/per) induciendo el cambio de isotipo de las Ig del linfocito S de
actúan sobre las cé lulas efectoras (T citotóxicos) y sobre los macrófagos, dando
IgM a IgG, A, E.
lugar a las reacciones de hipersensibilidad retardada.
Como resultado fina l de la respuesta T-dependiente, se genera un gran número
Los linfocitos T cit otóxicos (TC) reconocen el antígeno en asociación con las
de células secretoras de anticuerpos específicos y linfocitos B memoria, que
moléculas CPH en la membrana celular de otras cé lulas y, una vez activadas,
permitirán la respuest a secundaria tras subsigu ientes contactos con el mismo
lisan dichas células (células d iana).
antígeno.
El principal papel biológico de los linfocitos TC es interven ir en la elimina-
Antígenos T-independientes
ción de las células infectadas por v irus y cé lulas no infectadas, pero que son
detectadas como extrañas, tales como las tumorales o las de los órganos
Existe un pequeño número de sustancias, conocidas como antígenos T-in-
trasplant ados. La mayor parte de los linfocit os TC son CD8+, pero también
dependientes, que son capaces de inducir la respuesta de anticuerpos sin
existe cierta proporción de linfocitos T C04+ citotóxicos con especificidad
necesidad de la cooperación de los linfocitos T. Entre ellos están:
restringida a molécu las CPH de clase 11.
•
Lipopol isacárido (LPS) de la endotoxina bacteriana de Gram (-).
•
Flagelina polimérica microbiana.
Generación de linfocitos T citotóxicos. Al igual que la respuesta de anticuer-
•
Polisacáridos: dextrano, levano, entre otros.
pos, obedece a los mismos principios vistos con anterioridad:
•
Polímeros de D-aminoácidos.
•
Selección por el Ag de los escasos linfocitos específicos existentes antes
del estímulo antigén ico.
•
Se caracterizan por ser estructuras poliméricas en las que los determinantes
Amplificación clonal de los linfocitos seleccionados med iante un proceso
de proliferación selectiva. El número incrementado de linfocitos T C08+
antigénicos se repiten muchas veces, además de por ser resistentes a la degradación metabólica, por no ser presentados a través de las molécu las del sistema
específicos garantiza que la respuesta secundaria sea más potente y rápida.
HlA. Frente a estos antígenos, la respuesta siempre tiene características de respuesta primaria, aunque se hayan tenido contactos previos con el antígeno: se
La respuesta citotóxica, se desarrolla en tres etapas:
producen sólo anticuerpos IgM y no existe memoria inmunitaria, ya que la inte-
•
Reconocim iento del Ag. Los linfocitos T cit otóxicos reconocen el Ag unido
racción entre el linfocito S y el linfocito T es necesaria para generar el cambio de
a moléculas CPH propias, o bien reconocen exclusivamente (sin necesi-
isotipo de inmunoglobulina y para generar la memoria inmunológica S. Se debe
dad de que presenten ningún antígeno) las moléculas CPH presentes en
recordar que existe una subpoblación de linfocitos B que expresan el marcador
células alogénicas (por ejemplo, en el caso de un t rasplante de órganos de
C05, muy implicada en la respuesta de anticuerpos timoindependiente. Esta
un donante no HLA idéntico).
•
población celu lar no genera memoria, pero puede producir cierta cantidad de
Activación. Se activan y expresan receptores de Il- 2. Para que puedan
proliferar y manifestar su función citolítica, requieren que otras células
los estimu len con IL-2 (suelen ser linfocitos TH1 próximos).
IgG. Su funcionalidad es mínima en niños, especialmente menores de 2-3 años.
Es posible incrementar la inmunogen icidad de los antígenos pol isácaridos
•
Destrucción de las células diana. Como respuesta a la IL- 2, los linfocitos
conjugándose con un carrjer proteico, de modo que se cons iga una res-
citotóxicos proliferan y se activan de modo que, al entrar en contacto
puesta T-dependiente. Esta estrategia es la que siguen las nuevas vacunas
con las células diana que expresan el antígeno, inducen su apoptosis
conjugadas (MIR 07-08, 244) contra los meningococos.
(principalmente vía perforinas/caspasas). Una vez han destru ido la
La mayor parte de los linfocitos S productores de anticuerpos contra antíge-
cé lula, pueden seguir ejerciendo su efecto citotóxico sobre otras células,
nos T-independ ientes se encuentra en el bazo. Tras una esplenectomía, se
ya que la acción lítica es específica contra la diana y no existe daño con -
producen respuestas deficientes frente a ese tipo de antígenos.
tra la propia célula efectora de la respuest a.
Linfocitos T colaboradores
Los linfocitos T colaboradores modu lan la respuesta inmunitaria ofreciendo
Respuestas de las células T.
su colaboración, en forma de citocinas, a otras células del sistema inmunitario. Los linfocitos T pueden ser funcionalmente colaboradores, independien-
Cooperación y citotoxicidad
temente de que sean C04+ o CD8+. Estas cé lulas se clasificaban clásicamente
en diversas categorías determinadas por el patrón de citocinas que son capaces de producir (Figura 12 V Tabla 5):
•
Tras la activación específica de antígeno del li nfocit o T en la sinapsis inmuno-
TH1, producen Il -2 e IFN -y. Controlan las reacc iones de inmunidad celular; que son especia lmente útiles en infecciones por microorganismos de
lógica (véase el apartado 3.1. Unfocjtos T), se pueden presentar diferentes
17
ERRNVPHGLFRVRUJ
INMUNOLOGÍA
crecimiento intracelular o que son capaces de resistir dentro de las células
•
(mico bacterias). Aportan citoc inas que potencian la actividad de li nfocitos
reconocer el antígeno. Si es Il -12, se convertirá en THl, y si, por el contrario, es
1l-4, se convertirá en TH2; un microambiente rico en TGF-(3, Il-lO e IL- 2 favo-
T citotóxicos, NK V macrófagos.
rece la inducción de Treg y TGF-(3 e 1l-6 hacia TH-17 (MIR 13--14, 35).
TH2, producen IL-4, IL-5, IL-6 Y colaboran en las reacciones de inmunidad humoral (anticuerpos) mediante su capacidad de actuar sobre LB y
células plasmáticas, fundamentales pa ra neutralizar toxinas e infeccio-
Interviene
nes por gérmenes de crecimiento extracelular.
•
TH1
TH3, producen IL- lO y TGF-~, Y tienen funciones reguladoras o supre -
Il-2,IFN-y,
Implicaci6n
Inmunidad celular
Infecciones por microorganismos
de crecim iento intracelular
TNF-~a
soras. Expresan los marcadores CD25 y FOXP3 (además de CD3 y CD4).
THl
Il-4, IL-5, IL-6
Inmunidad humoral
Infecciones de crecimiento
extrarelular. Respuestas IgE
(mastodtos y eosinófilos)
THl
IL-l0, TGF-~
Funciones reguladoras
TH17, producen entre otras Il -17 e Il -22. Participan de forma importante
Regulación de la inhibición
de respuesta inmunitaria.
en la defensa f rente a infecciones por hongos, como Candida, V frente a
Regulación linfocitos autorreactivos
Se han descrito alteraciones en el número y/o función de las cé lulas T
reguladoras (Treg) en grupos de pacientes con diferentes enfermedades
inmunomediadas (enfermedad inflamatoria intestinal, espondiloartro patías, uveítis no infecciosas ... ).
•
algunas infecciones bacterianas. la disregulación de la respuesta inmu-
TH 17
nitaria hacia esta vía parece estar detrás de la fis iopatogenia de algunas
IL-17,IL-22
Infiamaoon, infeíciones
Infecciones fúngicas y bacterianas
Características de los linfocitos TH
enfermedades de base inmunológica como la psoriasis, espondiloartropa tías y enfermedad inflamatoria intestina l, entre otras, que hasta ahora se
consideraban mediadas principalmente por una disregulación TH1.
Alorreactividad
IFN-y
IL-2
TNF-a
La alorreactividad (o alorreconocimiento) es el hecho de que una gran proporción de los linfocitos T de un ind ividuo reconocen, sin necesidad de inmu-
nización previa, las moléculas CPH a logénicas distintas a las propias (de otro
individuo genéticamente distinto de la m isma especie), es decir, las variantes
polim6rficas exp resadas por otras personas. Es importante comprender la alorreactividad para el posterior estudio de los fenómenos de trasp lante en el
rechazo de órganos y en la enfermedad injerto contra huésped. No se conocen
los mecanismos exactos del alorreconocimiento. Se cons ideran var ias posibi-
IL4
IL-S
lidades de reconoc im iento por parte del RCT, siendo la principal que las regio-
nes polimórlicas de las moléculas CPH alogénicas, no presentes en el individuo
receptor, son reconocidas como el CPH propio con un Ag incorporado. La exis-
TGF-¡3
tencia de una gran proporción de linfocit os T alorreactivos determina que
Il~
la respuesta a estos antígenos tras una estimulación primaria sea ya muy
considerable.
IL-17
IL-22
Tolerancia
TGF-¡3
IL-1O
Se trata de un estado de ausencia de reactividad específica para antígenos
concretos que se adqu iere de forma activa. La más importante es la autotolerancia, que permite que el sistema inmunitario de un individuo no ataque
a las células de su propio organismo. Los mecanismos de toleranc ia pueden
est ablecerse a nivel central, durante la génesis y d iferenciación de las células
Grupos efectores de TH. Se representan las principales interleucinas
implicadas en la activación de los diferentes grupos, así como
las producidas por ellos
(timo en células TV médula ósea en células B) y a nivel periférico, sobre células adultas. la to lerancia establecida a nivel centra l sobre los linfocitos B en
la médula ósea es menos efectiva que la realizada sobre los linfocitos T en el
El tipo de respuesta de linfocitos co laboradores que se desarrolle f rente a un
timo, de ta l modo que se cons idera que la presencia de un pequeño número
antígeno concreto es tremendamente importante y puede significar que el
de linfocitos B levemente autorreactivos es norma l.
desarrollo de la respuesta concluya en desenlaces tan opuestos como la cura-
RECUERDA
ción o la aparición de formas graves de enfermedad. El que un li nfocito virgen
TH se conv ierta en THl, TH2, Treg o TH17 depende de múltiples factores, tanto
En sangre periférica no debe existir ningún linfocito T autorreactivo.
genéticos como adqu iridos (muchos no están todavía bien caracterizados). El
mejor conocido es la citocina con la que se ha coestimulado en el momento de
18
ERRNVPHGLFRVRUJ
Manual CTO de Medicina y Cirugía, 10. 3 edición
05 . Respuesta Inmunitaria
No obst ante, estos permanecen inactivos por la falta de colaboración de los
atrofiarse después de la adolescencia y que, en la mitad de la edad adu lta,
linfocitos TH2. Se conocen varios mecanismos para establecer la toleranc ia:
sólo tiene un 15% de su tamaño original.
•
•
Deleción clona!. Representa el principa l mecanismo de la "tolerancia a
nivel central" por el que se el iminan las célu las autorreactivas. Gracias
la capacidad de detectar moléculas extrañas se va perd iendo con la edad,
a él, se garantiza que los linfocitos maduros que dejan los órganos lin-
lo que conlleva que la incidencia de infeccio nes y neoplasias se incremente.
fo ides y van hacia tejidos periféricos no respondan a antígenos propios.
los anticuerpos se elaboran de forma m ás lenta V menos efectiva, por lo
Anergia clonal. Pérdida de la capacidad de respuesta a su antígeno de
que el efecto protector de vacunas, como la de la gripe, a veces no se pro-
células concretas. Se produce cu ando la cé lula present adora de antígeno
duce Vlos resultados de las campañas de vacunación en la tercera edad no
conf ie re estimulación antigénica al lin focito TH con activación de la 1.8
suelen ser los esperados.
señal (CD3), en ausencia de coestimulación antigénica (2. 8 seña l).
•
•
Supresión activa . Inhibición de la actividad celu lar por interacción con
También es frecuente en el anciano la aparición de autoanticuerpos a títu -
otras cé lulas, básicamente mediante secreción de citocinas inh ibitorias
los bajos. Algunos estudios han observado que la preva lencia de anticuer-
como TGF-~ e Il-lO (población TH3).
pos antinucleares (ANA) positivos a título bajo en población mayor de 65
Desviación de la respuesta. Por ejemplo, al cambiar una respu esta de
años, podría encontrarse alrededo r del 15%. Sin embargo, la mayo ría de
TH1 a TH2.
las veces éstos no son patogén icos ni causan clín icamente la enfermedad
autoinmun itaria, pues hay que recordar que este tipo de enfermedades,
con alguna excepción como el pénfigo y el penfigoide, no son típicas de
ancianos.
Envejecimiento e inmunidad
PREGUNTAS ·
MIR
Al iniciarse la vida adu lta, com ienza una dism inución lenta y permanente en
,/ MIR 15-16, 49
,/ MIR 13-14, 35
,/ MIR 09-10, 216
,/ MIR 07-08, 244
la inmun idad. El primer cambio aparece en el timo, órgano que comienza a
,/ la respuesta inmunit aria (inmunidad adquirida o específica) consiste en
el conjunto de acciones específicas que conducen a la eliminación de la
infección o situación de pel igro para el organismo.
produce la maduración de la afinidad, por lo que, además de ser más
rápida, es más eficaz.
,/ la mayoría de respuestas de an ticuerpos son T-depend ientes.
,/ la respuesta inmunit aria se puede dividir en varias fases, entre las que
destacan: el reconocimiento del antígeno extraño, la expansión de los
linfocitos específicos para ese antígeno, y el desarrollo de la respuesta
efectora propiamente dicha.
,/ En la respuesta de anticuerpos T-independiente, la latencia es m ás corta
pero no se produce memoria inmunológica.
,/ las vacunas conjugadas están form adas por antígenos polisacáridos
y proteínas que aumentan la inmunogenicidad para obtener una res puesta T-dependiente.
,/ Existen dos tipos de respuesta efectora: 1) humoral (anticuerpos), desarrollada por los linfocitos B V coordinada por los TH2, V 2) celul ar, desarrollada básicamente por los linfocitos T citotóxicos.
,/ la alorreactividad consiste en que los lin foc itos T de un individuo reco nocen, sin necesidad de inmunización previa, las cé lulas de otra persona genéticamente distinta (moléculas CPH alogénicas).
,/ la respuesta primaria (tras el primer contacto con el antígeno) tiene un tiempo de latencia, entre su inicio y la respuesta efectora, de algo menos de una
semana. Al fina l de la respuesta primaria se generan células de memoria.
,/ Aunque los linfocitos T C08+ y los NK tienen diferentes modos de identificar a las células diana, el mecanismo de eliminación es el mismo:
inducción de apoptosis en dicha célula.
,/ la respuesta secundaria (los sucesivos contactos con el antígeno) es llevada a cabo por las cé lulas de memoria y tiene un tiempo de latencia
muy corto (horas).
,/ la tolerancia consiste en la falt a de res puesta frente a antígenos propios
,/ En la respuesta primaria de anticuerpos, la Ig elaborada es la IgM . En la
o inofensivos (a limentos ... ).
i ,1
elaborados son I
I ~~'---_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _)
19
ERRNVPHGLFRVRUJ
Complemento
Este es un tema que hovuelto a ser preg unt!dode formo dirl'{t! en el MIR
en los últimos años. A.demás, su tra nsvmalidod hoce que seo ne(es.J riotener
claros todos los CO rKeplos ~ra poder resolver preg untls de otros temas más
preguntados de la asignaturil, CO/110 son las inm unodeficil'ncias yel trasplo nte.
Vía clásica
El sistema del complemento consiste en una cascada de activación enzimática
de un conjunto de proteínas, cuya finalidad principal es la de producir la lisis bacteriana. Los componentes del comp lemento son más de 30 proteínas séricas, la
Se inicia por la unión del Clq al Fc de las inmunoglobulinas G V M (excepto
mayoría de ellas se sintetizan en el hepatocito. Este sistema se encuadra dentro
G4). Por tanto, esta vía es el nexo del sistema del complemento con la inmu-
de la inmunidad innata o inespecífica, aunque como se verá, posee un nexo con
nidad específica. Necesariamente, para que se active est a vía, el anticuerpo
la inmunidad específica o adaptativa gracias a una de sus vías de activación.
se debe encontrar unido específicamente a su antígeno, por lo que forma
entonces los denominados inmunocomplejos. En definitiva, la vía clásica se
activa cuando reconoce inmunocomplejos (antígeno -anticuerpo).
Vía alternativa
Funciones del complemento
La ruta alternativa se activa directamente sobre la superficie de muchos
microorgan ismos a través de los factores C3 y factor B (FB). El principa l des•
Lisis del microorganismo o célula diana.
encadenante es el reconocim iento de LPS (lipopolisacárido). Es, sobre todo
•
Actuar como anafilotoxinas, re clutando células para la respuesta infla-
en los primeros años de vida, el principal mecan ismo de defensa f rente a
matoria.
bacterias encapsu ladas (M IR 08-09, 245).
•
Amplificación de la respuesta humoral específica.
•
El iminación de los inm unocomplejos: función rea lizada por la vía clás ica.
Vía de las lectinas (MBL)
Su mecanismo de activación es, en esencia, equivalente al de la vía alt ernativa, siendo específicamente manosas los antígenos m icrobianos reconocidos y siendo lectinas las proteínas del complemento que inician esta vía.
vías de activación del complemento
Clásicamente, se describían dos vías de activación del complemento: la vía clá-
vía común
sica y la vía alternativa. Más recientemente se describió una tercera vía de
activación, la vía de las lectinas unidoras de manosa (en inglés, vía de las MBL),
que es importante conocer, puesto que en la actualidad ya se han caracterizado deficiencias causadas por alteraciones en proteínas propias de esta vía.
Independ ientemente de la vía que inicie la activación, todas convergen en la
Es priorita rio conocer las diferencias en la activación de cada vía (Figura 13).
formación de una C3 conve rtasa, punto desde el que se pone en m archa una
ruta común para la formación del complejo de ataque a la membrana, CAM
(en inglés, MAC). El CAM se forma por el ensamblaje de las proteínas C5, C6,
............
¡rotlg_.
iln tKWrpo
SuperncJes
de mlc r_,.n.lslI'IOs
I
I
Comp~Jos
C7, C8 Y C9 sobre la membrana microbiana, formando en ella un poro, cuyo
efecto esencial es producir un notable desequilibrio osmótico en el m icroorganismo, que conduce a su lisis.
Regulación del complemento
Mo'<I1IIZK!On de (~Iulis
Op5Of\ClKIOn
UmlnioOn
InflMn.l:wI
potPnGllción
el, pJI6g-«f\OS
dell r.god:Olll
El comp lemento, por su vía alternativa, sufre una activación permanente-
de a-gentu fU!6genoJ
mente por la hidról isis espontánea de C3, por lo que necesit a unas v ías de
regu lación finas y precisas que eviten la producción de daños tisulares al
Tabla del complemento
propio individuo por el estado de inflamación continuo.
20
ERRNVPHGLFRVRUJ
Manual CTO de Medicina y Cirugía, 10. 3 edición
06 . Complemento
Existen var ias formas de regulación del complemento:
•
•
Labilidad de las proteínas del complemento, es decir, se degradan fácilmente.
•
Cllnh, que se une e inactiva Clr y Cls del comp lejo Cl.
•
Proteínas de control del comp lemento (CCP), que inactivan la formación
En mastocitos. Provocan la degranulación, con el consecuente aumento
de la permeabilidad.
Circulación extracorpórea y complemento
de C3 convertasas.
Durante el paso de la sangre por circuitos extracorpóreos se produce una
notable producción de anafilotoxinas, con las cons igu ientes secuelas cl ínicas.
El cont acto del plasma con las membranas de los aparatos de hemodiál isis
produce la activación de la vía alternativa del comp lemento, lo que causa
Receptores para el complemento
una granulopenia transitoria durante los primeros minutos de la d iálisis.
Unos efectos similares, pero más intensos, ocurren durante el bypass car-
•
CRl (: C03S). Su ligando es, sobre todo, el componente C3b, yen menor
d iopulmonar; en este caso, también se activa la vía clásica.
medida, el iC3b, así como C4b. Sus principa les funciones son:
Receptor opsónico en fagocitos, med iante el que reconocen y engu llen mejor los microorganismos recub iertos con C3b (MIR 1&.17, 49).
Los eritrocitos y plaquetas captan a través de este receptor inmu·
Cascada de las cininas
nocomplejos opsonizados y los llevan a los fagocitos "carroñeros"
del sistema reticuloendote liaL
•
Su defecto se ha asociado a pacientes con lupus.
Son el tercer sistema de formación de mediadores en cascada del plasma; los
CR2 (: (021). Se une a var ios productos de degradación derivados del C3b
otros son el sistema del comp lemento y la cascada de la coagulación. En la vía
(como iC3b y C3dg). También puede ligarse con el virus de Epstein-Barr.
de las cininas, el Clinh inhibe la enzima kalicreína, que es la responsable de la
conversión del cininógeno en brad icinina, molécula que incrementa notablemente la permeabilidad vascular.
En los pacientes con edema angioneurótico fam iliar, está aumentada la acti-
Complemento e inflamación
vidad de la enzima por la deficiencia del Cl inhibidor, cuantitativa o cualit ativa (MIR 11-12, 218). La deficiencia de factores de complemento más
frecuente en Europa es la de Clinh (MIR 15· 16, 48).
El sistema del comp lemento interviene de forma decisiva en el desencadenam iento de la inflamación, debido a la actividad quimiotáctica de las ana-
PREGUNTAS ·
fi lotoxinas C3a, C4a y CSa. De ellas, la más potente es CSa. De su acción se
MIR
derivan los siguientes efectos:
•
En neutrófilos. Aumento de las molécu las de adhesión y potenciación
.1 MIR 1&.17, 49, 51
.1 MIR 15-16, 48
.1 MIR 11· 12, 218
.1 MIR 08· 09, 245
del estallido respiratorio (producción de radicales libres).
Ideasclave
.1 El comp lemento en un sistema multiproteico de activación en cascada
.1 La principal deficiencia de factores del complemento a esca la mund ial
es la de C9. En Europa es la de C1 inhibidor (Clinh).
cuya misión consiste en marcar a las células potencia lmente pel igrosas
para ser fagocitadas y, si fuese posible, llenar la membrana plasmática
de poros por donde entren agua e iones que produzcan la descompensación de la homeostasis intracitoplásmica.
.1 El déficit de los factores de la vía clásica se asocia a una mayor pre disposición a padecer enfermedades autoinmunitarias, mientras que el
déficit de los factores de la vía común produce mayor susceptibil idad a
infecciones (MIR 16-17, 51) .
.1 Existen tres vías de activación: la clásica, la alternativa y la de las lectinas.
21
ERRNVPHGLFRVRUJ
clínica
Este es untemo muy importa nte. En los últimos años, las ¡¡reguntas de inm unología
clínica se hiln (oosolidado, ylo cierto es que olgurili lOO tan díniGlS que equivalen a
las de otras asigrliltu ras ~nfffd<l505,. RetJmiltOOJía ...). Se ~ estudi.Jr el terrn (00
detel1imiento, sin JIilSiIrlo por alto.
Tipos de trasplante
La inmunología clínica comprende múltiples facet as, entre las que se encuentra el trasplante, la patología a lérgica, la autoinmunidad y las inmunodefi·
ciencias.
Según la pareja donante-receptor:
Xenotrasplante. El donante y e l receptor son de especies animales d is•
tintas.
•
Alogénico. Donante y receptor son de la misma especie, pero distintos
genéticamente.
•
Singénico. Donante y receptor son genéticamente idénticos.
•
Autólogo. De células o tejidos procedentes de l propio rece pto r.
Estas últimas, por ser un tema amplio e importante en el estud io del MIR,
serán t ra tadas en un capítu lo propio.
o
Trasplante de órganos
Según la topo logía del trasp lante:
•
Ortotópico. El injerto se coloca en e l receptor en su luga r anatóm ico original.
•
Heterotópico. La localización del injerto en e l receptor es diferente a su
lugar anatómico original.
Generalidades
Tipos de rechazo
En la práctica, antes de rea lizar un trasplante se deben tener en cuenta tres
elementos en la evaluación de la compatibilidad donante-receptor:
En primer lugar, e l grupo sangu íneo ABO (MIR 16--17, 141).
•
•
El grado de semejanza entre los fenotipos CPH ent re donante y
receptor. La influencia de la compatibilidad CPH entre donante y
receptor varía de unos trasplant es a otros. En e l t rasplante hepático, por razones aún no muy bien aclaradas, la importancia de la
compatibil idad CPH donante-receptor es inferior a la del resto de los
órganos sól idos.
Una pecu liaridad especial la representa el trasp lante de córnea, tejido
que, por no estar vascularizado, no es accesib le para los linfocitos T
citotóxicos en cond iciones norma les y, por tanto, la compatibilidad CPH
carece totalmente de impo rtancia. Asimismo, distintos HLA parecen
tener una im portancia d iferente en el rech azo de los injertos, atr ibuyéndose una mayor influencia al DR. La gradación de la importancia de la
compatibilidad sería en el sigu iente orden:
•
•
Rechazo hiperagudo. Apa rece a las pocas horas del trasplante. Está causado por la existencia de anticuerpos preformados en la sangre del receptor cont ra el CPH del donante que fi jan complemento sobre las células del
injerto, destruyéndolas ráp ida y mas ivamente. Es una de las peores complicaciones de un trasplante, pero se puede prevenir realizando una prueba
cruzada pretrasplante con suero del receptor y linfocitos del donante.
Rechazo agudo. Aparece a las pocas semanas del trasp lante de un órgano
CPH no compatible. La causa es la a lorreactividad de los li nfocitos T a tra vés de una respuesta primaria de los linfocitos T CD8 contra las mo léculas
CPH de clase I del injerto, así como por la activación de los linfocitos T CD4
contra las moléculas CPH de clase 11 expresadas por las células dendr íticas
y monocitos del donante que se hallan presentes en e l tejido trasplantado
y que son distintas a los HLA de l receptor (MIR 10-11, 207).
RECUERDA
DR>B>A > C
•
A mayor expresión de B7 por las células presentadoras del
antígeno del donante, mayor es la tasa de rechazo agudo.
La posible existencia, previa al trasplante, de anticuerpos en el recep tor que puedan estar dirigidos contra los antígenos CPH de l donante
(prueba cruzada).
RECUERDA
•
En el trasplante de órganos sólidos, lo primero que hay que
comprobar es la compatibilidad ABO.
RECUERDA
En e l trasplante de médu la ósea, la compatibilidad HLA
debe ser del 100%, pero no se tiene en cuenta la compatibilidad ABO.
22
El rechazo agudo ace lerado ocurre unos días después de l trasp lante, y
se debe a la reactivación de células T sensibi lizadas previamente (es una
respuesta secundaria).
Rechazo crónico. Aparece años después de l trasp lante y se manifiesta
bajo la forma de una arteriosclerosis ace lerada en e l órgano inje rtado
(arteritis obliterante), lo que hace que, por lo genera l, e l envejecimiento
de los órganos trasplantados tenga lugar de una forma siete veces más
rápida que el que se desencadena de forma natural. La causa está poco
clara, pero en trasplante renal parece tener una re lación clara con las
diferenc ias en HLA entre donante y receptor. No se conocen fármacos
para controlarlo (Tabla 6).
ERRNVPHGLFRVRUJ
Manual CTO de Medicina y Cirugía, 10. 3 edición
07. Inmunología clínica
Tabla 6
no re lacionados familiarmente, HLA-i dénticos, son similares a los obtenidos
Rechazo
Patogenia
Cnlnlco
Agudo
cuando el donante y el receptor son hermanos.
Arterioesderosis acelerada
(arteritis obliterante)
RHS
RHS
TIpo 11
Tipo IV
Tarda en
aparecer
Horas postrasplante
Meses
postrasplante
Años postrasplante
Comentario
Prevención: prueba
cruzada con suero
del re<eptor
y linfocitos
deldonante
Alorreactividad
• No existe tratamiento
• Envejecimiento
acelerado del órgano
Los mejores resu ltados se obtienen en los pacientes afectados de leucemia mieloide crón ica, y los peores, en pacientes con aplasia medu lar grave.
Cuando el receptor presenta diferencias CPH con el donante, el éxito del
t raspl ante es muy inferior.
En leucemias, se ha observado que los t raspl antes de médu la alogén icos dan
mejor resultado que los autotrasplantes, porque las recidivas son menos f recuentes. La explicación que se ha dado es que aparece una reacción de injerto
contra leucem ia (forma leve y beneficiosa de la enfermedad de injerto contra
Características de Los distintos tipos de rechazo
huésped), que reconoce como extrañas las cé lulas ma lignas y las destruye.
Prevención del rechazo
Enfermedad del injerto contra el huésped (EICH)
El órgano ideal para trasplante es aquél que comparte todas las moléculas
Se desarrolla cuando se trasplantan células inmunocompete ntes procedentes
CPH presentes en el receptor. Esta situación es extremadamente rara y la
del donante a un individuo inmunodeprimido HLA-incompatible. Las célu las
alternativa razonable es encontrar la mayor compatibilidad posible.
T del sujeto trasplantado no pueden reaccionar contra aquéllas y rechazar las
(por la inmunodepresión), mientras que las del donante reconocen a las del
RECUERDA
receptor como extrañas y atacan al endotelio vascu lar; tejidos y órganos.
Las diferencias entre las moléculas HLA de clase 11 inducen a
una respuesta alogénica más fuerte que la inducida por las
d iferencias en clase 1.
Constituye una grave compl icación del trasp lante alogénico de progen itores
hematopoyéticos, aunque también puede aparecer en otros injertos (trasplante intestinal). La EICH no aparece exclusivamente en los trasp lantes.
En el trasplante de órganos sólidos, la compatibil idad debe establecerse, en
También puede presentarse cuando se realizan transfus iones de sangre a
primer lugar, a nivel de las moléculas CPH de clase 11, especialmente DR, ya
un paciente inmunodeprimido o con una inmunodefic iencia celular. Si un
que dichas moléculas están directamente implicadas en la activación de la
paciente presenta un déficit inmunitario ce lular grave y precisa una transfu-
población mayoritaria de los linfocitos T helper (los CD4+).
sión, la sangre que se le vaya a administrar debe ser previamente irradiada
con el fin de im pedir que los linfocitos T alorreactivos proliferen y desarrollen
Trasplante de progenitores hematopoyéticos
la enfermedad.
Consiste en administrar al receptor del trasp lante la infusión intravenosa de
los progenitores del donante, que han podido ser obten idos de la médu la
Las manifestaciones clínicas de la EICH son más comunes en pacientes mayores y mimetizan un proceso autoinmunitario. Las más comunes son las alte-
ósea, de sangre de cordón umbilical y de sangre periférica.
raciones cutáneas, hepáticas (colangitis con colest asis), gastrointestinales
(malabsorción), artritis y bronqu io litis obliterante.
Pueden producirse varias complicaciones: principalmente el rechazo de la
médula ósea trasp lantada y la enfermedad de injerto contra huésped, en
Dentro de las man ifestaciones cutáneas, lo más característico es la presen-
ocasiones fata l. Ambos fenómenos se deben a las respectivas diferencias
cia de un rash maculopapular. En los casos más graves, aparece la necrólisis
genéticas que puedan existir entre el donante y el receptor. Por ello, la pareja
donante-rece ptor idónea es la formada por dos hermanos HLA-idénticos o el
epidérmica tóxica.
autotrasplante o trasp lante autólogo, siempre que sea posible.
Según el momento de aparición, la EICH se clasifica en:
•
Aguda. Se desarrolla dentro de los primeros 3 meses postrasplante
A pesar de el lo, en el caso del trasplante alogénico HLA idéntico, también se
•
producen rechazos y, en mayor o menor medida, cuadros de enfermedad
(generalmente entre los 15 y 30 primeros días).
Crónica. 5e cons idera que existe una EICH crón ica si ésta se produce
de injerto contra huésped, lo que sugiere la existencia de otros sistemas de
después del tercer mes del trasplante o si una aguda se prolonga más
histocompatibilidad menores, algunos de el los de herencia ligada al cromosoma y, por lo que se han observado d iferencias entre los trasplantes de
allá de los 3 primeros meses. Se considera que, a los 6 meses de sufrir un
progen itores hematopoyéticos en f unción del género de la pa reja donan-
man ifestación de EICH crónica. El pronóstico es peor en los pacientes
te -receptor.
que sufren inmunodeficiencias. En el resto de los pacientes, si se some-
trasplante alogénico, al menos el 20% de los pacientes presenta alguna
ten a un adecuado tratamiento inmunosupresor a largo plazo (2 -3 años),
Un test funcional disponible para evidenciar si existe compatibilidad entre
la enfermedad revierte en la mayor parte de el los.
donante y receptor es el cultivo m ixto de linfocitos, donde se cocu ltivan linfocitos de ambos. La activación y prol iferac ión de los linfocitos implica que se
han reco nocido células extrañas y, por tanto, hay incompatibilidad.
Reacciones de hipersensibilidad
En muchas ocasiones, el paciente que requiere un t raspl ante de médu la ósea
no d ispone de hermanos HLA-idénticos; en estos casos, se puede realizar un
trasplante haploidéntico de un hermano, o de los padres, o un trasp lante
de médula procedente de donantes voluntarios HLA-idénticos (banco de
Existe una reacción de hipersensibil idad cuando se desarrolla una res-
donantes). Los resu ltados obtenidos con médu la procedente de donantes
puesta inmunitaria dirigida contra elementos que no deberían ser consi-
23
ERRNVPHGLFRVRUJ
INMUNOLOGÍA
derados como extraños, o hacia elementos patógenos, pero de una forma
•
Vasod ilatación.
inadecuada.
•
Aumento de la permeabil idad vascular.
•
Contracción de la musculatura lisa.
Hay cuatro tipos de reacc iones de hipersensibilidad, descritos por Gell V Coombs:
•
Hipersecreción mucosa.
•
Tipo l. Hipersensibi lidad mediada por IgE. Se verá con extensión más
adelante (M IR 14-15, 218).
•
Acumu lación de infi ltrados inflamatorios.
•
Tipo 11. Anticuerpos citotóxicos. Existen anticuerpos circulantes que se
La sintomatología aparece de forma brusca en cuestión de 2 a 20 minutos,
unen a células diana. La lisis se produce por fijación del complemento
tras la exposición al alérgeno. Las manifestaciones pueden quedar circuns-
o por citotoxicidad med iada por anticuerpos (NK). Como consecuenc ia
critas a un órgano o territorio (por ejemplo, rinitis) o bien dar lugar a una
de la activación del complemento, se liberan fragmentos qu imiotácticos (como eSa), que provocan la infiltración de polimorfonucleares.
reacción sistémica (shock anafiláctico).
Alérgenos
Son ejemplos de este mecanismo la enfermedad hemolítica del recién
nacido (por incompatibil idad Rh) y el rechazo hiperagudo de trasplantes.
•
TIpo 111. Patología por depósito de inmunocomplejos. Los inmunocomplejos
Los antígenos que estimulan la formación de respuestas de Ac IgE causantes
son agregados de antígeno, a nticuerpos y complemento q ue normalmente
de las enfermedades atópicas se denominan alérgenos. Puede tratarse de
son retirados de la circu lación por fagocitosis directa o por transporte de
proteínas o glucoproteínas que forman parte de productos naturales o de
los mismos hacia órganos, como el bazo, donde también son fagocitados
sustancias químicas de natura leza haptén ica -que se denom inan haptenos-
por los monocitos-macrófagos. En el Capítulo 6, Complemento, se trata el
(por ejemplo, la penicilina) que, al unirse a una proteína portadora del orga-
tema de los inmunocomplejos con más extensión (ejemplos son la enfer-
nismo, se conv ierten en material inmunogénico.
medad del suero y algunas de las manifestaciones del LES).
•
TIpo IV. Son las reacc iones tardías mediadas por células. El prototipo es la
Existen tres tipos de alérgenos, según la v ía de contacto con el mismo:
reacción de Mantoux: se produce tras la administración de tubercul ina a
•
Inhalables (aeroalérgenos).
un paciente que previamente esté sensibilizado. La reacción aparece a las
•
Alérgenos por ingestión (medicamentos, alimentos ... ).
48-72 h como una induración en el área de inyección. Ejemplos de pato-
•
Alérgenos por inoculación (fármacos y venenos de p icaduras de
logía mediada por hipersensibilidad de tipo IV son el rechazo agudo de los
insectos).
trasplantes (no se debe confundir con el hiperagudo) y los granulomas.
Los aeroalérgenos son los que provocan con mayor frecuencia alergia atópica de las v ías resp iratorias (asma y rin itis alérgica). Forman parte de la composición del material partículado de la atmósfera normal.
Alergia
Entre el los destacan:
•
Pólenes.
•
Materia l desprendido o producido por anima les (descamación de piel,
pelo ... ).
Todos los individuos sanos desarrollan respuestas de IgE frente a componentes de helmintos; esta respuesta desempeña un papel fundamental en la
•
Partícu las fecales de ácaros microscópicos del polvo doméstico.
protección del huésped frente a dichas infestaciones.
•
Esporas fúngicas.
•
Productos de polvo industria l.
No obstante, los atópicos tienen una pred isposición genética a desarrollar respuestas de IgE frente a moléculas antigén icas presentes en materia l
Los d istintos materiales alergén icos son mezclas antigén icas comp lejas. El
usua lmente no infeccioso ni patógeno (pólenes, ácaros ... ), antígenos ino-
aislamiento y la identificación bioquím ica del componente que actúa como
cuos, contra los que la mayoría de la población no presenta ta les respuestas.
alérgeno es de gran importancia para lograr la máxima fiabilidad en la est andarización de los preparados que se usan en las pruebas diagnósticas y en la
Los términos alergia otópico o otopia se usan, de forma mayoritaria, para
in m u noterapia hi posensi bi lizante.
designar a todo tipo de reacc iones alérgicas med iadas por IgE. Sin embargo,
Factores genéticos y ambientales que controlan
la síntesis de IgE
en algunas de las manifestaciones cl ínicas de los individuos atópicos, existen
mecanismos inmunológicos que implican a componentes del sistema inmunológico, más allá de la IgE.
Cuando los dos padres tienen antecedentes de atopia, hay un 50% de pro-
Respuesta de IgE
babilidades de que los hijos desarrollen enfermedades atópicas. Cuando es
sólo uno de el los, la posibilidad baja al 30%.
Las IgE tienen la propiedad de unirse a la membrana de basófi los y mastocitos a través de receptores de alta afin idad para el Fc de la IgE. Si un individuo
Existen dos mecanismos de control genético de la síntesis de IgE, uno re lacio-
sensibilizado entra de nuevo en cont acto con el mismo alérgeno, éste inte-
nado con CPH y otro independiente de HLA.
raccionará con las IgE fijadas en la membrana de los mastocitos y basófi los.
Esta interacción induce en las células un estado de activación que determ ina
Ciertas condiciones ambient ales actúan como factores promotores de las
la rápida y brusca liberación de med iadores inflamatorios preformados que
respuestas de Ac IgE frente a alérgenos en ind ividuos genéticamente predis-
contienen en sus gránulos (histam ina y otros) y la síntesis de novo de otros
puestos. Tales condiciones incluyen polutos atmosféricos irritables (tabaco,
mediadores (prostaglandinas y leucotrienos).
NO" gases de combustión de motores d iesel). La exposición a los aeroa lérgenos durante la primera infancia y la exposición al alérgeno durante infeccio-
Son el los los que determinan la sintomatología cl ínica, al inducir en los teji-
nes víricas de las vías respirator ias favorecen el desarrollo de enfermedades
dos a los que acceden:
respiratorias atópicas.
24
ERRNVPHGLFRVRUJ
Manual CTO de Medicina y Cirugía, 10. 3 edición
07. Inmunología c línica
Basófilos y mastocitos
mientras que su metabolización por la vía de la lipooxigenasa genera
leucotrienos (en concreto, LTC4) a partir del que se obtienen otros como
Mediadores preformados
contenidos en los gránulos
LTD4 y LTB4. A los leucotrienos (LTC4 y LTD4) se les llamaba sustancia de
Los gránulos citoplásmicos de basófilos y mastocitos contienen numerosos
ción de la musculatura lisa (broncoconstricción), vasodilatación e hiper-
mediadores preformados, como ami nas vasoactivas, proteoglicanos, protea-
permeabil idad vascular, edema e hipersecreción mucosa, así como un
sas neutras, factores quimiotácticos, hidrolasas ácidas y enzimas oxidativas:
potente efecto qu imiotáctico para los neutrófilos (LTB4).
Factor activador de plaquetas (PAF). Los basófi los y mastocitos activa -
•
reacción lenta de la anafi laxia (SRS -A), debido a una mayor duración de
su acción. La acción inflamatoria de estos metabol itos incluye contrac-
•
Histamina. Es la principa l amina vasoactiva en el ser humano. En otras
especies, los gránulos de los mastocitos también contienen serotonina,
dos de diversas especies producen factor activador de plaquetas (PAF)
pero ésto no ocurre en mastocitos humanos; las plaquetas humanas sí
a partir de un precursor almacenado, que causa agregación plaquetaria
contienen serotonina.
con formación de microtrombos y secreción de med iadore s contenidos
La histamina actúa sobre las estructuras hísticas a través de los recepto res:
(como la serotonina, en el caso de las plaquetas humanas). El PAF tiene
H1 (contracción de la musculatura lisa bronquial y gastrointestinal,
también propiedades espasmogénicas, que al igua l que las de los leuco-
vasod ilatación y aumento de la permeabi lidad vascular).
trienos son mucho más prolongadas que las de la histamina. Además,
H2 (secreción de ácido por las células parieta les gástricas, permea-
los leucocitos atra ídos al lugar de la reacción por los factores quimiotác·
bi lidad incrementada de las barreras epit eliales y aumento de la
ticos liberados por los mastocitos pueden, a su vez, liberar mediadores
secreción de moco). Existe un receptor H3 de la histamina que está
que refuerzan y prolongan los cit ados efectos inflamatorios.
implicado, únicamente, en la síntesis y liberación de la misma.
Anafilaxia generalizada o shock anafiláctico
•
Heparina. Principal proteoglicano.
•
Proteasas neutras. Las más conocidas de los gránulos de los mastocitos
Se trata de una reacc ión sistémica, a menudo incluso de carácter explosivo
humanos son la qu imasa y la triptasa.
(minutos), que refleja la liberación masiva de mediadores, principalmente
Triptasa. Tiene actividad proteolítica sobre C3, generando C3b y C3a.
histamina y leucotrienos (SRS-A), por basófilos sangu íneos y mastocitos de
Quimasa. In vitro, convierte la angiotensina I en angiotensina 11 V
mú ltiples territorios. Las manifestaciones clínicas aparecen con gran rapidez
tiene la capacidad de degradar componentes de la membrana basa l
tras la exposición al alérgeno en cuestión.
de las un iones dermoepidérmicas.
Los primeros síntomas suelen ser angustia V ma lestar profundos, y mani•
Factores quimiotácticos. Incluyen el factor quim iotáctico de los eosi-
festaciones de rin itis y conjuntivitis aguda (estornudos, rinorrea, congestión
nófilos de la anafilaxia (ECF -A) y otro factor con actividad quimiotáctica
nasal, lagrimeo y escozor conjuntival), prurit o y eritema general izados, segui-
restringida para los neutrófilos (NCF).
dos de urticaria y angioedema en diversas regiones; es frecuente el edema
laríngeo, pueden aparecer vómitos, diarrea y dolor abdominal. En los casos
Fijación de la IgE a eosinófilos, basófilos y mastocitos
más graves, aparece broncospasmo, taquicardia, arritmias e hipotensión.
La IgE se fija en la membrana de estas célu las (mastocitos y basófilos) a tra-
Los signos de shock pueden constit uir la primera manifestación V causar la
vés de receptores de alta afinidad para el Fc de IgE, a los que se denomina
muerte en los primeros momentos.
de tipo I (FclgEI) pa ra distinguirlos de los de baja afinidad, o de tipo 11, (CD23)
pre sentes en la membrana principalmente de eosinófi los. El receptor está
Los principales alérgenos implicados son medicamentos, venenos inocula-
compuesto por una cadena a, una cadena f) V dos cadenas y idénticas. Una
dos por insectos, alimentos y, con menor frecuencia, caspas de an imales V
sola célula cebada o mastocito puede unir cientos de moléculas IgE con
gas de óxido de etileno en las membranas de hemodiál isis. Los síntomas sue-
especificidades distintas.
len desaparecer a las 2 horas, pero pueden reaparecer a las 8 horas, motivo
por el que se debe ingresar a los pacientes durante 24 horas. El tratam iento
La un ión de la IgE con su receptor en el eosinófi lo lleva a una forma especial
requiere de identificación precoz del cuadro, y de la adm inistración de adre-
de citotoxicidad mediada por anticuerpos; está mediada, entre otras, por la
na lina por vía intramuscular (MIR 12-13, 163).
proteína catiónica del eosinófi lo, que desencadena la muerte de las células
de los helm intos.
La activación de basófilos y mastocitos med iada por IgE da lugar a la libera-
Inmunidad tumoral
ción de mediadores.
La unión del alérgeno con los anticuerpos IgE fijados en los basófilos V mas-
Inmunovigilancia
tocitos desencadena la activación de estas cé lulas, lo que conduce a varios
tipos de respuesta celular:
•
•
Degranulación, al cabo de 30-40 segundos se produce la exocitosis de
Se conoce desde hace mucho tiempo el fenómeno de la desaparición de
los gránu los, con la liberación de med iadores preformados (histamina);
algunos tumores mal ignos de modo espontáneo. A principios del siglo xx,
producen los efectos antes mencionados.
Erhlich sugirió que las células malignas podían ser detect adas y elim inadas
Formación de productos del metabolismo del ácido araquidónico. El
por el sistema inmunitario. El principio de la inmunovigilancia sostiene que,
entrelazamiento de los receptores FclgEI determina la activación de la
cuando surgen células aberrantes, son reconocidas y eliminadas por las célu-
enzima fosfo lipasa A2, que libera ácido araquidónico a partir de los fos-
las del sistema inmunitario (fundamenta lmente por las células T y NK). Los
fo lípidos de membrana. La metabolización por la vía de la cicloox ige-
fallos en esta respuesta inmunitaria serían uno de los múltiples factores que
nasa origina prost aglandinas, principa lmente, PGD2 V tromboxano A2,
en último término llevarían a la aparición del tumor.
2S
ERRNVPHGLFRVRUJ
INMUNOLOGÍA
•
Al existir reconocimiento por parte del sistema inmunit ar io, ello implica que
deben existir antígenos en las células ma lignas, que no existen en el resto de
Selección de células que no expresan los antígenos. Ya se vio con anter ioridad.
•
las células del organismo.
Factores bloqueantes. La secreción de productos inmunosupresores
como histamina V citocinas (TGF-¡3) por parte de las células del tumor.
•
Esta teoría estuvo e n vigencia hace unos años, cayendo en el o lvido con poster ioridad. Últimamente, han aparecido una serie de estudios y evidencias
Tolerancia forzada, por ejemplo, por la ausencia de e)(presión de moléculas como CD8D (B7) con la consiguiente anergia por ausencia de señal
que han llevado a replantearse el estudio de la inmun idad tumoral. Dichas
coestimu ladora.
•
evidencias son, entre otras, la alta incidencia de tumores ma lignos en los
pacientes con inmunodeficiencias primarias (Iin famas, carcinoma gástrico),
Expresión de moléculas protectoras en la superficie celular. Algunos
tumores e)(presan una variedad mutante de la ICAM-l (proteína de
adquiridas (Kaposi, linfamas ... ) o iatrógenas por inmunosupresores (Iinfa-
adhesión celular), que tiene una gran homología con las proteínas regu ·
mas, carcinoma gástrico).
ladoras del complemento (pertenecen a la misma superfami lia) y protege a las cé lulas de la lisis mediada por complemento.
•
Otro acontecimiento reciente es la consecución de remisiones de tumores
ma lignos mediante inmunoterapia (en tumores como el carcinoma renal y
Expresión de fas-ligando (CD95l). Esta molécula induce la apoptosis de
los linfocitos que se acercan a intentar destru irlas.
melanoma con resultados equivalentes, incluso a veces superiores, a otras
técnicas terapéuticas como quim ioterapia o radioterapia).
Ademásde estos mecanismos, las cé lulas tumorales su elen aumentar su capa cidad de diseminación disminuyendo su adhesión, por ejemplo mediante la
Sistema inmunitario y tumor
inactivación de moléculas como la cadherina E (MIR 09-10, 211-GT).
Las biopsias y piezas de e)(tracción quirúrgica de tumores sólidos sue-
Antígenos oncofetales
len mostrar un infiltrado de células mononucleares entre las células de l
estroma y del tumor. Est as células son una mezcla de fagocitos, monoci-
Algunos tumores e)(presan antígenos que, si bien no son específicos de tumor,
tos, linfocitos B, T V NK, Y a veces células plasmáticas. El vo lumen de esas
no es normal que se e)(presen en células adultas diferenciadas, pu esto que
cé lulas llega a representar, en algunos casos, más de la mitad del tota l de l
sólo lo hacen de modo normal durante el desarrol lo embrionario.
tumor.
Los mejor caracterizados son:
•
La reacción del sistema inmunitario contra los tumores im plica que en ellos
debe haber antígenos que no se encuentran en tejidos normales.
a-fetoproteína. Constituye la primera globulina en el suero embrionario. Se com ienza a producir en el saco v itelina V luego en endodermo
e hígado. Tras el nacimiento, cesa su producción y es paulatinamente
Los detractores de la teoría de la inmunovigilancia sostienen que el sistema
sustitu ida por la albúmina. Niveles altos en el adulto implican una desdi-
inmun itario es ineficaz contra las células neoplásicas, basándose en el hecho
ferenc iación del tejido hepático hacia fo rmas embrionarias, característi-
de que los tumores humanos son heterogéneos en cuanto a la presenta-
cas asociadas a la presencia de un hepatocarcinoma o a la rege neración
ción de antígenos debido, sobre todo, a la inestabil idad genética propi a de
hepática (hepatitis, cirrosis ... ).
•
las células mal ignas. Como consecuencia de dicha inestabi lidad, aparecen
Antígeno carcinoembrionario. Es una proteín a de superficie presente
muchos tipos de e)(presión an tigénica, el sistema inmunitar io destruirá las
en la membrana de las células del intestino fetal. Los niveles altos en
células que e)(presen antígenos detectables y dejará intactas a aq uéllas que
un adulto se asocian, asimismo, a procesos de desd iferenciación celular
no los e)(presen; en otras palabras, se seleccionará a la población que carece
en tejidos endodérmicos: tumores y procesos de regeneración tras des-
del antígeno V, a la larga, todas las células presentes en el tumor eludirán el
trucc iones celulares de origen inflamatorio. Aunque no es un marcador
tumoral utilizable en d iagnóstico, una vez confirmada la e)(istencia del
sistema inmunit ario.
tumor, sirve para va lorar la masa tumora l y va lorar la evolución posope-
Mecanismos de escape a la respuesta inmunitaria
ratoria (recidivas, metást asis ... ).
El crecimiento de un tumor implica que las cé lulas m alignas consiguen eludir
la respuesta inmunitaria frente a ellas o, al menos, la modulan para que sea
menos intensa que la capacidad proliferativa del tumor. E)(isten varios meca-
PREGUNTAS ·
MIR
nismos que util izan las células malignas para evitar su destrucción.
Modulación antigénica. Los antígenos son modulados por la cé lula
•
mal igna, y ésta deja de e)(presarlos mientras le suponga una desventaja.
26
ERRNVPHGLFRVRUJ
.1 MIR 16-17, 141
.1 MIR 14-15, 218
.1 MIR 12-13, 163
.1 MIR 10-11, 207
.1 MIR mi-lO, 211-GT
Manual CTO de Medicina y Cirugía, 10. 3 edición
07. Inmunología c línica
de médula ósea, y su clínica es simi lar a un cuadro autoinmunitario:
alteraciones cutáneas, hepáticas (colangitis con colestasis), gastrointestinales (malabsorción), artritis y bronquiolitis obliterante.
./ El estudio de expresión de moléculas y la cuantificación de las pob laciones de linfocitos (B, T CD4, T CDS y NK) Y el coc iente CD4/CDS mediante citometría de flujo es una de las herramientas más utilizadas
para valorar e l grado de competencia inmunitaria.
./ La manifestación más específica de la EICH es la bronquiolitis obliterante, m ientras que lo más frecuente son las manifestaciones cutá neas.
./ En un trasplante de órganos, la primera compatibilidad que se debe
asegurar es la de grupo sanguíneo ABO, deb ido a la presencia en la
sangre de los anticuerpos naturales frente a los GS.
./ La influencia de la compatibi lidad CPH entre donante y receptor varía
de unos trasplantes a otros, siendo máximo en médula ósea y poco
importante en el trasplante hepático .
./ La hipersens ibi lidad consiste en el desarrollo de una respuesta inmunitaria dirigida contra elementos que no deberían ser considerados
como extraños, o hacia elementos patógenos, pero de una forma inadecuada.
./ Se ha determinado que la principal compatibilidad de CPH, de cara
a la compatibilidad de órgano, es la de los CPH de clase 11, concreta mente DR.
./ Se han descrito cuatro tipos de reacciones de hipersensibil idad (RHS):
I (anafiláctica), 11 (citotóxica), II I (por inmunocomplejos) y IV (mediada
por células).
./ Se han descrito tres tipos de mecan ismo de rechazo: hiperagudo (inmun idad humoral), agudo (inmun idad celular) y crónico (no se cono ce su causa).
./ El rechazo hiperagudo es una reacción de hipersensibilidad tipo II y el
rechazo agudo es de tipo IV.
./ La inmunovigilancia es una teoría que sostiene que, al surgir células
mutantes con potencialidad mal igna, éstas son reconoc idas y el iminadas por las células del sistema inmunitario (f undamentalmente, las
cé lulas T y NK). Los fallos en esta respuesta inmun itaria, como el retardo en la respuesta, llevarían a la aparición de tumores.
./ La enfermedad de injerto contra huésped (E ICH) es una situación
parecida al rechazo agudo de órganos en la que el rechazado es el
cuerpo del receptor del tejido y los atacantes los linfocitos T del donante. La casi total idad de 105 casos se producen tras un trasp lante
27
ERRNVPHGLFRVRUJ
Inmunodeficiencias
Una ¡¡re\juntl OOIig,¡d.J en el examen MIRes sobre lasinmuoodeficieno::ias, tanto
en forma de caso clínko COOlO (OO(~tual. Es untema complicado yextenso,
en el que hayque detenerse y¡¡riorizar. Se debe prestar espe<:ial ,¡tenóoo ,¡ los
microoo¡.lnismos"propios" de cada grupo de inmun<Xleliciencias yarasgos
silldrómicúS, que ayudarán al estudiante,¡ orientlrel caso clínito
•
•
Por esta razón, deficiencias en diferentes componentes del sistema inmunitario producirán una patología infecciosa d iferente en cada caso:
Concepto de inmunodeficiencia
•
Déficit de anticuerpos. Las infecciones y sepsis suelen ser por Haemo-
philus, neumococo y estafilococos. Las de origen fúngico son muy infrecuentes y las infecciones víricas están causad as cas i exclusivamente por
Las alteraciones cuantitativas o cua litativas en uno o más de los componen-
enterovirus. En niños aparecen infecciones pulmon ares, otitis y meningitis;
tes de la respuesta inmunit aria producen una descoord inación de las res-
también son frecuentes las diarreas por Giardia lamblia. En adolescentes y
puestas inmunitarias que se manifiesta a nivel clínico, por lo general, como
una mayor susceptibilidad a las infecciones.
adultos son características las neumonías de repetición, bronquitis crónica,
sinusitis crónica (enfermedad sinopulmonar crónica) y otitis media crónica.
Las anomalías intrínsecas (genéticas) de los componentes del sistema inmu-
Déficit de co mplemento. Presenta una elevada incidencia de infecciones de repetición por bacterias capsuladas, especialmente por el género
nitario, que a su vez pueden ser congén itas o adquiridas, son la causa del
Neisse,ia y enfermedades por depósito de inmunocomplejos cuando el
grupo de síndromes y enfermedades denominadas inmunodeficiencias
déficit afecta a alguno de los factores de la vía clásica. La sintomatolo-
primarias. Las inmunodeficiencias secundarias se producen por agentes o
gía infecciosa es especia lmente re levante durante los primeros años de
situaciones ajenos al sistema inmunit ario, pero que, al alterarlo, dan lugar a
vida, especialmente las men ingitis.
•
una respuesta inmunitaria deficitaria, algunos autores citan al síndrome de
•
Défidt de inmunidad celular. Presenta infecciones graves y recu rrentes por
Down y al síndrome de Turner, como causas congénitas de inmunodeficien-
virus latentes como el herpes simple o varicela zóster. Estos pacientes pre-
cia secundaria.
sentan infecciones por microorganismos que habit ualmente no son patógenos (oportunistas), como hongos, levaduras y Pneumocystis. La candidi asis
Las inmunodeficiencias más frecuentes son las secundarias, globalmente
mucocutánea aparece, prácticamente, en todos ellos. También pueden
en primer lugar, la malnut rición y en el mundo desarrollado las iatrógenas
presentar infecciones por otros microorganismos como las mico bacterias.
(inmunosupresores, esplenectomías ... ) y, en segundo lugar, el SIDA.
•
Inmunodeficiencias combinadas. Afectan tanto a la inmun idad celular (mediada por célu las T) como a la humoral (anticuerpos) y son, por
•
•
lo general, las más graves. Cua lqu ier microorgan ismo, inclu idos los no
patógenos, puede dar lugar a infecciones graves, sobre todo, los de crecimiento intracelular obligado.
Clínica de los defectos inmunitarios
Es tan bajo el nivel de inmunidad de estos pacientes que, si se real iza
una transfusión sanguínea, los escasos linfocitos presentes en la sangre del donante pueden desencadenar una enfermedad injerto contra
huésped mortal.
Las infecciones de repetic ión suelen ser el motivo que hace sospechar que se
•
est á ante una inmunodeficiencia aunque, a veces, otros signos y característi-
Déficit de células fago citicas. Los agentes infecciosos más frecuentes
cas sindrómicas del paciente como, por ejemplo, la facies típic a del síndrome
suelen ser bacterias pi6genas, en especial Staphylococcus aureus, así
de Di George, orientan hacia el diagnóstico antes de que aparezca el déficit
como bacterias de crecimiento intracelular obligado, si los monoci-
inmun itario.
tos-macrófagos están afectados. Otras infecciones comunes en estos
pacientes son las de o rigen fúngico (Tabla 7 V Tabla 8).
Además de l síndrome infeccioso de repetición, los pacientes con inmunodeTabla 7
fic iencias (ID) padecen una mayor incidencia de neoplasias, enfermedades
autoinmunitarias y atopia. Estas asociaciones son más frecuentes en las ID
Déficit inmunitario
primarias que en las secund arias.
Anticuerpos
Infeccione5 re5piratllrias: neumonías, Iltitis ysinusitis de repetición
Inmunidad celular
• Farmas graves de enfennedades par virus
• (andida.Opartunistas
FagllCitllsi5
Infe<cione5 por hongos ybacterias saprofita5 cornil estafilllcllCo
(!la gulasa-negativll
Complemento
• Patolllgía par inmunllCllmplejlls (dá5ica)
• Predispllsidón a infeccjllnes pllr bacterias encapsuladas
Como se vio en capítulos anteriores, cuando la agresión por un germen pone
en marcha la respuesta inmun itaria, ambas vari antes, específica e inespecífica, actúan en estrecha conexión; esta respuesta global y coord inada no es
arnlca
igua l para todos los gérmenes.
Tampoco los diferentes componentes del sistema inmun itario tienen la
Orientación diagnóstica de las inmunodeficiencias
misma importancia en la defensa cont ra un agente infeccioso determinado.
28
ERRNVPHGLFRVRUJ
08. Inmunodeficiencias
Manual CTO de Medicina y Cirugía, 10. 3 edición
Tabla 8
Microorganismo
Anticuerpos
Complemento
•
•
•
Estafilococos
Neumococos
Haemophi/us
Ntisserio
Inmunidad
celular
Inmunodeficiencias primarias
Fagocitosis
•
La mayor parte son de origen genético, por lo que su diagnóstico es más
frecuente en la infancia. Son entidades descritas cl ín icamente mucho antes
de su caracterización genética, por lo que la actualización y revisión de su
clasificación es constante.
•
•
•
•
•
Virus ADN
Brotes repetidos virus
El objetivo de este texto es facilitar una breve descripción de las inmunodeficiencias primarias (IDP) más representativas (Tabla 9), sacrificando
para una mayor simplificación la clasificac ión Nacadémica N de las mismas
•
Enterovirus
•
Hongos
Gioroia /amblio
(Figura 14).
•
•
Infecciones en sujetos inmunodeprimidos
Anticuerpos _ ,
(50%)
r
- Co mplemento
(2%)
Inmunodeficiencias secundarias
Son las causadas por agentes que alteran, de forma indi recta, un sistema
inm un itario previamente norma l, desencadenando un cuadro de inmunodeficiencia. Son mucho más frecuentes que las primarias y, por lo general,
menos selectivas en cuanto al componente del sistema inmunitario afectado.
Si se soluciona el problema que originó la inmunodeficiencia secundaria,
generalmente es posible recuperar la función del sistema inmunitario. Las
más frecuentes son las debidas a malnutrición, fármacos, diabetes mell itus,
uremia e infección VIH.
Fagocitosis _-1
(25%)
L_ Resto (23%)
(combinadas, otras )D ...)
Las iatrógenas (fármacos, incluyendo la corticoterapia prolongada) son las
más frecuentes en los países desarrollados y la malnutrición, en los países en
vías de desarrollo (M IR 08-09, 236).
Frecuencia de diferentes inmunodeficiencias primarias
Tabla 9
Enfennedad
Herencia
TIpo de déficit
linfocitos
Inmunoglobulina
Síndrome de Bruton
X
Anticuerpos
Ausenda de B
Muy bajas
Síndrome hiper-lgM
X
Anticuerpos
• Déficit de CD40l en linfocitos T
• IgM elevadas
• Déficit de CD40 en linfodtos B
• IgG, IgA e IgE
AR
Inmunodeficienda variable común
,.,.
Anticuerpos
Inmunodeficienda combinada grave
X
Combinada
Ausenda de T
Bajas
Inmunodeficienda combinada grave
AR
Combinada
Ausenda de Ty B
Bajas
Déficit de ADA
AR
Combinada
Descenso progresivo de Ty B
Bajas
Déficit de HLA de dase 11
AR
Combinada
Normal (LT([)4+ bajos)
Normales o bajas
Síndrome de Wiskotl- Aldrich
X
Complejo
Descenso progresivo de Ty B
• IgM baja
Bajas
• IgA e IgE elevadas
Síndrome de ataxia-telangiectasia
AR
Complejo
• IgM elevadas
• IgA e IgE bajas
Síndrome de Di George
Cr22q
Complejo (celular)
Enfermei:lad granulomatosa crónica
AR (30%)
Fagocitosis
Enfermei:lad granulomatosa crónica
X(70%)
Fagocitosis
Síndrome hiper-lgE
AOoAR
Fagocitosis
Descenso progresivo de Ty B
Normales o bajas
IgE elevadas
Principales inmunodeficiencias primarias
29
ERRNVPHGLFRVRUJ
INMUNOLOGÍA
Patología asociada
Clínica
Se trata de un s índrome infeccioso de repetición que lleva asociado, a con-
Las infecciones típicas de l déficit de anticuerpos sue len comenzar entre
secuencia del mismo, una serie de síndromes como mal nutrición, retraso
en el crecimiento, anem ia ferropénica, organopatías pu lmonares y cua -
los 6 meses y el año de v ida, cuando han desaparecido los anticuerpos
maternos. Estos pacientes pueden padecer un cuadro de artritis reuma -
dros diarreicos crónicos. Las IDP se asocian con mayor frecuenc ia de la
toide Iike causada frecuentemente por Mycoplasma spp. Otro cuadro que
esperada a:
•
Autoinmunidad. las enfermedades autoinmunitarias son más frecuentes en las ID que conservan, al menos de manera parcial, la formación
incide en estos pacientes es un síndrome similar a la dermatomiositis,
que evoluciona hacia una men ingoencefa litis generalmente fata l, y que
de anticuerpos, en tanto que su incidencia es menor en las formas com -
diarreas por G. lamblia, pero só lo e l 10% de los pacientes desarrollan
binadas graves. Las enfermedades autoinmunitarias más frecuentes son
aquéllas en las que la autoagresión se d irige hacia células sanguíneas:
anemias hemolíticas, trombocitopenias y neut ropen ias.
diarreas crónicas que, dada su edad, pueden hacer pensar en una enfer-
Las colagenos is son especialmente frecuentes en las deficiencias del
Diagnóstico
es producido por una infección por virus ECHO. Son muy frecuentes las
medad celíaca.
complemento. En los pacientes con inmunodeficiencias, además de
•
las enfermedades autoinmun itarias, también es típica la presencia de
Se requ ieren los siguientes datos: sexo mascu lino, comienzo en la edad infan-
autoanticuerpos sin significado cl ínico concreto. Los más frecuentes son
til, va lores de IgG sérica inferiores a 200 mgjdl, IgA e IgM séricas práctica-
los dirigidos contra la IgA: cardiolip ina, antígeno microsomal tiroideo,
mente indetectables. Es característica la ausencia de linfocitos B circulantes
factor reumatoide y ANA.
«
Neoplasias. La incidencia de tumores es de 10 a 100 veces mayor de
inmun idad mediada por cé lulas T es normal.
2% de LB) y de células plasmáticas en los tej idos (como en intestino). La
la esperada en grupos de edad similar. Son más frecuentes en las ID
asociadas a alteraciones de las células T, por ejemplo, en el síndrome de
El tratamiento de elección, como para la mayoría de las deficiencias de anti-
ataxia -teleangiectasia.
cuerpos, es la administración periódica de gammaglobu lina por vía parenteral.
Las neoplasias más frecuentes son las de origen linforreticular (I infoma
Síndrome hiper-lgM
no Hodgkin), seguidas por el carc inoma gástrico (en los déficits de anticuerpos).
•
Etiología
Atopia. Las enfermedades de origen atópico son especialmente frecuentes en las deficiencias de IgA. También es frecuente la aparición de
dermatitis similar a la de origen atópico en las ID combinadas, así como
Se debe a mut aciones en los genes de las moléculas que median el cambio
en las deficiencias de la función fagocítica.
de clase de la inmunoglobul ina, siendo los principales CD40 ligando (CD40L)
y C040. Las alterac iones en el gen CD40L (también llamado (0154) present an un patrón de herencia ligada al cromosoma X (regla mnemotécnica
Nligando-ligada N ), mientras que (040 tiene un patrón de herencia autosómica reces iva.
Inmunodeficiencias primarias
Clínica
humorales
Los pacientes presentan un cuadro clínico de déficit de inmunidad humora l
(neumonías, sinusitis, otitis ... ). También son muy sensibles a las infecciones
Son las IDP más frecuentes. Engloban a las entidades cuyo cuadro cl ínico
por Cryptosporidium. Los niveles de IgM están elevados, mientras que los de
evidencia un fal lo en la formación de anticuerpos específicos. La inmunidad
IgG, IgA e IgE están muy bajos y a diferencia de los pacientes con síndrome
mediada por las células T suele ser normal. La sintomatología más habitua l
consiste en infecciones de repetición causadas por bacterias extracelula-
de Bruton, los li nfocitos B se encuentran en número normal.
Inmunodeficiencia variable común
res en tractos mucosos naturales. La localización más frecuente de estas
infecciones es el tracto respirator io (neumonías y bronquitis de repetición),
seguido del digestivo (diarreas intermit entes).
Es la segunda IDP más frecuente (M IR 08-09, 246) Y la más frecuente con
sintomatología infecciosa. Se cons idera que, en realidad, es un síndrome
Otras infecciones frecuentes son sepsis, otitis, sinusitis, meningitis y piodermitis. Como enfermedades asociadas destacan las autoinmunitarias (citope-
que agrupa var ias enfermedades que aparecen después de la infancia,
cuya expresión primaria es una producción de anticuerpos solubles alte-
nias, gastritis atróficas), los eczemas y los tumores, en particular carcinomas
rada, en base a un defecto en la diferenciación de las célu las plasmáticas.
gástricos. Las complicaciones más frecuentes son las bronquiectasias, otitis y
Aunque puede haber niños afectos (> 4 años), suele debutar con más fre-
sinusitis crón icas, así como síndromes malabsortivos.
cuencia a partir de la adolescencia, siendo la edad de comienzo más hab itua l entre los 20 y los 30 años. Afecta tanto a varones como a mujeres. Los
Agammaglobulinemia ligada al sexo
(slndrome de Bruton)
hal lazgos inmunológicos son muy variables. Las cifras de IgG son bajas,
pueden oscilar entre su ausencia y 500 mgjd l; la IgM e IgA pueden tener
valores muy variables. Los linfocitos B suelen ser normales en número,
Etiología
aunque un pequeño porcentaje de pacientes puede tenerlos disminu idos
(MIR 07-08, 245).
Se debe a deleciones en el gen de la tiros ina-cinasa de Bruton (BTK), loca lizado en el cromosoma X. Esta deficiencia condiciona un NstopN en la diferen-
La etiología es, por lo general, desconocida (se han descrito mutaciones en
ciación de los linfocit os B a nivel intramedular.
genes relacionados con la d iferenciación de linfocitos B en el 20-40% de los
30
ERRNVPHGLFRVRUJ
08. Inmunodeficiencias
Manual CTO de Medicina y Cirugía, 10. 3 edición
Pronóstico
pacientes); existe un defecto intrínseco del linfocito B, que es incapaz de
madurar a célu la plasmática o, si lo hace, las células plasmáticas no segregan
las IG que producen. Un dato característico es la ausencia de células plasmá-
La enfermedad suele pasar desapercibida (el 85% de los pacientes está asin-
ticas en tejidos.
tomático). Si los pacientes son sintomáticos, el pronóstico depende de las
enfermedades asociadas (infecciones recurrentes bacterianas de vías altas
El cuadro clínico corresponde al reseñado para las deficiencias de anti-
resp iratorias, alergia, autoinmunidad).
cuerpos (bronquitis y sinusitis crón icas, neumonías de repetición ... ). Las
Diagnóstico diferencial
infecciones más frecuentes son las de senos paranasales y pulmonares,
seguidas por las intestina les. La incidencia de enfermedades autoinmunitarias es alta. Son muy frecuentes la anemia perniciosa y la aclorhi-
Debe tenerse en cuenta que algunos anticonvulsivos como las hidantoí-
dria gástrica que, probab lemente, tenga que ver con la alta incidencia
nas suprimen la producción de IgA. Los criterios d iagnósticos establecidos
de carcinomas gástricos en estos pacient es. Son tamb ién frecuentes los
(Sociedad Europea de Inmunodeficiencias) re quiere n que el paciente tenga
linfomas. En un subgrupo de pacientes pueden aparecer fenómenos
más de 4 años de edad.
inflamatorios granulomat osos (sarcoidosis, poliangeitis granulomat osa,
Tratamiento
... ) (MIR 14-15, 220).
RECUERDA
Las infecciones de re petición se tratarán con antibióticos; no debe emplearse
gammaglobulina. Las vacunas contra Haemophilus spp y neumococos pueden
Es una inmunodeficiencia de anticuerpos primaria y ad quirida.
ser de utilidad en pacientes con síndrome infeccioso de repetición. Se recom ienda portar identificación con el diagnóstico de deficiencia selectiva de IgA.
Deficiencia selectiva de subclases de IgG
El pronóstico (principa lmente infeccioso), bien tratada, es re lativamente
bueno, pud iendo llegar a la octava década, pero siendo muy variable en
Definición
función de las complicac iones a d iferentes niveles (bronquiectasias, hipertensión portaL) ya presentes al momento del diagnóstico. Una de las cau sas principales de morta lidad en estos pacientes es el linfoma. Los hijos de
Se define como la existencia de valores inferiores a los norma les de una o
enfermas (no tratadas) nacen con niveles bajos de inmunoglobulinas, que
más subclases de IgG, con norma lidad de la cantidad total de IgG sérica. Es
progresivamente van aument ando hasta llegar a igua larse con los de niños
frecuente la inmunodeficiencia conjunta de subclases IgG2 e IgG4 y en un
nacidos de madres san as.
importante número de casos asociado a déficit parcial o total de IgA. Suelen
pre sentar clínica infecciosa recurrente ORL y de vías altas respiratorias. Los
Algunos pacientes que han recibido tratamiento con rituximab (anti -CD20)
criter ios d iagnósticos requieren que la edad del paciente sea mayor de 7
presentan de forma prolongada hipogammaglobulinemia en rangos simi la-
años y dos subclases de IgG (GI, G2 o G3) disminuidas.
res a la inmunodeficiencia variable común, asociando además infecciones
típ icas de este tipo de inmunodeficiencia (MIR 16-17, 100-HM).
Tratamiento
Deficiencia selectiva de IgA
El tratamiento de la deficiencia debe efectuarse con gammaglobu li na, si
existe una clínica infecciosa suficientemente grave y deficiencia en la capaci-
La producción, niveles sanguíneos y funcionalidad del resto de inmunoglobu-
dad de producción de anticuerpos frente a antígenos proteicos y polisacári-
linas, así como la inmun idad celular, son normales.
dos.
Hipogammaglobulinemia transitoria
de la Infancia
Según las publicaciones al uso, la frecuencia es de 1:800 en ind ividuos caucásicos e inferior en japoneses y afroamericanos, y es la IDP más frecuente.
Dichos estud ios se suelen referir a EE.UU.
Durante los últimos meses de la gestación, la IgG materna pasa a través de
Clínica
la placenta med iante un mecanismo activo y persiste en la sangre del recién
nacido hasta los 6-8 meses de vida. La primera Ig que produce el niño es la IgM,
La gran mayoría de los pacientes est án asintomáticos, pero algunos presen-
seguida por IgG e IgA. La IgG adquirida pasivamente se cataboliza, mientras
tan infecciones respiratorias o digestivas de repetic ión, de origen general-
que el lactante inicia su propia producción de IgG.
mente bacteriano.
Como consecuencia, entre los 3 y 6 meses de vida, existe una hipogammagEn estos pacientes hay mayor incidencia de trastornos autoinmun itarios
lobu linemia fisiológica, con va lores de IgG bajos. En algunos niños, este défi-
(especialmente diabetes tipo 1), alergia y enfermedad celíaca que en la
cit fisiológico se prolonga hasta los 3 años, en cuyo caso se d ice que padecen
población genera l (uno de cada 200 alérgicos es deficiente en IgA).
una hipogammaglobu linemia transitor ia de la infancia (Figura 15). El origen
de este trastorno no est á aclarado, aunque parece existir una mayor frecuencia en niños con familiares afectos de inmunodeficiencias primari as de anti-
Se han descrito reacc iones graves tras la administración de hemoderivados.
cuerpos.
RECUERDA
Si la inmunodeficiencia cursa con clínica, hay que sospechar
e investigar otro déficit de anticuerpos asociado, como el
déficit de subclases de IgG2 e IgG4. Si se confirma, puede
recibir tratam iento con gammaglobu linas.
La hipogammaglobul inemia transitoria produce sintomatología muy raras
veces, y genera lmente los pacientes conservan una adecuada respuesta f rente
a vacunas. No está indicado administrar gammaglobulina, excepto en los casos
extraordinarios en que se producen infecciones muy graves y repetidas.
31
ERRNVPHGLFRVRUJ
INMUNOLOGÍA
Inmunodeficiencia combinada grave
Es un síndrome que agrupa varias enfermedades congénitas con ausencia
mg/dI
virtual t anto de inmunidad med iada por cé lulas T, como la subsigu iente alte1.200
ración en una correcta respuesta humora l.
IIlOO
Patogenia
Ig totale~
IgGmatema
800
La patogen ia de este síndrome es múltiple (heterogeneidad genética), aun-
.\.
que recientemente se van identificando muchos de los genes responsables
y se han defin ido, por tanto, entidades clínicas concretas. En la mayoría de
los casos, los linfocitos T est án ausentes o muy disminuidos. El timo de estos
19G del niño
pacientes suele ser de pequeño tamaño y no se visualiza en la rad iografía de
400
tórax (MIR 16-17, 52).
~
200
01234567
I,M
Diagnóstico
---1
Se establece ante una historia clínica sugerente. Entre los datos de laborato-
8 O 1 2 3 4 5 6 7 8 9 101 1 12
Nacimiellto
rio, destacan los sigu ientes:
M.~
•
Hipogammaglobulinemia intensa. Afecta por lo general a todas las IG
(aunque, en algún caso, la IgM puede estar conservada).
Niveles de inmunoglobulinas durante el periodo fetal y la lactancia
•
Linfopenia. Más intensa a medida que se deteriora el estado genera l
del paciente.
•
Ausencia de linfocitos funcionantes. Pueden carecer de linfocitos T
o haberlos en bajo número. Cuando existen linfocitos T, éstos presentan incapacidad, prácticamente tota l, de responder con prol iferac ión a
Inmunodeficiencias primarias
mitógenos como fítohemag lutinina o concanava lina.
combinadas
Tratamiento
•
Trasplante de médula ósea (MIR 07.<JS, 242):
Representan cerca del 25% de las IOP. Todas son hered itarias y están causa-
Inmunodeficiencia comb inada grave ligada al sexo. Es la más fre -
das por la ausencia o por una alteración funcional grave de los linfocitos T y
cuente. Se debe a mutaciones en e l gen de la cadena y común del
B. Cuando se conoce el gen responsable, es posible su diagnóstico prenata l
receptor de la IL-2 (situado en e l cromosoma X). Los linfocitos T
est udiando el genotipo tras obtener cé lulas por amniocentesis, y en algu -
de sangre periférica están ausentes o en número muy bajo; los
nos casos se ha real izado, incluso, el tratamiento con trasplante de médu la
linfocitos B suelen estar norma les o elevados.
intraútero.
Inmunodeficiencia combinada grave autosómica recesiva. Se trata
de un grupo de enfermedades con heterogeneidad genética. Los
La ausencia o la alteración grave de la inmun idad mediada por las células
linfocitos T están muy bajos o ausentes; los linfocitos B pueden
T, así como la escasa o nula producción de Ac, ocasiona cuadros clínicos de
extrema gravedad, que, en ausencia de tratam iento correctivo, suelen conducir al fallecimiento durante la infancia.
ser norma les, muy bajos o estar ausentes.
Deficiencia de adenosindesaminasa (ADA)
Aunque estos pacientes sufren todo tipo de infecciones, las más frecuentes
El cuadro clínico de esta enfermedad es el de la inmunodeficiencia combi-
suelen ser las de origen vírico, así como las producidas por hongos y por bac-
nada grave. La ausencia de esta enzima del metabolismo de las purinas ori-
ter ias de crecimiento intracelular (MIR 07-0S, 182).
gina la acumu lación, en los linfocitos T y S, de metabolitos (dATP), por lo
que se bloquea la síntesis de ADN y los linfocitos pierden su capac idad de
proliferar. La entidad se asocia con frecuencia a anomalías en los cartílagos.
Los datos que permiten sospechar una ID comb inada son:
•
Aparic ión temprana de infecciones graves repetidas y de difícil con tro l.
Se trata de una enfermedad autosómica recesiva que se debe a deleción, o
Anorexia intensa con paro o retraso en el desarrol lo ponderoestatural y
mutaciones puntuales, en el gen que codifica la enzima. Los homocigotos
síndrome d iarreico.
sólo presentan ID cuando la actividad de la enzima es inferior al 5% de la
•
Candidiasis oral resistente al tratamiento tópico.
normal. Los heterocigotos no presentan alteraciones de la respuesta inmu-
•
Neumonías intersticiales.
nitaria.
•
Historia familiar de fallecimientos tempranos.
•
Disgenesia reticular
Ante la sospecha de la tríada: candid iasis, diarreas y neumonías debe evitarse la vacunac ión con gérmenes v ivos. En el caso de tener que transfundir-
Es la más infrecuente y grave de las ID combinadas. Est os pacientes presen-
los, y para evitar la enfermedad del injerto contra el huésped, la sangre o sus
tan pancitopen ia: carecen de linfocitos T y S Y de células mielomonocíticas,
derivados deben ser frescos y previamente irradiados con objeto de eliminar
lo que provoca infecciones extraordinariamente graves desde los primeros
la func iona lidad de los linfocitos T.
d ías de vida. Sin trasplante de médu la ósea, fallecen en el primer tr imestre.
32
ERRNVPHGLFRVRUJ
08. Inmunodeficiencias
Manual CTO de Medicina y Cirugía, 10. 3 edición
ill;;'iJ;;:::=.=========
Déficit de adhesión leucocitaria (LAD,)
Defectos primarios de la función fagocítica
Se produce por mutaciones en el gen de la 132 integrina (CDl8). Provoca síntomas desde el nacimiento, con el dato característico de retraso en la caída
del cordón umbilical, onfa litis y alteración en la cicatrización. El pronóstico en
Constituyen un grupo de enfermedades en las que se alteran uno o varios
los casos de deficiencia grave de CD18 es deletéreo a los 2 años de vida sin
de los pasos secuencia les de la fagocitosis. Las anomalías del funcionalismo
tratamiento (trasplante de progenitores hematopoyéticos).
leucocit ario pueden afectar a la quimiotaxis, la fagoc itosis o a la capacidad
=====
ill;;~
:
bactericida; existen también trastornos de carácter mixto:
•
Trastornos de la quimiotaxis. Debidos a la deficiencia de adhesión leu•
coc itaria.
Trastornos de la capacidad bactericida. Destacan la deficiencia de glu-
Defectos primarios por disregulación
cosa 6-fosfato-deshidrogenasa, la deficiencia de mieloperoxidasa y la
inmunológica
enfermedad granulomatosa crónica.
Enfermedad granulomatosa crónica
Síndrome de Chediak-Higashi
Se caracteriza por la falta de capacidad bactericida de los granulocitos y la
falta de producción de radicales libres de oxígeno. La cl ínica infecciosa suele
Las células fagocíticas presentan gránu los gigantes, debido a la fusión anó-
comenzar durante el primer año y las localizaciones más frecuentes son las
ma la de lisosomas (cuerpos de inclusión gigantes en todas las células con
pulmonares, hepáticas, genit ourinarias, en ganglios linfáticos y óseas.
gránu los, dato patognomónico). El cuadro clínico se caracteriza por infecciones piógenas recurrentes con frecuente loca lización cutánea, nistagmo,
Los gérmenes más frecuentes son los que habitua lmente no son patógenos para personas sanas como est afilococos coagulasa (+) y (-), Escher;chla
fotofobia y albin ismo parcial. En ocasiones asocian diátesis hemorrágica y
co/I, Serratia marcescens y hongos. Son típicas las infecciones por bacterias
desde la administración de factores estimu lantes de granulocitos (G -CSF),
catalasa -positivas. Paradójicamente, las bacterias como el neumococo y el
hasta el trasplante de progen itores hematopoyéticos.
neuropatía. Su herencia es autosómica recesiva. El tratam iento comprende
estreptococo J3 -hemolítico rara vez infectan a estos pacientes, ya que producen su propio peróxido de hidrógeno, que acabará siendo leta l para ellos.
Síndrome hemofagocítico
Fisiopatología
El síndrome hemofagocítico es una condición clínica, que se caracteriza por
fiebre, adenopatías/esplenomegal ia, h ipertransa m insasem ia, h iperferriti-
A nivel molecular, existe un fal lo en la activación del complejo NADPH-oxi-
nemia, hipertrigliceridemia e hipofibrinogenemia, y citopenias sanguíneas,
dasa, enzima formada por cuatro cadenas peptídicas: el citocromo b558
además de poder observarse hemofagocitosis en médula ósea, bazo y/o
(heterodímero: p9l y p22) Y otras dos prote ínas. Su func ión norma l es cat a-
adenopatías. Existe una forma adquirida, secundaria a condiciones infeccio-
bolizar el paso de un electrón al oxígeno para formar el an ión superóxido;
sas (VEB, listeria, ... ), tumora les (Iinfomas) e inflamatorias (artritis idiopática
con posterioridad se forma peróxido de hidrógeno.
juvenil, lupus eritematoso sistémico, ... ) y otra fam iliar o primaria (ésta es la
La mieloperoxidasa de la célula utiliza el H,.0, para formar el anión hipocloroso,
inmunodeficiencia primaria), en la que se han caracterizado diversas altera-
que es tremendamente oxidantey microbicida. En el transcurso del metabolismo
ciones genéticas en genes re lacionados con la citotoxicidad T y NK, como en
microbiano, las bacterias producen peróxido de hidrógeno, y aquéllas que son
los genes de las perforinas. Como datos inmunológicos destaca la elevación
catalasa -positivas lo degradan inmediatamente. Sin embargo, las catalasa -ne-
de CD25s (CD25 soluble o cadena alfa del receptor de la IL2) y ausencia o
gativas no lo degradan y aportan a la cé lula enferma el peróxido de hidrógeno
marcado descenso de la citotoxicidad NK en test funcionales.
que necesitaba para poder activar la maquinaria de destrucción microbiana. Por
tanto, no suele haber problemas para eliminar bacterias cata lasa-negativas.
Este síndrome tiene una altísima mortal idad. Su tratamiento incluye el uso de
corticoides sistém icos, qu imioterapia (etopósido) e inmunosupresión (gene-
Genética
ralmente con ciclosporina, aunque se han util izado otros como rituximab,
abatacept ... ). El tratamiento curativo definitivo en el caso de la forma fam iliar
Es una enfermedad con heterogeneidad genética. El 70% de los casos se
o primaria confirmada, debe incluir la búsqueda inmediata de un donante
debe a alteraciones en el gen de la p9 1 del citocromo b588, situado en el
para trasp lante de progenitores hematopoyéticos (MIR 14-15, 219).
cromosoma X y, por tanto, con herencia ligada al sexo. Los otros genes del
complejo NADPH-oxidasa tienen herencia autosómica recesiva.
o
o
Diagnóstico
Síndromes bien definidos que cursan
El diagnóstico se determina ante la negatividad repetida de la prueba de reduc-
con inmunodeficiencia (primaria)
ción del nitro-azul de tetrazolio (NBT). En la actualidad, se han desarrollado
test que emplean la citometría de flujo para estudiar la capacidad oxidativa.
Tratamiento
Síndrome de Di George
Es sintomático. IFN-ycomo profilaxis infecciosa. En algunos casos, se ha logrado
Es una embriopatía causada por microdeleciones en el brazo largo del cro-
la corrección del defecto mediante trasplante alogénico de médula ósea.
mosoma 22, que afecta a los órganos derivados del tercer y cuarto arcos
33
ERRNVPHGLFRVRUJ
INMUNOLOGÍA
faríngeos. Suele ser esporádica. Los pacientes presentan una amplia gama
asociar, además, clínica compatible de enfermedades mediadas por inmu-
de anomalías en su fenotipo: ausencia total o parcia l de la glándula timica,
ausencia total o parcia l de paratiroides, facies con micrognatia, hipertelo-
nocomplejos.
rismo e im plantación baja de pabellones auriculares. La manifestación cl ínica
Además de las deficiencias en factores que intervienen en la activación del
más temprana es la tetan ia, debida a la hipoca lcemia por ausencia de paratiroides. Otras malformaciones frecuentes son las card íacas, en particular en
la sal ida de los grandes vasos. Pueden asociar ade más retraso menta l leve,
complemento, se han identificado múltiples defectos de proteínas reguladoras ("inhibidoras") del mismo, cuyos cuadros clín icos no cursan con infecciones sino con síntomas derivados de un "exceso de func ión", como el
déficit de atención e hiperactividad, alteraciones esqueléticas (polidactilia ... ).
síndrome hemolítico urémico atípico (factor I y factor H).
Es frecuente que el número de linfocitos T esté disminuido y que éstos pre-
Angioedema hereditario (AEH)
senten características de inmadurez. Las cifras de IG séricas suelen ser norma les o discretamente d isminuidas; en general, la producción de anticuerpos
Se trata de un angioedema med iado por bradiquinina, no por histamina (no
no está abol ida, pero sí alterada. El espectro de in fecciones depende del
subyace patología alérgica). De características clínicas típicamente no erite-
grado de afección del sistema inmunita r io de cada paciente, habiendo casos
matosas, no pru rig inosas, sin respuesta a tratamiento con antih istamínicos ni
sin sintomatología infecciosa. El pronóstico es diferente para cada paciente,
corticoides. Cursa típic amente en brotes en ocasiones preced idos de facto-
así como la estrategia terapéutica, incluyendo en los casos de síndrome com-
res desencadentes (traumatismos, intervenciones qu irúrg icas, procedim ien-
pleto (con afectación grave del sistema inmunológico) el trasplante de proge-
tos odontológicos). Si los brotes afectan localizaciones faciales y cervicales
nito res hematopoyéticos (MIR 12-13, 214; MIR 15-16, 44).
(angioedema lingual, de glotis ... ), pueden comprometer seriamente la vía
aérea, siendo potencialmente mortales si no re ciben el diagnóstico y trata-
Síndrome hiper-lgE
miento adecuados. Se han descrito tres tipos principa les de AEH, todos ellos
con herencia autosómica dominante (MIR 15-16, 48; MIR 16-17, 51):
•
Suelen ser casos esporádicos, aunque existe una form a autosómica dominante (síndrome de Job, con mutaciones en el gen STAT3) con penetrancia
Tipo l. Por mutaciones en el gen del Cl inhibidor, que condicionan una
d isminución en la concentración sérica del C1 inhibidor.
incompleta. Se caracteriza por derm atitis crónica pruriginosa e infecciones
•
Tipo 11 . Por mut aciones en el gen del Cl inh ibidor, que cond icionan una
bacterianas sinopulmonares y cutáneas, acompañadas de cifras elevadas de
d isminución en la función del Cl inhibidor, con concentrac iones séricas
IgE y eosinofilia. La manifestación clínica más típica es la aparición de abscesos cutáneos recidivantes por Staphylococcus aureus.
normales.
•
Tipo 111. Hasta hace pocos años se incluían aquellos AEH sin alterac ión
cuantitativa o funcional en Cl inhibidor. Rec ientemente se ha identi-
El síndrome de Job presenta además unos rasgos fenotípicos faciales y cor-
ficado una mutación en el gen del factor XII de la co agu lación (factor
pora les característicos (hiperte lorismo, ensancham iento del puente nasal,
de Hageman) como causante de AEH -III en parte de estos pacientes.
retenc ión de piezas de la dentadura primaria, hiperlaxitud ligamentosa,
El estímulo hormonal estrogénico parece desempeñar un papel como
escoliosis ... ).
desencadenante.
Síndrome de Wiskott-Aldrich
~1l~=========
Su herencia está ligada al cromosoma X (gen WASP). Cursa con eczema,
Síndromes autoinflamatorios
trombocitopenia e infecciones recurrentes. Con datos de diátesis hemorrágica (epistaxis, melenas, hematomas, petequias). Produce infecciones de
loca lización ORL, neumonías, sepsis y candidiasis. El defecto inmunológico
es complejo, con dism inución de IgM, aumento de IgE y disminución de la
Incluidos en la clasificación de las inmunodeficiencias primarias. Se deben a
respuesta proliferativa de lin foc itos T. El tratamiento curativo cons iste en el
mutaciones en genes que intervienen en la respuesta inmune innata. Se dife-
trasplante de progen itores hematopoyéticos (MIR 11-12, 139).
re ncian dos grandes grupos, los que se deben a defectos en el inflamosoma
y las no relacionadas con el inflamososma.
Síndrome de ataxia-telangiectasia
Entre las inflamosomopatías se encuentran la fiebre mediterránea fam iliar,
Se encuadra dentro del grupo de inmunodeficiencias primarias con defectos
el síndrome hiper-l gO V el síndrome de Muckle-Wells. De las no relac ionadas
en la reparación del AON. Su herencia es autosómica recesiva, con muta-
con el inflamosoma, destacan el síndrome periódico asociado al receptor
ciones en el gen ATM. Se caracteriza por ataxia cerebelosa, telangiectasias
cutáneo-mucosas (frecuentemente con local ización ocular), in fecc iones
del TNF (TRAPS), enfermedad inflamatoria intestinal de inicio precoz, el síndrome PAPA (pioderma gangrenoso, acné, artritis piogénica estéril), déficit
(sinopulmonares) V neoplasias. Presenta disminución de la concentrac ión
del ant agon ista del receptor de la IL-1 (síndrome OIRA).
sérica de IgA, linfopen ia T y aumento de alfafetoproteína (AFP).
De forma genera l, cursan con brotes recur re ntes de fiebre sin causa infecciosa o tumoral conocida, ni autoinmune. Se acompaña de elevación de los
reactantes de fase aguda, como la PCR (proteína C reactiva), entre otras, o la
SAA (sustancia amiloidea A) con normal ización generalmente entre brotes.
Deficiencias primarias del complemento
Cada entidad asocia otra sintomatología como eritema migrans, poliserositis, urticar ia, úlceras en mucosas, afectación neurológica, artr itis, ...
De forma general, las inmunodeficiencias de l complemento se caracter i-
El tratamiento de la mayor parte de ellos incluye el uso de colchicina, y el
zan por incremento a la susceptibilidad por infec ciones bacterianas por
bloqueo de la vía de la inter l eucina - l- ~, con canakinumab y/o anakinra. En
bacterias encapsuladas. Los déficits de los factores de la vía clásica pueden
general, los corticoides son muy poco eficaces en ellos.
34
ERRNVPHGLFRVRUJ
08. Inmunodeficiencias
Manual CTO de Medicina y Cirugía, 10. 3 edición
•
Anticuerpos. Las concentraciones de las diferentes clases de Ig se
alteran no sólo en los déficits de func ión humoral, sino también en
Evaluación de la inmunidad
los de función celular: las inmunodeficiencias primarias de función
celular casi siempre se acompañan de alteraciones de las Ig, y en el
SIDA existe hipergammaglobul inemia po liclonal. Ante un caso de sos -
Un diagnóstico correcto de una inmunodeficiencia (ID) comienza con una
pech a de déficit de inmunidad humoral con niveles de Ig norma les, se
historia clín ica/exploración y una serie de analíticas básicas; encabezadas por
deben cuantificar las subclases de IgG y realizar pruebas funcionales
fórmula, recuento y velocidad de sedimentación y cuantificación de inmu-
para evaluar la respuesta de anticuerpos tras vacunac ión con toxo ide
noglobul inas. Deben seguir pruebas más específicas, según la clín ica del
tetánico o virus gripal. Se deben cuantificar las concentraciones de
paciente.
anticuerpos específicos frente a los microorgan ismos más habituales,
así como la respuest a frente a vacunas de antígenos proteicos y poli-
Valoración de la inmunidad celular
sacarídicos. Hay que realizar cuantificación de LB en sangre, mediante
Se establece mediante cuantificación de las poblaciones de linfocitos T CD4,
citometría de flujo.
Complemento. Para determin ar si existe déficit de complemento, se
•
T CD8 y NK Y el cociente CD4/CD8, mediante citometría de flujo. Puede
rea liza la prueba CH50 y la cuantific ación de C3 y C4. El CH50 es un
orientar hacia un d iagnóstico de déficit de inmunidad ce lular, pero la norma-
test que cons iste en utilizar el complemento del su ero del paciente
lidad en el número y proporción de las cé lulas no descarta la existencia de
en un ensayo de hemólisis. Se enfrentan hematíes de carnero y un
una alteración funcional.
anticuerpo d irigido contra ellos (sólo pueden lisarse en presencia de
complemento), y luego se añaden diluciones del suero de l pac iente,
Pruebas funcionales
aportando comp lemento en concentraciones decrecientes. La CH50
es la dilución del su ero en la que se consigue el 50% de hemólis is.
Las pruebas más empleadas en la clínica son:
•
Pruebas cutáneas de hipersensibilidad retardada a antígenos como
En las fases agudas de enfermedades infecciosas o autoinmun itarias,
PPD, cand idina estreptocinasa y estreptodornasa (ADNasa). Al ser reco-
pl em ento. Si la sospecha clín ica persiste a pesar de un CH50 normal,
nocida la sustancia como extraña, los monocitos -m acrófagos secretan
se debe testar mediante estudios funcionales las vías alternativa y
citocin as que atraen a otras células y desencadenan la típica reacción
MBL.
puede haber cifras bajas de CH50 por el consumo de factores de com -
inflamatoria con induración del área afectada.
Valoración de la función fagocítica
Un individuo sano, mayor de 3 años, debe responder al menos a uno de
estos antígenos, puesto que a lo largo de su vida ha debido sufrir alguna
•
infección por estreptococos o Candida y debe tener memoria inmunita-
Las dos pruebas más empleadas son el test de reducc ión de azul de tetrazo-
ria de los citados antígenos. La tubercu li na (PPD) contiene, entre otros,
muramildipéptidos que activan a los macrófagos y célu las de Langerhans.
lio (NBT es una prueba funcional que indica la capacidad de est as célu las de
experimentar la #explosión metabólica") y el test de inhibición de la migra-
Respuesta proliferativa. Consiste en estimular in v itro los li nfocitos del
ción (MIT). Existen en la actua lidad pruebas desarrolladas para citometría
paciente con mitógenos tipo lectinas (como fitohemaglutinina o concana-
de flujo, para estud iar la combustión oxidativa, la capacidad fagocítica y la
valina A) o estimulando directamente el RCT con un anticuerpo anti -CD3,
quim iotaxis.
o con antígenos específicos. La proli fe ración obtenida en los li nfocitos del
paciente y las moléculas secretadas en los sobrenadantes de estos cultivos ce lulares se comparan con la de las célu las de personas sanas.
.1 MIR 16-17, 51, 52, 100· HM
.1 MIR 15· 16, 44, 48
.1 MIR 14--15, 219, 220
.1 MIR 12· 13, 214
.1 MIR 11· 12, 139
.1 MIR 08-09, 236, 246
.1 MIR 07· 08,182,242,245
Valoración de la inmunidad humoral
Se deben estudiar los anticuerpos y el complemento, puesto que existe déficit de comp lemento que tiene sintomatología parecida a algunos déficit de
anticuerpos.
.1 Las inmunodeficiencias (ID) son enfermedades que aparecen como
.1 Además de las infecciones, en las ID aparecen autoinmunidad (a ve-
consecuenc ia de una descoordinación de la respuesta inmunitaria.
ces es e l cuadro predominante), anafilaxia y tumores (sobre todo, el
linfoma no Hodgkin) .
.1 Se las divide en dos grandes grupos: primarias (fallo en el propio sistema inmun itario) y secundarias a otras patologías. Aunque las secundarias son las más f recuentes, las ID primarias son las más preguntadas en el MIR .
.1 Existen diversos grupos de ID primarias, destacando de forma prác-
.1 Las inmunodeficiencias primarias suelen ser de origen congén ito y las
.1 Las ID de anticuerpos se caracterizan por infecciones de repetición
tica las predominantemente de anticuerpos, las comb inadas, las del
comp lemento y las de la fagocitos is.
por bacterias de crecimiento extracelular que afectan al aparato respiratorio (neumonías, otitis, etc.) y digestivo (diarreas) .
secundarias sue len ser adqu iridas.
.1 La clínica que más destaca en las inmunodeficiencias es el aumento
en el número de infecciones.
3S
ERRNVPHGLFRVRUJ
INMUNOLOGÍA
./ En las ID combinadas destaca e l fal lo en la inmunid ad celular (infecciones por vi rus, hongos, parásitos u nicelula res y mico bacterias) que
conlleva una deficienci a de la inm unidad humoral (d e ahí el nombre
d e combina das).
,/ De forma genera l, el trat amient o de las deficiencias de anticuerpos
consiste en la admin istración susti tu tiva de inmunoglobul inas (con traind icación en déficit ai slad o de IgA).
,/ Generalmente, el t rat amient o de las deficiencias combina d as conl leva el traspl ante de progenit o res hematopoyéticos.
./ La ID p rima ria más f recuente es el déficit ai slado de IgA, asint omátic a
e n la mayoría de los pacientes. No requiere tratami en to espe cífi co,
pero se recomi en da segu imie nto. Se debe ten er precaución co n la
adm inistración de hemoderivados. La ID combinada más f recuent e es
,/ El sín d rome d e Bru ton es una deficiencia de anticuerpos (agammaglob uli nemia ligada al X) que se caracteriza por la ausencia de linfocitos B en la sangre periférica .
la ID combinad a grave ligada a l X.
Casosclínicos
Niño de 11 meses que, a los 2 meses de vida, empieza a tener mugue! de
Javier es un joven de 30 años que acude al médico porqu e desde hace
1 año viene presentando otitis y sinusitis con cada vez más frecu e ncia.
El paciente com e nta que nunca hasta ahora había sido propenso a este
tipo de infecciones. Al leer la histori a clínica del paciente, figura que
hace 4 m eses tuvo un episodio de encefalitis vírica que requirió ingre·
so. Ademá s, cuenta que hace 1 año tuvo una mononucleosi s infecc iosa
diagnosticada por serología, que le tuvo en cama 2 semanas y de la cual
no cree habe rse recupe rado : el paciente refiere que, desde entonces,
cada dos por t r es está enfermo. A la vista de los datos, ¿qué prueba
solicitaría para confirmar su diagnóstico de sospecha?
repetición, diarrea e incapacidad para ganar peso. A los 10 meses tuvo una
neumonía por Pneumocystis corinU. En la analítica, se objetiva hipogam maglobullnem ia, IInfopen ia grave con ausencia de linfocitos T y de célu las
NK, y linfocitos B elevados. ¿Cuál sería el diagnóstico?
1) Síndrome de hiper-lgM ligado al cromosoma X.
2) Infección por VIH.
3) Inmunodefkiencia comb inada grave ligada al cromosoma X.
4) Síndrome de Wiscott-Aldrich.
RC: 3
1) Cuantificación del comp lemento.
2) Cuantificación de inmunoglobul inas.
3) Determinación por citometría de flujo de CD40l en los linfocitos del paciente.
4) Punción de médu la ósea ante la sospecha de proceso hematológico ma·
ligno.
Un niño de 20 meses, con antecedentes de un hermano V un primo materno muertos por neumonía en la infancia, ha presentado, desde los 10
meses de vida, 2 neumonías y 5 episodios de otitis media. Se encuentra
marcada hipogammaglobulinemia con recuento y fórmula leucocitaria
normales. ¿Cuál de los siguientes estudios solicitaría, en primer lugar, en
el proceso diagnóstico del paciente?
RC: 2
1) Gammagrafía con captación de Ga.
2) Biopsia del tejido linfoide am igdal ino/adenoideo.
3) Cuantificación de linfocitos circulantes 1, B Y NK.
4) Biopsia de timo.
RC:3
36
ERRNVPHGLFRVRUJ
Inmunoterapia
De fOOllil aislada SI' han ~untado cuestiones re!acÍOOi!c!as con inmunotl'fapia.
Es Jll'a'SilOO t!'fll'f Urlil ideaglobal, lo que pueOe ser de ayuda también paraotras
asignaturas. La inm u notf'lil~ es unGlm~ muy am~i<l y cada vez dema)l)f
ltIeva ncii! dínKa.
Inhibidores de m-Tor
En la actua lidad, son múltiples las patologías con base inmunológica,
cuyos pacientes son susceptib les de recibir tratamientos con fármacos que
poseen la capacidad de modular y/o disminu ir diferentes componentes
Están impl icados en el desarrollo correcto del ciclo celular y, por lo tanto,
de la respuesta inmuno lógica. El manejo clínico correcto de estos fárma-
cuentan con efecto antiproliferativo. Everolimus y sirolimus son los dos fár-
cos requiere un conocim iento tanto de su mecanismo de acción, como de
macos principales de este grupo. Se uti li zan principalmente en diferentes
las vías inmuno lógicas a las que afectan, además de sus posibles efect os
pautas de prevención de rechazo de trasplante, aprovechando su menor
adversos V riesgos derivados de su uso, especialmente la susceptibilidad a
nefrotoxicidad, respecto a los inhibidores de la calcineurina.
ciertas infecciones.
Inhibidores principalmente antiproliferativos
Por tanto, se intentará plasmar de forma resumida V clara los principales
grupos de fármacos que se utilizan en inmunología. En muchos casos sus
Inh iben el metabolismo de los nucleótidos, mediante la interrupción de
ind icaciones aprobadas, especialmente en el caso de los biológicos, se van
la síntesis de pirimidinas o purinas. Por su mecanismo de acción, algunos
ampliando a lo largo de los años, V suelen situarse en esca lones terapéuti-
poseen mayor especificidad sobre la proliferación de los linfocitos T y B,
cos finales, tras el fracaso de otros fármacos V/o en comb inación con ellos
como el micofeno lato y la leflunam ida, mientras que otros son de acción
(con diferencias y matices en re lación con cada molécu la V con cada indicación, que no competen a este manua l). Su uso fuera de indicación, o como
más inespecífica sobre diversas estirpes celulares, como el metotrexato
y la azatioprina. Su uso es diverso en mú ltip les enfermedades de etio lo-
tratamiento compasivo no es infrecuente, atendiendo al posible beneficio
gía auto inmune, aunque principalmente el metrotexato es de amplio uso
en pacientes en los que las med icaciones habitualmente utilizadas no son
en enfermedades con afectación articu lar, V la azatioprina se emplea en
eficaces.
enfermedades autoinmunes e inflamatorias del aparato digestivo. El micofenolato parece ser úti l en enfermedades autoinmunes que cursan con
autoanticuerpos patogénicos.
Agentes alquilantes
Inmunosupresores clásicos
Su efecto es altamente inespecífico; sin embargo, a pesar de su potencial
toxic idad, son de uso frecuente en situaciones que pueden cursar con alta
Corticoides
morbimortalidad. Requ iere especial mon itorización la leucopenia que producen de forma habitu al. Los fármacos de este grupo más usados en enfer-
De acción inespecífica sobre varios componentes del sistema inmunológico.
medades de etiología inmunológica son la ciclofosfamida V el clorambucilo.
Tienen una potente acción antiinflamatoria al actuar sobre la producción de
Algunas situaciones de posible riesgo vital donde se emplean son vasculitis
factores proinflamatorios. En dosis altas, añaden un efecto inmunosupresor.
sistémic as (principalmente con afectación renal) V nefropatía lúpica grave.
Suelen utilizarse para el control rápido en fases agudas de inflamación. Su
RECUERDA
uso prolongado conl leva múltiples efectos adversos (a lteraciones metabólicas, entre otras), por lo que tienden a ser re emplazados por fármacos inmu-
No todos los inmunosupresores clásicos tienen el mismo
mecanismo de acción.
nomoduladores, que ayuden a disminuir su dosis y retirarlos, una vez que
han pasado las fases in iciales de la inflamación.
Inhibidores de la calcineurina
La calcineurina es una molécula que interviene, principalmente, en diver-
Anticuerpos monoclonales
sas seña les de activación intracitoplasmática del linfocito T. Los fármacos de
este grupo desarrol lan su acción inmunosupresora de forma muy específica
y proteínas de fusión
sobre los linfocitos T. Son principalmente la ciclosporina y el tacrolimus. Se
utilizan en mú ltiples enfermedades de base inmunológica, así como en la
prevención del rechazo de algunos órganos trasplantad os V médula ósea. Su
toxic idad es principalmente nefrológica (mayor en el caso de la ciclosporina)
Comprendidos dentro de los denominados fármacos biológicos, en la
y neurológica (MIR 12· 13, 213).
actualidad existen múltiples fármacos que se dirigen contra diferentes
37
ERRNVPHGLFRVRUJ
INMUNOLOGÍA
moléculas implicadas en la respuesta inmunológica, tanto citocinas,
como otras, como son algunas moléculas de adhesión leucocitaria V pro-
Otras moléculas
teínas presentes en las membranas linfocita ri as. Su uso no se lim it a a
•
las enfermedades inmunomediadas, sino que se uti lizan en otras pato-
Tocilizumab. Anti -I L-6R (antirreceptor de IL-6), con ind icación en artritis
reumatoide, artritis id iopática juvenil sistémica y poliarticular.
logías principalmente t umorales. Conceptualmente, su respuesta es más
espe cífica que la de los inmunosupreso res tradicionales, y potencialmente con menores reacciones adve rsas inespecíficas, al dirigirse contra moléculas concretas, inhibiendo acciones específi cas de la respuesta
•
Ustekinumab. Dirigida contra la subunidad p40, común a la IL-12 e
IL-23. Indicación en psori asis en placa y artritis psori ásica.
•
Belimumab. Oirigido contra la molécu la BAFF o BLys, implicada en la
supervivencia de los linfocitos B. Ind icación en lupus eritematoso sistémico.
inmunológica. Dest acan por la importancia en número y diversidad de
•
patologías aprobadas y pacient es a tratamiento, las molécu las dirigidas
Natalizumab. Anti -u4 integrina (molécula re lacionada con la migración
y adhesión leucocitaria), indicado en escleros is múltiple remitente-re-
contra el TNF.
cur rente.
RECUERDA
•
Eculizumab. Anti -CS (factor de l sistema del complemento), impide su
escisión en CSa y CSb y la formación del complejo de at aque a la mem-
Dentro de los fármacos biológicos se incluyen diferentes ti pos de moléculas.
bran a (CAM). Indicado en dos patologías relacionadas con deficiencias
de factores regu ladores del comp lemento, la hemoglobinuria paroxística nocturna y el síndrome hemolítico urémico atípico.
Anti-TNF
•
Omalizumab. Anti-lgE, indicado en asma med iado por IgE con sensibili dad al érgica demostrada. Fuera de indicación, parece ser útil en algunos
Infliximab, adalimumab y golimumab son anticuerpos monoclonales anti-
pacientes con otros trastornos dentro del espectro de la atopia, urticaria
TNF. Todas ellas son molécu las comp letas de anticuerpo, la primera de ellas
crónica id iopática y trastornos eosinofflicos.
con un componente quimérico murino y humano (y más antigua), mientras
•
Mepolizumab. Anti-ILS, indicado en asma atópico.
que las dos últimas son comp let amente human izadas.
•
Secukinumab e ixekizumab. Anti -IL17, con indicaciones en el ámbito de
la psoriasis y espondi loartritis. Se han descrito casos de re activación de
Otras molécu las que bloquean TNF son certolizumab-pegol y etanercept. En
enfermedad de Crohn en algunos pacientes tratados con secukinumab.
•
ambos casos, no son moléculas completas de anticuerpo. El etanercept, con-
Abatacept. Proteína de f usión formada por un fragmento Fe de IgGl
cretamente, es una proteína de fusión, formada por un fragmento Fc de una
humana y una molécu la CTLA4 humana, que inh ibiría la coestimulación
IgGl humana y una molécula de receptor soluble de TNF humano.
de los linfocitos T por med io de su receptor C028, compitiendo con las
molécu las C080;C086 (B7). Ind icacion es en artritis reumatoid e y artritis
Sus ind icaciones aprobadas se encuentran en el ámbito de las espondilo ar-
idiopática j uvenil poliarticul ar.
•
tropatías: artritis reumatoide, enferm edad inflamatoria intestinal, psoria-
Basiliximab. Anti -CD2S (cadena a del receptor de la IL-2), indicado en el
rechazo agudo de trasplante renal alogénico de novo.
sis..., pudiendo variar entre unas moléculas y otras, principalmente por su
d iferente antigüedad dentro del arsenal terapéutico, y los resultados obte-
•
Ranibizumab y bevacizumab. Anti -VEGF (vascular endotelial growth
nidos en ensayos en diferentes patologías. Adalimumab tien e indicación en
ficha técnica para su uso en uveítis no infecciosas y en hidradenitis (mode-
factor). Ranibizumab es una molécula incompleta de anticuerpo indi-
rada -grave). Es obligado, previo a la utilización de este grupo de fárm acos,
en su forma exudativa, edema macular secundario a ocl usión venosa
descartar la existencia de tuberculosis latente en el paciente, debido al alto
retitiniana). Bevacizumab es un anticuerpo completo con ind icación,
riesgo de reactivación de la infección.
entre otras, en cáncer de mama, colon y recto, metastásicos.
cado en DMAE neovascular (degeneración macu lar asociada a la edad
•
Anti-CD20
Muromonab (OKT3). Anti -C03 murino, produce depleción de linfocitos
T. Ha sido usado en el rech azo agudo de trasp lantes alogén icos, princi palmente rena l y ca rdíaco.
•
El ritu ximab es una anticuerpo completo con componente quimérico murino
Vedolizumab. Se d irige f re nte a a4[37 . Sus indicaciones actuales en
y humano, dirigido contra la molécula CD20, presente en cé lulas linfoides de
ficha técnica es enfermedad inflamatoria intestinal (EII), tanto col itis
estirpe B. Su uso inicial fue en neoplasias linfoides B, pero actualmente sus
ulcerosa (CU), como enfermedad de Crohn (EC) en pacientes adultos
ind icacion es aprobadas comp re nden enfermedades autoinmunes que son
que hayan ten ido una respuesta inadecuada, pérdida de respuesta o
linfoma no Hodgkin, leucemia linfática crónica, artritis reumato ide, granu-
sean intolerantes al tratam iento convenc iona l o con un antagonista del
lomatosis con poliangeítis y poliangeítis microscópica. Se destaca el riesgo
factor de necros is tumoral alfa (TNFa ).
de reactivación de VHB (virus hepatitis B) y de leucoencefa lopatía multifoca l
Es un antagonista de la integrina selectivo a nivel intestina l sin actividad
progre siva (virus JC) (MIR 14-15, 217; MIR 16-17, 100).
inmunosupresora sistém ica identificada.
Anti-IL-1a y p y SUS receptoreS
Anak inra y canakinumab pertenecen a este grupo, algunos de ellos se
Gammaglobulinas
di rigen contra receptores de IL- l y otros di rectament e contra IL-1 13- Su
uso gira, p rincipalmente, en torno a un grupo de patologías denominadas inflamosomopatías pertenecientes a los síndromes autoinflamato rios, incluidos en la clasificación de las inmunodeficiencias primari as
Son inmunoglobulinas obtenidas de la fracc ión no celul ar de la sangre de
(crioporinopatías). Anak in ra y canakinumab tienen d iversas indicaciones
donantes. En el las están representadas, por tanto, las especific idades más
en e l campo de la artritis reumatoide, síndromes periódicos asociados a
prevalentes en la población. Sufren procesos en la industria farmacéutica
criop irinopatías (CAPS), artritis idiopática Juvenil de comienzo sistémico
pa ra estabi lizar las inmunoglobu li nas, purificar la IgG y dejar la menor con-
y gota artrítica.
centración posible de otras clases de inmunoglobulinas (relacionad as con
38
ERRNVPHGLFRVRUJ
09 . Inmunoterapia
Manual CTO de Medicina y Cirugía, 10. 3 edición
•
un mayor número de reacciones adversas relac ionadas con la infusión) y
Interferones. IFN-(l con propiedades antivirales, usado en el trata-
cump lir con los contro les microbiológicos establecidos. Se utilizan principal-
miento de algunas hepatitis víricas (VHB, VHC). IFN- I} de uso en escle-
mente con dos finalidades: sustitutivas en inmunodeficiencias primarias y
rosis múltiple remit ente-recurrente. IFN - V usado en la enfermedad
secundarias, y como inmunomoduladoras en algunas enfermedades inmu-
granulomatosa crónica (inmunodeficiencia primaria) y la osteopetrosis
nomediadas (MIR 13-14, 53).
ma ligna grave.
En su uso como terapia sustitutiva existe, además de preparados para uso
Además de las citados en este capítulo, existen otras molécu las y tratamien-
intravenoso, algunas para adm inistración subcutánea.
tos que se utilizan en enfermedades inmunomediadas, como las terapias de
desensibilización en pacientes con hipersensibilidad alérgica, la plasmafére-
Entre las indicaciones inmunomoduladoras/antiinflamatorias, se encuentran
sis en enfermedades autoinmunes con anticuerpos patogén icos (vascu litis
la púrpura trombocitopén ica idiopática, el síndrome de Gui llain-Barré y la
ANCA positivos ... ) y leucocitoaféresis (granulocitoaféresis), con indicación
enfermedad de Kawasaki.
en enfermedad inflamatoria intestinal y enfermedad de Beh¡;:et ocular, entre
otras.
RECUERDA
Otras moléculas
Las inmunoglobulinas tienen indicaciones en inmunodefi·
ciencias y en patologías inflamatorias y/o autoinmunitarias.
Apremila st . Es una molécu la pequeña. Su mecanismo de acción se basa en
la inhibición de la fosfodiesterasa 4, implicada en el metabol ismo del AMPc
y la expresión de genes de citoquinas proinflamatorias y anti-i nflamatorias.
Reciente incorporación en el arsenal terapéutico frente a la forma de espond iloartritis de artritis psoriásica y psoriasis en placas.
Otros fármacos
Citocinas
PREGUNTAS .
•
MIR
IL-2. De ind icación en el carc inoma metastásico de células renales. Se
.1 MIR 16-17, 100
.1 MIR 14-15, 217
.1 MIR 13-14, 53
.1 MIR 12-13, 213
emplea fuera de indicación en el mela noma .
.1 La inmunoterapia es un campo muy amp lio que engloba desde los corticoides hasta modernos fármacos biológicos .
.1 Su aplicación es diversa y se extiende mayoritariamente en el campo de las enfermedades inmunomed iadas (autoinmunidad, inflamación, trasplante, atopia ... ).
.1 Los inmunosupresores clásicos poseen diferentes mecanismos que los hacen más efectivos frente a distintas dianas inmunológicas .
.1 Los fármacos biológicos no sólo engloban a los anticuerpos monoclonales (mab), sino también a otras molécu las como ciertas proteínas de fusión
(cept), entre otras .
.1 El uso de inmunoglobu linas puede ser no sólo sustitutivo (en el caso de las inmunodeficiencias), también pueden utilizarse como inmunomoduladores
I
~~,~"~t~o~io~m
~"~o~it~'é,i~'~'é'Ci~~~~~~
_______________________________________________________________'
39
ERRNVPHGLFRVRUJ
Bibliografía
....
Bibliografía
SEI (Sociedad Española de Inmunología). Actualización en Inmunoterapiaen EnfermedadesAutoinmunes. Barcelona. Elsevier, 2017.
Rich R. Clinical Immunology. Principies and Practice, 3rd ed. Phi ladelphia. Mosby Elsevier, 2009.
Abbas AK. Cellular and Molecular Immunology, 8" ed. Philadelphia. Elsevier, 2015
Roitt 1. Inmunología. Fundamentos, 12." ed. Panamericana, 2014.
Sánchez-Ramón S.lnmunodeficiencias. Madrid. Marbán, 2013.
Abbas AK. Inmunología celular y molecular, 7.a ed. Madrid. Elsevier,
2012.
Shoenfeld Y. Diagnostic Criterio in Autoimmune Diseases. Towota.
Humana Press, 2008.
Grupo eTO. Manual de Inmunología. 8.- ed. Madrid. eTC Editorial,
2012.
40
ERRNVPHGLFRVRUJ
ERRNVPHGLFRVRUJ
ISBN: 97B-84-1709S-16-1
9
ISBN: 978-84-17095-17-B
9
ISBN: 978-84-1 7095-OO-Q
9
ERRNVPHGLFRVRUJ