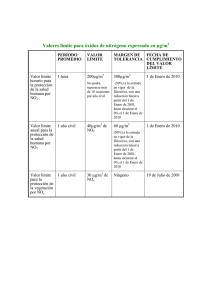
Óxidos de nitrógeno Por su tamaño, estas partículas son capaces de ingresar al sistema respiratorio, provocando potenciales daños a sus órganos principales. Mientras menor sea su diámetro, mayor será el potencial de daño a la salud humana. Las partículas de MP2,5 penetran hasta los alvéolos pulmonares e ingresan directamente al torrente sanguíneo, aumentando los riesgos de mortalidad prematura. El general, el MP puede también dañar a las plantas, inhibir el crecimiento de la vegetación y corroer materiales. Óxidos de nitrógeno (NOx): Estos gases se producen durante el quemado de maderas y combustibles fósiles, como gasolina, carbón y gas natural. El sector transporte constituye la fuente principal de emisión de NOx. El mayor desplazamiento en vehículos particulares por parte de la población en las grandes ciudades y el crecimiento sostenido del parque automotriz son una de las causas más importantes del aumento de las emisiones de este contaminante. Cabe tener presente que los vehículos con motor a diesel, emiten una mayor cantidad de contaminantes, que aquellos a gasolina, por lo que también es relevante considerar la composición del parque automotriz. Daño en la salud humana y el medio ambiente: Los NOx son responsables de importantes efectos sobre la salud y el medio ambiente, como problemas respiratorios o daño pulmonar, enfermedades en pulmones y bronquios, mayor susceptibilidad a las infecciones, daño celular, irritación ocular y pérdida de las mucosas. El NO2 puede reaccionar con la humedad presente en la atmósfera para formar ácido nítrico que puede ser causa de corrosión de las superficies metálicas y detener el crecimiento de plantas. Procedimiento para la determinación del NOx(Luminiscencia) Esta marcha se utiliza en general para todos los óxidos de nitrógeno, pero se hará el ejemplo para el NO2. 1. Principio El dióxido de nitrógeno es absorbido del aire por una solución acuosa de trietanolamina y el análisis posterior es realizado usando un reactivo que forma un compuesto azo–colorante de color púrpura rojizo por reacción de diazotación-copulación de sulfanilamida con diclorhidrato de N-(1-naftil)-etilendiamina (NEDA) a pH entre 2 y 2,5. El color producido por el reactivo es medido por espectrofotometría a 540 nm. 2. Sensibilidad La sensibilidad del método depende del reactivo de Griess - Saltzman. Para celda de trayectoria óptica de 1 cm y 0,1 unidades de absorbancia es equivalente a 0,14 μg/mL de NO2 en la solución de absorción. 3. Rango El rango de la concentración en el aire para el cual este método puede ser usado con confianza, está entre 10 a 1000 μg/m3 (0,005 a 0,50 ppm), tomando como base un periodo de muestreo de 24 horas. El comportamiento del método para niveles de NO2 por encima de 1000 μg/m3 (0,5ppm) aún no ha sido establecido. 4. Comparación Con La Norma Para No2 En la resolución 601 del año 2006 se establece la norma de aire o el valor máximo permisible de concentración de NO2 para periodos de tiempo iguales a 1 hora, 24 horas y anual. DEFINICIONES N. A. 5. Cuidados, salud y seguridad El cuidado que se debe tener con los reactivos utilizados en la práctica se encuentra relacionado en el anexo No 1 “Hojas de Seguridad” correspondientes. Antes de iniciar la práctica, revise los procedimientos. En el desarrollo de todos los análisis, utilice de manera obligatoria los siguientes implementos de seguridad: bata, guantes, respirador para vapores ácidos, gafas protectoras. 5.1. Limitaciones e interferencias El dióxido de azufre en concentraciones hasta de 2000 μg/m3 (0,7 ppm) no interfiere si se añade peróxido de hidrógeno después del muestreo y antes del desarrollo del color. El ozono no causa interferencias en los rangos de concentración atmosféricos (hasta de 1000μg/m3). El óxido nítrico en concentraciones hasta de 800μg/m3 (0,6 ppm) como promedio de 24 horas no causa interferencias. Los nitritos orgánicos y nitratos de peroxiacilo (PAN), que pueden estar presentes en el aire, podrían causar una interferencia positiva. No hay datos de la magnitud de la interferencia, sin embargo, en vista de las concentraciones promedio de 24 horas de los nitritos orgánicos y de los nitratos de peroxiacilo que han sido reportados en la literatura, parecería que esta interferencia podría ser despreciable. 5.2. Disposición de residuos Disponga los residuos de acuerdo con los procedimientos establecidos por el laboratorio de Ingeniería Ambiental. 6. Toma,transporte y preservación de la muestra La toma de la muestra realiza de acuerdo con el “procedimiento para la Toma de Muestra de tres gases (TME-003)” Luego de tomar las muestras, el transporte de estas al laboratorio se debe hacer en condiciones de temperatura menores o iguales a 4°C (+/- 2°C), para garantizar la composición de la muestra, para lo cual se emplean neveras de poliuretano refrigeradas mediante pilas de gel. Después del muestreo, la solución absorbente puede ser almacenada hasta por tres semanas sin pérdidas si se mantiene tapada en la oscuridad. Sin embargo, después de que se produzca el color por los reactivos adicionados, la muestra debe ser analizada en pocas horas. La absorbancia de la solución coloreada decrece cerca del 4% por día. Permita que las muestras se aclimaten una vez vaya a realizar el análisis. 7. Aparatos, reactivos y materiales. 7.1. Aparatos. 7.1.1. Espectrofotómetro: El instrumento usado debe ser capaz de medirla absorbancia de una solución a 540 nm. Una celda de trayectoria óptica de 1 cm es la más adecuada. 7.1.2. Filtro 7.1.3. Celdas del espectrofotómetro. 7.1.4. Dispositivo de control de temperatura. 7.1.5. Balanza analítica. 7.1.6. Nevera. 7.1.7. Absorbedor, equipo para medición de aire, líneas de muestreo y bomba de aire, se describen en el procedimiento de muestreo para NOx (TME-003) 7.2. REACTIVOS. 7.2.1. Agua Destilada (libre de nitritos) 7.2.2. Trietanolamina 7.2.3. N-butanol 7.2.4. Peróxido de Hidrógeno 7.2.5. Sulfanilamida 7.2.6. Acido fosfórico 85% 7.2.7. N-(1 NAFTIL) –Dihidrocloruro de Etilendiamina (NEDA) 7.2.8. Nitrito de sodio 7.3. MATERIALES. 7.3.1. Vidriería volumétrica clase A de varias capacidades para preparar y estandarizar reactivos y estándares y para distribuir soluciones durante el análisis. Dentro de este material, se incluyen pipetas, matraces aforados y buretas. - Balones aforados de 25, 50, 100, 250, 1000 mL - Vasos de precipitados de 100, 250 500 y 1000 ml - Pipetas aforadas de 1, 2,5,7,9, 10, 20, 25, 50 y 100 mL - Erlenmeyer 125mL - Frascos lavadores con agua desionizada - Pipeta Pasteur - probetas 7.3.2. Cronómetro 7.3.3. Termómetro 7.3.4. Termostato Verifique la existencia de los reactivos. 8. Procedimiento para la preparación de reactivos 8.1. PREPARACIÓN DE REACTIVOS Y ANÁLISIS DE LAS MUESTRAS. Los reactivos químicos usados deben ser grado analítico. El agua debe estar libre de nitritos. 8.1.1. Líquido absorbedor: Añada 15 g de trietanolamina aproximadamente a 500 mL de agua destilada; luego adicione 3 mL den-butanol. Mezcle bien y diluya hasta 1 L con agua destilada. El N-butanol actúa como un surfactante. Si se presenta formación de mucha espuma durante el muestreo, se debe disminuir la cantidad de n-butanol. El reactivo es estable por dos meses si se mantiene en una botella oscura y bajo refrigeración. 8.1.2. Peróxido de hidrógeno Diluya 0,2 mL de peróxido de hidrógeno al 30%hasta 250 mL con agua destilada. Precaución. El peróxido de hidrógeno es un oxidante fuerte y puede causar daño en la piel y en la ropa. 8.1.3. Reactivo de coloración. Disuelva 10 g de sulfanilamida en 400 mL de agua destilada, luego, adicione 100 mL de ácido fosfórico concentrado (85%) 1 gramo de N-(1 NAFTIL) – Dihidrocloruro de Etilendiamina (NEDA). Mezcle hasta disolver y complete a 1 litro con agua desionizada. Esta solución es estable por varios meses si se almacena en un frasco oscuro y hasta por 1 año si se mantiene refrigerada. 8.1.4. Solución Stock de nitrito. Disuelva 0,1363 g de nitrito de sodio en agua destilada y complete hasta 1 L. El nitrito de sodio debe ser de pureza conocida o analizada antes del uso. La solución así preparada contiene 90 μg de nitrito/mL, lo cual corresponde a 100 μg de NO2 (gas)/mL. Determine la concentración real de la solución stock mediante la siguiente ecuación: Donde: A= gramos de reactivo de Na2SO3 P= Pureza del reactivo Para un reactivo del 99% de pureza, pese 0,1363 g de reactivo para tener 0,135 g de nitrito de sodio. Antes de completar a volumen, preserve esta solución con 2 ml de cloroformo. Almacene la solución refrigerada a 4⁰C. La solución debe ser preparada una vez al mes. 9. CALIBRACIÓN ANALÍTICA 9.1. COMPATIBILIDAD DE LAS CELDAS DEL ESPECTROFOTÓMETRO Si se usan celdas de espectrofotómetro incompatibles, se debe determinar un factor de corrección de absorbancia de la siguiente manera: 9.1.1. Llene todas las celdas con agua destilada y designe la que tiene la absorbancia más baja a 548nm como la “celda de referencia”. (Esta celda de referencia debe ser marcada como9.1.2. Calibre el cero del espectrofotómetro con la celda de referencia. 9.1.3. Determine la absorbancia de cada una de las celdas restantes (Ac) en relación con la celda dereferencia y registre estos valores para un uso futuro. Marque todas las celdas de una maneraque identifique adecuadamente la corrección. La absorbancia corregida para futuros análisis usando cada celda previamente identificada y determinada su respectiva Ac, es determinada así: A = Aobs–Ac Donde: A = Absorbancia corregida Aobs = Absorbancia observada, sin corregir Ac= Corrección de la ceda 10. LIMPIEZA DE LA VIDRIERÍA Todo el material, incluyendo aquel utilizado para muestreo debe someterse a lavado siguiendo las instrucciones del procedimiento TLA 010 – Procedimiento para el lavado del material – Lave con jabón neutro y enjuague hasta fin de tensoactivos. Reserve la vidriería para análisis de NOx. Utilice únicamente el material al que se le haya efectuado el control de calidad. 11. PROCEDIMIENTO PARA LA PREPARACIÓN DE ESTÁNDARES PARA LAS CURVAS DE CALIBRACIÓN. La curva de calibración puede ser obtenida por dos métodos diferentes: El método más simple y conveniente es la estandarización con soluciones de nitrito. Sin embargo, mezclas exactas de gas conocidas darían estándares más reales para calibración de todo el sistema analítico. Las mezclas de gas conocidas pueden ser preparadas usando un sistema de dilución de flujo o tubos de permeación. 11.1. ESTANDARIZACIÓN CON SOLUCIÓN DE NITRITO 11.1.1. Solución de trabajo de nitrito. Transfiera 10 mL de solución de stock de nitrito a un balón volumétrico de 500mL y complete a volumen con solución de absorción. La solución de trabajo así obtenida contiene 1,8 μg NO2-/mL (que corresponde a 2 μg NO2 (gas)/mL). Determine el valor real de concentración de la solución de trabajo con la siguiente ecuación: Donde: V= volumen de solución stock de nitrito, ml 11.1.3. Prepare las soluciones Solución de trabajo de nitrito Volumen de solución de estándar de acuerdo con la (μg NO2/mL) trabajo tabla que aparece a ml continuación: Patrón NO2 en fase líquida NO2 en fase gaseosa μg NO2 totales μg NO2/mL* μg NO2 (gas) totales μg NO2 (gas)/mL 0 0 0 0 Blanco 0 1.08 0.0432 1,2 0.0480 0,18 6 1.8 0.0720 2,0 0.0800 0,18 10 5.4 0.216 6,0 0.240 1,8 3 9.0 0.360 10 0.400 1,8 5 16.2 0.648 18 0.720 1,8 9 C= Concentración de la solución stock de nitrito, μg de NO2/ml 11.1.2. Solución de trabajo de nitrito diluida (0,18 μg NO2-/mL). Transfiera a un balón de 100 ml, 10 ml de la solución de trabajo de 1,8 μg NO2-/mL y complete a volumen con solución de absorción del espectrofotómetro se dan en transmitancia, convierta en absorbancia utilizando la siguiente ecuación: Donde A = Absorbancia T = Transmitancia (0≤T≤1) 12.1.2. Filtro. De longitud de onda trazable a la Agencia Nacional de Estándares para verificar la calibración de la longitud de onda de acuerdo al procedimiento incluido con el filtro. La calibración de longitud de onda debe ser verificada durante el recibo inicial del instrumento y después de cada 160 horas de empleo normal o cada 6 meses, lo que ocurra primero. 12.1.3. Celdas del espectrofotómetro. Un juego de celdas de longitud de trayectoria de 1 cm conveniente para su uso en la región visible. Si las celdas son incomparables, se debe determinar un factor de corrección de comparación siguiendo los pasosdescritos en el numeral 9.1 del presente procedimiento. 13. PROCEDIMIENTO DE ANÁLISIS DE LAS MUESTRAS. 13.1. TOMA DE LA MUESTRA. Adicione 50 mL de la solución absorbente en el burbujeador. Conecte el burbujeador al tren de muestreo y prenda la bomba; ajuste la tasa de flujo rápidamente en 150 y 200 mL/min, si se usa válvula de aguja. Si se usa un orificio crítico verifique la tasa de flujo hasta determinar que esté conforme con la calibración, muestree por 24 horas y observe la tasa de flujo al final del periodo. Si la muestra está bien tapada puede ser almacenada por un periodo de 3 semanas antes del análisis sin pérdidas. 13.1.1. Remueva las muestras del contenedor de transporte. Si el período de transporte excede 12 horas después de la terminación de la muestra, verifique que la temperatura esté por debajo de 10 °C. 13.1.2. Compare el nivel de la solución con la señal de nivel temporal sobre el absorbedor. Si la temperatura está por encima de 10°C o hubo pérdida significativa (más de 10 ml) de la muestra durante el transporte, haga una anotación apropiada en el registro e invalide la muestra. 13.2. DESARROLLO DEL COLOR. 13.2.1. Complete cualquier pérdida en la solución absorbente llevándola a 50 mL, con agua destilada. 13.2.2. Con probeta o pipeta, tome una alícuota de 25 mL de la muestra a analizar; de manera similar tome una alícuota de 25 mL de la solución de absorción no expuesta (blanco), a un segundo recipiente (este puede ser un balón aforado de 25 mL o un Erlenmeyer de 125 ml), mida también este volumen para los estándares de control. 13.2.3. Adicione a cada muestra, al blanco y a los estándares de control 1 mL de solución de peróxido de hidrógeno diluido y mezcle bien. 13.2.4. Adicione 2 mL de reactivo de coloración, mezcle bien y permita que se desarrolle el color durante 20 min. 13.2.5. Mida la absorbancia contra el blanco a 540 nm. 13.2.6. Determine el contenido de NO2-gas/ml en los 50 ml de muestra interpolando directamente en la curva de calibración. 14. PROCESAMIENTO DE DATOS Y CÁLCULO DE RESULTADOS. 14.1. El volumen de aire muestreado debe corregirse a 25ºC y 760 mm de Hg. La desviación normal de estas condiciones adiciona solamente pequeñas correcciones. 14.2. La estandarización de la solución de nitrito requiere el uso de un factor empírico que relaciona los microgramos de NO2 (gas) con los microgramos de ión nitrito. El factor de conversión en este método es 0,90, esto es, el 90% de NO2 en la muestra de aire es eventualmente convertido a ión nitrito, el cual reacciona con el colorante produciendo el reactivo azo-colorante. Este factor ha sido incorporado en la solución stock de nitrito. 14.3. El cálculo de la concentración promedio de NO2 en el aire en un periodo de muestreo de 24 horas puede efectuarse como sigue: 14.3.1. Lea los μg de NO2-gas/ml directamente del gráfico. donde, C = concentración en μg NO2/m3 P = concentración en ppm de NO2 (μL/L) r = tasa de muestreo en L/min t = tiempo de muestreo en min V= Volumen de muestra recolectada en ml (50 ml) 0,532 = μL NO2 / μg NO2 a 25ºC y 760 mm de Hg 103 = factor de conversión de L a m3 NOMBRE DEL MÉTODO: Determinación de NOx en Aire por el Método de Espectrofotometría CODIGO DEL SOP: TLA-014 FECHA DEL INFORME DE VALIDACIÓN: Noviembre 14 de 2011 PARÁMETRO VALOR UNIDADES OBSERVACIÓN LIMITE DETECCION DE 0,0480 μg de NO2-gas/ml PRESICIÓN EN TERMINOS DE % CV 1,3 9,1 N.A. EXACTITUD EXPRESADO COMO % DE ERROR RELATIVO -5,71 -9,70 RANGO DE TRABAJO (Lectura Directa) INTERVALO DE APLICACIÓN DEL MÉTODO RECUPERACIÓN EXPRESADO COMO % 78 0,0480 – 0,0720 N.A. % % Corresponde al límite de cuantificación STD Bajo de 0,0480 μg de NO2gas/ml STD Alto de 0,400 μg de NO2-gas/ml STD Bajo de 0,0480 μg de NO2gas/ml STD Alto de 0,400 μg de NO2-gas/ml μg de NO2-gas/ml Sin dilución de la muestra 0,144 - 7,20 μg de NO2-gas/ml Para una dilución de 100 veces 72 % Nivel Bajo % de Adición Nivel de Adición Alto Se recomienda que una pequeña parte (<10 ml) de la muestra original sea analizada de nuevo (si es posible) si la muestra requiere una dilución mayor que 1:1. 17. SECCIÓN DE CONTROL DE CALIDAD Y ASEGURAMIENTO DE CALIDAD. Prepare la curva de calibración cada vez que prepare nuevos reactivos de nitrito, sulfanilamida o NEDA. Verifique que la sensibilidad de la curva de calibración (Pendiente) se encuentre en el orden que se reporta. Se debe verificar que los estándares de control se muevan dentro de los parámetros establecidos, de otra manera, deben revisarse tanto los estándares de calibración como los materiales y los blancos. Corrija y reanude el análisis. Los duplicados no deben igualmente mostrar diferencias mayores al 10%, en caso contrario, repetir el análisis. Se deben llevar registros de los estándares de control en cartas de control correspondientes Cuando los resultados se encuentren entre los límites de alarma y control, se debe revisar el procedimiento para hallar la causa. Si algún dato cae por fuera de los límites de control se debe reexaminar y si se considera necesario, repetir el análisis de todo el grupo de muestras.} Control y eliminación de los NOX Los óxidos de nitrógeno NOx constituyen uno de los principales contaminantes emitidos durante el proceso de combustión, la mayor parte de los cuales se relacionan con el transporte; para su control se pueden emplear una serie de procedimientos. Los automóviles y otros vehículos generan la principal fuente de emisiones de NOx; las plantas térmicas de producción de energía contribuyen con un cuarto de las emisiones globales; el NO es un gas incoloro, mientras que el pardo, que crea una pluma NO2 es un gas de color visible sobre la chimenea. Fig XXXIII.1.- Fuentes de emisión de NOx XXXIII. 1.- MECANISMOS DE FORMACIÓN DE LOS NOx Los NOx se refieren a un conjunto de emisiones de óxido nítrico NO, de dióxido nítrico NO 2 y trazas de otros, generados en la combustión. La combustión de cualquier combustible fósil produce un determinado nivel de NOx debido a las altas temperaturas y a la disponibilidad de oxígeno y nitrógeno, tanto en el aire comburente, como en el combustible. Las emisiones de NOx generadas en los procesos de combustión están constituidas por un 90÷ 95% de NO, y el resto por NO2; cuando los humos abandonan la chimenea, una gran parte del NO se oxida en la atmósfera, pasando a NO2. El NO2 presente en los humos crea el penacho grisáceo que se puede ver saliendo de la chimenea de una planta energética. Una vez en la atmósfera, el NO2 interviene en una serie de reacciones que forman contaminantes secundarios. El NO2 puede reaccionar con la luz solar y con radicales de hidrocarburos, para producir componentes fotoquímicos de huminiebla (presencia de humos y niebla en algunas ciudades) y de lluvia ácida. NOx térmico: Denomina al NOx formado, a alta temperatura, por la oxidación del nitrógeno que se encuentra en el aire comburente; su velocidad de formación depende de la temperatura y del tiempo de permanencia en la misma. Normalmente se forman cantidades significativas de NOx en condiciones de oxidación por encima de los 2200⁰F (1204⁰C), aumentando de forma exponencial con la temperatura; en esta situación, el N2 y el O2 moleculares presentes en la combustión, se disocian y pasan a su estado atómico, participando en una serie de reacciones, siendo uno de los productos el NO. La combustión, tiende a incrementar la formación del NOx térmico, por lo que se precisa de un cierto compromiso entre la combustión efectiva y la formación controlada de NOx. La formación del NOx térmico se controla reduciendo las temperaturas máxima y promedia de la llama, y se puede conseguir mediante una serie de modificaciones en el sistema de combustión: - Empleando quemadores de mezcla controlada, que reducen la turbulencia en la región de la llama próxima al quemador, retardando el proceso de combustión, lo que disminuye la temperatura de la llama, ya que de ella se extrae energía antes de que se alcance la temperatura máxima - Mediante el escalonamiento de la combustión, aportando inicialmente una parte del aire comburente, por lo que el combustible se oxida parcialmente quedando relativamente frío respecto al resto del aire comburente, que se añade posteriormente, para completar el proceso de combustión - Mezclando en el quemador parte de los humos generados con el aire comburente, configurando una recirculación de humos; de esta forma se incrementan los gases a calentar con la energía química del combustible, reduciéndose la temperatura de la llama Estas tecnologías se han utilizado eficientemente para reducir la formación de NOx quemando gases, aceites y carbones; su utilización, o una combinación de las mismas, depende de los costes, del combustible y de los requisitos reguladores. Para aquellos combustibles que no tienen cantidades significativas de nitrógeno estructural, como el gas natural, el NOx térmico se convierte en el principal contribuyente a las emisiones globales de NOx siendo las tecnologías citadas efectivas para su control. NOx del combustible.- La principal fuente de emisiones de NOx procedentes del nitrógeno estructural contenido en los combustibles, como parte de compuestos orgánicos en los carbones y aceites, se debe a la conversión del nitrógeno en NOx durante el proceso de combustión. El NOx del combustible contribuye aproximadamente al 50% de las emisiones totales incontroladas, cuando se queman aceites residuales, y a más del 80% en el caso de quemar carbones. Durante el proceso de combustión, el nitrógeno se libera como radical libre que, finalmente, forma NO o NO2. EFECTOS DEL NOx SOBRE LA SALUD Y EL MEDIO AMBIENTE Cuando el NOx sale a la atmósfera, comienza su participación en los fenómenos relativos a la formación de ozono/huminiebla fotoquímica, lluvia ácida, partículas sólidas y posibles productos cancerígenos que se encuentran suspendidos en el aire, con una significativa repercusión en la salud humana y en el medio ambiente. Ozono/huminiebla fotoquímica.- El ozono producido por la actividad del hombre, formado en las capas bajas de la atmósfera, se considera como un contaminante; este tipo de ozono, que se forma por la reacción de hidrocarburos y NOx en presencia de luz solar, es el componente principal de la huminiebla urbana fotoquímica, junto con el NO2 y gran variedad de otros compuestos. El NO contenido en los humos, una vez que se emite por la chimenea, se oxida para formar NO2 que es un gas oxidante de color amarillo-marrón. Lluvia ácida: El NOx y el SO2 contribuyen a la formación de la lluvia ácida, que incluye una disolución diluida de los ácidos nítrico y sulfúrico, con pequeñas cantidades de ácido carbónico y otros ácidos orgánicos. El NOx y el SO2 reaccionan con el vapor de agua para formar compuestos ácidos, que son los causantes de más del 90% de la lluvia ácida; la mayor parte de los controles de lluvia ácida se concentra en las contribuciones del SO2 imputando al NOx menos de un tercio. Partículas sólidas en suspensión.- El NOx puede contribuir a la existencia de partículas sólidas suspendidas en el medio ambiente. En la atmósfera, el NOx reacciona con otros productos químicos en suspensión, para producir nitratos. El NOx promueve también la transformación del SO2 en partículas de compuestos sulfatados. CONTROL DE LAS EMISIONES DE NOx EN GENERADORES DE VAPOR Como los NOx se forman durante el proceso de combustión, las investigaciones se han centrado sobre su control en la propia fuente; en la reducción de estas emisiones han resultado efectivas las aplicaciones que controlan las mezclas combustible + aire , y las puntas de temperatura de la llama. La tecnología de combustión basada en el control del NOx es el procedimiento de coste global más bajo, teniendo en cuenta que puede cumplimentar los requisitos de emisiones, tanto estatales como locales; cuando se necesita un control más severo, se añade alguna técnica de postcombustión. TRATAMIENTOS PREVIOS A LA COMBUSTIÓN.A diferencia de lo que ocurre con los componentes de S y de partículas presentes en el carbón, los compuestos de nitrógeno contenidos en el combustible no se pueden reducir o eliminar tan fácilmente. La opción más común para reducir los niveles de NOx como tratamiento previo a la combustión, consiste en cambiar a otro combustible con menor contenido de nitrógeno. En el caso de generadores de vapor capaces de quemar varios combustibles, con un mínimo impacto en el ciclo de vapor, el cambio del combustible suele ser la solución de mínimo coste. TÉCNICAS DE COMBUSTIÓN.- Los mecanismos específicos de reducción de NOx son: - La velocidad de la mezcla combustible-aire - La reducción de la disponibilidad de oxígeno en la zona de combustión inicial - La reducción de las puntas de temperatura de la llama. El desarrollo de sistemas de combustión específicos para reducir la formación de NOx incluyen: - Quemadores de bajo NOx, Fig XXXIII.4a.b.c.d - Técnicas de combustión escalonada - Recirculación de humos (FGR) Los sistemas más modernos pueden emitir menos de la tercera parte del NOx que el producido por unidades viejas. TÉCNICAS DE POSTCOMBUSTIÓN.- Muchas zonas requieren emisiones de NOx menores de las que económicamente es posible obtener modificando sólo la propia combustión; para alcanzar reducciones superiores, se aplican técnicas aguas abajo de la zona de combustión como la: - Reducción Selectiva No Catalítica (SNCR) - Reducción Selectiva Catalítica (SCR) En cada una de estas tecnologías, el NOx se reduce a N2 y H2 a través de una serie de reacciones con un agente químico que se inyecta en el flujo de humos. Los agentes químicos que se utilizan en las aplicaciones comerciales, son: - El amoniaco y la urea para los sistemas (SNCR) - El amoniaco para los sistemas (SCR) La mayoría de los sistemas que emplean amoniaco como agente de reducción le han utilizado en estado anhidro; sin embargo, debido a los riesgos inherentes al almacenamiento y manipulación del NH3, muchos sistemas han optado por el uso del amoniaco acuoso en concentraciones del 25÷ 28%. La urea, (NH2)2CO, se puede almacenar como sólido o, mezclada con agua, como una solución; un subproducto adicional de la inyección de urea es el CO2. a) Técnica de la reducción selectiva no catalítica (SNCR).- Se dispone de dos procesos básicos de reducción selectiva no catalítica: - El primero utiliza un agente reductor (reactivo) a base de nitrógeno, como el amoniaco, que ha desarrollado EXXON y que está patentado con el nombre Termal De-NO2 - El segundo utiliza una tecnología basada en la urea, que se ha desarrollado con el patrocinio de EPRI (Electric Power Research Institute) Técnica de la reducción selectiva catalítica (SCR) Consideraciones de diseño.- Los sistemas de reducción selectiva catalítica (SCR) reducen el NOx contenido en los humos, por vía catalítica, dando lugar a N2 y H2O, mediante la utilización de amoniaco como agente reductor. Esta tecnología constituye el método más eficiente de reducir las emisiones de NOx cuando se requieren altas eficiencias (70÷ 90%). Las reacciones de reducción del NOx tienen lugar cuando los humos atraviesan la cámara catalítica; antes de que éstos entren en el catalizador, se inyecta el amoniaco y se mezcla con el flujo de humos, Fig XXXIII.16. La técnica de reducción selectiva catalítica (SCR) se utiliza cuando se requieren altas eficiencias de eliminación de NOx en calderas industriales y energéticas, que quemen madera, gas, aceite o carbón. Los sistemas (SCR) (650ºC) eliminan el NOx de los gases de combustión mediante su reacción con el amoniaco inyectado (líquido o gaseoso); el NH3 se absorbe por la superficie del catalizador a base de óxidos metálicos, como por ejemplo, (V2O5 ó WO3 sobre TiO2) y reacciona con los NOx en presencia de O2, formando H2O y N2, de acuerdo con las reacciones: Conforme aumenta el contenido de S en los humos, el campo de temperaturas para poder eliminar la formación de sales de sulfato amónico en el lecho del catalizador es menor. Por encima de la temperatura máxima del citado campo, hay un determinado número de agentes catalizadores que tienden a ser menos efectivos, siendo la fórmula química del agente catalizador el punto clave para las características funcionales de un sistema (SCR). Aunque la fórmula de un catalizador suele ser secreta, el material catalizador normalmente se sitúa en uno de los grupos siguientes: Catalizadores de metales ordinarios y aleaciones. - La mayor parte de los catalizadores están constituidos por aleaciones de metales ordinarios; se componen de óxido de Ti con pequeñas cantidades de Va, Mo, y W, o combinaciones de otros productos químicos activos. Los catalizadores de metales ordinarios son selectivos y operan en el margen de temperaturas especificado. Su mayor inconveniente estriba en su potencial de oxidar el SO2 a SO3, grado de oxidación que depende de la formulación química del catalizador. Las cantidades de SO3 formadas pueden reaccionar con el NH3 arrastrado por el flujo de humos, dando lugar a sales de sulfato amónico; este problema se puede minimizar con un diseño del sistema y una formulación del catalizador adecuados. Catalizadores de zeolita. - Son relativamente nuevos en el control de las emisiones de NOx; son materiales a base de silicatos de Al, cuya función es muy parecida a la de los catalizadores de metales ordinarios. Una ventaja de las zeolitas radica en su más elevada temperatura de operación, 970ºF (521ºC). El catalizador de zeolitas puede oxidar el SO2 a SO3 y de ahí la necesidad de una cuidadosa coordinación con los componentes de los humos y con las temperaturas de operación. Catalizadores de metales preciosos. - Se fabrican a partir de platino y rodio. Al mismo tiempo que actúan en la reducción de NOx también pueden actuar como catalizadores oxidantes, convirtiendo CO en CO2, bajo unas adecuadas condiciones de temperatura. No obstante, la oxidación paralela del SO2 a SO3 y el coste elevado de los catalizadores de metales preciosos, los hacen poco atractivos para la reducción del NOx.La primitiva tecnología de los sistemas (SCR) utilizaba catalizadores granulados (en forma de nódulos), que se amontonaban configurando un lecho. Este material era efectivo para la reducción de NOx, pero su manipulación no era fácil y frecuentemente introducía en el propio sistema una significativa caída de presión, por lo que la tecnología de fabricación de catalizadores evolucionó hacia bloques uniformes y mayores de material catalizador; los sistemas más modernos de (SCR) utilizan catalizadores bloque fabricados con una configuración de placas paralelas o alveolar (en forma de panal), Fig XXXIII.20. La configuración tiene sus propias ventajas, como: - Cuando en el reactor de (SCR) se tratan flujos de humos cargados con partículas en suspensión el catalizador de placas presenta menor caída susceptible de de presión sufrir y es menos atoramientos y erosiones - El catalizador alveolar requiere menor volumen de reactor, para un área dada de superficie global. El catalizador va alojado en un reactor estratégicamente ubicado en el sistema, Fig. XXXIII.21, que les expone a las temperaturas de reacción para el sistema (SCR). El diseño del reactor incluye un sistema de sellado para impedir el bipaso de los humos y para facilitar un soporte interno del material del catalizador. La configuración del reactor puede ser vertical u horizontal, dependiendo: - Del combustible utilizado - Del espacio disponible - De la disposición del equipo que haya aguas arriba y aguas debajo de la unidad catalizadora. Bibliografía A., G. J. (1978). DETERMINACIÓN DE DIOXIDO DE NITRÓGENO EN LA ATMÓSFERA MEDIANTE MUESTREO CON FILTROS IMPREGNADOS. Madrid. Instituto de hidrologia, m. y. (2011). Procedimiento para la determinación del dióxido de nitrógeno en el aire. España: Universidad de La Salle. Patier, R. F. (2006). Metodología de la evaluación del aire. Madrid. http://libros.redsauce.net/ (2021) consultado 04/01/2020