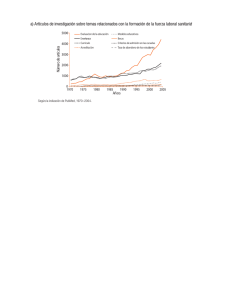
Questa, María. 2015 10 14 "Efectos del cultivo hipóxico sobre la
Anuncio

Efectos del cultivo hipóxico sobre la reprogramación de fibroblastos humanos a células madre pluripotentes inducidas Questa, María 2015 10 14 Tesis Doctoral Facultad de Ciencias Exactas y Naturales Universidad de Buenos Aires www.digital.bl.fcen.uba.ar Contacto: [email protected] Este documento forma parte de la colección de tesis doctorales y de maestría de la Biblioteca Central Dr. Luis Federico Leloir. Su utilización debe ser acompañada por la cita bibliográfica con reconocimiento de la fuente. This document is part of the doctoral theses collection of the Central Library Dr. Luis Federico Leloir. It should be used accompanied by the corresponding citation acknowledging the source. Fuente / source: Biblioteca Digital de la Facultad de Ciencias Exactas y Naturales - Universidad de Buenos Aires UNIVERSIDAD DE BUENOS AIRES Facultad de Ciencias Exactas y Naturales Departamento de Química Biológica Efectos del cultivo hipóxico sobre la reprogramación de fibroblastos humanos a Células Madre Pluripotentes Inducidas Tesis presentada para optar por el título de Doctor de la Universidad de Buenos Aires en el área Química Biológica Autora: Lic. María Questa Directora de tesis: Dra. Alejandra Guberman Consejera de estudios: Dra. Adalí Pecci Lugares de trabajo: Laboratorio de Biología del Desarrollo Celular, LIAN, Fundación para la Lucha contra las Enfermedad Neurodegenerativas de la Infancia Laboratorio de Regulación Génica en Células Madre, Departamento de Química Biológica, Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires Ciudad Autónoma de Buenos Aires, 2015. Fecha de defensa: 14 de octubre de 2015 Efectos del cultivo hipóxico sobre la reprogramación de fibroblastos humanos a Células Madre Pluripotentes Inducidas RESUMEN Las células somáticas pueden ser reprogramadas a células pluripotentes denominadas Células Madre Pluripotentes Inducidas (CMPi) y éstas, a su vez, pueden diferenciarse a cualquier tipo celular adulto. Esto es de gran importancia en los campos de la medicina regenerativa, las pruebas farmacológicas in vitro y la investigación de trastornos genéticos, dado que pueden obtenerse células pluripotentes sin enfrentar la barrera ética de la manipulación de embriones y con el valor agregado de ser genéticamente idénticas al donante de células somáticas. Por lo tanto, el estudio del proceso de reprogramación se ha vuelto cada vez más importante para mejorar los métodos en eficiencia, rapidez y seguridad. Teniendo en cuenta su interacción con factores de transcripción implicados en el estado pluripotente de la célula, los factores inducibles por hipoxia, HIF por su sigla en inglés, podrían ser de interés en el proceso de reprogramación. En el presente trabajo hemos aislado distintos tipos de células somáticas humanas, como fibroblastos, queratinocitos y células mononucleares de sangre periférica, los hemos cultivado, y hemos intentado reprogramarlos. Además estudiamos distintas condiciones de generación de CMPi, utilizando distintos vectores necesarios para la reprogramación, moléculas que modifican la estructura de la cromatina como Ácido Valproico y Butirato de Sodio, y diferentes protocolos, hasta llegar a un método confiable y reproducible. A partir de ello, hemos generado CMPi en condiciones de normoxia e hipoxia, y posteriormente las hemos cultivado a largo plazo también en ambas condiciones. Hemos validado la legitimidad bona fide de las líneas celulares generadas, evaluando la expresión de genes marcadores de estado indiferenciado y por otra parte, la pluripotencia, mediante protocolos de diferenciación in vitro, e in vivo, por formación de teratomas. Asimismo, encontramos que ambas líneas exhiben características morfológicas y proliferativas, y patrones de metilación del ADN propios de células madre pluripotentes. Estudiamos además el comportamiento de las líneas celulares en I RESUMEN cultivo en cuanto a eficiencia de reprogramación, expresión de factores del estado pluripotente, proliferación y apoptosis. Encontramos que la hipoxia aumentó la cantidad de colonias generadas pero éstas resultaron más pequeñas en tamaño, en comparación con las colonias generadas en normoxia. Además, fue menor la expresión de marcadores del estado pluripotente en colonias de CMPi evaluadas en estadios tempranos luego de ser establecidas en hipoxia, que en las colonias generadas en normoxia. Esta expresión no aumentó cuando las colonias fueron establecidas y cultivadas a largo plazo, y luego expuestas a hipoxia aguda, pero se indujo después de una hipoxia crónica. Por otro lado, encontramos una mayor tasa de proliferación celular en CMPi cultivadas en hipoxia sin diferencias en los niveles de apoptosis. Finalmente, a fin de sentar las bases para la continuación del proyecto, generamos vectores retrovirales para poder introducir los genes de los factores HIF- α y HIF- α en células a reprogramar, junto con los vectores de reprogramación. Los factores HIF clonados en los plásmidos retrovirales fueron wild type y también formas mutadas que le confieren a las proteínas HIF una mayor estabilidad, y en algunos casos también una mayor capacidad de activación de la transcripción. En último lugar, investigamos el efecto de los vectores generados, en células somáticas plausibles de ser reprogramadas, y encontramos un posible efecto tóxico, probablemente debido a la acumulación de altas cantidades de proteína HIF en la célula, o de una actividad transcripcional de los genes blanco tan alta que conlleva muerte celular. Dado que la expresión del gen introducido por el vector retroviral se puede controlar debido a que utilizamos un sistema modulable Tet-Off, encontramos que un tratamiento con el agente represor disminuye la muerte celular, y podría ser necesario a la hora de co-transducir con estos vectores y los vectores de reprogramación. Esperamos que los resultados y las herramientas generadas en este trabajo contribuyan a la comprensión de los mecanismos moleculares involucrados en la reprogramación y al desarrollo de protocolos para la generación de CMPi de alta calidad y con alta eficiencia. PALABRAS CLAVE células madre, células madre embrionarias humanas, células madre pluripotentes inducidas, hipoxia celular, reprogramación nuclear, pluripotencia, estado pluripotente, diferenciación II Effects of hipoxic culture on the reprogramming of human fibroblasts to induced pluripotent stem cells ABSTRACT Somatic cells can be reprogrammed into pluripotent cells called Induced Pluripotent Stem Cells (iPSC) and these, in turn, can be differentiated into adult cell type. This is of great importance in the fields of regenerative medicine, pharmacological in vitro testing and genetic disorder research, as pluripotent stem cells can now be obtained without facing the ethical barrier of embryo manipulation, and with the added value that these are genetically identical to the somatic cell donor. Hence, through the study of the reprogramming process itself, it has become increasingly important to improve reprogramming methods in terms of efficiency, speed and safety. Given their interaction with transcription factors involved in stemness, Hypoxia Inducible Factor, HIF, may be of interest in the reprogramming process. In the present work we isolated different types of human somatic cells, like fibroblasts, keratinocytes and peripheral blood mononuclear cells, we have cultured them and tried to reprogram them. Moreover, we have studied different conditions for the generation of iPSC, using different vector necessary for reprogramming, molecules that modify the structure of chromatin like Valproic Acid and Sodium Butyrate, and different protocols, until we obtained a reliable and reproducible method. Based on this, we have generated iPSC in normoxia and in hypoxia, and subsequently cultured them long-term in both conditions as well. We validated the bona fide legitimacy of the cell lines generated, by evaluating the expression of stemness marker genes and, on the other hand, by evaluating pluripotency, through in vitro differentiation protocols, and in vivo, by teratoma formation. Also, we have found that both lines exhibited morphological and proliferative characteristics and DNA methylation patterns typical of pluripotent stem cells. Furthermore, we studied the cell li es’ behavior in culture regarding reprogramming efficiency, expression of stemness factors, proliferation and apoptosis. We found that hypoxia increased the amount of colonies generated but these turned out to be III ABSTRACT smaller in size, when compared to colonies generated in normoxia. Moreover, stemness marker expression was lower in iPSC colonies evaluated in early stages after being established in hypoxia, than in colonies established in normoxia. This expression did not increase when colonies were established and cultured in the long-term, and then exposed to acute hypoxia. Nevertheless, these a ke s’ expression was up-regulated after chronic hypoxic culture. In addition, we also found a higher proliferation rate for stem cells in hypoxia and no differences in apoptosis levels. Finally, in order to provide a continuation to the project, we generated retroviral vectors to be able to introduce HIF-1α and HIF-2α genes in cells to be reprogrammed, together with the reprogramming vectors. The HIF cloned in the retroviral plasmids were wild type and also mutated forms, which provide the HIF proteins with more stability and in some cases, also increased transcriptional activation ability. Lastly, we investigated the effect of these vectors, on somatic cells capable of being reprogrammed, and we found a possible toxic effect, probably due to the accumulation of high amounts of HIF proteins in the cell, or such a an elevated target gene transcriptional activity, that it generated cell death. Given the fact that the gene introduced by the retroviral vector can be controlled, since we used a modulable Tet-Off system, we found that a treatment with the repressor agent decreases cell death, and might be necessary for the co-transduction with these vectors and the reprogramming vectors. We hope that the results obtained and the tools generated in this work will contribute to the understanding of the molecular mechanisms involved in reprogramming, and to the development of high quality and highly efficient iPSC generation protocols. KEYWORDS stem cells, human embryonic stem cells, induced pluripotent stem cells, cell hypoxia, nuclear reprogramming, pluripotency, stemness, differentiation IV AGRADECIMIENTOS Gracias a esos bellos humanos que forman parte de mi vida, sin los cuales nada o poco hubiera sido posible, incluyendo esta tesis: Mamá Silvia C. Chauchot Lalanne, Papá Fernando E. Questa, hermanillo Juan M. Questa, mi gorda hermosa Mia K. Ortiz, my love Kevin M. Ortiz, Guillermo F. Questa, Marta M. del P. Jantus, Ana Jantus, Carolina Jantus, Hernán J. Jantus, Hernán M. Jantus, Marisel Abel, Emilia Klappenbach, Josefina Feraud, Guadalupe Feraud, Celina Klappenbach, Vicente Klappenbach, Alejandro Klappenbach (Jr.), Alejandro Klappenbach, Juan Feraud, John P. Ortiz, Monica J. Ortiz, Sean P. Ortiz, Megan A. Mitchell, Brian P. Ortiz, C. Inés Enrique, M. Fernanda Grispo, Alejandro A. Serantes, Valentín Serantes, Gisela I. Mazaira, Pablo J. Negri, Ofelia Montans, Nadia S. Alén, Gabriela A. Ramirez, Lucía P. Palermo, Matías N. Palermo, María I. L. Betti, Julio A. Herrera, M. Inés Martinez-Oliver, Alejandro Morral, Mateo A. Morral, N. Soledad Minervini, Felipe Calderón, Mauro C. Gómez, M. Cristina Perasso, Susana E. Cajén, Susana Majdalani y Amanda Bíscaro. Los quiero mucho mucho. Gracias por compartir este largo camino conmigo, y por hacer que valga la pena. A los a i alitos del a o , si los uales la ida se ía e os ejo : De o ia, Le o ski, Je . A Santiago G. Miriuka, Alejandra S. Guberman y Gustavo Sevlever, por su dirección. Una mención especial para mis directores Santiago y Ale: por su apoyo, energía y por su enorme guía. Sin ellos, no existiría este trabajo, ni mi formación. ¡Gracias! Y gracias también por hacer el camino hacia un doctorado una experiencia no sólo profesional pero también humana. A José Cibelli y Steve Suhr, por recibirme en MSU y por sus enseñanzas en el CRL. A mis compañeros de laboratorio por su ayuda y apoyo. Agradecimientos especiales a Marcela Cañari y Leonardo Romorini. A Ariel Waisman, Claudia Solari, Gabriel Neiman, Noelia I. Losino, Carlos Luzzani, Carolina Blüguermann, Carolina P. García, Darío Fernández Espinosa, Diego A. Riva, Guillermo Videla Richardson, María E. Scassa, Nicolás Dimopoulos, Ximena Garate, MarieClaude Senut, Jiesi Luo, Eleni Beli, Beatriz Ferández Muñoz, Neli Ragina y Masha Savelieff. A otra gente linda que estuvo en el camino, como Graciela y Ana; Diana Radakoff, Brandon Brooks, Corina Rhett, Jessica Emelio, Holly Or, Ashii Vinod, Nana Tada, Rose Lazuardi y todas las Multicultural MuMs. V A mi Mia. VI ÍNDICE VII ÍNDICE ABREVIATURAS -1- INTRODUCCIÓN -4- CÉLULAS MADRE CÉLULAS MADRE EMBRIONARIAS CÉLULAS MADRE PLURIPOTENTES INDUCIDAS HIPOXIA Y CÉLULAS MADRE -5-6- 11 - 15 - HIPÓTESIS Y OBJETIVOS - 18 - MATERIALES Y MÉTODOS - 21 - AISLAMIENTO DE FIBROBLASTOS EMBRIONARIOS DE RATÓN CULTIVO Y AMPLIFICACIÓN DE FIBROBLASTOS EMBRIONARIOS DE RATÓN INACTIVACIÓN DE MEF MEDIANTE EXPOSICIÓN A RADIACIÓN Γ TRATAMIENTO DE PLACAS CON GELATINA BOVINA TRATAMIENTO DE PLACAS CON MATRIGEL CULTIVO DE CÉLULAS HEK-293T OBTENCIÓN, CULTIVO Y EXPANSIÓN DE FIBROBLASTOS HUMANOS OBTENCIÓN Y CULTIVO DE QUERATINOCITOS HUMANOS OBTENCIÓN DE QUERATINOCITOS DE EPIDERMIS (A PARTIR DE CUERO CABELLUDO) OBTENCIÓN DE QUERATINOCITOS DE FOLÍCULOS PILOSOS OBTENCIÓN Y CULTIVO DE CÉLULAS MONONUCLEARES DE SANGRE PERIFÉRICA NORMOXIA E HIPOXIA CONGELACIÓN DE CÉLULAS CONDICIONAMIENTO DE MEDIO PRODUCCIÓN DE VECTORES VIRALES GENERACIÓN DE CÉLULAS MADRE PLURIPOTENTES INDUCIDAS A PARTIR DE FIBROBLASTOS PROTOCOLO DE GENERACIÓN DE CMPI A PARTIR DE CMSP CULTIVO DE CÉLULAS MADRE PLURIPOTENTES AISLAMIENTO DE ARN, SÍNTESIS DE ADNC Y PCR CUANTITATIVA EN TIEMPO REAL REACCIÓN DE PCR A PUNTO FINAL INMUNOFLUORESCENCIA ANÁLISIS DE LA METILACIÓN DEL PROMOTOR DE POU5F1 EVALUACIÓN DE LA EXPRESIÓN DE LOS TRANSGENES STEMCCA - 22 - 23 - 23 - 24 - 25 - 25 - 25 - 26 - 26 - 27 - 28 - 29 - 29 - 29 - 30 - 31 - 34 - 35 - 36 - 37 - 39 - 41 - 42 - VIII ÍNDICE FORMACIÓN DE TERATOMAS Y ANÁLISIS HISTOLÓGICO WESTERN BLOT ACTIVIDAD DE FOSFATASA ALCALINA Y EFICIENCIA DE REPROGRAMACIÓN IN-CELL WESTERN BLOT REPROGRAMACIÓN EN PRESENCIA DE INHIBIDORES DE HIF ESTUDIO POR CITOMETRÍA DE FLUJO DE LA EXPRESIÓN DE NANOG DURANTE EL PROCESO DE REPROGRAMACIÓN ENSAYO DE AZUL DE TRIPÁN PARA VIABILIDAD CELULAR ENSAYO DE PROLIFERACIÓN CELULAR ANÁLISIS DE LA APOPTOSIS POR CITOMETRÍA DE FLUJO A TRAVÉS DE DOBLE TINCIÓN CON ANEXINA V / IODURO DE PROPIDIO CARIOTIPADO CONSTRUCCIÓN DE PLÁSMIDOS PARA REPROGRAMACIÓN CON FACTORES HIF TRANSDUCCIÓN CON YHIF10 Y YHIF20 TRATAMIENTO CON DOXICICLINA ANÁLISIS ESTADÍSTICO NORMAS ÉTICAS DETECCIÓN DE MICOPLASMA EXTRACCIÓN DE ADN GENÓMICO REACCIÓN DE PCR PARA MICOPLASMA - 42 - 43 - 44 - 45 - 46 - 46 - 47 - 47 - 48 - 48 - 49 - 53 - 53 - 54 - 54 - 55 - 55 - 56 - RESULTADOS - 57 - OBTENCIÓN DE CÉLULAS SOMÁTICAS PARA REPROGRAMACIÓN - 58 OBTENCIÓN DE FIBROBLASTOS SUBDÉRMICOS - 58 OBTENCIÓN DE QUERATINOCITOS - 59 OBTENCIÓN DE QUERATINOCITOS EPIDÉRMICOS - 59 OBTENCIÓN DE QUERATINOCITOS DE FOLÍCULOS PILOSOS - 63 OBTENCIÓN DE CÉLULAS MONONUCLEARES DE SANGRE PERIFÉRICA - 64 GENERACIÓN DE CMPI - 65 GENERACIÓN INICIAL DE CMPI EN EL CELLULAR REPROGRAMMING LABORATORY: - 66 EXPERIENCIAS DE GENERACIÓN DE CMPI EN LOS LABORATORIOS DE BUENOS AIRES: - 77 EFECTOS DE LA HIPOXIA SOBRE LA GENERACIÓN DE CMPI - 89 SE COMPROBÓ QUE LAS LÍNEAS DE FIBROBLASTOS REPROGRAMADAS EN NORMOXIA E HIPOXIA SON LÍNEAS DE CMPI BONA FIDE - 92 LOS FACTORES HIF FUERON ESTABILIZADOS EN NUESTRO MODELO DE HIPOXIA Y AUMENTÓ LA EXPRESIÓN DE SUS GENES BLANCO. EFECTOS SOBRE LA EXPRESIÓN DE MARCADORES DEL ESTADO PLURIPOTENTE. - 106 MODULACIÓN DE LA EXPRESIÓN DE GENES DE PLURIPOTENCIA POR CULTIVO HIPÓXICO. LOS MARCADORES DEL ESTADO PLURIPOTENTE SE INDUCEN A TIEMPOS DE CULTIVO HIPÓXICO A LARGO PLAZO. - 111 EFECTOS DEL CULTIVO HIPÓXICO SOBRE LA GENERACIÓN DE CMPI - 113 Efectos del cultivo hipóxico sobre la eficiencia de reprogramación. La hipoxia aumenta la eficiencia de reprogramación, pero las colonias formadas son más pequeñas - 113 - IX ÍNDICE La tasa de proliferación de CMPi es mayor en hipoxia. La menor expresión de marcadores del estado pluripotente en hipoxia es una consecuencia de un menor número de células en las colonias generadas en esta condición - 119 GENERACIÓN DE PLÁSMIDOS PARA LA GENERACIÓN DE VECTORES VIRALES PARA LA TRANSDUCCIÓN DE FACTORES HIF - 126 CLONACIÓN DE FACTORES HIF EN VECTORES VIRALES - 126 PUESTA A PUNTO DEL SISTEMA DE VECTORES RETROVIRALES HIF - 128 DISCUSIÓN - 133 - RECONOCIMIENTOS - 140 - BIBLIOGRAFÍA - 141 - X ABREVIATURAS ADN: ácido desoxirribonucleico ADNg: ácido desoxirribonucleico genómico AnV: Anexina V ARN: ácido ribonucleico bFGF: basic fibroblast growth factor CE: cuerpo embrioide CICUAL: Comisión Institucional para el Cuidado y Uso de Animales de Laboratorio CK: citoqueratina CM: célula madre CMA: célula madre adulta CME: célula madre embrionaria CMEh: célula madre embrionaria humana CMM: célula madre mesenquimal CMPi: células madre pluripotentes inducidas CMSP: células madre de sangre periférica ConA: concanavalina A DAPI: 4'-6-diamidino-2-fenilindol ddH2O: agua bidestilada Dm: doble mutación DMEM: Dul e o’s Modified Eagle Media DMSO: dimetilsulfóxido Dox: doxiciclina EDTA: ácido etildiaminotetraacético EGF: epidermal growth factor EHS: Engelbreth-Holm-Swarm ER: enzima de restricción FA: fosfatasa alcalina -1- ABREVIATURAS FGF: fibroblast growth factor GP: gag-Pol h: humano HDAC: histone deacetylase (deacetilasa de histonas) HIF: hypoxia inducible factor (factor inducible por hipoxia) IGF: insulin growth factor IL-2: interleukin 2 IP: ioduro de propidio iPSC: induced pluripotent stem cells iROCK: inhibidor de ROCK (Rho-associated protein kinase p160) KSR: knock-out serum replacement L-Glut: L-glutamina MCE: Masa celular externa MCI: macizo celular interno MCS: multiple cloning site (sitio múltiple de clonado) MEF: mouse embryonic fibroblasts (fibroblastos embrionarios de ratón) MEFi: fibroblastos embrionarios de ratón irradiados MOI: multiplicity of infection (multiplicidad de infección) MTA: material transfer agreement NaBu: butirato de sodio NEAA: non-essential aminoacids (aminoácidos no esenciales) NTE: buffer cloruro de sodio, tris, EDTA ODD/ODDD: oxygen dependent (degradation) domain (dominio de degradación dependiente de oxígeno) ORF: open reading frame (marco abierto de lectura) ORS: outer root sheath (vaina externa de la raíz) p: pasaje PBS: phosphate buffered saline (buffer fosfato salino) PBSA: buffer fosfato salino albúmina -2- ABREVIATURAS Pen/Strep: penicilina/estreptomicina SE: sangre entera SFB: suero fetal bovino SNC: suero normal de cabra TA: temperatura ambiente TAD: transactivation domain (dominio de transactivación) TGF-beta: transforming growth factor beta Tm: triple mutación TRF: tetracycline response element UV: ultravioleta VPA: ácido valproico wt: wild type (tipo salvaje) YFP: yellow fluorescent protein -3- INTRODUCCIÓN -4- INTRODUCCIÓN CÉLULAS MADRE Las células madre (CM) se definen como células clonogénicas capaces tanto de auto-renovación como de diferenciación a múltiples linajes [1]. La existencia de una CM, como el origen de la auto-renovación en tejidos que se auto-perpetúan, fue postulada por primera vez por Regaud, basándose en sus estudios sobre la espermatogénesis [2]. Junto con las observaciones de Ferrata sobre las células sanguíneas y su sugerencia de una posible célula troncal hematopoyética también al comienzo de los años 1900 [3], la comunidad científica llegó a la conclusión de que debía existir una célula ancestral con capacidad de auto-renovación. Históricamente, el concepto de CM y auto-renovación tisular estuvieron fuertemente asociados [2, 4]. Tradicionalmente, las CM han sido definidas como células indiferenciadas con varios grados de potencial, una medida del número de divisiones posibles y el rango de fenotipos a los que se puede diferenciar la célula. Al estudiar el desarrollo embrionario se han identificado células que son totipotentes, es decir que una sola célula puede, por sí misma, dar lugar a un organismo completo. Sin embargo la totipotencia es temporal y desaparece luego de las primeras divisiones del oocito fertilizado [2], para luego encontrar células pluripotentes en el Macizo Celular Interno (MCI); éstas pueden dar lugar a todas las células del organismo adulto completo, pero no son capaces de dar lugar al trofoblasto, como se describe más adelante. Existen varios tipos de CM que se han clasificado a lo largo del tiempo. En primera instancia, las CM se pueden dividir en Células Madre Pluripotentes o Células Madre Adultas (CMA). Entre las Pluripotentes encontramos a las Células Madre Embrionarias (CME), del MCI del blastocisto; a las Células Madre Germinales, provenientes de los esbozos gonadales fetales de aproximadamente 2 meses de edad [5]; y a las Células Madre Pluripotentes Inducidas (CMPi), que se obtienen a partir de la desdiferenciación de células somáticas. Entre las CMA, derivadas a través de procesos de aislamiento específicos de la mayoría de los órganos del cuerpo (médula ósea, hígado, páncreas, tejido adiposo, etc.), encontramos a las Células Madre de la Médula Ósea -las cuales comprenden a las Células Madre Hematopoyéticas y las Células Madre -5- INTRODUCCIÓN Mesenquimales (CMM), que fueron las primeras en ser introducidas en la clínica [6]-, las Células Madre de sangre de Cordón Umbilical, las Células Madre Progenitoras Endoteliales, y las Células Madre Tisulares, las cuales son sólo multipotentes, pudiendo dar lugar a distintos tipos celulares del mismo linaje. En este trabajo nos enfocaremos en las CME y las CMPi, en ambos casos de origen humano (h). CÉLULAS MADRE EMBRIONARIAS En mamíferos, luego del proceso de fecundación, el cigoto comienza la segmentación, la cual involucra varias divisiones celulares. Durante este tiempo, el embrión se traslada desde la trompa de Falopio hasta el útero. Una de las innovaciones importantes en la embriogénesis de los mamíferos es la formación del trofoblasto, tejido especializado que forma la interfaz trófica entre el embrión y la madre, durante el período de segmentación. La placenta representa la manifestación última de los tejidos trofoblásticos [7]. Hasta la etapa de 8 células, llamadas blastómeras, las células del embrión están agrupadas en forma poco compacta. Sin embargo, después de la tercera segmentación, los embriones de los mamíferos placentados entran en una fase llamada de compactación, generando que el contacto de las blastómeras entre sí sea máximo y formando un cúmulo compacto de células que se mantienen unidas por medio de uniones estrechas y comunicantes (tight y gap junctions) y pierden su identidad individual, como se observa en la Figura 1. Las células centrales de la mórula constituyen el MCI, y la capa circundante de células forma la Masa Celular Externa (MCE). El MCI origina los tejidos del embrión propiamente dicho y la MCE forma el trofoblasto, que más tarde contribuirá a formar la placenta y los tejidos extraembrionarios. -6- INTRODUCCIÓN Figura 1. Esquemas de las primeras fases de la segmentación en el embrión humano. Los esquemas de los estados de 58 y 107 células representan secciones a través del embrión [7]. Durante la formación del blastocisto, las blastómeras exteriores se asemejan a un epitelio, mientras que agua y sales se acumulan en los espacios entre las blastómeras internas. Este proceso se denomina cavitación y el espacio lleno de líquido se llama blastocele. En esta etapa, el embrión se conoce como blastocisto, como se observa en la Figura 2. Figura 2. Blastocisto humano. (A) Corte de un blastocisto humano de 107 células en el que se observan la masa celular interna y las células trofoblásticas. (B) Representación esquemática de un blastocisto humano. Las células de color azul corresponden a la masa celular interna o embrioblasto, y las de color verde al trofoblasto. (C) Representación esquemática de un blastocisto en el noveno día de desarrollo que muestra a las células trofoblásticas, situadas en el polo embrionario del blastocisto, penetrando en la mucosa uterina [8]. -7- INTRODUCCIÓN Tradicionalmente, las CMEh son aisladas a partir de embriones en etapa de blastocistos, donados mediante consentimiento informado por sus progenitores, los cuales no pueden ser implantados. Para la obtención de las células pluripotentes se utiliza en la mayoría de los casos la técnica de inmunocirugía [9], la cual permite remover selectivamente la capa trofoblástica del blastocisto, dejando un MCI intacto para ser cultivado. El aislamiento del MCI también puede lograrse mecánicamente por remoción del trofoblasto con agujas 27G o con pipetas, bajo lupa estereoscópica. Además, se pueden derivar líneas de CME sin remover el trofoblasto: el blastocisto en su totalidad es colocado sobre Fibroblastos Embrionarios de Ratón mitóticamente inactivados (MEFi, por su sigla en inglés), éste se adhiere y continúa su crecimiento junto con el trofoblasto circundante. Cuando el MCI alcanza el tamaño suficiente, es selectivamente extraído y propagado [10]. El desarrollo de CMEh fue descripto por primera vez en 1998 [11]. En estos trabajos se describió cómo desarrollar y cultivar indefinidamente las células pluripotentes originadas del MCI del blastocisto. En la Figura 3 se muestra una colonia de CMEh indiferenciadas en cultivo. Se puede observar que la colonia crece como una estructura en dos dimensiones con bordes definidos extendiéndose a la periferia, y que las células que la conforman son pequeñas, con una alta relación núcleo : citoplasma. Alrededor de la colonia pueden visualizarse células fibroblásticas, que componen la capa nutricia de MEFi. Figura 3. Colonia de CMEh, sobre MEFi. Línea HUES-5. Campo claro. Aumento 100X. -8- INTRODUCCIÓN Como se describió anteriormente, las CME poseen una capacidad ilimitada para auto-renovarse y proliferar in vitro, expresada por su capacidad de generar cientos de pasajes en cultivo, que no muestran signos de senescencia. Los pasajes o repiques en cultivo se refieren a la transferencia de células, con o sin dilución, de un recipiente de cultivo a otro, a fin de mantenerlas en estado proliferativo, amplificar su número y/o sembrarlas en recipientes adecuados para un dado experimento. E adela te se utiliza á el e o pasa sus conjugaciones para esta actividad. La capacidad proliferativa es prácticamente ilimitada gracias, en parte, a su actividad de telomerasa, en comparación con células normales y líneas humanas inmortales [11]. Es su capacidad de auto-renovarse durante un período prolongado de tiempo la que asegura que las poblaciones de CM se mantengan a lo largo de toda la vida de un organismo, sin extinguirse. Aunque in vivo las CMA deben ser capaces de auto-renovarse para mantener la homeostasis durante toda la vida, poblaciones aisladas de CMA, incluyendo las CM hematopoyéticas no son capaces de perpetuar la auto-renovación a largo plazo in vitro [12]. Esto hace imposible el mantenimiento de éstas poblaciones en cultivo por un período de tiempo prolongado, sin que las células atraviesen una significativa diferenciación, además de limitar la cantidad de expansión que se puede lograr [13]. Éste es un factor decisivo que limita el uso de las CMA, tanto para investigación como para aplicaciones clínicas, en especial cuando se trata de CMA específicas de paciente. Aunque la auto-renovación es la clave de la longevidad de las CM [14], los mecanismos que subyacen este proceso todavía no están del todo claros. Las CME proveen un excelente modelo para estudiar y dilucidar este proceso. Finalmente, su pluripotencia les aporta a las CME la capacidad de diferenciarse hacia todos los linajes celulares, excepto los tejidos extraembrionarios. Posteriormente al primer cultivo de CMEh, surgieron descripciones acerca de su diferenciación a precursores de las tres capas germinales, ectodermo, mesodermo y endodermo, y luego hacia distintos fenotipos celulares adultos. Se postuló que estas células podrían ser utilizadas con eficacia para regenerar tejidos dañados y posteriormente ser utilizadas en tratamientos de distintas patologías. Se cree que podrían tratarse disfunciones cardiovasculares [15-19], enfermedades neurodegenerativas y lesiones neurológicas [20], diabetes [21] y enfermedades de la sangre [22], entre otras patologías crónicas y prevalentes. Además, el cultivo de estas células resultó ser un excelente -9- INTRODUCCIÓN modelo para el análisis de las señales de diferenciación involucradas en el desarrollo embrionario. Por otro lado, las CMEh pueden considerarse una potencial herramienta para el estudio de enfermedades y para ensayos de drogas y toxicología [23, 24]. Algunos de los tejidos derivados de las tres capas germinales que se pueden derivar en cultivo se ejemplifican en la Figura 4 [25] y en la Figura 5, mostrando las distintas y complicadas vías que regulan los compromisos de linaje. Figura 4. Derivación y estrategias de diferenciación de CMEh. Luego de su obtención, las CMEh pueden ser diferenciadas a varios tipos celulares de los distintos linajes embrionarios, a través de técnicas como la formación de cuerpos embrioides, el co-cultivo con células somáticas y el cultivo en andamiajes tridimensionales [26]. - 10 - INTRODUCCIÓN Figura 5. Diferenciación de CME en cultivo. Este modelo ilustra la regulación de la formación de la línea primitiva, la inducción de las capas germinales y la especificación de tejidos, en un modelo murino, según la presencia y concentración de distintos morfógenos y activación de vías de señalización [27]. CÉLULAS MADRE PLURIPOTENTES INDUCIDAS Las CMPi fueron desarrolladas originalmente a partir de células de ratón por S. Yamanaka y colaboradores en 2006 [28]. Mediante la introducción de genes codificantes para factores de transcripción del estado pluripotente, como Oct3/4 (POU5F1), SOX2, KLF4, LIN-28 o C-MYC, en células terminalmente diferenciadas, fueron capaces de inducir un estado de pluripotencia en células somáticas de ratón que se asemejaba mucho al estado y propiedades de las CME [29], las cuales se mencionaron anteriormente. Estas CMPi, al igual que las CME, eran capaces de auto-renovación, poseían una alta tasa de proliferación, mostraban un patrón de marcadores del estado pluripotente similar al de CME –tales como la alta expresión de genes como NANOG, C-MYC, POU5F1, SOX2, LIN28; la alta actividad de fosfatasa alcalina (FA); o la presencia de marcadores de superficie como TRA-1-60 o TRA-1-81- y tenían la plasticidad para dar origen a - 11 - INTRODUCCIÓN células de las tres capas germinales, endodermo, mesodermo y ectodermo. La capacidad pluripotente se ha demostrado completamente, ya que las CMPi de ratón pueden generar ratones a término, mediante la obtención de quimeras por complementación tetraploide de blastocistos [30-34]. Las primeras CMPi humanas se generaron en 2007 a partir de fibroblastos humanos [35-38], en algunos casos incluso sin el uso del oncogén C-MYC [39], lo cual proporciona una forma de obtener células pluripotentes paciente-específico, y en el caso de no usar C-MYC, sin la introducción de un conocido oncogén en las células. Un esquema de la técnica de generación de CMPi se ilustra en la Figura 6 [40]. Figura 6. Generación de Células Madre Pluripotentes inducidas. Esquema de los pasos involucrados en la reprogramación de células somáticas a CMPi [40]. La obtención y el estudio de CMPi pueden ofrecer conocimientos sobre la biología de la reprogramación y el compromiso de linaje, así como proveer células para el modelado de enfermedades in vitro para una amplia variedad de patologías [41-50], pruebas farmacológicas [51-55] y potenciales tratamientos en medicina regenerativa, al igual que con las CME, pero sin las implicancias éticas por la utilización de embriones y evitando complicaciones debido al rechazo inmune por alogenicidad. Además se podrían estudiar in vitro cualidades específicas del paciente, en miras a terapias personalizadas. Estas aplicaciones se ilustran en la Figura 7 [36]. Aun así, la aplicación clínica de la tecnología de CMPi todavía se ve obstaculizada por la formación de teratomas [56-59], los cuales pueden generarse incluso cuando una población relativamente pequeña de células indiferenciadas queda en el implante, sumado al hecho de que el control de su diferenciación es sumamente complejo. - 12 - INTRODUCCIÓN Figura 7. Posibles aplicaciones de las CMPi. Esquemas de las posibles aplicaciones de CMPi en medicina regenerativa y modelos in vitro [36]. Desde el año 2006, han sido publicados innumerables artículos de investigación sobre la generación de CMPi [60-63], especialmente con el objetivo de aumentar la eficiencia de las técnicas de reprogramación, ya sea para obtener un mayor número o en cuanto a la velocidad de generación o la facilidad de la técnica, utilizando una amplia gama de modificaciones al método original, incluyendo el uso de distintas células adultas como punto de partida [60], el cambio de las condiciones de cultivo [64, 65], la introducción de moléculas potenciadoras [6669] y el uso de diferentes sistemas de introducción de genes [70, 71]. La reprogramación de células somáticas se logra por medio de la introducción en la célula de distintos factores de transcripción, típicamente OCT4, SOX2, KLF4 y C-MYC, aunque se han utilizados otros como Lin28. La secuencia codificante para estos factores de transcripción ha sido montada sobre diferentes vectores para ser introducida en las células, incluyendo a plásmidos retrovirales [28, 38], en especial lentivirales [37, 72-76]; adenovirales [77, 78] y episomáticos [79, 80], además de transposones [80]. También se ha logrado la reprogramación mediante la introducción de las proteínas [81] o de ARN mensajeros de dichos factores [82] , además de ciertas moléculas pequeñas que parecen inducir un estado de pluripotencia [83]. En la Figura 8 se muestran los - 13 - INTRODUCCIÓN distintos pasos de la reprogramación en los cuales se pueden introducir distintas variantes en la metodología [84]. Figura 8. Proceso de derivación de CMPi, incluyendo los distintos niveles donde la técnica puede variarse. Esquema de los pasos involucrados en la generación de CMPi, en los cuales puede introducirse variaciones de las técnicas o condiciones [84]. Sin embargo, todavía no se conocen completamente los mecanismos por los cuales los transgenes introducidos logran la desdiferenciación de una célula adulta. Se ha reportado que durante el proceso de reprogramación ocurre remodelado de la cromatina y posterior modificación de la expresión génica [85-88], así como reparación del ADN [89] y ajustes de la regulación del ciclo celular [90-93]. Varias vías celulares parecen estar implicadas; por lo tanto existe la posibilidad de manipular tales vías para modificar la eficiencia de reprogramación. Un factor que puede resultar fundamental en el proceso de generación de CMPi es la concentración de oxígeno en la cual se lleva a cabo la reprogramación, como se ha demostrado de forma preliminar en células de ratón [65] y humanas [64, 94]. - 14 - INTRODUCCIÓN HIPOXIA Y CÉLULAS MADRE La relevancia del oxígeno es una inducción lógica del hecho que el embrión naturalmente está inmerso en un nicho hipóxico [95, 96]. Se ha observado previamente que condiciones hipóxicas tienen un efecto sobre el cultivo de CME [97, 98], favoreciendo principalmente la pluripotencia in vitro de CME humanas (CMEh) [99-103] y contribuyendo a la preservación de un estado indiferenciado [104-107]. Sin embargo, se han publicado datos contradictorios sobre este tema, ya que también ha sido reportado que una baja tensión de O2 no es capaz de proporcionar ventajas significativas en el mantenimiento del estado indiferenciado sobre el estándar de cultivo en O2 atmosférico [104]. En particular, esto se ha visto con respecto a los factores HIF (Hypoxia Inducible Factors), ya que éstos juegan un papel central en la transducción de señales hipóxicas en la célula. Los factores HIF actúan como factores de transcripción y son dímeros compuestos por una subunidad de HIF-α u a su u idad de HIF-β o ARNT . E iste dife e tes variantes de HIF-α HIF-β, segú las o di io es tejidos, o o se e io a ás adela te, pero en todos los casos la subunidad HIF-β se e p esa constitutivamente y es estable en toda condición de oxígeno. Por el contrario, las subunidades HIF-α so eguladas í i a e te a nivel transcripcional, pero sufren degradación constitutiva por la vía del proteosoma, en condiciones de normoxia. Como se ilustra en la Figura 9, en dicha condición dos prolinas altamente conservadas del dominio de HIF-α depe die te de o íge o llamado ODD u ODDD (Oxygen Dependent Degradation Domain) son hidroxiladas por prolil-hidroxilasas dependientes de oxígeno; esta marca genera la degradación de HIF-α. Al disminuir los niveles de oxígeno las prolil-hidroxilasas son inhibidas y no hidroxilan las prolinas del ODDD, lo cual genera la estabilización de los HIF-α, su acumulación, y la posterior dimerización con la subunidad HIF-β, formándose el factor de transcripción activo que podrá translocarse a núcleo y cumplir su función sobre genes blanco. Éstos varían según de cuál HIF-α se t ate. - 15 - INTRODUCCIÓN Figura 9. Estructura de las subunidades de los factores HIF. Esquema de los factores HIF y sus dominios [96]. Se ha demostrado que estos factores tienen un efecto sobre las vías de señalización celular del estado pluripotente [108], y que los distintos factores HIF tienen un papel diferencial en su función en células madre [109]. Especialmente, se ha demostrado que HIF- α p o ue e la proliferación celular mediante la activación de la actividad de C-MYC [110, 111], un factor clave del estado pluripotente, mientras que HIF- α i hi e la a ti idad de este fa to [110, 112, 113] a través de la detención del ciclo celular [114]. Sin embargo estos dos factores inducibles por hipoxia parecen ser importantes en forma secuencial durante el proceso de reprogramación [115]. También se ha demostrado que es principalmente HIF- α o HIF- α, el ue se u e al promotor de POU5F1 e induce su expresión y actividad transcripcional [112]. Finalmente, HIFα i te a túa o Not h manteniendo un estado indiferenciado [116] y HIF- α tie e u ol directo en la regulación positiva de Sox2 [117], que controla la pluripotencia por modulación directa de los niveles de POU5F1, como se ha demostrado en CME de ratón [87, 118]. En general, ambos factores HIF tienen un papel en la transducción de señales de hipoxia en células madre, operando a diferentes niveles, distintos momentos y participando en diferentes vías. En síntesis, investigaciones previas han revelado en varias instancias los efectos de la hipoxia sobre - 16 - INTRODUCCIÓN la función de las células madre, el mantenimiento de la pluripotencia, la diferenciación y la reprogramación. La habilidad de comprender los mecanismos que regulan la reprogramación y las condiciones en las que se desarrolla, podrán asistir en la generación de CMPi más rápida y eficientemente, además de influir en la calidad y seguridad de las líneas resultantes. Si pudiéramos comprender a qué nivel la concentración de oxígeno regula y modifica el proceso, aportaremos valiosa información para la tecnología y su futura aplicación en medicina y farmacología. - 17 - HIPÓTESIS Y OBJETIVOS - 18 - HIPÓTESIS Y OBJETIVOS Considerando los antecedentes previamente mencionados, la hipótesis general de este trabajo es, basándose en la evidencia que rodea el papel de la hipoxia y los factores HIF sobre el estado pluripotente, que una concentración baja de oxígeno durante la reprogramación de células humanas a Células Madre Pluripotentes Inducidas no sólo podría modificar la eficiencia del proceso de reprogramación, sino también las propiedades de las células pluripotentes generadas. Con el fin de poner a prueba dicha hipótesis hemos planteado los siguientes objetivos: OBJETIVO GENERAL: El objetivo general del presente trabajo fue el desarrollo de CMPi humanas bajo normoxia e hipoxia y el posterior estudio de las características de las CMPi generadas en ambas condiciones, a fin de comprender los mecanismos moleculares involucrados en el proceso de desdiferenciación y en la posterior diferenciación, según el nivel de oxígeno; así como la influencia de los niveles de oxígeno durante la reprogramación, sobre las propiedades de las CMPi resultantes. OBJETIVOS PARTICULARES: Obtención de diferentes tipos de cultivos primarios de células somáticas para reprogramar. Generación de un protocolo confiable para la generación de CMPi humanas. Reprogramación de fibroblastos humanos a CMPi en condiciones de normoxia e hipoxia. Estudio de la eficiencia del proceso, en cuanto al número de colonias obtenidas, las características morfológicas, el patrón de expresión génica de las células resultantes, su - 19 - HIPÓTESIS Y OBJETIVOS pluripotencia y su comportamiento en cultivo, en cuanto a viabilidad, proliferación y apoptosis. Generación de vectores que permitan sobreexpresar los factores HIF en células a reprogramar. - 20 - MATERIALES Y MÉTODOS - 21 - MATERIALES Y MÉTODOS Aislamiento de Fibroblastos Embrionarios de Ratón Para los experimentos realizados en Argentina, ratones hembra preñadas (día 12 post-coito) de la cepa CF1 fueron obtenidas del Bioterio de la Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires. En el día 12.5 post-coito, se anestesiaron y se sacrificaron por dislocación cervical. Se procedió a limpiar el abdomen con etanol 70%, abrir la cavidad peritoneal y remover el útero, el cual contiene los embriones. Éstos se colocaron en una placa de Petri estéril y se lavaron 3 veces con buffer fosfato salino 1X (PBS: 8 mg NaCl, 200 mg KCl, 200 mg KH2PO4 y 1,14 mg Na2HPO4, por litro de solución, pH 7,4), siempre estéril, para remover toda la sangre. Posteriormente, se separaron los embriones de las placentas y de la membrana fetal y se colocaron en otra placa de Petri estéril, lavando nuevamente 3 veces con PBS. Bajo lupa estereoscópica se decapitaron los embriones y extirparon sus vísceras, descartando luego este material. El remanente se colocó en una nueva placa de Petri estéril y se lavó 2 veces con PBS. Se procesaron los embriones con bisturí y luego, agregando Tripsina/EDTA (Tripsina 0,25%, EDTA 0,53 mM, en PBS), se continuó el tratamiento de los mismos hasta disgregar completamente el tejido. Se agregó más Tripsina/EDTA a la placa y se incubaron los preparados durante 30 minutos en incubadora de cultivo celular (a 37° C con atmósfera humidificada y 5% de CO2, a menos que se indique otra condición). Pasado el tiempo de incubación, se colocaron los embriones procesados en un tubo cónico y se agregó Dul e o’s Modified Eagle’s Mediu (DMEM) (Gibco) suplementado con 10% de suero fetal bovino (SFB) (Gibco), aminoácidos no esenciales (NEAA) concentración final 0,1 mM (Gibco), Penicilina/Estreptomicina (Pen/Strep) (Penicilina concentración final 100 U/ml, Estreptomicina concentración final μg/ l (Gibco), L-Glutamina (L-Glut) concentración final 2 mM (Gibco) (DMEM SFB 10%), mezclando con pipeta de plástico de 5 ml. Se dejó decantar la preparación, recuperando luego el sobrenadante, que contiene la suspensión de fibroblastos embrionarios de ratón (MEF, por su sigla en inglés). El volumen obtenido de sobrenadante se dividió en X botellas de cultivo T75 (X = Número de Embriones Obtenidos/3), completando el volumen de las mismas con DMEM SFB 10%. Esta siembra de células se considera pasaje cero. Se mantuvieron las botellas T75 durante 2 ó 3 días - 22 - MATERIALES Y MÉTODOS en incubadora de cultivo celular. Cuando la monocapa de fibroblastos alcanzó un 100% de confluencia se procedió a su amplificación o a su congelamiento. Cultivo y Amplificación de Fibroblastos Embrionarios de Ratón Los MEF se cultivaron en medio DMEM 10% SFB. Una vez que la monocapa celular llega a 100% de confluencia, se debe realizar el pasaje de las células, dividiéndolas para su amplificación. Para una botella T75: se aspiró el medio de cultivo. Se realizó un lavado de la monocapa con 5 ml PBS, para eliminar la mayor parte de los detritos celulares y el remanente de suero. Se aspiró el PBS y se agregó 1 ml de Tripsina/EDTA, moviendo la botella para dispersar la enzima, y utilizando una pipeta de plástico de 5 ml para lavar las paredes, levantándose así los MEF. Una vez que se observaron al microscopio óptico los MEF completamente despegados del sustrato, se agregó 3 ml de medio con SFB para inactivar la tripsina. Luego se levantó la suspensión celular con pipeta de plástico y se transfirió a un tubo cónico de 50 ml. Se repitió este paso, lavando las paredes de la botella con 3 ml adicionales de medio y luego se transfirió el mismo al tubo cónico en uso. La suspensión de fibroblastos se centrifugó durante 5 minutos a 300 xg. Luego se aspiró el sobrenadante y se resuspendió el pellet en 6 ml de medio. Se sembraron 2 ml de la suspensión de MEF, en cada una de las 3 botellas T75, y se llevó a un volumen final de 10 ml con medio. Se mantuvieron los cultivos celulares 2 ó 3 días en incubadora de cultivo celular. Al llegar la monocapa a un 100% de confluencia, se debió realizar un nuevo pasaje de los MEF, hasta el pasaje 3 como máximo, dado que a partir del mismo se induce el proceso biológico de senescencia replicativa. Inactivación de MEF mediante Exposición a Radiación γ Los MEF irradiados se utilizan como capa nutricia, clásicamente llamada feeder layer por su denominación en inglés, dado que los factores de crecimiento secretados por ellos son requeridos para el cultivo, propagación y mantenimiento de la naturaleza pluripotente de las CM, y además aportan un soporte. Luego de su amplificación, y en el pasaje 3, los MEF se irradiaron según el siguiente protocolo: para una botella T75: se aspiró el medio de cultivo de la botella. Se lavaron las células con 2-3 ml de PBS. Se agregó 1 ml de Tripsina/EDTA y se incubó a - 23 - MATERIALES Y MÉTODOS 37° C entre 3 y 10 minutos, hasta que las células se despegaron de la placa. Se agregaron 9 ml de medio DMEM SFB 10%, se levantó la suspensión celular con pipeta de plástico y se colocó en un tubo cónico. La suspensión celular se homogeneizó para disgregar las células. Posteriormente las células fueron irradiadas (40-80 Gy, servicio provisto por CEBIRSA S.A., Buenos Aires, Argentina). La solución con MEF mitóticamente inactivados se centrifugó a 300 xg durante 5 minutos, descartándose luego el sobrenadante. El pellet celular se resuspendió en DMEM SFB 10%, en un volumen final tal que permita sembrar 1 x 106 células/placa de cultivo adherente de 10 cm de diámetro, previamente tratadas con una solución de gelatina bovina, como se indica más adelante. Se colocaron las placas sembradas sobre un agitador mecánico durante 5 minutos para lograr una distribución uniforme de las células y luego se colocaron en incubadora de cultivo celular. Los MEFi pueden utilizarse como capa nutricia para CM hasta 7 días después de haber sido irradiadas. Para los experimentos realizados en Estados Unidos, se utilizaron MEF irradiados comerciales (Globalstem, gamma irradiated MEF, 2 millones/vial, #GSC-6201G). Éstos se sembraron a razón de 1 x 106 células/placa de cultivo adherente de 10 cm de diámetro, previamente tratada con una solución de gelatina bovina. Se colocaron las placas sembradas sobre un agitador mecánico durante 5 minutos para lograr una distribución uniforme de las células y luego se colocaron en incubadora de cultivo celular. Estos MEFi pueden utilizarse como capa nutricia para CM hasta 7 días después de haber sido plaqueados. Tratamiento de Placas con Gelatina Bovina La superficie de las placas de cultivo debe recubrirse con una película de gelatina bovina 0,1% en PBS, para generar una matriz sobre la cual se adhieran los MEFi. La solución de gelatina bovina 0,1% en PBS se preparó a partir de una solución madre estéril de Gelatina Bovina 2% (Sigma), en PBS estéril. Luego la solución fue filtrada a través de un filtro (Corning) de diámetro de poro de 0,22 μm. Se colocó la solución de Gelatina Bovina 0,1%, moviendo la placa en varias direcciones para distribuir la solución, cubriendo toda la superficie. Se colocó la placa en incubadora de cultivo celular, durante al menos 1 hora. Se aspiró la solución remanente. - 24 - MATERIALES Y MÉTODOS Tratamiento de Placas con Matrigel En los experimentos donde debió evitarse la presencia de MEFi como capa nutricia, el cultivo de CM se realizó sobre una matriz de Matrigel (BD Biosciences). Para ello, la superficie de las placas de cultivo debió recubrirse para generar una película de Matrigel, una preparación de membrana basal solubilizada extraída del sarcoma de ratón Engelbreth-Holm-Swarm (EHS), un tumor rico en proteínas de matriz extracelular. Su mayor componente es la laminina, seguida por colágeno tipo IV, proteoglicanos heparán sulfato y entactina/nidógeno. Además, el Matrigel también contiene Transforming Growth Factor β (TGF-β), Epidermal Growth Factor (EGF), Insulin Growth Factor (IGF), Fibroblast Growth Factor (FGF) y otros factores naturalmente encontrados en el tumor EHS. El Matrigel debe descongelarse lentamente, en hielo y a 4° C, durante aproximadamente 16 horas. Para su almacenamiento se realizó una dilución 1:20 en medio DMEM/F12 y se conservó en alícuotas a -20° C. Para tratar las placas de cultivo se lo diluyó al medio (1:40). Luego de cubrir la superficie de las placas con la solución de Matrigel, éstas se incubaron a temperatura ambiente (TA) durante al menos una hora e, inmediatamente antes de sembrar las células, se aspiró la solución remanente. Cultivo de Células HEK-293T Células HEK-293T [119] utilizadas para la generación de vectores lentivirales se mantuvieron en placas de cultivo celular como monocapas hasta un 70-80% de confluencia, en DMEM 20%. Estos cultivos fueron expandidos por disociación con Tripsina/EDTA y replaqueados en una proporción 1:3. Las células se mantuvieron en incubadora de cultivo celular. Obtención, Cultivo y Expansión de Fibroblastos Humanos El cultivo primario de fibroblastos HFF-FM se estableció particularmente para y durante este estudio. Durante una cirugía de circuncisión rutinaria, se obtuvo una muestra de prepucio humano con previo consentimiento informado, y con un protocolo previamente aprobado por el Comité de Ética de FLENI. Después de la obtención de la muestra, el tejido fue cortado en secciones 0,5 cm2 con bisturí y pinzas. Las secciones de piel se colocaron en una botella de - 25 - MATERIALES Y MÉTODOS cultivo celular T75 tratada con SFB, como se describe en [120] y fueron cultivadas en medio DMEM SFB 20%. En todos los casos se realizó incubación en incubadoras de cultivo celular y el medio de cultivo fue cambiado cada 48 horas. Luego de que los fibroblastos crecieran lo suficiente para cubrir la superficie de la placa de cultivo, irradiando desde las secciones de tejido, éstas fueron transferidas a una nueva botella tratada con SFB y la monocapa restante de fibroblastos fue pasada con Tripsina/EDTA para expansión y congelamiento de células del nuevo cultivo celular primario establecido, llamado HFF-FM. El Laboratorio de Biología del Desarrollo Celular de FLENI contaba con el cultivo primario de fibroblastos HF. El laboratorio Cellular Reprogramming Laboratory de Michigan State University contaba con los cultivos primarios de fibroblastos JC-G, JC-S, HDNF y IN10. Todas establecidas con anterioridad. Todos los cultivos de fibroblastos utilizados en este estudio se mantuvieron en placas de cultivo celular como monocapas, en medio DMEM SFB 20%. Los cultivos fueron expandidos por disociación con Tripsina/EDTA y replaqueados en una proporción 1:3 en nuevas placas. Las células se mantuvieron en incubadora de cultivo celular. Obtención y Cultivo de Queratinocitos Humanos Para la obtención y cultivo de queratinocitos, se procedió basándose en antecedentes de cultivo descriptos en la bibliografía, como se detalla en [121]. Se utilizó un sistema de reactivos libre de componentes animales. Obtención de Queratinocitos de Epidermis (a partir de cuero cabelludo) Con previo consentimiento informado, y con un protocolo previamente aprobado por el Comité de Ética de FLENI, una muestra de cuero cabelludo de un paciente de 70 años de edad, fue disgregada en secciones con bisturíes, agitando con vórtex y pipeteando repetidamente con tip de pipeta automática de 1000 µl. Se separaron la sección epidérmica y dérmica con pelo de la sección subcutánea con tejido adiposo. Luego se trató la epidermis y dermis con Recombinant Trypsin/EDTA Solution (Invitrogen) durante 1 hora en incubadora de cultivo celular, tras lo cual se realizó una centrifugación a 300 xg y se plaquearon los remanentes de tejido y las células en suspensión de la sección de "epidermis y dermis con pelo" en placas de cultivo con tratamiento - 26 - MATERIALES Y MÉTODOS con matriz para queratinocitos Coating Matrix Kit Protein (Invitrogen) y medio de cultivo para queratinocitos EpiLife (Invitrogen) con suplemento para queratinocitos S7 (Invitrogen), constituyendo el pasaje 0 (p0). Las células comenzaron a proliferar e la pla a de epide is de is o pelo luego de 5 días de cultivo, la monocapa fue transferida con tratamiento con Recombinant Trypsin/EDTA Solution en las condiciones habituales para Tripsina/EDTA, a una relación 1:3 a placas tratadas con Coating Matrix Kit Protein, constituyendo el pasaje 1 (p1). Parte de las células del p0 fue congelada con el reactivo para congelamiento de queratinocitos Synth-a-freeze (Invitrogen). Las células del p1 fueron transferidas en una relación 1:1 nuevamente a placas tratadas, por incubación con Recombinant Trypsin/EDTA Solution en las condiciones habituales para Tripsina/EDTA, constituyendo el pasaje 2. Parte de las células p1 fue utilizada para realizar una inmunofluorescencia para citoqueratinas (total –CK total–, citoqueratina 7 –C7– y citoqueratina 20 –C20–). Las células del pasaje 2 se adhirieron, pero durante los siguientes días se fueron despegando de la matriz, hasta no encontrarse más células en adherencia. Obtención de Queratinocitos de Folículos Pilosos Con previo consentimiento informado, y con un protocolo previamente aprobado por el Comité de Ética de FLENI, se utilizaron pinzas para extraer pelo de cuero cabelludo y pubis de una donante sana de sexo femenino de 28 años de edad, y de un donante sano de sexo masculino de 33 años de edad, y se los colocó inmediatamente en una placa con PBS y Pen/Strep. Luego de realizar la extracción del pelo se comprobó la presencia de una Vaina Externa de la Raíz (ORS, por su sigla en inglés, Outer Root Sheath). Mientras el pelo se encontraba sumergido en PBS, se cortó su parte externa, dejando sólo el bulbo piloso con el ORS. Opcionalmente, en algunos casos, el pelo fue tratado durante 1 hora con Recombinant Trypsin/EDTA Solution, en las condiciones habituales para Tripsina/EDTA. A continuación se colocaron los pelos en placas de cultivo tratadas con Matrigel, y se los cultivó en medio DMEM/F12 condicionado por MEFi. Se hizo un esfuerzo por lograr que los folículos pilosos se adhirieran a la placa, ayudando a hundirlos con tips de pipeta automática, en caso de flotación. Durante los 3 días siguientes se - 27 - MATERIALES Y MÉTODOS agregó medio de cultivo fresco, sin remover el medio ya presente. Al día 4 se cambió al sistema de medio para queratinocitos EpiLife. A pesar de lo reportado, no se observó crecimiento celular a partir de los folículos pilosos. Obtención y Cultivo de Células Mononucleares de Sangre Periférica Para el aislamiento de células mononucleares de sangre periférica (CMSP), con previo consentimiento informado, y con un protocolo previamente aprobado por el Comité de Ética de FLENI, se realizó la extracción de 10 ml de sangre de vena periférica de una donante sana de sexo femenino de 30 años de edad, y de un donante sano de sexo masculino de 40 años de edad. Se mezcló la sangre entera (SE) con PBS libre de Ca2+, relación 1:1, y se sembró la mezcla SE-PBS cuidadosamente sobre Ficoll (Histopaque-1077, Sigma Aldrich) relación 1,5:1 SangrePBS:Ficoll. A continuación se realizó una centrifugación a 300 xg a 22ºC por 30 minutos, sin freno en desaceleración. Esto resulta en una clara separación entre los diferentes componentes de la sangre -de abajo hacia arriba-: los glóbulos rojos, luego el Ficoll, una interfase consistente de CMSP y un sobrenadante de plaquetas y plasma. La interfase de CMSP, con el aspecto de una nube blanca, se colectó usando una pipeta Pasteur estéril, con cuidado de no levantar el Ficoll subyacente. Las CMSP se resuspendieron en una solución buffer de PBS (sin Ca2+ ni Mg2+) + 2nM EDTA + 0.5% SFB. Se centrifugó a 300 xg por 10 minutos, se descartó el sobrenadante, y las CMSP se resuspendieron nuevamente en la solución buffer de PBS. Se centrifugó a 230 xg por 10 minutos, se descartó el sobrenadante (x2), y finalmente se resuspendieron las células en medio de cultivo RPMI SFB 15% + Concanavalina A (ConA) 10 µg/ml, y se colocaron en una placa de cultivo. La ConA es una lectina que actúa como mitógeno sobre linfocitos T, activándolos y consecuentemente estimulando su proliferación, lo cual permite la transducción por las partículas virales de reprogramación y la expresión de los transgenes. Los linfocitos permanecen en suspensión y los monocitos/macrófagos pueden adherirse. Fueron los linfocitos el tipo celular blanco elegido para reprogramar en este caso. En algunos casos las células fueron tratadas con 300 IU/ml de Interleuquina 2 (IL-2) humana recombinante (R&D Systems) y anticuerpos anti-CD3 solubles 10 ng/ml (R&D Systems, MAB100), antes de intentar la reprogramación, de acuerdo a [122], a fin de activar los clones y generar una alta tasa de - 28 - MATERIALES Y MÉTODOS duplicación celular. En otros casos, durante el protocolo de reprogramación, se realizaron selecciones de poblaciones celulares, como se detalla en la sección Protocolo de Generación de CMPi a partir de CMSP. Normoxia e Hipoxia Los cultivos normóxicos fueron realizados a niveles atmosféricos de O2. Para todas las instancias de hipoxia, las células fueron cultivadas en una incubadora de cultivo celular de hipoxia, con 5% de O2 y 5% de CO2, en ambiente humidificado y 37° C. Congelación de Células Luego de levantar la monocapa celular con las enzimas correspondientes, Tripsina/EDTA para MEF, HFF-FM y HEK-293T, y Colagenasa IV o Dispasa para CM, se centrifugó la suspensión celular a 300 xg durante 5 minutos y se descartó el sobrenadante. Luego se resuspendió la suspensión celular en un volumen final de medio de congelación (DMEM SFB 20% [DMEM suplementado con NEAA 1%, Pen/Strep 1%, L-Glut 2 mM, SFB 20%], dimetilsulfóxido (DMSO) (Sigma) 10%) igual al volumen total a congelar, aproximadamente 3-4 x 106 células por criotubo. El medio de congelación fue agregado a las células de forma lenta y mezclando suavemente. Se alicuotó la suspensión celular a razón de 1-1,8 ml por criotubo, rápidamente se los guardó en un contenedor de isopropanol a -80° C por 24 horas, permitiendo el descenso inicial de la temperatura a razón de 1° C/minuto, y finalmente se los almacenó a -140° C en un tanque conteniendo nitrógeno líquido. Condicionamiento de Medio Para el condicionamiento de medio a utilizar durante el cultivo de CM sobre Matrigel, 25 ml de DMEM/F12 suplementado con NEAA 1%, Pen/Strep 1%, L-Glut 2 mM, Knock-Out Serum Replacement (KSR) (Gibco) 5% y 2 ng/ml bFGF fueron incubados durante 24 horas en placas de cultivo de 100 mm de diámetro conteniendo una monocapa conformada por 3 x 10 6 MEFi. El medio condicionado fue luego congelado a -20º C. Luego del descongelamiento para su uso, el - 29 - MATERIALES Y MÉTODOS medio condicionado (incompleto) fue completado con una alícuota fresca de KSR 5% y bFGF 2 ng/ml, para alcanzar una concentración final de KSR 10% y bFGF 8 ng/ml. Producción de Vectores Virales El vector lentiviral OKSIM [123] fue generosamente proporcionado por el Dr. Steve Suhr y el Dr. José Cibelli del Cellular Reprogramming Laboratory, Departamento de Animal Science, de la Michigan State University, Michigan, EE.UU. La producción del vector fue: 1) Encargada como servicio comercial a la University of Michigan, sección Vector Core, Michigan. EE.UU. El vector fue generado por transfección con el plásmido OKSIM y el plásmido helper VSV-G y luego concentrado por ultracentrifugación. El concentrado fue luego almacenado en nitrógeno líquido a -140° C y utilizado dentro de 12 meses. Se evaluó la eficiencia de transducción del concentrado lentiviral en la línea de fibroblastos JC-G, defi ié dola o o: Nú e o de pa tí ulas le ti i ales/μl = Número de núcleos Oct4+ DAPI+ / μl de concentrado utilizado para el ensayo. 2) Generada por co-transfección con el plásmido OKSIM y los plásmidos helper (psPAX y VSV-G), introduciendo el sistema de tres plásmidos por co-precipitación con fosfato de calcio en células HEK-293T, como se describió anteriormente [123]. Brevemente, se combinó el ADN plasmídico en una proporción 2,5:1,5:1 OKSIM:psPAX:VSV-G, se agregó CaCl2 2M y buffer HBS 2X (NaCl 274 mM, KCl 10 mM, Na2HPO4 7H2O 1.4 mM, Glucosa 15 mM, Hepes 42 mM) en una relación 1:1 en la mezcla final, se mezcló y se dejó reposar durante 5-10 minutos. A continuación se agregó la mezcla a las células HEK-293T gota a gota y se incubó en incubadora de cultivo celular durante 8 horas, para luego cambiar el medio DMEM SFB 20% por medio fresco. Se recolectó el sobrenadante con vector lentiviral a las 48 y 72 horas post-transfección, se filtró a través de un filtro de poro de 0,45 µm de baja adhesión (low binding) y se concentró en una ultracentrífuga o con polietilenglicol 6000 (ver apartado Concentración de partículas lentivirales por precipitación con PEG). El concentrado fue alicuotado, almacenado a -80° C y utilizado dentro de 12 meses. Se evaluó la eficiencia de transducción del concentrado lentiviral en la línea de fibroblastos JC-G, defi ié dola o o: Nú e o de pa tí ulas le ti i ales/μl = Nú e o de ú leos Oct4+ DAPI+ / μl de o e t ado utilizado pa a el e sa o. - 30 - MATERIALES Y MÉTODOS Para la producción del vector lentiviral STEMCCA, el cassette lentiviral EF1a-hSTEMCCA-loxP (STEMCCA) o el cassette lentiviral EF1-hSTEMCCA-RedLight-loxp (STEMCCA RedLight, el cual no tiene el gen C-MYC, pero en su lugar tiene la proteína fluorescente roja mCherry) y los plásmidos helper (Tat, Rev, Gag/Pol y VSV-G) fueron generosamente proporcionados por el Dr. Gustavo Mostoslavsky y los vectores lentivirales fueron producidos como se describió anteriormente [73], con pequeñas modificaciones. Brevemente se introdujo el sistema de cinco plásmidos por transfección en células HEK-293T, usando el reactivo de transfección FuGene6 (Roche), según las recomendaciones del fabricante. La eficiencia óptima de transfección se obtuvo utilizando una relación de FuGene6:ADN de 6:1. Se recolectaron los sobrenadantes con vector lentiviral a las 48, 64 y 80 horas post-transfección, se filtraron a través de un filtro de poro de 0,45 µm de baja adhesión y se concentró en una columna Amicon Ultra 15 100.000 NMWL (EMD Millipore), según las instrucciones del fabricante. El concentrado fue alicuotado, almacenado en nitrógeno líquido a -140° C y utilizado dentro de 12 meses. Se evaluó la eficiencia de transducción del concentrado lentiviral en la línea de fibroblastos HFF-FM, definiéndola como: Nú e o de pa tí ulas le ti i ales/μl = Número de núcleos Oct4+ DAPI+ / μl de concentrado utilizado para el ensayo. Concentración de Partículas Lentivirales por Precipitación con PEG Al sobrenadante viral obtenido previamente se le agregó NaCl 5M, esterilizado con filtro de 0,22 µm, hasta una concentración final de NaCl 0,4M, revolviendo y manteniendo el sistema en hielo a una temperatura aproximada de 4° C. A continuación se agregó Polietilenglicol (PEG) 6000, esterilizado con filtro de 0,22 µm, hasta una concentración final de 8,5% (p/p), y se continuó revolviendo durante 1 hora a 4° C. Luego se colectó el precipitado por centrifugación a 7000 xg, durante 10 minutos a 4° C. Se descartó el sobrenadante y se disolvió el pellet obtenido en 1% del volumen original en buffer NTE (NaCl 100 mM, TrisCl, pH 7.4 10 mM, EDTA 1 mM) esterilizado con filtro de 0,22 µm. El concentrado fue alicuotado, almacenado en nitrógeno líquido a -140° C y utilizado dentro de 12 meses. Generación de Células Madre Pluripotentes Inducidas a partir de Fibroblastos Todas las líneas de CMPi se establecieron particularmente para y durante este estudio. - 31 - MATERIALES Y MÉTODOS Para la generación de CMPi con el vector lentiviral OKSIM, fibroblastos humanos JC-G, JC-S, HDNF (cultivos primarios residentes del laboratorio Cellular Reprogramming Laboratory) de bajo pasaje fueron plaqueados en placas multipocillo de 6 pocillos recubiertas con gelatina bovina 0.1%, a una concentración de 1,5 x 105-2,5 x 105 células por pocillo. Al día siguiente (día 0), o en su defecto en el día 1, dependiendo de la confluencia de la monocapa de fibroblastos, el medio de cultivo fue reemplazado con una nueva alícuota de 1 ml DMEM SFB 20% por pocillo, y se realizó la transducción viral a una Multiplicidad de Infección (MOI) de 1 (un tercio del volumen de 1 vial de concentrado viral producido por el Vector Core de la University of Michigan), con una concentración final de Polybrene (Sigma) de 10 µg/ml. El control negativo de transducción (simulacro) se realizó agregando el mismo volumen utilizado de concentrado viral, de medio celular. A las 24 horas post-transducción (día 1) las células fueron transferidas sobre MEFi GlobalStem a una relación 1:3, usando Tripsina/EDTA y plaqueando finalmente en medio DMEM 20% SFB. Al día siguiente (día 2), el medio fue reemplazado por DMEM/F12 suplementado con KSR 10%, además de los suplementos mencionados, β-mercaptoetanol 0,1 mM y 8 bFGF ng/ml (Protocolo 1). Alternativamente, en vez de tripsinizar y pasar las células el día 1, el cultivo fue mantenido en DMEM 20% SFB durante 7 días, y luego fue tripsinizado, retomando el protocolo a partir de la transferencia sobre MEFi a una relación 1:3 (Protocolo 2). Un esquema de los protocolos se muestra en la Figura 10. En algunos casos y cuando se lo indique particularmente se añadió Butirato de Sodio (NaBu), un remodelador de la cromatina [124-128], (Sigma-Aldrich) al medio de cultivo a partir del día 3 y hasta el día 14 incluido, a una concentración final de 0,5 mM - 32 - MATERIALES Y MÉTODOS Figura 10. Esquema de los protocolos de generación de CMPi con el vector OKSIM. Colonias de CMPi fueron visibles entre los días 7 y 10, como agregados celulares, y fueron manualmente seleccionadas con la ayuda de agujas 21G y tips de pipeta automática de 1000 µl a los 14 – 21 días post-transducción, basándose en el tamaño de la colonia, tasa de proliferación y morfología (nucléolos prominentes, material perinuclear brillante al contraste de fases, citoplasmas pequeños) y colocadas en una nueva capa nutricia de MEFi en placa multipocillo de 12 pocillos. Cada colonia fue plaqueada en un pocillo individual. Después de 1-3 más pasajes manuales, las colonias se amplificaron como se describió anteriormente para CM, a fin de establecer una línea celular. En todos los casos, el medio correspondiente fue reemplazado por medio fresco diariamente. Para la generación de CMPi con el vector lentiviral STEMCCA o STEMCCA RedLight, fibroblastos humanos HFF-FM de pasaje bajo fueron plaqueados en placas multipocillo de 6 pocillos recubiertas con gelatina bovina 0.1%, a una concentración de 1 x 105 células por pocillo. Al día siguiente (día 0), el medio de cultivo fue reemplazado con una nueva alícuota de 1,5 ml DMEM SFB 20% por pocillo, y se realizó la transducción a una MOI de 1, con una concentración final de Polybrene de 10 µg/ml y finalizando el proceso centrifugando la placa durante 55 minutos, a 700 xg, a 32° C (spinfection). El control negativo de transducción se realizó agregando el mismo volumen utilizado de concentrado pero de medio de cultivo. En este punto los cultivos celulares - 33 - MATERIALES Y MÉTODOS se colocaron en normoxia o hipoxia. A las 24 horas post-transducción (día 1) las células fueron transferidas sobre MEFi a una relación 1:3, usando Tripsina/EDTA y plaqueando finalmente en medio DMEM 20% SFB. El día 2, el medio fue reemplazado con DMEM/F12 suplementado con KSR 20%, además de los suplementos mencionados, β-mercaptoetanol 0,1 mM y 8 bFGF ng/ml. En algunos experimentos se añadió NaBu o Ácido Valproico (VPA), otro remodelador de la cromatina [69], al medio de cultivo a partir del día 3 y hasta el día 14 inclusive o 21 inclusive, respectivamente, a una concentración final de NaBu 0,5 mM, o VPA 0,75 mM. El protocolo final para la generación de CMPi, el cual fue utilizado para el establecimiento de las líneas de referencia en este estudio (iPS6.1, iPSC-FN2.1 e iPSC-FH2.1) incluye el agregado de NaBu 0,5M entre los días 3-14. Desde el día 14 en adelante el medio de cultivo fue reemplazado por DMEM/F12 suplementado con KSR 10%. Colonias de CMPi fueron visibles entre los días 7 y 10, como agregados celulares, y fueron manualmente seleccionadas con la ayuda de agujas 21G y tips de pipeta automática de 1000 µl a los 14 – 21 días post-transducción, basándose en el tamaño de la colonia, tasa de proliferación y morfología y colocadas en una nueva capa nutricia de MEFi en placa multipocillo de 12 pocillos, con la adición de inhibidor de la proteína Rhoassociated protein kinase p160 –ROCK- Y-27632 (inhibidor de ROCK) (R&D Systems) [129, 130] a una concentración final de 10 µM. Cada colonia fue plaqueada en un pocillo individual. Después de 1-3 más pasajes manuales, las colonias se amplificaron como se describió anteriormente para CM, a fin de establecer una línea celular. En todos los casos, el medio correspondiente fue reemplazado por medio fresco diariamente. En adelante, se hace referencia a líneas de CMPi esta le idas a largo plazo , indicando que los experimentos fueron realizados sobre la línea generada, luego de cultivarla y amplificarla durante al menos 5 pasajes. En contraposición, si se indica que los experimentos fueron realizados sobre líneas o colonias de CMPi jó e es o se hace referencia a colonias recién establecidas , se está hablando de colonias entre los pasajes 0 y 1. Protocolo de Generación de CMPi a partir de CMSP Para la generación de CMPi con el vector lentiviral STEMCCA o STEMCCA RedLight, los linfocitos aislados como se describió anteriormente fueron, en algunos casos, tratados con IL-2 y - 34 - MATERIALES Y MÉTODOS anticuerpos anti-CD3 a fin de activar e incentivar la proliferación de clones linfocitarios para reprogramación, el mismo día de la extracción sanguínea. Luego de la obtención de las CMSP ,y el tratamiento con IL-2 y anti-CD3 si correspondía, se realizó la transducción a una MOI de 1, en medio RPMI SFB 15% + ConA 10 µg/ml, con una concentración final de Polybrene de 10 µg/ml y finalizando el proceso con un paso de spinfection. El control negativo de transducción se realizó agregando el mismo volumen utilizado de concentrado pero de medio de cultivo. A fin de concentrar la población linfocitaria en el cultivo, a las 24 horas post-transducción (día 1) las células fueron centrifugadas a 300 xg, se descartó el pellet de monocitos/macrófagos y se replaqueó la suspensión celular de linfocitos en medio RPMI SFB 15% + ConA 10 µg/ml, el cual se cambió día por medio, hasta el día 5, momento en el cual se realizó una segunda transducción de la misma manera que el día 0. El día 6 se cambió el medio de la misma manera que el día 1. A partir del día 7 se cambió el medio a DMEM/F12 KSR 10%, el cual se cambió todos los días. Cuando se utilizó el vector STEMCCA-RedLight se agregó VPA 0,75 mM al medio entre los días 3-14. No se observaron colonias de CMPi en ninguno de los intentos realizados a partir de CMSP. Cultivo de Células Madre Pluripotentes La línea de CMEh WA-09 fue adquirida del Instituto de investigación WiCell (WI, Estados Unidos) en pasajes bajos (p15 a p20). Las líneas de CM se mantuvieron en capas nutricias de MEFi e edio o puesto po DMEM/Ha ’s F 2 1:1 (DMEM/F12) (Gibco) suplementado con KSR 10%, (NEAA) concentración final 0,1 mM, Penicilina concentración final 100 U/ml, Estreptomicina concentración final μg/ l, L-Glutamina concentración final 2 mM, β- mercaptoetanol 0,1 mM y bFGF 8 ng/ml (Invitrogen, PHG0263). Las CM fueron expandidas mediante la transferencia de colonias a través de incubación en Colagenasa IV 1mg/ml (Gibco, 17104019), disociación de la capa nutricia, división de las colonias y plaqueo sobre nuevas placas con MEFi, o tratadas con Matrigel (BD Biosciences, 354234) diluido (1/40) y mantenidas en medio condicionado por MEFi. En todas las instancias, las CM fueron cultivadas sobre capa nutricia de MEFi, excepto cuando se indique lo contrario. En estos últimos casos y cuando fue necesario, las CM fueron cultivadas sobre Matrigel y para su pasaje fueron tratadas con Acutasa - 35 - MATERIALES Y MÉTODOS (Invitrogen, A1110501) durante 20 minutos, obteniéndose una suspensión unicelular; dicha suspensión fue dividida y plaqueada en nuevas placas tratadas con Matrigel, con la adición de inhibidor de ROCK a una concentración final de 10 µM, y las células fueron crecidas hasta confluencia en medio condicionado. Para inducir la diferenciación, las colonias de CM fueron disociadas de la capa nutricia mediante tratamiento con Colagenasa IV 1 mg/ml. Las colonias fueron luego transferidas a placas de Petri no-adherentes con DMEM suplementado con 20% de SFB, además de los suplementos antes mencionados (DMEM 20%), utilizado como medio de diferenciación. Las colonias se incubaron en suspensión durante 7 días. Durante este período las células se agrupan en agregados llamados cuerpos embrioides (CE), los cuales son luego transferidos a microplacas de 24 pocillos tratadas con gelatina bovina 0.1 %, donde los CE se adhieren y son cultivados durante 7 días adicionales. Aislamiento de ARN, Síntesis de ADNc y PCR Cuantitativa en Tiempo Real El proceso de aislamiento de ARN, síntesis de ADNc y PCR Cuantitativa en Tiempo Real se define en su totalidad como RT-qPCR. ARN total fue extraído de los cultivos celulares con Trizol (Invitrogen) según recomendaciones del fabricante. El ADNc fue sintetizado a partir de 1 µg total de ARN con 13,33 ng/µl de random primers (Invitrogen), 0,67 mM de dNTP (Invitrogen) y Transcriptasa Reversa de MMLV (Promega), según las instrucciones del fabricante. En todos los casos, las muestras de ADNc se diluyeron 1/10, y la PCR Cuantitativa en Tiempo Real (qPCR) se realizó a una temperatura de annealing de 60° C. Todas las reacciones se mantuvieron dentro del rango lineal de amplificación. La detección y amplificación por qPCR se realizó en un equipo StepOne Plus Real Time PCR System (Applied Biosystems) y el análisis se realizó con el programa StepOne v2.1. Todas las reacciones se llevaron a cabo utilizando el reactivo de qPCR Sybr Green ER Super Mix (Invitrogen), siguiendo las instrucciones del fabricante en un volumen de reacción final de 20 µl. Un análisis de la curva de melting se realizó inmediatamente después de la amplificación a una tasa lineal de transición de temperatura de 0,3° C/s de 70 a 89° C con adquisición de fluorescencia continua, para confirmar la amplificación de amplicones individuales. Se muestra una lista de secuencias de los primers y el tamaño de los amplicones - 36 - MATERIALES Y MÉTODOS en la Tabla 1. En todos los experimentos en hipoxia la expresión del gen de housekeeping HPRT1 fue utilizada para normalizar los niveles de expresión. En todos los demás casos el gen de housekeeping GAPDH fue utilizado. Todos los primers fueron diseñados para detectar solamente ADNc y no ADN genómico (ADNg) y se revisaron para evitar la complementariedad con secuencias del ratón, dado el uso de MEFi en los cultivos celulares. Tabla 1. Primers utilizados para experimentos de qPCR y tamaño de amplicones correspondiente Gen / Secuencia Expresada AFP BNIP3 C-MYC GAPDH GFAP HPRT1 MESP-1 NANOG NKX2.5 PAX6 POU5F1 SOX17 SOX2 VEGF Primer Forward 5’- ’ Primer Reverse 5’- ’ Tamaño del Amplicón (pb) ACTATTGGCCTGTGGCGAGGGA AAGCATGGCCTCCTGTTGGC 132 ATCGCCGCAGTTGCCCTCTGG ATAGAAACCGAGGCTGGAACGCTG 138 CCTGGCTCCCCTCCTGCCTCGA GCTCCCTCTGCCTCTCGCTGGA 150 CGCTCTCTGCTCCTCCTGTTCG ACGACCAAATCCGTTGACTCCGAC 116 AGGAAGATTGAGTCGCTGGAG CGGTGAGGTCTGGCTTGG 136 GCGAACCTCTCGGCTTTCCCG CTGCGGGTCGCCATAACGGAG 132 TGAGGAGCCCAAGTGACAA CTCTTCCAGGAAAGGCAGTCT 111 TCCTTCCTCTCCCCTCCTCCCAT TAGGCTCCAACCATACTCCACCCTC 120 CCCACGCCCTTCTCAGTCAA GTAGGCCTCTGGCTTGAAGG 143 CAGGTGTCCAACGGATGTG GTCGCTACTCTCGGTTTACTAC 101 GCAGGCCCGAAAGAGAAAGCGA TGGCTGATCTGCTGCAGTGTGG 105 CGCCGAGTTGAGCAAGATG CTCTGCCTCCTCCACGAAG 82 AGCATGGAGAAAACCCGGTACGC CGTGAGTGTGGATGGGATTGGTGT 110 GTAGCTCGGAGGTCGTGGCG AATTCCAGCACCGAGCGCCC 112 Reacción de PCR a Punto Final Se extrajo ARN, y se sintetizó ADNc como se describe anteriormente. La reacción de PCR a pu to fi al se ealizó e u olu e fi al de 5 μl. Se colocaron los siguientes componentes en u tu o eppe do f de , l: ,5 μl de Buffe de PCR X Tris-HCl 200 mM [pH 8.4], KCl 500 mM), 0,75 μl de MgCl2 50 mM, 0,5 μl de mezcla de dNTPs 10 mM (10 mM cada uno: dATP, dGTP, dCTP y dTTP a pH neutro), 1,25 μl de Primer Forward 10 μM, 1,25 μl de Primer Reverse 10 μM, 0,2 μl de Polimerasa Platinum Taq DNA Polimerase 5 U/μl (Invitrogen) y 0,5 μl de ADNc dilución 1/10 obtenido en la reacción de retrotranscripción, completando el volumen a 25 μl - 37 - MATERIALES Y MÉTODOS con agua libre de ARNasas. En todos los casos se realizó un control negativo sin ADNc, para comprobar que no existan contaminantes en los reactivos. Se utilizaron los mismos primers que en la qPCR en Tiempo Real, además de los que se muestran en la Tabla 2. Tabla 2. Primers utilizados para experimentos de PCR a Punto Final, exclusivamente. Gen / Secuencia Expresada Primer Forward (5’- ’ Primer Reverse 5’- ’ βIIItubulina MyoD Nestina TGGGCGACTCGGACTTGC Tamaño del Amplicón (pb) CCACTCTGACCAAAGATGAAATTG 178 GCTGCCGCCGCTTTCCTTAAC CAGCTGGCGCACCTCAAGATG GCCGGAACTTTCTGCCGCTCG 133 AGGGAAGTTGGGCTCAGGACTGG 209 El programa de PCR utilizado fue el detallado a continuación: Desnaturalización inicial y activación de la polimerasa: 2 minutos a 94º C Desnaturalización: 30 segundos a 94° C Apareamiento: 30 segundos a 60° C 35 ciclos Extensión: 35 segundos a 72° C Extensión Final: 5 minutos a 72° C Al finalizar las reacciones de PCR, visualizamos los productos mediante electroforesis en gel de agarosa 2% , durante 5 minutos a 50 V y durante 1 hora a 110 V, en buffer TAE 1X (Tris-acético 0,04 M, EDTA 1 mM). Las muestras contenían 20 μl del p odu to de a plifi a ió 5 μl de buffer de siembra 5x Orange G [12, 5 ml Glicerol, 2,5 ml EDTA, 0,25 ml SDS 10%, 2,5 ml Orange G 2%, para un volumen final de 25 ml]. Una vez concluida la corrida electroforética, se incubó el gel en una solución de Bromuro de Etidio ( ,5 μg/ l en agua destilada, durante 30 minutos en oscuridad, se visualizó y se fotografió en un transiluminador con luz ultravioleta (UV) Fotodyne Modelo 602107CE acoplado a PC, controlado por programa Foto/Analyst PC Image versión 5.00. - 38 - MATERIALES Y MÉTODOS Inmunofluorescencia Los cultivos celulares se analizaron por inmunofluorescencia in situ. Brevemente, las células fueron lavadas con PBS frío y fijadas en PBS Albúmina de Suero Bovino 0.1% (PBSA) con formaldehído al 4% durante 45 minutos. Después de 3 lavados con PBSA las células fueron permeabilizadas con Triton X-100 0.1% en PBSA y bloqueadas con 10% de suero normal de cabra (SNC) durante 45 minutos, lavadas dos veces con PBSA y teñidas con los correspondientes anticuerpos primarios: anticuerpos monoclonales de ratón anti-CK7 (SC-23876), anti-CK total (SC-57004), anti-OCT-3/4 (SC-5279) (anti-POU5F1), anti-SSEA4 (SC-21704), anti-AFP (SC166325), anti-TRA1-60 (SC-21705), anti-TRA-1-81 (SC-21706), anti-GATA-4 (SC-25310), antiHIF2α 190b (SC-13596) (todos Santa Cruz Biotechnology); anticuerpos monoclonales de conejo anti-CK20 (SC-25725), anti-NANOG (Cell Signaling, #4903), anti-SOX2 (Cell Signaling, #3579); anticuerpos policlonales de conejo anti-NESTINA (Millipore, AB5922), anti-NKX2.5 (Santa Cruz Biotechnology, SC-14033), anti-C-MYC (Cell Signaling, #5605), anti-GFAP (Millipore, AB5804), anti-PAX6 (Abcam, AB5790), anti-ANP (Santa Cruz Biotechnology, SC-20158), según las instrucciones del fabricante, en PBSA SNC 10%, durante 2 horas a TA. Las diluciones utilizadas se observan en la Tabla 3. Las células luego se lavaron tres veces en PBSA y se utilizaron anticuerpos fluorescentes secundarios para localizar los complejos anticuerpo del antígeno primario: conjugados con Alexa Fluor 488 IgG de cabra anti-ratón (A11029), conjugados con Alexa Fluor 488 IgG de cabra anti-conejo (A11034), conjugados con Alexa Fluor 568 IgG de cabra anti-ratón (A11031) y conjugados con Alexa Fluor 568 IgG de cabra anti-conejo (A11036) (todos Molecular Probes/Invitrogen). Fueron diluidos en PBSA SNC 10%, según se indica en la Tabla 3, y utilizados según las instrucciones del fabricante, incubando durante 1 hora a TA. Las núcleos fueron también teñidos con 4'-6-diamidino-2-fenilindol (DAPI) (Molecular Probes/Invitrogen) según las instrucciones del fabricante y las células fueron examinadas y fotografiadas bajo un microscopio de contraste de fases invertido Nikon Eclipse TE2000-S. Las imágenes fueron adquiridas con una cámara digital Nikon DXM1200F, que fue controlada por el software EclipseNet (versión 1.20.0 build 61). - 39 - MATERIALES Y MÉTODOS Para la cuantificación de HIF- α e fi o lastos HFF-FM por inmunofluorescencia se utilizó el programa Metamorph, dado que provee un mejor algoritmo que el ImageJ para células ahusadas. Utilizando las herramientas de forma y a través de la función Region Measurement se seleccionó el área a medir y se cuantificó la fluorescencia, restando la señal del ruido de fondo. Tabla 3. Anticuerpos primarios y secundarios utilizados en inmunofluorescencia. Anticuerpo anti-AFP anti-ANP anti-CK total anti-CK20 anti-CK7 anti-C-MYC anti-conejo conjugado con Alexa Fluor 488 anti-conejo conjugado con Alexa Fluor 568 anti-GATA-4 anti-GFAP anti-HIF2α 190b anti-NANOG anti-NESTINA anti-NKX2.5 anti-OCT-3/4 anti-PAX6 anti-IgG de ratón conjugado con Alexa Fluor 488 anti-IgG de ratón conjugado con Alexa Fluor 568 anti-SOX2 anti-SSEA4 anti-TRA1-60 anti-TRA-1-81 Santa Cruz Biotechnology Inc. Santa Cruz Biotechnology Inc. Santa Cruz Biotechnology Inc. Santa Cruz Biotechnology Inc. Santa Cruz Biotechnology Inc. Cell Signaling N° de Catálogo SC-166325 SC-20158 SC-57004 SC-25725 SC-23876 #5605 Dilución empleada 1:200 1:200 1:200 1:200 1:200 1:200 Molecular Probes/Invitrogen A11034 1:400 Molecular Probes/Invitrogen A11036 1:400 Santa Cruz Biotechnology Inc. Millipore Santa Cruz Biotechnology Inc. Cell Signaling Millipore Santa Cruz Biotechnology Inc. Santa Cruz Biotechnology Inc. Abcam SC-25310 AB5804 SC-13596 #4903 AB5922 SC-14033 SC-5279 AB5790 1:200 1:200 1:250 1:200 1:200 1:200 1:200 1:200 Molecular Probes/Invitrogen A11029 1:400 Molecular Probes/Invitrogen A11031 1:400 Cell Signaling Santa Cruz Biotechnology Inc. Santa Cruz Biotechnology Inc. Santa Cruz Biotechnology Inc. #3579 SC-21704 SC-21705 SC-21706 1:200 1:200 1:200 1:200 Fabricante - 40 - MATERIALES Y MÉTODOS Análisis de la Metilación del Promotor de POU5F1 Se extrajo ADNg mediante el kit Wizard Genomic DNA (Promega). 200 ng de DNAg fueron convertidos utilizando el kit EZ DNA Methylation Gold (Zymo Research), según las instrucciones del fabricante. El kit utiliza la reacción del bisulfito de sodio con el ADN, la cual genera la desaminación de las citosinas, convirtiéndolas a uracilos, excepto en el caso de estar metiladas, en cuyo caso permanecen como citosinas. Luego de dicha conversión, las islas CpG en el promotor de POU5F1 fueron amplificadas usando los primers indicados en la Tabla 4 [131]. Los productos de PCR fueron entonces purificados mediante el kit ExoSAP-IT (Affymetrix) y secuenciados por Macrogen (Macrogen, Corea). Los resultados de la secuenciación permiten identificar citosinas, las cuales han de ser solamente citosinas metiladas en la secuencia original pre-conversión, ya que solamente éstas permanecen como tales, luego del tratamiento con bisulfito de sodio y amplificación. Se realizaron análisis semi-cuantitativos utilizando los programas BiQ Analyzer y BioEdit, midiendo las intensidades relativas de los picos de la curva para cada CpG en los electroferogramas. Para un análisis exhaustivo, se construyó un gráfico de intensidad donde las islas CpG 100% metiladas son representadas en rojo y las islas 100% demetiladas son representadas en color azul. Todos los valores intermedios se expresan en un gradiente de intensidades del color correspondiente. Tabla 4. Primers utilizados para el análisis de la metilación del promotor de POU5F1 Primer Forward 5’- ’ Primer Reverse 5’- ’ Cobertura de la secuencia relativa al TSS Temperatura de apareamiento (°C) TTTTTAGTTTTTTTTAGGTTTAA ATTTGTTTTTTGGGTAGTTAAAGGT GGATGTTATTAAGATGAAGATAGTTGG AATAGATTTTGAAGGGGAGTTTAGG TAGTTGGGATGTGTAGAGTTTGAGA AAGTTTTTGTGGGGGATTTGTAT GTTAGAGGTTAAGGTTAGTGGGTG TAAACAAAAAACCCATTCCC CCAACTATCTTCATCTTAATAACATCC CCTAAACTCCCCTTCAAAATCTATT TTCCTCCTTCCTCTAAAAAACTCA TAAACCAAAACAATCCTTCTACTCC CCACCCACTAACCTTAACCTCTA AAACCTTAAAAACTTAACCAAATCC -2995 a -2723 -2344 a -2126 -2136 a -1721 -1755 a -1574 -567 a -309 -215 a -29 -57 a +66 46 56 56 56 56 56 56 TSS: Transcription Start Site = Sitio de inicio de la transcripción. - 41 - MATERIALES Y MÉTODOS Evaluación de la Expresión de los Transgenes STEMCCA La qPCR fue realizada como se describió anteriormente, en cDNA generados a partir de la extracción de ARN (como se describió anteriormente) de líneas de CMPi establecidas entre pasaje 17 y pasaje 23, y del plásmido STEMCCA como control positivo, usando los primers Sox2bdg-cMyc y Oct4-bdg-Klf4 para detectar la expresión de parte de los genes junto a secuencias puente (bdg) en el plásmido STEMCCA. Estas secuencias bdg hacen de conexión entre dos de los cuatro transgenes (SOX2, C-MYC, OCT4 y KLF4), una situación que no se encuentra en ARNm endógeno/ADNc. Este diseño permite amplificar solamente el ADNc generado a partir de la expresión de ARNm exógena de estos genes, por apareamiento exactamente en los puentes que conectan los dos pares de genes [73]. Los primers fueron diseñados por el equipo del Dr. Mostoslavsky y sus secuencias y el tamaño de los amplicones se muestran en la Tabla 5. Tabla 5. Primers utilizados para experimentos de qPCR para la evaluación de la expresión de los transgenes STEMCCA y tamaño de amplicones correspondiente Gen / Secuencia Expresada Primer Forward 5’- ’ Primer Reverse 5’- ’ Sox2-bdg-c-Myc Oct4-bdg-Klf4 TTGGCTCCATGGGTTCGGTG CAACGAGAGGATTTTGAGGC AAGGGTGTGACCGCAACGTAGG ATCGTTGAACTCCTCGGTCTCTCT Tamaño del Amplicón (pb) 550 561 Formación de Teratomas y Análisis Histológico CM fueron cultivadas a alta confluencia sobre MEFi y disociadas de su capa nutricia con Colagenasa IV 1 mg/ml durante 1 hora en incubadora de cultivo celular. 4 x 106 células en una dilución 1/4 de Matrigel por dosis, fueron inyectadas por vía subcutánea en los flancos dorsales de ratones nude N:NIH(S)-Fox1nu macho de 6 a 8 semanas de edad. Cada ratón fue inyectado con una línea celular de CMPi cuya pluripotencia se deseaba evaluar, en un flanco y la línea control de CMEh WA-09 en el otro flanco, y cada experimento se realizó por duplicado. Los animales se mantuvieron en racks ventilados con agua y alimento ad libitum. La formación de tumores fue observada entre 3 a 8 semanas post-inyección, tras lo cual se mantuvieron los ratones por 20-90 días adicionales para permitir el crecimiento del tumor, hasta un tamaño tumoral de aproximadamente 1 cm. En este punto los animales fueron anestesiados en una - 42 - MATERIALES Y MÉTODOS cámara con atmósfera de CO2 y sacrificados por dislocación cervical. Los tumores fueron removidos quirúrgicamente después de la eutanasia y fijados en formaldehído al 4% en PBS. Posteriormente las muestras fueron embebidas en parafina y secciones de 4 mm fueron teñidas con tinción de Hematoxilina-Eosina (hematoxilina activada y solución acuosa de eosina 0.5%, Biopur, Argentina), según las instrucciones del fabricante y montadas en portaobjetos para observación, análisis y fotografía bajo un microscopio de luz directa Nikon Eclipse E400 equipado con una cámara digital Nikon Coolpix S4. El armado de los preparados fue realizado por el Dr. Marcelo Schultz y la Dra. Naomi Arakaki del Instituto FLENI, Buenos Aires. La identificación de tejidos derivados de las tres capas germinales fue realizada por el médico patólogo Dr. Gustavo Sevlever, del Instituto FLENI, Buenos Aires. Todos los procedimientos con animales se realizaron bajo lineamientos éticos locales y el protocolo fue aprobado por la Comisión Institucional para el Cuidado y Uso de Animales de Laboratorio (CICUAL) de la Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires. Ver apartado de Normas Éticas. Western Blot Se extrajo proteína total de células cultivadas en Matrigel durante 2 pasajes, en buffer de extracción de proteínas RIPA helado (Sigma-Aldrich, R0278) suplementado con tabletas cOmplete Mini Protease Inhibitor Cocktail (Roche Diagnostics). Los lisados se centrifugaron a 14.300 xg a 4° C por 10 minutos, y se descartó el pellet. La concentración de proteínas se determinó utilizando el kit Bicinchoninic Acid Protein Assay (Pierce). Las muestras se mantuvieron siempre en hielo o a 4° C durante todo el experimento. Se agregó Buffer de Carga 5X (0,32 M de Tris 0,32 M pH 6.8, 10% de SDS, 0,025% de Azul de Bromofenol, 50% de glicerol, 5% de 2-Mercaptoetanol) a las muestras de proteínas, las cuales fueron luego calentadas a 100° C por 5 minutos y cantidades iguales de proteína (60 µg por carril) fueron sembradas, junto con un marcador de peso molecular Li-Cor Odyssey Protein Molecular Weight Marker (Li Cor, 92840000) y corridas electroforéticamente durante 2 horas a 100 V en geles de poliacrilamida-SDS 8%. Luego se realizó una transferencia a una membrana de PVDF Immobilon-FL (Millipore, IPFL00010, tamaño de poro 0,45 µm) por electroblotting en buffer de transferencia con 20% de - 43 - MATERIALES Y MÉTODOS metanol, 0,19 M de glicina, 0,025 M de Tris-base (pH 8,3) a 250 mA, durante 80 minutos en un sistema de transferencia Mini Trans Blot Cell (BioRad). Las membranas fueron bloqueadas durante 1 hora a TA en buffer de bloqueo Li-Cor Odyssey (Li-Cor). Se realizaron incubaciones con anticuerpos primarios diluidos según las instrucciones del fabricante a TA, durante 15 minutos y luego durante la noche a 4 ° C, en buffer de bloqueo Li-Cor Odyssey, 0,1% de Tween 20 (PBS-Tween). Se utilizaron los siguientes anticuerpos primarios: policlonal de cabra antiactina I-19 (SC-1616) dilución 1:2500, monoclonal de ratón anti-HIF1α 28b dilución 1:200, y monoclonal de ratón anti-HIF2α 190b (SC-13596) dilución 1:200 (todos de Santa Cruz Biotechnology). La membrana se lavó 6 veces durante 5 minutos con PBS-Tween y luego se incubaron durante 90 minutos en una solución de buffer de bloqueo Li-Cor Odyssey, 0,1% de Tween 20, 0,01% de SDS y anticuerpos secundarios marcados con IRDye fluorescente IgG de cabra anti-conejo conjugado con IRDye 680RD (Li-Cor, 926-68071) y anti-ratón conjugado con IRDye 800CW (Li-Cor, 926-32210) (dilución de trabajo 1:10.000). Posteriormente se realizaron 5 lavados en PBS, se dejó secar la membrana e inmediatamente fue analizada en busca de bandas proteicas en un sistema de proyección de imágenes infrarrojas Odyssey Infrared Imaging System en los canales de 700 y 800 nm utilizando el programa Odyssey Imaging Software v3.0 en calidad de imagen Alta, intensidad de 4, resolución de 169 µm con un offset de 3,0 mm. Para la cuantificación de la señal, se midió la intensidad de las bandas mediante el programa ImageJ Software versión 1.48. Actividad de Fosfatasa Alcalina y Eficiencia de Reprogramación El día 21 post-transducción con el vector STEMCCA y posterior cultivo en normoxia o hipoxia, se analizaron cultivos reprogramados de fibroblastos HFF-FM sobre capa nutricia de MEFi para actividad de FA, utilizando el kit Leukocyte Alkaline Phosphatase (Sigma-Aldrich), según instrucciones del fabricante. Brevemente, las células en sus placas de cultivo fueron fijadas con una solución de ácido cítrico/acetona/paraformaldehído, lavadas con agua bidestilada (ddH2O) y teñidas con solución de FA (Nitrito de Sodio/solución FRV alcalina/solución alcalina naftol ASBI) a TA. Finalmente las células fueron lavadas con agua corriente, secadas al aire y fotografiadas. Un complejo de células color rojo sobre la capa de fibroblastos fue considerado - 44 - MATERIALES Y MÉTODOS como una colonia de CMPi derivada de un fibroblasto reprogramado, positiva para FA. La eficiencia de reprogramación se definió como el número de colonias de CMPi observadas al día 21 post-transducción positivas para actividad de FA (antes del primer pasaje) / número total de fibroblastos HFF-FM transducidos. Las colonias fueron fotografiadas como se describió anteriormente en microscopio invertido y la cantidad de colonias observadas y su área se calculó usando ImageJ software v1.48, utilizando el comando Analizar Partículas con un Tamaño de 50-Infinito y una Circularidad de 0,20-1,00. In-Cell Western Blot El ensayo fue realizado según las instrucciones del fabricante (Li-Cor Bioscience). Brevemente, en el día 21 post-transducción, cultivos de células reprogramadas en sus pocillos fueron lavados con PBS y luego fijados con formaldehído al 4% en PBS a TA durante 20 minutos. A continuación se realizaron 3 lavados con PBS-Triton X-100 0,1% para permeabilizar las células. Las células fueron bloqueadas con buffer de bloqueo Li-Cor Odyssey 1,5 horas a TA y marcadas durante 2 horas con los anticuerpos mencionados anteriormente, anti-NANOG y anti-SOX2 de conejo, anti-POU5F1 y anti-SSEA-4 de ratón, y anti-HIF- α a ti HIF- α diluidos en buffer de bloqueo Li-Cor Odyssey, en diluciones de trabajo como se indica en la Tabla 3, y según las instrucciones del fabricante. Los pocillos para utilizar para el control de la señal de fondo fueron tratados sólo con el buffer de bloqueo. Finalmente se realizaron 3 lavados con PBS-Tween 20 0,1%. Los anticuerpos secundarios marcados con IRdye fluorescente cercano al infrarrojo, IgG de cabra anti-conejo conjugado a IRDye 680RD e IgG de cabra anti-mouse conjugado a IRDye 800CW, se diluyeron a una concentración de 1: 200 y 1: 800 respectivamente en buffer de bloqueo Li-Cor Odyssey y las células se incubaron durante 1 hora. A continuación se realizaron 4 lavados con PBS-Tween 20 0,1% y, después del último lavado, la solución de lavado fue removida totalmente de los pocillos. Las placas se invirtieron y se golpearon suavemente sobre toallas de papel para eliminar los restos de solución de lavado. Finalmente, las placas fueron escaneadas inmediatamente en el sistema de imágenes infrarrojas Odyssey Infrared Imaging System, con detección en los canales de 700 y 800 nm, usando el programa Odyssey Imaging - 45 - MATERIALES Y MÉTODOS Software v3.0. Los parámetros de análisis utilizados fueron Calidad media, Resolución 169 µm con un Offset de 3,0 mm e Intensidad 5.0. Un algoritmo Li-Cor de remoción de ruido fue aplicado a los datos y se les restó la señal de fondo (background). Las señales de los anticuerpos fueron cuantificadas y las intensidades promedio integradas a partir de pozos duplicados. Reprogramación en Presencia de Inhibidores de HIF La reprogramación se realizó de la misma manera descripta anteriormente, con la adición de uno de los siguientes inhibidores: FM19G11 (EMD Millipore, 400089) [117], inhibidor de HIF1 (HIF1i) (Calbiochem, 400083) [132], DMOG (Calbiochem, 400091) [133] desde el día 1 hasta el día 14 post-transducción. En todos los casos, los inhibidores fueron utilizados según las instrucciones del fabricante: FM19G11 fue utilizado a una concentración final de 500 nM; HIF1i fue utilizado a una concentración final de 1,65 μM y DMOG a una concentración final de 1 mM. Estudio por Citometría de Flujo de la Expresión de NANOG durante el Proceso de Reprogramación Cultivos reprogramados de fibroblastos HFF-FM en MEFi, a diferente tiempos post-transducción fueron lavados con PBS e incubados con Tripsina/EDTA durante 5-10 minutos en incubadora de cultivo celular en normoxia o hipoxia, según las condiciones de cada experimento, hasta la disociación de las células de la placa y entre sí,. Las células en suspensión se fijaron con formaldehído al 4% en PBSA durante 45 minutos. Después de un lavado con PBS las células fueron permeabilizadas con PermBuffer III (BD Biosciences) durante 30 minutos en hielo, se lavaron nuevamente con PBS y se tiñeron con el anticuerpo primario monoclonal de conejo anti-NANOG dilución 1:200, durante 1 hora a TA. Las células luego se lavaron dos veces con PBS y se incubaron durante 1 hora a TA con el anticuerpo secundario de cabra anti-IgG de conejo, conjugado al fluoróforo Alexa Fluor 488 dilución 1:400. Finalmente, las células fueron lavadas y resuspendidas en PBS para su análisis en citómetro de flujo. La citometría de flujo se realizó en un citómetro BD Accuri (BD Biosciences) y la adquisición de datos de intensidad de fluorescencia y el análisis se realizó mediante el programa BD Accuri C6 v 1.0.264.21 (BD Biosciences). Al menos 5000 eventos fueron contados por muestra. - 46 - MATERIALES Y MÉTODOS Ensayo de Azul de Tripán para Viabilidad Celular Para el ensayo de exclusión de Azul de Tripán, las células fueron sembradas en placas de 6 pocillos a una densidad de 1 x 105 células/pocillo y cultivadas en normoxia o hipoxia durante 48 horas (día 2). En el día 2 tanto las células adherentes como las células no adheridas se recolectaron y se tiñeron con una solución de Azul de Tripán 0,4% (concentración final de Azul de Tripán 0.08%) durante 5 minutos a TA. Las células teñidas de azul (muertas) y las células sin tinción (vivas) fueron contaron en un hemocitómetro (cámara de Neubauer). Los conteos se realizaron al menos 4 veces para cada experimento independiente y se calculó un promedio para cada uno de 3 experimentos independientes. Se calcularon los porcentajes de células viables como la media del total del número de células vivas contadas, dividido por la media del número total de células contadas, multiplicado por 100. Ensayo de Proliferación Celular Para determinar la densidad celular de los cultivos a un tiempo inicial y un tiempo final, se utilizó el kit CyQUANT Cell Proliferation Assay (Invitrogen) en cultivos celulares cultivados sobre Matrigel. El colorante CyQUANT se une al ADN, por lo tanto se construyó una curva estándar de emisión fluorescente en función de la concentración de complejo ADN-CyQUANT, según las instrucciones del fabricante. En su segundo pasaje sobre Matrigel, una cantidad igual de células de cada línea, contadas en hemocitómetro, fue plaqueada en pocillos placa de 6 pocillos y se mantuvo en normoxia o en hipoxia. Después de 3 días de cultivo, se removió el medio de cultivo y se lavaron las células con PBS, el cual fue luego removido totalmente y las placas fueron colocadas a -80°C durante 2 horas, para luego ser descongeladas a TA. Una vez que los cultivos llegaron a TA, las células fueron lisadas y tratadas con RNasa A para deshacerse del ARN. A continuación se agregó la solución de colorante CyQUANT a cada muestra y se incubó durante 5 minutos a TA, para luego medir fluorescencia con filtros para 485 nm de excitación y emisión de 538 nm, en un fluorómetro de microplacas Fluoroskan Ascent FL 2.6 (Thermo Scientific) y usando el programa Ascent Software v2.6 (Thermo Scientific). Basándose en la curva estándar de ADN-fluorescencia construida anteriormente, se calculó el contenido de ADN - 47 - MATERIALES Y MÉTODOS en cada caso, lo cual es indicativo del nivel de proliferación de los cultivos después del plazo de 3 días, teniendo en cuenta una cantidad igual de células de partida. Análisis de la Apoptosis por Citometría de Flujo a través de doble tinción con Anexina V / Ioduro de Propidio La Anexina V (AnV) es un indicador de la presencia de fosfatidilserina expuesta en la membrana plasmática, un evento encontrado principalmente en procesos apoptóticos. La AnV se une a la fosfatidilserina, y cuando se la marca con una molécula fluorescente puede utilizarse para estudios de citometría de flujo. Se utilizó el kit FITC-Annexin V Apoptosis Detection I (BD Bioscience) para medir muerte celular y apoptosis, según instrucciones del fabricante. Brevemente, CM cultivadas sobre Matrigel en normoxia o hipoxia se lavaron con PBS y se incubaron con Tripsina/EDTA durante 5-10 minutos en incubadora de cultivo celular en normoxia o hipoxia, según las condiciones de cada experimento, a fin de obtener una suspensión unicelular. Luego se transfirieron las células a un tubo, se lavaron con PBS y se resuspendieron en Binding Buffer (0,01 M de HEPES pH 7,4, 0,14 M de NaCl y 2,5 mM de CaCl2). Se agregó FITC-AnV / Ioduro de Propidio (IP) a cada muestra conteniendo 1 x 105 células, se agitaron dichas muestras en vórtex y se incubaron durante 15 minutos a TA en oscuridad. Se agregó Binding Buffer a cada tubo e inmediatamente las muestras fueron analizadas por citometría de flujo. También se analizaron células sin tinción como control negativo. La citometría de flujo se llevó a cabo como se describió anteriormente para la cuantificación de Nanog. Se consideraron las células positivas para AnV y negativas para IP como apoptóticas tempranas, las células positivas para IP y negativas para AnV como necróticas y las células positivas para ambos marcadores como apoptóticas tardías. Cariotipado Las células fueron cultivadas en las condiciones anteriormente descritas, según el tipo de célula. Brevemente, para una placa de cultivo de 10 cm de diámetro y cuando el cultivo celular alcanzó una tasa de crecimiento exponencial, se agregó colchicina 0,1 mg/ml (Sigma-Aldrich) y se incubó por 1.5-3 horas. Los tiempos exactos se determinaron caso por caso, según los tiempos - 48 - MATERIALES Y MÉTODOS de crecimiento de cada línea. Las células fueron entonces disociadas con Tripsina/EDTA y transferidas a un tubo Eppendorf. Después de obtener un precipitado de células por centrifugación a 200 xg, el sobrenadante fue descartado, y se agregaron 1,5 ml de solución hipotónica de KCl 0,075M gota a gota. La muestra se incubó durante 20 minutos en incubadora de cultivo celular, y luego se centrifugó a 200 xg, a fin de obtener un pellet celular, el cual fue resuspendió en 1,5 ml de solución de fijación compuesta por ácido acético glacial:metanol 3:1 (Van Rossum y JT Baker). Seguidamente se realizó una incubación de 24 horas a 4° C. Después de este período, se prepararon muestras para microscopio colocando una gota de la muestra sobre un portaobjetos de microscopio, secando el portaobjetos en una campana de seguridad y realizando una tinción de Giemsa en solución de Giemsa al 5% (Biopur) en buffer fosfato pH 7, durante 20 minutos. Los portaobjetos fueron luego lavados dos veces en ddH2O, secados al aire y cubiertos con un cubreobjetos con medio de montaje. Las muestras fueron observadas en un microscopio Olympus BX60, a un aumento de 1000X, y fotografiadas utilizando una cámara Olympus NO C-5060. Los cariogramas fueron armados a partir de las fotografías utilizando el programa Adobe Photoshop CS5. Al menos 50 metafases fueron analizadas para cada línea celular. El armado de los preparados y el análisis de los cariotipos fue realizado por la Lic. Diana Radakoff del Instituto GENOS, Buenos Aires. Construcción de Plásmidos para Reprogramación con Factores HIF Se contaba con los siguientes plásmidos, generosamente provistos por la Dra. M. Celeste Simon [134] u obtenidos de Addgene, a través de un Material Transfer Agreement (MTA), para obtener cDNA de los factores HIF- α HIF- α, con distintas mutaciones: - HA-HIF1alpha-pcDNA3. Plásmido Addgene 18949. Plásmido wild type (wt). - HA-HIF2alpha-pcDNA3. Plásmido Addgene 18950. Plásmido wild type. - HA-HIF1alpha P402A/P564A-pcDNA3. Plásmido Addgene 18955. Plásmido Doble Mutado. - HA-HIF2alpha-P405A/P531A-pcDNA3. Plásmido Addgene 18956. Plásmido Doble Mutado. - pTRE2Hyg-HIF- α T . P o isto po la D a. Simón. Plásmido Triple Mutado. - pTRE2Hyg-HIF-2α Tm. Provisto por la Dra. Simón. Plásmido Triple Mutado. - 49 - MATERIALES Y MÉTODOS Como se mencionó anteriormente, los factores HIF poseen un dominio ODDD (Oxygen Dependent Degradation Domain) dependiente de oxígeno. Este dominio posee dos prolinas, las cuales son hidroxiladas en condiciones de normoxia, lo cual conlleva la degradación de HIF- α o HIF- α. E o di io es hipó i as di has p oli as o so hid o iladas, la p oteí a es esta ilizada y dimeriza con HIF- β, u plie do sus fu io es de regulación de la expresión de ciertos genes. Los plás idos e io ados a i a o tie e u a de t es fo as de los fa to es HIF α α : la forma salvaje, sin modificaciones, la forma Doble Mutadas (Dm), en la cual las prolinas están mutadas a alaninas, las cuales no pueden ser hidroxiladas, impidiendo la degradación de HIF y haciendo que la proteína esté activa tanto en hipoxia como en normoxia; y la forma Triple Mutada (Tm), en la cual no sólo las dos prolinas están mutadas a alaninas, sino que también el residuo asparagina del Dominio de Transactivación (TAD, por su sigla en inglés) está mutado a alanina, lo cual conlleva que tampoco pueda ser hidroxilado y consecuentemente esté siempre activo, interactuando positivamente con co-activadores, durante la regulación génica. La estructura de los factores HIF puede observarse en la Figura 9. En resumen, los HIF Dm tienen actividad en condiciones de hipoxia, pero también de normoxia; y los HIF Tm no sólo tienen actividad en ambas condiciones, pero su capacidad de activar la transcripción de sus genes blanco está aumentada. A partir de estos plásmidos se obtuvo la secuencia de los 3 tipos de tanto HIF- α o o HIF- α. Se utilizaron los primers indicados en la Tabla 6 para amplificar las secuencias de los factores HIF, a partir de los vectores pTRE2Hyg o pCDNA3, donde estaban clonados. Se utilizó el programa LaserGene SeqBuilder para abrir archivos de secuencias, diseñar primers y visualizar los resultados esperados de tratamientos con enzimas de restricción (ER). - 50 - MATERIALES Y MÉTODOS Tabla 6. Primers utilizados para amplificar las secuencias de los factores HIF wild type o con mutaciones, a partir de los plásmidos que los contienen Factor HIF HIF- α HIF- α Dirección Nombre del Primer del Primer Forward hif1aMA Reverse hif1aCB Secuencia 5'-3' GAGAGAGACGCGTGGCATGGAGGGCGCCGGCGGCGCGAAC Forward hif2aMA GAGAGAGACGCGTGGCATGACAGCTGACAA Reverse hif2aCB ACACACATCGATTCAGGTGGCCTGGTCCAGGGCTC ACACACATCGATTCAGTTAACTTGATCCAAAGCTC Las secuencias se clonaron en el plásmido de expresión NITSC, entre los sitios de restricción para MluI y ClaI en su Multiple Cloning Site (MCS). Este vector posee la secuencia para la proteína fluorescente YFP antes del MCS. Además el inserto queda bajo control Tet-Off, por lo que podemos apagar su expresión utilizando un agente modulador, como la Doxiciclina, un antibiótico de la clase tetraciclina. Un mapa básico del vector se muestra en la Figura 11. En la Tabla 7 se describe la conformación de la secuencia de los primers utilizados, a fin de que el amplicón de HIF esté flanqueado por los sitios de restricción apropiados, contenga la secuencia del factor HIF, y permita el libre movimiento de la proteína de fusión YFP-HIF. Tabla 7. Armado de las secuencia de los primers para amplificar los factores HIF. Secuencia GGC Descripción Secuencia para soporte de las ER. Se alterna purina y pirimidina para evitar formación de estructuras secundarias. Diferente en primer F y R, para evitar la formación de dímeros de primers. Código para el aminoácido Glicina, pequeño y no polar, para permitir movimiento libre entre la proteína YFP y la proteína inserto. Es el puente entre el sito para la enzima de restricción y el Marco Abierto de Lectura (ORF, por su sigla en inglés). XXX Secuencia codificante de factor HIF. ACGCGT Sitio para ER MluI ATCGAT Sitio para ER ClaI XXXXXX - 51 - MATERIALES Y MÉTODOS Figura 11. Mapa del vector NITSC. Neo: Resistencia para Neomicina. LTR: Long Terminal Repeats, utilizados por la particular viral para insertar su material genético en el genoma de la célula huésped. I: Promotor constitutivo para TTA. TTA: Tetracycline-controlled Transactivator. To: Promotor Tet-Off (TRE, Tetracycline Response Element). YFP: Proteína fluorescente YFP. Los vectores NITSC-YFP-HIF generados fueron los siguientes: AHIF1wt: HIF1α wt AHIF2wt: HIF2α wt AHIF1: HIF1α-Dm AHIF2: HIF2α-Dm YHIF10: HIF1α-Tm YHIF20: HIF2α-Tm Los vectores generados fueron secuenciados en sus extremos con los primers hif1aMA, hif1aCB, hif2aMA y hif2aCB; y luego en su totalidad para confirmar su identidad. Las muestras fueron preparadas en el laboratorio Cellular Reprogramming Laboratory (CRL) del Departamento de Animal Science de la Michigan State University (MSU) y la secuenciación fue realizada en el servicio de secuenciación del Genomics Core del Research Technology Support Facility de la MSU. En la Tabla 8 se indican los primers utilizados para las secuenciaciones totales. Los resultados de las secuenciaciones fueron analizados con el programa LaserGene SeqMan, y se confirmó la correcta clonación de las secuencias de los factores HIF wt o mutados, en el plásmido NITSC. - 52 - MATERIALES Y MÉTODOS Tabla 8. Primers utilizados para la secuenciación de los amplicones obtenidos a partir de los vectores iniciales, al amplificar las secuencias de los factores HIF. HIF- α Primer 5’- ’ ATGGAGGGCGCCGGCGGCGC ATTAACTCAGTTTGAACTAA ATGAAAGAATTACCGAATT AAGATACAAGTAGCCTCTTT ATAGTGATATGGTCAATGAA CCTACCCACATACATAAAGA AACAGAATGGAATGGAGCAA HIF- α Cobertura de bases del gen desde el TSS 1-20 363-383 767-785 1142-1161 1571-1590 1930-1949 285-2304 Primer 5’- ’ ATGACAGCTGACAAGGAGAA TTCGTGAGAACCTGAGTCTC AGCTGCTTGGCCGCTCAGCC CAAGGGGGCTGTGTCTGAGA ATGGACACAGAGGCCAAGGA ACCGGCCCATGTCCTCCATC ATGCCGGACAAGCCACTGAG Cobertura de bases del gen desde el TSS 1-20 428-447 812-831 1134-1153 1519-1538 1802-1821 2251-2270 Transducción con YHIF10 y YHIF20 Se realizó la transfección para la generación de vectores adenovirales con el plásmido YHIF y los plásmidos helper GP (Gag-Pol) y VSV-G, como se describió anteriormente por co-precipitación con fosfato de calcio en células HEK-293T. Brevemente se combinó el ADN plasmídico en una proporción 2,5:1,5:1 YHIF:GP:VSV-G, se agregó CaCl2 2M y buffer HBS 2X para una relación 1:1 en la mezcla final, se mezcló y se dejó reposar durante 5-10 minutos. A continuación se agregó la mezcla a las células HEK-293T gota a gota y se incubó en incubadora de cultivo celular durante 8 horas, para luego cambiar el medio DMEM SFB 20% por medio fresco. Se recolectó el sobrenadante con vector lentiviral a las 48 y 72 horas post-transfección, se filtró a través de un filtro de poro de 0,45 µm de baja adhesión y se concentró en una ultracentrífuga, o se utilizó sobrenadante fresco. El concentrado fue alicuotado, almacenado a -80° C y utilizado dentro de 12 meses. Se evaluó la eficiencia de transducción del concentrado lentiviral en las líneas de fibroblastos JC-G y IN10, definiéndola como: Número de partículas lentivirales/μl = Nú e o de núcleos Oct4+ DAPI+ / μl de o e t ado utilizado pa a el e sa o. Tratamiento con Doxiciclina Se plaquearon 250,000 fibroblastos JC-G o IN10 por pocillo, en una placa multipocillo de 6 pocillos. A las 24 horas (día 0), cada pocillo fue tratado con un vector distinto: el vector YFP que sólo lleva la proteína YFP clonada en el plásmido NITSC, el vector YHIF10 y el vector YHIF20. La - 53 - MATERIALES Y MÉTODOS generación de partículas virales se realizó como se describió anteriormente, en este caso en placas de cultivo de 100 mm de diámetro, y la transducción fue realizada con 2 ml de sobrenadante fresco sin concentrar. Luego (día 1), las células fueron tratadas durante 11 días con el antibiótico G418 50 mg/ml para seleccionar las células que fueron transducidas exitosamente. El día 4 las células fueron pasadas en una relación 1:3. A continuación (a partir del día 11) se dejaron transcurrir 6 días de recuperación sin ningún antibiótico, para permitir que las células seleccionadas se dividan. Finalmente se realizó un tratamiento con Doxiciclina 1 µg/ml de 13 días (a partir del día 17). Los pocillos Control sólo fueron transducidos con el plásmido correspondiente, sin recibir el tratamiento con Doxiciclina. Se contó el número total de células remanentes en cada pocillo al cabo de 30 días (el último día de tratamiento con Doxiciclina). A lo largo de todo el proceso, las células fueron pasadas según fue necesario cuando la confluencia alcanzó 80-90%. En todos los casos, se pasaron ambas condiciones, Control y Doxiciclina. Análisis Estadístico Todos los resultados se expresan como Media ± Error Estándar. En todos los casos se consideraron estadísticamente significativos valores p por debajo de 0,05. Se utilizó la prueba t de Student para determinar diferencias significativas entre las medias, excepto para el estudio de expresión del transgen STEMCCA, donde se aplicó una ANOVA de una vía, seguida de comparaciones múltiples de Dunnett; y para el estudio de eficiencia de reprogramación con el vector OKSIM bajo distintos Protocolos, la citometría de flujo de la expresión de NANOG durante la reprogramación; y el análisis de viabilidad de exclusión de azul de Tripán, donde se utilizó una ANOVA de dos vías, seguida de comparaciones múltiples de Fisher. Normas Éticas El Comité de Ética de FLENI revisó y aprobó todos los procedimientos que involucraban células humanas, utilizados en el presente estudio, incluyendo los protocolos de derivación de células y extracción y manipulación de material humano. Para los análisis de formación de teratomas, los experimentos se realizaron de acuerdo a normas éticas locales y en estricta conformidad con la - 54 - MATERIALES Y MÉTODOS guía para el cuidado y uso de animales de laboratorio (Guide for the Care and Use of Laboratory Animals) del National Institutes of Health, de los EE.UU. El protocolo fue aprobado y supervisado por la CICUAL de la Facultad de Ciencias Exactas y Naturales, Universidad de Buenos Aires. Se hicieron todos los esfuerzos para minimizar el sufrimiento. Detección de Micoplasma Como regla general se controló cada semana, la presencia de Micoplasma sp en todas las líneas celulares y cultivos primarios utilizados, dado que este microorganismo afecta considerablemente las propiedades y comportamiento de las CM. Para ello se realizó una extracción de ADN genómico de las células a analizar, seguida de una reacción de PCR con primers específicos para un gen característico de Micoplasma sp. Sólo se utilizaron células libres de micoplasma para todos los experimentos. Extracción de ADN genómico Se realizó la extracción de ADN genómico, utilizando el kit Wizard Genomic DNA Purification Kit (Promega). Brevemente, se cosecharon las células a controlar a partir de una placa de cultivo de 10 cm de diámetro, removiendo el medio, lavando con PBS y levantándolas de la placa con una espátula de plástico, transfiriendo luego una décima parte de la suspensión a un tubo eppendorf. Se centrifugó a 14.000 xg durante 10 segundos, se removió el sobrenadante y se resuspendió el pellet celular en PBS. El lavado se repitió 2 veces y finalmente se resuspendieron las células en Solución de Lisis de Núcleos, homogeneizado con pipeta automática. Luego se agregaron Solución de ARNasa al lisado y se homogeneizó nuevamente. Se incubó a 37° C durante 30 minutos y luego se enfrió a TA. A continuación se agregó Solución de Precipitación de Proteínas, mezclando con vórtex. Se dejó enfriar en hielo durante 5 minutos y se centrifugó a 14.000 xg durante 4 minutos. El sobrenadante fue transferido a un tubo nuevo conteniendo isopropanol a TA, mezclando suavemente por inversión. Se centrifugó a 14.000 xg durante 1 minuto y se descartó el sobrenadante, agregando luego etanol 70% a TA. Se mezcló suavemente por inversión y se centrifugó a 14.000 xg durante 1 minuto. Se removió el etanol y se dejó secar el pellet al aire por 30 minutos. Al cabo de este tiempo se rehidrató el ADN genómico en Solución de Rehidratación de ADN, incubando durante 1 hora a 65° C. - 55 - MATERIALES Y MÉTODOS Reacción de PCR para Micoplasma La reacción se ealizó e u olu e fi al de 5 μl. Se colocaron los siguientes componentes en u tu o de PCR: ,5 μl de Buffe de PCR Tris-HCl 200 mM [pH 8.4], KCl 500 mM), 1 μl de MgCl2 50 mM, 0,5 μl de mezcla de dNTPs 10 mM (10 mM cada uno: dATP, dGTP, dCTP y dTTP a pH neutro), 2,5 μl de Primer Forward 20 μM, 2.5 μl de Primer Reverse 20 μM, 0,2 μl de Polimerasa Taq DNA Polimerase 5 U/μl (Invitrogen) y 2 μl de ADN genómico, completando el volumen a 25 μl con agua libre de ARNasas. Las secuencias de los primers específicos para Micoplasma sp se especifican en la Tabla 9. El tamaño del amplicón es de aproximadamente 400 pb, dependiendo de la cepa de Micoplasma sp. Tabla 9. Secuencia de los primers específicos para Micoplasma sp. Secuencia del Primer 5’ a ’ Forward ACACCATGGGAGYTGGTAAT Reverse CTTCWTCGACTTYCAGACCCAAGGCAT El programa de PCR utilizado fue el que se detalla a continuación: Desnaturalización inicial y activación de la polimerasa: 5 minutos a 94° C Desnaturalización: 30 segundos a 94° C Apareamiento: 30 segundos a 55° C 35 ciclos Extensión: 40 segundos a 72° C Extensión Final: 5 minutos a 72° C Al finalizar las reacciones de PCR, se visualizaron los productos mediante electroforesis en geles de agarosa 0,8%, buscando la presencia o ausencia de bandas correspondientes a genes de Micoplasma en las muestras analizadas y en comparación con controles positivos comerciales Mycoplasma sp Co t ol DNA ATCC de distintas cepas del microorganismo. - 56 - RESULTADOS - 57 - RESULTADOS OBTENCIÓN DE CÉLULAS SOMÁTICAS PARA REPROGRAMACIÓN Con el propósito de reprogramar células humanas para la obtención de CMPi se decidió estudiar la factibilidad de obtener y reprogramar diferentes tipos celulares. Para ello el primer objetivo del presente trabajo fue la obtención de tejidos, para la derivación de cultivos primarios de células somáticas para desdiferenciar. A continuación se indican los diferentes tipos celulares que se obtuvieron: • Fibroblastos subdérmicos • Queratinocitos epidérmicos • Células Mononucleares de Sangre Periférica En cirugías de rutina y bajo consentimiento informado y protocolos aprobados por el Comité de Ética de FLENI, se obtuvieron muestras de biopsias de piel de donantes sanos y se establecieron fibroblastos subdérmicos y queratinocitos, como se describió en la sección de Materiales y Métodos La obtención de CMSP se realizó a partir de sangre entera fresca, extraída de donantes sanos. OBTENCIÓN DE FIBROBLASTOS SUBDÉRMICOS Cultivos primarios de fibroblastos obtenidos: 1. HFF-FM: Obtenida a partir de una muestra de piel de prepucio de un donante de sexo masculino de 3 años de edad. Los fibroblastos obtenidos se muestran en la Figura 12. - 58 - RESULTADOS Figura 12. Fibroblastos humanos de prepucio HFF FM. Se muestran imágenes de microscopía de contraste de fases de fibroblastos HFF-FM en el pasaje 0, antes de ser transferido y congelado por primera vez. Barras de escala: 200 µm. Los siguientes cultivos primarios fueron derivados durante el curso de este trabajo, pero no fueron utilizados para reprogramación. 2. HF-PM: Obtenidas a partir de una muestra de piel de brazo de una donante sana, de 36 años de edad, sexo femenino. 3. HF21F: Obtenidas a partir de una muestra de piel de tórax anterior de una donante con cardiomiopatía, de 69 años de edad, sexo femenino. 4. HF21M: Obtenidas a partir de una muestra de piel de tórax anterior de un donante sano, de 26 años de edad, sexo masculino. OBTENCIÓN DE QUERATINOCITOS Se intentó la obtención tanto de queratinocitos de epidermis, como queratinocitos de bulbos pilosos. Como se detalla a continuación y por las razones que se describen, en ningún caso se intentó generar CMPi a partir de los queratinocitos obtenidos. OBTENCIÓN DE QUERATINOCITOS EPIDÉRMICOS En el caso de los queratinocitos de epidermis, se obtuvieron células en cultivo a partir de la Muestra #1, que consistió de una muestra de cuero cabelludo, y a partir de la Muestra #4, que consistió de una muestra de cuero cabelludo (biopsia por punch) de un paciente de sexo y edad - 59 - RESULTADOS desconocidos; pero en ambos casos fue difícil su cultivo y amplificación, pudiéndose sólo amplificar las células durante 2 pasajes. Los queratinocitos obtenidos se observan en las Figuras 13 y 15. En el Día 15, justo antes del primer pasaje (del pasaje 0 al pasaje 1) y congelación para stock, se realizó una tinción para citoqueratinas sobre las células obtenidas a partir de la Muestra #1. Los resultados obtenidos se muestran en la Figura 14. Distintas citoqueratinas están presentes en diferentes epitelios, componiendo parte del citoesqueleto de los queratinocitos. Se encontró que las células derivadas son en efecto células queratinocíticas, debido a la presencia de estas proteínas. Sin embargo, la dificultad en su pasaje y expansión impuso una gran dificultad para obtener un número suficiente de células para reprogramar, razón por la cual se continuó en este trabajo con la reprogramación de fibroblastos subdérmicos. Para ambas muestras, las células del pasaje 2 se adhirieron, pero durante los siguientes días se fueron despegando de la matriz, hasta no encontrarse más células en adherencia. Se llegó a la conclusión de que estas células deberían ser transducidas en pasaje 0 ó 1, preferentemente en 0, dado que habría más actividad proliferativa. Las Muestras #2 y #3 no fueron capaces de generar células adheridas en cultivo. Ambas fueron muestras de cuero cabelludo de pacientes de sexo y edad desconocidos. Los cultivos se mantuvieron durante 30 días, antes de ser descartados. - 60 - RESULTADOS Figura 13. Queratinocitos de epidermis. Muestra #1. Se muestra una imagen de microscopía de contraste de fases correspondiente a queratinocitos en pasaje 0, a distintos tiempos post-plaqueo del tejido obtenido, antes de ser transferido y congelado por primera vez. Barras de escala: 200 µm. - 61 - RESULTADOS Figura 14. Detección de citoquinas mediante inmunofluorescencia en queratinocitos de epidermis 15 días post-plaqueo. Muestra #1. Se muestra una imagen de microscopía de contraste de fases (Fase), una tinción con DAPI y la citoqueratina (CK) correspondiente. Barras de escala: 200 µm. - 62 - RESULTADOS Figura 15. Queratinocitos de epidermis. Muestra #4. Se muestran imágenes de microscopía de contraste de fases de queratinocitos en pasaje 0, a distintos tiempos post-plaqueo del tejido obtenido, antes de ser transferido y congelado por primera vez. Barras de escala: 200 µm. OBTENCIÓN DE QUERATINOCITOS DE FOLÍCULOS PILOSOS Se obtuvieron alrededor de 100 pelos de distintas zonas del cuerpo, mediante extracción con pinza de depilar, como se describe en la sección de Materiales y Métodos, y se los procesó como se detalló anteriormente. En ninguno de los casos, ni de las variantes del protocolo, se obtuvieron células adheridas. Los cultivos fueron mantenidos por más de 30 días, antes de ser descartados. En la Figura 16 se muestran los pelos en cultivo. Figura 16. Pelos en cultivo. Se muestran imágenes de microscopía de contraste de fases de pelos en cultivo, luego de ser extraídos y tratados. Se observa parte de la Vaina Externa de la Raíz (ORS, por su sigla en inglés, Outer Root Sheath) adherida a la placa de cultivo. Barras de escala: 200 µm. - 63 - RESULTADOS OBTENCIÓN DE CÉLULAS MONONUCLEARES DE SANGRE PERIFÉRICA A partir de sangre periférica y mediante centrifugación en Ficoll-Hypaque, se obtuvieron células mononucleares en cultivo. Está reportado que un cultivo mitóticamente activo aporta más células capaces de ser reprogramadas [122], dado que una eficiente transducción precisa de un cultivo en división [135-137]. Por esta razón, posteriormente, en algunos casos las células fueron tratadas con IL-2 y anticuerpos anti-CD3, previo a la reprogramación, a fin de activar e incentivar la proliferación de clones linfocitarios, obteniendo un cultivo enriquecido en linfocitos. La preparación de CMSP/linfocitos se repitió 15 veces y se intentaron generar líneas de CMPi a partir de ellos, pero no se obtuvieron colonias en ningún intento, por lo cual todavía no hemos podido establecer líneas de CMPi a partir de CMSP, como se describe más adelante. En un caso, se observaron formaciones que podrían haber sido pre-CMPi, pero ninguna de ellas progresó hasta la formación de una colonia. Estas formaciones se muestran en la Figura 18. En las Figuras 17 y 18 se observan los cultivos de CMSP y linfocitos obtenidos. Los cultivos se mantuvieron durante 30 días, antes de ser descartados. Figura 17. Células Mononucleares de Sangre Periférica en cultivo. Se muestran imágenes de microscopía de campo claro de CMSP en cultivo, en el día 0, correspondiente al plaqueo post-extracción y procesamiento. CMSP: Células Mononucleares de Sangre Periférica. Barras de escala: 200 µm. - 64 - RESULTADOS Figura 18. Cultivo de células mononucleares de sangre periférica enriquecido en linfocitos. Se muestran imágenes de microscopía de contraste de fases de cultivos de CMSP luego del tratamiento para enriquecerlos en linfocitos, a distintos tiempos post-extracción y procesamiento. CMSP: Células Mononucleares de Sangre Periférica. Barras de escala: 200 µm. GENERACIÓN DE CMPi Al comienzo de este trabajo no se contaba con el conocimiento ni la experiencia para generar CMPi, ya que dichas habilidades no estaban desarrolladas en el país. En el transcurso de las investigaciones para este estudio, se experimentó con la generación de CMPi en distintas condiciones. Se utilizaron distintas células somáticas de partida, distintos tipos celulares, distintos vectores de reprogramación, distintos protocolos y moléculas modificadores de la cromatina. En algunos casos, la generación de CMPi no rindió frutos y no se observaron colonias. En otros casos se lograron generar colonias, pero no se las pudo expandir para establecer una línea. Dada la colaboración con el Dr. José Cibelli, investigador principal en la MSU, se realizó una pasantía en su laboratorio, el Cellular Reprogramming Laboratory (CRL) de - 65 - RESULTADOS la MSU en Michigan, EE.UU., la cual fue de invaluable ayuda para obtener las habilidades necesarias para llevar la técnica a cabo. El fin de dicha pasantía fue adquirir estas habilidades y volver a la Argentina, no solamente con el conocimiento, sino también con la experiencia para generar un protocolo confiable y reproducible para la generación de CMPi, mediante transducción con diferentes vectores [73]. Finalmente, luego de adquirir dicha experiencia, especialmente en cuanto a sortear los pequeños detalles del protocolo a los que el investigador se enfrenta al generar CMPi, como la identificación, selección y pasaje inicial de colonias jóvenes, o la manipulación inicial, se logró establecer líneas de CMPi a partir de las colonias generadas. A continuación se describen los intentos realizados y las líneas que se establecieron. GENERACIÓN INICIAL DE CMPi EN EL CELLULAR REPROGRAMMING LABORATORY: En primera instancia, se investigó la infectividad de los vectores virales a fin de encontrar la técnica más eficiente de generación de CMPi con el vector lentiviral OKSIM en cuanto a concentración y volumen de vector a utilizar, a través de la cuantificación de la inmunofluorescencia de POU5F1, uno de los genes codificados en este vector lentiviral, luego de la transducción. El vector lentiviral OKSIM lleva los genes POU5F1, KLF4, SOX2 y C-MYC, que serán expresados luego de la transducción a una célula eucariota [123]. Los plásmidos para la generación de las partículas lentivirales por transfección, fueron generosamente proporcionados por el Dr. Steve Suhr y el Dr. José Cibelli del CRL, junto con los plásmidos helper psPAX y VSV-G, necesarios para la formación de la partícula. Se estudiaron muestras del vector lentiviral generado y concentrado mediante ultracentrifugación en la University of Michigan (UofM); vector fresco producido por transfección con el plásmido OKSIM y los plásmidos helper en células HEK-293T para este experimento, ya sea sin concentrar o concentrado por ultracentifugación, y en distintos volúmenes. Los resultados se muestran en la Tabla 11. Se encontró que la realización de un paso de spinfection aumentó dramáticamente el porcentaje de células transducidas por el vector, por lo cual este paso no debería ser salteado nunca en el protocolo final. Además, se observó que el vector lentiviral fresco fue más efectivo sin concentrar, mientras que sea utilizado en un volumen mayor de 1 ml, aumentando también el porcentaje de células transducidas. Es posible que la concentración de otros factores o incluso - 66 - RESULTADOS de componentes del medio de cultivo durante la ultracentrifugación afecten los resultados. Además el virus producido por la UofM estaba cerca de los 12 meses de antigüedad, por lo cual quizás había perdido su infectividad, siendo posible que un virus nuevo concentrado por especialistas en virología haya sido más eficiente incluso que el sobrenadante fresco sin cocentrar. Lo que estos resultados nos indican es que es posible utilizar sobrenadante fresco sin concentrar en ultracentrífuga, obteniendo resultados de infectividad aceptables. Tabla 11. Infectividad del vector lentiviral OKSIM. Sin Spinfection (%) Control (sin vector lentiviral) 0 Spinfection (%) 0 UofM SM Concentrado SM No Concentrado 1 ml SM No Concentrado 4 ml 0.77 1.14 1.99 5.45 1.85 44.79 4.27 53.33 UofM: Vector producido por la University of Michigan. SM: Vector fresco producido para el experimento. Asimismo, se probó un protocolo de concentración viral nuevo para el laboratorio CRL, la concentración por precipitación con PEG, tal como se describe en el compendio Current Protocols in Molecular Biology, Unidad 9.12 Protocolo Alternativo 1 [138]. Se realizó un análisis de la infectividad del virus por inmunofluorescencia de POU5F1 post-transducción, y se encontró que la concentración con PEG pareció aumentar la eficiencia de transducción, en comparación con la utilización de sobrenadante viral fresco sin concentrar, tal como se muestra en la Figura 19. En síntesis, con el fin de generar el protocolo final de generación de CMPi, evaluamos diferentes alternativas para la transducción, utilizando sobrenadante conteniendo las partículas lentivirales fresco, congelado, sin concentrar o concentrado por distintos métodos. La conclusión obtenida fue que es posible concentrar el vector lentiviral por medios alternativos a la ultracentrifugación. - 67 - RESULTADOS Figura 19. Infectividad del vector lentiviral OKSIM concentrado por precipitación con PEG. Se muestran imágenes de microscopía de contraste de fases y fluorescencia de POU5F1 en fibroblastos JC-G, 24 horas post-transducción. Volumen de sobrenadante fresco sin concentrar: 2,5 ml. Volumen de sobrenadante concentrado con PEG: 50 µl en 2,5 ml de medio de cultivo. Barras de escala: 200 µm. Luego de haber comprobado la infectividad del vector OKSIM, se procedió a intentar la reprogramación de cultivos primarios con dicho vector, a fin de obtener experiencia en la técnica e investigar distintas condiciones de reprogramación y sus consecuencias sobre la eficiencia de reprogramación. Luego de la generación exitosa de varias líneas de CMPi, se estudió si las líneas obtenidas mantenían las propiedades fundamentales de CMP. Para ello se estudió la auto-renovación manteniendo el estado pluripotente y la pluripotencia. El estudio del estado pluripotente se realizó mediante el análisis por inmunofluorescencia de la presencia de marcadores del dicho estadío. Por otra parte, para el análisis de la pluripotencia, se estudió su capacidad de diferenciación a los tres linajes embrionarios, tanto mediante un protocolo in vitro y posterior análisis de marcadores, como mediante la obtención in vivo de teratomas por inyección subcutánea en ratones inmunosuprimidos y posterior análisis histológico. - 68 - RESULTADOS Finalmente, se integraron los resultados de las generaciones de CMPi en distintos medios de cultivo, con el fin de investigar cuál condición proveía una eficiencia de reprogramación más alta. Se seleccionó el medio de cultivo DMEM/F12 + NaBu para la futura generación de CMPi. A continuación se detallan las diferentes instancias de reprogramación realizadas y sus resultados. Líneas de CMPi establecidas: Vector lentiviral OKSIM – Línea establecida: iPSCRL4 Células somáticas reprogramadas: Fibroblastos HDNF Protocolos: Protocolo 1 (DMEM/F12), Protocolo 1 + NaBu (DMEM/F12 + NaBu) El butirato de sodio (NaBu) es un remodelador de la cromatina, que actúa a través de la inhibición de la actividad de las deacetilasas de histonas (HDAC), lo cual resulta en una hiperacetilación de las histonas [139, 140]. Esta modificación de las histonas promueve un estado relajado de la cromatina, asociado con altos niveles de transcripción [141-143]. Se ha reportado con anterioridad la utilización de NaBu durante la reprogramación, a fin de mejorar el proceso [124, 127, 128]. En la Figura 20 se observan esbozos de colonias, a medida que surgen, en Día 8 post-transducción. - 69 - RESULTADOS Figura 20. Colonias de CMPi iPSCRL4, en cultivo. Se muestran imágenes de microscopía de contraste de fases de colonias de CMPi iPSCRL4 a poco tiempo de surgir en cultivo, 8 días post-transducción. Barras de escala: 200 µm. Vector lentiviral OKSIM – Línea establecida: iPSCRL2 Células somáticas reprogramadas: Fibroblastos JC-S Protocolos: Protocolo 1 (DMEM/F12), Protocolo 1 + NaBu, Protocolo 2 (DMEM SFB 20%) + NaBu En la Figura 21 se muestran las colonias generadas, a los 14 días post-transducción, antes de ser pasadas individualmente. Luego de 36 días, los distintos clones establecidos comenzaron a generar una monocapa de células de aspecto pluripotente, es decir se observaron células pequeñas y refringentes con una alta relación núcleo : citoplasma, pero en monocapa. Por ello se descartaron los cultivos, dado que el crecimiento en monocapa no es una característica de CM, las cuales crecen en colonias como una estructura en dos dimensiones con bordes definidos extendiéndose a la periferia - 70 - RESULTADOS Figura 21. Colonias de CMPi iPSCRL1, en cultivo. Se muestran imágenes de microscopía de contraste de fases de colonias de CMPi iPSCRL1 a los 14 días post-transducción. Barras de escala: 200 µm. Vector lentiviral OKSIM – Línea establecida: iPSCRL3 Células somáticas reprogramadas: Fibroblastos JC-G Protocolos: Protocolo 1 (DMEM/F12), Protocolo 1 + NaBu, Protocolo 2 (DMEM SFB 20%) + NaBu En la Figura 22 se muestran colonias de iPSCRL3 generadas, a distintos tiempos posttransducción. - 71 - RESULTADOS Figura 22. Colonias de CMPi iPSCRL3, en cultivo. Se muestran imágenes de microscopía de contraste de fases de colonias de CMPi iPSCRL3 en cultivo, a distintos tiempos post-transducción. Barras de escala: 200 µm. La línea de CMPi iPSCRL3 fue caracterizada de forma preliminar, mostrando expresión de marcadores de pluripotencia evaluados por inmunofluorescencia, durante el pasaje 6. Además, con el propósito de estudiar la pluripotencia, también por inmunofluorescencia, al ser diferenciada durante el pasaje 13, la línea mostró expresión de marcadores de las tres capas germinales. Los resultados se pueden observar en las Figuras 23 y 24. - 72 - RESULTADOS Figura 23. Presencia de marcadores de pluripotencia evaluados por inmunofluorescencia en CMPi iPSCRL3. Se muestran imágenes de microscopía de contraste de fases y de inmunofluorescencia de tinción de DAPI y de marcadores, en colonias indiferenciadas de CMPi iPSCRL3 en cultivo. Barras de escala: 200 µm. Figura 24. Presencia de marcadores de las tres capas germinales evaluados por inmunofluorescencia en CE obtenidos por diferenciación de CMPi iPSCRL3. Se muestran imágenes de microscopía de contraste de fases de CE obtenidos a partir de CMPi en pasaje 11 (arriba) e imágenes de microscopía de contraste de fases e inmunofluorescencia de marcadores de las tres capas germinales y tinción de DAPI sobre CE derivados de CMPi en pasaje 13 (abajo). CE: cuerpo embrioide. p: pasaje. AFP: marcador del linaje endodérmico. MYH, VIM: marcadores del linaje mesodérmico. GFAP, PAX6, TUJ1: marcadores del linaje ectodérmico. Barras de escala: 200 µm. - 73 - RESULTADOS A continuación y a fin de cuantificar cuál de los Protocolos fue más efectivo utilizando el vector OKSIM, se investigó la eficiencia de generación de CMPi, bajo los tres distintos Protocolos estudiados. Se incluyeron los resultados de las generaciones de CMPi mencionadas anteriormente y se realizaron 4 repeticiones más a fin de tener un tamaño muestra apropiado. Los resultados se muestran en la Figura 25. Figura 25. Eficiencia de generación de CMPi. p0: Colonias generadas al Día 14 post-transducción (pasaje 0). p5: Colonias transferidas subsistentes luego de 5 pasajes. Se representa la media ± SEM de 2-5 experimentos independientes, según el protocolo. * = p < 0.05, con respecto al Protocolo 1 (DMEM/F12), diferencia significativa para el efecto Protocolo, por ANOVA de 2 vías. Los Protocolos Protocolo 1 (DMEM/F12) + NaBu y Protocolo 1 (DMEM SFB 20%) + NaBu, no resultaron significativamente distintos. A partir de lo observado en la Figura 25, se concluyó que a pesar de obtenerse una gran cantidad de colonias al no usar NaBu, muchas de estas colonias no progresan más allá de unos pocos pasajes (menos de 5). La utilización de NaBu, a pesar de disminuir la eficiencia inicial de formación de colonias de CMPi, probablemente debido a su efecto inhibitorio de la división celular, permite obtener más colonias con posibilidades de ser establecidas como líneas de CMPi, ya que las colonias formadas exhibieron una morfología más similar a la de CME, y - 74 - RESULTADOS mantuvieron características de CME a lo largo de los pasajes (después del pasaje 1 y hasta el pasaje 5). Es posible que el efecto de la relajación de la cromatina producido por el NaBu sea en parte responsable de la activación de la transcripción de genes endógenos del estado pluripotente, luego de la transducción y de la expresión de genes exógenos, y que esto sea lo que permite la generación de colonias bona fide. Al no haberse encontrado diferencias significativas entre los tratamientos con NaBu (DMEM/F12 versus DMEM SFB 20%), se tiende a favorecer el uso de DMEM/F12 por su amplia utilización en el cultivo de células madre. En conclusión, el Protocolo final involucrará el uso de DMEM/F12 como medio de cultivo y la utilización de NaBu. Una vez de vuelta en los laboratorios de Buenos Aires, y habiendo obtenido el plásmido OKSIM mediante la firma de un Material Transfer Agreement, se intentó la producción del vector lentiviral y la generación de CMPi con dicho vector, sin éxito. Se evaluó la infectividad del sobrenadante de transfección generado, encontrándose que la infectividad sobre fibroblastos HFF-FM era muy baja o nula, en todos los casos, como se muestra en la Figura 26. No se obtuvieron colonias en ningún intento. - 75 - RESULTADOS Figura 26. Infectividad del vector lentiviral OKSIM en los laboratorios de Buenos Aires. Evaluación de la eficiencia de transducción. Se muestran imágenes de inmunofluorescencia de POU5F1, y de DAPI, 24 horas post-transducción. Barras de escala: 200 µm. Por los problemas encontrados al tratar de generar CMPi con el vector OKSIM en Buenos Aires, se decidió continuar con el vector STEMCCA, ya disponible en los laboratorios del Instituto FLENI y del Departamento de Química Biológica de la UBA. El cassette lentiviral EF1ahSTEMCCA-loxP (STEMCCA) lleva los genes POU5F1, KLF4, SOX2 y C-MYC, que son expresados luego de la transducción a una célula eucariota; y el cassette lentiviral EF1-hSTEMCCA-RedLightloxp (STEMCCA RedLight) lleva los mismos genes excepto C-MYC, en cuyo lugar está la proteína fluorescente roja mCherry [73]. Los plásmidos para la generación de las partículas lentivirales por transfección, fueron generosamente proporcionados por el Dr. Gustavo Mostoslavsky, junto con los plásmidos helper (Tat, Rev, Gag/Pol y VSV-G), necesarios para la formación de dichas partículas. Está reportado que la expresión de este vector es silenciada luego de la reprogramación [73], como se detalla más adelante. Durante intentos previos utilizando el vector STEMCCA o STEMCCA-RedLight se habían podido generar colonias de CMPi, pero no establecer líneas. Ahora, utilizando la experiencia obtenida en el laboratorio CRL en USA con el - 76 - RESULTADOS vector OKSIM, se pudieron establecer líneas de CMPi con los vectores STEMCCA. A continuación se detallan los experimentos realizados. EXPERIENCIAS DE GENERACIÓN DE CMPi EN LOS LABORATORIOS DE BUENOS AIRES: A partir de este punto de la investigación se utilizó el protocolo de generación de CMPi con el vector STEMCCA descripto en los Materiales y Métodos, con ciertas diferencias indicadas en cada caso particular. Estas experiencias llevaron a la generación del protocolo final descripto. La utilización de las columnas Amicon en lugar del uso del sobrenadante viral fresco o la concentración por precipitación con PEG fue una modificación introducida a partir del Intento 1, que dio resultados satisfactorios, por lo cual fue utilizada para el protocolo final. Vector lentiviral STEMCCA-RedLight – Intento Preliminar 0 Fuente: Fibroblastos FH No hubo formación de colonias de CMPi. Como se muestra en la Figura 27, no se observó fluorescencia de la proteína mCherry, lo cual indica que la transducción no fue exitosa. No se observó formación de colonias, por lo que dichos cultivos fueron descartados en el día 21 post-transducción. El Ácido Valproico (VPA), al igual que el NaBu es un inhibidor de las deacetilasas de histonas, que promueve un estado laxo de la cromatina y consecuentemente elevada actividad transcripcional [144, 145]. Se ha reportado con anterioridad su uso en reprogramación a fin de mejorar el proceso [69, 146]. - 77 - RESULTADOS Tabla 12. Protocolo de generación de CMPi STEMCCA-RedLight Intento 0 Día 0 Día 1 Día 2 Día 3 Transducción 1 de fibroblastos HFF-FM sobre gelatina bovina en medio DMEM SFB 20%. Transducción 2 de fibroblastos HFF-FM sobre gelatina bovina en medio DMEM SFB 20%. Remoción medio DMEM SFB 20% con virus y reemplazo con medio DMEM SFB 20% sin virus Remoción medio DMEM SFB 20% y reemplazo por medio DMEM/F12 KSR 20% + VPA 0.75 mM. Día 4 Día 5 Día 6 Continúa con medio DMEM/F12 KSR 20% + VPA, cambio todos los días. Continúa con medio DMEM/F12 KSR 20% + VPA, cambio todos los días. Plaqueo de MEFi 1,5 x 6 10 /p100. Pasaje de las células transducidas a MEFi. Utilización de medio DMEM/F12 KSR 10%, Día 6 en adelante. Día 21 Pasar colonias mecánicamente con espátula y tipos, pasando cada una a 1 pocillo individual de placa multipocillo de 12 pocillos. Figura 27. Progreso del cultivo celular para la generación de CMPi a partir de fibroblastos FH, Intento 0, desde el día 2 hasta el día 7 post-transducción con el vector STEMCCA-RedLight. Se muestran imágenes de microscopía de contraste de fases y de fluorescencia de la proteína roja fluorescente mCherry. Barras de escala: 200 µm. - 78 - RESULTADOS Vector lentiviral STEMCCA-RedLight – Intento 1 Fuente: Fibroblastos HFF-FM Línea establecida: CMPi iPS1.5 El protocolo de generación utilizado fue el indicado en la Tabla 10. En la Figura 28 pueden observarse las colonias surgidas y su progreso, hasta el día 21 posttransducción, en el que fueron pasadas manual e individualmente. - 79 - RESULTADOS Figura 28. Progreso de las colonias de CMPi iPS1.5 desde el día 2 hasta el día 21 post-transducción con el vector STEMCCA-RedLight. (A) Imágenes de contraste de fases y de fluorescencia de mCherry de una monocapa de células HEK-293T productora de vector lentiviral y de fibroblastos HFF-FM transducidos, desde el día 2 hasta el día 6 post-transducción. Control: Sin transducción. Barras de escala: 200 µm. (B) Imágenes de contraste de fases de colonias selectas desde el día 13 hasta el día 21 post-transducción. Barras de escala: 200 µm. - 80 - RESULTADOS Luego de varios pasajes, se caracterizó la línea de CMPi iPS1.5 y se encontró que las colonias expresaban marcadores de pluripotencia y poseían alta actividad de fosfatasa alcalina. Los resultados se muestran en las Figuras 29 y 30. Además las células fueron capaces de diferenciarse in vitro, generando células de las tres capas germinales, como se observa en la Figura 31, demostrando ser pluripotente. Figura 29. Expresión de marcadores del estado pluripotente en colonias de la línea de CMPi iPS1.5. Se muestran imágenes de microscopía de contraste de fases y de fluorescencia de expresión de marcadores del estado pluripotente en colonias de CMPi en el día 21 post-transducción. Barras de escala: 200 µm. Figura 30. Actividad de fosfatasa alcalina en colonias de la línea de CMPi iPS1.5. Se muestran imágenes de microscopía de contraste de fases de inmunohistoquímica para fosfatasa alcalina en colonias de CMPi en el día 21 post-transducción. Barras de escala: 200 µm. - 81 - RESULTADOS Figura 31. Expresión de marcadores de las tres capas germinales en CMPi iPS1.5 diferenciadas. Se muestran imágenes de microscopía de contraste de fases y de fluorescencia de marcadores de las tres capas germinales en CE derivados de CMPi por diferenciación durante 14 días, a partir del día 21 posttransducción. GATA-4: marcador del linaje endodérmico. ANP: marcador del linaje mesodérmico. PAX6: marcador del linaje ectodérmico. Barras de escala: 200 µm. A partir de la colonia 11, la cual fue aislada, pasada individualmente y amplificada, se estableció la línea de CMPi iPS1.5. En la Figura 32 puede observarse una colonia de 51 días posttransducción (pasaje 5) de la línea establecida de CMPi iPS1.5. La línea expresó los marcadores de pluripotencia POU5F1 y NANOG, en un ensayo preliminar realizado por única vez por RT-PCR en Tiempo Real, como se muestra en la Figura 33. Como se muestra más adelante, sobre las líneas que fueron seleccionadas para continuar este trabajo, se realizaron las determinaciones por RT-qPCR en al menos tres réplicas biológicas. En todos los casos en los que se incluyó una condición de hipoxia, se utilizó el gen de housekeeping HPRT1 para normalizar los niveles de expresión, ya que éste no se ve afectado por la hipoxia. En todos los otros casos donde no hubo una condición de hipoxia, se utilizó el gen de housekeeping más ampliamente utilizado, GAPDH. - 82 - RESULTADOS Figura 32. Colonia establecida de 51 días post-transducción (pasaje 5) de la línea de CMPi iPS1.5. Se muestra una imagen de microscopía de contraste de fases. Barras de escala: 200 µm. Figura 33. Expresión de los marcadores de pluripotencia POU5F1 y NANOG en CMPi iPS1.5, por RTqPCR. Se grafica la expresión relativa a HFF-FM. HFF-FM: Fibroblastos HFF-FM. Ensayo preliminar. n = 1. Vectores lentivirales STEMCCA y STEMCCA-RedLight – Intento 2 Fuente: Fibroblastos HFF-FM No hubo formación de colonias de CMPi. - 83 - RESULTADOS Tabla 13. Protocolo de generación de CMPi STEMCCA y STEMCCA-RedLight Intento 2 Día 0 Día 1 Día 2 Día 3 Día 4 Día 5 Transducción 1 de fibroblastos HFF-FM sobre gelatina bovina en medio DMEM SFB 20%. Transducción 2 de fibroblastos HFF-FM sobre gelatina bovina en medio DMEM SFB 20%. Remoción medio DMEM SFB 20% con virus y reemplazo con medio DMEM SFB 20% sin virus Remoción medio DMEM SFB 20% y reemplazo por medio DMEM/F12 KSR 20% + VPA 0.75 mM. Continúa con medio DMEM/F12 KSR 20% + VPA, cambio todos los días. Continúa con medio DMEM/F12 KSR 20% + VPA, cambio todos los días. Día 8 Día 9 Día 21 Continúa con medio DMEM/F12 KSR 20% + VPA, cambio todos los días. Plaqueo de MEFi 2 x 6 10 /p100. Pasaje de las células transducidas a MEFi. Utilización de medio DMEM/F12 KSR 10%, Día 6 en adelante. Pasar colonias mecánicamente con espátula y tipos, pasando cada una a 1 pocillo individual de placa multipocillo de 12 pocillos. En la Figura 34 puede observarse el progreso del cultivo de fibroblastos HFF-FM transducidos con los vectores STEMCCA o STEMCCA-RedLight. Se dedujo que la transducción con el vector lentiviral STEMCCA-RedLight fue exitosa, dado que se observó fluorescencia de mCherry en los fibroblastos transducidos. Sin embargo, no se observaron colonias en los cultivos en ninguno de los dos casos, hasta el día 21 post-transducción, momento en el cual dichos cultivos fueron descartados. En conclusión, en este intento, si bien la transducción fue exitosa, no se obtuvo reprogramación. - 84 - RESULTADOS Figura 34. Progreso del cultivo celular para la generación de CMPi a partir de fibroblastos HFF-FM, Intento 2, desde el día 2 hasta el día 6 post-transducción con el vector STEMCCA o STEMCCA-RedLight. Se muestran imágenes de microscopía de contraste de fases y de fluorescencia de la proteína roja fluorescente mCherry. Barras de escala: 200 µm. Vector lentiviral STEMCCA-RedLight – Intento 3 Fuente: Fibroblastos HFF-FM No hubo formación de colonias de CMPi. El protocolo utilizado fue el mismo que se indica en la Tabla 13. En la Figura 34 puede observarse el progreso del cultivo de fibroblastos HFF-FM transducidos con el vector STEMCCARedLight. Se dedujo que la transducción con el vector lentiviral STEMCCA-RedLight fue exitosa, dado que se observó fluorescencia de mCherry en los fibroblastos transducidos. Sin embargo, no se observaron colonias en los cultivos hasta el día 21 post-transducción, momento en el cual dichos cultivos fueron descartados. - 85 - RESULTADOS Figura 35. Progreso del cultivo celular para la generación de CMPi a partir de fibroblastos HFF-FM, Intento 3, desde el día 3 hasta el día 4 post-transducción con el vector STEMCCA-RedLight. Se muestran imágenes de microscopía de contraste de fases y de fluorescencia de la proteína roja fluorescente mCherry. Barras de escala: 200 µm. Vector lentiviral STEMCCA – Intento 4 Fuente: Fibroblastos HFF-FM No se pudieron establecer líneas de CMPi No se observó formación de colonias hasta el día 21 post-transducción, momento en el cual dichos cultivos fueron descartados. Vector lentiviral STEMCCA y STEMCCA-RedLight – Intento 5 Fuente: CMSP frescas No hubo formación de colonias de CMPi. El protocolo utilizado se muestra en la Tabla 14. - 86 - RESULTADOS Tabla 14. Protocolo de generación de CMPi STEMCCA y STEMCCA-RedLight Intento 5 Día 2 Día 0 Día 1 Extracción 10 ml Sangre Entera -> Gradiente Ficoll --> CMSP. ___________________________ Transducción 1 de PBMC/linfocitos en suspensión Medio RPMI SFB 15% + ConA. Remoción Medio RPMI SFB 15% + ConA con virus y reemplazo con Medio RPMI SFB 15% + ConA sin virus. Cambio de medio por centrifugación a 300 xg durante 5 minutos. Día 3 Día 4 Continuar con Medio RPMI SFB 15% + ConA sin virus, cambio día por medio. Día 6 Día 7 Remoción Medio RPMI SFB 15% + ConA con virus y reemplazo con Medio RPMI SFB 15% + ConA sin virus. Replaqueo de las células transducidas en 2 placas: 2 p100 MEFi 2,5 x 106. Reemplazar Medio RPMI SFB 15% + ConA por medio DMEM/F12 KSR 10%, cambio todos los días. Día 5 Transducción 2 de PBMC/linfocitos en suspensión. Medio RPMI SFB 15% + ConA. En la Figura 36 pueden observarse las CMSP en los primeros días post-transducción. No se observó formación de colonias hasta el día 21 post-transducción, momento en el cual dichos cultivos fueron descartados. - 87 - RESULTADOS Figura 36. Progreso del cultivo celular de CMSP transducidas con el vector STEMCCA o STEMCCARedLight para la generación de CMPi, Intento 5, desde el día 0 hasta el día 2 post-transducción. Se muestran imágenes de microscopía de contraste de fases y de fluorescencia de la proteína roja fluorescente mCherry. Barras de escala: 200 µm. Se intentó la generación de CMPi a partir de CMSP en tres oportunidades más, con el protocolo indicado en la Tabla 14. En los dos últimos intentos se trató el cultivo con IL-2 y anticuerpos anti-CD3 para incentivar la proliferación de los clones linfocitarios, estimulando así la transducción y consecuentemente la reprogramación, según el protocolo indicado en la sección Materiales y Métodos. En ningún caso pudieron obtenerse colonias de CMPi a partir de CMSP. Vector lentiviral STEMCCA – Intento 7 Fuente: Fibroblastos HFF-FM No se observó generación de colonias de CMPi - 88 - RESULTADOS Hubo formación inicial de colonias, observadas al día 10 post-transducción, las cuales se muestran en la Figura 37; pero éstas no establecieron líneas de CMPi al ser pasadas individualmente. Figura 37. Colonias en el día 10 post-transducción, generadas a partir de HFF-FM transducidos con el vector STEMCCA para la generación de CMPi, Intento 7. Imágenes de contraste de fases. Barras de escala: 200 µm. A partir de éstas experiencias y con los conocimientos obtenidos, se estableció el protocolo final, descripto en la sección Materiales y Métodos, el cual se utilizó a continuación para el estudio de los efectos de la hipoxia sobre la generación de CMPi. EFECTOS DE LA HIPOXIA SOBRE LA GENERACIÓN DE CMPi Con el fin de estudiar los efectos de la hipoxia sobre la reprogramación de células adultas a un estado pluripotente, obtuvimos fibroblastos de la piel de un donante masculino sano (cultivo primario de fibroblastos HFF-FM) y los reprogramamos utilizando el vector STEMCCA a fin de - 89 - RESULTADOS generar varias líneas de CMPi. Las líneas iPSC-FN2.1 e iPS6.1 se generaron en normoxia (21% O2), y la línea iPSC-FH2.1 se generó en hipoxia (5% O2). Existen antecedentes en la bibliografía que indican que la hipoxia podría aumentar la eficiencia de reprogramación [99-107], sin embargo, ciertos estudios sobre el efecto de la hipoxia sobre CME son altamente contradictorios. El estudio más popular en el tema, realizado por Yoshida et. al. [65] sobre cómo la hipoxia aumenta la eficiencia de reprogramación fue realizado en células de ratón con un modelo limitado, con GFP como gen reportero del promotor de NANOG. Y los pocos estudios realizados en células humanas [64, 94, 102, 115, 147, 148], arrojan también conclusiones contradictorias. Por ello, se decidió estudiar el proceso en nuestro laboratorio y dilucidar qué ocurría en nuestro modelo de generación de CMPi, bajo normoxia e hipoxia. En primera instancia se verificó en un estudio preliminar de una sola repetición, si nuestro sistema de generación de una atmósfera hipóxica era efectivo. Se investigó si el cultivo de fibroblastos en una incubadora de cultivo celular con N2 gaseoso para generar una atmósfera de O2 al 5% generaba un aumento de la expresión de HIF- α. Pa a ello se a alizó la presencia y cantidad de la proteína HIF- α po cuantificación de imágenes de inmunofluorescencia en cultivos de fibroblastos HFF-FM mantenidos en hipoxia durante 7 días. Los resultados se muestran en la Figura 38. Como se observa, HIF-2α se estabilizó ante hipoxia, aumentando la cantidad de la proteína a partir del día 1, llegando a su máximo en el día 2. Luego comienzan a disminuir, manteniéndose siempre elevados hasta el día 7, en relación al día 0 (normoxia). - 90 - RESULTADOS Figura 38. Expresión de la proteína HIF- α en fibroblastos HFF-FM por inmunofluorescencia. Se muestra la cuantificación de la expresión de la proteína HIF- α po ua tifi a ió de i áge es de inmunofluorescencia, a distintos tiempos de exposición a Hipoxia. Día 0: Normoxia. Experimento preliminar, n=1. Normoxia= O2 al 21%. Hipoxia=O2 al 5%. Una vez validado el sistema para generar un estímulo de hipoxia, con el propósito de estudiar los efectos de la hipoxia en el proceso de reprogramación, se generaron las líneas de CMPi iPS6.1, CMPi iPSC-FN2.1 y CMPi iPSC-FH2.1 realizando el protocolo de reprogramación en hipoxia o normoxia, para comparar la eficiencia de reprogramación y las características de las líneas generadas en ambas condiciones. Además, posteriormente se realizó el cultivo de las líneas ya generadas bajo normoxia o hipoxia, bajo ambas condiciones. En el Intento 6 y 8 de generación de CMPi con el vector STEMCCA, se generaron la línea de CMPi iPS6.1, y las líneas iPSC-FN2.1 e iPSC-FH2.1, obtenidas bajo normoxia e hipoxia, respectivamente en forma paralela, con fines comparativos. En la Figura 39 pueden observarse colonias de las líneas establecidas en comparación con la línea de CMEh WA-09 que se utilizará como control. Todas las líneas fueron caracterizadas, como se describe más adelante. A continuación se describen en detalle las experiencias para el estudio de los efectos de la hipoxia sobre la reprogramación de fibroblastos humanos al estado pluripotente. - 91 - RESULTADOS Figura 39. Colonias de células pluripotentes. Se muestran imágenes representativas de microscopía de contraste de fases de las líneas de CMEh control WA-09, cultivada en normoxia; y líneas de CMPi iPSCFN2.1 e iPSC-FH2.1, cultivadas en normoxia o hipoxia respectivamente, por un período a largo plazo (por lo menos 5 pasajes, aproximadamente 3-4 semanas). Barras de escala: 200 µm. Se comprobó que las líneas de fibroblastos reprogramadas en normoxia e hipoxia son líneas de CMPi bona fide Las líneas de CMPi establecidas, las cuales fueron amplificadas durante por lo menos 5 pasajes, exhibieron características morfológicas clásicas de CM pluripotentes, similares a las de la línea control de CMEh WA-09; las células eran pequeñas, con una relación núcleo : citoplasma baja y organizadas en colonias con bordes definidos, que crecieron como estructuras tridimensionales, como se muestra en la Figura 39. Además, como se muestra en la Figura 40, la expresión de los marcadores del estado pluripotente POU5F1, NANOG y C-MYC en las líneas de CMPi establecidas, fue inducida cuando las células fueron reprogramadas, en comparación con los fibroblastos sin reprogramar. Esta expresión se midió para ambas líneas de CMPi, tanto en condiciones de cultivo normóxico como en cultivo hipóxico, mediante RT-qPCR. Observamos que la expresión de estos factores del estado pluripotente en condiciones de cultivo hipóxico mostró una ligera tendencia a disminuir, en comparación con el cultivo normóxico. La expresión de varios marcadores del estado pluripotente, incluyendo NANOG, POU5F1, SOX2, SSEA-4, TRA1-60, TRA-1-81 y C-MYC, fue estudiada también por inmunofluorescencia y se muestra en la Figura 41. Tanto las líneas establecidas en normoxia como la línea establecida en hipoxia exhibieron una expresión clara de estas proteínas, cuando fueron cultivadas tanto en normoxia como en hipoxia, de manera comparable con las CMEh y a diferencia de los fibroblastos de los que derivan. - 92 - RESULTADOS Figura 40. Análisis de la expresión de marcadores del estado pluripotente Estudio por RT-qPCR de la expresión de los marcadores POU5F1, NANOG y C-MYC sobre fibroblastos HFF-FM (control), CMEh control WA-09 y colonias de CMPi establecidas a largo plazo iPS6.1, iPSC-FN2.1 e iPSC-FH2.1, cultivados en normoxia o hipoxia durante 72 horas. Se grafica en barras la Expresión Relativa vs. control representando la media ± SEM de tres experimentos independientes. Todas las inducciones son estadísticamente significativas con respecto a los fibroblastos HFF-FM en la misma condición de O2, con p < 0.05. Se utilizó la expresión del gen de housekeeping HPRT1 para normalizar los niveles de expresión. - 93 - RESULTADOS Figura 41A. Imágenes representativas de contraste de fases e inmunofluorescencia de marcadores del estado pluripotente. Estudio en CMEh control WA-09 y colonias de CMPi establecidas a largo plazo iPS6.1, posteriormente cultivadas en normoxia o hipoxia crónica. Los núcleos fueron teñidos con DAPI. Barras de escala: 200 µm. - 94 - RESULTADOS Figura 41B. Imágenes representativas de contraste de fases e inmunofluorescencia de marcadores del estado pluripotente. Estudio en líneas de CMPi establecidas a largo plazo iPSC-FN2.1 e iPSC-FH2.1, posteriormente cultivadas en normoxia o hipoxia crónica. Los núcleos fueron teñidos con DAPI. Barras de escala: 200 µm. En CMPi, a diferencia de células terminalmente diferenciadas, las regiones promotoras de los genes de pluripotencia se encuentran principalmente demetiladas. Para evaluar si la reprogramación al estado pluripotente revirtió el estado de metilación, decidimos estudiar la - 95 - RESULTADOS metilación del promotor de gen POU5F1, dado que éste es uno de los genes tradicionales del estado pluripotente y un parámetro establecido del cambio del estado de metilación del ADN durante la desdiferenciación [87]. Para ello utilizamos la técnica de tratamiento del ADN con bisulfito de sodio, la cual genera la desaminación de las citosinas, convirtiéndolas a uracilos, excepto en el caso de estar metiladas, en cuyo caso permanecen como citosinas. Luego de dicha conversión, las islas CpG del promotor en estudio son amplificadas por PCR y secuenciadas, lo cual permite identificar citosinas, las cuales han de ser solamente citosinas metiladas en la secuencia original pre-conversión, ya que solamente éstas permanecen como tales luego del tratamiento con bisulfito de sodio y amplificación. Como se muestra en la Figura 42, encontramos que, a un nivel global, el promotor de POU5F1 se encuentra demetilado en varias islas CpG en las líneas establecidas de CMPi y CMEh control, en comparación con los fibroblastos de origen, en los cuales el promotor está ampliamente metilado. Esto indica un estado heterocromatínico inactivo de este promotor de un gen del estado indiferenciado en células terminalmente diferenciadas, como lo son los fibroblastos; y, contrariamente, un estado eucromatínico transcripcionalmente activo en las CMEh y en las CMPi. Más aún, es un ejemplo de cómo la reprogramación de fibroblastos a CMPi remodela la epigenética del promotor en estudio, apoyando la ocurrencia de una profunda remodelación de la expresión génica cuando una célula atraviesa una reprogramación nuclear al estado pluripotente. Vale la pena destacar que no se observaron diferencias entre las células reprogramadas en normoxia e hipoxia. - 96 - RESULTADOS Figura 42. Niveles de demetilación del promotor de POU5F1 en las líneas estudiadas. Estudio en la línea de fibroblastos HFF-FM, CMEh control WA-09 y líneas de CMPi establecidas a largo plazo iPS6.1, iPSC-FN2.1 e iPSC-FH2.1. Se utilizó la técnica del bisulfito de sodio, La secuenciación del promotor en islas CpG permitió identificar las citosinas remanentes, las cuales eran citosinas metiladas en la secuencia original. Cada rectángulo representa una isla CpG y cada sección delimitada por una línea negra representa un amplicón de PCR. Los números indican las posiciones de las base en relación con el sitio de inicio de transcripción. Niveles de demetilación de CMPi iPS6.1 realizados por el Dr. Carlos Luzzani. Se ha reportado que los transgenes insertados en el ADN cromosómico por el vector lentiviral STEMCCA se silencian luego del evento de reprogramación [73]. Asimismo, este silenciamiento es necesario para que la línea pueda diferenciarse ante los estímulos adecuados. Sin embargo, dicho silenciamiento depende de varios factores como la posición donde se insertó el transgen en el genoma, la naturaleza del transgen y el estado transcripcional y metabólico de la célula. Para estudiar la expresión endógena de los genes POU5F1, KLF4, SOX2 y C-MYC necesitábamos la seguridad de que la expresión de dichos genes exógenos llevados por el vector había sido silenciada; por lo tanto podríamos medir solamente la expresión endógena a partir del cDNA extraído a partir de mRNA total. Para poder diferenciar la expresión de los genes endógenos respecto de los exógenos aportados por el vector, una qPCR fue realizada como se describió anteriormente, sobre cDNA generado a partir de la extracción de ARN de líneas de CMPi establecidas entre los pasajes 17 y 23, y del plásmido STEMCCA como control positivo, utilizando los primers específicos Sox2-bdg-cMyc y Oct4-bdg-Klf4 que reconocen las secuencias unidas por puentes bdg de SOX2 y C-MYC y de POU5F1 y KLF4, únicamente contenidas en el mRNA derivado del vector. Esto permite la amplificación del ADNc generado únicamente a partir de la expresión exógena de estos genes. Como puede verse en la Figura 43, encontramos - 97 - RESULTADOS que en todos los casos el transgen estaba silenciado ya que no hubo expresión detectable de los transgenes, al realizar un estudio por RT-qPCR sobre cDNA de líneas de CMPi establecidas. Por lo tanto, las mediciones de los genes incluidos en el vector STEMCCA (SOX2, POU5F1 y CMYC) a lo largo de esta investigación, fueron de su expresión endógena. Figura 43. Análisis de la expresión de los transgenes insertados por el vector lentiviral STEMCCA. Estudio por RT-qPCR sobre CMEh WA-09 y líneas establecidas de CMPi iPS6.1, iPSC-FN2.1 e iPSC-FH2.1. El plásmido del vector STEMCCA se utilizó como control positivo de la reacción de PCR. Se representa la Expresión Relativa vs control en barras representando la media ± SEM de tres experimentos independientes. La represión de la expresión génica para CMEh y las líneas de CMPi es significativa con p < 0.05. Se utilizó la expresión del gen de housekeeping GAPDH para normalizar los niveles de expresión. Finalmente, con el propósito de estudiar si durante el proceso de reprogramación ocurrieron grandes rearreglos cromosómicos que pudieran ser perjudiciales para futuras aplicaciones, se analizó el cariotipo de las líneas generadas. Se encontró que todas las líneas celulares generadas y estudiadas poseían un cariotipo humano normal sin ninguna anomalía genética detectable por bandeo G, introducida por el proceso de reprogramación, ya sea en normoxia o en hipoxia. El armado y análisis de los preparados fue realizado por la Lic. Diana Radakoff del Instituto GENOS. Los cariogramas se muestran en la Figura 44. En conclusión no se encuentran diferencias perceptibles en el estado pluripotente entre las líneas de CMPi generadas en normoxia y entre éstas y una línea generada en hipoxia. - 98 - RESULTADOS Figura 44. Cariogramas de las líneas estudiadas. Se muestran los cariogramas de fibroblastos HFF-FM, y líneas CMEh WA-09, CMPi iPS6.1, iPSC-FN2.1 y iPSC-FH2.1. Los cariotipos son cariotipos humanos normales de 46 cromosomas. Las células fueron cultivadas en normoxia antes del análisis. La expresión de los marcadores del estado pluripotente que se inducen durante la reprogramación disminuye durante la diferenciación de las CMPi correspondientes. Con el propósito de estudiar la pluripotencia de las líneas generadas, las líneas de CM fueron inducidas a atravesar una diferenciación espontánea, a través del cultivo de CE en SFB en normoxia y, como se muestra en la Figura 45, la expresión de POU5F1, SOX2 y NANOG disminuyó entre el día 0 (estado indiferenciado) y el día 21 del proceso de diferenciación. Además, la expresión de - 99 - RESULTADOS marcadores de linaje de las capas germinales fue inducida durante la diferenciación, cuando las células se cultivaron en normoxia o hipoxia durante un período a largo plazo, como puede verse en la Figura 46. Los marcadores de endodermo temprano SOX17, de endodermo tardío AFP, de mesodermo temprano MESP-1, de mesodermo tardío NKX2.5, ectodermo temprano PAX6 y de ectodermo tardío GFAP fueron inducidos de una expresión basal en el día 0 a un aumento de la expresión en el día 7 a día 21, dependiendo de la cinética de cada gen. La variación interna de estos experimentos fue considerable como se esperaba, debido a la naturaleza enormemente heterogénea del método de diferenciación espontánea, en el cual el compromiso de linaje es arbitrario. Por lo tanto, pudimos observar resultados con un amplio desvío entre normoxia e hipoxia, e incluso dentro de la misma condición, entre diferentes líneas. Sin embargo, las experiencias demostraron que todas las líneas celulares fueron capaces de diferenciarse in vitro a las tres capas germinales, a un tiempo u otro, probando su pluripotencia. También se observó la presencia de proteínas marcadoras de diferenciación por inmunofluorescencia en CE en el día 21, como se muestra en la Figura 47. Los marcadores AFP, GATA-4 (endodermo), NKX2.5, PAX6, NESTINA (ectodermo) se expresaron en el día 21 post-diferenciación, tanto cuando se cultivó las células en normoxia o hipoxia. Figura 45. Análisis de la expresión de marcadores del estado pluripotente durante la diferenciación de células madre pluripotentes. Estudio por RT-qPCR de la expresión de los marcadores POU5F1, SOX2 y NANOG durante la diferenciación de CMEh control WA-09 y líneas de CMPi establecidas a largo plazo iPS6.1, iPSC-FN2.1 e iPSC-FH2.1, cultivadas en normoxia. D0: día 0 de diferenciación (control), D21: día 21 post-inducción de la diferenciación. Se grafica las Veces de Inducción vs. control en barras representando la media ± SEM de tres experimentos independientes. La represión es significativa con p < 0.05, para todos los genes y todas las líneas, dentro en la misma línea. Se utilizó la expresión del gen de housekeeping HPRT1 para normalizar los niveles de expresión. - 100 - RESULTADOS Figura 46A. Análisis de la expresión de marcadores de linaje de las tres capas germinales durante la diferenciación de células madre pluripotentes. Estudio por RT-qPCR de la expresión de los marcadores de endodermo SOX17 y AFP, marcadores de mesodermo MESP-1 y NKX2.1 y marcadores de ectodermo PAX6 y GFAP, en CE derivados de CMEh control WA-09 y de líneas de CMPi establecidas a largo plazo iPSC-FN2.1 e iPSC-FH2.1, cultivadas en normoxia o hipoxia durante un período de tiempo a largo plazo. D0: día 0 de diferenciación (estado indiferenciado, control), D7: día 7 post-inducción de la diferenciación, D21: día 21 post-inducción de la diferenciación. Se grafica las Veces de Inducción vs. control en barras representando la media ± SEM de tres experimentos independientes. * = p < 0.05. Se utilizó la expresión del gen de housekeeping HPRT1 para normalizar los niveles de expresión. - 101 - RESULTADOS Figura 46B. Expresión de marcadores de linaje de las tres capas germinales durante la diferenciación de células madre pluripotentes. Estudio por RT-PCR a punto final, de la expresión de los marcadores de endodermo α-FP y SOX17, marcadores de mesodermo MESP-1 y MYOD y marcadores de ectodermo NES y TUBB3, en CE derivados de CMEh control WA-09 y de la línea de CMPi establecida a largo plazo iPS6.1, cultivadas en normoxia o hipoxia durante un período de tiempo a largo plazo. D0: día 0 de diferenciación (estado indiferenciado, control), D4: día 4 post-inducción de la diferenciación, D21: día 21 post-inducción de la diferenciación. Resultados representativos de tres experimentos independientes. Se utilizó la expresión del gen de housekeeping HPRT1 para controlar la carga de DNA. - 102 - RESULTADOS Figura 47A. Expresión de marcadores de linaje de las tres capas germinales por inmunofluorescencia. Imágenes representativas de contraste de fases e inmunofluorescencia de los marcadores de endodermo AFP y GATA-4, el marcador de mesodermo NKX2.5 y los marcadores de ectodermo PAX6, GFAP y NES en CE derivados de CMEh control WA-09 y líneas de CMPi establecidas a largo plazo iPS6.1, cultivadas en normoxia o hipoxia durante un período a largo plazo. Los núcleos fueron teñidos con DAPI. Barras de escala: 200 µm. - 103 - RESULTADOS Figura 47B. Expresión de marcadores de linaje de las tres capas germinales por inmunofluorescencia. Imágenes representativas de contraste de fases e inmunofluorescencia de los marcadores de endodermo AFP y GATA-4, el marcador de mesodermo NKX2.5 y los marcadores de ectodermo PAX6, GFAP y NES en CE derivados de líneas de CMPi establecidas a largo plazo iPS-FN2.1 e iPSC-FH2.1, cultivadas en normoxia o hipoxia durante un período a largo plazo. Los núcleos fueron teñidos con DAPI. Barras de escala: 200 µm. - 104 - RESULTADOS Finalmente, las líneas de CMPi establecidas también mostraron pluripotencia in vivo, siendo capaces de generar teratomas en ratones inmunosuprimidos, siendo ésta la prueba máxima para estudiar pluripotencia en CMEh. Imágenes representativas de los tumores formados se muestran en la Figura 48. Los teratomas generados poseían células derivadas de las tres capas germinales, que fueron asignadas a diferentes tejidos y estructuras histológicas, como se muestra y describe en la Figura 49. Figura 48. Formación de teratomas en ratones inmunosuprimidos inyectados con células madre pluripotentes. Se muestran imágenes representativas de los individuos, luego de ser sacrificados, mostrando la formación tumoral formada; y los tumores luego de ser disecados y antes de ser embebidos en parafina para su análisis histológico. - 105 - RESULTADOS Figura 49. Histología de teratomas formados por inyección de células madre pluripotentes en ratones inmunosuprimidos. Imágenes representativas de microscopía de campo claro de cortes histológicos de teratomas, con tinción de hematoxilina-eosina, generados por las líneas celulares CMEh WA-09 y las líneas de CMPi establecidas a largo plazo iPS6.1, iPSC-FN2.1 e iPSC-FH2.1, en ratones inmunosuprimidos, mostrando la presencia de tejidos de las tres capas germinales. Barras de escala: 200 µm. Los factores HIF fueron estabilizados en nuestro modelo de hipoxia y aumentó la expresión de sus genes blanco. Efectos sobre la expresión de marcadores del estado pluripotente. Los factores HIF son dímeros compuestos por una subunidad de HIF-α u a su u idad de HIFβ, la ual es estable en toda condición de oxígeno. Por el contrario, la subunidad HIF-α suf e degradación constitutiva por la vía del proteosoma, en condiciones de normoxia. Al disminuir los niveles de oxígeno se genera la estabilización de HIF-α, su a u ula ió dimerización con la subunidad HIF-β, fo posterior á dose el fa to de t a s ip ió a ti o ue en esta - 106 - RESULTADOS condición puede cumplir su función sobre genes blanco, ya que los factores HIF son factores de transcripción con una señal de localización nuclear, para su exportación a núcleo postestabilización. Con el propósito de caracterizar la respuesta al estímulo de hipoxia respecto a los factores de transcripción reguladores de hipoxia, en nuestro modelo de CMEh WA-09, estudiamos la presencia de las proteínas HIF- α HIF- α po Western Blot. Como se muestra en la Figura 50, los i eles de HIF α pero no los niveles de HIF α estaban elevados durante la hipoxia. Éste fue un experimento representativo en una línea de células madre modelo, aunque no es raro observar una prevalencia de uno de los factores HIF, según el tejido o las condiciones [134, 149, 150]. Además se ha descripto anteriormente un papel secuencial de HIF- α HIF- α, donde predomina la expresión HIF- α du a te la hipoxia leve (5%) [115]. También estudiamos la expresión de las proteínas HIF- α HIF- α po In-Cell Western Blot, comparando las distintas líneas estudiadas, al ser cultivadas en normoxia o hipoxia. Como se muestra en la Figura 51, la expresión de HIF- α pa e e au e ta al e po e las élulas a hipo ia, e el aso de las CMEh estudiadas y de la línea de CMPi iPSC-FN2.1, generada en normoxia. En contraste, en el caso de la línea generada en hipoxia, iPSC-FH2.1, la expresión de HIF- α pa e e dis i ui . Co espe to a la expresión de HIF- α, ésta dis i u e pa a las CMEh la lí ea de CMPi ge e ada e hipo ia, pero aumenta para la línea generada en normoxia. Si bien estos últimos resultados no son estadísticamente significativos, la tendencia se observa claramente. - 107 - RESULTADOS Figura 50. Determinación de los niveles de factores HIF luego de hipoxia durante 16 horas. Las membranas fueron reveladas con anticuerpos contra HIF- α, HIF- α y actina. La cantidad de actina se utilizó como control de carga para la cuantificación de los factores HIF, lo cual se muestra en la parte inferior del gráfico. M: marcador de peso molecular (se observan bandas de 25, 37, 50, 75 y 100 kDa). Peso Molecular esperado para HIF- α: 132 kDa. Peso molecular esperado para HIF- α: 115 kDa. Peso molecular esperado para actina: 43 kDa. Figura 51. Detección de factores HIF durante la inducción de hipoxia. Estudio por In Cell-Western Blot de la expresión de las proteínas HIF- α y HIF- α en CMEh control WA-09 y líneas de CMPi establecidas a largo plazo iPSC-FN2.1 e iPSC-FH2.1, luego 24 horas post-inducción de hipoxia. Se grafica las Veces de Inducción vs. control en barras representando la media ± SEM de tres experimentos independientes. * = p < 0.05. - 108 - RESULTADOS A fin de verificar la inducción de los factores HIF después de una exposición a hipoxia aguda a corto plazo, estudiamos la expresión de los genes blanco utilizados como reporteros de ambos factores HIF, VEGF, BNIP3 y EPO. Como se muestra en la Figura 52, la expresión de dichos genes blanco aumentó en casi todos los casos al exponer las células a hipoxia, para luego disminuir y estabilizarse. El gen VEGF (blanco de HIF- α de HIF- α fue inducido al menos 2,5 veces después de 8 horas en hipoxia. Luego de este lapso de tiempo la expresión de VEGF pareció disminuir otra vez hasta alcanzar una estabilización alrededor de las 48 horas después de la inducción de hipoxia. Algo similar pareció ocurrir con la expresión de BNIP3, gen blanco de HIFα, aunque su cinética pareció ser un poco más lenta, estabilizándose luego de las 48 horas de hipoxia. Es posible que los efectos agudos de la hipoxia puedan ser contrarrestados en parte, luego de una exposición a largo plazo a tensiones bajas de oxígeno, como puede concluirse a partir del hecho de que el aumento de la expresión de VEGF y BNIP3 en hipoxia sólo fue transitorio. La expresión de EPO, blanco de HIF- α, presentó un comportamiento errático, aumentando inicialmente sólo para la línea de CMPi generada en hipoxia. Los resultados erráticos o contradictorios que se observan para HIF- α su ge la o EPO en las Figuras 51 y 52, respectivamente, podrían indicar alguna relación entre el cultivo hipóxico de líneas de CMPi ya establecidas a largo plazo, pero que fueron a su vez generadas inicialmente en condiciones hipóxicas. - 109 - RESULTADOS Figura 52. Análisis de la expresión de genes blanco de los factores HIF- α o HIF- α en líneas de CMP expuestas a hipoxia o en normoxia. Estudio por RT-qPCR de la expresión de los genes VEGF, BNIP3 y EPO en CMEh control WA-09 y líneas de CMPi establecidas a largo plazo iPS6.1, iPSC-FN2.1 e iPSC-FH2.1, luego de 0 (control), 8, 24 y 48 horas post-inducción de hipoxia. Se grafica las Veces de Inducción vs. control en barras representando la media ± SEM de tres experimentos independientes. * = p < 0.05. Se utilizó la expresión del gen de housekeeping HPRT1 para normalizar los niveles de expresión. - 110 - RESULTADOS Modulación de la expresión de genes de pluripotencia por cultivo hipóxico. Los marcadores del estado pluripotente se inducen a tiempos de cultivo hipóxico a largo plazo. Una vez comprobado que nuestro modelo de hipoxia respondía aumentando los niveles y actividad de los factores HIF, nuestro siguiente objetivo fue investigar si el cultivo hipóxico modulaba la expresión de marcadores del estado pluripotente y si había diferencias entre las distintas líneas celulares. La hipótesis propuesta es que las líneas generadas en normoxia o hipoxia podrían presentar diferencias intrínsecas en la señalización del estímulo y por esta razón podrían responder diferente entre sí al estímulo de hipoxia, además de responder diferente según si el estímulo es crónico o agudo. Las líneas de CMPi establecidas a largo plazo, fueron cultivadas en hipoxia y se tomaron muestras a las 0, 8, 24 y 48 horas (hipoxia aguda) y luego de 5 pasajes en hipoxia (hipoxia crónica o a largo plazo). Los resultados se muestran en la Figura 53. El marcador NANOG pareció disminuir en hipoxia aguda, pero luego aumentó en los cultivos a largo plazo en hipoxia crónica (p5). Lo mismo parece suceder con C-MYC, pero no con POU5F1. Este último marcador no mostró variaciones estadísticamente significativas a lo largo de la experiencia. También parece haber una resistencia a la disminución de la expresión de NANOG y C-MYC en las primeras horas de hipoxia para las líneas iPS6.1 e iPSC-FH2.1. Dado que las disminuciones en la expresión génica de NANOG y C-MYC no se dieron en todas las líneas de CMPi estudiadas, no se puede afirmar que la exposición aguda a hipoxia es responsable de este efecto, pero se observa una tendencia, que en los casos de la línea control WA-09 y la línea de CMPi iPSC-FN2.1 es significativa. Se llegó a la conclusión de que si la hipoxia genera la estabilización o aumento de la expresión de marcadores del estado pluripotente, este estado es alcanzado luego de varios pasajes en cultivo hipóxico, y no con una exposición aguda. - 111 - RESULTADOS Figura 53. Análisis de la expresión de genes del estado pluripotente en líneas de CMPi expuestas a hipoxia. Estudio por RT-qPCR de la expresión de los marcadores POU5F1, NANOG y C-MYC en CMEh control WA-09 y líneas de CMPi establecidas a largo plazo iPS6.1, iPSC-FN2.1 e iPSC-FH2.1, luego de 0 (control), 8, 24 y 48 horas post-inducción de hipoxia, y luego de cultivo en hipoxia crónica durante al menos 5 pasajes (p5). Se grafica las Veces de Inducción vs. control en barras representando la media ± SEM de tres experimentos independientes. * = p < 0.05. Se utilizó la expresión del gen de housekeeping HPRT1 para normalizar los niveles de expresión. - 112 - RESULTADOS Efectos del cultivo hipóxico sobre la generación de CMPi Los experimentos descriptos anteriormente fueron realizados en líneas de CMPi ya establecidas, y luego cultivadas bajo diferentes condiciones. Los antecedentes bibliográficos sobre los efectos de la hipoxia sobre el cultivo de CME son también mayormente basados en estudios sobre líneas establecidas [98, 103]. Es importante estudiar los efectos sobre la generación de CMPi, porque podrían no ser iguales que los efectos sobre el cultivo de líneas de CM pluripotentes ya establecidas y que llevan muchísimos pasajes en cultivo. Los siguiente experimentos se realizaron analizando los cultivos a 21 días post-transducción para estudiar la eficiencia del proceso de reprogramación a un dado punto final; y se evaluaron también los efectos de la hipoxia durante la generación de CMPi, ya que los antecedentes sobre los efectos de la hipoxia en la generación de CMPi fueron realizados en todos los casos a un punto final [64, 94, 148]. Efectos del cultivo hipóxico sobre la eficiencia de reprogramación. La hipoxia aumenta la eficiencia de reprogramación, pero las colonias formadas son más pequeñas A fin de evaluar los efectos de la hipoxia sobre la eficiencia de reprogramación transdujimos fibroblastos HFF-FM con el vector STEMCCA, en normoxia o hipoxia y calculamos la eficiencia de generación de CMPi mediante el recuento de la cantidad de colonias positivas para actividad de FA, en el día 21 post-transducción. El número de colonias generadas se vio aumentado en hipoxia, como puede verse en la Figura 54. Sin embargo, el tamaño de las colonias fue más pequeño para las CMPi generadas en condiciones de hipoxia, rindiendo un área cubierta por células reprogramadas significativamente menor en esta condición. La cuantificación se realizó con el programa ImageJ, utilizando el comando Analizar Partículas con un Tamaño de 50Infinito y una Circularidad de 0,20-1,00. Las imágenes, de las cuales se muestran imágenes representativas en la Figura 54, fueron analizadas por contraste y luminosidad, permitiendo identificar el borde de las colonias por contraste y color de las formaciones sobre la superficie de la placa de cultivo. Es posible que la hipoxia aporte las condiciones para la reprogramación de una mayor cantidad de células somáticas iniciales, pero no permita su proliferación o crecimiento, una vez establecido el evento de reprogramación. - 113 - RESULTADOS Con el propósito de estudiar si el efecto observado era debido a HIF-1α o a HIF- α, estudia os la expresión de BNIP3, un marcador de hipoxia, gen blanco de HIF- α, e presencia de diferentes inhibidores de los factores HIF. Como puede observarse en la Figura 55, BNIP3 se vio inducido cuando los cultivos celulares fueron expuestos a bajos niveles de oxígeno, y esta inducción fue inhibida al utilizar diferentes inhibidores de HIF- α. Utilizamos varios inhibidores de HIF, i lu e do FM G ue es u i hi ido de HIF α HIF α; HIF i ue es u i hi ido espe ífi o de HIF α DMOG ue es u i hi ido de la hid o ilasa p olil aspa agil de HIF; este último compuesto no sólo inhibió los efectos de ambos factores, sino que también tuvo un efecto devastador sobre los cultivos, como puede verse en la Figura 56. Este experimento demuestra que BNIP3 efectivamente se induce ante hipoxia, y que es HIF- α el espo sa le de dicha inducción, ya que al ser inhibido, la inducción no se da con significancia. Por otro lado, también se evaluó la eficiencia de reprogramación por In-Cell Western Blot estudiando la expresión de los marcadores del estado pluripotente NANOG, POU5F1, SOX2 y SSEA-4, en el día 21 post-transducción, en normoxia o hipoxia, como indicadores de la eficiencia del proceso de desdiferenciación. Como se muestra en la Figura 56, la expresión de los marcadores NANOG y POU5F1 en las colonias generadas bajo condiciones hipóxicas fue de aproximadamente la mitad de lo que se observó en las colonias generadas en normoxia. Cabe remarcar que esta es una correlación negativa entre la expresión de dichos marcadores y el efecto de la hipoxia sobre la eficiencia de reprogramación. Lo cual recuerda los resultados observados en la Figura 53, en los cuales se da una disminución inicial de la expresión de marcadores del estado pluripotente, durante las primeras horas de hipoxia. A pesar de que sólo se estudió el proceso hasta las 48 horas y luego en un pasaje 5, dicho pasaje involucra aproximadamente 4 semanas de cultivo, mientras que las mediciones por In-Cell Western Blot fueron realizadas a las 3 semanas post-transducción/comienzo de la hipoxia. Este resultado aparentemente contradictorio podría explicarse por el hecho de que la menor expresión de NANOG y POU5F1 en colonias derivadas en hipoxia puede deberse tanto a una menor cantidad de células por colonia, como se observó con anterioridad y en cuyo caso el resultado seguiría siendo contradictorio; o una menor expresión del marcador en cada célula, dado que se observó anteriormente que la eficiencia de reprogramación aumentó con la hipoxia, - 114 - RESULTADOS considerando el número de colonias generadas. Los efectos observados sobre la expresión de NANOG y POU5F1 no se observaron para la expresión de los marcadores SOX2 y SSEA-4, los cuales pueden no estar regulados en ninguna instancia por las vías de hipoxia, o no particularmente por los factores HIF. Hasta el momento no se ha reportado ninguna evidencia de regulación de SSEA-4, a través de mecanismos relacionados con la hipoxia o los factores HIF. SSEA-4 no es un factor de transcripción ni gen maestro que esté involucrado en los promiscuos mecanismos de mantenimiento de la pluripotencia, sino que es un marcador de superficie que está presente en células pluripotentes. Sería coherente que no esté controlado por eventos relacionados con la hipoxia celular. Sí se ha reportado que HIF- α regula positivamente a SOX2, el cual a su vez aumenta los niveles de expresión de POU5F1 [117], pero dado que este gen se encuentra río arriba de POU5F1 en la regulación del estado pluripotente, podría estar sujeto a una regulación negativa más estricta, y al haber niveles elevados de SOX2 exógeno por los genes reprogramadores transducidos por las partículas virales, los niveles endógenos podrían disminuir. Pudimos además observar que los efectos de la hipoxia fueron rescatados cuando la reprogramación se realizó en presencia de FM19G11, situación en la cual la eficiencia de reprogramación no resultó diferente en normoxia e hipoxia. También observamos que los efectos del tratamiento del cultivo celular durante el proceso de reprogramación con HIF1i no tuvieron ningún efecto estadísticamente significativo, lo cual sugiere que es un mecanismo que ocurre a través de HIF- α el que genera los efectos observados como consecuencia del tratamiento hipóxico durante la reprogramación al estado pluripotente. Desafortunadamente no se pudo conseguir un inhibidor específico de la actividad de HIF- α para comprobar esta hipótesis, por lo que este estudio será completado en el futuro. - 115 - RESULTADOS Figura 54. Eficiencia de reprogramación y área total de colonias. Determinación por recuento de colonias positivas para actividad FA y medición del área total de colonias en el día 21 post-transducción con el vector STEMCCA. Las barras representan la media ± SEM de cinco experimentos independientes. * = p < 0.05. El área total se representa en unidades arbitrarias. Los paneles son imágenes representativas de colonias positivas para actividad de FA en pocillos de placa de 12 pocillos. - 116 - RESULTADOS Figura 55. Análisis de la expresión del gen blanco del factor HIF- α, BNIP3. Estudio por RT-qPCR en fibroblastos HFF-FM, línea de CMEh control WA-09 y líneas de CMPi establecidas a largo plazo iPSCFN2.1 e iPSC-FH2.1, del gen BNIP3 durante la inducción de hipoxia durante 24 horas. H: hipoxia. Se representa la media ± SEM de tres experimentos independientes. * = p < 0.05. Se utilizó la expresión del gen de housekeeping HPRT1 para normalizar los niveles de expresión. - 117 - RESULTADOS Figura 56. Eficiencia de reprogramación determinada por In-Cell Western Blot de marcadores del estado pluripotente. Estudio por In-Cell Western Blot de los marcadores NANOG, POU5F1, SOX2 y SSEA4, en el día 21 post-transducción, en normoxia o hipoxia. –STEMCCA: Control sin transducción. Se muestran imágenes representativas de fluorescencia del marcador. N: Normoxia; H: hipoxia. Se grafica las Veces de inducción vs. control en barras representando la media ± SEM de cuatro experimentos independientes. * = p < 0.05. - 118 - RESULTADOS La tasa de proliferación de CMPi es mayor en hipoxia. La menor expresión de marcadores del estado pluripotente en hipoxia es una consecuencia de un menor número de células en las colonias generadas en esta condición Durante la reprogramación celular al estado pluripotente, se induce la expresión de marcadores del estado pluripotente, incluyendo la expresión de NANOG. Dado que dicho factor se comportó en las experiencias previas como el más estable y robusto, decidimos estudiar su patrón de expresión durante el proceso de reprogramación. En síntesis, hasta el momento encontramos que existió una mayor eficiencia de reprogramación en hipoxia y que marcadores del estado pluripotente como NANOG y C-MYC aumentan su expresión en hipoxia luego de ser cultivados a largo plazo en esta condición. Sin embargo también encontramos que los niveles de la proteína NANOG disminuyen en hipoxia al día 21 post-transducción. Con el propósito de identificar la causa de la aparente contradicción – en comparación a la eficiencia de reprogramación- del resultado anterior de expresión de NANOG por In-Cell Western Blot, se decidió analizar los niveles de este marcador en las células individuales mediante citometría de flujo. Así se esperó poder dilucidar si la menor expresión de NANOG y POU5F1 en colonias derivadas en hipoxia se debió a una menor cantidad de células por colonia o una menor expresión del marcador en cada célula. Se transdujeron cultivos de fibroblastos HFF-FM con el vector STEMCCA y se analizaron mediante citometría de flujo en los días 7, 14 y 21 post-transducción. Como se esperaba, los niveles de la proteína NANOG aumentaron en el tiempo, sin embargo, como puede verse en la Figura 57, éstos aumentaron aproximadamente 10 veces más en las células reprogramadas en normoxia, en comparación con las células reprogramadas en hipoxia, indicando un menor número de células positivas para NANOG en colonias generadas en hipoxia. Estos resultados se condicen con los ensayos por In-Cell Western Blot mostrados previamente, en los cuales es menor en hipoxia la eficiencia de reprogramación basada en la señal para los marcadores del estado pluripotente NANOG y POU5F1. Además lo observado fue consistente con la formación de colonias más pequeñas en hipoxia. En síntesis, durante la reprogramación en hipoxia se obtuvo un mayor número de colonias, sin embargo éstas expresaron menores niveles de NANOG que en normoxia, dado su tamaño. Se infiere que la cantidad de células que las componen fue menor. - 119 - RESULTADOS Figura 57. Análisis por citometría de flujo de la expresión de NANOG, en normoxia o hipoxia, a lo largo de la reprogramación. (a) Se muestran histogramas representativos, representando cell scatter vs la distribución de la señal fluorescente de NANOG. (b) Se grafica el porcentaje de células NANOG+ a los tiempos indicados post-transducción. * = p < 0.05. Las barras representan la media + SEM de 5 experimentos independientes. Con anterioridad, se observó que si bien se generaba mayor número de colonias de CMPi al reprogramar fibroblastos en hipoxia, eran menos las que persistían a largo plazo. Además se observó que las colonias generadas en hipoxia eran más pequeñas en tamaño y con menor cantidad de células. Por todo ello se pensó que es probable que la hipoxia estuviera permitiendo la reprogramación de una mayor cantidad de células iniciales, pudiendo estar afectada la supervivencia y la proliferación celular. Con el fin de evaluar la viabilidad de las líneas en estudio establecidas a largo plazo, cuando se las cultivó bajo normoxia o hipoxia, se analizó su capacidad de exclusión del colorante Trypan Blue, para calcular un porcentaje de viabilidad, después de 48 horas en normoxia o hipoxia. El Trypan Blue sólo se excluye de células viables, mientras que las células muertas no son capaces de expulsar este colorante. Como se muestra en la Figura 58, hubo diferencias mínimas entre las líneas celulares estudiadas, pero no se encontraron diferencias significativas entre el cultivo normóxico y el cultivo hipóxico, dentro de la misma línea celular. Sabiendo que no hay diferencias de viabilidad entre dichas condiciones, lo cual podría afectar otros resultados, y con la finalidad de determinar la tasa de proliferación de nuestros cultivos de líneas de CMPi establecidas a largo plazo -después de al - 120 - RESULTADOS menos 5 pasajes en cultivo en cada condición- cuantificamos el contenido de ADN en el día 3 de cultivo, partiendo de un número fijo de células iniciales (día 0). Como se observa en la Figura 59, encontramos que las líneas de CM estudiadas exhibieron una mayor tasa de proliferación en condiciones hipóxicas que en normoxia; este efecto no se observó en los fibroblastos HFF-FM. Finalmente, analizamos los niveles de apoptosis en las líneas celulares en estudio, a través de un análisis con Anexina V (AnV) / Ioduro de Propidio (IP), seguido por citometría de flujo. La AnV es un indicador de la presencia de fosfatidilserina expuesta en la cara externa de la membrana plasmática, un evento encontrado principalmente en procesos apoptóticos y necróticos. La AnV se une a la fosfatidilserina, y cuando se la marca con una molécula fluorescente puede utilizarse para estudios de citometría de flujo. El IP es un colorante que sólo ingresa a células necróticas o apoptóticas tardías, dado que la membrana plasmática íntegra es impermeable al mismo. Cultivamos las líneas celulares por 48 horas en normoxia o hipoxia, luego de lo cual se realizó la tinción para Anexina V, y con IP, y luego la citometría de flujo. Este análisis no reveló ninguna diferencia significativa entre las dos condiciones, para ninguna línea celular, como puede verse en la Figura 60, tanto para apoptosis como para necrosis. En conclusión, dado que no existieron diferencias significativas entre o dentro de las líneas estudiadas, tanto en normoxia como en hipoxia, para la viabilidad y los niveles de apoptosis/necrosis, pero se encontró que cada línea de CM es más proliferativa en hipoxia que en normoxia, se concluyó que no es posible que el menor tamaño y cantidad de células en las colonias de CMPi generadas en hipoxia, se deba a los niveles de proliferación intrínsecos de la línea o generados por la hipoxia. El hecho de que las colonias de CMPi generadas hayan sido más pequeñas ha de estar relacionado con otro factor. Es importante remarcar que los estudios de proliferación fueron realizados sobre líneas ya establecidas, dada la imposibilidad de evaluar dicha propiedad en células componentes de las colonias a medida que surgen, ya que el mero tiempo en cultivo es tiempo en el que las colonias cambian y se desarrollan. Pero es posible que los niveles proliferativos de las células que componen una colonia en los primeros días de formación, por ejemplo entre los días 1 y 21, no sea similar ni comparable a la proliferación de la línea establecida a partir de dicha colonia, como se evaluó en este estudio. Es posible que, de hecho, ocurra un cambio en las propiedades proliferativas durante la maduración de colonias - 121 - RESULTADOS jóvenes a colonias establecidas. Células somáticas, como los fibroblastos utilizados en este estudio, no parecen ver su proliferación afectada por los niveles de oxígeno, o al menos esta no aumenta. Figura 58. Viabilidad celular de las líneas de células madre. Se grafica el porcentaje de células viables, sobre el total de células contadas, por exclusión de Trypan Blue, a las 48 horas post-inducción de hipoxia en la línea de CMEh control WA-09 y las líneas de CMPi establecidas a largo plazo iPS6.1, iPSC-FN2.1 e iPSC-FH2.1. Este colorante sólo es excluido por células viables. Las barras representan la media ± SEM de tres experimentos independientes. No se encontraron diferencias significativas dentro de cada línea. - 122 - RESULTADOS Figura 59. Tasa de proliferación de las líneas en estudio. Análisis de la cuantificación de ADN en el día 3 de cultivo en normoxia o hipoxia, con un número fijo inicial de las células, en la línea de HFF-FM y las líneas de CMPi establecidas a largo plazo. Las barras representan la media ± SEM de tres experimentos independientes. * = p < 0.05. - 123 - RESULTADOS Figura 60A. Análisis de los niveles de apoptosis en las líneas de células madre estudiadas. Análisis por citometría de flujo de tinción de Anexina V (AnV) y con Ioduro de Propidio (IP), a las 0 horas (Normoxia) y 48 horas después de la inducción de hipoxia (Hipoxia) en la línea de CMEh control WA-09 y las líneas de CMPi establecidas a largo plazo. Células apoptóticas tempranas: IP-, AnV +; Células necróticas: IP +, AnV-; Células apoptóticas tardías: IP +, AnV +. (a) Se muestran histogramas representativos, representando señal fluorescente de la tinción con IP vs señal fluorescente de Anexina V. (b) Se grafica el porcentaje de células en apoptosis (vivas) o necrosis + apoptosis tardía (muertas), en barras representando la media ± SEM de dos experimentos independientes. No se encontraron diferencias significativas. - 124 - RESULTADOS Figura 60B. Análisis de los niveles de apoptosis en las líneas de células madre estudiadas. Se muestran histogramas representativos adicionales, graficando distintas variables, como forward/side cell scatter, conteo, señal de AnV y señal de IP. - 125 - RESULTADOS GENERACIÓN DE PLÁSMIDOS PARA LA GENERACIÓN DE VECTORES VIRALES PARA LA TRANSDUCCIÓN DE FACTORES HIF Clonación de factores HIF en vectores virales En instancias previas de este trabajo, se observó que la inhibición de los factores HIF tiene un efecto sobre la eficiencia de reprogramación de fibroblastos a CMPi, de modo que estos factores de transcripción están involucrados en el proceso. Dados los resultados observados anteriormente, y lo reportado en la bibliografía con respecto a los efectos de los factores HIF sobre el estado pluripotente [108-114, 117], y sobre la generación de CMPi [115, 148], y habiendo confirmado en este estudio que la hipoxia permite la generación de un mayor número de colonias de CMPi, se pensó que la reprogramación en presencia de los factores HIF puede modular la eficiencia del proceso. De hecho, es posible que el efecto de aumento de la reprogramación que tiene la hipoxia esté basado en dichos factores, y que al sobreexpresarlos individualmente se evite el resto de los efectos y consecuencias de la hipoxia. Dado nuestro interés en los efectos de la actividad de los factores HIF, como transductores de hipoxia, sobre la generación de CMPi, el objetivo de la presente sección fue, entonces, la generación de una herramienta para poder evaluar los efectos de los factores HIF, independientemente de los niveles de oxígeno, sobre la reprogramación al estado pluripotente. Nos interesó la generación de plásmidos de expresión eucariota, que una vez transfectados generen la producción de partículas virales capaces de introducir HIF- α o HIF- α en células a reprogramar, por el mismo método que los vectores OKSIM o STEMCCA, utilizados en este trabajo. Consecuentemente, a fin de evaluar la influencia de la actividad de HIF- α y HIF α en la generación de CMPi, utilizando la sobreexpresión de estos factores durante la reprogramación en reemplazo de la condición hipóxica, se clonó HIF- α o HIF α wild type (wt) y con distintas mutaciones en el plásmido retroviral NITSC, generosamente cedido por Steve Suhr del laboratorio CRL, generando un inserto de fusión con la proteína YFP, bajo un promotor TetOff. En el sistema TetOff, la expresión del gen clonado en el MCS del plásmido está regulada por un promotor eucariota constitutivo, de modo que en condiciones sin agente modulador, hay expresión del gen. Pero ante el agregado de dicho agente, Tetraciclina o Doxiciclina (Dox), al - 126 - RESULTADOS cultivo, el sistema se ve apagado -Off, por su denominación en inglés-, inhibiéndose la expresión de los genes controlados por el sistema TetOff. Se generaron las siguientes construcciones: Tabla 15. Plásmidos retrovirales generados para la transducción de factores HIF en células a desdiferenciar Número de Nombre referencia 1 AHIF1wt 2 AHIF2wt 3 AHIF1 4 AHIF2 5 YHIF10 6 YHIF20: Backbone Inserto NITSC NITSC NITSC NITSC NITSC NITSC HIF1A wt HIF2A wt HIF1A Dm (Doble mutación) HIF2A Dm HIF1A Tm (Triple mutación) HIF2A Tm Los HIF wt no contienen mutaciones. Los HIF Dm tienen actividad en condiciones de hipoxia, pero también de normoxia; y los HIF Tm no sólo tienen actividad en ambas condiciones, sino que también su capacidad de activar la transcripción de sus genes blanco está aumentada. En la sección Materiales y Métodos se detallan los elementos mutados que causan estos cambios moleculares. En la Figura 61 pueden observarse las fotografías de los geles de agarosa 1%, de los productos de ligación entre los insertos y el vector NITSC vacío, digeridos con las enzimas de restricción MluI y ClaI, para comprobar su identidad. El peso del vector vacío es de 9 kb, y el peso de los insertos 2,5 kb para HIF- α 2,6 kb para HIF- α. Así, se confirmó la obtención de los plásmidos diseñados, los cuales fueron secuenciados en sus extremos y luego en su totalidad para confirmar su identidad. Se prepararon las muestras para secuenciación con los primers utilizados en el clonado, y se enviaron a secuenciar al servicio de secuenciación del Genomics Core del Research Technology Support Facility de la MSU, según se describe en la sección Materiales y Métodos. Los resultados de la secuenciación indicaron que el clonado era correcto para cada construcción. - 127 - RESULTADOS En el futuro se plantea la idea de generar CMPi con el vector STEMCCA, u otro vector de generación de CMPi, y comparar la eficiencia de generación de colonias y su calidad, con la utilización del mismo vector en forma conjunta con uno de estos vectores HIF generados. Figura 61. Geles de agarosa de los plásmidos HIF generados, digeridos con ClaI y MluI. M: Marcador de peso molecular. 1: AHIF1wt. 2: AHIF2wt. 3: AHIF1. 4: AHIF2. 5: YHIF10. 6: YHIF20. Puesta a punto del sistema de vectores retrovirales HIF A fin de poner a punto el sistema, se generaron las partículas retrovirales de las nuevas construcciones 5 y 6, las cuales llevan HIF- α HIF- α, espe ti a e te, o la t iple uta ió . Luego se transdujeron distintos fibroblastos; en la Figura 62 se observan los resultados. A pesar de detectarse fluorescencia de YFP en las células HEK-293T transfectadas, se observó baja eficiencia de transducción y alta tasa de muerte celular en fibroblastos. Se eligieron estas construcciones en particular dado que son las que proveerían más sobre-expresión y/o actividad de HIF, ya sea a nivel de estabilización de la proteína o de regulación de la transcripción de genes blanco. Es posible que la sobre-expresión de HIF en alta cantidad haya resultado tóxica para la célula, dado que luego de un cierto tiempo de cultivo se encuentran evidencias como muerte celular, alta cantidad de células despegadas del sustrato, morfología ahusada, la cual usualmente indica estrés y disminución de la viabilidad en fibroblastos. Para - 128 - RESULTADOS evaluar esta hipótesis, se realizó una experiencia inicial de inhibición de la expresión de factores HIF por Doxiciclina (Dox), observándose preliminarmente que disminuyendo la cantidad expresada de proteínas HIF, se reduce la muerte celular, como se observa en la Figura 63. Figura 62. Transfección de células HEK-293T y transducción de fibroblastos JC-G con los vectores YHIF10 y YHIF20. Se muestran imágenes representativas de contraste de fases y de fluorescencia de YFP. PT: post-transducción. YHIF10: Vector NITSC con HIF- α Triple Mutado fusionado a YFP. YHIF20: Vector NITSC con HIF- α Triple Mutado fusionado a YFP. Barras de escala: 200 µm. - 129 - RESULTADOS Figura 63. Transducción de fibroblastos JC-G y fibroblastos IN10, con los vectores YHIF10 y YHIF20, en presencia de Doxiciclina. (a) Se grafica el número de células remanentes luego de 30 días de cultivo en Doxiciclina. Dox: Condición con Doxiciclina 1 µg/ml. (b) Se muestran imágenes representativas de contraste de fases, en ausencia (Control) y presencia de Doxiciclina. PT: post-transducción. YHIF10: Células transducidas con el vector YHIF10. YHIF20: Células transducidas con el vector YHIF20. Barras de escala: 200 µm. Para comprobar que la expresión de las proteínas YFP-HIF disminuye con el tratamiento con Dox, como se espera en un sistema TetOff, y a fin de relacionar el rescate de la alta muerte celular por este antibiótico, realizamos una tinción para HIF-1 y HIF-2 en las células tratadas con - 130 - RESULTADOS Dox, al final del tratamiento. Como se observa en la Figura 64, las células sin tratar expresan los factores HIF, mientras que en las células tratadas no se detectó dicha expresión, situación que podría indicar, junto con los resultados observados en la Figura 62, que era la alta expresión de estas proteínas la causa de la alta muerte celular y falta de proliferación, lo cual a su vez explicaría la baja tasa de transducción. Figura 64. Detección de los factores HIF por inmunofluorescencia en fibroblastos JC-G y fibroblastos IN10 transducidos con los vectores YHIF10 y YHIF20. Se muestran imágenes representativas de contraste de fases, tinción con DAPI e inmunofluorescencia de HIF-1α y HIF-2α, según corresponda. – Dox: Sin Doxiciclina. +Dox: Con Doxiciclina. YHIF10: Células transducidas con el vector YHIF10. YHIF20: Células transducidas con el vector YHIF20. Barras de escala: 200 µm. - 131 - RESULTADOS Por otra parte, en muchos casos se observó una localización subcelular extra-nuclear de los factores HIF, lo cual podría estar indicando una sobre-acumulación, dado que la localización de estas proteínas es nuclear, ya que son factores de transcripción con una señal de localización nuclear, que normalmente son degradadas por el proteosoma, pero la hipoxia causa su estabilización y exportación a núcleo, como se mencionó anteriormente. Finalmente, HIF-2α parece no poseer un efecto deletéreo tan marcado como HIF- α, ya que no fue tan alta la muerte celular observada en las células remanentes al final del experimento en condiciones sin Dox, ni tanta la diferencia observada entre esta situación y la inhibición de la expresión del gen exógeno con Dox. En síntesis, se generaron una serie de plásmidos retrovirales con los genes de las proteínas HIFα. Estas herramientas serán de utilidad en el futuro, a fin de dilucidar el mecanismo de acción de los factores HIF sobre la reprogramación al estado pluripotente. Dados los resultados preliminares obtenidos, se propone la posibilidad de que la expresión de factores HIF con la triple mutación es tóxica, posiblemente por una sobreexpresión y acumulación de HIF. Se debe buscar una dosis de Dox intermedia que permita la expresión de HIF-, sin llegar a generar muerte celular. Para generar CMPi se propone realizar la transducción de las células a reprogramar con los plásmidos HIF, luego realizar un pre-tratamiento con una concentración de Dox apropiada para evitar la sobre-expresión de YFP-HIF-α, tratar con G418 para seleccionar las células transducidas con HIF-α y finalmente realizar la transducción con los plásmidos reprogramadores. - 132 - DISCUSIÓN - 133 - DISCUSIÓN El desarrollo de las CMPi revolucionó el campo de las células madre, entre otras razones, por la posibilidad que presentan de generar células pluripotentes paciente-específicas para su diferenciación y posterior tratamiento de diversas patologías mediante el aporte de células o tejidos funcionales, como enfermedades cardiovasculares [15-19], enfermedades neurodegenerativas y lesiones neurológicas [20], diabetes [21] y enfermedades de la sangre [22]. Además, el cultivo de estas células provee un modelo para el análisis de las señales de diferenciación involucradas en el desarrollo embrionario y una herramienta para el estudio in vitro de enfermedades y para ensayos de drogas y toxicología [23, 24]. En este trabajo nos propusimos obtener CMPi humanas y estudiar el efecto de la hipoxia, tanto sobre la eficiencia en su obtención como en las propiedades de las células generadas. En primera instancia, nos enfocamos en la obtención de cultivos celulares primarios a partir de tejidos frescos. En particular, a pesar de haber podido obtener células queratinocíticas, se observó que las posibilidades de expandir el número de queratinocitos a través de pasajes en cultivo fueron bajas o nulas, lo cual no aporta las condiciones necesarias para la generación de CMPi, para lo cual se precisa un número mínimo de células en división. Además, el fin último del presente proyecto era la generación de CMPi reproducible y en repetidas instancias a partir del mismo cultivo celular, a fin de estudiar el proceso en sí y las líneas generadas, por lo cual era necesario un cultivo primario de células somáticas proliferativo y expandible, que no resultó ser el caso de los queratinocitos obtenidos con el método y condiciones utilizadas. Por esta misma razón no se llegó a la instancia de reprogramación con queratinocitos, no sólo por imposibilidad, sino también porque el cultivo celular no cumplía con las características necesarias para el presente estudio. Con respecto a las CMSP, se logró obtener un alto número de células, pero las reprogramaciones no fueron exitosas, incluso cuando se utilizaron moléculas para estimular la división de los clones linfocitarios, haciéndolos receptivos de transducción. Es posible que sea necesario un protocolo más específico para generar las condiciones apropiadas para la reprogramación de este tipo celular. En el pasado, los protocolos de obtención de queratinocitos más exitosos solían utilizar una batería de medios y reactivos comerciales que soportan la derivación de este tipo celular más específicamente [121]. Por otro lado, la - 134 - DISCUSIÓN obtención de fibroblastos fue no solamente simple, sino que también generó un gran número de células capaces de ser expandidas en cultivo por varios pasajes, al menos hasta el pasaje 15. Además, los fibroblastos primarios obtenidos fueron reprogramados a CMPi en numerosas instancias, proveyendo un modelo confiable y consistente para el estudio de las condiciones de generación de estas células pluripotentes, de la eficiencia del proceso y de las líneas celulares obtenidas. Las pruebas realizadas en cuanto a las condiciones de reprogramación y moléculas utilizadas a fin de mejorar el proceso fueron suficientes para establecer un protocolo consistente de generación de CMPi, el cual puede utilizarse para dichas investigaciones. Los resultados sobre los efectos de la hipoxia sobre la reprogramación al estado pluripotente son insuficientes o contradictorios. Por ejemplo, el popular estudio de Yoshida et. al. [65] es un modelo de reportero de un único gen realizado en células de ratón. En el estudio de Iida et. al. [64] se reporta la formación de mayor número de colonias en hipoxia, al comparar con normoxia, pero la expresión de NANOG en particular no cambia, o lo hace de forma dependiente de la línea en estudio; en el trabajo de Shimada et. al. [94] sólo se reporta una formación de colonias de CMPi en hipoxia acelerada en el tiempo, pero no un aumento de la eficiencia en dicha condición; los trabajos de Mathieu et. al. [102, 115] son mecanísticos o hablan de una re-entrada en la pluripotencia de células ya derivadas de CME o CMPi; en Bae et. al. [147] se habla del proceso contrario a la reprogramación, indicando que la hipoxia aumentaría la eficiencia del proceso de generación de células de retina a partir de CMPi; y en Hasegawa et. al. [148] se habla de la manutención de CMPi en cultivo, y cómo la hipoxia apoyaría el mantenimiento del estado pluripotente. Por ello nos pareció necesario aportar más información al estudio de los efectos de la hipoxia sobre la generación de CMPi. De acuerdo con estudios previos realizados tanto en células de ratón [65] como en células humanas [64, 115], en este trabajo mostramos que al final del proceso de reprogramación se observó una mayor eficiencia de reprogramación bajo condiciones hipóxicas, aunque esto no se tradujo en una diferencia funcional entre las líneas de CMPi establecidas bajo dichas condiciones, ya que todas las líneas obtenidas poseían propiedades similares y cumplían con iguales condiciones de pluripotencia. Los datos mostraron que una línea de CMPi generada en - 135 - DISCUSIÓN hipoxia al 5% O2 tuvo morfología de colonias y características funcionales similares a una línea control de CMEh generada en normoxia. Además dichas CMPi exhibieron similar metilación del promotor de POU5F1 y pluripotencia in vitro e in vivo, que la línea de CMEh control y líneas de CMPi generadas en normoxia. Sin embargo, si bien las líneas obtenidas en ambas condiciones, normoxia e hipoxia, se asemejaban en todos esos aspectos mencionados, presentaron diferentes características en la expresión de marcadores del estado pluripotente. Se observaron diferencias en particular a lo largo del proceso de reprogramación estudiado por citometría de flujo, en colonias de CMPi jóvenes, y en colonias de CMPi establecidas por un período de tiempo correspondiente a al menos 5 pasajes en cultivo. Nuestros datos sugieren que colonias de CMPi jóvenes, recientemente generadas, exhiben igual o menor expresión de estos marcadores de pluripotencia, en particular una expresión más baja en los casos de los genes NANOG y C-MYC, cuando son generadas en hipoxia en comparación con sus pares generados en normoxia. Algunos estudios previos no han mostrado diferencias significativas en la expresión de marcadores del estado pluripotente en CMEh cuando éstas fueron cultivadas en hipoxia o en normoxia [104]. Además, en ciertos trabajos anteriores sobre la generación de CMPi no hubo diferencias significativas o hubo una fuerte dependencia de línea en la expresión de marcadores del estado pluripotente al generar CMPi en normoxia o hipoxia [64], pero esto fue evaluado a un punto final luego de la generación de las líneas. En nuestro estudio se observó una disminución en la expresión de marcadores del estado pluripotente, destacando que esta disminución se observó en colonias de CMPi jóvenes y en colonias recién establecidas expuestas a hipoxia aguda. La mayoría de los estudios previos sobre eficiencia de reprogramación ha medido solamente la cantidad de colonias generadas y la expresión de marcadores en líneas ya establecidas a largo plazo. Nuestros datos para líneas de CMPi establecidas a largo plazo -generadas en hipoxia, amplificadas durante por lo menos 5 pasajes y posteriormente cultivadas en hipoxia crónica durante al menos 5 pasajes- mostraron en contraste, un aumento en la expresión de marcadores del estado pluripotente, en comparación con nuestros datos para colonias jóvenes o colonias establecidas en hipoxia aguda. Esto se condice con el trabajo de Yoshida et al. [65] y ciertos antecedente sobre el cultivo de CMEh - 136 - DISCUSIÓN bajo bajas tensiones de O2 [98, 105, 107, 108]. Es claro que la literatura es, al menos, contradictoria con respecto al comportamiento de marcadores del estado pluripotente en CMPi generadas en hipoxia. Es posible que estas diferencias reportadas a lo largo de la literatura, se deban tanto a la variabilidad intrínseca entre líneas, como a los diferentes protocolos utilizados y a distintas formas de medir dichos parámetros. El cultivo de células madre requiere de mucho trabajo y es, a veces también, muy dependiente del operador. Finalmente, esto podría indicar que el efecto no es tan contundente si la variabilidad entre líneas es mayor que la diferencia entre tratamientos o es difícil de detectar de acuerdo a las condiciones de análisis. Finalmente, también encontramos que las colonias de CMPi generadas bajo hipoxia son mucho más pequeñas en tamaño, según lo determinado por el área que cubren. Esto también se condice con resultados anteriores para el cultivo de CMEh, en los cuales las colonias cultivadas en hipoxia son de menor tamaño [98, 101, 105]. La inducción de hipoxia en fibroblastos adultos comprometidos podría no permitir una inducción de la expresión de marcadores del estado pluripotente endógenos o un aumento de la tasa de proliferación en colonias de CMPi jóvenes. Esto fue evidenciado por el pequeño tamaño de las colonias generadas bajo hipoxia y la menor expresión de marcadores del estado pluripotente NANOG y C-MYC en muestras de CMPi generadas recientemente, como se observó tanto en el número de células que expresaban los marcadores por citometría de flujo y en los niveles de expresión generales estudiados por In-Cell Western Blot. Pero a medida que la reprogramación progresa y las colonias se van estableciendo, la hipoxia podría estar induciendo la expresión de genes del estado pluripotente, como se describió anteriormente en la literatura, generando una mayor tasa de proliferación, como se observó en las líneas establecidas cultivadas bajo hipoxia crónica. Esto podría ser una posible consecuencia de un cambio desde una baja expresión de marcadores del estado pluripotente a una alta expresión cuando las células maduran en esta condición de bajo oxígeno. Con respecto a los niveles de proliferación, los estudios previos en CMEh son contradictorios encontrándose casos en los que la proliferación aumenta en hipoxia, y casos en los que disminuye [98, 108]. Pero si, de hecho, las tasas de proliferación son homogéneamente más altas entre diferentes líneas de CMPi cuando se establecen a largo plazo, en comparación con colonias de CMPi jóvenes, esto estaría de - 137 - DISCUSIÓN acuerdo con un cambio de una identidad parcial de CM a un estado de célula pluripotente bona fide, a medida que las CMPi maduran. Los factores HIF son clave en la señalización del estímulo de hipoxia. Estos son estabilizados frente a dicha señal mediante un mecanismo de hidroxilación de prolinas en normoxia y degradación por proteosoma, que no se da en hipoxia. Con el propósito de estudiar si estos factores estaban involucraos en nuestro sistemas estudiamos el efecto de la hipoxia sobre la eficiencia de reprogramación a través de la expresión de NANOG, y más específicamente utilizamos inhibidores de los factores HIF a fin de dilucidar cuál factor estaba involucrado en los efectos observados. Nuestros resultados utilizando tales inhibidores sugieren que es el factor HIF- α el que genera las diferencias observadas en las colonias jóvenes de CMPi, entre los estados de hipoxia y normoxia. Un estudio reciente ha demostrado cómo los HIF poseen roles específicos de fase durante la reprogramación [115], en la cual ocurre un cambio metabólico temprano dependiente de HIF- α y HIF- α, pero luego HIF- α parece perder su rol previo, convirtiéndose en una molécula inhibitoria del proceso de reprogramación. Esta secuencia de acontecimientos puede proporcionar una explicación para los diferentes efectos aparentes de los HIF en la disminución de la expresión de NANOG en condiciones de hipoxia, al generar CMPi. En cuanto a los plásmidos con las proteínas HIF-α generados y los resultados preliminares obtenidos con su uso, se propone la posibilidad de que la expresión de factores HIF con la triple mutación es tóxica para fibroblastos humanos primarios y células HEK-293T, posiblemente por una sobre-expresión y acumulación, y consecuentemente una alta actividad de HIF sobre sus genes blanco. Además se propone la posibilidad de que la hipoxia real induzca además del aumento de la expresión de genes blanco de los factores HIF, mecanismos adicionales que protejan a las células de los efectos nocivos de una exagerada actividad de HIF, funcionando como reguladores negativos. Un exceso de HIF durante normoxia podría ir en detrimento de las vías activadas por hipoxia, dado que no estarían activas otras vías que deberían regular los excesos de factores HIF o de los genes que éstos regulan. Se debe buscar un nivel intermedio de Doxiciclina, el represor de este sistema de sobreexpresión, que permita la expresión de HIF-, - 138 - DISCUSIÓN sin llegar a generar muerte celular, realizando una curva de concentración de Doxiciclina entre 0,01–1 μg/ l, el rango que clásicamente se utiliza y que los fabricantes recomiendan. Los estudios futuros deberán centrarse en el mecanismo de acción de los factores HIF sobre marcadores del estado pluripotente, más allá de lo que ya sabemos sobre su rol en la activación del promotor de POU5F1 [103, 112] y la interacción diferencial con C-MYC [110, 113] en CMEh y CMPi. Estos resultados pueden ayudar a desentrañar los papeles de los factores HIF durante y después del proceso de reprogramación y contribuir a elucidar cuándo la hipoxia es beneficiosa o perjudicial en la reprogramación y en el cultivo de CM. Creemos que estos resultados, en su totalidad, contribuirán al progreso del conocimiento sobre la generación de CMPi, sobre las condiciones que la afectan y al entendimiento de los mecanismos que subyacen el proceso. - 139 - RECONOCIMIENTOS Al Dr. José Cibelli y al Dr. Steve Suhr del Cellular Reprogramming Laboratory, Michigan State University, Michigan, EE.UU., por sus enseñanzas respecto a la generación de CMPi. A la Lic. Diana Radakoff de Genos, Buenos Aires, Argentina por su invaluable aporte en la preparación e identificación de cariotipos. A la Dra. Naomi Arakaki, el Dr. Marcelo Schultz y el Dr. Gustavo Sevlever del Laboratorio de Neuropatología de FLENI, Buenos Aires, Argentina por su ayuda con los preparados histológicos y el análisis de tejidos derivados de teratomas. A la Dra. Susana Acevedo del Departamento de Genética del Instituto de Investigaciones Hematológicas, Academia Nacional de Medicina, Buenos Aires, Argentina por su aporte con la preparación y análisis del cariotipo de la línea de CMEh WA-09. El Dr. Carlos Luzzani realizó el estudio de los niveles de demetilación de CMPi iPS6.1. La Dra. Carolina Blüguermann realizó la expansión inicial de la línea de CMPi iPS6.1, a partir del pasaje 0. - 140 - BIBLIOGRAFÍA 1. Gronthos S, Brahim J, Li W, Fisher LW, Cherman N, Boyde A, et al. Stem cell properties of human dental pulp stem cells. Journal of dental research. 2002;81(8):531-5. Epub 2002/07/31. PubMed PMID: 12147742. 2. Robey PG. Stem cells near the century mark. The Journal of clinical investigation. 2000;105(11):1489-91. Epub 2000/06/07. doi: 10.1172/JCI10256. PubMed PMID: 10841501; PubMed Central PMCID: PMC300867. 3. Baserga A, Zavagli G. Ferrata's stem cells: an historical review on hemocytoblasts and hemohistioblasts. Blood cells. 1981;7(3):537-45. Epub 1981/01/01. PubMed PMID: 7039727. 4. Leblond CP. Classification of Cell Populations on the Basis of Their Proliferative Behavior. National Cancer Institute monograph. 1964;14:119-50. Epub 1964/05/01. PubMed PMID: 14147128. 5. Odorico JS, Kaufman DS, Thomson JA. Multilineage differentiation from human embryonic stem cell lines. Stem Cells. 2001;19(3):193-204. Epub 2001/05/22. doi: 10.1634/stemcells.19-3-193. PubMed PMID: 11359944. 6. Kassem M. Stem cells: potential therapy for age-related diseases. Annals of the New York Academy of Sciences. 2006;1067:436-42. Epub 2006/06/29. doi: 10.1196/annals.1354.062. PubMed PMID: 16804023. 7. Carlson BM. Human embryology and developmental biology. 4th ed. Philadelphia, PA: Mosby/Elsevier; 2009. xvi, 541 p. p. 8. Sadler TW, Langman J. Langman's medical embryology. 11th ed. Baltimore, Md.: Lippincott William & Wilkins; 2009. p. p. 9. Solter D, Knowles BB. Immunosurgery of mouse blastocyst. Proc Natl Acad Sci U S A. 1975;72(12):5099-102. Epub 1975/12/01. PubMed PMID: 1108013; PubMed Central PMCID: PMC388883. 10. Amit M, Itskovitz-Eldor J. Derivation and maintenance of human embryonic stem cells. Methods Mol Biol. 2006;331:43-53. Epub 2006/08/03. doi: 10.1385/1-59745-046-4:43. PubMed PMID: 16881508. 11. Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145-7. Epub 1998/11/06. PubMed PMID: 9804556. 12. Wang L, Menendez P, Cerdan C, Bhatia M. Hematopoietic development from human embryonic stem cell lines. Experimental hematology. 2005;33(9):987-96. Epub 2005/09/06. doi: 10.1016/j.exphem.2005.06.002. PubMed PMID: 16140146. 13. Trounson A. The production and directed differentiation of human embryonic stem cells. Endocrine reviews. 2006;27(2):208-19. Epub 2006/01/26. doi: 10.1210/er.2005-0016. PubMed PMID: 16434509. 14. Wobus AM, Boheler KR. Embryonic stem cells: prospects for developmental biology and cell therapy. Physiological reviews. 2005;85(2):635-78. Epub 2005/03/25. doi: 10.1152/physrev.00054.2003. PubMed PMID: 15788707. 15. Menard C, Hagege AA, Agbulut O, Barro M, Morichetti MC, Brasselet C, et al. Transplantation of cardiac-committed mouse embryonic stem cells to infarcted sheep myocardium: a preclinical study. Lancet. 2005;366(9490):1005-12. Epub 2005/09/20. doi: 10.1016/S0140-6736(05)67380-1. PubMed PMID: 16168783. 16. Rosenstrauch D, Poglajen G, Zidar N, Gregoric ID. Stem cell therapy for ischemic heart failure. Tex Heart Inst J. 2005;32(3):339-47. Epub 2006/01/06. PubMed PMID: 16392214; PubMed Central PMCID: PMC1336704. - 141 - BIBLIOGRAFÍA 17. Yoon YS, Lee N, Scadova H. Myocardial regeneration with bone-marrow-derived stem cells. Biology of the cell / under the auspices of the European Cell Biology Organization. 2005;97(4):253-63. Epub 2005/03/15. doi: 10.1042/BC20040099. PubMed PMID: 15762847. 18. Yoon YS, Wecker A, Heyd L, Park JS, Tkebuchava T, Kusano K, et al. Clonally expanded novel multipotent stem cells from human bone marrow regenerate myocardium after myocardial infarction. J Clin Invest. 2005;115(2):326-38. Epub 2005/02/04. doi: 10.1172/JCI22326. PubMed PMID: 15690083; PubMed Central PMCID: PMC546424. 19. Itescu S, Schuster MD, Kocher AA. New directions in strategies using cell therapy for heart disease. J Mol Med. 2003;81(5):288-96. Epub 2003/04/17. doi: 10.1007/s00109-003-0432-0. PubMed PMID: 12698252. 20. Battista D, Ferrari CC, Gage FH, Pitossi FJ. Neurogenic niche modulation by activated microglia: transforming growth factor beta increases neurogenesis in the adult dentate gyrus. The European journal of neuroscience. 2006;23(1):83-93. Epub 2006/01/20. doi: 10.1111/j.1460-9568.2005.04539.x. PubMed PMID: 16420418. 21. Furth ME, Atala A. Stem cell sources to treat diabetes. J Cell Biochem. 2009;106(4):507-11. Epub 2009/01/09. doi: 10.1002/jcb.22000. PubMed PMID: 19130494. 22. Niwa A, Umeda K, Chang H, Saito M, Okita K, Takahashi K, et al. Orderly hematopoietic development of induced pluripotent stem cells via Flk-1(+) hemoangiogenic progenitors. Journal of cellular physiology. 2009;221(2):367-77. Epub 2009/06/30. doi: 10.1002/jcp.21864. PubMed PMID: 19562687. 23. Steel D, Hyllner J, Sartipy P. Cardiomyocytes derived from human embryonic stem cells characteristics and utility for drug discovery. Curr Opin Drug Discov Devel. 2009;12(1):133-40. Epub 2009/01/20. PubMed PMID: 19152222. 24. Reppel M, Igelmund P, Egert U, Juchelka F, Hescheler J, Drobinskaya I. Effect of cardioactive drugs on action potential generation and propagation in embryonic stem cell-derived cardiomyocytes. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2007;19(5-6):213-24. Epub 2007/05/15. doi: 10.1159/000100628. PubMed PMID: 17495462. 25. Questa M. Cardiomiocitos derivados de células madre embrionarias humanas como modelo experimental para el estudio de infecciones por Virus Coxsackie B. Buenos Aires: Universidad de Buenos Aires; 2010. 26. Hyslop LA, Armstrong L, Stojkovic M, Lako M. Human embryonic stem cells: biology and clinical implications. Expert reviews in molecular medicine. 2005;7(19):1-21. Epub 2005/09/21. doi: 10.1017/S1462399405009804. PubMed PMID: 16171533. 27. Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132(4):661-80. Epub 2008/02/26. doi: 10.1016/j.cell.2008.02.008. PubMed PMID: 18295582. 28. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663-76. Epub 2006/08/15. doi: 10.1016/j.cell.2006.07.024. PubMed PMID: 16904174. 29. Takahashi K, Okita K, Nakagawa M, Yamanaka S. Induction of pluripotent stem cells from fibroblast cultures. Nature protocols. 2007;2(12):3081-9. Epub 2007/12/15. doi: 10.1038/nprot.2007.418. PubMed PMID: 18079707. 30. Boland MJ, Hazen JL, Nazor KL, Rodriguez AR, Gifford W, Martin G, et al. Adult mice generated from induced pluripotent stem cells. Nature. 2009. Epub 2009/08/13. doi: 10.1038/nature08310. PubMed PMID: 19672243. - 142 - BIBLIOGRAFÍA 31. Kang L, Wang J, Zhang Y, Kou Z, Gao S. iPS cells can support full-term development of tetraploid blastocyst-complemented embryos. Cell stem cell. 2009;5(2):135-8. Epub 2009/07/28. doi: 10.1016/j.stem.2009.07.001. PubMed PMID: 19631602. 32. Meissner A, Wernig M, Jaenisch R. Direct reprogramming of genetically unmodified fibroblasts into pluripotent stem cells. Nat Biotechnol. 2007;25(10):1177-81. Epub 2007/08/29. doi: 10.1038/nbt1335. PubMed PMID: 17724450. 33. Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448(7151):313-7. Epub 2007/06/08. doi: 10.1038/nature05934. PubMed PMID: 17554338. 34. Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448(7151):318-24. Epub 2007/06/08. doi: 10.1038/nature05944. PubMed PMID: 17554336. 35. Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451(7175):141-6. Epub 2007/12/25. doi: doi: 10.1038/nature06534. PubMed PMID: 18157115. 36. Yamanaka S. A fresh look at iPS cells. Cell. 2009;137(1):13-7. Epub 2009/04/07. doi: 10.1016/j.cell.2009.03.034. PubMed PMID: 19345179. 37. Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917-20. Epub 2007/11/22. doi: 10.1126/science.1151526. PubMed PMID: 18029452. 38. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861-72. Epub 2007/11/24. doi: 10.1016/j.cell.2007.11.019. PubMed PMID: 18035408. 39. Nakagawa M, Koyanagi M, Tanabe K, Takahashi K, Ichisaka T, Aoi T, et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol. 2008;26(1):1016. Epub 2007/12/07. doi: 10.1038/nbt1374. PubMed PMID: 18059259. 40. Yamanaka S, Blau HM. Nuclear reprogramming to a pluripotent state by three approaches. Nature. 2010;465(7299):704-12. Epub 2010/06/11. doi: 10.1038/nature09229. PubMed PMID: 20535199; PubMed Central PMCID: PMC2901154. 41. Chun YS, Chaudhari P, Jang YY. Applications of patient-specific induced pluripotent stem cells; focused on disease modeling, drug screening and therapeutic potentials for liver disease. International journal of biological sciences. 2010;6(7):796-805. Epub 2010/12/24. PubMed PMID: 21179587; PubMed Central PMCID: PMC3005346. 42. Itzhaki I, Maizels L, Huber I, Zwi-Dantsis L, Caspi O, Winterstern A, et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature. 2011;471(7337):225-9. Epub 2011/01/18. doi: 10.1038/nature09747. PubMed PMID: 21240260. 43. Tiscornia G, Vivas EL, Izpisua Belmonte JC. Diseases in a dish: modeling human genetic disorders using induced pluripotent cells. Nat Med. 2011;17(12):1570-6. Epub 2011/12/08. doi: 10.1038/nm.2504. PubMed PMID: 22146428. 44. Yi F, Liu GH, Izpisua Belmonte JC. Human induced pluripotent stem cells derived hepatocytes: rising promise for disease modeling, drug development and cell therapy. Protein & cell. 2012;3(4):24650. Epub 2012/03/24. doi: 10.1007/s13238-012-2918-4. PubMed PMID: 22441839. 45. Drawnel FM, Boccardo S, Prummer M, Delobel F, Graff A, Weber M, et al. Disease modeling and phenotypic drug screening for diabetic cardiomyopathy using human induced pluripotent stem cells. Cell reports. 2014;9(3):810-21. Epub 2014/12/02. doi: 10.1016/j.celrep.2014.09.055. PubMed PMID: 25437537. - 143 - BIBLIOGRAFÍA 46. Liu EY, Scott CT. Great expectations: autism spectrum disorder and induced pluripotent stem cell technologies. Stem cell reviews. 2014;10(2):145-50. Epub 2014/02/04. doi: 10.1007/s12015-014-9497-0. PubMed PMID: 24488263. 47. Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321(5893):1218-21. Epub 2008/08/02. doi: 10.1126/science.1158799. PubMed PMID: 18669821. 48. Ebert AD, Yu J, Rose FF, Jr., Mattis VB, Lorson CL, Thomson JA, et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457(7227):277-80. Epub 2008/12/23. doi: 10.1038/nature07677. PubMed PMID: 19098894; PubMed Central PMCID: PMC2659408. 49. Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134(5):877-86. Epub 2008/08/12. doi: 10.1016/j.cell.2008.07.041. PubMed PMID: 18691744; PubMed Central PMCID: PMC2633781. 50. Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, Cook EG, et al. Parkinson's disease patientderived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136(5):964-77. Epub 2009/03/10. doi: 10.1016/j.cell.2009.02.013. PubMed PMID: 19269371. 51. Egawa N, Kitaoka S, Tsukita K, Naitoh M, Takahashi K, Yamamoto T, et al. Drug screening for ALS using patient-specific induced pluripotent stem cells. Science translational medicine. 2012;4(145):145ra04. Epub 2012/08/03. doi: 10.1126/scitranslmed.3004052. PubMed PMID: 22855461. 52. Guo L, Abrams RM, Babiarz JE, Cohen JD, Kameoka S, Sanders MJ, et al. Estimating the risk of drug-induced proarrhythmia using human induced pluripotent stem cell-derived cardiomyocytes. Toxicological sciences : an official journal of the Society of Toxicology. 2011;123(1):281-9. Epub 2011/06/23. doi: 10.1093/toxsci/kfr158. PubMed PMID: 21693436. 53. Ko HC, Gelb BD. Concise review: drug discovery in the age of the induced pluripotent stem cell. Stem cells translational medicine. 2014;3(4):500-9. Epub 2014/02/05. doi: 10.5966/sctm.2013-0162. PubMed PMID: 24493856; PubMed Central PMCID: PMC3973715. 54. Sinnecker D, Goedel A, Dorn T, Dirschinger RJ, Moretti A, Laugwitz KL. Modeling long-QT syndromes with iPS cells. Journal of cardiovascular translational research. 2013;6(1):31-6. Epub 2012/10/19. doi: 10.1007/s12265-012-9416-1. PubMed PMID: 23076501. 55. Sirenko O, Crittenden C, Callamaras N, Hesley J, Chen YW, Funes C, et al. Multiparameter in vitro assessment of compound effects on cardiomyocyte physiology using iPSC cells. Journal of biomolecular screening. 2013;18(1):39-53. Epub 2012/09/14. doi: 10.1177/1087057112457590. PubMed PMID: 22972846. 56. Knoepfler PS. Deconstructing stem cell tumorigenicity: a roadmap to safe regenerative medicine. Stem Cells. 2009;27(5):1050-6. Epub 2009/05/06. doi: 10.1002/stem.37. PubMed PMID: 19415771; PubMed Central PMCID: PMC2733374. 57. Li JY, Christophersen NS, Hall V, Soulet D, Brundin P. Critical issues of clinical human embryonic stem cell therapy for brain repair. Trends Neurosci. 2008;31(3):146-53. Epub 2008/02/08. doi: 10.1016/j.tins.2007.12.001. PubMed PMID: 18255164. 58. Liu Z, Tang Y, Lu S, Zhou J, Du Z, Duan C, et al. The tumourigenicity of iPS cells and their differentiated derivates. Journal of cellular and molecular medicine. 2013;17(6):782-91. Epub 2013/05/29. doi: 10.1111/jcmm.12062. PubMed PMID: 23711115; PubMed Central PMCID: PMC3823182. 59. Ratajczak MZ, Jadczyk T, Pedziwiatr D, Wojakowski W. New advances in stem cell research: practical implications for regenerative medicine. Polskie Archiwum Medycyny Wewnetrznej. 2014;124(7-8):417-26. Epub 2014/06/24. PubMed PMID: 24956404. - 144 - BIBLIOGRAFÍA 60. Byrne JA, Nguyen HN, Reijo Pera RA. Enhanced generation of induced pluripotent stem cells from a subpopulation of human fibroblasts. PloS one. 2009;4(9):e7118. Epub 2009/09/24. doi: 10.1371/journal.pone.0007118. PubMed PMID: 19774082; PubMed Central PMCID: PMC2744017. 61. Okita K, Yamanaka S. Induction of pluripotency by defined factors. Experimental cell research. 2010;316(16):2565-70. Epub 2010/04/28. doi: 10.1016/j.yexcr.2010.04.023. PubMed PMID: 20420827. 62. Takahashi K, Yamanaka S. Induced pluripotent stem cells in medicine and biology. Development. 2013;140(12):2457-61. Epub 2013/05/30. doi: 10.1242/dev.092551. PubMed PMID: 23715538. 63. Tanabe K, Takahashi K, Yamanaka S. Induction of pluripotency by defined factors. Proceedings of the Japan Academy Series B, Physical and biological sciences. 2014;90(3):83-96. Epub 2014/03/14. PubMed PMID: 24621955; PubMed Central PMCID: PMC3997808. 64. Iida K, Takeda-Kawaguchi T, Hada M, Yuriguchi M, Aoki H, Tamaoki N, et al. Hypoxia-enhanced derivation of iPSCs from human dental pulp cells. Journal of dental research. 2013;92(10):905-10. Epub 2013/08/22. doi: 10.1177/0022034513502204. PubMed PMID: 23962749. 65. Yoshida Y, Takahashi K, Okita K, Ichisaka T, Yamanaka S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell. 2009;5(3):237-41. Epub 2009/09/01. doi: 10.1016/j.stem.2009.08.001. PubMed PMID: 19716359. 66. Li W, Tian E, Chen ZX, Sun G, Ye P, Yang S, et al. Identification of Oct4-activating compounds that enhance reprogramming efficiency. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(51):20853-8. Epub 2012/12/06. doi: 10.1073/pnas.1219181110. PubMed PMID: 23213213; PubMed Central PMCID: PMC3529047. 67. Li Z, Rana TM. A kinase inhibitor screen identifies small-molecule enhancers of reprogramming and iPS cell generation. Nature communications. 2012;3:1085. Epub 2012/09/27. doi: 10.1038/ncomms2059. PubMed PMID: 23011139; PubMed Central PMCID: PMC3658009. 68. Mohseni R, Shoae-Hassani A, Verdi J. Reprogramming of endometrial adult stromal cells in the presence of a ROCK inhibitor, thiazovivin, could obtain more efficient iPSCs. Cell biology international. 2014. Epub 2014/12/11. doi: 10.1002/cbin.10411. PubMed PMID: 25490878. 69. Teng HF, Li PN, Hou DR, Liu SW, Lin CT, Loo MR, et al. Valproic acid enhances Oct4 promoter activity through PI3K/Akt/mTOR pathway activated nuclear receptors. Molecular and cellular endocrinology. 2014;383(1-2):147-58. Epub 2013/12/24. doi: 10.1016/j.mce.2013.12.008. PubMed PMID: 24361750. 70. Li Z, Rana TM. Using microRNAs to enhance the generation of induced pluripotent stem cells. Current protocols in stem cell biology. 2012;Chapter 4:Unit 4A Epub 2012/03/15. doi: 10.1002/9780470151808.sc04a04s20. PubMed PMID: 22415842. 71. Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell stem cell. 2010;7(5):618-30. Epub 2010/10/05. doi: 10.1016/j.stem.2010.08.012. PubMed PMID: 20888316; PubMed Central PMCID: PMC3656821. 72. Miyoshi H, Blomer U, Takahashi M, Gage FH, Verma IM. Development of a self-inactivating lentivirus vector. Journal of virology. 1998;72(10):8150-7. Epub 1998/09/12. PubMed PMID: 9733856; PubMed Central PMCID: PMC110156. 73. Sommer CA, Stadtfeld M, Murphy GJ, Hochedlinger K, Kotton DN, Mostoslavsky G. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells. 2009;27(3):543-9. Epub 2008/12/20. doi: 10.1634/stemcells.2008-1075. PubMed PMID: 19096035. 74. Shao L, Feng W, Sun Y, Bai H, Liu J, Currie C, et al. Generation of iPS cells using defined factors linked via the self-cleaving 2A sequences in a single open reading frame. Cell research. 2009;19(3):296306. Epub 2009/02/25. doi: 10.1038/cr.2009.20. PubMed PMID: 19238173; PubMed Central PMCID: PMC2821711. - 145 - BIBLIOGRAFÍA 75. Lowry WE, Richter L, Yachechko R, Pyle AD, Tchieu J, Sridharan R, et al. Generation of human induced pluripotent stem cells from dermal fibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(8):2883-8. Epub 2008/02/22. doi: 10.1073/pnas.0711983105. PubMed PMID: 18287077; PubMed Central PMCID: PMC2268554. 76. Carey BW, Markoulaki S, Hanna J, Saha K, Gao Q, Mitalipova M, et al. Reprogramming of murine and human somatic cells using a single polycistronic vector. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(1):157-62. Epub 2008/12/26. doi: 10.1073/pnas.0811426106. PubMed PMID: 19109433; PubMed Central PMCID: PMC2629226. 77. Zhou W, Freed CR. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells. 2009;27(11):2667-74. Epub 2009/08/22. doi: 10.1002/stem.201. PubMed PMID: 19697349. 78. Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322(5903):945-9. Epub 2008/09/27. doi: 10.1126/science.1162494. PubMed PMID: 18818365; PubMed Central PMCID: PMC3987909. 79. Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin, II, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324(5928):797-801. Epub 2009/03/28. doi: 10.1126/science.1172482. PubMed PMID: 19325077; PubMed Central PMCID: PMC2758053. 80. Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, Woltjen K. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458(7239):771-5. Epub 2009/03/03. doi: 10.1038/nature07864. PubMed PMID: 19252477; PubMed Central PMCID: PMC2667910. 81. Zhou H, Wu S, Joo JY, Zhu S, Han DW, Lin T, et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell stem cell. 2009;4(5):381-4. Epub 2009/04/29. doi: 10.1016/j.stem.2009.04.005. PubMed PMID: 19398399. 82. Warren L, Wang J. Feeder-free reprogramming of human fibroblasts with messenger RNA. Current protocols in stem cell biology. 2013;27:Unit 4A 6. Epub 2014/02/11. doi: 10.1002/9780470151808.sc04a06s27. PubMed PMID: 24510287. 83. Feng B, Ng JH, Heng JC, Ng HH. Molecules that promote or enhance reprogramming of somatic cells to induced pluripotent stem cells. Cell stem cell. 2009;4(4):301-12. Epub 2009/04/04. doi: 10.1016/j.stem.2009.03.005. PubMed PMID: 19341620. 84. Maherali N, Hochedlinger K. Guidelines and techniques for the generation of induced pluripotent stem cells. Cell stem cell. 2008;3(6):595-605. Epub 2008/12/02. doi: 10.1016/j.stem.2008.11.008. PubMed PMID: 19041776. 85. Fragola G, Germain PL, Laise P, Cuomo A, Blasimme A, Gross F, et al. Cell reprogramming requires silencing of a core subset of polycomb targets. PLoS genetics. 2013;9(2):e1003292. Epub 2013/03/08. doi: 10.1371/journal.pgen.1003292. PubMed PMID: 23468641; PubMed Central PMCID: PMC3585017. 86. Koche RP, Smith ZD, Adli M, Gu H, Ku M, Gnirke A, et al. Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell stem cell. 2011;8(1):96-105. Epub 2011/01/08. doi: 10.1016/j.stem.2010.12.001. PubMed PMID: 21211784; PubMed Central PMCID: PMC3220622. 87. Luzzani C, Solari C, Losino N, Ariel W, Romorini L, Bluguermann C, et al. Modulation of chromatin modifying factors' gene expression in embryonic and induced pluripotent stem cells. Biochemical and biophysical research communications. 2011;410(4):816-22. Epub 2011/06/28. doi: 10.1016/j.bbrc.2011.06.070. PubMed PMID: 21703227. 88. Mansour AA, Gafni O, Weinberger L, Zviran A, Ayyash M, Rais Y, et al. The H3K27 demethylase Utx regulates somatic and germ cell epigenetic reprogramming. Nature. 2012;488(7411):409-13. Epub 2012/07/18. doi: 10.1038/nature11272. PubMed PMID: 22801502. - 146 - BIBLIOGRAFÍA 89. Fan J, Robert C, Jang YY, Liu H, Sharkis S, Baylin SB, et al. Human induced pluripotent cells resemble embryonic stem cells demonstrating enhanced levels of DNA repair and efficacy of nonhomologous end-joining. Mutation research. 2011;713(1-2):8-17. Epub 2011/07/02. doi: 10.1016/j.mrfmmm.2011.05.018. PubMed PMID: 21718709; PubMed Central PMCID: PMC3872739. 90. Kareta MS, Gorges LL, Hafeez S, Benayoun BA, Marro S, Zmoos AF, et al. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell stem cell. 2015;16(1):39-50. Epub 2014/12/04. doi: 10.1016/j.stem.2014.10.019. PubMed PMID: 25467916. 91. Lin YC, Murayama Y, Hashimoto K, Nakamura Y, Lin CS, Yokoyama KK, et al. Role of tumor suppressor genes in the cancer-associated reprogramming of human induced pluripotent stem cells. Stem cell research & therapy. 2014;5(2):58. Epub 2014/08/27. doi: 10.1186/scrt447. PubMed PMID: 25157408; PubMed Central PMCID: PMC4056745. 92. McLenachan S, Menchon C, Raya A, Consiglio A, Edel MJ. Cyclin A1 is essential for setting the pluripotent state and reducing tumorigenicity of induced pluripotent stem cells. Stem cells and development. 2012;21(15):2891-9. Epub 2012/04/17. doi: 10.1089/scd.2012.0190. PubMed PMID: 22500553; PubMed Central PMCID: PMC3464069. 93. Menendez JA, Vellon L, Oliveras-Ferraros C, Cufi S, Vazquez-Martin A. mTOR-regulated senescence and autophagy during reprogramming of somatic cells to pluripotency: a roadmap from energy metabolism to stem cell renewal and aging. Cell Cycle. 2011;10(21):3658-77. Epub 2011/11/05. doi: 10.4161/cc.10.21.18128. PubMed PMID: 22052357. 94. Shimada H, Hashimoto Y, Nakada A, Shigeno K, Nakamura T. Accelerated generation of human induced pluripotent stem cells with retroviral transduction and chemical inhibitors under physiological hypoxia. Biochemical and biophysical research communications. 2012;417(2):659-64. Epub 2011/12/17. doi: 10.1016/j.bbrc.2011.11.111. PubMed PMID: 22172948. 95. Mohyeldin A, Garzon-Muvdi T, Quinones-Hinojosa A. Oxygen in stem cell biology: a critical component of the stem cell niche. Cell Stem Cell. 2010;7(2):150-61. Epub 2010/08/05. doi: 10.1016/j.stem.2010.07.007. PubMed PMID: 20682444. 96. Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nature reviews Molecular cell biology. 2008;9(4):285-96. Epub 2008/02/21. doi: 10.1038/nrm2354. PubMed PMID: 18285802; PubMed Central PMCID: PMC2876333. 97. Guo CW, Kawakatsu M, Idemitsu M, Urata Y, Goto S, Ono Y, et al. Culture under low physiological oxygen conditions improves the stemness and quality of induced pluripotent stem cells. Journal of cellular physiology. 2013;228(11):2159-66. Epub 2013/04/17. doi: 10.1002/jcp.24389. PubMed PMID: 23589166. 98. Lim HJ, Han J, Woo DH, Kim SE, Kim SK, Kang HG, et al. Biochemical and morphological effects of hypoxic environment on human embryonic stem cells in long-term culture and differentiating embryoid bodies. Molecules and cells. 2011;31(2):123-32. Epub 2011/02/25. doi: 10.1007/s10059-011-0016-8. PubMed PMID: 21347709; PubMed Central PMCID: PMC3932683. 99. Abaci HE, Truitt R, Luong E, Drazer G, Gerecht S. Adaptation to oxygen deprivation in cultures of human pluripotent stem cells, endothelial progenitor cells, and umbilical vein endothelial cells. American journal of physiology Cell physiology. 2010;298(6):C1527-37. Epub 2010/02/26. doi: 10.1152/ajpcell.00484.2009. PubMed PMID: 20181925; PubMed Central PMCID: PMC2889635. 100. Ezashi T, Das P, Roberts RM. Low O2 tensions and the prevention of differentiation of hES cells. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(13):4783-8. Epub 2005/03/18. doi: 10.1073/pnas.0501283102. PubMed PMID: 15772165; PubMed Central PMCID: PMC554750. - 147 - BIBLIOGRAFÍA 101. Hawkins KE, Sharp TV, McKay TR. The role of hypoxia in stem cell potency and differentiation. Regenerative medicine. 2013;8(6):771-82. Epub 2013/10/24. doi: 10.2217/rme.13.71. PubMed PMID: 24147532. 102. Mathieu J, Zhang Z, Nelson A, Lamba DA, Reh TA, Ware C, et al. Hypoxia induces re-entry of committed cells into pluripotency. Stem Cells. 2013;31(9):1737-48. Epub 2013/06/15. doi: 10.1002/stem.1446. PubMed PMID: 23765801; PubMed Central PMCID: PMC3921075. 103. Zachar V, Prasad SM, Weli SC, Gabrielsen A, Petersen K, Petersen MB, et al. The effect of human embryonic stem cells (hESCs) long-term normoxic and hypoxic cultures on the maintenance of pluripotency. In vitro cellular & developmental biology Animal. 2010;46(3-4):276-83. Epub 2010/02/24. doi: 10.1007/s11626-010-9305-3. PubMed PMID: 20177991. 104. Chen HF, Kuo HC, Chen W, Wu FC, Yang YS, Ho HN. A reduced oxygen tension (5%) is not beneficial for maintaining human embryonic stem cells in the undifferentiated state with short splitting intervals. Hum Reprod. 2009;24(1):71-80. Epub 2008/09/30. doi: 10.1093/humrep/den345. PubMed PMID: 18819965. 105. Narva E, Pursiheimo JP, Laiho A, Rahkonen N, Emani MR, Viitala M, et al. Continuous hypoxic culturing of human embryonic stem cells enhances SSEA-3 and MYC levels. PloS one. 2013;8(11):e78847. Epub 2013/11/16. doi: 10.1371/journal.pone.0078847. PubMed PMID: 24236059; PubMed Central PMCID: PMC3827269. 106. Prasad SM, Czepiel M, Cetinkaya C, Smigielska K, Weli SC, Lysdahl H, et al. Continuous hypoxic culturing maintains activation of Notch and allows long-term propagation of human embryonic stem cells without spontaneous differentiation. Cell proliferation. 2009;42(1):63-74. Epub 2009/01/16. doi: 10.1111/j.1365-2184.2008.00571.x. PubMed PMID: 19143764. 107. Szablowska-Gadomska I, Zayat V, Buzanska L. Influence of low oxygen tensions on expression of pluripotency genes in stem cells. Acta neurobiologiae experimentalis. 2011;71(1):86-93. Epub 2011/04/19. PubMed PMID: 21499329. 108. Forristal CE, Wright KL, Hanley NA, Oreffo RO, Houghton FD. Hypoxia inducible factors regulate pluripotency and proliferation in human embryonic stem cells cultured at reduced oxygen tensions. Reproduction. 2010;139(1):85-97. Epub 2009/09/17. doi: 10.1530/REP-09-0300. PubMed PMID: 19755485; PubMed Central PMCID: PMC2791494. 109. Hu CJ, Iyer S, Sataur A, Covello KL, Chodosh LA, Simon MC. Differential regulation of the transcriptional activities of hypoxia-inducible factor 1 alpha (HIF-1alpha) and HIF-2alpha in stem cells. Molecular and cellular biology. 2006;26(9):3514-26. Epub 2006/04/14. doi: 10.1128/MCB.26.9.35143526.2006. PubMed PMID: 16611993; PubMed Central PMCID: PMC1447431. 110. Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer cell. 2007;11(4):335-47. Epub 2007/04/10. doi: 10.1016/j.ccr.2007.02.006. PubMed PMID: 17418410; PubMed Central PMCID: PMC3145406. 111. Huang LE. Carrot and stick: HIF-alpha engages c-Myc in hypoxic adaptation. Cell death and differentiation. 2008;15(4):672-7. Epub 2008/01/12. doi: 10.1038/sj.cdd.4402302. PubMed PMID: 18188166. 112. Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, et al. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes & development. 2006;20(5):557-70. Epub 2006/03/03. doi: 10.1101/gad.1399906. PubMed PMID: 16510872; PubMed Central PMCID: PMC1410808. 113. Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1alpha induces cell cycle arrest by functionally counteracting Myc. The EMBO journal. 2004;23(9):1949-56. Epub 2004/04/09. doi: 10.1038/sj.emboj.7600196. PubMed PMID: 15071503; PubMed Central PMCID: PMC404317. 114. Jeong CH, Lee HJ, Cha JH, Kim JH, Kim KR, Yoon DK, et al. Hypoxia-inducible factor-1 alpha inhibits self-renewal of mouse embryonic stem cells in Vitro via negative regulation of the leukemia - 148 - BIBLIOGRAFÍA inhibitory factor-STAT3 pathway. The Journal of biological chemistry. 2007;282(18):13672-9. Epub 2007/03/16. doi: 10.1074/jbc.M700534200. PubMed PMID: 17360716. 115. Mathieu J, Zhou W, Xing Y, Sperber H, Ferreccio A, Agoston Z, et al. Hypoxia-inducible factors have distinct and stage-specific roles during reprogramming of human cells to pluripotency. Cell stem cell. 2014;14(5):592-605. Epub 2014/03/25. doi: 10.1016/j.stem.2014.02.012. PubMed PMID: 24656769; PubMed Central PMCID: PMC4028142. 116. Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Developmental cell. 2005;9(5):617-28. Epub 2005/11/01. doi: 10.1016/j.devcel.2005.09.010. PubMed PMID: 16256737. 117. Moreno-Manzano V, Rodriguez-Jimenez FJ, Acena-Bonilla JL, Fustero-Lardies S, Erceg S, Dopazo J, et al. FM19G11, a new hypoxia-inducible factor (HIF) modulator, affects stem cell differentiation status. The Journal of biological chemistry. 2010;285(2):1333-42. Epub 2009/11/10. doi: 10.1074/jbc.M109.008326. PubMed PMID: 19897487; PubMed Central PMCID: PMC2801260. 118. Masui S, Nakatake Y, Toyooka Y, Shimosato D, Yagi R, Takahashi K, et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nature cell biology. 2007;9(6):625-35. Epub 2007/05/23. doi: 10.1038/ncb1589. PubMed PMID: 17515932. 119. Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. The Journal of general virology. 1977;36(1):59-74. Epub 1977/07/01. PubMed PMID: 886304. 120. Meng G, Liu S, Krawetz R, Chan M, Chernos J, Rancourt DE. A novel method for generating xenofree human feeder cells for human embryonic stem cell culture. Stem cells and development. 2008;17(3):413-22. Epub 2008/06/03. doi: 10.1089/scd.2007.0236. PubMed PMID: 18513158. 121. Aasen T, Raya A, Barrero MJ, Garreta E, Consiglio A, Gonzalez F, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nature biotechnology. 2008;26(11):1276-84. Epub 2008/10/22. doi: 10.1038/nbt.1503. PubMed PMID: 18931654. 122. Brown ME, Rondon E, Rajesh D, Mack A, Lewis R, Feng X, et al. Derivation of induced pluripotent stem cells from human peripheral blood T lymphocytes. PloS one. 2010;5(6):e11373. Epub 2010/07/10. doi: 10.1371/journal.pone.0011373. PubMed PMID: 20617191; PubMed Central PMCID: PMC2894062. 123. Ross PJ, Suhr ST, Rodriguez RM, Chang EA, Wang K, Siripattarapravat K, et al. Human-induced pluripotent stem cells produced under xeno-free conditions. Stem cells and development. 2010;19(8):1221-9. Epub 2009/12/25. doi: 10.1089/scd.2009.0459. PubMed PMID: 20030562. 124. Kang SJ, Park YI, So B, Kang HG. Sodium butyrate efficiently converts fully reprogrammed induced pluripotent stem cells from mouse partially reprogrammed cells. Cellular reprogramming. 2014;16(5):345-54. Epub 2014/08/06. doi: 10.1089/cell.2013.0087. PubMed PMID: 25093667. 125. Trokovic R, Weltner J, Manninen T, Mikkola M, Lundin K, Hamalainen R, et al. Small molecule inhibitors promote efficient generation of induced pluripotent stem cells from human skeletal myoblasts. Stem cells and development. 2013;22(1):114-23. Epub 2012/06/08. doi: 10.1089/scd.2012.0157. PubMed PMID: 22671711. 126. Zhang Q, Yang Y, Zhang J, Wang GY, Liu W, Qiu DB, et al. Efficient derivation of functional hepatocytes from mouse induced pluripotent stem cells by a combination of cytokines and sodium butyrate. Chinese medical journal. 2011;124(22):3786-93. Epub 2012/02/22. PubMed PMID: 22340242. 127. Zhang Z, Wu WS. Sodium butyrate promotes generation of human induced pluripotent stem cells through induction of the miR302/367 cluster. Stem cells and development. 2013;22(16):2268-77. Epub 2013/03/29. doi: 10.1089/scd.2012.0650. PubMed PMID: 23534850; PubMed Central PMCID: PMC3730377. 128. Zhang Z, Xiang D, Wu WS. Sodium butyrate facilitates reprogramming by derepressing OCT4 transactivity at the promoter of embryonic stem cell-specific miR-302/367 cluster. Cellular - 149 - BIBLIOGRAFÍA reprogramming. 2014;16(2):130-9. Epub 2014/02/27. doi: 10.1089/cell.2013.0070. PubMed PMID: 24568633; PubMed Central PMCID: PMC3967376. 129. Croze RH, Buchholz DE, Radeke MJ, Thi WJ, Hu Q, Coffey PJ, et al. ROCK Inhibition Extends Passage of Pluripotent Stem Cell-Derived Retinal Pigmented Epithelium. Stem cells translational medicine. 2014;3(9):1066-78. Epub 2014/07/30. doi: 10.5966/sctm.2014-0079. PubMed PMID: 25069775; PubMed Central PMCID: PMC4149306. 130. Ichikawa H, Yoshie S, Shirasawa S, Yokoyama T, Yue F, Tomotsune D, et al. Freeze-thawing single human embryonic stem cells induce e-cadherin and actin filament network disruption via g13 signaling. Cryo letters. 2011;32(6):516-24. Epub 2012/01/10. PubMed PMID: 22227712. 131. Freberg CT, Dahl JA, Timoskainen S, Collas P. Epigenetic reprogramming of OCT4 and NANOG regulatory regions by embryonal carcinoma cell extract. Molecular biology of the cell. 2007;18(5):154353. Epub 2007/02/23. doi: 10.1091/mbc.E07-01-0029. PubMed PMID: 17314394; PubMed Central PMCID: PMC1855029. 132. Lee K, Ban HS, Naik R, Hong YS, Son S, Kim BK, et al. Identification of malate dehydrogenase 2 as a target protein of the HIF-1 inhibitor LW6 using chemical probes. Angew Chem Int Ed Engl. 2013;52(39):10286-9. Epub 2013/08/13. doi: 10.1002/anie.201304987. PubMed PMID: 23934700. 133. Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295(5556):858-61. Epub 2002/02/02. doi: 10.1126/science.1068592. PubMed PMID: 11823643. 134. Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Molecular and cellular biology. 2003;23(24):9361-74. Epub 2003/12/04. PubMed PMID: 14645546; PubMed Central PMCID: PMC309606. 135. Park F, Ohashi K, Chiu W, Naldini L, Kay MA. Efficient lentiviral transduction of liver requires cell cycling in vivo. Nature genetics. 2000;24(1):49-52. Epub 1999/12/30. doi: 10.1038/71673. PubMed PMID: 10615126. 136. Naldini L, Blomer U, Gage FH, Trono D, Verma IM. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(21):11382-8. Epub 1996/10/15. PubMed PMID: 8876144; PubMed Central PMCID: PMC38066. 137. Miyoshi H, Takahashi M, Gage FH, Verma IM. Stable and efficient gene transfer into the retina using an HIV-based lentiviral vector. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(19):10319-23. Epub 1997/09/18. PubMed PMID: 9294208; PubMed Central PMCID: PMC23360. 138. Cepko C. Large-scale preparation and concentration of retrovirus stocks. Current protocols in molecular biology / edited by Frederick M Ausubel [et al]. 2001;Chapter 9:Unit9 12. Epub 2008/02/12. doi: 10.1002/0471142727.mb0912s37. PubMed PMID: 18265280. 139. Candido EP, Reeves R, Davie JR. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell. 1978;14(1):105-13. Epub 1978/05/01. PubMed PMID: 667927. 140. Davie JR. Inhibition of histone deacetylase activity by butyrate. The Journal of nutrition. 2003;133(7 Suppl):2485S-93S. Epub 2003/07/04. PubMed PMID: 12840228. 141. Verdone L, Agricola E, Caserta M, Di Mauro E. Histone acetylation in gene regulation. Briefings in functional genomics & proteomics. 2006;5(3):209-21. Epub 2006/08/01. doi: 10.1093/bfgp/ell028. PubMed PMID: 16877467. 142. Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389(6649):349-52. Epub 1997/10/06. doi: 10.1038/38664. PubMed PMID: 9311776. 143. Kruh J. Effects of sodium butyrate, a new pharmacological agent, on cells in culture. Molecular and cellular biochemistry. 1982;42(2):65-82. Epub 1982/02/05. PubMed PMID: 6174854. - 150 - BIBLIOGRAFÍA 144. Gurvich N, Tsygankova OM, Meinkoth JL, Klein PS. Histone deacetylase is a target of valproic acid-mediated cellular differentiation. Cancer research. 2004;64(3):1079-86. Epub 2004/02/12. PubMed PMID: 14871841. 145. Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. The Journal of biological chemistry. 2001;276(39):36734-41. Epub 2001/07/27. doi: 10.1074/jbc.M101287200. PubMed PMID: 11473107. 146. Zhai Y, Chen X, Yu D, Li T, Cui J, Wang G, et al. Histone deacetylase inhibitor valproic acid promotes the induction of pluripotency in mouse fibroblasts by suppressing reprogramming-induced senescence stress. Experimental cell research. 2015. Epub 2015/06/27. doi: 10.1016/j.yexcr.2015.06.003. PubMed PMID: 26112217. 147. Bae D, Mondragon-Teran P, Hernandez D, Ruban L, Mason C, Bhattacharya SS, et al. Hypoxia enhances the generation of retinal progenitor cells from human induced pluripotent and embryonic stem cells. Stem cells and development. 2012;21(8):1344-55. Epub 2011/08/31. doi: 10.1089/scd.2011.0225. PubMed PMID: 21875341. 148. Hasegawa Y, Tang D, Takahashi N, Hayashizaki Y, Forrest AR, Suzuki H. CCL2 enhances pluripotency of human induced pluripotent stem cells by activating hypoxia related genes. Scientific reports. 2014;4:5228. Epub 2014/06/25. doi: 10.1038/srep05228. PubMed PMID: 24957798; PubMed Central PMCID: PMC4067614. 149. Mohindra V, Tripathi RK, Singh RK, Lal KK. Molecular characterization and expression analysis of three hypoxia-inducible factor alpha subunits, HIF-1alpha, -2alpha and -3alpha in hypoxia-tolerant Indian catfish, Clarias batrachus [Linnaeus, 1758]. Molecular biology reports. 2013;40(10):5805-15. Epub 2013/09/26. doi: 10.1007/s11033-013-2685-1. PubMed PMID: 24065526. 150. Xiang ZL, Zeng ZC, Fan J, Tang ZY, Zeng HY, Gao DM. Gene expression profiling of fixed tissues identified hypoxia-inducible factor-1alpha, VEGF, and matrix metalloproteinase-2 as biomarkers of lymph node metastasis in hepatocellular carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(16):5463-72. Epub 2011/06/30. doi: 10.1158/10780432.CCR-10-3096. PubMed PMID: 21712445. - 151 -