Mosaicismo diploide-triploide con epilepsia y retraso mental
Anuncio

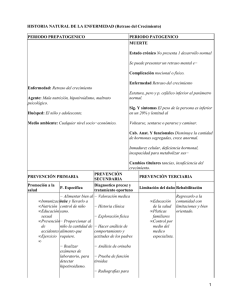
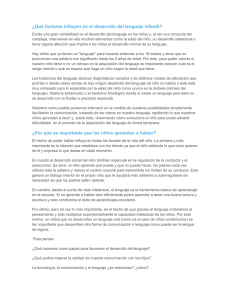
COMUNICACIONES BREVES El estudio patológico demostró, en la corteza afecta, la presencia de astrocitoma anaplásico con rasgos gemistocíticos, y en la sustancia blanca del centro semioval, astrocitoma de bajo grado, tipo gemistocítico, en transición a alto grado. El estudio inmunohistoquímico de la muestra biópsica presentó p16 negativo y p53 inferior al 5%. Los astrocitomas son tumores histológicamente heterogéneos, por lo que las biopsias focales pueden no ser representativas. Los hallazgos de PET sugieren que la afectación corticoespinal corresponde a una progresión tumoral y no a una degeneración de la vía piramidal. Esta técnica, en cambio, no aclara inequívocamente el grado tumoral [4-6]. La fusión de las imágenes de RM y PET y la ayuda de un neuronavegador permiten obtener muestras de áreas sugestivas de mayor malignidad histológica [7]. Esto es especialmente útil en regiones próximas a áreas elocuentes de la corteza cerebral. Esta mejor localización contribuye a mejorar el rendimiento diagnóstico de las biopsias [8]. Como se sabe, una parte importante de los astrocitomas de bajo grado de tipo gemistocítico sufren degeneración maligna. Dicha variante gemistocítica, por tanto, constituye un signo de mal pronóstico porque la progresión a un tumor de alto grado es mucho más frecuente que en las variantes fibrilar o protoplasmática [9]. La variante gemistocítica se asocia a mayor frecuencia de mutaciones en el gen p53 [10-12]. Asimismo, la pérdida de función de los genes p16 y p53 se ha relacionado también con la malignización [13]. Este caso ilustra también que la progresión tumoral de los gliomas por la sustancia blanca no afecta exclusivamente a tractos intrahemisféricos o interhemisféricos, sino también a vías largas, como el haz corticoespinal o el tracto espinotalámico, aunque de modo más excepcional [14]. M. Riverol-Fernández, D. Lázaro-Blázquez, B. Bejarano-Herruzo, M. Manrique-Smela Recibido: 10.06.02. Aceptado: 08.07.02. Departamento de Neurología y Neurocirugía. Clínica Universitaria de Navarra. Pamplona, España. Correspondencia: Dr. Bartolomé Bejarano. Departamento de Neurocirugía. Clínica Universitaria de Navarra. Avenida Pío XII, 36. E-31008 Pamplona. E-mail: [email protected] Fujita H, Murakami M, et al. Cerebral glioma: evaluation with methionine PET. Radiology 1993; 186: 45-53. 5. Sasaki M, Kuwabara Y, Yoshida T, Nakagawa M, Fukumura T, Mihara F, et al. A comparative study of thallium-201 SPET, carbon-11 methionine PET and fluorine-18 fluorodeoxyglucose PET for the differentiation of astrocytic tumours. Eur J Nucl Med 1998; 25: 1261-9. 6. Hustinx R, Smith RJ, Benard F, Bhatnagar A, Alavi A. Can the standardized uptake value characterize primary brain tumors on FDGPET? Eur J Nucl Med 1999; 26: 1501-9. 7. Coenen VA, Krings T, Mayfrank L, Polin RS, Reinges MH, Thron A, et al. Three-dimensional visualization of the pyramidal tract in a neuronavigation system during brain tumor surgery: first experiences and technical note. Neurosurgery 2001; 49: 86-92. 8. Vaquero J, Martínez R, Manrique M. Stereotactic biopsy for brain tumors: is it always necessary? Surg Neurol 2000; 53: 432-8. 9. Watanabe K, Tachibana O, Yonekawa Y, Kleihues P, Ohgaki H. Role of gemistocytes in astrocytoma progression. Lab Invest 1997; 76: 277-84. 10. Watanabe K, Peraud A, Gratas C, Wakai S, Kleihues P, Ohgaki H. p53 and PTEN gene mutations in gemistocytic astrocytomas. Acta Neuropathol (Berl) 1998; 95: 559-64. 11. Kosel S, Scheithauer BW, Graeber MB. Genotype-phenotype correlation in gemistocytic astrocytomas. Neurosurgery 2001; 48: 187-93. 12. Reis RM, Hara A, Kleihues P, Ohgaki H. Genetic evidence of the neoplastic nature of gemistocytes in astrocytomas. Acta Neuropathol (Berl) 2001; 102: 422-5. 13. Nozaki M, Tada M, Kobayashi H, Zhang CL, Sawamura Y, Abe H, et al. Roles of the functional loss of p53 and other genes in astrocytoma tumorigenesis and progression. Neuro-Oncol 1999; 1: 124-37. 14. Spuler A, Atkinson JL. Images in clinical medicine. Astrocytoma following the pyramidal tract. N Engl J Med 2000; 343: 1702. Mosaicismo diploide-triploide con epilepsia y retraso mental: supervivencia excepcional hasta la edad adulta BIBLIOGRAFÍA 1. Tynninen O, Aronen HJ, Ruhala M, Paetau A, Von Boguslawski K, Salonen O, et al. MRI enhancement and microvascular density in gliomas. Correlation with tumor cell proliferation. Invest Radiol 1999; 34: 427-34. 2. Roberts HC, Roberts TP, Brasch RC, Dillon WP. Quantitative measurement of microvascular permeability in human brain tumors achieved using dynamic contrast-enhanced MR imaging: correlation with histologic grade. Am J Neuroradiol 2000; 21: 891-9. 3. Jackson A, Kassner A, Annesley-Williams D, Reid H, Zhu XP, Li KL. Abnormalities in the recirculation phase of contrast agent bolus passage in cerebral gliomas: comparison with relative blood volume and tumor grade. Am J Neuroradiol 2002; 23: 7-14. 4. Ogawa T, Shishido F, Kanno I, Inugami A, 198 La triploidía (3n) es un trastorno cromosómico letal en humanos, siendo de meses la supervivencia postnatal máxima registrada en la literatura [1-4]. El síndrome triploide en neonatos se caracteriza por la presencia de retraso en el desarrollo prenatal y asimetría en el crecimiento (con desproporción cabeza-tronco), colobomas oculares, hipertelorismo, orejas incompletamente formadas, micrognatia, paladar hendido, sindactilia parcial del tercer y cuarto dedos y, con menos frecuencia, malformaciones congénitas cardíacas y genitourinarias, mielomeningocele y labio leporino [5]. Este mismo fenotipo puede aparecer, en parte, en el síndrome de Russell-Silver, cuyo origen se debe a una disomía materna en el cromosoma 7 [6]. La triploidía supone entre un 15 y un 20% de los abortos espontáneos por anormalidades cromosómicas y se asocia con cambios molares y degeneración quística de la placenta, factor que se ha postulado como causa de aborto en los embarazos triploides. Los casos de supervivencia prolongada, superior a meses, que se han descrito en la literatura han sucedido en individuos con mosaicismo diploide-triploide [5,8-11]. Existen evidencias de que algunas epilepsias pueden tener un componente genético significativo [12]. Ciertas anomalías cromosómicas se han asociado con epilepsia y retraso mental; ocho de ellas tienen una alta asociación con epilepsia [13]: el síndrome de Wolf-Hirschchorn (4p-), el síndrome de Miller-Dieker (deleción 17p13.3), el síndrome de Angelman (deleción 15q11-q13), el síndrome de inversión-duplicación del cromosoma 15, las deleciones terminales 1q y 1p del cromosoma 1, y los cromosomas en anillo 14 y 20 [13-15]. Debido a su baja frecuencia, los síndromes de poliploidía o mixoploidía se han estudiado menos en relación con la presencia de crisis epilépticas. Presentamos el caso de una paciente con mosaicismo 46,XX/ 69,XXX, con clínica de retraso mental y epilepsia generalizada, y una supervivencia excepcional hasta la edad adulta. Caso clínico. Mujer de 36 años de edad, admitida en nuestro servicio por presentar retraso mental grave y crisis convulsivas, iniciadas a los 8 meses de vida. En la actualidad se encontraba en tratamiento con fenobarbital y fenitoína. Nació de parto normal y su gestación sucedió sin intercurrencias; los padres no eran consanguíneos. A los 8 meses de vida presentó crisis convulsivas, en ausencia de fiebre, que la madre caracterizaba como tonicoclónicas generalizadas. La paciente tuvo un retraso psicomotor: se sentó sin apoyo a los 5 años, adquirió la marcha independiente a los 6,5 años y apenas desarrolló el lenguaje. Utiliza en la actualidad palabras simples del tipo ‘padre’ o ‘madre’, señala los objetos sin denominarlos y no ha consiguido construir frases; la comprensión para órdenes simples está preservada. Recibió un diagnóstico de hipotiroidismo congénito e inició tratamiento con levotiroxina (50 mg/día). Las crisis se controlaron a los 18 años de edad (hasta ese momento se consideró como una epilepsia de difícil control, e incluso tenía antecedentes de un status epiléptico a los 18 años); desde ese momento mantuvo tratamiento con fenitoína y fenobarbital. En el examen físico general era llamativo una baja estatura, obesidad, facies sindrómica, macroglosia, tendencia a mantener la boca abierta con la lengua protrusa, una moderada escoliosis y presencia de lesiones maculopapulosas en el tronco. En el examen neurológico, la paciente mantenía una cierta comprensión para órdenes simples (abrir la boca, apuntar al techo o la ventana) y emitía palabras aisladas sin sintaxis. La fuerza muscular era grado 5/5 en las extremidades. Los reflejos profundos eran presentes y simétricos, y el reflejo cutáneo plantar, flexor. No fue posible explorar la sensibilidad ni el fondo de ojo por falta de colaboración. Los pares craneanos eran normales, así como la coordinación de sus movimientos y el equilibrio. REV NEUROL 2002; 35 (2) COMUNICACIONES BREVES Figura 2. Resonancia magnética de encéfalo que muestra atrofia cortical y cerebelar moderadas, aumento de la neumatización de los senos frontales y un quiste aracnoideo asociado. Figura 1. Cariotipo de leucocitos de sangre periférica que muestra triploidía. Examenes complementarios: el estudio bioquímico básico, incluyendo hemograma, función renal y hepática, gasometría, amoníaco, lactato, glucosa, colesterol, fósforo, calcio, parathormona, estudio de errores innatos de metabolismo y de enfermedades lisosomales de depósito, cromatografía de aminoácidos plasmáticos y urinarios, y fenilalanina, fueron normales. La hormona tiroestimulante (TSH) estaba elevada (10,87 uUI/mL; valor de referencia: 0,27-4,20), y la T4 libre era normal (1,1 ng/dL; valor de referencia: 0,931,70). Un cariotipo de sangre periférica, sobre un total de 100 células, mostró 46,XX en 76 células, 69,XXX,(t (2:7) (q21.1; q 22.1) en 21 células, un número modal próximo a tetraploide en dos células, y un 45,X en otra célula (Fig. 1). Una ecografía tiroidea evidenció una glándula aumentada de volumen, de ecotextura heterogénea con múltiples nódulos y textura isoecogénica en relación al parénquima, situados en ambos lóbulos tiroideos y en istmo. El trazado electroencefalográfico obtenido, tanto en vigilia como en sueño espontáneo, mostró descargas con puntas de proyección frontal derecha y temporal izquierda. Un electromiograma fue normal. Una resonancia magnética de encéfalo mostró atrofia cortical y cerebelar moderada, aumento de la neumatización de los senos frontales y un quiste arecnoideo asociado en la fisura silviana derecha y otro en la fosa posterior (Fig. 2). La trisomía 16, la monosomía 45,XX y la triploidía son las anomalías cromosómicas más frecuentes en los abortos espontáneos [7,16]. La triploidía aparece en un 1-3% de los embarazos precoces, pero casi todos ellos (más del 99%) se pierden como abortos en el primer trimestre, o como muerte fetal uterina en el segundo [7]. El contaje cromosómico en la triploidía es 3n= 69, siendo doble (2n) la contribución cromosómica doble de uno de los padres, bien de la madre (diginia), bien del padre (diandria). La diandria es consecuencia tanto de la fertili- REV NEUROL 2002; 35 (2) zación simultánea del huevo por dos espermatozoides (dispermia), como de la fecundación de un espermatozoide diploide procedente de una disyunción incompleta en la espermatogénesis. La mayoría de los triploides diándricos se originan por dispermia, y sólo un 8,3% de ellos se producen por esperma diploide procedente de errores meióticos [16]. La diginia puede deberse a una disyunción incompleta (tanto en la primera como en la segunda división meiótica, en la oogénesis), a la retención de un corpúsculo polar o a la fecundación de un oocito primario ovulado. Un error en la primera meiosis sucede en los casos de triploidía asociados con traslocación materna. Los escasos triploides diándricos que sobreviven al segundo trimestre muestran un fenotipo placentario clásico de mola hidatidiforme; en estos casos, el feto es microcefálico. Daniel et al [17] propusieron una barrera de supervivencia para los fetos diándricos precozmente en la gestación, basado en la proporción de villi placentarios vascularizados. Los fetos triploides digínicos (McFadden/Kalousek tipo 2) suceden con más frecuencia en el segundo trimestre y dan lugar a fetos con grave retraso en el crecimiento, con marcada desproporción cabeza-cuerpo y placenta pequeña no molar. La supervivencia excepcional en el tercer trimestre de gestación se asocia con muerte perinatal [7,17]. Estudios clásicos basados en polimorfismo citogenético indican un predominio diándrico en el origen de la triploidía; sin embargo, el trabajo de Baumer et al [18] ha mostrado una relación inversa materna/paterna de 4:1. Los casos con supervivencia intrauterina mayor suelen ser de origen digínico. Un cigoto inicialmente triploide puede autocorregirse genéticamente por extrusión de un pronúcleo en la primera mitosis, y luego reincorporarlo en una división celular subsiguiente [19]. El feto afecto puede sobrevivir en el útero debido al efecto moderador de la línea celular diploide [8]. En la mayoría de los casos, la línea triploide no se observa en los análisis sanguíneos, por lo que el cultivo de fibroblastos es necesario para determinar ese estado de mosaicismo. En nuestra paciente, sin embargo, el estudio del cariotipo en leucocitos de sangre periférica fue suficiente para detectar esta anormalidad. Los casos clínicos descritos en la literatura sobre mosaicismo diploide/triploide han sido en niños. Carakushansky et al describieron un mosaicismo diploide/triploide en una niña de 5 años con retraso psicomotor grave [8]. Graham et al relataron el caso de una niña de 19 meses con retraso en el crecimiento, asimétrico, y sindactilia [5]. Meinecke et al describieron un niño con triploidía incompleta, retraso mental grave, corta estatura, asimetría corporal e hipogenitalismo [9]. Nuestra paciente sufría hipotiroidismo congénito, hallazgo descrito también en otra paciente mixoploide 46,XX/69,XXX junto con pubertad precoz [10]. En la edad adulta, los casos descritos similares al nuestro son muy infrecuentes. Existe un caso descrito por Fryns et al en una mujer de 21 años con retraso mental profundo y mosaicismo 2n/3n [11]. Existen algunos otros casos de mosaicismo con retraso mental y supervivencia prolongada; así, Edwards et al describieron dos casos de mixoploidía diploidetetraploide en dos mujeres de 11 y 21 años de edad, que clínicamente presentaban retraso mental profundo, epilepsia y displasia pigmentaria de la piel [20]. En conclusión, recomendamos la realización de cariotipo en aquellos pacientes con fenotipo de retraso mental moderado o grave asociado a epilepsia, incluso en adultos, y resaltamos la supervivencia excepcional de nuestra paciente en la edad adulta, asociada a este estado de mosaico. F.J. Carod-Artal, T.V. Fernandes da Silva Recibido: 21.06.02. Aceptado: 09.07.02. Servicio de Neurología. Hospital Sarah. Brasilia DF, Brasil. 199 CRÍTICA DE LIBROS Correspondencia: Dr. Francisco Javier CarodArtal. Servicio de Neurología. Hospital Sarah. SMHS quadra 501 conjunto A. CEP 70335-901. Brasilia DF. Brasil. E-mail: [email protected]; [email protected] BIBLIOGRAFÍA 1. Fryns JP, Van de Kerckhove A, Goddeeris P, Van den Berghe H. Unusually long survival in a case of full triploidy of maternal origin. Hum Genet 1977; 22: 147-55. 2. Cassidy SB, Whitworth T, Sanders D, Lorber CA, Engel E. Five month extrauterine survival in a female triploid (69,XXY) child. Ann Genet 1977; 20: 277-9. 3. Niemann-Seyde SC, Rehder H, Zoll B. A case of full triploidy (69,XXX) of paternal origin with unusually long survival time. Clin Genet 1993; 43: 79-82. 4. Arvidsson CG, Hamberg H, Johnsson H, Myrdal U, Anneren G, Brun A. A boy with complete triploidy and unusually long survival. Acta Paediatr Scand 1986; 75: 507-10. 5. Graham JM Jr, Hoehn H, Lin MS, Smith DW. Diploid-triploid mixoploidy: clinical and cytogenetic aspects. Pediatrics 1981; 68: 23-8. 6. Bernard LE, Penaherrera MS, Van Allen MI, Wang MS, Yong SL, Gareis F, et al. Clinical and molecular findings in two patients with Russell-Silver syndrome and UPD7: compar- ison with non-UPD7 cases. Am J Med Genet 1999; 87: 230-6. 7. Gardner RJM, Sutherland GR, eds. Chromosome abnormalities ad genetic counseling. 2 ed. New York: Oxford University Press;1996. 8. Carakushansky G, Teich E, Ribeiro MG, Horowitz DD, Pellegrini S. Diploid/triploid mosaicism: further delineation of the phenotype. Am J Med Genet 1994; 52: 399-401. 9. Meinecke P, Engelbrecht R. Abnormalitiesretardation syndrome caused by incomplete triploidy. Monatsschr Kinderheilkd 1988; 136: 206-9. 10. Jarvela IE, Salo MK, Santavuori P, Salonen RK. 46,XX/69,XXX diploid-triploid mixoploidy with hypothyroidism and precocious puberty. J Med Genet 1993; 30: 966-7. 11. Fryns JP, Vinken L, Geutjens J, Marien J, Deroover J, Van den Berghe H. Triploid-diploid mosaicism in a deeply mentally retarded adult. Ann Genet 1980; 23: 232-4. 12. Berkovic SF, Scheffer IE. Genetics of the epilepsies. Epilepsia 2001; 42 (Suppl 5): 1623. 13. Singh R, McKinlay-Gardner RJ, Crossland KM, Scheffer IE, Berkovic SF. Chromosomal abnormalities and epilepsy: a review for clinicians and gene hunters. Epilepsia 2002; 43: 127-40. 14. Zankl A, Addor MC, Maeder-Ingvar MM, Schorderet DF. A characteristic EEG pattern in 4p- syndrome: case report and review of the literature. Eur J Pediatr 2001; 160: 123-7. 15. Serrano-Castro PJ, Aguilar-Castillo MJ, Olivares-Romero J, Jiménez-Machado R, Molina-Aparicio MJ. Cromosoma 20 en anillo: ¿una canalopatía epiléptica? Rev Neurol 2001; 32: 237-41. 16. Egozcue S, Blanco J, Vidal F, Egozcue J. Diploid sperm and the origin of the triploidy. Human Reprod 2002; 17: 5-7. 17. Daniel A, Wu Z, Bennetts B, Slater H, Osborn R, Jackson J, et al. Kariotype, phenotype and parental origin in 19 cases of triploidy. Prenat Diag 2001; 21: 1034-48. 18. Baumer A, Balmer D, Binkert F, Schinzel A. Parental origin and mechanisms of formation of triploidy: a study of 25 cases. Eur J Hum Genet 2000; 8: 911-7. 19. McFadden DE, Kwong LC, Yam IYL, Langlois S. Parental origin of triploidy in human fetuses: evidence for genomic imprinting. Hum Genet 1993; 92: 465-9. 20. Edwards MJ, Park JP, Wurster-Hill DH, Graham JM Jr. Mixoploidy in humans: two surviving cases of diploid-tetraploid mixoploidy and comparison diploid-triploid mixoploidy. Am J Med Genet 1994; 52: 324-30. CRÍTICA DE LIBROS Evolución cerebral y psicopatología T. Crow, O. Gonzálcz, L. Puelles, F. Rodríguez, C. Salas, J. Sancho-Rof , J. Sanjuán Madrid : Triacastela Esta obra aporta una revisión de la influencia que han tenido las teorías evolucionistas, desde su aparición hasta hoy, sobre la psiquiatría y la psicología. Nos acerca a los conocimientos actuales sobre el origen del cerebro y de la mente, de la inteligencia y de las emociones. Retoma el concepto de medicina psicosomática para el estudio de las interacciones entre los fenómenos fisiológicos y psicológicos. Describe el origen filogenético de las emociones, la conducta, la percepción y la inteligencia, y su correlación con la anatomía. Establece un paralelismo entre determinados niveles evolutivos y la aparición de los grandes síndromes psicopatológicos: la ansiedad, la depresión y la psicosis. El interés de la teoría de la evolución en la comprensión de la mente humana y sus trastornos se plantea en un doble sentido: por un lado, es complementaria de los modelos psicopatológicos tradicionales (biológico, cognitivo, social, psicoanalítico...), y por otro, permite aunar las aportaciones de distintas disciplinas sobre la evolución cerebral y el origen de las 200 enfermedades mentales. Además, añade el concepto de ‘desadaptación’ a la definición de enfermedad El libro se divide en siete capítulos. En el primero, Julio Sanjuán, editor de la obra, hace un repaso histórico de las teorías evolucionistas en psiquiatría. Describe la situación actual centrándose en la sociobiología y la psicología evolucionista, y las controversias que ambas disciplinas han suscitado. En el capítulo 2, Fernando Rodríguez y Cosme Salas hablan de la evolución dcl aprendizaje y sus bases anatómicas. El capítulo 3, a cargo de Luis Puelles, se refiere al origen filogenético de la inteligencia. El capítulo 4 versa sobre ‘Evolución, neuroinmunoendocrinología y emociones’; José Sancho-Rof y Olga González hacen hincapié en las complejas relaciones entre los sistemas nervioso, inmunitario y endocrino, en el efecto modelador dcl medio sobre cl cerebro y la conducta, y exponen la necesidad de crear una nueva medicina psicosomática. En el capítulo 5, ‘Orígenes filogenéticos de las emociones y sus trastornos’, Julio Sanjuán establece la relación entre la evolución cerebral y la adquisición de capacidades adaptativas que conllevan el desarrollo de las emociones y de sus trastomos. En el capítulo 6, Timothy Crow presenta su teoría de ‘la esquizofrenia como precio por el lenguaje’. Esta teoría concluye que la predisposición a la psicosis y la aparición del lenguaje como ca- rácter que define a nuestra especie son fenómenos interrelacionados, propiciados por el cambio genético que dio origen al Homo sapiens. En el último capítulo, Julio Sanjuán resume el interés práctico del enfoque evolucionista. Este libro es el tercero de la colección ‘Psicopatología’ de la editorial Triacastela. Editado en formato de bolsillo, su presentación resulta adecuada. Cada capítulo dispone de una bibliografía extensa y actualizada. Cuenta con un índice analítico y un índice onomástico muy completos. El prólogo es obra de Carmen Leal Cercós. Los autores desarrollan su exposición de un modo estructurado y hacen asequible y amena la lectura, lo que despertará la curiosidad de neurólogos, psiquiatras, psicólogos e investigadores interesados en el funcionamiento de la mente humana, en el porqué de las emociones. Para los iniciados en las teorías evolucionistas de la mente, la obra constituye una excelente puesta al día, y para los legos en la materia será sin duda una buena forma de tomar contacto con estos planteamientos, que suponen un paso más en la comprensión de la fisiopatogenia de las enfermedades mentales. M.T. Frutos-Alegría Servicio de Neurología. Hospital General de Alicante. Alicante, España. REV NEUROL 2002; 35 (2)