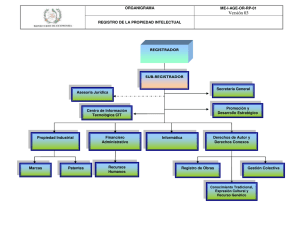
Consejo genético en demencias degenerativas de
Anuncio

Jornada sobre la malaltia de Huntington i demències degeneratives d’inici precoç. Organització assistencial i línies d’investigació Consejo genético en demencias degenerativas de inicio precoz Dra Raquel q Sánchez-Valle Programa de información y consejo genético para demencias familiares Unidad de Alzheimer y otros trastornos cognitivos Hospital Clínic Barcelona, divendres 23 d’octubre. Hotel Alimara Asesoramiento genético Sociedad Americana de Genética Humana define asesoramiento genético como: “Proceso de comunicación que se ocupa de los problemas humanos asociados a la presentación o riesgo de presentación de un trastorno genético en una familia familia” Este proceso consiste en el hecho de que diferentes especialistas procuren ayudar al individuo o a la familia a: 1)) comprender p el diagnóstico, g , el posible p curso de la enfermedad y de los tratamientos disponibles; 2) conocer la manera en que la herencia contribuye al trastorno y al riesgo de recurrencia en determinados pacientes/familias; 3) entender las alternativas para hacer frente al riesgo de recurrencia; 4) elegir una forma de actuación adecuada a su visión del riesgo, a sus objetivos j familiares y a sus normas éticas y religiosas, g y actuar de acuerdo con esta decisión, ó y 5) llevar a cabo la mejor adaptación posible al trastorno en el miembro afectado y/o al riesgo de recurrencia. Demencias genéticamente determinadas • ± 0,1 0 0,11-15 15% % d de casos d de llas demencias d i d degenerativas ti están tá genéticamente determinadas • Herencia autosómica dominante • Penetrancia P t i prácticamente á ti t completa l t • Fenotipo habitualmente similar a las formas esporádicas • Con frecuencia, a edades más precoces que las formas esporádicas • Pero, según la enfermedad y el gen implicado: -p presentación mayoritariamente y familiar o no - presentación siempre presenil o no • No existen tratamientos curativos ni modificadores del curso de la enfermedad PICOGENPICOGEN -Hospital H pit l Clíni Clínic Objetivo: prestar asesoramiento genético a pacientes con demencias neurodegenerativas y a sus familiares • • 1 fase 1º fase: 4 4º trimestre 2001 2001- 2003 Financiación privada (Caixa Catalunya-Editorial Planeta) Dirigido especialmente a sujetos a riesgo de enfermedad de Alzheimer Aprox. p 300 sujetos, j , de los cuales,, 10% tributarios de estudios g genéticos 2º fase: junio 2006Financiación p privada ligada g a proyectos p y de investigación g (Fundación Pfi Pfizer, FIS) Enfocado a demencias neurodegenerativas familiares: enfermedad de Alzheimer, degeneración lobular frontotemporal y enfermedades por priones Asesoramiento genético en demencias: niveles 1) Sujeto afecto • ¿Puede tratarse de una demencia determinada genéticamente? estudio fenotipo y árbol genealógico • ¿Podemos ¿P d id identificar tifi lla alteración lt ió genética éti responsable de la enfermedad? • ¿Qué tipo de estudios genéticos están indicados en el caso? • Interpretación de resultados genéticos (positivas: causalidad?; negativas: ausencia?) Recomendaciones: sujeto afecto • El estudio de cribado de mutaciones conocidas puede ser realizado en sujetos con demencia familiar autosómica dominante o con determinados fenotipos f p (EFNS,2007, recomendación tipo C). • Este estudio se debe realizar tras asesoramiento adecuado del paciente y/o familiares y tras su consentimiento explícito (recomendación tipo C). • El papel que un cambio génico juega en la génesis de la enfermedad ha de ser evaluada en estudios de segregación, asociación y/o implicación biológica (recomendación tipo C). Enfermedad de Alzheimer genéticamente determinada 0,1% casos de EA (5% de los casos de EA presenil) Presentación familiar, HAD E excepcional Es i l la l presentación t ió esporádica ádi Habitualmente presenil 3 genes implicados: •APP (cr 21q21.2), 32 mut. (17% EA gen.), rango edad 33- 65 años •PSEN1 (cr 14q23.3), 177 mut. (78% % EA gen.), rango 27-71a •PSEN2 (1q31-q42), 14 mut. (5% EA gen.), rango 40-71a Indicaciones I di i d de estudio t di genético éti en EA EA: PICOGEN • EA de inicio presenil con HAD de EA de inicio presenil • EA de inicio presenil sin historia familiar fiable • EA de inicio senil con HAD de inicio presenil en algunos miembros (PSEN2) • Datos neuropatológicos p g que q apoyen p y un origen g genético g de la EA • No existen evidencias que apoyen que la EA senil aún a pesar s de d patrón t ó HAD ssea de d origen i monogénico é i • No está indicado el estudio genético de APOE con fines de consejo genético Degeneración lobular frontotemporal determinada genéticamente DLFT es la segunda causa de demencia presenil neurodegenerativa 40% sujetos j t refieren fi hi historia t i f familiar ili Genéticamente heterogénea 9 Gen MAPT 9Gen PGRN 9 Gen VCP 9Gen CHMP2B 9 Gen DCN1 9 Varios locus en estudio DLFT y mutaciones en gen MAPT • Cromosoma 17 • 5-15% 5 15% Fenotipo F ti conductual d t l (DFT) (DFT), rara presencia i de d enfermedad de motoneurona • Presentación familiar familiar, HAD • Penetrancia prácticamente 100% a los 65 años • Depósitos D pósit s de d la l proteína p t ín asociada s i d a microtúbulos mi túb l s ttau en n el estudio anatomopatológico DLFT y mutaciones en gen PGRN • Cromosoma 17; HAD • 5-15% casos DLFT ; fenotipo DFT, afectación del lenguaje prominente • AP: AP DLFT sin i depósitos d ó i de d tau, síí inclusiones i l i con ubicuitina bi i i (intranucleares). No hay depósitos de progranulina • Excepcional la presencia de enfermedad de motoneurona •Edad de presentación variable (48 a 83 años) •Penetrancia clínica incompleta (ocasionalmente esporádicos) Brain, 2006 Indicaciones de estudio genético en DLFT: PICOGEN • El estudio t di d dell gen MAPT estaría t í iindicado di d en pacientes i t con DLFT de inicio precoz y patrón autosómico dominante, especialmente si se objetivan depósitos de tau en el estudio patológico • El estudio del gen de PGRN podría estar indicado en todos los casos de DLFT y patrón autosómico dominante independientemente d d d de lla edad, d d especialmente l en presencia d de inclusiones de ubiquitina intranucleares (recomendación tipo B) • Dada la baja prevalencia de mutaciones en los genes CHMP2B y VCP el estudio de estos genes se debería reservar a fenotipos p (recomendación ( tipo p C). ) específicos Enfermedades priónicas genéticamente determinadas • Descritas > 25 mutaciones en gen PRNP (cr 20) • HAD • 3 fenotipos mutaciones puntuales : -Enf. de Creutzfeldt-Jakob familiar (ECJf) -Insomnio familiar letal (IFL) -Enf. de Gertsmann-Sträussler-Scheinker (GSS) • Inserciones (1 a 9 OPRI) : fenotipo variable • Hasta 2/3 de los casos de enfermedades por priones genética NO refieren antecedentes familiares y presentan variabilidad en edad de presentación Recomendación de estudio genético en enfermedades priónicas El estudio genético de PRNP estaría indicado d d en todo d caso de d enfermedad f d d priónica independiente p p de Hº familiar o edad de presentación PICOGEN: resultado de estudio en sintomáticos • • 80 pacientes índices fueron candidatos a estudio genético 20 con historia familiar autosómica dominante de inicio presenil (HADp) • • 21 pacientes índice presentaban una mutación patogénica Enfermedad de Alzheimer genética (9 familias): PSEN1 (E120G, (E120G M139T M139Tx2, 2 P264L P264Lx2, 2 L286P L286P, K239N) PSEN2 (T430M) T430M) APP (I716F) Todos excepto un caso presentaban HADp Todos, Degeneración lobar frontotemporal (4 familias) MAPT (P301L x4) Enfermedades por priones genética (8 familias) PRNP (E200K x2;D178N x4; 9 OPRI; 4 OPRI) Sólo un 50% presentaban HADp • • • • En 16 (80%) de los casos HADp se identificaron mutaciones patogénicas En 4 (20%) casos HADp no se encontró ninguna mutación patogénica, 3 clasificadas como EA y 1 como DFT Asesoramiento genético : niveles 1. Sujeto j afecto 2 Sujeto asintomático a riesgo 2. R Recomendaciones: d i sujeto j t asintomático i t áti • El estudio st di genético néti presintomático p sint máti o predictivo se puede realizar en sujetos mayores de edad con clara historia familiar de demencia y mutación conocida en sujetos afectos (recomendación tipo C). • Se recomienda d para ell diagnóstico d ó presintomático la aplicación de las guías de diagnóstico de enfermedad de Huntington dentro de un equipo multidisciplinar con experiencia p en asesoramiento genético g y siguiendo i i d protocolos l específicos ífi pre y post– test (recomendación tipo C). Algoritmo de actuación PICOGEN (estudio presintomático, mutación patogénica conocida) Visita basal con neurólogo (evaluación e información) 3 meses “asimilación de la situación” 2º visita con neurólogo g Valoración psicológica/ psiquiátrica No candidato Candidato Estudio genético Entrega de resultados Negativo Positivo Seguimiento (PSQ/PS y NRL) Estudio de presintomáticos PICOGEN • 50 sujetos asintomáticos de familias con mutación patogénica (0-9 casos/familia) acudieron a consejo genético individual • 21 (42%) decidieron realizar el estudio genético presintomático g p • 9 (43%) resultaron portadores de la mutación patogénica • Razones esgrimidas: 1- iniciar un tratamiento precoz futuro 2- disminuir su ansiedad ante la incerteza 3- planificar su futuro 4 informar 4i f a sus hijos hij • Consecuencias en portadores: 50% portadores ↓ ansiedad 25% portadores ↑ ansiedad transitoriamente (discretos cambios en estilo de vida) 25% no cambios apreciables Seguimiento a 30 meses: no efectos psicológicos negativos • Todos los NO-portadores experimentaron ↓ ansiedad Conclusión: El estudio genético predictivo es seguro a corto y medio plazo con protocolos de actuación pre-test y programas de seguimiento adecuados Asesoramiento g genético : niveles 1. Sujeto j afecto 3. 3 Nivel prenatal Recomendación • Los individuos a riesgo de transmitir una enfermedad genética a sus descendientes debieran ser informados de este hecho y de las posibilidades existentes según l conocimientos los i i t y lla legislación l i l ió vigente i t para evitar it dicha transmisión, ayudándoles de esta forma a elegir una f un forma rm de actuación u n adecuada u a su u visión n del riesgo, a sus objetivos familiares y a sus normas éticas y/o creencias, y actuar de acuerdo con esta decisión (recomendación tipo C). C) Posibilidades actuales para evitar la transmisión de enfermedades genéticas • Diagnóstico prenatal: biopsia coriónica 99,9% fiabilidad diagnóstica (60% éxito) • Diagnóstico preimplantacional: FIV y selección preimplantacional de embriones-mórulas sanos 95% fiabilidad diagnóstica (15% éxito) Nueva ley l de d reproducción d ó asistida: d autorización individual • Donación de gametos sanos Líneas de investigación Búsqueda de nuevos genes candidatos: 11% EA con HADp p y 37% de los casos con DLFT con HAD no presentan mutaciones en genes candidatos conocidos Correlaciones genotipo-fenotipo …. DeKosky, y, Science 2003 • Enero 2009 • FIS080036-ISCIII IP: R. Sánchez-Valle • Estudio E t di de d sujetos j t de d f familias ili con dif diferentes t tipos de demencia genética: evaluaciones clínicas y cognitivas m en LCR y plasma p m análisis de biomarcadores neuroimagen estructural y funcional con MRI • Aprobado por el comité ético local • Todos los sujetos incluidos conocían su posición d riesgo de i • Iniciamos el proyecto estudiando las familias con mutaciones en PSEN1 • 19 sujetos j t de d 4 familias f ili diferentes dif t (L286P (L286P, M139T x 2 2, K239N) • 14 sujetos aceptaron participar en el estudio de LCR : 8p portadores (4 ( CDR 0,, 2 CDR =0,5, , , 1 CDR=1,, 1 CDR=2)) 6 no portadores • LCR fue recogido en tubos de polypropylen tubes, preprocesado según protocolo estándar y conservados a -80 -80ºC C • El estudio de biomarcadores en LCR se realizó mediante kits comerciales (Innogenetics®, Gent, Belgium) • 3T3T MRI • Los análisis estadísticos se realizaron con SPSS, version 16.0 • Todos los estudios se realizaron tras la firma de consentimiento informado Results. CSF biomarkers and clinical correlations in MC CSF Aβ1Aβ1-42 levels and adjusted age showed an inverse correlation CSF t-tau and CSF p-tau levels showed a positive correlation with CDR-SOB and a negative correlation with MMSE Resting functional MRI Default mode network map Default Mode Network Control group Default Mode Network asymptomatic carriers Default Mode Network symptomatic carriers Conclusiones • Los niveles en LCR de amiloide β1-42 correlacionaron i inversamente t con lla edad d d relativa l ti d dell sujeto j t en portadores t d d de mutaciones en PSEN1, sugiriendo que estos niveles descienden en los portadores a medida que se acercan a la enfermedad. Sujetos a más de una década del inicio de la enfermedad presentan niveles normales • Los niveles en LCR de β1-42 amiloide se encuentraban reducidos portadores de mutaciones en PSEN1 desde los p primeros en p síntomas de la enfermedad y se mantienen estables a partir de entonces • Los niveles de tau-total y fosfo-tau en LCR sólo se encontraron elevados l d en sujetos j t claramente l t sintomáticos, i t áti correlacionando l i d con la severidad clínica • El estudio t di d de D Default f lt mode d network t k objetiva bj ti alteraciones lt i funcionales en portadores asintomáticos de mutaciones en PSEN1 de forma previa a la detección de anomalías estructurales Gracias por su atención rsanchez@clinic ub es [email protected]