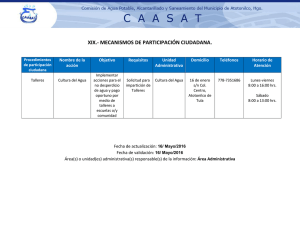
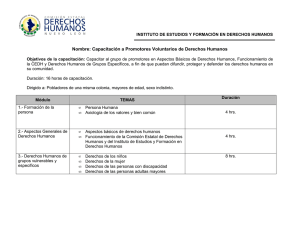
2do Congreso Anual de Patólogos Mexicanos • 5 León, Guanajuato
Anuncio

2 do Congreso Anual de Patólogos Mexicanos León, Guanajuato • 2016 Resúmenes LVIII CONGRESO ANUAL DE LA ASOCIACIÓN MEXICANA DE PATÓLOGOS A.C. XV CONGRESO ANUAL DE LA FEDERACIÓN DE ANATOMÍA PATOLÓGICA DE LA REPÚBLICA MEXICANA A.C. ÍNDICE DOMINGO 1º DE MAYO PATOLOGÍA ORAL ............................................................................................... 5 CITOPATOLOGÍA.................................................................................................. 8 INMUNOHISTOQUÍMICA....................................................................................... 11 LUNES 2 DE MAYO NEUROPATOLOGÍA.............................................................................................. 12 GINECOPATOLOGÍA............................................................................................. 21 PATOLOGÍA DE CABEZA Y CUELLO.................................................................. 28 OFTALMOPATOLOGÍA......................................................................................... 31 PATOLOGÍA DE LA GLÁNDULA MAMARIA........................................................ 36 PATOLOGÍA PEDIÁTRICA.................................................................................... 43 MARTES 3 DE MAYO PATOLOGÍA DE TEJIDOS BLANDOS.................................................................. 49 PATOLOGÍA GASTROINTESTINAL..................................................................... 58 NEFROPATOLOGÍA.............................................................................................. 69 PATOLOGÍA ÓSEA................................................................................................ 71 HEMATOPATOLOGÍA............................................................................................ 81 UROPATOLOGÍA................................................................................................... 89 MIÉRCOLES 4 DE MAYO DERMATOPATOLOGÍA......................................................................................... 96 PATOLOGÍA MOLECULAR................................................................................... 98 PATOLOGÍA PULMONAR..................................................................................... 99 PATOLOGÍA FORENSE........................................................................................ 104 PATOLOGÍA POSMORTEM.................................................................................. 105 PATOLOGÍA INFECCIOSA.................................................................................... 108 PATOLOGÍA VETERINARIA.................................................................................. 109 PATOLOGÍA QUIRÚRGICA DIVERSA.................................................................. 111 DOMINGO 1° DE MAYO PATOLOGÍA ORAL renciales y otorgar el diagnóstico definitivo; además, el caso reportado es relevante por su localización infrecuente en el maxilar INSTITUCIÓN: Hospital Civil de Culiacán ESTADO: SINALOA Mampara: 11 B Fecha y hora Instalación: 1 Mayo 13:00 hrs. Fecha y hora retiro: 1 Mayo 19.00 hrs. Mampara: 11 A Fecha y hora Instalación: 1 Mayo 13:00 hrs. Fecha y hora retiro: 1 Mayo 19.00 hrs. TC-56 AUTORES : DRA. ANA ISABEL VALDEZ , Dra. Cynthia Marina Urias Barreras , Dr. Efren Ríos Burgueño , Dra. Sara Rodríguez Carrillo, Dra. Dinorah Bátiz Salazar , Dra. Ana Cristina Benítez Morales. TITULO: FIBROMA OSIFICANTE CENTRAL INTRODUCCIÓN: Es un tumor fibro-óseo benigno de cre- cimiento lento,destructivo y deformante.En 1872, Menzel dio la primera descripción de una variante de fibroma osificante y lo llamó fibroma cemento-osificante(FCO).En 1927 Makek sugirió el término periodontoma benigno.En 1971, la OMS clasifica al FCO dentro de las lesiones formadoras de cemento.En 2005,el término se redujo a fibroma osificante(FO) OBJETIVOS: Revisión de la literatura y descripción de un caso de fibroma osificante de maxilar. MATERIAL: Femenino de 46 años de edad, presentó aumento de volumen en paladar, movilidad de molares de maxilar izquierdo, con tiempo de evolución desconocido. En los estudios de imagen se evidenció una lesión bien delimitada, de apariencia mixta radiolúcida/radiopaca con expansión de corticales, con desplazamiento dental y leve reabsorción radicular. Se tomó biopsia incisional a nivel alveolar y apical de maxilar izquierdo. RESULTADOS: Al estudio microscópico se observó una lesión neoplásica benigna bien delimitada de tejido fibroso hipercelular, con múltiples estructuras calcificadas formando trabéculas óseas y material tipo cemento. El diagnóstico emitido fue de fibroma osificante central. CONCLUSIONES: Este tipo de lesión fibro-ósea benigna, requiere la estricta correlación de datos clínicos, imagenológicos e histopatológicos para descartar diagnósticos dife- TC-61 AUTORES: DRA. DIANA BRISA SEVILLA, Dra. Ana Graciela Puebla Mora, Dra. Ana Maria Cano Valdez , Dra. Judith Rebeca Dávila Rodríguez, Dra. Claudia Isela Apreza González. TITULO : FIBROMATOSIS AGRESIVA EN ESPACIO PARAFARINGEO INTRODUCCIÓN: La fibromatosis agresiva (Tumor Desmoide) es un proceso neoplásico de tejido conectivo originado de la fascia y de los tejidos músculo-aponeuróticos con un comportamiento agresivo local. Representa menos del 3% de todos los tumores de tejidos blandos, se presenta con mayor frecuencia en extremidades y abdomen. Solamente el 10% se presentan en la región de cabeza y cuello. En general los tumores del espacio parafaríngeo son raros. Corresponden aproximadamente al 0.5% de tumores de cabeza y cuello. Los más frecuentes en esta localización son los de tipo glándula salival, los tumores de nervio periférico y los paragangliomas. A la fecha no existen reportes de Fibromatosis del espacio parafaríngeo. OBJETIVOS: Presentación de dos casos del Instituto Nacional de Cancerología MATERIAL: Se presentan dos casos de Fibromatosis agresiva del espacio parafaríngeo identificados en una revisión retrospectiva de diez años en el Instituto Nacional de Cancerología, enfatizando en el diagnóstico diferencial. RESULTADOS: La fibromatosis agresiva en la región de cabeza y cuello es rara. Usualmente se presenta en estadios avanzados. Tiene comportamiento localmente agresivo y a menudo con efectos mutilantes por destrucción de estructuras neurovasculares. La radioterapia es el tratamiento de elección, con el cual se puede lograr el control prolongado en la mayoría de los pacientes. Si la terapia sistémica está indicada, se puede considerar la combinación de Tamoxifeno y Sulindac. Otras opciones terapéuticas son el Imatinib y el Sorafenib, aunque su papel todavía no está claro. 2do Congreso Anual de Patólogos Mexicanos • 5 León, Guanajuato • 2016 CONCLUSIONES: La fibromatosis es una neoplasia localmente agresiva que puede confundirse morfológicamente con lesiones benignas o de comportamiento clínico indolente. Por ello, es fundamental su reconocimiento. El estudio inmunohistoquímico es útil para descartar otros sarcomas de bajo grado y diferentes condiciones benignas. La expresión nuclear de la beta-catenina es clave para el diagnóstico. INSTITUCIÓN: Centro Médico de Occidente del IMSS, Instituto Nacional de Cancerologia, Hospital Fray Antonio Alcalde. ESTADO:DISTRITO FEDERAL RESULTADOS: El SAPB puede presentar metástasis tempranas (pulmón, hueso y cerebro), aún sin recurrencias, por lo que su reconocimiento en sitios inusuales es fundamental para planear el tratamiento post-operatorio. CONCLUSIONES: Se presenta el primer caso de SAPB del espacio parafaríngeo, enfatizando en el diagnóstico diferencial con otros tumores de células epitelioides más comunes en la región. INSTITUCIÓN: Centro Médico de Occidente del IMSS, Instituto Nacional de Cancerología, Hospital Fray Antonio Alcalde. Hospital General Regional 270 del IMSS. ESTADO: DISTRITO FEDERAL Mampara: 12 A Fecha y hora Instalación: 1 Mayo 13:00 hrs. Fecha y hora retiro: 1 Mayo 19.00 hrs. Mampara: 12 B TC-62 Fecha y hora Instalación: 1 Mayo 13:00 hrs. Fecha y hora retiro: 1 Mayo 19.00 hrs. AUTORES: DRA. DIANA BRISA SEVILLA, Dra. Ana Graciela Puebla Mora, Dra. Ana Maria Cano Valdez, Dra. Claudia Isela Apreza González. Dra. Lorena Borja Márquez TC-64 TITULO: SARCOMA ALVEOLAR DE PARTES BLANDAS DEL ESPACIO PARAFARÍNGEO. REPORTE DE UN CASO. rio Rocha, Dra. Lidia Citlali Frutos Sierra INTRODUCCIÓN: El Sarcoma Alveolar de Partes Blan- das (SAPB) es una neoplasia rara compuesta por células epitelioides con citoplasma eosinófilo granular abundante dispuestas en nidos sólidos y / o estructuras alveolares. En los adultos ocurre con mayor frecuencia en los tejidos blandos profundos de extremidades inferiores. En niños es más frecuente en cabeza y cuello (lengua y órbita). Aunque se han reportado casos en múltiples localizaciones, no existen casos previos de SAPB del espacio parafaríngeo. OBJETIVOS: Presentación del Caso: Mujer de 42 años de edad que acudió al Instituto Nacional de Cancerología con historia de un año de evolución. Refirió que posterior a una extracción dental presentó dolor y aumento de volumen progresivo en la región parotídea izquierda. MATERIAL: Recibió tratamiento anti-inflamatorio sin mejoría. Seis meses después se agregó dificultad para la apertura bucal y aumento en la intensidad del dolor. La exploración física reveló la presencia de un tumor mal definido en la región parotídea, con aparente infiltración a la mucosa del trígono retromolar izquierdo, desviación de la úvula y fosa amigdalina izquierda. Los estudios de imagen mostraron que el tumor se encontraba en el espacio parafarín. AUTORES: DR. GERARDO NIETO, Dr. Fernando TenoTITULO: Schwanoma en dorso de lengua INTRODUCCIÓN: Neurolemoma, neoplasia benigna. Masculino de 28 años, sin antecedentes personales patológicos, ni heredofamiliares de interés para el padecimiento actual, presenta aumento de volumen precedido a traumatismo hace aproximadamente 6 años en vértice lingual, de crecimiento lento y progresivo, asintomática, del mismo color de la mucosa adyacente, firme y fluctuante a la palpación de 2 cm de diámetro. OBJETIVOS: Describir las características clíncas de esta neoplasia. Describir los patrones histológicos más comunes de los neurilemomas - Determinar los marcadores de inmunohistoquímca para estas neoplasias MATERIAL: Se describe nódulo en dorso lingual el cual se extirpa, al especímen quirúrgico se le realiza tinción de rutina , así como inmunomarcadores específicos S-100, Vimentina y PGAF. RESULTADOS: Se confirma del diagnóstico de Schwannoma (neurolemoma) y se describe el patrón celular. CONCLUSIONES: A pesar de que estas lesiones son consideradas como benignas y de escasa recurrencia es importante su identificación, diagnóstico y tratamiento opor- 2do Congreso Anual de Patólogos Mexicanos • 6 León, Guanajuato • 2016 tuno, para lo cual es de relevancia conocer las características tanto clínicas como histológicas. INSTITUCIÓN: Escuela Nacional de Estudios Superiores Unidad León Rinocerebral crónica puede presentarse con síntomas atípicos. Se requiere un alto grado de sospecha clínica para eldiagnóstico correcto y el pronto inicio de un tratamiento adecuado. INSTITUCIÓN: HOSPITAL GENERAL DE LEÓN ESTADO: GUANAJUATO ESTADO: GUANAJUATO Mampara: 13 A Mampara: 13 B Fecha y hora Instalación: 1 Mayo 13:00 hrs. Fecha y hora retiro: 1 Mayo 19.00 hrs. Fecha y hora Instalación: 1 Mayo 13:00 hrs. Fecha y hora retiro: 1 Mayo 19.00 hrs. TC-97 TC-110 AUTORES: Paulina Berenice Crespo-Morfin1, Ma.Dolores Hernández-Padilla1, Raul Chavira-Guerrero3, Rosalba García-Ramirez4 AUTORES: Dra. Denisse Fuentes Gutiérrez, Dra. Martha vasiva causada por hongos filamentosos de la familia Mucoraceae. La forma rinocerebral de la enfermedad representa la variante más común y tiene dos entidades clínicas distintas. La presentación más frecuente consiste en una infección rápidamente progresiva con alta tasa de mortalidad, mientras que la otra es una infección crónica con una menor mortalidad, pero muy rara y pocos casos reportados en literatura. benignos, que usualmente se encuentran en la nasofaringe de adolescentes del sexo masculino. La morbilidad de éste tumor generalmente se encuentra relacionada a su amplia vascularidad y a su crecimiento agresivo. Su histología es característica, siendo un tumor fibrovascular que tiene predilección de origen en la pared posterolateral de la cavidad nasal en donde el proceso esfenoidal del hueso palatino se une con el ala horizontal del vómer y el proceso pterigoides. Los angiofibromas constituyen en 0.5% de todos los tumores de cabeza y cuello. Aún más raros son los angiofibromas de localización extranasofaríngea. Los cuales generalmente han sido reportados en mujeres jóvenes.Reportamos el caso de un angiofibroma con localización en la amígdala palatina. Leticia Llamas Ceras, Dra María Irene Rivera Salgado TITULO: Angiofibroma atípico en la amígdala TITULO: Mucormicosis Rinocerebral crónico, palatina, presentación de caso. reporte de caso INTRODUCCIÓN: Los angiofibromas son tumores altaINTRODUCCIÓN: La Mucormicosises una infección in- mente vascularizados, no encapsulados, histológicamente OBJETIVOS: Presentar un caso de Mucormicosis crónica atendido en nuestro hospital, su presentación clínica e histopatológica. MATERIAL: Material y Métodos Masculino 57 años con Diabetes Mellitus tipo 2, de 16 años de diagnóstico, mal apego al tratamiento. Cursó con una historia de 2 meses de evolución con dolor en hemicara derecha, tipo urente, intenso, localizada en zona malar y en paladar duro, valorado por odontólogos, se extrajo muela y se indicó antibióticos sin respuesta, por lo que acude a la consulta de nuestro hospital. El desarrollo de lesión ulcerosa de la mucosa del p RESULTADOS: La segunda Biopsia reportó Infección por hifas no septadasmorfológicamente compatibles con Mucor (mucormicosis), con necrosis extensa e inflamación aguda y crónica moderadas, con afectación a hueso trabecular (tinciones de Grocott y PAS positivas). El paciente fue sometido a un estricto control glucémico, tratado con anfotericina B 1mg/kg/día y se realizó Hemimaxilectomía total radical. El cultivo resultó negativo a las 3 semanas. CONCLUSIONES: Conclusiones: La Mucormicosis OBJETIVOS: Presentación de un caso de Angiofibroma atípico. MATERIAL: Paciente femenina de 23 años, ocupación estudiante. Antecedentes de múltiples traumatismos en extremidades superiores e inferiores entre 2003-2010; Sx de ovario poliquístico en 2006, sin tratamiento; Fumadora desde los 19 años (12 cigarrillos/día), hasta la fecha. En abril de 2013 presenta hematemesis aislada, por lo que se realiza panendoscopía teniendo como hallazgo un pólipo pediculado de 1cm en región amigdalina derecha. RESULTADOS: Se determina clínicamente un tumor benigno de la amígdala y se mantiene a la paciente en vigilancia con nasofibrolaringoscopía cada 4 meses, sin reportar crecimiento de la lesión. En julio del 2015 la paciente solicita la resección quirúrgica. En el servicio de patología se recibe 2do Congreso Anual de Patólogos Mexicanos • 7 León, Guanajuato • 2016 espécimen esférico de 1 cm de diámetro, color rosa claro, firme, al corte con apariencia fibrosa y suave al tacto. Se diagnostica como: -angiofibroma atípico. bajo anestesia general y se realiza estudio histopatotológico al espécimen obtenido.Los cortes histológicos fueron teñidos con tinción de rutina H&E para su estudio. CONCLUSIONES: Los angiofibromas de cabeza y RESULTADOS: Histológicamente se encontraron múlticuello son tumores no encapsulados que ocurren principalmente en la nasofarínge, siendo poco comunes las localizaciones extranasales. Son tumoraciones benignas consideradas agresivas dependiendo de la edad del paciente y su localización. Llama la atención su incidencia, típica en adolescentes masculinos, por lo cual no se descartan factores hormonales implicados en su génesis. Los angiofibromas en localizaciones atípicas (extranasosinusales), son poco frecuentes con predominio en adultos jóvenes del sexo femenino como se describe en éste caso. El tratamiento de elección consiste en resección quirúrgica que generalmente es curativa. ples estructuras polipoides o papilares cubiertas de epitelio escamoso estratificado paraqueratinizado , con un estroma sub epitelial de tejido linfoide: así como una cavidad revestida por epitelio cúbico simple que contienen un material proteinaseo en su interior. CONCLUSIONES: La Hiperplasia Linfoide Polipoide es una alteración poco frecuente el mayor número de casos se ha descrito en el continente asiático y de acuerdo a la literatura solo 3 casos en occidente es por eso la relevancia del caso. Aunado a esto se identificó un quiste de inclusión y que no ha sido reportada la presencia de estas dos alteraciones simultáneamente. INSTITUCIÓN: HOSPITAL CENTRAL SUR DE ALTA ESPECIALIDAD DE PETRÓLEOS MEXI- INSTITUCIÓN: Escuela Nacional de Estudios CANOS Superiores Unidad León, UNAM ESTADO: DISTRITO FEDERAL ESTADO: GUANAJUATO Mampara: 14 A CITOPATOLOGÍA Fecha y hora Instalación: 1 Mayo 13:00 hrs. Fecha y hora retiro: 1 Mayo 19.00 hrs. TC-166 AUTORES: DRA. NANCY PÉREZ, Mtro. Fernando Te- norio Rocha, Dr. Francisco José Paz Gómez, Mtro. Javier de la Fuente Hernández Esp.Karla Mayela Avelar Juárez, Mtro. Jacinto Armando Díaz Acevedo. TITULO: Hiperplasia Linfoide Polipoide. Presentación de un caso INTRODUCCIÓN: La hiperplasia linfoide polipoide de la amígdala palatina es una condición poco frecuente que se caracteriza por la formación de proyecciones digitiformes sobre la superficie de la mucosa que recubre las amígdalas, corresponde a una excesiva formación del tejido linfoide subepitelial. La patogénesis sigue siendo incierta, sin embargo se han sugerido varios factores causales. OBJETIVOS: Presentar el caso de una mujer de 28 años con diagnóstico de hiperplasia linfoide polipoide en tonsila izquierda MATERIAL: Paciente femenina de 28 años de edad sin antecedentes de relevancia para el padecimiento actual, que presenta lesión nodular de color amarillo en tonsila palatina izquierda con tiempo de evolución desconocido, sintomatología espontánea de ardor. Se realiza biopsia excisional Mampara: 16 B Fecha y hora Instalación: 1 Mayo 13:00 hrs. Fecha y hora retiro: 1 Mayo 19.30 hrs. TC-33 AUTORES: Irving Daniel Ortiz Sánchez, Ramiro Sandoval Macías, Angélica Grajales Rodríguez, Candelaria Córdova Uscanga, Ana Lilia Remírez Castellanos, Fernando Candanedo González. TITULO: ANÁLISIS CLÍNICO PATOLÓGICO DE LINFOMAS EN MEDIASTINO Y EMPLEO DE BIOPSIA CON AGUJA DE CORTE GUIADA POR IMAGEN INTRODUCCIÓN: En México, Santillán en 2006 reportó que los linfomas representan el 26% de todas las neoplasias mediastinales. La biopsia guiada por imagen es cada vez más importante en el diagnóstico de linfoma en mediastino, ya que es un estudio confiable, poco invasivo y que reduce costos. OBJETIVOS: Analizar las características clínico patoló- 2do Congreso Anual de Patólogos Mexicanos • 8 León, Guanajuato • 2016 gicas de pacientes con linfoma en mediastino y empleo de la biopsia guiada por imagen en el Hospital de Oncología. Cancerología. MATERIAL: Se realizó estudio retrospectivo de pacientes cipalmente un estudio de tamizaje en busca de lesiones escamosas intraepiteliales y carcinoma de células escamosas; su sensibilidad para detectar lesiones glandulares está limitada por problemas relacionados con la obtención de la muestra y su interpretación. El porcentaje esperado de diagnósticos de AGC (Células glandulares atípicas) en un laboratorio de citología es de 1.8 al 2% del total de los estudios diagnosticados. El término AGC (Células glandulares atípicas) según el Sistema Bethesda se utiliza cuando las células endocervicales presentan atipia nuclear que excede los cambios reactivos o reparativos pero carece de características certeras de adenocarcinoma. con diagnótico de linfoma en mediastino en el periodo de 2014-2015, en el Hospital de Oncología de CMN Siglo XXI. Se revisaron los informes de patología del archivo de quirúrgicos del departamento de patología. RESULTADOS: Se identificaron un total de 60 pacientes con neoplasia mediastinal, de los cuales 9 correspondieron a linfomas en mediastino 5 mujeres y 4 hombres. La evolución de la enfermedad tuvo una duración promedio de 9.5 meses. Y el promedio de la supervivencia, hasta el momento que terminó el estudio, de 12 meses. En un paciente, se realizaron 4 biopsias, 2 de ellas guiadas por TC, pero ninguna fue concluyente, por lo tanto, se realizó mediastinoscopía, y finalmente terminó en toracotomía con obtención de material adecuado. Microscópicamente, 5 casos correspondieron a linfomas difusos de células grandes (LDCG) de estirpe B, 2 a linfomas de Hodking y un caso de linfoma linfoblástico. CONCLUSIONES: En nuestra serie los linfomas de me- diastino no son infrecuentes (15%). No se observaron diferencias en género. Los linfomas en mediastino en nuestra población de estudio se presentaron con mayor frecuencia en pacientes jóvenes (Promedio 41 años). Los LDCG de estirpe B fueron los más frecuentes (56%). El sitio de afección más frecuente fue mediastino anterior (44%). El síntoma principal fue dolor (67%). La biopsia guiada por TC puede ser de utilidad para establecer el diagnóstico de linfoma, pero es necesario incluir un mayor número de pacientes para confirmar su certeza diagnóstica. INTRODUCCIÓN: El estudio citológico cervical es prin- OBJETIVOS: Las células glandulares atípicas pueden deberse a diversos procesos benignos o malignos, como un cepillado enérgico, endometriosis, pólipos endocervicales, lesiones escamosas de alto grado con penetración glandular, adenocarcinomas, entre otros, debido a esto, las guías de la sociedad americana de colposcopia y patología cervical recomiendan para el manejo de mujeres con diagnóstico de AGC la colposcopía como primen paso. Si una enfermedad invasiva no se identifica durante la colposcopia inicial, se recomiendan procedimientos excisionales de diagnóstico, como la conización. Si no se identifica lesión, se recomienda que la mujer sea seguida usando un programa de citología cervical repetida a intervalos de 4 a 6 meses hasta obtener 4 resultados citológicos consecutivos “negativos”, después de lo cual la mujer puede retornar a detección de rutina. MATERIAL: Se realizó una búsqueda en la base de da- INSTITUCIÓN: Departamento de Patología, Hospital de Oncología de Centro Médico Nacional Siglo XXI. tos del Instituto Nacional de Cancerología de citologías con diagnóstico de AGC para determinar el porcentaje de los mismos y la correlación con los hallazgos de la biopsia de estas pacientes en el periodo comprendido entre enero del 2010 a diciembre del 2014. ESTADO: ESTADO DE MÉXICO RESULTADOS: De un total de 66,480 citologías se iden- Mampara: 17 A Fecha y hora Instalación: 1 Mayo 13:00 hrs. Fecha y hora retiro: 1 Mayo 19.30 hrs. TC-53 AUTORES: Gabriela Lucero Ramírez García, María Delia Pérez-Montiel Gómez, David Francisco Cantú De León, Lidia Faridi Villegas, Lorena Flores Hernández, Mónica L. Serrano Arévalo. TITULO: Correlación cito-histológica de AGC. Experiencia de 5 años del Instituto Nacional de tificaron 307 casos diagnosticados como AGC (0.45%). A 260 (85%) se les realizo biopsia o fueron sometidas las pacientes a procedimiento quirúrgico con obtención de tejido para estudio histopatológico. De estas pacientes en el estudio histopatológico 28% (74) casos fueron diagnosticadas como adenocarcinomas en la biopsia, 28% (74) el resultados fue inflamatorio y/o normal, 14% (36) lesiones benignas (pólipos, un leiomioma), 10% (26) fueron carcinoma epidermoide, 9% (22) Neoplasia Intraepitelial de Bajo grado, 6% (15) otras neoplasias malignas (carcinomas serosos de ovario, sarcomas uterinos, metástasis), 3% (7) Neoplasia intraepitelial del alto grado, 2% (6) fueron insuficientes para diagnóstico. Del 28% de los casos que fueron diagnosticas en biopsia como adenocarcinomas, se revisaron nuevamente las citologías con el fin de identificar las características citomorfológicas que impidieron emitir el diagnostico de adenocarcinoma, encontrando principalmente que en el 50% de los casos los frotis eran intensamente hemorrágicos e inflamatorios (en más del 75% del frotis), en un 73% 2do Congreso Anual de Patólogos Mexicanos • 9 León, Guanajuato • 2016 la cantidad de células era escaso oscilando de 1 grupo de células atípicas hasta 6 grupos. CONCLUSIONES: La citología cervicovaginal es un método de tamizaje principalmente para lesiones escamosas, su sensibilidad para detectar lesiones glandulares es limitada, debido a la obtención de la muestra, sin embargo el porcentaje de AGC que en el seguimiento son relacionadas con lesiones escamosas de alto grado 10 al 40%, no hace nada despreciable su significado, en nuestro trabajo encontramos un porcentaje que parece elevado de adenocarcinomas 28%, sin embargo en la revisión las características morfológicas que limitaron la interpretación de la citología fueron 2 principalmente: los frotis intensamente hemorrágicos e inflamatorios y la escasa cantidad de células glandulares anormales encontradas en las muestras. INSTITUCIÓN: INSTITUTO NACIONAL DE CANCEROLOGIA MEXICO ESTADO: DISTRITO FEDERAL Mampara: 18 A Fecha y hora Instalación: 1 Mayo 13:00 hrs. Fecha y hora retiro: 1 Mayo 19.30 hrs. RESULTADOS: Se revisaron los reportes de anormalidades en el epitelio glandular de las citologías cervicales diagnosticadas en el Laboratorio de Citología del Hospital General de León de enero a diciembre de 2015. Se revisaron los diagnóstico histopatológico y el seguimiento de las mismas en la plataforma del Programa de Prevención y Control del Cáncer del Cuello Uterino. CONCLUSIONES: Nuestro Laboratorio de Citología tiene un bajo porcentaje de detección de anormalidades de células glandulares cervicales. Una buena proporción de ellas se confirmaron como verdaderas neoplasias de origen glandular sin embargo las biopsias negativas en su mayoría fueron insuficientes para diagnóstico. INSTITUCIÓN: Hospital Regional de León Departamento de Medicina y Nutrición Universidad de Guanajuato ESTADO: GUANAJUATO Mampara: 18 B Fecha y hora Instalación: 1 Mayo 13:00 hrs. Fecha y hora retiro: 1 Mayo 19.30 hrs. TC-86 TC-80 AUTORES: Dra. Patricia Ortega-González, Dra. Clau- AUTORES: Dra. María José González Salazar, Dr. Luis dia Acosta-Mata, Dr. Luis Raul Chavira-Guerrero, Dra. Ana Ruth Alonso Ibarra. Antonio Sepúlveda Rodríguez, Dra. Gloria Madrid Valero, Dr. Miguel Ángel Paz, Dr. Diego Rodríguez Mendoza, Dr. Horacio Decanini Arcaute. TITULO: Anormalidades en el epitelio glandular cervical. Experiencia del Laboratorio de Citología del HGL en el 2015. TITULO: VIRUS HERPES SIMPLEX TIPO 1 ASOCIADO A NEUMONÍA: REPORTE DE UN CASO DETECTADO EN LAVADO BRONQUIAL INTRODUCCIÓN: La importancia de las lesiones origi- INTRODUCCIÓN: Se trata de paciente masculino de 85 nadas en la porción glandular del canal endocervical ha ido en aumento en los últimos años. Los instrumentos para la toma de la citología cervical permiten un mejor muestreo de esta zona además de que los criterios citológicos son cada vez más precisos. OBJETIVOS: Describir la incidencia, correlación histopatológica y seguimiento de las citologías cervicales con anormalidades en el epitelio glandular. MATERIAL: Se revisaron los reportes de anormalidades en el epitelio glandular de las citologías cervicales diagnosticadas en el Laboratorio de Citología del Hospital General de León de enero a diciembre de 2015. Se revisaron los diagnóstico histopatológico y el seguimiento de las mismas en la plataforma del Programa de Prevención y Control del Cáncer del Cuello Uterino. años de edad, con antecedente de hipertensión arterial sistémica, síndrome de apnea obstructiva y enfermedad renal crónica. Cinco días previos a su ingreso inicia con ataque al estado general, hipertermia de 38°C y tos en accesos con expectoración mucopurulenta. Al presentar exacerbación de su sintomatología, acude a urgencias. OBJETIVOS: A la exploración física se encuentra somnoliento, se auscultan estertores roncantes bilaterales e hipoventilación basal derecha, presenta edema de extremidades inferiores. Se detecta alcalosis respiratoria con acidosis metabólica concomitante. MATERIAL: A su ingreso se solicita radiografía con áreas de infiltrado mixto apical y basal con borramiento del seno costodiafragmático derecho, en relación a derrame pleural, concluyendo un proceso neumónico, que persiste durante 2do Congreso Anual de Patólogos Mexicanos • 10 León, Guanajuato • 2016 su estancia intrahospitalaria a pesar del tratamiento de soporte y antibioticoterapia. Se realizan pruebas para influenza, micoplasma y BAAR resultando negativos. El cultivo reporta escasos cocos gram positivos y levaduras. RESULTADOS: Se realiza broncoscopía y se recibe líquido del lavado bronquial. Los extendidos evaluados muestran fondo con detritus celulares, inflamación de predominio agudo y células que tienden a la citólisis. Se observan células bronquiales de características reactivas, además de presencia de células multinucleadas, con núcleos despulidos citológicamente consistentes con inclusiones virales de herpes de tipo Cowdry A. Se corrobora mediante inmunohistoquímica utilizando el marcador para Herpes Virus Simplex tipo I.El paciente progresó a insuficiencia respiratoria. Durante su transcurso no recibió antiretrovirales. A los 20 días desde su internamiento presenta paro cardiorrespiratorio. CONCLUSIONES: El Virus Herpes Simplex tipo 1, es prevalente en la población e infecta la superficie mucocutánea, al sistema nervioso central y órganos viscerales. Existe controversia en su rol patogénico en el tracto respiratorio, debido a que se aísla de la faringe de adultos asintomáticos (hasta 5%). Se encuentra asociado con neumonía en pacientes inmunocomprometidos e inmunocompetentes, a uso de ventilación mecánica. No se ha demostrado efectividad con el tratamiento con Aciclovir. En algunos estudios, se asocia a peor evolución en pacientes en enfermedad crítica, con un aumento de mortalidad de 25%. INSTITUCIÓN: UNIVERSIDAD DE MONTERREY HOSPITAL CHRISTUS MUGUERZA ALTA ESPECIALIDAD ESTADO: NUEVO LEÓN INMUNOHISTOQUÍMICA Mampara: 18 A Fecha y hora Instalación: 1 Mayo 13:00 hrs. Fecha y hora retiro: 1 Mayo 19.00 hrs. DAD DE NESTINA SE ASOCIA CON ESTADIOS AVANZADOS Y METÁSTASIS EN ADENO EL INCREMENTO EN LA DENSIDAD DE NESTINA SE ASOCIA CON ESTADIOS AVANZADOS Y METÁSTASIS EN ADENOCARCINOMA DE PÁNCREAS. INTRODUCCIÓN: El adenocarcinoma de páncreas(AP) es una enfermedad altamente letal, la cual es usualmente diagnosticada en estados avanzados,las características que definen al AP son la invasión local y la diseminación temprana del tumor, confieriendo un comportamiento agresivo y una tasa de sobrevida pobre a los pacientes. Se ha demostrado que la expresión de nestina esta relacionada con la invasión tumoral. OBJETIVOS: Comparar los niveles de expresión de la proteína nestina en adenocarcinoma de páncreas (AP) y adenocarcinoma de la ámpula de váter (ACAV) y evaluar su asociación con invasión, metástasis y estadio clínico. MATERIAL: Se incluyeron 22 casos de AP Y 29 de ACAV. El protocolo fue aprobado por el comité de ética local y financiado por la Coordinación de Investigación en Salud (FIS/ IMSS/PROT/G14/1310).Se construyeron cinco microarreglos de tejidos a partir de bloques de parafina de todos los casos, y se evaluó por inmunohistoquímica la expresión de nestina. La expresión de nestina fue cuantificada en células tumorales y tejido total con un software de imagen. RESULTADOS: Encontramos que ambos tumores so- breexpresan nestina en comparación con el tejido normal (páncreas residual), las células tumorales de AP expresan mayor densidad de nestina que su contraparte ACAV (p=0.032). Nestina se expresa en células tumorales pobremente diferenciadas y no en grados bien diferenciados, así como en el estroma y vasos.La expresión de nestina total y por tipo celular no tuvo asociación con invasión vascular y perineural, sin embargo la presentación de metástasis en tumor de AP con respecto a nestina total, fue estadísticamente significativa (p=0.047). Finalmente se observó asociación en estadio clínico de nestina total (p=0.039) y por células tumorales (p=0.010) en AP. CONCLUSIONES: El AP no tuvo una mayor expresión de TC-36 nestina total que el de ACAV, pero sí de nestina en células tumorales. La expresión elevada de nestina total se asocia con la presencia de metástasis y estadios avanzados en AP. AUTORES: DAMIÁN SÁNCHEZ, Dra. Anna Karen Félix INSTITUCIÓN: Unidad de Investigación Médica Félix,Dr. Fernando Candanedo González,Dra. Sara Huerta Yepez,Dra.Isabel Alvarado Cabrero,Dra.Patricia Piña Sánchez,Dr Hector Mayani Viveros, Dra. Eugenia Flores Figueroa. TITULO: EL INCREMENTO EN LA DENSI- en Enfermedades Oncológicas.Departamento de Patología. Servicio de Sacromas. Hospital de Oncología, Centro Médico Nacional Siglo XXI. IMSS. Hospital Infantil Federico Gómez. Ciudad de México. 2do Congreso Anual de Patólogos Mexicanos • 11 León, Guanajuato • 2016 ESTADO: ESTADO DE MÉXICO INSTITUCIÓN: Martha Lilia Tena-Suck, Daniel LUNES Rembao Bojorquez, Miguel Fernando Salazar, Citlaltepetl salinas Lara. Departamento de Neuropatología, Instituto Nacional de Neurología y Neurocirugía. Ciudad de México. Hospital General de México. 2 DE MAYO NEUROPATOLOGÍA Mampara: 1 B Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. Mampara: 1 A Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. ESTADO: DISTRITO FEDERAL 7:30 hrs. 7:30 hrs. TC-11 AUTORES: Martha Lilia Tena-Suck, Miguel Fdo. Salazar Morales, Citlaltepelt Salinas Lara, Noemi Gelista. Carlos Sánchez Garibay TC-9 AUTORES Martha Lilia Tena-Suck, Daniel Rembao Bojorquez, Miguel Fernando Salazar, Citlaltepetl salinas Lara. Departamento de Neuropatología, Instituto Nacional de Neurología y Neurocirugía. Ciudad de México. Hospital General de México. TITULO: Schwanoma intraventricular reporte de un caso INTRODUCCIÓN: Neurilemomas son neoplasias benig- nas rara vez que se pueden originar en el sistema nervioso central. Por otra parte, su localización intraventricular se ha notado con menos de 30 casos reportados, reporteamos un caso que correspondería al quinto de la población de América Latina. OBJETIVOS: Reporte de un raro caso de tumor intraventricular MATERIAL: Caso Clínico: Hombre joven de 16 años de edad, sin datos clínicos significativos últimos debutó con dolor de cabeza y la ceguera del ojo izquierdo progresiva durante seis meses. Las imágenes de RNM mostraron una lesión multilobulada, un tumor voluminoso, intraventricular que emerge del cuerno posterior del ventrículo lateral izquierdo. Microscópicamente, la lesión estaba formada por células schwannian, fusiformes dispuestas en ambas áreas compa RESULTADOS: Discusión: La expresión de GFAP fue generalizada e inesperadamente visto, la proteína s-100, vimentina, desmina fueron positivas. ENE, sinaptofisina, EMA, citoqueratinas fueron negativas. Como aconsejado anteriormente por algunos autores, la transformación neoplásica de una célula madre multipotenciales (stem cells) puede explicar el hallazgo ocasional de estos tumores en compartimentos intracraneales no convencionales. TITULO: Condrosarcoma desdiferenciado vs mixoide secundario a astrocitoma de bajo grado radiado. Reporte de un caso INTRODUCCIÓN: El condrosarcoma es un tumor mesenquimal maligno que sólo en raras ocasiones en los huesos del cráneo, su localización más frecuente es la del etmoides y el seno esfenoidal. Menos del 5% de todos los condrosarcoma se encuentra en la cabeza y el cuello y corresponde a un tumor con baja tasa de malignidad con crecimiento lento. Se presenta un caso raro de condrosarcoma con áreas de tipo mixoide y áreas de tipo indiferenciadas, supratentorial secundaria y el tumor de después de la irradiación en un paciente con astrocitoma de bajo grado. MATERIAL: Caso clínico: Hombre de 26 años quien fue tratado con radioterapia de un astrocitoma de bajo grado, hace 7 años y después de encontrarse asintomático, desarrollo de un tumor que fue considerada como la recurrencia. Este segundo tumor que correspondió a una neoplasia diferente a la primera formada por células fusiformes inmersas en el redondeo estroma mixoide por los astrocitos gigantes y atípicos. Las células tumorales fueron positivas para v CONCLUSIONES: Se presenta un caso raro de condrosarcoma mixoide e indiferenciado inducido por la radioterapia es un tumor raro con esta asociación. Muestro caso además presenta astrocitos gigantes y atípicos que imitan astrocitoma. INSTITUCIÓN: Departamento de Neuropatología, Instituto Nacional de Neurología y Neurocirugía. Ciudad de México. Hospital General de México. ESTADO: DISTRITO FEDERAL 2do Congreso Anual de Patólogos Mexicanos • 12 León, Guanajuato • 2016 TITULO: Expresión de transportadores de Glucosa y de proteínas de choque térmico en Fecha y hora Instalación: 2 mayo 7:30 hrs. craniofaringiomas. Mampara: 2 A Fecha y hora retiro: 2 mayo 13:00 hrs. INTRODUCCIÓN: Craniofaringioma es un tumor sólido TC-12 AUTORES: Martha Lilia Tena-Suck, Armando Ruiz Treviño, Daniel Rembao Bojorquez, Miguel Fernando Salazar, Citlaltepetl Salinas Lara y quístico de la región selar, tumor benigno, tiene un comportamiento agresivo con frecuentes secuelas neurológicas y endocrinas con alta incidencia a recidivar, suele asociarse a trastornos metabólicos tales como diabetes insípida y mellitus, obesidad y síndrome metabólico. TITULO: Enfermedad de Lhermitte-Duclos o OBJETIVOS: Correlación clínico patológico y de IHQ en gangliocitoma desmoplasico cerebeloso, tres craniopafingiomas. Estudio descriptivo, retrospectivo, observacional, serie de casos. casos. INTRODUCCIÓN: Enfermedad de Lhermitte-Duclos es una enfermedad rara consistente en un crecimiento lento de un tumor en el cerebelo, a veces considerado un hamartoma, caracterizado por una hipertrofia difusa del estrato granuloso del cerebelo. A menudo suele asociarse con síndrome de Cowden con inicio en la etapa adulta, pueden ser atribuidos a mutaciones en el gen PTEN (Phosphatase and Tensin Homolog). OBJETIVOS: Caso Clínico: Presentamos 3 casos. Dos hombre una mujer, edad de 21, 30 y 37 años. Todos se presentaron cono tumor cerebeloso. 2 recidivaron y uno recibió radioterapia. Histológicamente se observan neuronas displasicas y heterotopias neuronales aisladas en la sustancia blanca apoyan un trastorno migratorio, además de que las neuronas pierden su territorialidad y tienden a agruparse. Fueron intensamente positivas para Neu-N, sinaptofisina, DARP32, dos fueron positivos para PTEN y el índice mib1 (ki67) fue bajo e incluso en las recidivas). CONCLUSIONES: No se consideran verdades neoplasias sino lesiones hamartomatosas a pesar de mostrar atipias celulares y de ser tumores altamente residivantes. MATERIAL: Estudiamos la expresión de lao transportadores de glucosa (GLU1, 3 y 4), en asociación al factor de crecimiento de insulina (IGF1a) y las proteínas de choque termino (HSP27, 70, 90 y 94) como factor pronóstico de los craniofaringiomas. RESULTADOS: Se estudiaron 28 casos. 12(43%) mujeres y 16(57%) hombres, con edad de 17 a 55 años (media de 31.43± 10.057). 26(93%) de tipo adamantinomatous y 2(7%) de tipo papilar. Recurrencia fueron 12(46%). GLUT1, 3 y IGF1a fueron positive en el porción externa del epitelio externo, en las capa basal de la queratina, húmeda, en las calcificaciones distróficas y en los arreglos. Mientras que las HSP27, y 70 fueron positivas en el tejido cerebral adyacente y las HSP90 y 94 fueron positivas tanto en el EE como en el retículo estrellado. CONCLUSIONES: Por lo sugerimos que los CF son metabólicamente activos en las QH, CD y en los arreglos y que el tejido cerebral adyacente expresa proteínas de choque térmico. Existiendo áreas de coexpression en el EE lo que sugiere que al romperse las estructuras quísticas promueve la expresión de la HSP. INSTITUCIÓN: Departamento de Neuropatolo- INSTITUCIÓN: Departamento de Neuropatología, gía, Instituto Nacional de Neurología y Neuro- Instituto Nacional de Neurología y Neurocirugía. cirugía. Ciudad de México. Hospital General de Ciudad de México. Hospital general de México. México. ESTADO: DISTRITO FEDERAL ESTADO: DISTRITO FEDERAL Mampara: 3 A Mampara: 2 B Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. TC-14 TC-13 AUTORES: Martha Lilia Tena-Suck, Miguel Fernando Salazar, Daniel Rembao Bojorquez, Citlaltepetl salinas Lara. AUTORES: Martha Lilia Tena-Suck, Miguel Fdo. Salazar, Francisca Fernández-Valderde 2do Congreso Anual de Patólogos Mexicanos • 13 León, Guanajuato • 2016 TITULO: Cambios Oncociticos en tumores de plexos coroides. INTRODUCCIÓN: Tumores del plexo coroideo son los tumores cerebrales epiteliales raros, de acuerdo a la OMS se clasifican en papilomas, papilomas atipicos y carcinomas, Son más frecuentes en niños que en adultos. Histológicamente se ha reportado cambios de metaplasia histológicamente, así como cambios oncocíticas y la deposición de amiloide, sin embargo poco se sabe al respecto, y de qué manera estos hallazgos contribuyen a la transformación maligna de estos tumores. OBJETIVOS: El objetivo de estudiar los cambios oncociticos en los TPC. MATERIAL: Se trata de una serie retrospectiva, descriptiva y de los casos con enfoque en técnica de 42 tumores del plexo coroideo primario y recurrente se incluyeron en este estudio, de los cuales 28 (47%) fueron papiloma de plexos coroides, grado I, 8 (13%), papilomas atípicos, grado II y 6 (10%) fueron carcinomas, grado III según la OMS. 37 eran mujeres (62% 9) y 2 y de un análisis de microscopía electrónica, correlación clínico patológica y de IHQ. Mampara: 3 B Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. TC-24 AUTORES: Dra. Brenda Esther Vázquez López, Dr. Jorge Edgardo Márquez Escalante. TITULO: HIPOFISITIS XANTOMATOSA. PRESENTACIÓN DE UN CASO Y REVISIÓN DE LA LITERATURA. INTRODUCCIÓN: Los trastornos inflamatorios de la hipófisis son un grupo poco frecuentes de padecimientos relacionados con la disfunción de dicha glándula que pueden simular una neoplasia hipofisiaria. Los casos de hipofisitis pueden ser secundarios a diversos padecimientos o de causa desconocida. OBJETIVOS: Presentar una patología poco frecuente. Revisar la literatura reciente. Analizar los hallazgos histológicos con lo reportado en la literatura. RESULTADOS: 42 tumores del plexo coroideo primario y recurrente se incluyeron en este estudio, de los cuales 28 (47%) fueron papiloma de plexos coroides, grado I, 8 (13%), papilomas atípicos, grado II y 6 (10%) fueron carcinomas, grado III según la OMS. 37 eran mujeres (62% 9) y 23 varones (42%). Se observaron cambios oncocíticas en 14 (33%). cambios papilas degenerativa o hidrópica en 18 (43%) casos, 11 (26%) mostraron cuerpos de psammoma, 15 (36%), depósito de amiloide en 14 (33%) y cambios oncocíticas en 14(33%), estos cambios fueron positivos para fascin, CD1a, IL2, IL6, TNF, CD68 y HIF1? y las células epiteliales normales fueron negativas, que corresponde por EM a las células dendríticas, y no se observaron mitocondrias hiperplasicos como en otros tumores oncocíticas. CONCLUSIONES: Por lo que en los tumores de plexos coroides corresponden a hiperplasia de células dendríticas. Las células dendríticas expresan altos niveles de clase I y moléculas de MHC II, IL1, IL2, IL10IL-6, IL-8, IL-l0, TNFa, y TNF?, así como las proteínas inflamatorias de macrófagos (MIPl? y MIP-1 beta). Estas citoquinas pueden participar en la respuesta de células dendríticas para la presentación de antígenos. MATERIAL: Paciente femenino de 52 años de edad, que inicia en 2012 de forma insidiosa con disminución de la visión, valorada por oftalmología, con diagnóstico y tratamiento para glaucoma. La sintomatología progresa, acude con oftalmólogo del medio privado. Se realiza TAC, encontrando datos de expansión supraselar de 4.3 x 3 x 1.7 cm, con erosión de la silla turca y densidad heterogénea de la lesión. El laboratorio reporta disminución de FSH y LH. RESULTADOS: Ingresa a quirófano con diagnóstico de Macroadenoma de HIpófisis. En el servicio de Anatomía Patológica, se recibieron múltiples fragmentos que en conjunto medían 2 x 1.4 x 0.7 cm. Irregulares, café claros, hemorrágicos y blandos. El análisis microscópico de los fragmentos mostró un escaso parénquima hipofisiario remanente, mezclado con un intenso infiltrado inflamatorio crónico a expensas de linfocitos y macrófagos, algunos de ellos de aspecto espumoso, así como células gigantes multinucleadas de tipo cuerpo extraño y abundantes espículas de colesterol, sin presencia de granulomas ni necrosis. CONCLUSIONES: La hipofisitis es un trastorno inflama- infrecuente que debido a su cuadro clínico, fácilmenINSTITUCIÓN: Departamento de Neuropatología, torio te puede ser confundido con otras neoplasias hipofisiarias Instituto Nacional de Neurología y Neurocirugía. Ciudad de México. Hospital general de México. ESTADO: DISTRITO FEDERAL como adenomas, o quistes de Ratke. Es importante mencionar que si bien, los cristales de colesterol no se consideran dentro de la descripción histológica clásica de la hipofisitis xantomatosa, muchos casos se desarrollan posteriormente a otra patología, por lo que no es posible descartar alguna alteración previa de la hipófisis en esta paciente. INSTITUCIÓN: UMAE #25, IMSS. Monterrey, NUevo León. 2do Congreso Anual de Patólogos Mexicanos • 14 León, Guanajuato • 2016 ESTADO: NUEVO LEÓN Mampara: 4 A Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. TC-31 AUTORES: DIANA BRISA SEVILLA, Dr. Hugo Domínguez Malagon, Dr. Leonardo Saúl Lino Silva. TITULO: Ependimoma de células gigantes de filum terminale: una entidad poco conocida con aspecto histológico maligno pero comportamiento indolente. Presentación de un caso con correlación clínica, radiológica y ultraestructural; revisión de la literatura. INTRODUCCIÓN: Dentro de las neoplasias del sistema nervioso central los ependimomas (EP) son raros, pero una variante excepcional es el ependimoma de células gigantes (ECG), caracterizado por la presencia de células gigantes pleomorfas en ausencia de necrosis y actividad mitótica. Característico del ECG es la discrepancia entre su morfología preocupante y su conducta biológica generalmente indolente, es importante enfatizar sobre sus características morfológicas y establecer el diagnóstico diferencial con otras neoplasias pleomorfas de localización similar. OBJETIVOS: Se presenta un caso de ECG extra-axial ubicado en la región sacra lo cual hasta la fecha no ha sido informado, se describen sus características clínico-patológicas y ultraestructurales, se discute el diagnóstico diferencial y se revisa la literatura. MATERIAL: Se revisó el archivo del Departamento de Patología del INCan, entre 2007 y 2015, se encontraron 47 EP. De estos, 22 fueron de tipo convencional, 6 de tipo mixopapilar, 3 de tipo papilar, 2 de células claras, 7 de tipo tanicitico, 3 de tipo anaplásico, uno de tipo celular, uno no clasificable, y dos de ECG (uno localizado en fosa posterior y otro en la región sacra), este último se estudia en el presente trabajo. RESULTADOS: Aunque las neoplasias del FT pueden de los casos de hasta 35 meses no se han reportado recurrencias, sin embargo el número de casos publicados de ECG en filum terminale es limitado y se requiere un mayor tiempo de seguimiento, sin embargo, por definición es una lesión de comportamiento indolente. INSTITUCIÓN: INSTITUTO NACIONAL DE CANCEROLOGIA ESTADO: DISTRITO FEDERAL Mampara: 4 B Fecha y hora Instalación: 2 mayo 7:30 hrs. Fecha y hora retiro: 2 mayo 13:00 hrs. TC-44 AUTORES: SILVIA JUDITH HERNANDEZ, Dra. Karina Larios, Dra. Yolanda Martínez, Dr. Eduardo González, Dra. Karina Piña, Dr. Edmundo Castelán, Dra. Julia de Lieja, Dr. Louis Valéry Carius Estrada, Dr. Víctor Manuel Romero Vazquez TITULO: Hamartoma Hipotalámico en la edad pediátrica: Presentación de tres casos y revisión de la literatura INTRODUCCIÓN: El hamartoma hipotalámico es una malformación congénita originada del hipotálamo y el tuber cinereum con una prevalencia de 1-2 casos/100,000 habitantes. Los pacientes pueden cursar asintomáticos, con crisis gelásticas o alteraciones endocrinológicas. Se componen de neuronas distribuidas individualmente o en acúmulos dentro de un fondo fibrilar y en algunos casos con células ganglionares. OBJETIVOS: Describir las características clínicas, radiológicas e histopatológicas en pacientes pediátricos con diagnóstico de hamartoma hipotalámico. encontrarse cercanas a la región sacra, nuestro caso es el primer ECG de localización extra-axial que afecta el tejido blando y subcutáneo de dicha región sin conexión con el neuroeje como se demostró durante la cirugíaEl ECG no ha sido gradificado por la OMS. Su característica principal es la discrepancia entre su morfología preocupante y su conducta biológica generalmente indolente. MATERIAL: Se revisaron retrospectivamente las historias CONCLUSIONES: El caso descrito en el presente tra- RESULTADOS: Caso 1, Fem, 2 años, crisis gelásticas bajo se localizaba en el tejido subcutáneo de la región sacra la cual es un sitio cercano al FT. ECG del FT constituye una entidad extremadamente rara descrita por primera vez por Zec en 1996.La remoción completa del tumor es el factor pronóstico más significativo, con periodos de seguimiento clínicas, los estudios de imagen y las laminillas teñidas con H&E de tres pacientes con diagnóstico de hamartoma hipotalámico realizados en los últimos 5 años en la UMAE 25, Monterrey, N.L. Las biopsias fueron obtenidas mediante craneotomía posterior, frontoorbitocigomatica derecha y frontoparietal derecha, respectivamente. y pubertad precoz. Caso 2, Fem, 6 años, pubertad precoz. Caso 3, Fem, 7 años, pubertad precoz e hipertensión endocraneal. La resonancia magnética: Caso 1, lesión de 3.3 cms, ovalada, hipointensa que desplaza la protuberancia y mesencéfalo. Caso 2, lesión de 1 cms, redondeada, pediculada, anterior a los cuerpos mamilares. Caso 3, lesión 2do Congreso Anual de Patólogos Mexicanos • 15 León, Guanajuato • 2016 de 2.7 cms, lobulada y pediculada, posterior a región selar. Caso 1, resección del 40%, BTO (lesión glial de bajo grado), caso 2, resección total, BTO (gliosis reactiva), caso 3, resección del 20%, BTO (tejido glial normal). Todos presentaron neuronas pequeñas en un fondo glial y solo 1 presentó células ganglionares. CONCLUSIONES: La pubertad precoz fue la manifesta- ción inicial en 2 pacientes y en el tercer caso se agregó de manera posterior lo cual difiere con lo reportado en la literatura mundial. La RMN es el estudio de imagen de elección. Los tratamientos quirúrgicos descritos son: 1. Craneotomía y resección/desconección, 2. Resección endoscópica/desconección, 3. Técnicas esteriotácticas. El diagnóstico diferencial principal es con tejido cerebral normal y posteriormente con gangliogliomas. Es crítico tener los hallazgos neuroradiológicos y la discusión con el neurocirujano para ofrecer un diagnóstico certero. pañada de vómitos, debilidad generalizada, acentuada en miembros pélvicos, así como episodios de desorientación. Las alteraciones del líquido cefalorraquídeo y engrosamiento difuso de leptomenínges en imagen de RMN hicieron sospechar meningitis crónica. Se realizó biopsia meníngea y durante la cirugía se evidencio pigmentación café oscura difusa. RESULTADOS: La clasificación definitiva de las neoplasias melanocíticas primarias en sistema nervioso central precisa integración de criterios histológicos y clínicos. Es indispensable realizar diagnóstico diferencial con neoplasias primarias del SNC pigmentadas no melanocíticas y con neoplasias metastásicas. Aunque sin criterios citohistológicos de malignidad, la melanocitosis meníngea tiene pronóstico desfavorable y carece de tratamiento efectivo. CONCLUSIONES: La clasificación definitiva de las neo- INSTITUCIÓN: Instituto Mexicano del Seguro plasias melanocíticas primarias en sistema nervioso central Social, Unidad Médica de Alta Especialidad, precisa integración de criterios histológicos y clínicos. Es indispensable realizar diagnóstico diferencial con neoplaHospital #25, Monterrey, N.L. México sias primarias del SNC pigmentadas no melanocíticas y con neoplasias metastásicas. Aunque sin criterios citohistológicos de malignidad, la melanocitosis meníngea tiene pronóstico desfavorable y carece de tratamiento efectivo. El caso que presentamos resulta relevante por su baja frecuencia. El diagnóstico de certeza tiene implicaciones pronósticas. ESTADO: NUEVO LEÓN Mampara: 5 A Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. TC-93 AUTORES: Dra. Paola Yolotzin Valenzuela Torres1, Dr Luis Moreira Ponce 2, Dra. Teresa Cristina Cuesta Mejías3, Dr Rafael Mendizabal Guerra4 TITULO: Melanocitosis leptomeníngea. Dificultades diagnósticas. A propósito de un caso. INTRODUCCIÓN: Las neoplasias melanocíticas primarias del sistema nervioso central son infrecuentes, incidencia estimada de 1/10 millones por año. Incluyen lesiones localizadas, difusas, benignas y malignas. La proliferación difusa de melanocitos leptomeningeos define a la melanocitosis. Describimos los hallazgos clínicos, imagenológicos, quirúrgicos, histológicos e inmunofenotipicos que establecieron el diagnostico de melanocitosis meníngea en adulto joven, con diagnóstico inicial de meningitis crónica. OBJETIVOS: Revisar las dificultades para el diagnóstico diferencial y clasificación precisa de melanocitosis meníngea MATERIAL: Hombre de 42 años de edad, quien 6 meses previos a su internamiento refirió cefalea progresiva, acom- REFERENCIAS: 1.- Brat DJ, Perry A. Melanocytic le- sions. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO classification of tumours of the central nervous system. Lyon: IARC; 2007. p. 181?3 2.-Heidi V.N. Küsters-Vandevelde, Benno Küsters y cols. REVIEW Primary Melanocytic Tumors of the Central Nervous System: a Review with Focus on Molecular Aspects. Brain Pathology 25(2015) 209?226 3.-Honigberg, M.C., Papavassiliou, E. y cols. Case report Primary leptomeningeal melanocytosis presenting as chronic meningitis. Journal of clinical neuroscience. Vol 21, Issue 6, June 2014, pp 1056?1058. INSTITUCIÓN: Medico residente del 3er año de Anatomía Patológica del Hospital Juárez de México, 2 Medico residente del 4to año de Neurgocirugía del HJM, 3 Medico Adscrito del servicio de Anatomía Patológica del HJM, 4 Medico Adscrito del servicio de Neurocirugía del HJM ESTADO: DISTRITO FEDERAL Mampara: 5 B Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. TC-102 AUTORES: Dra. Alejandra Toledo Manriquez, Dra. Veró- 2do Congreso Anual de Patólogos Mexicanos • 16 León, Guanajuato • 2016 TITULO: ASTROCITOMA SUBEPENDIMARIO DE CÉLULAS GIGANTES ASOCIADO A COMPLEJO DE ESCLEROSIS TUBEROSA. PRESENTACIÓN DE CASO año de los cuales 240 tendrán complejo de esclerosis tuberosa. Histológicamente bien delimitados, pueden tener minina infiltración, compuesto por células grandes poligonales de citoplasma eosinófilo. Ocasional pleomorfismo nuclear y atipia, con raras mitosis, necrosis y proliferación vascular. Inmunohistoquímica, diferenciación astrocítica positiva a GFAP, S100, diferenciación neuronal positiva a sinaptofisina, cromogranina, NSE y neurofilamentos INTRODUCCIÓN: El complejo de esclerosis tubero- REFERENCIAS: 1.Louis DN, Ohgaki H, Wiestler OD, et OBJETIVOS: Presentación de caso de astrocitoma INSTITUCIÓN: HOSPITAL CENTRAL SUR DE ALTA ESPECIALIDAD DE PETRÓLEOS MEXICANOS nica Bautista Piña, Dra. Rosa Maria Vicuña Gonzalez, Dra. María Irene Rivera Salgado. sa comprende un trastorno autosómico dominante que se caracteriza por alteraciones en la migración, diferenciación y proliferación celulares con formación de múltiples tumores benignos (hamartomas), que afectan principalmente piel, encéfalo, riñón, ojo, corazón y pulmón. Incidencia de 1/5,000-10,000 nacidos vivos. El astrocitoma subependimario de células gigantes (ASGC) se considera un glioma grado I de la OMS, de crecimiento lento, tumor más frecuente del sistema nervioso central en pacientes con complejo de esclerosis tuberosa, en el 5-20% de los casos. La mayoría de los pacientes muestran la sintomatología entre los 8-19 años. Se localizan sobre la pared de los ventrículos laterales, son únicos aunque pueden ser bilaterales hasta en un 21%. subependimario de células gigantes asociado a complejo de esclerosis tuberosa en el Hospital Central Sur de Alta Especialidad de Petróleos Mexicanos. al. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathologica. 2007;114(2):97109. doi:10.1007/s00401-007-0243-4 2.Shepherd CW, Gómez MR, Lie JT, Crowson CS. Causes of death in patients with tuberous sclerosis. Mayo Clinic Proc.1991;66:792?6. 3.Boesel CP, Paulson GW, Kosnick EJ, Earle KM. Brain hamartomas and tumors associated with tuberous sclerosis. Neurosurgery. 1979;4:4107. 4.Ruiz M, Sánchez S, Medina A, Rea A, Sanromán R, García J et al. Astrocitoma subependimario de células gigantes asociado a complejo de esclerosis tuberosa: recomendaciones para el diagnóstico oportuno y tratamiento. Acta Pediatr Mex 2014;35(Sup 1):S1-S25. ESTADO: DISTRITO FEDERAL RESULTADOS: Paciente femenino de 15 años de edad, carga genética para hipertensión arterial, diabetes mellitus, tía paterna padece esclerosis tuberosa y padre en estudio para descartar la misma enfermedad. Inició padecimiento actual desde los 3 años de edad con convulsiones, se diagnosticó como esclerosis tuberosa (Síndrome de Bourneville Pringle). Se realizó estudio de control en 11/2013, demostró lesiones intraventriculares, nódulos subependimarios y múltiples tubers corticales, se realizó resección completa del tumor intraventricular con diagnóstico histopatológico de astrocitoma subependimario de células gigantes (Grado I de la OMS). El servicio de genética ha integrado su estudio con la participación de diferentes servicios y emite diagnóstico de esclerosis tuberosa con angiofibromas faciales, placas fibrosas en cuero cabelludo, fibroma gingival, fibromas ungueales, placa de shagreen, rabdomioma en ventrículo izquierdo. El ultrasonido renal con imágenes sugestivas de angiomiolipomas derechos. CONCLUSIONES: El astrocitoma subependimario de células gigantes es un tumor benigno (Grado I OMS), de lento crecimiento. La esclerosis tuberosa es una facomatosis autosómica dominante de alta penetrancia y variabilidad fenotípica, asociado a alteración de los genes TSC1 y TSC2, caracterizado por hamartomas generalizados. Típicamente se presentan durante la infancia y adolescencia, se localizan en la pared de los ventrículos laterales, cercano a los agujeros de Monro. Incidencia de uno en 5 000 a 10 000 nacidos vivos, existen en México alrededor de 15000 pacientes con complejo de esclerosis tuberosa, se espera el nacimiento de alrededor de dos millones de mexicanos por Mampara: 6 A Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. TC-121 AUTORES: Dra. Hersilia Aide Hernandez Zamonsett, Dr. Eduardo Ruiz Holguin, Dr. Marco Antonio Ponce Camacho, Dra. Ivett Miranda Maldonado, Dra. Oralia Barboza Quintana, Dra. Raquel Garza Guajardo. TITULO: Carcinoma de plexo coroideos en un paciente de la edad pediatrica, Reporte de un caso y revision de la literatura. INTRODUCCIÓN: Los carcinomas de plexos coroideos están clasificados por la Organización Mundial de la Salud como una neoplasia de grado III; Comprenden del 0,3 a 0,6% de todos los tumores intracraneales. Son tumores muy raros que se presentan en menores de 2 años y tienen alta agresividad y mal pronóstico. OBJETIVOS: Lactante de 11 meses de edad, con antecedente de Ictericia neonatal. Inició su padecimiento con crisis convulsivas, se realiza TAC de cráneo, encontrado tumor intracraneal parietal intraventricular. Presenta deterioro neurológico, con 8 puntos de ECG, se realiza ventriculos- 2do Congreso Anual de Patólogos Mexicanos • 17 León, Guanajuato • 2016 tomia y se reseca 50% de lesión y se recibe para estudio transoperatorio MATERIAL: Se realizan cortes de tejido congelado, el res de primarios de SNC. La clasificación de la OMS los gradifica en tres: I benigno; II atípico; III anaplasico/maligno. La mayoría son grado I 80.6%; grado II 10.4% y grado III 8.1%. tejido restante se fija con formaldehido y se incluye en parafina, realizandose cortes con microtomo, y tiñendose con la tecnica de hematoxilina y eosina, ademas se realizan tinciones de inmunohistoquimica a base de citoqueratina, INI-1, CD30 y Proteina serica fibrilar glial. OBJETIVOS: El propósito de este estudio fue revisar las RESULTADOS: En los cortes histologicos, se obser- MATERIAL: Los pacientes fueron identificados en la base van una lesion con presencia de pequeñas papilas, y areas solidas, las cuales estan cubiertas de celulas columnares las cuales tienen nucleos redondos, hipercromaticos, con áreas de extensa anaplasia y presencia de abundantes mitosis aberrantes, además de focos de necrosis. Se realizaron tinciones de inmunohistoquimica las cuales resultaron Positivas para CITOQUERATINA e INI-1 y negatividad para CD30, y PAFG. Se diagnostica como carcinomade plexos coroideos. CONCLUSIONES: En el diagnostico diferencial es imoportante considerar un papiloma de plexos coroideos, sin embargo la extensión extraventricular hacia el parénquima cerebral circundante por invasión tumoral, la heterogeneidad de señal (necrosis, hemorragia y quistificación) y la presencia de edema vasogénico en la sustancia blanca periventricular, son datos que inclinan hacia el diagnóstico de carcinoma.Ademas en el caso de nuestro paciente, la positividad para los marcadores de inunohistoquimica y la importante anaplasia que presenta la lesion, son confirmatorios de este diagnostico. INSTITUCIÓN: Departamento de Anatomia Patologica y Citopatologia, Hospital Universitario “Dr. Jose Eleuterio Gonzalez” de datos del archivo de patología. Se incluyeron pacientes pediátricos de 18 años o menos en la primera resección quirúrgica, con el diagnostico de meningioma. Se revisaron los expedientes clínicos, los hallazgos radiológicos mediante el sistema IMPAX del Hospital de Pediatría de CMN S. XXI y el resultado definitivo emitido en el servicio. RESULTADOS: Se identificaron un total de 19 pacientes (10 hombres y 9 mujeres). La edad promedio al momento del diagnostico fue de 10.2 años (rango de 4 meses-16 años). La cefalea, crisis convulsivas e hipertensión intracraneal fueron las manifestaciones mas frecuentes de presentación. La variedad histológica mas frecuentemente encontrada fue meningioma transicional (52.6%). El 73.6% de los casos corresponde a grado I de clasificación de la OMS; Grado II de la OMS 10.5%; Grado III de la OMS 5.2%. CONCLUSIONES: Los meningiomas son extremadamente raros en la población pediátrica; La mayor parte de ellos corresponde a meningiomas de comportamiento benigno, esta es una de las series mas grande informada en el país. INSTITUCIÓN: Hospital de Pediatría Centro Medico Nacional Siglo XXI ESTADO: DISTRITO FEDERAL ESTADO: NUEVO LEÓN Mampara: 7 A Mampara: 6 B Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. características clínicas, radiológicas y patológicas de todos los meningiomas pediátricos tratados quirúrgicamente durante el periodo de 2005 a 2015 en el Hospital de pediatría CMN S. XXI. 7:30 hrs. Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. TC-162 TC-126 AUTORES: Alejandro Santana González, Alicia Georgina Siordia Reyes AUTORES: LUIS ENRIQUE MANOLA, Dra. Laura G. Chávez Macías, Dr. Erick Gómez Apo, Dra. Verónica Velasco Vales, Dr. Valentín González Flores, Dra. Janeet Garduño Becerra. TITULO: Meningiomas. Serie de casos en una población pediátrica. TITULO: QUISTES EPIDERMOIDES EN SISTEMA NERVIOSO CENTRAL, UN HALLAZGO ININTRODUCCIÓN: Los meningionas son un grupo de FRECUENTE PERO MORTAL. REVISIÓN DE 11 neoplasias con morfología, inmunohistoquimica y ultraes- CASOS POST MORTEM tructura semejante a la célula meningotelial, raro en edad pediátrica, la incidencia es de 0.4 a 4.6% de todos los tumo- 2do Congreso Anual de Patólogos Mexicanos • 18 León, Guanajuato • 2016 INTRODUCCIÓN: Los quistes en SNC se clasifican por el origen embrionario; la prevalencia es baja, los más comunes son aquellos derivados del neuroectodermo. Los derivados del ectodermo como quistes epidermoides y dermoides son infrecuentes, pueden causar graves complicaciones y secuelas neurológicas y en algunos casos, la muerte; por lo que su diagnóstico y tratamiento temprano es imprescindible. OBJETIVOS: Describir las características clínicas y anatomopatológicas de 11 casos posmortem con diagnóstico final de quiste epidermoide en SNC, y mencionar los principales diagnósticos diferenciales y localización MATERIAL: Se revisaron los casos posmortem del archivo de Anatomía Patológica del Hospital General de México en un período de 39 años (19772016) donde se obtuvo información acerca de la edad, sexo, localización y diagnóstico clínico de la lesión. RESULTADOS: Se encontraron 11 casos de quistes epidermoides en SNC; nueve (81.8%) correspondieron a hombres entre los 21 y los 73 años; y dos (18.1%) fueron en mujeres de 24 y 43 años. La localización más frecuente es el ángulo pontocerebeloso (63.3%), el resto en la región sillar, parasillar, cerebelo y lóbulo temporal. El tiempo de evolución fue entre ocho meses y cinco años, la mayoría fueron diagnosticadas premortem como meningiomas y astrocitomas de alto grado por las manifestaciones clínicas y radiológicas. Tres casos fueron hallazgos posmortem. AUTORES: Dra. Ma. Verónica Velasco Vales, Dr. Erick Gómez-Apo, Dr. Víctor Hugo Ramos Pacheco, Dr. Leoncio Alberto Tovar Romero, Dra. Celene Martínez Ruiz, Dr. Eduardo Flores Álvarez, Dra. Laura G. Chávez-Macías. TITULO: ASTROCITOMA ANAPLÁSICO SUPRASILLAR: Informe de un caso INTRODUCCIÓN: Los tumores neuroepiteliales que se originan en la región sillar y suprasillar son raros y se originan de la neurohipofisis y del infundíbulo. Además, existen los gliomas hipotalámicos o del quiasma óptico, que presentan un reto diagnóstico; son lesiones raras, que pueden hacer prominencia hacia la región sillar. En el examen radiológico se presentan como lesiones suprasillares o intrasillares MATERIAL: Hombre de 40 años. El padecimiento actual inició con hemianopsia temporal bilateral. Ante la persistencia y progresión acudió con médico facultativo quien lo envío al HGM. Se efectuó RM, se identificó una lesión suprasillar, multilobulada, con componente quístico hiperintenso, que desplazaba la porción ventral del hipotálamo en sentido dorsal, en sentido lateral la carótida interna izquierda y ventralmente a la hipófisis. Los niveles séricos horm RESULTADOS: En patología quirúrgica se recibió tejido multifragmentado. Microscópicamente corresponde a lesión neoplásica de crecimiento difuso, formada por células pleomórficas indiferenciadas y escasa malla fibrilar, negativas para CKAE1/3 y RP; positivas para GFAP y P53; Ki67 >5%. Se concluyó el diagnóstico de astrocitoma anaplásico. CONCLUSIONES: Los quistes epidermoides en SNC representan <1% de las lesiones intracraneanas. Afectan más a hombres entre la 3ª y 7ª década. La localización principal es ángulo pontocerebeloso, región suprasillar y piso del IV ventrículo. Las manifestaciones clínicas dependen de la localización, sin embargo el incremento de la presión intracraneana es el común denominador. El comportamiento clínico es benigno, sin embargo, depende del tamaño y la localización puede ocasionar complicaciones como ruptura con meningitis química y recurrencia en casos de resección incompleta.> CONCLUSIONES: Conocer el origen de estas lesiones es complicado, ya que se pueden originar de las células gliales modificadas de la neurohipófisis e infiltrar el hipotálamo; como se pueden originar de la vía óptica e infiltrar la región sillar. Los gliomas de la región sillar y los gliomas ópticos son lesiones que presentan un reto diagnóstico para el cirujano, imagenólogo y patólogo; por lo que es muy importante poder lograr una correlación adecuada y diagnosticarlas de manera correcta INSTITUCIÓN: Hospital General de México “Dr. INSTITUCIÓN: Hospital General de México Eduardo Liceaga” “Eduardo Liceaga” y Facultad de Medicina, UNAM ESTADO: DISTRITO FEDERAL ESTADO: DISTRITO FEDERAL Mampara: 8 A Mampara: 7 B Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. TC-163 7:30 hrs. Fecha y hora Instalación: 2 mayo 7:30 hrs. Fecha y hora retiro: 2 mayo 13:00 hrs. TC-164 AUTORES: Dra. Ma. Verónica Velasco Vales, Dr. Luis E. Manola Aguilar, Dr. Emanuel I. Juarez Zapata, Dr. Erick Gómez-Apo, Dra. Laura G. Chávez Macías 2do Congreso Anual de Patólogos Mexicanos • 19 León, Guanajuato • 2016 TITULO: OLIGOASTROCITOMAS: Revisión de 11 casos Marco Durán Padilla Dr. Erick Gómez Apo Dra. Laura G. Chávez Macías INTRODUCCIÓN: El oligoastrocitoma es un glioma in- TITULO: TUMORES MEDULARES CON AFECCIÓN EXTENSA: PRESENTACION DE TRES CASOS DEL HOSPITAL GENERAL DE MEXICO “DR. EDUARDO LICEAGA” filtrante, mezcla de la proliferación de dos tipos de células neoplásicas con fenotipo oligodendroglial y astrocítico. Pueden ser grado II y III. Corresponden a una zona gris de los gliomas infiltrantes. Fueron descritos por Cooper en 1935. Se presentan principalmente en individuos entre 35 y 45 años, con mayor afección al género masculino (1.3:1) OBJETIVOS: Describir las características clínicas e histológicas de 11 casos de oligoastrocitomas MATERIAL: Estudio retrospectivo y analítico de 11 casos de oligoastrocitomas en el Hospital General de México “Dr. Eduardo Liceaga” desde 2000 a 2015. De cada caso se obtuvieron las características clínicas de los informes de patología, además de las laminillas y bloques de parafina, solamente disponibles en seis casos. Se efectuó la revisión histológica y se clasificaron de acuerdo a la variante bifásica y mezclada. RESULTADOS: La edad promedio de presentación es 38 años; con mayor afección en el género femenino (7/4). El sitio principal de afección fue el lóbulo temporal (72%) con predominio izquierdo. El 45% fueron oligoastrocitomas anaplásicos. El 66% variante bifásico y 34% mezclado. CONCLUSIONES: Los oligoastrocitomas son neoplasias con fenotipo convincente astrocítico y oligodendroglial en la misma neoplasia, grado II o III según las características morfológicas. El porcentaje mínimo de los componentes para hacer el diagnóstico es un campo a 100X oligodendrogial o astrocítico. En la mayoría de los casos es complicado poder relacionar la extensión precisa de cada uno de los componentes. Existen dos variantes 1) bifásicos, son raros y con áreas distintivas de los tumores yuxtapuestas y 2) mezclados, son más comunes, ambos componentes se encuentran intercalads. INSTITUCIÓN: Hospital General de México, “Dr. Eduardo Liceaga” ESTADO: DISTRITO FEDERAL INTRODUCCIÓN: Los tumores de la médula espinal son poco frecuentes y la afección extensa en forma holocorda es extraordinaria. Los tumores intramedulares más comunes son los ependimomas, astrocitomas y hemangioblastomas. Debido a la poca incidencia, crecimiento lento y manifestaciones inespecíficas iniciales, es difícil detectarlos y algunos casos tienen diagnóstico tardío. OBJETIVOS: Estudiar los casos de los pacientes con lesión en medula espinal. MATERIAL: Se presentan tres casos (uno posmortem y dos especímenes quirúrgicos) estudiados en el Hospital General de México ?Dr. Eduardo Liceaga? de lesiones con afección medular extensa. RESULTADOS: 1. Niño de 3 años, con dolor cérvico-to- rácico y debilidad muscular. Lesión medular C2?T10, con siringobulbia y siringomielia. Diagnóstico: astrocitoma pilocítico. 2. Mujer de 29 años, con alteraciónes sensitivas y motoras en miembros torácicos. Lesión extensa en médula espinal, con siringomielia y siringobulbia. Diagnóstico: ependimoastrocitoma. 3. Hombre de 43 años, con disminución de la fuerza en mano derecha y alteraciones en la marcha. Lesión medular con afección de varios segmentos , heterogénea, con siringomielia proximal y distal. Diagnóstico: ependimoma CONCLUSIONES: Los tumores holocorda tienen un curso clínico inespecífico, por lo que la detección de la neoplasia se realiza meses o años después de las manifestaciones iniciales; lo cual permite que adquieran el crecimiento tan extenso. Clínicamente debutan con alteraciones motoras y sensitivas de manera bilateral; se asocian a NF-1, NF-2 y VHL. La siringomielia en ocasiones permite la adecuada resección y en ocasiones con recuperación favorable posquirúrgica. INSTITUCIÓN: HOSPITAL GENERAL DE MEXICO “DR. EDUARDO LICEAGA” Mampara: 8 B Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. ESTADO: DISTRITO FEDERAL 7:30 hrs. Mampara: 9 A TC-165 Fecha y hora Instalación: 2 mayo AUTORES: Dra. Ma. Verónica Velasco Vales Dra. Mara Fecha y hora retiro: 2 mayo 13:00 hrs. Cárdenas Escudero Dra. Jimena Trillo Tinoco Dra. Tania Garibay Huarte Dr. Marco Antonio Rodríguez-Florido Dr. TC-171 2do Congreso Anual de Patólogos Mexicanos • 20 León, Guanajuato • 2016 7:30 hrs. AUTORES: Itzel María Montoya, Dra. Maria Del Rocio Estrada Hernandez , Dra. Ana Lilia Morales Leite, Dra Sara Parraguirre Martinez GINECOPATOLOGÍA TITULO: Displasia fibromuscular de arterias TC-17 cerebrales. reporte de un caso de autopsia en AUTORES: Dra. Brenda Esther Vázquez López, Dra. joven de 15 años. INTRODUCCIÓN: La displasia fibromuscular (dfm), primero descrita por leadbetter y burkland en 1938, es una enfermedad arterial que afecta típicamente las arterias renales en 85% de pacientes.la arteria carótida interna es la segunda localización más común, afecta principalmente a mujeres en edades medias de la vida con una relación 3:1 respecto al hombre. OBJETIVOS: Presentar el caso de autopsia realizada a un hombre de 15 años , que sufre de muerte no esperada en su domicilio , sin sintomatología previa , ni antecedentes de importancia para el padecimiento final. con diagnosticos de ingreso de evento cerebral vascular isquémico,infarto cerebral frontoparietal temporoccipital izquierdo y edema cerebral severo. MATERIAL: Se realiza autopsia parcial unicamente con extraccion de encefalo por limitaciones propias de los familiares. RESULTADOS: Microscopicamente se observa en ar- teria cerebral media derecha de manera segmentaria: engrosamiento de la adventicia, reduplicación de la lámina elástica interna y fibrosis de la íntima, en los segmentos del vaso que presentan estas alteraciones, hay formación de trombo de fibrina. Existen segmentos de la arteria que tienen características normales. Con tinción de hematoxilina acida fosfotumsica (ptah) se hace evidente la malla de fibrina firmemente adherida a la pared de una arteria. Cabe destacar que en la arteria cerebral media izquierda la pared en algunos segmentos esta discretamente engrosada a expensas de hiperplasia de la capa muscular, la elástica interna se encuentra desprendida de la luz y tiene aspecto festoneado, en otras áreas los eritrocitos irrumpen entre la íntima y la elástica interna disecándola. CONCLUSIONES: Es una patología no ateroesclerótica ni inflamatoria que se caracteriza por lesiones estenosantes en serie intercaladas por aneurismas murales.La presentación clínica varía desde una condición asintomática hasta una enfermedad multisistémica que imita una vasculitis necrotizante, dependiendo del segmento arterial involucrado, el grado de estenosis y el tipo de displasia fibromuscular. Esta angiopatía compromete diferentes arterias musculares de mediano calibre siendo las arterias renales las más frecuentemente afectadas. Olga Esquivel Hernández, Dr. Jorge Edgardo Marquez Escalante. TITULO: DISPLASIA MESENQUIMAL PLACENTARIA. REPORTE DE CASO. INTRODUCCIÓN: La Displasia mesenquimal placentaria (DMP) es una anomalía vascular de baja incidenciaque se caracteriza por placentomegalia y múltiples vesículas en “racimos de uvas”. Frecuentemente se diagnostica como embarazo molar en ultrasonido. La DMP generalmente presenta un feto viable de cariotipo normal. El diagnostico final es histopatológico, sin las alteraciones características de enfermedad molar. OBJETIVOS: - Presentar un reporte de caso de baja incidencia. - Revisión de la literatura. MATERIAL: Femenina de 17 años, con antecedente de un aborto un año previo al embarazo actual. Cursa con embarazo normoevolutivo. Durante ultrasonido de rutina se detectan datos de Retraso de crecimiento intrauterino (29.6 SDG por FUR y de 26.3 por US), oligohidramnios (ILA 7.9), anemia fetal, placentomegalia así como múltiples quistes de 13 mm en promedio en mesénquima placentario sin flujo. Por lo que se inicia inductores de la maduración pulmonar. RESULTADOS: En el servicio de Anatomía Patológica, se recibió una placenta de 19 x 19 x 7 cm, con un peso de 450 gr. Por su cara fetal se observó un cordón trivascular de inserción central con vasculatura prominente, con pared de 0.2 cm y con datos de trombosis extensa. Por su cara materna se observaron cotiledones café obscuros de aspecto fragmentado con abundantes vesículas de 0.8 cm en promedio. El análisis microscópico mostró vellosidades de aspecto edematoso, sin datos de proliferación trofoblastica que alternaban con vellosidades de aspecto normal. CONCLUSIONES: En conclusión, la DMP es una patología rara que fácilmente puede ser confundida con una enfermedad molar. Sin embargo ante la presencia de una placentomegalia con un feto vivo con cariotipo normal, el uso de los valores séricos de la AFP y la hGCH, en combinación con los hallazgos de ultrasonido, permiten sospechar de una DMP. Y una vez confirmado el diagnostico por anatomopatologia, realizar la búsqueda intencionada de otras alteraciones fetales para descartar el Síndrome de Beckwith-Wiedeman. INSTITUCIÓN: Hospital General Dr. Manuel INSTITUCIÓN: UMAE #23, IMSS. Monterrey, Gea Gonzalez Nuevo León. ESTADO: DISTRITO FEDERAL 2do Congreso Anual de Patólogos Mexicanos • 21 León, Guanajuato • 2016 ESTADO: NUEVO LEÓN ESTADO: NUEVO LEÓN TC-25 TC-42 AUTORES: Dra. Hersilia Aide Hernandez Zamonsett, AUTORES: Dra. Lucía Alemán Meza, Dra. Oralia BarboDra. Gabriela Sofia Gomez Macias, Dra. Raquel Garza Guajardo, Dra. Oralia Barboza Quintana. za Quintana, Dra. Raquel Garza Guajardo. Dra. Gabriela Sofía Gómez Macías. TITULO: Adenosarcoma Mulleriano Con Diferenciacion Heterologa De Tipo Osteoide, Reporte De Un Caso. TITULO: Tumor De Cordones Sexuales De Tipo Tubulos Anulares Ovarico Bilateral Asociado A Hiperplasia De Celulas De Leydig: Reporte De Un Caso Y Revisión De La Literatura. INTRODUCCIÓN: El Adenosarcoma mulleriano pertenece al grupo de los tumores mullerianos mixtos, son neoplasias uterinas poco frecuentes, con elementos glandulares benignos y malignos del estroma que puede los cuales pueden tener diferenciación homóloga o heteróloga (cartílago, músculo estriado, hueso, etc.). OBJETIVOS: Paciente femenina de 49 años de edad, diabética tipo II, se presenta con sangrado irregular, dolor abdominal y sensación de cuerpo extraño en vagina. El examen ginecológico reveló un tumor que sobresale de la vagina (9x7cm), blando, liso con bordes dentados, a la palpación bimanual se evidencia un pedículo proveniente de cuello uterino, que al girar en sentido contrario a las manecillas del reloj, se reseca, sin sangrado. En especuloscopia la vagina mostró rastros de hemorragia, cuello del útero de 2 cm, indurado. El tumor fue enviado para su evaluación patológica. INTRODUCCIÓN: El tumor de cordones sexuales con túbulos anulares es una neoplasia ovárica benigna rara (6% de los tumores de cordones sexuales) y únicamente 1% de todas las neoplasias ováricas. Hasta un 30% de ellos se asocia al síndrome de Peutz Jeghers. Otras asociaciones son con el adenoma maligno del cérvix y con el síndrome de Turner. OBJETIVOS: Presentamos el caso de una mujer de 71 años de edad con historia de sangrado transvaginal de 2 meses de evolución. El ultrasonido que revela un pólipo endometrial, asi como quistes ováricos bilaterales el derecho de 5.9 x 3.9 cm y el izquierdo de 2 x 1.8 cm. El Ca19-9 elevado en 66 U/mL (normal 0-30) y el Ca 19.9 en 11 U/mL (normal 0-37). MATERIAL: Se realizó histerectomía con salpingooforecMATERIAL: Macroscópicamente es una lesión polipoide friable de 9x7x4cm; superficie externa ulcerada. A la seccion heterogéneo, carnoso, de color rosado a gris marrón. A la microscopia compuesto por componente glandular endocervical y un componente de estromal sarcomatoso. El componente estromal presenta un índice mitótico con 3 mitosis/10 campos. Se identificaron áreas osteoides heterólogas entre el tejido con grandes áreas de necrosis. RESULTADOS: Se realizó un diagnóstico de adenosarcoma Mülleriano con elementos heterólogos osteoides. CONCLUSIONES: Sólo el 2% de MA se presenten en el cuello uterino, con menos de dos docenas de casos documentados hasta el momento en la literatura hasta el año 2014. La edad media de aparición es de 31 años (rango 11-65 años). El diagnóstico diferencial incluye benigna (adenofibroma, pólipo endocervical atípico, y adenomioma del cuello uterino) y malignos (adenosarcoma uterino con participación secundaria del cuello del útero, carcinosarcoma, y rabdomiosarcoma embrionario. La aparicion de elementos heterólogos se ha relacionado con un peor pronóstico. INSTITUCIÓN: Servicio De Anatomia Patologica Y Citopatologia, Hospital Universitario “Dr. Jose Eleuterio Gonzalez” tomía bilateral. RESULTADOS: El útero presentó un pólipo endometrial de 1.3 cm, el ovario derecho de 8 x 5 cm, quístico con pequeños nódulos en su cara interna, el ovario izquierdo de 3 x 1 cm quístico. Microscopicamente se observó una neoplasia formadora de patrones de túbulos anulares simples y complejos. Con túbulos en forma de anillos compuestos por células con núcleo orientado periféricamente y alrededor de un cuerpo hialino central, con una zona citoplasmica anuclear formando el componente mayor del anillo. Los túbulos complejos se observan formando estructuras redondas compuestas de anillos intercomunicantes orientados alrededor de cuerpos hialinos. Así como una proliferación adyacente de células de Leydig. CONCLUSIONES: Se realizó inmunohístoquímica para inhibina y calretinina, las cuales fueron positivas. A la paciente se le considera en curación sin tratamiento adyuvante. Se trata de un tumor raro con una importante asociación al síndrome de Peutz-Jeghers y al adenoma maligno del cérvix (por lo cual debe muestrearse extensamente el espécimen). Asi como la importancia del diagnóstico diferencial entre un tumor de Sertoli o de la granulosa con los que puede compartir características, pero este tiene una histopatología única con los clásicos túbulos repetitivos. 2do Congreso Anual de Patólogos Mexicanos • 22 León, Guanajuato • 2016 INSTITUCIÓN: Servicio de Anatomía Patológi- INSTITUCIÓN: Hospital Universitario “José ca del Hospital Universitario “Dr. José Eleute- Eleuterio González” rio González” de la Universidad Autónoma de Nuevo León. Monterrey N.L. ESTADO: NUEVO LEÓN ESTADO: NUEVO LEÓN TC-83 TC-82 AUTORES: Dr. Gabriel Juan Mandujano Alvarez, Dra. AUTORES: Dra. Med. Oralia Barboza Quintana, Dra. Med. Raquel Garza Guajardo, Dr. Alvaro Barbosa Quintana, Dra. Gabriela Sofía Goméz Macias Meztli Guadalupe Gómez Morales, Dra. Clara Magdalena Martínez Hernández, M. en E.M. Dra. Yaneth Pérez Méndez, Dr. Gabriel Manuel Mandujano Vera. TITULO: CONCORDANCIA DE LA INDICACION TITULO: Carcinoma mucinoso de ovario con DE HISTERECTOMÍA OBSTÉTRICA CON EL nódulo mural con Carcinoma Anaplásico y ESTUDIO HISTOPATOLÓGICO EN EL HOSPIAdenoma Adrenal a nivel de ligamento ancho: TAL REGIONAL DE ALTA ESPECIALIDAD DE Reporte de un caso LA MUJER. TABASCO. INTRODUCCIÓN: Los tumores mucinosos representan un 10-15% de los tumores ovaricos.. Los tumores mucino- INTRODUCCIÓN: La Histerectomía Obstétrica (HO) es sos del ovario ya sean benignos, malignos o borderline pueden contener nódulos murales. Los nódulos pueden presentarse con histología benigna o maligna, los benignos como sarcoma- like o leiomioma; mientras los malignos, son de tipo carcinoma anaplásico, sarcoma o carcinosarcoma. OBJETIVOS: El objetivo es presentar el caso de una paciente femenina de 49 años de edad que se le diagnosticó un carcinoma mucinoso de ovario derecho con áreas de carcinoma anaplásico con cápsula íntegra. MATERIAL: Se recuperó la información de la paciente, junto con su historial clínico, así como su material de laminillas y bloques de parafina para estudiar a fondo el caso y documentarlo. RESULTADOS: Se recibió anexo derecho con un tumor en ovario derecho que pesa 13,650 gramos y que mide 38 x30x30 cm, con cápsula íntegra de donde sale material hemático y en su interior presentaba áreas sólidas, heterogéneas que miden 10x7x3 cm. A nivel del ligamento ancho un tumor de 15 gr y de 3 cms de color amarillento separado del ovario. A nivel histológico y con marcadores de inmunohistoquímica se logró diagnosticar un carcinoma mucinoso de ovario derecho con características intestinales y endocervicales y con áreas de carcinoma anaplásico de 38 cm de diámetro mayor con cápsula íntegra y un adenoma adrenal izquierdo de 3 cm de diámetro mayor originado en restos adrenales en el ligamento ancho. CONCLUSIONES: En cuestión de las neoplasias mucinosas de ovario Es necesario el análisis histológico e inmunohistoquímico minucioso de los nódulos murales para la determinación del tratamiento y el pronóstico. Así mismo sólo está descrito un caso en la literatura mundial un adenoma adrenal originado en el ligamento ancho. una cirugía urgente la cual se debe considerar como última medida para controlar la hemorragia obstétrica cuando el tratamiento conservador no ha sido eficaz, se necesitan medidas más radicales para evitar el choque hipovolémico, ya que la hemorragia obstétrica es la responsable del 2-11% de las muertes maternas alrededor del mundo. OBJETIVOS: Objetivos específicos. 1. Describir las características clínicas de las pacientes con histerectomía obstétrica. 2. Identificar los factores de riesgo asociados a histerectomía obstétrica. 3. Identificar la relación entre la indicación de la histerectomía y el estudio histopatológico. MATERIAL: El estudio realizado es de tipo retrospectivo, transversal y observacional, donde se analizaron en el Laboratorio de Patología del Hospital Regional de Alta Especialidad de la Mujer en Villahermosa, Tabasco; las piezas uterinas producto de histerectomía posparto. Durante el periodo del 1° de Enero del 2010 al 31 de Diciembre 2013. El universo estuvo conformado por 127 pacientes atendidas vía vaginal, cesárea o LAPE por ectópico. RESULTADOS: Los hallazgos histopatológicos se clasificaron en los grupos de: atonía uterina, desgarro/ruptura uterina, embarazo ectópico, hemorragia/hematoma desecante, placentación anormal, útero con cuadro infeccioso, útero obstétrico sin cambios anatómicos. De acuerdo a los valores establecidos por Landis y Koch, se obtuvo una concordancia moderada con un valor de Kappa de 0.481 y una p=0.000. Por lo que se acepta la hipótesis alterna Hi: Existe concordancia entre el diagnóstico de indicación de histerectomía obstétrica con el estudio histopatológico. CONCLUSIONES: Las complicaciones fueron choque hipovolémico, hemorragia, hematoma y lesión vesical. El objetivo es lograr su prevención, así como protocolizar medidas alternativas a la histerectomía cuando sea inevitable 2do Congreso Anual de Patólogos Mexicanos • 23 León, Guanajuato • 2016 su desarrollo. La elevada frecuencia de placentación anormal, los datos relacionados con el tono uterino y con desgarros de este órgano, constituyeron las principales causas de histerectomía posparto, reportadas a nivel histopatológico. La indicación de histerectomía por clínica y el estudio histopatológico presentaron una concordancia buena. INSTITUCIÓN: Hospital Regional del Alta Especialidad de la Mujer. Secretaria de Salud. Villahermosa, Tabasco. Universidad Juárez Autónoma de Tabasco. ESTADO: TABASCO dermicos). CONCLUSIONES: En contraste con el testículo, donde la combinación más común es el de carcinoma embrionario y teratoma, en el ovario, tumor del saco vitelino (YST) y disgerminoma representa una mezcla más común en la familia de los tumores de células germinales mixtos, lo que representa 11 de 30 tumores en una serie. Hay casos reportados de quistes dermoides con un componente de tumor de senos endodermicos, sin embargo no se encontraron casos reportados, en los que hubiera un teratoma maduro, con áreas tan complejas como el que observamos, alternando con tumor de senos endodermicos. INSTITUCIÓN: Servicio de Anatomia Patologica y Citopatologia, Hospital Universitario “Dr. Jose Eleuterio Gonzalez” TC-123 AUTORES: Dra. Hersilia Aide Hernandez Zamonsett, ESTADO: NUEVO LEÓN Dra. Gabriela Sofia Gomez Macias, Dra. Oralia Barboza Quintana, Dra. Raquel Garza Guajardo. TITULO: Tumor germinal mixto ovari- TC-132 co con componente de teratoma maduro AUTORES: ANGELICA RODRIGUEZ, Dra. Mercedes y senos endodermicos. Reporte de Caso. Hernández González INTRODUCCIÓN: Los tumores de células germinales del ovario malignos representan aproximadamente el 1-2% de todos los tumores malignos de ovario, aparecen en mujeres jóvenes, con un pico de incidencia alrededor de los 20 años. TITULO: “Análisis clínico-patológico de sarcomas uterinos en muestras quirúrgicas del Hospital General de México “Dr. Eduardo Liceaga” OBJETIVOS: Femenina de 27 años con dolor y aumento de perimetro abdominal y saciedad temprana.En exploracion bimanual se palpa tumor anexial derecho,movil, irregular. Tac reporta tumor pelvico, redondeado, de 11.5x8.4cm con delgada capsula, contenido heterogeneo y calcificaciones. CA125:757 u/ml, CA19-9:50.2 u/ml. Se realiza salpingooforectomia izquierda. MATERIAL: Se recibe tumor ovárico de 12x9x5cm, de 450 gramos y oviducto de 7x0.7x0.7cm. Tumor ovárico cubierto por capsula lisa, café claro y vasos congestivos. A la sección heterogéneo, con áreas complejas de consistencia pétrea y color café blanquecino, alternando con áreas blandas, solido- quísticas de color café claro, con presencia de material de aspecto adiposo y áreas extensas de necrosis, se realizan cortes histológicos. RESULTADOS: Microscópicamente se observa un componente maduro con tejido oseo, cartilaginoso, epitelio respiratorio, tejido dérmico y epidérmico, alternando con áreas con estroma hipocelular y mixoide, presencia de células tumorales con citoplasma claro, nucleo hipercromatico , irregular, nucléolo visible y una ligera actividad mitótica, presencia de cuerpos de schiller- duval, estas áreas mostraron positividad para alfafetoproteina, y fueron negativas para cd30, ck7 y ck20. se diagnostica como tumor germinal mixto (70% teratoma maduro, 30% tumor de senos endo- INTRODUCCIÓN: Los sarcomas uterinos son un gru- po diverso de tumores, representan 3-7% de las neoplasias uterinas malignas. El diagnóstico diferencial entre los subtipos de sarcomas uterinos se basa en parámetros histológicos, los cuales pueden ser subjetivos y poco reproducibles, por lo que el uso de un panel amplio de IHQ ayuda en la clasifican de subtipos histológicos de sarcomas uterinos. OBJETIVOS: Evaluar el uso de un panel amplio de inmunohistoquímica de utilidad en el diagnóstico de sarcomas uterinos y conocer su comportamiento clínico-patológico. MATERIAL: Se realizo un estudio retrospectivo, descriptivo, transversal no experimental. Se analizaron los casos de sarcomas uterinos recibidos en el servicio de patología quirúrgica del Hospital general de México en el periodo comprendido del 1/1/09 al 30/6/15. Se estudiaron las características clínico-patológicas y se les realizó IHQ: AML, CD10, CKAE1/AE3, RE, RP, Vimentina, p53 y ki67. RESULTADOS: Se captaron un total de 99 casos, de los cuales se excluyeron 33 posterior a la revisión de material histológico y expediente clínico. Dentro de la reevaluación se reclasificaron 5 casos de sarcoma uterino. La edad y tamaño promedio en la muestra fue de 53 años y 8.8 cm, 2do Congreso Anual de Patólogos Mexicanos • 24 León, Guanajuato • 2016 el 67% de la muestra exhibió invasión miometrial mayor al 50%, el 25% de la muestra exhibió invasión al cérvix, l 44% de la muestra presentó invasión angiolinfática y el 66% de la muestra se diagnóstico en etapa histológica I. Dentro de los subtipos histológicos el 30% correspondió a leiomiosarcoma, el 33% a carcinosarcoma, el 20% a SEE, el 11% a adenosarcoma y el 6% a adenofibroma. CONCLUSIONES: Los sarcomas uterinos son un grupo heterogéneo de tumores, los cuales son difíciles de abordar debido a su amplio espectro histológico. Al igual que en la bibliografía, en el HGM se tiene una prevalencia del 6% de sarcomas uterinos. Las características clínico-patológicas expuestas en este trabajo coinciden con lo reportado en la literatura. Aunque ninguno de los marcadores presentados en este estudio son específicos para un subtipo de sarcoma, la utilización de un panel amplio de IHQ puede ayudar en la correcta clasificación de subtipos específicos de sarcomas. Las células forman nidos sólidos con distribución en empalizada de las células periféricas y retracción del estroma circundante. Estas células fueron positivas a cromogranina y sinaptofisina. El diagnóstico fue de tumor carcinoide insular. CONCLUSIONES: Los carcinoides son tumores endocrinos asociados a menudo a un síndrome característico. Siempre son unilaterales y, si es bilateral, hay que considerar que es metástasico y buscar el tumor primario, el diagnóstico diferencial se debe hacer con el tumor de células de la granulosa (los cuerpos característicos de Call-Exner se pueden confundir con un acino carcinoide), tumor de células de Sertoly-Leydig, Si el tumor está limitado a un solo ovario, la supervivencia parece ser excelente con tratamiento quirúrgico y deben ser tratados como tumores ováricos de baja malignidad. INSTITUCIÓN: HOSPITAL GENERAL DE LEÓN SSA INSTITUCIÓN: Hospital General de México, ESTADO:GUANAJUATO “Dr. Eduardo Liceaga” ESTADO: ZACATECAS TC-159 AUTORES: Dra. María del Carmen Vázquez Castro, Dra. TC-144 AUTORES: Dr. Mario Murguía Peréz TITULO: TUMOR CARCINOIDE DE OVARIO INTRODUCCIÓN: El tumor carcinoide de ovario es raro (1%) es una neoplasia de crecimiento lento, se origina en células del sistema neuroendocrino, se ha descrito en cualquier órgano del endodermo primitivo. Pueden permanecer sin diagnóstico durante meses o años antes de dar metástasis. Según el patrón histológico se divide en: insular, trabecular, estromal, mucinoso y asociados o no a teratoma maduro Karla Judith González Colunga, Dr. Erik Noé Gudiño Ruiz, Dr. Sergio Ravindranath Domínguez Cortes, Dr. Antonio Isaías Rocha Herrera. TITULO: CORANGIOMA GIGANTE Y CORANGIOSIS PLACENTARIA, REPORTE DE UN CASO Y REVISIÓN DE LA LITERATURA. INTRODUCCIÓN: El Corioangioma es un tumor no tro- caso de tumor carcinoide de ovario foblástico de incidencia real desconocida, se asocia con complicaciones materno-fetales cuando su tamaño es mayor de 4 cm. La Corangiosis es una hipervascularidad vellosa capilar. Ambos son causa de mortalidad perinatal, se asocian a condiciones de hipoperfusión y decremento de oxígeno, lo que puede explicar que se encontraran en una misma placenta. MATERIAL: Paciente femenina de 26 años de edad sin OBJETIVOS: Se describe el caso de una paciente que OBJETIVOS: Presentación anatomopatologica de un antecedentes personales patológicos de importancia, quien acude con clínico por aumento del perímetro abdominal sin otros síntomas agregados, confirmando por ultrasonido, tumoración quistica en ovario, con elevación de CA 125 de forma discreta, se somete a laparotomia exploradora. RESULTADOS: Macroscópicamente se observó una tumoración con dimensiones de 17 x 17 x 16.5 cms de consistencia renitente,superficie lisa blanquecina al corte quística con nódulo de 11 x 9 x 9 cms de color amarillo anaranjado, granular. Microscópicamente se observan células redondas y cilíndricas con núcleos ovales, regulares y uniformes, ausencia de nucleolos y excepcionales inclusiones intranucleares citoplasmáticas; el citoplasma celular es eosinófilo. presentó Diabetes Gestacional y su hijo detención del crecimiento y Anemia aguda por Transfusión Feto-Materna secundarios a Corangioma gigante y Corangiosis placentaria. Su incidencia real se desconoce por la baja solicitud de estudio histopatológico de las placentas. Se realiza revisión de la bibliografía. MATERIAL: Primigesta de 36 años portadora de diabetes gestacional bien controlada. Inició su control prenatal desde el 1er trimestre, en la semana 34 se detecta detención del crecimiento, Acude a urgencias por referir hipomotilidad fetal, durante su revisión se realiza registro tococardiográfico que mostró desaceleración hasta 65 x min con recuperación lenta, el Ultrasonido muestró oligohidramnios. Se decide la interrupción mediante cesárea. 2do Congreso Anual de Patólogos Mexicanos • 25 León, Guanajuato • 2016 RESULTADOS: Se obtuvo recién nacido único vivo, sexo masculino, 37 semanas, pesó 2300 gr, APGAR 7/8, talla 47 cm. Presentó dificultad respiratoria severa, requirió intubación e internamiento en terapia, su hemoglobina de 3 gr/dl y se integró el diagnóstico; Anemia severa por Transfusión Feto-Materna. Egresó por mejoría. El estudio histopatológico mostró; Un Corangioma con patrón angiomatoso de 5.5 cm y Corangiosis en áreas adyacentes. Se identificaron 2 dilataciones venosas en el cordón de 6 y 5 cm de longitud por 1 cm de diámetro a 2.5 y 10 cm de su inserción. Se considera que el tumor y las dilataciones venosas secuestraron aproximadamente 59 ml de sangre fetal, explicando la anemia aguda del RN. CONCLUSIONES: Ambas entidades son causa de Mor- talidad Perinatal y complicaciones obstetricas agudas. Aunque su etiología es desconocida, hay diversos factores maternos, fetales y placentarios implicados en su desarrollo, en los que existe un estado de hipoperfusión placentaria crónica y la transferencia de oxígeno al feto está disminuida, lo que explicaría su presencia en la misma placenta. En nuestro caso se asoció a Diabetes Gestacional y malformaciones del cordón. Aunque el tumor no fue detectado previamente por ultrasonido, Se considera que el tumor y las dilataciones venosas secuestraron aproximadamente 59 ml de sangre fetal, explicando la anemia aguda del RN.Sobrevivió gracias a la atención oportuna REFERENCIAS: 1.-Ramírez Arreola L, Nieto Galicia, Gómez García E, Cerda López JA. Corioangioma gigante y sus complicaciones perinatales. Reporte de un caso. Ginecol Obstet Mex 2007; 75: 104-10. 2.-Pérez-García GE, Sierra-Avendaño JA, Rangel-Navia E, Fuentes-Porras JS. Corangioma placentario: enfoque clínico-patológico de un caso descrito en Colombia. Ginecol Obstet Mex 2013; 81: 109-114. 3.-González Santacruz M., Tarazona Fargueta J.L.,Alenda González C. y Jiménez Cobo B. Muerte perinatal en una gestación múltiple asociada a corangiosis placentaria. An Pediatr (Barc). 2006; 65 (6): 626-42 4.-Shubhangi Vinayak A., Deepti Dayanand D., Case Reports Chorangiosis of Placenta. Bombay Hospital Journal 2009; 5(2): 251252 5.-Gonzalez AB, Viñuales AI, Juárez F. Corangioma placentario asociado a hemangiomatosis neonatal difusa. A propósito de un caso. Rev Esp Patol. 2010;43(4):217?219. 6.-Guschmann M. Henrich W, Entezami M, Dudenhausen J., Chorioangioma new insights into a well-Know problem. I. Results of a clinical and morphological study of 36 cases. J. Perinat Med 2003; 31(2):163-9. 7.-Acharya S., Pringle S., A Case Of Placental Chorioangioma With The Review Of Literature. The Internet Journal of Gynecology and Obstetrics. 2004 Volume 5 Number 1. INSTITUCIÓN: HOSPITAL GENERAL CADEREYTA DE PEMEX AUTORES: Sánchez-García Citlalli Edith, Peña-Zepeda Claudia. TITULO: DISPLASIA MESENQUIMATOSA PLACENTARIA INTRODUCCIÓN: La displasia mesenquimal placentaria es una rara entidad patológica, caracterizada por placentomegalia y malformaciones vasculares placentarias con apariencia macroscópica y ecográfica de una mola parcial de la cual con frecuencia es confundida. Se asocia a RCIU, óbito fetal, tumores fetales y síndrome de Beckwith-Wiedemann. OBJETIVOS: Conocer esta rara entidad patológica e identificar los hallazgos macroscópicos y microscópicos. MATERIAL: Mujer de 34 años de edad con embarazo de 32.2 sdg y diagnóstico de preeclampsia severa con resolución de embarazo médiate cesárea con evolución favorable sin complicaciones. La placenta es recibida en laboratorio de anatomía patológica para estudio histopatológico. RESULTADOS: La placenta tenia las siguientes características: monocorionica monoamniotica de 850g, disco corial de 16x15x5cm, la cara materna con cotiledones anfractuosos, heterogéneos, con áreas de aspecto esponjoso alternando con vesículas pequeñas de paredes delgadas, áreas hemorrágicas, vasos trombosados, la cara fetal café grisácea, lisa con trayectos vasculares visibles y prominentes, el aminios es café claro, opaco, el segmento de cordon umbilical de 13x1cm, trivascular. El tejido fue procesado y en los cortes histológicos se observa una displasia de estirpe mesénquimal constituida por una difusa degeneración hidrópica de las vellosidades coriales (grandes y edematosas) y ausencia de trofoblasto CONCLUSIONES: La displasia mesenquimatosa de placenta es una alteración vascular placentaria caracterizada por una placenta grande con dilataciones quísticas, de etiología desconocida, su diagnóstico definitivo es mediante estudio histopatológico de la placenta. Su prevalencia estimada es de 0.02 a 0.2%; sin embargo, su diagnóstico es subestimado debido a que no en todos los casos se envía la placenta a estudio patológico. INSTITUCIÓN: Hospital Central ¨Dr. Ignacio Morones Prieto¨, San Luis Potosí, SLP. ESTADO: SAN LUIS POTOSÍ ESTADO: NUEVO LEÓN TC-183 TC-172 AUTORES: Dra. Ana Karen Soto Sanudo, Dr. Aldo Antonio Alcaraz Wong 2do Congreso Anual de Patólogos Mexicanos • 26 León, Guanajuato • 2016 TITULO: Adenocarcinoma Endometrial Originado en un Adenomioma Extrauterino Parasalpingeario: Reporte de Caso INTRODUCCIÓN: Los adenomiomas son tumores be- nignos compuestos por músculo liso y endometrio. Estos tumores generalmente se desarrollan en el útero presentándose raramente en lugares extrauterinos. La degeneración quística es aun mas rara y se han reportado casos con adenocarcinoma endometrioide sin invasión estromal. OBJETIVOS: Presentación de Caso MATERIAL: Se recibe salpinge de 3cms de longitud la cual presenta tumoración nodular extramural de 2cms por 1.3cms de consistencia firme. A los cortes presenta formaciones quísticas las cuales miden 0.2 a 0.4cms en promedio, con paredes lisas. A la microscopia, se observa tumor circunscrito, compuesto por estructuras glandulares tipo endometrial revestidas por epitelio neoplásico y rodeadas por músculo liso. RESULTADOS: Se diagnostica un adenocarcinoma de tipo endometrioide grado I, no invasivo, intraquístico, de 0.6cms originado de un adenomioma extrauterino parasalpingeario de 2cms de diámetro mayor, sin permeación vascular ni necrosis. CONCLUSIONES: Los adenomiomas extrauterinos son tumores poco frecuentes que pueden presentarse en el ligamento ancho, salpinge, ovario y otras localizaciones. Se han descrito dos teorías para determinar su etiología. La primera es un defecto en la fusión del conducto Mulleriano y la segunda es una transformación del mesenquima subcelomico. En nuestro caso, se observa restos mesonefricos los cuales pudieran tener una asociación con el desarrollo de esta neoplasia. INSTITUCIÓN: Centro Medico Nacional de Occidente Instituto Mexicano del Seguro Social ESTADO: JALISCO TC-192 AUTORES: Dr. Saulo Mendoza Ramirez, Dr. Felipe de Jesus Navarro Cordoba, Dr. Adrian Antonio Lopez Vera. TITULO: Rotura uterina espontanea y hemoperitoneo como primera manifestacion de tumor mixto mülleriano maligno. Informe de un caso de autopsia. INTRODUCCIÓN: La rotura uterina (RU) es un evento que amenaza la vida, dando lugar a complicaciones graves que pueden llevar a la muerte. La incidencia es de 5.1/1000 alumbramientos, es más frecuente en mujeres multíparas, y el riesgo aumenta si se asocia a ruptura de cicatriz uterina. El riesgo tambien esta presente en mujeres con anomalias del ducto mülleriano, otras causas menos comunes son la gangrena gaseosa uterina y las neoplasias. OBJETIVOS: Las tumores uterinos son neoplasias frecuentes en las mujeres, y pueden derivarse del endometrio, estroma endometrial, músculo liso y peritoneo. El tumor mixto mülleriano maligno (TMMM) es una neoplasia bifásica compuesta de elementos epiteliales y mesenquimatosos malignos distintivos y separados, pero mezclados, y acontece cerca de la mitad de todos los sarcomas uterinos. La presentacion con RU y hemoperitoneo es muy rara, por lo que presentamos un caso con esa asociacion. MATERIAL: Mujer de 75 años, sin antecedentes de importancia, con historia de 3 semanas con síndrome anémico descompensado y dificultad respiratoria. En la exploracion fisica se observo palidez generalizada y se palpo tumor abdominal. Los marcadores tumorales fueron negativos. El US mostro un tumor pelvico probablemente dependiente de anexos. Subitamente presento datos de choque hipovolemico y fallecio. RESULTADOS: En el estudio de autopsia se encontro hemoperitoneo de 4000 ml, con peritonitis generalizada de asas intestinales. El utero presento ruptura de la pared en la cara anterior del cuerpo y en el fondo, donde se observo un tumor dependiente del utero que midio 16x14x12cm, al corte de consistencia blanda, mal delimitado, de superficie heterogenea, con areas necrohemorragicas. Tambien se observaron varias lesiones nodulares intramurales y subserosas con calcificaciónes. Microscopicamente se identifico una neoplasia maligna con extensas areas de necrosis, con celulas fusiformes con alto grado nuclear y conteo mitosico elevado. Ademas se identificaron areas con formacion de osteoide y cartilago maligno, y celulas pleomorficas con nucleos grandes e irregulares. En las areas adyacentes al sitio de ruptura se observo necrosis asociada con celulas neoplasicas. Se concluyo como TMMM con componentes heterologos. CONCLUSIONES: La RU es una condicion grave que ocurre con mayor frecuencia en mujeres embarazadas y asociadas a uso inapropiado de oxitocina. En cuanto a las neoplasias, se asocian con mayor frecuencia a leiomiomas. Otras neoplasias que pueden ocasionar RU, con mucho menos frecuencia, son las neoplasias trofoblásticas gestacionales, carcinoma endometrial y leiomiosarcoma. La asociacion con TMMM es extraordinariamente rara. Nuestro caso reune las caracteristicas clinicas e histopatologicas de un TMMM con RU y hemoperitoneo, causantes de la muerte de la paciente. Ante la presencia de cualquier neoplasia uterina es necesario descartar la presencia de esta complicacion. INSTITUCIÓN: Unidad Medica de Alta Especialidad N° 1 IMSS Leon, Hospital Medica Campestre, Hospital General de Mexico, Hospital General Regional IMSS Nº 200, Hospital 2do Congreso Anual de Patólogos Mexicanos • 27 León, Guanajuato • 2016 General Regional IMSS Nº 196 ESTADO: GUANAJUATO PATOLOGÍA DE CABEZA Y CUELLO CONCLUSIONES: Los melanomas que se originan en la cavidad nasal y en los senos paranasales, corresponden al 1% de todos los melanomas. Los patrones microscópicos más comúnmente encontrados son:células pequeñas,fusiformes,epitelioides y pleomórficas. La actividad de unión está presente solo en un tercio de los casos, debido a la ulceración de la superficie. En el caso de melanomas amelanóticos es importante la realización de Fontana Masson para detectar la presencia de melanina en el citoplasma de las células tumorales, o el uso de técnicas de Inmunohistoquímica como proteína S-100, Melan- A y HMB-45. Mampara: 18 A INSTITUCIÓN: HOSPITAL CIVIL DE CULIACÁN Fecha y hora Instalación: 2 mayo 7:30 hrs. Fecha y hora retiro: 2 mayo 13:00 hrs. ESTADO: SINALOA TC-3 Mampara: 18 B AUTORES: DINORAH MARITZA BÁTIZ, Dra. Ana Guadalupe Ruelas Perea, Dra. Ana Cristina Benitez Morales, Dra. Ana Isabel Valdez Flores, Dra. Sara Rodríguez Carrillo, Dr. Eri Peña Martínez, Dr. Efrén Rafael Ríos Burgueño TITULO: MELANOMA EN CAVIDAD NASAL: LOCALIZACIÓN INFRECUENTE. Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. TC-10 AUTORES: Martha Lilia Tena-Suck, Miguel Fdo. Salazar INTRODUCCIÓN: El melanoma de cavidad nasal y se- Morales, Citlaltepelt Salinas Lara, Noemi Gelista. Carlos Sánchez Garibay nos paranasales es una neoplasia maligna extremadamente rara que representa menos del 1% de los tumores de esta localización. Su sintomatología es inespecífica, lo que dificulta el diagnóstico. Su pronóstico es malo, con bajas tasas de supervivencia. TITULO: leiomiosarcoma nasal con diferenciación rabdomioblastica (tumor tritón), reporte de un caso OBJETIVOS: Presentamos el caso de femenino de 44 OBJETIVOS: Leiomiosarcomas nasales y paranasales años de edad que acudió a consulta por epistaxis de tres semanas de evolución y tumor en fosa nasal izquierda, obstruyéndola por completo. son lesiones extremadamente raras. Se reportó como tumor tritón a la combinación de tumores de vaina nerviosa más células con diferenciación rabdomioblastica. MATERIAL: Se realizó biopsia excisional; la muestra fue MATERIAL: En este caso presentamos un leiomiosarco- enviada al departamento de Anatomía Patológica y se fijó en formol al 10%. Los fragmentos tomados fueron incluidos en bloques de parafina, se realizaron cortes de 3 a 4 micras y se tiñeron con Hematoxilina y Eosina. RESULTADOS: Al examen macroscópico se observó un fragmento polipoide de tejido de 3.5x3.3x2.5cm, así como múltiples fragmentos irregulares que midieron en conjunto 2x1.4cm, café oscuros, con adherencias de fibrina y consistencia semifirme. Microscópicamente, se identificó epitelio respiratorio de revestimiento, ulcerado, cuyo estroma subyacente presentó una lesión nodular sólida, de células uniformes que se disponen alrededor de los vasos sanguíneos. Las células son de citoplasma escaso, eosinófilo, con núcleos grandes, irregulares, de cromatina fina, con nucléolo prominente eosinófilo, algunas con presencia de pigmento en su citoplasma; con hasta 24 mitosis en 10 campos de alto poder. ma con diferenciación rabdomioblastica. RESULTADOS: Mujer de 74 años con hipertensión, diabetes mellitus de 10 años, fumadora desde los 15 por 21 años. Inicio con hiposmia, y aumento progresivo de la nariz, hasta llegar a ser una lesión de le deformaba la cara, además presentaba perdida de la visión en forma progresiva y cefalea así como hipoacusia. A la exploración física mostraba una lesión nasal y una lesión en oído derecho. Se biopsiaron ambas lesiones y se reportaron como leiomiosarcoma. Posteriormente se realizó exesión parcial de la lesión. Histológicamente se identifica una neoplasia de células largadas fusocelulares con ocasionales figuras de mitosis, con áreas hialinizadas y presencia de células grandes eosinofilas con núcleos grandes redondos con nucléolo prominente, estas células mostraban citoplasma abundante con áreas de aspecto desgarrado fibrilar. La IHQ fue positiva para vimentina, desmina en las células alargadas y el antígeno de musculo liso, miogenina y miocina, siendo positiva 2do Congreso Anual de Patólogos Mexicanos • 28 León, Guanajuato • 2016 en las células grandes eosinofilas y CAM 5.2 y negativas para EMA, CD34, S100, CK20, ENE, sinaptofisina, Neu-N, PSA, CDX-2, CD45, GFAP, and C-KIT (CD117). El índice Mib-1(Ki-67) fue de 5%. CONCLUSIONES: Las células grandes eosinofilas semejan neuronas por el nucléolo prominente sin embargo, se descarta la posibilidad de diferenciación neuronal al ser negativas para ENE, sinaptofisina y Neu-N. H Y E Se Identifico Una Lesión No Delimitada Que Sustituía Parcialmente A La Glándula Salival Con Un Patrón De Crecimiento Difuso Con Formaciones Quísticas Separadas Por Finos Septos Fibrovasculares Y Abundante Moco En El Interior De Los Quistes Con Nidos Solidos De Células Epiteliales Flotando En El Moco.las Células De Los Nidos Son Redondas A Cubicas Con Perdida De La Relación Núcleo Citoplasma,Nucleomegalia Con Presencia De 1 A 2 Nucléolos, Cromatina Fina. En Otros Campos Se Logro Apreciar Células Neoplásicas Dispuestas Alrededor De Los Nervios. con Estos Hallazgos El Diagnostico Inicial Fue De Un Adenocarcinoma Mucinoso. INSTITUCIÓN: Departamento de Neuropatología, Instituto Nacional de Neurología y Neurocirugía. Ciudad de México. Hospital General de CONCLUSIONES: De la revisión de esta patología poMéxico. ESTADO: DISTRITO FEDERAL Mampara: 19 A Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. demos concluir que el carcinoma mucinoso primario de la glándula parótida es una entidad poco frecuente, representa un reto diagnostico para el patólogo, no solo por lo raro en esta localización, además por que las características morfológicas pueden simular un amplio espectro de lesiones , todas y cada una de ellas tienen una evolución y pronostico diferente que enfatiza la necesidad de un diagnostico correcto por el impacto para el enfoque terapéutico y seguimiento de la paciente. TC-37 INSTITUCIÓN: HOSPITAL DE ONCOLOGIA CENTRO MEDICO NACIONAL SIGLO XXI IMSS AUTORES: Itzel María Montoya, Dra. Ivone Cuadra Gar- ESTADO: DISTRITO FEDERAL cia Medico Adscrito Al Servicio De Patologia Del Hospital De Oncologia Del Centro Medico Nacional Siglo Xxi Del Imss TITULO: Carcinoma Coloideo Primario De Glandula Parotida. Entidad Poco Frecuente. Reporte De Caso. INTRODUCCIÓN: El carcinoma coloide primario de la glándula parótida es una neoplasia maligna poco frecuente que se presenta en personas mayores de 5o años. La oms define a este tumor como una lesión compuesta de células epiteliales dispuestas en grupos inmersos en grandes lagos extracelulares de mucina, el componente de mucina por lo general ocupa mas del 50 % del tumor. Mampara: 19 B Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. TC-87 AUTORES: Dra. Lucia Nereyda Melgarejo Ortega. Dr. Miguel Ángel Paz González. Dr. Luís Sepúlveda Rodríguez. Dra. Gloria Madrid Valero, Dra. Marcela Madrid Ballesteros, Dr. Horacio Decanini Arcaute. OBJETIVOS: Presentamos un caso de carcinoma coloide de la glándula parótida y revisión de esta patología ya que es un tumor poco frecuente, representa un reto diagnostico por su baja incidencia y por que histológicamente son simuladores de otros tumores que exhiben componente mucoide. TITULO: CARCINOMA PAPILAR CON PATRÓN FOLICULAR, VARIANTE ESCLEROSANTE DIFUSA INTRODUCCIÓN: Desde el año 2005 al 2015 en el MATERIAL: El servicio de anatomía patológica del hospital de oncología del cmn siglo xxi imss se recibió para estudio histopatológico el producto de parotidectomia superficial de lado derecho que midió 8.0 X 4.0 X 2.0 Cm,por la superficie externa era café claro, irregular con una zona nodular periférica, en la superficie de corte se identifico un nódulo delimitado de 2.5 X 2 x 1.8 Cm, blanco grisáceo, con aspecto mixoide, homogéneo y solido. RESULTADOS: En Los Cortes Examinados Teñidos Con Hospital Christus Muguerza de Alta Especialidad, se han realizado 190 tiroidectomías totales de las cuales se diagnosticaron 73 carcinomas papilares y solo 3 han sido de la variante esclerosante difusa. OBJETIVOS: Se presenta el caso de un femenino de 15 años con antecedente de hipotiroidismo. Inició a los 10 años con taquicardia, pérdida de peso, polifagia y aumento de volumen en el cuello. Tratada con Propanolol, Tiamazol, Iodo radiactivo y Levotiroxina. El motivo de ingreso fue un nódulo de 1.0 cm en el lado izquierdo del cuello, no móvil. 2do Congreso Anual de Patólogos Mexicanos • 29 León, Guanajuato • 2016 MATERIAL: Se realiza tiroidectomía total con radical de cuello bilateral. Se recibe en fresco lóbulo tiroideo izquierdo de 3.1 x 2.5 x 1.5 cm. A la sección, se identifica nódulo de 1.0 cm, café, con áreas blanquecinas, firme y delimitado. A los cortes por congelación se reporta ?Lesión papilar a menos de 0.1 cm del borde de resección?. Posteriormente se recibe en formol el lóbulo tiroideo derecho e istmo de 9.0 g y de 3.5 x 2.0 cm. A la sección es fibroso RESULTADOS: Se observan estructuras microfoliculares, revestidas por células de citoplasma acidófilo con núcleos irregulares, pseudoinclusiones intranucleares, barras de cromatina despulida y mitosis. Infiltran difusamente el resto del tejido tiroideo, presentando extensas zonas de fibrosis y esclerosis e incluso de manera focal en tejidos blandos peritiroideos. Mostró positividad para Galectina-3, Mesotelina y Citoqueratina 19. Se diagnosticó como carcinoma papilar con patrón folicular, variante esclerosante, multifocal, con 72 ganglios linfáticos negativos para metástasis, márgenes focalmente involucrados, invasión linfovascular presente y perineural no identificada. pTNM: pT3 pN0.? CONCLUSIONES: El carcinoma papilar constituye del 70 al 80% de todos los tumores tiroideos. Se han reconocido 14 variantes siendo la esclerosante difusa, una de las mas agresivas. Constituye el 2% de todos los carcinomas papilares de tiroides, se caracteriza por afectar a uno o ambos lóbulos sin formar una masa dominante. Histológicamente se observan pequeñas estructuras papilares dentro de espacios linfovasculares dilatados con metaplasia escamosa extensa, numerosos cuerpos de psamoma, infiltración linfocítica importante de manera difusa y fibrosis estromal. intestino delgado, divertículo de Meckel, tracto biliar, vesícula, páncreas, ombligo, apéndice, recto, conducto vitelino, conducto torácico y canal raquídeo. Este tipo de lesiones pueden presentarse de forma quística o sólida. OBJETIVOS: El objetivo es presentar el caso de una infante de 1 mes de vida que se presentó por aumento de volumen del piso de la boca y terminó siendo diagnosticada con esta entidad. MATERIAL: Se busco el expediente clínico de la infante, así mismo sus estudios paraclínicos y su historial quirúrgico para poder documentar el caso. RESULTADOS: Paciente femenina de 1 mes de edad que acude a consulta de otorrinolaringología por aumento de volumen en el piso de la boca. Los familiares relatan que la lesión esta desde el nacimiento y que era más fácil de visualizar al momento del llanto. Cabe destacar que la lesión no impedía a la paciente al momento del alimento. La impresión diagnóstica inicial fue de una glándula salival con hiperplasia, por lo que el servicio de otorrinolaringología decidió extirparla, por lo que a nuestro departamento de patología nos llegó una tumoración bien circunscrita de 3 cm que al seleccionarla salió líquido mucinoso de su interior y en la histología presenta epitelio gastrointestinal heterotópico. CONCLUSIONES: El diagnóstico del quiste gastrointestinal es eminentemente microscópico y su descripción sólo pretende ser un aporte al conocimiento de patologías poco frecuentes y enriquecer el diagnóstico diferencial de las lesiones estomatológicas. INSTITUCIÓN: Hospital Christus Muguerza INSTITUCIÓN: Hospital Universitario “José Alta Especialidad, Monterrey, Nuevo León Eleuterio González” ESTADO: NUEVO LEÓN ESTADO: NUEVO LEÓN Mampara: 20 B Mampara: 20 A Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. TC-120 Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. TC-128 AUTORES: Dra. Erika Gpe. Castañeda Angeles 1, Dra. AUTORES: BÁRBARA SÁENZ, Dr. Med. Marco Antonio Ponce Camacho, Dra. Med. Oralia Barboza Quintana. TITULO: Quiste Gastrointestinal Heterotópico: Reporte de un caso y revisión de la literatura. INTRODUCCIÓN: La presencia de epitelio gástrico aberrante en diversas zonas del tracto digestivo se encuentra bien documentada en la literatura, y es así como se describen heterotopias en epiglotis, esófago, pared gástrica, Sara Parraguirre Martínez 1, Dr. Héctor Manuel Prado Calleros 2. TITULO: Condroma (hamartoma condroide) de primer anillo traqueal. Reporte de caso. INTRODUCCIÓN: Los hamartomas de tráquea son poco frecuentes, crecen lentamente, no tienen potencial maligno, pueden obstruir la vía aérea, y es importante la detección temprana. En la broncoscopía son lesiones polipoides que pueden obstruir la vía aérea. El hamartoma tra- 2do Congreso Anual de Patólogos Mexicanos • 30 León, Guanajuato • 2016 queal también se ha denominado mesenquimoma benigno y hamartoma condroide. OBJETIVOS: Presentar un caso benigno de tráquea poco frecuente que se extirpó en etapa temprana. MATERIAL: Mujer de 54 años, con tabaquismo positivo de 19 años de evolución (media cajetilla al día). En julio de 2014 presentó faringodinia y disfonía; acudió a consulta de otorrinolaringología, realizaron laringoscopía que reportó lesión subglótica anterior de escasos milímetros, se decide vigilancia. En febrero de 2015 en el estudio control notaron aumento de tamaño y decidieron resecar primer anillo traqueal, se envía a Patología. RESULTADOS: Se recibe anillo traqueal de 3x1x0.3 cm, blanquecino, firme, aspecto mucoso, en el espesor de la parte anterior presenta lesión elevada de 0.6 cm, lisa, blanca. En el estudio histológico, es hipocelular con abundante matriz condroide y material mixoide rodeado de pequeños racimos de condrocitos, algunos binucleados y otros con núcleo redondo y grande, sin evidencia de anaplasia ni mitosis, la mucosa adyacente presenta inflamación crónica. Emitimos el diagnóstico de condroma de primer anillo traqueal sin lesión en bordes quirúrgico CONCLUSIONES: El condroma es más frecuente en cartílago cricoides (75%, incidencia de 0.07-0.2%), en tráquea hay escasos reportes y hasta la actualidad se han descrito 20 casos. La sintomatología depende de la localización, puede ser disfonía, obstrucción progresiva de vías aéreas superiores, disnea o disfagia. Se presenta en la tercera y séptima décadas de la vida, es más común en hombres. El diagnóstico diferencial incluye condrometaplasia y condrosarcoma de bajo grado, la presencia de calcificaciones sugiere cambios malignos. INSTITUCIÓN: Hospital General “Dr. Manuel Gea González”, 1. División de Anatomía Patológica, 2 División de Otorrinolaringología. ESTADO: DISTRITO FEDERAL INTRODUCCIÓN: El carcinoma de células claras, sin especificar, es una neoplasia epitelial con diferenciación ductal, en riñón, pulmón, ovario y cavidad oral. El primario nasal carece de características de aquellos de glándula salival. En 2005 se agregó a la clasificación de los tumores de cabeza y cuello (OMS). Existen pocos datos clínicos y rara vez se presenta en cavidad nasal. OBJETIVOS: Dar a conocer un caso de carcinoma de células claras en cavidad nasal, cuya localización es rara y en el que se pudo descartar neoplasia primaria en otro sitio. MATERIAL: Se revisó el expediente clínico e imagenológico del paciente en cuestión. Se realizó análisis morfológico de la resección del tumor en laminillas teñidas con hematoxilina y eosina, además de reacciones de inmunohistoquímica con CKAE1/3, CK 19, Cromogranina, sinaptofisina y CD10. RESULTADOS: Hombre, 47 años, obstrucción nasal, epistaxis, hiposmia-anosmia y exoftalmos derecho, 4 meses. Fosa nasal derecha lesión con extensión a la izquierda. TAC cráneo tumor en cavidad nasal, infiltra piso anterior. Maxilectomia derecha, sangrado 4000cc; estudio histológico de múltiples fragmentos, con neoplasia en nidos y septos fibrovasculares, células claras con nucléolo evidente. CKAE1/3 y CK19 positivas, cromogranina, sinaptofisina y CD10 negativas. Ki-67 30%. Se diagnosticó carcinoma de células claras; sugiriendo descartar neoplasia metastásica. TAC cráneo: tumor residual, desplaza la vaina óptica; descartó neoplasia primaria en TAC tóracoabdominopélvica. Actualmente quimio y radioterapia. CONCLUSIONES: Los adenocarcinomas de células claras primarios sinonasales son infrecuentes; por lo que es indispensable descartar neoplasia primaria en otro sitio para hacer el diagnóstico, principalmente riñón, pulmón y ovario. Por la baja frecuencia de la enfermedad y los pocos casos reportados, no se conoce el comportamiento y pronóstico de esta neoplasia. INSTITUCIÓN: Hospital Juárez de México ESTADO: ESTADO DE MÉXICO Mampara: 21 A Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. TC-147 OFTALMOPATOLOGÍA AUTORES: Dra. Ana Lirio Ramírez Ávila, Dra. Maria Elena Arroyo Mendoza, Dr. Juan Manuel García Salas, Dr. Del Bosque Mampara: 1 A Fecha y hora Instalación: 2 de mayo 13:00 TITULO: Carcinoma de células claras primario Fecha y hora retiro: 2 de mayo 18.30 nasal. Reporte de caso. TC-48 2do Congreso Anual de Patólogos Mexicanos • 31 León, Guanajuato • 2016 AUTORES: Dolores Ríos y Valles Valles, Ivette Hernández Ayuso, Abelardo A. Rodríguez Reyes, Héctor A. Rodríguez Martínez, Elizmara L. Aguilar Ayala, Ariel Ceriotto García, Paulina Nuñez Rivera de México. ESTADO: ESTADO DE MÉXICO TITULO: Hemangioma intraóseo primario de la órbita. Presentación de un caso. Mampara: 1 B INTRODUCCIÓN: El hemangioma intraóseo es una neoplasia benigna muy poco frecuente y representa menos del 1% de todos los tumores óseos. En más del 50% de los casos se presenta afectando vértebras o huesos del cráneo. El involucro del esqueleto facial y en particular los huesos que conforman la órbita es extremadamente raro. OBJETIVOS: Presentar los hallazgos clínicos, imagenológicos e histopatológicos de un caso de hemangioma intraóseo primario de la órbita en una mujer de 43 años MATERIAL: Mujer de 43 años que consulta con dolor en párpado inferior de 3 meses de evolución y masa sensible en reborde orbitario inferior derecho. En la exploración oftalmológica se palpó lesión indurada, no móvil, dolorosa, en esta localización acompañada de proptosis e hiperglobo derecho. El resto de la exploración se encontró normal. La TAC demostró lesión intraósea expansil heterogénea. Se realizó biopsia orbitaria con diagnóstico de osteosarcoma. RESULTADOS: En el estudio macroscópico se encontraron varios fragmentos de hueso (30 x 15 x 3 mm) de forma y tamaño variable, color café grisáceo y aspecto poroso. El estudio histopatológico mostró varios fragmentos de tejido compuestos por delgadas trabéculas de hueso compacto maduro entre las cuales se identificaron numerosos canales vasculares de amplias luces con contenido hemático y paredes irregulares, revestidos por células endoteliales planas. No se encontraron datos de malignidad. El diagnóstico histopatológico fue de hemangioma cavernoso intraóseo del piso orbitario. CONCLUSIONES: Toynbee en 1845 describió el primer caso de hemangioma intraóseo en los huesos del cráneo. El primer caso con afección orbitaria fue descrito por Rowbotham en 1942 y desde entonces sólo 45 casos han sido informados en la literatura. La patogénesis es incierta; algunos casos se han relacionado con un antecedente traumático. Afecta a mujeres entre la cuarta y quinta década y es más común en el reborde orbitario superior. Los estudios de imagen describen una lesión con aspecto en panal de abeja. Microscópicamente hay 2 tipos: Capilar y cavernoso. Este último es el tipo más frecuente. REFERENCIAS: Madge SN, Simon S, Abidin Z, Ghabrial R, Davis G, McNab A, et al. Primary orbital intraosseous haemangioma. Ophthal Plast Reconstr Surg 2009;25:37-41 INSTITUCIÓN: Asociación para Evitar la Ceguera en México I.A.P. Hospital “Dr. Luis Sánchez Bulnes”, Depto. Medicina Experimental,Facultad de Medicina, UNAM, Hospital General Fecha y hora Instalación: 2 de Fecha y hora retiro: 2 de mayo 18.30 mayo 13:00 TC-49 AUTORES: Dra. María de los Angeles Hernández Cueto, Dra. Elvira González Bojórquez, Dr. Arturo Carrasco Quiróz y Dra. Karla Verdiguel Sotelo TITULO: INMUNOFENOTIPO DE CORNEAS CON FINES DE TRANSPLANTE CON SEROLOGIA POSITIVA A TORCH. EXPERIENCIA DE CMN LA RAZA, IMSS INTRODUCCIÓN: El trasplante de órganos y tejidos se acompaña del potencial peligro de transmisión de enfermedades, por lo que la Asociación Americana de Bancos de Ojos (EBAA) y la FDA de EUA, así como el Servicio de Oftalmología han establecido un panel que incluye VIH, VHC, VHB, CMV, sífilis, herpes, toxoplasma y rubeola, por lo que las córneas positivas a cualquiera de estos marcadores no se trasplanta. OBJETIVOS: Describir el inmunofenotipo de córneas procuradas con fines de trasplante y reporte serológico positivo a TORCH, pero con características físicas apropiadas para transplante y así comprobar o descartar la presencia de estos microorganismos, pues cada córnea que se descarta, es la pérdida de oportunidad de un paciente de recuperar la visión. MATERIAL: Se estudiaron cinco donadores de córneas pareadas a través de microarreglos de tejidos (MAT) en la que se incluyeron 4 spots de tejido corneal avascular y 2 spots de limbo esclero-corneal vascular, así como 3 d control positivo para CMV, HV, Rubéola y Toxoplasma g. Previa comprobación de la integridad de todos los spots por histoquímica, se buscó la expresión con anticuerpos específicos contra CMV, HV y Rubeola, y Giemsa para Toxoplasma g. RESULTADOS: Las córneas examinadas con microscopía especular mostraban buena calidad para transplante, tanto en el conteo endotelial, como por su hexagonalidad y coeficiente de variación. Sin embargo, la serología de los donadores dieron positivo para TORCH, por lo que se destinaron para MAT y búsqueda con anticuerpos específicos contra los microorganismos de interés. En los cortes del MAT teñidos con histoquímica e inmunohistoquímica se efectuó análisis morfométrico de la expresión en color café. Dicho análisis arrojó expresión nula y definitiva en córnea 2do Congreso Anual de Patólogos Mexicanos • 32 León, Guanajuato • 2016 avascular y en limbo esclero-corneal en ambas córneas, tanto en núcleo como citoplasma de epitelio, lámina de Bowman, estroma, membrana de Descemet y endotelio, al compararse con los controles positivos. CONCLUSIONES: La expresión nuclear negativa en las córneas de MAT para CMV, HV y Rubeola indican que no replicación viral y de igual forma la expresión citoplásmica negativa para Toxoplasma g. indica la ausencia de infestación por el parásito. Por tanto los títulos serológicos deben evaluarse cuidadosamente, sobre todo porque la integridad clínica apoya la ausencia ante la búsqueda intencionada de estos microorganismos. INSTITUCIÓN: Laboratorio Central de Epidemiología, DLVIE, CMN La Raza, IMSS Servicio de Oftamología del Hospital General Dr. Gaudenco González Garza, CMN La Raza, IMSS ESTADO: DISTRITO FEDERAL Mampara: 2 A Fecha y hora Instalación: 2 de Fecha y hora retiro: 2 de mayo 18.30 RESULTADOS: Macroscópicamente la masa midió 21 x 18 x 17 mm, de forma nodular, color blanco grisáceo y consistencia media. Microscópicamente, estaba compuesta por nidos sólidos de células de apariencia epitelial con abundante citoplasma, muchos de ellos claros y núcleos redondos a ovales con numerosas mitosis típicas y atípicas. La tinción de PAS confirmó la presencia de glucógeno. Los nidos estaban rodeados por finos septos fibrovasculares, mezclados con extensas zonas de necrosis. Las reacciones de vimentina, proteína S-100, calponina, AE1/AE3 y CEA-M resultaron positivas en el citoplasma de las células neoplásicas. El diagnóstico final fue de CME de la glándula lagrimal. CONCLUSIONES: El CME es una neoplasia muy poco frecuente de la glándula lagrimal, macro y microscópicamente comparte características similares a las que se presentan en las glándulas salivales. El estudio de inmunohistoquímica es definitivo para confirmar su diagnóstico y para diferenciarlo de neoplasias como mioepitelioma, sarcomas, carcinoma epitelial-mioepitelial, plasmocitoma y otras neoplasias de células claras. El CME se considera un tumor de mediano o alto grado, generalmente cursa con recidivas frecuentes y da metástasis a ganglios linfáticos regionales. REFERENCIAS: Iida K, Shikishima K, Okido M, Sato S, mayo 13:00 TC-67 AUTORES: Abelardo A. Rodríguez Reyes, Héctor A. Rodríguez Martínez, Martín José González Benito, Dolores Ríos y Valles Valles, Ivette Hernández Ayuso, Elizmara Leslie Aguilar Ayala, Armando Medina Cruz. TITULO: Carcinoma Mioepitelial de la Glándula Lagrimal: Presentación de un caso INTRODUCCIÓN: El carcinoma mioepitelial (CME) tam- bién conocido como mioepitelioma maligno es una neoplasia poco frecuente de las glándulas salivales, pero es aun más raro en la glándula lagrimal. Corresponden a menos del 2% de las neoplasias malignas de glándulas salivales y aproximadamente el 50% de ellos se desarrollan a partir de un adenoma pleomorfo preexistente o de un mioepitelioma. OBJETIVOS: Conocer las características clínicas, imagenológicas y anatomopatológicas de un carcinoma mioepitelial (mioepitelioma maligno) originado en la glándula lagrimal. MATERIAL: Se presenta el caso de una mujer de 63 años de edad sin antecedentes de importancia, quien cursó con exoftalmos, ptosis mecánica de párpado superior izquierdo y disminución de visión ipsolateral de 2 meses de evolución. La tomografía computada demostró masa intraconal hiperdensa de bordes bien delimitados, sin destrucción de la pared ósea. Se realizó resección quirúrgica de la masa. Masuda Y. A case of malignant myoepithelioma in the lacrimal gland. Nippon Ganka Gakkai Zasshi. 2001; 105:42-6. INSTITUCIÓN: Asociación Para Evitar la Ceguera en México, Hospital “Dr. Luis Sánchez Bulnes”; Depto. de Medicina Experimental, Facultad de Medicina, UNAM y Hospital General de México; Instituto de Oftalmología F.A.P. Conde de Valenciana ESTADO: DISTRITO FEDERAL Mampara: 2 B Fecha y hora Instalación: 2 de Fecha y hora retiro: 2 de mayo 18.30 mayo 13:00 TC-74 AUTORES: Ivette Hernández Ayuso, Elizmara L. Aguilar Ayala, Dolores Ríos y Valles Valles, Hector A. Rodríguez Martínez, Abelardo A. Rodríguez Reyes. TITULO: Carcinoma basocelular periocular en pacientes jóvenes. Una serie de casos en 10 años. INTRODUCCIÓN: El carcinoma basocelular es el cán- cer más frecuente de piel, el 16% se localiza en párpado, siendo la principal neoplasia de esta región. Clásicamente se presenta en adultos mayores con piel blanca y exposición solar, el tipo sólido es el más frecuente. Raramente se 2do Congreso Anual de Patólogos Mexicanos • 33 León, Guanajuato • 2016 ve en pacientes menores de 40 años regularmente asociado a síndrome de Gorlin-Goltz y xeroderma pigmentoso. OBJETIVOS: Determinar el número y características histopatológicas de los casos con diagnóstico de carcinoma basocelular de la región periocular en pacientes menores de 40 años, confirmado por biopsia en un hospital de referencia oftalmológica en un periodo de 10 años. jido mesenquimatoso primitivo que no dan metástasis. Microscópicamente presentan células fusiformes y estelares sobre un estroma rico en ácido hialurónico. El mixoma es el tumor primario más común del corazón. Otros sitios de afección son hueso, piel y músculo estriado. La afección ocular es rara pero documentada en órbita, córnea, párpado y conjuntiva. OBJETIVOS: Conocer la frecuencia del mixoma primario MATERIAL: Revisión de casos con diagnóstico histopatológico de carcinoma basocelular en el periodo de enero 2005 a diciembre 2015. Se seleccionaron los pacientes menores de 40 años con diagnóstico confirmado de carcinoma basocelular con o sin expediente de la Institución. Se registraron las características demográficas, clínicas e histopatológicas de los casos obtenidos. RESULTADOS: Se recopilaron 231 casos con diagnóstico histopatológico de carcinoma basocelular de la región periocular, se excluyeron 8 casos por edad desconocida. De 223 casos, 5 casos fueron menores de 40 años, 4 hombres y 1 mujer. El tipo sólido se observó en 4, 1 de ellos con presencia de variedad superficial y otro con variedad pigmentada, 1 caso adenoideo. Solo un caso se asoció a síndrome de Gorlin-Goltz CONCLUSIONES: El carcinoma basocelular de la re- gión periocular en jóvenes es raro, representa el 2.2% de nuestros casos, la mayoría género masculino, la variedad más frecuente fue el tipo sólido, la mayoria de los pacientes no tuvieron asosiación con otros síndromes. INSTITUCIÓN: Asociación para Evitar la Ceguera en México I.A.P. Dr. Luis Sánchez Bulnes. Depto. de Medicina Experimental, Facultad de Medicina UNAM, Hospital General de México. ESTADO: DISTRITO FEDERAL de conjuntiva en la población Mexicana. Describir las características clínicas, histopatológicas e inmunohistoquímicas de los casos de mixoma primario de conjuntiva MATERIAL: Se revisaron los casos con diagnóstico de Mixoma Conjuntival (MC), de los archivos del laboratorio de patología oftálmica de nuestro hospital en un período de 58 años (1957-2015). Se reevaluaron las características clínicas, histopatológicas, histoquímicas e inmunohistoquímicas en todos los casos. RESULTADOS: Se obtuvieron 6 casos de MC de un total de 5923 biopsias conjuntivales. La edad media de presentación fue 41 años y no hubo predominio de sexo. No se encontró enfermedad sistémica asociada en ningún caso. El ojo izquierdo fue el más afectado y la conjuntiva bulbar la localización más común. El diagnóstico clínico más empleado fue quiste conjuntival y el diagnóstico de MC se sospechó en 1 caso. Microscópicamente, se observaron lesiones compuestas por células fusiformes y estelares dispuestas sobre un estroma mucinoso rico en ácido hialurónico con escasos vasos sanguíneos. La inmunohistoquímica resultó positiva para VIM y negativa para PS-100, SMA y MSA. CONCLUSIONES: Los MC son lesiones poco frecuentes. Grossniklaus, en un estudio de 2455 lesiones conjuntivales encontró 4 casos (0.002%) de MC. Sólo 26 casos en esta localización han sido informados en la literatura. Se presentan como masas bien delimitadas, translúcidas, amarillentas, localizadas en la conjuntiva bulbar. El estudio histopatológico es indispensable para el diagnóstico certero y para excluir otras lesiones como neurofibroma mixoide, linfangioma y nevo amelanótico entre otras. REFERENCIAS: Demirici H, Shield CL, Eagle RC, Shiels Mampara: 3 A 13:00 JA. Report of a conjunctival myxoma case and review of the literature. Arch Ophthalmol 2006;124:735-738 AUTORES: Dra. Dolores Ríos y Valles Valles, Dr. Abelar- INSTITUCIÓN: Asociación para Evitar la Ceguera en México I.A.P. Hospital “Dr. Luis Sánchez Bulnes”, Depto. Medicina Experimental, Facultad de Medicina UNAM, Hospital General de México Fecha y hora Instalación: 2 de Fecha y hora retiro: 2 de mayo 18.30 mayo TC-76 do A. Rodríguez Reyes, Dra. Ivette Hernández Ayuso, Dr. Héctor A. Rodríguez Martínez, Dra. Elizmar L. Aguilar Ayala, Dra. Vianhi López Rioja ESTADO: ESTADO DE MÉXICO TITULO: Mixoma Primario de la Conjuntiva. Serie de Casos y Revisión de la Literatura. Mampara: 3 B INTRODUCCIÓN: Los mixomas son neoplasias de te- Fecha y hora Instalación: 2 de 2do Congreso Anual de Patólogos Mexicanos • 34 León, Guanajuato • 2016 mayo 13:00 Fecha y hora retiro: 2 de mayo 18.30 Mampara: 4 A TC-108 Fecha y hora Instalación: 2 de Fecha y hora retiro: 2 de mayo 18.30 mayo 13:00 AUTORES: BÁRBARA SÁENZ, Dr. Luis Angel Ceceñas Falcón, Dra. Med. Oralia Barbosa Quintana. TC-119 TITULO: Melanoma de localización coroi- AUTORES: BÁRBARA SÁENZ, Dr. Luis Angel Ceceñas Falcón, Dra. Med. Oralia Barboza Quintana dea: reporte de 4 casos INTRODUCCIÓN: Los tumores primarios del tracto uveal más frecuentes son los melanomas; el melanoma de coroides es el tumor intraocular primario más frecuente en adultos, con un predominio en el sexo masculino y en la raza blanca, aparece entre la quinta y la octava década de la vida. Es de interés el estudio y reporte de estos tumores que aunque son poco frecuentes tienen alta malignidad y mal pronóstico. La importancia en el estudio y reporte de estos casos clínicos, está dada por lo infrecuentes que son estos tumores y por sus características agresivas. OBJETIVOS: Presentar los casos diagnosticados como melanoma coroideo en el Hospital Universitario ?José Eleuterio González? desde el 2013 a la fecha. MATERIAL: Se busco en la base de datos del Hospital Universitario “José Eleuterio González” todas las exanteraciones y/o enucleaciones orbitarias desde el 2013, y se encontraron 4 casos diagnosticados como melanoma coroideo, se buscaron sus historiales clínicos, radiológicos y se revisó nuevamente su histología para documentarlos. RESULTADOS: De la base de datos del Hospital Univer- sitario “José Eleuterio González”, se encontraron 80 enucleaciones/exanteraciones, de las cuales sólo 4 casos se diagnosticaron como melanoma coroideo. Tres de los pacientes son de sexo masculino y la edad oscila entre los 37 a los 81 años. Dos de los casos presentaron un melanoma coriodeo de tipo fusocelular, mientras que otro tuvo un predominio epitelioide y el cuarto de tipo mixto, pero con predominio epitelial. Así mismo existió un predomio de afección del lado izquierdo (3 de 4). El pronóstico de 3 de los pacientes se encontraba en etapa 4 y el cuarto paciente en etapa 2. Todos los casos se encontraban libres de lesión a nivel del nervio óptico. TITULO: Rabdomiosarcoma alveolar ocular con diferen- ciación neuroendócrina: Reporte de un caso y revisión de la literatura INTRODUCCIÓN: El rabdomiosarcoma es el tumor ma- ligno primario más común de la órbita en los niños, sin embargo también puede ocurrir en adultos. Se debe sospechar en un caso de proptosis rápidamente progresiva en adultos. En la literatura se encuentran descritos estas variedades de Rabdomiosarcoma alveolar, las cuales manifiestan marcadores para células neuroendócrinas (Cromogranina, sinaptofisina, S100 y CD 56) así como también los marcadores para células musculares ( Miogenina y Desmina). OBJETIVOS: Presentar el caso de un paciente con un rabdomiosarcoma alveolar orbital con diferenciación neuroendócrina. MATERIAL: Se recupero la historia clínica del paciente, así como sus estudios de imagen y paraclínicos para poder documentar el caso. RESULTADOS: Se recibió el producto de una exanteración ocular de una paciente femenina de 41 años, que presentaba proptosis y dolor ocular en ojo derecho. A la sección se identificó un tumor carnoso, blando de 3 cm de diámetro mayor, que infiltraba tejidos blandos perioculares infiltrando la conjuntiva bulbar y con bordes de resección quirúrgicos positivos. A nivel histológico se identificó una neoplasia de células pequeñas, redondas y azules de aspecto primitivo, y se demostró la expresión aberrante de marcadores epiteliales y neuroendocrinas en subgrupos significativos de los rabdomiosarcomas alveolares, incluyendo casos confirmados genéticamente. CONCLUSIONES: El melanoma en la localización a CONCLUSIONES: Este tipo de tumores no son comunivel de la coroides, se sigue diagnosticando, en nuestro hospital sigue apareciendo en pacientes de la 3 a 8 década de la vida. Así mismo se sigue presentando en etapas no iniciales, afectando directamente en el pronóstico y culminando con un tratamiento agresivo que es la enucleación. nes en pacientes adultos, tienden a ser agresivos y comúnmente con extensión a ganglios linfáticos cervicales, pulmones y los huesos. Así mismo la enfermedad metastásica en el momento de presentación y la mala respuesta a la quimioterapia están fuertemente asociados con un mal pronóstico. INSTITUCIÓN: Hospital Universitario José INSTITUCIÓN: Hospital Universitario José Eleuterio Gonzalez Eleuterio Gonzalez ESTADO: NUEVO LEÓN ESTADO: NUEVO LEÓN 2do Congreso Anual de Patólogos Mexicanos • 35 León, Guanajuato • 2016 Mampara: 4 B Fecha y hora Instalación: 2 de Fecha y hora retiro: 2 de mayo 18.30 mayo 13:00 PATOLOGÍA DE LA GLÁNDULA MAMARIA TC-173 Elizmara L. Aguilar-Ayala, Dolores Rios Y Valles-Valles, Ivette Hernandez-Ayuso, Abelardo A. Rodriguez-Reyes, Hector A. Rodriguez-Martinez AUTORES: TITULO: CARCINOMAS METASTASICOS EN ORBITA Y GLOBO OCULAR INTRODUCCIÓN: Los carcinomas metastásicos son pocos frecuentes, sin embargo pueden presentarse en carcinomas de mama, pulmón y próstata varios años después del tratamiento o resección del tumor primario. Es importante saber los antecedentes personales patológicos del paciente, así como identificar mediante la clínica las probabilidades diagnósticas de estos tumores en órbita y globo ocular, para corroborarlo por estudio histopatológico. OBJETIVOS: Conocer la incidencia de los tumores metastásicos en un centro oftalmológico de referencia nacional, así como el tumor primario más frecuente de estas metástasis. MATERIAL: Se realizó una revision retrospectiva de 10 años de casos con diagnostico histopatológico de tumor metastásico, de los cuales solo se seleccionaron aquellos que resultaron carcinomas metastásicos en órbita y globo ocular. Se analizaron los tumores primarios más frecuentes, el género y la edad de los pacientes, así como el tiempo de evolución de la enfermedad. RESULTADOS: En un período de 10 años (2005 a 2015), se recopilaron 14 casos con diagnóstico histopatológico de carcinoma metastásico en órbita y/o globo ocular, siendo los más frecuentes en mujeres carcinoma de mama y en hombres carcinomas de pulmón y próstata. CONCLUSIONES: La incidencia de tumores metastásicos es poco frecuentes, sin embargo es muy importante tener encuenta los antecedentes personales patológicos de los pacientes y analizar el tipo de tumor primario que presentan, el tiempo de evolución y tratamiento. Se debe realizar una adecuada correlación clinico-patológica para considerar como diagnóstico una probable metastasis al globo ocular y a la órbita. INSTITUCIÓN: Apec, Hospital “Dr. Luis Sanchez Bulnes”; Medicina Experimental, Hospital General De Mexico; Issste Delegacion Regional Zona Norte ESTADO: DISTRITO FEDERAL Mampara: 6 A Fecha y hora Instalación: 2 de Fecha y hora retiro: 2 de mayo 18.30 mayo 13:00 TC-5 AUTORES: Dra. Diana Brisa Sevilla, Dra. Claudia Haydee Sarai Caro Sánchez. Dra. Claudia Isela Apreza Gonzalez TITULO: CÁNCER DE MAMA TRIPLE NEGATIVO CON METÁSTASIS A SISTEMA NERVIOSO CENTRAL: UNA PERSPECTIVA CLÍNICO-PATOLÓGICA Y PERFIL GENÉTICO. INTRODUCCIÓN: El termino cáncer de mama triple negativo (CMTN) es utilizado para identificar el cáncer de mama sin expresión de receptores de estrógeno (RE) ni de progesterona (RP) y que no exhiban amplificación del gen del receptor 2 del factor de crecimiento epidérmico humano (HER2) que corresponde al 12-17% de los tumores malignos de la mama. En México se ha reportado una mayor proporción de este subtipo tumoral (23.1%) OBJETIVOS: Los CMTN se caracterizan por una histo- ria clínica agresiva al presentar un riesgo incrementado de recaída y muerte, y se asocian a una mayor probabilidad de desarrollo de enfermedad metastásica a SNC. A pesar de múltiples esfuerzos de investigación para el diagnóstico y tratamiento, tanto de esta neoplasia y sus complicaciones, no existen herramientas para la prevención del desarrollo de metástasis cerebrales ni para la mejoría del pronóstico de las pacientes que presentan enfermedad metastásica a SNC. Con la Subclasificación por anticuerpos subrogados de los CMTN se pretende distinguir a un subgrupo de pacientes que pudiesen beneficiarse de radioterapia craneal profiláctica, para intentar un impacto positivo en su pronóstico y calidad de vida. MATERIAL: Se emplearon casos y controles de pacientes con cáncer de mama TNG, en estadio III?IV.? Los casos se dividieron en tres grupos principales aquellos con metástasis a SNC, con metástasis a NO SNC y sin metástasis; realizo y analizó el perfil de inmunohistoquímica, se calculo su supervivencia global y se evaluó de manera individual cada marcador de acuerdo a su evolución clínica y supervivencia libre de progresión. RESULTADOS: Con el análisis de las características generales de las pacientes se obtuvieron los siguientes da- 2do Congreso Anual de Patólogos Mexicanos • 36 León, Guanajuato • 2016 tos: del total de 69 muestras analizadas 31 (44.92%) corresponden a casos con metástasis a SNC, 16 (23.18%) a pacientes con metástasis a otros sitios y 22 (31.88%) sin metástasis a algún sitio al inicio del estudio (casos controles), por tanto tenemos a un 55.07% (38 pacientes) que no presentaron metástasis en SNC en este periodo. Con un seguimiento de 2 años (61.4 meses) posterior a la fecha de diagnóstico inicial de cada paciente. Las pacientes que presentaron metástasis a SNC durante el seguimiento de 2 años todas fueron negativas a los siete marcadores de inmunohistoquímica, es decir permanecieron siendo CMTN sin poder etiquetarse en algún subgrupo molecular. Para el análisis de inmunohistoquímica, encontramos 34 casos que fueron negativos a todos los marcadores 49.27%, el resto (35 casos/50.72%) presentaron una positividad para uno ó más marcadores. Los positivos a EGFR fueron 9(13%), a CK5/6 11(16%), actina 14 (21%), RA 5(7%), p63 4(6%), CK17 13(19%), CK14 fueron 15 casos positivos (22%). De los CMTN que tuvieron progresión hacia SNC con expresión de EGFR 6/9, CK5-6, 6/11, actina 5/14, p63 2/4 , CK17 8/13, CK14 10/15. Los pacientes que expresaron CK14 (15 pacientes) el 100% fallecieron, 10 de ellos con metástasis a SNC (p 0.001)y a hígado (p 0.028), con una SLP más corta. CONCLUSIONES: Existe relevancia en estudiar la expresión de citoqueratinas en los tumores primarios de mama, como factor pronóstico y para lograr una terapia adaptada para los tumores con esta expresión subrogada de marcadores en búsqueda de oportunidades de tratamiento desde etapas tempranas para lograr la cura del cáncer de mama en el futuro. Es necesario, en un futuro corto, correlacionar la expresión génica de estos casos de manera individual para que la inmunohistoquímica sea una herramienta para encontrar de manera indirecta estos genes los cuales probablemente son cruciales para permitir el paso de las células neoplásicas a SNC y la célula tumoral es capaz de vencer la barrera Hemato-encefálica y depositarse en el tejido cerebral; los cuales se encuentran sobre expresados en una paciente con metástasis a otros sitios. INSTITUCIÓN: Instituto Nacional De Cancerologia. Ciudad De Mexico, Mexico Hospital Fray Antonio Alcalde. Guadalajara, Jalisco ESTADO: DISTRITO FEDERAL TITULO: Inmunofenotipos del Cáncer de Mama en México utilizando una plataforma automatizada. INTRODUCCIÓN: Los marcadores para el cáncer de mama son necesarios para la clasificación de estos tumores, y decidir el manejo que se dará, en México se encuentran documentadas las características de expresión de los mismos, aunque con la tipificación del HER2-neu existe heterogeneidad en los resultados de los estudios. OBJETIVOS: El objetivo del presente estudio es describir dichas características inmuno-fenotípicas y compararlo con la literatura mexicana e internacional. MATERIAL: Estudio prospectivo y descriptivo. Tomando en cuenta los casos referidos a nuestra institución, desde diversas regiones de México del año 2014 y 2015 diagnosticados con cáncer de mama. Se realizaron tinciones de inmunohistoquímica en una plataforma automatizada para receptores de estrógenos, progesterona y HER2. Se clasificaron en subtipos según su fenotipo según la guía de la ASCO. Se utilizó prueba de X2 para el análisis de los datos obtenidos. RESULTADOS: De los 5359 casos, 442(8.9%) fue- ron inadecuados por tejido, se analizaron 4917; el 29.99% fueron HER2 positivos, 67.92% positivo para estrógenos, y 61.74% positivo para progesterona. La prueba de ISH dio positiva para el 36.51% de los casos HER2 2+. El subtipo más frecuente fue RE(+) RP(+) HER2() con 45.54% de los casos; los tumores triple negativos se asociaron a una edad menor de 50 años con respecto al resto de los tumores al momento del diagnóstico (p <0.001); los tumores HER2 no se asocian a una edad de presentación en específico (p=0.1471).> CONCLUSIONES: Los estudios sobre las característi- cas inmunofenotípicas en México tienen muchas variaciones entre laboratorios en cuanto al diagnóstico de HER2, variando desde un 19.7% a un 36.1% con la utilización de una plataforma manual, a diferencia de las frecuencias en Estados Unidos que reportan entre 25% y 30% donde se utilizan plataformas automatizadas, lo que evita que se subdiagnostiquen o sobrediagnostiquen tumores con HER2 positivos en menor proporción. Mampara: 6 B INSTITUCIÓN: Servicio de Anatomía Patológica y Citopatología del Hospital Universitario Fecha y hora Instalación: 2 de mayo 13:00 “Dr. José Eleuterio González” Fecha y hora retiro: 2 de mayo 18.30 ESTADO: NUEVO LEÓN TC-18 AUTORES: Dr. Alfredo Elizondo-Montemayor, Dra. med Raquel Garza-Guajardo, Dra. Natalia Vilches-Cisneros, dr. Enrique Delgadillo-Esteban, dr. med Juan Pablo Flores-Gutiérrez. Mampara: 7 A Fecha y hora Instalación: 2 de Fecha y hora retiro: 2 de mayo 18.30 2do Congreso Anual de Patólogos Mexicanos • 37 León, Guanajuato • 2016 mayo 13:00 TC-26 Mampara: 7 B AUTORES: Dr. Hersilia Aide Hernandez Zamonsett, Dr. Fecha y hora Instalación: 2 de mayo 13:00 Juana Elizabeth Tadeo Gonzalez, Dr. med. Marco Antonio Ponce Camacho, Dr. med. Raquel Garza Guajardo, Dr. med. Oralia Barboza Quintana Fecha y hora retiro: 2 de mayo 18.30 TC-50 TITULO: Carcinoma Renal De Celulas Claras AUTORES: Luis Gerardo Jacobo Rivera. Álvaro BarboMetastasico A Glandula Mamaria, Reporte De sa Quintana. Luis Eduardo Cuervo Pérez. Gabriela Sofía Gómez Macías. Ireán García Hernández. Claudia Martínez Un Caso Amador. INTRODUCCIÓN: Las metástasis a glándula mamaria ocupan del 0.5-2% de los carcinomas mamarios, el diagnostico diferencial con un tumor primario es mandatorio. Solo el 3% de los carcinomas de células renales tiende a metastatizar a glandula mamaria, hasta el 2014 25 casos han sido reportados. TITULO: Carcinoma lobulillar invasor de la mama con mucina extracellular. Reporte de un caso OBJETIVOS: Paciente con antecedente de carcinoma segunda neoplasia maligna más común de la mama, suma entre el 5-15% de los casos.La secresión de mucina extracelular es una característica del carcinoma ductal.Hay pocos casos reportados.Todos ellos comparten características histológicas. Nuestro caso es de una mujer de 60ª de edad en la cual se confirma la presencia de mucina extracelular. renal de células claras, identifica tumor en mama derecha mamografía reporta imagen nodular, isodensa, irregular de 20mm cuadrante superoexterno. Mama izquierda en intercuadrantes externos identifican imagen nodular, ovalada, isodensa, bien circunscrita de 10 mm.Se realiza biopsia por trucut de ambas glándulas mamarias previo consentimiento informado. MATERIAL: Se realizaron secciones de bloques de parafina que contenían el tejido a evaluar, se tiñen con la técnica de hematoxilina y eosina. Además se realizaron secciones de tejido congelado para su evaluación con inmunohistoquímica a los cuales se les aplicaron anticuerpos monoclonales para CD10, VIMENTINA, GCFP15. RESULTADOS: Los hallazgos histopatológicos demuestran una neoplasia compuesta por nidos y cordones de células con un citoplasma claro, en un estroma fibrovascular. La inmunohistoquimica mostro positividad para CD10 y Vimentina en células tumorales, y negatividad para GCDFP15. El 94% de las neoplasias renales expresan CD10, la negatividad para la proteína de la leche (GCFP15) descarta el origen mamario de la lesión, con este inmunofenotipo se confirmo el diagnostico de carcinoma renal de células claras metastasico. CONCLUSIONES: En diferentes series reportan una incidencia de metástasis de carcinoma renal de células claras a glándula mamaria del 3%, con tan solo 24 casos reportados hasta el 2014. El curso del carcinoma de células renales es extremadamente variable e impredecible tanto en su desarrollo clínico y la presencia de metástasis INSTITUCIÓN: Departamento de Anatomia Patologica y Citopatologia, Hospital Universitario “Dr. Jose Eleuterio Gonzalez”. ESTADO: NUEVO LEÓN INTRODUCCIÓN: El carcinoma lobulillar invasor es la OBJETIVOS: Reporte de un caso MATERIAL: Recibimos un ganglio centinela derecho y un fragmento de glándula mamaria derecha marcada por arpón guiado, producto de mastectomía parcial con disección de ganglio centinela, con diagnóstico previo en biopsia por aguja gruesa de carcinoma lobulillar invasor, motivo por el cual es intervenida quirúrgicamente. El fragmento de glándula mamaria es de color amarillento, lobulado y de consistencia blanda. En el sitio del arpón hay un área blanquecina RESULTADOS: Es un carcinoma lobulillar invasor con mucina extracelular de 9 mm. Se le asigna un 3 en formación glandular, 1 de núcleos y un 1 de índice mitótico ya que solamente se contaron 3 mitosis en 10 CAP en un diámetro de 0.65 por lo que se clasifica como bien diferenciado utilizando la escala de Nottinghamm/SBRM (Score 5 en escala de 3-9). También se encontraron áreas de carcinoma in situ de tipo sólido con grado nuclear de 2 y necrosis central presente. Los biomarcadores se reportaron de la siguiente manera: RE: Positivos, score de Allred (5 de proporción y 2 de intensidad), con control interno presente. RP: Negativos. HER 2: Negativo 0+ y Cadherina-E Negativa en el carcinoma invasor e in situ. CONCLUSIONES: No existen los suficientes casos como para poder predecir el comportamiento de estos tumores, si bien es cierto que el Carcinoma ductal invasor con mucina extracelular es de mejor pronóstico que el de tipo convencional y el carcinoma lobulillar invasor todavía más que el ductal, es necesario estudiar más estos casos aislados para poder predecir la conducta de estas lesiones. INSTITUCIÓN: Instituto Tecnológico y de Estudios Superiores de Monterrey 2do Congreso Anual de Patólogos Mexicanos • 38 León, Guanajuato • 2016 REFERENCIAS: 1. Wachter DL, Hartmann A, Beckmann ESTADO: NUEVO LEÓN Mampara: 8 A Fecha y hora Instalación: 2 de Fecha y hora retiro: 2 de mayo 18.30 mayo 13:00 TC-60 AUTORES: Dr. Cesar Iván Peña Ruelas, Dr. Julio Ce- sar Sánchez Venegas, Dr. Jorge Arturo Sánchez Garza, Dr. Hugo M. Vázquez García, Dra. Elsa Edith de la Rosa Vélez, Dra. Sarai Páez Moreno. TITULO: Carcinoma neuroendocrino de mama. INTRODUCCIÓN: El carcinoma neuroendocrino representa cerca del 1% de todas las neoplasias malignas de mama.(1) La primera descripción se realizó en 1963 por Feyrter y Hartmann, fue incluido en la clasificación de la OMS en 2003.(2-3) OBJETIVOS: Presentar las características clínicas, imagenológicas y patológicas de un caso de carcinoma neuroendocrino de mama. MATERIAL: Caso clínico: Mujer de 82 años quien inició su padecimiento actual al identificarse en mamografía de tamizaje una alteración en la arquitectura en CSE de mama derecha. El ultrasonido mostró una imagen nodular de 10 mm, con vascularidad dominante. RESULTADOS: En transoperatorio se recibió tejido mamario de 5x4x1.5 cm, a la sección se identificó un tumor de 1.5x1.0 cm, de bordes poco definidos, blanco-amarillento con zonas rojizas y consistencia firme. La citología mostró células distribuidas individualmente con núcleos redondos, excéntricos, con cromatina gruesa y granular, citoplasma moderadamente acidófilo, algunos grupos con amoldamiento intercelular. El estudio histológico mostró una neoplasia infiltrante, con áreas solidas, cordones y formación de algunas rosetas, así como mucina en menos del 25%. La IHQ mostró CgA + (100% de las células), RE+, RP+, HER2/neu-, Ki67 <2%, SYP-. El diagnóstico fue de Carcinoma Neuroendocrino Bien Diferenciado.> CONCLUSIONES: Desde el 2003 la OMS define al car- cinoma neuroendocrino como una entidad específica que expresa marcadores neuroendocrinos (cromogranina A y/o sinaptofisina) en más del 50% de las células tumorales.(3) Generalmente se presenta en pacientes de edad avanzada con una media de 70 años, el tamaño del tumor varía de 0.6 a 6 cm.(3) El pronóstico de esta neoplasia depende en gran medida del grado histológico. Debido a que el perfil molecular corresponde a un subtipo luminal A se espera que el pronóstico sea favorable.(1-3) MW, et al. Expression of neuroendocrine markers in different molecular subtypes of breast carcinoma. Biomed Research International DOI 10.1155/2014/408459. 2. Ghanem S, Kabaj H, Naciri S, et al. Primary neuroendocrine carcinoma of the breast: a rare and distinct entity. J Cancer Res Exp Oncol 2011;3(5):50-54. 3. Rovera F, Lavazza M, La Rosa S, et al. Neuroendocrine breast cancer: retrospective analysis of 96 patients and review of literature. International Journal of Surgery 2013;11(51):579-83 INSTITUCIÓN: Hospital General de Zona No. 6 del IMSS. Delegación Nuevo León. ESTADO: NUEVO LEÓN Mampara: 8 B Fecha y hora Instalación: 2 de Fecha y hora retiro: 2 de mayo 18.30 mayo 13:00 TC-131 AUTORES: Dr. Luis Enrique Cortés Hdz,Est.Veronica Pravia Mtz,Est.Mauricio Romero Garcia,Dra.med.Raquel Garza Guajardo,Dra.med.Oralia Barboza Quintana,Dra. Natalia Vilches Cisneros,Dr.Enrique Delgadillo,Dr.med.Juan Pablo Flores Gutierres. TITULO: Relación de la expresión del Receptor de andrógenos y Ki67 en cáncer de mama INTRODUCCIÓN: El Receptor de Androgenos(AR) está relacionado con vías proliferativas de los receptores de estrógenos(RE) y progesterona(RP) en cáncer de mama y la expresión de Ki67 está directamente relacionada con la proliferación tumoral. OBJETIVOS: El objetivo es medir la expresión de RA con relación a Her2/RE/RP y Ki67, identificando su relación. MATERIAL: Se buscaron casos de adenocarcinoma ductal infiltrante con IHQ(RE/RP/Her2) en nuestra institución retrospectivamente de 1/2007-3/2015 y prospectivamente de 4/2015 a 12/2015. Se excluyeron casos sin material o datos. 2 patólogos revaluaron usando escala Bloom-Richardson modificada y se seleccionó área para histoarreglos. Se hizo IHQ para RA y Ki67 en una plataforma automatiza. Se evaluó RA en cuanto a intensidad y porcentaje y se uso consenso de St.Gallen para Ki67, se utilizo estadística univariada. RESULTADOS: De 80 casos, 32.5% fueron RA- y 67.5% fueron RA+, en Ki67 encontramos 39% con expresión <20% y 61% con expresión ?20-29%. No se encontró diferencia de expresión entre RA con Ki67, así como tampoco con grado histológico o estadio clínico(p>0.05) Los casos con RA 2do Congreso Anual de Patólogos Mexicanos • 39 León, Guanajuato • 2016 elevado presentaron un mayor porcentaje de ki67(p <0.05). Encontramos una relación directa en la expresión de los RA y los RE/RP, así como AR con Her2(p><0.05). > contaron con estudios de inmunohistoquimica que confirmaron el diagnostico. RESULTADOS: En el periodo de estudio evaluado se encontraron 3 CNEM de celulas grandes (1.21%). El rango CONCLUSIONES: Concluyendo, la expresión del RA de edad de las pacientes oscilo entre 52 y 81 años (media se encuentra directamente relacionada a la expresión de RE/RP y con Her2, además los RA que presentan una expresión mayor en porcentaje muestran menor índice proliferativo. REFERENCIAS: 1. Fioretti FM, Bevan CL, Brooke GN. Revising the role of the androgen receptor in breast cancer. 2001. 2. Coates AS, Winer EP, Goldhirsch A, et al. Tailoring therapies - improving the management of early breast cancer: St GallenInternational Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann Oncol . May 2015. INSTITUCIÓN: Servicio de Anatomía Patológica y Citopatología, Hospital Universitario “José Eleuterio González” U.A.N.L. ESTADO: NUEVO LEÓN 66.7 años). Las tres pacientes clinicamente se clasificaron como BIRADS III, con crecimiento progresivo de la lesion. La localizacion mas comun fue cuadrante superior externo de mama derecha, el diagnostico realizado en la biopsia por trucut fue de carcinoma ductal infiltrante NOS grado 2 (2 casos) y 3 (1 caso). El tamaño promedio de las lesiones fue de 3.1 cm de eje mayor, con margenes expansivos, bien delimitados, de color blanco grisáceo. Ninguno presento metastasis ganglionares. Histológicamente el 100% de los casos se caracterizo por presentar patron de crecimiento organoide, conformadas por células neoplásicas grandes con escaso citoplasma y cromatina granular con patrón en “sal y pimienta”, ninguno presento necrosis asociada al tumor, tampoco permeacion linfatica. Por inmunohistoquimica los 3 casos fueron positivos para cromogranina en mas del 50% de las celulas neoplasicas, dos fueron positivos para sinaptofisina con el mismo porcentaje, con Ki67 mayor al 70%, y positivos para receptores de estrogenos y progesterona. Todos los casos fueron negativos para Her2. CONCLUSIONES: Los CNEM presentan un curso clíni- Mampara: 9 A Fecha y hora Instalación: 2 de mayo 13:00 hrs. Fecha y hora retiro: 2 de mayo 18.30 hrs. TC-138 Dr. Mario Murguía Pérez, Dra. Teresa Lopez Gaytan, Dr. Lazaro Ariel Ramirez Balderrama, Dr. Felipe Navarro Córdoba, Dr. Adrián Antonio López Vera, Dr. Saulo Mendoza Ramirez AUTORES: co mas agresivo que los carcinomas ductales NOS, con una elevada propension a presentar metastasis, y con pobre supervivencia a 5 años. El tratamiento de estas neoplasias es el mismo que el de los carcinomas ductales NOS. Es importante reconocer esta entidad y excluir una metastasis de otro sitio primario para un tratamiento correcto. INSTITUCIÓN: Unidad Medica de Alta Especialidad No. 1 IMSS Leon, Hospital Medica Campestre, Hospital General de Mexico ESTADO: GUANAJUATO TITULO: Carcinoma neuroendocrino de mama. Presentacion de tres casos. Mampara: 9 B INTRODUCCIÓN: El carcinoma neuroendocrino de mama (CNEM) es una neoplasia maligna poco comun, representa entre el 2 y 5% de todas las neoplasias mamarias, histologicamente recuerda a su contraparte pulmonar o de tracto gastrointestinal, y debe presentar positividad para algun marcador neuroendocrino en al menos 50% de las celulas neoplasicas. Fecha y hora Instalación: 2 de mayo 13:00 hrs. Fecha y hora retiro: 2 de mayo 18.30 hrs. OBJETIVOS: Presentamos tres casos identificados en AUTORES: Mariela Sánchez Claudio, Jocabed Martínez MATERIAL: Se realizo una busqueda en el archivo de pa- TITULO: DIFERENCIACIÓN OSTEOSARCOMATOSA Y CONDROSARCOMATOSA EN TUMOR PHYLLODES MALIGNO. CASO CLÍNICO Y REVISIÓN DE LA LITERATURA. nuestra institucion con los criterios necesarios para clasificarlos en esta entidad. tologia de la UMAE No. 1 IMSS Leon, en el periodo que abarco del 1 de enero del 2013 al 31 de diciembre del 2015 (3 años) de CNEM y se investigaron datos clinicos de importancia en el expediente. Los tres casos identificados TC-156 Olivares 2do Congreso Anual de Patólogos Mexicanos • 40 León, Guanajuato • 2016 INTRODUCCIÓN: El tumor phyllodes representa del 0.3 INTRODUCCIÓN: El linfoma primario de la glándula tosa y condrosarcomatosa en un tumor phyllodes maligno y realizar una revisión de la literatura mamaria es un raro pero bien definido subtipo de Linfoma no Hodgkin, con una incidencia que va desde el 0.04 al 0.5% de todos los tumores malignos de mama. La estirpe histológica predominante es de células B Difuso. Se han reportado casos de linfoma anaplásico de células grandes T, que se presenta en las mujeres portadoras de implantes mamarios. MATERIAL: Se revisó el expediente clínico, estudios de OBJETIVOS: Reportar un caso de linfoma T difuso de al 1 % de todos los tumores de la mama y del 2 al 3 % de las lesiones fibroepiteliales. De acuerdo a la OMS, se clasifica según su grado histológico en benigno, limítrofe o maligno. OBJETIVOS: Describir la diferenciación osteosarcoma- imagen y el material enviado al servicio de Patología del Hospital de Ginecología y Obstetricia No. 3 CMN La Raza. celulas grandes en una paciente que no es portadora de protesis mamarias. RESULTADOS: Mujer de 50 años de edad, inicia con MATERIAL: Cortes histopatologicos, estudio de inmuno- un nódulo mamario izquierdo de 8 meses de evolución de rápido crecimiento. Para estudio transoperatorio se recibió espécimen en fresco, que se reportó como lesión fibroepitelial con necrosis extensa a clasificar en estudio definitivo. Posteriormente se recibió espécimen ovoide de 14x10.5 cm, con extensa necrosis y hemorrágia. Histológicamente se identifica tumor phyllodes maligno con diferenciación osteosarcomatosa y condrosarcomatosas extensas y límites quirúrgicos afectados por la neoplasia. CONCLUSIONES: El tumor phyllodes maligno representa el 10% de todos los tumores phyllodes, estos pueden contener elementos heterólogos como liposarcoma, osteosarcoma, condrosarcoma, rabdomiosarcoma, pero la diferenciación osteosarcomatosa ocurre sólo en el 1.3% de los casos. La importancia de esta diferenciación radica en los diagnósticos diferenciales como el carcinoma ductal infiltrante metaplásico, carcinoma con células gigantes parecidas a osteoclastos, metástasis de osteosarcoma o sarcomas primario de glándula mamaria. histoquimica RESULTADOS: Linfoma T anaplasico difuso de celulas grandes de glandula mamaria. CONCLUSIONES: El presente caso constituye una forma inusual de presentación de linfoma no Hodgkin anaplásico de células T primario de glándula mamaria, pues, como bien se mencionó, está relacionado con mujeres que tienen implantes mamarios, sin embargo la paciente no cuenta con dicho antecedente. Esta condición es muy poco usual, lo cual es demostrado por la limitada bibliografía encontrada. Según la mayoría de los autores, los linfomas difusos de células grandes son los más frecuentes (40 a 70%), los más raros son los que muestran un inmunofenotipo T que representan un 3% de los linfomas mamarios. INSTITUCIÓN: HOSPITAL CENTRAL IGNACIO MORONES PRIETO. Departamento de anatomia patologica. INSTITUCIÓN: HGO 3 Dr. Víctor Manuel Espi- ESTADO: SAN LUIS POTOSÍ nosa de los Reyes Sánchez CMN “La Raza”, Hospital de Especialidades “Antonio Fraga Mouret” Centro Médico Nacional “La Raza” ESTADO: ESTADO DE MÉXICO Mampara: 10 B Fecha y hora Instalación: 2 de mayo 13:00 Fecha y hora retiro: 2 de mayo 18.30 Mampara: 10 A Fecha y hora Instalación: 2 de mayo 13:00 hrs. Fecha y hora retiro: 2 de mayo 18.30 hrs. TC-177 AUTORES: Patricia Anchondo, Dr. Cuauhtémoc Oros Ovalle, Dra Claudia Peña Zepeda, Dra Gabriela Perez Coronado TC-190 AUTORES: Dr. José Samuel Almeida Navarro, Dra. Clau- dia Haydee Sarahi Caro Sanchez, Dra. Roxana Barajas, Dr. José Alberto Mejia TITULO: METASTASIS EN GLÁNDULA MAMARIA: DESCRIPCIÓN DE 10 CASOS. INTRODUCCIÓN: La metástasis en la glándula mama- TITULO: LINFOMA T DIFUSO DE CELULAS GRANDES DE GLANDULA MAMARIA. PRESENTACION DE UN CASO ria de neoplasias no hematológica corresponde a una lesión rara con una incidencia entre 0.5 al 2 %. Comúnmente involucra el tejido celular subcutáneo y clínicamente no produce retracción del pezón. Las neoplasias no hematológicas que 2do Congreso Anual de Patólogos Mexicanos • 41 León, Guanajuato • 2016 metastatizan a la glándula mamaria en orden de incidencia son: Melanoma, sarcoma, próstata, pulmón, estomago, ovario, etc. OBJETIVOS: El objetivo de este estudio es evaluar la incidencia de las metastasis, asi como sitio primario y los diferentes factores asociados en el Instituto Nacional de Cancerología en un periodo de 26 meses. MATERIAL: Se realizó estudio retrospectivo, observacio- nal en una cohorte de pacientes con diagnóstico de metástasis de órgano solido a la glándula mamaria en el Instituto Nacional de Cancerología (INCan) entre enero de 2014 y febrero de 2016 (26 meses). Se incluyeron todos aquellos que contaran datos clínicos dentro del expediente clínico INCAUTE y se excluyeron las neoplasias hematopoyeticas. RESULTADOS: Se identificaron 10 pacientes con carci- noma metastasico a la glándula mamario en el periodo de 26 meses. Todos los casos correspondían a pacientes del sexo femenino con una mediana de edad de 54 años (rango 41-66), cinco presentaban metástasis en mama izquierda, dos en mama derecha y tres metástasis bilaterales. El sitio primario más frecuente fue carcinoma seroso papilar de ovario en tres casos (30%) y todos los siguientes con un caso: Pulmón, estomago, cervix, estomago, paneras, vía biliar y liposarcoma de células pequeñas. Tres pacientes fallecieron dentro del primer año, y el resto se encontraban en tratamiento paliativo durante la mediana de seguimiento de 11 meses. CONCLUSIONES: Metastasis a la glandular mamaria es una patologia rara (nuestro estudio correspondió a una incidencia del 0.13%), con órgano primario más frecuente al ovario seguido de Pulmón, estomago, cervix, estomago, paneras, vía biliar y liposarcoma de células pequeñas INSTITUCIÓN: Instituto Nacional de Cancerologia ESTADO: DISTRITO FEDERAL Mampara: 11 A Fecha y hora Instalación: 2 de mayo 13:00 hrs. Fecha y hora retiro: 2 de mayo 18.30 hrs. TC-195 AUTORES: MARIO MURGUIA,Dr. Saulo Mendoza Ramirez, Dr. Felipe de Jesus Navarro Cordoba, Dr. Adrian Antonio Lopez Vera INTRODUCCIÓN: El carcinoma de mama con celulas gigantes tipo osteoclasto (CMCGO) fue descrito por Robb en 1958, y posteriormente por Haagensen en 1971, y Agnantis en 1979, quienes la describieron como una neoplasia de mal pronostico, sin embargo, hay autores que difieren sobre esta conducta biologica. OBJETIVOS: Son tumores raros, constituyendo del 0.5% a 1.2% de los carcinomas de mama. Informamos las características clínicas, histologicas e inmunohistoquimicas de un CMCGO. MATERIAL: Mujer de 37 años, sin antecedentes de importancia. Menarca a los 13 años, IVSA a los 21 años, , gesta 2, para 2, sin tratamiento con hormonales. Noto un nodulo en CSE de mama izquierda de 2x2cm, movil, bien delimitado, no doloroso, sin alteraciones de la piel y sin telorrea. Por USG se sospecho fibroadenoma, por lo que se realizo biopsia excisional. RESULTADOS: El especimen resecado consistio en una lesion neoplasica bien circunscrita, que midio 3.5x3.3cm, de consistencia aumentada, color cafe marron, macroscopicamente sin lesion en los bordes quirurgicos. Histológicamente se observo un carcinoma ductal infiltrante grado 1, SBR de 3. Se identificaron celulas gigantes multinucleadas tipo osteoclasto (CGO) tanto entre las celulas epiteliales de los tubulos como en el estroma entre las formaciones tubulares. No se observaron areas de fibrosis, tejido de granulacion y/o macrofagos asociados a las CGO. Por IHQ se observo positividad para AE1/E3 y negatividad para CD68, RP y Her2/neu. Las CGMO fueron positivas para CD68 y negativas para el resto de los marcadores. Se concluyo como CMCGO. CONCLUSIONES: Los CMCGO son tumores muy in- usuales. La presencia de CGO en el carcinoma de mama ha sido atribuidos a una reaccion inusual del estroma del carcinoma invasor, una transformacion de los monocitos de sangre periferica mediante un virus presente en los monocitos de pacientes con cancer de mama, o una respuesta osteoclastogenica de los macrofagos de sangre periferica a citocinas localizadas producidas por las celulas tumorales en el proceso de estimulacion angiogenica. Algunos autores consideran que este tipo de tumor tiene pobre pronóstico, sin embargo, no existe un consenso, ya que otros autores sugieren que el comportamiento de la neoplasia se da en base al tipo histológico y grado de malignidad. INSTITUCIÓN: Unidad Medica de Alta Especialidad N° 1 IMSS Leon, Hospital Medica Campestre, Hospital General de Mexico, Hospital General Regional IMSS Nº 200, Hospital General Regional IMSS Nº 196 ESTADO: GUANAJUATO TITULO: Carcinoma de mama con celulas gigantes tipo osteoclasto. Informe de un caso de una variante rara. 2do Congreso Anual de Patólogos Mexicanos • 42 León, Guanajuato • 2016 PATOLOGÍA PEDIÁTRICA Mampara: 13 A desmoplasico es negativo mientras el Osteosarcoma de bajo grado es positivo. El fibroma desmoplasico es localmente agresivo representa menos del 1 % de las lesiones primarias de hueso tiene una tasa de recurrencia de 42 % si se trata con curetaje más que si se realiza resección en bloque. INSTITUCIÓN: *Anatomopatologa Adscrita a Fecha y hora Instalación: 2 de mayo 13:00 Hospital de Pediatría Centro Médico Nacional Siglo XXI. **Anatomopatologa Adscrita a HosFecha y hora retiro: 2 de mayo 18.30 pital de Especialidades Centro Médico Nacional La Raza. *** Médico Laboratorio Estatal de Hidalgo. TC-73 AUTORES: *Dra. Alicia Rodríguez Velasco, *Dra. Marisi Meza Caballero, ***Dra. Georgina Siordia Reyes **Dra. Blanca Sinahi García Aguilar, ****Dr. Abel Meza Morales ESTADO: DISTRITO FEDERAL TITULO: Fibroma Desmoplasico Intramedular Mampara: 13 B Una Entidad Poco Frecuente. Reporte De Un Fecha y hora Instalación: 2 de mayo 13:00 hrs. Caso Y Revisión De La Literatura. Fecha y hora retiro: 2 de mayo 18.30 hrs. INTRODUCCIÓN: Paciente de 15 años masculino que cursa con una lesión osteolitica en diáfisis de fémur derecho con una imagen radiológica con involucro intramedular sin infiltrar tejidos blandos. Los hallazgos radiográficos conforma a una lesión osteolítica heterogénea que involucra diáfisis de fémur con reacción cortical sin aparente infiltración a tejidos blandos. TC-77 OBJETIVOS: Se recibe producto de resección en blo- TITULO: PERFIL DE EXPRESIÓN DE MICRORNAS EN ASTROCITOMAS PEDIÁTRICOS que de fémur distal con 20 x 4 x 4 cm al corte se identifica una lesión intramedular de bordes regulares de 3.5 x 2 x 2 cm blanco gris blanco gris, consistencia blanda. El examen microscópico muestra una lesion de células fusiformes con núcleo normocromatico extensa colagenización sin atipia ni mitosis. MATERIAL: Se realizó inmunomarcación B-catenina positivo en 10% de las células, Actina musculo liso positivo, Ki67 -1%, CD117 negativo. RESULTADOS: El fibroma desmopásico es un tumor poco frecuente que morfológiamente semeja a la fibromatosis de tipo desmoide de tejidos blandos, ocurre en las primeras tres décadas de vida. Los sitios más afectados son huesos pélvicos, huesos largos y mandíbula es localmente agresivo presentandose como lesión osteolítica expansiva sin formación de matriz ósea ni mineralización. Microscópicamente se compone de una población de células fusiformes con núcleos normocromaticos sin atipia con áreas hipocelulares. Las mitosis son extremadamente raras, existe presencia de canales vasculares similares a los encontrados en fibromatosis de tipo desmoide con un patrón infiltrativo. CONCLUSIONES: El diagnóstico diferencial principal es con displasia fibrosa y un osteosarcoma de bajo grado. Inmunohistoquimica: positividad a B-catenina, MDM-2 Y CDK4 o la ampliación molecular de MDM-2 ya que el fibroma AUTORES: Eguía-Aguilar P, Pérezpeña-Díazconti M,Retana-Contreras C, Chico Ponce de León F,Gordillo-Domínguez L, González-Carranza V, Arenas-Huertero F INTRODUCCIÓN: En niños los astrocitomas ocupan el segundo lugar de incidencia de neoplasias malignas después de las leucemias y linfomas. Recientemente se ha comprobado el papel de los microRNAs (miRNAs) en el desarrollo de diferentes tipos de cáncer. Las alteraciones de los miRNAs observadas en los astrocitomas de adultos son distintas a las descritas en población pediátrica. OBJETIVOS: En los astrocitomas de adulto hay un amplio grupo de microRNAs descritos, en los tumores de los niños la información al respecto es escasa, por lo que nos preguntamos si la expresión de los microRNAs puede caracterizar a los distintos grados de astrocitomas pediátricos, alto grado y bajo grado. MATERIAL: De manera inicial se analizó la expresión de ocho miRNAs en astrocitomas GI y GIV pediátricos. Posteriormente se eligieron tres microRNAs, miR-124-3p, miR128-3p y miR-221-3p y se cuantificaron los niveles de expresión de las formas precursoras y maduras en los cuatro grados de astrocitomas pediátricos. Se incluyeron 13 muestras de corteza cerebral no neoplásica. Las diferencias se analizaron con t-student y ANOVA RESULTADOS: En astrocitoma GIV la expresión de miR- 124-3p y -128-3p descendió de manera significativa, 407 y 1469 veces, respectivamente. En cuanto a la expresión de 2do Congreso Anual de Patólogos Mexicanos • 43 León, Guanajuato • 2016 las formas precursoras de miR-128 y -221 se observó que en GIV su expresión fue mayor (3.5 veces) en comparación con GI. CONCLUSIONES: De manera general en los casos GIV recurrentes que fallecieron y en los de bajo grado que recurrieron, los miR-124-3p y miR-128-3p siempre disminuyeron su expresión a la mitad con respecto a los niveles observados en los casos que no recurrieron y siguen vivos. la baja expresión de los tres microRNAs caracteriza a los astrocitomas de GIV y su alta expresión puede constituirse como un factor pronóstico favorable para el paciente. INSTITUCIÓN: Laboratorio de Biología Molecular, Departamento de Patología Clínica y Experimental y Departamento de Neurocirugía. Hospital Infantil de México Federico Gómez ESTADO: DISTRITO FEDERAL Mampara: 14 A RESULTADOS: Se estudiaron 42 ARMS de los cuales 27 (64%) fueron PAX3 positivos, 2 (5%) PAX7 positivos y 13 (31%) fueron negativos a la fusión, también se incluyeron 13 controles. La expresión de miR-221 no mostró diferencias entre los grupos estudiados y el control, al comparar los ARMS con translocación contra los ARMS negativos a fusión, se observó una mayor expresión de miR-221 en estos últimos. Este mismo comportamiento se observó para miR-29b y -29c CONCLUSIONES: Nuestros resultados muestran que la expresión de los miRNAs estudiados se ve más afectada en los ARMS con translocación PAX3/FOXO1 o PAX7/ FOXO1, que en los casos de ARMS que no muestran translocación. Este método es una herramienta útil para apoyar la presencia o ausencia de translocación INSTITUCIÓN: Laboratorio de Biología Molecular, Departamento de Patología Clínica y Experimental. Hospital Infantil de México Federico Gómez ESTADO: DISTRITO FEDERAL Fecha y hora Instalación: 2 de mayo 13:00 Fecha y hora retiro: 2 de mayo 18.30 Mampara: 14 B TC-78 Fecha y hora Instalación: 2 de AUTORES: Pérezpeña-Díazconti Mario, Eguía-Aguilar Fecha y hora retiro: 2 de mayo 18.30 Pilar, Retana-Contreras Carmen, Arenas-Huertero Francisco mayo 13:00 TC-92 TITULO: Expresión de miR-221, miR-29b y miR-29c en rabdomiosarcoma alveolar positivo para PAX3/FOXO1, PAX7/FOXO1 o negativo a fusión INTRODUCCIÓN: La expresión de oncoproteínas de fusión generadas por translocaciones cromosomales, representan un mecanismo de carcinogénesis en diferentes tipos de cáncer. En el rabdomiosarcoma alveolar se conocen dos translocaciones t(2;13)PAX3/FOXO1 y t(1;13)PAX7/FOXO1 exclusivas. Los microRNAs participan en la regulación de varios procesos biológicos como proliferación celular, apoptosis, diferenciación y cáncer OBJETIVOS: En ARMS miR-29b/c y miR-221 se encuen- tran alterados. Sin embargo, se desconoce si hay diferencias en la expresión de estos miRNAs en relación al tipo de translocación. En este estudio se exploró la expresión de miR-29b/c y -221 en ARMS con translocación PAX3, PAX7 o negativos a fusión y su correlación MATERIAL: Se utilizó PCR para determinar el tipo de translocación y PCR en tiempo real para estudiar la expresión de los miRNAs, como controles endógenos se usaron GAPDH y RNU-48, respectivamente. Los casos se obtuvieron del archivo de bloques de parafina del Departmento de Patología AUTORES: Dra. María José González Salazar, Dra. Gloria Madrid Valero, Dr. Luis Antonio Sepúlveda Rodríguez, Dr. Miguel Ángel Paz, Dr. Diego Rodríguez Mendoza, Dr. Horacio Decanini Arcaute. TITULO: HEMANGIOMA CAVERNOSO DEL TESTÍCULO: REPORTE DE UN CASO. INTRODUCCIÓN: Se trata de un paciente masculino de 1 mes de edad, con diagnóstico prenatal a las 30 semanas de gestación de hidronefrosis renal izquierda y estenosis ureterovesical izquierda. Fue producto pretérmino por cesárea de un embarazo normoevolutivo. OBJETIVOS: Al nacer se le realiza ultrasonido identifi- cando testículo izquierdo de 3 cm de tamaño, localizado en cavidad abdominal. Al mes de edad se le realiza ultrasonido de seguimiento evidenciado aumento del tamaño testicular hasta 4 cm, heterogéneo y de vascularidad aumentada. Se ingresa a esta unidad para realización de orquidectomía radical izquierda. MATERIAL: En el departamento de patología se recibe testículo que pesa 21 g y mide 5.0 x 3.0 x 2.5 cm. Se encuentra cubierto por túnica vaginal de superficie lisa, de co- 2do Congreso Anual de Patólogos Mexicanos • 44 León, Guanajuato • 2016 lor violáceo, y con red vascular prominente. A la sección se observa salida de líquido hemático, el testículo es de color café oscuro y presenta áreas quísticas. Se identifica epidídimo de 3.0 x 1.2 x 0.3 cm, café y blando. RESULTADOS: Los cortes histológicos realizados muestran luces vasculares congestivas, dilatadas y de pared delgada, sin datos de atipia, algunos corresponden a vasos sanguíneos arteriales. En la periferia se identifica testículo y epidídimo de histología conservada. Se realiza inmunohistoquímica, resultando positivo para los marcadores CD34 y Factor VIII. CONCLUSIONES: Los hemangiomas cavernosos son considerados como malformaciones vasculares, correspondiendo a una alteración congénita en la morfogénesis de los vasos sanguíneos. Actualmente en la literatura, se han reportado alrededor de 20 casos de anomalías vasculares en el testículo, siendo la variante más común el hemangioma cavernoso. El rango de edad varía entre 17 semanas a 77 años de edad. Se ha reportado un caso previo de hemangioma capilar asociado a criptorquidia. INSTITUCIÓN: UNIVERSIDAD DE MONTERREY “ HOSPITAL CHRISTUS MUGUERZA ALTA ESPECIALIDAD. ESTADO NUEVO LEÓN Mampara: 15 A Fecha y hora Instalación: 2 de mayo 13:00 hrs. Fecha y hora retiro: 2 de mayo 18.30 hrs. TC-111 vivencia en comparación con la cirugía única. Es quimiorresistente, con pobre pronóstico. Presentamos caso en centro pediátrico de tercer nivel. OBJETIVOS: Reporte de caso MATERIAL: Realizamos estudio de inmunohistoquímica con los marcadores TFE3, Ki67, desmina, AE1-AE3, Miogenina, distrofina, actina de músculo liso, CD10, S100, CD31, así como histoquimica con PAS, PAS-D y estudio de microscopía electrónica. RESULTADOS: Hombre, 15 años, nódulo único en región temporal derecha, siete meses de evolución.Seis meses posteriores, desarrolló lesión en brazo izquierdo, referido a INP, se realizó biopsia de ésta que reportó sarcoma alveolar de partes blandas; la RMN de cabeza demuestra lesión nodular lobulada en tejidos blandos en región temporal derecha, durales en región parietal izquierda, subcortical en región precentral derecha, reforzamiento nodular central y edema circundante.TAC pulmonar y PET-CT con actividad en pulmón, hombro izquierdo, brazo izquierdo, pulmones e hígado.El tratamiento fue quimioterapia y radioterapia paliativa.El diagnóstico es morfológico con inmunotinción con TFE-3 y ultraestructura. CONCLUSIONES: Este caso refleja la presentación clínica, evolución, comportamiento, histopatología, inmunofenotipo y características ultraecturales de esta entidad, con lo que al igual que en la literatura corroboramos origen miógeno desmina positivo así como la presencia de la expresión nuclear de TF3 y se corroboró ausencia de primario renal. INSTITUCIÓN: 1Residente III año INCMNSZ 2Residente II año Patología Pediátrica 3MA Departamento Patologia Instituto Nacional de Pediatría ESTADO: DISTRITO FEDERAL AUTORES: Dr. Alejandro Francisco Cruz1, Dra. Laura Rosario Cárdenas Mastrascusa2, Dr. Celso Tomás Corcuera Delgado3 Mampara: 15 B TITULO: SARCOMA ALVEOLAR DE PARTES BLANDAS EN PEDIATRIA: PRESENTACIÓN DE UN CASO Fecha y hora Instalación: 2 de Fecha y hora retiro: 2 de mayo 18.30 INTRODUCCIÓN: El sarcoma alveolar de partes blan- TC-117 das fue descrito en 1950. Afecta principalmente extremidades inferiores (glúteos y muslo) siendo el 40% de los casos. En niños ocurre con en cabeza y cuello. Es un tumor maligno que representa el 0.2-0.9% de los sarcomas de partes blandas, en pacientes entre 15 y 35 años; con proporción mujer:hombre de 2:1. Curso clínico indolente, con tendencia a metástasis por vía hematógena. El tratamiento del tumor y metástasis pulmonares es quirúrgico, pero no curativo, por lo que la radioterapia tiene un papel importante en el tratamiento de la enfermedad residual. Se ha observado que la RT adyuvante para pacientes con enfermedad locorregional en el momento del diagnóstico, se asocia con mejor super- mayo 13:00 AUTORES: DRA. BÁRBARA SÁENZ, Dra. Eirali Garcia Chapa, Dr. Med. Marco Antonio Ponce Camacho, Dra. Med. Oralia Barboza Quintana. TITULO: Hemangioendotelioma infintil retroesternal: reporte de un caso INTRODUCCIÓN: El término hemangioendotelioma fue acuñado por Mallory (1908) para describir todos los tumores que derivan del endotelio de los vasos sanguíneos. Este tér- 2do Congreso Anual de Patólogos Mexicanos • 45 León, Guanajuato • 2016 mino es utilizado para nombrar los tumores vasculares que muestran un comportamiento biológico borderline. En el caso del tipo infantil, corresponde a un subtipo compuesto por capilares, que ocurren con en la infancia. Aun y cuando esta descrito que pueden llegar presentar regresión, el tratamiento debe ser individualizado ya que depende de la localización y lo rápido del crecimiento. blación pediátrica, sin embargo es una neoplasia muy poco frecuente que a menudo se asocia con elevación de alfa-fetoproteína. Existe una ligera predilección hacia los varones y ocurre casi invariablemente en la primera década de la vida con una edad media de 4.5 años, se trata de un tumor sólido, de localización variable en el páncreas. OBJETIVOS: Describir la presentación clínica y hallazOBJETIVOS: Presentar el caso de un paciente femenino gos radiológicos del pancreatoblastoma en una edad infrede 1 año de edad con una tumoración retroesternal con este diagnóstico. MATERIAL: Se recuperó el historial clínico de la paciente, así como los estudios de imagen y gabinete para poder documentar el caso. RESULTADOS: Es una paciente femenina de 1 año de edad hospitalizada desde los 8 días de vida por dificultad de respiratoria que requirieron intubación orotraqueal, debido a un tumor definido por tomografía como una masa hipercaptante al medio de contraste mal definida que se extiende desde subglotis rodeado estructuras a nivel de cuello hasta mediastino. En la resección se resecan múltiples fragmentos que a nivel histológico forman múltiples canales vasculares rodeados de células endoteliales de aspecto benigno, sin embargo con bordes positivos para neoplasia. En la inmunohistoquímica fue Glut-1 y CD 34 positivo y D240 negativo. cuente. MATERIAL: Paciente femenino de 12 años de edad, originaria de León Guanajuato, inició su padecimiento actual el 15.07.14 con ictericia, astenia, adinamia, artralgias, llegando a incapacitarla para sus actividades con diagnóstico de probable Hepatitis. Acudió al Hospital General León el 11.08.2014 por dolor en flanco dere RESULTADOS: Resultados. En el laboratorio de pa- tología se realizaron cortes histológicos del tumor el cual presenta un aspecto embrionario con diferenciación ácinar, algunas zonas mejor diferenciadas que recuerdan cordones de hepatocitos y zonas amplias de necrosis, fue reportado como hepatoblastoma puro epitelial tipo fetal, sin embargo la paciente no tuvo buena respuesta al tratamiento con quimioterapia por lo cual se realizó resección completa del tumor, se envió pieza a patología el cual se obtuvo resultado definitivo de Pancreatoblastoma. CONCLUSIONES: Los tumores vasculares presentes CONCLUSIONES: Conclusión: La rareza de esta neo- en infantes, aunque se han descrito que la gran mayoría tienen regresión, existe un bajo porcentaje como en el caso de nuestro paciente en que la vida se ve amenazada por comprimir las vías aéreas, por lo que se necesita extrema vigilancia de este tipo de tumores sin importar la edad. plasia y su cercanía con el hígado no permitieron diferenciar su origen desde el inicio, por otra parte, desde el punto de vista histológico tanto el pancreatoblastoma como el hepatoblastoma comparten algunas características morfológicas dado su origen embrionario. INSTITUCIÓN: Hospital Universitario José Eleuterio Gonzalez INSTITUCIÓN: HOSPITAL GENERAL DE LEON ESTADO: NUEVO LEÓN ESTADO: GUANAJUATO Mampara: 16 B Mampara: 16 A Fecha y hora Instalación: 2 de mayo 13:00 hrs. Fecha y hora retiro: 2 de mayo 18.30 hrs. TC-122 Fecha y hora Instalación: 2 de mayo 13:00 hrs. Fecha y hora retiro: 2 de mayo 18.30 hrs. TC-139 AUTORES: Dr. Mario Murguía Pérez, Dra. Teresa Lopez AUTORES: Dra. Elsa Yzayhat Flores Salazar, Dra. Paulina Montaño Ascencio, Dra.Juana Rosalba García Ramírez Gaytan, Dr. Lazaro Ariel Ramirez Balderrama, Dr. Jorge Israel Muñoz Torres, Dr. Felipe Navarro Córdoba, Dr. Adrián Antonio López Vera, Dr. Saulo Mendoza Ramirez TITULO: PANCREATOBLASTOMA EN LA TITULO: Hamartoma fibroso de la infancia. Reporte de un caso. EDAD PEDIATRICA, REPORTE DE UN CASO INTRODUCCIÓN: Introducción. El pancreatoblastoma es la neoplasia maligna de páncreas más común en la po- INTRODUCCIÓN: El hamartoma fibroso de la infancia (HFI) es un tumor benigno, fibroso, de crecimiento rapido, cuya frecuencia es mayor durante los primeros dos años de 2do Congreso Anual de Patólogos Mexicanos • 46 León, Guanajuato • 2016 la vida y muestra predileccion por el torax, axila, hombro y region inguinal. Aunque representa el 0.02%de todos los tumores fibrosos benignos, es una de las neoplasias fibrosas mas comunes en niños. OBJETIVOS: Presentamos un HFI tratado en nuestra unidad, el cual incialmente fue abordado como un sarcoma de tejidos blandos. Fecha y hora retiro: 2 de mayo 18.30 hrs. TC-154 AUTORES: Mariela Sánchez Claudio, Jocabed Martínez Olivares, Víctor Manuel Monroy Hernández. TITULO: Nefroma Quístico Y Nefroblastoma Quístico Parcialmente Diferenciado En Edad MATERIAL: Se trato de niño de 9 años de edad, sin antecedentes de importancia, que acudio al servicio de cirugia Pediátrica: Serie De 4 Casos Diagnósticados pediatrica con un tumor de gran tamaño ubicado en hombro En El Hospital De Especialidades Centro Méizquierdo, con dificultad para la elevacion de la extremidad dico Nacional La Raza. superior. En la tomografia computada de cuello y torax se aprecio una lesion hiperdensa, bien delimitada, encapsulada, sin invasion a otras estructuras y sin datos de necrosis y hemorragia. RESULTADOS: Se realizo una biopsia por trucut en la cual se identifico un tumor benigno caracterizado por una mezcla de fasciculos fibroblasticos bien diferenciados, tejido adiposo maduro y escasas areas con celulas mesenquimales de apariencia primitiva, y areas hialinizadas con artificios de ruptura. Por lo anterior se diagnostico como HFI. Se realizo excision completa de la lesion con margenes amplios, recibiendo en el servicio de patologia una lesion de 11.7 x 10.3 x 6 cm, bien delimitada, semilisa, de color cafe grisaceo amarillento, al corte era homogenea, solida, con formacion de septos finos de tejido conectivo, y areas adiposas y mixoides, sin zonas de necrosis y hemorragia. Microscopicamente se observaron las mismas caracteristicas identificadas en la biopsia por trucut, con mayor predominio de tejido adiposo y nodulos de celulas mesenquimales de apariencia primitiva, con margenes quirurgicos negativos. Por lo anterior, se emitio el diagnostico de HFI. CONCLUSIONES: Aunque el diagnostico de esta lesion se realiza con criterios morfologicos, en ocasiones puede realizarse CD34 para diferenciarlo de un fibroblastoma de celulas gigantes. Tambien es necesario descartar dermatofibrosarcoma protuberans, lipofibromatosis y fibroma aponeurotico calcificante. El tratamiento de estas lesiones es la excision completa de la lesion, como ocurrio en nuestro caso. A un año de su diagnostico y tratamiento, actualmente el paciente no presenta recidivas, y recupero la movilidad de la extremidad al 100%. A pesar de su nombre, estas lesiones representan tumores benignos mas que verdaderos hamartomas. INSTITUCIÓN: Unidad Médica de Alta Especialidad No. 1 IMSS Leon, Hospital Medica Campestre, Hospital General de Mexico INTRODUCCIÓN: El nefroma quístico y el nefroblastoma quístico parcialmente diferenciado son tumores renales multiquísticos, benignos, raros, que pertenecen a un espectro de tumores que incluye al nefroblastoma (Tumor de Willms). Son frecuentes en niños menores de 2 años, predominantemente en hombres. La presentación clínica más frecuente es como una masa incidental abdominal. OBJETIVOS: Seleccionar los tumores diagnósticados como nefromas quísticos y nefroblastomas quísticos parcialmente diferenciados en personas menores de 15 años para describir y analizar las características clínicas e histopatológicas de éste espectro de tumores. MATERIAL: Estudio retrospectivo, observacional, des- criptivo y comparativo. Recolectamos 80 casos de tumores renales en pacientes menores de 15 años, de 1993 al 2013 en el servicio de Anatomía Patológica del Hospital de Especialidades “Antonio Fraga Mouret, en CMN “La Raza” y se seleccionaron los casos diagnósticados como nefroma quístico y nefroblastoma quístico parcialmente diferenciado con su historia clínica y su seguimiento clínico correspondiente. RESULTADOS: Se identificaron cuatro casos con com- ponente quístico, dos de ellos diagnosticados como nefroma quístico y dos como nefroblastoma quístico parcialmente diferenciado. Todos fueron hombres, en un rango de 1 a 2 años de edad, dos izquierdos y dos derechos, en todos los casos, clínicamente con incremento de volumen en los flancos; macroscópicamente, multiquísticos y bien delimitados. Microscópicamente, los quistes revestidos por epitelio plano, cúbico o en “tachuela”, con tubulos maduros atrapados entre los septos y algunos focos de blastema sin anaplasia. Todos con nefrectectomía total y con seguimiento de cinco años, sin complicaciones. CONCLUSIONES: Éste espectro de tumores quísticos ESTADO: GUANAJUATO Mampara: 17 A Fecha y hora Instalación: 2 de mayo 13:00 hrs. representa el 5% del total de los tumores renales recolectados. La importancia de identificar estas lesiones, radica en el pronóstico y tratamiento del paciente y en hacer el diagnóstico diferencial con riñones poliquísticos. En general, el pronóstico de éstos tumores es favorable posterior a la nefrectomía sin requerir quimioterapia. Se ha encontrado que el 10% de éstas neoplasias, pueden estar asociadas a blastomas pleuropulmonares, lo cual repercute en un peor 2do Congreso Anual de Patólogos Mexicanos • 47 León, Guanajuato • 2016 pronóstico; en nuestra serie de casos no se encontró tal asociación. CONCLUSIONES: Nuestros hallazgos histológicos y ultraestructuales fueron los característicos de le enfermedad (LALD) que debe de buscarse en todos aquellos pacientes con esteatosis microvesicular y una cuadro clínico sugestivo. INSTITUCIÓN: HGO 3 Dr. Víctor Manuel Espinosa de los Reyes Sánchez Centro Médico Nacional “La Raza”, Hospital de Especialidades “Antonio Fraga Mouret” Centro Médico Nacio- INSTITUCIÓN: HOSPITAL INFANTIL DE MÉXICO, FEDERICO GÓMEZ nal “La Raza”. ESTADO: ESTADO DE MÉXICO ESTADO: DISTRITO FEDERAL Mampara: 17 B Mampara: 18 A Fecha y hora Instalación: 2 de mayo 13:00 Fecha y hora retiro: 2 de mayo 18.30 Fecha y hora Instalación: 2 de mayo 13:00 hrs. Fecha y hora retiro: 2 de mayo 18.30 hrs. TC-157 TC-160 AUTORES: Dr. Fernando Villavicencio1, Dr. Pedro Va- AUTORES: 1Eguía-Aguilar P, 1Cruz-Contreras Luis, lencia Mayoral1. Dra. Alejandra Consuelo2; Dra. Penélope Ortal2; Dr. Carlos Serrano Bello1; 1,- Departamento de patología; 2.- Departamento de gastroenterología TITULO: DEFICIENCIA DE LIPASA ACIDA LISOSOMAL EN BIOPSIAS HEPÁTICAS. REPORTE DE 10 PACIENTES DEL HOSPITAL INFANTIL DE MÉXICO FEDERICO GÓMEZ INTRODUCCIÓN: La deficiencia de lipasa acida lisosomal (LALD) es una enfermedad metabólica con incidencia de 1 en 300000 nacimientos, se caracteriza por ausencia total de la enzima (enfermedad de Wolman) o parcial de la enzima (Enfermad por almacenamiento de esteres de colesterol) lo que lleva al depósito intra lisosoma de lípidos. OBJETIVOS: Describir la frecuencia y características de la enfermedad por deficiencia de lipasa ácida lisosomal (LALD) en pacientes pediátricos del Hospital Infantil de México. MATERIAL: Revisamos los casos con diagnóstico de enfermedad de Wolman o de almacenamiento de esteres de colesterol (LALD), diagnosticados en el departamento del patología del hospital. RESULTADOS: Obtuvimos 10 casos con edad media de 52 meses. Todos presentaron hepato-esplenomegalia, hipertensión portal, elevación de las amino trasferasas, colesterol LDL elevado y HDL bajo. Los hallazgos microscópicos presentan, esteatosis micro vesicular difusa, cristales de colesterol intracitoplásmicos en células de Kupfer y hepatocitos con refringencia a la luz polarizada y macrófagos de citoplasma espumoso en espacios porta. La microscopia electrónica contribuyo a demostrar el depósito de lípidos intralisosomales y de los cristales de colesterol. La determinación enzimática demostró niveles por debajo de 0.01 mMol. 1PérezPeña-DíazConti M, 1Serrano-Bello Carlos, 1García-Quintana J, 1Retana-Contreras Carmen, 1Arenas-Huertero F, 1Valencia-Mayoral P TITULO: Expresión de miR-100 y miR-221 en tumor rabdoide/teratoide atípico del sistema nervioso central INTRODUCCIÓN: Introducción. En niños los tumores de sistema nervioso ocupan el segundo lugar de incidencia después de las leucemias; el tumor rabdoide teratoide atípico (TR/TA) representa del 1 a 2 % de todos los tumores pediátricos de sistema nervioso central, es un tumor de comportamiento agresivo y de mal pronóstico a corto plazo. La característica más frecuente de estos tumores es la mutación o pérdida en la expresión del gen INI1 (hSNF5/ SMARCB1) localizado en el cromosoma 22q11.2. Otros mecanismos moleculares se han explorado en estos tumores, como la regulación post-transcripcional ejercida por los microRNAs. Varios estudios muestran que la expresión alterada de los microRNAs en diferentes tipos de tumores se relaciona con su desarrollo y progresión. OBJETIVOS: Determinar el nivel de expresión de los microRNAs miR-100 y miR-221 en tumores rabdoides tetratoides atípicos en pacientes del HIMFG MATERIAL: Las muestras fueron obtenidas del banco de tejidos del Departamento de Patología del HIMFG. Abarcaron los años 2003 al 2014. Se incluyeron 13 muestras de TR/TA y 8 muestras de cerebro de autopsias de pacientes libres de tumor. Se excluyeron 9 casos con diagnóstico TR/ TA por no contar con material suficiente. Se analizó la expresión de miR-100 y miR-221 por PCR en tiempo real utilizando sondas TaqMan. Se realizó cuantificación relativa RESULTADOS: En las muestras estudiadas, se obtuvo RNA en cantidad y calidad suficiente para ser estudiadas, 2do Congreso Anual de Patólogos Mexicanos • 48 León, Guanajuato • 2016 tomando como control RNU48, gen constitutivo. En los tumores TR/TA, el mir-100 se observó 14 veces menos expresado (P= 0.0005) y miR-221, 43 veces (P <0.0001) con respecto al control no neoplásico> ca tumor germinal variante senos endodermicos en un 90% y tumor de los cordones sexuales sertoli “ Leydig. El estudio de inmunohistoquimica fue positivo a: CK AE1/AE3, Inhibina, alfa feto proteína, Vimentina y positivo focal para EMA. CONCLUSIONES: En los casos que estudiamos los CONCLUSIONES: Los tumores mixtos de ovario (seniveles de miR-100 tuvieron niveles bajos, como se ha referido en la literatura, expresión baja mostrada en otros tumores como cáncer de vejiga, ovario y gliomas. miR-100 actúa como supresor tumoral a través de regular blancos importantes como mTOR. Por su parte los niveles de miR221 que encontramos fueron bajos, al igual que en casos de glioblastoma multiforme. En otras neoplasias fue reportado con expresión alta; sus implicaciones en el desarrollo de este tipo de tumores dependerán de los blancos que este regulando nos endodermicos + tumor sertoli - leydig), son tumores raros que requieren especial estudio mediante tinciones especiales. INSTITUCIÓN: Hospital Central ¨Dr. Ignacio Morones Prieto¨, San Luis Potosí, S.L.P. Universidad Autonoma de San Luis Potosí, SLP. Departamento de Anatomia Patologica ESTADO: SAN LUIS POTOSÍ INSTITUCIÓN: 1Laboratorio de Biología Molecular, Departamento de Patología Clínica y Experimental; Hospital Infantil de México Federico Gómez. ESTADO: DISTRITO FEDERAL MARTES 3 DE MAYO Mampara: 18 B Fecha y hora Instalación: 2 de mayo 13:00 hrs. Fecha y hora retiro: 2 de mayo 18.30 hrs. TC-179 AUTORES: Sánchez García Citlalli Edith, Oros Ovalle Cuauhtemoc PATOLOGÍA DE TEJIDOS BLANDOS TITULO: Tumor De Ovario Mixto (Tumor Germinal Variedad Senos Endodermicos + Tumor De Mampara: 1 A Los Cordones Sexuales Sertoli-Leydig). PreFecha y hora Instalación: 3 de mayo 7:30 hrs sentacion De Un Caso Pediatrico. Fecha y hora retiro: 3 de mayo 13:00 hrs INTRODUCCIÓN: Se presenta un caso de una niña de 1 año y 7 meses con un tumor de ovario mixto: germinal variedad senos endodermicos y tumor de los cordones sexuales sertoli “ Leydig. Se revisa la literatura sobre esta rara presentación. OBJETIVOS: Conocer e identificar este infrecuente tumor de ovario con los hallazgos macroscópicos y microscópicos. MATERIAL: Se analiza espécimen de ooferectomia derecha de 9.0 x 7.5 x 5.5cm, al corte se identifica tumor blanco rosado con áreas amarillentas, necrosis y microhemorragia, sólido y de aspecto mucinoso. RESULTADOS: En los cortes histológicos y mediante método convencional con hematoxilina y eosina, se identifi- TC-32 AUTORES: Irving Daniel Ortiz Sánchez, Ramiro Sandoval Macías, Angélica Grajales Rodríguez, Candelaria Córdova Uscanga, Ana Lilia Remírez Castellanos, Fernando Candanedo González. TITULO: ANÁLISIS CLÍNICO PATOLÓGICO DE PACIENTES CON TUMORES DE TEJIDOS BLANDOS EN MEDIASTINO INTRODUCCIÓN: Los tumores de tejidos blandos en mediastino son muy poco frecuentes, representan 2 a 6%, en su mayoría son benignos. El cuadro clínico es inespecífico. La biopsia con aguja de corte guiada por imagen permite 2do Congreso Anual de Patólogos Mexicanos • 49 León, Guanajuato • 2016 establecer un diagnóstico oportuno y su resección quirúrgica. OBJETIVOS: Analizar las características clínico patológicas de pacientes con tumores de tejidos blandos en mediastino en el Hospital de Oncología. MATERIAL: Se realizó estudio retrospectivo de pacien- tes con diagnóstico de tumor de tejidos blandos en mediastino en el periodo de Enero de 2014 a Diciembre de 2015, en el Hospital de Oncología de CMN Siglo XXI. Se revisaron los informes de patología del archivo de quirúrgicos del departamento de patología. RESULTADOS: Se identificaron un total de 60 pacientes con neoplasia mediastinal, de los cuales 5 pacientes correspondieron a tumores de tejidos blandos. Se identificó un solo caso que infiltraba pulmón, mientras que otro caso comprimía parcialmente algunos vasos de gran calibre y en otro caso infiltraba al esófago. El tamaño promedio de los tumores fue de 17 cm de eje mayor. En todos los casos se realizó resección total del tumor mediante toracotomía. Sólo en 2 pacientes fue necesaria la quimioterapia post-resección. Microscópicamente, se encontraron 3 Schwannomas, un sarcoma indiferenciado y un liposarcoma.La supervivencia de los tumores malignos fue de 12 meses hasta el momento que duró el estudio. TITULO: Sarcoma Fibroblastico Mixoinflamatorio: Presentacion De Un Caso Y Revision De La Literatura. INTRODUCCIÓN: El sarcoma fibroblástico mixoinflamatorio es un tumor maligno raro de bajo grado que característicamente afecta a varones y mujeres entre los 40 y 50 años de edad. Y que usualmente afecta manos y manos. Este tumor tiene una amplia gana de diagnósticos diferenciales, desde un proceso inflamatorio reactivo y con tumores de mayor potencial de malignidad. OBJETIVOS: Presentamos el caso de un paciente masculino de 80 años de edad con antecedente de traumatismo 2 meses previos a su valoración, quien posteriormente desarrolla una lesión exofìtica de 4x4cm en el 5t dedo de la mano izquierdo, la radiografía demostró una lesión dependiente de tejidos blandos. Se realiza biopsia por sacabocado RESULTADOS: Microscopicamente se observa una lesión localizada en la dermis y tejido celular subcutáneo, con un componente fusocelular, infiltrado inflamatorio y nódulos mixoides, dentro de las células neoplásicas se encuentran algunas con citoplasma abundante, núcleo agrando y núcleolo visible, las cuales se describen como “virocitos-like”, los nódulos mixoides muestran la presencia de múltiples células neoplásicas con inclusiones de mucina que les otorgan CONCLUSIONES: En nuestra serie los tumores de tejidos blandos en mediastino representan 8% de todas las neoplasias de mediastino. Fue más frecuente en el género femenino, y se presentó con mayor frecuencia en pacientes jóvenes con edad promedio de 44 años. Los tumores de tejidos blandos benignos que se encontraron con mayor frecuencia fueron los Schwannomas. El sitio más frecuente fue el compartimiento posterior (3/5 pacientes del estudio. Los tumores malignos mostraron una evolución más corta (en promedio 4.5 meses), que los benignos (en promedio 9 meses). INSTITUCIÓN: Departamento de Patología, Hospital de Oncología de Centro Médico Nacional Siglo XXI. IMSS. Ciudad de México. ESTADO: ESTADO DE MÉXICO Mampara: 1 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-43 AUTORES: DR. LUIS ARTURO ACOSTA, Dra. Lucía Alemán Meza, Dra. Oralia Barboza Quintana, Dra. Ivette Miranda Maldonado CONCLUSIONES: El diagnóstico diferencial debe in- cluir un proceso inflamatorio reactivo, dentro de los imitadores a descartar es el fibromixosarcoma, fibrosarcoma mixoide de bajo grado, tumor de células gigantes tenosinovial localizado y sarcoma epiteloide, ninguno de los tumores antes mencionado presenta áreas mixoides y fusocelulares entremezcladas con infiltrado inflamatorio mixto, la positivad para INI-1 descarta el diagnóstico de sarcoma epiteloide. Se han reportado 128 casos en la literatura, es una entidad recientemente descrita por lo que se debe de incluir en el espectro de las lesiones mixoides. INSTITUCIÓN: Servicio de Anatomía Patológica del Hospital Universitario “Dr. José Eleuterio González” de la Universidad Autónoma de Nuevo León. Monterrey N.L. ESTADO: NUEVO LEÓN Mampara: 2 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-96 2do Congreso Anual de Patólogos Mexicanos • 50 León, Guanajuato • 2016 AUTORES: MA. GPE. JAZMÍN DE ANDA, Dra. Blandina Hernández Cruz, Dra. Sonia Tavares García, Dra. Liliana García Ilizaliturri, caso que se presenta pertenece al grupo de los PEComas malignos. INSTITUCIÓN: Hospital de Oncología de Centro Médico Nacional Siglo XXI. TITULO: Tumor de células epitelioides perivasculares en la pared uterina (PEComas), reporte ESTADO: DISTRITO FEDERAL de caso y revisión de la literatura. INTRODUCCIÓN: Los PEComas (neoplasias de células epitelioides perivasculares) han sido definidos por la Organización mundial de la Salud como tumores mesenquimatosos compuestos por células epitelioides perivasculares histológicamente e inmunohistoquímicamente distintos. En esta familia de tumores encontramos el angiomiolipoma, tumor de células claras “sugar tumor” del pulmón y la linfangioleiomiomatosis. Este tipo de tumores se relaciona con el complejo de esclerosis tuberosa. Pueden presentarse en el tracto genitourinario (riñón, vejiga, próstata, útero ovario y vulva). Histológicamente forman nidos o fascículos con apariencia epitelioide o fusocelular con citoplasma claro a granular con un núcleo central, redondo a oval, nucléolo no evidente y atipia leve en caso de que exista. Expresan HMB45, HMSA-1, Melan A, Mart1, y factor de transcripción de microoftalmía y marcadores miogénicos como actina y desmina. OBJETIVOS: Mujer de 42 años de edad con antecedente de esclerosis tuberosa y estenosis subaórtica corregida; inicia su padecimiento actual con alteraciones del ciclo menstrual, dolor abdominal de intensidad moderada; ultrasonido con lesión dependiente de la pared uterina solido quística, probable leiomioma con degeneración quística. Se realizó histerectomía. Histerectomía con salpingooforectomía derecha demostró lesión subserosa dependiente de la pared uterina anterior y fondo uterino de 13 x 11 cm, heterogénea, solido-quística, las áreas solidas nodulares blanquecinas, necrosis, y hemorragia. El área quística con un recubrimiento café obscuro anfractuoso y opaco. Histológicamente se observó una neoplasia con patrón en nidos y fascículos débilmente infiltrante, compuesto por células epitelioides y células fusocelulares, con citoplasma amplio, eosinófilo, finamente granular. Núcleo ovoide, con nucléolo prominente; áreas de necrosis, e índice mitótico de 10 mitosis por 10 campos de alto poder. Los resultados de inmunohistoquímica mostraron positividad para PS100, HMB45, Actina de Musculo liso de manera focal y Melan A. Negatividad para desmina, H-caldesmon y vimentina. CONCLUSIONES: Los PEcomas Uterinos son neoplasias poco frecuentes, afectan a mujeres de entre 40 a 75 años de edad, pueden tener un amplio espectro morfológico diferente, con inmunoexpresión variable de HMB45 y actina y desmina. Aquellos asociados a el complejo de esclerosis tuberosa tienen una conducta mas agresiva. Folpe y cols, proponen un sistema de clasificación para los PEComas del tracto ginecológico, los dividen en benignos, potencial maligno incierto y maligno. Estos autores se basaron en el tamaño del tumor mayor a 5 cm, patrón de crecimiento infiltrante, grado nuclear alto, necrosis, actividad mitótica mayor a 1 en 50 campos de alto poder. De acuerdo a lo anterior el Mampara: 2 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-99 AUTORES: SONIA TAVARES, Blandina Hernández Cruz, Jazmín de Anda González, Liliana García Ilizaliturri, Alejandra Mantilla Morales TITULO: Angiomatosis difusa la gran simuladora de sarcomas de tejidos blandos.Presentaciòn de 4 casos y revisión de la literatura. INTRODUCCIÓN: La angiomatosis o hemangioma difuso es una lesión vascular rara benigna localizada en los tejidos blandos. Afecta el tronco, sin embargo también se puede localizar en las extremidades y menos frecuente en cabeza y cuello. La angiomatosis puede presentarse como una lesión vertical extensa afectando hipodermis, tejido adiposo, musculo y hueso o involucrar un solo tejido de manera extensa. La lesión se caracteriza por una proliferación de vasos sanguíneos mezclados con grandes vasos venosos cavernosos pero también se puede presentar como una gran proliferación de vasos capilares en los tejidos blandos Los diagnósticos diferenciales se deben de hacer con el lipoma, angiomiolipoma y el liposarcoma. RESULTADOS: Sexo Edad Localización Tamaño Dx clínico Mujer 34 Muslo derecho 23 cm Liposarcoma Mujer 33 Muslo izquierdo 17 cm Sarcoma Hombre 32 Muslo derecho 11 cm Liposarcoma Hombre 27 Retroperitoneo 23 Tumor de vaina de nervio periferico Histologicamente en tres de los casos se observo una proliferación vascular, con vasos de gran tamaño infiltrando los tejidos blandos, sin embargo en un caso se identifico una proliferacion capilar adyacente a los vasos de mayor tamaño CONCLUSIONES: La angiomatosis es una lesion poco frecuente que se confunde de manera constante con neoplasias mesenquimatosas malignas de las cuales difiere en pronóstico y tratamiento de manera radical. INSTITUCIÓN: Hospital de Oncología, Centro Médico Nacional Siglo XXI ESTADO: DISTRITO FEDERAL 2do Congreso Anual de Patólogos Mexicanos • 51 León, Guanajuato • 2016 Mampara: 3 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-103 of the gastrointestinal tract. ACG Case Reports Journal 2014;1(4):193-5. 2. McGhan LJ, Wasif N, Young SW, et al. Granular cell tumor of the anterior abdominal wall. Radiology Case Reports 2012;7(3):1-4. 3. Machado I, Cruz J, Lavernia J, y cols. Lesiones ocupantes de espacio en pared abdominal (n herniaria). La visión del patólogo. Rev Hispanoam Hernia 2015;3(3):85-94. AUTORES: Dr. Julio C. Sánchez Venegas, Dr. Cesar I. INSTITUCIÓN: UMAE Hospital de Especialidades Peña Ruelas, Dr. Edmundo Castelán Maldonado, Dra. Julia De Leija P., Dr. Carlos Garza Lizano, Dr. Jorge A. Sánchez Garza, Dr. Luis R. Villalba Monrreal. TITULO: Tumor de Células Granulares en pared abdominal. INTRODUCCIÓN: El tumor de células granulares (TCG) es poco frecuente, el primer caso fue descrito por Abrikossoff en 1926.(1) Pueden localizarse en cualquier sitio, la mayoría ocurre en la cabeza y cuello, seguido por la piel y los tejidos blandos.(2-3) OBJETIVOS: Presentar un caso de tumor de células granulares de localización abdominal, así como la estadística de los casos diagnosticados en nuestra unidad entre 2010 y 2015. MATERIAL: Caso clínico: Mujer de 46 años de edad con antecedente de 2 cesáreas. A la exploración se observa un tumor localizado a nivel de hipogastrio, dependiente de tejidos blandos, indurado, irregular, fijo a la piel. RESULTADOS: Se recibió un espécimen que midió 5 x 4 x 3 cm, de superficie lobulada y cubierto parcialmente por un huso de piel; a la sección se identificó un tumor de 2.5 x 2.0 cm, de bordes estrellados, color café-blanquecino y de consistencia firme. El estudio histopatológico evidencio la presencia de una neoplasia mal delimitada, no encapsulada, constituida por células grandes, poligonales o alargadas con membranas mal definidas que formaban un sincitio; el citoplasma fue granular y eosinófilo, los núcleos centrales y pequeños. La tinción de PAS resaltó el patrón granular del citoplasma. La IHQ demostró positividad difusa e intensa para S100 y CD68 en las células tumorales. CONCLUSIONES: En nuestra unidad se realizó el diag- nóstico de cuatro casos, se identificó una relación hombre mujer de 1:1. El rango de edad fue de 46 a 75 años con un promedio de 50 años. Las localizaciones fueron: mastoides, submaxilar, inguinal y el caso reportado en región abdominal. El tamaño vario de 0.7 a 10 cm, con una media de 2.43 cm. el único criterio de malignidad reconocido son las metástasis, ya que hay reportes de casos que no presentaron ninguno de los criterios y que presentaron metástasis ganglionares o a distancia. El tratamiento es la resección quirúrgica completa. REFERENCIAS: 1. Lipkin-Moore Z, Thomas RM, Rothstein RD. Multifocal synchronous granular cell tumors No. 25, IMSS. Monterrey, Nuevo León. ESTADO: NUEVO LEÓN Mampara: 8 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-116 AUTORES: Susana Elizabeth Martinez, Dra. Claudia Haydee Sarai Caro Sánchez, Dr. Hugo Ricardo Dominguez Malagón ,Dr. Giovanni Socra Chafre Dr. Leonardo Saul Lino Silva TITULO: Tumor fibroso solitario INTRODUCCIÓN: El tumor fibroso solitario en una neo- plasia de tejidos blandos , poco frecuente, con un comportamiento que va desde incierto hasta maligno; desde fque ue descrito, se han tratado de encontrar sus características tanto morfológicas como inmunoreactivas, debido a que la imagen morfológica puede confundirse con otros tumores. Para hacer el diagnóstico se ha descrito como marcador específico el STAT-6 con especificidad y sensibilidad de hasta 98%. El diagnóstico diferencial obligado entre muchos otros es el sarcoma sinovial monofásico. OBJETIVOS: Documentar la sensibilidad y especificidad del marcador de inmunohistoquímica STAT-6 como marcador específico del tumor fibroso solitario en comparación con el sarcoma sinovial. MATERIAL: Estudio descriptivo, retrospectivo y observacional en pacientes con diagnóstico de tumor fibroso solitario y sarcoma sinovial del Instituto Nacional de Cancerología del año 2009 al 2014 con tejido suficiente para realizar microarreglos de tejido. RESULTADOS: Se obtuvieron un total de 19 casos con diagnóstico de tumor fibroso solitario y 20 casos con diagnóstico de sarcoma sinovial en un lapso de 7 años, en el Instituto Nacional de Cancerología. El grupo de pacientes con diagnóstico de tumor fibroso solitario consistió en 11 mujeres (57.8%) y 8 hombres (42.10%) con una edad media de 49.21 años (rango 18 - 86). El segundo con diagnóstico de sarcoma sinovial consistió 55 eran hombres (52%) y 9 2do Congreso Anual de Patólogos Mexicanos • 52 León, Guanajuato • 2016 mujeres (45%) con una media de edad de 36.9 años (rango 16 - 72). Entre los grupos no hubo predominio de sexo y la edad media fue similar. CONCLUSIONES: En nuestro instituto, la expresión del marcador de inmunohistoquímica STAT-6 como pauta diagnóstica para el tumor fibroso solitario tiene especificidad DEL 100% pero sensibilidad DEL 34% en comparación con el sarcoma sinovial. Este estudio demostró que la inmunoreactividad positiva del STAT-6 para el tumor fibroso solitario es poco sensible en comparación con la literatura mundial, por lo tanto sugerimos se amplie el campo de estudio de este marcador, ya que en nuestro instituto este marcador no es una herramienta útil diagnóstica como marcador específico. INSTITUCIÓN: INSTITUTO NACIONAL DE CANCEROLOGIA ESTADO: PUEBLA rio y sarcoma sinovial del Instituto Nacional de Cancerología del año 2009 al 2014 con tejido suficiente para realizar microarreglos de tejido. Se realizaron cinco marcadores de inmunohistoquímica (CD34, CD99, BCL-2, TLE-1, STAT-6). Los marcadores se evaluaron por separado según cada reacción. Las reacciones de inmunohistoquímica fueron evaluadas. RESULTADOS: Se obtuvieron un total de 19 casos con diagnóstico de tumor fibroso solitario y 20 casos con diagnóstico de sarcoma sinovial en un lapso de 7 años, en el Instituto Nacional de Cancerología. El grupo de pacientes con diagnóstico de tumor fibroso solitario consistió en 11 mujeres (57.8%) y 8 hombres (42.10%) con una edad media de 49.21 años (rango 18 - 86). El segundo con diagnóstico de sarcoma sinovial consistió 55 eran hombres (52%) y 9 mujeres (45%) con una media de edad de 36.9 años (rango 16 - 72). Entre los grupos no hubo predominio de sexo y la edad media fue similar. CONCLUSIONES: En nuestro instituto, la expresión Mampara: 3 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-118 AUTORES: Dra. Susana Elizabeth Martínez Moreno, Dra. Claudia Haydee Sarai Caro Sánchez, Dr. Hugo Ricardo Dominguez Malagón ,Dr. Giovanni Socra Chafre Dr. Leonardo Saul Lino Silva del marcador de inmunohistoquímica STAT-6 como pauta diagnóstica para el tumor fibroso solitario tiene especificidad DEL 100% pero sensibilidad DEL 34% en comparación con el sarcoma sinovial. Este estudio demostró que la inmunoreactividad positiva del STAT-6 para el tumor fibroso solitario es poco sensible en comparación con la literatura mundial, por lo tanto sugerimos se amplie el campo de estudio de este marcador, ya que en nuestro instituto este marcador no es una herramienta útil diagnóstica como marcador específico. INSTITUCIÓN: INSTITUTO NACIONAL DE CANCEROLOGÍA ESTADO: PUEBLA TITULO: ANÁLISIS DE LA EXPRESIÓN DEL MARCADOR DE TRANSCRIPCIÓN NAB2STAT6 EN TUMOR FIBROSO SOLITARIO EN Mampara: 4 A COMPARACIÓN CON SARCOMA SINOVIAL. Fecha y hora Instalación: 3 de mayo 7:30 hrs. INTRODUCCIÓN: El tumor fibroso solitario en una neo- plasia de tejidos blandos , poco frecuente, con un comportamiento que va desde incierto hasta maligno; desde fque ue descrito, se han tratado de encontrar sus características tanto morfológicas como inmunoreactivas, debido a que la imagen morfológica puede confundirse con otros tumores. Para hacer el diagnóstico se ha descrito como marcador específico el STAT-6 con especificidad y sensibilidad de hasta 98%. El diagnóstico diferencial obligado entre muchos otros es el sarcoma sinovial monofásico. OBJETIVOS: Documentar la sensibilidad y especificidad del marcador de inmunohistoquímica STAT-6 como marcador específico del tumor fibroso solitario en comparación con el sarcoma sinovial. MATERIAL: Estudio descriptivo, retrospectivo y observacional en pacientes con diagnóstico de tumor fibroso solita- Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-133 AUTORES: Lazos Ochoa Minerva, M.B. / Corredor Alonso Guillermo R A.P. TITULO: RABDOMIOSARCOMA PLEOMÓRFICO DE GLÚTEO DERECHO: INFORME DE UN CASO DE AUTOPSIA INTRODUCCIÓN: El rabdomiosarcoma representa del 2 al 5% de los sarcomas. Existen tres subtipos: alveolar , embrionario y pleomórfico. Los dos primeros representan el 80% , se presentan en menores de 20 años y se localizan en cabeza, cuello y tracto genitourinario ; el pleomórfico corresponde al 20%, ocurre después de la sexta década de la 2do Congreso Anual de Patólogos Mexicanos • 53 León, Guanajuato • 2016 vida y se localiza en tejidos blandos profundos de miembros pélvicos OBJETIVOS: Presentar los hallazgos anatomopatológicos distintivos del rabdomiosarcoma pleomorfo, como variante poco frecuente de los Rabdomiosarcomas y elicitar sus diagnósticos diferenciales MATERIAL: Estudio de autopsia en el que se observó una neoplasia en el glúteo derecho que midió 24 x 20 x 19 cm (fig.1), irregularmente ovoide, rodeado por tejido adiposo y músculo esquelético, de bordes empujantes, color rosa pálido, aspecto carnoso, consistencia semiblanda, con áreas de necrosis y hemorragia. En los pulmones se observaron múltiples metástasis RESULTADOS: Sarcoma pleomorfo con morfología e inmunohistoquímica positiva para rabdomiosarcoma (miogenina, vimentina y desmina) CONCLUSIONES: Los rabdomiosarcomas pleomorfos tienen una amplia gama de diagnósticos diferenciales que incluyen carcinomas y linfomas anaplásicos, melanoma, leiomiosarcoma pleomorfo, liposarcoma pleomorfo, tumor de triton y sarcoma pleomorfo indiferenciado. Para su diagnóstico es necesario realizar un panel amplio de reacciones de inmunohistoquímica que incluya CK, EMA, PS100, HMB-45, CD30, ALK, SMA, desmina y miogenina. En este caso la edad, localización, características macroscópicas, microscópicas e inmunofenotípicas corresponden al diagnóstico de rabdomiosarcoma pleomórfico. bilidad de entre 63% a 90%, con 48% de tinción fuerte. Un anticuerpo con alta sensibilidad y especificidad para SS es TLE-1. La especificidad de ambos ha sido corroborada con estudios moleculares. OBJETIVOS: Comunicar el análisis clinicopatológico e inmunohistoquímico de 55 casos de sarcoma sinovial, con énfasis en la expresión de FLI-1. MATERIAL: Estudio descriptivo, retrospectivo y observacional de pacientes con diagnóstico de Sarcoma Sinovial del Instituto Nacional de Cancerología del año 2005 al 2015, tejido suficiente para realizar microarreglos de tejido. RESULTADOS: De los 54 pacientes, 28 fueron hombres y 26 mujeres, con edades entre 20 y 68 años. La distribución del tumor coincidió con la literatura, más frecuentemente se concentró en las extremidades (24/54), glúteo (6/54), las cinturas escapular y pélvica (6/54), y retroperitoneo (5/54). Otras localizaciones fueron: cuello, ingle, región paravertebral entre otras. Acorde con la literatura, TLE-1 resultó positivo nuclear en 96% de los casos (52/54), BCL-2 fue positivo en 98%. EMA en (46%). CD56 fue positivo en 71% de los casos (40/54), CD99 resultó positivo en 70% de los casos (38/54), con expresión citoplasmática de intensidad variable, y sólo 6 en membrana. CONCLUSIONES: FLI-1 es un marcador de inmunohis- INSTITUCIÓN: Hospital General de México/ Universidad Nacional Autónoma de México”Facultad de Medicina toquímica poco específico para Tumor Neuroectodérmico primitivo, pues resultó positivo en 91% de sarcomas sinoviales. En un medio en el que los recursos para el estudio inmunohistoquímico y molecular de estas neoplasias es limitado, el uso de FLI-1 y su interpretación debe ser tomada con reservas, y echar mano de combinaciones con otros marcadores clásicos del sarcoma sinovial (BCL-2, EMA). ESTADO: DISTRITO FEDERAL INSTITUCIÓN: Instituto Nacional de Cancerología ESTADO: DISTRITO FEDERAL Mampara: 4 B Fecha y hora Instalación: 3 de mayo 7:30 hrs.Fecha y hora retiro: 3 de mayo 13:00 hrs. Mampara: 5 A TC-136 Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. AUTORES: Dra. Raquel Navarro Alvarado, Dr. Hugo Ri- TC-140 cardo Domínguez Malagón TITULO: EXPRESIÓN POR INMUNOHISTOQUÍMICA DE LA PROTEÍNA FLI-1 EN SARCOMA SINOVIAL. ESTUDIO DE 54 CASOS. INTRODUCCIÓN: El marcador inmunohistoquímico FLI-1 ha sido utilizado en el diagnóstico diferencial de sarcomas de células pequeñas redondas, con una especificidad comunicada importante para TNEP/SE (65%), y sensi- AUTORES: Dr. Mario Murguía Pérez, Dr. Luis Tinoco Té- llez, Dra. Bertha Alicia Moreno Pérez, Dra. Cindy Sharon Ortiz Arce, Dr. Juan Reyes Nava, Dr. Marco Vinicio Alonso Briones, Dr. Juan Carlos Orozco Oregón, Dr. David Medina Pineda, Dr. Jorge Israel Muñoz Torres, Dr. Saulo Mendoza Ramírez TITULO: Carcinoma mioepitelial de partes blandas. Informe de un caso. 2do Congreso Anual de Patólogos Mexicanos • 54 León, Guanajuato • 2016 INTRODUCCIÓN: Los tumores mioepiteliales de tejidos blandos son similares a su contraparte de glandula salival en el aspecto morfologico e inmunohistoquimico. Dependiendo del componente celular predominante es el nombre dado a la lesion, el mioepitelioma esta compuesto exclusivamente por celulas mioepiteliales. Cuando presentan criterios de malignidad, como atipia nuclear, indice mitosico elevado y/o necrosis, se trata de un carcinoma mioepitelial (CME) de tejidos blandos. OBJETIVOS: Los tumores mioepiteliales de partes blandas son muy poco comunes, unicamente informandose como casos aislados o series cortas. Nosotros informamos un caso de un carcinoma mioepitelial de antebrazo izquierdo de crecimiento rapido en un paciente joven. MATERIAL: Hombre de 19 años, sin antecedentes de importancia, acude a consulta por presentar una lesion dependiente de tejidos blandos del antebrazo derecho, de crecimiento rapido y expansivo en 4 meses. Los estudios de laboratorio fueron normales. En la tomografia computada se aprecio una lesion dependiente de tejidos blandos, sin afeccion del radio y cubito, con margenes expansivos, areas hipo e hiperdensas y osificacion multifocal central RESULTADOS: Se realizo una biopsia incisional de la lesion, en la cual se observo una neoplasia maligna constituida por celulas de tamaño varible, en su mayoria grandes, de aspecto ovalado y epitelioide, con citoplasma eosinofilo palido a claro, nucleos grandes, algunos lobulados, con cromatima grumosa, nucleolos evidentes y mitosis atipicas, inmersas en su gran mayoria en un estroma de aspecto condromixoide y con zonas de diferenciacion osea. Se realizaron estudios de inmunohistoquimica, con positividad para calponina, p63, GFAP y osteonectina, negativo para citoqueratinas y PS100. Se concluyo se trato de un carcinoma mioepitelial de tejidos blandos. El paciente fue sometido a quimioterapia neoadyuvante y radioterapia, con disminucion del tamaño tumoral, y actualmente programado para reseccion de la lesion. CONCLUSIONES: Debido al escaso numero de casos informados resulta dificil conocer la verdadera evolucion natural de los mioepiteliomas de tejidos blandos, la mayoria de los casos se comportan de manera indolente, aunque esta claro que pueden dar lugar a recidivas locales. los CME tienen una mayor tasa de recidiva, y metastasis frecuentes a pulmon, mediastino, columna, hueso y sistema nervioso central. Algunos autores indican que la presencia de atipia celular al menos moderada justifica la clasificacion del tumor como carcinoma mioepitelial. Los nuevos estudios citogeneticos muestran que estos tumores presentan rearreglos del gen EWSR1, similar a su contraparte de glandula salival, por lo que a pesar de que su curso clinico es distinto, puede ayudar a dilucidar nuevas terapias blanco contra estas neoplasias. INSTITUCIÓN: Unidad Medica de Alta Especialidad N° 1 IMSS Leon - Comite de tumores, Hospital Medica Campestre, Hospital General de Mexico ESTADO: GUANAJUATO Mampara: 7 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-142 AUTORES: DRA. ITZEL MARÍA MONTOYA, Dra.Alejandra Mantilla Morales Dr. Rafael Silva TITULO: Histiocitoma fibroso angiomatoide con prominente estroma mixoide. Presentación de un caso y revisión de la literatura INTRODUCCIÓN: El histiocitoma fibroso angiomatoide (HFA) fue descrito por el Dr. Franz Enzinger en 1979.Se trata de una neoplasia mesenquimal de origen incierto y malignidad intermedia. Su incidencia es muy escasa y constituye el 0.3% de todos los tumores de partes blandas.Desde el punto de vista clínico el HFA afecta preferentemente a niños y adultos jóvenes y muestra ligera predilección por el sexo femenino. OBJETIVOS: Se presenta el caso de una mujer de 45 años con cuatro meses de evolución con aumento de volumen en antebrazo derecho que le ocasionaba dolor y parestesias en mano derecha. Se realiza revisión de esta infrecuente neoplasia,haciendo hincapié en su morfología, histogénesis y características moleculares así como su diagnóstico diferencial. MATERIAL: Se realiza revisión de laminillas al recibir tratamiento (resección amplia) fuera de la unidad. y se emite el diagnostico tras los hallazgos histologicos de Histiocitoma fibroso angiomatoide con prominente estroma mixoide. RESULTADOS: Histologicamente se observo una lesión unifocal delimitada por una pseudocapsula fibrosa.Las células neoplásicas con patrón de crecimiento estoriforme , estas son fusiformes en estrecha relación con áreas centrales hemorrágicas sin endotelizar(pseudoquiste).Al detalle celular con moderada cantidad de citoplasma eosinófilo y núcleos vesiculares con pequeños nucleolos y escasez de mitosis.Todas estas características de las células neoplásicas estaban inmersas en abundantes áreas de aspecto mixoide en aproximadamente mas del 50% del tumor.Esas zonas eran relativamente hipocelulares con células neoplásicas sueltas o en pequeños nidos.Se realizo inmunohistoquimica resultado positiva principalmente para desmina. CONCLUSIONES: El origen celular del HFA es desconocido, aunque se han postulado diferentes teorias: a partir de células endoteliales en formación, de células histiocitarias, o de células estromales de estirpe mioide del ganglio 2do Congreso Anual de Patólogos Mexicanos • 55 León, Guanajuato • 2016 linfático. Sin embargo ninguna de estas teorías han sido concluyentes. Desde el punto de vista pronóstico, y aunque ha sido considerado durante años como una neoplasia fíbrohistiocitaria maligna, hoy se reconoce como una neoplasia de malignidad intermedia, ya que aunque muestra capacidad de recidiva (15-20%), su incidencia metastásica es casi nula (< 5%). INSTITUCIÓN: Hospital de oncología del Instituto Mexicano del Seguro Social CMN siglo XXI ESTADO: DISTRITO FEDERAL de nervio periférico maligno. CONCLUSIONES: El tumor Tritón ocurre en el 5% de todos los tumores de vaina de nervio periférico maligno. La localización mas frecuente es cabeza y cuello. La patogénesis es desconocida. Tiene un comportamiento biológico agresivo, se ha observado una sobrevida a 5 años del 11%, en contraste con el 39% de sobrevida de pacientes con tumor de vaina de nervio periférico maligno. La histopatología e inmunohistoquímica siguen siendo el pilar de la investigación para establecer el diagnóstico. INSTITUCIÓN: Centro Medico Nacional de Occidente ESTADO: DISTRITO FEDERAL Mampara: 7 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. Mampara: 8 A TC-143 Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. AUTORES: Dr. Alejandro Santana González, Dr. Gerónimo M. Tavares Macías TC-145 TITULO: Tumor tritón. Una serie de casos en AUTORES: DRA. ANGELA DAYANA WILLIAMS, Dra. Alejandra Zárate Osorno, Dr. José Mauricio Erosa González población mexicana TITULO: Angiofibroma del cordón espermátiINTRODUCCIÓN: El tumor tritón es un subgrupo histológico de tumores de vaina de nervio periférico maligno con co. Reporte de un caso y revisión de la literaelementos de diferenciación rabdomioblastica. Ocurre en el tura. 5% de los tumores de vaina de nervio periférico maligno. El 50-70% de los casos se asocia a NF1, principalmente en hombres jóvenes. Tienen un pronostico pobre en contraste con tumor de vaina de nervio periférico maligno. OBJETIVOS: El propósito de este estudio fue revisar las características clínicas e histopatológicas de todos los casos con diagnostico de tumor tritón, tratados quirúrgicamente durante el periodo de 2000 a 2013 en Centro Medico Nacional de Occidente. MATERIAL: Los pacientes fueron identificados en la base de datos del archivo de patología de Centro Medico Nacional de Occidente con diagnostico de tumor tritón en un periodo de 13 años (2000-2013). Se revisaron los expedientes clínicos y los hallazgos histopatológicos fueron revisados nuevamente para confirmar el diagnostico. RESULTADOS: Se identifico un total de 12 pacientes (6 hombres y 6 mujeres). La edad promedio al momento del diagnostico fue de 39 años (rango de 1-62 años). El 83% de los casos eran mayores de 18 años. La localización mas frecuente fue en miembros torácicos (41%), la menos frecuente en tronco (15%). El 83% de los casos se presento como un tumor de vaina de nervio periférico maligno con diferenciación rabdomioblastica, mientras que el 17% se presento como rabdomiosarcoma con diferenciación a vaina INTRODUCCIÓN: El angiofibroma celular o tumor tipoangiomiofibroblastoma fue descrito en 1997 por Nucci y son neoplasias que se originan en la región inguinal, peritoneo y escroto en los hombres y vulva en las mujeres. Son tumores raros cuya histogénesis se cree de células madres perivasculares con capacidad de diferenciación miofibroblástica y a tejido adiposo. OBJETIVOS: Descripción de las características histológicas e inmunohistoquímicas de este tipo de neoplasias, asi como sus principales diagnósticos diferenciales. MATERIAL: Se observó una lesión neoplásica bien delimitada, de consistencia suave y aspecto adiposo con dimensionaes de 6.0 x 5.0 x 5.0 cm. Una vez obtenidos los cortes histológicos se observó una neoplasia constituida por células fusiformes dispuestas en fascículos, sin atipia ni mitosis con vasos de pequeño calibre de paredes hialinizadas e infiltrado inflamatorio crónico linfocítico focal. RESULTADOS: Se realizaron estudios de inmunohistoquímica siendo positivos recetores estrogénicos y androgénicos y negativos CKAE1/2, WT-1, STAT-6, H-caldesmona, actina músculo liso, calretinina, CD-34, PS-100 y beta catenina, con un índice de prodilifeción del 5%. Con los anticuer- 2do Congreso Anual de Patólogos Mexicanos • 56 León, Guanajuato • 2016 pos realizados se descartó el tumor adenomatoide. CONCLUSIONES: El angiofibroma celular es una neoplasia mesenquimatosa rara localizada en la región genital de ambos sexos. Histológicamente es una neoplasia rica en fibroblastos y de origen vascular. Se ha descrito relación con lipoma de células fusiformes y miofibroblastoma de tipo mamario.Su diagnóstico inicial es difícil debido a su localización, naturaleza y relación con otras estructuras. Es de importancia crucial diferenciarlo de otras neoplasias como angiomixoma agresivo y otras neoplasias de células fusiformes debido a que éstas tienen componente maligno pueden presentar recurrencias y metástasis. INSTITUCIÓN: Hospital Regional de Alta especialidad de la Península de Yucatán ESTADO: YUCATÁN Mampara: 9 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-180 AUTORES: DR. CESAR ANTONIO FLORES, Dra. Patricia Soto Márquez; Dr. Guillermo Valero Elizondo TITULO: Tumor Maligno De Vaina De Nervio Periférico (Tmvnp) Mediastinal Con Diferenciación Heteróloga A Rabdomiosarcoma (Tumor Tritón) Y Condrosarcoma otros con hipercelularidad perivascular a manera de manguitos, ; se observaron hasta 7 mitosis tanto típicas y atípicas por campo de alto poder. Existen áreas de fibrosis hialinizada, necrosis tumoral en parches, y transición a zonas con diferenciación a cartílago maligno y rabdomioblastos. IHQ : S-100 +, débil a moderada. ENE: + intensa y difusa; CD 34, Bcl-2: + en parches; Mioglobina, Actina sarcomérica y Actina Musculo Especifica: + en rabdomioblastos. CONCLUSIONES: Los TMVNP (antes erróneamente denominados sarcomas neurogénicos o schwannomas malignos) son una familia de neoplasias derivadas de estructuras ectodérmicas, expertos como el Dr. Christopher D.M. Fletcher recomienda que el término apropiado para esta familia es Tumores Neuroectodermicos Periféricos. La proteína S-100 es positiva focal en menos de la mitad de los casos, por tanto la negatividad o debilidad a la misma no descarta el diagnóstico. Del 10 al 15 % de TMVNP originados en neurofibromas de pacientes con NF1 tienen diferenciación heterologa. REFERENCIAS: Diagnostic histopathology of tumors. Christopher D.M. Fletcher. 4th ed. Vol. 1 Chapter 27 Peripheral Neuroectodermal Tumors, pp 2047-2050. -Schaefer Inga-Marie, Fletcher Christopher D.M., Malignant Peripheral Nerve Sheath Tumor (MPNST) Arising in Diffuse-type Neurofibroma . Am J Surg Pathol [1] Volume 39, Number 9, September 2015. -Kitamura Masanori, et al, Malignant peripheral nerve sheath tumor associated with neurofibromatosis type 1, with metastasis to the heart: a case report. Diagnostic Pathology 2010, 5:2 INSTITUCIÓN: INSTITUTO MEXICANO DEL SEGURO SOCIAL. CENTRO MÉDICO NACIONAL SIGLO XXI. HOSPITAL DE CARDIOLOGÍA ESTADO: DISTRITO FEDERAL INTRODUCCIÓN: Presentamos el caso de un hombre de 26 años con neurofibromatosis tipo I, diagnosticada desde nacimiento, antecedente de padre con la misma enfermedad, inició padecimiento con tos, disfagia, edema facial y braquial, en protocolo diagnóstico se detectó lesión en mediastino medio sólida, multilobulada, adherida a pericardio, pleura visceral y vena innominada, fue sometido a resección quirúrgica. MATERIAL: Se recibió espécimen nodular de 13.0 x 11.5 x Mampara: 6 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-181 8.3 cm superficie gris amarilla, de contornos lobulados lisos con zonas anfractuosas y escaso tejido fibroconectivo. Al corte seriado la superficie era sólida blanco amarilla, lobulada con nódulos de diversos tamaños con bordes empujantes y cápsula discontinua de grosor menor a 0.1 cm. Mostró zonas laxas de aspecto mixoide que alternaban con otras de aspecto fibroso con necrosis y hemorragia. AUTORES: Dra. Baena-Ocampo Leticia del Carmen, Dr. RESULTADOS: Histologicamente la neoplasia es sólida de tejidos blandos con morfología benigna y clínicamente indolente pero metastásica. Predomina en jóvenes, en extremidades, es una neoplasia maligna de bajo grado, fibroblástica, variante del fibrosarcoma, de aspecto benigno e compuesta por fascículos alargados y alternantes (hipo e hipercelulares), no intersectantes, de células fusiformes con aspecto neuroide, ondulado y tendencia a la empalizada, en Juan Carlos Ramírez. TITULO: SARCOMA FIBROMIXOIDE DE BAJO GRADO. PRESENTACION DE UN CASO RECURRENTE. INTRODUCCIÓN: Descrito por Evans, como neoplasia 2do Congreso Anual de Patólogos Mexicanos • 57 León, Guanajuato • 2016 infiltrativa, morfológicamente puede ser indistinguible de lesiones mixoides benignas, su correlación clínico-radiológica es basica. OBJETIVOS: Presentar un caso de sarcoma fibromixoide de bajo grado diagnosticado originalmente como neurofibroma que recurio cuatro años posterior a tratamiento quirurgico. Destacando la importancia del correcto diagnostico histopatologico inicial. MATERIAL: Femenino de 7 años, con antecedente qui- rúrgico de resección de neurofibroma en región dorso lateral de pie derecho a los 3 años. Actualmente presenta tumor de tejidos blandos en mismo sitio. En exploración física: Piel con cambios de coloración, a la palpación tumor subcutáneo bien delimitado, móvil, no fijo a planos profundos y doloroso. En Rx con aumento leve de los tejidos blandos regionales; en RMN se observó neoformación heterogenea, en imágen resaltada con gadolinio, identificándose tumor bien delimitado. Se programo para reseccion quirurgica con estudio transoperatorio. RESULTADOS: En estudio transoperatorio, se identificó lesión fibromixoide de 1.5 x 1 x 1 cm, regularmente delimitada de bordes empujantes, En impronta se observó lesión mixoide de aspecto benigno constituida por estroma fibroso con finos vasos sanguíneos ramificados y curvilíneos. En los cortes histológicos: lesión neoplásica con leve celularidad compuesta por células fusiformes con patrón arremolinado con finos vasos sanguíneos curvilíneos y ramificados de pared delgada sin mitosis atípicas ni pleomórfismo. diagnosticandose posteriormente en estudio definitivo como: Sarcoma Fibromixoide de bajo grado. CONCLUSIONES: El Sarcoma fibromixoide es una lesión maligna de bajo grado de origen fibroblástico. Morfológicamente presenta aspecto benigno con poca a moderada celularidad. Macroscópicamente es delimitada, pero puede infiltrar el tejido adyacente. La correlación clínico-radiológica es importante ya que su reconocimiento es util para establecer un adecuado tratamiento quirúrgico con margenes amplios, así como seguimiento clínico del paciente a largo plazo, debido a la evolución metástasica que puede presentarse muchos años después del diagnóstico inicial. REFERENCIAS: Fletcher CDM, Unni KK, Fletcher F (Eds.),World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon; 2002. Intramuscular Low-grade Fibromyxoid Sarcoma: A Case Report J Med Sci. 2009;25(8):448-54. INSTITUCIÓN: Hospital Angeles Acoxpa. Ciudad de México ESTADO: DISTRITO FEDERAL PATOLOGÍA GASTROINTESTINAL Mampara: 11 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-4 AUTORES: JULIA MARIANA COUTIÑO, Jorge González Angulo, Ulises Ángeles Garay, Gustavo Adolfo Segovia Cuevas, Pablo Ramírez Mendoza. TITULO: Evaluación histopatológica de gastritis atrófica INTRODUCCIÓN: La identificación histológica de atrofia y metaplasia es decisiva para detener la potencial secuencia gastritis->carcinoma en los pacientes con gastritis crónica. OBJETIVOS: Comparar la concordancia de las clasificaciones del sistema Sidney y del Operative Link on Gastritis Assesmennt (OLGA) para diagnóstico de atrofia gástrica. MATERIAL: 120 biopsias gástricas consecutivas fueron analizadas por un grupo de patólogos generales, quienes las clasificaron de acuerdo con el sistema Sidney actualizado. Todas fueron revisadas por un segundo patólogo, quien aplicó el sistema OLGA. Se utilizó el índice de kappa para evaluar la concordancia diagnóstica entre ambas clasificaciones. RESULTADOS: Las manifestaciones clínicas en los pacientes fueron dispepsia (94%), dolor abdominal (50%), reflujo gastroesofágico (30%), sangrado de tubo digestivo alto (24%) y presencia de Helicobacter pylori (47.5%). Con el sistema Sidney se hizo diagnóstico de atrofia en cuatro casos y con OLGA en 26 casos, para una concordancia débil entre los sistemas (kappa 0.22, p=0.05). CONCLUSIONES: La concordancia para el diagnóstico de atrofia fue baja entre los sistemas Sidney y OLGA, debido probablemente a la inclusión de la atrofia metaplásica en el sistema OLGA. Dado que la descripción de la atrofia no metaplásica es equivalente en ambos sistemas, los patólogos generales podrían identificar mayor número de casos de atrofia gástrica con el sistema OLGA. INSTITUCIÓN: UMAE Hospital de Especialidades Centro Médico Nacional La Raza ESTADO: DISTRITO FEDERAL 2do Congreso Anual de Patólogos Mexicanos • 58 León, Guanajuato • 2016 Mampara: 11 B INSTITUCIÓN: Instituto Tecnológico y de Estudios Superiores de Monterrey Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. ESTADO: NUEVO LEÓN TC-6 Mampara: 12 A AUTORES: LUIS GERARDO JACOBO, José Eduardo Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. Pérez Saucedo. Ireán García Hernández. Claudia Martínez Amador. Álvaro Barbosa Quintana. Luis Eduardo Cuervo Pérez. Josafat Vázquez Soria. TC-20 TITULO: Neoplasia intraductal papilar mucinosa (NIPM) con neoplasia intraepitelial pancreática (NIP). Reporte de un caso. AUTORES: DR. ENRIQUE DELGADILLO, Dra. Natalia INTRODUCCIÓN: Actualmente se conoce poco sobre TITULO: Serie de casos de segundas neoplasias en cáncer de colon de inicio temprano. Análisis histopatológico, inmunohistoquímico y mutacional por BRAF. la naturaleza de las lesiones precursoras del cáncer pancreático.Dos de ellas son las NIPM y NIP,las son motivo de debate entre los expertos. Algunos creen que son parte de un mismo espectro de lesiones precursoras y otros creen que se tratan de diferentes vías. Nosotros presentamos un caso en el que estas dos lesiones se presentaron de manera sincrónica. Vilches Cisneros,Dra. Oralia Barboza Quintana,Dra. Raquel Garza Guajardo,Dr. Luis Enrique Cortés Hernández, Dr. Juan Pablo Flores Gutiérrez. INTRODUCCIÓN: El cáncer de colon es la tercera neo- OBJETIVOS: Reporte de un caso. plasia maligna más común en México. Pocos estudios en México han descrito las segundas neoplasias en cáncer de colon. MATERIAL: Recibimos un páncreas, bazo y segmento OBJETIVOS: El objetivo del presente estudio es conocer de intestino delgado de un paciente de 56 años, el cual ingresó por un cuadro de dolor abdominal, además de pérdida de peso de 10 kg en 6 meses y esteatorrea. En tratamiento por el endocrinólogo por insuficiencia pancreática, así como tener el antecedente de quistes pancreáticos diagnosticados en el 2001. El páncreas peso 375.0 g y midió 21.5 x 3.0 cm. El páncreas es de color amarillento café y de superficie RESULTADOS: Se trata de una NIPM de fenotipo intestinal la cual nace del sistema de conductos pancreáticos principales. El epitelio presenta zonas de displasia de bajo y alto grado pero sin llegar a invadir el estroma pancreático, por lo que la neoplasia se subclasifica como no invasora. En los conductos pancreáticos menores se observa NIP, desde la cabeza hasta la cola. En el resto del parénquima pancreático hay datos de pancreatitis crónica. Se disecaron un total de 29 ganglios linfáticos todos ellos sin evidencia de metástasis. CONCLUSIONES: Se necesitan más estudios para esclarecer las vías y el tiempo que tardan estas lesiones a progresar al cáncer, ya que se ha reportado un incremento en la aparición de estas y en la actualidad no contamos con la suficiente información sobre la conducta a seguir en pacientes a los que se les diagnostica una lesión precursora. Nuestro paciente fue tratado por un año con insuficiencia pancreática exócrina y solamente tuvo seguimiento, hasta que se intervino quirúrgicamente. Actualmente el paciente es tratado por endocrinología y no presenta enfermedad metastásica. la prevalencia de segundas neoplasias en pacientes jóvenes. MATERIAL: Se tomaron en cuenta en la base de datos los casos con diagnóstico de adenocarcinoma de colon en menores de 50 años con una segunda neoplasia asociada. Se analizó la expresión de proteínas de reparación en equipo automatizado Ventana (MSH2, MLH1, MSH6, PMS2) y la presencia de mutación en BRAFV600 en PCR de tiempo real. RESULTADOS: De los 334 casos, sólo 18(16.78) cumplieron los criterios de inclusión. De este grupo, los casos se localizaron en colon, endometrio, estómago, ovario, piel, glándula suprarrenal y páncreas. De los tumores, 2(11.1) no presentaron pérdida de la expresión en ninguna proteína de reparación, 12 en una (66.6%) y 4(22.2%) en dos proteínas de reparación. Todos los casos fueron negativos para BRAFV600. La ausencia de expresión de MLH1 fue la alteración más frecuente en esta casuística. CONCLUSIONES: Nuestros hallazgos sugieren que la casuística seleccionada tiene síndrome de Lynch. Futuros estudios con inmunohistoquímica y BRAFV600 serán necesarios para determinar la frecuencia del síndrome de Lynch en un contexto nacional. INSTITUCIÓN: Servicio de Anatomía Patológica y Citopatología. Hospital Universitario José Eleuterio González 2do Congreso Anual de Patólogos Mexicanos • 59 León, Guanajuato • 2016 ESTADO: NUEVO LEÓN (in situ o invasor). La mayoría no presentan síntomas, solamente un vago dolor abdominal, acompañado de sensación de plenitud y en algunas ocasiones fiebre e ictericia. Mampara: 12 B INSTITUCIÓN: Carpermor. Laboratorio de Referencia Internacional Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. ESTADO: DISTRITO FEDERAL TC-22 AUTORES: Dra. Fabiola Navidad Cervera, Dr. Adalberto Pineda Zapata Mampara: 13 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TITULO: Tumor Mucinoso Quístico Del Páncreas Con Displasia De Alto Grado (Cistade- TC-38 nocarcinoma Mucinoso In Situ). Reporte De AUTORES: DR. JORGE SÁNCHEZ, Biol. Damián SánUn Caso. INTRODUCCIÓN: Se presenta el caso de un Tumor mucinoso quístico del páncreas con displasia de alto grado, en una paciente femenina de 60 años de edad, con dolor abdominal, obstrucción intestinal y pancreatitis de 3 meses de evolución. Las neoplasias quísticas del páncreas son lesiones poco frecuentes que suponen el 1% de los tumores primarios del páncreas exocrino y el 10% de los quistes pancreáticos. OBJETIVOS: Presentación de un caso de Tumor mucinoso quístico del páncreas que representa una lesión poco frecuente y Revisión de la literatura. MATERIAL: Se recibe espécimen que mide 13x12x10 cm y pesa 1,025 g, es de superficie externa lisa, de color café; con tejido pancreático residual de 2.0 cm de eje mayor y de consistencia renitente. Al corte es quístico, con salida de líquido café mucinoso y con presencia de áreas papilares en la superficie interna. Se realiza estudio Histopatológico, que se tiñe con Hematoxilina y Eosina; así como Técnicas de Histoquímica (PAS/Azul alciano). RESULTADOS: Microscópicamente se trata de un Tumor mucinoso quístico del páncreas, con displasia de alto grado (Cistadenocarcinoma mucinoso in situ) se observa tejido pancreático, con quiste revestido de epitelio cilíndrico con citoplasma eosinófilo a vacuolado y núcleo irregular con atipia nuclear y pérdida de la polaridad; se identifican formaciones papilares de crecimiento exofítico y algunas calcificaciones de tipo psammomatoso. La pared no muestra infiltración por las papilas. CONCLUSIONES: El tumor mucinoso quístico del páncreas representa el 1% de los tumores del páncreas y el 10% de los quistes pancreáticos. Se presentan mayormente en mujeres, en una edad media de 45 años y se ubican en el cuerpo o en la cola del páncreas en el 95% de los casos. Son clasificados histológicamente por la OMS como Cistadenomas, Tumores limítrofes y Cistadenocarcinomas chez Ramírez, Dra. Anna Karen Félix Félix, Dra. Eugenia Flores Figueroa, Dr. Fernando Candanedo González TITULO: Análisis clinicopatológico del Adenocarcinoma de Ampolla de Vater por Subtipos Intestinal versus Pancreatobiliar. INTRODUCCIÓN: En 1994, Kimura et al, propusieron dividir a los adenocarcinomas de la Ampolla de Vater en los subtipos intestinal y pancreatobiliar, dado que muestran diferencias morfológicas distintivas. En 2001 Albores et al confirmaron diferencias en el comportamiento biológico siendo de peor pronóstico el subtipo pancreatobiliar. Sin embargo, no existen estudios en México que comparen ambos grupos. OBJETIVOS: El objetivo de este estudio es comparar las características clinico patológicas de una serie de pacientes con adenocarcinoma de la ampolla de Vater de tipo intestinal versus pancreato biliar. MATERIAL: Se analizó una cohorte histórica de pacientes sometidos a resección de Whipple por adenocarcinoma de Ampolla de Vater entre 2007 y 2012 en el Hospital de Oncología del CMN Siglo XXI. RESULTADOS: Se analizaron 58 pacientes, 34 mujeres y 24 hombres con edad promedio de 59 años (rango 3386). El tiempo de evolución fue de 4 meses promedio (rango 0.5-21). Cuarenta pacientes presentaron ictericia (76%), 36 pérdida de peso (69%), 29 dolor abdominal (55%) y 2 con tumor palpable (3%). Se encontró ACE de 4.3 mcg/L (rango 1.52-11.7), Ca 19.9 de 342.3 UI/ml (rango 1.42-1654.17), bilirrubinas totales (BT) de 7.58 mg/dL (rango 0.25-32). El tamaño tumoral promedio fue 3.2 cm (rango 0.4-12). El subtipo intestinal se observó en 39 pacientes (67%) versus 19 con pancreatobiliar (33%). En 9 casos hubo invasión linfovascular, 15 invasión pancreática, 13 ganglios positivos y 19 metástasis. Sólo 29 pacientes (50%) se encontraron en estadio I-II, con supervivencia promedio de 14.28 meses 2do Congreso Anual de Patólogos Mexicanos • 60 León, Guanajuato • 2016 (rango 0.5-57). CONCLUSIONES: El carcinoma ampular en nuestra población es más frecuente en mujeres. El tiempo de evolución promedio fue de 4 meses. Los signos y síntomas más frecuentes son ictericia, pérdida de peso y dolor abdominal. El subtipo intestinal es el más frecuente asociado a estadios tempranos (I-II), en contraparte con el subtipo pancreatobiliar que a pesar de tener valores elevados de BT de manera significativa se detecta en estadios avanzados. INSTITUCIÓN: Unidad de Investigación Médica Oncológica y Departamento de Patología. Hospital de Oncología de Centro Médico Nacional Siglo XXI. IMSS. Ciudad de México. Centro Médico Nacional de Occidente, Guadalajara ESTADO: DISTRITO FEDERAL Mampara: 13 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-39 AUTORES: DRA. KARLA ALEJANDRA LÓPEZ, Dr. Gámez Castro I., Dr. Aristi Urista G. TITULO: Trombosis aguda del eje venoso espleno-portal asociada a colecistitis crónica litiásica. INTRODUCCIÓN: Cuando la trombosis portal se extien- de a las venas esplénica o mesentéricas se denomina trombosis del eje venoso espleno-portal (TEEP). Es diagnosticada raramente, aunque se ha informado en 1% de autopsias. La TEEP no asociada a cirrosis o neoplasias hepáticas es la segunda causa de hipertensión portal y se relaciona con trastornos protrombóticos sistémicos y procesos inflamatorios locales. Existe la forma aguda y crónica (“cavernomatosis portal”). OBJETIVOS: Presentamos un caso de autopsia de trombosis aguda del eje venoso espleno-portal en una mujer de 57 años cuya manifestación principal fue dolor abdominal. MATERIAL: Se documentó anemia hipocrómica microcí- a la mesentérica superior y esplénica, bazo congestivo, un segmento de yeyuno con infarto hemorrágico transmural y vesícula biliar dilatada, con pared engrosada, fibrosa y calcificada, adherida al lecho hepático, con un cálculo único de tipo mixto enclavado en el cuello y microscópicamente un proceso fibroinflamatorio que afectaba toda la pared. CONCLUSIONES: Es posible que la TEEP sea más frecuente de lo que suponemos, pues el diagnóstico clínico requiere gran intuición y empleo de técnicas de imagen; y el patólogo sólo puede diagnosticarla mediante autopsia en la que efectúe una disección cuidadosa del eje venoso espleno-portal. En 70% de los pacientes se identifica un factor etiológico; lo más común son enfermedades trombogénicas sistémicas (principalmente neoplasias mieloproliferativas crónicas y otras más raramente). En segundo lugar se encuentran procesos inflamatorios regionales: pancreatitis, colecistitis, colangitis, apendicitis, onfalitis; o lesión transquirúrgica del eje venoso espleno-portal. En este caso particular, el factor asociado fue la colecistitis crónica, pero no es posible determinar con certeza si existió algún otro trastorno protrombótico asociado. En todo paciente con diagnóstico de TEEP debe investigarse la presencia subclínica de todas las enfermedades asociadas. Existen dos formas clínico-patológicas de TEEP, la aguda y la crónica. Las manifestaciones de la etapa aguda son inespecíficas y predomina el dolor abdominal y la fiebre; el infarto intestinal es raro (2% de los pacientes), las lesiones producidas en el hígado son mínimas o nulas, y el pronóstico es bueno. En caso contrario, se desarrolla la etapa crónica (“cavernomatosis portal”) en la que predominan las manifestaciones de hipertensión venosa portal. INSTITUCIÓN: Servicio y Departamento Universitario de Anatomía Patológica, Hospital General de México y Universidad Nacional Autónoma de México. ESTADO: DISTRITO FEDERAL Mampara: 14 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-40 tica, plaquetas normales; tiempos de coagulación y pruebas de función hepática normales. El ultrasonograma hepático demostró datos de colecistitis crónica litíasica, hidrocolecisto, esplenomegalia y “hallazgos compatibles con trombosis de la vena porta”. En la laparotomía concluyeron que era un carcinoma de vesícula biliar avanzado, no efectuaron colecistectomía, ni revisión del sistema venoso portal. AUTORES: IAN MARCO LUCIANO MARTÍNEZ, Miguel RESULTADOS: En la autopsia, encontramos trombosis INTRODUCCIÓN: Las neoplasias quísticas del pán- reciente con oclusión total de la vena porta que se extendía Ángel Ríos Torres, Dr. Luis Raúl Chavira Guerrero, Dra. J. Rosalba García Ramírez TITULO: Cistoadenocarcinoma Mucinoso In-Situ Pancreático, Presentación de un caso. creas son raros y constituyen aproximadamente el 0,5% de todas las neoplasias de páncreas. Dentro de la clasificación 2do Congreso Anual de Patólogos Mexicanos • 61 León, Guanajuato • 2016 histológica de las neoplasias quísticas del páncreas se encuentra el tipo mucinoso generalmente macroquistico, que es potencialmente maligno. Existe mayor incidencia en mujeres entre los 40 y 50 años de edad. poco conocida por clínicos y patólogos y comúnmente diagnosticada erróneamente. OBJETIVOS: Revisar la presentación anatomo-clínica del cistoadenocarcinoma mucinoso In-situ pancreático. jer de 71 años, diagnosticada clínica y endoscópicamente como “pólipo hiperplásico”, tratada mediante gastrectomía subtotal. MATERIAL: Paciente femenino de 32 años de edad. In- MATERIAL: Se recibió una gastrectomía subtotal (13 gresó por aumento de volumen abdominal con 10 meses de evolución, dolor en epigastrio e hipocondrio izquierdo leve y opresivo, niega pérdida de peso. A la exploración abdomen globoso depresible a expensas de ascitis,la tomografía reportó masa dependiente de páncreas. Se sometió a pancreatectomía distal, se extrajo quiste de 3 kg de peso. El espécimen fue enviado para estudio histologico. RESULTADOS: En el laboratorio de anatomía patoló- OBJETIVOS: Presentamos un caso de EM en una mu- x 10 cm) con una lesión que abarcaba toda la superficie mucosa y tenía pliegues gástricos elevados, redundantes, con aspecto similar a las circunvoluciones cerebrales, por engrosamiento mucoso, con gran hiperplasia foveolar, formación de quistes; atrofia grave con desaparición de las glándulas y muy escasa inflamación asociada. RESULTADOS: Se trata de un caso característico de gica se recibió un espécimen quístico de 25.0 cm de eje mayor, la superficie lisa de color blanco y al corte con material mucinoso, se incluyeron cortes para su análisis al microscopio de luz donde se observó carcinoma in-situ en dos focos microscópicos, con bordes libres sin invasión vascular o linfática. Posterior al resultado se inició tratamiento complementario con quimioterapia. EM no reconocida por los clínicos, ni los endoscopistas, diagnosticada erróneamente como “pólipo hiperplásico”. Los diagnósticos diferenciales más importantes, en este caso particular, fueron adenoma foveolar y adenoma tubular (tipo intestinal), que fueron descartados al evaluar, en conjunto, todas las características macro y microscópicas de la lesión. Desconocemos si la paciente tenía hipoclorhidria e hipoproteinemia. CONCLUSIONES: El cistoadenocarcinoma mucinoso in CONCLUSIONES: La EM se caracteriza por pliegues situ del páncreas es una entidad poco común, su diagnóstico histopatológico permite dar tratamiento complementario con quimioterapia y seguimiento estrecho de la paciente. gástricos prominentes, hiperplasia foveolar, hipoclorhidria y pérdida protéica. Hay variantes infantil (autolimitada y asociada a Helicobacter pylori y citomegalovirus) y adulta (asociada a sobreexpresión de Factor de Crecimiento Transformante-alfa). Puede ser regional (afectar sólo el cuerpo gástrico) y semejar múltiples pólipos. El diagnóstico en biopsia puede ser difícil (es necesaria una biopsia que abarque todo el espesor de la mucosa para diagnosticarla correctamente) y puede confundirse con otras enfermedades que cursan con hiperplasia foveolar: pólipos hiperplásicos, gastropatía química, gastritis crónicas, otras gastropatías hiperplásicas y síndrome de Cronkhite-Canada. En adultos la EM es crónica y de difícil control. Se trata farmacológicamente y los casos refractarios con gastrectomía total o subtotal. INSTITUCIÓN: Departamento de Medicina y nutrición, Universidad de Guanajuato Hospital General León ESTADO: GUANAJUATO Mampara: 14 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. INSTITUCIÓN: Servicio y Departamento Universitario de Anatomía Patológica, Hospital Fecha y hora retiro: 3 de mayo 13:00 hrs. General de México y Universidad Nacional Autónoma de México TC-45 AUTORES: Dra. García Romero B, Dra. Martínez Ruiz C, ESTADO: DISTRITO FEDERAL Dr. Aristi Urista G. TITULO: Enfermedad de Ménetriér en gastrectomía subtotal. Mampara: 15 A INTRODUCCIÓN: La enfermedad de Ménetriér (EM) fue Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. descrita por el patólogo francés Pierre Eugéne Ménetriér en una serie de autopsias (“polyadenomes en nappe” “poliadenomas en mantel-). Forma parte de las enfermedades que originan pliegues gástricos prominentes. Junto con el Síndrome de Zollinger Ellison, son las gastropatías hiperplásicas más frecuentes. Sin embargo, es una enfermedad TC-51 AUTORES: Dra. Brenda Esther Vázquez López, Dra. 2do Congreso Anual de Patólogos Mexicanos • 62 León, Guanajuato • 2016 Petra Yuridia Lizeth Alvarado Beltran, Dra. Olga Hernandez Esquivel, Dr. Marco Antonio Ponce Camacho. Mampara: 15 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. TITULO: Fibromixoma Plexiforme. Descripción Fecha y hora retiro: 3 de mayo 13:00 hrs. de un caso y revisión de la literatura INTRODUCCIÓN: El Fibromixoma Plexiforme (FP) es una entidad recientemente reconocida como una neoplasia mesenquimal benigna del estroma gastrointestinal, con predilección por el antro gástrico, la cual se presenta como una masa mural nodular, que se puede extender a la luz gástrica. Tiene un patrón de crecimiento multinodular con abundante estroma mixoide. El diagnóstico diferencial es con el GIST. OBJETIVOS: Describir una patología poco frecuente. Revisar la literatura actual. MATERIAL: Se trata de un paciente masculino de 67 años de edad, el cual cuenta con historia de 3 meses de evolución con evacuaciones melénicas y pérdida de peso de 6 a 8 kg. En el servicio de Patología se recibe producto de gastrectomía parcial con una lesión nodular en la pared de 5 cm de diámetro mayor, la cual se extendía a la luz gástrica y ulceraba la mucosa. RESULTADOS: Los cortes histológicos muestran una lesión nodular que se extiende a la luz gástrica y ulcera la mucosa, la cual muestra un patrón de crecimiento multinodular con moderada cantidad de estroma mixoide, abundantes vasos de paredes delgadas y células de fusiformes a ovales, con núcleos hipercromáticos y citoplasma claro. No se identifica invasión vascular. Los bordes de resección quirúrgica son negativos para neoplasia. Se disecaron dos ganglios linfáticos los cuales muestran hiperplasia reactiva del tejido.Se realizaron estudios de inmunohistoquímica, los cuales fueron negativos para DOG 1, CD117 y S100; y positivos para Antígeno de Músculo Específico y Caldesmón. CONCLUSIONES: El FP es una neoplasia mesenquima benigna recientemente reconocida, poco frecuente y cuyo principal diagnóstico diferencial es el GIST. En la literatura sólo se encuentran reportados 32 casos. Se ha descrito como una masa en forma de domo en la pared que puede ulcerar la mucosa, con una superficie de corte multinodular. La imagen histológica se caracteriza por un patrón de crecimiento nodular, conformada por células fusiformes de citoplasma claro en un estroma mixoide o fibromixoide con red de capilares. Expresa un inmunofenotipo miofibroblástico, particularmente negativo para CD117 y DOG1. INSTITUCIÓN: Doctors Hospital, Monterrey, Nuevo León. ESTADO: NUEVO LEÓN TC-55 AUTORES: Dra. Analy Mendoza Gómez , Dra. Luz María Gómez Jiménez , Dra.Katia Hop García. TITULO: HEMANGIOMA CAVERNOSO EN ESÓFAGO REPORTE DE UN CASO INTRODUCCIÓN: Los hemangiomas son tumores benignos de origen vascular. Se identifican 3 tipos: cavernoso, hamartomatoso y malformación arteriovenosa. La localización esofágica es infrecuente, son únicos y se localizan en el tercio inferior. Pueden causar síntomas de tipo obstructivo y/o hemorrágico. Su diagnóstico en la edad adulta es raro, solo existen publicados casos aislados OBJETIVOS: Reportar el caso de un hemangioma cavernoso en esófago MATERIAL: Mujer de 29 años con disfagia moderada e intermitente, la endoscopia mostro tumor gris violáceo submucoso y por su gran tamaño requirió tratamiento quirúrgico. La imagen histológica corresponde a tumor cubierto por mucosa normal localizado en lámina propia y submucosa formado por vasos sanguíneos grandes. Se reportó como hemangioma cavernoso CONCLUSIONES: Es un tumor extremadamente raro en el adulto, que puede ser tratado endoscópicamente de forma eficaz mediante diferentes modalidades (escleroterapia, láser, resección mucosa endoscópica), reservando la cirugía para aquellos casos en que no sea posible dicha modalidad terapéutica INSTITUCIÓN: Servicio de Anatomía Patológica, Hospital de Especialidades Dr. Bernardo Sepúlveda CMN Siglo XXI ESTADO: DISTRITO FEDERAL Mampara: 16 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-63 AUTORES: María Celina Briz Sánchez Residente de 2 año de Anatomía Patológica CMNSXXI, Dra. Marely López Jiménez. Médico Adscrito al servicio de Anatomía Patológica CMNSXXI. 2do Congreso Anual de Patólogos Mexicanos • 63 León, Guanajuato • 2016 TITULO: CISTADENOCARCINOMA BILIAR INTRAHEPÁTICO. A PROPOSITO DE UN CASO INTRODUCCIÓN: De todos los quistes hepáticos solo el 5% son neoplasias (1) por lo que la incidencia del cistadenoma y cistadenocarcinoma biliar intrahepático es baja (2). El diagnóstico de esas neoplasias quísticas suele dificultarse debido a que los datos clínicos e imagenológicos son inespecíficos mas aún para diferenciar uno del otro, ya que ambos son quistes multiloculados, septados con proyecciones papilares o nódulos murales (3) por lo que sigue siendo necesario el estudio histopatológico minucioso y un muestreo adecuado para clasificar adecuadamente estas neoplasias quísticas debido a la diferencia en el manejo clínico (3). Hasta el 85% de los cistadenomas biliares se presentan en mujeres proponiéndose una etiología hormonal (1) mientras que el cistadenocarcinoma sucede de igual manera entre hombres y mujeres con edad entre 50-60 años (2). El diagnóstico preoperatorio es todo un reto y la poca utilidad con la aspiración con aguja delgada dificultan la decisión terapéutica preoperatoria (3). Es necesario tener en mente estas dos neoplasias y sus diferencias para poder proponer un amplio panorama de diagnósticos diferenciales con un análisis detallado de las características clínicas, imagenológicas y anatomopatológicas. OBJETIVOS: Presentar un caso de cistadenocarcinoma biliar intrahepático enviado para estudio transoperatorio. MATERIAL: Masculino de 69 años,evolucición de 1 mes con distención abdominal,pérdida de peso (5 Kg)y aumento de la circunferencia abdominal.En TC abdominal,se observa quíste heterogéneo en hígado que abarca cavidad abdominal izquierda rodeando bazo y fosa iliaca derecha.Se recibe fragmento de pared de quiste de 8cm de eje mayor con superficie interna granular blanco amarillo con improntas que muestran estructuras papilares con células malignas. RESULTADOS: Inmunomarcación: CK 19 positiva difusa, intensa CONCLUSIONES: A pesar de ser consideradas neoplasias hepáticas de baja frecuencia en presentación, es necesario tenerlas en mente para ampliar el rango de diagnósticos diferenciales y poder realizar un análisis minucioso de los datos clínicos, imagenológicos y anatomopatológicos que integrados nos lleven a un diagnóstico certero y así proporcionar el tratamiento adecuado y beneficioso para el paciente. Mampara: 16 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-65 AUTORES: KARINA JUDITH LARIOS, Edmundo Erbey Castelán Maldonado, Silvia Judith Hernández Martínez, Yolanda Martínez Flores. TITULO: Tumores Neuroendocrinos: Relación de casos y seguimiento en dos años de los pacientes de la Unidad Médica de Alta especialidad No 25 del Instituto Mexicano del Seguro Social Monterrey, Nuevo León. INTRODUCCIÓN: Los tumores neuroendocrinos constituyen un conjunto de enfermedades neoplásicas de una gran heterogeneidad clínica, bioquímica y biológica. Comparten una serie de características histopatológicas comunes si bien se originan en células neuroendocrinas de amplia distribución anatómica incluyendo células de la cresta neural, glándulas endocrinas, islotes endocrinos. Afectan con más frecuencia el páncreas y el tracto gastrointestinal dentro de éstos la localización más frecuente es el intestino delgado. La edad media al diagnóstico está en la quinta década de vida y su pronóstico suele ser mejor que el de otros cánceres. El diagnóstico suele confirmarse con la presencia en las células tumorales de los marcadores inmunohistoquímicos sinaptofisina y/o cromogranina A. Su curso clínico viene dado tanto por la diversa localización del tumor primario y su heterogéneo comportamiento biológico. Pueden ser de comportamiento indolente y cursar sin síntomas, como es el caso de los tumores diagnosticados de manera incidental en procedimientos quirúrgicos o autopsias practicadas por otras causas, o cursar con síntomas derivados del efecto mecánico del propio tumor o por el potencial de algunos de ellos de sintetizar y excretar distintas hormonas polipeptídicas al torrente circulatorio que causan síndromes clínicos específicos, en cuyo caso se denominan tumores neuroendocrinos funcionales. OBJETIVOS: Realizar una estadística de los casos con Diagnostico de Tumor Neuroendocrino, del Departamento de Anatomía Patológica de la UMAE No. 25 en un periodo de dos años así como darles seguimiento. MATERIAL: Se revisaron los archivos del Departamento INSTITUCIÓN: INSTITUTO MEXICANO DEL SE- de Anatomía Patológica buscando los Tumores NeuroendoGURO SOCIAL CENTRO MÉDICO NACIONAL crinos diagnosticados en un periodo de 2 años a la fecha SIGLO XXI, HOSPITAL DE ESPECIALIDADES (2014-2015). Se incluyeron todos los casos con Diagnostico ESTADO: DISTRITO FEDERAL de Tumor Neuroendocrino, independientemente del grado histológico, localización o si eran metástasis. Todos tenían marcadores de inmunohistoquimica Cromogranina, Enolasa, Sinaptofisina y Ki67. RESULTADOS: Se encontraron 38 casos de Tumores Neuroendocrinos con un promedio de edad de 52 años, 2do Congreso Anual de Patólogos Mexicanos • 64 León, Guanajuato • 2016 22 fueron del sexo femenino y 16 del sexo masculino. Un caso fue pediátrico con 11 años de edad. En 14 casos el sitio primario fue tracto gastrointestinal, en 10 casos el sitio primario de la lesión fue páncreas, 2 tenían primario en hígado, 1 caso con primario glándula mamaria, 1 caso con primario en vejiga y 1 caso con primario en próstata. En el resto de los casos el sitio primario fue desconocido. En 20 casos (52.6%) ya se encontraban metástasis. Los sitios de metástasis más comunes fueron ganglios linfáticos, hígado y tejido óseo. 18 casos fueron carcinomas neuroendocrinos de alto grado, 15 fueron tumores neuroendocrinos de bajo grado y 5 fueron de grado intermedio. Del total de pacientes 3 han fallecido independiente del grado histológico, (2 tumores neuroendocrinos de bajo grado, 1 carcinoma neuroendocrino de alto grado) dos tenían metástasis a otros órganos, el resto se han manejado con cirugía, 19 pacientes se están manejando con análogos de la somatostatina. Los Tumores neuroendocrinos tienen un comportamiento biológico muy heterogéneo. En nuestro centro el sitio primario de localización más frecuentes es tracto gastrointestinal (colon, intestino delgado y estomago), seguido del páncreas. 35 casos están en seguimiento de los cuales 13 solo tienen vigilancia, 19 se están manejando con análogos de la somatostatina, 3 casos se están manejando con cisplatino y radioterapia. El 52.6% de los casos ya mostraban algún tipo de metástasis al momento del diagnóstico como lo muestra la literatura. CONCLUSIONES: INSTITUCIÓN: Departamento de Anatomía Patológica de la Unidad Médica de Alta Especialidad (UMAE) No 25 del Instituto Mexicano del Seguro Social, Monterrey Nuevo León. OBJETIVOS: El objetivo de este estudio fue determinar los factores de pronóstico en un grupo de pacientes con adenocarcinoma del sistema Vateriano, operados en el Instituto Nacional de Cancerología MATERIAL: Del archivo de Patología Quirúrgica, se obtuvieron en el período de 2005 a 2015, los casos de adenocarcinoma del sistema Vateriano. Se seleccionaron los casos en los que se realizó pancreatoduodenectomía y se registró la información demográfica y patológica relacionada. El análisis estadístico uni y multivariado se realizó mediante la regresión de Cox y de Log-rank, para la supervivencia RESULTADOS: Se estudiaron 53 casos. La afección ganglionar fue la única variable que mostró diferencias estadísticamente significativas en la supervivencia global de los individuos estudiados (p=0.19). Las metástasis ganglionares se correlacionaron con menor grado de diferenciación e invasión perineural. Los adenocarcinomas con fenotipo intestinal estaban mejor diferenciados, tuvieron menor tamaño, así como menos invasión vascular linfática y perineural, con tendencia a mostrar mejor supervivencia global, sin alcanzar la significación estadística CONCLUSIONES: El factor pronóstico más significativo en los adenocarcinomas del sistema Vateriano operables, es la presencia de metástasis ganglionares INSTITUCIÓN: Instituto Nacional de Cancerología, México ESTADO: DISTRITO FEDERAL ESTADO: NUEVO LEÓN Mampara: 17 B Mampara: 17 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-81 TC-71 AUTORES: Dr. Fernando De la Torre Rendón, Dra.Xóchitl AUTORES: ROBERTO HERRERA, Samuel Almeida-Navarro, Giovanny Soca-Chafre, Norma Hernández-Pedro TITULO: FACTORES PRONOSTICO EN ADENOCARCINOMA DEL SISTEMA VATERIANO INTRODUCCIÓN: Los adenocarcinomas del sistema Vateriano (ampolla de Vater) representan el 0.2% de la neoplasias malignas del tracto gastrointestinal y del 15?20% de los pacientes afectados, son candidatos a cirugía. Se han propuesto como factores de pronóstico posquirúrgicos, la profundidad de invasión, la afección ganglionar, la razón de ganglios linfáticos metastásicos, la invasión vascular sanguínea y linfática, la diferenciación de la neoplasia y los bordes quirúrgicos positivos, entre otros García-Samper, Dr. Rafael Rodriguez Rojas TITULO: ENTEROCOLITIS AUTOINMUNE DEL ADULTO CON ACTIVIDAD INTRAEPITELIAL POR LINFOCITOS CD8+ INTRODUCCIÓN: La enterocolitis autoinmune es una forma rara de enteropatía asociada a auto anticuerpos.se presenta como diarrea y pérdida de peso. Afecta a colon,estómago, páncreas, riñón y tiroides. microscópicamente hay atrofia de vellosidades, Inflamación intensa de la lámina propia, con infiltración de linfocitos CD4+ al epitelio,y ausencia de células caliciformes, células de Paneth. OBJETIVOS: mostrar mediante un caso clínico una forma 2do Congreso Anual de Patólogos Mexicanos • 65 León, Guanajuato • 2016 rara de enteropatía asociada a auto anticuerpos y a linfocitos CD8(+). positivos.El diagnóstico diferencial debe hacerse con GIST. MATERIAL: presentamos el caso de una mujer de 37 hipotiroidismo en control, e hipertensión arterial. Inicia su padecimiento con malestar general, nausea, dolor abdominal e intolerancia a la vía oral. La endoscopía con lesión submucosa en cuerpo y antro gástrico, la TAC señaló tumor de origen en la capa submucosa dependiente de pared posterior y cuerpo gástrico, se le realizó gastrectomía total. La gastrectomía total demostró tumor en la pared posterior y curvatura menor, submucoso, de 4.5 x 4 cm, al corte es sólido, café claro y gris con una calcificación central no encapsulado. Histológicamente es fusocelular, con atípia citológica leve, extenso infiltrado inflamatorio compuesto por linfocitos y células plasmáticas. La inmunohistoquímica: positividad para PS100, y vimentina y negatividad para DOG1, CD34, y actina de musculo liso. Caso numero 2: Mujer de 80 años con hipertensión arterial, asintomatica en laparotomía por colecistectomía se detectó tumor gástrico. La TAC con tumor en curvatura mayor, intramural de crecimiento exofítico expansivo, no infiltrante. La endoscopia con lesión submucosa probable diagnóstico de GIST, se le realizo gastrectomía total la cual mostró en curvatura mayor y pared posterior, tumor de 5.5 x 5 cm, no encapsulado, ovoide, amarillo, cubierto por mucosa intacta, con una cicatriz central gris blanquecina. Histológicamente les fusocelular con áreas hipercelulares que alternan con áreas hipocelulares y el borde de la neoplasia se observa un anillo denso de células plasmáticas y linfocitos. No se observó atipia citológica ni necrosis. PS100 y vimentina positivos y DOG-1, actina de musculo liso y CD34 negativos. años, que acudió a consulta el 05 de noviembre del 2014 por diarrea. con antecedente Asma de larga evolución y alergia a la penicilina. se reportó inició con diarrea de forma súbita, fue multitratada si mejoría del cuadro clínico, entrando a protocolo de investigación con múltiples pruebas de laboratorio y gabinete negativas. se realizo toma de biopsia de intestino delgado y pruebas serológicas. RESULTADOS: Las biopsias de intestino delgado que se interpretaron como Enteritis crónica intensa con aplanamiento de vellosidades intestinales compatibles con enfermedad celiaca o enteritis autoinmune. Se realizó inmunohistoquimica mostrando positividad para CD3, CD4, CD8 y CD 15 con penetración predominante de linfocitos CD8 al epitelio. y las pruebas serológicas reportaron positividad para anticuerpos antinucleares positivos (3.5), anti DNA de doble cadena positivo con elevación de IgE e IgG. El diagnóstico final fue Enteropatía autoinmune. CONCLUSIONES: La mayoría de los pacientes con enterocolitis autoinmune tienen la participación de auto anticuerpos (típicamente anti-enterocitos), participación preferencial intestino delgado,y predominantemente infiltrados CD3+ CD4+. con la presentación de este caso se demuestra que los linfocitos T Cd3+ y CD4+ no son los únicos involucrados en los casos de enterocolitis autoinmune. RESULTADOS: Caso numero 1: Mujer de 68 años con Los schwannomas son neoplasias INSTITUCIÓN: Hospital Regional Lic. Adolfo CONCLUSIONES: benignas muy raras y es importante hacer el diagnóstico lópez Mateos. ISSSTE. ESTADO: DISTRITO FEDERAL diferencial con tumor del estroma gastrointestinal, ya que el tratamiento es diferente. INSTITUCIÓN: Hospital de Oncología Centro Médico Nacional Siglo XXI Mampara: 18 A ESTADO: DISTRITO FEDERAL Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. Mampara: 18 B TC-90 AUTORES: DRA. MA. GPE. JAZMÍN DE ANDA, Dra. Blandina Hernández Cruz, Dra. Sonia Tavares García, Dra. Liliana García Ilizaliturri, TITULO: Schwannomas gástricos, reporte de dos casos Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-107 AUTORES: Dora Maria Marin, Dr Carlos Serrano Bello TITULO: Adenocarcinoma gastrico de tipo inINTRODUCCIÓN: Los Schwannomas son neoplasias testinal y tumor neuroendocrino de intestino benignas, que se originan de las células de Schwann, son tumores de lento crecimiento y raros en el tracto gastroin- delgado, tumores sincronicos. Reporte de un testinal y el sitio mas frecuente es el estómago. Los sínto- caso. mas son inespecíficos como tumor palpable, dolor epigástrico y sangrado de tubo digestivo.Son PS100 y vimentina 2do Congreso Anual de Patólogos Mexicanos • 66 León, Guanajuato • 2016 INTRODUCCIÓN: La relacion entre el tumor neuroendocrino de intestino delgado y adenocarcinoma gastrico es infrecuente, no se ha establecido una relacion directa entre estos dos tipos de tumores asi como su comportamiento biologico y su pronostico clinico. OBJETIVOS: Informar el caso clinico de un paciente masculino de 75 años con diagnostico sincronico de adenocarcinoma gastrico y tumor neuroendocrino de intestino delgado MATERIAL: Por endoscopia se realizaron biopsias de dos lesiones: la primera en estomago y la segunda en intestino delgado. RESULTADOS: Al analizar las biopsias endoscopicas de estomago se observo una neoplasia maligna consistente con adenocarinoma de tipo intestinal moderadamente diferenciado. La biopsia correspondiente a la lesion de intestino delgado mostraba un patron distinto a la lesion del estomago, la cual estaba caracterizada por nidos de celulas pequeñas de nucleos hipercromaticos y de escaso citoplasma eosinofilo,con presencia de menos de 2 mitosis en 10hpf, divididos por finos tabiques de tejido conectivo. Las cuales fueron positivas para cromogranina y sinaptofisina con un ki <5%, se pudo establecer el diagnostico de doble lesion primaria (adenocarcinoma gastrico de tipo intestinal y tumor neuroendocrino gi)> CONCLUSIONES: Son pocos los casos reportados en- tre estos tipos de neoplasia, no se sabe a ciencia cierta la relacion que existe entre la histogenesis y el comportamiento biologico de ambas lesiones, probablemente el tumor de intestino delgado sea un hallazgo y no afecte el pronostico de este paciente. Se requiere investigar a mayor profundidad si existe asociacion directa entre este tipo de tumores INTRODUCCIÓN: Introducción. El hígado graso no alcohólico (HGNA) es una enfermedad crónica del hígado cada vez más común en todo el mundo con un amplio espectro histopatológico que va desde la esteatosis simple (ES) sin inflamación significativa a esteato hepatitis (EHNA) con diferentes etapas de la fibrosis, cirrosis y carcinoma hepatocelular. OBJETIVOS: Objetivo. Identificar la incidencia de esteatosis hepática no alcohólica en pacientes con litiasis vesicular programados para colecistectomía MATERIAL: Se realizó un estudio prospectivo transversal y prospectivo, en pacientes programados para colecistectomía laparoscópica con diagnóstico de colelitiasis que aceptaron participar. El estudio se llevo a cabo en el periodo de tiempo del 1 de marzo del 2015 al 30 noviembre del 2015. A los pacientes que aceptaron participar se les realizaron pruebas de funcion hepática, medidas antropometricas y toma de biopsia de higado de por lo menos 2.0 cc RESULTADOS: 70 pacientes que cumplieron con nuestros criterios de inclusión; desde el punto de vista de estudios de imagen, los ultrasonidos realizados mostraron que 25.7% de los pacientes presentan alguna alteración sonográfica en relación a hígado graso no alcohólico y de este grupo más del 95% fue reportado como esteatosis leve, mientras que solo el 10% de nuestros pacientes presentaron alguna alteración en los marcadores bioquímicos. Sin embargo desde el punto de vista histopatológico el 82.9 mostró alguna alteración que vario de esteatosis simple a cirrosis y el 17.1% se reporto como hígado normal. CONCLUSIONES: Existe una incidencia alta (82.9%) de alteraciones histológicas hepáticas en pacientes con diagnóstico de colelitiasis en nuestra población. INSTITUCIÓN: HOSPITAL JUAREZ DE MEXICO INSTITUCIÓN: Hospital General León ESTADO: MICHOACÁN ESTADO: GUANAJUATO Mampara: 19 A Mampara: 19 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-112 TC-148 AUTORES: DRA. JUANA ROSALBA GARCIA, Dra.Ore- AUTORES: Dr. Roberto De Leon Caballero, Dr. Alberto gel Aguilar Viridiana, Dr. Jórdan Pérez Benjamín, Dr. Garnelo Cabañes Serafín, Dra. Juana Rosalba García Ramírez. Ibarra Moedano TITULO: Incidencia De Higado Graso No AlTITULO: Teratoma Maduro en Cabeza de Pancoholico En Pacientes Con Colelitiasis En El creas: reporte de caso en México Hospital General Leon 2do Congreso Anual de Patólogos Mexicanos • 67 León, Guanajuato • 2016 INTRODUCCIÓN: Los teratomas son tumores derivados de las células germinales capaces de generar tejidos del endodermo, mesodermo y ectodermo. Pueden clasificarse en maduros o inmaduros debido a la presencia de elementos neuroectodérmicos; los tipos maduros se clasifican en sólidos o quísticos. Comúnmente están en ovario, testículo, retroperitoneo, vejiga y cráneo. El sitio más raro de presentación es el páncreas OBJETIVOS: Reportar el caso de una paciente femenino con diagnóstico de teratoma maduro en cabeza de páncreas. MATERIAL: Se trata de paciente femenino de 31 años, residente de Hermosillo, sin antecedentes de importancia en relación a su padecimiento actual, el cual lo inicio con lumbalgia de dos meses de evolución constante, localizada, sin irradiación ni predominio de horario, que cede parcialmente a la ingesta de AINES (ketorolaco y tramadol). A la exploración física no se encontraron datos de importancia en relación al padecimiento. RESULTADOS: Se realiza tomografía axial computada que reporta presencia de tumoración quística a nivel de la cabeza de páncreas. Se realiza biopsia por aspiración con aguja fina sin reportarse células para diagnóstico. Se decide realizar intervención de tipo Whipple, obteniéndose producto de pancreatoduodenectomía con: teratoma quístico maduro (6.5 cm) en cabeza de páncreas; segmento de duodeno con inflamación crónica leve; segmento de estómago con inflamación crónica moderada; vesícula y conducto biliar sin alteraciones histopatológicas de importancia; 3 de 3 ganglios linfáticos peripancreáticos con hiperplasia linfoide (mayor de 1.5 cm). CONCLUSIONES: El teratoma quístico del páncreas puede presentar un diagnóstico preoperatorio difícil. Es necesaria una correlación clínica, radiológica y anatomopatológica para establecer el diagnóstico correcto y ofrecer el mejor tratamiento disponible evaluando el riesgo/beneficio que éste conlleve. INSTITUCIÓN: HOSPITAL GENERAL DEL ESTADO DE SONORA na Benemelis. TITULO: METAPLASIA ÓSEA EN SÍNDROME POLIPÓSICO JUVENIL. REPORTE DE UN HALLAZGO INUSUAL INTRODUCCIÓN: Los síndromes polipósicos hamartomatosos son entidades poco frecuentes que a diferencia de los pólipos solitarios, tienen una predisposición a desarrollar cáncer colorrectal y suelen presentarse en personas que cursan en edades pediátricas y durante la segunda década de la vida. La localización puede estar a lo largo de todo el tubo digestivo y su tamaño es variable. El síndrome de poliposis juvenil es una enfermedad autosómica dominante caracterizado por 5 o más pólipos, generalmente localizados en el recto y se manifiestan con sangrado rectal, dolor abdominal, ocasionalmente con diarrea y en casos de larga evolución con anemia y prolapso del pólipo. La formación de hueso heterotópico o metaplasia ósea es un cambio que se ha descrito en numerosos tejidos neoplásicos y no neoplásicos, sin embargo, en pólipos intestinales es un fenómeno raro descrito en 19 reportes de casos, la mayoría encontrados en pólipos aislados. En el presente trabajo, reportamos un caso de Síndrome polipósico juvenil con metaplasia ósea en un hombre de 18 años de edad, siendo el segundo caso en la literatura descrito en un síndrome polipósico. OBJETIVOS: Reportar un hallazgo inusual de metaplasia ósea en un paciente de 18 años con síndrome poliposico juvenil. CONCLUSIONES: La metaplasia ósea es un hallazgo raro descrito en pólipos del tracto gastrointestinal, encontrandose en la literatura 19 reporte de casos, la mayoría en pólipos solitarios sugiriendo diversos mecanismos de desarrollo. En el presente trabajo la metaplasia ósea se reporta en un síndrome polipósico, siendo el segundo en la literatura, sin reconocer un significado clínico, pero atrayendo la curiosidad del patólogo. INSTITUCIÓN: LABORATORIO DE PATOLOGIA QUIRURGICA Y CITOLOGIA DE PUEBLA SC. ESTADO: PUEBLA ESTADO: SONORA Mampara: 20 B Mampara: 20 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-155 AUTORES: Dr. Mario Murguía Pérez, Dr. Luis Raul Cha- TC-151 AUTORES: Dr. Oscar Xavier Hernández Rodríguez, Dr. Sergio Sánchez Sosa, Dra Julieta Garcia Gutierrez, Dra Iri- vira, Dr. Luis Tinoco Tellez, Dra. Bertha A. Moreno Perez, Dr. Juan Carlos Orozco Oregon, Dra. Cindy Sharon Ortiz Arce, Dr. Jose Reyes Nava, Dr. Marco Vinicio Alonso Briones, Dr. David Medina Pineda, Dr. Saulo Mendoza Ramírez 2do Congreso Anual de Patólogos Mexicanos • 68 León, Guanajuato • 2016 TITULO: Tumor solido pseudopapilar. Estudio pital General de Mexico de 10 casos con analisis morfologico y de inESTADO: GUANAJUATO munohistoquimica. INTRODUCCIÓN: El tumor solido pseudopapilar de pancreas (TSP) es una neoplasia poco frecuente, representa menos del 3% de todas las neoplasias malignas pancreaticas, y entre los tumores quisticos de dicho organo, corresponde a la variedad menos frecuente. Se considera que esta neoplasia es de bajo grado de potencial maligno, con un pronóstico favorable con resecciones completas. Su histogenesis permanece incierta. OBJETIVOS: En Mexico, se han informado algunos casos aislados, sin embargo, no existe una serie de casos con datos clinico-patologicos en un determinado periodo de tiempo. El objetivo de este estudio es conocer la frecuencia del TSP en casos de patologia quirurgica en un periodo de 5 años; asi tambien, señalar algunos aspectos epidemiologicos, histologia, inmunofenotipo, y su seguimiento. MATERIAL: Se realizo un estudio descriptivo, retrospec- tivo, longitudinal y observacional, en el que se revisaron los informes del archivo del Departamento de Patologia Quirurgica del archivo de patologia de la UMAE Nº 1 IMSS Leon en un periodo de tiempo comprendido del 1º de febrero del 2011 al 31 de enero del 2016. De cada caso se obtuvieron datos epidemiologicos, se reviso el material de cada caso y se realizaron estudios de inmunohistoquimica complement RESULTADOS: En el periodo de tiempo estudiado se en- contraron 10 casos de TSP. 7 de los 10 casos encontrados correspondieron a mujeres. El sintoma mas frecuente fue aumento del perimetro abdominal.La localización mas frecuente fue cuerpo y cola (50%). En ningun caso se realizo la sospecha clinica diagnostica. En 5 casos se optó por realizar pancreatectomía distal (50%), 3 casos con pancreatectomía subtotal (30%), 1 caso con pancreatoduodenectomía (10%), y 1 caso con diagnóstico por biopsia con trucut guiada por tomografia (10%). Ninguno de los pacientes desarrolló metástasis. La mayor parte de los tumores fueron solidos. Histologicamente se observaron patrones solido, trabecular y pseudopapilar, sin mitosis. Por inmunohistoquimica se aprecio positividad para vimentina, a1-AT, a1-AQT, ENE, receptores de progesterona y CD10, con intensidades variables, y con indice de proliferacion menor al 2%. La sobrevida actual de los pacientes es de 31.1 meses, y sin presentar recidivas. CONCLUSIONES: El TSP es una neoplasia de pancreas poco frecuente, con muy buen pronóstico. Es importante para el patologo estar familiarizado con sus caracteristicas clinicas, histopatologicas e inmunohistoquimicas. Tal conocimiento es esencial para diferenciarlo de otras neoplasias pancreaticas potencialmente mas agresivas. INSTITUCIÓN: Unidad Médica de Alta Especialidad Nº1 IMSS Leon, Hospital General Regional de Leon, Hospital Medica Campestre, Hos- NEFROPATOLOGÍA Mampara: 25 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-21 AUTORES: DRA. HERSILIA AIDE HERNANDEZ ZA- MONSETT, DRA. JUANA ELIZABETH TADEO GONZALEZ, DRA. MED. RAQUEL GARZA GUAJARDO, DRA. MED. ORALIA BARBOZA QUINTANA, DRA. GABRIELA ALARCON GALVAN TITULO: NEFROPATIA POR DEPOSITOS DE IgA EN UN PACIENTE CON VIH, REPORTE DE UN CASO INTRODUCCIÓN: De las nefropatías relacionadas a los pacientes con VIH, la mas común es la glomeruloesclerosis focal y segmentaria, también se ha visto asociada a la nefropatía clásica asociada a el VIH(HIVAN), Enfermedad renal mediada por complejos inmunes(HIVICK y Microangiopatia trombotica entre otras. Series de informes de nefropatía IgA en pacientes infectados por el VIH,revelan una prevalencia del 1,1%. OBJETIVOS: Masculino de 40 años de edad,VIH positivo en tratamiento. Inicia con disnea progresiva, tos productiva y fiebre. A la exploración física con crepitantes difusos. Urianalisis: abundantes eritrocitos/campo, >10 leucos/campo, proteínas 1628mg/dl, Creatinina sérica 4.2mg/dl, Cuantificación de proteínas en orina/24 hrs. 1190ml 4.79gr/24hr. MATERIAL: Se realiza biopsia renal previo consentimiento informado. Se realizaron secciones de los bloques de parafina que contenían el tejido a evaluar, estos se tiñeron con la técnica de hematoxilina y eosina, Acido peryodico de Schiff y metenamina de plata. Además se realizaron secciones de tejido congelado para su evaluación con la tecnica de inmunofluorescencia con anticuerpos para albumina, IgG, IgM, IgA, C3c, C1q, kappa y lambda. RESULTADOS: Se evaluo la biopsia renal la cual resulto adecuada con 19 glomerulos renales evaluables, observándose nefritis del intersticio renal de predominio linfocitico y atrofia tubular leve , los glomerulos mostraron una apariencia lobulada, con hipercelularidad endocapilar difusa y aumento de la matriz mesangial a expensas de depósitos inmunes, glomeruloesclerosis del 10% del tejido evaluado 2do Congreso Anual de Patólogos Mexicanos • 69 León, Guanajuato • 2016 . En la inmunofluorescencia se observo positividad 4 cruces para IgA y C3c únicamente. La microscopia electrónica reporto un marcado incremento con la matriz mesangial y presencia de moderadas estructuras electrodensas mesangiales compatibles con complejos inmunes. traron una biopsia renal adecuada, semilunas celulares en el 50% de los glomérulos, y esclerosis en el 50% de los glomérulos restantes, asociado a nefritis intersticial moderada y atrofia tubular. La inmunofluorescencia mostro positividad lineal capilar, intensidad 4+ para IgG. CONCLUSIONES: Los hallazgos histopatológicos encontrados en nuestro paciente son consistentes con una nefropatía por depósitos inmunes de tipo IgA, la positividad en la inmunofluorescencia para IgA y C3c son confirmatorios del diagnóstico. La patogenia de la nefropatía IgA se relaciona con la formación de complejos inmunes, sin embargo el antígeno subyacente suele ser desconocido. La verdadera prevalencia de esta nefropatía en pacientes infectados por el VIH puede estar subestimada debido al curso indolente que tiene la misma lo que probablemente conduce a un menor número de tomas de biopsias renales. RESULTADOS: En nuestro paciente la positividad lineal INSTITUCIÓN: DEPARTAMENTO DE ANATOMIA PATOLOGICA Y CITOPATOLOGIA, HOSPITAL UNIVERSITARIO “DR. JOSE ELEUTERIO GONZALEZ” ESTADO: NUEVO LEÓN de la pared capilar de con intensidad de 4+ para IgG, apoya el diagnóstico de anti-GBM. CONCLUSIONES: Diferentes series estiman que la fre- cuencia varia de 0,5-1 casos por millón al año, se informo que la enfermedad anti membrana basal glomerular es responsable de 1-5% del total de las glomerulonefritis inducidas por anticuerpos y es la causa de 10 a 20% de los casos de glomerulonefritis en semilunas. La imagen histológica muestra expansión mesangial en hipercelularidad, que evoluciona a glomerulonefritis focal y segmentaria con infiltración por leucocitos. Se acompaña de necrosis segmentaria. Hay extensa formación de semilunas en asociación con la destrucción de la membrana basal glomerular. INSTITUCIÓN: Hospital Universitario. Hospital Universitario “Dr. José Eleuterio González” ESTADO: NUEVO LEÓN Mampara: 25 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. Mampara: 26 A TC-115 Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. AUTORES: Dra. Juana Elizabeth Tadeo González. Dra. TC-196 Hersilia Aide Hernandez Zamonsett. Dra. Gabriela Alarcon Galvan. Dra. Raquel Garza Guajardo. Dra. Oralia Barboza Quintana. , Dr. Marco Antonio Ponce Camacho TITULO: Enfermedad anti-membrana basal glomerular. Presentación de un caso y revisión de la literatura INTRODUCCIÓN: Enfermedad anti-membrana basal glomerular es una enfermedad autoinmune, en la que los anticuerpos circulantes se dirigen contra la cadena alfa-3 de colágeno tipo IV presente en la membrana basal glomerular y los alvéolos pulmonares. Es rara causa de glomerulonefritis y se define por la presencia de anticuerpos en el suero y/o unión de IgG lineal detectada por inmunofluorescencia directa. OBJETIVOS: Reportar el caso de una mujer de 22 años, comienza con alteración de la conciencia, hematuria, edema e hipertensión. Posterior presenta crisis convulsivas y hemorragia pulmonar.Las cadenas Kappa/lambda, determinación de ANA, ANCA,anticuerpos antifosfolípidos, niveles de anticardiolipina y del complemento fueron negativos. Se realizó biopsia renal MATERIAL: Se recibieron dos biopsias. Los cortes mos- AUTORES: Dra. Martha Arisbeth Villanueva Pérez. Dra. Ana Karen Soto Sañudo, Dr. Renato Parra Michel. TITULO: Nefropatía Membranosa y Glomerulonefritis Necrosante con medias lunas asociada a ANCA’s en paciente con infección por VIH INTRODUCCIÓN: El deterioro de la función renal en pa- cientes con VIH es multifactorial; entre los mecanismos de daño renal se encuentran la infección directa del virus a las estructuras renales, infecciones oportunistas que afectan el parénquima renal, toxicidad relacionada a fármacos o bien procesos autoinmunes. Las glomerulopatías en pacientes con infección por VIH son la principal causa de síndrome nefrótico-nefrítico. OBJETIVOS: Presentar un caso de Nefropatía Membra- nosa y Glomerulonefritis Necrosante con medias lunas asociada a ANCA’s en paciente con infección por VIH. Describir los procesos de autoinmunidad como parte del daño renal en estos pacientes. MATERIAL: Caso clínico de paciente con VIH y nefropa- 2do Congreso Anual de Patólogos Mexicanos • 70 León, Guanajuato • 2016 tía, correlacionar con histopatología de biopsia renal. RESULTADOS: Ficha clínica: Hombre de 23 años, solte- ro, escolaridad secundaria, de ocupación mesero, originario de Veracruz y residente en Guadalajara, Jalisco. Antecedentes de importancia: No antecedentes heredofamiliares de importancia para el padecimiento. Niega toxicomanías. Padecimiento actual: Inició un mes previo a su ingreso con dolor abdominal en región epigástrica, acompañado de malestar general y fiebre de hasta 39-40 oC , sin predominio de horario,sin foco infeccioso sin irradiaciones, aunado a aparición de lesiones verrucosas en región perianal, se diagnostica enfermedad acidopéptica y condiloma acuminado, realizando ELISA para HIV que resultó positivo. Se agrega la presencia de orina de color marrón, se documenta anemia y elevación de niveles de azoados por lo que se interconsulta al servicio de nefrología. Exploración física: Signos vitales: TA: 110/70 mmHg, Peso: 63 kgs, Talla: 1.61 m, FC: 94 latidos por minuto, FR: 18 por minuto, Temperatura: 37°C. Exploración sin datos patológicos; abdomen blando, depresible, con regular panículo adiposo, con leve dolor a la palpación profunda en epigastrio, no rebote, no visceromegalias ni tumoraciones, con ruidos peristálticos normales en intensidad y frecuencia. Auxiliares de diagnóstico: Citometría hemática: leucocitos 7900/ul, linfocitos 1872 /ul, plaquetas 342,000/ul, hemoglobina 9.6 g/dl, hematocrito 27.7 % . Química sanguínea: urea 94 mg/dl, creatinina 4.49 mg/dl, colesterol 159 mg/dl, albúmina: 2.7 mg/dl. Examen general de orina: Densidad de 1.020, Ph 6, Proteinas >300mg/dl, leucocitos 3/campo, células epiteliales escasas, no cilindros. Proteínas en 24 gr: 6.9g. Serológicos: Acs Anti VIH reactivo, perfil para hepatitis B y C negativos. ANAs (+) 1:320 Anti, C3 101 mg/dl C4: 17.4 mg/dl, p-ANCA(-) c-ANCA (+++), Acs anti DNA (+).Gabinete: Ultrasonido abdominal y renal. Presencia de líquido libre en cavidad de ecogenicidad homogénea, probable ascítico. Riñones de bordes regulares, con hipercogenicidad cortical, tamaño de 12x5.5 cms ambos, seno renal hiperecogénico, adecuada relación corticomedular, sin dilatación del sistema pielocalicial, no litos, sin colecciones. Vejiga con escasa repleción, sin evidencia de litos. Se realizo biopsia renal percutánea con los siguientes hallazgos: Glomerulos 19, de los cuales 17 presentan necrosis fibrinoide y proliferación extracapilar con lesiones activas y crónicas. El mesangio sin expansión; los capilares con luces abiertas y rígidas, con membranas gruesas sin irregularidades, no proliferación endocapilar. Túbulos con cilindros hemáticos y granulosos, atrofia del 20%. Intersticio con fibrosis del 20% e infiltrado mononuclear difuso. Arterias de mediano y pequeño calibre sin necrosis y con luces permeables. Inmunofluorescencia mostro positividad para C3c e IgG con patrón granular epimembranoso. Conclusión diagnóstica: Nefropatía Membranosa y Glomerulonefritis Necrosante con medias lunas asociada a ANCA’s lógicas en estos pacientes es probable encontrar enfermedades concomitantes con diferente fisiopatogénia. La vasculitis pauciinmune asociada a ANCAs y la glomerulopatía membranosa tienen diferente mecanismo de daño y pueden de forma índependiente asociarse a otros factores como la infección por VEB. REFERENCIAS: Colvin R. Diagnostico-patología Enfermedades renales. Marban 2014 2(124). Kidney International, Vol. 67 (2005), pp. 1381?1390 INSTITUCIÓN: Servicio de Anatomía Patológica, Centro Médico Nacional de Occidente IMSS. Servicio de Nefrología Hospital regional No. 46 IMSS ESTADO: JALISCO PATOLOGÍA ÓSEA Mampara: 27 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-79 AUTORES: Dra. Julieta Y. Peralta Serna, Dr. Fernando De la Torre Rendón,Dr. David Cruz Guillen,Dr. Rafael Rodríguez Rojas. TITULO: Condromatosis sinovial bilateral de la rodilla con presentación intra y extra articular asociados a depósitos óseos de dihidrato de pirofosfato de calcio (CPPD). INTRODUCCIÓN: La condromatosis sinovial es una rara condición, que consiste en una proliferación de nódulos irregulares de cartílago hipercelular en el tejido sinovial estos pueden presentar calcificación y osificación secundaria. La etiología es desconocida. OBJETIVOS: mediante un caso clínico se propone mostrar las características poco comunes en la presentación de un caso de condromatosis sinovial siendo una presentación bilateral y extra-articular, ademas de depósitos óseos de dihidrato de pirofosfato de calcio (CPPD). CONCLUSIONES: En nuestro medio las lesiones glomerulares más frecuentes en pacientes con VIH son las asociadas a complejos inmunes, entre ellas la glomerulonefritis membranoproliferativa, la nefropatía “lupus like” y la glomerulopatía membranosa; la nefropatía colapsante (Nefropatía por VIH) es una entidad que se presenta principalmente en pacientes afroamericanos y es poco común en nuestra población. Como parte de alteraciones inmuno- MATERIAL: mediante un caso clínico de una femenina de 64 años que presenta en pierna izquierda aumento de volumen, dolor y problemas con la deambulación; se explora y mediante rayos X, resonancia magnética se observa lesiones nodulares en ambas rodillas, se realiza toma de biopsia y se hace una revisión actualizada del tema y las 2do Congreso Anual de Patólogos Mexicanos • 71 León, Guanajuato • 2016 diferentes formas de presentación de la condromatosis sinovial. RESULTADOS: Se obtienen mediante biopsia lesiones de bordes regulares, localizadas alrededor del fémur distal y lateral, de 2, 2.5 y 3.5 cm respectivamente; y muestra de tibia proximal, las hallazgos microscópicos corresponden a tejidos óseos y fibrosos reparativos con calcificación extensa, necrosis parcial e inflamación crónica compatible con depósitos de dihidrato de pirofosfato de calcio (CPPD) y 2) nódulos cartilaginosos compatibles con condromatosis de localizacion intra y extraarticular. CONCLUSIONES: la condromatosis sinovial es una entidad de etología desconocida, con un recidiva local de 15% luego de la escisión. la presentación aunque solo se conoce intraarticular puede manifestarse en sitios extrarticulares. RESULTADOS: En este estudio, se obtuvieron mediante Resonancia Magnética Nuclear de 1H los perfiles metabonómicos del suero de pacientes con encondroma, con el fin de determinar las alteraciones metabólicas significativas asociadas con este tumor condroide. Los resultados demuestran una clara diferencia entre los perfiles de pacientes con encondroma respecto a controles sanos, como se evidenció por medio de el análisis de mínimos cuadrados parciales discriminante-ortogonal (OPLS-DA). CONCLUSIONES: Los análisis metabonómicos sugieren alteraciones en el metabolismo del propanoato y en el metabolismo de la metionina, dichos cambios se observaron como el aumento de concentraciones séricas de estos metabolitos en pacientes diagnosticados con encondroma. INSTITUCIÓN: Escuela Nacional de Medicina y Homeopatía, Instituto Politécnico Nacional, Hos- INSTITUCIÓN: Hospital Regional Lic. Adolfo pital de Ortopedia “Dr. Victorio de la Fuente Narváez”, Instituto Mexicano del Seguro Socia. López Mateos. ISSSTE ESTADO: DISTRITO FEDERAL ESTADO: DISTRITO FEDERAL Mampara: 27 B Mampara: 28 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-149 TC-153 AUTORES: LILIANA ISABEL LOPEZ, D.C. Angel E. Ba- AUTORES: Francisco-Javier Martínez López, Angel E. ñuelos Hernández,D.C. Elvia Becerra-Martínez,D.C. Nury Pérez Hernández,D.C. Elizabeth Pérez-Hernández Bañuelos Hernández, Elvia Becerra-Martínez, Elizabeth Pérez Hernández, Nury Pérez-Hernández TITULO: Estudio Metabonomico del Suero de TITULO: Estudio Metabonómico del Tejido del Tumor de Celulas Gigantes de Hueso Pacientes con Encondroma INTRODUCCIÓN: El encondroma representa el 2.6% de los tumores óseos benignos. Este tumor condroide es asintomático y puede afectar a personas de cualquier edad, afectando los huesos largos de manos y pies. Las características radiográficas son la calcificación punteada y festoneado endostal. El diagnóstico exacto es esencial para distinguir entre un encondroma y un tumor maligno de bajo grado. INTRODUCCIÓN: El tumor de células gigantes de hue- OBJETIVOS: Identificar el perfil metabonomico de los tu- OBJETIVOS: Identificar el perfil metabolómico del TCGH mores óseos condrogénicos con énfasis en la búsqueda de biomarcadores asociados a la evolución y comportamiento clínico. MATERIAL: 1. 200 uL suero and 400 uL NaCl 0.9% 2. Desproteinización 3. 100 uL Agua deuterada y 0.1 mM TSP (Trimethylsilyl propanoic acid) 4. Resonancia Magnetica Nuclear 5. Análisis y cuantificación de metabolitos 6. Análisis multivariado 7. Análisis de impacto de vías so (TCGH)suma el 20% de los tumores óseos benignos. Aparece en la 3a década de la vida en la epífisis de los huesos largos. Aunque el TCGH se considera benigno es de un curso impredecible, con recidivas del 20-40% y metástasis pulmonar de 6%. La patogénesis molecular se desconoce y debe de ser estudiada para el desarrollo de tratamientos o diagnósticos tempranos. mediante la técnica de Resonancia Magnetica Nuclear con énfasis en la búsqueda de biomarcadores asociados al comportamiento biológico MATERIAL: 1. Pulverización del tejido y extracción de los metabolitos polares. 2. Preparación de la muestra para Resonancia Magnetica Nuclear de 1H, mediante la disolución de los metabolitos en agua deuterada. 4. Obtención de los espectros, identificación y cuantificación de los metabolitos. 2do Congreso Anual de Patólogos Mexicanos • 72 León, Guanajuato • 2016 5. Análisis multivariado. 6. Análisis de impacto de vías. RESULTADOS: En este estudio, la elaboración de perfiles metabolómicos de tejido del TCGH se abordó mediante la técnica de Resonancia Magnética Nuclear de 1H y dirigida a determinar alteraciones metabólicas significativas. Los resultados demuestran diferencias metabonómicas claras entre los grupos de TCGH y control evidenciadas por el análisis de mínimos cuadrados parciales discriminante-ortogonal (OPLS-DA). Los metabolitos diferenciales identificados sugieren un metabolismo alterado en el TCGH caracterizado por un aumento en la producción de metionina y en la biosíntesis de fosfolipidos. CONCLUSIONES: se seleccionaron pacientes con diagnóstico de osteocondroma en el esqueleto apendicular intervenidos quirúrgicamente entre el 1 de enero del 1995 al 31 de Diciembre del 2015. Los diagnósticos asociados a malignidad fueron corroborados por análisis de histopatología y se correlacionaron con el grosor de la capa cartilaginosa expresada en milímetros. RESULTADOS: El tamaño de la capa cartilaginosa del ostecondroma en el esqueleto apendicular mayor a 1 cm acorde con lo reportado en la literatura representa riesgo de malignidad con una frecuencia del 2%. CONCLUSIONES: La detección de una capa mayor de 1 cm en la pieza quirúrgica del osteocondroma representa un factor de riesgo asociado a una incidencia elevada para presentar transformación condrosarcomatosa y requiere una vigilancia estrecha así como también sugerimos se realice una resección cuidadosa tomando en cuenta el espesor de manera preoperatoria, para determinar la amplitud de los bordes de resección. INSTITUCIÓN: Escuela Nacional de Medicina y Homeopatía, Instituto Politécnico Nacional, México, D. F.,07320 Mexico. Hospital de Ortopedia “Dr. Victorio de la Fuente Narváez”, Instituto Mexicano del Seguro Social, 07760, México D.F.,Mexico INSTITUCIÓN: UMAE DE TRAUMATOLOGIA, ORESTADO: DISTRITO FEDERAL TOPEDIA Y REHABILITACION “DR. VICTORIO DE LA FUENTE NARVAEZ”, IMSS ESTADO: MICHOACÁN Mampara: 28 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. Mampara: 29 A TC-161 Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. AUTORES: Dr. C. F. Aguirre Parra, Dra. E. Pérez Hernán- TC-187 dez, Dra. A. Atencio Chan, Dra. G.M. Rangel Díaz, Dr. J. M. Pérez Atanacio, Dr. R. Amaya Zepeda AUTORES: Dra. Elizabeth Pérez-Hernández, Dr. Gerardo Gutiérrez-Sánchez, Dr. Romeo Tecualt-Gómez, Dr. TITULO: Grosor de la capa cartilaginosa como Guillermo Pérez-Ishiwara, Dra. Adriana Atencio Chan, Dra. factor de malignidad en pacientes con osteo- Nury Pérez-Hernández. condromas en esqueleto apendicular. TITULO: PROTEINAS ONCOGENICAS ASOINTRODUCCIÓN: En la Ortopedia y específicamente en CIADAS AL TUMOR DE CELULAS GIGANTES los tumores óseos, los osteocondromas representan el tu- DE HUESO mor más común, con incidencia de malignidad entre el 1-2% en forma de condrosarcomas, con reportes en la literatura donde las mediciones del grosor de la capa cartilaginosa mayor de 1 cm suele correlacionar con malignidad. El riesgo exacto de cambio condrosarcomatoso en los pacientes es desconocido. OBJETIVOS: Identificar si el Grosor de la capa cartilaginosa es un factor de riesgo para la malignidad en pacientes con osteocondromas en el esqueleto apendicular MATERIAL: Se diseñó un estudio de casos y controles, INTRODUCCIÓN: El tumor de células gigantes de hueso (TCGh) representa el 20% de los tumores benignos del esqueleto. El curso clínico es impredecible, recurre en el 20-40% de los casos posterior a la resección intralesional o marginal y el 1-6% desarrollan diseminación pulmonar. El TCGh es ligeramente más frecuente en mujeres, más agresivo cuando se asocia al embarazo. OBJETIVOS: Búsqueda de marcadores moleculares relacionados con la patogénesis del TCGh y su posible relación con el comportamiento biológico. 2do Congreso Anual de Patólogos Mexicanos • 73 León, Guanajuato • 2016 MATERIAL: Se obtuvieron muestras de TCGh en fresco MATERIAL: Las membranas periimplante se obtuvieron RESULTADOS: Se colectaron 9 muestras de pacientes RESULTADOS: Se colectaron 89 membranas periim- y embebidas en parafina de piezas quirúrgicas del Departamento de Anatomía Patológica del HOVFN. Las primeras se emplearon en análisis proteómicos diferenciales a partir de geles 2D e identificación mediante espectrometría de masas. Además se realizaron ensayos de inmunoperoxidasa complementarios (ciclina D1, receptores de estrógenos y progesterona, p53 y Ki-67). con TCGh con una edad promedio de 28.2 años, clasificadas en 3 grupos (hombres, mujeres no gestantes y mujeres gestantes). Se identificaron 11 proteínas diferenciales (ausentes en varones y mujeres no gestantes). CONCLUSIONES: Se identificaron 3 proteínas candidatos de la oncogénesis del TCGh (Wnt16, SNIP1 y SUZ12) posiblemente relacionadas al sexo y la evolución clínica. INSTITUCIÓN: UMAE de Traumatología, Ortopedia y Rehabilitación “Dr. Victorio de la Fuente Narvaéz”; Escuela Nacional de Medicina y Homeopatía IPN;Complex Carbohydrate Research Center, University of Georgia, Athens, Georgia, US de pacientes sometidos a retiro material de osteosíntesis. El tejido se procesó para análisis histopatológico y se clasificó de acuerdo a los criterios de Morawietz. El desarrollo bacteriológico se obtuvo de cultivos para microorganismos aeróbicos. La velocidad de sedimentación globular, proteína C reactiva y cuenta leucocitaria se incluyeron en el protocolo de estudio preoperatorio. plante obtenidas de prótesis cementadas (49.4%), prótesis no cementadas (14.6%), clavos centromedulares (22.4%), sistemas DHS (4.4%), placas DCP (7.8%) y alambre (1.1%). El 65.1% correspondieron a tipo I ó inducida por partículas de desgaste, 20.2% a tipo II o infecciosa, 11.2% a tipo III ó combinada y 33.3% a tipo IV ó indeterminada. Se identificó un predominio de membranas tipo I en prótesis cementadas (50%), tipo II en prótesis cementadas y clavos centromedulares (83.2%), tipo III en prótesis cementadas (60%) y tipo IV en clavos centromedulares (66.6%), con una p=0.0027. Los cultivos reportaron S. aureus en membranas tipo II (27.7%) y III (50%), con una p<0.0001. > CONCLUSIONES: La clasificación histopatológica de ESTADO:DISTRITO FEDERAL las membranas correlaciona significativamente con el tipo de implante y el microorganismo aislado. No existe correlación entre el tipo de membrana y los marcadores biológicos de la inflamación. Mampara: 30 A INSTITUCIÓN: Hospital de Ortopedia Dr. Victorio de la Fuente Narvaez, Distrito Federal Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. ESTADO: DISTRITO FEDERAL TC-194 AUTORES: Dra. Adriana Ivette Castañeda Martínez, Dr. Mampara: 11 A José Antonio Hernández García, Dra. Elizabeth Pérez Hernández, Dr. Adrián Miguel Pérez, Dr. José Jesus Pérez Correa, Dra. Adriana Atencio Chan, Dr. José Alberto Sánchez Cañas Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. TC-17 TITULO: CARACTERIZACIÓN HISTOPATOLÓGICA Y BACTERIOLÓGICA DE LAS MEMBRANAS PERIIMPLANTE EN PACIENTES SOMETIDOS A RETIRO DE MATERIAL DE OSTEOSÍNTESIS Y SU CORRELACIÓN CLÍNICA INTRODUCCIÓN: Las membranas periprotésicas cubren la interfase hueso-metal o hueso-cemento, son un sitio de alojo de microorganismos donde se mantienen protegidos de la respuesta inmune y los tratamientos antimicrobianos. OBJETIVOS: Caracterizar las membranas periimplante mediante análisis histopatologico y bacteriológico y establecer su relación con marcadores biológicos de la inflamación. AUTORES: Dra. Brenda Esther Vázquez López, Dra. Olga Esquivel Hernández, Dr. Jorge Edgardo Marquez Escalante. TITULO: DISPLASIA MESENQUIMAL PLACENTARIA. REPORTE DE CASO. INTRODUCCIÓN: La Displasia mesenquimal placentaria (DMP) es una anomalía vascular de baja incidenciaque se caracteriza por placentomegalia y múltiples vesículas en “racimos de uvas”. Frecuentemente se diagnostica como embarazo molar en ultrasonido. La DMP generalmente presenta un feto viable de cariotipo normal. El diagnostico final es histopatológico, sin las alteraciones características de enfermedad molar. 2do Congreso Anual de Patólogos Mexicanos • 74 León, Guanajuato • 2016 OBJETIVOS: - Presentar un reporte de caso de baja incidencia. - Revisión de la literatura. MATERIAL: Femenina de 17 años, con antecedente de un aborto un año previo al embarazo actual. Cursa con embarazo normoevolutivo. Durante ultrasonido º se detectan datos de Retraso de crecimiento intrauterino (29.6 SDG por FUR y de 26.3 por US), oligohidramnios (ILA 7.9), anemia fetal, placentomegalia así como múltiples quistes de 13 mm en promedio en mesénquima placentario sin flujo. Por lo que se inicia inductores de la maduración pulmonar. RESULTADOS: En el servicio de Anatomía Patológica, se recibió una placenta de 19 x 19 x 7 cm, con un peso de 450 gr. Por su cara fetal se observó un cordón trivascular de inserción central con vasculatura prominente, con pared de 0.2 cm y con datos de trombosis extensa. Por su cara materna se observaron cotiledones café obscuros de aspecto fragmentado con abundantes vesículas de 0.8 cm en promedio. El análisis microscópico mostró vellosidades de aspecto edematoso, sin datos de proliferación trofoblastica que alternaban con vellosidades de aspecto normal. CONCLUSIONES: En conclusión, la DMP es una patología rara que fácilmente puede ser confundida con una enfermedad molar. Sin embargo ante la presencia de una placentomegalia con un feto vivo con cariotipo normal, el uso de los valores séricos de la AFP y la hGCH, en combinación con los hallazgos de ultrasonido, permiten sospechar de una DMP. Y una vez confirmado el diagnostico por anatomopatologia, realizar la búsqueda intencionada de otras alteraciones fetales para descartar el Síndrome de Beckwith-Wiedeman. INSTITUCIÓN: UMAE #23, IMSS. Monterrey, Nuevo León. ESTADO: NUEVO LEÓN nece al grupo de los tumores mullerianos mixtos, son neoplasias uterinas poco frecuentes, con elementos glandulares benignos y malignos del estroma que puede los cuales pueden tener diferenciación homóloga o heteróloga (cartílago, músculo estriado, hueso, etc.). OBJETIVOS: Paciente femenina de 49 años de edad, diabética tipo II, se presenta con sangrado irregular, dolor abdominal y sensación de cuerpo extraño en vagina. El examen ginecológico reveló un tumor que sobresale de la vagina (9x7cm), blando, liso con bordes dentados, a la palpación bimanual se evidencia un pedículo proveniente de cuello uterino, que al girar en sentido contrario a las manecillas del reloj, se reseca, sin sangrado. En especuloscopia la vagina mostró rastros de hemorragia, cuello del útero de 2 cm, indurado. El tumor fue enviado para su evaluación patológica. MATERIAL: Macroscópicamente es una lesión polipoide friable de 9x7x4cm; superficie externa ulcerada. A la seccion heterogéneo, carnoso, de color rosado a gris marrón. A la microscopia compuesto por componente glandular endocervical y un componente de estromal sarcomatoso. El componente estromal presenta un índice mitótico con 3 mitosis/10 campos. Se identificaron áreas osteoides heterólogas entre el tejido con grandes áreas de necrosis. RESULTADOS: Se realizó un diagnóstico de adenosarcoma Mülleriano con elementos heterólogos osteoides. CONCLUSIONES: Sólo el 2% de MA se presenten en el cuello uterino, con menos de dos docenas de casos documentados hasta el momento en la literatura hasta el año 2014. La edad media de aparición es de 31 años (rango 1165 años). El diagnóstico diferencial incluye benigna (adenofibroma, pólipo endocervical atípico, y adenomioma del cuello uterino) y malignos (adenosarcoma uterino con participación secundaria del cuello del útero, carcinosarcoma, y rabdomiosarcoma embrionario. La aparicion de elementos heterólogos se ha relacionado con un peor pronóstico. INSTITUCIÓN: SERVICIO DE ANATOMIA PATOLOGICA Y CITOPATOLOGIA, HOSPITAL UNIVERSITARIO “DR. JOSE ELEUTERIO GONFecha y hora Instalación: 2 mayo 7:30 hrs. ZALEZ” Mampara: 11 B Fecha y hora retiro: 2 mayo 13:00 hrs. ESTADO: NUEVO LEÓN TC-25 AUTORES: DRA. HERSILIA AIDE HERNANDEZ ZAMONSETT, DRA. GABRIELA SOFIA GOMEZ MACIAS, DRA. RAQUEL GARZA GUAJARDO, DRA. ORALIA BARBOZA QUINTANA. Mampara: 12 A Fecha y hora Instalación: 2 mayo 7:30 hrs. Fecha y hora retiro: 2 mayo 13:00 hrs. TITULO: ADENOSARCOMA MULLERIANO CON DIFERENCIACION HETEROLOGA DE TC-42 TIPO OSTEOIDE, REPORTE DE UN CASO. AUTORES: LUIS ARTURO ACOSTA, Dra. Lucía Alemán INTRODUCCIÓN: El Adenosarcoma mulleriano perte- Meza, Dra. Oralia Barboza Quintana, Dra. Raquel Garza 2do Congreso Anual de Patólogos Mexicanos • 75 León, Guanajuato • 2016 Guajardo. Dra. Gabriela Sofía Gómez Macías. Mampara: 12 B TITULO: TUMOR DE CORDONES SEXUALES Fecha y hora Instalación: 2 mayo 7:30 hrs. DE TIPO TUBULOS ANULARES OVARICO BI- Fecha y hora retiro: 2 mayo 13:00 hrs. LATERAL ASOCIADO A HIPERPLASIA DE CELULAS DE LEYDIG: REPORTE DE UN CASO Y TC-82 REVISIÓN DE LA LITERATURA. AUTORES: BÁRBARA SÁENZ, Dra. Med. Oralia BarboQuintana, Dra. Med. Raquel Garza Guajardo, Dr. Alvaro INTRODUCCIÓN: El tumor de cordones sexuales con za Barbosa Quintana, Dra. Gabriela Sofía Goméz Macias túbulos anulares es una neoplasia ovárica benigna rara (6% de los tumores de cordones sexuales) y únicamente 1% de todas las neoplasias ováricas. Hasta un 30% de ellos se asocia al síndrome de Peutz Jeghers. Otras asociaciones son con el adenoma maligno del cérvix y con el síndrome de Turner. OBJETIVOS: Presentamos el caso de una mujer de 71 años de edad con historia de sangrado transvaginal de 2 meses de evolución. El ultrasonido que revela un pólipo endometrial, asi como quistes ováricos bilaterales el derecho de 5.9 x 3.9 cm y el izquierdo de 2 x 1.8 cm. El Ca19-9 elevado en 66 U/mL (normal 0-30) y el Ca 19.9 en 11 U/mL (normal 0-37). MATERIAL: Se realizó histerectomía con salpingooforectomía bilateral. RESULTADOS: El útero presentó un pólipo endometrial de 1.3 cm, el ovario derecho de 8 x 5 cm, quístico con pequeños nódulos en su cara interna, el ovario izquierdo de 3 x 1 cm quístico. Microscopicamente se observó una neoplasia formadora de patrones de túbulos anulares simples y complejos. Con túbulos en forma de anillos compuestos por células con núcleo orientado periféricamente y alrededor de un cuerpo hialino central, con una zona citoplasmica anuclear formando el componente mayor del anillo. Los túbulos complejos se observan formando estructuras redondas compuestas de anillos intercomunicantes orientados alrededor de cuerpos hialinos. Así como una proliferación adyacente de células de Leydig. CONCLUSIONES: Se realizó inmunohístoquímica para inhibina y calretinina, las cuales fueron positivas. A la paciente se le considera en curación sin tratamiento adyuvante. Se trata de un tumor raro con una importante asociación al síndrome de Peutz-Jeghers y al adenoma maligno del cérvix (por lo cual debe muestrearse extensamente el espécimen). Asi como la importancia del diagnóstico diferencial entre un tumor de Sertoli o de la granulosa con los que puede compartir características, pero este tiene una histopatología única con los clásicos túbulos repetitivos. INSTITUCIÓN: Servicio de Anatomía Patológica del Hospital Universitario “Dr. José Eleuterio González” de la Universidad Autónoma de Nuevo León. Monterrey N.L. ESTADO: NUEVO LEÓN TITULO: Carcinoma mucinoso de ovario con nódulo mural con Carcinoma Anaplásico y Adenoma Adrenal a nivel de ligamento ancho: Reporte de un caso INTRODUCCIÓN: Los tumores mucinosos representan un 10-15% de los tumores ovaricos.. Los tumores mucinosos del ovario ya sean benignos, malignos o borderline pueden contener nódulos murales. Los nódulos pueden presentarse con histología benigna o maligna, los benignos como sarcoma- like o leiomioma; mientras los malignos, son de tipo carcinoma anaplásico, sarcoma o carcinosarcoma. OBJETIVOS: El objetivo es presentar el caso de una paciente femenina de 49 años de edad que se le diagnosticó un carcinoma mucinoso de ovario derecho con áreas de carcinoma anaplásico con cápsula íntegra. MATERIAL: Se recuperó la información de la paciente, junto con su historial clínico, así como su material de laminillas y bloques de parafina para estudiar a fondo el caso y documentarlo. RESULTADOS: Se recibió anexo derecho con un tumor en ovario derecho que pesa 13,650 gramos y que mide 38 x30x30 cm, con cápsula íntegra de donde sale material hemático y en su interior presentaba áreas sólidas, heterogéneas que miden 10x7x3 cm. A nivel del ligamento ancho un tumor de 15 gr y de 3 cms de color amarillento separado del ovario. A nivel histológico y con marcadores de inmunohistoquímica se logró diagnosticar un carcinoma mucinoso de ovario derecho con características intestinales y endocervicales y con áreas de carcinoma anaplásico de 38 cm de diámetro mayor con cápsula íntegra y un adenoma adrenal izquierdo de 3 cm de diámetro mayor originado en restos adrenales en el ligamento ancho. CONCLUSIONES: En cuestión de las neoplasias mucinosas de ovario Es necesario el análisis histológico e inmunohistoquímico minucioso de los nódulos murales para la determinación del tratamiento y el pronóstico. Así mismo sólo está descrito un caso en la literatura mundial un adenoma adrenal originado en el ligamento ancho. INSTITUCIÓN: Hospital Universitario “José Eleuterio González” 2do Congreso Anual de Patólogos Mexicanos • 76 León, Guanajuato • 2016 ESTADO: NUEVO LEÓN Mampara: 13 A Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. TC-83 AUTORES: Dr. Gabriel Juan Mandujano Alvarez, Dra. Meztli Guadalupe Gómez Morales, Dra. Clara Magdalena Martínez Hernández, M. en E.M. Dra. Yaneth Pérez Méndez, Dr. Gabriel Manuel Mandujano Vera. didas alternativas a la histerectomía cuando sea inevitable su desarrollo. La elevada frecuencia de placentación anormal, los datos relacionados con el tono uterino y con desgarros de este órgano, constituyeron las principales causas de histerectomía posparto, reportadas a nivel histopatológico. La indicación de histerectomía por clínica y el estudio histopatológico presentaron una concordancia buena. INSTITUCIÓN: Hospital Regional del Alta Especialidad de la Mujer. Secretaria de Salud. Villahermosa, Tabasco. Universidad Juárez Autónoma de Tabasco. ESTADO: TABASCO TITULO: CONCORDANCIA DE LA INDICACION DE HISTERECTOMÍA OBSTÉTRICA CON EL ESTUDIO HISTOPATOLÓGICO EN EL HOSPI- Mampara: 13 B TAL REGIONAL DE ALTA ESPECIALIDAD DE Fecha y hora Instalación: 2 mayo 7:30 hrs. LA MUJER. TABASCO. Fecha y hora retiro: 2 mayo 13:00 hrs. INTRODUCCIÓN: La Histerectomía Obstétrica (HO) es una cirugía urgente la cual se debe considerar como última medida para controlar la hemorragia obstétrica cuando el tratamiento conservador no ha sido eficaz, se necesitan medidas más radicales para evitar el choque hipovolémico, ya que la hemorragia obstétrica es la responsable del 2-11% de las muertes maternas alrededor del mundo. OBJETIVOS: Objetivos específicos. 1. Describir las características clínicas de las pacientes con histerectomía obstétrica. 2. Identificar los factores de riesgo asociados a histerectomía obstétrica. 3. Identificar la relación entre la indicación de la histerectomía y el estudio histopatológico. MATERIAL: El estudio realizado es de tipo retrospectivo, transversal y observacional, donde se analizaron en el Laboratorio de Patología del Hospital Regional de Alta Especialidad de la Mujer en Villahermosa, Tabasco; las piezas uterinas producto de histerectomía posparto. Durante el periodo del 1° de Enero del 2010 al 31 de Diciembre 2013. El universo estuvo conformado por 127 pacientes atendidas vía vaginal, cesárea o LAPE por ectópico. RESULTADOS: Los hallazgos histopatológicos se clasificaron en los grupos de: atonía uterina, desgarro/ruptura uterina, embarazo ectópico, hemorragia/hematoma desecante, placentación anormal, útero con cuadro infeccioso, útero obstétrico sin cambios anatómicos. De acuerdo a los valores establecidos por Landis y Koch, se obtuvo una concordancia moderada con un valor de Kappa de 0.481 y una p=0.000. Por lo que se acepta la hipótesis alterna Hi: Existe concordancia entre el diagnóstico de indicación de histerectomía obstétrica con el estudio histopatológico. CONCLUSIONES: Las complicaciones fueron choque hipovolémico, hemorragia, hematoma y lesión vesical. El objetivo es lograr su prevención, así como protocolizar me- TC-123 AUTORES: Dra. Hersilia Aide Hernandez Zamonsett, Dra. Gabriela Sofia Gomez Macias, Dra. Oralia Barboza Quintana, Dra. Raquel Garza Guajardo. TITULO: Tumor germinal mixto ovarico con componente de teratoma maduro y senos endodermicos. Reporte de Caso. INTRODUCCIÓN: Los tumores de células germinales del ovario malignos representan aproximadamente el 1-2% de todos los tumores malignos de ovario, aparecen en mujeres jóvenes, con un pico de incidencia alrededor de los 20 años. OBJETIVOS: Femenina de 27 años con dolor y aumento de perimetro abdominal y saciedad temprana.En exploracion bimanual se palpa tumor anexial derecho,movil, irregular. Tac reporta tumor pelvico, redondeado, de 11.5x8.4cm con delgada capsula, contenido heterogeneo y calcificaciones. CA125:757 u/ml, CA19-9:50.2 u/ml. Se realiza salpingooforectomia izquierda. MATERIAL: Se recibe tumor ovárico de 12x9x5cm, de 450 gramos y oviducto de 7x0.7x0.7cm. Tumor ovárico cubierto por capsula lisa, café claro y vasos congestivos. A la sección heterogéneo, con áreas complejas de consistencia pétrea y color café blanquecino, alternando con áreas blandas, solido- quísticas de color café claro, con presencia de material de aspecto adiposo y áreas extensas de necrosis, se realizan cortes histológicos. RESULTADOS: microscópicamente se observa un componente maduro con tejido oseo, cartilaginoso, epitelio respiratorio, tejido dérmico y epidérmico, alternando con 2do Congreso Anual de Patólogos Mexicanos • 77 León, Guanajuato • 2016 áreas con estroma hipocelular y mixoide, presencia de células tumorales con citoplasma claro, nucleo hipercromatico , irregular, nucléolo visible y una ligera actividad mitótica, presencia de cuerpos de schiller- duval, estas áreas mostraron positividad para alfafetoproteina, y fueron negativas para cd30, ck7 y ck20. se diagnostica como tumor germinal mixto (70% teratoma maduro, 30% tumor de senos endodermicos ). CONCLUSIONES: En contraste con el testículo, donde la combinación más común es el de carcinoma embrionario y teratoma, en el ovario, tumor del saco vitelino (YST) y disgerminoma representa una mezcla más común en la familia de los tumores de células germinales mixtos, lo que representa 11 de 30 tumores en una serie. Hay casos reportados de quistes dermoides con un componente de tumor de senos endodermicos, sin embargo no se encontraron casos reportados, en los que hubiera un teratoma maduro, con áreas tan complejas como el que observamos, alternando con tumor de senos endodermicos. INSTITUCIÓN: Servicio de Anatomia Patologica y Citopatologia, Hospital Universitario “Dr. Jose Eleuterio Gonzalez” ESTADO: NUEVO LEÓN RESULTADOS: Se captaron un total de 99 casos, de los cuales se excluyeron 33 posterior a la revisión de material histológico y expediente clínico. Dentro de la reevaluación se reclasificaron 5 casos de sarcoma uterino. La edad y tamaño promedio en la muestra fue de 53 años y 8.8 cm, el 67% de la muestra exhibió invasión miometrial mayor al 50%, el 25% de la muestra exhibió invasión al cérvix, l 44% de la muestra presentó invasión angiolinfática y el 66% de la muestra se diagnóstico en etapa histológica I. Dentro de los subtipos histológicos el 30% correspondió a leiomiosarcoma, el 33% a carcinosarcoma, el 20% a SEE, el 11% a adenosarcoma y el 6% a adenofibroma. CONCLUSIONES: Los sarcomas uterinos son un grupo heterogéneo de tumores, los cuales son difíciles de abordar debido a su amplio espectro histológico. Al igual que en la bibliografía, en el HGM se tiene una prevalencia del 6% de sarcomas uterinos. Las características clínico-patológicas expuestas en este trabajo coinciden con lo reportado en la literatura. Aunque ninguno de los marcadores presentados en este estudio son específicos para un subtipo de sarcoma, la utilización de un panel amplio de IHQ puede ayudar en la correcta clasificación de subtipos específicos de sarcomas. INSTITUCIÓN: Hospital General de México, “Dr. Eduardo Liceaga” Mampara: 14 A Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. quirúrgica del Hospital general de México en el periodo comprendido del 1/1/09 al 30/6/15. Se estudiaron las características clínico-patológicas y se les realizó IHQ: AML, CD10, CKAE1/AE3, RE, RP, Vimentina, p53 y ki67. 7:30 hrs. ESTADO: ZACATECAS TC-132 AUTORES: ANGELICA RODRIGUEZ, Dra. Mercedes Hernández González TITULO: “Análisis clínico-patológico de sarcomas uterinos en muestras quirúrgicas del Hospital General de México “Dr. Eduardo Liceaga” INTRODUCCIÓN: Los sarcomas uterinos son un grupo diverso de tumores, representan 3-7% de las neoplasias uterinas malignas. El diagnóstico diferencial entre los subtipos de sarcomas uterinos se basa en parámetros histológicos, los cuales pueden ser subjetivos y poco reproducibles, por lo que el uso de un panel amplio de IHQ ayuda en la clasifican de subtipos histológicos de sarcomas uterinos. OBJETIVOS: Evaluar el uso de un panel amplio de inmunohistoquímica de utilidad en el diagnóstico de sarcomas uterinos y conocer su comportamiento clínico-patológico. MATERIAL: Se realizo un estudio retrospectivo, descriptivo, transversal no experimental. Se analizaron los casos de sarcomas uterinos recibidos en el servicio de patología Mampara: 16 B Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 hrs. TC-144 AUTORES: LUIS RAUL CHAVIRA, Dr. Mario Murguía Peréz TITULO: TUMOR CARCINOIDE DE OVARIO INTRODUCCIÓN: El tumor carcinoide de ovario es raro (1%) es una neoplasia de crecimiento lento, se origina en células del sistema neuroendocrino, se ha descrito en cualquier órgano del endodermo primitivo. Pueden permanecer sin diagnóstico durante meses o años antes de dar metástasis. Según el patrón histológico se divide en: insular, trabecular, estromal, mucinoso y asociados o no a teratoma maduro OBJETIVOS: Presentación anatomopatologica de un caso de tumor carcinoide de ovario 2do Congreso Anual de Patólogos Mexicanos • 78 León, Guanajuato • 2016 MATERIAL: Paciente femenina de 26 años de edad sin antecedentes personales patológicos de importancia, quien acude con clínico por aumento del perímetro abdominal sin otros síntomas agregados, confirmando por ultrasonido, tumoración quistica en ovario, con elevación de CA 125 de forma discreta, se somete a laparotomia exploradora. RESULTADOS: Macroscópicamente se observó una tu- moración con dimensiones de 17 x 17 x 16.5 cms de consistencia renitente,superficie lisa blanquecina al corte quística con nódulo de 11 x 9 x 9 cms de color amarillo anaranjado, granular. Microscópicamente se observan células redondas y cilíndricas con núcleos ovales, regulares y uniformes, ausencia de nucleolos y excepcionales inclusiones intranucleares citoplasmáticas; el citoplasma celular es eosinófilo. Las células forman nidos sólidos con distribución en empalizada de las células periféricas y retracción del estroma circundante. Estas células fueron positivas a cromogranina y sinaptofisina. El diagnóstico fue de tumor carcinoide insular. CONCLUSIONES: Los carcinoides son tumores endocrinos asociados a menudo a un síndrome característico. Siempre son unilaterales y, si es bilateral, hay que considerar que es metástasico y buscar el tumor primario, el diagnóstico diferencial se debe hacer con el tumor de células de la granulosa (los cuerpos característicos de Call-Exner se pueden confundir con un acino carcinoide), tumor de células de Sertoly-Leydig, Si el tumor está limitado a un solo ovario, la supervivencia parece ser excelente con tratamiento quirúrgico y deben ser tratados como tumores ováricos de baja malignidad. INSTITUCIÓN: HOSPITAL GENERAL DE LEÓN SSA ESTADO: GUANAJUATO OBJETIVOS: Se describe el caso de una paciente que presentó Diabetes Gestacional y su hijo detención del crecimiento y Anemia aguda por Transfusión Feto-Materna secundarios a Corangioma gigante y Corangiosis placentaria. Su incidencia real se desconoce por la baja solicitud de estudio histopatológico de las placentas. Se realiza revisión de la bibliografía. MATERIAL: Primigesta de 36 años portadora de diabetes gestacional bien controlada. Inició su control prenatal desde el 1er trimestre, en la semana 34 se detecta detención del crecimiento, Acude a urgencias por referir hipomotilidad fetal, durante su revisión se realiza registro tococardiográfico que mostró desaceleración hasta 65 x min con recuperación lenta, el Ultrasonido muestró oligohidramnios. Se decide la interrupción mediante cesárea. RESULTADOS:Se obtuvo recién nacido único vivo, sexo masculino, 37 semanas, pesó 2300 gr, APGAR 7/8, talla 47 cm. Presentó dificultad respiratoria severa, requirió intubación e internamiento en terapia, su hemoglobina de 3 gr/dl y se integró el diagnóstico; Anemia severa por Transfusión Feto-Materna. Egresó por mejoría. El estudio histopatológico mostró; Un Corangioma con patrón angiomatoso de 5.5 cm y Corangiosis en áreas adyacentes. Se identificaron 2 dilataciones venosas en el cordón de 6 y 5 cm de longitud por 1 cm de diámetro a 2.5 y 10 cm de su inserción. Se considera que el tumor y las dilataciones venosas secuestraron aproximadamente 59 ml de sangre fetal, explicando la anemia aguda del RN. CONCLUSIONES: Ambas entidades son causa de Mampara: 14 B Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. llosa capilar. Ambos son causa de mortalidad perinatal, se asocian a condiciones de hipoperfusión y decremento de oxígeno, lo que puede explicar que se encontraran en una misma placenta. 7:30 hrs. TC-159 AUTORES: Dra. María del Carmen Vázquez Castro, Dra. Karla Judith González Colunga, Dr. Erik Noé Gudiño Ruiz, Dr. Sergio Ravindranath Domínguez Cortes, Dr. Antonio Isaías Rocha Herrera. TITULO: CORANGIOMA GIGANTE Y CORANGIOSIS PLACENTARIA, REPORTE DE UN CASO Y REVISIÓN DE LA LITERATURA. INTRODUCCIÓN: El Corioangioma es un tumor no trofoblástico de incidencia real desconocida, se asocia con complicaciones materno-fetales cuando su tamaño es mayor de 4 cm. La Corangiosis es una hipervascularidad ve- Mortalidad Perinatal y complicaciones obstetricas agudas. Aunque su etiología es desconocida, hay diversos factores maternos, fetales y placentarios implicados en su desarrollo, en los que existe un estado de hipoperfusión placentaria crónica y la transferencia de oxígeno al feto está disminuida, lo que explicaría su presencia en la misma placenta. En nuestro caso se asoció a Diabetes Gestacional y malformaciones del cordón. Aunque el tumor no fue detectado previamente por ultrasonido, Se considera que el tumor y las dilataciones venosas secuestraron aproximadamente 59 ml de sangre fetal, explicando la anemia aguda del RN.Sobrevivió gracias a la atención oportuna REFERENCIAS: 1.-Ramírez Arreola L, Nieto Galicia, Gómez García E, Cerda López JA. Corioangioma gigante y sus complicaciones perinatales. Reporte de un caso. Ginecol Obstet Mex 2007; 75: 104-10. 2.-Pérez-García GE, Sierra-Avendaño JA, Rangel-Navia E, Fuentes-Porras JS. Corangioma placentario: enfoque clínico-patológico de un caso descrito en Colombia. Ginecol Obstet Mex 2013; 81: 109-114. 3.-González Santacruz M., Tarazona Fargueta J.L.,Alenda González C. y Jiménez Cobo B. Muerte perinatal en una gestación múltiple asociada a corangiosis placentaria. An Pediatr (Barc). 2006; 65 (6): 626-42 4.-Shubhangi 2do Congreso Anual de Patólogos Mexicanos • 79 León, Guanajuato • 2016 Vinayak A., Deepti Dayanand D., Case Reports Chorangiosis of Placenta. Bombay Hospital Journal 2009; 5(2): 251252 5.-Gonzalez AB, Viñuales AI, Juárez F. Corangioma placentario asociado a hemangiomatosis neonatal difusa. A propósito de un caso. Rev Esp Patol. 2010;43(4):217?219. 6.-Guschmann M. Henrich W, Entezami M, Dudenhausen J., Chorioangioma new insights into a well-Know problem. I. Results of a clinical and morphological study of 36 cases. J. Perinat Med 2003; 31(2):163-9. 7.-Acharya S., Pringle S., A Case Of Placental Chorioangioma With The Review Of Literature. The Internet Journal of Gynecology and Obstetrics. 2004 Volume 5 Number 1. INSTITUCION: HOSPITAL GENERAL CADEREYTA DE PEMEX ESTADO:NUEVO LEÓN umbilical de 13x1cm, trivascular. El tejido fue procesado y en los cortes histológicos se observa una displasia de estirpe mesénquimal constituida por una difusa degeneración hidrópica de las vellosidades coriales (grandes y edematosas) y ausencia de trofoblasto CONCLUSIONES: La displasia mesenquimatosa de placenta es una alteración vascular placentaria caracterizada por una placenta grande con dilataciones quísticas, de etiología desconocida, su diagnóstico definitivo es mediante estudio histopatológico de la placenta. Su prevalencia estimada es de 0.02 a 0.2%; sin embargo, su diagnóstico es subestimado debido a que no en todos los casos se envía la placenta a estudio patológico. INSTITUCIÓN: Hospital Central ¨Dr. Ignacio Morones Prieto¨, San Luis Potosí, SLP. ESTADO: SAN LUIS POTOSÍ Mampara: 15 A Fecha y hora Instalación: 2 mayo 7:30 hrs. Fecha y hora retiro: 2 mayo 13:00 hrs. Mampara: 15 B TC-172 Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. AUTORES:Sánchez-García Citlalli Edith, Peña-Zepeda TC-183 7:30 hrs. Claudia. TITULO: DISPLASIA MESENQUIMATOSA PLACENTARIA INTRODUCCIÓN: La displasia mesenquimal placentaria es una rara entidad patológica, caracterizada por placentomegalia y malformaciones vasculares placentarias con apariencia macroscópica y ecográfica de una mola parcial de la cual con frecuencia es confundida. Se asocia a RCIU, óbito fetal, tumores fetales y síndrome de Beckwith-Wiedemann. OBJETIVOS: Conocer esta rara entidad patológica e identificar los hallazgos macroscópicos y microscópicos. MATERIAL: Mujer de 34 años de edad con embarazo de 32.2 sdg y diagnóstico de preeclampsia severa con resolución de embarazo médiate cesárea con evolución favorable sin complicaciones. La placenta es recibida en laboratorio de anatomía patológica para estudio histopatológico. RESULTADOS: La placenta tenia las siguientes características: monocorionica monoamniotica de 850g, disco corial de 16x15x5cm, la cara materna con cotiledones anfractuosos, heterogéneos, con áreas de aspecto esponjoso alternando con vesículas pequeñas de paredes delgadas, áreas hemorrágicas, vasos trombosados, la cara fetal café grisácea, lisa con trayectos vasculares visibles y prominentes, el aminios es café claro, opaco, el segmento de cordon AUTORES:Dra. Ana Karen Soto Sanudo, Dr. Aldo Antonio Alcaraz Wong TITULO: Adenocarcinoma Endometrial Originado en un Adenomioma Extrauterino Parasalpingeario: Reporte de Caso INTRODUCCIÓN: Los adenomiomas son tumores be- nignos compuestos por músculo liso y endometrio. Estos tumores generalmente se desarrollan en el útero presentándose raramente en lugares extrauterinos. La degeneración quística es aun mas rara y se han reportado casos con adenocarcinoma endometrioide sin invasión estromal. OBJETIVOS: Presentación de Caso MATERIAL: Se recibe salpinge de 3cms de longitud la cual presenta tumoración nodular extramural de 2cms por 1.3cms de consistencia firme. A los cortes presenta formaciones quísticas las cuales miden 0.2 a 0.4cms en promedio, con paredes lisas. A la microscopia, se observa tumor circunscrito, compuesto por estructuras glandulares tipo endometrial revestidas por epitelio neoplásico y rodeadas por músculo liso. RESULTADOS: Se diagnostica un adenocarcinoma de tipo endometrioide grado I, no invasivo, intraquístico, de 0.6cms originado de un adenomioma extrauterino parasalpingeario de 2cms de diámetro mayor, sin permeación vas- 2do Congreso Anual de Patólogos Mexicanos • 80 León, Guanajuato • 2016 cular ni necrosis. CONCLUSIONES: Los adenomiomas extrauterinos son tumores poco frecuentes que pueden presentarse en el ligamento ancho, salpinge, ovario y otras localizaciones. Se han descrito dos teorías para determinar su etiología. La primera es un defecto en la fusión del conducto Mulleriano y la segunda es una transformación del mesenquima subcelomico. En nuestro caso, se observa restos mesonefricos los cuales pudieran tener una asociación con el desarrollo de esta neoplasia. INSTITUCIÓN: Centro Medico Nacional de Occidente Instituto Mexicano del Seguro Social ESTADO: JALISCO Mampara: 16 A Fecha y hora Instalación: 2 mayo Fecha y hora retiro: 2 mayo 13:00 hrs. 7:30 dominal. Los marcadores tumorales fueron negativos. El US mostro un tumor pelvico probablemente dependiente de anexos. Subitamente presento datos de choque hipovolemico y fallecio. RESULTADOS: En el estudio de autopsia se encontro hemoperitoneo de 4000 ml, con peritonitis generalizada de asas intestinales. El utero presento ruptura de la pared en la cara anterior del cuerpo y en el fondo, donde se observo un tumor dependiente del utero que midio 16x14x12cm, al corte de consistencia blanda, mal delimitado, de superficie heterogenea, con areas necrohemorragicas. Tambien se observaron varias lesiones nodulares intramurales y subserosas con calcificaciónes. Microscopicamente se identifico una neoplasia maligna con extensas areas de necrosis, con celulas fusiformes con alto grado nuclear y conteo mitosico elevado. Ademas se identificaron areas con formacion de osteoide y cartilago maligno, y celulas pleomorficas con nucleos grandes e irregulares. En las areas adyacentes al sitio de ruptura se observo necrosis asociada con celulas neoplasicas. Se concluyo como TMMM con componentes heterologos. hrs. TC-192 AUTORES: MARIO MURGUIA, Dr. Saulo Mendoza Ramirez, Dr. Felipe de Jesus Navarro Cordoba, Dr. Adrian Antonio Lopez Vera TITULO: Rotura uterina espontanea y hemoperitoneo como primera manifestacion de tumor mixto mülleriano maligno. Informe de un caso de autopsia. INTRODUCCIÓN: La rotura uterina (RU) es un evento que amenaza la vida, dando lugar a complicaciones graves que pueden llevar a la muerte. La incidencia es de 5.1/1000 alumbramientos, es más frecuente en mujeres multíparas, y el riesgo aumenta si se asocia a ruptura de cicatriz uterina. El riesgo tambien esta presente en mujeres con anomalias del ducto mülleriano, otras causas menos comunes son la gangrena gaseosa uterina y las neoplasias. CONCLUSIONES: La RU es una condicion grave que ocurre con mayor frecuencia en mujeres embarazadas y asociadas a uso inapropiado de oxitocina. En cuanto a las neoplasias, se asocian con mayor frecuencia a leiomiomas. Otras neoplasias que pueden ocasionar RU, con mucho menos frecuencia, son las neoplasias trofoblásticas gestacionales, carcinoma endometrial y leiomiosarcoma. La asociacion con TMMM es extraordinariamente rara. Nuestro caso reune las caracteristicas clinicas e histopatologicas de un TMMM con RU y hemoperitoneo, causantes de la muerte de la paciente. Ante la presencia de cualquier neoplasia uterina es necesario descartar la presencia de esta complicacion. INSTITUCIÓN: Unidad Medica de Alta Especialidad N° 1 IMSS Leon, Hospital Medica Campestre, Hospital General de Mexico, Hospital General Regional IMSS Nº 200, Hospital General Regional IMSS Nº 196 ESTADO: GUANAJUATO OBJETIVOS: Las tumores uterinos son neoplasias frecuentes en las mujeres, y pueden derivarse del endometrio, estroma endometrial, músculo liso y peritoneo. El tumor mixto mülleriano maligno (TMMM) es una neoplasia bifásica compuesta de elementos epiteliales y mesenquimatosos malignos distintivos y separados, pero mezclados, y acontece cerca de la mitad de todos los sarcomas uterinos. La presentacion con RU y hemoperitoneo es muy rara, por lo que presentamos un caso con esa asociacion. HEMATOPATOLOGÍA Mampara: 16 A Fecha y hora Instalación: 3 de Mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. MATERIAL: Mujer de 75 años, sin antecedentes de importancia, con historia de 3 semanas con síndrome anémico descompensado y dificultad respiratoria. En la exploracion fisica se observo palidez generalizada y se palpo tumor ab- TC-1 AUTORES: ROSALINDA PEÑALOZA, Dr. Eleazar Her- 2do Congreso Anual de Patólogos Mexicanos • 81 León, Guanajuato • 2016 nández-Ruiz,Dra. Elena Elsemini García-Pedro, Dr. Fabian Tafoya- Ramírez ,Dr. Jorge Luis Aquino-Salgado TITULO: LINFOMA PLASMABLASTICO DE OVARIO PRESENTACIÓN DE UN CASO INTRODUCCION:El linfoma plasmablástico inicialmente se diagnosticó en” cavidad oral. La” localización extra-oral” se observa hasta en 52% de los casos diagnosticados. Representa el 2.6 % de los linfomas no hodgkin en pacientes inmunodeprimidos. La mayoría de los pacientes son VIH positivos y están co-infectados por el virus de Epstein Barr (VEB)? hasta en el? 74%. OBJETIVOS: En el Hospital Regional de Alta Especialidad de Oaxaca la incidencia de pacientes diagnosticados con neoplasias hematológicas va en aumento. Este tipo de linfomas” infrecuentes “ se asocian a co-infección de los” virus Epstein Barr y VIH” por lo que en este caso se determinara la asociación vírica.” MATERIAL: Femenina de 28 años, originaria de Tehuan- tepec, diagnosticada con VIH a los 25 años con esquema antirretroviral (Atripla). Inicio hace 5 meses con dolor abdominal, sensación de plenitud temprana, nauseas, vomito, fiebre e intolerancia a la vía oral así como aparición de una masa abdominal de 15x 10 cm” en flanco y fosa iliaca derecha.” En la Tomografía axial se encontró tumoración dependiente de ovario derecho, hepatomegalia y adenopatías múltipl TC-23 AUTORES: Dra. Petra Yuridia Lizeth Alvarado Bernal, Dra. Brenda Esther Vázquez López, Dra. Olga Esquivel Hernández. TITULO: Linfoma MALT primario de glándula Tiroides. Presentación de un caso. INTRODUCCIÓN: Los linfomas malignos de la glándula tiroides (LMGT) están asociados principalmente con tiroiditis linfocítica crónica y tiroiditis de Hashimoto (TH). Entre el 80% a 100% de los pacientes con LMGT muestran tiroiditis linfocítica crónica en el resto del tejido. La probabilidad de desarrollar LMGT en los pacientes con tiroiditis linfocítica crónica se incrementa significativamente. OBJETIVOS: Presentar una patología poco frecuente. Revisar diagnósticos diferenciales de los infiltrados linfoides en glándula Tiroides. MATERIAL: Paciente femenino de 53 años de edad, antecedentes patológicos negados, cuadro clínico de 3 años de evolución con aumento de volumen paulatino en región cervical derecha, disfagia y disnea, sin otra sintomatología. La TAC reporta lóbulo tiroideo derecho aumentado de tamaño, con un tumor sólido, con calcificaciones, compatible con Neoplasia tiroidea. Se reciben laminillas y bloques de parafina para Revisión del producto de la tiroidectomía tota. RESULTADOS: El Linfoma difuso de células grandes, RESULTADOS: En los cortes histológicos se observa un linfoma de Burkitt, linfoma primario de serosas y el linfoma”plasmablástico”son los más frecuentes en pacientes diagnosticados con VIH. La localización ovárica es poco frecuente y se deben cumplir los criterios propuestos por Fox y Langley. Según la literatura el pronóstico es bueno si está confinado al ovario. CONCLUSIONES:En este caso la paciente presento la lesión primaria en anexos derechos, infiltración? a útero y anexos izquierdos así como a tejido celular subcutáneo de la región pélvica. Sin embargo respondió al? esquema de quimioterapia? (esquema CDE Ciclofosfamida, doxorrubicina? y etopósido en infusión continua) y no hay evidencia de infiltración a medula ósea. Actualmente a 6 ciclos de quimioterapia se encuentra en remisión completa. INSTITUCIÓN: HOSPITAL REGIONAL DE ALTA ESPECIALIDAD DE OAXACA ESTADO: OAXACA infiltrado linfoide con patrón folicular, con extenso daño linfoepitelial y áreas con diferenciación plasmocitoide, distorsionando la arquitectura de la glándula; también se identifica tiroiditis de Hashimoto en el resto del tejido. Se realizaron marcadores de inmunohistoquímica, los cuales mostrando positividad para CD20 y negatividad para CD3 y CD138, se observó predominio de cadenas ligeras para Kappa en las células con diferenciación plasmocitoide. Con lo anterior se diagnosticó un Linfoma de la zona marginal extranodal del tejido linfoide asociado a mucosas, con tiroiditis de Hashimoto en el resto del tejido tiroideo. CONCLUSIONES: Los linfomas MALT representan alrededor del 4% de los linfomas primarios de la tiroides. El diagnóstico diferencial incluye a los infiltrados linfocíticos benignos, los cuales muestran folículos linfoides reactivos y cambios oncocíticos en las células foliculares tiroideas. El linfoma folicular es histológica e inmunofenotípicamente similar al linfoma MALT, pero con enfermedad extratiroidea. El pronóstico de estos pacientes es relativamente bueno. Sin embargo estos pacientes requieren seguimiento clínico por el riesgo de presentar recurrencias o diseminación. Mampara: 16 B INSTITUCIÓN: DOCTOR´S HOSPITAL, MONTERREY, NUEVO LEÓN. Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. ESTADO: NUEVO LEÓN 2do Congreso Anual de Patólogos Mexicanos • 82 León, Guanajuato • 2016 Mampara: 17 A Mampara: 17 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-41 TC-57 AUTORES: LUCIA ALEMAN, Dra. Natalia Vilches Cisne- AUTORES: NICOLÁS ROMERO SALINAS (1), J. RO- ros, Dra. Oralia Barbosa Quintana, Dra. Raquel Garza Guajardo, Dr. Juan Pablo Flores Gutierrez TITULO: TSPAN33 COMO BIOMARCADOR EN LINFOMA DE HODGKIN INTRODUCCIÓN: TSPAN33 es un gen de la superfami- SALBA GARCÍA RAMÍREZ(2) TITULO:LINFOMA NO HODGKIN ANAPLÁSICO. PRESENTACIÓN DE UN CASO INTRODUCCIÓN: El linfoma anaplásico de células lia de las tetraspaninas implicado en funciones de proliferación, adhesión, motilidad y diferenciación celular. Estudios previos han demostrado que está implicado en la hematopoyesis y se ha demostrado su expresión exclusiva en células B activadas y neoplásicas. grandes representa cerca del 2 % de todos los linfomas no Hodgkin, se presenta en varones (70%) y jóvenes (33 años). Se diagnostica con el reconocimiento de la imagen morfológica típica y demostración del inmunofenotipo de linfocito T o de célula nula junto con la positividad del CD30. Se confirma cuando se comprueba la sobreexpresión de la proteína ALK. OBJETIVOS: Evaluar el estatus de expresión del anti- OBJETIVOS: Describir la presentación clínica de un lin- cuerpo TSP33 en las células de Reed-Sternberg y en el resto de células presentes en Linfomas de Hodgkin y No-Hodgkin. MATERIAL: Se obtuvieron bloques de parafina del archivo del Hospital Universitario, de los cuales se realizaron microarreglos incluyendo 30 ganglios linfáticos con diagnostico de Linfoma de Hodgkin, y 10 tejidos con diagnóstico de Linfoma No Hodgkin, posteriormente se realizó inmunohistoquímica para el anticuerpo TSPAN 33, donde se evaluó la positividad o negatividad del anticuerpo y su patrón de expresión. RESULTADOS: En los microarreglos realizados se encontró la expresión de TSPAN33 mediante inmunohistoquímica, exclusivamente en las células de Reed-Sternberg, las cuales demostraron positividad fuerte membranosa. El patrón de expresión incluye a las células Reed Sternberg clásicas, mononucleares y en menor medida a las pleomórficas y apoptóticas. Siendo el resto de la población de células inflamatorias no neoplásicas del microambiente del gánglio linfático, negativas. CONCLUSIONES: Presentamos a TSPAN33 como un biomarcador sensible y específico para demostrar células de Reed- Sternberg en el Linfoma de Hodgkin. Siendo una posible herramienta diagnóstica importante y como un potencial blanco terapéutico en el desarrollo de terapias biológicas para Linfomas de Hodgkin foma no Hodgkin anaplásico en una paciente femenina de 2 años de edad. MATERIAL: Paciente femenino de 2 años de edad con 9.4kg de peso, comienza su padecimiento actual en noviembre del 2015 con la presencia de un aumento de masa en la región axilar izquierda acompañada de tos seca. Acude a médico particular quien receta amoxicilina y penicilina sin mejoría, posteriormente le realiza ultrasonido que reporta adenomegalias en regiones axilares y hemicuello izquierdo de aspecto inflamatorio necrótico y algunos con sugerente infil RESULTADOS: La pieza extirpada es enviada a patología donde se procesa y se realizan cortes histológicos los cuales muestran una neoplasia maligna formada por células no cohesivas que forman nidos difusos grandes, las células son anaplásicas y tienen núcleos gigantes e hipercromàticos, con citoplasma abundante y eosinofilo. Se le realiza también perfil de inmunohistoquímica el cual es positivo para ALK +++, CD30 +++, CD45+++ y negativo para CD20 y CD3 con lo anterior se concluye diagnóstico de Linfoma no Hodgkin Anaplásico. CONCLUSIONES: La presentación de linfoma anaplàsico en edad pediátrica es poco frecuente, sin embargo dado el rápido crecimiento del conglomerado ganglionar, su extirpación y análisis histológico fue posible establecer el diagnóstico. Es importante tener en cuenta todas las posibilidades diagnósticas en caso de una adenopatía y realizar los estudios necesarios, dado que el diagnóstico certero dará pauta al tratamiento y pronóstico del paciente. INSTITUCIÓN: Servicio de Anatomía Patológica del Hospital Universitario “Dr. José Eleuterio González” de la Universidad Autónoma de Nuevo León. Monterrey N.L. INSTITUCIÓN: 1LABORATORIO DE MORFOLOGÍA, DEPARTAMENTO DE MEDICINA Y NUESTADO: NUEVO LEÓN TRICIÓN. UNIVERSIDAD DE GUANAJUATO, 2do Congreso Anual de Patólogos Mexicanos • 83 León, Guanajuato • 2016 LEÓN. 2HOSPITAL GENERAL DE LEÓN, LEÓN. variante celularidad mixta. Por otro lado 5 casos (7%) preESTADO: GUANAJUATO Mampara: 18 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-66 AUTORES: Dr. Eduardo Mora Tinajero, Dra. Rosa María Vicuña González, Dra.María Irene Rivera Salgado TITULO: LINFOMAS DE HODGKIN EN EL HOSPITAL CENTRAL SUR DE ALTA ESPECIALIDAD EN UN PERIODO DE 16 AÑOS, A PROPOSITO DE UN CASO DE LINFOMA DE HODGKIN VARIEDAD DEPLECION LINFOCITARIA INTRODUCCIÓN: Se calculan 8,500 casos nuevos y 1,120 defunciones por linfoma de Hodgkin en los Estados Unidos para el 2016. En México hasta el 2003 se reportaron 935 casos, con mayor incidencia en el grupo de varones de 15 a 19 años y en mujeres igual incidencia en los grupos de 15 a 19 y de 20 a 24 años. En el Instituto Nacional de Cancerología (INCan) hasta el 2004 representó el 0.8% de los linfomas, con 162 casos diagnosticados de los cuales 88 fueron hombres y 74 mujeres. OBJETIVOS: Presentar un caso de autopsia de Linfoma de Hodgkin variedad depleción linfocitaria, y revisar la casuística de los linfomas de Hodgkin en el Hospital Central Sur de Alta Especialidad (HCSAE) de Petróleos Mexicanos en los últimos 16 años. MATERIAL: Paciente masculino de 69 años de edad, con antecedentes de Diabetes mellitus tipo 2 de 6 meses de diagnóstico, Hipertensión Arterial de 3 años de evolución, y cardiopatía isquémica tratada con revascularción en 2002, con injerto de venas safenas. Inició su padecimiento 5 meses antes de su defunción con fiebre intermitente, sin predominio de horario, posteriormente se agregó diaforesis nocturna y pérdida de 12 kg de peso en los últimos 3 meses. RESULTADOS: En el servicio de patología del HCSAE se encontraron 72 casos de Linfomas de Hogkin (LH) en un período de 16 años (1999-2015) de los cuales 41 (55%) correspondieron a linfoma de Hodgkin clásico (LHC) variante celularidad mixta, 12 (16%) a LHC variante rico en linfocitos, 10 (14%) a LHC variante esclerosis nodular, 5 (7%) a LHC variante depleción linfocitaria y 6 (8%) a LH de predominio linfocitico. Fueron 41 hombres y 31 mujeres, con una edad promedio de 47.1 y 42.4 años respectivamente, y un rango entre los 8 y 89 años. Únicamente 5 casos (7%) presentaron recurrencia, la variante involucrada fue el LHC sentaron infiltración extraganglionar, 3 correspondieron al LHC variante rico en linfocitos y 2 a la variante celularidad mixta. Los sitios afectados fueron bazo, intestino delgado, higado, páncreas y tejidos blandos. De los casos de LHC variante depleción linfocitaria 3 de ellos fueron en hombres y 2 mujeres, con rango de edad entre 21 y 73 años. CONCLUSIONES: El linfoma de Hodgkin variedad depleción linfocitaria es la forma menos frecuente de las variedades de este (menos del 5% de los LH). Es común en pacientes adultos y con VIH. Se presenta con mayor frecuencia en hombres (60-75%). Clínicamente es común la presencia de síntomas B (fiebre, sudores nocturnos y pérdida de peso). Los sitios más comúnmente infiltrados por esta neoplasia son los órganos abdominales, ganglios retroperitonealesy médula ósea. Las características histológicas de esta patología comprenden el borramiento completo de la arquitectura ganglionar convencional y la presencia de células Reed-Sternberg las cuales presentan positividad a los marcadores inmunohistoquímicos CD15 (+) y CD30 (+++). Además de las manifestaciones clínicas típicas. INSTITUCIÓN: Hospital Central Sur de Alta Especialidad de Petróleos Mexicanos ESTADO: DISTRITO FEDERAL Mampara: 18 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-69 AUTORES: Dr. Cesar Iván Peña Ruelas, Dr. Edmundo E. Castelán Maldonado, Dr. Julio Cesar Sánchez Venegas, Dra. Julia O. De Leija Portilla, Dr. Ana Lourdes Rincón Rodríguez, Dr. Jorge Arturo Sánchez Garza. TITULO: Sarcoma de Kaposi Ganglionar sin afección cutánea. INTRODUCCIÓN: El sarcoma de Kaposi (SK) fue descrito inicialmente en 1872, es un tumor vascular de bajo grado asociado al herpesvirus humano 8.(1) El compromiso de órganos internos (hígado, bazo, pulmón) y ganglio ocurren en menos de 10% de los casos.(2) OBJETIVOS: Presentación de un caso de sarcoma de Kaposi ganglionar en un paciente VIH positivo que no presento afección cutánea. MATERIAL: Caso clínico: Hombre de 31 años de edad, a quien en junio de 2014 se realizó diagnóstico de VIH. Inició el padecimiento actual al identificar conglomerado ganglionar en región cervical de lado derecho; no se identificaron 2do Congreso Anual de Patólogos Mexicanos • 84 León, Guanajuato • 2016 lesiones cutáneas. La cuenta de CD4 fue menor a 40 células/mm3 y la carga viral de 279,000 copias/mL. Se realizó biopsia excisional de uno de los ganglios ante la sospecha de infección oportunista o neoplasia linfoproliferativa. RESULTADOS: En Anatomía Patológica se recibió un nódulo que midió 1x1 cm, ovoide, con superficie de corte de color rojo vinoso. El estudio histopatológico mostró una proliferación de células fusiformes dispuestas en nódulos, formación de hendiduras vasculares y múltiples vasos capilares neoformados. La IHQ mostró positividad para vimentina y CD34 en las células tumorales, mientras el CD20 y CD45Ro fueron positivos en el tejido linfoide residual. CONCLUSIONES: El SK se clasifica en cuatro varian- tes: clásico, endémico, iatrógenico y epidémico.(1) Con el aumento del VIH SIDA la forma epidémica es la más frecuente.(1) Se ha descrito la afectación ganglionar por SK tanto en pacientes VIH+ y VIH-. (2-3) Cuando los ganglios linfáticos están afectados, la infiltración tumoral puede ser uni o multifocal, y el ganglio puede estar completamente afectado.(4) La IHQ permite identificar el origen endotelial del tumor, principalmente con CD31 o CD34, así como la tinción nuclear con HHV8. El pronóstico y evolución depende del estadio clínico y el compromiso inmunológico.(1-2) REFERENCIAS: 1. Revilla-López J, Mendoza-Fabián R, Anampa-Guzmán A, et al. Sarcoma de Kaposi endémico en un paciente VIH negativo. Rev Peru Med Exp Salud Publica 2015;32(4):808-12. 2. Mohanna S, Sánchez J, Ferrutino JC, y cols. Sarcoma de Kaposi clásico ganglionar. Comunicación de tres casos. Rev Méd Chile 2007;135:1166-70. 3. Hernández-Ruiz E, García-Herrera A, Ferrando J. Sarcoma de Kaposi. Med Cutan Iber Lat Am 2012;40(2):39-48. 4. Turcu M, Cotoi OS, Chira L, et al. Secondary involvement of lymph nodes in Kaposi sarcoma. Rom J Morphol Embryol 2011;52(3):943-946. INSTITUCIÓN: UMAE Hospital de Especialidades No. 25 del IMSS. Delegación Nuevo León. ESTADO: NUEVO LEÓN Mampara: 19 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. INTRODUCCIÓN: Paciente femenino de 35 años de edad sin antecedentes médicos relevantes. Inicia 4 meses previos a su ingreso con dermatosis maculopapular limitada a extremidades inferiores. Se le realizó panel para enfermedades autoinmunes el cual resultó negativo. 2 meses previos se le realiza tomografía en la cual se observan múltiples nódulos pulmonares, renales, hepáticos, pancreáticos y subcutáneos. OBJETIVOS: Se le realizó biopsia de piel de una de las lesiones subcutáneas. Posteriormente se le tomó una biopsia excisional de riñón, los fragmentos enviados eran de color café oscuro con áreas blanquecinas. También se le realizó una biopsia por aspiración de aguja fina a los nódulos pancreáticos. MATERIAL: Los cortes histológicos realizados a los fragmentos de piel muestran un infiltrado linfoide atípico dispuesto en patrón angiocéntrico asociado a extensas zonas de necrosis. El tejido renal se encontró involucrado por un infiltrado linfoide atípico y difuso con una distribución en predominio angiocéntrico y angiodestructivo, compuesto por linfocitos atípicos de gran tamaño y con núcleos irregulares. RESULTADOS: Se realizaron tinciones de inmunohistoquímica al tejido renal, observando positividad en los linfocitos atípicos para CD20, PAX5, CD30, MUM-1 y Ki-67 en un 95%. Las células son negativas para CD10, BCL-2, BCL-6. Se realizó además hibridación in situ para RNA de Epstein-Barr (EBER), la cual resultó positiva en más de 50 células por campo de alto poder, por lo que se concluyó que se trata de un caso de Linfomatosis Granulomatoide grado 3 de 3. Los hallazgos observados a nivel de la piel y páncreas son compatibles con este diagnóstico. CONCLUSIONES: La linfomatosis granulomatoide corresponde a un desorden linfoproliferativo extranodal de células B asociado al virus de Epstein-Barr (EBV). Se caracteriza por un infiltrado linfoide polimórfico angiocéntrico y angiodestructivo, la cual se gradúa dependiendo del número de células positivas para EBV y la cantidad de necrosis. El sitio más afectado es el pulmón (en 90% de los casos). Otros sitios que pueden verse afectados son la piel, el riñón, el hígado y el cerebro. La mayoría de los casos se asocia a inmunodeficiencia, incluyendo VIH, enfermedades autoinmunes y trasplantes. INSTITUCIÓN: Universidad de Monterrey - Hospital Christus Muguerza Alta Especialidad ESTADO: NUEVO LEÓN TC-85 AUTORES: Dra. Michelle Figueroa Andere, Dr. Miguel Ángel Paz González, Dra. Gloria Madrid Valero, Dr. Luis Antonio Sepúlveda Rodríguez, Dr. Carlos Barboza Chávez, Dr. Horacio Decanini Arcaute TITULO: Granulomatosis linfomatoide con afectación mul- Mampara: 19 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. tisistémica, reporte de un caso. 2do Congreso Anual de Patólogos Mexicanos • 85 León, Guanajuato • 2016 TC-94 AUTORES: Dra. Ana Lirio Ramírez Avila, Dra. Nelly Alejandra Rangel Rodríguez. TITULO: Sarcoma de células dendríticas foliculares asociado a Enfermedad de Castleman hialino-vascular. Presentación de un caso INTRODUCCIÓN: Las células dendríticas foliculares constituyen junto a las células de Largenhans, un elemento esencial de los folículos de linfocitos B, ejerciendo la labor de presentación de antígenos y la generación y regulación de la respuesta de los centros germinales. El sarcoma de células dendríticas es una patología muy infrecuente, asociado o no a otras enfermedades, como es en el seno de una enfermedad de Castleman, participando como precursor de la misma. Se presenta con mayor frecuencia en los nódulos linfáticos, aunque existe una importante proporción que se manifiesta de manera extranodal. Presentamos el caso de una mujer joven previamente sana, quien fue diagnosticada inicialmente como LNHCB. Sin embargo por morfología y hallazgos inmunohistoquimicos se confirma el diagnóstico de Sarcoma de células dendríticas foliculares asociado a Enfermedad de Castleman hialino-vascular. OBJETIVOS: Establecer la dificultad diagnostica del sarcoma de células dendríticas foliculares asociado a enfermedad de castleman, con otras entidades clínico-patológicas que afectan a los nódulos linfáticos. MATERIAL: Mujer de 40 años que presenta un nódulo cervical derecho, indoloro, no adherido a planos profundos. Apoyo diagnostico con microscopia óptica e inmunohistoquimica. RESULTADOS: En el estudio microscópico se identifica nódulo linfático residual, con pérdida parcial de su arquitectura, revelando una neoplasia maligna conformada por una proliferación homogénea de células fusiformes, con citoplasma eosinofilo, y limites mal definidos, alternando con otras células de aspecto epitelioide con nucléolo evidente. Se realiza inmunohistoquimica, siendo relevante la positividad de CD21, PS100 en células neoplásicas y Ki 67 del 20%. Se identifican además folículos grandes con centros germinales en regresión, así como anillos concéntricos de linfocitos en la zona del manto y aumento de la proliferación vascular en zonas interfoliculares. CONCLUSIONES: El sarcoma de células dendríticas foliculares asociado a enfermedad de Castleman, constituye una entidad clínico-patológica poco frecuente. Sus características clínicas lo hacen difícilmente distinguible de otras neoplasias que afectan a los nódulos linfáticos. El diagnóstico es complicado, siendo necesario tanto el estudio mediante microscopía óptica como el inmunohistoquímico para determinarlo. Se comportan como neoplasias de bajo grado de agresividad en general. El tratamiento es la excisión quirúrgica completa y el uso de terapia adyuvante es un tema de discrepancia en el momento actual. La etiología de la Enfermedad de Castleman es desconocida, se asocia a la desregulación del factor de crecimiento endotelial, y a la displasia de células dendríticas foliculares como precursor. El pronóstico es excelente y la extirpación quirúrgica suele ser curativa. REFERENCIAS: ANALES DE LA SOCIEDAD OTORRINOLARINGOLÓGICA ANDALUZA (15795810 )- 2002, n.5p.31-40. Diagnostic pathology. Lymph nodes and spleen with extranodal lymphomas L.Jeffrey Medeiros, 2014, 3:40 Sofía Ornelas Arroyo,* Salomón Aguilar Medina,* Iliana Mac Kinney Novelo. Neoplasia de células dendríticas foliculares. Rev Invest Med Sur Mex, Enero-Marzo 2011; 18 (1): 18-2 INSTITUCIÓN: Hospital Juárez de México ESTADO: ESTADO DE MÉXICO Mampara: 20 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-101 AUTORES: Dr. Miguel Enrique Cuellar Mendoza, Dra. Jazmin de Anda Gonzalez, Dr. Eladio Salas Torrez TITULO: Linfoma intracardiaco de celulas grandes B. Reporte de un caso y revision de la literatura. INTRODUCCIÓN: Los linfomas son tumores intracardiacos raros y usualmente fatales.Representan del 1.3 al 2% de los tumores primarios en este sitio y 0.5% de los linfomas extranodales. Este tipo de tumores se reconoce raramente antes de la muerte de los pacientes. Los linfomas de celulas grandes B son los mas comunes, usualmente en cavidades derechas y pericardio. OBJETIVOS: Informar una localizacion poco frecuente del linfoma de celulas grandes B MATERIAL: Paciente masculino de 35 años de edad con diagnostico de VIH, el cual ingresa al hospital con datos de bajo gasto cardiaco. El estudio de imagen mostro masa intracardiaca por lo que se programo para cirugia diagnostica. Se realizo estudio transoperatorio de la lesion la cual se reporto como neoplasia hematologica. En estudio definitivo se caracterizo como linfoma de celulas grandes B. RESULTADOS: Las laminillas de hematoxilina y eosina muestran neoplasia de origen hematologico de crecimiento difuso que reemplaza estructuras normales. Se realizo estudio de inmunohistoquimica para caracterizar la lesion. 2do Congreso Anual de Patólogos Mexicanos • 86 León, Guanajuato • 2016 CONCLUSIONES: Los linfomas intracardiacos son raros y usualmente fatales. En esta localizacion tienden a progresar rapidamente y su retraso diagnostico se asocia a un mal pronostico. Los pacientes pueden encontrarse asintomaticos o tener sintomas por extension del tumor o embolos tumorales. Este tipo de tumores se suele encontrar en pacientes inmunocomprometidos. Las opciones de tratamiento incluyen radioterapia y quimioterapia INSTITUCIÓN: Instituto Nacional de Ciencias Medicas y Nutricion “Salvador Zubiran” ESTADO: DISTRITO FEDERAL Mampara: 20 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. con núcleo de cromatina dispersa, otras son grandes con núcleo rechazado a la periferia y abundante citoplasma, se encuentran embebidas en una malla de aspecto fibrilar. Se realiza inmunohistoquímica con proteína acida fibrilar glial, la cual resulta positiva en la población polimórfica. CONCLUSIONES: Hasta el año 2005 se han reportado 19 casos de metástasis de tumores primarios de sistema nervioso central a medula ósea, de los cuales 13 corresponden a glioblastoma. El rango de presentación es muy amplio, va de los 11 a los 60 años; desde que se realiza el diagnóstico intracerebral, el tiempo de presentación de metástasis es de 4 a 12 meses, la presentación clínica es de citopenias (anemia, trombocitopenia, pancitopenia), así como dolor de espalda. Se ha sugerido que la metástasis extra craneal probablemente sea parte del curso natural de la enfermedad en estos tumores de alto grado. INSTITUCIÓN: Hospital General “Dr. Manuel Gea González”. ESTADO: DISTRITO FEDERAL TC-129 AUTORES: Dra. Erika Gpe. Castañeda Angeles, Dra. María del Rocío Estrada Hernández, Dra. Ana Lilia Morales Leyte. Mampara: 21 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. TITULO: Metástasis de glioblastoma a médula Fecha y hora retiro: 3 de mayo 13:00 hrs. ósea. INTRODUCCIÓN: El glioblastoma es la neoplasia ma- ligna primaria más frecuente del sistema nervioso central, se presenta de los 45 a los 75 años, es de rápida invasión a estructuras cercanas. Las metástasis extra craneales son raras, con una incidencia reportada en la literatura <2%, entre los diversos sitios de metástasis hay pocos casos reportados en hueso (médula ósea) con sintomatología.> OBJETIVOS: Describir un caso poco frecuente de metástasis extra craneal (médula ósea).de un tumor cerebral primario. MATERIAL: Hombre de 71 años, tabaquismo positivo TC-158 AUTORES: JACOBO IVAN GUERRERO, DR. REYNALDO FALCON ESCOBEDO TITULO: SARCOMA GRANULOCITICO. INFORME DE UN CASO PEDIATRICO CON PRESENTACION CENTROFACIAL. INTRODUCCIÓN: Se muestra la solucion diagnostica de un complejo caso de tumor centrofacial de celulas pequeñas (una cajetilla al día). Neurocirugía en mayo de 2015, resecaron tumor de SNC con reporte de glioblastoma. En julio de 2015 presentó alteración del estado de alerta, episodios de somnolencia, agresividad, alteraciones de lenguaje y dolor lumbar (7/10) irradiado a miembros pélvicos, BH con reporte de anemia y trombocitopenia (citopenias) motivo por el cual se toma biopsia de médula ósea y se envía a Patología. OBJETIVOS: Muestra de ruta diagnostica RESULTADOS: Se recibe fragmento de tejido (2.5 x 0.2 RESULTADOS: Sarcoma granulocitico x 0.2 cm), cilíndrico, café oscuro, poroso, de consistencia dura. Al estudio histológico se observa médula ósea con doce espacios medulares evaluables, celularidad y relación mieloide eritroide no valorables, debido a que los linajes se encuentran sustituidos por fibrosis, necrosis extensa, grupos de células neoplásicas polimórficas, algunas son pequeñas MATERIAL: Tejido fijado con formaldehido al 10%, embebido en parafina, cortado y teñido con tecnica convencional de hematoxilina y eosina asi como tambien inmunorreaccion con anticuerpos. CONCLUSIONES: Es una entidad rara e interesante INSTITUCIÓN: HOSPITAL CENTRAL DE SAN LUIS POTOSI “DR. IGNASIO MORONES PRIETO”. 2do Congreso Anual de Patólogos Mexicanos • 87 León, Guanajuato • 2016 ESTADO: TAMAULIPAS Mampara: 22 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. Mampara: 21 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-174 AUTORES: ALFREDO SEGURA Dra. Elsa Acosta Jiménez, Dra. Maria de los Angeles Macias Clavijo, Dra. Alejandra Zárate Osorno TC-169 AUTORES: Sánchez-García Citlalli Edith, Peña-Zepeda Claudia, Ortiz-Juan José TITULO: Leucemia linfoblastica aguda de precursores B,como efecto secundario del tratamiento de sarcoma de Ewing. INTRODUCCIÓN: El sarcoma de Ewing es una neoplasia altamente maligna constituida por una proliferación uniforme de células redondas, pequeñas y azules que afecta a niños y adultos jóvenes. La leucemia linfoblastica aguda es la principal neoplasia maligna en niños y es un trastorno heterogéneo, por su variada presentación clínica, morfológica, inmunofenotípica y citogenética. La asociación del sarcoma de Ewing y la leucemia linfoblastica aguda es extremadamente rara. OBJETIVOS: Conocer la frecuencia y asociación entre estas dos entidades patológicas MATERIAL: Niña de 7 años de edad con diagnóstico de Sarcoma de Ewing no metastasico de hace 1 año, con antecedente de resección de tumor y quimioterapia. Actualmente se recibió en el departamento de anatomía patológica biopsia de medula ósea, coagulo de medula ósea y líquido cefalorraquídeo y se identifica infiltración por neoplasia blastica de células pequeñas redondas y azules. RESULTADOS: Se procedió a realizar estudio de inmunohistoquimica a la biopsia de medula osea y fue positiva para TDT, Fli-1, Cd79a, PAX-5 y negativa para Cd33, Cd117, Mieloperoxidasa, Cd20, Sinaptofisina y Cromogranina. Se descartó la posibilidad de TNP/Sarcoma de Ewing en el material estudiado. CONCLUSIONES: La LLA como resultado a tratamiento post quimioterapia por sarcoma de Ewing es extremadamente rara e infrecuente. No se encontro caso reportado en la literatura. TITULO: LINFOMA DE CÉLULAS T SUBCUTANEO PANICULITICO EN NIÑOS: PRESETACION DE DOS CASOS Y DISCUSIÓN DEL INMUNOFENOTIPO. INTRODUCCIÓN: El linfoma de células T subcutáneo paniculítico (LCTSP) fue descrito por González y cols, como una distinta forma de linfoma T cutáneo, es particularmente raro en niños y sus características clínico e histológicas pueden simular otras entidades. MATERIAL: PRESENTACIÓN DE LOS CASOS: Caso 1. Niña de 3 años de edad con lesiones nodulares de 6 meses de evolución que afectaba región lumbosacra, glúteo y brazo izquierdo, diagnostico inicial de eritema nodoso; posterior incremento del tamaño y numero de lesiones obligo a la toma de una segunda biopsia. Caso 2. Niña de 13 años de edad con lesiones papulares en piel de tronco y extremidades, con diagnósticos de paniculitis y eritema nodoso. Posterior RESULTADOS: DISCUSIÓN: Clínicamente el LCTSP se presenta como múltiples nódulos o placas localizados en tronco y extremidades inferiores, menos común en extremidades superiores o cara. Histológicamente es un infiltrado linfohistiocitico en tejido subcutáneo y adiposo con tendencia a rodear a los adipocitos, similar a la paniculitis lobular, este es uno de los diagnósticos diferenciales, además de una variedad de leucemias y linfomas. Esta entidad afecta a niños y adultos jóvenes, los niños tienen un mejor pronóstico con remisión en el 50%. En pacientes pediátricos con infiltración a médula ósea y síndrome hemofagocitico tienen un peor pronóstico. CONCLUSIONES: El LCTSP fue un término confuso antes de la clasificación WHO-EORTC, actualmente es confinado al inmunofenotipo de células T alfa /beta, típicamente positivo BF1, CD3,CD8 y proteínas citotóxicas. REFERENCIAS: 1. Kong Y., MD. et al. Subcutaneous INSTITUCIÓN: Hospital Central ¨Dr. Ignacio Panniculitis-like T-cell Lymphoma. A clinicopathologic, Immunophenotypic, and molecular study of 22 asian cases Morones Prieto¨San Luis Potosí, SLP. according to WHO-EORTC classification. Am J Surg Pathol. ESTADO: SAN LUIS POTOSÍ 2008: 32(10): 1495-1502. 2. YeonLim G., Tae Hahu S., Gyun Chung N., Ki Kim H. Subcutaneous paniculitis-like T-cell lymphoma in a child: whole-body MRI in the initial and follow-up evaluations.Pediatr Radiol. 2009: 39:57-61. 3. shani Adir A., et al. Subcutaneous paniculitis-like T-cell lymphoma in chil- 2do Congreso Anual de Patólogos Mexicanos • 88 León, Guanajuato • 2016 dren. Response to combination therapy with cyclosporine and chemoterapy. J AM ACAD DERMATOL: 2004;50(2);: 518-522. determinar criterios diagnósticos reproducibles e identificar mediante estudios moleculares sus diversas asociaciones. INSTITUCIÓN: Centro Médico Nacional de OcINSTITUCIÓN: Instituto Mexicano del Seguro cidente, Instituto Mexicano del Seguro Social Social ESTADO: JALISCO ESTADO: ESTADO DE MÉXICO UROPATOLOGÍA Mampara: 22 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-185 AUTORES: Dra. Martha Arisbeth Villanueva Perez Dr. Luis Carlos Martínez Ascencio TITULO: Linfoma T/NK testicular: Reporte de un caso y revisión de la literatura. INTRODUCCIÓN: Los linfomas T/NK se presentan en casi todos los casos de manera extranodal, con preferencia por la via aerodigestiva, presentándose extranasalmente en piel, tejidos blandos, tracto gastrointestinal y testícul. OBJETIVOS: Presentación de caso MATERIAL: Paciente masculino de 38 años de edad que inicia 6 meses previos a su ingreso con aumento de volumen testicular izquierdo, acompañado de adenopatías inguinales y cervicales. Manejado con orquiectomía en hospital general de zona, posteriormente enviándose el material histopatológico a esta unidad para revisión de laminillas. RESULTADOS: Para revisión de laminillas se reciben seis bloques de parafina y seis preparaciones histopatológicas teñidas con técnica de hematoxilina y eosina, identificados como 7621-15 1 a 6, referidas como producto de orquiectomía radical izquierda con: -Neoplasia linfoide difusa con patrón infiltrante intersticial de células medianas, infiltra el 90% del tejido analizado, respeta túnica albugínea y vaginal, no se reciben elementos del cordón espermático. -Inmunorreacción positiva para CD3 y CD56 citoplasmatico. Negativa para CD4, CD10, CD20, CD138, MUM1, Kappa, Lambda, CK y PLAP. CONCLUSIONES: Debido a los hallazgos morfológicos e inmunohistoquímicos se concluye el caso como un linfoma difuso de células medianas, inmunofenotipo T, compatible con un linfoma T/NK extranodal. Se requieren estudios genéticos-moleculares para hacer la diferencia específicamente con el linfoma T periférico. La importancia de la revisión y presentación de estos casos radica en la poca frecuencia y caracter agresivo de las neoplasias T/NK, para Mampara: 1 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-2 AUTORES: DRA. ELENA ELSEMINI GARCIA, Dra. Ro- salinda Peñaloza Ramirez , Dr. Fabian Tafoya Ramírez , Dr. Edgar Gaytán Escobar TITULO: LEIOMIOSARCOMA RENAL PRESENTACIÓN DE UN CASO INTRODUCCIÓN: El leiomiosarcoma renal constituye el 0.12% de todos las neoplasias renales, la presentación clínica es variable,se presenta a los 60 años, Clínicamente surge como una masa que deriva del riñón, bien delimitada, con densidad e intensidad variable, difícil de diferenciar del carcinoma renal de tipo sarcomatoide o carcinosarcoma, siendo ùtiles estudios de inmunohistoquimica. OBJETIVOS: El tratamiento fue discutible, en nuestro caso quedándose únicamente en vigilancia clínica posterior a la nefrectomía radical, debido al grado histológico desfavorable, la naturaleza rara del tumor marca discusión en cuestión a la terapéutica a seguir. MATERIAL: Masculino de 61 años, con diarrea, caquexia, dolor en fosa renal izquierda y masa palpable. En la TAC abdominopélvica con tumor quístico de 13 cm, sin adenopatías retroperitoneales. Estudios de laboratorio: Leucocitos 3.8 x 10 9/L, Hb 6.4 gr/dL, Hto 18.6%, Glucosa 94 mg/dl, BUN 26 mg/dL, UREA 50 mg/dL, Creatinina 0.8 mg/ dL. Se realizó nefrectomía radical izquierda, encontrando neoplasia con células fusiformes de citoplasma eosinófilo y pleomorfi RESULTADOS:El leiomiosarcoma en el riñón constituye aproximadamente 2 a 3% de todas las neoplasias malignas, son tumores agresivos con recurrencia local y metástasis temprana hematógena, la literatura solo refiere informes y casos de algunas series. El diagnóstico diferencial incluye el leiomiosarcoma retroperitoneal, carcinoma sarcomatoide 2do Congreso Anual de Patólogos Mexicanos • 89 León, Guanajuato • 2016 renal, etc. A pesar de tener un pronóstico adverso, la opción terapéutica primaria es la resección quirúrgica, que puede ser complementada con radioterapia o quimioterapia. CONCLUSIONES: En este caso por ser un tumor muy poco frecuente, así como por presentar signos inespecíficos de malestar abdominal, y por las características histológicas desfavorables, junto con otros criterios clínicos, la paciente no es candidata a quimioterapia, quedándose únicamente con cuidados paliativos hasta el momento. INSTITUCIÓN: HOSPITAL REGIONAL DE ALTA ESPECIALIDAD DE OAXACA ESTADO: OAXACA Mampara: 1 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. les numerosas y citoplasma granular abundante. Se aprecia permeación angiovascular y áreas extensas de necrosis. La neoplasia midió 10.0 cm de eje mayor y afecto los bordes quirúrgicos. También se encontró metástasis de carcinoma en conglomerado ganglionar peri cólico y en epiplón. CONCLUSIONES: La presentación clínica habitual de este tipo de tumores consiste del dolor abdominal, masa palpable y hematuria macroscópica. Este caso inició con metástasis en un ganglio del cuello, debido a lo inusual de esta presentación el tumor evoluciono durante 1 año hasta manifestar crecimiento abdominal. En retrospectiva podemos enfatizar que ante la presencia de un tumor papilar en cuello es necesario precisar el origen de la neoplasia para un adecuado tratamiento. INSTITUCIÓN: Laboratorio de Morfología, Departamento de Medicina y Nutrición, Universidad de Guanajuato Laboratorio de Anatomía Patológica, Hospital General León. ESTADO: GUANAJUATO TC-34 AUTORES: DR. MIGUEL ÁNGEL RIOS, Dra. J. Rosalba García Ramírez Mampara: 2 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TITULO: Presentación inusual de Carcinoma Papilar de Células Renales en Edad Pediátrica, TC-52 Reporte de un Caso. AUTORES: DR. ALDO ELEAZAR VAZQUEZ, Dr. I EstraINTRODUCCIÓN: Las neoplasias malignas del riñón da-Moscoso, Dr. AE Vazquez-Ortega representan el 2% de los cánceres. El carcinoma papilar de células renales se caracteriza por papilas al menos en un 50% del tumor. Hay dos tipos de carcinoma papilar renal; los tumores tipo 1 están formados por células cúbicas con poco citoplasma, dispuestas en una sola capa, mientras que el tipo 2 contienen células seudoestratificadas con un mayor grado nuclear y citoplasma eosinofilo. OBJETIVOS: Describir la presentación clínica e histopatológica de un carcinoma papilar de células renales en edad pediátrica. MATERIAL: Paciente Femenino de 13 años. Inicio su padecimiento un año antes presentando un tumor en el cuello de 3 cm de longitud, 6 meses después se extirparon 2 ganglios de cuello izquierdo donde se reportó un carcinoma metastàsico de patrón papilar compatible con primario de tiroides, se realizó TAC de cuello la cual reportó lifadenomegalia supra clavicular izquierda por lo cual se realizó una segunda biopsia del ganglio en cuello, la cual resultó neg RESULTADOS: El estudio histopatológico del riñón derecho muestra una neoplasia conformada por papilas que son revestidas por varias capas células renales, las células malignas tienen pleomorfismo moderado, mitosis anorma- TITULO: Reporte de caso: Carcinoma epidermoide y carcinoma papilar urotelial de pelvis renal sincrónico ipsilateral. INTRODUCCIÓN: Los reportes tumores sincrónicos de la pelvis renal ipsilaterales son poco frecuentes en la literatura, siendo tumores de distinto origen celular estos constituidos principalmente de tumores renales de células transicionales con tumores renales de células claras. Se hace la presentación del caso de un carcinoma epidermoide de manera sincrónica con un carcinoma papilar urotelial. OBJETIVOS: Realizar la descripción y el primer reporte de caso de una neoplasia de la pelvis renal sincrónica ipsilateral. MATERIAL: Hombre de 52 años, con hematuria de 4 me- ses de evolución, dolor lumbar crónico, náusea, vómito, pérdida de 15 kg en un mes, hiporexia y fiebre no cuantificada. Tabaquismo de 20 años de evolución con índice tabáquico de 10, alcoholismo de 12 años de evolución, con ingesta mensual. Uro-Tomografía con diagnóstico de litiasis renal y tumor en pelvis renal. Se realiza nefrectomía radical izquierda con resección de glándula suprarrenal. 2do Congreso Anual de Patólogos Mexicanos • 90 León, Guanajuato • 2016 RESULTADOS: En producto de nefrectomía se observaron dos lesiones independientes; la primera, asociada a un lito coraliforme impactado en pelvis renal, la cual afecto grasa perirenal y sustituye el parénquima del polo inferior y la segunda separada por 8 cm de la previa en una cavidad situada en el polo superior de la pelvis exofítica, de aspecto papilar. Al estudio histológico la primera lesión presenta epitelio plano estratificado moderadamente diferenciado, nidos de queratina y marcadores de inmunohistoquímica CKAE1/ AE, EMA y p63 positivos, CK20 y CK7 negativos; la segunda con células transicionales, un patrón papilar e inmunoreacción a CK7, CKAE1/AE3 positivos, y CK20, EMA y p63 negativos. sano en parafina, obteniéndose cortes de 4 micras que se tiñeron con hematoxilina y eosina. El estudio inmunohistoquímico se realizo empleando anticuerpos contra calretinina, inhibina, vimentina y AME. RESULTADOS: Macroscópicamente se identificó un nódulo de 2 cm de diámetro, circunscrito, homogéneo, de consistencia carnosa, coloración amarilla. Histológicamente se observó una neoplasia formada por nidos de células poligonales, citoplasma eosinófilo, con núcleo redondo y nucléolo prominente, se identifican algunos cristales de Reinke. Inmunohistoquímicamente expreso positividad para Calretinina, Inhibina y Vimentina. CONCLUSIONES: Se trata del primer caso reportado CONCLUSIONES: Se trata de un tumor testicular poco en la literatura de un tumor sincrónico ipsilateral de pelvis renal con dos tipos histológicos diferentes con el estudio inmunohistoquímico apoyando este diagnóstico, por lo que consideramos es necesario reportarlo para futuras referencias. frecuente. Destaca su buena evolución. INSTITUCIÓN: HOSPITAL CIVIL DE CULIACAN ESTADO: SINALOA INSTITUCIÓN: Hospital General “Dr Manuel Gea Gonzalez” ESTADO: DISTRITO FEDERAL Mampara: 3 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. Mampara: 2 B TC-70 Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. AUTORES: Dra. Judith Leticia Acosta Piña, Dr. Carlos TC-54 Sanchez Lara, Dr.Rafael Rodriguez Rojas, Dra. Gigiola Garibaldi Garcia AUTORES: Dra. Sara Rodríguez, Dr. Ricardo Ochoa, TITULO: Tumor de células de Leydig en testículo asociado a adenocarcinoma intestinal TITULO: TUMOR DE CÉLULAS DE LEYDIG INTRODUCCIÓN: Los tumores de células de Leydig y la Dra. Ana Isabel Valdez, Dra. Dinorah Batiz, Dra. Ana Cristina Benitez,Dr.Rafael Efrén Ríos. INTRODUCCIÓN: Es una neoplasia derivada de células no germinales, con una incidencia de 1-3 % de todos los tumores de testículo. El 80% aparece en adultos de entre 20 y 60 años de edad. En el 85% de los casos son menores de 5 cm de diámetro aproximadamente. Histológicamente se encuentran cristales de Reinke en un 30%. El tratamiento de elección es la orquiectomia radical por vía inguinal. OBJETIVOS: Presentar estudio histopatológico e inmu- hiperplasia de células de Leydig representan del 1 al 3% de las neoplasias testiculares. La hiperplasia de las células de Leydig es una condición benigna que puede tener dificultades en el diagnóstico diferencial con un tumor de células de Leydig. La causa de la HCL no se entiende bien, probablemente se deba a una retroalimentación negativa defectuosa del eje hipotálamo-hipofisario-testicular, además se ha visto asociada a caquexia, y enfermedades crónicas como la tuberculosis, la sífilis, la anemia perniciosa, carcinomas y alcoholismo. nohistoquímico de un caso. Paciente de 45 años de edad, con aumento progresivo de testículo izquierdo de aproximadamente un año de evolución. Destacaba entre sus antecedentes personales; Linfoma No Hodgkin en 1992, tratado con quimioterapia por 18 meses. Actualmente libre de enfermedad. OBJETIVOS: El objetivo de este trabajo es presentar un MATERIAL: Pieza quirúrgica se fijo con solución de for- tico de adenocarcinoma de tipo intestinal moderadamente diferenciado del colon, candidato a terapia anti EGFR con 8 ciclos de quimioterapia. Cuenta con tac que mostró nódulo maldehído al 10% y se incluyeron cortes de tumor y tejido caso histopatológico e inmunohistoquímico de un paciente de 67 años con un nódulo testicular derecho e historia clínica de adenocarcinoma de colon de un año de evolución. MATERIAL: Paciente de 67 años de edad con diagnós- 2do Congreso Anual de Patólogos Mexicanos • 91 León, Guanajuato • 2016 sólido en el testículo derecho y USG testicular con imagen hipoecoica de bordes bien delimitados de 7.2 x 5.6 mm con vascularidad periférica y vaso intranodular. Se realiza orquiectomía radical derecha por nódulo sospechoso para metást RESULTADOS: El examen histológico de la pieza quirúrgica reveló un tumor de células de Leydig (0.5 cm) e hiperplasia difusa de células de Leydig. Imunohistoquímicamente las células de Leydig expresaron positividad para alfa inhibina y calretinina con un índice de proliferación celular bajo. CONCLUSIONES: El interés de este caso es mostrar tanto la escasa frecuencia de esta entidad, la probable asociación con carcinomas y por último establecer un diagnóstico diferencial histológico con otras condiciones neoplásicas como la hiperplasia de células de Leydig, tumor de células de Sartori calcificante de células grandes, la malacoplaquia, el seminoma y la metástasis por un carcinoma prostático. ción de la OMS de 1973 y 2004. RESULTADOS: Se incluyeron 132 pacientes de los cuales 37 fueron mujeres (28%) y 95 fueron hombres (72%). 60 (45.5%), 47 (35.6%), y 25 (18.9%) de los casos fueron diagnosticados como grado 1, 2 y 3 según la clasificación de la WHO/1973, respectivamente. De estos 73 (55.3%) fueron clasificados como bajo grado y 59 (44.7%) como alto grado según la clasificación de la WHO/2004. El 100% (60%) de los pacientes con diagnóstico de G1 se catalogaron como de bajo grado, al igual que los pacientes G3, en donde el 100% (25) se catalogó de alto grado. En la clasificación G2, 27.65% (13) fue diagnosticado como bajo grado y el 72.34% (34) como alto grado. La concordancia entre el diagnostico 2004 vs 1973 es de 0.464. CONCLUSIONES: Nosotros consideramos que la mala concordancia se debe a variabilidad de los pacientes con grado nuclear 2, ya que no se observan cambios es estadiaje en pacientes con grado 1 y 3. INSTITUCIÓN: Instituto de Seguriad y Servi- INSTITUCIÓN: Instituto Nacional de Ciencias cios Sociales de los Trabajadores del Estado Médicas y Nutrición Salvador Zubiran. ISSSTE ESTADO: DISTRITO FEDERAL ESTADO: DISTRITO FEDERAL Mampara: 3 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. Mampara: 4 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-88 TC-75 AUTORES: Valdivieso-Sánchez Gloria; Castillejos-Molina Ricardo; Herrera-Cacere Omar; Martínez- Braulio TITULO: Comparación de la clasificación 1973/OMS con 2004/OMS en carcinoma urotelial no musculo invasor. INTRODUCCIÓN: El carcinoma urotelial es la neoplasia más común de la vejiga. Más de la mitad de los carcinomas uroteliales se presentan como tumores de bajo grado, tumores superficiales, no invasores (pTa) o invasores a lamina propia (pT1). De acuerdo con las guías de la EAU y la AUA, se recomienda usar las 2 clasificaciones de la OMS para cáncer vesical no musculo invasor desde 2004 OBJETIVOS: Comparar la clasificación 1973/OMS con 2004/OMS para carcinoma urotelial, y determinar la concordancia entre ellas. MATERIAL: Se analizó una cohorte histórica de 132 pacientes con diagnóstico patológico inicial de Ta y T1 tratados con RTUV entre 1984-2015. Se excluyeron a pacientes con diagnóstico inicial de T2, carcinoma in situ o papiloma invertido. Se reclasificó las laminillas utilizando la clasifica- AUTORES: Dra. Michelle Figueroa Andere, Dra. Gloria Madrid Valero, Dr. Miguel Ángel Paz González, Dr. Luis Antonio Sepúlveda Rodríguez, Dr. Carlos Barboza Chávez, Dr. Horacio Decanini Arcaute TITULO: Neoplasia mucinosa originada en remanente del uraco, reporte de un caso. INTRODUCCIÓN: Paciente femenina de 43 años de edad, con antecedente histerectomía y salpingooforectomía bilateral por hiperplasia endometrial. Actualmente refiere hematuria de 2 semanas de evolución. Se le realizan estudios de imagen en los cuales se observa una masa que involucra la porción anterosuperior de la vejiga, la cual es resecada. OBJETIVOS: Se recibe un fragmento sacular de tejido de 5.0 x 5.0 x 3.8 cm, de color café oscuro. En su porción inferior se encuentra cubierto por un fragmento de mucosa vesical de 2.7 x 2.4 cm, lisa y de color café claro. A la sección se observa una lesión quística que mide 4.5 x 4.0 cm, de contenido mucinoso. MATERIAL: Los cortes teñidos con hematoxilina y eosina 2do Congreso Anual de Patólogos Mexicanos • 92 León, Guanajuato • 2016 muestran la superficie revestida por mucosa vesical. En la capa muscular se observa material de aspecto mucinoso el cual diseca la pared, hasta llegar a la serosa. Entre estos lagos de moco se observan estructuras glandulares revestidas por epitelio cilíndrico simple mucosecretor, los núcleos presentan estratificación, con atipia moderada y nucléolos inconspicuos. RESULTADOS: No se observan zonas de desmoplasia adyacentes a las glándulas neoplásicas. Se realizaron tinciones de inmunohistoquímica, las cuales resultaron negativas para receptores de estrógeno y progesterona, mientras que mostraron positividad para Citoqueratina 7, Citoqueratina 20 y CDX2, inmunofenotipo compatible con una lesión originada en remanentes del uraco, por lo que el diagnóstico en este caso fue de una neoplasia mucinosa de potencial maligno incierto. CONCLUSIONES: Los tumores malignos del uraco son poco frecuentes, representan menos del 1% de los tumores de vejiga. La mayoría siendo adenocarcinomas del tipo mucinoso. El principal síntoma es hematuria, por la infiltración de la cúpula vesical. Entre los criterios diagnósticos se encuentran la localización en la cúpula vesical o línea media vesical, una clara diferenciación entre el tumor y la superficie del epitelio transicional normal, así como la exclusión de un adenocarcinoma primario de otro órgano. Además muestra positividad para CK7, CK20, CDX2, trombomodulina y con ausencia de ?-catenina. INSTITUCIÓN: Universidad de Monterrey Hospital Christus Muguerza Alta Especialidad ESTADO: NUEVO LEÓN Mampara: 4 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-100 AUTORES: MA. GPE. JAZMÍN DE ANDA, Blandina Hernández Cruz, Sonia Tavares García, Liliana García Ilizaliturri, TITULO: Metástasis de coriocarcinoma Presentación inusual en sitios poco frecuentes INTRODUCCIÓN: Los tumores de testiculo son más frecuentes en pacientes entre la segunda y tercera decada de la vida, el más común es el tumor germinal mixto en un 69%, sin embargo, el coriocarcinoma puro es poco frecuente y representa menos de 1% en pacientes de 20 a 39 años de edad con niveles sèricos altos de B-gonadotropina coriònica humana. El coriocarcioma tiene predilecciòn por dar metàstasis en primer lugar al pulmón, higado y cerebro, sin embargo, existen otros sitios poco habituales de metastasis como la piel, el corazón y otros. RESULTADOS: Presentación del caso 1: Hombre de 25 años de edad con nòdulo en piel cabelluda de 6 cm de eje mayor de color café violaceo, friable, el cual fue resecado y donde se diagnòstico metastasis de coriocarcioma. Presentación del caso 2: Hombre de 19 años de edad acudió al servicio de oftalmología por pèrdida de la vision de ojo derecho y aparición de un nòdulo en el pàrpado inferior ipsilateral que midió 2 cm de eje mayor. El nòdulo se resecò y fuè diagnosticado como coriocarcinoma metastàsico a piel. En ambos casos despuès del diagnostico de coriocarcinoma metastàsico se realize busqueda de la neoplasia primaria encontràndose en testìculo. CONCLUSIONES: El coriocarcinoma es una neoplasia germinal muy agresiva con metastasis frecuentes a pulmòn y cerebro. Las metastasis a piel cabelluda y ojo son raras en un porcentaje menor al 1%. Es importante reconocer el patròn morfològico por la relevancia en el diagnostico clinico y terapeutico posterior para bùsqueda de sitio primario y quimioterapia subsecuente. INSTITUCIÓN: HOSPITAL DE ONCOLOGIA DE CENTRO MEDICO NACIONAL SIGLO XXI ESTADO: DISTRITO FEDERAL Mampara: 5 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-141 AUTORES: DRA. ITZEL MARÍA MONTOYA, Dra.Sara Parraguirre Martinez, Dr. Elio German Recinos Carrera, Dra. Lorena Troncoso Vazquez TITULO: Paraganglioma de vejiga (“Feocromocitoma”).Reporte de un caso. INTRODUCCIÓN: El paraganglioma de vejiga urinaria cuyo sinónimo es “feocromocitoma de vejiga” es definido por la OMS como una neoplasia derivada de las células de los paraganglios de la pared vesical, en la que el único criterio de malignidad es la presencia de metástasis.Tiene una incidencia muy baja representando <0.5% de las neoplasias propias de vejiga.La edad de presentación es amplia de 10 a 90 años> OBJETIVOS: Reporte de caso de hombre de 61 años de edad evolución de tres meses con disuria, tenesmo vesical,fiebre no cuantificada e intermitente,escalofríos y hematuria macroscópica formadora de coágulos.Hallazgo por tomografía de lito intramural en vejiga de 0.6 cm, mal definido 2do Congreso Anual de Patólogos Mexicanos • 93 León, Guanajuato • 2016 y dependiente de pared anterolateral izquierda. MATERIAL: Se recibió en nuestro departamento de patología el producto de resección transuretral de vejiga etiquetado como pared anterior vesical y tumor vesical múltiples fragmentos de tejido irregulares (2.0 x 2.0 x1.0 cm.).Se incluyeron en su totalidad para su estudio microscópico. RESULTADOS: Por histología se evidencio la presencia de células neoplásicas dispuestas en nidos principalmente “nidos de zelballen”, en algunas áreas mostrando un patrón mas difuso.Al detalle celular se observaron células redondas a poligonales con abundante citoplasma eosinofilo granular y en algunas áreas las células con citoplasma basofilo. Los núcleos de estas células son centrales con cromatina vesicular.Cabe destacar que se encontraron fragmentos de urotelio residual con escasa inflamación y artificio por daño térmico.Se realizo complemento diagnostico con inmunohistoquimica resultando positivas estas células neoplásicas en citoplasma para cromogranina y sinaptofisina. CONCLUSIONES: Es importante hacer revisión de esta entidad ya que existen pocos casos descritos en la literatura por su baja incidencia.Se plantea tres principales problemas para el patologo , el primero de ellos es desconocer por completo la entidad,el segundo es que la morfología puede en ocasiones semejar un carcinoma urotelial invasivo lo que dificulta el reconocimiento entre estas dos entidades y por ultimo el tercer reto para el patologo es reconocer las variantes histologicas que el paraganglioma puede mostrar. Es importante destacar que es una neoplasia de comportamiento benigno. INSTITUCIÓN: HOSPITAL GENERAL “DR. MANUEL GEA GONZALEZ” SERVICIO DE ANATOMÍA PATOLÓGICA. OBJETIVOS: Presentación de caso clínico y revisión de la literatura. MATERIAL: Presentamos el caso de una mujer de 55 años con antecedente clínico de hematuria de al menos 4 meses de evolución y dolor en flanco izquierdo. Se realiza ultrasonido renal en el que se identifica tumor renal de 30x30mm localizado en la porción media de riñón izquierdo, realizándose nefrectomía ipsilateral que es remitida al laboratorio para su estudio histopatológico. RESULTADOS: Los cortes histológicos realizados al riñón evidenciaron la presencia de una neoplasia maligna conformada por conductos con contornos irregulares y patrón de crecimiento infiltrante que se encuentran inmersos en un estroma desmoplásico. Las células que los componen muestran un alto grado (4 en la clasificación de Fuhrman). Se identifica de manera multifocal la presencia de células en “estoperol” que revisten algunos de los túbulos neoplásicos, la lesión presenta importante infiltrado inflamatorio. No se identifican zonas con células claras o formación de papilas. Todos los hallazgos histológicos son consistentes con el diagnóstico de carcinoma renal de los conductos colectores. CONCLUSIONES: El carcinoma de los conductos colectores es una neoplasia rara, pero de importante distinción diagnóstica ante otros tumores renales, debido principalmente a su comportamiento clínico agresivo debida a la presencia de enfermedad metastásica hasta en el 50% de los casos y una tasa de sobrevida media de 24 meses tras el diagnóstico inicial. INSTITUCIÓN: Laboratorio Carpermor ESTADO: ESTADO DE MÉXICO ESTADO: DISTRITO FEDERAL Mampara: 6 A Mampara: 5 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-175 TC-146 AUTORES: DR. ALFREDO SEGURA, Dra. Maria de los Angeles Macias Clavijo AUTORES: DRA. ANA CECILIA BELTRAN, Dr. Adalberto Pineda Zapata, Dra. Fabiola Navidad Cervera TITULO: Carcinoma renal de los conductos colectores de Bellini: Presentación de un caso y revisión de la literatura INTRODUCCIÓN: El carcinoma renal de los conductos colectores (de Bellini) es una neoplasia maligna de alto grado que constituye menos del 1% de los carcinomas renales. Presenta un pronóstico clínico desfavorable. TITULO: LEIOMIOMA RENAL. PRESENTACIÒN DE DOS CASOS. INTRODUCCIÓN: Los leiomiomas renales pueden detectarse en autopsia, incidentalmente en estudios de imagen. La incidencia de los leiomiomas renales es desconocida, predominan en mujeres, en una edad promedio de 63 años, se encuentra por igual en ambos riñones. El diagnóstico diferencial principal es con el Angiomiolipoma (AML) pobre en adipocitos. 2do Congreso Anual de Patólogos Mexicanos • 94 León, Guanajuato • 2016 MATERIAL: CASO 1. Masculino de 14 años de edad, inició 6 meses previos con dolor testicular bilateral, estreñimiento y cólicos, acudió con particular quien encuentra tumor en abdomen. Se le realizó TAC con tumor de suprarrenal. Los hallazgos quirúrgicos son: tumor de riñón izquierdo con peso de 4.5Kg. Caso 2. Masculino de 26 años con antecedente de esclerosis tuberosa, con dolor abdominal y hematuria macroscópica. Macroscopicamente con tumor dependiente RESULTADOS: El leiomioma se define como una neo- plasia benigna, originada del músculo liso, raro en riñón. Macroscópicamente miden en promedio 2.9 cm, bien circunscritos, sólidos, blancos, superficie arremolinada, ocurren predominantemente en la proximidad de la cápsula renal y pelvis. CONCLUSIONES: Los criterios histológicos para su diagnostico son: celularidad moderada de células fusiformes eosinofílicas dispuestas en fascículos, con núcleos elongados, con cromatina fina y ausencia de atipia, mitosis, necrosis, células vacuoladas, adipocitos o células tumorales con arquitectura perivascular, pueden presentarse con degeneración quística, calcificación o hemorragia focal. Inmunohistoquimicamente la desmina, HMB-45, catepsina K, receptores de estrógeno y receptores de progestágeno, son útiles para distinguirlo de un AML pobre en adipocitos, miolipoma y fibroma medular. REFERENCIAS: 1.Pallavi A. Patil, MD, Jesse K. McKen- ney, MD, Kiril Trpkov, MD, et al. Renal Leiomyoma. A Contemporary Multi-institution Study of and Infrequent and Frequently Misclassified Neoplasm. Am J Surg Pathol. 2015: 39(3):349-356. 2.Tsujimura A., Miki T., Sugao H., Takaha M., Takeda M., Kurata A. Renal Leiomyoma Associated with Tuberous Sclerosis. Urol Int 1996;57:192-193. INTRODUCCIÓN: Las neoplasias vesicales, no epiteliales ocupan el 0.5%-2%. La mayoria son malignas (rabdomiosarcoma) mas que benignas. El TMI vesical es un tumor mesenquimatoso benigno con similitud a neoplasias malignas que confiere alto grado de dificultad diagnóstica. Los estudios IHQ son basicos en el diagnóstico final. El tratamiento de elección es la resección transuretral amplia y/o cistectomía parcial. OBJETIVOS: Presentar un caso exitoso de diagnostico y tratamiento quirurgico de Tumor inflamatorio miofibroblastico de vejiga en una paciente de 18 meses, que debuto inicialmente con cuadro de abdomen agudo y septicemia. MATERIAL: Femenino de 18 meses de edad sin antecedentes patológicos previos con cuadro de abdomen agudo, abultamiento y cambios de coloración de región umbilical, leucocitosis con diagnóstico clínico de hernia umbilical encarcelada sometida, es sometido a tratamiento quirúrgico identificándose tumor dependiente de domo de vejiga con extensión a tejidos y protrusión a cicatriz umbilical, se realizó resección de tumor y cistectomía parcial. RESULTADOS: Espécimen quirúrgico: tumor ovoide ahusado de 6 x 5 x 4 cm, superficie convexa, erosionada centralmente, con solución de continuidad, hemorragia y necrosis con áreas mucosas café claro y edematoso, finamente granular. Microscópica: mostraron neoplasia fribrohistiocitica con patrón de crecimiento infiltrativo, con miofibroblastos de núcleo estrellado y citoplasma mal definido (Actina, ALK-1 +), sin atipia nuclear, inmersas en estroma colagenoso que infiltra la pared vesical con abundante infiltrado linfoplasmocitario difuso eosinofilos y agregados linfoides. La mucosa urotelial presenta cambios reactivos y proliferación epitelial. INSTITUCIÓN: Instituto Mexicano del Seguro CONCLUSIONES: El TMI (pseudotumor inflamatorio o pseudosarcoma) es un tumor mesenquimatoso benigno, la Social localización vesical es muy poco común en la infancia, es ESTADO: ESTADO DE MÉXICO Mampara: 6 B Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-176 AUTORES: DRA. LETICIA DEL CARMEN BAENA-OCAMPO, DRA. CARLA LORENA ECHEVARRIA CESPEDES, DR. JORGE IVAN VALENCIA MONCADA importante su diagnóstico diferencial con neoplasias malignas debido a la similitus morfologica con sarcomas, el estudio de IHQ es basico para su diagnostico final siendo el ALK-1 cleve para su diagnostico morfologico. En la presentación vesical el tratamiento consiste en resección transuretral, con buen pronóstico a largo plazo, aunque se ha determinado que aquellos casos que presenta ALK-1 + Tienden apresentar un comportamiento mas agresivo REFERENCIAS: Fletcher CDM, Unni KK, Fletcher F (Eds.),World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Soft Tissue and Bone. Lyon; 2002. Abascal Junquera, J.M.; Trilla Herrera, E.; Esquena Fernández, S. y cols.: “Pseudotumor inflamatorio vesical”. Actas Urol. Esp., 28: 62, 2004. INSTITUCIÓN: HOSPITAL ANGELES ACOXPA. TITULO: TUMOR MIOFIBROBLASTICO INFLA- SERVICIO DE ANATOMIA PATOLOGICA, SERMATORIO DE VEJIGA. PRESENTACION DE UN VICIO DE PEDIATRIA CASO PEDIATRICO ESTADO: DISTRITO FEDERAL 2do Congreso Anual de Patólogos Mexicanos • 95 León, Guanajuato • 2016 Mampara: 7 A Fecha y hora Instalación: 3 de mayo 7:30 hrs. Fecha y hora retiro: 3 de mayo 13:00 hrs. TC-186 AUTORES: DR. MARIO MURGUIA, Dr. Jorge Israel Muñoz Torres, Dr. Jorge A. Muñoz Lopez, Dr. Luis Raul Chavira, Dr. Felipe Navarro Cordoba, Dr. Adrian Antonio Lopez Vera, Dr. Saulo Mendoza Ramirez. TITULO:Angiomiolipoma epitelioide renal. Informe de un caso. INTRODUCCIÓN: El angiomiolipoma renal (AML) usual o clasico es un tumor benigno, compuesto por una proporcion variable de tejido graso, músculo liso y vasos sanguineos anormales de pared delgada. Al momento de su diagnostico, estas lesiones suelen tener un tamaño muy grande y debido a su comportamiento local agresivo, suele diagnosticarse clinicamente y por imagen con un carcinoma renal. El AML epiteliode (AMLE) es una variante rara de angiomiolipoma que consiste en presentar al menos el 80% de células epitelioides y constituyen alrededor del 4.6% de todos los AML resecados. OBJETIVOS: Existe evidencia inequivoca de que el AMLE exhibe un comportamiento maligno, establecido en varias series largas de casos. En los ultimos 15 años se han informado unicamente informes de casos aislados. Al ser poco frecuentes, el patologo que no conoce esta entidad suele confundir el diagnostico con un carcinoma renal de celulas claras. Nosotros presentamos un caso tipico de AMLE que cumple con los criterios histologicos e inmunofenotipicos que inicialmente fue abordado clinicamente como un carcinoma renal. MATERIAL: Hombre de 68 años, diabetico de larga evolucion, insulinodependiente. Acude por presentar sindrome anemico y disnea de medianos esfuerzos. Se realizo un examen general de orina, que evidencio hematuria macroscopica. En la exploracion fisica se encontro palido, con perdida de peso de 4 kg en 4 meses, y a la palpacion con masa en flanco derecho. La tomografia computada evidencio una masa de gran tamaño en riñon derecho. RESULTADOS: Se realizo nefrectomia radical derecha con diagnostico clinico de carcinoma renal. En el laboratorio de patologia se recibio riñon derecho que midio 16x13x10cm, y tamaño tumoral de 13x10x8.6cm, con margenes expansivos, areas de aspecto fibroadiposo y necrosis. Microscopicamente se observo una neoplasia ricamente vascularizada constituida por celulas epitelioides con citoplasma claro y eosinofilo palido, con aitpia nuclear y nucleolos evidentes centrales, algunas de estas celular con morfologia ameboide y de tipo celulas gigantes multinucleadas, con disposicion perivascular, con necrosis tumoral. No se observon afeccion del seno renal, El tumor fue positivo intensamente para HMB-45 y negativo para AE1/E3. Se concluyo como AMLE. CONCLUSIONES: Los factores pronosticos que se asocian con recaida y/o metastasis son presencia de esclerosis tuberosa junto con el tumor, tamaño mayor a 7 cm, patron morfologico parecido a carcinoma, afectacion de la grasa perinefrica y/o vena renal, y presencia de necrosis. Sin embargo, otras series largas mencionan que la proporcion de pacientes que caen en esta categoria es bastante pequeña de acuerdo a series largas, si se aplican los criterios de la OMS del 2013. Nuestro paciente cumple 3 de 5 criterios, por lo que se mantiene en vigilancia estrecha. Es necesario conocer y tener en cuenta este tumor, ya que su comportamiento biologico es parecido al de un sarcoma, por lo que son inutiles las terapias convencionales para carcinomas. INSTITUCIÓN: Unidad Medica de Alta Especialidad N°1 IMSS Leon, Hospital Medica Campestre, Hospital General Regional ISSSTE Leon, Hospital General Regional Leon, Hospital General de Mexico ESTADO: GUANAJUATO MIÉRCOLES 4 DE MAYO DERMATOPATOLOGÍA Mampara: 1 A Fecha y hora Instalación: 4 de mayo 7:30 hrs. Fecha y hora retiro: 4 de mayo 13:00 hrs. TC-89 AUTORES: Dra. Lucia Nereyda Melgarejo Ortega, Dr. Luís Sepúlveda Rodríguez, Dr. Miguel Ángel Paz González, Dra. Gloria Madrid Valero, Dr. Horacio Decanini Arcaute. Dra. Marcela Madrid Ballesteros. TITULO: BLASTOMYCES INTRODUCCIÓN: La blastomicosis es una enfermedad infecciosa granulomatosa, causada por un hongo dimorfo Blastomyces dermatitidis. Predomina en Estados Unidos de América y su entrada al organismo es a través de la inhalación o inoculación de conidios siendo los sitios mas fre- 2do Congreso Anual de Patólogos Mexicanos • 96 León, Guanajuato • 2016 cuentes la piel, huesos, el tracto genitourinario y el sistema nervioso. OBJETIVOS: Se presenta el caso de un masculino de 65 años de edad inmunocomprometido, con antecedentes de carcinoma hepatocelular y trasplante hepático, originario de Monterrey Nuevo León con una dermatosis crónica en la extremidad inferior izquierda MATERIAL: Evolución de 4 meses con dolor punzante de moderada intensidad asociado a una dermatosis, hiperqueratósica, exofítica, coliforme, neovascularizada, con una úlcera central de 5.0 x 5.0 cm en la cara lateral del talón izquierdo. Presenta discreta elevación de azoados y de pruebas de función hepática, hemoglobina, 9.3 g/dl, leucocito 4.17 K/uL, plaquetas 113 K/Ul. Resonancia magnética: lesión heterogénea en talón de 5.0 x 2.0 cm con edema sobre mús RESULTADOS: La tinción de hematoxilina y eosina, PAS y Grocott; muestran epidermis con hiperplasia pseudoepiteliomatosa y áreas ulceradas, con presencia de un intenso infiltrado inflamatorio de tipo mixto, con formación de microabscesos y reacción granulomatosa, presencia de células gigantes multinucleadas de tipo Langhans, con presencia de estructuras micóticas en su interior, caracterizadas por ser estructuras levaduriformes de 10 a 15 micrometros, las cuales presentan gemación, se observan además hifas delgadas y septadas. Se diagnostica patológicamente como: Proceso inflamatorio crónico granulomatoso con presencia de estructuras micóticas morfológicamente consistente con Blastomyces. CONCLUSIONES: El periodo de incubación es de 3 semanas a 3 meses después de la exposición. La imagen histológica de una lesión cutánea muestra hiperplasia epidermal pseudocarcinomatosa, con acantosis moderada; en la dermis se observan granulomas y microabscesos compuestos por células gigantes multinucleadas, células epiteliodes, neutrófilos, linfocitos y células esféricas de levaduras gemantes, con paredes gruesas refractarias de 7 a 15 ?m, que se encuentra en el centro de los abscesos. que TITULO: Carcinoma basocelular con metástasis ganglionar y pulmonar. Presentación de un caso. INTRODUCCIÓN: El carcinoma basocelular es la neo- plasia dermatológica más común a nivel mundial, originada a partir de células pluripotenciales en la epidermis y los folículos pilosos. Generalmente, localizada en cabeza y cuello, así como zonas expuestas; es localmente destructivo, rara vez produce metástasis que se estiman en 0.0028 ? 0.5% de los casos en un tiempo promedio de 9.6 años. OBJETIVOS: Presentar un caso clínico con metástasis ganglionares y pulmonares de baja frecuencia que debe considerarse en pacientes con carcinoma basocelular recurrente después de varios años de seguimiento libres de enfermedad. MATERIAL: Se revisa expediente clínico, archivo de imagen y archivo de Anatomía Patológica del Hospital Juárez de México para obtener material de revisión y resecciones previas de laminillas te;idad con H/E. Se realiza estudio comparativo en la biopsia pulmonar actual, a la que se le realizan reacciones de inmunohistoquímica con CD10, p63 y CK5/6. RESULTADOS: Mujer, sesenta años, diez años previos con diagnóstico de carcinoma basocelular resecado en el 2000. Recaída en 2003, 2007 y 2008, resección de la lesión y radioterapia con seguimiento hasta 2014, con disnea de medianos esfuerzos, radiografía AP de tórax y TAC de tórax con imágenes sugestivas de metástasis pulmonares. Se realiza biopsia por aguja de corte con neoplasia conformada por células de aspecto basaloide y escasas mitosis atípicas. El material previo con mismas mismas características en oreja residual, conducto auditivo externo, tejidos blandos adyacentes y disección ganglionar yugular. Reacciones inmunohistoquímica positivas para CD10, p63 y CK5/6, que confirman el diagnóstico. INSTITUCIÓN: Hospital Christus Muguerza CONCLUSIONES: El carcinoma basocelular es la neoplasia cutánea maligna más frecuente. Generalmente Alta Especialidad, Monterrey, Nuevo León. su evolución es indolente. La frecuencia de metástasis a ESTADO: NUEVO LEÓN Mampara: 1 B distancia es 0.0028% a 0.5%. Para determinar histológicamente la metástasis de un carcinoma basocelular la lesión primaria debe sere en piel, localizarse en un sitio distante a la misma, con una apariencia morfológica similar y sin la presencia de células escamosas (Lattes and Kessler). El pronóstico en estos casos es pobre a corto plazo y presentan mutación en PTCH1. Fecha y hora Instalación: 4 de mayo 7:30 hrs. Fecha y hora retiro: 4 de mayo 13:00 hrs. INSTITUCIÓN: Hospital Juárez de México. TC-95 ESTADO: ESTADO DE MÉXICO AUTORES: Dra. Ana Lirio Ramírez Ávila, Dra. Maria Elena Arroyo Mendoza, Dr. Oscar Rosas Guerra, Dr. Del Bos- 2do Congreso Anual de Patólogos Mexicanos • 97 León, Guanajuato • 2016 Mampara: 2 A Fecha y hora Instalación: 4 de mayo 7:30 hrs. Fecha y hora retiro: 4 de mayo 13:00 hrs. TC-125 AUTORES: Ana Cristina Benitez, Dr.francisco Javier Me- rino Ramirez,Dra. Paulina Leticia Alfaro Orozco,Jesus Sebastian Rodriguez Gutierrez, Dra.dinorah Batiz Salazar,Dra. ana Isabel Valdéz Flores,Dra.sara Rodríguez Carrillo vez realizado el diagnóstico clínico de sospecha, los estudios de histopatología y posteriormente de inmunohistoquímica son fundamentales para integrar un diagnóstico más certero y así poder otorgar el tratamiento de forma oportuna a los pacientes. INSTITUCIÓN: CENTRO DE INVESTIGACIÓN Y DOCENCIA EN CIENCIAS DE LA SALUD, HOSPITAL CIVIL DE CULIACAN ESTADO: SINALOA TITULO: MICOSIS FUNGOIDE EN NIÑOS PATOLOGÍA MOLECULAR INTRODUCCIÓN: La micosis fungoide es el subtipo más común de los linfomas cutáneos de células T. La edad más frecuente de presentación es entre los 40 y 60 años de edad, y rara vez se describe en la niñez. La frecuencia de micosis fungoide en la edad pediátrica es del 4 a 5% del total de casos. Histológicamente, el dato más característico es la extensión de las células mononucleares hasta la epidermis. OBJETIVOS: Masculino de 12 años de edad, acude a la consulta de Dermatología por presentar múltiples manchas en la espalda, de 6 años de evolución, las cuales eran eritematosas y de 2 a 3 semanas evolucionaron a manchas hipocrómicas; le indican manejo tópico para micosis, sin mejoría y posteriormente se generaliza a tórax, extremidades, asintomáticas. MATERIAL: Se realizó biopsia incisional de lesiones cutáneas localizadas en espalda y pierna; posteriormente son enviadas al departamento de Anatomía Patológica, se fijó en formol al 10%. Los fragmentos tomados fueron incluidos en bloques de parafina, se realizaron cortes de 3 a 4 micras y se tiñeron con Hematoxilina y Eosina. Se realizaron nuevos cortes para estudios de inmunohistoquímica. RESULTADOS: Macroscópicamente se observa un huso de piel de 0.8x0.4 cm y 0.9x0.4 cm, café claro y de consistencia blanda. Microscópicamente se identificó epidermis con espongiosis con alineación de linfocitos en la unión dermoepidérmica y tendencia hacia el epidermotropismo así como formación de colecciones de linfocitos medianos, algunos de ellos con halo perinuclear y otros formando cúmulos. La membrana basal muestra discreta vacuolización, en la dermis papilar linfocitos dispuestos en forma lineal así como caída de pigmento. En dermis reticular superficial hay infiltrado linfocítico perivascular. El reporte de inmunohistoquímica incluyó CD20(pan B)-, CD4+ 80%, CD7-, Ki67 10%, CD3 (pan T)+, CD8+ 20% CONCLUSIONES: Es importante tener en mente esta patología en niños aunque represeta sólo 4-5% de los casos y la prevalencia es mayor en adultos. Debe considerarse como posible diagnóstico diferencial en los casos de lesiones hipocrómicas en piel y es importante tomar biopsia de estas lesiones para estudios histopatológicos ya que una Mampara: 5 A Fecha y hora Instalación: 4 de mayo 7:30 hrs. Fecha y hora retiro: 4 de mayo 13:00 hrs. TC-127 AUTORES: Juan Pablo Marquez, +*Fernando Durazo, +* Jose Antonio Matute TITULO: IMPLEMENTACIÓN DE UNA CUANTIFICACIÓN DEL INMUNOCONTEXTO CON IMPLICACIONES EN LAS DECISIONES DIAGNÓSTICAS, PRONÓSTICAS Y TERAPÉUTICAS EN PACIENTES ONCOLÓGICOS INTRODUCCIÓN: El conocimiento del crucial rol del sistema inmune sobre el cáncer humano ha abierto un nuevo campo de tratamiento e investigación a nivel mundial la inmunooncologia. Por lo que es imperativo contar con un sistema diagnostico objetivo del inmunoxontexto que nos determine el estado basal del paciente,selección tratamientos inmunomoduladores, presencia de marcador celular de buen o mal pronostico OBJETIVOS: implementar un estudio inmuno-onco-patologico integral con cuantificacion celular individual en margen tumor y centro tumoral, valoracion del estroma tumoral, integrar el indice KM, inmunoescore tradicionales, para tener una valoración amplia y completa de todo el microambiente tumoral. MATERIAL: Todos los pacientes oncologicos atendidos en el CICS y OMA de abril 2014 a Enero 2016, se elije una laminilla que muestre claramente una zona central y periferica tumorales y de una mayor densidad optica del infiltrado 2do Congreso Anual de Patólogos Mexicanos • 98 León, Guanajuato • 2016 inmune; se realiza un conteo objetivo campo por campo, de los distintos componentes celulares del inmuncontexto,se emplean otros parametros de cuantificacion y tecnicas especiales de inmunohistoquimica. RESULTADOS: Se han realizado 225 estudios, siendo los mas prevalentes cancer de colon, mama y ovario, hemos tenido pacientes con mas de una biopsia lo que nos ha servido para verificar los cambios en el inmunocontexto tras los tratamientos inmunomoduladores y tras la administracion de farmacos como ipilumimab. Además estudios han servido para planificar los tratamientos anti estroma tumoral, diseño de tratamiento inmunomodulares personalizados, vacunas peptidicas , terapias adyuvantes y han sido base para estudios moleculares de pronostico mas especifico individualmente para cada paciente. CONCLUSIONES: Las complejas interacciones entre las células tumorales, respuesta inmune y el microambiente tumoral, requieren el uso de sistemas de evaluación y estandarización de los mismos. inmunocontexto ha demostrado ser un parámetro pronóstico y supervivencia global. Es imperativo comenzar a reconocer el valor del estado inmune en el desarrollo, progresión y evolución de los pacientes con cáncer. Por lo que debemos reconocer y utilizar todas las herramientas posibles para un diagnóstico integral y las mejores opciones de tratamiento para nuestros pacientes INSTITUCIÓN: ONCOLOGIA MOLECULAR AVANZADA (OMA) Y CENTRO PARA LA INVESTIGACIÓN DEL CÁNCER SONORA (CICS) ESTADO:SONORA bronquial, un tumor extremadamente raro, solitario, bien circunscrito, multiquístico y predominantemente exofítico. La lesión se clasifica como un tumor epitelial benigno del pulmón.Se origina de las glándulas mucosas de los bronquios más grandes, lobares o segmentarios.Esta compuesto de acinos que contienen moco revestidos por células cuboidales sin pleomorfismo. OBJETIVOS: Presentación de un caso que se presento en la unidad médica de alta especialidad 25 en Monterrey N.L. MATERIAL: Masculino de 24 años de edad con antecedente de consumo de marihuana y drogas intravenosas,con cuadro de dolor torácico derecho y disnea, sin tos ni fiebre. Se maneja como neumonía sin mejoría; se sospecha tuberculosis y se realiza BAAR con resultado negativo. Se somete a biopsia con RHP Tuberculosis pulmonar. Se inicia tratamiento por 6 meses sin mejoria y una TAC de control que reporta en bronquio inferior derecho imagen nodular de 1.3x2.2 cm. RESULTADOS: Se sometio a lobectomía abierta con una masa referida de 4cm y ganglios con fibrosis. La imagen histológica muestra glándulas dilatadas revestidas con células secretoras mucosas columnares, cúbicas o aplanada con núcleos sin pleomorfismo en un estroma compuesto por tejido conectivo hialino en el cual se observan fibroblastos y células plasmáticas y linfocitos ocasionales. No se observan figuras mitóticas, áreas de necrosis ni permeación vascular concluyendo adenoma mucinoso bronquial. El resto del parerénquima con hemorragia intraparéquimatosa. Se disecaron 6 ganglios linfáticos con hiperplasia folícular reactiva. La inmunohistoquímica realizada resulto postiiva para CK5/6, CK7, EMA, ACE Y S-100 y negativa para TTF1. CONCLUSIONES: El adenoma de las glándulas muco- TITULO: Adenoma de glándulas mucosas bronquial sas es un raro tumor benigno de las glándulas secretoras mucosas de las vías respiratorias. Se describio por primera vez en 1882 por Muller como una entidad patológica separada del carcinoma de pulmón. La mayoria de los casos se observan del bronquio pero también se han descrito en la tráquea o las vías aéreas periféricas. El tumor se presenta con igual frecuencia en hombres y mujeres y en cualquier edad (media de 52) incluidos niños. Los síntomas principales son tos, fiebre y neumonía recurrente como resultado de la obstrucción bronquial por el tumor. La tos crónica, sibilancias unilaterales, hemoptisis o infecciones pulmonares pueden estar presentes antes del diagnóstico correcto. Inmunohistoquimicamente las células que revisten los espacios quísticos son positivas para marcadores epiteliales y expresan citoqueratinas de alto peso molecular, negativas para TTF-1 y positividad focal S-100, EMA Y ACE indican un componente mioepetilial. El diagnóstico diferencial debe hacerse con el carcinoma mucoepidermoide de bajo grado los cuales son infiltrantes, tienen atipia citológica, mitosis, necrosis y y una fuerte positividad para TTF-1 en las glándulas neoplásicas. PorlLo anterior nos concluye que se trata de esta entidad. INTRODUCCIÓN: El adenoma de glándulas mucosas INSTITUCIÓN: Instituto Mexicano del Seguro PATOLOGÍA PULMONAR Mampara: 10 A Fecha y hora Instalación: 4 de mayo 7:30 hrs. Fecha y hora retiro: 4 de mayo 13:00 hrs. TC-8 AUTORES: DRA. ANA PAULINA SÁNCHEZ, Dra. Silvia Judith Hernández Martínez, Dra. Yolanda Martínez Flores, Dr. Juan José Treviño González, Dra. Karina Larios Cárdenas 2do Congreso Anual de Patólogos Mexicanos • 99 León, Guanajuato • 2016 Social. Unidad Médica de Alta Especialidad 25 Mampara: 10 B ESTADO: NUEVO LEÓN Fecha y hora Instalación: 4 de mayo 7:30 hrs. Fecha y hora retiro: 4 de mayo 13:00 hrs. Mampara: 10 B Fecha y hora Instalación: 4 de mayo 7:30 hrs. Fecha y hora retiro: 4 de mayo 13:00 hrs. TC-114 AUTORES: DR. JESÚS CIENFUEGOS, Dr. Isaías Estrada Moscoso, Dr. Alberto Aranda, Dra. Sara Parraguirre TITULO: Leiomiosarcoma primario de aurícula derecha: Reporte de un caso. TC-106 AUTORES: DRA. SONIA TAVARES, Dra. Blandina Her- INTRODUCCIÓN: TITULO: HEMANGIOMA ESCLEROSANTE. PRESENTACION DE UN CASO. Los sarcomas primarios cardiacos representan el 25% de las neoplasias malignas cardiacas. El leiomiosarcoma es una neoplasia maligna rara en esta localización, representando el 8% de estas lesiones. Presentamos un caso de estudio postmortem parcial (sólo corazón), de un hombre con leiomiosarcoma primario de la aurícula derecha. INTRODUCCIÓN: El neumocitoma (hemangioma es- OBJETIVOS: Describir el caso clínico-patológico de un nández Cruz, Dra. Jazmín de Anda González, Dra. Liliana García Ilizaliturri, clerosante pulmonar), es una neoplasia benigna descrita en 1956 por Liebow y Hubbel. Se clasifica como tumor epitelial con diferenciación a neumocitos tipo II. Neoplasia de baja prevalencia, predomina en mujeres y en la raza asiática. La edad promedio de presentación es de 46 años; clínicamente es un nódulo pulmonar solitario asintomático. Puede llegar a medir hasta 7cm. Se han descrito casos de nódulos múltiples. Histológicamente puede presentar cuatro patrones: papilar, sólido, esclerótico y hemorrágico, presenta dos componentes, de células poligonales y de células cuboidales; con inmunoexpresión para marcadores epiteliales y neuroendocrinos. El diagnóstico diferencial se plantea con adenocarcinoma con patrón lepídico, carcinoide, adenoma papilar, angiosarcoma y mesotelioma. La resección quirúrgica es el tratamiento de elección. RESULTADOS: Paciente femenino de 57 años de edad, sin antecedentes de importancia. Inicia hace un año al diagnosticarse adenocarcinoma moderadamente diferenciado de duodeno, tratado con cirugía y adyuvancia. Durante el seguimiento se identifica nódulo pulmonar solitario en lóbulo inferior derecho de 4x3cm; sometida a bilobectomia derecha con estudio transoperatorio, reportado como neoplasia epitelioide con patrón papilar. Se realizaron cortes histológicos definitivos con hematoxilina ? eosina, y tinciones de inmunohistoquímica diagnosticándose como hemangioma esclerosante. CONCLUSIONES: Presentamos una neoplasia de baja incidencia, que puede ser de difícil diagnostico en el estudio transoperatorio por los cuatro patrones morfológicos, por lo que es importante y necesario realizar la correlación clínico-radiológica-patológica. hombre de 33 años de edad con diagnóstico post-mortem de leiomiosarcoma primario de la aurícula derecha. MATERIAL: Se realizó estudio post-mortem parcial (sólo corazón) a un hombre de 33 años de edad. Se revisó el historial clínico y variables histopatológicas y de inmunohistoquímica. RESULTADOS: Se encontró un tumor de 7.0 x 6.5 x 6.0 cm que ocupaba la totalidad de la aurícula derecha, con permeación a aurícula izquierda a través de un foramen oval permeable. Al estudio histopatológico se evidenció una neoplasia maligna de origen mesenquimal con un patrón fusiforme, conformado por células con núcleos ahusados de bordes romos con citoplasma eosinófilo y células pleomórficas atípicas, mitosis frecuentes (3/10 HPF), atipia nuclear marcada. La inmunohistoquímica fue positiva para actina de músculo liso, actina de músculo específico, vimentina y desmina. CONCLUSIONES: El leiomiosarcoma es una neoplasia maligna de origen mesenquimatoso con baja incidencia en corazón. Su presentación en aurícula derecha es aún más raro, presentándose hasta en 16% de los casos. La clínica es inespecífica. Son tumores de crecimiento rápido con pronóstico desfavorable. INSTITUCIÓN: Hospital General Dr. Manuel Gea González ESTADO: DISTRITO FEDERAL INSTITUCIÓN: Hospital de Oncologia de Centro Medico Nacional Siglo XXI ESTADO: DISTRITO FEDERAL 2do Congreso Anual de Patólogos Mexicanos • 100 León, Guanajuato • 2016 Mampara: 11 B Fecha y hora Instalación: 4 de mayo Fecha y hora retiro: 4 de mayo 13:00 hrs. Mampara: 12 A 7:30 hrs. Fecha y hora Instalación: 4 de mayo 7:30 hrs. Fecha y hora retiro: 4 de mayo 13:00 hrs. TC-124 TC-150 AUTORES: Dra. Juana Elizabeth Tadeo González. Dra. AUTORES: DRA. JENIFFER CALDERON, Dra. Gutié- Hersilia Aide Hernandez Zamonsett, Dra. Natalia Vilches Cisneros. Dra. Raquel Garza Guajardo, Dra. Oralia Barboza Quintana, Dr. Juan Pablo Flores Gutierrez. Dra.Gabriela Alarcon Galvan. rrez Gil Mary Carmen, Dr. Pinedo Onofre Javier. TITULO: Tumor Fibroso Solitario Gigante De Pleura TITULO: Mesotelioma Papilar Bien Diferencia- INTRODUCCIÓN: El TFSP descrito por primera vez do Pleural. Presentación de un caso y revisión en 1767 por Lietaud; es una entidad rara. Corresponde al 8% de las patologías intratorácicas y al 10% de los tumode la Literatura. INTRODUCCIÓN: El mesotelioma papilar bien diferenciado de la pleura es raro. Se presenta mas comúnmente en peritoneo, los casos pleurales se reducen a 50 reportados. Se caracteriza por una arquitectura papilar con características citologicas benignas. Su morfología, clínica y pronostico son separadas del mesotelioma maligno. OBJETIVOS: Presentar el caso de un paciente masculino de 42 años de edad, que inicia con cuadro de un mes de evolución con disnea progresiva, tos seca perdida de peso, a su ingreso con derrame pleural masivo izquierdo, por tomografia axial computada se evidencia masa en pleura mediastinal. Se tomaron biopsias pleurales. MATERIAL: Se realizo el estudio histologíco de 5 biopsias pleurales, con tinción de hematoxilina y esosina, se evidencio una proliferacion celular que se originaba a partir de la superficie mesotelial de la pleura, a expensas de papilas rodeadas por una sola capa de células cubicas, sin atipia mitosis o invasión, con centros fibrovasculares y mixoides, ademas se realizaron tinciones de inmunohistoquimica WT1 y Calretinina las cuales fueron positivas. RESULTADOS: Con los datos clínicos e histopatológicos se hizo el diagnóstico de Mesotelioma papilar bien diferenciado pleural. CONCLUSIONES: El mesotelioma papilar es un tumor indolente, asociado con largo periodo de supervivencia, el sitio mas frecuente es el peritoneo, no se a encontrado estrecha relación con la exposicion con asbesto, se ha relacionado con la mutación de la linea germinal BAP1, el diagnostico se realiza mediante citomorfologia e inmunohistoquimica, los diagnosticos diferenciales incluyen mesotelioma epiteliode con crecimiento papilar e hiperplasia papilar mesotelial. INSTITUCIÓN: Hospital Universitario “Dr. José Eleuterio González” ESTADO: NUEVO LEÓN res pleurales. Por su comportamiento e histomorfología se pueden clasificar en benignos y malignos. Clínicamente son asintomáticos y ocasionalmente solo presenta tos, disnea y dolor torácico. OBJETIVOS: Mostrar un caso de Tumor Fibroso Solitario de Pleura en un paciente masculino de la quinta dé vida, el cual fue intervenido quirúrgicamente por el Servicio de Cirugía Torácica de este hospital, estudiado por el Servicio de Patología con microscopia de luz y confirmado por medio de la inmunohistoquímica correspondiente. MATERIAL: Se estudió por medio de microscopia de luz convencional, en primera instancia realizándose tinciones de hematoxilina & eosina. Posteriormente se obtuvieron laminillas con diversos anticuerpos de inmunohistoquímica para su confirmación, siendo las siguientes: CD34, BCL2, Actina de Músculo Liso, Ki-67 RESULTADOS: En el servicio de patología se recibió espécimen tisular de 22 x 19 x 13 cm y peso de 2720g. Presentó pseudocápsula con superficie café violácea, lisa y brillante. Al corte: sólido, amarillo claro con áreas de aspecto hemorrágico y mixoide. Histológicamente se observó neoplasia mesenquimatosa benigna constituida por células fusiformes, dispuestas en distintos patrones; focos de colagenización, vasos sanguíneos en forma de “asta de venado” y baja actividad mitótica. Se realizó inmunohistoquímica: CD34 (+), BCL2 (+), AML (-), Ki-67 (+) <1%, lo cual confirma el diagnóstico de TFSP, descartando el de TFSP maligno> CONCLUSIONES: Siendo esta entidad del grupo de patologías que se llegan a diagnosticar incidentalmente, a causa del estado asintomático de la mayoría de los individuos con esta patología, es de extrema importancia tanto la sospecha clínica como el diagnóstico certero que ofrece la histopatología con la ayuda de los métodos de inmunohistoquímica correspondientes a este tipo de entidad. Igual de importante se convierte la determinación del índice de proliferación celular, para así poder categorizar benignidad y malignidad de esta entidad, lo cual repercute en la supervivencia. INSTITUCIÓN: Hospital Central “Dr. Ignacio 2do Congreso Anual de Patólogos Mexicanos • 101 León, Guanajuato • 2016 Morones Prieto”, San Luis Potosí, San Luis Potosí, México ESTADO: SAN LUIS POTOSÍ Mampara: 12 B Fecha y hora Instalación: 4 de mayo 7:30 hrs. Fecha y hora retiro: 4 de mayo 13:00 hrs. TC-167 AUTORES: DR. GUILLERMO VALERO, Dr. César A. Flores García, Dra. Patricia Soto Márquez. es recomendable efectuar un panel de IHQ que incluya 3 a favor de mesotelioma (calretinina, WT-1, CK 5/6, HBME-1,D2-40, mesotelina, etc) y 3 a favor de adenocarcinoma (TTF-1, Napsin-A, MOC 31, ACE, EMA, etc). La variedad o componente sarcomatoide usualmente solo llega a expresar queratinas. INSTITUCIÓN: UMAE Hospital de Cardiología CMN SXXI, IMSS. ESTADO: DISTRITO FEDERAL Mampara: 13 A Fecha y hora Instalación: 4 de mayo 7:30 hrs. Fecha y hora retiro: 4 de mayo 13:00 hrs. TITULO: Mesotelioma maligno (MM) difuso pleural. Revisión de 5 años TC-168 INTRODUCCIÓN: El MM de la pleura constituye actual- mente un problema de salud ocupacional con importante repercusión familiar y social. Se describe exposición a asbestos hasta en el 90% de hombres y alrededor del 20% de las mujeres. La mayoría se presentan después de los 60 años de edad, con relación H:M de 4:1 y un periodo de latencia de menos de 15, hasta 30-40 años. OBJETIVOS: Determinar la frecuencia y distribución por edad y género del MM pleural en los casos revisados en el Hospital de Cardiología. Conocer los principales tipos y subvariedades del MM pleural. Identificar los marcadores de IHQ de mayor utilidad para el diagnóstico diferencial con adenocarcinoma. MATERIAL:Registros histopatológicos del servicio de Anatomía de la UMAE Hospital de Cardiología CMN SXXI, IMSS del 1 enero 2011 al 31 diciembre 2015. Determinar el número de biopsias pleurales y seleccionar aquellas con diagnóstico de mesotelioma maligno. Cuantificaron las diferentes variedades histológicas. Identificar los anticuerpos de IHQ mas frecuentemente utilizados. RESULTADOS: Se revisaron 1,587 biopsias pleurales. En total se encontraron 201 casos de MM difuso pleural: 170 epitelioides,23 bifásicos y 8 sarcomatoides (2 desmoplásicos). 148 fueron hombres vs 53 mujeres (relación 3:1). La mayoría después de los 60 años edad. Los Ac. mas frecuente utilizados fueron: CK 5/6, calretinina, WT-1 y HBME1 para mesotelioma y MOC-31, ACE,TTF-1 y napsina-A para adenocarcinoma (relacionado a la disponibilidad del recurso). AUTORES: DR. CESAR ANTONIO FLORES, Dra. Patricia Soto Márquez, Dr. Guillermo Valero Elizondo TITULO: NEUMOCITOMA ESCLEROSANTE. REPORTE DE UN CASO. INTRODUCCIÓN: Presentamos el caso de una mujer de 60 años, asintomática, sin antecedentes de importancia , inició su padecimiento actual con detección fortuita de lesión nodular única, en lóbulo superior pulmonar izquierdo de 3 cm de diámetro, sólida, bien delimitada. Se realizó biopsia incisional a cielo abierto. MATERIAL: En el servicio de Patología recibimos dos fragmentos de tejido de 1.2x1.1x1.0 cm y 1.2x0.7x0.2 cm respectivamente, ambos con pleura visceral gris opaca, el mayor era café blanquecino al corte sólido, con puntillo violáceo, opaco de consistencia blanda, el menor era laminar café oscuro, esponjoso, se incluyeron totalmente. RESULTADOS: En los cortes histológicos se observó neoplasia predominantemente sólida y en menor proporción papilar, con formaciones retiformes y canales de aspecto vascular, posee dos poblaciones celulares (neumocitos tipo II y células estromales redondas), el intersticio presentó calcificaciones distróficas. Se realizó inmunohistoquímica con los anticuerpos: CD 34: negativo en células neoplásicas; mientras que CK AE1/AE3(citoplásmico), Napsina A(citoplásmico) y TTF-1 (nuclear): positivos, intensos y difusos en neumocitos tipo II, EMA(citoplásmico) y Vimentina(citoplásmico): positivos en células estromales redondas. CONCLUSIONES: Es un tumor con predominio en muCONCLUSIONES: El MM epitelioide puede presentar jeres con reportes desde 11 a 80 años, su incidencia es alta una diversidad de patrones histológicos, los mas frecuentes son: túbulopapilar, glandular, y sólido. Conviene conocer el espectro morfológico para identificar a los poco usuales. Para el dx. diferencial con metástasis de adenocarcinoma, en el Este de Asia y baja en países occidentales. Se consideró como neoplasia de linaje vascular, ahora se considera que deriva de epitelio respiratorio primitivo. Clásicamente es lesión única bien delimitada y periférica. Histológicamente 2do Congreso Anual de Patólogos Mexicanos • 102 León, Guanajuato • 2016 posee dos poblaciones neumocitos tipo II y células estromales redondas. En la 4a edición OMS 2015 se considera benigno, aunque existen contados casos reportados en la literatura con metástasis en ganglios linfáticos siendo en realidad una neoplasia de bajo potencial maligno. REFERENCIAS: -William D. Travis, Elisabeth Brambilla, Allen P. Burke, Alexander Marx, Andrew G. Nicholson: WHO Classification of Tumours of Lung, Pleura, Thymus and Heart , 4th edition , IARC: Lyon 2015. pp 110-111. -Miyagawa-Hayashino A, Tazelaar HD, Langel DJ, et al. Pulmonary sclerosing hemangioma with lymph node metastases: report of 4 cases. Arch Pathol Lab Med 2003, 127:321-5. estratificación del riesgo de complicaciones y el estudio familiar, son indispensables en el tratamiento de los pacientes con cardiomiopatía hipertrófica. Esta entidad es una causa de muerte súbita en pacientes jóvenes. INSTITUCIÓN: UMAE Hospital de Cardiología, CMN SXXI, IMSS. ESTADO: DISTRITO FEDERAL Mampara: 14 A INSTITUCIÓN: INSTITUTO MEXICANO DEL SEGURO SOCIAL. CENTRO MÉDICO NACIONAL Fecha y hora Instalación: 4 de mayo 7:30 hrs. SIGLO XXI. HOSPITAL DE CARDIOLOGÍA Fecha y hora retiro: 4 de mayo 13:00 hrs. ESTADO: DISTRITO FEDERAL TC-188 AUTORES: DR. CESAR ANTONIO FLORES, Dra. Patri- Mampara: 13 B Fecha y hora Instalación: 4 de mayo 7:30 hrs. Fecha y hora retiro: 4 de mayo 13:00 hrs. TC-170 AUTORES: DRA. PATRICIA SOTO, Dr. César Antonio Flores García; Dr. Guillermo Valero Elizondo TITULO: CARDIOMIOPATÍA HIPERTRÓFICA SEPTAL ASIMÉTRICA CON INFARTO AGUDO DEL MIOCARDIO. REPORTE DE UN CASO. INTRODUCCIÓN: Masculino de 37 años: Inició con deterioro de su clase funcional, ecocardiograma mostró FEVI 80%, miocardiopatia hipertrófica asimétrica septal, obstructiva, coronariografía sin alteraciones significativas. Sometido a implante valvular aórtico y mitral y miomectomía septal del VI, debido a su evolución en abril se realiza trasplante cardiaco ortotópico bicaval. OBJETIVOS: Presentar un caso de cardiomiopatía hipertrófica septal asimétrica asociado a infarto agudo del miocardio. RESULTADOS: Producto de explante cardíaco de 500 g, espesor pared libre de VD 0.5 cm, VI 1.5 cm, septo IV 3.0 cm. Histológicamente muestra hipertrofia individual de miocitos, con formas y ramificaciones anómalas y el característico desarreglo de fibras, fibrosis intersticial de moderada a severa, las ramas de las arterias intramiocárdicas con severa hipertrofia irregular de la media. Con infarto agudo del miocardio de 12 a 48 hrs. de evolución histológica. CONCLUSIONES: El diagnóstico precoz correcto, la cia Soto Márquez, Dr. Guillermo Valero Elizondo TITULO: EXCRECENCIAS CARDIACAS INCIDENTALES MESOTELIALES/MONOCÍTICAS (MICE) COMPLICADAS CON ENDOCARDITIS AGUDA BACTERIANA INTRODUCCIÓN: Presentamos el caso de un hombre de 23 años que inició su padecimiento actual 15 días con fiebre, dolor en epigastrio e hipocondrio izquierdo, náusea y vómito. Recibió tratamiento no especificado, sin mejoría. Evolucionó con astenia, adinamia y disnea. Se detectó soplo diastólico en foco aórtico. Ecocardiograma transtorácico mostró válvula aórtica con insuficiencia severa y vegetación de 17 mm, siendo sometido a cirugía para colocación de implante valvular aórtico. MATERIAL: Recibimos en el servicio dos valvas semilu- nares nativas que midieron 2x1.2x0.1 cm y 2.1x1.1x0.1 cm, respectivamente con superficie gris blanca, lisas, opacas y ahuladas. En una de ellas se identifica formación exofítica adherida al velo valvular de aspecto vegetante midió 1.4x1x0.5 cm, café amarilla, opaca al corte es blanquecina , sólida, opaca y blanda, cubierta por material fibrinoso friable. RESULTADOS: En los cortes histológicos se observan vegetaciones compuestas por fibrina, eritrocitos, restos celulares con incontables neutrófilos, y destrucción tisular valvular, formación de abscesos en el espesor valvular con colonias de bacterias cocoides Gram positivas; inmediatamente se observan mas fibrina con macrófagos inmersos, alternando con adipocitos y de forma multifocal grupos de células mesoteliales dispuestas en hileras y papilas. Se realizó inmunohistoquímica con los anticuerpos: CD 31 (membrana) y CD 68(citoplásmico): positivos en macrófagos; calretinina (citoplasma/núcleo), WT-1 (nuclear) y CK 5/6 (citoplásmico): positivos intensos y difusos en células 2do Congreso Anual de Patólogos Mexicanos • 103 León, Guanajuato • 2016 mesoteliales. fuera del bronquio provocando atrapamiento aéreo. CONCLUSIONES: Las M.I.C.E. consisten en una le- RESULTADOS: Se realiza cepillado y lavado bronquial sión benigna incidental de etiología incierta localizada en cámaras cardiacas, válvulas o pericardio. Se compone en cantidades variables por células mesoteliales, histiocitos, fibrina, adipocitos. Existen 3 teorías: Reactiva que atañe a intervencionismo previo (cirugía, cateterismo); Por artificio: dice que las bombas de circulación extracorpórea siembran los diversos tipos celulares entre pacientes y la mas actual dice que existen Células Multipotenciales derivadas de Monocitos circulantes (MOMCs) capaces de diferenciarse a cualquier linaje celular. REFERENCIAS: -Veinot John, Tazelaar Henry, et. al. Mesothelial/Monocytic Incidental Cardiac Excrescences: Cardiac MICE. Modern Pathology 1994, Vol 7 no 1, pag 9-16. -Zhong-Liang Hu, Hui Lü, et. al. A Case of mesothelial/ monocytic incidental cardiac excrescence and literature review. Diagnostic Pathology 2010, 5:40 negativo para malignidad con cambios inflamatorios severos. Se decide realizar lobectomía superior derecha por presencia de tumor invasivo bronquial. El resultado histopatológico reportó lóbulo superior de pulmón derecho con hamartoma condroide de 0.8 cm y focos de atelectasia. Clinicamente la paciente presentó mejoria de su padecimiento, sin sintomatología pulmonar residual. CONCLUSIONES: Los hamartomas pulmonares son tumores infrecuentes localizados en los bronquios pulmonares causando dificultad diagnóstica. Los estudios complementarios de citología y biopsia pueden ayudar a diagnosticar estos casos, evitando la cirugía. Sin embargo, el acto quirúrgico puede ser necesario. INSTITUCIÓN: HOSPITAL GENERAL DEL ESTADO DE SONORA INSTITUCIÓN: INSTITUTO MEXICANO DEL SE- ESTADO: SONORA GURO SOCIAL. CENTRO MÉDICO NACIONAL SIGLO XXI. HOSPITAL DE CARDIOLOGÍA ESTADO: DISTRITO FEDERAL PATOLOGÍA FORENSE Mampara: 14 B Fecha y hora Instalación: 4 de mayo 7:30 hrs. Fecha y hora retiro: 4 de mayo 13:00 hrs. TC-191 AUTORES: DR. ALBERTO IBARRA, DR. ROBERTO DE LEON CABALLERO Mampara: 16 A Fecha y hora Instalación: 4 de mayo 7:30 hrs. Fecha y hora retiro: 4 de mayo 13:00 hrs. TC-104 AUTORES: Dr. Roberto Cuauhtémoc Mendoza Morales, TITULO: Hamartoma Condroide: Reporte de Caso Dra. María Yolotzin Valdespino Vázquez, Dr. Fernando García Dolores, Dr. Eduardo Escamilla Mondragón, H.T. Francisco Javier Alvarado Martínez INTRODUCCIÓN: Hamartomas pulmonares son tumo- TITULO: Mioglobinuria secundaria a rabdomiólisis ocasio- res benignos que derivan del mesénquima peribronquial, siendo los tipos histológicos más frecuentes el condromatoso y el lipoide. El origen endobronquial de los hamartomas es poco frecuente y supone alrededor de un 3% de los hamartomas pulmonares. OBJETIVOS: Reportar el caso de una paciente con hamartoma condroide intrabronquial MATERIAL: Se trata de paciente femenino de 59 años con tabaquismo activo a razon de 2 cajetillas al dia que incia su padecimiento 3 meses previos por presentar fiebre de predominio vespertino, escalofrios, perdida de peso de 15 kg; se le encuentra nodulo pulmonar superior derecho. La tomografía de tórax reporta lesión intrabronquial en el segmento anterior del lóbulo superior derecho que se extiende nada por traumatismos múltiples: Reporte de un caso INTRODUCCIÓN: La Rabdomiólisis es secundaria a diversos factores que incluyen traumatismo muscular directo, actividad muscular excesiva, isquemia entre otros. La rabdomiólisis asociada a falla renal aguda representa entre el 6-8% de los casos. Se reporta un caso de un hombre de 21 años con múltiples contusiones que desarrollo insuficiencia renal aguda secundaria a rabdomiólisis que lo llevo a la muerte. OBJETIVOS: Se reporta el caso de un hombre de 21 años, que presentó lesión renal aguda con datos de rabdomiólisis secundaria a múltiples contusiones. MATERIAL: Hombre de 21 años recluso, que sufrió trau- 2do Congreso Anual de Patólogos Mexicanos • 104 León, Guanajuato • 2016 matismos múltiples por pelea en el reclusorio 2 dias antes. Es llevado al servicio de urgencias inconsiente, en estado de choque, con sindrome de dificultad respiratoria, taquicardia y edema en extremidades inferiores. Por los estudios de laboratorio se determina lesión renal aguda AKIN3 y era necesario tratamiento sustitutivo. Se realiza colocación de catéter peritoneal y fallece en quirófano. RESULTADOS: Se realiza necropsia con los siguientes hallazgos: Pulmones con datos de edema pulmonar. Riñones de aspecto congestivo. Microscópicamente en riñón se identificó necrosis tubular aguda extensa, con vacuolización citoplasmática fina del epitelio tubular, edema intersticial, congestión glomerular, así como la presencia de abundantes cilindros granulares amórfos principalmente en túbulos distales. Se realizó inmunotinción para mioglobina la cual resultó positiva multifocal tanto intratubular como intracitoplasmática. Por lo que se determinó como causa de muerte edema pulmonar agudo e insuficiencia renal aguda ocasionada por el depósito de mioglobina en los túbulos renales. CONCLUSIONES: La rabdomiólisis es un síndrome clínico de daño muscular asociado con mioglobinuria, anormalidades en los electrolitos y daño renal agudo. La mioglobina es el constituyente muscular primario que contribuye a la lesión renal en la rabdomiolisis. El diagnóstico es confirmado por la elevación de los niveles de CPK, como fueron corroborados nuestro caso. La mortalidad varía de 1.7 a 46%, dependiendo las causas, el tratamiento hidroelectrolítico, asi como el reemplazo sustitutivo de la función renal. En nuestro caso no fue posible un tratamiento adecuado debido a los días de evolución. REFERENCIAS: 1.Bosch X, Poch E, Grau JM. Rhabdo- myolysis and acute kidney injury. N Engl J Med.2009;361:6272, 2. Zager RA. Rhabdomyolysis and myohemoglobinuric acute renal failure. Kidney Int.1996;49:314-326. 3 Holt SG, Moore KP. Pathogenesis and treatment of renal dysfunction in rhabdomyolysis.Intensive Care Med.2001;27:803-811. INSTITUCIÓN: Instituto de Ciencias Forenses de la Cuidad de México, Hospital General de México ESTADO: DISTRITO FEDERAL PATOLOGÍA POSMORTEM Mampara: 7 A Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. TC-46 AUTORES: DRA. SARAI PAEZ, Dra. Ana Elizabeth Balderas Cordero, Dra. Amalia Castro Rodríguez TITULO: Angiosarcoma primario cardiaco en aurícula de- recha con extensión a vena cava inferior y sus tributarias: Reporte de un caso en estudio Post-Mortem INTRODUCCIÓN: De las neoplasias cardiacas primarias, el angiosarcoma es la neoplasia maligna más frecuente. La localización más común es en aurícula derecha; en pacientes de la tercera a quinta década de la vida, sin predilección de sexo. La mayoría son casos esporádicos, sin embargo se han reportado casos con incidencia familiar. Los síntomas son inespecíficos. OBJETIVOS: Se trata de mujer de 74 años, con antecedentes de tiroidectomía por cáncer tiroideo de estirpe histológica no conocida, diabetes mellitus tipo 2, hipertensión arterial sistémica y tabaquismo positivo con índice tabáquico de 30 paquetes por año, este último durante 60 años. Inicia su padecimiento con dolor abdominal tipo cólico, aumento del perímetro abdominal, distensión y plenitud postprandial, además de cambio de coloración y edema de miembros inferiores, con pérdida de peso no intencionada de 8 a 9 kg en un mes. En TAC abdomino pélvico se documenta imagen sugestiva de trombo que se extiende de la cava inferior desde nivel hepático hacia aurícula derecha; se complementa con ecocardiograma transtorácico en el que se documenta masa intrauricular derecha de 30x11 mm dependiente del techo. Se realiza toma de biopsia de dicha lesión. Posterior a procedimiento presenta datos de falla renal y hepática con cuadro de acidosis metabólica refractaria a manejo médico; la paciente fallece y se solicita autopsia. Dentro de los hallazgos macroscópicos relevantes se documenta tumor intrauricular derecho de 4.5x4 cm nodular, violáceo, adherido a pared auricular por pedículo de 0.7 cm; el tumor se continúa hacia la vena cava inferior, ocupándola y dilatándola hasta bifurcación de venas ilíacas primitivas, además de involucrar venas tributarias. Histológicamente observamos una neoplasia poco diferenciada, que infiltra la pared de aurícula derecha; con extensas zonas de necrosis y hemorragia; con células dispuestas de manera aleatoria o formando hendiduras ocupadas por eritrocitos; las células muestran marcado pleomorfismo, multinucleación e hipercromatismo. Por inmunohistoquímica las células neoplásicas son positivas para vimentina, CD31 y Factor VIII, y negativas para desmina, citoqueratina y actina músculo específico. Con los 2do Congreso Anual de Patólogos Mexicanos • 105 León, Guanajuato • 2016 hallazgos histopatológicos y de inmunohistoquímica se concluye el diagnóstico de angiosarcoma primario de aurícula derecha con extensión a vena cava inferior y tributarias. cífica el diagnóstico de los angiosarcoma cardíacos suele realizarse en etapas avanzadas de la enfermedad. Uno de los mecanismos que provoca las manifestaciones clínicas es la obstrucción al flujo como ocurrió en nuestro caso. El diagnóstico por estudios de imagen es en base a ecocardiograma, TAC o RMN; sin embargo el estudio histopatológico con inmunohistoquímica de cualquier tumor cardíaco es mandatorio para el diagnóstico. El tratamiento en todos los tumores intracardiacos es la resección quirúrgica, en el caso de los sarcomas se utilizan también la quimioterapia y radioterapia; aunque de todos modos el pronóstico en estos tumores es muy pobre. personales patológicos de importancia. Dentro de sus antecedentes ginecoobstétricos destacan: Gesta 6, Partos 4, Abortos 1, FUM 18-01-2015. Acudió al servicio de ginecobstetricia para conducción de trabajo de parto con diagnóstico de embarazo de 40.2 SDG por FUM. Refirió adecuada cinética fetal, negando otra sintomatología. La exploración física se encontró sin alteraciones y signos vitales dentro de parámetros normales. Diez horas posteriores a su ingreso presentó paro cardiorrespiratorio de forma súbita sin obtener respuesta a las maniobras de reanimación avanzada. Se realizó autopsia y se encontró únicamente alteración en los cortes microscópicos de pulmón, que mostraron múltiples microémbolos formados por escamas anucleadas provenientes del lanugo del feto y material mucinoso de origen meconial, corroborados con estudio de inmunoperoxidasa (citoqueratina AE1/AE3) e histoquímica (tinción Azul alciano) respectivamente. INSTITUCIÓN: UMAE 25 IMSS, UMAE 34 IMSS CONCLUSIONES: Se han propuesto varios factores CONCLUSIONES: Debido a su sintomatología inespe- ESTADO: NUEVO LEÓN Mampara: 7 B Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. TC-72 AUTORES: DR. JOSE CARLOS ZACATE ROJAS, DRA. LINA URANIA HERNANDEZ ROMAN TITULO: EMBOLISMO PULMONAR POR LIQUIDO AMNIOTICO. REPORTE DE UN CASO DE AUTOPSIA. INTRODUCCIÓN: El embolismo de líquido amniótico es una complicación obstétrica poco frecuente que se presenta en 1 por cada 25, 000 embarazos. Es una afección no prevenible ni predecible que una vez instaurado presenta una mortalidad materna de hasta el 80%. Es considerada la causa número uno de muerte súbita en embarazadas, por lo que su diagnóstico clínico de certeza es difícil de realizar y su comprobación diagnostica solo se confirma con el estudio anatomopatológico. OBJETIVOS: Presentar un caso de autopsia poco frecuente, donde el estudio postmortem fue crucial para establecer el diagnóstico. MATERIAL: Se realizó el estudio de autopsia a una paciente obstétrica con un embarazo de término en conducción del trabajo de parto que presentó muerte súbita. que pudieran predisponer el desarrollo de embolismo por líquido amniótico como son: edad avanzada, multiparidad, desgarro de cérvix, rotura prematura de membranas y líquido amniótico teñido con meconio. Aunque no se consideran condicionantes para el desarrollo del mismo. La etiopatogenia no está del todo clara, por lo que se han propuesto diversas teorías que tienen como factor común la penetración de líquido amniótico en los vasos venosos del lecho placentario y los vasos uterinos adyacentes. La presencia de líquido amniótico en el pulmón no produce cambios macroscópicos evidentes. Microscópicamente los émbolos en su mayoría están conformados por células epidérmicas escamosas anucleadas formando un material laminar que se corrobora por medio de inmnohistoquimica con citoqueratinas de bajo peso molecular como la CK 5/6. La tinción con azul Alcian es otro método sensible para detectar derivados mucinosos del meconio en la circulación materna. El estudio anatomopatológico en este caso fue crucial para confirmar el diagnóstico, debido a que no hay ningún dato clínico ni de laboratorio que por sí mismo pueda hacer sospechar o excluir el diagnóstico de embolismo de líquido amniótico. INSTITUCIÓN: HOSPITAL GENERAL “DR. MIGUEL SILVA” ESTADO: MICHOACÁN Mampara: 8 A Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. TC-91 AUTORES: DR. MIGUEL ENRIQUE CUELLAR, Dra. Norma Ofelia Uribe Uribe, Dra Yadira Gandhi Mata Mendoza RESULTADOS: Paciente femenina de 38 años de edad TITULO: Asociación entre telangiectasia hesin antecedentes familiares de interés ni antecedentes 2do Congreso Anual de Patólogos Mexicanos • 106 León, Guanajuato • 2016 morrágica hereditaria (enfermedad de Rendu-Osler-Weber)y poliposis juvenil. Presentación de un caso de autopsia. INTRODUCCIÓN: El síndrome de Osler Weber Rendu es un trastorno autosómico dominante de penetrancia variable que afecta a múltiples órganos y se suele caracterizar por telangiectasias, malformaciones arteriovenosas y aneurismas.Del 15 al 22% de los pacientes con mutaciones de SMAD 4 tienen un síndrome de sobre posición entre telangiectasia hereditaria hemorrágica y poliposis juvenil. OBJETIVOS: El reporte de un caso de la asociación entre la enfermedad de Rendu-Osler-Weber y la poliposis juvenil MATERIAL: Mujer de 29 años de edad, comenzó su padecimiento final dos años previos con rectorragia, dolor abdominal, ascitis y pérdida de peso no cuantificada. La paciente ingreso al área de urgencias donde se encontró caquéctica, con ascitis y caput medusae, deteriorando rápidamente y falleció en nuestra institución; por lo que se realizó autopsia. RESULTADOS: Cadáver caquéctico,con abdomen en batracio y dedos hipocráticos con carcinomatosis peritoneal, en pulmones se observó malformaciónes arterio-venosas. Hay tumor de aspecto lobulado que infiltra pared, blanco amarillento y blando al corte.Se observan polipos en toda la mucosa.A la apertura de cavidad craneana hay puntilleo hemorrágico. En intestido delgado, colon, utero, vejiga y ovarios hay neoplasia de origen epitelial con citoplasma claro (anillos de sello). Coexistiendose observan pólipos juveniles. En la sustancia blanca hay vasos que varían en forma y tamaño, en los que el endotelio se encuentra adelgazado, con congestión vascular, neuronas con citoplasma eosinófilo y núcleos picnóticos. INTRODUCCIÓN: Cocccidiodomicos es una infección que generalmente es letal en pacientes que se encuentran inmunodeprimidos, se presenta en zonas endémicas aridas en el norte y sur de México; el caso se hace de interés al encontrar imagen en panal de abeja en el parenquima pulmonar y además que el paciente no era residente de un lugar endémico. OBJETIVOS: Informar un caso de coccidiodomicosis diseminada MATERIAL: Estudio de autopsia realizado en Hospital Infantil de México “Federico Gómez RESULTADOS: Los cortes muestran parénquima pulmonar, tiroideo, esplénico, pericardico, hepático, páncreático, medula ósea y encéfalo con granulomas constituidos por histiocitos epiteliodes, células gigantes multinucleadas tipo Langhans y tipo cuerpo extraño, además se observan extensas áreas de necrosis con numerosas estructuras esféricas con reforzamiento de la membrana, algunas de ellas contienen numerosas estructuras esféricas de menor tamaño. Panal de abeja CONCLUSIONES: Los casos diseminados representan menos del 1% de los casos. Predominan en el género masculino, usualmente, en un contexto de inmunosupresión. Afectan comúnmente la piel, las meninges y huesos, así como sitios menos comunes como la glándula tiroides, la conjuntiva y el pericardio. INSTITUCIÓN: Hospital Infantil de México “Féderico Gómez” ESTADO: DISTRITO FEDERAL INSTITUCIÓN: Instituto Nacional de Ciencias Mampara: 9 A Médicas y Nutrición “Salvador Zubirán” Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. ESTADO: DISTRITO FEDERAL TC-152 Mampara: 8 B Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. TC-98 AUTORES: DRA. ATZIN ANDREA ANGELES, Dra. Sara Rodríguez, Dr. Guillermo Ramón García. TITULO: Coccidioidomicosis diseminada, en un caso de autopsia AUTORES: Dra. Mojica González Z., Dra. Garduño Becerra J., Dr. Salazar Morales M., Dra. Soto Abraham V., Dr. Aristi Urista G. TITULO:Deficiencia de enzima ?-galactosidasa A (enfermedad de Fabry), reporte de un caso de autopsia. INTRODUCCIÓN: La enfermedad de Fabry, causada por la deficiencia genética alfa-galactosidasa A (aGal), situado en el gen Xq22.1, lleva al almacenamiento de globotriaosilceramida (GL3) en muchos tipos celulares. Afecta la función renal, cardiaca, de glándulas sudoríparas y nervios. 2do Congreso Anual de Patólogos Mexicanos • 107 León, Guanajuato • 2016 Los signos y síntomas varían de acuerdo a la mutación y consecuente nivel funcional de aGal. OBJETIVOS: Presentamos un caso de autopsia de enfermedad de Fabry en un hombre de 45 años, cuya manifestación principal fueron síntomas isquémicos tanto cardiacos como neurológicos. MATERIAL: Se realizó el diagnóstico de enfermedad re- nal crónica en diciembre del 2014, así como enfermedad de Fabry confirmada en abril del 2015. Presentó crisis convulsivas en noviembre del 2014 de tipo tónico-clónicas, de 1 min de duración, con periodo post-ictal de 1 hora, acompañada de astenia, adinamia, con posterior deterioro neurológico, por lo que se le realizó resonancia magnética documentado enfermedad multiinfarto. Se documentó elevación del seg RESULTADOS: En la autopsia encontramos ateroesclerosis en todas las ramas del polígono de Willis, así como infartos cerebrales múltiples y con diferente fase de evolución, y microscópicamente la presencia de material grumoso con leve afinidad tintorial para PAS, PAS diastasa y luxol fast blue en células meningoteliales, neuronas de la corteza parahipocampica derecha, células acinares de lóbulo pituitario anterior y edema intracelular de leiomiocitos en túnica media de vasos arteriales. El ventrículo izquierdo con hipertrofia concéntrica de su pared, así como infarto transmural oclusión total por una placa calcificada. Así como endocarditis bacteriana con trombo mural en ventrículo izquierdo. CONCLUSIONES: Hay más de 500 mutaciones en aGal, 5% de las cuales son espontáneas. Hay 2 tipos principales de presentación clínica: la tipo 1 que se da en varones, niños o adolescentes, con acroparestesias, hipohidrosis, angioqueratomas y distrofia corneal, con desarrollo progresivo tardío de disfunción renal, cardiaca y cerebrovascular; y la tipo 2 que se presenta en varones después de la cuarta década con enfermedad cardiaca y renal. De acuerdo a las características clínicas que se presentan en la historia del caso de autopsia muy probablemente se trata del tipo 1, con nula o muy poca actividad aGal. Arisbeth Villanueva Pérez TITULO: Displasia Renal Bilateral Multiquística: Reporte de Caso INTRODUCCIÓN: La displasia renal multiquística aparece tras un desarrollo anormal o maduración inadecuada. Se cree que no es una patología hereditaria pero se ha encontrado relacionada con antecedentes heredo familiares. La displasia renal multiquística representa la patología del tracto urinario congénita mas frecuente y por lo general se presenta como parte del Síndrome de Potter. OBJETIVOS: Reporte de caso MATERIAL: Paciente masculino de 31.6 semanas de gestación el cual ingresa para estudio postmortem con antecedente de enfermedad renal poliquística bilateral diagnosticada a las 27 semanas de gestación por ultrasonido obstétrico. Nace vivo permaneciendo en anuria e insuficiencia renal secundaria. Se obtiene bloque renal con presencia de múltiples quistes de contenido seroso y paredes lisas con escaso parénquima renal residual. Ureteros no permeables. RESULTADOS: Se concluye como enfermedad principal una displasia renal bilateral multiquística con uropatía obstructiva. CONCLUSIONES: Los casos de displasia renal multiquística bilateral progresan a muerte fetal secundario a oligohidramnios y se acompañan de malformaciones en el tracto urinario. En nuestro caso, se realiza diagnostico prenatal de enfermedad multiquística bilateral con confirmación histológica postmortem de displasia renal multiquistica bilateral sin encontrar componente metaplasico cartilaginoso y con uropatía obstructiva en ambos ureteres. INSTITUCIÓN: Anatomía Patológica de CMNO IMSS ESTADO: JALISCO INSTITUCIÓN: Servicio y Departamento Universitario de Anatomía Patológica, Hospital General de México y Universidad Nacional Autónoma de México ESTADO: DISTRITO FEDERAL PATOLOGÍA INFECCIOSA Mampara: 9 B Mampara: 10 A Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. TC-193 TC-182 AUTORES: Dra. Ana Karen Soto Sanudo, Dra. Martha AUTORES: Dr. Jacobo Ivan Guerrero, Dr. Cuauhtemoc 2do Congreso Anual de Patólogos Mexicanos • 108 León, Guanajuato • 2016 Oros Ovalle. MATERIAL: Se presenta a consulta en el Hospital Veterinario de la FMVZ-UNAM, un canino, hembra, mestiza de 9 TITULO: TRIQUINOSIS. PRESENTACION DE años de edad, no castrada con historia clínica de epistaxis, anosmia, cambios de comportamiento e hiporexia. Se deUN CASO AUTOPSICO. INTRODUCCIÓN: La triquinosis es una enfermedad infectocontagiosa poco frecuente en nuestros dias y en medio urbano que todo patologo debe sospechar con hallazgos como los que se muestran. OBJETIVOS: Identificar el parasito con tinciones convencionales. MATERIAL: Tejido Obtenido Por Autopsia, Fijado Con Formaldehido Al 10%, Embebido Con Parafina, Cortado Y Teñido Con Tinciones Convencionales. RESULTADOS: Triquinosis. CONCLUSIONES: Se Diagnostico Triquinosis Sistemica. INSTITUCIÓN: HOSPITAL CENTRAL “DR IGNASIO MORONES PRIETO” SAN LUIS POTOSI, SAN LUIS POTOSI. ESTADO: TAMAULIPAS PATOLOGÍA VETERINARIA Mampara: 16 A Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. TC-35 cide realizar rinoscopía y lavado nasal en donde se tomó muestra para su estudio citológico e histopatológico. La paciente no presenta mejoría y se decide realizar eutanasia, el cadáver es enviado para su estudio post mortem. RESULTADOS: En la citología se observó un fondo de aspecto proteináceo con numerosas células redondas sueltas, de bordes celulares pacialmente definidos, escaso citoplasma, núcleo redondo central de cromatina fina. En los cortes histológicos se apreció un tejido de nueva formación compuesto por numerosas células poliédricas, que se disponen formando rosetas y pseudorosetas delimitadas por fino estroma fibrovascular, dichas células son de bordes celulares bien definidos, escaso citoplasma, núcleo redondo central de cromatina fina con un nucléolo evidente, se aprecia moderada anisocitosis, anisocariosis y pleomorfismo celular, se realiza inmunohistoquímica en donde fue positivo para Proteína S-100, Sinaptofisina, Enolasa Neurona Específica, GFAP, Cromogranina y negativo para Vimentina y a VIP. Dando como diagnóstico un neuroblastoma olfatorio. CONCLUSIONES: Las neoplasias nasales y de los senos paranasales se presentan entre el 52% de todos los tumores del tracto respiratorio canino. Los tumores nasales suelen aparecer en la cavidad nasal con una extensión secundaria hacia los senos frontales y otros a los senos paranasales. La mayoría de los tumores nasales son de comportamiento maligno, localmente infiltrativos y poco frecuente que cursen con metástasis. La edad de presentación en el perro varía de los 2 a los 16 años, con una edad promedio de 10 años. En un estudio reciente, la raza no era un factor predictivo. INSTITUCIÓN: Departamento de Patología, FMVZ-UNAM. Veterinary Medicine College, Auburn University, Alabama, EUA. AUTORES: DR. JULIO CESAR OSORIO, Dra. Gisela ESTADO: HIDALGO TITULO: Neuroblastoma olfatorio en un canino, reporte de un caso Mampara: 17 A Martínez Romero, MVZ.Esp. Luis Henrique García Rodríguez INTRODUCCIÓN: El neuroblastoma olfatorio es una neoplasia poco común en animales, derivada de neuroblastos de las ramificaciones nasales del sistema sensorial olfatorio cuyo asiento es el epitelio respiratorio neuroectodérmico, En el presente trabajo se describe a un canino, hembra, de 9 años que fue diagnosticada por citología, biopsia y necropsia con neuroblastoma olfatorio. OBJETIVOS: Describir las lesiones micro y macroscópicas de un caso de neuroblastoma olfatorio en un canino. Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. TC-47 AUTORES: Rodríguez-Martínez HA, Rodríguez-Reyes AA, García-Sánchez GA, Ríos y Valles-Valles D, Maldonado-Hernández G, Hernández-Ayuso I, Pérez-Olvera O, Hernández R, Medina-Cruz A TITULO:Macroadenoma hipofisiario no funcionante ectó- 2do Congreso Anual de Patólogos Mexicanos • 109 León, Guanajuato • 2016 pico en el reborde orbitario inferoexterno en una perra poodle INTRODUCCIÓN: Panhipopituitarismo e hiperpitutarismo pueden ocurrir en animales. La causa principal del hiperpituitarismo son los adenomas funcionantes secretores de ACTH que producen síndrome de Cushing (SC). Se presenta un macroadenoma hipofisiario no funcionante en una perra poodle de 10 años, el cual se manifestó como una masa en el reborde orbitario inferoexterno. MATERIAL: Clínicamente, durante 10 meses se desa- rrolló una masa sobre el reborde orbitario inferoexterno del OS; el ultrasonido confirmó que había un quiste o un tumor quístico en el mismo sitio. Se extirpó quirúrgicamente. El tumor era semiovoide, sólido, de consistencia media, de color grisáceo, café y amarillento, medía 25X18X14 mm. Se practicaron tinciones de HE, PAS c/s diastasa, hierro coloidal, tricrómico de Masson, methenamina de plata y Grimelius. Se prepararon inmunotinciones contra HACT, HC, Prol, HET, HEF y HL; así como contra marcadores paneuroendócrinos, vimentina, citoqueratinas, CD31 y CD34 RESULTADOS: Septos conectivo-vasculares dividían al tumor en lóbulos. Las células eran poliédricas y rectangulares, estaban dotadas de núcleos redondos, sin atipias ni actividad mitótica. Las células se adosaban ordenadamente a la superficie exterior de numerosos vasos delgados, adoptando un patrón endócrino. Los citoplasmas eran acidófilos y claros, alojaban a múltiples gránulos de glucógeno y a otros que se hacían aparentes con tricrómico de Masson y methenamina de plata. Las inmunoreacciones para hormonas, paneuroendócrinos y citoqueratinas resultaron negativas, dos veces. La vimentina, CD31 y CD34 solamente sirvieron para hacer más aparente la vascularización. CONCLUSIONES: Se han sido descrito adenomas hipofisiarios en perros, caballos, gatos, pericos, ratas y ratones. Entre 65 y 85% se manifiestan por pd-pu y/o SC, o sea por polifagia, aumento de peso, abdomen abalonado, hepatomegalia, pioderma recurrente, adelgazamiento de la piel y alopecia. En los seres humanos, se presentan tanto ectopias como duplicaciones de la hipófisis. Las ectopias y duplicaciones más frecuentes ocurren sobre los restos de la bolsa de Rathke y asociadas a malformaciones congénitas graves del tallo hipotálamo-hipofisiario y craneofaciales. Las ectopias pueden asociarse a una silla turca vacía. En animales ambas malformaciones son muy raras. El caso que aquí se presenta, probablemente corresponde a una ectopia, en un sitio no informado hasta ahora, y una duplicación de la glándula hipófisis. REFERENCIAS: Kollias SS, Ball WS, Prenger EC. Review of the embryologic development of the pituitary gland and report of a case of hypophyseal duplication detected by MRI. Neuroradiology 1995;37:3-12 INSTITUCIÓN: Laboratorio de Investigaciones Anatomopatológicas “Roberto Ruiz Obregón”, Unidad de Investigación en Medicina Experimental, Fac de Med, UNAM y Hosp Gral de Méx. Patolo- gía Oftálmica, Asoc para Evitar la Ceguera en Méx, IAP, Hospital “Dr. Luis Sánchez Bulnes”. Bronson Animal Disease Diagnostic Laboratory, Kissimmee, Fl, USA ESTADO: ESTADO DE MÉXICO Mampara: 18 A Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. TC-137 AUTORES: KARINA FABIOLA MARTINEZ, MVZ. Sergio Elguera. Dra. Elizabeth fuentes luiton TITULO: Tumor mixto maligno de glandula mamaria en canino de 11 años. Informe de un caso y revision de la literatura. INTRODUCCIÓN: El tumor mixto benigno mamario es de los mas comunes en caninos femeninos no asi su contraparte maligna, estos casos se asocian a lesiones de larga evolucion asi como a alteraciones moleculares como perdida de p63, cadherina y cateninas. estas lesiones representan tambien un mal pronostico y poca sobrevida. OBJETIVOS: nuestro objetivo es la presentacion de caso clinico veterinario asi como una revision de la literatura para tener informacion de datos estadisticos e histopatologicos ya que es muy similar el estudio de la glandula mamaria canina y humana. MATERIAL: Informe de caso: se trata de canino femenino de 11 años que inicia padecimiento de 7 meses de evolucion con formacion de tumor en region mamaria toracica, misma que es extraida quirurgicamente y posteriormente con mala evolucion y crecimiento de numerosos nodulos mamarios. se realizo mastectomia radical encontrandose tumor mixto maligno con componente mixto, epitelial y mesenquimatoso de tipo osteoide y cartilaginoso.con grado histologico II. RESULTADOS: Revision de la literatura: hasta 2014 se realizo una revision de casos malignos caninos que fueron divididos en seis grupos, 1) hiperplasia 2)adenocarcinoma 3)carcinoma complejo 4) carcinoma simple 5)tumor mixto benigno y 6) tumor mixto maligno, de los 15 casos encontrados y revisados 3 de ellos fueron tumores mixtos malignos (20%), uno de localizacion de mama inguinal y dos de ellos toracica. de los tres casos, dos de ellos tenian un grado histologico II y uno de ellos grado histologico III. en los tres casos revisados el componente mesenquimatoso correspondio a fibrosarcoma. CONCLUSIONES: El estudio histopatologico de la glan- 2do Congreso Anual de Patólogos Mexicanos • 110 León, Guanajuato • 2016 dula mamaria canina es muy similar al de la glandula mamaria humana. en caninos el tumor maligno mas frecuente es el adenocarcinoma, la localizacion mas frecuente es en el area inguinal canina y el grado histologico II fue el mayor observado, la lesion es mas frecuente en un rango de edad de 6 a 14 años y su pronostico es malo. en nuestro caso varió la localizacion no asi el grado histologico sin embargo los componentes osteoide y cartilaginoso maligno son muy raros en las descripciones de la literatura ya que el componente fibrosarcoma es el mas frecuente. INSTITUCIÓN: Fundacion Mexicana para la Planeacion Familiar A.C, posterior a la resección quirúrgica de la neoplasia y de la realización del transplante corneal se volvió a evaluar al paciente encontrando que A los 7 días, había desaparecido el blefaroespasmo y la epífora, aunque se mantenía la hiperemia conjuntival, el edema corneal se localizaba especialmente en la zona del trasplante .El rango de presiones fue 7-20 mmHg, el reflejo de amenaza +. A los 20 días la córnea en la periferia presentaba edema, existía vascularización superficial corneal, el rango de presiones fue de 9-20 mmHg. A los 45 días se observó solamente edema en la zona transplantada en todos los perro así como fluoresceína -, presentaban vasos superficiales corneales y el reflejo de amenaza +. El rango de presiones osciló entre 9-18 mmHg. ESTADO: DISTRITO FEDERAL CONCLUSIONES: Los resultados obtenidos permiten Mampara: 19 A Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. considerar al transplante corneal y mandil conjuntival como una técnica segura y satisfactoria, para el tratamiento de melanoma limbal, ya que aporta buenos resultados para preservar las estructuras oculares y la visión. INSTITUCIÓN: OFTALVET,Asociacion para evitar la ceguera en mexico APEC ESTADO: DISTRITO FEDERAL TC-189 AUTORES: DRA. ROMINA HERNANDEZ, Dr.Gustavo Adolfo García, Dr. Abelardo Rodriguez, Dra Mayela Carranza PATOLOGIA QUIRÚRGICA DIVERSA TITULO: Transplante corneal y mandil conjuntival como tratamiento para melanoma limbal Mampara: 1 A primario en un perro INTRODUCCIÓN: Las neoplasias melanociticas representan el 3% del total de neoplasias y más del 7% de los tumores malignos en el canino. Dentro de las neoplasias primarias el melanoma ocular representa la neoplasia primaria más frecuente en el perro, sin embargo no hay estudios que correlacionen la aparición clínica e histopatológica , con los resultados del tratamiento. OBJETIVOS: El propósito de este informe es comenzar un estudio preliminar para correlacionar los hallazgos histopatológicos con el comportamiento biológico tumoral en respuesta al transplante corneal y mandil conjuntival como alternativa de tratamiento MATERIAL: El paciente fue derivado a Oftalmología, donde se le realizó un examen oftalmológico completo, así mismo se hizo la resección quirúrgica de la neoplasia reformando la superficie corneal afectada con transplante corneal parcial y mandil conjuntival. El estudio histopatológico de la lesión del ojo derecho demostró la presencia de melanocitos hipercromáticos suprabasales con infiltración a esclera. Posterior a la cirugía se realizaron evaluaciones RESULTADOS: En la primera evaluación oftalmológica se observó blefaroespasmo, epifora e hiperemia conuntival, Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. TC-58 AUTORES: Dr. César Iván Peña Ruelas, Dr. Edmundo Erbey Castelan Maldonado, Dr. Julio César Sánchez Venegas, Dra. Julia O. De Leija Portilla, Dra. Sarai Páez Moreno Dra. Elsa Edith de la Rosa Vélez, Dr. Jorge Arturo Sánchez Garza. TITULO: Mielolipoma suprarrenal INTRODUCCIÓN: El mielolipoma adrenal es un tumor benigno no funcionante que representa de 3 a 5% de los tumores adrenales.(1) La primera descripción fue realizada por Gierke en 1905 y fue denominado mielolipoma por Oberling en 1929.(2) OBJETIVOS: Presentar las características clínicas, imagenológicas y patológicas de un caso de mielolipoma adrenal, así como la estadística de los casos diagnosticados entre 2010 y 2015 en nuestro hospital. MATERIAL: Material y Metodos: Caso Clínico: Mujer de 2do Congreso Anual de Patólogos Mexicanos • 111 León, Guanajuato • 2016 47 años a quien en 2011 se detectó en forma incidental en estudios de imagen un tumor en glándula suprarrenal izquierda de 6 cm, redondo, con densidad grasa, se optó por una conducta expectante. En el 2015 la paciente presentó dolor a nivel de fosa renal izquierda; se realizó TC que mostró un tumor de 10 cm, mayormente isodenso, así como un hematoma perirrenal ipsilateral. En Anatomía Patológica se recibieron múltiples fragmentos que en conjunto midieron 15 x 15 cm, de color amarillo y aspecto adiposo. El estudio histológico mostró que el tumor estaba constituido por tejido adiposo maduro y elementos hematopoyéticos, identificándose remanente adrenal. RIOR CON DIFERENCIACIÓN MÜLLERIANA (QUISTE DE HATTORI). RESULTADOS: En nuestra unidad de 2010 a 2015 se MATERIAL: Se revisa con hematoxilina y eosina mos- diagnosticaron 11 casos de mielolipoma adrenal, el 81.8% correspondió a mujeres, lo cual representa una razón 4.5 mujeres por cada hombre. El rango de edad fue de 39 a 56 años, con una mediana de 50 años. El 66.6% de los casos afectó al lado derecho. El tamaño vario de 5 a 15 cm, con una media de 7.9 cm. trando características particulares de quiste con revestimiento de tipo cilíndrico alto ciliado a cubico bajo, con pared delgada y sin algún otro componente histológico (glándulas, cartílago, etc). Se realiza tinciones especiales de inmunohistoquímica para corroborar su estirpe histológica. INTRODUCCIÓN: Múltiples lesiones quísticas en mediastino han sido reportadas en la literatura con un rango amplio de etiologías. En particular son un reto diagnóstico dependiendo su localización y su histogénesis. OBJETIVOS: Presentar el caso de esta entidad rara de localización mediastinal posterior, en una mujer de 49 años, asintomática, como hallazgo incidental. RESULTADOS: Se revisó la inmunohistoquímica resulCONCLUSIONES: La teoría más aceptada de la etiología de los mielolipomas es que son una consecuencia de la metaplasia de las células adrenocorticales, en respuesta a estímulos como estrés, infección o necrosis.(2) La localización más frecuente del mielolipoma es la glándula suprarrenal pero se han descrito casos extra-adrenales en pulmón, mediastino, hígado, estómago, leptomeninges y región presacra.(3) Se recomienda resección quirúrgica en los casos sintomáticos o en tumores con tamaño mayor a 6 cm.(4) REFERENCIAS: 1. Joy PS, Marak CP, Nashed NS, et al. Giant adrenal myelolipoma masquerading as heart failure. Case Rep Oncol 2014;7:182-187. 2. Martínez-Cornelio A, Hernández-Toriz N, Alvarado-Cabrero P, et al. Mielolipoma suprarrenal, revisión de casos en México. Rev Mex Urol 2008;68(6):334-340. 3. Olivar-Roldan J, Molina-Baena B, Pavón-de Paz I, y cols. Mielolipoma adrenal gigante: revisión a propósito de un caso. 4. Gershuni VM, Bittner JG, Moley JF, et al. Adrenal myelolipoma: operative indications and outcomes. J Laparoendosc Adv Sug Tech A 2014;24(1):8-12. INSTITUCIÓN: UMAE Hospital de Especialidades No. 25 del IMSS. Delegación Nuevo León. tando positivo citoplasmático para CK AE1/AE3 y CK7, positivo nuclear difuso e intenso para WT-1 y para RE, así como RP positivo nuclear ++/+++. Negativo para CK 5/6, Calretinina, TTF-1 y Napsina A. Por debajo del epitelio de revestimiento presenta una capa de músculo liso que se corrobora con actina de músculo liso. Con esta inmunohistoquímica se concluye que este quiste presenta diferenciación mülleriana con RE, RP y WT-1 positivos, descartando quistes de origen mesotelial o broncogénicos los cuales son sus principales diagnósticos diferenciales. El quiste de Hattori fue descrito recientemente (2005) y hasta la fecha han sido reportados solo 16 casos. CONCLUSIONES: Es una entidad subdiagnosticada por lo que es conveniente su conocimiento general para realizar con precisión la misma. Es interesante ya que este quiste aparece en sitios que no corresponden con tracto urogenital femenino, entre ellos mediastino posterior. Está abriéndose paso a próximas investigaciones embriológicas al respecto de estos remanentes müllerianos ya que su histogénesis no es definitiva. INSTITUCIÓN: Instituto Mexicano del Seguro Social. UMAE Hospital de Cardiología CMN Siglo XXI. ESTADO: NUEVO LEÓN ESTADO: DISTRITO FEDERAL Mampara: 1 B Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. Mampara: 2 A TC-68 Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. AUTORES: Dra. Margarita Carolina Moreno Gómez; Dra. Gemali Soto Vidal TC-113 TITULO: QUISTE DE MEDIASTINO POSTE- AUTORES: DRA. BÁRBARA SÁENZ, Dra. Hersilia Aide Henandez Zamonsett , Dra. Juana Elizabeth Tadeo Gonzá- 2do Congreso Anual de Patólogos Mexicanos • 112 León, Guanajuato • 2016 lez, Dr. Luis Angel Ceceñas Falcón, Dra. Med. Oralia Barbosa Quintana TITULO: Mielolipoma: Reporte de un caso y revisión de la literatura INTRODUCCIÓN: El mielolipoma de la glándula suprarrenal es un tumor benigno, infrecuente, no funcionante, caracterizado por la presencia de tejido adiposo y elementos de la médula ósea. Habitualmente es descubierto de manera incidental. La mayoría de ellos son asintomáticos. TITULO: Tumor carcinoide atípico tímico productor de ACTH asociado a síndrome de Cushing de tipo cíclico. INTRODUCCIÓN: El síndrome de Cushing secundario a secreción ectópica de ACTH se ha descrito en asociación a tumores neuroendocrinos en 10-15% (1). El síndrome de Cushing de tipo cíclico, es una variante caracterizada por una fluctuación rítmica en la secreción de ACTH; ocasionando cuadros clínicos cortos e intermitentes de hipercortisolismo. (359) OBJETIVOS: Presentar el caso de un paciente de 51 OBJETIVOS: Analizar las características clinicopatológiaños con un mielolipoma localizado en la glándula adrenal izquierda. cas de un hombre de 24 años con tumor carcinoide atípico de timo asociado a síndrome de Cushing de tipo cíclico. MATERIAL: Presentamos el caso de un paciente de 51 MATERIAL: Se revisó el expediente clínico, laminillas te- años de sexo femenino, se obtuvo su historial clínico, sus estudios radiológicos y de gabinete para complementar el hallazgo patológico. RESULTADOS: El paciente femenino con historial de hipertensión controlada, presenta síntomas de diarrea y dolor abdominal y al realizarle un ultrasonido corroborado por un TAC, se encontró una tumoración retroperitoneal de contenido graso en topografía de la glándula suprarrenal. Posteriormente, se recibió la resección de la glándula suprarrenal izquierda con un tumor de 965 gramos y de 18 cm de diámetro mayor con una histología constituida mayoritariamente por tejido adiposo y celularidad hematopoyética, haciendo el diagnóstico de un mielolipoma de 18 cm. ñidas con H-E y marcadores de inmunohistoquímica (IHQ). RESULTADOS: Hombre de 24 años inicio su padeci- miento desde la niñez con obesidad centrípeta, facies de luna llena; se manifestaron en forma intermitente con periodos de remisión y reactivación. Ingreso con hipertensión arterial e hipocalemia refractaria a tratamiento. Una tomografia de tórax confirmó la presencia de un tumor en mediastino anterosuperior de 15x18 cm, con bordes bien definidos y que desplazaba la aorta y arterias pulmonares izquierdas. Se realizó una biopsia del tumor, que mostró un tumor carcinoide atípico con necrosis; por IHQ positivo para cromogranina, sinaptofisina, CD56, citoqueratina y antígeno de membrana epitelial. Anti ACTH resulto positivo con marca citoplasmática. CONCLUSIONES: El mielolipoma es un tumor infrecuente constituido por elementos hematopoyéticos en diferentes estadios madurativos y sin alteraciones histológicas, combinados con tejido adiposo maduro. La mayoría son hallazgos incidentales durante exploraciones radiológicas complementarias. El tratamiento quirúrgico está indicada cuando la lesión sea mayor de 6 cm, funcionante y/o sintomática. INSTITUCIÓN: Hospital Universitario “José Eleuterio González” CONCLUSIONES: Los carcinoides atípicos del timo son tumores poco frecuentes. Su asociación con síndrome de Cushing de tipo ciclico con producción de ACTH es aún más rara. Por lo que es importante reconocer esta variante y considerarla dentro de los diagnósticos diferenciales de síndrome de Cushing. Este caso ejemplifica como un tumor carcinoide productor de ACTH ectópico puede mostrar secreción de la hormona durante un periodo de tiempo prolongado de manera intermitente. ESTADO: NUEVO LEÓN INSTITUCIÓN: 1. Hospital de Oncología, Centro Médico Nacional Siglo XXI, I.M.S.S. 2. Facultad de Medicina, Universidad Nacional Autónoma de México. Mampara: 2 B ESTADO: DISTRITO FEDERAL Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. TC-135 AUTORES: Ana Cristina Torres Pérez (2), Erick Antonio Osorio López (2). Irving Daniel Ortiz Sanchez(2), Fernando Candanedo González (1). Mampara: 3 A Fecha y hora Instalación: 4 de mayo 13:00 hrs. Fecha y hora retiro: 4 de mayo 19:00 hrs. TC-178 2do Congreso Anual de Patólogos Mexicanos • 113 León, Guanajuato • 2016 AUTORES: Dra. Ana Karen Soto Sanudo, Dr. Gerónimo Martin Tavares Macías TITULO: PARAGANGLIOMAS UNA SERIE DE 92 CASOS ESTUDIO RETROSPECTIVO 20002015 SERVICIO DE ANATOMIA PATOLOGICA UMAE, HE, CMNO IMSS INTRODUCCIÓN: Los paraganglios son un sistema especializado de celulas de la cresta neural asociados a ganglio autonómico con funciones especializadas. Los cuerpos carotideo y aórtico son quimiorreceptores especializados en detectar cambios del Ph y presión de oxigeno, son cromafin negativos, se arreglan en pequeños grupos pequeños (zellballen) y sus tumores son usualmente no funcionales. OBJETIVOS: Presentar los hallazgos epidemiológicos e histopatológicos de una serie de paragangliomas vistos en UMAE, HE, CMNO, IMSS MATERIAL: Se revisaron en forma retrospectiva los archivos de Anatomía Patológica durante el periodo de enero del 2000 a diciembre de 2015 en busca de casos diagnosticados como paragangliomas convencionales, variantes, atípicos y malignos, incluyendo su edad, sexo, localización, hallazgos como: diametro, invasión capsular, invasión a tejido blandos, permeacion vascular, necrosis, hemorragia,etc. RESULTADOS: Durante el periodo del 2000 al 2015 se realizaron aproximadamente 272, 000 estudios histopatológicos de los cuales 92 casos correspondieron a Paragangliomas es decir 0.0.3 %. La edad fue en 70 casos: 2 casos < 18 a. 47 casos (67.1%) 18 a 60 a. 22 casos > 60 a. El sexo fue: 72 femeninos (78.3%), 20 masculinos (21.7%). La localizaciones mas frecuente fue: Cuerpo Carotideo 70 casos (76%), Yugulo Timpánicos 7 casos (7.6%) y Retroperitoneales 5 casos (5.4 %). El tipo histológico mas frecuente fue: Convencionales 88 casos (95.6%), Atípicos 2 casos (2.2%), Fusocelulares 1 caso (1%) y Oncocitico 1caso (1%). No hubo casos malignos. CONCLUSIONES: Nuestros hallazgos epidemiológicos y histopatológicos encontrados en nuestra serie de 92 casos son similares a los reportados en la literatura, excepto en lo referente a la frecuencia de casos con histología maligna, ya que no encontramos ningún caso en nuestra serie. INSTITUCIÓN: Centro Medico Nacional de Occidente Instituto Mexicano del Seguro Social ESTADO: JALISCO -- El autor(es) de los resúmenes tiene a su cargo la responsabilidad legal del contenido del material enviado; el comité organizador del congreso, la AMP, A.C. y la FEDPATMEX, A.C. se deslindan de cualquier responsabilidad al respecto. -- La información para la elaboración de constancias y publicación de los trabajos aceptados se obtuvo de los datos capturados por el autor(es) en el sistema -redacción, edición, ortografía, etc.- 2do Congreso Anual de Patólogos Mexicanos • 114 León, Guanajuato • 2016