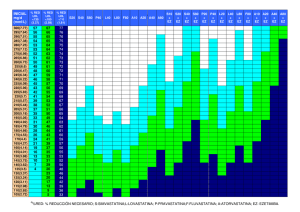
universidad autónoma metropolitana unidad iztapalapa ciencias
Anuncio

UNIVERSIDAD AUTÓNOMA METROPOLITANA UNIDAD IZTAPALAPA CIENCIAS BIOLÓGICAS Y DE LA SALUD LICENCIATURA EN BIOLOGÍA EXPERIMENTAL ANALISIS MOLECULAR DEL RECEPTOR DE LA LIPOPROTEINA DE BAJA DENSIDAD EN PACIENTES CON DIABETES TIPO 2 MARTHA ELVIRA MEJIA GARCIA ASESORAS: DRA. SOCORRO DURAN VARGAS M. EN ISP. GLORIA RUIZ GUZMAN MEXICO,D.F. DICIEMBRE DE 2003 AGRADECIMIENTOS v Gracias a Dios por ser bueno conmigo. v A mis Padres Luis y Josefina por todo el apoyo que día a día me brindaron para poder concluir la carrera. Los quiero. v A mis hermanos Gaby y Dany. A mi tía Sofy. Y a quien me motiva a seguir adelante, la razón de mi vida mi niña Luisita TE AMO. v A la Doctora Socorro Durán, por la paciencia que me tuvo y por toda la dedicación que me dio. No pude haber estado en mejor laboratorio Gracias. v A la Maestra Gloria Ruiz, le agradezco todas su atenciones para conmigo, gracias por ese apoyo que fue tan esencial en el desarrollo de este proyecto. v A todos mi compañeros del Laboratorio Giselita, Osvaldo, Angy, Víctor y Alejandro. Por sacarme de dudas y ayudarme siempre. v A mejor maestra de Bioquímica que he conocido Martita Ruiz. v A todas las personas que han sido de gran apoyo en mi vida, especialmente en los momentos difíciles. A Gaby y Eladio gracias por estar siempre cerca. A mi amiga Marisol eres lo máximo. A la maestra Rocío Barcena a quien debo el amor a la biología. Gracias. INDICE GENERAL Pagina INTRODUCCIÓN. 1. Lípidos. 1 2. Transporte de lípidos. 2 3. Metabolismo de Lipoproteínas. 6 4. Dislipidemias. 8 4.1. Hipercolesterolemia Familiar. 9 4.1.1. Vía del receptor LDL. 11 4.1.2. Genética molecular del receptor LDL. 12 4.2. Diabetes tipo 2. 4.2.1. Lipoproteínas en DT 2. 5. Aterogénesis en HF y DT 2. 15 16 18 JUSTIFICACIÓN. 20 OBEJTIVO GENERAL. 21 Objetivos específicos. 21 MATERIAL Y MÉTODO. 22 RESULTADOS Y DISCUSIÓN. 29 Exones 3, 4, y 16. 29 Exón 17. 30 CONCLUSIONES. 35 PERSPECTIVAS. 36 REFERENCIAS. 37 Indice de figuras. Paginas Figura A. Estructura de triglicéridos. 1 Figura B. Colesterol. 2 Figura C. Lipoproteína de baja densidad (LDL). 4 Figura D. Grafica de composición en porcentaje de las lipoproteínas. 5 Figura E. Vía endógena y exógena del metabolismo de lípidos. 7 Figura F. Dominio que componen el receptor LDL. 10 Figura G. Gen del receptor LDL. 10 Figura H. Vía del receptor LDL. 11 Figura I. Clasificación de las mutaciones en el gen del receptor LDL. 14 Figura J. Formación de células espumosas. 19 Figura K Programa de termocilcador. 24 Figura L. Gel de agarosa al 1.8% con producto de PCR. 29 Figura M. SSCP del exón 17. 30 Figura N. Histograma de secuencia sentido del exón 17 del paciente 105. 32 Indice de tablas. Paginas. Tabla 1. Características de las lipoproteínas en cuanto a densidad, 5 diámetro, composición y apo asociadas. Tabla 2. Características comunes entre la HF y DT 2. 20 Tabla 3. Oligonucleótidos sentido y antisentido, utilizados para la amplifcación 22 en los exones 3, 4, 16 y 17. Tabla 4. Temperaturas de alineación. 25 Tabla 5. Reactivos para preparar un gel de poliacrilamida al 6 %. 26 Tabla 6. Mutaciones silenciosas. 31 Tabla 7. Mutaciones con sentido erróneo. 33 INTRODUCCIÓN. 1. Lípidos. Los lípidos son biomoléculas orgánicas formadas básicamente por carbono hidrogeno y oxigeno, pero en porcentajes mucho más bajos. Además, los ácidos grasos se dividen en saturados, los que tienen enlaces simples entre los átomos de carbono; y los insaturados que se los que contienen uno o varios dobles enlaces a lo largo de su cadena. Los lípidos más sencillos obtenidos a partir de los ácidos grasos son los triacilgliceroles o triglicéridos y las grasas o grasas neutras. Los triacilgliceroles están compuestos de tres ácidos grasos en enlace éster con un solo glicerol1 (Fig. A). Figura. A. Estructura de Triglicéridos. Los esteroles son lípidos estructurales que se hallan presentes en la mayoría de células eucarióticas. Su estructura característica es la del núcleo esteroide que consiste en cuatro anillos fusionados, tres de ellos con seis carbonos y uno con cinco. Los esteroles son los precursores de diversos productos con actividades biológicas específicas; como los ácidos biliares, actúan como detergentes en el intestino, emulsionando las grasas de la dieta para hacerlas mas accesibles a las lipasas 1 digestivas. El colesterol es el principal esterol en los tejidos animales, es anfipático (Fig. B). Diversas hormonas esteroides también se producen a partir del colesterol1. Fig. B. Colesterol. 2. Transporte de lípidos. El transporte de triacilgliceroles y colesterol esta a cargo de estructuras especializadas llamadas lipoproteínas, que son agregados moleculares de proteínas transportadoras específicas denominadas apolipoproteínas (apo) con diversas combinaciones de fosfolípidos, colesterol, ésteres de colesterol y triacilgliceroles. Las apo se combinan con agregados esféricos con lípidos hidrofóbicos en el núcleo central y las cadenas laterales hidrofílicas de aminoácidos de la proteína en la superficie. Combinaciones variables de lípido y proteína producen partículas de densidades diferentes que van desde lipoproteínas de muy baja densidad a lipoproteínas de alta densidad, que se pueden separar por centrifugación y visualizar al microscopio electrónico2. 2 Las lipoproteínas son un grupo heterogéneo de partículas con una composición diferente de lípidos y proteínas, además de tamaño, existen cuatro diferentes lipoproteínas, que se describen a continuación (ver tabla 1): QUILOMICRONES. Son las lipoproteínas más grandes en tamaño y de menor densidad, y en su composición tienen una gran cantidad de triacilgliceroles (ver figura D). Se sintetizan en el retículo endoplasmatico de las células epiteliales que forran al intestino delgado y se trasladan a través del sistema linfático entrando en el torrente circulatorio a través de la vena subclavia izquierda. Se encargan del transporte de ácidos grasos obtenidos en la dieta al tejido en el que han de ser consumidos o almacenados como combustible3. LIPOPROTEÍNA DE MUY BAJA DENSIDAD (VLDL). Los ácidos grasos ingeridos en la dieta y que se necesitan inmediatamente como combustibles, son convertidos en triacilgliceroles en el hígado y se incorporan en apolipoproteínas específicas formando lipoproteínas de muy baja densidad (ver figura D). Estas lipoproteínas se transportan por la sangre desde el hígado hasta el músculo y el tejido adiposo en donde la activación de la lipoproteína lipasa por la apoC-II libera ácidos de los triglicéridos de la VLDL3. LIPOPROTEÍNA DE BAJA DENSIDAD (LDL). La pérdida de triacilgliceroles convierte a la VLDL en lipoproteína de baja densidad (ver figura D). Estas moléculas son ricas en colesterol y presentan apoB-100 como única apolipoproteína (ver figura C). Las LDL transportan colesterol a los tejidos periféricos con receptores específicos de superficie que reconocen a la apoB-1003. 3 Figura C Lipoproteína de baja densidad (LDL). LIPOPROTEÍNA DE ALTA DENSIDAD (HDL). Estas se forman en hígado e intestino delgado son partículas ricas en proteína que contiene relativamente poca cantidad de colesterol y nada de sus ésteres (ver figura D). Las HDL contiene apoC-I y apoC-II entre otras apo, la enzima lecitina-colesterol acil transferasa (LCAT), que cataliza la formación de ésteres de colesterol a partir de lecitina y colesterol. La LCAT en la superficie de las partículas nacientes de HDL convierte esta fosfatidilcolina y colesterol así como los restos de quilomicrones y VLDL en ésteres de colesterol, que empiezan a formar un núcleo, transformando la HDL naciente que tiene forma de disco en una partícula esférica de HDL madura. Esta lipoproteína rica en colesterol retorna ahora al hígado en donde se descarga el colesterol. Parte de este colesterol se convierte en sales biliares3. 4 Tabla 1. características de las lipoproteínas en cuanto a densidad, diámetro, composición y apo asociada2. Lipoproteína Rango de Diámetro densidad (nm) Lípidos Apolipoproteínas Triacilgliceroles de B48, AI, AII, CI, la dieta diaria CII,CIII, E. Triacilgliceroles B100, CI, CII, CIII, endógenos (hígado) E. Colesterol y B100. (g/ml) Quilomicrones VLDL <0.950 0.950- 80-1000 30-80 1.006 LDL 1.019- 20-22 1.063 colesterol esterificado HDL 1.0631.090 9-15 Colesterol AI, AII, CI, CII, CIII esterificado y E. fosfolípidos Figura D. Grafica de composición en porcentaje de las lipoproteínas. 5 3. Metabolismo de las lipoproteínas. Las diferentes lipoproteínas, participan en dos vías distintas en donde cada una de las moléculas juega un papel importante. Estas dos vías son la endógena y exógena. La vía exógena empieza en el intestino y termina en el hígado, esta vía transporta las grasas de la dieta (ver figura E). Los triacilgliceroles y el colesterol son absorbidos y re-esterificados en las células del intestino delgado, en donde son secretadas las partículas de quilomicrones, de la vía linfática a la circulación; los quilomicrones adquieren apo C II, que activa a la lipoproteín lipasa, y también facilita el paso a través de los capilares. En la circulación, los quilomicrones sufren lipólisis; pierden triacilgliceroles y adquieren esteres de colesterol de otras lipoproteínas. Interaccionan con otras partículas, y algunas apolipoproteínas más pequeñas que pasan de una partícula a otra, como la apo E la cual los convierte en remanentes de quilomicrones2,3,4. En la vía endógena los triacilgliceroles del hígado se distribuyen a otros tejidos (ver figura E). En el hígado se secretan las VLDL, las cuales contienen triacilgliceroles, colesterol esterificado, apo B100, apo E y apo C. Las LDL tienen únicamente apo B100. En el hígado apoB-100 se combina con varios lípidos para dar lugar a la VLDL, los triacilgliceroles que contiene esta lipoproteína se hidrolizan para dar lugar a la IDL. Algunos de los remanentes de VLDL, también conocidos como IDL se remueven de la circulación mediante el hígado a través de un proceso mediado por apoE, mientras que otros se hidrolizan dando lugar a la LDL. La LDL tiene la función de transportar el colesterol a diferentes tejidos periféricos, mientras que el resto de colesterol, el cual es mayoría, se remueve del torrente sanguíneo de nuevo a través del hígado. Dicha remoción se lleva a cabo cuando la única molécula de apoB-100 que contiene la LDL se une al receptor hepático de la LDL2,3,4. 6 Figura. E Vía endógena y exógena del metabolismo de lípidos. 7 4. Dislipidemias Las dislipidemias son alteraciones en los valores normales de lípidos (o grasas de la sangre), que son fundamentalmente colesterol y triglicéridos. Los defectos genéticos del metabolismo de las lipoproteínas ocasionan las siguientes enfermedades llamadas comúnmente hiperlipoproteínemias: hipocolesterolemia, hipoalfalipoproteínemia hipertrigliceridemia, hipercolesterolemia y otras a las cuales se les llama mixtas. Las hiperlipoproteinemias se pueden clasificar en 2 grupos, las primarias y las secundarias. Dentro de las primarias tenemos: (extremadamente raras), hipercolesterolemia familiar, hiperquilomicrolemias hiperlipidemia familiar combinada, disbetalipoproteinemia, hipertrigliceridemia familiar, hipercolesterolemia esporádica e hipertrigliceridemia esporádica. Mientras que las dislipidemias secundarias son aquellas alteraciones en el metabolismo de los lípidos o de las lipoproteínas que se producen como consecuencia de otra entidad patológica como las neuropatías, la diabetes mellitus, la obesidad entre otros4. En este trabajo nos enfocaremos en dos dislipidemias una primaria, la hipercolesterolemia Familiar y una secundaria que se desarrolla a consecuencia de DT 2 por tener características similares en el perfil de lípidos. 8 4.1 Hipercolesterolemia Familiar. La hipercolesterolemia Familiar es una enfermedad autosomica dominante, caracterizada por mutaciones en el receptor LDL. En pacientes heterocigotos hay disminución a la mitad de los receptores para apoB-100, mientras que para homocigotos se demuestra la ausencia total de los receptores para apoB-100. La enfermedad prevalece en homocigotos 1/1 000 000 y en heterocigotos 1/5004. La anormalidad principal de esta patología es la elevación de colesterol LDL como consecuencia de las mutaciones en el receptor LDL o en apoB-100, que lleva a un defecto de su catabolismo, ya que la interacción entre el receptor LDL y la apoB-100 hace posible que un alto porcentaje de las LDL sean eliminadas del plasma5,6. El receptor de la LDL es una glicoproteína de superficie de membrana, que es sitio de unión específico, para el paso y degradación de lipoproteínas plasmáticas que contengan apoB-100. La proteína esta constituida de 839 aminoácidos, en su paso por el aparato de golgi reduce su peso molecular de 160 a 120 KDa. El receptor LDL maduro contiene numerosos puentes disulfuro que confieren estabilidad de el ligando al sitio de unión. El receptor LDL esta constituido de 5 dominios: dominio de unión al ligando, dominio homologo al precursor de EGF (factor de crecimiento epidermal), dominio de glicosilación, dominio transmembranal y el dominio citoplasmático5,6 (ver Fig. .F). 9 Figura F. Dominios que componen al receptor LDL. El gen ldlr, se localiza en posición distal del brazo corto del cromosoma 19 (p13.1p13.3) se compone aproximadamente de 45 Kb (kilobases), consta de 18 exones y 17 intrones5,6(ver figura G). 45 kb 5 ´ Exón 3 1 2 3 4 5 6 11 12 13 14 15 Homólogo al precursor del Factor de Crecimiento Epidermal (EGF) Secuencia Señal Promoto r 7 8 9 10 Unión al Ligando RNAm = 5.3 kb 16 17 18 Transmembrana l Unión a Cadenas de Carbohidratos Citoplasmátic o Figura G. Gen del receptor LDL 10 El exón 1 codifica para una secuencia señal de 22 aminoácidos, del exón 2 al 6 codifican para el dominio de unión al ligando, los exones 7 al 14 para el precursor homologo EGF, el 15 para el dominio de unión de azucares, el exón 16 y el extremo 5’ del 17 codifican para el dominio transmembranal, y por último, el extremo 3’ del exón 17 y el extremo 5’ del exón 18 codifican para el dominio citoplasmático5,6. El resto del exón 18 se refiere a la región 3’ del ARNm de 2.6 Kb la cual no se traduce. 4.1.1. Vía del receptor LDL. La proteína precursora del receptor LDL (120 kDa) se produce en el retículo endoplásmico, para madurar pasa al complejo de Golgi después se transporta a la superficie celular para unirse a su ligando. Las partículas LDL circundante en el torrente sanguíneo contienen apoB-100 que es reconocida por las proteínas receptoras de superficie (receptor LDL), de las células que necesitan captar colesterol. La fijación de LDL a un receptor de LDL inicia la endocitosis que lleva la LDL y su receptor asociado al interior de la célula dentro de un endosoma. Este endosoma se fusiona finalmente con un lisosoma que contiene enzimas que hidrolizan los ésteres de colesterol liberando colesterol y ácidos grasos al citosol. La apoB-100 de la LDL también se degrada dando aminoácidos que se liberan al citosol, pero el receptor LDL escapa de la degradación y vuelve a la superficie celular en donde puede funcionar una vez más en la captación de LDL3 (ver figura H). 11 Figura H. Vía del receptor LDL. 4.1.2 Genética molecular del receptor LDL. Las mutaciones en el gen que codifica para el receptor LDL, da como fenotipo la Hipercolesterolemia Familiar. Actualmente se han reportado más 800 mutaciones que afectan al gen ldlr, entre ellas hay inserciones y deleciones. Se han clasificado las mutaciones del receptor LDL en 5 categorías de acuerdo a las consecuencias funcionales de a mutación (Fig. I). Clase 1. Alelos Nulos. Es una falla completa para producir receptor inmunoprecipitable. Son las mutaciones que reducen en mayor medida el número de receptores LDL funcionales5. 12 Clase 2. Alelos con defecto en el transporte. Es el tipo de mutación más común. El defecto esta en el transporte de la proteína del receptor entre el RE y el sitio en donde se efectúa la glucosilación (AG). Existen dos tipos 2A se produce una proteína que no puede ser transportad fuera del RE. En el tipo 2B una parte del receptor maduro es expresado en la superficie celular5. Clase 3. Alelo con defecto en la unión al ligando. Es un defecto de unión a la apoB de la LDL al receptor. La proteína del receptor es transportada normalmente a la superficie celular pero solo une lipoproteínas ricas en apo-E como lo es VLDL y en una cantidad disminuida y variable de LDL, esto es dependiendo de la mutación. Estos defectos se presentan en los exones 7 al 14 5. Clase 4. Alelos con defecto de internalización. Existen alteraciones de 50 aminoácidos del dominio citoplasmático. Existen 2 subtipos, el 4-A en el que la mutación se encuentra en el dominio citoplasmático de receptor y afecta las secuencias indispensables para la migración de complejo receptor-lipoproteína hacia vesículas recubiertas. En las 4-B la alteración se encuentra en los dominios del receptor que facilitan la unión a la membrana celular, como consecuenciales receptores muestran pobre adherencia a la superficie celular. Los receptores se encuentran de manera difusa en la superficie celular y no son concentrados en vesículas y por lo tanto no pueden internalizar LDL. Todas las deleciones se encuentran desde el intrón 15 a la región no codificada del exón 185. Clase5. Alelo con deficiencia en el reciclaje. Defecto en la disociación ácido-dependiente del receptor y el ligando en el endosoma, evento que es esencial en el reciclaje del receptor. El defecto se encuentra en las repeticiones idénticas al EGF5. 13 Figura I. Clasificación de las mutaciones en el gen del receptor LDL. 14 4.2 Diabetes tipo 2. Es una enfermedad crónica generalizada, con predisposición genética y participación de diversos factores ambientales que en conjunto interfieren con el metabolismo de carbohidratos y lípidos7. La enfermedad se caracteriza por hiperglucemia persistente ya sea por la deficiencia en la producción o acción de la insulina, que afecta el metabolismo de carbohidratos, proteínas y grasas. La Diabetes mellitus se ha clasificado en dos grandes grupos, donde la tipo 2 es la más frecuente con una prevalencia mayor al 90% de todos los casos reportados8. La diabetes de tipo 2 (DT 2) es una afección caracterizada por una marcada ineficiencia del cuerpo utilizando insulina. Los mecanismos por los cuales esta resistencia a la insulina se origina son muy variados. En su mayoría se deben a una combinación de factores genéticos asociados con factores de estilo de vida como obesidad y hábitos dietéticos inapropiados4. La DT 2 es un problema de salud mundial se estima en más de 150 millones el número de afectados y de estos, 19 millones viven en América Latina8. Se estima que 11.7 millones de mexicanos aproximadamente tendrán diabetes para el año 20259. Según la Encuesta Nacional de Enfermedades Crónicas (ENEC), el 10% de la población mexicana entre los 20 y 69 años padece diabetes. La prevalencia tiende a aumentar en adultos. La Diabetes es la causa más común de muerte en México, al año se registran más de 300,000 nuevos casos y la tasa de incidencia se ha estimado en 300/ 100,000 habitantes, con una mortalidad asociada a diabetes que es considerada entre las cinco más elevadas del mundo8. Se ha demostrado que adultos mexicanos que padecen DT 2 han tenido alta prevalencia de factores de riesgo que contribuyen a complicaciones macro y microvasculares9. Otros factores de riesgo son: obesidad, niveles elevados de triglicéridos, niveles bajos de colesterol HDL, colesterol LDL elevado, resistencia a la acción de la insulina y presión arterial elevada8. 15 4.2.1 Lipoproteínas en DT 2. Durante la DT2 se incrementan los niveles de triacilgliceroles, dando como resultado el aumento en plasma de lipoproteínas ricas en triacilgliceroles (TRL’s), que se asocian con el desarrollo de potencial aterogenico además de ser un factor de riesgo de enfermedades de las coronarias. Lo anterior se debe a una alteración en el metabolismo de lípidos durante la DT 2, principalmente en las VLDL LDL y HDL. Existen dos componentes cruciales en la dislipidemia diabética, que son el incremento en plasma de triglicéridos y las bajas concentraciones de colesterol HDL10. Las TRL’s se caracterizan por la diferencia en la densidad y la composición de apo, derivada de los quilomicrones del intestino y de las VLDL del hígado. En este padecimiento, las partículas VLDL endógenas se dividen en dos clases: las VLDL1 que son partículas más grandes de lo normal (Sf 60-400) y las VLDL2 (Sf 20-60)que son pequeñas y densas. El metabolismo y la síntesis de las TRL’s es regulado por dos enzimas: la lipoproteín lipasa (LPL) y la lipasa hepática (HL). La hidrólisis de los triglicéridos de las lipoproteínas esta a cargo de la LPL; en varios estudios se ha reportado que la actividad de la LPL frecuentemente se encuentra baja en pacientes con DT 2. Por otro lado, la HL es responsable de la conversión de IDL a LDL; hay evidencias que indican que la actividad de la HL comúnmente se incrementa en la DT 2, amplificando la conversión de IDL a LDL. Las VLDL1 y VLDL2 tienen diversas vías para producir las IDL y las LDL. Las VLDL1 por acción de la CETP son precursoras de las LDL pequeñas y densas10. Las LDL pequeñas y densas se definen como partículas con diámetro menor a 25.5nm, estas lipoproteínas contienen más colesterol esterifcado de lo normal, lo cual las hace mas densas. Las LDL pequeñas y densas, por su composición química 16 tienden a tener mayor afinidad por los proteoglicanos. Estudios indican que las LDL pequeñas y densas, están presentes en pacientes con DT 2 en un porcentaje de 40 a 50%. Estas partículas se correlacionan con la resistencia a la insulina y el control de triacilgliceroles. Estos cambios en la conformación de lípidos de estas lipoproteínas esta mediado por la vía de la CETP. Por otro lado el incremento en HL y apo B-100 en pacientes con DT 2 favorece la formación de las partículas LDL pequeñas y densas. Estas no son reconocidas por su receptor LDL, es decir pierden afinidad, de manera que aumenta su concentración en el plasma10. 17 5. Aterogénesis en HF y DT 2. La hipercolesterolemia familiar se caracteriza por la elevación del colesterol LDL en el torrente sanguíneo, este es el mecanismo más frecuente sobre el proceso aterógeno. Para que las LDL puedan expresar su potencial aterógeno, primero deben ser modificadas en su estructura química, principalmente mediante la oxidación de sus lípidos y de la apoproteína B-100 estos cambios químicos permiten que estas lipoproteínas ingresen a los macrófagos, transformándolos en células espumosas. La participación de un receptor diferente al de las LDL nativas fue sugerida por Goldstein y Brown, quienes describieron en detalle el funcionamiento del receptor de estas lipoproteínas y el hecho, aparentemente paradójico, de los pacientes con hipercolesterolemia familiar homocigótica que están desprovistos del mismo por completo y presentan depósitos masivos de lípidos en el reticuloendotelio, en particular en los macrófagos de las placas de ateroma y de los tejidos blandos (Xantomas). El receptor LDL nativas exhibe un fenómeno de “retroalimentación a la baja” que impide el cúmulo de colesterol en las células provistas del mismo, como en hepatocitos y células de la estirpe monolito/macrófago que poseen este tipo de receptor pueden acumular colesterol en grandes cantidades4,11. Es poco probable que la oxidación de las LDL ocurra en la circulación, por la presencia en la sangre de numerosas sustancias antioxidantes. Sin embargo, cuando las LDL penetran a través del endotelio a la pared arterial, sobre todo de las arterias afectadas por factores capaces de lesionarlas, se exponen a radicales tóxicos de oxígeno como el radical superóxido y otras formas reactivas de oxígeno. En este caso se ponen en marcha la oxidación de los componentes de la LDL, que parecen iniciarse con la autooxidación de los ácidos grasos poliinsaturados de los fosfolípidos contenidos en las LDL. La lecitina se transforma en lisolecitina y los ácidos grasos en aldehídos reactivos (alquenantes). Las LDL oxidadas (LDLox) tienen una acción quimiotáctica hacia los monolitos circulantes, quimiostática para los macrófagos del 18 espacio subendotelial y citotóxica para las células endoteliales. Los cambios químicos en la apo B-100 de las LDL permiten el reconocimiento de la misma por los receptores de los macrófagos y dan origen a epitopos que al ser desconocidos por el sistema inmunológico evocan a la formación de anticuerpos (anti-LDLox)4. Small and dense Small and dense Figura J. Formación de células espumosas. En la DT 2 las LDL son transformadas a LDL pequeñas y densas, de las cuales recientemente se ha comprobado que tienen potencial aterogénico. Se considera que el tamaño pequeño de estas partículas favorece a la entrada a la capa interna de la arteria, en donde se exponen a estrés oxidativo. Las LDL pequeñas y densas son oxidadas, fagocitadas por macrófagos formando células espumosas, las cuales intervienen en la formación de la placa ateromatosa10. 19 JUSTIFICACIÓN. Se sabe que los pacientes con Hipercolesterolemia Familiar, presentan la característica de tener una concentración elevada de colesterol LDL y una disminución de colesterol HDL y también en pacientes con DT 2 se ha observado un patrón similar en cuanto a colesterol LDL y HDL. Lo anterior nos motivo a realizar un estudio entre ambas dislipidemias. Como se menciono anteriormente la HF se debe a mutaciones en el gen del LDLR, por lo que propusimos que los altos niveles de C-LDL en DT 2 podrían presentarse mutaciones en el gen ldlr. Tabla 2. Características comunes entre la HF y la DT 2. hipercolesterolemia familiar Diabetes tipo 2 C- LDL C-LDL C-HDL C-HDL Desarrollo prematuro de aterosclerosis Desarrollo prematuro de aterosclerosis 20 OBJETIVO GENERAL Ø Determinar si las mutaciones en el gen del receptor LDL, están relacionadas con la hiperlipidemia presente en pacientes con Diabetes mellitus tipo 2. Objetivos específicos. Ø Amplificación de los exones 3, 4, 16 y 17 de gen del receptor LDL a partir de DNA genómico mediante la técnica de PCR. Ø Determinar si existen cambios conformacionales en las muestras para los exones a analizar. Ø Elección de posibles candidatos con cambios en la secuencia. Ø Análisis de la secuencia nucleotídica de los candidatos a mutación en la base de datos Gene Bank. Ø Determinar posible correlación entre los niveles de C-LDL y las mutaciones presentes en el receptor LDL. 21 MATERIAL Y MÉTODO. Obtención de las muestras. Se trabajo con el DNA de 126 pacientes diagnosticados con DT 2 que presentaran niveles de colesterol LDL por arriba de 130mg/dL y de colesterol HDL por debajo de 65 mg/dL. Las muestras de DNA fueron obtenidas de la colaboración que hay con el Dr. Miguel Cruz del la Unidad de Investigación Bioquímica del IMSS. Reacción en Cadena de la Polimerasa (PCR: Polymerase Chain Reaction). A partir del DNA de los pacientes se amplificaron los exones 3, 4, 16 y 17 del gen LDLR, mediante la técnica de PCR12,13, para esto se utilizaron los siguientes oligonucleótidos específicos, para cada exón. OLIGONUCLEÓTIDOS5. EXÓN OLIGO SENTIDO OLIGO ANTISENTIDO PRODUCTO 3 5’-TGA CAG TTC AAT CCT GTC TCT TCT G -3’ 5’-ATA GCA AAG GCA GGG CCA CAC TTA C -3’ 176 pb 4 5’-GTT GGG AGA CTT CAC ACG GTG ATG G -3’ 5’-ACT TAG GCA GTG GAA CTC GAA GGC C -3’ 356 pb 16 5’-CCT TCC TTT AGA CCT GGG CC -3’ 5’-CAT AGC GGG AGG CTG TGA CC -3’ 173 pb 17 5’-CAC GGA GCT GGG TCT CTG GTC -3’ 5’- TCG GCC TGG TCC CTT GAG G -3’ 302 pb Tabla 3. Oligonucleótidos sentido y antisentido, utilizados para la amplificación de los exones 3, 4, 16 y 17. 22 En cada tubo de PCR se depositaron 2 l de ADN de cada paciente y cada reacción fue de 15 l, la mezcla se preparo con los siguientes reactivos: Para los exones 3, 4 y l6 se usaron las siguientes condiciones. MEZCLA Reactivo 1 Reacción Concentración fina/reacción. Buffer 1.5 l 1X DMSO 1.5µl 10% dNTP’s 2.4µl 0.2µM MgCl2 0.45µl 1.5mM Taq Pol 0.13µl 0.5 unidades Oligo Sentido 100ng/µl 100ng Oligo Antisentido 100ng/µl 100ng Marca [α32P] dCTP 0.086µl DNA 2µl 200 ng H2O 5.3µl Aforar hasta 15 l Para el exón y 17, se usaron las siguientes condiciones. MEZCLA Reactivo 1 Reacción Concentración fina/reacción Buffer 1.5 l 1X DMSO 1.5µl 10% dNTP’s 2.4µl 0.2µM MgCl2 0.45µl 1.5mM 23 Taq Pol 0.075µl 0.375 unidades Oligo Sentido 100ng/µl 100ng (150 para exón 14) Oligo Antisentido 100ng/µl 100ng (150 para exón 14) Marca [α32P] dCTP 0.075µl DNA 2µl 200 ng H2O 5.3µl Aforar hasta 15 l Nota: la marca radioactiva solo se utilizo en PCR para análisis SSCP Programa de Termociclador El programa utilizado en el termociclador, consta de 3 fases, la primera de inicio en la que las muestras se mantienen a 95ºC durante 4 minutos con 30 segundos, esto es para desnaturalizar la hebra de DNA. La segunda fase es la de los ciclos, las reacciones se harán a 30 ciclos, los cuales van a consistir en 95ºC durante 30 segundos, la temperatura de alineación (Tm) de los oligos (ver tabla 4) durante 30 segundos y por último a 72ºC durante 30 segundos. La tercera fase se compone de 7 minutos a 72ºC y posteriormente a 4ºC ∞ (ver figura K). 95ºC 95ºC 72ºC 72ºC TmºC 4ºC ∞ 30 ciclos 1ª Fase 2ª Fase 3ª Fase Figura K. Programa de termociclador. 24 Cada oligonucleótido tiene una temperatura de alineación (Tm) especifica. EXÓN TmºC 3 58º 4 54º 16 54º 17 56º Tabla 4 . Temperaturas de alineación. Polimorfismo Conformacional de Cadena Sencilla. (SSCP: Single-Strand Conformation Polymorphism)11. CARGADO Y CORRIMIENTO DE LOS GELES. Se realizó electroforesis de los productos de PCR marcados radiactivamente, diluidos y desnaturalizados en geles de poliacrilamida de 30 x 40 cm. y 0.4 mm de grosor. Diluciones y desnaturalización. Muestras de pacientes y controles. a) l de la reacción de PCR más 120µl de solución desnaturalizante (EDTA 10mM y SDS 0.1%). b) l de la dilución a más 5 l de colorante formamida (Formamida 95%, Xylen Cyanol 0.25% y Azul de bromofenol 0.25%). Se calentaron las muestras de la dilución b a 95º durante 5 minutos, inmediatamente se pasaron a hielo y posteriormente se depositaron en el gel. 25 Muestras sin desnaturalizar. Se escogieron dos muestras al azar y se realizó la siguiente dilución. a) l de la reacción de PCR más 120 l de buffer TE (Tris-HCl 10mM y EDTA 0.1%). b) l de la dilución a mas 5 l de colorante-glicerol (glicerol 30% en agua, Xylen cyanol 0.25% y azul de bromofenol 0.25%). Cargar de 2.5 a 2.8 µl de las diluciones b de las muestras de pacientes, controles y controles sin desnaturalizar, en el gel de poliacrilamida al 6% en condiciones nativas (ver tabla 4). Gel de Poliacrilamida al 6% 120ml Agua 84 Acrilamida/Bisacrilamida 29:1% 24 TBE 10X 12 Persulfato de amonio al 10% 400 l Temed. 90 l Tabla 5. Reactivos para preparar un gel de poliacrilamida al 6%. Los geles se corrieron a voltaje constante de 4 watts. Con una solución amortiguadora TBE 0.5X, durante un periodo de 14 horas. Se transfirió el gel a papel de cromatografía Wattman y se puso a secar a 60ºC durante 45 minutos. Exposición de la placa y el revelado. En este caso solo para el exón 17, se tuvieron que utilizar casetes de exposición de 5 días debido a que el analizador de imágenes departamental, permanece ocupado por periodos prolongados. 26 • Se coloco el papel Wattman viendo hacia arriba dentro del cassette para que queden igual, guiarse por los controles (manchas mas grandes). • En el cuarto oscuro se puso una placa de revelado encima del papel filtro y cerrar el casete. Dejo el casete a -72ºC durante 5 días a temperatura ambiente. • Se revelo la placa y se analizo el patrón de bandas obtenido. BIO-RAD FX IMAGING. Una vez obtenidos los geles de los exones 3, 4 y l6 fueron analizados utilizando equipo departamental; el analizador de imágenes FX de Bio-Rad. El cual consiste en exponer los geles a una pantalla Kodak sensible a radioisotopos como lo es el durante 24 horas aproximadamente. 32 P, Posteriormente se lee en el analizador de imágenes el cual detectara las bandas radioactivas, dándonos el patrón de bandas esperado. La imagen se graba en un disco de 31/2 y se checa la migración de las muestras. Purificación del producto de PCR. Los posibles candidatos a mutación seleccionados, se enviaron a secuenciar, siendo purificados previamente mediante el Kit “Rapid PCR Purification System” de Gibco. • Se hizo una PCR de 100µl. • A 100µl de reacción de PCR se adiciono 400µl de solución de unión H1 (guanidina hidrodotada concentrada, EDTA, Tris-HCl e isopropanol), mezclar bien con vortex y pasarlo a la columna, la cual debe de estar dentro del tubo de lavado de 2 ml. • Centrifugar a 12000 rpm a temperatura ambiente en microfuga durante 1 minuto, desechar la solución recuperada. 27 • Se añadio 700µl de buffer de lavado (NaCl, EDTA, Tris-HCl, etanol absoluto) a la columna, la cual de nuevo debe de estar en el mismo tubo de lavado. • Centrifugar a 12000 rpm a temperatura ambiente en microfuga durante 1 minuto, desechar la solución recuperada y centrifugar una vez más en las mismas condiciones. • Transferir la columna a un tubo eppendorf y adicionar 80µl de agua bidestilada estéril precalentada a 70ªC. Incubar a temperatura ambiente durante 5 minutos y centrifugar a 12000 rpm a temperatura ambiente en microfuga durante 2 minutos. • En hormo de vacío a 50ºC concentrar la muestra a un volumen final 20µl. • Correr 4µl de la muestra para verificar los productos en un gel de agarosa al 1.8%. El producto de PCR se llevo a secuenciar, y se analizaron las secuencias candidatas a mutación. 28 RESULTADOS Y DISCUSION. En el análisis de los exones 3, 4, 16 y 17 en los 126 pacientes y lo 10 controles sanos se obtuvieron los siguientes resultados. Los exones amplificados corresponden al tamaño esperado. El tamaño del exón 3 es de 176pb, el del exón 4 es de 356pb, el exón 16 de 173pb y el exón 17 consta de 302pb (ver figura L). Marcador Exón 3 Exón 4 Exón16 Exón 17 Marcador 400pb 200pb Figura L. Gel de agarosa al 1.8% con producto de PCR y marcador de peso molecular Low DNA Mass Ladder. Exones 3 y 4. Los exones 3 y 4, pertenecen al dominio de unión al ligando, el cual consta de 7 repeticiones de 40 aminoácidos, cada repetición contiene 6 residuos de cisteínas que forman 3 puentes disulfuro entre ellas. Es de gran importancia para la unión al ligando. El análisis por SSCP para los exones 3 y 4 no mostró diferencia en la migración de bandas entre los 126 pacientes, tampoco en comparación con los 10 controles sanos. 29 Exón 16. El exón 16 pertenece al dominio transmembranal, que esta constituido por 22 residuos de aminoácidos hidrofóbicos interacción con la membrana celular. El análisis por SSCP del exón 16 no mostró diferencias en la migración de bandas entre los 126 pacientes y los 10 controles sanos. Exón 17 El extremo 5’ del exón 17 codifica para una parte del dominio transmembranal, mientras que el extremo 3’ de este mismo exón codifica para el dominio citoplasmático. Este último dominio juega un papel importante en la internalización del complejo lipoproteína-receptor. El análisis por SSCP del exón 17 de los 126 pacientes y los 10 controles sanos, mostró solamente una diferencia en la migración de la banda perteneciente al paciente 105 (ver figura M), la banda migro por debajo de las bandas correspondientes a los 125 pacientes restantes y aún de los 10 controles sanos. Fig M. SSCP de exón 17, fecha roja muestra candidato a mutación. 100 105 110 111 120 121 126 C-1 C-10 Al encontrar esta diferencia en el paciente 105, se amplifico nuevamente, pero sin marca, se purifico y se envió a secuenciar en dos ocasiones; lo anterior fue debido a que la primera secuencia obtenida al ser comparada con la secuencia original del exón, mostró una homología del 90%. Por esta razón se decidió corroborar estos 30 resultados, enviando nuevamente a secuenciar el producto de PCR de este paciente (105). Se obtuvo el histograma de secuencia de la figura N. Se comparo la secuencia normal del exón 17 con la obtenida del paciente 105 para el exón 17, mediante el programa DNA MAN y se obtuvo una homología del 90%. Al analizar se encontraron varios cambios en la secuencia del paciente 105. Las mutaciones encontradas en el paciente 105 en el exón 17, fueron 9 silenciosas y 6 mutaciones con sentido erróneo, las cuales no habían sido reportadas con anterioridad y se muestran en las siguientes tablas junto con la nomenclatura13,14. Tabla 6. Mutaciones silenciosas, es decir, cambio de nucleótido en la tercera base del triplete. núm. no. de nO. de codón codon aminoácido cambio NOMENC nucleotido codón original presente codificado 1 2391 776 GTG GTA VAL G>A 2391 G>T 2 2397 778 CTC CTT LEU C>T 2397 C>T 3 2400 779 GTC GTT VAL C>T 2400 C>T 4 2406 781 CTT CTC LEU T>C 2406 T>C 5 2424 787 CTT CTC LEU T>C 2424 T>C 6 2427 788 CTA CTC LEU A>C 2427 A>C 7 2484 807 TAT TAC TRY T>C 2484 T>C 8 2487 808 CAG CAA GLN G>A 2487 G>A 9 2496 811 ACA ACG THR A>G 2496 A>G LATURA .Las mutaciones silenciosas que se muestran en la tabla 6 hay cambios de base en la tercera posición de los codones, por lo que no cambia el aminoácido, sino los nucleótidos 31 Figura N 32 Histograma de secuencia sentido del exón 17 del paciente 105. Al exón 17 los componen 53 codones, de los cuales los primeros 14 forman parte del dominio transmembranal, en esta región se encontraron 6 mutaciones silenciosas, que son las siguientes: 2391 G>T, 2397 C>T, 2400 C>T, 2406 T>C, 2424 T>C y 2427 A>C. Los 39 codones restantes del exón 17, codifican para el dominio citoplasmático, dentro de esta región se encontraron 3 mutaciones silenciosas (ver tabla 6) las cuales son: 2484 T>C, 2487 G>A y 2496 A>G, que no cambia el aminoácido. Tabla7. Mutaciones con sentido erróneo. Cambio de aminoácido. num. no. de no. de codon codon cambio de cambio de NOMEN nucleotido codon original presente aminoácido nucleotido CLATUR A 1 2410-2412 783 CTG GTC LEU > VAL C>G y L 783 V G>C 2 2417 785 GTC GCC VAL > ALA T>C V 785 A 3 2431 790 AAG AGG LYS > ARG A>G K 790 R 4 2450 796 AAC AGC ASN > SER A>G N 796 S 5 2452 797 ATC GTC ILE > VAL A>G I 797 V 6 2519 819 CAC CGC HIS > ARG A>G H 819 R Cuando hay un cambio de nucleótido en la primera o segunda base del triplete, cambia el aminoácido, a este tipo de mutaciones se les conoce como mutaciones con sentido erróneo. Los cambios de aminoácidos encontrados en el este exón para este paciente, son por otro del mismo grupo, es decir con las mismas características fisicoquímicas. La región transmembranal presento 2 cambios de aminoácidos que son L78 V y V785A, estos aminoácidos leucina, valina y alanina están dentro del Grupo R apolares alifáticos los cuales son importantes para las interacciones hidrofóbicas entre la proteína y la membrana citoplasmática1. 33 En la región del dominio citoplasmático se encontraron 4 mutaciones con sentido erróneo (ver tabla 7), la cuales son: K790R, N796S, I797V y H819R. En el caso de la mutación K 790 R y la H 819 R, los aminoácidos involucrados son lisina-arginina e histidina-arginina, que pertenecen al grupo R cargados positivamente, por lo que el cambio no es tan relevante. En la mutación N796S, en el que hay un cambio de asparagina por serina, estos aminoácidos pertenecen al grupo R polares sin carga. En la bibliografía se han reportado que existe un secuencia altamente conservada de gran importancia en el dominio citoplasmático para la internalización , la cual va desde el codón 804 al 807, los cuales codifican para los aminoácidos asparagina, prolina, valina y tirosina ( NPXY), en esta secuencia la valina puede ser sustituida por alguno otro aminoácido. Esta secuencia es de gran importancia, porque cuando hay un cambio en alguno de estos residuos, se ve afectado la formación del poro recubierto (coated pit), para la internalización del complejo lipoproteína receptor. Se realizaron experimentos de mutagénesis dirigida en la cual cambiaban ciertos residuos de aminoácidos a lo largo de todo el exón, sin embargo esta región es la que resulto ser más sensible a cambios de aminoácidos, es decir que si influye sobre la actividad normal del receptor en la internalización15. Los codones 804 al 807 pertenecientes al exón 17 en este paciente encuentran normales, es decir no se mostré ningún cambio en esta región, que es de gran importancia. 34 CONCLUSION. Después de analizar los resultados obtenidos al practicar SSCP de las 126 muestras de pacientes con DT2, para los exones 3, 4, 16 y 17, no se encontraron mutaciones como probable factor responsable de los niveles elevados de colesterol LDL en la DT2. Sin embargo al analizar el exón 17, se encontró un paciente con cambios conformacionales en este exón. En este sujeto se encontraron 15 cambios de nucleótidos que no se han reportado hasta la fecha. Estos cambios consisten en 9 mutaciones silenciosas y 6 de mutaciones con sentido erróneo. A pesar de lo anterior, cabe la posibilidad de los niveles elevados de LDL en este padecimiento se deba algún defecto en el gen de la apoB-100, ya que en HF cambios en la estructura de esta apo impiden la unión de esta última al receptor. Por lo anterior, concluimos que los niveles elevados de colesterol LDL en pacientes con diabetes tipo 2, no están relacionado con las mutaciones en el gen de este receptor. 35 PERSPECTIVAS. v Búsqueda del factor responsable de la hiperlipidemia LDL en la DT 2. v Escaneo del gen de la apo B-100, la cual es el ligando al receptor LDL, y cambios en ella impiden esta unión. v Análisis molecular de los familiares del paciente 105 que presento cambios conformacionales en el exón 17. 36 REFERENCIAS. 1. Lenhinger A., Nelson N., y Cox M., 1995. Principios de Bioquímica. Segunda Edición. Editorial. Omega. Págs. 240-266, 359-788, 479-504, 642-686. 2. Keith N. Frayn. 1996. Metabolic Regulation. A Human Perspective. Lipoproteín metabolism. Portland Press. Pags. 197-217. 3. Howard Barbara V., 1987. LIpoprotein metabolism in diabetes mellitus. Journal of lipid Research. Vol. 28. Pags. 613-628. 4. Gómez, P. F., Aguilar, S. C. HIPERLIPOPROTEINEMIAS PRIMARIAS. Posadas R. C. Dislipidemias y Aterosclerosis. 1995. Editorial Interamericana Mc. GrawHill. Págs. 87-104. 5. Hobbs, H.H., Brown, M.S., and Goldstein, J. L. 1992. Molecular Genetics of the LDL receptor Gene in Familial Hypercholesterolemia. Human Mutation 1: 445-466 6. Aguilar, S. C. 2001. Hipercolesterolemia Familiar. Revista de Investigación Clínica. Vol. 53, Núm. 3. Págs. 254-265. 7. Cruz Miguel, Emilio Espinoza-Simón, Jaime García MENA, Socorro Duran, Niels Wacher Rodarte, Jesús Kumate. Genes candidatos en Diabetes tipo 2 y poblaciones en riesgo de padecer la enfermedad. Comunicación personal. 8. Cruz Miguel, Montoya Carlos, Gutiérrez Margarita, Wacher Niels H., y Kumate Jesús. 2002. Polimorfismos de genes relacionados con diabetes tipo 2. Rev. Med. IMSS. Vol. 40 (2). Págs. 113-125. 37 9. Aguilar Salinas C., Molina Cuevas V., Velásquez Monroy O., Rull-Rodrigo J., Gómez Pérez F., Tapia Conyer R., González Chávez A., Lara Esqueda A. and ENSA (Encuesta Nacional de Salud) 2000 Group. 2003. Characteristics of patients with type 2 Diabetes in México. Diabetes Care. July. Vol. 26. Núm. 7. Pags. 2021-2026. 10. Taskinen M. R., 2003. Diabetic dyslipidaemia: from basic research to clinical practice. Diabetologia. Vol. 46. Págs. 733-749. 11. Larhiri, D. and Nurnberger, J. Jr. (1991). A rapid non-enzymatic method for the preparation of HMW DNA from blood for RFLP studies. Nuc Acids Res. 19:5444. 12. Humphries, S. E., Gudnason, R. W., and Ian N. M. 1997. Single-strand conformation polymorphism analysis with high throughput modifications, and its use in mutation detection en familial hypercholesterolemia. Clinical chemistry 43:3 pgs. 427-435. 13. Humphries, S. E., Gudnason, R. W., and Ian N. M. 1997. Single-strand conformation polymorphism analysis with high throughput modifications, and its use in mutation detection en familial hypercholesterolemia. Clinical chemistry 43:3 pgs. 427-435. 14. Humphries, S. E., Gudnason, R. W., and Ian N. M. 1997. Single-strand conformation polymorphism analysis with high throughput modifications, and its use in mutation detection en familial hypercholesterolemia. Clinical chemistry 43:3 pgs. 427-435. 38 15. Chen W.J., Goldstein J.L. and Brown M.S. 1990. NPXY, a sequence often found in cytoplasmic tails, is requiered for Coated pit-mediated internalization of the low density lipoprotein receptor. The Journal of biological chemistry. Vol. 265. No. 6. Pags. 3116-3123. 16. Peeters A.V., Thiart R., de Villiers J.N., Jensen H. K., Van Gaal L.F. and Kotze M. J. 1999. Intronic mutations at splice junctions in the low-density lipoprotein receptor gene. Molecular and cellular probes. Núm. 13. Pags. 257-260. 39