practicas en - Inicio - Universidad de Salamanca
Anuncio

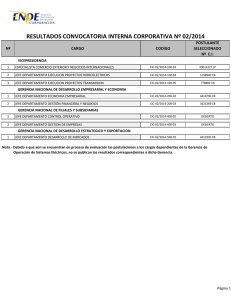
PRACTICAS EN:
Sergio Luquero Bueno
71132703-M
Facultad de Biología.
Salamanca.
Prácticas en CIC
Índice Introducción.3 El CIC.4 Isidro Sánchez García.6 Breve descriptiva previa sobre la familia Snail.7 PCR.9 ELECTROFORESIS EN GEL DE AGAROSA.13 CULTIVOS CELULARES.15 MEDIOS DE CULTIVO.15 PHOENIX Y MEF’s.16 Preparación de células Phoenix.16 CONSERVACIÓN DE PHOENIX.17 CONGELAR CÉLULAS MEF SIN TRATAR.17 FORMACIÓN DE cDNA.18 ESTUDIO EN PLACAS DE DAÑO EN ADN.19 EXTRACCIÓN PROTEICA.19 WESTERN‐BLOT.20 Tecnología de ADN recombinante.25 MINIPREPs.26 ENZIMAS DE RESTRICCIÓN.27 Purificación proteica.28 Southern‐Blot.30 Seguimiento del trabajo de otros compañeros.33 Otras Actividades.34 Conclusiones.36 Bibliografía.37 2 2
Prácticas en CIC
Introducción: Este verano, tras cursar quinto curso, tuve la oportunidad de realizar las prácticas
de mi carrera, Biología, en el Centro de Investigación del Cáncer dentro del laboratorio de
Isidro Sánchez García. El periodo de realización fue del 1 de Julio al 14 de Agosto de
2008.
Ante la decisión de dónde realizar las prácticas, y tras el conocimiento por parte
del Vicedecano Fernado Leal de la posibilidad de realizarlas en CSIC gracias al convenio
entre éste y la facultad de biología de Salamanca decidí realizarlas en el Centro de
investigación de Cáncer.
La elección vino dada por mi interés en realizar prácticas de laboratorio, la
investigación en el cáncer me resultaba atrayente y por último la decisión de la selección
del tutor vino dada tras la lectura de las interesantes líneas de investigación presentes y
pasadas del Dr. Isidro Sánchez García.
En esta memoria de prácticas expongo brevemente la enriquecedora experiencia
que ha supuesto este periodo en el centro y las técnicas dadas en la carrera que he podido
poner en práctica.
Sergio Luquero Bueno.
3 3
Prácticas en CIC
El CIC: El Centro de investigación del Cáncer está situado en Salamanca, en el campus
Miguel de Unamuno. Su estructura organizativa está centrada alrededor del Instituto de
Biología Molecular y Celular del Cáncer (IBMCC), que tiene carácter de Instituto
Universitario Mixto, patrocinado por la Universidad de Salamanca y el Consejo
Superior de Investigaciones Científicas: USAL-CSIC. Está también reconocido como
Centro Sanitario (N° Registro 5-14-0001) por el Sistema de Salud de Castilla y León
(SACYL).
El IBMCC es un centro integrado de investigación del cáncer cuya constitución
fue aprobada en 1997 que, respondiendo al modelo de los Comprehensive Cancer
Center norteamericanos, se caracteriza por integrar investigación competitiva y de
excelencia sobre cáncer en tres niveles distintos: básico, clínico y aplicado o
traslacional. Para ello está formado por un Instituto mixto dependiente de la
Universidad de Salamanca y el Consejo Superior de Investigaciones Científicas así
como por diversas unidades dependientes del Sistema de Salud de Castilla y León.
La idea inicial de formar un centro especializado en investigación sobre el cáncer en la
Universidad de Salamanca, surgió ante la constatación de la presencia en la misma de
un núcleo importante de investigadores de los procesos básicos y clínicos del cáncer que
si bien estaban desconectados entre sí por estar dispersos entre varios departamentos de
la Universidad de Salamanca, resultaba muy significativo en calidad y número en
comparación con otras universidades del país.
De esta forma surgió el Centro de Investigación del Cáncer, con una contribución
científica local y regional a través de la Universidad de Salamanca y una dimensión
nacional mediante el CSIC, quien manifestó su deseo explícito de participar creando el
Instituto Mixto de Biología Molecular y Celular del Cáncer del CIC, para permitir el
acceso de investigadores y científicos especializados de todo el territorio nacional a este
Centro. Las Consejerías de Sanidad y de Educación de la Junta de Castilla y León han
asumido una participación directa en el CIC a través de su presencia en el Patronato de
la FICUS (Fundación para la Investigación del Cáncer de la Universidad de Salamanca)
y la promoción de esta institución como Centro Sanitario del sistema autonómico
SACYL. El Instituto de Salud Carlos III del Ministerio de Sanidad participa en el CIC a
través de la designación de este centro como uno de los nodos de su Red Temática de
Investigación cooperativa de Cáncer a nivel nacional y de la adscripción de
Investigadores FIS a este centro.
4 4
Prácticas en CIC
Objetivos del centro:
1. Realizar investigación puntera en cáncer a nivel básico, aplicado y clínico.
2. Favorecer el trasvase bidireccional de información entre la ciencia biomédica básica
y la aplicada, para fomentar la sinergia de los tres tipos de investigación y así mejorar la
productividad.
3. Constituirse como un centro científico de excelencia capaz de competir en igualdad
de condiciones con otros centros internacionales.
4. Fomentar la conexión del CIC con redes temáticas de investigación oncológica tanto
nacionales como internacionales.
5. Potenciar la creación de riqueza y servicios que reviertan en el bienestar social y
desarrollo económico a nivel regional y nacional.
Finalidad:
El CIC se concibe como un elemento de aglutinación de los mejores recursos humanos
existentes en el campo de la investigación del cáncer a nivel regional (Castilla y León) y
nacional. Ser referencia por su calidad, tanto a nivel nacional como internacional.
5 5
Prácticas en CIC
Isidro Sánchez García: Dr. Isidro Sánchez-García. Investigador Científico, CSIC. Lab. 13. Es uno de
los investigadores principales del CIC en el área de Investigación Aplicada. Las líneas
generales de su proyecto de investigación son la generación de modelos genéticos
animales para el estudio de tumorigénesis y hematopoyesis, las alteraciones
cromosómicas en tumores hematopoyéticos y terapia génica.
Líneas de investigación:
“Aunque la mayor parte de los pacientes con cáncer responden a la terapia, tan sólo una
minoría son curados. Datos recientes que sugieren el origen de muchas enfermedades en
una población poco común de células responsables exclusivamente de conservar la capacidad
de auto-renovación y mantenimiento del tumor (es decir, ?células stem cancerígenas?)
podrían explicar la paradoja de que respuesta y supervivencia no siempre van unidas. En
este contexto, una pequeña población de células cancerígenas adquiriría propiedades
características de células stem. En algunos casos, las células stem cancerígenas (CSC)
podrían derivar directamente de las células stem normales de un tejido. En cualquiera de las
situaciones el resultado final sería el mismo, conduciendo a las CSC como células diana en el
desarrollo de terapias moleculares y farmacéuticas para el tratamiento y prevención del
cáncer humano. Este podría representa un nuevo paradigma en el tratamiento del cáncer,
alejado del tratamiento dirigido a las células blásticas y orientado a las CSC. El desafío para
esta nueva aproximación es encontrar una forma de atacar específicamente estas CSC sin
que sea tóxica para las células normales. En este sentido, nuestro laboratorio se sirve de la
genética y genómica del ratón para diseccionar los mecanismos moleculares y procesos
biológicos que gobiernan el papel de las CSC en la biología tumoral y en la Oncología
Translacional. Con este objetivo hemos desarrollado los siguientes programas de
investigación:
a) Identificación y caracterización de las células stem cancerígenas en varios tipos de tumor.
b) Atlas genómico de las CSC: genética y epigenética.
c) Biología Celular y de sistemas de las células stem cancerígenas (Análisis funcional de los
genes Slug y Snail en el desarrollo y mantenimiento de la célula stem cancerígena).
d) Modelos animales para el Estudio de la Célula Stem Cancerígena.
e) Aplicaciones diagnósticas y pronósticas del Estudio de la Célula Stem Cancerígena
(Imagen Molecular).
f) Identificación y evaluación de dianas terapéuticas en las células stem cancerígenas.
g) Descubrimiento y desarrollo de inhibidores selectivos de las CSC.
Esperamos que esta investigación no sólo redunde en nuevos conocimientos en la biología y
desarrollo del cáncer, sino que proporcione las bases para el desarrollo de nuevas estrategias
en la terapia frente al cáncer y nuevos métodos para valorar tratamientos eficaces.”
Isidro Sánchez García.
6 6
Prácticas en CIC
Experiencia personal en el CIC: Mi trabajo dentro del Laboratorio de Dr. Isidro Sánchez García, el laboratorio
número 13 dentro del instituto del Cáncer , vino determinado por el miembro del equipo
que Isidro seleccionó para tutelarme y que fue variando en el tiempo, permitiéndome
así conocer y participar en las múltiples líneas de investigación que posee dicho equipo.
En primer lugar estuve colaborando con Katja Gutsche, bióloga de origen
alemán que basa su investigación en el estudio de la función del gen Snai3 en procesos
en respuesta al daño en el ADN. Snai3 forma parte de la familia de genes Snail.
Breve descriptiva previa sobre la familia Snail:
Miembros de la superfamilia Snail donde también se aúna la familia Scratch.
Los genes de la familia Snail de vertebrados se organizan en Subfamilias: Snai1, Snai2
y Snai3. Todos ellos formados por una región carboxi-terminal muy conservada que
contiene entre 4 y 6 dominios en dedos de Zinc y una región amino-terminal mucho más
variable. Los dedos de Zinc son C2H2 (2 cisteínas y 2 histidinas), estos son importantes
para la unión a secuencias específicas de ADN, importantes por tanto para su función
como represores transcripcionales.
Funciones de las proteínas de la familia Snail:
-
Transición epitelio-mesénquima: fundamental para el desarrollo normal y
tumoral. Los genes de la familia Snail actúan reprimiendo marcadores epiteliales
y aumentando los mesenquimales, participando además en el cambio de forma
celular y en propiedades invasivas. Snai1 y Snai2 reprimen la expresión del gen
E-caderina.
- Decisiones del destino celular y supervivencia celular.
- División celular (inhibición de la mitosis).
- Asimetría derecha-izquierda en desarrollo embrional y neural.
- Evidencias de su intervención en la protección de las células ante muerte celular.
o Snai 2: Antiapoptótico; inducido a nivel transcripcional por p53.
o Snai 1: Factor de supervivencia; células con resistencia a señales
apoptóticas directas (ante daño en ADN,…).
Ante Cáncer: estas funciones, tan importantes en desarrollo embrionario, en desarrollo
patológico, como cáncer, facilitarán el desarrollo del tumor y su metástasis. Actúan
como marcadores de malignidad y potencial metastásico.
Snai3 (Smuc) : se encuentra presente en el cromosoma 16 humano, se expresa en
desarrollo embrionario y en diversos tejidos adultos.
7 7
Prácticas en CIC
Se han realizado estudios sobre su secuencia, estructura y su expresión en tejidos pero
muy poco sobre su función.
Dadas las evidencias de participación de los miembros de la superfamilia Snail en
supervivencia celular Katja Gutsche estudia la función de Snai 3 en respuesta a daño en
el ADN.
(Podría poseer funciones diferenciales con respecto a los demás miembros de la familia
Snail).
La razón de ese desconocimiento funcional está relacionado con la baja expresión de
Snai3, lo que dificulta su estudio.
Gracias a dicho estudio pude poner en práctica diversas técnicas:
9
9
9
9
8 PCR y electroforesis.
Cultivos celulares.
Extracción proteica.
Western blot.
8
Prácticas en CIC
PCR: La reacción en cadena de la polimerasa, PCR, es una técnica que permite
replicar entre cientos de miles y millones de veces, en el transcurrir de pocas horas e in
vitro, pequeñas cantidades de ADN. La técnica se basa en la replicación del ADN
realizada por la ADN polimerasa. Estas enzimas realizan la síntesis de una cadena
complementaria de ADN en el sentido 5´-> 3´ usando un molde de cadena sencilla, pero
a partir de una región de doble cadena. Para crear esta región doble cadena se usan los
denominados iniciadores (primers). Son una pareja de oligonucleótidos sintetizados de
manera que sean complementarios a cada uno de los extremos del fragmento de ADN
que se desea amplificar.
Partiendo de este principio, la Reacción en Cadena de la Polimerasa se basa en la
repetición de un ciclo básico formado por tres etapas:
1ª
Desnaturalización del ADN doble cadena:
la doble hélice de ADN se separa en dos hebras. Para ello se realiza una
incubación de la muestra a altas temperaturas (93-97ºC). (La renaturalización se
producirá cuando la temperatura disminuya).
2ª
Hibridación de los iniciadores a la zona 3´ específica de cada una de las hebras:
(50-65º C) la temperatura se reduce para permitir el apareamiento o hibridación
de cada una de dos cadenas cortas de nucleótidos (oligonucleótidos) con cada una de las
9 9
Prácticas en CIC
hebras separadas del ADN molde. Se trata de segmentos de ADN de cadena simple,
sintetizados en el laboratorio y diseñados de manera tal que permiten definir los límites
del tramo de ADN que se desea replicar. Obviamente, para que se pueda producir el
apareamiento, cada uno de estos oligonucleótidos, a los que también denominan
"iniciadores", debe ser complementario del tramo al que tienen que unirse en las
cadenas separadas del ADN molde.
3ª
Extensión del cebador por actuación de la ADN polimerasa:
Elongación, se produce la síntesis de una cadena sencilla (produciéndose un
fragmento de doble cadena por la complementariedad) en la dirección 5´-> 3´ mediante
la enzima ADN polimerasa, la cual incorpora siguiendo la cadena molde los
desoxinucleótidos fosfato (dNTPs) agregados a la mezcla de reacción. La temperatura a
la que se realiza el tercer paso está condicionada por aquélla a la cual trabaja la enzima
ADN polimerasa. Al cabo del primer ciclo de tres reacciones (desnaturalización,
apareamiento, extensión) el tramo de ADN elegido se ha duplicado y el doble de su
cantidad original se encuentra disponible para ser nuevamente replicado en un segundo
ciclo. El resultado de la aplicación de numerosos ciclos "en cadena" da lugar a la
amplificación geométrica del segmento de ADN delimitado por los primers.
10 10
Prácticas en CIC
Hemos llevado a cabo en el laboratorio la técnica PCR semicuantitativa, que incluye
diluciones de cada muestra de cDNA de 1/5 y 1/25 ya que nuestras PCR´s tenían en su
gran mayoría una finalidad comparativa entre los patrones de expresión de Snai3 o
Smuc en distintas variantes génicas y mutantes, con estas diluciones controlamos y
observamos mejor las variaciones.
Mix por cada eppendorf/muestra por cada 10 µl de cDNA:
19’5 µl MPW.
5 µl MgCl2 5mM.
5 µl dNTPs 2mM.
Previamente: Baño 30º
5 µl 10x Taq Buffer.
2’5 µl Oligo 1 (1/100).
Direcciones contrarias
2’5 µl Oligo 2 (1/100).
0’5 µl Taq polimerasa.
Hielo (para evitar pérdida de
actividad)
Realmente lo que realizamos es un Mix conjunto para todas las muestras, además
debemos incluir un eppendorf sin muestra, sólo con mix para utilizarlo como control
evitando posibles contaminaciones.
Taq. ADN polimerasa: enzima recombinante purificada en Scherichia Coli DH1
(mediante el buffer se ajusta la [Mg+2], eliminando la necesidad de optimización de
este componente crítico).
El cDNA utilizado en estos estudios procede de MEF’s (murine embryonic fibroblast) y
Timocitos de ratones.
Variantes utilizadas:
-
-
11 wt.
wt iR(2,5 Gy) (irradiado gamma).
p53 -/-.
p53 -/- iR(2,5 Gy).
CombiSmuc x p53 -/-.
CombiSmuc x p53 -/- iR.
CombiSmuc.
CombiSmuc iR.
11
Prácticas en CIC
-
-
Hemos utilizado también marcadores de gen Slug (Snai2) para conocer la
posible interferencia.
cDNA + retrovirus vacío.
cDNA + retrovirus vacío dañado con Doxorrubicina.
cDNA con siARN (pequeño de interferencia).
cDNA con siARN y dañado con Doxorrubicina.
Los programas utilizados para llevar a cabo la PCR de nuestras muestras en la
termocicladora han sido Actina y GAPdh con un número variable de ciclos.
12 12
Prácticas en CIC
ELECTROFORESIS EN GEL DE AGAROSA: Estudia la migración de las partículas coloidales en el seno de un campo eléctrico
Consiste en la separación de los fragmentos de distinto tamaño de ADN en el gel de
agarosa, en el que dichos fragmentos migraran a diferente velocidad según su peso
molecular al ser sometidos a una corriente eléctrica.
La agarosa es un polisacárido (originalmente obtenido de algas, como el agaragar, pero de composición homogénea), cuyas disoluciones (típicamente de 0.5 a 2 %)
poseen la propiedad de permanecer liquidas por encima de 50 ºC y formar un gel
semisólido al enfriarse. Este gel esta constituido por una matriz o trama tridimensional
de fibras poliméricas embebida en gran cantidad de medio líquido, que retarda el paso
de las moléculas, se usa para separar moléculas grandes de alrededor de 20.000
nucleótidos.
Dependiendo del número de muestras y, por tanto, de pocillos necesarios hemos
utilizado dos tamaños distintos de cámaras electroforéticas horizontales para las que
realicé los geles con esta composición y características:
-
250 / 50 ml de TBE 1x.
4’5 / 0’9 gr. de Agarosa (1’8%).
5 / 1’5 µl de Bromuro de etidio: colorante que se intercala entre las bases
nitrogenadas de ADN y emite fluorescencia ante luz ultravioleta (carcinógeno).
140 / 55 Voltios durante una hora.
Tras la realización de las mezclas para los geles, colocación de peines de formación
de pocillos y la solidificación del gel debemos cubrirlo para evitar su deshidratación con
solución TBE 1x.
13 13
Prácticas en CIC
Previa a la introducción de las muestras obtenidas por PCR a los pocillos mediante
pipeta, debemos aplicar a cada muestra (cDNA+Mix) una solución “Loading Buffer”, es
el tampón de carga:
-
-
50% Glicerol.
0.1% Azul de Bromofenol: quedando así marcada la muestra a simple vista.
200mM EDTA pH=8.
Gracias a esta solución monitorizamos la migración del ADN.
Con estos estudios de PCR y electroforesis y tras el revelado del gel con el
transiluminador hemos ido ponderando las concentraciones de cDNA de las muestras
para unificar los resultados expresados por las bandas para luego realizar los pertinentes
estudios PCR de expresión diferencial del gen Smuc (Snai3).
14 14
Prácticas en CIC
CULTIVOS CELULARES: MEDIOS DE CULTIVO. Mantenemos Placas Petri en crecimiento con los siguientes cultivos:
-
-
-
8 placas con MEF’s wt ≠ 9 (embrión 9) con retrovirus vacío.
8 placas con MEF’s Smuc.
8 placas con MEF’s Smuc A45-128.: mutante de Smuc con supresión de
fracción de aminoácidos 45-128 para reconocer si actúa de la misma manera
sobre p21.
1 placa control de MEF sin modificación alguna.
Cambios de medio de Cultivo:
Se realizan los cambios de medio cada 3 días aproximadamente dependiendo de las
necesidades celulares.
Para realizar dicho cambio acudimos a la sala de cultivos y utilizamos la cámara de
cultivos tras apagar la luz ultravioleta y esterilizar todo el material utilizable con etanol.
-
-
Desechamos el medio de cultivo mediante aspiración.
Echamos PBS para eliminar restos del medio anterior y facilitar así la acción de
la Tripsina.
Utilizamos Tripsina: 3ml por placa para despegar los MEF’s que se encuentran
adheridas a la placa. Dejamos que actúe durante 5 minutos.
Recogemos en los tubos Falcon las muestras celulares recogidas gracias a la
Tripsina y unimos el nuevo medio de cultivo.
Medio de cultivo:
o 500 ml DMEM.
o 10% Suero bovino (50 ml) filtrado previamente (0’45µm)
o 5’5 ml Glutamina.
o 5’5 ml Penicilina//Estreptomicina.
-
15 Centrifugación del Falcon a 1200 rpm. Durante 10 minutos, obteniendo un pellet
más o menos marcado(Tripsina ya inactivada por el medio).
A continuación se introduce en el medio de cultivo Puromicina a razón de 2
µg/ml; la utilizamos para seleccionar las células marcadas debido al cassette
asociado al retrovirus, de esta forma, en el falcon control que sólo posee células
MEF’s se provocará la muerte celular en presencia de puromicina.
15
Prácticas en CIC
-
Tras resuspender el pellet con el medio + puromicina y dependiendo de las
condiciones celulares observadas debemos determinar si pasamos nuestro
cultivo al mismo número de placas, a menos o a más.
PHOENIX Y MEF’s. Los MEF’s fueron infectados utilizando retrovirus producidos mediante la
transfección pasajera de células Phoenix. Una vez trasfectadas con ADN retroviral,
estas células codifican para las proteínas de la cápsida de los retrovirus, que se van a
liberar al medio de cultivo.
Los vectores retrovirales utilizados para transfectar phoenix y así obtener retrovirus
para infectar los MEF’s son varios y entre ellos está pQCXIP, que es un vector de
expresión bicistrónico diseñado para expresar un determinado gen bajo la acción del
promotor de citomegalovirus (CMV), junto con un marcador de selección de
puromicina.
Preparación de células phoenix: En medio DMEM (Dulbecco’s modified Eagle’s suplementado con un 10% de
suero bovino fetal inactivado con calor (30 min. a 56ºC) y filtrado por 0’45 µm, 2
mM L-Glutamina, 50µg/ml Estreptomicina.
Preparación del ADN: Por cada placa Phoenix a transfectar 16 µg de ADN
retroviral.
Preparación de MEF’s: Plaqueamos 1x106 céls/placa 10cm diámetro.
Infección de las MEF’s:
Para infectarlas se recogió el medio de cultivo de las Phoenix que contendrá los
retrovirus correspondientes en cada caso liberados en el medio. El sobrenadante de
cada placa Phoenix filtrado sirve para infectar dos placas MEF. Se realizarán tres
infecciones consecutivas por placa MEF.
(Los retrovirus generados con la cápsida y el fragmento de ADN que nos interesa se
encuentran en el sobrenadante de Phoenix, no pegado a la placa).
(Ver gráfico en página siguiente)
16 16
Prácticas en CIC
Placa de Phoenix transfectada.
Se introduce CaCl2 para mejorar la entrada del vector.
12ml
Sobrenadante
6 ml.
6ml.
X3 : Se realiza la infección de
MEFs desde el mismo Phoenix al que se va añadiendo medio de
cultivo y hacia los dos mismos placas sin retirarles el sobrenadante
(finalmente serán 18 ml.)
CONSERVACIÓN DE PHOENIX SIN SER TRANSFECTADOS. -
-
En la campana de cultivo retiro el medio de placa Phoenix mediante aspiración.
Vierto 3 ml. de Tripsina en placa para despegar las Phoenix del fondo: 5 min.
incubando.
En este caso (retrovirales) no es necesario echar antes PBS para que los restos
del medio no afecten a la tripsina.
Tras la tripsinización queremos hacer tres placas con Split en proporción 1/15;
para ello utilizo un falcon de 12 ml de medio sin puromicina y junto a los 3 ml.
de tripsina que contiene las células en suspensión, nos da 15 ml.
De esos 15 ml. tomaremos 1 ml. para cada placa con las células (proporción
1/15) y echamos el resto de medio hasta completar volumen.
CONGELAR CÉLULAS MEF SIN TRATAR: Para ello quitamos el medio y usamos tripsina, las pasamos a Falcon con medio y tras
centrifugar, el pellet obtenido se resuspende en suero, del cual verteremos en el dial de
congelación 0’5 ml. y otros 0’5 ml. de suero + DMSO que mantiene la forma celular.
El dial rápidamente se pone a 4ºC debido al suero utilizado que es frío, lo pasamos a 20ºC en el congelador aproximadamente una media hora y después lo pasamos a -80ºC
y finalmente se mantiene en N2 Líquido ( es importante esta congelación gradual).
17 17
Prácticas en CIC
FORMACIÓN DE cDNA: El Adn copia lo realizaremos a partir de 5 µg de ARN con un volumen total de
12 µl. Deberemos conocer la concentración de ARN por µl para tomar los microlitros
necesarios hasta los 5 µgramos de ARN y llevar hasta 12 µl con agua.
Ejemplo: [ARN]= 0’64 µg/µl.
7’8 µl ARN
4’2 µl MPW (el H2O utilizado
para esta técnica se debe mantener exclusivamente para esta técnica y tapón cerrado con
parafilm).
RNAsa-libre tubo eppendorff
hielo
+ 7 µl RNA-MPW.
+ 1 µl RNAsin (inhibidor RNAasa).
+ 5 µl RNA.
5 minutos a 65ºC.
Spin
Transferir a Temperatura Ambiente
+ 1 µl Oligo dt.
+ 1 µl ARN sin.
+ 4 µl 5x buffer (-20ºC).
+ 1 µl RT (Transcriptasa Inversa).
Mix
1 hora a 42ºC.
Almacenamiento
a -20ºC.
18 Mix
+ 80 µl RNA-MPW
18
Prácticas en CIC
ESTUDIO EN PLACAS DE DAÑO EN ADN: Sobre los cultivos hacemos pruebas con doxorrubicina en tres tipos de placas:
-
wt ≠ 9.
wt ≠ 9 Smuc.
proteico.
wt ≠ 9 Smuc A45-128.
Para observar el daño y efectos mediante análisis
La doxorrubicina la dejamos actuar en las placas a distintos tiempos, en algunas 12
horas, en otras 24 horas; tras lo cual:
- Quitamos el medio de cultivo.
- Lavado con PBS.
- Tripsinizamos.
- Resuspensión en medio de cultivo.
- Centrifugación.
- Nos quedamos con el pellet tras quitar el sobrenadante y conservamos a -80ºC.
EXTRACCIÓN PROTEICA: -
-
19 Resuspenderemos el pellet obtenido tras la aplicación de doxorrubicina a las
placas anteriormente citadas con solución de lisis previamente preparada, esta
solución lisa la membrana celular y nuclear y tiene como componentes Trix e
inhibidores de proteasas.
La solución de lisis utilizada varía entre 100-200 µl dependiendo del tamaño del
pellet. Se agita cada eppendorf y los mantenemos en hielo durante 15 minutos.
Centrifugación a 4ºC, 14000 rpm durante 30 minutos. De esta forma el extracto
proteico quedará en el sobrenadante y los restos celulares en el fondo.
La concentración de proteína obtenida la mediremos mediante espectofotometría
utilizando una recta patrón a base de BSA por el método Bradford utilizando el
reactivo comercial facilitado.
19
Prácticas en CIC
WESTERN‐BLOT: La transferencia de proteínas o 'blotting' supone la inmovilización de las
proteínas sobre membranas sintéticas, seguido de la detección empleando sistemas
especialmente diseñados para la tinción de 'blots'. El método más potente es el
denominado 'Western blot' en el que las proteínas son separadas en primer lugar
mediante electroforesis en geles de poliacrilamida y posteriormente se transfieren
mediante la aplicación de un campo eléctrico perpendicular al gel a una membrana.
Todo procedimiento de 'blotting' consta de 5 etapas :
•
Inmovilización de proteínas sobre la membrana mediante transferencia
electroforética.
•
Saturación de todos los lugares de unión de proteínas de la membrana no
ocupados para evitar la unión no específica de anticuerpos, que son proteínas.
•
Incubación del 'blot' con anticuerpo primarios contra la/s proteína/s de interés.
•
Incubación del 'blot' con anticuerpos secundarios, o reactivos que actúan de
ligando del anticuerpo primario unidos a enzimas u otros marcadores.
•
Las bandas de proteínas marcadas con enzimas se hacen visibles por incubación
con los sustratos apropiados para formar productos coloreados insolubles en el
lugar donde se encuentran las bandas de proteína.
El gel:
-
-
20 Gel al 12%.
6 ml Acrilamida (actúa como espesante).
3’75 ml 1’5M Tris pH 8’8.
5’05 ml H2O.
0’15 ml 10% SDS.
0’2 ml 25% Ampersulfato (hace solidificar la acrilamida).
0’03 ml TEMED (tetrametiletildiamina).
20
Prácticas en CIC
El gel diferencial:
En la parte superior del gel, coincidiendo con la zona de inclusión del peine formador de
pocillos se dispone otro tipo de gel:
- -
Marcado con 1’38 ml. de Bromofenol dando apariencia azulada para observar la
diferencia.
0’5 ml Acrilamida.
Tris a 0’5M pH 6’8.
3 ml Agua.
0’15 ml 10% SDS.
0’2 ml 25% Ampersulfato.
0’02 ml. TEMED.
Este gel será utilizado para depositar las
muestras en los pocillos y será separado
fácilmente del gel por el que correrán las bandas
antes de transferencia a membrana gracias a su
menor estabilidad y solidificación.
El grosor del gel es un factor que influye
notablemente en la transferencia, siendo muy
ineficiente a partir de geles de 2 mm de espesor,
y tanto más eficiente cuanto más fino es el gel,
con un límite debido a los problemas de
manipulación en torno a los 0.4 mm.
El gel se deja correr en la cubeta electroforética durante
2:15 horas a 85 Voltios.
Tras la extracción del gel lo colocamos en una cubeta con la
solución de transferencia durante 10 minutos (balanceo).
Solución de transferencia:
21 21
Prácticas en CIC
-
14’4 gr. Glicina.
3’03 gr. Tris (Trihidroximetil aminometano en polvo).
200 ml. Metanol.
Lo ajusto hasta un litro con agua destilada.
Disolución y
conservamos a 4ºC.
hasta que se use.
Preparamos la membrana de nitrocelulosa a la que pasaremos las proteínas del gel para
un correcto estudio con anticuerpos. Dicha membrana tiene como medidas 6x8cm.
Para su utilización pasamos la membrana a una cubeta con metanol durante un minuto y
luego mantendremos la misma en solución de transferencia cinco minutos.
Transferencia electroforética ('electroblotting'):
En este método, descrito por Towbin, las proteínas son transferidas desde el gel
a la membrana electroforéticamente.
La ventaja de este proceso es su
corta duración (de 30 min a pocas
horas), lo que reduce notablemente
el efecto de difusión de las bandas.
El procedimiento se inicia apilando
sucesivamente sobre una esponja
plana papel de filtro empapado en
tampón de transferencia, el gel, la
membrana en contacto directo con el gel, más papel de filtro y finalmente una esponja
plana. Este conjunto se recoge entre dos capas
de plástico perforado y se introduce en un
tanque en el que se encuentra una solución
salina (tampón de transferencia) y dos
electrodos planos (diseñados para conseguir
un campo uniforme en toda la superficie del
gel). Se dispone de forma que el gel quede
hacia el ánodo (-) y la membrana hacia el
cátodo (+). La carga neta de las proteínas en
el caso de los geles de SDS-PAGE es positiva
debida a la carga del SDS.
Debemos además tener en cuenta a la hora de preparar esta transferencia de
echar solución de transferencia al colocar cada una de las capas así como alisarlas para
que quede bien prensado y sin burbujas.
Una vez montado se inserta dentro de la cubeta en el rail facilitado a tal efecto
acompañado de un depósito para hielo necesario para mantener la temperatura baja. En
el fondo de la cubeta principal colocamos un imán (“estéril”) ya que el tiempo que
22 22
Prácticas en CIC
tengamos la cubeta electroforética conectada la mantendremos encima de un rotatorio
para que la solución de transferencia se encuentre en movimiento gracias a dicho imán.
Dejamos la cubeta conectada toda la noche a 35 Voltios en la cámara fría de 4ºC de la
que disponemos.
Al día siguiente tras extraer el gel de la cubeta obtenemos la membrana ya marcada;
debemos secarla para lo que es suspendida en metanol en dos ciclos consecutivos de un
minuto.
Una vez realizada la transferencia la membrana se puede analizar inmediatamente o
bien conservarla en frio (2 a 8ºC) durante meses.
Tras el secado utilizamos solución Ponceau, un marcador granate que nos permite
observar a simple vista todas las fracciones proteicas en membrana según el peso
molecular dado en KD. (el Ponceau no lo desechamos sino que es reutilizable).
Pasamos la membrana por una serie de lavados mediante solución de lavado TBST
cambiándola cada 5 minutos (3 ciclos).
Tras estos lavados preparamos una nueva solución, esta vez de leche en polvo libre de
grasa con TBST, se utiliza para rellenar los espacios libres de la membrana sin proteína
con la proteína de la leche y así evitar un contacto inespecífico entre la membrana y los
anticuerpos primarios que insertaremos. Dejamos por ello la membrana durante 2 horas
en hibridación con la leche en el balanceador.
Además, cada membrana que estudiamos la cortamos dividiéndola (por los Kda de las
fracciones proteicas mediante el patrón de estudio) en los distintos fragmentos que
recibirán un tratamiento diferencial dependiendo de los estudios a realizar: en los
distintos western hemos realizado tratamiento para estudiar p53, p21, caspasas activas e
inactivas, Smuc,…
Utilizamos Anticuerpos específicos primarios con una concentración que se conoce de
antemano que funciona, la mayoría 1:500 aunque alguno es 1:1000. Dejamos que actúe
durante dos horas sobre la membrana.
Lavamos con TBST 6 veces tras la aplicación del primario, 3 lavados cada 5 minutos y
3 lavados cada 10.
23 23
Prácticas en CIC
El Anticuerpo secundario lo dejamos actuar durante una hora tras lo cual se realizan los
lavados con TBST igual que en el caso anterior.
Dado que todos los trabajos que realizamos son comparativos es importante que no
existan errores de nuestra manipulación en los pocillos y por tanto una seña que nos
permita conocer que los resultados son comparables, poder relacionar el régimen de
bandas; para ello realizaremos sobre las muestras tratadas con los anticuerpos un
posterior tratamiento con anticuerpos para Actina (marcador de tamaño). El protocolo
de actuación es igual al anterior comenzando por secar con metanol la membrana, lavar
con TBST, bloquear con leche,… Esta segunda señalización de la membrana con
anticuerpos es posible debido a la distinta naturaleza entre los anticuerpos utilizados.
Tras estos tratamientos se utiliza la solución de western comercial que constituye una
reacción enzimática con el anticuerpo secundario y revelaremos por Rayos X
manteniendo el cassette dentro de la cámara oscura.
24 24
Prácticas en CIC
Por un lado el trabajo con Isabel Lara (Bióloga y bioquímica) me ha permitido conocer
técnicas utilizadas en tecnología de ADN recombinante y por otro conocimientos sobre
asilamiento y purificación de proteínas para su posterior cristalización.
Tecnología de ADN recombinante: Vector Ply 6 – CRE ert2:
Realizamos la separación de un fragmento de ADN con el que queremos realizar
un vector; tras la digestión previa hemos realizado un gel y gracias a la luz ultravioleta y
mediante una cuchilla hemos tomado el fragmento de ADN con el tamaño que interesa.
25 25
Prácticas en CIC
Mediante un kit comercial se separa el ADN de la Agarosa, este proceso conlleva
separación del ADN mediante filtro, centrifugaciones y distintos buffer.
Finalizada la extracción del fragmento se quiere ligar a otro plásmido y para ello es
necesario utilizar ligasas y buffer en distintas proporciones. Las proporciones que
hemos utilizado en este caso son las siguientes:
-
7 (fragmento) : 1 (plásmido)
13(fragmento) : 3 (plásmido)
1 Buffer + 1 enzima Ligasa = 10 µl.
2 Buffer + 2 enzima Ligasa = 20 µl.
Toda la noche
a 16ºC.
Para su conservación al día siguiente tras una corta centrifugación se guarda a -20ºC.
La fracción 13:3 la utilizamos para hacer Transformación, utilizamos células
competentes EColi que asimilarán el plásmido y lo harán actuar.
-
200 µl de células competentes + 10 µl de 13:3. Lo metemos en hielo media hora.
Lo pasamos a 40ºC durante un par de minutos y de nuevo a hielo provocando así
el choque térmico que hará que la célula asimile el plásmido.
Echamos medio de cultivo y lo dejamos crecer a 37ºC y después Plaqueamos.
Placas Petri con medio LB Agar con Ampicilina (células competentes resistentes
a ampicilina).
Realizamos 6 placas de Ply6 CRE ert2 utilizando para cada una 100 µl que
esparcimos con el asa de siembra hasta que la superficie quede seca.
Para comprobar las ligaciones y adquisición del vector se realiza un gel al 0’8% de
Agarosa:
- 50 ml TBE.
- 0’4 gr. Agarosa.
- 1 µl de Bromuro de Etidio.
El contenido del pocillo:
o 4 µl de muestra purificada con el kit Adn con buffer de elución de la
fracción 13:3.
o 4 µl Loading Buffer.
o 16 µl de MPW.
-
En cubeta a 60 Voltios durante 1:30 h.
MINIPREPs: -
26 Echamos en 6 tubos 2 ml. de medio 2xYT y 4 µl de Ampicilina.
26
Prácticas en CIC
-
-
-
Tomamos con punta de pipeta una colonia al azar de cada placa Ply6 CRE ert2,
la sembramos en una placa común control y la punta la metemos en el tubo con
2xYT-AMP y lo ponemos en estufa en agitación a 37ºC hasta el día siguiente.
Las placas Ply6 CRE ert2 utilizadas las conservamos a 4ºC y la nueva placa
control se pone a crecer a 37ºC.
Vortex y centrifugación de los tubos durante 1 minuto.
Resuspender pellet en 250 µl de STETL (STET + Lisozima (2’5 µl)).
Tubos en baño de agua hirviendo.
Spin durante 10 minutos a 12000 r.p.m.
Eliminación del pellet mediante un palillo.
Añadimos unos 210 µl de isopropanol y lo dejamos 5 minutos tras lo cual
realizamos un spin a temperatura ambiente, decantamos, spin y quitamos restos
de sobrenadante.
Finalmente se resuspende en 30 µl de TE con RNasa (50 µg/ml), se deja 10
minutos a temperatura ambiente y finalmente se congela.
ENZIMAS DE RESTRICCIÓN: Las enzimas o endonucleasas de restricción son proteínas que reconocen secuencias
particulares del ADN, uniéndose a ellas y cortando en sitios específicos siendo
empleado en el laboratorio para cortar moléculas de ADN. Las secuencias reconocidas
por las enzimas de restricción son generalmente de cuatro o seis pares de bases y
palindrómicas, es decir que la secuencia de bases leída de 5’ a 3’ en ambas hebras es la
misma. Las enzimas de restricción de tipo II cortan en una posición específica del ADN
dentro de su secuencia de reconocimiento, denominada sitio de restricción.
Algunas enzimas de restricción cortan cada cadena del ADN en diferente sitio, dando
lugar a extremos cohesivos, mientras otras cortan en el mismo sitio ambas cadenas,
dando lugar a extremos romos. Cada enzima trabaja bajo condiciones apropiadas de
concentración salina, pH y temperatura.
Una molécula de ADN cortada por una determinada enzima de restricción originará una
serie de fragmentos cuyo número y tamaño dependerá de la cantidad y la localización de
los sitios de reconocimiento que existan para esa enzima en esa molécula de ADN. Los
fragmentos generados por la digestión con enzimas de restricción pueden unirse por la
acción de enzima ADN ligasa, formando enlaces fosfodiester entre extremos 5’ y 3’ de
los nucleótidos. De esta forma, los extremos romos pueden unirse independientemente
de la secuencia que contengan, mientras que los extremos cohesivos sólo se unirán si
existe complementariedad entre las porciones de cadena.
En nuestro caso realizamos la digestión con BamH1, que nos fragmentará el plásmido
en 3 zonas y observando el tamaño de los fragmentos sabremos si ha sido efectivo. 30
minutos a 37ºC con BamHI de digestión.
Tras la digestión y el revelado del gel observamos que la colonia 5 encaja con los
fragmentos de la digestión que buscábamos.
27 27
Prácticas en CIC
Para comprobar que realmente es un positivo realizaremos la digestión con Eco RI / Not
I. (se dio buena digestión).
Con los resultados obtenidos en el gel (que se hizo con: Marcador / Ply6 CRE ert2BamHI / Ply6 CRE ert2 – EcoRI / NotI.) se debe realizar un Southern blot para
hibridarlo. (Técnica Southern será explicada más adelante). Es importante el marcaje
con la sonda para conocer si el fragmento está colocado correctamente ya que los
extremos no son cohesivos por lo que puede ponerse al derecho o al revés (sens o
antisens).
Finalmente las pruebas de hibridación no dieron los resultados esperados y se tuvo que
comenzar el proceso de introducción del vector de nuevo.
Purificación proteica: Con respecto a sus estudios proteicos, con Isabel pude conocer el proceso de
aislamiento de las proteínas mSlug y mSnail tanto en formato full length como
fragmentos de extremos amino y carboxi terminal.
Exponiendo brevemente todo el proceso que llevaba a cabo: aislando los fragmentos de
gen en PCR y tras realizar ligaciones en plásmidos, realizaba turbos para conocer las
colonias positivas de las que realizaba minis y midipreps. Las transformaba a BL21
conociendo mediante western si realmente expresaban sus proteínas tras lo cual obtenía
extractos proteicos que pasaba por columna de agarosa con níquel separando gracias al
brazo de 5 histidinas a cada una de sus proteínas en diferentes fracciones de imidazol,
siendo estos fragmentos obtenidos estudiados mediante Western y Comassie dializando
fracciones donde hay proteína, purificando así por diálisis y urea en distintas
concentraciones hasta llevarlo a PBS y a partir de ahí separar por centrifugación la parte
soluble que finalmente se mantiene a -70ºC para su posterior Cristalización. El
conocimiento de la estructura molecular tridimensional de un compuesto es a menudo la
condición sine qua non para avanzar en determinadas líneas de investigación por lo que
los estudios realizados permitirán avanzar en el estudio de los genes de la familia Snail.
Pude seguir la diálisis
28 28
Prácticas en CIC
La muestra conteniendo la proteína se introduce en una bolsa o tubo de membrana
semipermeable y se cierran los extremos como se muestra en la figura.
Cuando esta bolsa se suspende en un volumen suficientemente grande de solución
tamponada de fuerza iónica apropiada, la membrana permite el intercambio de sales y
tampón entre el interior y el exterior de la bolsa, pero no el de proteína.
Preparación de la tripa de diálisis:
-
-
-
Se corta membrana semipermeable en trozos de
10-20 cm con precaución de no doblarlos ni
pincharlos.
Hervir 10 minutos en un volumen grande de
bicarbonato de sodio 2%/ 1 mM de EDTA pH = 8.
Colocamos matraz sobre las tripas para que no
floten.
Aclarar abundantemente por fuera y por dentro con
ayuda de pipeta con MPW.
Hervir durante otros 10 minutos en 1mM de
EDTA pH 8.
Dejar enfriar y guardar a 4ºC.
Se lava con MPW.
Para realizar la solución
membranas para diálisis:
para
introducir
las
o 500 ml MPW.
o 10 gr. de NaHCO3.
o 1 ml EDTA.
Es una solución necesaria para la hidratación de las membranas. La solución
posterior sólo posee EDTA.
También he podido seguir los estudios Western para las fracciones dializadas y la
detección de la fracción final soluble.
29 29
Prácticas en CIC
Gracias a la Técnica Esther he tenido la posibilidad de conocer el registro diario que se
lleva a cabo con los ratones del animalario y la actualización puntual de sus fichas en la
base informática.
Además tuve la posibilidad de pasar un par de días con ella para conocer de forma
práctica la técnica Southern-Blot utilizada para genotipar a los nuevos ratones
procedentes de los cruces en el animalario.
Southern‐Blot: Southern detecta la presencia de una secuencia de ADN en una mezcla compleja de
este ácido. Para ello, emplea la técnica de electroforesis en gel de agarosa con el fin de
separar en base a la longitud de los fragmentos de ADN y, después, una transferencia a
una membrana en la cual se efectúa la hibridación de la sonda.
-
-
30 Trituramos los fragmentos de cola de ratón con Formace y se deja toda la noche
a 55ºC.
A la mañana siguiente se realiza spin y se añaden 450 µl de fenol a cada
Eppendorf.
Agitación y centrifuga a 12000 rpm durante 10 minutos. Tras ello nos quedan
dos fases: la superior transparente y la inferior amarillenta.
El fase superior se pone en otro eppendorf con 500 µl de cloroformo.
El nuevo sobrenadante tras la centrifugación con cloroformo se pone en un
nuevo eppendorf con 980 µl de etanol 100%. Se precipita por spin y vertemos
fuera el etanol quedando el precipitado.
Utilizamos ahora etanol al 70%.
Tras un nuevo lavado y spin con el etanol, este se elimina secando bien el
precipitado.
Resuspensión en agua (unos 45 µl MPW aunque depende de la resolución de la
digestión). Se deja 30 minutos.
Preparación de la digestión, por cada eppendorf:
o 3 µl DTT.
o 3 µl S.
o 3 µl 10x solución de Carlos.
o 1 µl EcoRI.
30
Prácticas en CIC
-
Lo realizo común a todas las muestras y voy echando 10 µl en cada tubo
eppendorf + 20 µl de la suspensión de ADN anterior.
En baño a 37ºC durante 4 horas.
-
Los geles los realizamos al 0’8% de Agarosa con AGB como Buffer.
-
Tras cargar las muestras se pone el gel a 35 Voltios durante 12 horas.
-
-
Al día siguiente el gel que ha corrido se pasa a recipientes de plástico con el
buffer en el que ha corrido y se añaden tres gotas de Bromuro de Etidio con
pipeta pasteur y lo dejamos actuar durante unos 5 minutos.
Lavado del gel con agua destilada.
Revelado en la cámara ultravioleta.
-
A continuación realizamos en estos geles lavados (en el balanceador):
-
o 45
minutos
con
solución
desnaturalizante (500 ml/gel).
o Lavado con agua destilada.
o 45
minutos
con
solución
neutralizante (500 ml/gel).
o Lavado con agua destilada.
Realizamos
el
montaje
de
la
transferencia a membrana, que en el caso
del southern es de nailon.
o Peso (botella).
o Cristal.
31 31
Prácticas en CIC
Toallas absorbentes.
3 filtros Whatman. (18x18 cm).
Membrana nailon (18x18 cm).
Gel.
Filtros Whatman que se encuentran en contacto con la solución.
(36x20cm).
o Cristal.
o Bandeja contenedora de 600 ml de 20xSSCC.
Se deja toda la noche.
- Al día siguiente se desmonta el aparataje de transferencia y antes de retirar el gel
marcamos con un lápiz los pocillos en la membrana.
- Las membranas se someten a un UVcrosslink para fijar así el ADN a su
superficie.
- Se cortaran las membranas separando los pocillos dependiendo de la sonda
utilizada.
- A continuación se dará la hibridación:
o Preparación para cada tubo de hibridación de 250 µl de esperma de
salmón y 250 µl de Tris y 45 ml de Solución de Hibridación. El esperma
de salmón es de cadena simple y se une a todo el ADN y competirá con
la sonda de forma que hará la unión más específica.
o Se introduce en el horno con rotores cilíndricos a 65ºC y se comprueba
que no existan burbujas entre la membrana y el cristal del tubo.
Permanece con la solución de hibridación 2 horas.
o
o
o
o
o
o A continuación 250 µl de la sonda a cada tubo de hibridación y se deja
actuar toda la noche.
32 32
Prácticas en CIC
-
A la mañana siguiente se lava la membrana con solución de lavado (1xSSCC,
0.1 de SDS + MPW).
Secado parcial.
Se pone a exponer la membrana tras cubrirla con film transparente hasta el día
siguiente; y si los resultados de exposición no son suficientes ha de dejarse unos
5 días.
Seguimiento del trabajo de otros compañeros: Junto a mi compañera Alejandra he seguido sus estudios en la digestión de un plásmido,
en este caso Bluescript, para observar la posible correcta introducción del Vector
EML4-ALK (Equinodermic microtubulic likefour – Anaplastic lynphoma quinasa).
También he colaborado con ella en la realización de Turbos (Turboscreening) para la
identificación de colonias marcadas raciactivamente así como en el protocolo de cultivo
de ES-CELLs.
Junto a mi compañera Inés he podido observar como llevaba a cabo la toma de muestras
de los ratones del animalario para los experimentos del laboratorio dándome la
posibilidad de conocer crecimientos tumorales en los roedores.
Junto a la técnica Beatriz he podido seguir de primera mano el tratamiento de muestras
de los tejidos animales, básicos para los estudios de tumorigénesis que lleva a cabo el
laboratorio. Para ello he realizado visitas a la unidad de Anatomía patológica en el
Hospital Clínico Universitario, que es donde se tratan dichas muestras.
El tratamiento de las muestras comienza con su inmersión en formol, se pasa la muestra
por el Tecnicom después de haberlas tallado, así se realizan baños en xilol para que se
eliminen los excesos sanguíneos y se deje la muestra en condiciones óptimas para su
paso por parafina mediante cassette.
Tras el enfriamiento y solidificación en parafina he llevado a cabo el calibrado mediante
desbaste con el micrótomo para posteriormente realizar los cortes deseados.
Tras realizar los cortes:
- Si vamos a usar una tinción hematoxicilina-eosina se pasa directamente los
cortes a una máquina que automáticamente realiza el lavado en distintas
concentraciones de xilol (quita restos de parafina), hematoxicilina, xilol y
eosina.
- Si deseamos realizar inmunohistoquímica se dejaran los cortes en baño a 65ºc
toda la noche para quitar la parafina y después la inmunohistoquímica, mediante
33 33
Prácticas en CIC
la introducción de los parámetros necesarios en un ordenador, se realiza de
forma completamente automática.
Finalmente protegemos los portas introduciéndolos en una máquina que los cubre
con goma arábiga.
Otras Actividades Reuniones semanales: A nivel de laboratorio se realiza una exposición común ante Isidro y los demás
compañeros del equipo de los pasos dados en los distintos proyectos, así como las
imperfecciones, fallos y posibles soluciones mediante un debate común que permite a
los investigadores estar al día del trabajo global que se realiza en el laboratorio 13.
Journal Club: Cada dos semanas en laboratorio organiza esta actividad que consiste en la selección
por parte de uno de los compañeros de un artículo científico publicado que le interese
(normalmente de temática relacionada con la desarrollada en el laboratorio) y lo expone
a sus compañeros.
Además de los beneficios prácticos de la exposición pública me resultó muy
constructivo conocer como se analiza de forma común el artículo desde el punto de vista
técnico, práctico y formal gracias a que los presentes poseen con una anterioridad de
una semana aproximadamente el artículo para leerlo y analizarlo siendo muy fructífera
su puesta en común.
Artículos seleccionados durante mi estancia:
Generation of a function mammary gland from single stem cell.
Purification and unique properties of mammary epithelial stem cells.
GATA-3 links tumor differentiation and dissemination in a luminal breast cancer
model.
34 34
Prácticas en CIC
Conferencias: El CIC organiza una gran cantidad de conferencias que se realizan en el propio centro y
permiten, tras la exposición del tema, una interacción entre los investigadores del centro
y los invitados conferenciantes de otros centros favoreciendo el flujo de técnicas, ideas
y descubrimientos.
Asistí a las siguientes:
Wisdom from worms on the nature of cells and tissues: fusing cells, fusing photons and
pharmacology.
William A. Mohler. Dept. of genetics and developmental biology. University of
Connecticut health center, Michigan.
TGFB: from cytostosis to tumor progression and metastasis.
Roger Gomis. ICREA researcher, Institute for research in biomedicine IRB, Barcelona.
35 35
Prácticas en CIC
Conclusiones: Estas prácticas han sido un gran complemento para mi formación académica, he
podido poner en práctica muchos conocimientos y adquirir nuevos. Gracias a la
amplitud de investigaciones que se dan en el laboratorio y la posible movilidad dentro
del equipo he podido conocer el funcionamiento global del mismo y los proyectos
individuales. Cabe destacar la gran cantidad de medios no sólo humanos sino también
recursos técnicos con que cuenta este laboratorio y que juntos permiten que el equipo
funcione tan bien.
Además he tenido la gran suerte de encontrarme un perfecto grupo de trabajo en el
laboratorio de Dr. Isidro Sánchez García, gente motivada por sus quehaceres que han
sabido y querido transmitirme sus conocimientos y me han hecho sentir uno más.
Gracias a todos ellos por estas prácticas que recomiendo fervientemente a todos mis
compañeros de titulación.
36 36
Prácticas en CIC
Bibliografía: www.cicancer.org.
Function of the Zinc-Finger transcription factor SNAI2 in Cancer and development.
Isidro Sánchez García, César Cobaleda (et.al)
Annual review of Genetics.2007.41: 41-61.
SLUG in cáncer Development.
Isidro Sánchez García (et.al)
Oncogene (2005) 24, 3073-3082.
http://users.ugent.be/~avierstr/principles/pcr.html
http://www.javeriana.edu.co/Facultades/Ciencias/neurobioquimica/libros/celular/electro
foresis.html
http://www.ub.es/biocel/wbc/tecnicas/westernblot.htm#arriba
http://upload.wikimedia.org/wikipedia/commons/a/ae/Western_blot.jpg
37 37
38 Prácticas en CIC
38