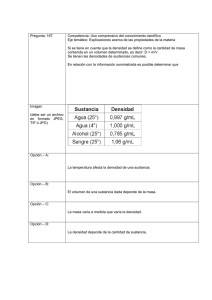
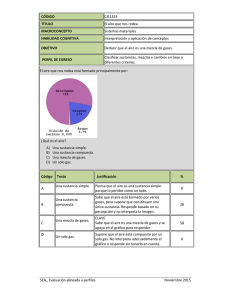
compuestos y composiciones farmaceuticas para el
Anuncio

19 OFICINA ESPAÑOLA DE PATENTES Y MARCAS 11 Número de publicación: 2 225 834 51 Int. Cl. : C07H 15/203 7 C07H 15/04 C07H 23/00 A61K 31/70 A61P 31/04 ESPAÑA 12 TRADUCCIÓN DE PATENTE EUROPEA T3 86 Número de solicitud europea: 95902653 .5 86 Fecha de presentación: 18.11.1994 87 Número de publicación de la solicitud: 0730601 87 Fecha de publicación de la solicitud: 11.09.1996 54 Título: Compuestos y composiciones farmacéuticas para el tratamiento y profilaxis de infecciones bacteria nas. 30 Prioridad: 18.11.1993 US 154035 73 Titular/es: Washington University Box 8013, 724 South Euclide Avenue Saint Louis, Missouri 63110, US Siga Technologies, Inc. 45 Fecha de publicación de la mención BOPI: 16.03.2005 72 Inventor/es: Hultgren, Scott; Kuehn, Meta; Xu, Zheng; Ogg, Derek; Harris, Mark; Lepisto, Matti; Jones, Charles, Hal y Kihlberg, Jan 45 Fecha de la publicación del folleto de la patente: 74 Agente: Carpintero López, Francisco ES 2 225 834 T3 16.03.2005 Aviso: En el plazo de nueve meses a contar desde la fecha de publicación en el Boletín europeo de patentes, de la mención de concesión de la patente europea, cualquier persona podrá oponerse ante la Oficina Europea de Patentes a la patente concedida. La oposición deberá formularse por escrito y estar motivada; sólo se considerará como formulada una vez que se haya realizado el pago de la tasa de oposición (art. 99.1 del Convenio sobre concesión de Patentes Europeas). Venta de fascículos: Oficina Española de Patentes y Marcas. C/Panamá, 1 – 28036 Madrid ES 2 225 834 T3 DESCRIPCIÓN Compuestos y composiciones farmacéuticas para el tratamiento y profilaxis de infecciones bacterianas. 5 Campo de la invención 10 La presente invención se refiere a sustancias para uso en el tratamiento y/o profilaxis de enfermedades provocadas por bacterias que forman pilus que se adhieren al tejido por interacción con la unión entre las subunidades de pilus y las chaperonas periplásmicas. La invención también se refiere a procedimientos para identificar/diseñar sustancias capaces de interactuar con las chaperonas periplásmicas y a procedimientos para identificar los sitios de unión en las chaperonas periplásmicas. Finalmente, la invención se refiere a nuevas sustancias capaces de interactuar con las chaperonas periplásmicas así como a preparaciones farmacéuticas que comprenden sustancias capaces de interactuar con las chaperonas periplásmicas. 15 Antecedente de la invención La bacteria patogénica Gram-negativa provoca diversas afecciones patológicas tales como bacteremia, diarrea relacionada con bacterias, meningitis y (muy comúnmente) infecciones del tracto urinario, es decir, pielonefritis, cistitis, uretritis, etc. 20 25 30 35 Las infecciones del tracto urinario son una de las causas principales de morbididad en las mujeres. A pesar de la gran importancia de las infecciones del tracto urinario en las mujeres, se han hecho pocos esfuerzos para aplicar nuevas estrategias con el fin de tratar y/o prevenir estas enfermedades. Frecuentemente, se usan antibióticos convencionales para tratar estas infecciones, tales como el tratamiento con penicilinas, cefalosporinas, aminoglicósidos, sulfonamidas y tetraciclinas; en el caso especial de infecciones del tracto urinario, también se emplean antisépticos urinarios tales como nitrofuranotoína y ácido nalidíxico. Sin embargo, en el futuro la resistencia emergente a los antibióticos dificultará la capacidad para tratar satisfactoriamente las infecciones del tracto urinario. Está aumentando la resistencia a múltiples antibióticos entre estos uropatógenos. Se ha estimado que el coste anual para la evaluación y el tratamiento de mujeres con infecciones del tracto urinario supera el billón de dólares. Además, aproximadamente un cuarto del coste anual de 4 billones de dólares atribuidos a infecciones nosocomiales es una consecuencia de las infecciones del tracto urinario. Entre los agentes que causan las infecciones del tracto urinario, Escherichia coli predomina claramente entre las bacterias Gram-negativas. Las bacterias patogénicas gram negativas, notablemente Escherichia coli, Haemophilus influenzae, Salmonella enteriditis, Salmonella typhimurium, Bordetella pertussis, Yersinia pestis, Yersinia enterocolitica Helicobacter pylori y Klebsiella pneumoniae deben parte de su infectabilidad a su capacidad para adherirse a diversos tejidos epiteliales. De esta manera, por ejemplo E. coli se adhiere a las células epiteliales en el tracto urinario superior a pesar del efecto aclarador del flujo unidireccional de orina de los riñones. 40 Como se ha indicado anteriormente, las bacterias mencionadas anteriormente están implicadas en diversas enfermedades: Infecciones del tracto urinario (E. coli), diarrea aguda (E. coli, Y. eterocolitica y Salmonella spp), meningitis (E. coli y H. influenzae), tos convulsiva (B. pertussis), peste (Y. pestis), neumonía y otras infecciones del tracto respiratorio (K. pneumoniae, H. influenzae) y úlcera péptica (H. pylori). 45 Se cree que el inicio y persistencia de muchas infecciones bacterianas tales como las descritas anteriormente requieren la presentación de adhesinas en la superficie del microbio en configuraciones accesibles que promueven los eventos de unión que dictan si se producirá la colonización extracelular, internalización u otras respuestas celulares. Las adhesinas normalmente son componentes de los anexos proteicos heteropoliméricos largos, finos y filamentosos conocidos como pili, fimbrias o fibrillas (estos tres términos se usarán indistintamente en este documento). El evento de unión bacteriana normalmente es el resultado de un ajuste estereoquímico entre una adhesina frecuentemente localizada en la punta del pilus y las arquitecturas de un receptor específico en las células huésped, que comprende normalmente estructuras carbohidrato en glicoconjugados asociados a la membrana. 50 55 60 65 Las cepas uropatogénicas de E. coli P expresan los pili P y de tipo 1 que se unen a los receptores presentes en células uroepiteliales. La adhesina presente en la punta del pilus P, PapG (polipéptido G asociado al pilus), se une al resto Galα(1-4)Gal presente en las globoseries de glicolípidos, mientras que la adhesina de tipo 1, FimH, se une a la D-manosa presente en glicolípidos y glicoproteínas. Los pili P adhesivos son determinantes virulentos asociados con cepas pielonefríticas de E. coli mientras que los pilus de tipo 1 parecen ser más comunes en E. coli provocando cistitis. Al menos once genes están implicados en la biosíntesis y expresión de los pili P funcionales; se ha determinado la secuencia de ADN del grupo del gen pap entero. Los pili P están compuestos de fibras heteropoliméricas compuestas de fibrillas adhesivas flexibles unidas de extremo a extremo a los bastoncillos del pilus. El bastoncillo del pilus se compone de subunidades proteicas PapA repetidas organizadas en un cilindro helicoidal a mano derecha. Se descubrió que las fibrillas terminales que se extienden de los extremos distales de cada bastoncillo del pilus están compuestas mayormente de subunidades repetidas de PapE organizadas en una conformación helicoidal abierta. La adhesina PapG se localizó en los extremos distales de las fibrillas terminales, un lugar que se asume que maximiza su capacidad para reconocer los receptores de glicolípidos en células eucariotas. Dos componentes minoritarios del pilus, PapF y PapK, son proteínas de adaptoras especializadas que se encuentran en la fibrilla terminal. PapF se une al residuo adhesina en la fibrilla mientras que PapK une la fibrilla al bastoncillo del pilus. La composición de la arquitectura de la fibra del 2 ES 2 225 834 T3 5 10 15 20 pilus P revela la estrategia usada por E. coli uropatogénico para presentar la adhesina PapG a los receptores eucariotas. El bastoncillo PapA rígido extiende la adhesina lejos de la interferencia provocada por LPS y otros componentes en la superficie celular bacteriana mientras que la fibrilla flexible permite a PapG una libertad estérica para reconocer y unirse al residuo digalactósido en el uroepitelio. Con unas pocas excepciones, la organización estructural de los pili de tipo 1 es muy similar a la descrita para los pili P. En los pili de tipo 1, la manosa que se une a la adhesina terminal fibrilar se conoce como FimH. La unión de los pili asociados con virulencia en patógenos Gram-negativos requiere la función de chaperonas periplásmicas. Las chaperonas moleculares son componentes vitales de todas las células vivas, procariotas y eucariotas. Las chaperonas ofrecen diversas funciones celulares incluyendo plegamiento, importación y exportación de proteínas en diversos compartimentos celulares (Gething y Sambrook, 1992). De esta manera, una chaperona periplásmica es una chaperona molecular que ejerce su acción en el espacio periplásmico en las bacterias. PapD y FimC son las chaperonas periplásmicas que median la unión de los pili P y de tipo 1, respectivamente. Los análisis estructurales detallados han revelado que PapD es el miembro prototipo de una familia conservada de chaperonas periplásmicas en bacterias Gram-negativas. Estas chaperonas tienen una función que es parte de una estrategia general usada por bacterias para tapar y repartir las subunidades interactivas importadas en el espacio periplásmico en los complejos de componentes de unión, sin realizar interacciones productivas desfavorables. La determinación de la estructura tridimensional de PapD revela que ésta se compone de dos dominios de tipo inmunoglobulina orientados en un configuración en forma de bumerang de tal manera que se forma una hendidura. PapD se une a cada uno de los tipos de subunidad de pilus ya que emergen de la membrana citoplásmica y las acompaña en el componente de unión, conformaciones de tipo natural de la membrana citoplásmica a los sitios de unión exterior de la membrana compuestos de PapC. PapC se refiere a un acomodador molecular ya que recibe los complejos chaperona-subunidad e incorpora, o hace pasar, las subunidades del complejo de chaperonas en el pilus creciente en un orden definido. 25 30 35 40 45 50 55 60 65 Con la excepción de la clase de tipo IV de los pili, todos los otros sistemas del pilus bien caracterizados genéticamente en procariotas Gram-negativas contienen un gen análogo a PapD (Normark y col., 1986; Hultgren y col., 1991); véase también la tabla A. FanE, faeE, sfaE, ClpE y f17-D se han secuenciado (Lintermans, 1990; Schmoll y col., 1990; Bertin Y y col., 1993; Bakker y col., 1991) y codifica las chaperonas del pilus requeridas para la unión de los pili K99, K88, S y f17, respectivamente, en E. coli. La unión de los pili Klebsiella pneumoniae de tipo 3 y pili Haemophilus influenzae de tipo B requiere los productos genéticos mkrb y hifB, respectivamente (Gerlach y col., 1989; Allen y col., 1991). Las relaciones entre estructura-función de todas estas chaperonas se ha analizado usando sus secuencias de aminoácidos y la información de la estructura cristalina de PapD (Anders Holmgren y col., 1992). Los resultados han proporcionado una nueva percepción de las complejidades moleculares que se han conservado evolutivamente en esta clase de proteínas y sugieren similitudes estructurales significativas con las inmunoglobulinas. De esta manera, PapD es el miembro prototipo de una familia de proteínas chaperona periplásmicas que son necesarias para la correcta unión supramolecular de los pili bacterianos. Las chaperonas tales como PapD en E. coli se requieren para unir las proteínas del pilus mencionadas anteriormente importadas en el espacio periplásmico, para repartirlas en los complejos de unión competentes y para prevenir la agregación no productiva de las subunidades en el periplasma (Dodson y col., 1993). En ausencia de una interacción con la chaperona, las subunidades de pilus se agregan y se degradan proteolíticamente (Kuehn, Normak y Hultgren, 1991). Se ha descubierto recientemente (Strauch, Johnson y Beckwith, 1989) que la proteasa DegP es responsable en gran medida de la degradación de las subunidades de pilina en ausencia de la chaperona. Este descubrimiento ha permitido la aclaración del destino de las subunidades de pilus expresadas en presencia o ausencia de la chaperona usando antisuero monoespecífico en transferencias de western de la membrana citoplásmica, de la membrana exterior y de las proteínas periplásmicas preparadas de acuerdo con los procedimientos convencionales. La expresión de papG o papA en la cepa degP41 (una cepa DegP− de E. coli) en ausencia de una chaperona fue tóxica para la bacteria debido a la acumulación de estas proteínas en la membrana citoplásmica, lo que sugiere que la chaperona se requirió para importar la subunidad en el espacio periplásmico. La gravedad del defecto en el crecimiento se relacionó con el nivel de expresión de la subunidad de pilina, y generalmente fue más dramático con PapG que con PapA. La co-expresión de PapD bajo el control del promotor de arabinosa inducible recuperó el defecto de crecimiento asociado con la expresión de la subunidad en la cepa ddegP41 y permitió que se importase PapG en el periplasma. Hasta la fecha, se ha aclarado poco sobre los motivos del reconocimiento molecular de las chaperonas. Las chaperona citoplásmicas tales como SecB, GroEL y DNaK se unen a un grupo diverso de proteínas diana no plegadas en una secuencia de manera independiente (Gething y Sambrook, 1992). Recientemente, se ha sugerido que DNaK se une principalmente mediante el esqueleto peptídico de la diana (Landry y col., 1992) y que GroEL puede depender más de la hidrofobicidad de cadena lateral y de la capacidad de la secuencia diana para formar una hélice α anfipática (Landry y Gierasch, 1991). PapD difiere, sin embargo, en que parece que se une a sus proteínas diana en conformaciones plegadas (Kuehn y col., 1991). La estructura tridimensional de PapD se ha resuelto previamente (Holmgren y Branden, 1989). Esto ha demostrado que PapD consta en dos dominios globulares posicionados de tal forma que la configuración global de la molécula se parece a un bumerang con una hendidura entre los dos dominios. Cada dominio es una estructura de barril β formado por dos láminas plegadas β antiparalelas, envasadas fuertemente para formar un núcleo hidrófobo, con una topología 3 ES 2 225 834 T3 similar a la de un pliegue de inmunoglobulina. El dominio del extremo N de PapD se parece mucho a un dominio de variable Ig mientras que el dominio del extremo C de PapD se parece a CD4 (Wang y col., 1990, Ryu y col., 1990) y al receptor de la hormona de crecimiento humano (de Vos y col., 1992). 5 10 15 Una alineación estructural entre PapD y varias chaperonas periplásmicas que se pronostica que tienen una estructura similar de tipo inmunoglobulina ha identificado residuos invariantes, altamente conservados y variables en esta familia proteica (Holmgren y col., 1992). Los residuos más conservados parecen participarse manteniendo la estructura y orientación globales de unos dominios hacia otros. Sin embargo, dos residuos conservados, ARg-8 y Lys-112 son de superficie expuesta y orientada hacia la hendidura entre los dominios. La mutagénesis dirigida al sitio del aminoácido Arg-8 ha demostrado que forma al menos parte de la cavidad que se une a la subunidad de pilus (Holmgren y col., 1992; Kuehn y col., 1993). A partir de un análisis secuencial de varias proteínas de la subunidad de pilus mencionadas anteriormente, se ha observado que poseen diversas características comunes incluyendo homologías en los extremos C (véase también el ejemplo 2). Se cree que estas similitudes en la secuencia pueden ser responsables de alguna función común a todas las proteínas del pilus, tales como la unión a su chaperona periplásmica. De hecho, ya se ha demostrado que la región C-terminal de la adhesina de los pili P PpaG es importante en la unión in vivo a PapD (Hultgren y col., 1989). La Tabla A enumera 16 proteínas periplásmicas, todas implicadas en la unión de las estructura celulares superficiales en bacterias patogénicas y todas con homología significativa con PapD. 20 TABLA A 25 30 35 40 45 50 55 60 65 * Esta chaperona está presente en todas las Enterobacateriaceae, ya que todos los miembros de esta familia producen pili de tipo 1. 4 ES 2 225 834 T3 En resumen, se conocen la estructura tridimensional de PapD así como la función de PapD y otras chaperonas periplásmicas, mientras que se sigue desconociendo el motivo exacto de la unión entre PapD y las subunidades de pilus. 5 10 15 20 25 30 35 Descripción de la invención De acuerdo con la presente invención, se ha comprendido que las características mencionadas anteriormente de PapD y de otras chaperonas relacionadas las hacen dianas interesantes para los fármacos diseñados principalmente para reducir la patogenicidad de bacterias que se adhieren mediante pili; por ejemplo, un fármaco que bloquea la unión entre una chaperona y las subunidades de pilus (interfiriendo, por lo tanto, con el ensamblaje del pilus intacto) interferirá con la formación de los pili intactos, reduciendo por lo tanto la capacidad bacteriana para adherirse al epitelio del huésped. Con el fin de diseñar tal fármaco es de gran valor que se conozca con detalle el motivo de la unión entre la chaperona y la(s) proteína(s) del pilus, con el fin de desarrollar el procedimiento para identificar eficazmente los compuestos capaces de bloquear esta unión. Un aspecto de la presente invención se refiere a una sustancia para uso en el tratamiento y/o profilaxis de enfermedades provocadas por bacterias que forman pilus que se adhieren al tejido, que comprende prevenir, inhibir o mejorar la unión entre al menos un tipo de subunidad de pilus y al menos un tipo de chaperona molecular en bacterias que forman pilus, cuya chaperona molecular se une a las subunidades de pilus durante el transporte de estas subunidades de pilus a través del espacio periplásmico y/o durante el procedimiento de ensamblaje de pilus intacto, donde el sitio de unión es un sitio de unión al que se puede unir la parte carboxilo terminal de una subunidad de pilus, y que comprende puntos del sitio sustancialmente idénticos a los residuos invariantes Arg-8 y Lys-112 en PapD, y un fragmento polipeptídico que puede interactuar con una cadena β de la parte carboxilo terminal de la subunidad de pilus estabilizando, por lo tanto, la unión de dicha subunidad en los puntos del sitio Arg-8 y Lys-112 del sitio de unión. Un aspecto especialmente preferido de la invención es un procedimiento como se ha descrito anteriormente, donde el lado de unión es el sitio de unión a la proteína G de PapD como se describe en este documento. Como se usa en este documento, el término “pilus”, “fimbria” o “fimbrilla” se refiere a estructuras heteropoliméricas fibrilares incrustadas en la membrana externa de muchas bacterias patogénicas que se adhieren a tejidos, notablemente la bacteria patogénica gram negativa. En la presente memoria descriptiva, los términos pilus, fibrilla y fimbria se usarán indistintamente. Un pilus, como se ha explicado anteriormente, se compone de varias “subunidades de pilus”, que constituyen distintas partes funcionales del pilus intacto. Una subunidad de pilus muy importante es la “adhesina”, la subunidad de pilus que es responsable de la capacidad de unión a tejidos de la bacteria. 45 Por el término “chaperona molecular” se entiende una molécula que en las células vivas tiene la responsabilidad de unirse a los péptidos con el fin de madurar los péptidos de varias formas. Muchas chaperonas moleculares están implicadas en el procedimiento de plegamiento de péptidos en su conformación nativo mientras que otras chaperonas moleculares están implicadas en el procedimiento de exportación o importación de la célula de los péptidos. Las chaperonas moleculares especializadas son “chaperonas periplásmicas”, que son chaperonas moleculares bacterianas que ejercen sus acciones principales en el “espacio periplásmico” (el espacio entre la membrana bacteriana interior y exterior). Las chaperonas periplásmicas están implicadas en el procedimiento de corregir la unión de los pili intactos. Como se usa en este documento, el término simple “chaperona” designa una chaperona periplásmica molecular si no se indica otra cosa. 50 Cuando se usa la frase “un tipo de” se quiere decir que la subunidad de pilus o la chaperona en cuestión es de una especie distinta. Sin embargo, especialmente el hecho de que haya una amplia homología entre las diferentes especies de chaperonas periplásmicas moleculares hace probable que la interferencia con un tipo de chaperona usando por ejemplo un compuesto haga posible también el uso del compuesto en la interferencia con otras chaperonas. 40 55 60 65 La frase “prevenir, inhibir o mejorar la unión entre las subunidades de pilus y al menos una chaperona molecular en las bacterias que forman pilus” indica que la interacción normal entre una chaperona y su ligando nativo, es decir, la subunidad de pilus, está afectada por estar completamente o sustancialmente completamente prevenida, o por inhibirse o por expresarse de otra manera, reducida a tal punto que la unión de las subunidades de pilus a la chaperona es menos medible que en el caso en el que la chaperona interactúa con la subunidad de pilus en condiciones sustancialmente idénticas (con respecto al pH, concentración de iones y otras moléculas) a las condiciones naturales en el espacio periplásmico. De igual forma, la mejora de la unión entre la chaperona y la subunidad de pilus debe ser tal que la unión de las subunidades de pilus a la chaperona sea más medidle que en el caso en el que la chaperona interactúa con la subunidad de pilus en condiciones que son sustancialmente idénticas (con respecto a pH, concentración de iones y otras moléculas) a las condiciones naturales en el espacio periplásmico. La medición del grado de unión puede determinarse in vitro mediante procedimientos conocidos para el especialista en la técnica (microcalorimetría, radioinmunoensayos, inmunoensayos basados en enzimas, etc.). Debería estar claro, en función de lo explicado anteriormente, que la prevención o inhibición de la interacción normal entre una subunidad de pilus y una chaperona debería tener un efecto sustancialmente limitante en el ensamblaje de pilus. Sin embargo, también puede probarse que una mejora de la unión entre las subunidades de pilus y las chaperonas es devastadora para una bacteria. Como aparecerá a partir del ejemplo 2, diferentes subunidades de pilus 5 ES 2 225 834 T3 se unen a PapD con diferentes afinidades y afectar a este sistema por poco equilibrado también puede provocar una limitación en la velocidad y eficacia del ensamblaje de pilus. 5 10 Se cree que incluso cambios leves en la unión entre las subunidades de pilus y las chaperonas pueden tener un impacto dramático en la eficacia del ensamblaje de pilus, y por tanto en la capacidad de las bacterias para adherirse. Por ejemplo, si el cambio en la unión entre una chaperona y una subunidad de pilus es tal que el orden normal de afinidades entre las chaperonas y las subunidades de pilus que se unen normalmente a ellas se altera, entonces la unión normal del pilus debería alterarse, ya que el orden de ensamblaje de pilus puede depender i.a. de las afinidades entre las subunidades de pilus y la chaperona: Las subunidades de pilus con más afinidades a la chaperona pueden incorporarse antes de otras subunidades de pilus con menos afinidades. De esta manera, la prevención, inhibición o mejora de la unión entre las subunidades de pilus y una chaperona periplásmica molecular tienen un efecto de alteración del ensamblaje de pilus, por lo que se reduce la infectividad del microorganismo que expresa normalmente los pili. 15 20 25 30 La prevención, inhibición o mejora de la unión entre las subunidades de pilus puede realizarse de diversas formas. Un procedimiento preferido de acuerdo con la invención de tratamiento y/o profilaxis de enfermedades provocadas por bacterias que forman pilus que se adhiere a tejidos es administrar una cantidad eficaz de una sustancia a un sujeto en necesidad de la misma, siendo la sustancia capaz de interactuar con al menos un tipo de chaperona molecular que se une a las subunidades de pilus durante el transporte de estas subunidades de pilus a través del espacio periplásmico y/o durante el procedimiento de ensamblaje de pilus intacto, de tal manera que la unión de las subunidades de pilus a la chaperona molecular se previene, inhibe o mejora. La sustancia puede ser cualquier compuesto que tenga uno o más de los efectos mencionados anteriormente en la interacción entre chaperonas y subunidades de pilus y por lo tanto sobre el ensamblaje de pilus. Las sustancias especialmente interesantes son las que es probable que interactúen con la parte de la chaperona que se une a la subunidad de pilus, aunque la interacción con otros sitios en las chaperonas también puede provocar la prevención, inhibición o mejora de la unión entre las subunidades de pilus y la chaperona. Esto puede tener un efecto de bloqueo estérico directo de la unión normal entre la subunidad y la chaperona, pero también puede tener un efecto de cambio conformacional en la chaperona. Más adelante se describe un procedimiento para identificar sustancias para usar en el procedimiento de la invención. La interacción entre la sustancia y la chaperona puede ser una unión covalente así como una unión no covalente a la chaperona por parte de la sustancia. 35 Por el término “sujeto en necesidad del mismo” se entiende, en el presente contexto, un sujeto, que puede ser cualquier animal, incluyendo un ser humano, que está infectado con, o que es probable que esté infectado con, bacterias que forman pilus que se adhieren a tejidos que se cree que son patogénicas. 40 45 Por la expresión “una cantidad eficaz” se entiende una cantidad de la sustancia en cuestión que tendrá en la mayoría de los pacientes el efecto de curar la enfermedad provocada por bacterias patogénicas o, si la sustancia se ha dado profilácticamente, el efecto de prevenir la manifestación de la enfermedad. La expresión “una cantidad eficaz” también implica que la sustancia se da en una cantidad que sólo provoca efectos leves o no adversos en el sujeto al que se administra, o que los efectos adversos pueden tolerarse desde un punto de vista médico y farmacéutico en vista de la gravedad de la enfermedad para la que se ha suministrado la sustancia. La vía de administración de la sustancia podría ser cualquier vía de administración convencional, es decir, oral, intravenosa, intramuscular, intradérmica, subcutánea, etc. Se prefiere la vía oral. 50 55 60 Se espera que la dosificación de tal sustancia sea la dosificación que se emplea normalmente cuando se administran fármacos antibacterianos a pacientes o animales, es decir, 1 µg-1000 µg por kilogramo de peso corporal al día. La dosificación dependerá parcialmente de la vía de administración de la sustancia. Si se emplea la vía oral, la absorción de la sustancia será un factor importante. Una absorción lenta supondrá que el tracto gastro-intestinal serán necesarias concentraciones más altas, y por tanto serán dosificaciones más altas. Además, la dosificación de tal sustancia cuando se tratan infecciones del sistema nervioso central (SNC) dependerá de la permeabilidad de la barrera hematoencefálica para la sustancia. Como se bien se sabe en el tratamiento de la meningitis bacteriana con penicilina, son necesarias dosificaciones muy altas con el fin de obtener concentraciones eficaces en el SNC. Se entenderá que la dosificación apropiada de la sustancia deberá evaluarse adecuadamente realizando ensayos en modelos animales, donde el nivel de dosis eficaz (por ejemplo, DE50 ) y el nivel de dosis tóxica (por ejemplo, DT50 ) así como el nivel de dosis letal (por ejemplo, DL50 o DL10 ) se establecen en modelos animales adecuados y aceptables. Además, si se ha probado que una sustancia es eficaz en tales modelos animales, deberán realizarse ensayos clínicos controlados. Es innecesario establecer que tales ensayos clínicos deberán realizarse de acuerdo con los patrones de la Práctica Clínica Adecuada. 65 Aunque la vía preferida de prevención, inhibición o mejora de la unión entre las subunidades de pilus y las chaperonas es administrar una sustancia con los efectos mencionados anteriormente en la chaperona, también son posibles otras vías. Por ejemplo, las sustancias que interactúan con un tipo de subunidad de pilus también podrían tener los 6 ES 2 225 834 T3 efectos descritos anteriormente, y por las mismas razones. Sin embargo, como es probable que la interacción con la chaperona ejerza efectos en la unión en el pilus de la mayor parte, si no de todas, las subunidades de pilus que constituyen el pilus intacto, se espera que la interacción con la chaperona sea lo más eficaz en términos de dificultar la infectividad bacteriana. 5 10 15 20 25 30 35 Como será obvio a partir de los ejemplos mostrados más adelante, la mayor parte de los datos sobre la unión entre las chaperonas y las subunidades de pilus se han obtenido estudiando la interacción entre la chaperona PapD de E. coli. Sin embargo, como se ha descubierto que muchas bacterias que se adhieren a tejidos expresan los pili que comparten homologías sustanciales en su parte C-terminal, y como se ha demostrado las homologías sustanciales entre las diversas chaperonas periplásmicas que se habían aislado hasta ahora (véase la tabla), está justificado asumir que alguna sustancias y clases de sustancias serán capaces de interactuar con la mayor parte de las chaperonas periplásmicas existentes y de esta manera ser útiles en el tratamiento y/o profilaxis de enfermedades provocadas por las bacterias que esconden estas chaperonas cuando la sustancia se administra al paciente infectado con las bacterias. De esta manera, la sustancia de la invención para uso en el tratamiento y/o profilaxis se desea especialmente en pacientes que están infectados por bacterias seleccionadas entre el grupo compuesto por Haemophilus spp, Helicobacter spp, Pseudomonas aeruginosa, Mycoplasma spp y todos los miembros de la familia Enterobacteriacieae, incluyendo Escherichia spp, Salmonella spp, Bordetella spp, Yersinia spp, Proteus spp y Klebsiella spp. A este respecto, especialmente las bacterias seleccionadas entre el grupo compuesto por E. coli, Y. pestis, Y. enterocolitica, B. pertussis, K. pneumoniae, S. typhimurium, S. typhi, S. paratyphii, Helicobacter pylori, Proteus mirabilis y Haemophilus influenzae se consideran como infectantes que provocan infecciones que pueden tratarse y/o prevenirse mediante el uso del procedimiento de acuerdo con la invención. Por consiguiente, en aspectos importantes de la invención, se previene, inhibe o mejora la unión de una subunidad de pilus con una chaperona seleccionada entre el grupo compuesto por PapD, FimC, SfaE, FaeE, FanE, Cs3-1, F17D, ClpE, EcpD, Mrkb, FimB, SefB, HifB, MyfB, PsaB, PefD, YehC, MrpD, CssC, NfaE, AggD y Caf1M. Se prefiere especialmente que esté afectada la unión de PapD con al menos una subunidad de pilus. Como se ha indicado anteriormente, en una realización preferida de la invención, la prevención, inhibición o mejora de la unión se realiza por la interactuación con, en la chaperona molecular, un sitio de unión que normalmente está implicado en la unión a las subunidades de pilus durante el transporte de estas subunidades de pilus a través del espacio periplásmico y/o durante el procedimiento de la unión al pilus. Como se ha mencionado, con relación a la presente invención, se ha determinado el motivo de la unión entre PapD y un péptido que constituye los 19 aminoácidos del extremo C-terminal de PapG (G1’-19’), una subunidad de pilus. Como se describe con detalle en este documento, otras chaperonas comparten homologías sustanciales con PapD en este sitio de unión. De esta manera, tal sitio de unión es de gran interés como diana para fármacos que pretenden interactuar con las chaperonas periplásmicas. Por lo tanto, en una realización preferida de los procedimientos descritos anteriormente de la invención, el sitio de unión que está afectado es uno que une G1’-G19’. 40 45 50 55 60 65 El término “punto del sitio” se refiere a un grupo químico con características físicas/químicas bien definidas tales como el tamaño, carga, hidrofobicidad/hidrofilicidad, polaridad, dirección de los enlaces hidrógeno así como una posición tridimensional (distancia y ángulo) relativa a otro de tales grupos químicos. De esta manera, los puntos del sitio que son “sustancialmente idénticos” a Arg-8 y Lys-112, son grupos químicos que comparten sustancialmente las mismas características físicas/químicas bien definidas de estos dos aminoácidos. El término “residuos invariantes” se refiere a residuos aminoácidos que pueden encontrarse en varias proteínas sin que haya ninguna variación con respecto al tipo preciso del aminoácido y sin que haya ninguna variación sustancial en su función en las proteínas. La presencia de residuos invariantes en un gran número de proteínas relacionadas normalmente es una indicación de la importancia biológica de tales residuos, ya que las mutaciones que carecen de residuos aparentemente carecen también de la función de la proteína intacta. Como se ha descrito en este documento, se ha descubierto que todas las chaperonas periplásmicas moleculares comparten los residuos aminoácidos que son equivalentes a Arg-8 y Lys-112. Por lo tanto, se cree que estos dos residuos son de considerable importancia para las bacterias que forman pilus. “Un fragmento de polipéptidos capaz de interactuar con una cadena β de la parte carboxilo terminal de una subunidad de pilus” indica que parte de la chaperona (que también es parte del sitio de unión) es capaz de interactuar con una cadena β de la subunidad de pilus. Esta interacción sirve como factor estabilizante en la unión entre la subunidad de pilus y la chaperona y se considera una parte muy importante del motivo total de la unión entre la chaperona y la subunidad del pilus. Además, recientemente, se ha hecho verosímil por los inventores que la cadena β sirve como plantilla para el correcto plegamiento de la subunidad de pilus (véase el ejemplo 10). Como se explica en este documento, la parte C-terminal de muchas, si no de todas, las subunidades de pilus conocidas, comparte homologías sustanciales, que es otro indicio de la importancia de la estructura tridimensional de la subunidad de pilus así como también de la chaperona con el fin de que tenga lugar la unión y de que sea estable. Como se observa a partir de los ejemplos, se ha identificado otro sitio de unión que reside en el dominio 2 de PapD. Este sitio de unión interactúa con la proteína de fusión MBP-G1’-140’ así como con un péptido corto compuesto por 7 ES 2 225 834 T3 los residuos del aminoácido C-terminal de 125’ a 140’ de PapG. De esta manera, este sitio de unión es también de gran interés como diana para los fármacos que pretenden interactuar con las chaperonas periplásmicas. Por lo tanto, una realización preferida de los procedimientos descritos anteriormente de la invención es un procedimiento en el que el sitio de unión afectado es uno que une cualquiera de los dos péptidos descritos anteriormente. 5 Se entenderá que las sustancias descritas anteriormente para uso en el tratamiento y/o prevención de enfermedades depende de la identificación o del diseño de novo de sustancias que son capaces de ejercer los efectos que conducirán a la prevención, inhibición o mejora de la interacción entre las subunidades de pilus y las chaperonas periplásmicas moleculares. También es importante que estas sustancias tengan una gran oportunidad de ser terapéuticamente activas. 10 De esta manera, un aspecto de la invención se refiere a un procedimiento para identificar una sustancia potencialmente terapéuticamente útil capaz de interactuar con una chaperona periplásmica molecular, que por lo tanto, previene, inhibe o mejora la interacción entre una chaperona periplásmica molecular y una subunidad de pilus, comprendiendo el procedimiento al menos una de las siguientes etapas: 15 1) ensayar una sustancia candidata en un ensayo en el que la posible prevención, inhibición o mejora mediante la sustancia de la interacción entre la chaperona periplásmica molecular y la subunidad de pilus se determine mediante 20 25 30 35 40 45 50 55 60 65 a) la adición de la sustancia a un sistema que comprende la chaperona periplásmica molecular o un análogo de la misma en una forma inmovilizada y la subunidad de pilus o un equivalente de la misma en una forma solubilizada y la determinación del cambio en la unión entre la subunidad de pilus o equivalente de la misma y la chaperona periplásmica molecular o el análogo de la misma provocado por la adición de la sustancia, o b) la adición de la sustancia a un sistema que comprende la subunidad de pilus o un equivalente de la misma en una forma inmovilizada y la chaperona periplásmica molecular o un análogo de la misma en una forma solubilizada y la determinación del cambio en la unión entre la subunidad de pilus o el equivalente de la misma y la chaperona periplásmica molecular o el análogo de la misma provocado por la adición de la sustancia, o c) la adición de la sustancia a un sistema que comprende la subunidad de pilus o un equivalente de la misma así como la chaperona periplásmica molecular o un análogo de la misma en una forma solubilizada y la determinación del cambio en la unión entre la subunidad de pilus o el equivalente de la misma y la chaperona periplásmica molecular o el análogo de la misma provocada por la adición de la sustancia, o d) la adición de la sustancia a un sistema que comprende la subunidad de pilus o un equivalente de la misma así como la chaperona periplásmica molecular o un análogo de la misma en forma solubilizada y la medición del cambio en la energía de unión provocado por la adición de la sustancia, y la identificación de la sustancia como potencialmente terapéuticamente útil si se observa un cambio en la energía de unión entre la subunidad de pilus o el equivalente de la misma y la chaperona periplásmica molecular o un análogo de la misma, e identificar la sustancia como potencial y terapéuticamente útil si se observa un cambio significativo en la unión o en la energía de la unión entre la subunidad de pilus o el equivalente de la misma y la chaperona periplásmica molecular o el análogo de la misma; 2) ensayar una sustancia candidata en un ensayo en el que la posible prevención, inhibición o mejora de la interacción entre la chaperona periplásmica molecular y la subunidad de pilus se determine mediante la adición de la sustancia a un sistema que comprende bacterias vivas que forman pilus que se adhieren a tejidos seguido de la determinación de la velocidad de crecimiento de las bacterias, siendo indicativo de prevención, inhibición o mejora de la unión entre la chaperona periplásmica molecular y la subunidad de pilus una reducción en el crecimiento en comparación con un sistema correspondiente en el que la sustancia no se ha añadido, o la adición de la sustancia a un sistema que comprende bacterias que forman pilus que se adhieren adhesión a tejidos seguido de una determinación de la adhesión al tejido de las bacterias, siendo indicativo de prevención, inhibición o mejora de la unión entre la chaperona periplásmica molecular y la subunidad de pilus una reducción en la adhesión al tejido en comparación con un sistema correspondiente en el que no se ha añadido la sustancia, e identificar la sustancia como potencialmente terapéuticamente útil si se observa una reducción de la velocidad de crecimiento o de adhesión al tejido después de la adición de la sustancia; y 3) identificar si una sustancia es capaz de interactuar in vivo con una chaperona periplásmica molecular mediante el uso de una sustancia, donde dicha sustancia se ha establecido in vitro para prevenir, inhibir o mejorar la interacción entre una chaperona periplásmica molecular y una subunidad de pilus, para la administración a un animal experimental, donde las bacterias que forman pilus que se adhieren a tejidos del animal se ha inoculado antes, a la vez o después de la administración de la sustancia, para la identificación de si la sustancia es capaz de prevenir y/o curar y/o aliviar la enfermedad provocada por las bacterias. 8 ES 2 225 834 T3 5 10 Por la expresión “un equivalente de una subunidad de pilus” se entiende un compuesto que se ha establecido que se une a la chaperona de manera que es comparable con la forma en la que se une la subunidad de pilus a la chaperona, por ejemplo, mediante la demostración de la subunidad de pilus y el equivalente que compite por la unión con la chaperona. Los equivalentes preferidos de las subunidades de pilus son G1’-19’WT, MBP-G1’-140’ y G125’-140’, que se describen con detalle en este documento. La expresión “un análogo de una chaperona” indica cualquier sustancia que puede unirse a al menos una subunidad de pilus de manera que corresponda con al unión de dicha chaperona a una subunidad de pilus. Tal análogo de la chaperona puede ser una forma truncada de la chaperona intacta (por ejemplo, uno de los dos dominios de PapD) o puede ser una forma modificada de la chaperona que puede, por ejemplo, acoplarse a una sonda, marcador u otro residuo. Finalmente, el análogo de la chaperona puede ser un sitio de unión aislado, pero parcialmente del sitio de unión completamente funcional, de la chaperona o una sustancia sintética que imite a tal sitio de unión. 15 La inmovilización mencionada anteriormente puede ser una unión no covalente simple a una superficie de adhesión o una molécula huésped o receptora tal como un anticuerpo, o una unión covalente a una molécula espaciadora tal como un polímero o un péptido. 20 En la etapa 1a) mencionada anteriormente, la subunidad de pilus o el equivalente de la misma que se une a la chaperona periplásmica molecular o a un análogo de la misma puede detectarse de diversas formas, por ejemplo marcando la subunidad de pilus o el equivalente de la misma, o por medio de un ligando marcado (tal como un anticuerpo) capaz de reaccionar con la subunidad de pilus o el equivalente de la misma, o por medio de una determinación basada en el índice refractivo de la extensión de la unión, tal como el ensayo Pharmacia BioCore®. 25 30 35 Por consiguiente, en la etapa 1b) la chaperona periplásmica molecular o el análogo de la misma que se une a la subunidad de pilus o al equivalente de la misma puede detectarse marcando la chaperona periplásmica molecular o el análogo de la misma, por medio de un ligando marcado (por ejemplo anticuerpo) capaz de reaccionar con la chaperona periplásmica molecular o el análogo de la misma, o por medio de una determinación basada en el índice refractivo de la extensión de la unión, tal como el ensayo Pharmacia BiaCore®. En la etapa 1c) la chaperona periplásmica molecular o el análogo de la misma que se une a la subunidad de pilus o al equivalente de la misma puede detectarse por la separación de los complejos subunidad de pilus/chaperona (por ejemplo, por ultracentrifugación/ultrafiltración, cromatografía líquida, tal como cromatografía por oclusión del tamaño, o electroforesis). A continuación se describe un procedimiento que depende de los cambios en la fluorescencia de un fragmento corto de PapG cuando este fragmento se une a PapD. Este procedimiento es un ensayo preferido en el procedimiento de la invención. La determinación de la energía de unión en la etapa 1c) se realiza preferiblemente en un sistema microcalorimétrico usando la técnica bien conocida de microcalorimetría. 40 45 50 55 60 Las etapas indicadas anteriormente sirven para 3 propósitos. Los tipos de ensayo de la etapa 1) pretenden arrojar luz sobre la capacidad de la sustancia candidata de interactuar con la chaperona. En los ejemplos en los que se usan sustancias, chaperonas o anticuerpos marcados, el marcador podría ser un marcador radiactivo, un marcador fluorescente o un marcador que absorba la luz, una enzima tal como peroxidasa de rábano picante, un ligando tal como biotina o cualquier otro sistema de marcación convencional conocido para un especialista en la técnica. La detección del compuesto marcado depende de la elección del marcador: la radiactividad puede medirse en un contador de escintilación líquida, un contador gamma, o cualquier otro sistema de detección conveniente para la radiactividad, los marcadores enzimáticos se detectan por la presencia o ausencia de un sustrato específico para la enzima (ensayo de densidad óptica, reactividad química del sustrato restante o del producto, etc.), los marcadores fluorescentes pueden detectarse por microscopía de fluorescencia o por la simple medición de la emisión de fluorescencia, los marcadores que absorben la luz pueden detectarse mediante la medición de la absorción de la luz de una longitud de onda característica y la biotina puede detectarse por su unión a estreptavidina. La separación de los altos complejos moleculares por ultracentrifugación o ultrafiltración en 1) puede detectarse mediante uno de los componentes del complejo que se marca como se ha descrito anteriormente; de esta manera, es posible detectar la relación entre la subunidad/equivalente del pilus no unido y unida, pero la etapa de detección también puede depender de la unión de los anticuerpos a uno de los componentes del complejo, y la posterior detección de este anticuerpo. Puede emplearse cualquier técnica cromatográfica convencional (HPLC, FPLC, exclusión del tamaño, etc.). La separación por electroforesis puede realizarse por ejemplo por electroforesis capilar. Todos los ensayos de la etapa 2) están relacionados con los efectos de la sustancia candidata en la actividad bacteriana in vitro. La demostración de una reducción en la velocidad de crecimiento de las bacterias o una demostración de la menor adherencia a las células o a superficies sintéticas en un ensayo no podrá contribuir únicamente, por supuesto, al efecto de la interacción con las chaperonas, sino que una demostración de este tipo podría proporcionar una buena estimación de la gran utilidad terapéutica de tal sustancia. 65 La determinación de la velocidad de crecimiento puede realizarse contando las colonias en placas de ágar sólidas destiladas con las bacterias, contando la densidad bacteriana en el medio de crecimiento líquido (determinación de DO600 ), midiendo la fluorescencia de sustancias tales como NAD(P)H, ATP o aminoácidos, que están contenidos 9 ES 2 225 834 T3 5 10 15 20 únicamente en células bacterianas, o mediante cualquier otro sistema de detección conveniente conocido para un especialista en la técnica. La determinación de la adherencia de las bacterias puede realizarse de una forma similar después de que se hayan aislado las bacterias de adhesión. Una determinación de la adherencia se realiza preferiblemente midiendo la capacidad de las bacterias para aglutinar glóbulos rojos o perlas de látex recubiertas con receptor, midiendo la adhesión bacteriana a las placas de microtitulación recubiertas con receptor, o midiendo la adhesión bacteriana a otras superficies sintéticas. En una realización preferida del procedimiento descrito anteriormente, las bacterias vivas que forman pilus que se adhieren a tejidos son de una cepa deficiente de proteasa, siendo la proteasa una que al menos es parcialmente responsable de la degradación de las subunidades de pilus. Un tipo de cepa especialmente preferido es la cepa degP41 de E. coli. Como se describe en este documento, la cepa degP41 carece de la actividad de la proteasa DegP que es responsable de la degradación de las subunidades de pilus en la E. coli cuando éstas no se recuperan en el espacio periplásmico por PapD, y de esta manera, las cepas degP41 son especialmente sensibles a los cambios en la eficacia de papD, ya que la acumulación de las subunidades de pilus es tóxica para la célula. Se cree que existen proteasas equivalentes en otras bacterias que expresan pilus. El estudio animal en la etapa 3) se realiza con el fin de demostrar la gran utilidad terapéutica de la sustancia candidata in vivo. Además, como ya se ha mencionado anteriormente, tales estudios animales también deben establecer unos valores a priori con respecto a la dosificación eficaz y a la toxicidad antes de que la sustancia candidata se ensaye finalmente en seres humanos en ensayos clínicos controlados. Los estudios con animales también deben proporcionar información con respecto a la formulación conveniente de la sustancia en una preparación farmacéutica así como a la vía preferida de administración, ya que es posible obtener, a partir del modelo animal, datos para la absorción de la sustancia así como datos para el metabolismo y excreción de la sustancia. El animal experimental es preferiblemente un ratón, una rata, un gato, un perro, un mono, un caballo, una cabra, un cerdo o un pollo. 25 La expresión “adecuadamente capaz de interactuar con una chaperona molecular” pretende indicar que una sustancia, aparte de ser capaz de interactuar con una chaperona molecular, también es capaz de ejercer efectos en un sistema in vivo, es decir, que la sustancia además de su capacidad de unión también muestra una compatibilidad con un sistema biológico, i.a. un paciente. 30 35 40 45 50 55 60 Aunque los estudios in vivo indicados anteriormente, especialmente los experimentos en modelos animales, son los mejores indicadores de la gran utilidad terapéutica de una sustancia en la prevención, inhibición o mejora de la unión entre una chaperona y una subunidad de pilus, no debería olvidarse que los ensayos in vitro indicados anteriormente sirven como indicaciones importantes cuando se desarrollan compuestos con un potencial terapéutico. Si se confía sólo en los ensayos in vivo, es muy probable que los compuestos que de hecho muestran el efecto deseado sobre la interacción chaperona/subunidad de pilus sean seleccionados por ensayos in vivo, ya que estos compuestos podrían carecer por ejemplo de la capacidad de penetrar las membranas biológicas. Cuando se usan los ensayos in vivo, se mantiene una oportunidad mucho mayor de encontrar un compuesto de indicación. La evaluación del efecto de una sustancia ensayada en los ensayos in vitro descritos en este documento (véanse a este respecto especialmente los ejemplos) depende de varios factores. Se entenderá por parte del especialista en la técnica que puede añadirse una pequeña molécula en concentraciones molares más altas con el fin de ejercer un efecto sobre la interacción chaperona/subunidad de pilus (e incluso entonces la pequeña molécula puede seguir siendo un interesante compuesto de indicación), al tiempo que las moléculas más grandes pueden ejercer efectos marcados incluso en concentraciones molares bajas. En general, cuando cualquier ensayo in vitro descrito en este documento se considera que tiene un resultado positivo cuando se ensaya una sustancia candidata (es decir, que la sustancia ensayada muestra un efecto “significativo”), deberá considerarse la siguiente condición: El compuesto deberá ejercer un efecto significativo en la interacción subunidad de pilus/chaperona (o sobre una interacción en un sistema equivalente que se correlacione bien con la interacción subunidad de pilus/chaperona), siendo el efecto significativo tal que no pueda atribuirse ninguna duda a la interacción entre la sustancia y la chaperona y que no sea una interacción inespecífica entre la chaperona y la sustancia (debido a, por ejemplo, cambios radicales en el medio físico y químico cuando se añade la sustancia). Una manera de excluir las interacciones inespecíficas como razón para el efecto ejercido es usar al menos un control que es una sustancia químicamente comparable (con respecto a la masa molecular, carga/polaridad y conformación del grosor tridimensional (globular, fibrilar, etc.). Si el control no da como resultado sustancialmente el mismo efecto en el ensayo que la sustancia, puede concluirse que la sustancia debería considerarse como una sustancia positiva para ensayo. Los ensayos descritos en los ejemplos son todos buenos ejemplos de tipos de ensayo, que podrían servir como sistema de ensayo en el procedimiento de la invención descrito anteriormente. Sin embargo, se prefiere que en la etapa 1c) se use el procedimiento descrito en el ejemplo 10 que emplea una variante marcada fluorescente de una subunidad de pilus. Este ensayo puede describirse brevemente como se indica a continuación: - añadir la sustancia a un primer sistema que comprende la chaperona periplásmica molecular o un análogo de la misma, 65 - posteriormente, añadir una subunidad de pilus o un equivalente de la misma que se ha marcado con una sonda fluorescente medioambientalmente sensible, 10 ES 2 225 834 T3 - determinar la emisión de fluorescencia a una longitud de onda particular que indica la cantidad de unión entre la chaperona periplásmica molecular o el análogo de la misma y la subunidad de pilus o el equivalente de la misma, y 5 - comparar la emisión de fluorescencia determinada con la emisión de fluorescencia determinada en un segundo sistema correspondiente que contiene sustancialmente las mismas concentraciones de la chaperona molecular o del análogo de la misma y la subunidad de pilus o el equivalente de la misma pero sustancialmente sin sustancia, una diferencia significativa en la emisión de fluorescencia entre el primer y el segundo sistema que indica la interacción entre la chaperona periplásmica molecular o el análogo de la misma y la sustancia. 10 15 La ventaja de este ensayo es que puede emplearse para determinaciones cuantitativas del efecto de la sustancia ensayada en el sistema chaperona/subunidad de pilus. Usando este ensayo, los inventores han determinado por ejemplo la constante de unión entre un análogo de PapG y PapD. Las determinaciones cuantitativas pueden realizarse realizando la determinación de la emisión de fluorescencia en el segundo sistema muchas veces a relaciones molares variantes entre la subunidad de pilus o el equivalente de la misma y la chaperona periplásmica y el equivalente de la misma, con lo cual la constante de unión entre la subunidad de pilus o el equivalente de la misma y la chaperona periplásmica molecular o el análogo de la misma se valora a partir de los datos de emisión de fluorescencia determinados. A partir de los datos obtenidos de esta manera también es posible determinar la constante de unión de la sustancia de manera paralela, lo que se verá a partir de la reivindicación 11. 20 25 30 Se entenderá que el procedimiento indicado anteriormente para identificar una sustancia potencial y terapéuticamente útil depende de la presencia real de la sustancia. Normalmente, es necesario purificar o sintetizar la sustancia candidata antes de que se someta al procedimiento mencionado anteriormente. Sin embargo, como es probable que muchas de tales sustancias candidatas se ensayen antes de que se identifique una sustancia que puede, de forma adecuada, interactuar con una chaperona, es de interés identificar tales sustancias antes de que se sometan al procedimiento anterior, disminuyendo por lo tanto las fuentes agotadas en las etapas de purificación y/o síntesis. Por lo tanto, la invención también se refiere a un procedimiento para identificar y/o diseñar una sustancia, X, capaz de interactuar con una chaperona, por ejemplo de unirse a la chaperona, con una energía de unión pronosticada igual a o mejor que un valor umbral predeterminado, donde el procedimiento comprende 1) seleccionar una sustancia, A, que podría interactuar en gran medida con un sitio en la chaperona, y proporcionar una representación estructural tridimensional de la misma, 35 2) predecir la energía libre de unión entre la sustancia A y el sitio en la chaperona, 3) si la energía de unión pronosticada entre la sustancia A y el sitio en la chaperona es igual a o mejor que el valor umbral predeterminado, entonces identificar la sustancia A como la sustancia X, 40 4) si la energía libre de unión pronosticada entre la sustancia A y el sitio en la chaperona no es igual a o mejor que el valor umbral predeterminado, entonces modificar la representación estructural tridimensional, entonces modificar la representación estructural tridimensional y predecir la energía libre de unión entre la sustancia modificada de esta manera, B, y el sitio en la chaperona, y 45 5) repetir la etapa 4 hasta que la energía libre de unión pronosticada determinada entre la sustancia resultante, X, y el sitio en la chaperona sea igual a o mejor que el valor umbral predeterminado. 50 Es posible ampliar el procedimiento mencionado anteriormente con dos etapas más, donde se determina la energía libre de unión exacta, con el fin de establecer que la energía libre de unión experimental también es mejor que el valor umbral predeterminado. Realizando las siguientes dos etapas 55 6) proporcionar una muestra de la sustancia química X y una muestra de la chaperona y medir la energía libre de unión entre la sustancia química X y la chaperona (por ejemplo, por microcalorimetría como se ha mencionado anteriormente), y establecer que la energía libre de unión medida entre la sustancia química X y la chaperona es igual a o mejor que el valor umbral predeterminado, y opcionalmente 60 65 7) someter la sustancia X al procedimiento mencionado anteriormente para identificar una sustancia que puede, de forma adecuada, interactuar con una chaperona, con el fin de verificar que la sustancia X es una sustancia potencial y terapéuticamente útil capaz de interactuar con una chaperona, de esta manera se verifica que la energía libre de unión entre la sustancia candidata y la chaperona realmente es mejor que el valor umbral predeterminado. La etapa 7) además establece que la sustancia candidata tiene muchas oportunidades de ser terapéuticamente útil. La frase “predecir la energía libre de unión” pretende implicar que la energía libre de unión se determina calculando en lugar de realizando el tratamiento experimental que determina la energía libre de unión real. Una manera (teórica) de predecir la energía libre de unión es realizar cálculos de perturbación de la energía libre (FEP) sobre las sustancias que interactúan, pero a causa de la gran cantidad de cálculos que esta aproximación conllevaría como resultado, se prefiere emplear el procedimiento aproximativo empírico descrito más adelante. 11 ES 2 225 834 T3 5 El término “mejor que” pretende significar que la energía libre de unión tiene un valor que es superior a de la energía libre de unión que se ha elegido como valor umbral, lo que significa que ∆G es numéricamente mayor que el valor umbral seleccionado. O en otras palabras: El término pretende significar que la unión entre la sustancia y la chaperona es energéticamente más favorable que la situación en la que la sustancia y la chaperona se suspenden independientemente en solución. Con el fin de predecir la energía de unión en el procedimiento indicado anteriormente, de acuerdo con la invención se prefiere especialmente usar el siguiente procedimiento: 10 15 20 Valorar la diferencia de la energía media, <∆Vel X−s >, definida como <Vel X−s >B - <Vel X−s >A , entre la contribución de las interacciones polares a la energía potencial, entre la sustancia química X y la que la rodea (denominada S) en dos estados, un estado (A) en donde la sustancia química está rodea por el disolvente y el otro estado (B) en donde la sustancia química, unida a una chaperona periplásmica molecular o a un análogo de la misma, está rodea por el disolvente, ensayar la diferencia de la energía media, <∆Vvdw X−s > definida como <∆Vvdw X−s >B - <∆Vvdw X−s >A , entre la contribución de las interacciones no polares a la energía potencial entre la sustancia química X y la que la rodea (denominada S) en dos estados, un estado (A) en donde la sustancia química está rodea por el disolvente y el otro estado (B) en donde la sustancia química, unida a una chaperona periplásmica molecular o a un análogo de la misma, está rodea por el disolvente, y calcular la energía libre de unión absoluta como combinación ajustada de las dos diferencias de la energía media mencionadas anteriormente. 25 30 35 40 45 En las ecuaciones matemáticas que se muestran en este documento, el símbolo < > significa media dinámica molecular. El índice X-s significa compuesto-disolvente (o compuesto-agente que rodea), la letra “X” indica la sustancia química X. Normalmente, la sustancia X funcionará como inhibidor de la unión entre la chaperona periplásmica y las subunidades de pilus, pero como se ha analizado en este documento, también existe la posibilidad de que el compuesto o fármaco afecte a la chaperona de tal forma que se mejore la unión entre las subunidades de pilus y la chaperona. El superíndice “el” indica la energía polar o electrostática, mientras que el superíndice “vdw” indica “van der Waals”, otra denominación para las interacciones no polares. El símbolo ∆ indica que la cantidad en el estado A se resta de la cantidad en el estado B. En el presente contexto, por la frase “un análogo de una chaperona periplásmica molecular” deberá entenderse, en un sentido amplio, cualquier sustancia que imite (con respecto a las características de unión) a una parte interesante de una chaperona periplásmica molecular (por ejemplo, la parte o las partes que se unen a la subunidad de pilus) y cuya interacción con una sustancia química o un grupo o varias sustancias químicas, por ejemplo candidatos a fármacos, se va a estudiar. De esta manera, el análogo puede ser simplemente cualquier otro compuesto químico considerado capaz de interactuar con la sustancia química de manera que imite la unión entre la chaperona y una subunidad de pilus in vivo, pero de forma más frecuente el análogo será una molécula relativamente grande, en otras palabras una macromolécula tal como una proteína o un oligonucleótido, que es relativamente grande en comparación con la sustancia química; aunque la sustancia química que interactúa con el análogo, en sí misma es, por supuesto, una macromolécula. En el presente contexto, la chaperona periplásmica molecular o análogo de la misma es preferiblemente la chaperona periplásmica o un análogo de la misma que muestra al menos una característica de unión interesante relevante para la unión de los pili. La base del enfoque indicado anteriormente para determinar la energía libre de unión se explica a continuación: 50 Como punto de partida se toma la aproximación de la respuesta lineal para fuerzas electrostáticas que, como resultado, para soluciones polares produce funciones cuadráticas de energía libre en respuesta al desarrollo de las cargas. Esto es, por ejemplo, el resultado común de la teoría de Marcus de reacciones de transferencia de electrones (Marcus, 1964). Para un sistema con dos estados, A y B, dados por dos funciones de energía potencial VA y VB uno obtiene la relación en la aproximación de las funciones armónicas de energía libre de igual curvatura (véase Lee y col., 1992 y las referencias en ese documento): 55 λ =< VB − VA >A −∆GAB =< VA − VB >B +∆GAB 60 donde ∆GAB es la diferencia de energía libre entre B y A, λ es la energía de reorganización correspondiente y < >I indica una media evaluada cerca del mínimo del potencial i. De esta manera, ∆GAB 1 /2 (< ∆V >A + < ∆V >B ) 65 (a) (b) donde ∆V ahora indica la diferencia de energía VB - VA . Si se considera la hidración de un único ión, esto puede mostrarse para dar ∆Gel sol = ½ <Vel X−s >, es decir, que la contribución electrostática a la energía de solvatación es igual a la mitad de la energía de interacción del ión-disolvente correspondiente (Warshel y Russell, 1984; Roux y col., 1990). Volviendo ahora al problema de la unión, este resultado puede interpretarse de la siguiente manera: Para cada 12 ES 2 225 834 T3 5 10 15 20 25 30 procedimiento de solvatación, es decir, solvatación de la sustancia en agua y dentro de la proteína, se consideran dos estados en los que el primero tiene la sustancia al vacío y una cavidad no polar (dada, por ejemplo, por el potencial de Lennard-Jones) ya fabricada en el medio dado. El segundo estado se corresponde con la sustancia intacta rodeada por agua o la proteína solvatada. Después, la aproximación de la respuesta lineal volverá a dar que ∆Gel unión ½ <Vel X−s >, donde Vel X−s es el término electrostático del soluto-disolvente. Por tanto, la contribución electrostática a la energía libre de unión puede aproximarse mediante ∆Gel unión ½ Vel X−s > (donde ∆ ahora se refiere a la diferencia entre la proteína y el agua) y de esta manera puede obtenerse a partir de dos simulaciones MD de la sustancia solvatada y del complejo sustancia-proteína. Se ha confirmado la validez de los resultados de respuesta lineal en el caso de la solvatación iónica, por ejemplo en el estudio de Roux y col. (1990). También se realizaron más cálculos sobre sistemas simples que corroboran la aproximación de la ecuación b. Estos ensayos se realizaron comparando la energía libre obtenida de las simulaciones FEP/MD de cargar iones Na+ y Ca2+ en un sistema de agua esférico (Aqvist, 1990) con el correspondiente <Vel X−s > a partir de trayectorias MD de 75 ps. Esto produjo factores que relacionaban <Vel X−s > con ∆Gel sol de 0,49 para Na+ y 0,52 para Ca2+ , siendo ambos valores afines al resultado pronosticados de ½. Un ensayo similar sobre la carga de una molécula de metanol, dado para el potencial OPLS (Jorgensen, 1986) en agua dio una relación ∆Gel sol /<Vel X−s > de 0,43. Una cuestión crucial es cómo cuantificar la contribución de las interacciones no polares y de los efectos hidrófobos a la energía libre de unión que se denominó ∆Gvdw unión . En el caso ideal, debería ser posible estimar esta contribución a partir de energías de interacción no polares (o van der Waals). Las teorías sobre líquidos de Chandler y colaboradores (Chandler y col., 1983; Pratt y Chandler, 1997) se han usado satisfactoriamente para analizar los efectos hidrófobos y para calcular las energías libres de transferencia para algunas moléculas no polares (Pratt y Chandler, 1977), pero no parece posible el tratamiento analítico de ese tipo para la solvatación en un medio no homogéneo tal como un sitio activo de una proteína. Sin embargo, se ha observado que la energía libre experimental de la solvatación para diversos compuestos de hidrocarburos, tales como n-alcanos, depende aproximadamente linealmente de la longitud de ambas cadena de carbono en sus propios líquidos así como en agua (Ben-Naim y Marcus, 1984). Se han realizado simulaciones MD de n-alcanos solvatados en agua y en van der Waals no polar, lo que indica que también las energías de interacción medias soluto-disolvente varían aproximadamente linealmente con el número de carbonos en la cadena (siendo las relaciones diferentes en disolventes diferentes, por supuesto). De esta manera, parece posible que una aproximación lineal sencilla de ∆Gvdw unión a partir de <∆Vvdw X−s > pueda ser capaz de justificar la contribución de unión no polar. Por ejemplo, si σ se considera una medición apropiada del tamaño del soluto y si las energías de interacción van der Waals soluto-disolvente y las correspondientes contribuciones de energía libre no polar (ambas en agua y proteína) dependen linealmente de σ, de tal forma que 35 < Vvdw p >= αp σ, < Vvdw w >= σw σ, ∆Gvdw p = βp σy∆Gvdw w = βw σ 40 45 Ya que parece difícil derivar un factor que relacione entonces se obtiene las dos cantidades de una manera fiable a partir de consideraciones puramente teóricas, el enfoque se toma para intentar determinar empíricamente tal relación que es capaz de producir los datos de unión experimentales. De esta manera, la energía libre de unión se aproxima en una realización de la invención mediante ∆Gunión = 1 /2 < ∆Vel X−s > +α < ∆Vvdw X−s > 50 (1) determinándose el parámetro α por calibración empírica. Aunque, como se ha analizado anteriormente, una predicción teórica del coeficiente para <∆Vel X−s > es ½, también puede ser prácticamente útil tratar este coeficiente como un parámetro empírico. Esto conduciría a que la energía libre de unión se aproximase mediante 55 ∆Gunión = β < ∆Vel X−s > +α < ∆Vvdw X−s > (1b) donde ambos parámetros, α y β, se determinan por calibración empírica. 60 Finalmente, en algunos casos, parece adecuado añadir un término constante más a la Ecuación 1, para que la ecuación se convierta en 65 ∆Gunión = 1 /2 < ∆Vel X−s > +α < ∆Vvdw X−s > +c (2) donde c es una constante que refleja la extrapolación a tamaño cero de la sustancia química, esto es, donde la regresión lineal se compensa claramente desde el origen cuando se mueve hacia el tamaño cero de la sustancia química. El 13 ES 2 225 834 T3 5 parámetro c también puede usarse para corregir posibles errores sistemáticos debidos a, por ejemplo, la negligencia de la polarización inducida, posibles deficiencias en el campo de fuerza, etc. En estos casos, c asumirá normalmente un valor entre -10 y 10 kcal/mol, típicamente entre -3 y 3 kcal/mol, de tal forma que esté entre -2 y 2 kcal/mol, por ejemplo entre -1 y 1 kcal/mol. Sin embargo, se anticipa que en muchos casos, c puede establecerse adecuadamente a cero, ya que el alcance de la derivación será de menor importancia para la utilidad de los valores pronosticados. Si el coeficiente electrostático i también se trata como un parámetro empírico, la aproximación de la energía libre de unión asume su forma más general, denominada 10 ∆Gunión = β < ∆Vel X−s > +α < ∆Vvdw X−s > +c (2b) donde ahora α, β y c se determinan por calibración empírica. 15 20 25 30 35 40 45 50 55 60 65 Aunque el disolvente usado en el procedimiento anterior es adecuadamente y más normalmente un disolvente acuoso tal como agua, está dentro del alcance de la invención tomar cualquier otro disolvente adecuado como punto de partida, incluyendo, por ejemplo, metanol, etanol, acetona, acetonitrilo, cloroformo, hexano, etc., o mezclas de los mismos o combinaciones de tales disolventes o mezclas de los mismos con agua. La selección del disolvente será de poca importancia para los valores pronosticados siempre que el disolvente sea capaz de disolver o solvatar la molécula receptora y la sustancia (en el presente contexto esto significa que una cantidad suficiente de la chaperona periplásmica molecular o el análogo de la misma puede mezclarse homogéneamente con el disolvente sin precipitación para permitir la determinación de las energías de unión mediante algún procedimiento adecuado), pero puede haber casos en los que sea ventajoso modificar el medio disolvente (por ejemplo, modulando la fuerza iónica) donde tendrá lugar la interacción de la sustancia y la molécula receptora. Si el medio en el que va a tener lugar la interacción entre la sustancia química, tal como un fármaco, y una chaperona periplásmica molecular o un análogo de la misma en el uso real del fármaco es el cuerpo humano, debería ser particularmente adecuado imitar por ejemplo el plasma humano como disolvente. En la Solicitud de Patente Internacional Nº PCT/IB94/00257 y en Aqvist y col., 1994 puede encontrase un análisis minucioso del procedimiento indicado anteriormente para determinar la energía libre de unión entre dos moléculas. Estos dos documentos se incorporan en este documento por referencia. El procedimiento indicado anteriormente para determinar la energía libre de unión se ha empleado en el ejemplo 3 con el fin de identificar compuestos que tienen una elevada probabilidad de unirse al sitio de unión de PapD; esto significa que los cálculos como los descritos anteriormente se han realizado como la última etapa teórica antes de que los compuestos se hayan sintetizado realmente. Se entenderá que los procedimientos mencionados anteriormente para identificar sustancias capaces de interactuar con chaperonas probarán ser especialmente eficaces en la identificación de sustancias que son de gran valor farmacéutico si se sabe que el sitio al que se unen está implicado en el ensamblaje de pilus. Por lo tanto, se prefiere que el sitio con el que puede interactuar potencialmente la sustancia, y del que se pronostica la energía libre de unión, sea la parte de unión a la subunidad de pilus de una chaperona molecular, tal como el sitio de unión a la subunidad de pilus de una chaperona molecular seleccionada entre el grupo compuesto por PapD, FimC, SfaE, FaeE, FanE, Cs3-27, F17D, ClpE, EcpD, Mrkb, FimB, SefB, HifB, MyfB, PsaB, PefD, YehC, MrpD, CssC, NfaE, AggD y Caf1M, o un análogo de tal sitio de unión a la subunidad de pilus, ya que los sitios de unión a la subunidad de pilus en estas chaperonas muestran amplias homologías. Se prefiere especialmente que el sitio de unión sea el sitio de unión a la subunidad de pilus de PapD o un análogo del mismo. Como se verá a partir de los ejemplos, se ha descubierto una parte importante del motivo de unión a la chaperona y un péptido correspondiente a este motivo se ha sintetizado y co-cristalizado con PapD para proporcionar una base estructural para el mecanismo de acción de PapD. Los detalles moleculares de la interfaz de reconocimiento de PapD-adhesina demuestran claramente la función de la hendidura conservada en toda la superfamilia de chaperonas con pilus en unión de la subunidad y en el traslado de los determinantes de virulencia a la superficie de bacterias patogénicas. La estructura cristalina de PapD-péptido representa esencialmente una “instantánea” de un procedimiento fundamental en la patogénesis bacteriana: la interacción de una adhesina con una chaperona, que es un prerrequisito para la presentación de la adhesina en la superficie microbiana. De esta manera, los inventores de la presente invención han aclarado mediante el uso de cristalografía de rayos X el mecanismo de unión entre PapD y la subunidad de pilus PapG identificando, por lo tanto, una parte esencial de un sitio de unión definido responsable de la unión entre las subunidades de pilus y sus chaperonas periplásmicas, y proporcionando de esta manera un procedimiento para permitir el diseño de fármacos de chaperonas que inhiben los compuestos anti-bacterianos. Habiendo determinado la localización de un sitio prometedor de unión para ligandos inhibidores como se ha descrito anteriormente (véanse detalles en los ejemplos 1 y 2), se han usado los programas informáticos “PLIM” y “PLIM_DBS” (desarrollados por Symbicom AB) para encontrar plantillas para las familias de compuestos capaces de unirse al sitio de unión. 14 ES 2 225 834 T3 5 10 15 PLIM es un modelador de la interacción proteína-ligando que construye supuestos ligandos para una proteína usando criterios termodinámicos. Este programa calcula la energía de interacción entre la proteína y las sondas de muestra que se colocan sucesivamente en diferentes puntos sobre una cuadrícula red alrededor de la molécula. Para cada posición y orientación se calcula la energía de interacción entre la sonda y los átomos de la proteína. Las energías se almacenan, y se escriben las mejores posiciones para una sonda particular (los cálculos básicos se describen por Goodford (1985) y Boobyer (1989) y se aplican en el programa GRID disponible en el mercado; La aplicación de PLIM es algo diferente en el sentido de que los valores energéticos se convierten en puntos discretos que se asocian con la sonda química, permitiendo un fácil rendimiento para, por ejemplo, programas que buscan bases de datos). Después, el programa construye el ligando incorporando átomos de sonda seleccionados en posiciones de mínima energía sobre la red. El usuario selecciona los átomos y grupos que deberían usarse como sondas, y los criterios que deberían usarse para determinar los que se incorporarán en el ligando. La energía se calcula como la suma de Van der Waals electrostático y las contribuciones de la unión a hidrógeno como se describe en este documento. Los datos de PLIM dan como resultado un número de posiciones y orientaciones sugeridas de grupos químicos favorables en la región cercana al sitio de unión. Estos grupos que tienen propiedades físicas como carga, direccionalidad de unión a hidrógeno y radio atómico amplio, se denominarán en lo sucesivo “puntos del sitio”. Después se realiza una búsqueda de ligandos potenciales buscando una base de datos para estructuras moleculares conocidas que cuadran con las posiciones de estos grupos de los puntos del sitio, usando PLIM_DBS. 20 25 30 35 40 El núcleo de PLIM_DBS es un algoritmo para el isomorfismo subgráfico (véase Ullman (1976) y Brint (1987)), en el que se representan tres puntos del sitio como una matriz de distancia (“la matriz patrón”). El programa busca el patrón de distancia en la matriz de distancia formada a partir de cada entrada de la base de datos. Si se encuentra el patrón, la entrada se superpone en los puntos del sitio y si los tipos de átomo correspondientes cuadran con la entrada su orientación se guarda en una lista de aciertos. Además de este esquema básico pueden usarse diversas opciones con respecto a la superficie complementaria, es decir, sólo se guardan las entradas que cuadran con la superficie de la proteína con respecto a las propiedades hidrófobas y estéricas. De esta manera, PLIM_DBS es un buscador de bases de datos que busca a través de un grupo de lotes coordinados de moléculas tridimensionales, buscando entradas que contienen un cierto patrón de átomos. Este patrón se especifica en términos de tipo de átomo y de posición y orientación espacial; por ejemplo, puede realizarse una búsqueda para compuestos que contengan un átomo de carbono sp3 que está a 4,2 Å de un oxígeno sp2 y a 5,1 Å de un grupo hidroxilo que a su vez está a 5,6 Å del oxígeno. El usuario puede ajustar la rigurosidad de la búsqueda variando la tolerancia de los criterios de distancia y la correspondencia con el tipo de átomo, determinando por ejemplo, si un carbono sp2 que es un poco más de 4,2 Å de uno de oxígeno debería considerarse como acierto. Esos aciertos que se han encontrado después se clasifican de acuerdo con una puntuación que refleja cómo de bien se superponen los átomos diana en la molécula real, y también cómo es de complementaria la superficie de la molécula del compuesto para la cavidad de unión de la proteína. El resultado de una búsqueda PLIM_DBS es una lista de estructuras moleculares y sus coordenadas atómicas, superpuestas en los puntos del sitio, y a las que se les da una puntuación (“calidad de ajuste”). El procedimiento no intenta optimizar el posicionamiento de las estructuras, ni tampoco realiza cualquier cálculo mecánico o dinámico molecular. Tanto la proteína como las estructuras extraídas se tratan como cuerpos rígidos. 45 50 55 60 65 Las estructuras de la búsqueda en la base de datos se muestran en el contexto de la proteína y su superficie en un sistema de gráficos usando un grupo de modelado molecular habitualmente disponible. Normalmente, las estructuras muestran algunas interacciones desfavorables con la proteína, o carecen de grupos para completarse, por ejemplo, cavidades hidrófobas. Por lo tanto, las estructuras de la búsqueda en la base de datos se consideran plantillas, que se modificarán y mejorarán por un químico orgánico. Este procedimiento también implica elegir compuestos que sean fáciles de sintetizar, lo que es de particular interés si la capacidad de síntesis es limitada. De esta manera, los mejores de estos aciertos en la base de datos se examinan visualmente usando un sistema de modelación gráfica por ordenador, y los más prometedores de éstos se seleccionan en función de la abundancia de razonamiento físico-químico. Las plantillas se modifican usando un constructor en 3D de moléculas pequeñas (MacMimic). Cada plantilla da lugar a una clase de compuesto, por ejemplo, denominado “hdo”. A cada modificación se le asigna un número específico (por ejemplo hdo_3) y las coordenadas y una descripción se almacenan en una estructura en forma de árbol, usando el programa ARVS-JAKT desarrollado por Symbicom. El diseño se realiza en colaboración entre los expertos de estructura proteica y químicos orgánicos, con el fin de proporcionar las mejores herramientas posibles para los químicos que realmente sintetizarán los compuestos. La eficacia de estas modificaciones se evalúa finalmente usando cálculos dinámicos moleculares de energía libre como se describen en este documento para estudiar la estabilidad del complejo proteína-ligando (Aqvist y col., 1994; Aqvist y Medina, 1993). Con el fin de maximizar la eficacia de los procedimientos mencionados anteriormente para identificar/diseñar sus15 ES 2 225 834 T3 tancias que sean capaces de interactuar con una chaperona molecular, se prefiere que la sustancia A sea probablemente una sustancia capaz de unirse al sitio de unión seleccionado. 5 10 15 En vista del modus operandi descrito anteriormente para seleccionar sustancias que deben interactuar con chaperonas como PapD, se puede realizar, de acuerdo con la invención, cuando la sustancia A se selecciona mediante la realización de las siguientes etapas: - co-cristalizar la chaperona periplásmica molecular o el análogo de la misma con un ligando capaz de interactuar con un sitio en la chaperona periplásmica molecular o el análogo de la misma y establecer la conformación tridimensional de la chaperona periplásmica molecular o del análogo de la misma y el ligando cuando interactúan por medio de cristalografía de rayos X, - usar la conformación establecida anteriormente de la chaperona periplásmica molecular o del análogo de la misma para establecer la representación tridimensional del sitio en la chaperona periplásmica molecular o el análogo de la misma que interactúa con el ligando durante la unión, - seleccionar varios grupos químicos distintos, X1, y determinar las posibles distribuciones espaciales de los grupos químicos X1 que maximizan la energía libre de unión entre los grupos químicos y el sitio en la chaperona o el análogo que interactúa con el ligando, 20 - extraer, de una base de datos que comprende representaciones tridimensionales de moléculas, una molécula que tenga los grupos químicos X1 en las posibles distribuciones espaciales determinadas anteriormente, 25 - modificar opcionalmente la representación tridimensional de la molécula extraída de la base de datos, e identificar la molécula opcionalmente modificada como la sustancia A. De acuerdo con la invención, las etapas indicadas anteriormente se prefieren especialmente cuando el ligando es una subunidad de pilus o una parte de la misma con la que la chaperona actúa normalmente durante el transporte de la subunidad de pilus a través del espacio periplásmico y/o durante la unión al pilus. 30 Mediante el uso del procedimiento mencionado anteriormente para identificar sustancias capaces de interactuar con las chaperonas periplásmicas, se han identificado varias clases de sustancias que han demostrado ser prometedoras en la fase de diseño. 35 40 45 50 55 60 65 Los esfuerzos en el diseño de fármacos para inhibidores/mejoradores de PapD se han concentrado en la región de la molécula en la que se observa que se une el péptido G. Esta región ahora se describirá con detalle, usando uno de los supuestos inhibidores (bpy_9, véase más adelante) como estructura de referencia. El sitio de unión se domina por las cadenas laterales cargadas centrales de Arg-8 y Lys-112, que se unen al resto sulfato de bpy_9. Adyacente a éste está una pequeña cavidad hidrófoba poco profunda formada por las cadenas laterales de Ile-154, Ile-194 y Thr-7, frente al grupo 2-etilo de los paquetes bpy_9 (Cavidad 1). Un grupo tan largo como el anillo fenilo podría acomodarse aquí, unirse a la posición 2 del azúcar y con posibles sustituyentes que podrían recibir o donar un enlace hidrógeno a Thr-7, o donar el esqueleto carbonilo de 198. También hay una gran cavidad que comprende los residuos Leu-4, Ile-111, Thr-7 y Thr-109, en los que el anillo fenilo de bpy_9 se acomoda (Cavidad 2). Un grupo tan grande como naftaleno podría estar sustituido en la posición 6 del estrado carbohidrato para rellenar este sub-sitio, con sustituyentes que podrían formar un enlace hidrógeno a Thr109, o a cualquiera de los átomos del esqueleto polar de los residuos Leu-4, Arg-6 o Lys-110. Entonces, hay un gran parche llano que incluye Tyr-87, y las regiones alifáticas de Lys-110 y Lys-112, que podrían acomodar un sistema tricíclico tal como 2-fenantrilo sustituido en la posición 3 del azúcar (Parche 3). Pueden considerarse los sustituyentes del hidrógeno unido a Tyr-87 o el esqueleto de Lys-110, así como grupos cargados negativamente para complementar las cadenas laterales de Lys-110 y Lys-112. Tyr-87 es un donador de transferencia de carga potencial a un sistema π deficiente de electrones tal como nitroarilo. Se han fabricado modelos de varias chaperonas homólogas (SfaE, MrkB, HifB y FimC) a partir de la estructura de PapD, y las diferencias entre los modelos estructurales han influenciado el diseño de inhibidores, de tal forma que los ligandos propuestos deben unirse a todas las estructuras examinadas. Por ejemplo, es tentador complementar la cadena lateral cargada de Arg-200 con un grupo ácido en el ligando, pero como dos de las otras estructuras tienen un Asp en esta posición, el residuo no se considera un buen candidato. Arg-8 se conserva completamente, como Lys-112, Thr7 e Ile-11. Tyr-87 se convierte en Trp en tres estructuras, pero la naturaleza global del parche 3 no se carga mediante éste. PapD está realmente sólo con su secuencia Thr-Lys en 109-110, siendo las otras cuatro estructuras Ser-Arg aquí, pero de nuevo, estos cambios conservativos no alteran de forma significativa los criterios de diseño. Aunque las sustancias que interactúan con el sitio de unión responsable de la unión a las proteínas G son candidatos obvios como inhibidores/mejoradores de las chaperonas periplásmicas, se entenderá que las moléculas capaces de interactuar con otros sitios en chaperonas periplásmicas también son interesantes en este aspecto. Es muy posible que una interacción con otro sitio diferente de una proteína de unión G en i.e. PapD pueda hacer que se prevenga, inhiba o mejore PapD en su acción como chaperona periplásmica. 16 ES 2 225 834 T3 Como se ha mencionado anteriormente, una familia de sustancias (denominadas en este documento familia bpy) es un aspecto importante de la invención. De esta manera, la invención se refiere a nuevos compuestos de fórmula general 5 10 15 en la que 20 V1 es O, S, SO, SO2 , CH2 , C(OH)H, CO o CS; W1 es O, S, SO2 , SO3 , CH2 o NH; 25 R1 es H; alquilo C1−24 , alquenilo C1−24 o alquinilo C1−24 , donde el alquilo, alquenilo y alquinilo pueden estar sustituidos con uno o más sustituyentes seleccionados entre OH, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO2 NH2 y -SO2 NH2 ; acilo; o -(CH2 CH2 O)s -H, donde s = 1, 2, 3; R2 es un grupo de la fórmula 30 35 40 en la que 45 A es -CH-(CH2 )n - o -CH=CH-(CH2 )n−1 - (n > 0) o -CH-(CH2 )n -; k Y2 B es -(CH2 )m - o =CH-(CH2 )m−1 - (m > 0); 50 X2 es N, CH o C (donde B es =CH-(CH2 )m−1 -; e Y2 es O, S, NH, H2 o H (n = 1); y 4 > m + n > 0, n < 3 y m < 3; 55 o R2 es un grupo de fórmula 60 65 17 ES 2 225 834 T3 en la que 5 A es -CH-(CH2 )n - o -CH=CH-(CH2 )n−1 - (n > 0) o -CH-(CH2 )n -; k Y2 B es -(CH2 )m - o =CH-(CH2 )m−1 - (m > 0); y X’2 es O, NH, CH2 o S (cuando p = 0); N o CH (p = 1); 10 o C (cuando p = 1 y B es =CH-(CH2 )m−1 -); 15 V2 , Z2 y W2 son independientemente H, OH, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO2 NH, -SO2 NH2 , o V2 y Z2 , o Z2 y W2 juntos forman -NHC(O)NH-, -C(O)NHC(O)-, -NHS(O2 ) NH-, -C(O)NHO-, -C(S)NHO-, -S(O2 )NHO- o -S(O2 )NHC(O)-; 4 > m + n > 0, n < 3 y m < 3; 20 25 o R2 es un grupo -W5 -(alquilo C1−5 o alquenilo C2−5 o alquinilo C2−5 ) donde W5 es un enlace o se selecciona entre -O-, -S-, -SO2 - y -NHC(O)- y el resto alquilo C1−5 , alquenilo C2−5 o alquinilo C2−5 puede estar sustituido con hasta tres grupos seleccionados independientemente entre OH, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, NHCONH2 , -NHCSNH2 , -NHSO2 NH2 y -SO2 NH2 ; -Z1 -R3 es -SO2 (OH), -PO(OH)2 , -OSO2 (OH), -NHSO2 (OH), -NH-CO-COOH, -SPO(OH)2 , -CH2 COOH, tetrazol5-ilo o tetrazol-5-ilmetilo, o sales de los mismos; o Z1 es -O-, -S-, -NH- o -CH2 - y R3 es un grupo de fórmula: 30 35 40 45 50 55 D es -CH2 -, -CO-, -SO2 -, -NH-SO2 -, -NH-CO-, -O-PO(OH)- o una sal de los mismos; 60 65 Z3 es H, OH, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO2 NH2 , SO2 NH2 , -SO2 (OH), -PO(OH)2 , -OSO2 (OH), -NHSO2 (OH), -COOH, tetrazolil-5-ilo o tatrazolil-5-ilmetilo o una sal de los mismos, con la condición de que cuando D sea -CH2 , -CO-, -SO2 -, -NHSO2 - o -NHCO-, entonces Z3 sea -SO2 (OH), -PO (OH)2 , -OSO2 (OH), -NHSO2 (OH), -COOH, tetrazolil-5-ilo o tetrazolil-5-ilmetilo o una sal de los mismos; X3 e Y3 son independientemente H, NO2 , SO2 NH2 , CONH2 , CF3 o F; y 18 ES 2 225 834 T3 U4 -W4 es -CHCH-, -CH2 CH2 -, -C(OH)CH-, -CHC(OH)-, -CH(OH)CH2 -, -CH2 CH(OH)-, -CH(OH)CH(OH)-, -C (O)NH-, -NHC(O)-; Y1 es -OH- o -S-; 5 10 R4 es H o, cuando Y1 es S, S(CH2 )q N(R9 )3 + y q es un número entero de 2-4, donde R9 es H o CH3 ; R5 es H; alquilo C1−6 , alquenilo C2−6 o alquinilo C2−6 , y el reto alquilo C1−6 , alquenilo C2−6 o alquinilo C2−6 puede estar sustituido con OH, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO2 NH2 o -SO2 NH2 ; o arilo, aril-alquilo (C1−2 ), heterociclilo o heterociclil-alquilo (C1−2 ) que pueden estar opcionalmente sustituidos en los restos arilo o heterociclilo con uno, dos o tres sustituyentes seleccionados independientemente entre OH, F, Cl, NH2 , CONH2 , NHCOH y SO2 NH2 ; X1 es -O-, -S- o -NH-; 15 R6 es H o, cuando X1 es NH, acilo, HOCNH-Val-Met-, HOCNH-Ile-(S,S)-dioxo-metionil- o HOCNH-Val-(piran4-on-2-il)-alanil-; o una sal de tal compuesto; 20 También se ha sintetizado otra familia de sustancias llamadas familia hdo. Por tanto, la invención también se refiere a nuevos compuestos de fórmula general 25 30 35 en la que X es O, P, P(O), S, SO, SO2 , CH2 , C(OH)H o un grupo NQ11 , donde Q11 es H, OH, acilo C1−24 o alquilo C1−24 ; 40 45 Z11 es un enlace, O, CH2 , S, SO, SO2 o un grupo NQ1 , donde Q12 es H, acilo C1−24 o alquilo C1−24 ; R11 es H; alquilo C1−24 , alquenilo C2−24 o alquinilo C2−24 , que puede estar sustituido con uno o más sustituyentes seleccionados independientemente entre -OH, -COOH, -F, -Cl, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO3 NH2 y -SO2 NH2 ; acilo; o -(CH2 CH2 O)s -H, donde s = 1, 2 ó 3; o R11 es CH=CH-(CH2 )n0 -Q13 o -(CH2 )n0 -Q13 , donde Q13 es un grupo arilo o heteroarilo sustituido con -OH, -COOH, -F, -Cl, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO3 NH2 y -SO2 NH2 , y donde n’ ≥ 0; 50 55 R12 y R13 son independientemente OH, H, F, Cl, OW11 u O(CO)W11 , donde W11 es alquilo C1−24 , alquenilo C2−24 o alquinilo C2−24 , o un grupo arilo o heteroarilo sustituido con -OH, -COOH, -F, -Cl, -CNH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO3 NH2 y -SO2 NH2 ; Z12 es un enlace, O, S o CH2 ; R14 es -(CH2 )n00 -Q14 , donde Q14 es un grupo arilo o un grupo heteroarilo sustituido con -OH, -COOH, -F, -Cl, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO3 NH2 y -SO2 NH2 , y donde n” = 0, 1, 2 ó 3; 60 65 Z13 es un enlace, O, CH2 , S, SO, SO2 , NQ14 Q15 , donde Q14 es H, acilo C1−24 o alquilo C1−24 , y Q15 es CO o -C(O) W12 , donde W12 es O o NW13 , donde W13 es H, OH, acilo C1−24 o alquilo C1−24 ; R15 es H; alquilo C1−24 , alquenilo C2−24 o alquinilo C2−24 , donde el alquilo, alquenilo o alquinilo pueden estar sustituidos con uno o más sustituyentes seleccionados independientemente entre -OH, -COOH, -F, -Cl -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO3 NH2 y -SO2 NH2 ; acilo o -(CH2 CH2 O)s -H, donde s = 1, 2 ó 3; 19 ES 2 225 834 T3 o R15 es CH=CH-(CH2 )n0 -Q13 o -(CH2 )n0 -Q13 , donde Q3 es como se ha definido anteriormente y donde n’ ≥ 0; o una sal de tal compuesto. 5 10 15 20 En el presente contexto, los términos “alquilo C1−5 , C1−6 y C1−24 ” pretenden significar grupos alquilo con 1-5, 1-6 y 1-24 átomos de carbono, respectivamente, que pueden ser lineales, ramificados o cíclicos tales como metilo, etilo, propilo, isopropilo, butilo, isobutilo, terc-butilo, hexilo, octilo, dodecilo, ciclopentilo, ciclohexilo, etc. Además, como se usan en este documento, los términos “alquenilo C2−5 , C2−6 y C2−24 ” pretenden significar grupos alquilo mono- o poliinsaturados con 2-5 y 2-24 átomos de carbono, respectivamente, que pueden ser lineales, ramificados o cíclicos donde pueden estar presentes dobles enlaces en cualquier sitio de la cadena o del anillo, por ejemplo, vinilo, 1-propenilo, 2-propenilo, hexenilo, decenilo, 1,3-heptadienilo, ciclohexenilo, etc. Algunos de los sustituyentes existen tanto en la configuración cis como en trans. El alcance de estas invenciones comprende las formas cis y trans. En el presente contexto, los términos “alquinilo C2−5 , C2−6 y C2−24 ” pretenden significar un grupo alquilo lineal o ramificado con 2-5 y 2-24 átomos de carbono, respectivamente, e incorporan uno o más triples enlaces, por ejemplo, etinilo, 1-propinilo, 2-propinilo, 2-butinilo, 1,6-heptadinilo, etc. Los términos “alcoxi C1−6 y C1−24 ” indican grupos alquilo como se han definido anteriormente que comprenden una función oxi. En el presente contexto, el término “arilo” pretende significar fenilo y naftilo. El término “heteroarilo” pretende significar un sistema aromático cíclico, en el que al menos un átomo que no es carbono contribuye al sistema de unión π. 25 Los ejemplos de grupos arilo sustituidos son: 3-nitrofenilo, 3-hidroxifenilo, 4-hidroxifenilo, 3,4-dihidroxifenilo, 3carboxamidofenilo, 3-formamidilfenilo, 3-acetamidilfenilo, 3-fluoronaftil-2-ilo, 7-fluoronaftilo, 3,7-difluoronaftilo, 3hidroxinaftilo, 7-hidroxinaftilo, 3,7-dihidroxinaftilo, 3-fluoro-7-hidroxinaftilo, 7-fluoro-3-hidroxinaftilo, 4-fluoronaft2-ilo, 6-fluoronaft-ilo, 8-fluoronaft-2-ilo, 4,6-difluoronaft-2-ilo, etc. 30 35 40 45 Los ejemplos de grupos heterocíclicos y heteroarilo son pirrolilo, furanilo, 2,3-dihidrofuranilo, tetrahidrofuranilo, tienilo, 2,3-dihidrotienilo, tetrahidrotienilo, imidazolilo, oxazolilo, tiazolilo, pirazolilo, indolilo, pirazinilo, dioxolanilo, dioxanilo, 1,3,5-trioxanilo, tetrahidrotiopiranilo, ditiolanilo, pirazolidinilo, iminazolidinilo, sim-triazinilo, simtetrazinilo, quinazolinilo, pteridinilo, isoindolilo, 1,2,4-triazolilo, 1,2,3-triazolilo, bencimidazolilo, indazolilo, benzofuranilo, isobenzofuranilo, benzotiofenilo, tienotiofenilo, isoxazolilo, 1,2,5-oxadiazolilo, isotiazolilo, 1,3,4-tiadiazolilo, benzoxazolilo, benzotiazolilo, azaindolilo, oxoindolilo, hidroxiindolilo, N-oxiisoquinolilo, etc. En el presente contexto, el término “acilo” (por ejemplo, acilo C1−24 ) pretende indicar el residuo acilo de un resto ácido carboxílico o un ácido sulfónico que comprende un grupo carbonilo o sulfonilo y un resto orgánico. Los ejemplos de grupos acilo incluyen alcanoílo C1−24 (por ejemplo, formilo, acetilo, propionilo, butirilo, isobutirilo, valerilo, isovalerilo, pivaloílo y hexanoílo), alquenoílo C1−24 (por ejemplo, acriloílo, metacriloílo, crotonoílo, iso-crotonoílo, 2-pentenoílo, 3-pentenoílo, 2-metilpentenoílo, 3-pentenoílo, 3-fenilpropenoílo, 2-fenil-trans-propenoílo, 2,4-hexadienoílo), alquinoílo C1−24 (por ejemplo, propionoílo, 2-butinoílo, 3-butinoílo, 2-metil-3-butinoílo, 2,2-dimetilbutinoílo, 2-pentinoílo, 3-pentinoílo, 2-pentin-4-trans-enoílo), alcoxicarbonilo C1−24 (por ejemplo, metoxicarbonilo, etoxicarbonilo, propoxicarbonilo, butoxicarbonilo y t-butoxicarbonilo), alqueniloxicarbonilo C1−24 (por ejemplo cis-2-buteniloxicarbonilo, 1-metil-2-propeniloxicarbonilo, 1,1-dimetil-2-propeniloxicarbonilo, trans-2-buteniloxicarbonilo), aroílo (por ejemplo, benzoílo), heterociclilcarbonilo (por ejemplo, 2-furoílo, 3-furoílo, 2-furanoílo, 3-furanoílo, 2-pirrolcarboxilo, 3-pirrolcarboxilo, 2-tenoílo, 3-tenoílo, 2-indolcarboxilo, 3-indolcarboxilo, 1-naftanoílo y 2-naftanoílo), etc. 50 55 El término “sal” pretende comprender una sal tal como una sal de adición de ácidos orgánicos (por ejemplo, acetato, valerato, salicilato, galacturonato, gluconato, tannato, trifluoroacetato, maleato, tartrato, metanosulfonato, bencenosulfonato, formiato, tiocianato y toluenosulfonato), una sal de adición de ácidos inorgánicos (por ejemplo, clorhidrato, bromhidrato, yodhidrato, diclorhidrato, dibromhidrato, diyodhidrato, sulfato, hidrogenosulfato, halosulfato tal como yodosulfato, nitrato, fosfato y carbonato) o una sal con un aminoácido (por ejemplo, arginina, ácido aspártico y ácido glutámico) o una sal metálica tal como una sal de metal alcalino (por ejemplo, sal sódica y sal potásica) y una sal de metal alcalinotérreo (por ejemplo, sal de calcio y sal de magnesio), una sal amónica, una sal álcali orgánica (por ejemplo, sal trimetilamina, sal trietilamina, sal piridina, sal picolina, sal diciclohexilamina y sal N,N’-dibenciletilenodiamina), y los hidratos de las mismas. 60 Cuando el sustituyente R5 indica heterociclilo, se prefiere que el sustituyente indique un grupo heterociclilo de fórmula 65 20 ES 2 225 834 T3 5 10 15 20 en la que ’X es -CH-, -CH2 -, -O-, -N-, -S-, -S→O, -N→O o -CO-, Y es -CH- o -NH-, y Z es -CH-, -CH2 -, -O-, -S-, -N-, -CO-, -S→O o -N→O, especialmente un grupo seleccionado entre el grupo compuesto por inden-7-ilo, benzofuran-4-ilo, isobenzofuran-4-ilo, tionaften-4-ilo, isotionaften-4-ilo, 2-oxo-inden-7-ilo, 2-oxo-inden-4-ilo, inden-4ilo, benzofuran-7-ilo, isobenzofuran-7-ilo, tionaften-7-ilo, isotionaften-7-ilo, 1-oxo-tionaften-4-ilo, 1-oxo-tionaften-7ilo, antran-4-ilo, antran-7-ilo, tioantran-4-ilo, tioantran-7-ilo, benzotiozol-4-ilo, benzotiozol-7-ilo, 2H-2-isobenzo-1,3dion-7-ilo, isobenzofuran-5-ilo, isobenzofuran-6-ilo, 3H-2-oxo-benzofuran-5-ilo, 3H-2-oxo-benzofuran-6-ilo, 3H-2oxotionaften-5-ilo, 3H-2-oxotionaften-6-ilo, indol-5-ilo, indol-6-ilo, 3H-2-oxoindol-5-ilo, 3H-2-oxoindol-6-ilo, 3H-2oxobenzoxazol-5-ilo, 3H-2-oxobenzoxazol-6-ilo, benzotiazol-5-ilo, benzotiazol-6-ilo, 2-oxobenzo-1,3-ditiol-5-ilo, 2oxobenzo-1,3-ditiol-6-ilo, 3H-2-oxobencimidazol-5-ilo, 3H-2-oxobencimidazol-6-ilo, benzoxatiol-5-ilo, benzoxatiol6-ilo, 3H-2-oxobenzotiazol-5-ilo y 3H-2-oxobenzotiazol-6-ilo. También se prefiere que el sustituyente R5 sea un grupo de fórmula 25 30 o que R5 sea un grupo de la fórmula 35 40 45 50 55 60 Con respecto al sustituyente R2 que es heterociclilo, se prefiere especialmente que se seleccione entre el grupo compuesto por isobenzofuran-5-ilo, isobenzofuran-6-ilo, 3H-2-oxo-benzofuran-5-ilo, 3H-2-oxo-benzofuran-6-ilo, 3H-2oxotionaften-5-ilo, 3H-2-oxotionaften-6-ilo, indol-5-ilo, indol-6-ilo, 3H-2-oxoindol-5-ilo, 3H-2-oxoindol-6-ilo, 3H-2oxobenzoxazol-5-ilo, 3H-2-oxobenzoxazol-6-ilo, benzotiazol-5-ilo, benzotiazol-6-ilo, 2-oxobenzo-1,3-ditiol-5-ilo, 2oxobenzo-1,3-ditiol-6-ilo, 3H-2-oxobencimidazol-5-ilo, 3H-2-oxobencimidazol-6-ilo, benzoxatiol-5-ilo, benzoxatiol6-ilo, 3H-2-oxobenzotiazol-5-ilo y 3H-2-oxobenzotiazol-6-ilo. El número exacto de sustituyentes presentes sobre un resto alquilo, alquenilo o alquinilo R1 dependerá de la longitud de la cadena de carbonos, siendo el propósito de los sustituyentes hacer que el grupo entero R1 sea compatible con agua ya que, en el complejo chaperona-ligando tal como el complejo PapD-ligando, el grupo R1 se extenderá en el medio acuoso que lo rodea. De esta manera, para una cadena de carbono bastante corta tal como hasta de cuatro carbonos, se contempla que será suficiente uno de los sustituyentes polares anteriores, en particular cuando el sustituyente se encuentra en el extremo mientras que, para las cadenas más largas, puede requerirse un gran número de sustituyentes, tales como un sustituyente para cada átomo de carbono. Los glicósidos 4,6-O-(4’-metoxi)fenilmetiliden-α-D-glucohexopiranósido o 4,6-O-(4’-metoxi)fenilmetiliden-β-Dglucohexopiranósido: 65 21 ES 2 225 834 T3 5 10 15 20 25 30 35 (se usan aquí como ejemplos preferidos, pero también pueden usarse otros arilmetiliden o viniliden acetales) pueden prepararse como se indica a continuación: La glucosa peracilada se hace reaccionar con, por ejemplo, bromuro de hidrógeno o cloruro de hidrógeno en un disolvente adecuado tal como, por ejemplo, ácido acético o diclorometano, formando bromuro o cloruro de glicosilo per-O-acilado (O-acilación y síntesis de haluro de glicosilo: M. L. Wolfrom y A. Thompson, Methods in Carbohydrate Chemistry, Vol. 2, 211-215, ed. por R. L. Whistler y L. Wolfrom, Academic Press, Nueva York, 1963; G. Hewit y G. Fletcher Jr., ibid, 226-228; y R. U. Lemieux, ibid, 223-224). El alcohol o tiol de aglicona protegido de forma adecuada, cuando es necesario (H-W1 R1 -PG1 o H-W1 R1 ) (grupos protectores: Protective Groups in Organic Synthesis, Editores T. W. Greene y P. G. M. Wuts, John Wiley y Sons, Inc., Nueva York, 1991) se hace reaccionar con la glucosa per-O-acilada usando un ácido de Lewis tal como eterato de trifluoruro de boro (R. J. Ferrier y R. H. Furneaux, Carbohydr. Res., 52 (1976) 63-68; J. Dahmén, T. Frejd, G. Gronberg, T. Lave, G. Magnusson y G. Noori, Carbohydr. Res 116 (1983) 303-307) o trifluorometanosulfonato de trimetilsililo (T. Ogawa, K. Beppu, S. Nakabayashi, Carbohydr. Res., 93 (1981) C6-C9) como promotores. La reacción se realiza en un disolvente adecuado tal como cloroformo, diclorometano o tolueno. Cuando el derivado de monosacárido en cuestión es un bromuro o cloruro de glicosilo per-O-acilado, pueden usarse promotores tales como trifluorometanosulfonato de plata o sales de mercurio (II) (H. Paulsen, Angew. Chem. Int. Ed. Engl., 21 (1982) 155-173) y las reacciones se realizan en un disolvente adecuado tal como diclorometano o tolueno. La glucosa W1 R1 o los glicósidos W1 R1 PG1 se obtienen después de la des-O-acilación usando metóxido sódico (A. Thompson, M. L. Wolfrom y E. Pascu, páginas 215-220, Methods in Carbohydrate Chemistry, Vol. II, Editores: R. L. Whistler y M. L., E. Wolfrom, Academic Press, Nueva York, 1963) en metanol en un co-disolvente que contiene metanol tal como diclorometano o tetrahidrofurano. Después, se obtienen los 4,6-(4’-metoxi)bencilideno acetales por reacción con 4-metoxibenzaldehído dimetil acetal y ácido en un disolvente polar no prótico tal como por ejemplo dimetilformamida, acetonitrilo o tetrahidrofurano (J. J. Patroni y col., Aust. J. Chem. 1988, (41), 91-102; para otros procedimientos de formación de acetales, véase por ejemplo A. N. de Belder, 1979, adv. Carbohydr. Chem. Biochem., 34, 179 y las referencias citadas en ese documento). Después, se obtienen los epóxidos B1 o B2 40 45 50 55 60 a través de ésteres sulfonato: Los manno epóxidos B1 pueden prepararse haciendo reaccionar el derivado de glucósido A con hidruro sódico y p-toluenosulfonilimidazol en dimetilformamida (D. Hicks y B. Fraser-Reid; Synthesis 1974, 203) o con hidruro sódico y cloruro de p-toluenosulfonilo en tetrahidrofurano (V. S. Murthy y col., Synthetic Commun. 1993, 23 (3), 285-289). Los allo epóxidos B2 pueden prepararse haciendo reaccionar el derivado de glucósido A con metilsulfonilo o cloruro de p-toluenosulfonilo en piridina y tratando el diéster de metilsulfonato resultante con etóxido sódico en etanol (Y. Ali, A. C. Richardson, Carbohydrate Res. 1967, 5, 441-448; N. Richtmeyer, Methods in Carbohydrate Chemistry, Vol. 1, 107). Los epóxidos B1 o B2 pueden hacerse reaccionar con reactivos nucleófilos adecuados, produciendo las allo hexopiranósidos diaxialmente sustituidas C1 y C2 65 22 ES 2 225 834 T3 5 10 (para referencias generales sobre el uso de epóxidos, véase por ejemplo J. Gorzynski Smith, Synthesis, 1984, 8, 629656 Masamune S., Choy W., Petersen J y Sita L R, Angew. Chem. Int. Ed. Engl., 1985, 24, 1-76; A. S. Rao y col., Tetrahedron Lett., 1983, 39, 2323). 15 Cuando el primer átomo de R2 y Z1 (como se ha definido anteriormente) conectado al resto carbohidrato en el producto final deseado es un nitrógeno (= el átomo nucleófilo), entonces el nucleófilo preferido Nu1 o Nu2 es azida (N3 − ). El epóxido se trata con azida sódica y cloruro amónico en ebullición 2-metoxi-etanol (R. D. Guthrie y D. Murphy; J. Chem. Soc. 1963, 5288-5294). 20 Cuando el átomo nucleófilo es oxígeno o azufre, el procedimiento general preferido de apertura del epóxido implica tratamiento con un alcohol o tiol protegido adecuadamente en presencia de alúmina neutra en éter (G. H. Posner y D. Z. Rogers, J. Am. Chem. Soc. 1977, 99, 8208; G. H. Posner, D. Z. Rogers y A. Romero, Isr. J. Chem. 1979, 18, 259; y G. W. Posner, M. Hulce y R. K. Rose, Synth. Commun. 1981, 11, 737). 25 Cuando el átomo nucleófilo es carbono, los reactivos usados más comúnmente son compuestos de organomagnesio, organolitio, organocobre, organoaluminio y organoboro (J. Gorzynski Smith, Synthesis, 1984, 8, 629-656 y las referencias citadas en ese documento). 30 Cuando el producto es una allo hexopiranósido C1, la función 2-hidroxi puede bloquearse con un grupo protector que permite la introducción de la funcionalidad R2 en la última etapa (se prefiere si R2 es un éster, no mostrado en la figura) o se introduce la funcionalidad R2 protegida adecuadamente, cuando es necesario, para producir D1 35 40 45 50 55 60 Por ejemplo, los grupos OH a éteres o ésteres (Protective Groups in Organic Synthesis, Editores T. W. Greene y P. G. M. Wuts, John Wiley y Sons, Inc., Nueva York, 1991); grupos OH a carbonatos (J. March, Advanced Organic Chemistry-Reaction Mechanisms, and Structure, 3ª ed. John Wiley y Sons, Nueva York, 347 (1985) y las referencias citadas en ese documento); grupos OH a carbamatos (J. March, Advanced Organic Chemistry-Reaction Mechanisms, and Structure, 3ª Ed. John Wiley y Sons, Nueva York, 791-792 (195) y las referencias citadas en ese documento); grupos OH a grupos alquilo mediante derivados de exometileno y posterior hidrogenación o mediante otras vías (H. O. H. House, Modern Synthetic Reactions, 2ª Ed. W. A. Benjamin, Inc., Menlo Park, C. A., 1-130 (1972) y las referencias citadas en ese documento; J. Yoshimura, Adv. Carbohydr. Chem. Biochem., 42 (1984) 69-134); e intercambio de grupos heterocíclicos por grupos OH, mediante diferentes vías (A. R. Katritzky, Handbook of Heterocyclic Chemistry, Pergamon Press, Oxford, 1985). Cuando el producto es una allo hexopiranósido C2, la función 3-hidroxi se bloquea con un grupo protector que permite la introducción de la funcionalidad Z1 -R3 en la última etapa dando como resultado intermedios de tipo D2 65 23 ES 2 225 834 T3 5 10 (Protective Groups in Organic Synthesis, Editores T. W. Greene y P. G. M. Wuts, John Wiley y Sons, Inc., Nueva York, 1991) o se transforman en un intermedio de manno hexopiranósido D3 15 20 25 30 35 40 45 50 donde Nu1 es una forma protegida o enmascarada de la funcionalidad Z1 . Grupos OH a éteres o ésteres (Protective Groups in Organic Synthesis, Editores T. W. Greene y P. G. M. Wuts, John Wiley y Sons, Inc., Nueva York, 1991); grupos OH a grupos azido: J. March, Advanced Organic Chemistry-Reaction Mechanism, and Structure, 3ª Ed. John Wiley y Sons, Nueva York, 380, (1985) y las referencias citadas en ese documento; H. H. Baer, Pure Appl. Chem., 61 (7) (1989) 1217-1234 y las referencias citadas en ese documento; grupos OH a grupos amino mediante azidas u otras vías (March, Advanced Organic Chemistry-Reaction Mechanisms, and Structure, 3ª Ed. John Wiley y Sons, Nueva York, 1106, 798-800 (1985) y las referencias citadas en ese documento; H. H. Baer, Pure Appl. Chem., 61 (7) (1989) 1217-1234 y las referencias citadas en ese documento). Después, la función 4,6-O-acetal se abre reductivamente, produciendo la función R6 en el caso en el que R6 es un éter, o los intermedios F1, F2 o F3 con una función hidroxi en la posición 6 (apertura reductiva de acetales, véase Garegg P J y Hultberg H, Carbohydr. Res. 1981, 93, c10-11; Garegg P J, Hultberg H y Wallin S, Carbohydr. Res. 1982, 108, 97-101; Liptak A, Jodal I, Nanasi P, Carbohydr. Res. 1975, 44, 1-11; Baker D C, Horton D, Tindall C G, Methods in Carbohydr. Chem., 1976 Vol. 6, 3-6; Mikami T, Asano H, Mitsunobu O, Chem. Lett. 1987, 10, 2003-2036; Ed M, Garegg P J, Hulberg H, Oscarsson S, J. Carbohydr. Chem. 1983, 2, 305-311; Hunter R, Bartels B, Michael J P, Tetrahedron Lett. 1991, 32, 1095-1098; Rao S P, Grindley T B, Carbohydr. Res. 1991, 218, 83-93; Hunter, R. Bartels, B. J. Chem. Soc. Chem. Commun. 1991, 2887-2888). Por ejemplo, los intermedios de tipo D1, D2 o D3 se tratan con cianoborohidruro sódico y clorotrimetilsilano en acetonitrilo, (R. Johansson y B. Samuelsson, J. Chem. Soc. Perkin Trans. 1, 1984, 2371-2374) o complejo de boranotrimetilamina y tricloruro de aluminio. El resultado regioquímico de la reacción normalmente depende del disolvente (Ek M, Garegg P J, Hultberg H, Oscarsson S, J. Carbohydr. Chem. 1983, 2, 305-311). Los intermedios aldehído de tipo G1, G2 o G3 55 60 65 se obtienen por oxidación de los correspondientes intermedios de 6-alcohol de tipo D1, D2 o D3, preferiblemente mediante el procedimiento de Swern. (Mancuso A J, Swern D, Synthesis, 1981, 165-185; Tidwell T, Synthesis, 1990, 857-870; para otros procedimientos de oxidación, véase A. H. Haines, 1988, Methods for the Oxidation of Organic Compounds, Capítulo 2, Academic Press, San Diego y las referencias citadas en ese documento). 24 ES 2 225 834 T3 En la siguiente etapa se añade un nucleófilo de carbono a la función aldehído de los intermedios de tipo G1, G2 o G3. Preferiblemente, se añade un reactivo de alquillitio o arillitio protegido adecuadamente o un reactivo de grignard al aldehído en un disolvente de éter o hidrocarburo, produciendo los alcoholes secundarios H1, H2 o H3: 5 10 15 Para las reacciones de los aldehídos con compuestos de organolitio y organomagnesio, véase J. March, Advanced Organic Chemistry-Reaction Mechanisms, and Structure, 3ª Ed. John Wiley y Sons, Nueva York, 347 (1985) y las referencias citadas en ese documento. 20 25 30 35 Para las reacciones de los aldehídos con organolitio y organomagnesio y otros nucleófilos de carbono, véase Evans D A, Aldrichim. Acta, 1982, 15, 23 y las referencias citadas en ese documento. Para ejemplos específicos sobre el uso y preparación de reactivos fenillitio sustituidos con arilo y de grignard, véase Ames M M, Castagnoli Jr. N, J. Labelled Compd., 1974, 10 (2), 195-205; documento DE 3807910 A1; Mills R J, Snieckus V, Polynucl. Aromat. Hydrocarbons, [Pap. Int. Symp.], 8ª, Meeting 1983, 913-24. Editado por: Cooke M y Dennis A J, Battelle Press 1985: Columbus, Ohio; Iriye R, Furukawa K, Nishida R, Kim C, Fukami H, Biosci. Biotechnol. Biochem. 1992, 56 (11), 1773-5; Comber M F, Sargent M V U, J. Chem. Soc., Chem. Commun., 1991, (3), 190-2; Hirai T, Yoshizawa A, Nishiyama I, Fukumasa M, Shiratori N, Yokoyama A, documento EP 341686 A2; Leeson P D, Emmet J C, Shah V P, Showell G A, Novelli R, Prain H D, Benson M G, Ellis D, Pearce N J, Underwood A H, J. Med. Chem. 1989, 32 (2), 320-36); Meltzer P C, Liang A Y, Brownell A L, Elmaleh D R, Madras B K, J. Med. Chem., 36 (7), 855-62; Willard A K, Novello F C, Hoffman W F, Cragoe Jr E J., documento US 4.459.423. Para reactivos de fenillitio sustituidos que también pueden elaborarse en compuestos heterocíclicos, véase: Lang H J, Muschaweck R, documento AU 514406 B2; Lang H J, Muschaweck R, Hropot M, documento HU 19761; Lang H J, Muschaweck R, Hropot M, documento DE 2737195. Para reactivos de arillitios heteroaromáticos y de grignard, véase: Yang Y, Martin A R, Nelson D L, Regan J, Heterocycles, 1992, 34 (6), 1169-75. 40 45 Para ejemplos de otros reactivos organometálicos para la síntesis estereoselectiva de alcoholes secundarios a partir de aldehídos: Homoalilalcoholes con complejos de crotilmolibdeno: Faller J W, John J A, Mazzieri M R, Tetrahedron Lett. 1989, 30, 1769-1772. Homoalilalcoholes con complejos de titanio: Riediker M, Duthaler R O, Angew. Chem. Int. Ed. Eng., 1989, 28, 494-495. 50 3-Pirrolilalcoholes con 3-litiopirrol protegido con sililo: Bray B L, Mathies P H, Naef R, Solas D R, Tidwell T T, Artis D R, Muchowski J M, J. Org. Chem., 1990, 55, 6317-6328. Alcoholes alílicos con E-vinilalano: A P Kozikowski y Jiang-Ping Wu, Tetrahedron Lett. 190, 30, 43094312 y las referencias citadas en ese documento. 55 Dioles trans-alílicos con vinilestanos: Corey E J, Wollenberg R H, J. Org. Chem., 1975, 40, 2265-2266. Carbinoles de pirrolidina con α-litio pirrolidina amidinas: Sanner M A, Tetrahedron Lett. 1989, 30, 19091912. 60 Reactivos de Organocinc: Furstner A, Synthesis, 1989, 571-590 y las referencias citadas en ese documento. En lo sucesivo, el sustituyente de la posición 3 (Nu1 u OPG1 ) se transforma en un nucleófilo con el fin de instalar la funcionalidad cargada negativamente Z1 -D-R3 . 65 Los 6-alcoholes secundarios H1, H2 y H3 se protegen primero, (Protective Groups in Organic Synthesis, Editores T. W. Greene y P. G. M. Wuts, John Wiley y Sons, Inc., Nueva York, 1991) o se transforman en funciones amino en los casos en los que X1 -R6 forma una funcionalidad peptídica (grupos OH a grupos amino mediante azidas u otras vías: 25 ES 2 225 834 T3 Véase por ejemplo March, Advanced Organic Chemistry-Reaction Mechanisms, and Structure, 3ª Ed., John Wiley y Sons, Nueva York, 1106, 798-800 (1985) y las referencias citadas en ese documento; H. H. Baer, Pure Appl. Chem., 61 (7) (1989) 1217-1234, y las referencias citadas en ese documento). 5 Para la síntesis de péptidos, véase Gross y Meienhofer, The Peptides, vol. 3, Academic Press, Nueva York, 19791981; Grant G A y col., Synthetic Peptides, A Users Guide, 1991, W. A. Freeman y Compañía, Nueva York, y las referencias citadas en ese documento. La posición 3 se desprotege para un intermedio de alcohol, tiol o amina I1, I2 o I3 10 15 20 (Protective Groups in Organic Synthesis, Editores T. W. Greene y P. G. M. Wuts, John Wiley y Sons, Inc., Nueva York, 1991, y las referencias citadas en ese documento). 25 30 Por ejemplo, el tratamiento de I1, donde R2 -PG1 es una combinación de funciones éter y Q1 es azuda, con sulfito de hidrógeno gaseoso en piridina y agua (T. Adashi, Y. Yamada, I. Inoue y M. Saneyoshi, Synthesis, 1977, 45). Para otros procedimientos de reducciones de azida, véase Poopeiko N E, Pricota T I, Mikhailopulo I A, Synlett, 1991, 5, 342; Samano M C, robins M J, Tetrahedron Lett. 1991, 32, 6293-6296; Rakotomanomana N, Lacombe J-M, Pavia A, Carbohydr. Res., 1990, 197, 318-323; Malik A A, Preston S B, Archibald T G, Cohen M P, Baum K, Synthesis, 1989, 450-451; Maiti S, Spevak P, Reddy N, Synt. Commun. 1988, 18, 1201-1206; Bayley H, Standring D N, Knowles J R, Tetrahedron Lett., 1978, 39, 3633-3634). Los intermedios I1, I2 o I3 se sulfatan con complejo trióxido de azufre-piridina o -trietilamina, obteniendo los intermedios J2, J4 y J6 35 40 45 50 (véase, por ejemplo J. Basten, G. Jaurand, B. Olde-Hanter, M. Petitou y C. A. A. van Boeckel, Bioorg. Med. Chem. Lett., 1992, 2 (9), 901-904 y las referencias citadas en ese documento; J. Basten, G. Jaurand, B. Olde-Hanter, P. Duchaussoy, M. Petitou y C. A. A. van Boeckel, Bioorg. Med. Chem. Lett., 1992, 2 (9), 905-910 y las referencias citadas en ese documento; Bocker, T. Lindhorst, TK. Thiem, J. Vill, V. Carbohydr. Res. 1992, 230, 245-256) o se acoplan para formar los intermedios fosfoéster J1, J3 y J5 55 60 65 Como se sabe que el enlace fósforo-nitrógeno es ácido lábil, se prefieren los intermedios que conducen a productos finales fosfodiéster (M. Selim y T. N. Thanh, C. R. Seances Acad. Sci, 1960, 250, 2377). 26 ES 2 225 834 T3 5 10 Por ejemplo, los intermedios alcohol I1, I2 o I3 se tratan con 2-clorofenilfosfocloridato de 2,2,2-tricloroetilo en cloroformo y piridina para formar los triésteres fosfato. El grupo 2,2,2-tricloroetilo se retira por tratamiento con polvo de cinc y el diéster fosfato resultante se activa con 3-nitro-1-(2,4,6-triisopropilbencenosulfonil)-1,2,4-triazol y se acopla con el alcohol R3 -OH para formar los intermedios J1, J3 y J5. El grupo 2-clorofenilo se retira por tratamiento con piridina-2-aldoxima y N,N,N,N-tetrametilguanidina en piridina húmeda (véase por ejemplo P. J. Garegg, R. Johansson, I. Lindh y B. Samuelsson, Carbohydr. Res. 1986, 150, 285-289). Como alternativa, mediante el enfoque del triéster fosfito, los intermedios alcohol I1, I2 o I3 se tratan con fenilcloro-N,N-diisopropilfosforamidito en acetonitrilo para formar fosfoamiditos carbohidrato. Después de la purificación por cromatografía, éstos se exponen a un alcohol R3 -OH en presencia de un ácido medio, tal como p-toluenosulfonato de piridinio y se tratan con hidroperóxido de terc-butilo para formar los diésteres fosfato J1, J3 y J5 (PG3 = H) (H-N. Caro, M. Martín-Lomas y M. Bernabé, Carbohydr. Res. 1993, 240, 119-131 y las referencias citadas en ese documento). 20 Para una revisión de fosfodiésteres en síntesis de ADN, véase Narang S, Tetrahedron, 1983, 39, 1-22 y D.W. Hutchinson, 1991, Capítulo 3 en Chemistry of Nucleosides and Nucleoties, vol. 2, ed. B. Townsend, Plenum Press, Nueva York, y todas la referencias citadas en ese documento. Para otros ejemplos sobre síntesis del fosfodiéster carbohidrato, véase por ejemplo Ichikawa Y, Sim M M, Wong C H, J. Org. Chem. 1992, 57, 2943-2946 en Schmidt R R, Braun H, Jung K-H, Tetrahedron Lett. 1992, 33, 1585-1588. Para la síntesis de engarces fosfodiéster modificados, véase R.S. Varma, 1993, Synlett, 621-636, y las referencias citadas en ese documento. 25 Para obtener ésteres de ácido arilfosfónico y amidas J1, J3 y J5, donde R3 es un grupo alquilo o aromático, el ácido arilfosfónico R3 -PO3 H2 se acopla a los intermedios de alcohol y I1, I2 o I3, por ejemplo, con un reactivo carboodiimida, o mediante del tratamiento de los dicloruros fosfónicos con los intermedios de alcohol I1, I2 o I3 en piridina (T. H. Siddal III y C. A. Prohaska, J. Am. Chem. 1962, 84, 3467). 15 Los triésteres alquilfosfónicos se forman a partir de trialquilfosfitos y haluros de alquilo mediante la reacción de Arbuzov (Arbuzov, Pure Appl. Chem. 1964, 9, 307-335, J. March, Advanced Organic Chemistry-Reaction Mechanisms, and Structure, 3ª Ed. John Wiley & Sons, Nueva York, 347 (1985), y referencias citadas en ese documento). 30 Para la preparación de triésteres aril fosfónicos mediante tricloruro de fósforo, véase por ejemplo: K. Sasse, Methoden der Organichem Chemie (Houben-Weyl), 4ª ed., Vol. 12/1, Georg Thieme, Stuttgart, 1963, p. 314 y p. 392 y referencias citadas en ese documento; G. M. Kosolopoff, Org. React. 6, 273 (1951), y referencias citadas en ese documento; L.D. Freedman y G. O. Doak, Chem. Rev. 1957, 57, 479, y referencias citadas en ese documento. 35 Para la preparación de triésteres arilfosfónicos mediante reactivos de organomagnesio u organolitio, véase por ejemplo: K. Sasse, Methoden der Organichen Chemie (Houben-Weyl), 4ª ed. Vol. 12/1, Georg Thieme, Stuttgart, 1963, p. 372, y referencias citadas en ese documento; G. M. Kosolapoff, Org. React. 6, 273 (1951), y referencias citadas en ese documento. 40 Los intermedios J1, J2, J3, J4, J5 y J6 se desprotegen para formar los productos finales (Protective Groups in Organic Synthesis, Editors T. W. Greene y P. G. M. Wuts, John Wiley & Sons, Inc., Nueva York, 1991, y referencias citadas en ese documento) y se transforman en sales sódicas o potásicas. 45 50 Esta justificado asumir que los compuestos descritos anteriormente pueden interactuar con sitios en PapD y otras chaperonas periplásmicas. Para establecer que éste es realmente el caso, pueden realizarse ensayos parecidos a los descritos en los ejemplos. De esta forma, un compuesto preferido de la invención es un compuesto como se describe más adelante, que provoca la prevención, inhibición o mejora de la unión de G1’-19’WT a PapD y/o provoca la prevención, inhibición o mejora de la unión del péptido de fusión MBP-Gl’-140’ a PapD y/o provoca la prevención, inhibición o mejora de la unión del péptido G125’-140’ (que tiene la secuencia SEC ID Nº: 20) a PapD y/o pueden inhibir la restauración del complejo PapD-PapG normalmente provocado por la adición de PapD de acceso. 55 El ensayo usado para establecer que una sustancia muestra uno de los efectos anteriores es preferiblemente uno de los ensayos descritos en los ejemplos que se muestran en este documento. Por supuesto, pueden utilizarse otros ensayos como los analizados anteriormente en los procedimientos de la invención para establecer que el compuesto realmente puede inhibir la unión al pilus. 60 Un punto importante que debería tenerse en cuenta cuando se lleva a cabo un ensayo, es el papel que desempeñan la chaperonas in vivo. Se unen a las subunidades de pilus durante el transporte a lo largo de la membrana celular de la subunidades y por lo tanto se asume que las subunidades de pilus están más o menos sin plegar, (es decir, no en su conformación plegada final) cuando se unen a la chaperona. Se ha observado por parte de los inventores que la cinética de la unión entre subunidades de pilus completamente plegadas (o análogos de las mismas) y la chaperona PapD está un procedimiento lento, aunque se sabe que el procedimiento de la unión al pilus es relativamente rápido in vivo. Para acelerar la velocidad de unión del complejo chaperona/subunidad de pilus in vitro, se contempla que podrían imponerse condiciones desnaturalizantes más o menos severas en las subunidades de pilus (o análogos de las mismas) antes del ensayo. Tal desnaturalización podría obtenerse sometiendo las subunidades de pilus a una fuerza 65 27 ES 2 225 834 T3 física (por ejemplo a una temperatura elevada, cambios de presión, radicación, etc). o a cambios en el medio químico (por ejemplo cambios en la fuerza iónica, cambios en el pH o la adición de compuestos desnaturalizantes o compuestos que reducen la cantidad de disulfuro). 5 10 La expresión “compuesto desnaturalizante” se refiere a un compuesto que cuando está presente en forma de uno de los solutos en una fase líquida que comprende moléculas polipeptídicas puede desestabilizar los estados plegados de las moléculas polipeptídicas lo que lleva a un no plegamiento completo o parcial de las cadenas polipeptídicas. El efecto desnaturalizante ejercido por un compuesto desnaturalizado aumenta cuando aumenta la concentración del compuesto desnaturalizante en la solución, pero además puede mejorarse o moderarse debido a la presencia de otros solutos de la solución, o mediante cambios en los parámetros físicos, por ejemplo temperatura o presión. Como ejemplos de compuestos desnaturalizantes adecuados para uso pueden mencionarse urea, guanidina-HCl, di-alquilformamida C1 -C6 tales como dimetilformamida y di-alquilsulfonas C1 -C6 . 15 20 25 30 35 Son ejemplos de compuestos que reducen el nivel de disulfuro, disulfuro de glutation il-2-tiopiridilo, disulfuro de 2-tiocolil-2-tiopiridilo, disulfuro de 2-mercaptoetanol-2-tiopiridilo y disulfuro de mercaptoacetato-2-tiopiridilo. Una serie de observaciones confirman la suposición de que las subunidades de pilus están más o menos sin plegar aunque están unidas a la chaperona: como se describen en Kuehn y col., 1991, es posible restaurar el complejo PapDPapG después de la desnaturalización añadiendo un exceso de PapD. Además, los resultados preliminares obtenidos por electroforesis capilar demuestran que la desnaturalización de la proteína de fusión MBP-G1’-140’ proporciona una forma sencilla de la proteína de fusión, mientras que las dos formas de la proteína de fusión se observan en la electroforesis capilar antes de la desnaturalización; una importante que no puede interactuar con PapD y una inferior que puede interactuar con PapD es capaz de interactuar con PapD. Por lo tanto se contempla que la proteína de fusión desnaturalizada puede servir como sustrato superior en los diferentes ensayos competitivos descritos en este documento igual que lo hace la forma no desnaturalizada de la proteína de fusión. De esta forma, en una realización preferida de la invención, el compuesto de la invención provoca la prevención, inhibición o mejora de la unión de una forma desnaturalizada de una subunidad de pilus o de un análogo de la misma a PapD, y/o provoca la prevención, inhibición o mejora de la unión de la forma desnaturalizada de MBP-G1’-140’ a PapD y/o provoca la prevención, inhibición o mejora de la unión de una forma desnaturalizada de G125’-140’ a PapD. Como se observará a partir de los ejemplos, los compuestos que pueden interactuar con PapD y otras chaperonas ya se han identificado y sintetizado. Estos compuestos que son todos piranósidos también son una parte importante de la invención: 2,3-O-Dibenzoil-4-O-bencil-1-tio-β-D-glucohexopiranósida de etilo; 6-O-Acetil-2,3-O-dibenzoil-4-O-bencil-1-tio-β-D-glucohexopiranósido de etilo; 40 6-O-Acetil-2,3-O-dibenzoil-4-O-bencil-β-D-glucohexopiranósido de metilglicolilo; 4-O-Bencil-β-D-glucopiranósido de 2-(hidroxi) etilo; 45 4-O-Bencil-β-D-glucohexopiranósido de glicolil sódico; 2-O-Etil-4,6-O-(4’-metoxi)fenilmetilen-α-D-mannohexopiranósido de metilo; 2-O-Etil-3-O-dimetil-t-butilsilil-4,6-O-(4’-metoxi)fenilmetilen-α-D-mannohexopiranósido de metilo; 50 2-O-etil-3-O-dimetil-t-butilsilil-4,-O-(4’-metoxi)-bencil-α-D-mannohexopiranósido de metilo; 2-O-Etil-3-O-dimetil-t-butilsilil-6-O-(4’-metoxi)-bencil-α-D-mannohexopiranósido de metilo; 55 2-O-Etil-3-O-dimetil-t-butilsilil-4,-O-(4’-metoxi)-bencil-6(S)-fenil-α-D-mannohexopiranósido de metilo; 2,3-Anhidro-4,6-O-p-metoxibenciliden-α-D-mannopiranósido de metilo; 3-Azido-4,6-O-p-metoxibenciliden-α-D-altropiranósido de metilo; 60 3-Azido-2-O-etil-4,6-O-p-metoxibenciliden-α-D-altropiranósido de metilo; 3-Azido-3-desoxi-2-O-etil-4-O-p-metoxibencil-α-D-altropiranósido de metilo; 65 3-Azido-6-O-benzoil-3-desoxi-2-O-etil-4-O-p-metoxi-bencil-α-D-altropiranósido de metilo; Sal sódica de 6-O-benzoil-3-desoxi-2-O-etil-4-O-p-metoxibencil-3-sulfamino-α-D-altropiranósido de metilo; 28 ES 2 225 834 T3 Sal de amonio de 6-O-benzoil-3-desoxi-2-O-etil-3-sulfamino-α-D-altropiranósido de metilo; 3-Azido-6-O-pivaloil-3-desoxi-2-O-etil-4-O-p-metoxi-bencil-α-D-altropiranósido de metilo; 5 Sal sódica de 6-O-pivaloil-3-desoxi-2-O-etil-4-O-p-metoxibencil-3-sulfamino-α-D-altropiranósido de metilo; Sal de amonio de 6-O-pivaloil-3-desoxi-2-O-etil-3-sulfamino-α-D-altropiranósido de metilo; 6-O-pivaloil-3-desoxi-2-O-etil-4-O-p-metoxibencil-3-t-butiloxamido-α-D-altropiranósido de metilo; 10 Sal de amonio de 6-O-pivaloil-3-desoxi-2-O-etil-3-oxamido-α-D-altropiranósido de metilo; 3-Azido-6-O-pirrol-3’-ilcarboxil-3-desoxi-2-O-etil-4-O-p-metoxibencil-α-D-altropiranósido; y 15 Sal de amonio de 6-O-pirrol-3’-ilcarboxil-3-desoxi-2-O-etil-3-sulfamino-α-D-altropiranósido de metilo. Por supuesto, también son posibles otros compuestos en forma de interactores con sitios de chaperonas. Como es evidente a partir del ejemplo 7, los péptidos modificados también pueden ser útiles en los procedimientos de la invención. 20 Como será evidente a partir de lo indicado anteriormente, la identificación de un sitio en la chaperona que puede estar afectado de forma interfiere con la agrupación de pilus es un punto de partida importante en los procedimientos descritos en este documento para la identificación, aislamiento y síntesis de compuestos que pueden interactuar con chaperonas periplásmicas. 25 De esta forma la invención también se refiere a un procedimiento para identificar un sitio de unión en una chaperona molecular, que comprende: 30 co-cristalizar la chaperona molecular periplásmica o un análogo de la misma con un ligando de unión a la chaperona molecular periplásmica o al análogo de la misma, resolver la estructura tridimensional de la interacción chaperona/ligando, resolviendo por tanto la estructura tridimensional de la chaperona molecular periplásmica o el análogo de la misma cuando se une al ligando, 35 determinar los puntos de los sitios implicados en la interacción intermolecular entre la chaperona molecular periplásmica o el análogo de la misma y el ligando, e identificar los puntos de los sitios determinados de esta forma de la chaperona molecular periplásmica o del análogo de la misma como sitio de unión en la chaperona molecular periplásmica o análogo de la misma. 40 45 Por el término “ligando”, como se usa en este documento se entiende una sustancia que muestra la unión a un huésped o molécula receptora (en su conexión con una chaperona). La unión no es una interacción no específica, lo que significa que existe un motivo de unión entre el ligando y el huésped o molécula receptora. En otra palabras, cuando se ponen en contacto una muestra del ligando y una muestra del huésped o molécula receptora, los complejos formados entre el ligando y el huésped o molécula receptora reflejarán sustancialmente todos las mismas interacciones intermoleculares. Como se ha mencionado anteriormente, una realización de la invención es administrar una sustancia que pueda prevenir, inhibir o mejorar la unión entre las subunidades de pilus y las chaperonas moleculares. 50 55 60 65 Por consiguiente, la invención se refiere a una composición farmacéutica, que comprende, como compuesto activo, una sustancia que puede interactuar con al menos un tipo de chaperona molecular periplásmica que une las subunidades de pilus durante el transporte de esas subunidades de pilus a lo largo del espacio periplásmico y/o durante el procedimiento de unión de los compuestos de pilus intacto, de tal forma que la unión de las subunidades de pilus a la chaperona molecular periplásmica se previene, inhibe o mejora, en combinación con al menos un vehículo o excipiente farmacéuticamente aceptable. Preferiblemente, tal sustancia es una sustancia de acuerdo con la invención o una sustancia identificada/diseñada de acuerdo con los procedimientos de la invención. De esta forma los agentes farmacéuticos analizados en este documento son, de acuerdo con la invención, para el tratamiento y/o profilaxis de las mismas afecciones que las analizadas cuando se describe el procedimiento de tratamiento de la invención y provocadas por las mismas especies bacterianas. Por lo tanto, una composición farmacéutica, que comprende, como compuesto activo, un sustancia usada en los procedimientos terapéuticos de la invención, una sustancia de acuerdo con la invención o una sustancia identificada de acuerdo con los procedimientos de la invención, en combinación con al menos un vehículo excipiente farmacéuticamente aceptable, es una parte de la invención. Tales composiciones farmacéuticas de la invención también podrían comprender al menos una sustancia farma29 ES 2 225 834 T3 céutica adicional, que entre otros podrían mejorar los efectos farmacéuticos ejercidos por la sustancia de la invención o la sustancia identificada de acuerdo con la invención. 5 Las sustancias farmacéuticas adicionales podrían ser hormonas esteroideas, desinfectantes, anti-piréticos, etc. Preferiblemente tal sustancia adicional podría ser un agente anti-bacteriano. Tal agente antibacteriano podría seleccionarse convenientemente entre el grupo compuesto por penicilinas, cefalosporinas, aminoglicósidos, sulfonamidas, tetraciclinas, cloranfenicol, polimixinas, fármacos antimicobacterianos y antisépticos urinarios. 10 15 20 La invención también se refiere a una sustancia empleada en los procedimientos de la invención así como a una sustancia de la invención y a una sustancia identificada de acuerdo con los procedimientos de la invención para uso como agente farmacéutico. Se prefiere que el agente farmacéutico sea para tratamiento anti-bacteriano y/o profilaxis, especialmente tratamiento y/o profilaxis de enfermedades causadas por bacterias que forman subunidades de pilus que se adhieren al tejido, y se prefiere especialmente que el agente farmacéutico sea para el tratamiento y/o profilaxis de la infección del tracto urinario. Finalmente, la invención se refiere al uso de una sustancia empleada en los procedimientos de la invención así como a una sustancia de la invención y a una sustancia identificada de acuerdo con los procedimientos de la invención para la preparación de una composición farmacéutica para el tratamiento y/o profilaxis de una infección bacteriana. Leyendas de las figuras 25 Fig. 1: PapD se une a los péptidos relacionados con subunidades de pilus recubiertos en placas de microtitulación ELISA y en solución. Las secuencias de los péptidos purificados sintéticos usados en este estudio se describen por separado en Flemmer K, Walse B. Roy S, y Kihlberg J. A y B: los péptidos de la tabla 1 se recubrieron en pocillos de microtitulación y se ensayaron con respecto a su capacidad para unirse a PapD en un ensayo ELISA descrito en el ejemplo 2. Se disolvieron péptidos insolubles en agua en ácido acético (AA) al 2,5% que no tuvo efecto en la unión de G1’-19’WT a PapD en este ensayo. Cada gráfico representa la media de pocillos duplicados. 30 Fig. 2: La unión de péptidos relacionados con subunidades de pilus proporciona protección contra proteolisis enzimática y bloquea la unión de PapD a PapG. 35 40 45 50 A Superior: PapD (15 µg) se incubó con PBS (calle D) o con 1,5 µg de tripsina (calle D y Tr) a 37ºC durante 20 minutos y se aplicó a SDS-PAGE al 20%. Se identificaron bandas teñidas con azul de Coomassie correspondientes a PapD de longitud completa (PapD), tripsina (Tr, el fragmento NH2 -terminal de PapD (N), y el fragmento COOHterminal de PapD que comienza el residuo 100 (C). A Inferior: el porcentaje de escisión de tripsina de PapD disminuyó después de la preincubación con los péptidos G1’-19’WT, G1’-16’WT o K1’-19’WT, pero el péptido G2’-21’amida no tuvo efecto. Se preincubaron 50 µg de PapD purificado durante 15 minutos a 25ºC con un exceso molar de 20 veces de péptidos G1’-19’-WT, G1’-16’WT, K1’19’Wt o G2’-21’amida (cualquier sitio de escisión de tripsina en estos péptidos se produce en sus extremos amino donde se pronosticó que no interferían con PapD), o un equivalente molar de agua. Después, cada muestra se incubó a 37ºC con 3,2 µg de tripsina. Las alícuotas se retiraron después de 0,5, 10, 20, 30 y 40 minutos y se hirvieron en un tampón de muestra SDS-PAGE para interrumpir la digestión. Las muestras se aplicaron a geles SDS-PAGE al 15% y la cantidad de PapD de longitud completa restante se determinó por densitometría de bandas teñidas con azul de Coomassie correspondientes a PapD de longitud completa que se muestran. B: PapD incubado con la G1’-19’WT, G1’-16’WT, K1’-19’WT y G2’-21’amida se añadió a PapD-PapG desnaturalizada reducida y la cantidad del complejo PapD-PapG restaurado en cada muestra se cuantificó como se describe en el ejemplo 2. El % de inhibición representa la cantidad del complejo PapD-PapG restaurado con PapD tratado con péptido en comparación con la cantidad del complejo PapD-PapG restaurado con PapD no tratado. El gráfico representa la media de 4 experimentos. PapD y el complejo PapD-PapG se prepararon como se describe en F. Lindberg y col, J. Bacteriol. 171, 6052 (1989) y S. J. Hultgren y col., Proc. Natl. Acad. Sci. Estados Unidos 86, 4357 (1989). 55 60 Fig. 3 Vista estereoscópica de la estructura tridimensional de PapD co-cristalizada con el péptido G1-19WT determinado a una resolución de 3,0 Å. El péptido se une en una conformación extendida a lo largo de la cadena G1 β en la hendidura de PapD con el grupo carboxilato terminal formando enlaces de hidrógeno con restos Arg-8 y Lys-112 de PapD. Fig. 4 Vista estereoscópica de la densidad de electrones a una resolución de 3,0 Å del extremo COOH del péptido G1-19WT y los restos PapD cercanos superpuestos en la estructura refinada. El mapa de densidad de electrones se calculó con coeficientes (2 |Fo|-|Fc|) y se contabilizó en 1s. 65 Fig. 5 Vista estereoscópica de un complejo PapD-péptido que muestra su interacción con un complejo o no cristalográficamente relacionado dos veces. 30 ES 2 225 834 T3 Fig. 6 Superposición de los dominios COOH-terminal de PapD nativo (líneas gruesas) y PapD complejado (líneas finas) que muestra 13º de diferencia en el ángulo de plegamiento en bisagra entre las dos estructuras. Las estructuras se superpusieron usando el programa O opción LSQ. Los rms resultantes para átomos de 98 Ca del dominio COOH terminal fueron de 0,66 Å. 5 10 15 Fig. 7: Capacidad de mutantes Arg-8, Lys-112 y Thr-7 de PapD para unir PapG y restaurar el complejo PapD-PapG in vitro. Se redujeron 0,4 µg del complejo PapD-PapG y se desnaturalizó como se describe en la fig. 2B. Después, el complejo PapD-PapG desnaturalizado se diluyó con 0-630 ng de PapD purificado mutante o de tipo silvestre (WT). La cantidad de PapD-PagG restaurada en las muestras se determinó como en la fig. 2B y se estableció en un gráfico como porcentaje de la cantidad más grande PapD-PapG restaurado. Los valores indicados en el gráfico representan la media de 3 experimentos. PapD mutante o de tipo silvestre y el complejo PapD-PapG se purificaron como se describe en F. Lindberg y col, J. Bacteriol. 171, 6052 (1989) y S. J. Hultgren y col., Proc. Natl. Acad. Sci. Estados Unidos 86, 4357 (1989). Fig. 8: 6-hidroxidopamina, hdo_0, y otros miembros de la familia hdo. Se usó hdo_0 como punto de partida para la construcción de una familia de compuestos que pretenden unirse al sitio de unión de PapD. hdo_4: “Arilo” indica 3,4-dihidroxifenilo. hdo_6: “Arilo” es un grupo 3,4-dihidroxifenilo. 20 hdo_7: “Arilo” es un grupo 3-hidroxifenilo. hdo_8: “Arilo” es un grupo 3-nitrofenilo. 25 hdo_9: “Arilo” es un grupo fenilo. Fig. 9 La familia hdo y su interacción con PapD. 30 35 A: “Arilo” es un grupo fenilo, un anillo heteroaromático, o un grupo fenilo sustituido con sustituyentes polares no simétricos para obtener por un lado contacto hidrófobo con la proteína y por otro lado interacción con el agua del disolvente. B: Ejemplos de “arilo” donde R es hidrógeno o alquilo; “X” es un átomo de oxígeno, un átomo de azufre, o un grupo amino; y “CG” es un grupo cargado negativamente, tal como un grupo carboxilo, un grupo tetrazoílo, un grupo fosfato, un éster fosfato o un grupo sulfato. C: Ejemplos del grupo CG. Fig. 10: Esquema de reacción general para la producción de diversos miembros de la familia hdo. 40 Fig. 11: Esquema de reacción general para la producción de diversos miembros de la familia bpy. Fig. 12: Esquema de reacción general para la producción de diversos miembros de la familia bpy. 45 Fig. 13: Esquema de reacción general para la producción de diversos miembros de la familia bpy. Fig. 14: Esquema de reacción general para la producción de diversos miembros de la familia bpy. Fig. 15: Esquema de reacción general para la producción de diversos miembros de la familia bpy. 50 Fig. 16: Esquema de reacción general para la producción de diversos miembros de la familia bpy. Fig. 17: Gráfico sobre el efecto inhibidor de diferentes longitudes de los fragmentos del péptido C-terminal de PapG en la unión entre PapD y la proteína de fusión MBP-G1’-140’. 55 El fragmento G1’-8’ muestra un efecto inhibidor significativamente más alto en la unión que los fragmentos G1’6’ y G1’-7’ más cortos. 60 Fig. 18: Gráficos sobre el efecto inhibidor de diferentes análogos de G1’-8’ (donde los 8 residuos aminoacídicos a la vez se han reemplazado por serina (S) 0 alanina (A)) en la unión entre PapD y la proteína de fusión MBP-Gl’-140’. El efectos de los reemplazamientos de los restos 4’, 5’ y 6’ en G1’-8’ revela que los restos son importantes en la interacción con PapD, ya que el reemplazo de estos residuos da lugar a péptidos menos inhibidores. 65 Fig. 19: Gráficos sobre el efecto inhibidor de diferentes análogos de G1’-8’ (donde los 8 residuos aminoacídicos a la vez se han suprimido simultáneamente con la adición de una serina N-terminal) en la unión entre PapD y la proteína de fusión MBP-G1’-140’. 31 ES 2 225 834 T3 También en este experimento, el significado de los restos de aminoácido 4’, 5’ y 6’ se enfatizan, ya que las deleciones de estas posiciones dan lugar a péptidos que son menos inhibidores. Fig. 20: Construcciones de la fusión MBP/G, péptidos sintéticos y truncados con PapG usados en el ejemplo 10. 5 10 El cuadrado abierto indica la secuencia primaria de PapG. Se muestran las posiciones de los cuatro restos Cys. La barra sombreada representa MBP. Los restos del principio y del final de PapG fusionado en el extremo COOH de MBP se indican para cada fusión. El resto del final para cada PapG-truncado también se indica. Se indican los nombres de las proteínas de fusión MBP/G. En la parte más baja de la figura, se indican los recuadros rellenos que localizan los sitios interactivos de PapD y las secuencias de los cuatro péptidos usadas en el ejemplo (véase también SEC ID Nº: 19-22). Fig. 21: Interacciones de PapD-MBP/G in vivo. 15 20 25 Se sometieron extractos periplásmicos que contienen PapD y cada fusión de MBP/G a cromatografía por afinidad de amilosa. Los eluidos se analizaron en A) un gel de poliacrilamida SDS teñido con azul de Coomassie al 12,5%; B) una transferencia de western usando un antisuero anti-PapD; o C) un gel IEF teñido con plata. En la figs. 21A, B y C, las muestras se purificaron a partir de extractos periplásmicos que contenían PapD y MBP (calle 1), PapD y MBP-G1’-19’ (calle 2), PapD y MBP-G1’-81’ (calle 3), PapD y MBP-G1’-140’ (calle 4). Se indica la posición de PapD co-purificado. MBP sola y truncada de fusión de MBP/G también se co-purificaron con las fusiones de MBP/G. El peso molecular de las bandas de migración más bajas en SDS-PAGE corresponden a cada longitud de la proteína de fusión MBP/G. En el gen IE F (C) pueden observarse diversas bandas para las mismas reacciones. Se detectó una única banda pI 5,2 en la fig. 2C, calle 4. Esta banda se extrajo, se hirvió en tampón de muestra SDS y se analizó mediante transferencia de western con antisuero anti-PapD y anti-MBP. Estaba compuesto tanto de MBP-G1’-140’ como de PapD (Fig. 2D, calle 1). Fig. 22: Inhibición de la función de la chaperona mediante la expresión de fusiones MBP/G in vivo. 30 35 Las cepas que llevan pFJ22 (papDJKEFGA) y plásmidos que codifican las fusiones MBP/G se indujeron con IPTG 1 mM. Los extractos periplásmicos que contenían subunidades de pilus y MBP (calle 1), o MBP-G1’-19’ (calle 2), o MBP-G1’-81’ (calle 3), o MBP-G1’-140’ (calle 4) se analizaron mediante transferencia de western usando un antisuero anti-PapA (A), o un antisuero anti-PapD-PapG (B), o un anti-suero contra fibrillas terminales (C). En este ensayo, la presencia de la subunidad indica que las interacciones entre chaperona-subunidad producidas a partir de las subunidades se degradan en ausencia de una interacción con la chaperona. Obsérvese que la presencia de las subunidades PapA, PapG y PapF disminuye significativamente cuando se co-expresan con MPB-G1’-140’. Fig. 23: Unión de PapD a proteínas de fusión MBP/G in vitro. 40 45 (A) PapD se incubó con 1 µg de MBP purificado con afinidad a amilosa (calle), MBP-G1’-19’ (calle 2), MBPG1’-81’ (calle 3), o MBP-G1’-140’ (calle 4) y la formación del complejo se evaluó en un gel IEF teñido con plata. Se indican las posiciones de las fusiones MBP/G, PapD y el complejo PapD-MBP-G1’-140’. (B) Se recubrieron proteínas de fusión MBP/G purificadas con afinidad a la amilosa en pocillos de placa de microtitulación. Se indica la concentración de las proteínas MBP/G en los pocillos. La unión de 50 pmol/50 ml de PapD a las proteínas inmovilizadas se determinó mediante el ensayo ELISA usando un antisuero anti-PapD. Fig. 24: Identificación del complejo PapD-PapG2 truncado mediante electroforesis de gel nativo ácido. 50 El periplasma que contiene PapD y PapG, PapG2 o PapG3 (calle 2, 3 y 4 respectivamente) se sometió a cromatografía Gal α(1-4) Gal y se analizaron ocho eluidos en electroforesis de gel nativo ácido después de la transferencia de western usando antisueros anti-PapD (A) y anti-PapD (B). El complejo PapD- PapG purificado se cargó en la calle 1. Fig. 25: Caracterización de un segundo sitio en PapG reconocido por PapD. 55 Se recubrieron cuatro péptidos sintéticos que superponían la región PapG del resto 156’ a 120’ (indicado en la Fig. 20) en pocillos de placa de microtitulación). Se indica la concentración de cada péptido en los pocillos. La unión de 200 pmol/50 ml de PapD al péptido inmovilizado se determinó mediante el ensayo ELISA usando antisuero anti-PapD. Ejemplos 60 Ejemplo 1 Identificación del motivo de unión entre PapD y G1’-19’WT 65 Materiales y procedimientos PapD se preparó como se describe por A. Holmgren y col. en J. Mol. Biol. 203, 279 (1988) y se obtuvo a partir del Departamento de Microbiología Molecular, Washington University School of Medicine, St. Louis, Estados Unidos. 32 ES 2 225 834 T3 El péptido G1’-19’WT se preparó usando síntesis en fase sólida de Fmoc seguido de purificación por HPLC de fase inversa. El péptido se obtuvo del Departament of Chesmistry, University of Lund, Lund, Suecia. La tabla 1 indica los péptidos usados en el ensayo de unión en esta solicitud. 5 10 15 En la presente solicitud, los restos de aminoácido o fragmentos peptídicos que se originan a partir de otros péptidos en lugar de PapD (por ejemplo de PapG o de PapK) se indicaran con ’, por ejemplo “G1’-19’WT” o “Pro-1’ ”, para distinguir tales restos de los restos de aminoácido de PapD. Además, la numeración es del extremo C-terminal del péptido que no es PapD, es decir G1’-19’ indica los 19 restos de aminoácido de terminal de PapG. Cristalización del complejo PapD-péptido Se obtuvieron cristales del complejo PapD-péptido mediante difusión de vapor contra PEG8000 al 20%, tampón cacodilato 0,1 M a pH 5,0, y acetato de calcio 0,2 M. Las gotas de cristalización contenían los mismos volúmenes del depósito y de las soluciones proteicas. La solución proteica (17 mg/ml) contenía una relación molar 1:1 entre PapD y el péptido en MES 20 mM (ácido 2-[N-morfolino]etano sulfónico) a pH 6,5 con glucósido β-octilo al 1,0%. Cristalografía de rayos X 20 25 Los cristales obtenidos del complejo entre PapD y la proteína se colocaron dentro de tubos capilares de cuarzovidrio e inicialmente se caracterizaron examinándolos en una cámara de precisión de rayos X obteniendo por lo tanto imágenes del modelo de difracción de rayos X en la película fotográfica que representa imágenes espacialmente no distorsionadas del enrejado recíproco de los cristales. Usando análisis convencionales de las imágenes, se determinó que los cristales tenían un grupo de espacio monoclínico, C2, con dimensiones celulares: a = 130,7 Å, b = 83,5 Å, c = 59,2 Å y β= 117,2º, dos moléculas en la unidad asimétrica y que presentan difracción a una resolución de 2,9 Å en una fuente de rayos X de laboratorio con un ánodo giratorio y objetivo de Cu Kα . Recopilación y procesamientos de datos experimentales 30 35 Para resolver la estructura atómica de moléculas mediante cristalografía de rayos X, tienen que medirse las posiciones e intensidades de la máxima de difracción. Los datos de intensidad para los cristales de PapD-péptido se recogieron en un sistema de detector de área multicable Huong-Hamlin (Xuong y col., 1985). Todos los datos se obtuvieron a partir de un único cristal e inicialmente el procesamiento se realizó usando el paquete de software MADNES (Messerschmit y Pflugrath, 1987). La fusión y la ampliación a escala de los datos se realizó usando ROTAVATA y AGROVATA a partir del paquete CCP4 (véase CCP4, The SERC (UK) Collaborative Project No.4, A Suite of Programs for Protein Crystallography, Darebury Laboratory, UK). El conjunto de datos finales contenían 9592 reflexiones independientes con un Rsym del 6,8% para los datos entre 20,0 y 3,0 Å de resolución (Rsym = SS 40 (|Ihi | − |Ih |) , Shi Ihi donde Ihi e Ih son las intensidades de los factores individuales y de estructura media, respectivamente). Solución de la estructura tridimensional 45 50 La estructura del complejo PapD-péptido se resolvió usando el programa XPLOR que realiza el procedimiento convencional de reemplazo molecular. El modelo de búsqueda usado fue la estructura de resolución refinada de 2,0 Å de PapD. Usando los datos de resolución entre 8,0 Å y 4,0 Å, la función de auto-rotación proporcionó un eje doble plegado no cristalográfico transparente, y los dos picos más altos de la interrotación dieron la solución exacta que se mejoró usando el refinamiento de correlación de Patterson (PC). La solución correcta también se obtuvo a partir los picos más altos en las funciones de traducción, siendo el factor R del 39,0% para los datos de resolución de 8,0 a 4,0 Å (el factor R definido como factor R = 55 S(|Fo| − |Fc|) , SFo El refinamiento se obtuvo con un posterior refinamiento del cuerpo rígido en el que los cuatro dominios de las dos moléculas PapD en la unidad asimétrica se dejaron refinar dando lugar de forma independiente a un factor R del 36,4% para los mismos datos. 60 El examen de un mapa de densidad de electrones |Fo| - |Fc| a este nivel usando el programa de gráficos 0 mostró una clara densidad correspondiente al péptido en la hendidura de PapD y a lo largo de la superficie de la proteína. La orientación del péptido se determinó fácilmente a partir de la densidad del electrón, pero inicialmente sólo pudo modelarse en cuanto a la densidad 10 aminoácidos C-terminal finales del péptido. 65 Refinamiento y análisis de estructura En esta etapa se inició un refinamiento de templado simulado con XPLOR. Se realizaron diversos ciclos adicio33 ES 2 225 834 T3 5 10 15 20 25 30 35 40 nales del modelo de construcción y refinamiento añadiéndose 4 aminoácidos peptídicos adicionales para producir un factor R para el actual modelo del 18,2% para datos de resolución de 8,0 a 3,0 Å. El modelo en la presente etapa de refinamiento (que no contiene moléculas de agua y que no incluye los 5 primeros aminoácidos N-terminal del péptido) tiene derivaciones de media cuadrática (rms) de geometría ideal de 0,020 para longitudes de enlaces y de 4,2º para ángulos de enlaces. Se observa que el péptido se une en una conformación amplia con la pro-1’ C-terminal anclada en la hendidura del interdominio y sitio de unión de la subunidad propuesto (6). Los enlaces de hidrógeno se forman entre los extremos carboxi peptídicos y dos restos cargados positivamente invariantes de PapD, Arg-8 y Lys-112. Ahora la mutagénesis dirigida al sitio ha confirmado que Arg-8 y Lys-112 son esenciales para la unión de subunidades de pilina tanto in vitro como en in vivo (cf. ejemplo 2). La cadena lateral Pro-1’ hace contactos van der Waals en la hendidura con restos de ambos dominios de PapD: Thr-7, Thr-152, Ile-154, Thr-170 y Ile-194. El resto peptídico próximo, Phe-2’, se encuentra en una cavidad poco profunda formada entre las lámina β y el dominio N-terminal y realiza interacciones hidrófobas con Leu-4, Thr-7, Thr-109 e Ile-111. Después, el péptido recorre la superficie del dominio N-terminal, formando una interacción paralela de cadena β con la cadena G1. De esta manera, se forman 7 enlaces de hidrógeno de cadena principal entre Met-8’ y Phe-2’ del péptido y entre Gln-104 y Lys-110 de PapD, ampliándose de esta forma la lámina β de PapD en el péptido. Además de los restos C-terminal Phe-2’ y Pro-1’, hay relativamente pocos contactos entre las cadenas laterales del péptido y PapD. Las interacciones principales se proporcionan mediante enlaces de hidrógeno de cadena principal a la cadena G1. Sin embargo, hay diversas interacciones hidrófobas en la lámina β, en particular entre Met-8’ y Met-6’del péptido con Leu-103, Ile-105 y Leu-107 de cadena G1. Los cálculos revelaron que las cuatro cadenas laterales peptídicas hidrófobas de los restos 2’, 4’, 6’ y 8’ aportan el 20% del área superficial oculta total (582 Å2 ) entre el péptido y la proteína. Por lo tanto, incluso aunque la principal estabilización del complejo se proporciona mediante la unión de hidrógeno, las interacciones hidrófobas no son insignificantes y se cree que proporcionan parte de la explicación para la especificidad de PapD para péptidos y subunidades relacionados con pilus. La base experimental de esta teoría se posiciona mediante la unión reducida de PapD al péptido G2’-21’ amida en comparación con el péptido G1’-19’WT (Fig. 1B y tabla C). La unión de hidrógeno del extremo COOH de la G2’-21’ amida (que carece de Pro-1’) a Arg-8 y Lys-112 de PapD permite la unión de hidrógeno de cadena principal, pero trastoca las cuatro cadenas laterales hidrófobas en el péptido a partir de sus subsitios en PapD, dando lugar a una reducción de la resistencia de la unión. En el cristal, la lámina β PapD-péptido se amplió incluso aún más como resultado de una simetría plegada dos veces no cristalográfica que colocó un segundo complejo PapD-péptido adyacente al primero de forma que la dos cadenas peptídicas unidas interactuaron como cadenas β antiparalelas. En el presente modelo, se forman ocho enlaces de hidrógeno de cadena entre los dos péptidos; véase la siguiente tabla: TABLA B 45 50 55 60 65 34 ES 2 225 834 T3 5 10 15 20 25 30 35 40 45 50 55 60 65 De esta forma, se crea una lámina β mezclada entre los dos complejos y alcanza más de 10 cadenas β (Fig. 5). No se observan contactos entre las dos moléculas PapD no relacionadas cristalográficamente, las cuales se colocaron en medios similares en el cristal y poseían un número similar de contactos intermoleculares. El área superficial oculta calculada entre los dos péptidos no relacionados cristalográficamente fue de 520 Å2 , un valor similar al del área superficial oculta entre la proteína y el péptido. Esta “dimerización” parece ser una consecuencia del envasado de cristal, ya que todas las pruebas muestran que PapD forma complejos monoméricos con péptidos y con PapG intacto en solución. El modelo de unión de hidrógeno entre PapD y el péptido se rompe en Ser-9’, aunque el péptido permanece en contacto van der Waals con PapD hasta Ser-11’ donde el péptido se dirige más allá del bucle F1-G1, pero el hidrógeno permanece unido al péptido no relacionado cristalográficamente hasta Glu-13’ (tabla B). El último aminoácido resuelto del péptido es Gly-14’ que se coloca cerca de la hendidura de unión del PapD no relacionado cristalográficamente. Los primeros cinco aminoácidos terminales, incluyendo tres restos cargados positivamente, no tuvieron densidad y por lo tanto deben de haberse desordenado en la estructura de cristal. De acuerdo con esto, el extremo amino del péptido no es importante para la unión a PapD en la solución, ya que se ha descubierto que el péptido G1’-7’WT que carece de los doce aminoácidos amino terminal es un inhibidor eficaz de la unión de PapD al péptido G1’-19’WT inmovilizado (tabla C y ejemplo 2). Las estructuras de los dominios PapD individuales en el complejo peptídico son esencialmente las mismas que PapD nativo (Holmgren y Branden, 1989). Sin embargo, existe un movimiento significativo de los dominios con respecto a los dominios entre sí haciendo el movimiento de plegamiento en bisagra de 13º o el cierre en mandíbula que el ángulo del bumerang de PapD sea más agudo (Fig. 6). Independientemente de que este cambio conformacional sea o no el resultado de la unión del péptido o de diferentes cristales, el envasado entre las dos estructuras de cristal no está claro. En la estructura PapD natural, la densidad del electrón obtenida para el bucle F1-G1 largo es escasa entre los residuos 96 y 102, lo que sugiere que es poco flexible y está desordenado en el cristal. En el complejo peptídico, sin embargo, este bucle se resuelve mejor, lo que indica que la unión del péptido hace que este bucle sea más rígido. La superposición de los dominios NH2 -terminal de PapD nativo y el complejo peptídico demuestra que también existe una diferencia significativa en la posición del bucle F1-G1 entre las dos estructuras (rms para los 110 átomos Cα NH2 terminal es de 1,84 Å, con un movimiento de cadena principal máximo de aproximadamente 9 Å para Leu-103). En el complejo peptídico, se observa que el bucle se enrolla en un extremo lejos del barril β del dominio NH2 -terminal facilitando de esta forma un contacto más amplio entre la cadena G1 y el péptido. Como con el plegamiento en bisagra de los dos dominios, todavía no posible decir con exactitud si el cambio del bucle es una consecuencia de la unión peptídica o del envasado del cristal; la gran conformación abierta del bucle F1-G1 sugiere que puede ser en gran parte el último. Sin embargo, se proporcionan pruebas de que la interacción similar entre PapD y péptidos o subunidades de pilus se produce en solución mediante experimentos de protección de proteasa después de que se proteja el bucle F135 ES 2 225 834 T3 G1 de PapD de la escisión tríptica por la unión tanto de PapG nativo como del péptido G1’-19’WT (ejemplo 2 y Fig. 2A). Ejemplo 2 5 Unión entre PapD y el extremo carboxilo de otros péptidos Pap 10 Se sintetizaron péptidos G1’-19’WT, E1’-19’WT, F1’-19’WT, K1’-19’WT y H1’-19’WT (véase tabla 1) que correspondían a los 19 residuos carboxilo terminal de proteína de subunidad de pilus P Papg, PapE, PapF, PapK y PapH, respectivamente (véase: Grant y col., 1992, y referencias citadas en ese documento). TABLA 1 15 20 25 30 35 40 45 Los residuos en los péptidos se enumeraron comenzando con el residuo carboxilo terminal como 1, y terminando con el residuo amino terminal. Los péptidos también se sintetizaron que se desviaron en cuanto a longitud (G1’-16’WT, G1’-11’-WT, G1’-7’WT) o secuencia (G2’-21’amida, G1’-19’SV, G1’-19’NH2 ) de la secuencia carboxi terminal PapG de tipo silvestre. Usando un ensayo inmunoabsorbente ligado a enzima (ELISA), se ensayó la capacidad de PapD para unirse a cada péptido recubierto en pocillos de placas de microtitulación: 50 55 60 65 Se diluyeron 5 mg/ml de soluciones madre de péptidos en agua o ácido acético al 50% a una concentración de 2,5 pmol/50 µl en PBS. Se recubrieron 50 µl de la solución peptídica durante una noche en pocillos de microtitulación (Nunc-Immuno Plate Maxisorp) a 4ºC. Las soluciones en las placas se desecharon, y los pocillos se bloquearon con 200 µl de albúmina de suero bovino al 3% (BSA, Sigma) en PBS (NaCl 120 mM/KCl 2,7 mM/sales de tampón fosfato 10 mM, pH 7,4) durante 2 horas a 25ºC. Las placas se lavaron vigorosamente tres veces con PBS y se incubaron con 50 µl de la cantidad indicada de PapD purificado (Lindberg y col, 1989). Después de 3 lavados con PBS, los pocillos se incubaron con una dilución 1:500 de antisuero de conejo anti-PapD (Lindberg y col, 1989) en BSA al 3%/PBS durante 45 minutos a 25ºC. Después de 3 lavados con PBS, los pocillos se incubaron con una dilución 1:1000 de IgG de cabra anti-conejo acoplada a fosfatasa alcalina (Sigma) en BSA al 3%/PBS durante 45 minutos a 25ºC. Después de 3 lavados con PBS y 3 lavados con tampón de revelado (dietanolamina 10 mM, pH 9,5, MgCl2 0,5 mM), se añadieron 50 µl de sustrato de p-nitrofenil fosfato filtrado (Sigma) en tampón de revelado (10 mg/ml). La absorbancia a 405 nm se leyó después de 60 minutos de incubación en la oscuridad a 25ºC. Además, se ensayó la capacidad de los péptidos solubles en agua para inhibir la unión de PapD a pocillos recubiertos con G1’-19’WT en un ensayo ELISA de inhibición soluble, ya que las conformaciones peptídicas pueden haber sido afectadas por la unión de la unión a las placas de microtitulación de plástico: Se recubrieron pocillos de microtitulación durante una noche a 4ºC con 50 µl de 2,5 pmol/50 µl del péptido G1’36 ES 2 225 834 T3 5 10 15 19’WT. Los pocillos se lavaron con PBS y se bloquearon con albúmina de suero bovino al 3% (BSA). Se pre-incubó un exceso molar de 25 veces de cada péptido de ensayo con 100 pmoles de PapD durante 30 minutos y después se añadió la solución PapD-péptido a los pocillos recubiertos y se incubó a 25ºC durante 45 minutos en presencia de BSA al 3%/PBS. El anticuerpo primario posterior, anticuerpo secundario y etapas de revelado se han descrito anteriormente. La capacidad de los péptidos para inhibir la unión de PapD al péptido G1’-19’WT se calculó dividiendo la cantidad de la unión de PapD en presencia del péptido con la cantidad de la unión de PapD en presencia de agua. La inhibición al 0% incluye valores en los que la unión fue superior a la de PapD pre-incubado con H2 O. Los resultados de la unión de PapD a los péptidos de longitud variable y diferente de tipo silvestre se muestran en la Figs. 1A, B y C. Como puede observarse, los péptidos se unieron bien al péptido PapG, moderadamente a PapE, PapF y PapK, al tiempo que no hubo unión a PapH y el péptido hidrófobo aleatorio, MS. (Fig. 1A). Estos resultados sugieren que la chaperona reconoce PapG, PapE, PapF y PapK en parte por la unión al extremo carboxilo de estas subunidades. La incapacidad de PapD para interactuar con el péptido PapH indica que PapD se une de forma diferente a PapH, posiblemente debido a la función de PapH como terminador de polimerización (Baga y col., 1987). La capacidad del péptido para inhibir la unión de PapD a G1’-19’WT inmovilizado generalmente se correspondía con la afinidad que PapD tenía para el péptido inmovilizado respectivo (Fig. 1, % de inhibición), lo que argumenta que la interacción de PapD con péptidos solubles inmovilizados es similar. 20 25 30 35 Como puede observarse a partir del ejemplo 1, las bases moleculares de las interacciones PapD-péptido se han estudiado mediante co-cristalización de PapD con el péptido G1’-19’WT. En resumen, el péptido de 19 aminoácidos se ancla en la hendidura de la chaperona mediante enlaces de hidrógeno entre el extremo carboxi del péptido y Arg-8 y Lys-112 que son invariantes en todas las chaperonas periplásmicas (Holmgren y col., 1992). El péptido se unió a la cadena G1 de PapD en forma de una cadena β paralela, formando al menos 10 enlaces de hidrógeno en el esqueleto, y dio lugar a una ordenación del bucle F1 a G1. El reemplazo de la prolina carboxilo terminal en el péptido con una amida (G2’- 21’ amida) suprimió la unión a PapD en solución y redujo la unión de PapD al péptido inmovilizado en aproximadamente un 75% (Fig. 1B). Por el contrario, al sustituir sólo el grupo carboxilato con una amida para crear G1’-19’NH2 , el péptido no afectó a la unión a PapD en los ensayos de ELISA de inhibición soluble e inmovilizada (Fig. 1B). Estos resultados indican que la prolina terminal probablemente se requiera para colocar el grupo carboxilato de forma que pueda formar enlaces de hidrógeno con el Arg-8 invariante y los residuos de la hendidura Lys-112. PapD también se une a los péptidos más cortos inmovilizados G1’-16’WT y G1’-11’WT y los péptidos G1’16’WT y G1’-7’WT inhibieron la unión de PapD al péptido G1’-19’WT inmovilizado en solución. La tabla C muestra la capacidad de un exceso molar de 25 veces de los péptidos solubles en agua para inhibir la unión de 100 pmol/pocillo de PapD a pocillos recubiertos con G1’-19’WT. El % de inhibición representa el porcentaje de unión de PapD en presencia de agua y son la media del experimento realizados por duplicado: 40 TABLA C 45 50 55 60 65 PapD no se une al péptido G1’-7’WT inmovilizado, probablemente porque este péptido es demasiado corto para unirse a los pocillos de microtitulación así como a PapD. De esta forma, parece que al menos se necesitan 7 residuos carboxilo terminal para la unión de PapD de un péptido. Conjuntamente, estos resultados avalan un modelo, además de la interacción del anclaje del grupo carboxilato en la hendidura PapD, se requiere 7 residuos carboxilo terminal para el péptido “en cremallera” junto con la cadena G1 del PapD durante la formación del complejo PapD-péptido. Se sustituyeron residuos conservados de fenilalanina y residuos de glicina en las posiciones 2 y 14 de los extremos carboxilo mediante serina y valina, respectivamente, para crear el péptido G1’-19’SV y se disminuyó la unión a PapD en un 36%. Esto pudo dar lugar a un recubrimiento menos eficiente o un recubrimiento o presentación menos eficiente en la placa de microtitulación ya que G1’-19’ SV era un inhibidor de PapD soluble tan eficiente como el péptido G1’37 ES 2 225 834 T3 19’WT (Fig. 1B). Como estos residuos son importantes para la incorporación de PapG en la subunidad de pilus, se cree que pueden ser más importantes para la interacciones de polimerización entre subunidades y la unión de subunidades de pilus que en las interacciones de las subunidad PapD in vivo. 5 10 15 20 25 30 35 40 45 50 55 60 65 La digestión parcial con tripsina escinde PapD en el bucle F1-G1 en los residuos Leu-103 y Lys-99, respectivamente: Se digirieron parcialmente 400 µg de PapD por incubación con 4,5 µg de tripsina o 0,45 µg de quimotripsina en PBS durante 20 minutos a 37ºC. Los productos de digestión de PapD se aplicaron a una columna de HPLC C-18 (Beckman) y se eluyeron 2 fragmentos principales con un gradiente de acetonitrilo de 0-100% en ácido trifluoroacético al 0,01%. Los fragmentos de PapD se identificaron por su peso molecular en SDS-PAGE y por secuenciación aminoterminal. Las secuencias de aminoácidos N-terminales de los fragmentos trípticos y quimiotrípticos de aproximadamente 14 kDa se identificaron como los residuos 100-108 y 104-109 de PapD, respectivamente, correspondientes a la escisión después de Lys-99 (en el caso de la tripsina) y Ley-103 (en el caso de la quimotripsina). Las secuencias Nterminales de la bandas de 11 y 12 kDa en los productos de digestión fueron idénticas a la secuencia N-terminal de PapD. Los péptidos G1’-19’WT, G1’-16’WT y K1’-19’WT, pero no el péptido G2’-21’amida, redujeron la velocidad de escisión tríptica de PapD a lo largo del tiempo (Fig. 2A). Estos datos indican que la unión de PapD al péptido alteraba el contacto del bucle y por lo tanto la protección para formar la escisión. Este efecto puede estar relacionado con el orden del bucle F1-G1 de PapD observado en la estructura cristalina del péptido PapD y sugiere, que en solución, los dos péptidos se extienden a lo largo de la cadena β de G1 e interaccionan con el bucle F1-G1 de PapD. Como se describe en Kuehn et al., Proc. Natl. Acad. Sci. ESTADOS UNIDOS 88, 10586 (1991), el PapD nativo puede unirse a PapG desnaturalizado o reducido y restaurar el complejo PapD-PapG in vitro. Este ensayo de reconstitución se usó para determinar la capacidad de los péptidos para inhibir la actividad de PapD in vitro. La solubilidad limitada del péptido G1’-7’WT desafortunadamente ha impedido el ensayo de este péptido en el análisis. Cantidades crecientes de los péptidos G1’-19’WT, G1’-16’WTy K1’-19’WT inhiben la restauración del complejo PapD-PapG por PapD, mientras que el péptido G2’-21’-amida no tiene ningún efecto (Fig. 2B). La capacidad de los péptidos G1’-19’WT, G1’-16’WT y K1’-19’WT para impedir la unión de PapD a PapG indicó que estos péptidos se unen al sitio de unión de la subunidad de PapD. La capacidad de los péptidos para inhibir la restauración del complejo PapD-PapG por PapD se ensayó como se indica a continuación: se redujeron 0,3 µg de complejo PapD-PapG y se desnaturalizaron por incubación a 25ºC durante 20 minutos con urea 4 M/ditiotreitol (DTT) 10 mM, 1,2 µg (50 pmol) de PapD se incubaron a 25ºC durante 10 minutos con 5-14,5 µg (2,5-7,25 mmol) de péptido. Después, la solución de PapD con péptido se añadió a PapDPapG desnaturalizado reducido, se incubó a 25ºC durante 10 minutos y se aplicó a un gel IEF 3-9 (Pharmacia). La cantidad de PapD-PapG restaurado en cada muestra se cuantificó por densitometría de la banda de IEF teñida con plata correspondiente al pI del complejo DG. Se construyeron mutaciones de localización dirigida en residuos escindidos estrictamente conservados de PapD que, según se había previsto, eran críticas en la interacción PapD-péptido para ensayar si la estructura cristalina de PapD-péptido es una reflexión de parte de la interfaz de interacción de PapD-subunidad de pilus (las posiciones de la mutaciones se indican en la Fig. 3). La Thr-7 muy conservada se cambió a una valina (Thr-7-Val) para ensayar si su grupo hidroxilo formaba enlaces de hidrógeno críticos para la unión de PapD a las subunidades. Esta mutación retiró el grupo hidroxilo mientras que mantenía el volumen estérico de la cadena lateral. Se diseñaron mutaciones en Lys-112 y Arg-8 para ensayar si la formación de enlaces de hidrógeno con el grupo carboxilado terminal de la subunidades de pilus es una característica crítica del procedimiento de reconocimiento de chaperona. El residuos Lys-112 invariantes se cambiaron a una alanina (Lys-112-Ala) para retirar la cadena lateral cargada y a una metionina (Lys-112 -Met) para reemplazar el grupo cargado con un grupo hidrófilo mientras se mantenía el empaquetamiento de la cadena lateral. Previamente se ha demostrado que se requiere el mantenimiento de la Arg-8 para que PapD pueda unirse a la subunidades y mediar el ensamblaje del pilus in vivo (Slonim y col, 1992). Glu-167/E167, un residuo variable en el dominio 2 de PapD (Fig. 3), no parece estar implicado en la interacción entre PapD y el péptido en la estructura cristalina y se ha demostrado que las mutaciones en este residuo tienen un efecto pequeño o nulo sobre la función de PapD in vivo (Slonim y col, 1992). E167 se cambió a histidina (E167H) para ensayar si este residuo cargado negativamente en el borde del dominio 2 tiene algún papel en la interacción PapD-péptido. Todos los mutantes se secretaron en el espacio periplásmico como proteínas estables similares a PapD de tipo silvestre. Además, el perfil de elución forma una columna de FPLC de intercambio catiónico y las propiedades electroforéticas de los PapD mutantes purificados fueron similares a las de la proteína del tipo silvestre, soportando la predicción de que esta mutaciones no afectarían a la estructura global de PapD (datos no mostrados). La capacidad de las chaperonas mutantes para unirse a subunidades de pilus y modular el ensamblaje del pilus in vivo se correlacionó con su capacidad de unirse al péptido G1’-19’WT y PapG in vitro. El PapD de tipo silvestre unido al péptido G1’-19’-WT produce un cambio de movilidad hacia el electrodo negativo en un ensayo de gel de poliacrilamida nativo (Lam y Calderwood, 1992), probablemente debido a un aumento de carga positiva neta en el complejo PapD-péptido. Por el contrario, cuando se incubaron mutantes de PapD Arg-8-Gly, Arg-8-Ala y Lys-11238 ES 2 225 834 T3 Ala con péptido G1’-19’WT no produjeron un cambio de movilidad en el ensayo PAGE-nativo, lo que indica que esta mutaciones en PapD anulaban la unión del péptido. De forma similar, las mutaciones en Arg-8 y Lys-112 anulaban la capacidad de PapD de unirse a PapG y reconstituir el complejo de PapD- PapG in vitro (Fig. 7) y de unirse a la subunidades de pilus y mediar el ensamblaje de pilus in vivo (véase la tabla 2). 5 TABLA 2 10 15 20 25 30 35 40 * La titulación de hemaglutinación (HA) se cuantificó después de la inducción con IPTG 0,01 mM. La titulación de HA representa la mayor dilución bacteriana que aún aglutina eritrocitos. HB101/pPAP37 que expresa PapD de tipo silvestre tiene una titulación de HA de 128. 45 50 + La cantidad de pili ensamblados se liberaron de las células y se cuantificaron después de la inducción con IPTG 0,01 mM. “-” indica que no se detectaron pili; el grado de formación o de liberación de pili de HB101/pPAP37 que expresa PapD de tipo silvestre corresponde a “++++”. # La estabilización periplásmica ayudada por chaperona de subunidades de pilus PapA, PapE, PapF, PapK y PapG se determinó como se ha descrito anteriormente (Slonim y col., 1992); “-” indica que no hay estabilización de la subunidad; el número de símbolos + indica el grado de estabilización de la subunidad en comparación con PapD de tipo silvestre. ND indica no determinado. § X indica mutaciones en His, Asp, Thr o Gly. 55 La notable consistencia de los resultados in vitro e in vivo y la estructura cristalina indican que el anclaje de la subunidades de pilus por residuos conservados Arg-8 y Lys-112 es una parte crítica de la interacción entre chaperonas de tipo PapD y la biogénesis de pilus. 60 Se realizó una mutagénesis de localización dirigida usando el Bio-Rad en el kit de mutagénesis in vitro siguiendo las instrucciones de los fabricantes. Para introducir mutaciones en el gen papD (SEC ID Nº: 1) se usaron los siguiente cebadores (cadena no codificante): Thr-7-Val 5’-GTCAAACACCGCCGGAACTCGTCCAGGCGA-3’ SEC ID Nº: 3 65 Lys-112-Ala 5’CGGGCGATAAAAAAGAGCTATTTTGGTCTG-3’ SEC ID Nº: 4 Lys-112-Met 5’-GCGATAAAAAAGCATTATTTTCCTCTG-3’ SEC ID Nº: 5 39 ES 2 225 834 T3 5 10 15 20 25 Las mutaciones en papD se confirmaron por secuenciación y los genes PapD alterados se clonaron en el vector pMMB91 bajo el promotor Ptac inducible como se ha descrito previamente para otras mutaciones de papD (Slonim y col, 1992). Los plásmidos se denominaron pThr-7-Val, pLys-112-Ala y pLys-112-Met de acuerdo con el número de residuos y los aminoácidos mutados. El plásmido pLS101 es una construcción isogénica que contiene el gen papD de tipo silvestre; los plásmidos pE167H, pR8G y pR8A contienen genes que codifican PapD con mutaciones puntuales que cambian Glu-167 a His y Arg-8 a Gly y Ala, respectivamente. Los datos presentados en este ejemplo y en el ejemplo 1 demuestran que las interacciones PapD-péptido encontradas en la estructura cristalina reflejan la interacción PapD-subunidades de pilus que reflejan la interfaz de interacción PapD-subunidad de pilus in vivo. Estas interacciones definen por primera vez un mecanismo por el cual la chaperonas utilizan sus dominios de tipo inmunoglobulina en un nuevo paradigma de reconocimiento para unirse a un grupo variado de proteínas. Los residuos Arg-8 y Lys-112 de PapD, localizados en cadenas β en el fragmentos escindidos entre los dos dominios, juegan papeles cruciales en el reconocimiento de chaperona sirviendo como anclajes moleculares para el extremo carboxilo de las proteínas de los pili. El hecho de que Arg-8 y Lys-112 estén muy conservadas entre las chaperonas periplásmicas indica que probablemente tengan un papel universal en la formación de “sitio activo” que se une al grupo carboxilato terminal de otras subunidades proteicas. Los enlaces de hidrógeno de esqueleto a lo largo de la cadena β de PapD posteriormente proporcionan interacciones fuertes independientes de la secuencia a lo largo del extremo carboxilo de la subunidad. La interacción “en cremallera” de los residuos hidrófobos alternos conservados del extremo carboxilo de la subunidad del pilus con los residuos hidrófobos alternos conservados en la cadena β de G1 de la chaperona añade resistencia y especificidad a la unión. Por lo tanto, a diferencia de otras proteínas parecidas a inmunoglobulina (Amit y col, 1988 y de Vos y col, 1992), PapD utiliza la cadena β y las características de hendidura interdominio de su estructura parecida a la inmunoglobulina para proporcionar un mecanismo de reconocimiento para unirse a varias proteínas diferentes. Los residuos variables en las regiones del bucle de los anticuerpos proporcionan la especificidad exquisita necesaria de un amplio repertorio de unión de anticuerpos. De forma similar, los residuos en el bucle F1-G1 de PapD pueden proporcionar especificidad por la unión de chaperona, ya que se ha descubierto que estos residuos varían en longitud y composición entre los diversos miembros de la familia de chaperonas periplásmicas (Holmgren y col, 1992). Ejemplo 3 30 Diseño de compuestos capaces de unirse al sitio de unión de PapD 35 40 45 Habiendo determinado la localización de un sitio de unión prometedor para ligandos inhibidores como se describe en los ejemplos 1 y 2, se usaron los programas informáticos ’PLIM’ y ’PLIM_DBS’ (creados por Symbicom AB) para encontrar plantillas para familias de compuestos capaces de unirse al sitio de unión (véase la descripción general anterior). PLIM se ejecutó en una caja de 20 Å alrededor de la región Arg-8 de la estructura de alta resolución de PapD, buscando posiciones de unión de baja energía para sondas NH, NH2 , O, OH y C. Las ejecuciones de PLIM dieron como resultado varias posiciones sugeridas y orientaciones de grupos químicos favorables (puntos de sitio) en la región cerca de Arg-8 y Lys-112. Se identificaron aproximadamente 50 sitios y después se seleccionaron tripletes y cuadrupletes de estos puntos de sitio con respecto a su compatibilidad mutua, es decir debían tener relaciones geométricas apropiadas - distancias de orientaciones que podrían encontrarse en una molécula real. Después se realizó una búsqueda de ligandos potenciales buscando en una base de datos estructuras moleculares conocidas que correspondieran a las posiciones de esos grupos de puntos de sitio, usando PLIM_DBS. 50 55 60 Los mejores de estos aciertos de la base de datos se examinaron visualmente usando un sistema de formación de modelos gráficos por ordenador, y los más prometedores de estos se seleccionaron según la abundancia de razonamiento físico-químico. Se realizaron modificaciones como se ha descrito anteriormente para mejorar la unión y simplificar la síntesis. Por ejemplo, se añadieron anillos de fenilo para proporcionar una mejor complementariedad con la superficie hidrófoba del sitio de unión, se añadieron grupos cargados para unirse a los grupos cargados presentes en la proteína que habían estado fuera del alcance de Plim, se retiraron sub-estructuras frecuentemente tóxicas y se reemplazaron grupos por grupos funcionalmente similares por razones de síntesis. Se realizaron muchos de tales juicios, dando como resultado dos familias de compuestos (hdo y bpy) con diferentes combinaciones de características. La eficacia de estas modificaciones finalmente se evaluó usando cálculos de energía libre de dinámica molecular como se describe en este documento para estudiar la estabilidad del complejo proteína-ligando (Åqvist y col., 1994). 65 Posteriormente, dos estructuras de una familia y una de la segunda familia de consideraron dignas de síntesis y ensayo. El desarrollo de varios de los miembros de estas familias de compuestos se describirá a continuación con detalle. 40 ES 2 225 834 T3 La primera familia (denominada en este documento hdo) se obtuvo a partir de una base de datos para 6-hidroxidopamina, denominada hdo_0 (Fig. 8). La molécula se une a las cadenas laterales de PapD de Arg-8 y Lys-112, y al esqueleto de Lys-110 a través de una donación de enlaces de hidrógeno desde los grupos hidroxilo. Los enlaces de hidrógeno del grupo amino primario se unen a la cadena lateral de Thr-170. 5 10 15 A partir de esta estructura básica se creó el derivado hdo_1 (Fig. 8) reemplazando un grupo hidroxilo por un ácido carboxílico que puede tener una interacción de carga con Arg-8 y Lys-112, y reemplazando el hidroxilo en posición para con respecto a esta posición por un nuevo anillo de fenilo, para rellenar la hendidura hidrófoba en la proteína formada por los residuos Ile-111, Leu-4 y parte de Thr-7. Se creó hdo_4 (Fig. 8) a partir de hdo_1 reemplazando el anillo aromático original por un azúcar, uniendo los sustituyentes de hidroxilo, la amina primaria y ácido carboxílico de tal forma que se mantuviera geometría relativa similar, y después añadiendo grupos hidroxilo a un lado del nuevo anillo de benceno para que presentara una cara hidrófila al disolvente en esta región. En este punto, la estructura parecía suficientemente convincente como para invertir tiempo en una simulación de dinámica molecular, cuyos resultados sugirieron que el grupo amino efectivamente no se unía al carbono gamma de Thr-7 de PapD, sino que preferentemente se disolvería, satisfaciendo sus requisitos de unión de hidrógeno con moléculas de agua en lugar de con la cadena lateral de la proteína. De esta manera, el grupo amina se reemplazó por el grupo hidroxilo donante individual, una etapa que también simplificaba la síntesis, siendo el resultado la estructura hdo_6 (Fig. 8). 20 Debido a la probabilidad percibida de la toxicidad debido al modelo de sustitución de hidroxilo en el anillo de benceno, el grupo para-hidroxilo se retiró para dar una estructura hdo_7 (Fig. 8) y el grupo hidroxilo restante en este anillo se reemplazó por un grupo nitro para facilitar adicionalmente la síntesis (hdo_8, Fig. 8). 25 También se consideró una posibilidad un compuesto final con un compuesto final en el que el grupo nitro previo se había dejado fuera, y se denominó hdo_9 (Fig. 8). 30 Se realizó otra simulación de dinámica molecular sobre hdo_9, cuyos resultados sugirieron que ahora, podrían realizarse las interacciones previstas y mantenerse en solución. Un análisis estadístico de las trayectorias con el programa “Miss_Fit” demostró que la conformación del ligando no cambiaba sustancialmente durante el procedimiento, y los cambios que se produjeron se compensaron por desplazamientos complementarios en la estructura proteica que mantenían todas las interacciones significativas. 35 La simulación dinámica de hdo_8 se complicó por la falta de disponibilidad de parámetros de mecánica molecular adecuados para el anillo sustituido con nitro en posición meta, lo cual hizo que fuera más difícil basarse en la evaluación por medio de simulaciones de dinámica molecular, y esto influye sobre la decisión de realizar un seguimiento. El intervalo de posibilidades que se consideraron durante la fase de diseño de la familia hdo se resumen en la Fig. 9. 40 La segunda familia, conocida como bpy, se derivó de una entrada de la base de datos para (metil-O,N,N-azoxi)metil-β-D-glucopiranósido-cicasina (Kawaminami y col., 1981) denominada bpy_0 en este documento: 45 50 55 60 65 La conformación almacenada en la base de datos no fue óptima para la unión al sitio PpaD, aunque se aproximó lo suficiente como para ser recogida por la búsqueda de Plim_DBS. Los pequeños ajustes realizados para los ángulos de torsión internos mejoraron de alguna manera el patrón de unión. Es interesante observar que hay precedentes para el enfoque de usar un carbohidrato central como estrado al que se unen las funcionalidades como para imitar un péptido (Hirschmann y col., 1992). Después, se creó el derivado bpy1-benzopiran-3-carboxílico añadiendo un anillo fenilo a la posición 6, con el fin de rellenar la hendidura hidrófoba de la proteína. El grupo 4-OH debería formar una interacción dipolar favorable con 41 ES 2 225 834 T3 el centro rico en electrones del grupo 6-fenilo, y junto con el hidrógeno intramolecular 6-OH..O5 debería estabilizar el rotámero C5-C6 deseado. 5 10 Se decidió que las cadenas laterales Lys-112 y Arg-8 que deben interactuar con el grupo hidroxilo en la posición 2 del ligando lo hagan más con un grupo cargado, y por lo tanto se creó bpy_2 reemplazando este hidroxilo con un sulfato, bpy_3 reemplazándolo con un fosfato cíclico y bpy_5 reemplazándolo con un fosfato no cíclico. Se creó bpy_4 retirando el grupo 3-hidroxilo de BPY_2 para que el grupo sulfato tuviera una mejor oportunidad de interactuar con la cadena lateral Arg-8, y se creó de forma análoga bpy_6 retirando el grupo 3-hidroxilo de bpy_5. La siguiente fórmula muestra bpy_1-bpy_6: 15 20 25 30 35 40 Después, se tomó en consideración una pequeña cavidad hidrófoba y parches o residuos hidrófobos expuestos al disolvente cerca de la posición 1 del ligando. Se buscó una aglicona que pudiera contactar con estas partes de la proteína y preferiblemente también interactuar con el disolvente. Estas consideraciones condujeron al reemplazo del grupo azoxi en la posición 1 con un grupo 2-hidroxi-4-metilciclopentilo, dando como resultado bpy_7: 45 50 55 60 65 En este punto, se hicieron realizaciones moleculares dinámicas, cuyos resultados sugirieron que la estructura se enlazó bien, pero que los contactos hidrófobos esperados entre el ciclopentano y la proteína no se mantuvieron. De esta manera, se creó bpy_9 a partir de bpy_7 con el grupo ciclopentano retirado, reemplazado con un grupo α-metilo para la estabilidad interna. En cambio, se intentaron contactos hidrófobos adicionales mediante la adición de un grupo etoxi axial en la posición 2. La carga se movió de la ecuatorial 2 al axial 3, un movimiento establecido por la inclusión de un enlace NH que puede donar un enlace hidrógeno al oxígeno en el axial 1. La reorganización estereoquímica de las funcionalidades en las posiciones 1-3 ahora confiere rigidez conformacional a la estructura, así como una simplificación de la síntesis. 42 ES 2 225 834 T3 5 10 15 Se hicieron más realizaciones dinámicas, esta vez sugiriendo que bpy_9 formaría un complejo estable con la proteína, similar a la manera en la que se preveía durante el diseño. 20 Desarrollos adicionales 25 Usando el razonamiento en línea con lo descrito anteriormente, se han diseñado más compuestos (a partir de la familia hdo y de la familia bpy). Los compuestos de interés son los descritos por las fórmulas generales I y II en este documento. Se han sintetizado los compuestos más prometedores que resultan de los análisis, cf. ejemplo 4. 30 En el desarrollo adicional de los compuestos diseñados usando los procedimientos descritos anteriormente, se ha descubierto de forma interesante que la estabilidad de la unión entre los compuestos diseñados y PapD parece depender únicamente de la interacción entre el compuesto y el residuo Arg-8, incluso aunque Arg-8 es esencial para la función in vivo de PapD. Por otra parte, la estabilidad de la interacción entre los compuestos diseñados y PapD aún depende mucho de la interacción con Lys-112 así como con la cadena β. Estas observaciones confirman la sorprendente observación de que la unión estable entre PapD y las subunidades de pilus depende mucho más de la interacción entre las láminas β de las dos moléculas. 35 Ejemplo 4 Síntesis de compuestos de la familia hdo capaces de unirse al sitio de unión de PapD Procedimientos generales 40 Los espectros 1 H y 13 C RMN se registraron en CDCl3 a 300 y 75 MHz respectivamente en un espectrómetro Varian Gemini 300 a menos que se indique otra cosa. Las señales del disolvente no deuterado a 7,25 y 77,0 ppm respectivamente se usaron como señales de referencia interna. Se midieron las rotaciones ópticas usando un polarímetro Perkin Elmer 241. 45 La cromatografía de capa fina se realizó en un Merck DC-Fertig-platten (Kiselgel 60 F254 0,25 mm) y las manchas se visualizaron pulverizándolas con ácido sulfúrico al 10% seguido de carbonización a temperatura elevada y/o pulverización con ácido molibdatofosfórico hidrato/Ce(SO4 )2 /H2 SO4 diluido seguido de calentamiento (ácido molibdatofosfórico hidrato = H3 [P(Mo3 O10 )4 ] x H2 O, E. Merck, Darmstadt, Alemania). 50 2,3-O-Dibenzoil-4-O-bencil-1-tio-β-D-glucohexopiranósido de etilo 55 60 Se disolvió 2,3-O-dibenzoil-4,6-O-benciliden-1-tio-β-D-glucohexopiranósido de etilo (150 mg,0,29 mmol) junto con complejo de borano-dimetilamina (68 mg, 1,16 mmol) en tolueno seco (20 Å). Se añadieron tamices moleculares de 4 Å en polvo (180 mg) y la mezcla se agitó a temperatura ambiente durante 20 minutos. Se añadió tricloruro de aluminio (154 mg, 1,16 mmol) y después de la desaparición del material de partida (aprox. 10 mn, ccf sobre sílice con 4:1 de tolueno/acetonitrilo) la mezcla se filtró, se trató con resina de intercambio iónico Dowex (H+ ) hasta que la solución fue transparente y se filtró de nuevo. El filtrado se concentró y se co-evaporó dos veces con metanol, produciendo 200 mg de residuo que después de la cromatografía sobre gel de sílice (8:1 de tolueno-acetonitrilo) dio 60 mg, 40% de 2,3-O-dibenzoil-4-O-bencil-1-tio-β-D-glucohexopiranósido de etilo. H RMN (CDCl3 , δ): 8,0-7,2 (mm, 15H), 5,75 (t, 9,4 Hz, 1H, H3), 5,37 (t, 9,8 Hz, 1H, H2), 4,74 (d, 9,8 Hz, 1H, H1), 4,61 (s, 2H) 4,02-3,95 y 3,85-3,76 (2 m a, 2H, 2H6), 3,94 (t, 9,5 Hz, 1H, H4), 3,63 (ddd, 9,7 Hz, 3,9 Hz, 2,6 Hz, 1H, H5), 2,74 (2c, 7,5 Hz, 2H), 1,25 (t; 7,5, 3H). 1 65 43 ES 2 225 834 T3 6-O-Acetil-2,3-O-dibenzoil-4-O-bencil-1-tio-β-D-glucohexopiranósido de etilo 5 10 15 20 25 Se disolvió 2,3-O-dibenzoil-4-O-bencil-1-tiol-β-D-glucohexopiranósido de etilo (200 mg, 0,38 mmol) en una mezcla enfriada con hielo de piridina y anhídrido acético (1:1, 6 Å). La mezcla se agitó y se puso a temperatura ambiente durante una noche. La mezcla se diluyó con diclorometano, se lavó con carbonato ácido sódico sat. ac. y agua y se secó sobre sulfato sódico. Los disolventes se retiraron a presión reducida y el residuo se sometió a cromatografía ultrarrápida sobre gel de sílice (6:1 de tolueno-acetonitrilo), produciendo 204 mg, 93% de 6-O-acetil-2,3-O-dibenzoil4-O-bencil-1-tio-β-D-glucohexopiranósido de etilo. 1 H RMN (CDCl3 , δ): 8,0-7,2 (mm, 15H), 5,75 (t, 9,2 Hz, 1H, H3), 5,40 (t, 9,8 Hz, 1H, H2), 4,71 (d, 10,0 Hz, 1H, H1), 4,59 y 4,55 (2d, 10,8 Hz, 2H, bzl-CH2 ), 4,44 (dd, 12,0 Hz, 1,9 Hz, 1H, H6), 4,27 (dd, 12,2 Hz, 4,6 Hz, 1H, H6’), 3,87 (t, 9,5 Hz, 1H, H4), 3,77 (ddd, 9,7 Hz, 4,6 Hz, 1,9 Hz, 1H, H5), 2,73 (m, 2H),2,09 (s, 3H), 1,26 (t, 3H). 6-O-Acetil-2,3-O-dibenzoil-4-O-bencil-β-D-glucohexopiranósido de metilglicolilo Se disolvieron 6-O-acetil-2,3-O-dibenzoil-4-O-bencil-1-tio-β-D-glucohexopiranósido (600 mg, 1,04 mmol) y Nyodosuccinimida (468 mg, 2,08 mmol, secada durante 3 h, < 0,1 mbar) en acetonitrilo seco (7 Å, pasado a través de alúmina) a temperatura ambiente. Se añadió glicolato de metilo (161 µl, 2,08 mmol) y la mezcla se agitó durante 25 min a temperatura ambiente y después se enfrió en un baño de hielo. Se añadió cuidadosamente ácido trifluorometilsulfónico (18 µl, 0,21 mmol). La completa conversión del material de partida tuvo lugar después de 10 minutos (ccf: sílice 4:1 de tolueno-acetonitrilo). La reacción se inactivó mediante la adición de trietilamina y la mezcla se diluyó con diclorometano, se lavó con agua y carbonato ácido sódico y se secó sobre sulfato sódico. Los disolventes se retiraron a presión reducida y el residuo se sometió a cromatografía sobre sílice (11:1 de tolueno-acetonitrilo), produciendo 560 mg, 86% de 6-O-acetil-2,3-O-dibenzoil-4-O-bencil-1-tio-β-D-glucohexopiranósido de etilo. 2,3-O-Dibenzoil-4-O-bencil-1-tio-β-D-glucohexopiranósido de etilo (hdo_9:1) 30 35 Se disolvió 2,3-O-dibenzoil-4,6-O-benciliden-1-tiol-β-D-glucohexopiranósido de etilo (Chemical Abstracts, RN 149563-08-6 o Hallgren y Widmalm, 1993) junto con complejo de borano-dimetilamina (68 mg, 1,16 mmol) en tolueno seco (20 ml) (reacción “a” en la Fig. 6). Se añadieron tamices moleculares 4 Å en polvo (180 mg) y la mezcla se agitó a temperatura ambiente durante 20 minutos. Se añadió tricloruro de aluminio (154 mg, 1,16 mmol) y después de la desaparición del material de partida (aprox. 10 min, la ccf sobre sílice, 4:1 de tolueno/acetonitrilo) la mezcla se filtró, se trató con resina de intercambio iónico Dowex (H+ ) hasta que la solución fue transparente y se filtró de nuevo. El filtrado se concentró y se co-evaporó dos veces con metanol, produciendo 200 mg de residuo que después de la cromatografía sobre gel de sílice (8:1 de tolueno:acetonitrilo) dio 60 mg, 40% de hdo_9:1. H RMN (CDCl3 , δ): 8,0-7,2 (mm, 15H), 5,75 (t, 9,4 Hz, 1H, H3), 5,37 (t, 9,8 Hz, 1H, H2), 4,74 (d, 9,8 Hz, 1H, H1), 4,61 (s, 2H), 4,02-3,95 y 3,85-3,76 (2 m a, 2H, 2H6), 3,94 (t, 9,5 Hz, 1H, H4), 3,63 (ddd, 9,7 Hz, 3,9 Hz, 2,6 Hz, 1H, H5), 2,74 (2c, 7,5 Hz, 2H), 1,25 (t; 7,5 Hz, 3H). 1 40 6-O-Acetil-2,3-O-dibenzoil-4-O-bencil-1-tio-β-D-glucohexopiranósido de etilo (hdo_9:2) 45 50 55 60 65 Se disolvió 2,3-O-dibenzoil-4-O-bencil-1-tio-β-D-glucohexopiranósido de etilo (hdo_9:1) (200 mg, 0,38 mmol) en una mezcla enfriada con hielo de piridina y anhídrido acético (1:1, 6 ml). La mezcla se agitó y se dejó alcanzar la temperatura ambiente durante una noche. La mezcla se diluyó con diclorometano y se lavó con carbonato ácido sódico saturado acuso y agua y se secó sobre sulfato de magnesio. Los disolventes se retiraron a presión reducida y el residuo se sometió a cromatografía ultrarrápida sobre gel de sílice (6:1 de tolueno-acetonitrilo), produciendo 204 mg, 93% de hdo_9:2. H RMN (CDCl3 , δ): 8,0-7,2 (mm, 15H), 5,75 (t, 9,2 Hz, 1H, H3), 5,40 (t, 9,8 Hz, 1H, H2), 4,71 (d, 10,0 Hz, 1H, H1), 4,59 y 4,55 (2d, 10,8 Hz, 2H, bzl-CH2), 4,44 (dd, 12,0 Hz, 1,9 Hz, 1H, H6), 4,27 (dd, 12,2 Hz, 4,6 Hz, 1H, H6’), 3,87 (t, 9,5 Hz, 1H, H4), 3,77 (ddd, 9,7 Hz, 4,6 Hz, 1,9 Hz, 1H, H5), 2,73 (m, 2H), 2,09 (s, 3H), 1,26 (t, 3H). 1 6-O-Acetil-2,3-O-dibenzoil-4-O-bencil-β-D-glucohexopiranósido de metilglicolilo (hdo_9:3) Se disolvieron 6-O-acetil-2,3-O-dibenzoil-4-O-bencil-1-tio-β-D-glucohexopiranósido (hdo_9:2) (600 mg, 1,04 mmol) y N-yodosuccinimida (hdo_9:2) (468 mg, 2,08 mmol, secada durante 3 h, < 0,1 mbar) en acetonitrilo seco (7 ml, pasado a través de alúmina) a temperatura ambiente. Se añadió glicolato de metilo (161 µl, 2,08 mmol) y la mezcla se agitó durante 25 min a temperatura ambiente y después se enfrió en un baño de hielo. Se añadió cuidadosamente ácido trifluorometilsulfónico (18 µl, 0,21 mmol). La completa conversión del material de partida tuvo lugar después de 10 minutos (ccf: sílice, 4:1 de tolueno-acetonitrilo). La reacción se inactivó mediante la adición de trietilamina y la mezcla se diluyó con diclorometano, se lavó con agua y carbonato ácido sódico y se secó sobre sulfato sódico. Los disolventes se retiraron a presión reducida y el residuo se sometió a cromatografía sobre gel de sílice (11:1 de tolueno-acetonitrilo), produciendo 560 mg, 86% de (hdo_9:3). 44 ES 2 225 834 T3 Glicolil-4-O-bencil-β-D-glucohexopiranósido sódica (hdo_9) 5 10 15 20 Se disolvió 6-O-acetil-2,3-O-dibenzoil-4-O-bencil-β-D-glucohexopiranósido de metilglicolilo (hdo_9:3) (350 mg, 0,59 mmol) en metóxido sódico 25 mM 7 Å (metanol) y se agitó a temperatura ambiente durante 20 h o hasta que sólo se pudo detectar un pequeño punto de movimiento lento por ccf mediante carbonación de ácido sulfúrico (sílice, 4:1 de tolueno-acetonitrilo). Se retuvo una pequeña muestra de esta solución (= i) como referencia de ccf antes de la hidrólisis con glicolato de metilo. Se añadieron hidróxido sódico (15 mg) y agua (200 µl) y la agitación se continuó durante 8 h o hasta que se consumió toda la “i” (ccf sobre sílice, 8:3:1 de acetato de etilo-etanol-ácido acético). La reacción se inactivó llevando el pH a 4,2 con ácido acético. Los disolventes se retiraron a presión reducida y el residuo (650 mg) se sometió a cromatografía ultrarrápida sobre gel de sílice (8:3:1 de acetato de etilo-etanol-ácido acético). El material en el intervalo Rf de 0,15-0,25 se recogió y se concentró, produciendo 178 mg, 92% de un sólido blanco amorfo. 1 H RMN (CD3 OD, δ relativo a metanol a 3,31 ppm): 7,45 (m, 5H), 4,65 (d, 11,0 Hz, 1H), 4,36 (d, 16,0 Hz, 1H), 4,32 (d, 7,9 Hz, 1H, H1), 4,11 (d, 15,8 Hz, 1H), 3,82 (dd, 2,0 Hz, 12,1 Hz, 1H, H6), 3,65 (dd, 4,8 Hz, 12,1 Hz, 1H, H6), 3,59 (t, 9,0 Hz, 1H, H3 o H4), 3,40 (t, 9,1 Hz, 1H, H3 o H4). Este material aún contenía mucho ácido acético, así que se disolvieron de nuevo 50 mg en agua y se liofilizaron, dando 30 mg de sustancia libre de ácido acético pero con un contenido de agua (1 H RMN) de 24,5 equiv. mol. Este material se redisolvió en agua y se le añadió 1 equivalente de hidróxido sódico y la solución se liofilizó, dando 22 mg del compuesto del título. H RMN (CD3 OD, δ relativo a metanol a 3,31 ppm): 7,40-7,22 (m, 5H), 4,95 (d, 10,8 Hz, solapamiento por HDO residual a 4,93), 4,65 (d, 11,0 Hz, 1H), 4,34 (d, 16,0 Hz, 1H), 4,32 (d, 7,5 Hz, 1H, H1), 4,10 (d, 15,8 Hz), 3,82 (dd, 1,8 Hz, 11,9 Hz, 1H, H6), 3,66 (dd, 4,8 Hz, 12,1 Hz, 1H, H6), 3,59 (t, 8,8 Hz, 1H, H3), 3,41 (t, 9,7 Hz, 1H, H4), 3,153,06 (m, 2H, H5, H2). 1 25 30 13 C RMN (CD3 OD, δ relativo a metanol a 49,0 ppm): 176,1, 140,0, 129,3, 129,1, 128,6, 104,3, 79,1, 78,1, 77,2, 75,7, 75,2, 68,4, 62,3. 4-O-Bencil-β-D-glucopiranósido de 2-(hidroxi)etilo (hdo_23) 35 40 45 Una mezcla de N-yodosuccinimida (0,109 g, 0,484 mmol) y ácido tríficlo (6,6 µl, 0,0744 mmol) en diclorometanoéter dietílico (2 ml, 1:1) se añadió a una mezcla agitada de 6-O-acetil-2,3-O-benzoil-4O-bencil-1-tio-β-D-glucopiranósido de tioglicosidoetilo (hdo_9:2) (0,210 g, 0,372 mmol), etilen glicol (0,125 ml, 2,232 mmol) y tamices moleculares (0,3 g, 4 Å) en diclorometano-éter dietílico (3 ml, 1:2) a temperatura ambiente durante 5 min. Después de 25 min, la CCF mostró una conversión de aprox. el 20% del tioglicósido, así que se añadió más N-yodosuccinimida (0,109 g, 0,484 mmol) y ácido tríflico (6,6 µl, 0,0744 mmol) en diclorometano-éter dietílico (2 ml, 1:1) durante 5 min. Después de 30 min más, la mezcla de reacción se filtró a través de una capa de Celite en una solución acuosa de carbonato ácido sódico y bisulfito sódico. La fase orgánica se separó, se lavó con agua, se secó (Na2 SO4 ) y se concentró. El residuo se coevaporó dos veces con tolueno, se disolvió en metanol que contenía metóxido sódico (10 ml, 0,2 M), se mantuvo durante 1 h a 50ºC y se evaporó. La cromatografía en columna (SiO2 , cloroformo-metanol-agua, 100:15:1) del residuo dio hdo_23 (33 mg, 30%). [α]22 D + 0,9º (c 1,4, metanol) CCF: Rf 0,29 (cloroformo-metanol-agua, 100:15:1). 50 C RMN (CD3 OD, δ: relativo a metanol a 49,0 ppm) δ: 139,9, 129,2, 129,0, 128,6, 104,4 (C-1), 79,2, 78,2, 77,0, 75,7, 75,4, 72,3, 62,3, 62,3 ppm. 13 H RMN (CD3 OD, δ: relativo a metanol a 3,31 ppm) δ: 7,45-7,25 (m, 5H), 4,95 (d, 11,0 Hz, 1H, bencílico), 4,65 (d, 11,0 Hz, 1H, bencílico), 4,29 (d, 7,7 Hz, 1H, H-1) ppm. 1 55 Ejemplo 5 Síntesis de compuestos de la familia bpy capaces de unirse al sitio de unión de PapD 60 4’-Metoxifenilmetilen-α-D-mannohexopiranósidos de metilo, mezcla de 4,6-, 2,3-endo y 2,3-exo-monoacetales (Véase: Patroni y col., 1988). 65 Se disolvieron α-D-mannohexopiranósido de metilo (38,8 g, 200 mmol) y ácido p-toluenosulfónico (200 mg) en dimetilformamida (300 ml) y se agitaron a 100ºC en una atmósfera de nitrógeno. Se añadió gota a gota 4-metoxibenzaldehído dimetil acetal (37,7 g, 240 mmol) en dimetilformamida (300 ml) durante 3 h. Como aún quedaba material de partida (ccf, sílice, acetato de etilo), la agitación y el calentamiento se continuaron durante 2 h. La composición 45 ES 2 225 834 T3 5 de la mezcla de reacción no mostró cambios visibles en ese momento (ccf, sílice, acetato de etilo) y la reacción se inactivó con carbonato potásico (2,15 g). El disolvente se retiró a presión reducida y el residuo se filtró a través de sílice (acetato de etilo) para retirar los monoacetales depositados (1 H RMN) tras la trituración con metil t-butil éter (11 g, 18%), que se recogió por filtración. Los filtrados se sometieron a cromatografía ultrarrápida sobre gel de sílice, produciendo 20 g de la mezcla monoacetal (rendimiento del 51%) y 17 g de los 2,3;4,6-diacetales (endo/exo 1:1). 2-O-Etil-4,6-O-(4’-metoxi)fenilmetileno-α-D-mannohexopiranósido de metilo (bpy_9:1) 10 15 20 Se disolvieron 4’-metoxifenilmetileno-α-D-mannohexopiranósido de metilo, mezcla de 4,6-, 2,3-endo y 2,3-exomonofenilmetilenacetales (11,3 g, 36,2 mmol), yoduro de etilo (4,35 ml, 54,3 mmol) e hidrogenosulfato de tetrabutilaminio (2,48 g, 7,24 mmol) en cloruro de metileno (500 ml). A esto se le añadió una solución de hidróxido sódico (2,5 g, 140 mmol) en agua (50 ml) y la mezcla se calentó a reflujo durante 4 días, con adiciones diarias de yoduro de etilo (2,4 ml) e hidróxido sódico al 20% (1 ml). Después de esto, no podrían detectarse cambios visibles en la composición de la mezcla de reacción por ccf (2:1 de tolueno-acetato de etilo). La mezcla se dejó enfriar, las fases se separaron y la fase orgánica se lavó con agua (3 x) y se secó sobre sulfato sódico. El disolvente se retiró a presión reducida y el residuo se sometió a cromatografía ultrarrápida sobre gel de sílice (6:1 de tolueno-acetato de etilo). Las fracciones que contenían el componente con Rf = 0,4 tras la ccf (2:1 de tolueno-acetato de etilo) se concentraron y recromatografiaron como antes, produciendo 3,15 g, 26% del compuesto del título. Se descubrió que las fracciones que contenían los componentes con Rf = 0,32 tras la ccf consistían en una mezcla de regioisómeros monofenilmetilenacetal monoetilados (1 H RMN). [α]20 D = +11,2º (c 1,02, CHCl3 ) H RMN (CDCl3 , δ): 7,42 (m. sim, 2H), 6,88 (m. sim, 2H), 5,53 (s, 1H), 4,77 (d, 1,3 Hz, 1H, H1), 4,24 (dd, 4,2 Hz, 9,7 Hz, 1H, H6), 4,02 (dt, 4,0 Hz, 8,8 Hz, 1H, H5), 3,83 (dd, 1,8 Hz, 9,7 Hz, 1H, H6), 3,80-3,69 (s y solapamiento de m, 6H, 1-OMe, H3, H4, 2-O-CHHCH3 ), 3,69 (s, 3H, Ar-OMe), 2,48 (d, 1H, OH), 1,26 (t, 7,0 Hz, 3H; 2-O-CH2 CH3 ). 1 25 30 1 H RMN (CDCl3 , con Cl3 CCONC añadido, δ): 8,54 (s, 1H, acilcarbamato NH), 7,38 (m. sim, 2H), 6,88 (m. sim, 2H), 5,52 (s, 1H), 5,27 (dd, 3,5 Hz, 10,3 Hz, 1H, H3), 4,76 (d, 1,6 Hz, 1H, H1), 4,32-4,20 (m, 1H), 4,13 (m. sim, 1H, H5), 3,97 (dd, 1,8 Hz, 3,5 Hz, 1H, H2), 3,91-3,80 (m, 1H), 3,79 (s, 3H, 1-OMe), 3,77-3,54 (m, 3H), 3,41 (s, 3H, ArOMe), 1,23 (t, 7,0 Hz, 3H). 13 35 C RMN (CDCl3 , δ): 160,1, 129,9, 127,6, 113,6, 102,0, 99,3, 79,4, 78,8, 68,7, 68,3, 67,2, 63,2, 55,2, 54,9, 15,4. Calc. para C17 H25 O7 : C, 59,8 H, 7,38, 2-O-Etil-3-O-dimetil-t-butilsilil-4,6-O-(4’-metoxi)fenilmetileno-α-D-mannohexopiranósido (bpy_9:2) 45 Se disolvió 2-O-etil-4,6-O-(4’-metoxi)fenilmetileno-α-D-mannohexopiranósido de metilo (bpy_9:1) (0,98 g, 2,88 mmol) en cloruro de metileno seco (18 ml) junto con trietilamina (600 µl, 4,32 mmol) y la mezcla se enfrió con hielo. Se añadió gota a gota trifluorometilsulfonato de t-butildimetilsililo (795 µl, 3,45 mmol) en cloruro de metileno (2 ml) y la mezcla de reacción con el baño de enfriamiento se dejó a temperatura ambiente durante una noche (ccf sobre sílice, 4:1 de tolueno-metil t-butil éter). El exceso de reactivo de sililación se destruyó mediante la adición de metanol (3 ml), la mezcla se concentró a presión reducida y el residuo se sometió a cromatografía ultrarrápida sobre gel de sílice (40:1 de tolueno-metil t-butil éter), produciendo 1,40 g, 100% de (bpy_9:2). 50 1 H RMN (CDCl3 , δ): 7,40 (m. sim, 2H), 6,87 (m. sim, 2H), 5,53 (s, 1H), 4,70 (d, 1,3 Hz, 1H, H1), 4,21 (dd, 4,6 Hz, 9,9 Hz, 1H, H6), 4,10 (dd, 3,3, Hz, 9,7 Hz, 1H, H3), 3,91 (t, 9,5 Hz, 1H, H4), 3,90-3,79 (s solapado con m, 5H, H6, 2-OCHHCH3 ; 1-OMe), 3,76-3,63 (m, 2H, H5, 2-OCHHCH3 ), 3,55 (dd, 1,5 Hz, 3,3 Hz, 1H, H2), 3,67 (s, 3H, ArOMe), 1,24 (t, 7,0 Hz, 3H), 0,88 (s, 9H), 0,08 (s, 3H), 0,03 (s, 3H). 40 13 C RMN (CDCl3 , δ): 159,9, 130,3, 127,5, 113,4, 101,8, 101,2, 79,9, 79,1, 70,3, 68,8, 68,2, 64,1, 55,2, 54,8, 25,8, 18,3, 15,6, -4,4, -4,9. 55 2-O-Etil-3-O-dimetil-t-butilsilil-4,-O-(4’-metoxi)-bencil-α-D-mannohexopiranósido de metilo (bpy_9:3) y 2-O-etil-3-O-dimetil-t-butilsilil-6-O-(4’-metoxi)-bencil-α-D-mannohexopiranósido de metilo (bpy_9:8) 60 65 Se disolvió 2-O-etil-3-O-dimetil-t-butilsilil-4,6-O-(4’-metoxi)fenilmetilen-α-D-mannohexopiranósido de metilo (bpy_9:2) (3,61 g, 7,95 mmol) en acetonitrilo (150 ml, secado sobre tamices moleculares 4 Å), se añadieron tamices moleculares 3 Å en polvo (6 g) y la mezcla se enfrió a -30ºC en una atmósfera de nitrógeno y se agitó durante 15 min. Se añadió cianoborohidruro sódico (3,04 g, 47,7 mmol) y se añadió gota a gota una solución de clorotrimetilsilano (7,95 ml, 47,7 mmol) en acetonitrilo seco (50 ml, secado sobre tamices moleculares 4 Å) en 1 h. La agitación se continuó durante 2 h a -30ºC, tras lo cual la temperatura se dejó aumentar a 0ºC. Después de 1 h, la ccf (4:1 de tolueno:acetato de etilo) mostró la completa conversión del material de partida. La mezcla se filtró a través de una 46 ES 2 225 834 T3 capa de Celite® y el Celite® se lavó minuciosamente con acetato de etilo. Los filtrados combinados se lavaron con carbonato ácido sódico ac. sat. y cloruro sódico ac. sat., se concentraron a presión reducida, se redisolvieron en tolueno y se concentraron a presión reducida. El residuo se sometió a cromatografía ultrarrápida sobre gel de sílice (4:1 de tolueno-acetato de etilo, después 1:1), obteniendo 578 mg de bpy_9:8 (= bpy_21:1), 16% y 2,34 g de (bpy_9:3), 65%. 5 bpy-9:3 (algunas tareas de COSY, HETCOR): 10 1 H RMN (CDCl3 , δ): 7,25 (m. sim, 2H), 6,87 (m. sim, 2H), 4,81 (d, 11,0 Hz, 1H), 4,66 (d, 1,8 Hz, 1H, H1), 4,51 (d, 10,8 Hz, 1H), 4,02 (dd, 3,1 Hz, 5,9 Hz, 1H, H3), 3,80 (s, 3H, 1-OMe), 3,79-3,60 (m, 5H; H4, 2H6, 2-OCH2 CH3 ), 3,54 (ddd, 2,9 Hz, 4,8 Hz, 9,7 Hz, 1H, H5), 3,46 (dd, 2,0 Hz, 3,3 Hz, 1H, H2), 3,33 (s, 3H, Ar-OMe), 2,210 y 1,86 (2 s muy a, 1H, 6-OH), 1,23 (t, 6,8 Hz, 3H), 0,95 (s, 9H), 0,12 (s, 6H). H RMN (CDCl3 , con Cl3 CCONC añadido, δ): 8,40 (s, 1H, acilcarbamato NH), 7,24 (m. sim, 2H, Ar H3’,5’), 6,86 (m. sim, 2H, Ar H2’, H6’), 4,82 (d, 11,2 Hz, 1H), 4,67 (d, 2,0 Hz, 1H, H1), 4,43-4,32 (m, 2H, 2H6), 4,08-3,98 (m, 1H, H3), 3,78 (s, 3H, 4,43-4,32 (m, 2H, 2H6), 4,08-3,98 (m, 1H, H3), 3,78 (s, 3H, 1-OMe), 3,77-3,61 (m, 4H, H5, 2OCH2 CH3 , H4), 3,46 (dd, 1,8 Hz, 3,1 Hz, 1H, H2), 3,34 (s, 3H, Ar-OMe), 1,22 (t, 7,0 Hz, 3H), 0,96 (s, 9H), 0,14 (s, 3H), 0,13 (s, 3H). 1 15 20 13 C RMN (CDCl3 , δ): 159,1, 130,7, 129,3, 113,8, 99,8, 79,5, 75,7, 74,7, 73,0, 72,2, 67,4, 62,5, 55,2, 54,7, 18,0, 15,7, -4,3, -4,6. C RMN (CDCl3 , con Cl3 CCONC añadido, δ): 159,3 (Ar C4’), 157,4, 149,3, 130,2, (Ar C2’), 129,6 (Ar C3’,C5’), 113,8 (Ar C2’,C6’), 99,6 (C1), 79,2 (C2), 74,56* (4-OCH2 Ar o 2-CH2 CH3 ), 74,54* (4-OCH2 Ar o 2-CH2 CH3 ), 73,2 (C3), 69,7 (C5), 67,2 (C4), 66,3 (C6), 55,2 (1-OMe), 54,9 (Ar-OMe), 25,9 (CMe3 ), 18,0 (CMe3 ), 15,6, -4,3, -4,7. 13 25 bpy_9:8: 30 1 H RMN (CDCl3 , δ): 7,28 (m. sim, 2H), 6,86 (m. sim, 2H), 4,70 (d, 1,8 Hz, 1H, H1), 4,55 (d, 11,7 Hz, 1H, H6), 4,51 (d, 11,6 Hz, 1H, H6), 3,89-3,55 y 3,79 (m y s, 7H, H3, H4, H5, 2-OCH2 CH3 , 1-OMe), 3,44 (dd, 1,8 Hz, 2,9 Hz, 1H, H2), 3,56 (s, 3H, Ar-OMe), 3,44 (dd, 1,8 Hz, 2,9 Hz, 1H, H2), 3,56 (s, 3H, Ar-OMe), 2,29 (d, 2,0 Hz, 1H, 4-OH), 1,20 (t, 7,0 Hz, 3H), 0,92 (s, 9H), 0,13 (s, 3H), 0,12 (s, 3H). C RMN (CDCl3 , δ): 159,1, 130,3, 129,3, 113,7, 99,7, 78,9, 73,19*, 73,16*, 71,3, 70,3, 69,1, 67,2, 55,2, 54,8, 25,8, 18,2, 15,6, -4,5, -4,6. 13 35 *resuelto multiplicando FID con una función de peso gaussiana. 2-O-Etil-3-O-dimetil-t-butilsilil-4,-O-(4’-metoxi)-bencil-6(S)-fenil-α-D-mannohexopiranósido de metilo (bpy_9:4) 40 45 50 (Oxidación ref. D. F. Taber y col., J. Org. Chem. 52, 5621-2, 1987) Se disolvieron 2-O-etil-3-O-dimetil-t-butilsilil4,-O-(4’-metoxi)bencil-α-D-mannohexopiranósido de metilo (bpy_9:3) (200 mg, 0,438 mmol) y dimetilsulfóxido (64,0 µl, 0,876 mmol) en diclorometano (2,5 ml, secado sobre tamices moleculares 4 Å) y la solución se enfrió en un baño de hielo. Se añadió rápidamente pentóxido de fósforo (124 mg, 0,876 mmol) y se dejó que la suspensión agitada resultante alcanzara la TA (1 h). La mezcla se enfrió de nuevo con hielo y se le añadió trietilamina (214 µl, 1,53 mmol) provocando una inmediata disolución de la suspensión de tipo gel. El baño de hielo se retiró y la mezcla se agitó a TA durante 1 h, se diluyó con acetato de etilo, se lavó con dihidrogenofosfato sódico 0,2 M ac., se secó sobre sulfato sódico y se concentró a presión reducida (<0,02 mbar). El residuo se disolvió en tetrahidrofurano (2 ml, se secó sobre tamices moleculares 4 Å), se agitó y se enfrió a -20ºC. Se añadió gota a gota cloruro de fenilmagnesio en tetrahidrofurano (345 µl de una solución al 25% en peso, 0,657 mmol, Janssen, Bélgica) (3 min). La reacción se controló con ccf (4:1 de tolueno-acetato de etilo) y se inactivó con sulfato amónico ac. al 10% (3 ml). La mezcla se diluyó con acetato de etilo y se lavó dos veces con agua, se secó sobre sulfato sódico y se concentró a presión reducida. El residuo se sometió a cromatografía ultrarrápida sobre gel de sílice (20:1 de tolueno-metil t-butil éter). Las fracciones que contenían el componente con un Rf = 0,44 sobre ccf (4:1 de tolueno-acetato de etilo) se reunieron y se concentraron a presión reducida, produciendo 65 mg, 30% del compuesto del título. 55 60 Los cálculos de mecánica molecular con MM2(91) (Burkert y Allinger, 1982) de los diastereómeros R y S y los posteriores cálculos con la ecuación de Karplus (Bothner-By, 1965) predijeron que la 1 H RMN J5−6 sería de 4,8 y 6,2 respectivamente. Esta diferencia es demasiado pequeña para ser sólo una indicación de la estereoquímica. Además, la señal H6 se resuelve pobremente y la señal H5 se solapa severamente por otras resonancias. El pico cruzado 1 HCOSY H4-H5, sin embargo, contiene la suma de J4−5 + J5−6 y ya que J4−5 puede observarse en la resonancia de H4, a J5−6 se le puede asignar el valor de 6,8 Hz. Esto indica que el producto aislado tiene la estereoquímica 6-(S). Después de la retirada del grupo 4-metoxibencilo, las constantes de acoplamiento calculadas se convirtieron en 5,5 Hz y 12,9 Hz, respectivamente. 65 H RMN (CDCl3 , δ): 7,44-7,19 (m, 7H), 6,89 (m. sim, 2H), 5,00 (m a, 1H, H6), 4,90 (d, 10,8 Hz, 1H), 4,66 (d, 10,8 Hz, 1H), 4,58 (d, 1,8 Hz, 1H, H1), 4,04 (dd, 3,1 Hz, 9,0 Hz), 3,99 (t, 9,2 Hz, 1H, H4), 3,81 y 3,82-3,56 (s y m, 1 47 ES 2 225 834 T3 6H, 1-OMe, 2-CH2 CH3 ), H5), 3,44 (dd, 2,0 Hz, 2,9 Hz, 1H, H2),2,84 (s, 3H, Ar-OMe), 1,24 (t, 3H), 1,97 (s, 9H), 0,15 (s, 3H), 0,13 (s, 3H). C RMN (CDCl3 , δ): 159,0, 14 2,5, 130,8, 129,2, 127,7, 126,7, 1 26,6, 125,7, 113,7, 99,5, 79,8, 76,1, 74,5, 73,0, 70,9, 67,9, 55,1, 54,1, 25,8, 17,9, 15,4, -4,5, -4,8. 13 5 2,3-Anhidro-4,6-O-p-metoxibenciliden-α-D-mannopiranósido de metilo (bpy_9:9) 10 15 A una suspensión agitada de hidruro sódico (2,6 g, 65 mmol, dispersión al 65% en aceite mineral) en N,Ndimetilformamida (150 ml) se le añadió con agitación una solución de 4,6-O-p-metoxibenciliden-α-D-glucopiranósido (9,36 g, 30 mmol) en N,N-dimetilformamida (65 ml). La mezcla se agitó durante 45 minutos y se le añadió ptoluenosulfonilimidazol (7,24 g, 33 mmol). La agitación se continuó durante 2 horas y la mezcla se vertió en agua enfriada con hielo. El precipitado se retiró por filtración y se secó al vacío, dando bpy_9:8 bruto (7,7 g). Recristalización dos veces en metanol (250 ml dieron bpy_9:9 (3,59 g). La cromatografía (SiO2 , acetato de etiloheptano, 2:3) del agua madre seguida de cristalización en metanol (150 ml) dio más bpy_9:9 (1,87 g). El rendimiento total de bpy_9:9 fue de 5,45 g (61%). p.f. 152-153,5ºC (metanol) 20 [α]22 D : 96,4º (c 1,8, cloroformo) 25 1 H RMN (CDCl3 ) δ: 7,43 y 6,91 (patrón AB, acoplado adicionalmente, 2H, JAB = 8,7 Hz), 5,53 (s, 1H, ArCH), 4,90 (s, 1H, H-1), 4,30-4,19 (sim. m, 1H), 3,82 (s, 3H, CH3 OAr), 3,78-3,64 (3H), 3,49-3,46 (4H), 3,48 (s, 3H, CH3 O), 3,18 (d, 1H, J = 3,7 Hz, H-2/H-3) ppm. 13 C RMN (CDCl3 ) δ: 160,3, 129,6, 127,5, 113,7, 102,4, 96,9, 74,8, 69,4, 61,7, 55,7, 53,8 y 50,5 ppm. 3-Azido-4,6-O-p-metoxibenciliden-α-D-altropiranósido de metilo (bpy_9:10) 30 35 Se agitaron 2,3-anhidro-4,6-O-p-metoxibenciliden-α-D-mannopiranósido de metilo (bpy_9:9, 2,35 g, 8 mmol), azida sódica (2,08 g, 32 mmol) y cloruro amónico (0,86 g, 16 mmol) durante 5 horas a 110ºC en una mezcla de 2metoxietanol (25 ml) y agua (5 ml). La suspensión se disolvió gradualmente, dando una solución turbia. Después, la mezcla se repartió entre acetato de etilo y NaOH acuoso (0,25 M, 100 ml). La fase acuosa se extrajo dos veces con acetato de etilo y las fases orgánicas combinadas se lavaron dos veces con agua y otras tantas con NaCl saturado acuoso, se secaron (Na2 SO4 ), se filtraron y se concentraron. La cromatografía (SiO2 ) con acetato de etilo-heptano, 2:32:1) dio bpy_9:10 (1,97 g, 73%). Se cristalizó una muestra analítica en acetato de etilo-heptano. p.f. 91-93ºC (EtOAc/Heptano) 40 [α]22 D : +26,1º (c 1,1, cloroformo) 45 1 H RMN (CDCl3 ) δ: 7,44 y 6,91 (patrón AB acoplado adicionalmente, 2H, JAB = 8,6 Hz), 5,57 (s, 1H, ArCH), 4,56 (s, 1H, H-1), 4,43-4,21 (2H), 4,16-4,08 (1H), 4,03 (dd sin resolver, 1H, J1 = J2 = 3 Hz), 3,91 (s a, 1H, H-2), 3,83-3,74 (4H), 3,80 (s, 3H, CH3 O), 3,43 (s, 3H, CH3 O), 2,36 (s a, 1H, OH) ppm. 13 50 C RMN (CDCl3 ) δ: 160,3, 129,5, 127,5, 113,7, 102,3, 101,4, 75,8, 69,8, 69,0, 60,1, 59,0, 55,7 y 55,3 pm. La acetilación (anhídrido acético-piridina, 3:5) de una muestra de bpy_9:10 dio un acetato que tenía el siguiente espectro 1 H RMN (las asignaciones se confirmaron por el correspondiente espectro mononuclear COSY): 1 H RMN (CDCl3 ) δ: 7,43 y 6,91 (patrón AB acoplado adicionalmente, 2H, JAB = 8,8 Hz), 5,59 (s, 1H, ArCH), 4,97 (dd, 1H, J = 2,2 y 0,9 Hz, H-2), 4,57 (s, 1H, H-1), 4,36 (2H, H-6 y H-4), 4,09-4,01 (2H, H-6 y H-3), 3,84-3,74 (2H, CH3 O y H-5), 3,81 (s, 3H, CH3 O), 3,44 (s, 3H, CH3 O), 2,15 (s, 3H, CH3 CO) ppm. 55 3-Azido-2-O-etil-4,6-O-p-metoxibenciliden-α-D-altropiranósido de metilo (bpy_9:11) 60 65 Se agitaron 3-azido-4,6-O-p-metoxibenciliden-α-D-altropiranósido de metilo (bpy_9:10, 1,7 g, 5 mmol), óxido de bario (3,0 g, 19,6 mmol) y yoduro de etilo (5 ml, 62 mmol) en dimetilsulfóxido (5 ml). Se añadió agua (10 µl) y la agitación se continuó durante 16 horas. Después, la mezcla se repartió entre acetato de etilo y agua. La fase acuosa se extrajo dos veces con acetato de etilo y las fases orgánicas combinadas se lavaron dos veces con agua y después con NaCl saturado acuoso, se secaron (Na2 SO4 ), se filtraron y se concentraron. La cromatografía (SiO2 , acetato de etiloheptano, 1:3) dio bpy_9:11 en forma de un aceite (1,7 g, 92%). [α]22 D : 19,1º (c 1,6, cloroformo). 1 H RMN (CDCl3 ) δ: 7,44 y 6,91 (patrón AB, acoplado adicionalmente, 2H, JAB = 8,6 Hz), 55,57 (s, 2H, ArCH), 48 ES 2 225 834 T3 4,63 (s a, 1H, H-1), 4,14-4,06 (2H), 3,85-3,75 (4H), 3,81 (s, 3H, CH3 O), 3,73-3,56 (sim. m, 2H), 3,52 (dd, 1H, J = 2,4 y 0,9 Hz, H-3), 3,34 (s, 3H, CH3 O), 1,25 (t, 3H, J = 6,9 Hz, CH3 CH2 ) ppm. 13 5 C RMN (CDCl3 ) δ: 160,1, 129,6, 127,4, 113,6, 102,1, 99,6, 77,0, 76,1, 69,0, 66,5, 58,7, 58,1, 55,5, 55,2 y 15,3 ppm. 3-Azido-3-desoxi-2-O-4-O-p-metoxibencil-α-D-altro-piranósido de metilo (Cf. R. Johansson y B. Samuelsson; J. Chem. Soc. Commun. 1984, 201-202). 10 15 20 25 Se agitaron 3-azido-3-desoxi-2-O-etil-4,6-O-p-metoxibenciliden-α-D-altropiranósido de metilo (bpy_9:11) (2,41 g, 6,6 mmol), cianoborohidruro sódico (2,5 g, 39,6 mmol) y tamices moleculares triturados (5 g) en acetonitrilo (130 ml) a 0ºC. Se añadió una solución de cloruro de trimetilsililo (5,0 ml, 40 mmol) en acetonitrilo (45 ml) durante 5 min. Después de 1,5 h más de agitación, el baño de refrigeración se retiró y la agitación se continuó durante 21 h. La mezcla se filtró a través de celite y el filtrado se repartió entre acetato de etilo y NaHCO3 saturado acuoso. La fase orgánica se lavó dos veces con NaHCO3 saturado acuoso y después con NaCl saturado acuoso, se secó (Na2 SO4 +NaHCO3 ), se filtró y se evaporó. La cromatografía dos veces (SiO2 , 2:7 → 2:1 de acetato de etilo-heptano y 1:2 → 2:1 de acetato de etilo-tolueno) dio el compuesto del título (2,23 g, 92%) en forma de un aceite. [α]22 D : +113,6º (c 1,4, cloroformo) Datos de 1 H RMN (CDCl3 ) δ: 7,30 y 6,89 (patrón AB, acoplado adicionalmente, 4H, J = 8,8 Hz), 4,62 y 4,56 (patrón AB, 2H, J = 11,3 Hz, H bencílico), 4,58 (d sin resolver, acoplado virtualmente, 1H, J <2 Hz, H-1), 4,00-3,84 (3H, H-3, H-4 y H-5), 3,81 (s, 3H, CH3 OAr), 3,84-3,7 (2H, H-6), 3,62-3,43 (3H, CH2 CH3 y H-2), 3,38 (s, 3H, CH3 O), 1,99 (s a, 1H, OH) y 1,19 (t, 3H, J = 7,0 Hz, CH2 CH3 ) ppm. Datos de 13 C RMN (CDCl3 ) δ: 159,6, 129,9, 129,6, 113,9, 99,9, 76,9, 72,2, 71,9, 67,7, 66,3, 62,4, 58,6, 55,4, 55,3 y 15,4 ppm. 30 35 40 45 50 3-Azido-6-O-benzoil-3-desoxi-2-O-etil-4-O-p-metoxi-bencil-α-D-altropiranósido de metilo Una solución de 3-azido-3-desoxi-2-O-etil-4-O-p-metoxi-bencil-α-D-altro-piranósido de metilo (1,47 g, 4 mmol) en piridina (35 ml) se agitó a 0ºC. Se añadió cloruro de benzoílo (1,2 ml, 10,3 mmol) y la temperatura se aumentó a temperatura ambiente. La agitación se continuó durante 1,5 h y después la mezcla se enfrió a 0ºC. Se añadió metanol (10 ml) y la mezcla se agitó durante 20 min más a temperatura ambiente, se evaporó y se repartió entre acetato de etilo y agua. La fase orgánica se lavó posteriormente con agua, NaHCO3 saturado acuoso, agua y NaCl saturado acuoso, se secó (Na2 SO4 ), se filtró y se evaporó. La cromatografía (SiO2 , 1:4 de acetato de etilo-heptano) dio el benzoato (1,64 g, 87%). [α]22 D : +0,95º (c 1,0, cloroformo) Datos de 1 H RMN (CDCl3 ) δ: 8,04-7,98 (m, 2H), 7,56 (tt, 1H, J = 7,5 y 1,3 Hz), 7,43 (t, acoplado adicionalmente, 2H, J = 7,5 Hz), 7,26 y 6,83 (patrón AB, acoplado adicionalmente, 4H, J = 8,6 Hz), 4,62 y 4,50 (patrón AB, 2H, J = 11,3 Hz, ArCH2 ) 4,63 (d, acoplado adicionalmente, J = 1,3 Hz, H-1), 4,54 (parte A de un sistema ABX, 1H, J = 11,8 y 2,7 Hz, H-6a ), 4,47 (parte B de un sistema ABX, 1H, J = 11,8 y 5,5 Hz, H-6b ), 4,27 (ddd, 1H, J = 8,4, 5,5 y 2,7 Hz, H-5), 3,99-3,92 (2H, H-3 y H-4), 3,74 (s, 3H, CH3 OAr), 3,67-3,48 (3H, CH2 CH3 y H-2) y 1,22 (t, 3H, J = 7,0 Hz, CH2 CH3 ) ppm. Datos de 13 C RMN (CDCl3 ) δ: 166,2, 159,5, 132,9, 130,1, 129,9, 129,6, 129,2, 128,3, 113,9, 99,6, 76,72, 72,0, 71,7, 62,3, 62,2, 64,1, 58,5, 55,4, 55,2 y 15,4 ppm. Sal sódica de 6-O-benzoil-3-desoxi-2-O-etil-4-O-p-metoxibencil-3-sulfamido-α-D-altropiranósido de metilo (Cf. H. P. Wessel; J. Carbohydr. Chem. 11 (8), (1992), 1039-1052). 55 60 65 A una solución agitada de 3-azido-6-O-benzoil-3-desoxi-2-O-etil-4O-p-metoxibencil-α-D-altopiranósido de metilo (620 mg, 1,31 mmol) en tetrahidrofurano (40 ml) se le añadieron agua (265 ml) y trifenilfosfina (1,7 g, 6,5 mmol). La solución se agitó durante 24 h y después se concentró hasta 12 ml. El residuo se diluyó con metanol (10 ml) y agua (3 ml). Después, a la solución agitada se le añadió complejo de trimetilamina-trióxido de azufre (370 mg, 2,7 mmol). El pH se ajustó a 8,5 mediante NaOH acuoso 1 M. Después de unos minutos se formó un precipitado que se disolvió añadiendo tetrahidrofurano (3 ml) y metanol (4 ml). La mezcla de reacción se agitó durante 0,5 h, después de lo cual se añadió más complejo de trimetilamina-trióxido de azufre (86 mg). La agitación se continuó durante 15 min. Durante el transcurso de la reacción, el pH se mantuvo a 8-9 mediante la adición de NaOH acuoso 1 M. Después, la mezcla se diluyó con agua (7 ml) y los disolventes orgánicos se evaporaron a presión reducida. Se añadió más agua (5 ml) y la suspensión se liofilizó y se cromatografió (SiO2 , 100:15:1 → 70:30:5 de cloroformo-metanol-agua), dando el compuesto sulfamino, presumiblemente en forma de sal sódica (668 mg, 92%) después de la liofilización en agua. Se cristalizó una muestra en acetona acuosa. 49 ES 2 225 834 T3 p.f. 112-115ºC (acetona acuosa). [α]22 D : +156,4º (c 0,5 agua) 5 10 Datos de 1 H RMN (CD3 OD) δ: 7,94-7,89 (m, 2H), 2H, 7,64-7,56 (m, 1H), 7,44 (m, 2H), 7,28 y 6,73 (patrón AB, acoplado adicionalmente, 4H, J = 8,6 Hz), 4,71 y 4,49 (patrón AB, 2H, J = 11,4 Hz, ArCH2 ), 4,68 (s a, 1H, H-1), 4,51 (dd, 1H, J = 11,6 y 2,4 Hz, H-6a ), 4,44 (dd, 1H, J = 11,6 y 4,4 Hz, H-6b ), 4,09 (ddd sin resolver, 1H, J 4, 3 y 1 Hz, H-3), 4,00-3,92 (2H, H-5 y H-2), 3,86 (dd, 1H, J = 9,9 Hz y 4,2 Hz, H-4), 3,71-3,54 (5H, CH2 CH3 y CH3 OAr), 3,65 (s, CH3 OAr), 3,40 (s, 3H, CH3 O) y 1,17 (t, 3H, J = 7,0 Hz, CH2 CH3 ) ppm. Datos de 13 C RMN (CD3 OD) δ: 167,8, 160,8, 134,2, 131,4, 130,9, 130,6, 129,5, 114,7, 101,6, 77,03, 70,02, 69,2, 67,0, 66,4, 65,2, 55,6, 55,5, 50,6 y 15,8 ppm. Sal amónica de 6-O-benzoil-3-desoxi-2-O-etil-3-sulfamido-α-D-altropiranósido de metilo (bpy_30) 15 20 Se hidrogenó la sal sódica de 6-O-benzoil-3-desoxi-2-O-etil-4-O-p-metoxibencil-3-sulfamino-α-D-altro-piranósido de metilo (334 mg, 2,0 mmol) durante 6 h en ácido acético glacial (55 ml) a 0,23 MPa y a temperatura ambiente, usando paladio al 5% sobre carbón como catalizador. El filtrado y la liofilización dieron un residuo que se cromatografió (SiO2 , 100:15:1 → 70:30:5 de cloroformo-metanol-agua). El producto se disolvió en agua y se pasó a través de una resina de intercambio de cationes (BIO-REX® 70, malla 200-400, en forma de amonio) y se liofilizó, dando el compuesto del título (222 mg, 86%). [α]22 D : +55,7º (c 1,3, agua) 25 30 35 Datos de 1 H RMN (piridina-d5 ) δ: 8,23-8,17 (2H), 7,52-7,45 (1H), 7,42-7,35 (2H), 5,09 (dd, 1H, J = 11,65 y 1,54 Hz, H-6a ), 4,82 (dd, 1h, J = 11,43 y 6,81 Hz, H-6b ), 4,82 (s, 1H, H-1), 4,66 (dd sin resolver, 1H, J 3,5 y 3,5 Hz, H-3), 4,36 (dd, 1H, J = 10,11 y 3,96 Hz, H-4), 4,32-4,23 (1H, H-5), 4,18 (d, 1H, J = 3,08 Hz, H-2), 3,60-3,44 (2H, CH2 CH3 ), 3,16 (s, 3H, CH3 O) y 1,00 (t, 3H, J = 7,0 Hz, CH2 CH3 ) ppm. Datos de 1 H RMN (D2 O, ref. acetona a 2,35 ppm) δ: 8,22-8,17 (2H), 7,83 (tt, 1H, J = 7,5 y <2 Hz), 7,72-7,65 (2H), 4,97 (HDO), 4,93 (s, 1H, H-1), 4,81 (dd, acoplado virtualmente, 1H, J = 12 y 1,7 Hz, H-6a ), 4,65 (dd, acoplado virtualmente, 1H, J = 12 y 5,5 Hz, H-6b ), 4,18-4,15 (2H, H-4 y H-5), 4,05 (dd, 1H, J = 3,3 y 1,4 Hz, H-2), 3,953,75 (sim. m, 2H, CH2 CH3 ), 3,94-3,91 (m, 1H, H-3), 3,54 (s, 3H, CH3 O) y 1,35 (t, 3H, J = 7 Hz, CH2 CH3 ) ppm (las asignaciones de las señales del espectro 1 H RMN se hicieron sobre la base de un experimento COSY). Datos de 13 C RMN (D2 O, ref.: acetona a 33,19 ppm) δ 171,4, 136,9, 132,5, 132,1, 131,7, 102,5, 78,4, 69,9, 69,1, 67,4, 65,7, 58,1, 56,0 y 17,5 ppm. 3-Azido-6-O-pivaloil-3-desoxi-2-O-etil-4-O-p-metoxibencil-α-D-altopiranósido de metilo 40 45 50 55 Una solución de 3-azido-3-desoxi-2-O-etil-4-O-p-metoxi-bencil-α-D-altro-piranósido de metilo (476 mg, 1,3 mmol) en piridina (8 ml) se agitó a 0ºC. Se añadió cloruro de pivaloílo (390 µl, 3,25 mmol) y la temperatura se aumentó a temperatura ambiente. La agitación se continuó durante 1,5 h y después la mezcla se enfrió a 0ºC. Se añadió metanol (10 ml) y la mezcla se agitó durante 20 min más a temperatura ambiente, se evaporó y se repartió entre acetato de etilo y agua. La fase orgánica se lavó posteriormente con agua, NaHCO3 saturado acuoso, agua y NaCl saturado acuoso, se secó (Na2 SO4 ), se filtró y se evaporó. La cromatografía (SiO2 , 1:4 de metil terc-butil éter-heptano) y (C-18 Lobar, Merck, 4:1 de agua con acetonitrilo) dio el pivaloato (431 mg, 75%). Datos de 1 H RMN (CDCl3 ) δ: 7,28 y 6,89 (patrón AB, acoplado adicionalmente, 4H, J = 8,6 Hz,), 4,60 y 4,49 (patrón AB, 2H, J = 11,0 Hz, ArCH2 ), 4,57 (s, 1H, H-1), 4,39 (dd, 1H, J = 14,5 y 5,3 Hz, H-6a ), 4,57 (s, 1H, H-1), 4,39 (dd, 1H, J = 14,5 y 5,3 Hz, H-6a ), 4,13 (2H, H-6b y H-4), 3,93 (m, 1H, H-3), 3,84-3,78 (4H, H-5 y MeOAr), 3,64-3,45 (3H, CH2 CH3 y H-2) 3,39 (s, 3H, 1-OMe) y 1,22-1,15 (s y t, 12H, J = 7,0 Hz, C(CH3 )3 y CH2 CH3 ) ppm. Datos de 13 C RMN (CDCl3 ) δ: 178,1, 159,6, 129,9, 129,3, 113,9, 99,5 (C-1), 76,7 (C-2), 72,5 (C-5), 71,9 (ArCH2 ), 66,4 (C-4), 66,2 (CH2 CH3 ), 63,6 (C-6), 58,5, 55,3, 55,2, 38,8, 27,2 y 15,4 ppm. Sal sódica de 6-O-pivaloil-3-desoxi-2-O-etil-4-O-p-metoxibencil-3-sulfamino-α-D-altropiranósido de metilo (Cf. H. P. Wessel; J. Carbohydr. Chem. 11 (8), (1992), 1039-1052). 60 65 A una solución agitada de 3-azido-6-O-pivaloil-3-desoxi-2-O-etil-4-O-p-metoxibencil-α-D-altopiranósido de metilo (300 mg, 0,683 mmol) en tetrahidrofurano (20 ml) se le añadieron agua (150 ml) y trifenilfosfina (0,90 g, 3,3 mmol). La solución se agitó durante 24 h y después se concentró hasta 5 ml. El residuo se diluyó con metanol (5 ml) y agua (1 ml). Una mitad de esta solución se usó para preparar el correspondiente oxamato (véase 48,182). A la mitad restante de la solución agitada (0,342 mmol) se le añadió después complejo de trimetilamina-trióxido de azufre (99 mg, 0,719 mmol). El pH se ajustó a 8,5 mediante NaOH acuoso 1 M. Después de unos minutos se formó un precipitado que se disolvió mediante la adición de tetrahidrofurano (0,7 ml) y metanol (1 ml). La mezcla de reacción se agitó durante 0,5 h, después de lo cual se añadió más complejo de trimetilamina-trióxido de azufre (45 mg). La 50 ES 2 225 834 T3 5 agitación se continuó durante 15 min. Durante el transcurso de la reacción el pH se mantuvo a 8-9 mediante la adición de NaOH acuoso 1 M. Después, la mezcla se diluyó con agua (2 ml) y los disolventes orgánicos se evaporaron a presión reducida. Se añadió más agua (5 ml) y la suspensión se liofilizó y se cromatografió (SiO2 , 100:15:1 → 80:20:1 de cloroformo-metanol-agua), dando el compuesto sulfamino, presumiblemente en forma de la sal sódica (161 mg, 80%) después de la liofilización en agua. 10 Datos de 1 H RMN (dmso-D6 ) δ: 7,24 y 6,87 (patrón AB, acoplado adicionalmente, 4H, J = 8,8 Hz), 4,69 y 4,24 (patrón AB, 2H, J = 10,8 Hz, ArCH2 ), 4,58 (s, 1H, H-1), 4,29 (dd, 1H, J = 11,4 y 1,7 Hz, H-6a ), 4,05-3,96 (2H, H-6b y NH), 3,83-3,76 (m, 2H, H-2 y H-3), 3,76-3,73 (4H, s y m, H-5 y MeOAr), 3,54-3,40 (3H, CH2 CH3 y H-4), 3,27 (s, 3H, 1-OMe) y 1,12-1,02 (s y t, 12H, J = 7,0 Hz, C(CH3 )3 y CH2 CH3 ) ppm. Sal amónica de 6-O-pivaloil-3-desoxi-2-O-etil-3-sulfamino-α-D-altropiranósido de metilo (bpy_37) 15 20 25 Se hidrogenó la sal sódica de 6-O-pivaloil-3-desoxi-2-O-etil-4-O-p-metoxibencil-3-sulfamino-α-D-altro-piranósido de metilo (153 mg, 0,259 mmol) durante 6 h en ácido acético glacial (8 ml) a 0,23 MPa y a temperatura ambiente, usando paladio al 5% sobre carbono como catalizador. El filtrado y la liofilización dieron un residuo que se cromatografió (SiO2 , 80:20:1 de cloroformo-metanol-agua con NH3 al 0,1%), dando el compuesto del título (77,2 mg, 74%). Datos de 1 H RMN (D2 O, ref. acetona a 2,35 ppm) δ: 4,83 (HDO), 4,78 (s, 1H, H-1), 4,43 (dd, acoplado virtualmente, H, J = 11,5 y 1,1 Hz, H-6a ), 4,30 (dd, acoplado virtualmente, 1H, J = 11,5 y 5,1 Hz, H-6b ), 3,95-3,91 (2H, H-4 y H-5), 3,89 (dd, 1H, J = 3,3 y 1,3 Hz, H-2), 3,80-3,62 (m, 3H, H-3 y CH2 CH3 ), 3,42 (s, 3H, CH3 O) y 1,24-1,21 (s y t, 12H, J = 7 Hz, t Bu y CH2 CH3 ) ppm (las asignaciones de las señales del espectro 1 H RMN se hicieron sobre la base de un experimento COSY). Datos de 13 C RMN (D2 O, ref.: acetona a 33,19 ppm) δ 188,3, 106,2, 82,2, 73,8, 72,8, 70,6, 69,3, 61,9, 59,8, 45,6 37,1 y 21,3. 6-O-Pivaloil-3-deoxi-2-O-etli-4-O-p-metoxibencil-3-t butiloxamido-α-D-altropiranósido de metilo 30 35 40 45 50 Se enfrió cloruro de oxalilo (120 µl, 1,37 mmol) disuelto en tetrahidrofurano (1 ml) a -26ºC. Se añadió gota a gota una solución de t-butanol (131 mg, 1,77 mmol) y piridina (154 µl, 1,91 mmol) en tetrahidrofurano (2 ml) y la mezcla se agitó a -26ºC durante 15 min, mientras el precipitado amarillento formado inicialmente se volvía blanco. Los disolventes se retiraron a presión reducida de la solución en tetrahidrofurano a partir de la reducción de 3azido-6-O-pivaloil-3-desoxi-2-O-etil-4-O-p-metoxibencil-α-D-altropiranósido (150 mg, 0,342 mmol). El residuo se disolvió de nuevo en tetrahidrofurano (2,5 ml) y piridina (154 µl) y se añadió gota a gota a la solución enfriada anterior y se agitó a -26ºC durante 1 h. Se añadió agua (2 ml) y la mezcla se diluyó con t-butil metil éter (15 ml) y se lavó con agua, carbonato ácido sódico saturado y salmuera. La solución orgánica se secó sobre sulfato sódico y los disolventes se retiraron a presión reducida. El residuo se cromatografió (SiO2 , 1:8 de t-butil metil éter-tolueno), produciendo 187 mg, 100% del compuesto del título. Datos de 1 H RMN (CDCl3 ) δ: 8,25 (d, 1H, 10,1 Hz, 3-NH), 7,26 y 6,85 (patrón AB, acoplado adicionalmente, 4H, J = 8,6 Hz), 4,82-4,71 (m y d, 2H, 10,5 Hz, H-3 y ArCH2 ), 4,69 (s, 1H, H-1), 4,42 (dd, 1H, 1,7 y 11,7 Hz, H-6a ), 4,31 (d, 1H, 10,5 Hz, ArCH2 ), 4,16 (dd, 1H, 6,2 y 11,7 Hz, H-6b ), 3,88 (ddd, 1H, 1,8, 5,9 y 10,3 Hz, H-5), 3,81-3,75 (dd y s, 4H, H-4 y ArOMe), 3,64-3,49 (m, 2H, CH2 CH3 ), 3,47-3,43 (dd y s, 4H, 1,3 y 3,1 Hz, H-2 y 1-OMe), 1,55 (s, 9H, oxamato C(CH3 )3 ), 1,19 y 1,18 (s y t, 12H, 7 Hz, 6-O pivaloato t Bu y CH2 CH3 ) ppm. Datos de 13 C RMN (CDCl3 ) δ: 178,1, 159,4, 159,1, 157,4, 130,3, 129,4, 113,8, 98,9, 84,2, 77,2, 75,9, 70,8, 69,5, 65,8, 65,7, 63,4, 55,2, 55,1, 45,7, 38,8, 27,7, 27,1, 15,3 ppm. Sal amónica de 6-O-pivaloil-3-desoxi-2-O-etil-3-oxamido-α-D-altropiranósido de metilo (bpy_54) 55 60 65 Se disolvió 6-O-pivaloil-3-desoxi-2-O-etil-4-O-p-metoxibencil3-t butiloxamido-α-D-altropiranósido de metilo (180 mg, 332 µmol) en una mezcla de ácido acético-etanol (1:1, 6 ml), se le añadió paladio sobre carbono (5%, 250 mg) y se sometió a hidrogenolisis a 268,895 kPa (39 psi) durante una noche. El catalizador se filtró sobre una capa de celite y se lavó con etanol. El filtrado se concentró a presión reducida y se redisolvió en diclorometano (2,4 ml). Se añadió ácido trifluoroacético (960 µl) y la mezcla se agitó durante 4,5 h a temperatura ambiente. Se añadieron agua (3 ml) y amoniaco (3 M, 300 µl) y la mezcla de reacción se concentró a presión reducida y se liofilizó. El residuo se cromatografió (SiO2 , 80:20:1 de cloroformo-metanol-agua con NH3 al 0,1%), dando el compuesto del título, 117,1 mg, 89%. Datos de 1 H RMN (D2 O, ref. acetona a 2,35 ppm) δ: 4,80 (HDO), j 4,78 (s, 1H, H-1), 4,45-4,35 (m, 2H, H-3 y H6a ), 4,26 (dd, 1H, 4,4 y 12,1 Hz, H-6b ), 4,07-3,96 (m, 2H, H-4 y H-5), 3,77-3,58 (m, 3H, CH2 CH3 y H-2), 3,45 (s, 3H, 1-OMe), 1,20-1,13 (s y t, 12H, 6-O-pivaloato t Bu y CH2 CH3 ) ppm. Datos de 13 C RMN (D2 O, ref.: acetona a 33,19 ppm) δ: 188,1, 171,9, 171,4, 105,7, 82,4, 73,7, 72,9, 70,3, 69,7, 62,0, 56,1, 45,9, 37,0, 21,2 ppm. 51 ES 2 225 834 T3 3-Azido-6-O-pirrol-3’-ilcarboxil-3-deoxi-2-O-etil-4-O-p-metoxibencil-α-D-altropiranósido de metilo 5 Una solución de 3-azido-3-desoxi-2-O-etil-4-O-p-metoxi-bencil-α-D-altro-piranósido de metilo (476 mg, 1,3 mmol) en piridina (496 µl) y diclorometano (2 ml) se agitó a 0ºC. Se añadió cloruro de metanosulfonilo (380 µl, 4,90 mmol) y la temperatura se aumentó a temperatura ambiente. La agitación se continuó durante 5 h y después la mezcla se enfrió a 0ºC. Se añadió agua (1 ml) y la mezcla se agitó durante 20 min más a temperatura ambiente, se evaporó y se repartió entre acetato de etilo y agua. La fase orgánica se lavó posteriormente con agua, NaHCO3 saturado acuoso, agua y NaCl saturado acuoso, se secó (Na2 SO4 ), se filtró y se evaporó. La cromatografía (SiO2 , 1:9 de metil terc-butil éter-tolueno) produjo 216 mg (79%) del mesilato en forma de un aceite amarillento. 10 Se disolvieron ácido N-triisopropilsilil-pirrolo-3-carboxílico (484 mg, 1,81 mmol) y 1,8-diazabiciclo[5,4,0]undec7-eno (255 µl) en dimetilformamida (1 ml) y se añadieron a una solución del mesilato anterior en dimetilformamida (1 ml). La mezcla se calentó a 90ºC durante una noche y se sometió a cromatografía (Lober C-8 de 210 g, Merck, 3:2 de acetonitrilo:agua), produciendo 30 mg, 16% del éster de pirroloílo. 15 20 Datos de 1 H RMN (CDCl3 ) δ: 8,73 (s a, 1H, pirrol NH), 7,39 (m. sim, 1H, pirrol H-4), 7,27 y 6,85 (patrón AB, acoplado adicionalmente, 4H, J = 8,8 Hz), 3,74 (m. sim, 1H, pirrol H-2), 3,64 (m. sim, 1H, pirrol H-5), 4,63-4,49 (s y 2 d, 3H, H-1 y ArCH2 ), 4,48-4,36 (m, 2H, 2 H-6), 4,22 (m. sim, 1H, H-5), 3,94-3,84 (m, 2H, H-3 y H-4), 3,78 (s, 3H, ArOMe), 3,68-3,55 (m, 3H, CH2 CH3 y H-2), 3,40 (s, 3H, 1-OMe), 1,20 (t, 3H, 6,8 Hz, CH2 CH3 ) ppm. Datos de 13 C RMN (CDCl3 ) δ: 164,6, 159,5, 129,9, 129,4, 123,6, 11,7, 116,1, 113,9, 109,9, 99,9, 76,9, 72,5, 72,0, 66,8, 66,3, 63,0, 58,9, 55,3, 55,2, 15,4 pm. Sal amónica de 6-O-pirrol-3’-ilcarboxil-3-desoxi-2-O-etil-3-sulfamino-α-D-altropiranósido de metilo (bpy_40) 25 30 35 40 45 50 55 A una solución agitada de 3-azido-6-O-pivaloil-3-desoxi-2-O-etil-4-O-p-metoxibencil-α-D-altropiranósido de metilo (30 mg, 61 µmol) en tetrahidrofurano (1 ml) se le añadieron agua (11 µl) y trifenilfosfina (83 mg, 305 µmol). La solución se agitó durante 24 h y se diluyó con agua (1 ml). Después, se añadió complejo de trimetilamina-trióxido de azufre (9 mg, 0,72 µmol). El pH se ajustó a 8,5 mediante NaOH acuoso 1 M. Después de unos minutos, se formó un precipitado que se disolvió mediante la adición de tetrahidrofurano (0,7 ml) y metanol (1 ml). La mezcla de reacción se agitó durante 0,5 h, después de lo cual se añadió más complejo de trimetilamina-trióxido de azufre (9 mg). La agitación se continuó durante 15 min. Durante el transcurso de la reacción, el pH se mantuvo a 8-9 mediante la adición de NaOH acuoso 1 M. Después, la mezcla se diluyó con agua (2 ml), se lavó con t-butil metil éter, se liofilizó y se cromatografió (SiO2 , 80:20:1 de cloroformo-metanol-agua), dando el compuesto sulfamino, presumiblemente en forma de sal sódica, (30,6 g, 93%) después de la liofilización en agua. Datos de 1 H RMN (CD3 OD) δ: 7,35 (dd sin resolver, x parte del patrón ABX, 1H, JAX +JBX = 3,5 Hz, pirrol H-2), 7,29 y 6,79 (patrón AB, acoplado adicionalmente, 4H, J = 8,6 Hz), 6,75 (dd, 1H, 2,0 y 2,9 Hz, pirrol H-4), 6,51 (dd, 1H, 1,3 y 2,9 Hz, pirrol H-5), 4,71 y 4,46 (patrón AB, 2H, J = 11,4 Hz, ArCH2 ), 4,66 (s a, 1H, H-1), 4,39 y 4,33 (2 dd, 2H, 2,6, 5,1 y 11,7 Hz, 2 H-6), 4,04 (m, 1H, H-3), 3,97-3,87 (m y dd, 2H; 1,1 y 3,1 Hz, H-5 y H-2), 3,80 (dd, 1H, 4,4 y 10,1 Hz, H-4), 3,73 (s, 3H, ArOMe), 3,68-3,52 (m, 2H, CH2 CH3 ), 3,39 (s, 3H, 1-OMe), 1,15 (t, 3H, 7,0 Hz, CH2 CH3 ) ppm. Datos de 13 C RMN (CD3 OD) δ: 167,32, 160,9, 131,3, 131,0, 125,3, 120,1, 116,3, 114,7, 110,2, 101,7, 77,2, 10,4, 69,9, 67,2, 66,4, 55,6, 55,5, 50,9 y 15,8 ppm. El residuo se disolvió en metanol (15 ml), se le añadió paladio sobre carbono (5%, 50 mg) y la mezcla se hidrogenó a 206,842 (30 psi) durante 3 h. El catalizador se retiró por filtración, se redisolvió en amoniaco (2%) en metanol-agua (1:1, 0,6 ml) y se cromatografió (Supelco C-18 de 3 ml, 2:3 de metanol-agua). El metanol se retiró a presión reducida de las fracciones reunidas que contenían el producto y el residuo se liofilizó, dando 6,2 g del compuesto del título en forma de sal amónica. Datos de 1 H RMN (CD3 OD) δ: 7,35 (m, 1H, pirrol H-2), 6,75 (dd, 1H, 2,0 y 2,9 Hz, pirrol H-4), 6,55 (dd, 1H, 1,3 y 11,4 Hz, H-6a ), 4,31 (dd, 1H, 5,9 y 11,6 Hz, H-6b ), 3,86-3,76 (m, 4H, H-2, H-3, H-4, H-5), 3,75-3,56 (m, 2H, CH2 CH3 ), 3,40 (s, 3H, 1-OMe), 1,21 (t, 3H, Hz, CH2 CH3 ) ppm. Ejemplo 6 Ensayos de chaperona y el uso de los mismos para ensayar compuestos para la inhibición de la unión de chaperona 60 65 Para identificar de forma concluyente péptidos o miméticos de péptidos que se unan fuertemente a PapD, se requiere un buen ensayo. Los inventores han obtenido por ingeniería genética la proteína de unión a maltosa (MBP) de forma que se reconoce por PapD. Se usó el plásmido disponible en el mercado pMAL-P2 que codifica MBP con un brazo de engarce que codifica una secuencia fusionada al extremo 3’ del gel. MBP se secreta en el espacio periplásmico y se purifica fácilmente por cromatografía de afinidad usando una resina de amilosa. MBP se eluye de la columna con maltosa 20 mM. PapD no se co-eluye con MBP cuando se co-expresa con MBP, mientras que la fusión del extremo COOH de PapD a MBP da como resultado la unión de PapD a MBP para formar un complejo que se co-eluye a partir de la columna de afinidad. Usando esta estrategia, se ha descubierto que la fusión de los aminoácidos carboxi 52 ES 2 225 834 T3 5 10 15 20 25 30 35 40 45 50 55 60 65 terminales 140 (MBP-G1’-140’) y 134 (MBP-G134’) de PapD a MBP se obtiene la formación de un complejo PapDMBP fuerte que se purifica por cromatografía de afinidad de maltosa. Se ha demostrado que el complejo entre la fusión de MBP y PapD es estable en urea hasta 3 m similar a la estabilidad del complejo PapD- PapG, lo que indica que los aminoácidos COOH-terminales 134 de PapG probablemente tienen la mayor parte del motivo de reconocimiento de chaperona. Además, la unión a la proteína de fusión MBP ha resultado estar dependiente de los residuos de anclaje de chaperona universales, Arg-8 y Lys-112, como se describe en este documento, ya que la mutaciones en estos dos residuos anulaban la unión de PapD a la proteína de fusión MBP. También es interesante ensayar la capacidad de fusiones MBP-G para inhibir el ensamblaje del pilus y de esta manera la unión de bacterias por co-expresión de las proteínas de fusión en células productoras de pili P. Si PapD se une a la fusión MBP-G, es titulará a partir de subunidades de pilus y de esta manera reducirá o anulará la formación del pilus. Esta técnica será un mecanismo para validar el concepto de que los péptidos de unión a chaperona pueden anular la virulencia de un patógeno impidiendo el ensamblaje de las adhesinas localizadas en la superficie. Usando este análisis, se ha descubierto que en cepas productoras de pili P, la co-expresión de la fusión MBP-G1’-140’ y MBPG134’ anula completamente la capacidad de las bacterias para unirse a los glóbulos rojos y producir hemaglutinación. Además, los inventores han demostrado recientemente que después de la co-expresión de la fusión MBP-G1’-140’ en bacterias HB101/pPAP5, no fue posible detectar ningún signo visual de pili sobre la superficie de las bacterias por microscopía electrónica. Esto es una evidencia de la teoría expresada en esta invención, de que la prevención/inhibición de la unión entre las subunidades del pilus y las chaperonas moleculares también previene el ensamblaje de los pili intactos. Estos resultados confirman la relevancia biológica de la estructura cristalina descrita en la Fig. 3, que demuestra que pueden obtenerse por ingeniería genética otras proteínas para ser reconocidas por PapD por fusión del sitio de reconocimiento carboxi terminal sobre esa proteína. Se han purificado las fusiones MBP-G134’ y MBP-G1’-140’ para establecer ensayos in vitro para medir interacciones con PapD. Las proteínas de fusión purificadas se aplican como un recubrimiento sobre pocillos de placas de microtitulación y la capacidad de PapD de unirse a las proteínas de fusión se ensaya en un procedimiento ELISA. Este ensayo es crítico ya que permite ensayar la capacidad de los compuestos para inhibir la unión de PapD al dominio en PapG reconocido por la chaperona. Además, las proteínas de fusión purificadas pueden usarse en el ensayo Pharmacia BiaCore® para cuantificar la unión a PapD y establecer ensayos de inhibición para compuestos de la invención. Ya se ha sugerido que ciertos péptidos carboxi terminales corresponden a parte del motivo de reconocimiento de la chaperona. De esta manera, se puede investigar si los péptidos carboxi terminales pueden inhibir la unión de PapD a la fusión MBP-G1’-140’ en el ensayo ELISA. El desarrollo de este ensayo de alto rendimiento hace que sea posible seleccionar bibliotecas peptídicas, bibliotecas químicas, compuestos nativos y miméticos de péptidos con respecto a su capacidad de inhibir la unión de chaperona. Además, este ensayo puede usarse para ensayar si las chaperonas periplásmicas conocidas utilizan paradigmas de reconocimiento comunes. Además de ensayar los compuestos con respecto a su capacidad de inhibir la unión a la fusión MBP-G, se han establecido otros ensayos que miden las interacciones PapD-péptido. Por ejemplo, se ha creado un ELISA para medir la unión de PapD a péptidos aplicados como recubrimientos sobre pocillos de placas de microtitulación. En este ensayo, el péptido octamérico G1’-8’WT fue un inhibidor igualmente eficaz que el 19-mero G1’-19’WT, revelando que el octámero era un punto de partida óptimo para el diseño de compuestos modificados con una mayor afinidad para el sitio de unión de PapD (véase el Ejemplo 7). Además, como se ha descrito previamente, el PapD nativo puede unirse a PapG desnaturalizado reducido y restaurar el complejo de PapD-PapG in vitro en un ensayo de reconstitución (Kuehn y col., 1991). Este ensayo se ha propuesto para reflejar la fusión de reconocimiento de PapD in vivo y se usará para determinar la capacidad de los compuestos para inhibir la unión de PapD a PapG in vitro. Por ejemplo, se ha demostrado que el péptido PapG carboxi terminal inhibe la unión de PapD a PapG en este ensayo, estableciendo que ocupa el sitio de unión de la seguridad de pilus de PapD. Otra forma de localizar las interacciones PapD-péptido es ensayar la capacidad de los compuestos para reducir la proporción de acontecimientos de escisión proteolítica bien definidos. Por ejemplo, la digestión parcial con tripsina escinde PapD en el bucle de P1-G1 en el residuo Lys-99 (véase el sitio “T” indicado en la Fig. 3). La velocidad de escisión tríptica de PapD se redujo por preincubación de PapD con péptidos PapG y PapK (véase el ejemplo 2). La protección observada de PapD por lo péptidos unidos puede deberse a un cambio en la conformación local del bucle F1-G1, o debido al contacto físico del bucle por el péptido. De esta manera, estos ensayos pueden usarse como investigación inicial de la capacidad de nuevos compuestos para unirse a PapD e interferir con su función de reconocimiento. Los compuestos de unión fuerte que inhiben la unión de PapD a la fusión MBP-G1’-140’ se co-cristalizaran con PapD para proporcionar la base estructural de la superficie de reconocimiento usada por PapD. Como se dispone de nuevos datos cristalográficos, la relevancia de las interacciones críticas PapD-inhibidor PapD-potenciador se ensayará determinando el efecto de mutaciones de localización dirigida sobre la capacidad de la chaperona para unirse a subunidades de pilus y mediar el ensamblaje del pilus. Esta importante información conducirá a definir cómo las chaperonas reconocen la subunidades y la función de las interacciones chaperona-subunidad. También se ha desarrollado un ensayo cuantitativo de unión de chaperona: se ha sintetizado un péptido PapG modificado, donde la Ser-9’ está sustituida con una Cys. A partir de la estructura cristalina del PapD unido al péptido, no se predijo que la cadena lateral de Ser-9’ interaccionaría con PapD. En su lugar, esta cadena lateral se orientaba hacia el disolvente, pero la Ser-9’ esta adyacente al último aminoácido de la cremallera interactiva entre PapD y el péptido. Después, la sonda fluorescente sensible ambientalmente 5-IAF (5-yodoacetamidofluoresceína) se acopló covalentemente al péptido a través del grupo sulfhidrilo en el residuo Cys y el péptido marcado se purificó; por supuesto, 53 ES 2 225 834 T3 5 10 15 20 25 puede emplearse cualquier otra sonda fluorescente ambientalmente sensible adecuada. Se descubrió que la adición de PapD al péptido produce una notable reducción en la intensidad de la fluorescencia y un cambio en el máximo de emisión. Esta información se usó para calcular una constante de unión para la interacción PapD-péptido. Se determinó el cambio en la intensidad de fluorescencia a 514 nm después de la adición de concentraciones recientes de PapD. Representando las intensidades de fluorescencia frente a las concentraciones de PapD, se calculó una constante de unión de 2,5 x 10−6 M (Los cálculos se realizaron en el programa informático disponible en el mercado “Kaleidagraph”). De esta manera, es posible evaluar las constantes de unión de otras sustancias que se unen a PapD de una manera cuantitativa, ya que la adición de una sustancia que se une competitivamente a PapD dará como resultado una mayor fluorescencia en comparación con una situación en la que está presente menos sustancia competitiva, o no está presente nada de dicha sustancia en el sistema. Se desarrollará un sistema de ensayo similar usando MBP-G1’-140’ en lugar del péptido PapD como se ha descrito en este documento. Como se ha descubierto que esta proteína de fusión interacciona con dos sitios de unión en PapD, será posible 1) cuantificar la afinidad de unión con el segundo sitio de unión (en el dominio 2) y 2) seleccionar compuestos que interaccionen con cualquiera de los dos sitios de unión y cuantificar la interacción. Por supuesto, el marcaje del péptido del sitio 2 con una sonda ambientalmente sensible permitiría la determinación de la constante de unión usando la misma metodología que se ha descrito anteriormente. El descubrimiento de que la proteasa DegP es responsable en gran medida de la degradación de las subunidades de pilina en ausencia de una chaperona hace que la cepa degP41 sea un candidato interesante para un sistema de ensayo in vivo de los efectos ejercidos sobre PapD por compuestos de la invención. Cuando se administra una sustancia que previene, inhibe o mejora la unión entre la chaperona y la subunidades de pilus a un sistema que contiene la cepa de degP41, la sustancia, incluso en pequeñas cantidades, debería ser tóxica para la bacteria, ya que la bacteria DegP− será incapaz de degradar las subunidades de pilus que se acumulan. Debe indicarse que se usó una cepa degP41 para identificar un sitio de unión desconocido hasta ahora en el dominio 2 de PapD, véase el ejemplo 10. 35 Sin embargo, tal ensayo requiere que la sustancia ensayada pueda entrar en el espacio periplásmico o antes de que pueda ejercer su efecto; este hecho hace que este tipo de ensayo sea menos adecuado como ensayo de selección para compuestos de plomo, ya que “ignorará” sustancias que tienen los efectos deseados sobre la interacción chaperonasubunidad, pero que, por ejemplo, son demasiado hidrófilos para entrar en el espacio periplásmico. Por otra parte, el sistema será adecuado para evaluar el potencial clínico de sustancias que ya han resultado satisfactorias en los ensayos in vitro descritos en este documento. 40 Una vez que está en su sitio el sistema modelo PapD, por supuesto, los experimentos descritos anteriormente pueden expandirse para incluir los otros miembros de la familia de tipo PapD de las chaperonas. De esta forma, será posible establecer los requisitos generales para el reconocimiento de chaperona y la base molecular del paradigma de reconocimiento de chaperona en bacterias gram negativas. 30 En el Ejemplo 10 se describen con detalle algunos ensayos específicos para determinar las interacciones con chaperonas moleculares. 45 Ejemplo 7 Diseño de péptidos y miméticos de péptidos capaces de unirse al sitio de unión de PapD y otras chaperonas de ensamblaje de pilus 50 55 60 Para proporcionar más información sobre las interacciones chaperona-subunidad, se contempla la síntesis de péptidos solapantes con la proteína PapG entera y otras subunidades de pilus. Además, debe realizarse un sondeo de péptidos que se unen a chaperonas disponibles usando tanto una biblioteca de péptidos químicos como una biblioteca de presentación de fagos. Las dos bibliotecas son complementarias ya que los péptidos se presentan en diferentes medios que podrían influir de manera importante sobre la unión a las chaperonas. En la práctica, la biblioteca química está limitada a todos los hexapéptidos posibles y a hepta- y octapéptidos si algunos de los aminoácidos nativos se excluyen o si uno o dos residuos se mantienen constantes. Esta biblioteca puede evaluarse por unión directa de la chaperona a las perlas de resina y secuenciación de los péptidos interesantes. La biblioteca de presentación de fagos contiene aproximadamente 1010 péptidos en la proteína PIII del denominado “fago de fusión” que retiene la función del fago y presenta los péptidos extraños en la superficie. La capacidad de los fagos que contienen péptidos de unirse a PapD aplicado como un recubrimiento en pocillos de placas de microtitulación puede detectarse en un ELISA usando las técnicas descritas en el Ejemplo 6. Los fagos de unión positiva se amplificarán, purificarán y se volverán a ensayar con respecto a su capacidad de unirse a PapD. Se determinará la secuencia del inserto de péptido de fagos de unión positiva y los correspondientes péptidos se sintetizarán y se ensayarán con respecto a su capacidad para inhibir la unión de PapD a la fusión MBP-G como se describe en el Ejemplo 6. 65 Los resultados de los estudios indicados anteriormente serán de máxima importancia para el diseño y evaluación de ligandos que actúan como “inhibidores de chaperona”. A pesar de esto, la estructura cristalina del complejo peptídico 19-mero PapD- PapG proporciona una perspectiva suficiente en las interacciones de chaperona-ligando para iniciar los 54 ES 2 225 834 T3 estudios sistemáticos de pequeños péptidos y miméticos de péptidos como inhibidores de chaperona que ya están en el presente estado. El alto poder inhibidor del péptido 8-mero PapG a PapD (igual péptido 19-mero PapG, cf. Ejemplo 6) revela la viabilidad de tales estudios y las características conservadas del sitio de unión propuesto en las chaperonas indica que tales inhibidores podrían tener una amplia especificidad. 5 10 Se ha sugerido que la especificidad en el reconocimiento del 19-mero PapG por PapD se proporciona por el anclaje del extremo carboxilo del péptido a los residuos Arg-8 y Lys-112 de la hendidura de PapD y las posteriores interacciones “de cremallera” entre residuos hidrófobos alternos en el péptido y residuos hidrófobos complementarios en PapD. Los siguientes experimentos confirman esta hipótesis. En primer lugar, las mutaciones en los residuos de anclaje Arg-8 y Lys-112 han anulado la unión de la subunidad in vivo. En segundo lugar, la deleción del residuo Cterminal en el 19-mero PapG conduce a una reducción sustancial de la unión a PapD ya que las interacciones “de cremallera” hidrófobas entre el péptido y PapD después se pusieron fuera de registro cuando el extremo C-terminal del péptido de deleción se ancló a Arg-8 y Lys-112. La hipótesis “de cremallera” para la especificidad en la unión entre PapD y péptidos (o subunidades de pilus) se ha investigado preliminarmente realizando los siguientes experimentos: 15 20 25 30 35 Se preparó una serie de longitudes de péptidos que forman el extremo C de PapG que consta de G1’-5’, G1’-6’, G1’-7’, G1’-8’, G1’-11’, G1’-16’ y G1’-19’. También se sintetizó una serie de reemplazo y una serie de deleción de G1’-8’. En la serie de reemplazo se reemplazaron Pro-1’, Phe-2’, Leu-4’, Val-5’, Met-6’ y Met-8’ por Ser, mientras que Ser-3’ y Thr-7’ se reemplazaron por Ala (es decir los aminoácidos hidrófobos se reemplazaron por la Ser polar y los aminoácidos hidrófilos se reemplazaron por Ala). En la serie de deleción, un residuo en un momento en G1’8’ se delecionó con la adición simultanea de serina (se encuentra en la posición 9’ en PapG nativo) para mantener la longitud del péptido a 8 aminoácidos. Todos los péptidos se sintetizaron por la estrategia en fase sólida Fmoc y se purificaron por HPLC de fase inversa. Después se investigó la capacidad de los péptidos para inhibir la unión de PapD a la proteína de fusión MBP-G1’140’ usando el siguiente ensayo ELISA. Se diluyeron soluciones madre de proteínas MBP-G1’-140’ en PBS a 0,1 µM con PBS. Los pocillos de placas de microtitulación se recubrieron con 50 µl de las soluciones de proteína durante una noche a 4ºC. Los pocillos se lavaron con PBS y se bloquearon con 200 µl de albúmina de suero bovino (BSA) al 3% en PBS durante 2 horas a 25ºC. Las placas se lavaron vigorosamente tres veces con PBS y se incubaron con 50 µl de proteínas PapD 1-5 µM en BSA-PBS al 3% durante 45 minutos a 45ºC. El PapD se preincubó con cada uno de los péptidos a una relación 1:25 durante 30 minutos antes de añadirse a los pocillos. Después de tres lavados con PBS, los pocillos se incubaron con una dilución 1:500 de antisuero anti- PapD de conejo en BSA-PBS al 3% durante 45 minutos a 25ºC. Después de tres lavados con PBS, los pocillos se incubaron con una dilución 1:1000 de antisuero de cabra contra IgG de conejo acoplado a fosfatasa alcalina en BSA-PBS al 3% durante 45 minutos a 25ºC. Después de 3 lavados con PBS y 3 lavados con el tampón revelado (dietanolamina 10 mM, MgCl2 0,5 mM), se añadieron 500 µl de p-nitrofenil fosfato a 1 mg/ml filtrado en tampón de revelado, la reacción se incubó durante 1 hora o más si era necesario en la oscuridad a 25ºC y se leyó la absorbancia de 405 nm. 40 45 50 55 60 65 En las Figs. 17-19 se presentan las potencias inhibidoras de los péptidos en las tres series y en las figuras se proporciona el número de experimentos realizados con cada serie. Las líneas verticales para cada péptido en las figuras son intervalos de confianza del 95% obtenidos después de un análisis estadístico de los datos experimentales. Como se revela por la evaluación de la serie de longitud, los péptidos G1’-8’, G1’-11’ y G1’-16’ son significativamente más potentes que los péptidos G1’-6’ y G1’-7’ más cortos (Fig. 17). Esta observación se ajusta muy bien con la estructura cristalina de PapD complejado con G1’-19’ que demuestra que los 8 residuos C-terminales en G1’-19’ presentan unión de hidrógeno a PapD. Los péptidos G1’-6’ y G1’-7’ más cortos no pueden satisfacer este modelo de unión de hidrógeno y, por lo tanto, son péptidos inhibidores menos activos (Fig. 18). La serie de reemplazo revela que los residuos 4’, 5’ y 6’ en G1’-8’ forman contactos importantes con PapD, ya que su reemplazo produce péptidos inhibidores menos activos (Fig. 18). La serie de deleción de nuevo indicó un papel importante para los residuos 4’, 5’ y 6’ en G1’-8’ para la formación del complejo con PapD (Fig. 19). Sin embargo, los resultados obtenidos con la serie de deleción no confirmaron la hipótesis “de cremallera” según la cual los miembros de la serie de deleción mostrarían una potencia inhibidora creciente ya que la deleción se mueve desde el extremo C hacia el extremo N de G1’-8’. Como parece a partir de estos resultados preliminares, los resultados obtenidos en el ensayo presentan grandes desviaciones de la media en cada experimento. Como se analiza en este documento, una razón de esto podría ser la lenta cinética de unión entre PapD y proteínas de subunidad de pilus plegadas correctamente y análogos de tales proteínas de subunidad plegadas correctamente. Por lo tanto, se contempla modificar el ensayo introduciendo influencias desnaturalizantes suficientemente poderosas como para desplegar al menos parcialmente la subunidad de pilus (análogos). Es de esperar que esto reduzca las desviaciones en los resultados de ensayo. Otra razón de las grandes desviaciones podría ser la unión de los péptidos a BSA usados en el ELISA. Por lo tanto, se investigará el reemplazo de BSA con otras macromoléculas. En el caso de que pueda confirmarse el “mecanismo de cremallera” para la unión de péptidos a PapD, la unión del péptido chaperona puede optimizarse usando una biblioteca de péptidos sintéticos limitada en la que Pro-1’, Phe2’, Leu-4’, Met-6’ y Met-8’ del 8-mero PapG se reemplazan por los aminoácidos hidrófobos Val, Leu, Ile, Met, Phe, Trp, Tyr e His. También se incorporarán en la biblioteca química aminoácidos no nativos tales como D-aminoácidos y aminoácidos N-metilados, así como aminoácidos que contienen cadenas laterales alifáticas y diversas cadenas la55 ES 2 225 834 T3 terales aromáticas. En la síntesis de los inhibidores de chaperona descritos más adelante en este ejemplo se usaran combinaciones de aminoácidos óptimas para estas cinco posiciones. Si el “mecanismo de cremallera” no se confirma, las estrategias indicadas se aplicarán a residuos que se han demostrado que son importantes para la unión a PapD. 5 10 Se crearán inhibidores de chaperona que forman enlaces covalentes con la chaperona después del acoplamiento en su sitio activo. La estructura cristalina del complejo PapD-péptido demuestra que el grupo carboxilo C-terminal del péptido forma enlaces de hidrógeno con Lys-112 y Arg-8 en la chaperona y que la cadena lateral de Val-5’ en el péptido está próxima al grupo amino de la cadena lateral de otra Lys en PapD. La introducción de grupos reactivos tales como haluros de alquilo, aldehídos, haluros de ácido y ésteres activos en estas posiciones del péptido 8-mero optimizado conducirá a la formación de enlaces covalentes con las lisinas en PapD, y los derivados de péptidos constituyen, de esta manera, inhibidores de alta afinidad. Los inhibidores se basan en péptidos nonaméricos o más cortos en los que el residuo 5 (desde el extremo C) y/o el grupo carboxilo C-terminal se han modificado como se indica: Reemplazos para el residuo 5 del extremo C-terminal 15 20 25 30 35 40 45 50 Reemplazos para el grupo COOH C-terminal -COX, X como se ha indicado anteriormente 55 -CH2 Y, Y como se ha indicado anteriormente Tales interacciones entre aldehídos y cadenas laterales de lisina han proporcionado potentes candidatos de fármaco para la anemia de células facilformes. 60 65 En el cristal del péptido PaPD, el péptido forma una extensión de un lámina β en la chaperona. Las restricciones que proporcionan al péptido un conformación de tipo lámina β por lo tanto darán como resultado un cambio favorable en la entropía de unión. Se prepararán péptidos conformacionalmente restringidos que constituyen láminas βcíclicas miniatura o péptidos que tienen las cadenas laterales de aminoácidos separadas por un aminoácido unido covalentemente a otro. Los péptidos con cadenas laterales unidas covalentemente tendrán los tres aminoácidos consecutivos reemplazados por los fragmentos mostrados a continuación. 56 ES 2 225 834 T3 5 10 15 20 AA = cualquier aminoácido nativo o no nativo Ra , Rb = todas las combinaciones posibles de H, Me, Et, C3 H7 , C4 H9 , C6 H5 25 30 35 40 AA = cualquier aminoácido nativo o no nativo Ra , Rb = todas las combinaciones posibles de H, Me, Et, C3 H7 , C4 H9 , C6 H5 45 50 Los inhibidores interesantes se co-cristalizarán con chaperonas y los complejos también se investigarán por espectroscopía de RMN. Los péptidos y miméticos de péptidos usados como fármacos pueden metabolizarse rápidamente por enzimas proteolíticas que escinden los enlaces peptídicos. La quimotripsina escinde selectivamente el enlace peptídico en el lado carboxilo de aminoácidos con cadenas laterales aromáticas e hidrófobas largas. En 8-mero PapG, los enlaces amida de met-8’, Thr-7’, Met-6’, Val-5’ y Phe-2’ Pro-1’ por lo tanto, son especialmente sensibles a la proteolisis y deben reemplazarse por isósteros peptídicos metabólicamente estables. A continuación se proporcionan ejemplos de tales isósteros peptídicos: 55 Reemplazos para el fragmento : 60 65 57 ES 2 225 834 T3 5 10 15 20 25 -CH2 CH2 Ejemplo 8 30 El papel de chaperonas periplásmicas en la infectividad de bacterias que se adhieren por medio de pili 35 40 El papel específico del ensamblaje del pilus ayudado por chaperona en la virulencia puede determinarse comparando la adherencia y patogenia del tipo silvestre con pili y mutantes isogénicos que no presentan pili debido a mutaciones de localización dirigidas en el sitio activo de la chaperona. En el cromosoma bacteriano de la cepa de aislado clínico DS17 pueden recombinarse mutaciones en residuos tales como Arg-8 y Lys-112 que constituyen el sitio de unión a la subunidad de papG usando el mismo procedimiento que ha sido satisfactorio para introducir mutaciones puntuales en el gen papG en el cromosoma de la misma cepa. La cepa DS17 de E. coli se propaga epidérmicamente dentro de una sala de neonatos, produciendo varios casos de pielonefritis. Además, se ha descubierto que la cepa produce infecciones renales agudas en un modelo de pielonefritis de mono cynomolgus. DS17 contiene un agrupamiento de gen papG con un gen papG que expresa una adhesina papG típica. También expresa pili típicos de tipo 1 que contienen la adhesina de unión a manosa, FimH. Se ha sugerido que los pili de tipo I son determinantes de la virulencia importantes en la cistitis. Por lo tanto, para ensayar este concepto también se generarían mutantes de localización dirigida en el sitio de unión a la subunidad de la chaperona FimC. 45 50 55 60 65 Se contempla que un compuesto que se une al sitio activo de la chaperona bloquearía el ensamblaje del pilus y de esta forma prevendría la unión. Si este concepto es válido, los mutantes de chaperona bien definidos tales como en Arg-8 y Lys-112 anularían también la unión al receptor. La actividad de unión al receptor de diversos mutantes de chaperona debe medirse en los ensayos ELISA de unión a receptores descritos en los ejemplos 2 y 6. Para medir la unión mediada por pilus de tipo 1, se unirá manosa a los pocillos de la placas de microtitulación. La adherencia de diversos mutantes de chaperona FimC y PapD a los receptores inmovilizados se cuantificará usando anticuerpos contra E. coli DS17 en un experimento ELISA. Además, se ensayará la capacidad de pequeños péptidos de unión a PapD para bloquear el ensamblaje del pilus en DS17 y, de esta manera, para anular la unión del receptor. La cepa DS17 crecerá en presencia de péptidos cortos que se unen o que no se unen a PapD y después se ensayará con respecto a su capacidad para unirse al receptor en el experimento ELISA. Estos experimentos realizarán pasos importantes en la validación del concepto de que los inhibidores anti-chaperona prevendrían la unión bacteriana in vitro. Entonces, se establecerá el papel de las adhesinas ensambladas a chaperona en la aparición de enfermedades. La causalidad entre E. coli que expresa pili P y pielonefritis hasta hace poco se ha basado únicamente en datos epidemiológicos. Se ha demostrado que la cepa DS17 con pili P produce pielonefritis en el tracto urinario normal de monos cynomolgus. Se generó una mutación en el gen PapG de la cepa DS17 por reemplazo alélico para crear la cepa derivada DS17-8 para ensayar el requisito de la adhesina PapG en la producción de la enfermedad. DS17-8 contiene una deleción de un par de bases después del codón 37 en PapG dando como resultado la expresión de pili P que carecen de la adhesina terminal PapG. Este mutante no podía unirse al receptor de globósido in vitro aunque no podía unirse al tejido renal de seres humanos o monos cynomolgus en el modelo de unión in situ de los presentes solicitantes. Se comparó la virulencia entre DS17 y su mutante de PapG DS17-8 en el mono cynomolgus. Para estudiar el papel de la adhesina PapG en la pielonefritis, se infectaron cinco monos con la cepa DS17 de E. coli mientras que 58 ES 2 225 834 T3 5 10 15 seis monos recibieron la cepa mutante DS17-8 a través de un catéter uretral insertado citoscópicamente y se demostró que había una diferencia significativa entre los dos grupos. Los monos que recibieron la cepa DS17 de tipo silvestre tuvieron una bacteruria media de 21 días en comparación con los 6,8 días de los monos que recibieron la cepa mutante DS17-8. El aclaramiento renal también fue significativamente menor para la cepa de tipo silvestre y la función renal se redujo significativamente los monos que recibieron la cepa de tipo silvestre, pero no en los monos que recibieron la cepa mutante de PapG isogénica. La evaluación patológica del mono infectado confirmó los estudios funcionales que demostraban una inflamación de cambios patológicos significativamente menores en el grupo mutante en comparación con los monos que recibieron el tipo silvestre. Por lo tanto, se concluye que la adhesina PapG de unión a Galα1-4Gal en extremo de pili P es necesaria para que se produzca pielonefritis en el tracto urinario normal de primate. Hasta ahora, no se ha realizado ninguna demostración directa del papel de una adhesina asociada a pilus en una infección bacteriana específica. Resulta interesante que en los experimentos descritos anteriormente no se encontraron pruebas de que la adhesina PapG fuera necesaria para la colonización del tracto urinario inferior o para el desarrollo de la cistitis aguda. Las dos cepas colonizaron la vagina, persistieron en el intestino y produjeron infección de vejiga. Aunque PapG era un determinante de virulencia crítico en la aparición de pielonefritis, no parecía ser crucial en la cistitis. Sin embargo, esto no excluyó la posibilidad de que otros componentes del pilus P o que otro tipo de pilus, tales como el tipo 1, fueran necesarios para la cistitis. 20 25 Estas hipótesis se investigarán ensayando la virulencia de cepas mutantes de DS17 isogénicas que contienen mutaciones de localización dirigida en Arg-8 de PapD o FimC. Los mutantes presumiblemente serán defectuosos en su capacidad de ensamblar pili P y de tipo 1. Si el mutante de papD es no virulento en el modelo de cistitis de mono, puede indicar que algún componentes del pilus P distinto de PapG es esencial para E. coli para generar infección de vejiga. Por ejemplo, se ha demostrado que el componente principal de la fribilla del extremo, PapE, se une a fribonectina y la fibronectina podría ser un factor importante en la cistitis. De forma similar, si el mutante fimC no es virulente, confirmaría el papel de pili de tipo de unión a manosa en la aparición de la cistitis. La capacidad estas mutaciones puntuales de anular la virulencia de DS17 en la aparición de pielonefritis o cistitis validaría los potenciales terapéuticos para un inhibidor de chaperona. 30 Ejemplo 9 Identificación del motivo de unión entre PapD y K1’-19’WT 35 40 Para confirmar la generalizad del modelo de unión para péptidos C-terminales de subunidades de pilina a PapD como se observa en la estructura cristalina PapD-G1’-19’WT (véase el Ejemplo 1) se investigó un segundo complejo PapD-péptido por cristalografía de rayos X. Como se ha observado una unión fuerte para el péptido de derivado de los 19 aminoácidos C-terminales de tipo silvestre de PapK (K1’-19’WT, SEC ID Nº: 18 y numerado en este documento des de la Arg-1’ C-terminal hasta las Lys-19’ N-terminal), se decidió usar éste como péptido para el segundo complejo de PapD. Obtención del material; proteína y péptido 45 Se preparó PapD como se ha descrito previamente (Holmgren y col., 1988) y se obtuvo a partir del Dr. Scott Hultgren, Departamento de Microbiología Molecular, Escuela de Medicina de la Universidad de Washington, St. Louis Estados Unidos. El péptido K1’-19’WT se preparó por síntesis en fase sólida Fmoc, se purificó por HPLC de fase inversa y se obtuvo a partir de Dr. Jan Kihlberg, Departamento de Química, Universidad de Lund, Lund, Suecia. Cristalización del complejo PapD-péptido 50 55 60 Después de haber explorado varias condiciones experimentales diferentes alrededor de las usadas previamente para obtener cristales PapD-G1’-19’WT, los mejores cristales del complejo PapD-K1’-19’WT se desarrollaron por difusión de vapor contra PEG8000 al 20%, MES 0,1 M pH 6,5. La gota de cristalización contenía volúmenes iguales de solución de depósito de proteína. La solución de proteína (15 mg/ml) contenía una relación molar 1:1 entre PapD a péptido en MES 20 mM, p 6,5 con β-octilo (β-OG) al 1,0%. Estos cristales se montaron dentro de tubos capilares de cuarzo-vidrio cerrados herméticamente y se caracterizaron inicialmente examinándolos en una cámara de precisión de rayos X. A partir de análisis convencionales de tales imágenes, se determinó que los cristales mencionados anteriormente tienen un grupo espacial ortorrómbico, C2221 (difiriendo de esta manera de los cristales PapD-G1’-19’WT que eran C2), con dimensiones de celda A=57,1 Å, b=153,2 Å, c=135,4 Å y α=β=γ=90º, 2 moléculas en la unidad asimétrica y presentaban una difracción a una resolución de 2,7 Å en una fuente de rayos X de laboratorio con ánodo giratorio y objetivo de Cu Kα . Recogida y procesamiento de datos experimentales 65 Los datos de intensidad para los cristales de PapD-K-péptido se recogieron en un sistema detector de área R-AXIS II (de R-AXIS) en Symbicom Ab., Uppsala. Todos los datos se obtuvieron a partir de un solo cristal y se procesaron inicialmente con el paquete de software DENZO (Otwinsky, 1993). La fusión y la ampliación a escala de los datos, 59 ES 2 225 834 T3 sin embargo, se realizó usando ROTAVATA y AGROVATA del paquete CCP4 (CCP4, 1979). La serie de datos finales contenía 15.989 reflexiones independientes con un Rsym del 8,7% para los datos con una resolución entre 20,0 y 2,7 Å. 5 10 15 20 Solución de la estructura tridimensional La estructura del complejo se solucionó por el procedimiento convencional de reemplazo molecular usando el programa XPLOR (Brunger, 1992). El modelo de búsqueda usado fue la estructura de resolución refinada de 2,0 Å de PapD (Holmgren y Branden, 1989). Usando datos de resolución de 8,0 a 4,0 Å, la función de auto-rotación de nuevo proporcionó un eje doble plegado no cristalográfico claro. Los picos superiores en las funciones de traducción también proporcionaron las soluciones correctas. Después de las funciones de traducción, el factor R era del 36,7% para los datos de resolución de 8,0 a 4,0 Å. El refinado de cuerpos rígidos posterior en el que se permitió que se refinaran los cuatro dominios de las dos moléculas de PapD en la unidad asimétrica independientemente dio como resultado un factor R del 33,6% para los mismos datos. El examen de un mapa de densidad electrónica |Fo| - |Fc| en esta fase usando el programa de gráficas O (Jones y Kjeldgaard, 1994) demostró una clara densidad correspondiente al péptido K en la hendidura de PapD y que corría a lo largo de la superficie de la proteína de una manera análoga a la encontrada para el péptido G. La orientación del péptido se determinó fácilmente a partir de la densidad electrónica, pero inicialmente sólo pudieron moderarse en densidad los 12 aminoácidos C-terminales del péptido. Refinado y análisis de la estructura 25 30 35 40 45 50 55 Se inició un refinado de templado simulado con XPLOR (Brunger, 1992) en esta etapa. Se realizaron varios ciclos adicionales de construcción de modelos y refinado añadiéndose dos aminoácidos adicionales del péptido al extremo N-terminal del péptido para producir un factor R para el modelo actual del 19,2% para los datos de resolución de 8,0 a 2,7 Å. El modelo en la presente fase de refinado (que no contiene ninguna molécula de agua y no incluye los primeros cinco aminoácidos N-terminales del péptido) tiene desviaciones de raíz-mínimos cuadrados (rms) de la geometría ideal de 0,019 para longitudes de enlaces y 3,8º para ángulos de enlace. A pesar de que los dos complejos de PapD-péptido se solucionaban en diferentes grupos espaciales, se descubre que las estructuras globales son muy similares con el péptido que interacciona con PapD de una manera esencialmente idéntica a la observada previamente para la estructura PapD-G1’-19’WT. De esta manera, se ha observado de nuevo que el péptido K1’-19’WT se une en una conformación extendida con la Arg-1’ C-terminal anclada dentro de la hendidura interdominio y el sitio de unión a la subunidad. También se forman enlaces de hidrógeno entre el extremo carboxilo del péptido y dos residuos cargados positivamente invariantes de PapD, Arg-8 y Lys-112. El péptido K1’19’WT después corre a lo largo de la superficie del dominio N-terminal, formando una interacción de cadena β paralela para la cadena G1. De esta forma, entre 9 cadenas principales se forman enlaces de hidrógeno entre los residuos 10’ a 2’ del péptido y Val-102 a Lys-110 de PapD y de esta manera se extiende la lámina β de PapD dentro del péptido. Además, se observa una asociación dimérica entre los dos complejos PapD-péptido dentro de la célula unitaria similar a la observada en la estructura cristalina de PapD-G1’-19’WT. De nuevo, la lámina β se extiende como resultado de una simetría plegada dos veces no cristalográfica que pone un segundo complejo PapD-péptido adyacente al primero de tal forma que las dos cadenas peptídicas unidas interaccionan como cadena β antiparalelas con una lámina β mixta de nuevo que se crea entre los dos complejos implicados en un total de 10 cadenas β. La única diferencia importante entre los dos complejos PapD-péptido es el hecho de que en la estructura PapD-K1’-19’WT los péptidos de los complejos relacionados no cristalográficamente estén colocados dos residuos más cerca del extremo COOH de su homólogo. De esta manera, aunque se forman ocho enlaces de hidrógeno entre los péptidos en el complejo PapD-G1’19’WT, se observa un total de 10 en PapD-K1’-19’WT. Además de los residuos C-terminales Try-2’ y Arg-1’, hay relativamente pocos contactos entre las cadenas laterales del péptido y PapD. Las interacciones principales se proporcionan por los enlaces de hidrógeno de la cadena principal con la cadena G1. Sin embargo, hay varias interacciones hidrófobas dentro de la lámina β, en particular entre la Tyr6’ del péptido con Ile-105 y Leu-107 de la cadena G1. Ejemplo 10 Identificación de un segundo sitio de unión en PapD y desarrollo de nuevos ensayos 60 65 Introducción Se desarrolló un ensayo de unión de chaperona para delinear interacciones PapD-PapG usando fusiones MBP/G, que contenían longitudes crecientes del extremo COOH de PapG. También se examinó la capacidad de PapD para unirse a proteínas truncadas PapG que carecen de longitudes crecientes del extremo COOH de PapG. 60 ES 2 225 834 T3 Procedimientos experimentales Cepas bacterianas 5 10 15 20 25 30 35 En los estudios en los que están implicadas las proteínas de fusión MBP/G se usó la cepa de E. coli HB101 (Maniatis y col., 1982) como cepa hospedadora. Como hospedador para construir proteínas de fusión MBP/G (Hanaban, 1983) y truncados PapG se usó la cepa de DH5α. KS474 (degP::kan) (Strauch y col., 1989) se proporcionó amablemente por J. Beckwith y se usó para la expresión de las proteínas truncadas PapG. Construcción de plásmidos Para la construcción de pMAL-p4, pMAL/G1’-19’, pMAL/G1’-81’ y pMAL/G1’-140’ se usó el plásmido de expresión de E. coli pMAL-p2 (New England Biolabs, Beverly, MA, Estados Unidos) usando procedimientos esencialmente de Maniatis (Maniatis y col., 1982). El gen maIE codificado por pMAL-p2 tiene una secuencia lacZα fusionada al extremo 3’. El plásmido pMAL-p2 se digirió con SalI y se rellenó con Klenow (Maniatis y col., 1982). El fragmento se volvió a unir y el plásmido resultante se denominó pMAL-p4 y tenía un codón stop entre las secuencias maIE y lacZα. El plásmido p-MAL-p4 se digirió con HindIII y se rellenó con Klenow y el fragmento resultante se unió con un engarce ClaI, 5’CCATCGATGG·’ (New England Biolabs, Bervely, MA, Estados Unidos) para producir el plásmido pMALp5. Para las Reacciones en Cadena de la Polimerasa (PCR) se usaron los cebadores 23969 (5’CCCCCCTGCAGATCAGATTAAGCAGCTACCTGC3’ SEC ID Nº: 23) y el cebador 23557 (5’CCCCTGCAGTAAAAATATCTCTGCTCAGAAATAC3’, SEC ID Nº: 24) y el cebador 23559 (5’CCATCGATGAACAGCCAGTCAGATAATC3’, SEC ID Nº: 25) usando pPAP5 (Hull y col., 1981; Lindberg y col., 1984) como plantilla. Los fragmentos de ADN que codificaban la región de los residuos aminoacídicos 1’-81’ y 1’-140’ de PapG se obtuvieron por las reacciones de PCR usando los cebadores 23557 y 23559; y los cebadores 23969 y 23559, respectivamente. Los fragmentos de ADN amplificados se purificaron y se restringieron con PstI y ClaI y se unieron al fragmento de pMAL-p5 sometido a restricción con PstI y ClaI para construir pMAL/G1’-81’ y pMAL/G1’-140’, respectivamente. Para crear pMAL/G1’-19’, se sintetizaron químicamente dos oligómeros de nucleótidos (5’GGAAAGAGAAAACCCGGGGAGCTATCTGGTTCTATGACTATGGTTCTGAGTTTCCCCTGAT3’, SEC ID Nº: 26 y 5’ CGATCAGGGGAAACTCAGAACCATAGTCATAGAACCAGATAGCCCCGGGTTTTCTCTTTCCTGCA3’, SEC ID Nº: 27) y se templaron. El fragmento de ADN templado contenía la secuencia de los residuos aminoacídicos 1’-19’ carboxi terminales de PapG se unió al fragmento PstI-ClaI de pMAL-p5 para producir pMAL/G1’-19’. Todas las construcciones usadas en este estudio se confirmaron por análisis de la secuencia de ADN usando Sequenase Version 2,0 de acuerdo con las instrucciones del fabricante (USB, Cleveland, OH, Estados Unidos). pHJ14 (PapG3) se creó por digestión de pHJ8 (PapG wt en el plásmido del promotor Ptac , pMMB66) con BamHI y BglII, eliminación de aproximadamente 500 nucleótidos, purificación del fragmento grande y unión de nuevo. pHJ23 (PapG 2) se creó clonando primero papG en pUC19 en EcoRI y BamHI, digiriendo con HincII para retirar los últimos aproximadamente 300 nucleótidos y volviendo a unir. El fragmento EcoRI HindII después se clonó en pMM66 para crear pHJ23. Inducción y purificación parcial de fusiones MBP/G 40 45 50 Se indujeron cepas que llevaban plásmidos de MBP o plásmido del gen de fusión MBP/G en cultivo con caldo de LB con isopropil tiogalactósido (IPTG) 1 mM se prepararon extracto periplásmicos como se describe (Slonim y col., 1992). Se añadió un décimo de volumen de solución salina tamponada con fosfato 10 X (PBS, NaCl 120 mM, KCl 2,7 mM, Na2 HPO4 10 mM y KH2 PO4 2 mM, pH 7,4) al extracto periplásmico. Después se añadió resina de amilosa a los extractos periplásmicos a una relación de 1:5. La mezcla se agitó a 4ºC durante una noche y las perlas posteriormente se lavaron 5 veces con PBS. MBP o las proteínas de fusión MBP/G se eluyeron con maltosa 20 mM en PBS por agitación a 4ºC durante 1 hora. Las concentraciones de proteína se determinaron usando el kit de ensayo de proteínas Bio-Rad DC (Bio-Rad Laboratories, Hercules, CA, Estados Unidos). La concentración de proteína de fusión de longitud completa se cuantificó adicionalmente por el gel SDS-PAGE teñido con Coomassie con concentraciones conocidas de albúmina bovina (BSA) como patrones seguido de exploración por densitometría. Caracterización de interacciones entre PapD y proteínas de fusión MBP/G 55 Las preparaciones de MBP o de fusión MBP/G de HB101/pLS101/pMAL-p4, HB101/pLS101/pMAL/G1’-19’, HB101/pLS101/pMAL/G1’-81’ y BH101/pLS101/pMAL/G1’-140’ se aplicaron a geles pI 3-9 en enfoque isoeléctrico (IEF) (Pharmacia Phast System, Pharmacia, Suecia) o SDS-PAGE al 12,5% seguido de tinción con plata, tinción con azul de Coomassie o inmunotransferencia con antisuero anti-PapD como se ha descrito (Kuehn, y col., 1991). 60 La estabilidad del complejo PapD-MBP/G1’-140’ en urea se determinó incubando 1 µg de preparación MBP/G1’140’ parcialmente purificada procedente de HB101/pLS101/pMAL-G140 en urea 0,5 M durante 5 minutos a 25ºC (Kuehn, y col., 1991). Las proteínas se analizaron por IEF (pI 3-9) con tinción con plata. El complejo PapD-MBP/G1’140’ en el gel IEF se cuantificó por densitometría (Digital Imaging System, Is-1000). 65 La interacción entre PapD y la fusión de MBP/G in vitro se ensayó incubando PapD purificado (1 µg) y proteína de fusión MBP/G purificada por afinidad en perlas de amilosa (1 µg) en 4 µl de PBS durante 30 minutos y después aplicándolos a IEF (pI 3-9), seguido de tinción con plata. Otro ensayo in vitro que es más cuantitativo es ELISA descrito más adelante. 61 ES 2 225 834 T3 Caracterización de la interacción entre PapD y truncados de PapG 5 10 15 Se co-expresaron PapD y los truncados de PapG en KS474, los truncados se indujeron a DO600 de 0,6 con IPTG 0,5 mM y PapD bajo el control del promotor de arabinosa se indujo con arabinosa al 0,2%. Después de una hora de inducción, se prepararon extractos periplásmicos y se agitaron con perlas de Gal α (1-4) Gal durante una noche a 4ºC y se eluyeron con 15 µg/ml de solución Gal α (1-4) Gal-TMSET como se ha descrito previamente (Hultgren y col., 1989; Kuehn y col., 1991). Los eluidos se analizaron por electroforesis en gel nativo ácido (Striker y col., 1994) seguido se transferencia de western usando antisuero anti-PapD y anti-PapG. Titulaciones de hemaglutinación HB101/pFJ3 que llevaba diversos plásmidos de fusión MBP/G se pasó sobre Agar Tripticasa de Soja (TSA; Becton Dickinson, Cockeysville, MD, Estados Unidos) tres veces para inducir la expresión de pili P. En el último pase, la expresión de las fusiones MBP/G se indujo en TSA que contenía IPTG 100 µM. Después, se recogieron las células de diferentes cepas y se determinaron las titulaciones de HA como se ha descrito (Jacob-Dubuisson y col., 1993b). Preparación y cuantificación de pili 20 25 30 35 40 Se recogieron las células obtenidas a partir de placas de TSA. Se prepararon pili a partir de las mismas cantidades de células usando el procedimiento descrito previamente (Jacob-Dubuisson, y col., 1993b). Estas preparaciones de pili se hirvieron en tampón de muestra de Lammeli con urea 4 M, y después se analizaron en geles de SDS-poliacrilamida con azul de Coomassie. La cantidad relativa de formación de pili se cuantificó por exploración citométrica de las bandas de PapA. Ensayo ELISA Se diluyeron soluciones madre de proteínas de fusión MBP/G en PBS a 40 pmol/50 µl con PBS. Las soluciones de las proteínas de fusión MBP/G 40 pmol/50 µl se diluyeron en serie en PBS y se usaron 50 µl de cada solución para recubrir los pocillos de una Nunc Immunoplate de 96 pocillos (Inter Med, DK-4000 Roskilde, Dinamarca) durante una noche a 4ºC. Los pocillos después se lavaron con PBS y se bloquearon con 200 µl de BSA al 3% en PBS durante 2 horas a 25ºC. Las placas se lavaron vigorosamente tres veces con PBS y se incubaron con 50 µl de PapD diluido en 50 pmol de PapD en 50 µl de BSA-PBS al 3% durante 45 minutos a 25ºC. Después de tres lavados con PBS, los pocillos se incubaron con una dilución 1:500 de antisuero de conejo anti-PapD en BSA-PBS al 3% durante 45 minutos a 25ºC. Después de tres lavados con PBS, los pocillos se incubaron con una dilución 1:1000 de antisuero de cabra contra IgG de conejo acoplada a fosfatasa alcalina en BSA-PBS al 3% durante 45 minutos a 25ºC. Después de tres lavados con PBS y tres lavados con tampón de desarrollo (dietanolamina 10 mM, MgCl2 0,5 mM), se añadieron 50 µl de pnitrofenil fosfato a 1 mg/ml filtrado (Sigma, St. Louis, MO, Estados Unidos) en tampón de revelado. La reacción se incubó durante 1 hora en la oscuridad a 25ºC y se leyó la absorbancia de 405 nm. Para ensayar los péptidos del segundo sitio, todos los péptidos se disolvieron en dimetil sulfóxido (DMSO, Sigma, St. Louis, MO, Estados Unidos) hasta una concentración final de 2 mM. Las soluciones madre se diluyeron a una concentración apropiada en PBS y se aplicaron como un recubrimiento en pocillos de microtitulación a 4ºC. Todas las soluciones de péptidos se ajustaron a la misma concentración de DMSO. Las etapas posteriores siguieron el procedimiento descrito anteriormente. 45 Resultados Fusiones MBP/G 50 55 Para identificar determinantes sobre PapG esenciales para la interacción con PapD, los presentes solicitantes construyeron tres fusiones en fase entre el gen MalE, que codifica la proteína de unión a maltosa, y secuencias que codifican los residuos COOH-terminales 1’-19’, 1’-81’ y 1’-140’ de PagG (Fig. 20). Las proteínas quiméricas resultantes se denominaron MBP/G1’-19’, MBP/G1’-81’ y MBP/G1’-140’, respectivamente, para indicar las regiones de PapG presentes en las fusiones MBP/G. Se creó MBP/G1’-19’ ya que recientemente se había mostrado que PapD se unía a un péptido que constaba de los 19 aminoácidos C-terminales de PapD in vitro (Kuehn y col., 1993). MBP/G1’-81’ y MBP/G1’-140’ se crearon para ensayar el requisito del enlace disulfuro (C196-C228 en PapG) para el reconocimiento por PapD. Prácticamente en todas las subunidades de pilus se encuentra un enlace disulfuro localizado de forma similar (Simons y col., 1990). MBP/G1’-81’ los dos restos de cisteína mientras que MBP/G1’-140’ contiene el enlace disulfuro (Fig. 20). 60 Interacciones PapD-MBP/G in vivo 65 La capacidad de PapD para unirse a las fusiones MBP/G se investigó usando cromatografía de afinidad de amilosa (Kellermann y Ferenci, 1982). Se co-expresó PapD a partir del plásmido pLS101 (Slonim y col., 1992) con cada una de las fusiones MBP/G. Se sometieron extractos periplásmicos de cada cepa que contenía las proteínas MBP/G y PapD a cromatografía de afinidad de amilosa y los eluidos se analizaron por SDS-PAGE (Fig. 21A) y por transferencia de western con antisuero anti-PapD (Fig. 21B). En este ensayo, la co-elución de PapD significaba la capacidad de PapD para interaccionar con la proteína MBP/G. La transferencia de western reveló que PapD co-eluía con las tres 62 ES 2 225 834 T3 5 10 15 proteínas de fusión (Fig. 21B, calles 2, 3 y 4) pero no con el control MBP (Fig. 21A y 21B, calle 1). Sin embargo, PapD interaccionaba muchos más fuertemente con la fusión MBP/G1’-140’ como puede verse en la transferencia de western (Fig. 21B, calle 4) así como en el gel teñido con azul de Coomassie de los eluidos (Fig. 21A, calle 4). De esta manera, PapD interaccionaba fuertemente con la proteína MBP/G1’-140’ y sólo débilmente con las proteínas MBP/G1’-19’ y MBP/G1’-81’. El análisis de los eluidos en geles de enfoques isoeléctrico (IEF) teñidos con plata reveló que el complejo PapDMBP/G1’-140’ migraba en un punto isoeléctrico (pI) de 5,2 (Fig. 21C, calle 4) que era intermedio entre los pI de MBP/G1’-140’ (∼4,4) y PapD (∼9,1). Se confirmó que esta banda contenía tanto PapD como MBP/G1’-140’ por escisión de la banda no teñida, aplicación de los materiales a SDS-PAGE y análisis de los mismos por transferencia de western usando antisueros anti-PapD y anti-MBP (Fig. 21D). No fue posible detectar complejos estables en los eluidos de MBP, MBP/G1’-19’ o MBP/G1’-81 cuando se co-expresaron con PapD (Fig. 21C, calles 1, 2 y 3). La estabilidad del complejo PapD-MBP/G1’-140’ se midió como una función de la concentración de urea en presencia de DTT 15 mM. Tanto el complejo PapD-PapG como el complejo PapD-MBP/G1’-140’ se comportaron de manera similar en estas condiciones y se disociaron después de la incubación en urea 2 M (datos no mostrados). Por estos criterios, el complejo PapD-MBP/G1’-140’ era tan estable como el complejo PapD-PapG. La expresión de proteínas MBP/G inhibe la formación del pilus 20 25 30 PapD es esencial para ensamblaje del pilus p (Hultgren y col., 1991). Se ha demostrado que una reducción en la concentración de PapD en el periplasma produce una reducción concomitante en la formación de pili (Slonim y col., 1992). Se ensayó la capacidad de las fusiones MBP/G para bloquear la formación del pilus por inhibición de la formación del complejo chaperona-subunidad cuando se co-expresaba en trans con el operon pap. Se utilizaron plásmidos pMAL-p4, pMAL/G1’-19’, pMAL/G1’-81’ y pMAL/G1’-140’ para transformar la cepa HB101/pFJ3 que contiene el operon pap bajo el control de su propio promotor en el vector pACYC184 (Jacob-Dubuisson y col., 1993b). Cada proteína de fusión MBP/G se localizó en el periplasma como se determina por SDS-PAGE y transferencia de western con antisuero anti-MBP (datos no mostrados). Se realizaron ensayos de hemaglutinación (HA) sobre las células que co-expresaban el operon pap con MBP (pMAL-p4) o con cada una de las fusiones MBP/G. La co-expresión de las fusiones MBP/G1’-140’ con el operon pap redujo la titulación de HA 30 veces en comparación con el control de MBP (véase la tabla 3). 35 40 a 45 50 55 60 65 Se utilizaron plásmidos pMAL-p4, pMAL/G1’-19’, pMAL/G1’-81’ y pMAL/G1’- 140’, que codificaban MBP, MBP-G1’-19’, MBP-G1’-81’ y MBP-G1’-140’ respectivamente, para transformar la cepa HB1010/pFJ3 y la expresión del operon pap y cada una de las fusiones MBP/G se indujo en cada cepa b Máxima dilución de suspensión de células que produce HA detectable c Las preparaciones de pili se analizaron en geles teñidos con azul de Coomassie y las bandas PapA se exploraron por un densitómetro. La formación de pili relativa se determinó igualando el valor densitométrico de PapA al 100% del control (HB101/pFJ3/pMAL-p4). La co-expresión de las fusiones MBP/G1’-81’ y MBP/G1’-19’ con el operon pap sólo tuvo efectos débiles sobre la titulación de HA (tabla 3). La co-expresión de MBP/G1’-140’ con el operon pap redujo la cantidad de pili que podía purificarse a partir de las células en un 90%, mientras que MBP/G1’-19’ y MBP/G1’-81’ redujeron la cantidad de pili formados en aproximadamente 50% en comparación con MBP solo (tabla 3). La microscopía electrónica confirmó que las células que expresaban la fusión MBP/G1’-140’ tenían pocos o ningún pilus en comparación con las células con pili completos que co-expresaban MBP (datos no mostrados). Los presentes solicitantes sugirieron la hipótesis de que la co-expresión de la fusión MBP/G1’-140’ con el operon pap inhibía la formación del pilus por titulación de PapD lejos de las subunidades, llevando de esta manera a las subunidades a rutas muertas de agregación y degradación proteolítica (Hultgren y col., 1989); Holmgren y col., 1992). Esta hipótesis se ensayó como se indica a continuación. Se utilizaron pMAL-p4, pMAL/G1’-19’, pMAL/G1’-81’ o pMAL/G1’-140’, para transformar BH101 que llevaba pFJ22 (Jacob-Dubuisson y col., 1994). El plásmido pFJ22 codifica papDJKEFGA bajo el control del promotor Ptac . En HB101/pFJ22, las subunidades del pilus se acumulan en el espacio periplásmico debido a la ausencia del acomodador, PapC. El efecto de la expresión de cada fusión sobre el destino de cada tipo de subunidad del pilus se determinó por transferencia de western (Fig. 22). Se detectó una 63 ES 2 225 834 T3 5 reducción significativa en la cantidad de PapG, PapA y PapF en células que co-expresaban la fusión MBP/G1’-140’, lo que indica que la fusión MBP/G1’-140’ bloqueaba la formación del complejo chaperona/subunidad por interacción con PapD y las separaba de las subunidades del pilus. No hubo ningún efecto sobre la estabilidad de PapK y PapE por razones que no se entienden. Sin embargo, la fusión pudo bloquear la formación del pilus por interferencia con la formación de complejos críticos chaperona-subunidad. Ensayos de unión a chaperona in vitro 10 15 20 25 30 35 40 La unión de PapD a las proteínas de fusión MBP/G se investigó adicionalmente usando dos ensayos in vitro diferentes. En el primer ensayo, se incubaron proteínas de fusión MBP/G purificadas o MBP con PapD y se analizó la formación de complejos en geles IEF teñidos con planta. PapD se unión a la proteína de fusión MBP/G1’-140’ y formó un complejo estable que migraba a un pI idéntico (5,2) que el observado para el complejo formado in vivo (Fig. 23A, calle 4). Por el contrario, no se detectaron complejos entre PapD y MBP/G1’-81’, MBP/G1’-19’ o el control de MBP (Fig. 23A, calle 1, 2 y 3). En el segundo ensayo, los presentes solicitantes investigaron la capacidad de PapD para unirse a las tres proteínas de fusión MBP/G diferentes inmovilizadas en pocillos de microtitulación y se cuantificaron las interacciones en un ensayo de inmunoabsorbente con enzima unida (ELISA) usando antisuero antiPapD (Fig. 23B). PapD se unión débilmente a MBP/G1’-19’, ligeramente mejor a MBP/C1’-81’ pero muy fuertemente a MBP/G1’-140’. PapD no se unió al control de MBP. El aumento dramático en la afinidad de PapD por la fusión MBP/G1’-140’ en comparación con las fusiones MBP/G1’-81 y MBP/G1’-19’ sugirieron que los residuos 81’ a 140’ en PapG era críticos para la fuerte unión a PapD. Se ha demostrado que los residuos de la hendidura invariantes Arg-8 y Lys-112 en PapD forman un anclaje molecular en la hendidura de chaperona necesario para la unión a las subunidades del pilus (Slonim y col., 1992; Kuehn y col., 1993). Las mutaciones en estos residuos anulaban o reducían en tal medida la capacidad de PapD para unirse a las subunidades y mediar el ensamblaje del pilus (Kuehn y col., 1993). Las proteínas PaPD de mutantes en Arg-8 A y Lys-112A demostraron una capacidad muy reducida para unirse a la proteína de fusión MBP/G1’-140’ tanto in vivo como in vitro (datos no mostrados). Estos resultados indican que las interacciones entre PapD y la fusión MBP/G1’140’ son biológicamente relevantes. El sitio cadena arriba es un determinante de unión independiente Los presentes solicitantes ensayaron la capacidad de PapD para unirse a tres proteínas truncadas COOH terminales PapG (mostradas en la Fig. 20) para delinear los límites del sitio de unión de PapD cadena arriba y para ensayar si podía funcionar como sitio independiente. Las proteínas truncadas se co-expresaron con PapD en la cepa KS474 (Strauch y col., 1989) que lleva un cassette de canamicina en el locus degP (mutante degP41). Tanto el truncado PapG2 como el truncado PapG3 se expresaron y fueron estables en KS474, sin embargo el truncado PapG1 (retirando los últimos 14 residuos) se sometió una degradación limitada (datos no mostrados). La formación de complejos se ensayo analizando extractos periplásmicos en geles de poliacrilamida nativos ácidos (Striker y col., 1994). Se usaron antisueros anti-PapD y anti-PapG para detectar los complejos PapD-PapG después de transferencia de western (Fig. 24). Los complejos PapD-PapG se resuelven a partir de PapD solo en estos geles debido a sus diferencias en tamaño, forma y carga (Striker y col., 1994). PapD formó un complejo con PapG de longitud completa así como con el truncado PapG2 (Fig. 24A, calles 2 y 3). La banda compleja también se reconoció por el antisuero anti PapG (Fig. 24B, calles 2 y 3). PapD no formo un complejo con el truncado PapG3 (Fig. 24A y B, calle 4) que termina en el aminoácido 145. Esta información de línea un punto final para el segundo sitio de unión de PapD (como se indica en la Fig. 20). 45 50 55 60 65 La combinación de los datos del truncado PapG y la fusión MBP/G (mostrados en la Fig. 20) sugirieron que el segundo sitio en PapG reconocido por PapD residía en una región entre los residuos 117’ a 141’ de PapG. Se sintetizaron cuatro péptidos solapantes correspondientes a la región entre los residuos 120’ a 156’ y se ensayaron con respecto a su capacidad para unirse a PapD en un ensayo ELISA. Tal estrategia resultó satisfactoria en el estudio del sitio de unión a PapD COOH-terminal (Kuehn y col., 1993). El péptido correspondiente a los residuos 125’ a 140’, pero no los otros tres péptidos, se unieron a PapD en el ELISA (Fig. 25). Estos datos indican que PapD reconoce dos superficies en PapG. PapD forma una interacción de cremallera de cadena β con el extremo COOH, pero también reconoce una región que contiene los residuos 125’-140’. Discusión y conclusiones En conclusión, se creó un ensayo de unión de chaperona usando fusiones del extremo carboxilo de PapG a la proteína de unión a maltosa (fusiones MBP/G) para investigar si la formación del complejo chaperona-subunidad requiere interacciones adicionales. PapD se unía fuertemente a una fusión MBP/G que contenía los 140 aminoácidos C-terminales de PapG (MBP-G1’-140’), pero sólo se unía débilmente a MBP-G1’-81’, lo que indica que la región entre los residuos C-terminales 81’ y 140’ contiene información adicional que se requiere para interacciones fuertes PapDPapG. Se ha demostrado adicionalmente que PapD interacciona con el truncado C terminal de PapG que contiene los residuos 117’-314’ (correspondientes a un truncado que consta de los primeros 198 residuos aminoacídicos N terminales de PapG), pero no con un truncado que contiene los residuos 170’-314’ (correspondiente a un truncado que consta de los primeros 145 residuos aminoacídicos N terminal de PapG). Estos resultados conjuntamente sugieren que existe un segundo sitio interactivo de PapD independiente que no interacciona con el extremo C de la subunidad del pilus. Esta suposición se confirma adicionalmente por el hecho de 64 ES 2 225 834 T3 que uno de los cuatro péptidos que solapaban con la región del segundo sitio de PapG se reconoció por PapD. Este último resultado también indica que la función del sitio cadena arriba era ejercer efectos secundarios sobre el sitio de unión de chaperona COOH terminal ya que esta región era independientemente capaz de facilitar la unión de PapD. 5 10 15 20 25 A diferencia de lo que ocurre en el caso de las subunidades nativas, se demostró que MBP-G1’-140’ es capaz de plegarse incluso en ausencia de PapD y permanece soluble en el espacio periplásmico; este plegamiento implica la formación de un enlace disulfuro intramolecular. Esto es coherente con los datos previos que demuestran que las propias subunidades están muy plegadas cuando se unen a PapD (Kuehn y col., 1991; Striker y col., 1994). sin embargo, aún no se sabe si PapD se une a las subunidades in vivo después de plegarse o si el plegamiento se produce en el contexto de una interacción con PapD. Finalmente, las expresión de las fusiones MBP/G en células que producen pili P inhibía el ensamblaje de pilus en diversas medidas por la unión a PapD e impedía la formación del complejo chaperona-subunidad. La capacidad de las fusiones para bloquear el ensamblaje del pilus aumentó según aumentaba la longitud de la proteína de fusión. MBP-G1’-140’ era un fuerte inhibidor del ensamblaje de pilus mientras que MBP-G1’-81’ y MBP-G1’-19’ eran inhibidores débiles. Estos es coherente con el hallazgo de que los residuos 140’-81’ contienen una región necesaria para interacciones fuertes entre PapD y PapG. Una investigación reciente por los inventores ha revelado adicionalmente que la expresión simultánea in vivo de PapG y un polipéptido PapD modificado que carece del dominio 1 en una cepa degP41 (KS474) que expresa este PapD truncado da como resultado 1) la supresión de la toxicidad de PapG en esta línea celular y 2) una división eficaz en el espacio periplásmico de PapG. Sin embargo, la subunidad del pilus no se pliega correctamente después de la liberación desde PapD. De esta manera, los inventores sugieren que la unión entre subunidades del pilus y PapD tiene lugar por la unión de las subunidades de pilus al dominio 1 y 2 de PapD. La unión entre la subunidad y el dominio 1 es importante para el plegamiento correcto de la subunidad, y la unión entre la subunidad y el dominio 2 es importante para el transporte de la subunidad al interior del periplasma. No se sabe si el dominio 2 de PapD también participa en el plegamiento de las subunidades. 30 Como los resultados anteriores son indicadores fuertes de la existencia de al menos un sitio de unión a parte del que implica la Arg-8 y Lys-112 de PapD, y como parece ser que este nuevo sitio de unión también es muy importante en la interacción lenta entre PapD y la subunidades del pilus, se contempla que también este sitio de unión tendrá los efectos antibacterianos descritos en este documento. 35 De esta manera, este es el plan para esclarecer el motivo de unión entre este segundo sitio de unión de PapD de una manera similar a las descritas en este documento, y adicionalmente es el plan para diseñar/identificar compuestos de interaccionar con este segundo sitio de unión para sintetizar finalmente compuestos capaces de interaccionar con este sitio de tal manera que se impida, inhiba o mejore el ensamblaje de pili intactos. 40 Referencias Allen B L y col., 1991, J. Bacteriol., 173, 916-920. 45 Amit A G y col., 1988, Science, 230, 747-753. Baga M y col., 1987, Cell, 49, 241. Bakker y col., 1991., Molec. Microbiol.,5, 875. 50 Ben-Naim y Marcus, 1984, J. Chem. Phys., 81, 2016-2027. Bertin Y y col., 1993, FEMS Microbiol. Lett., 108, 59. 55 Blond-Elguindi S y col., 1993, Cell, 75, 717-728. Bock, K. y col, 1985, J. Biol. Chem., 620, 8545-8551 Boobbyer, 1989, J. Med. Chem., 32, 1083-1094. 60 Bothner-By A A, 1965, Adv. Magn. Resonance, 1, 195 Brint A T y Willet P, 1987, J. Mol. Graphics, 5, 49-56. 65 Brunger A T, 1992, X-PLOR Manual, Versión 3.0, Yale Univ., New Haven. Burkert U y Allinger N L, 1982, Molecular Mechanics, ACS Monographs, Washington D. C. 65 ES 2 225 834 T3 CCP4, The SERC, UK Colalborative Project Nº 4, A Suite of Programs for Protein Crystallography, Darebury Laboratory, UK. Chandler D y col., 1983, Science, 220, 787-794. 5 Clouthier S C y col., 1993, J. Bacteriol., 175, 2523. Dodson y col., 1993, Proc. Natl. Acad. Sci., 90, 3670-3674. 10 Galyov E E y col., 1991, FEBS Lett., 286, 79. Gerlach G D, Clegg, S. y Allen B L, 1989, J. Bacteriol., 171, 1262-70. Gething M-J y Sambrook J, 1992, Nature, 355, 33. 15 Goodford, 1985, J. Med. Chem., 1985, 28, 849-857. Grant G A y col., 1992, Synthetic Peptides, A Users Guide, W. A. Freeman y Compañía, Nueva York. 20 Hanahan D, J. Mol. Biol., 1983, 166, 557-580. Hirschmann R y col., 1992, J. Am. Chem. Soc., 114, 9217-9218. Holmgren A y col., 1988, J. Mol. Biol. 203, 279. 25 Holmgren A y Branden C I, 1989, Nature, 342, 248. Holmgren A y col., 1992, The EMBO Journal, 11, 4, 1617-1622. 30 Hultgren S J y col., 1989, Proc. Natl. Acad. Sci. USA, 86, 4357. Hull R A y col., 1981, Infect. and Immun., 33, 933-938. Hultgreen S J y col., 1993, Cell, 73, 887-901. 35 Hultgren S J y col., 1989, Proc. Natl. Acad. Sci. USA, 86, 4357-61. Hultgren S J y col., 1991, Annu. Rev. Microbiol., 45, 383-415. 40 Hultgren S J y col., 1993, Cell, 73, 887-901. Hallgren C y Widmalm G, 1993, J. Carbohydr. Chem., 12 83), 309-333. Iriarte y col., 1993, Mol. Microbiol., 9, 507. 45 Jacob-Dubuisson F y col., 1993a, Trends Microbiol, 1, 50-55. Jacob-Dubuisson F y col., 1993b, EMBO J., 21, 837-847. 50 Jacob-Dubuisson F y col., 1994, J. Biol. Chem., 269, 12447-12455. Jalajakumari M B y col., 1989, Mol. Microbiol., 3, 1685. Jones A T y Kjeldgaard M, 1994, O Version 59, Dept. Molec. Biol. Uppsala. Suecia. 55 Jorgensen W L, 1986, J. Phys. Chem., 90, 1276-1284. Kawaminami M y col., 1981, Acta Crystallogr. Sección B 107 (37), 2026. 60 Kellermann O K y Ferenci T, 192, Methods in Enzymology, 90, 459-463. Klann A G y col., 1994, J. Bacteriol., 176, 2312-2317. Klemm P y col., 1992, Res. Microbiol., 143, 831. 65 Kuehn M. J., Normark S y Hultgren S J., 1991. proc. Natl. Acad. Sci. 88, 10586-10590. Kuehn M. J. y col., 1993, Science, 262, 1234-1241. 66 ES 2 225 834 T3 Lam K T y Calderwood S K, 1992, Biochem y Biophys., Res. Comm. 184, 167. Landry S J y Gierasch L M, 1991, Biochemistry, 30, 7359. 5 Landry S J y col., 1992, Nature, 355, 455. Lee F S y col., 1992, Prot. Eng., 5, 215-228. Lindberg F y col., 1984, EMBO J., 3, 1167-1173. 10 Lindberg F y col., 1987, Nature, 328, 84-87. Lindberg F y col., 1989, J. Bacteriol. 171, 6052. 15 Lindler y col., 1993, Genbank, acceso Nº: M86713. Lintermans P, 1990, Thesis, Rijksuniversiteit Ghent, Bélgica. Locht C y col., 1992, EMBO J., 11, 3175. 20 Lund B y col., 1987, Proc. Nat. Acad. Sci. USA, 84, 5898. Marcus R A, 1964, Ann. Rev. Phys. Chem., 15, 155-196. 25 Maniatis T y col., 1982, Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, NY. Messerschmit A y Pflugrath J W, 1987, J. Appl. Crystalogr., 20, 306. 30 Normark S y col., Genetics and Biogenesis of Escherichia coli Adhsin. In Microbial lecctins and Agglutinins: Properties and Biological Activity, D. Mirelman (ed.), Wiley Interscience, Nueva York, 113-143. Otwinowski Z, 1993, Proc. CCP4 Study Weekend 29-30 Jan 1001. Data Collection and Processing. SERC Daresbury Lab. 35 Patroni J J y col., 1988, Aust. J. Chem., 41, 91-102. Pratt L R y Chandler D, 1977, J. Chem. Phys, 67, 3683-3704. Raina S y col., 1993, J. Bacteriol., 175, 5009. 40 Roux B y col., 1990, J. Phys. Chem., 94, 4683-4688. Ryu S-E y col., 1990, Nature, 348, 419. 45 Schmoll y col., 1990, Microb. Pathog., 9, 331. Simons B L y col., 1990, FEMS Microbiol. Lett., 67, 107-112. Slonim L y col., 1992, EMBO J. 11, 4747-56. 50 Strauch K L, Johnson K y Beckwith J., 1989. J. Bacteriol. 171, 2689-2696. Striker R y col., 1994, J. Biol. Chem., 269, 4747-56. 55 Stromberg N y col., 1990, EMBO J., 9, 2001-2101. Stromberg N y col., 1991, Proc. Natl. Acad. Sci. USA, 88, 9340-9344. Ullman J R, 1976, J. Ass. Comp. Machinery, 23, 31-42. 60 de Vos A M y col., 1992, Science, 255, 306. Wang J y col., 1990, Nature, 348, 411. 65 Warshel A y Russell S T, 1984, Q. Rev. Biophys., 17, 283-422. Willems R J L y col., 1992, Molec. Microbiol., 6, 2661. 67 ES 2 225 834 T3 Xuong N-H y col., 1985, J. Appl. Crystallogr., 18, 342. Aqvist J, 1990, J. Phys. Chem., 94, 8021-8024. 5 Aqvist J y Medina C, 1993, 206th ACS National Meeting, Phys: 124, Catalogue of Abstracts. Aqvist J y col., 1994, Protein Engineering, 7 (3), 385-391. 10 15 20 25 30 35 40 45 50 55 60 65 68 ES 2 225 834 T3 REIVINDICACIONES 5 10 1. Un procedimiento para identificar y/o diseñar una sustancia, X, capaz de interactuar con una chaperona molecular periplásmica, por ejemplo uniéndose a la chaperona molecular periplásmica, con una energía de unión libre pronosticada igual a o mejor que un valor umbral predeterminado, comprendiendo el procedimiento 1) seleccionar una sustancia, A, que podría interactuar en gran medida con un sitio en la chaperona molecular periplásmica, siendo dicho sitio el sitio de unión de la subunidad de pilus de dicha chaperona molecular, y proporcionar una representación estructural tridimensional de la misma, 2) predecir la energía libre de unión entre la sustancia A y el sitio en la chaperona molecular periplásmica o sitio análogo a tal dicho, siendo dicho sitio, el sitio de unión de la subunidad de pilus de dicha chaperona molecular, 15 20 25 30 3) si la energía libre de unión pronosticada entre la sustancia A y el sitio en la chaperona molecular periplásmica o el sitio análogo es igual a o mejor que el valor umbral predeterminado, entonces identificar la sustancia A como la sustancia X, 4) si la energía libre de unión pronosticada entre la sustancia A y el sitio en la chaperona molecular periplásmica o el sitio análogo no es igual a o mejor que el valor umbral predeterminado, entonces modificar la representación estructural tridimensional y predecir la energía libre de unión entre la sustancia modificada de esta manera, B, y el sitio en la chaperona, y 5) repetir la etapa 4 hasta que la energía libre de unión pronosticada determinada entre la sustancia resultante, X, y el sitio en la chaperona molecular periplásmica sea igual a, o mejor que, el valor umbral predeterminado. 2. Un procedimiento para identificar y/o diseñar una sustancia, Y, capaz de interactuar con una chaperona molecular periplásmica, por ejemplo uniéndose a la chaperona molecular periplásmica, con una energía de unión libre igual a, o mejor que, un valor umbral predeterminado, comprendiendo el procedimiento 1) identificar una sustancia, X, de acuerdo con el procedimiento de la reivindicación 1, y 35 40 45 50 55 60 65 2) proporcionar una muestra de la sustancia química X y una muestra de la chaperona molecular periplásmica o un análogo de la misma y medir la energía libre de unión entre la sustancia química X y la chaperona molecular periplásmica o análogo de la misma, y establecer que la energía libre de unión medida entre la sustancia química X y la chaperona molecular periplásmica o análogo de la misma es igual a o mejor que el valor umbral predeterminado, y entonces, identificar la sustancia X como la sustancia Y. 3. Un procedimiento de acuerdo con la reivindicación 2, en el que el procedimiento someter la sustancia Y a al menos una de las siguientes etapas del procedimiento para verificar que la sustancia Y es una sustancia potencial y terapéuticamente útil capaz de interactuar con una chaperona molecular periplásmica: 1) ensayar una sustancia candidata en un ensayo en el que la posible prevención, inhibición o mejora mediante la sustancia de la interacción entre la chaperona periplásmica molecular y la subunidad de pilus se determine mediante la adición de la sustancia a un sistema que comprende la chaperona periplásmica molecular o un análogo de la misma en una forma inmovilizada y la subunidad de pilus o un equivalente de la misma en una forma solubilizada y la determinación del cambio en la unión entre la subunidad de pilus o equivalente de la misma y la chaperona periplásmica molecular o el análogo de la misma provocado por la adición de la sustancia, o la adición de la sustancia a un sistema que comprende la subunidad de pilus o un equivalente de la misma en una forma inmovilizada y la chaperona periplásmica molecular o un análogo de la misma en una forma solubilizada y la determinación del cambio en la unión entre la subunidad de pilus o el equivalente de la misma y la chaperona periplásmica molecular o el análogo de la misma provocado por la adición de la sustancia, o la adición de la sustancia a un sistema que comprende la subunidad de pilus o un equivalente de la misma así como la chaperona periplásmica molecular o un análogo de la misma en una forma solubilizada y la determinación del cambio en la unión entre la subunidad de pilus o el equivalente de la misma y la chaperona periplásmica molecular o el análogo de la misma provocada por la adición de la sustancia, o la adición de la sustancia a un sistema que comprende la subunidad de pilus o un equivalente de la misma así como la chaperona periplásmica molecular o un análogo de la misma en forma solubilizada y la medición del cambio en la energía de unión provocado por la adición de la sustancia, y la identificación de la sustancia como potencialmente terapéuticamente útil si se observa un cambio en la energía de unión entre la subunidad de pilus o el equivalente de la misma y la chaperona periplásmica molecular o un análogo de la misma, 69 ES 2 225 834 T3 e identificar la sustancia como potencial y terapéuticamente útil si se observa un cambio significativo en la unión o en la energía de la unión entre la subunidad de pilus o el equivalente de la misma y la chaperona periplásmica molecular o el análogo de la misma; 5 10 15 2) ensayar una sustancia candidata en un ensayo en el que la posible prevención, inhibición o mejora de la interacción entre la chaperona periplásmica molecular y la subunidad de pilus se determine mediante la adición de la sustancia a un sistema que comprende bacterias vivas que forman pilus que se adhieren a tejidos seguido de la determinación de la velocidad de crecimiento de las bacterias, siendo indicativo de prevención, inhibición o mejora de la unión entre la chaperona periplásmica molecular y la subunidad de pilus una reducción en el crecimiento en comparación con un sistema correspondiente en el que la sustancia no se ha añadido, o la adición de la sustancia a un sistema que comprende bacterias vivas que forman pilus que se adhieren a tejidos seguido de una determinación de la adhesión al tejido de las bacterias, siendo indicativo de prevención, inhibición o mejora de la unión entre la chaperona periplásmica molecular y la subunidad de pilus una reducción en la adhesión al tejido en comparación con un sistema correspondiente en el que no se ha añadido la sustancia, e identificar la sustancia como potencial y terapéuticamente útil si se observa una reducción de la velocidad de crecimiento o de adhesión al tejido después de la adición de la sustancia; y 20 25 3) identificar si una sustancia es capaz de interactuar in vivo con una chaperona periplásmica molecular mediante el uso de una sustancia, donde dicha sustancia se ha establecido in vitro para prevenir, inhibir o mejorar la interacción entre una chaperona periplásmica molecular y una subunidad de pilus, para la administración a un animal experimental, donde las bacterias que forman pilus que se adhieren a tejidos del animal se ha inoculado antes, a la vez o después de la administración de la sustancia, para la identificación de si la sustancia es capaz de prevenir y/o curar y/o aliviar la enfermedad provocada por las bacterias. 4. Un procedimiento de acuerdo con la reivindicación 3, en el que el ensayo en la etapa 1 comprende 30 - añadir la sustancia a un primer sistema que comprende la chaperona periplásmica molecular o un análogo de la misma, - posteriormente, añadir una subunidad de pilus o un equivalente de la misma que se ha marcado con una sonda fluorescente medioambientalmente sensible, 35 - determinar la emisión de fluorescencia a una longitud de onda particular que indica la cantidad de unión entre la chaperona periplásmica molecular o el análogo de la misma y la subunidad de pilus o el equivalente de la misma, y 40 - comparar la emisión de fluorescencia determinada con la emisión de fluorescencia determinada en un segundo sistema correspondiente que contiene sustancialmente las mismas concentraciones de la chaperona molecular o del análogo de la misma y la subunidad de pilus o el equivalente de la misma pero sustancialmente sin sustancia, una diferencia significativa en la emisión de fluorescencia entre el primer y el segundo sistema que indica la interacción entre la chaperona periplásmica molecular o el análogo de la misma y la sustancia. 45 50 5. Un procedimiento de acuerdo con la reivindicación 4, en el que la determinación de la emisión de fluorescencia en el segundo sistema se realiza muchas veces a diversas proporciones molares entre la subunidad de pilus o el equivalente de la misma y la chaperona periplásmica y el equivalente de la misma, después de lo cual, la constante de unión entre la subunidad de pilus o el equivalente de la misma y la chaperona molecular periplásmica o análogo de la misma se evalúa a partir de los datos de emisión fluorescente determinados. 55 6. Un procedimiento de acuerdo con la reivindicación 3 ó 4, en el que la determinación de la emisión fluorescente en el primer sistema se realiza muchas veces a diversas proporciones molares entre la sustancia y la chaperona periplásmica y el equivalente de la misma, después de lo cual, la constante de unión entre la sustancia y la chaperona molecular periplásmica o análogo de la misma se evalúa a partir de los datos de emisión fluorescente determinados. 7. Un procedimiento de acuerdo con cualquiera de las reivindicaciones 3-6, en el que el equivalente de la subunidad de pilus es G1’-19’WT, MBP-G1’-140’, o G125’-140’ y la chaperona molecular periplásmica es PapD. 60 65 8. Un procedimiento de acuerdo con cualquiera de las reivindicaciones 3-7, en el que las bacterias vivas que forman pilus que se adhieren al tejido son de una cepa deficiente de proteasa, siendo la proteasa una que es al menos responsable de la degradación de las subunidades de pilus. 9. Un procedimiento de acuerdo con la reivindicación 8, en el que la cepa deficiente de proteasa es una cepa degP41. 10. Un procedimiento de acuerdo con cualquiera de las reivindicaciones 1-9, donde la energía de unión libre se pronostica 70 ES 2 225 834 T3 5 10 evaluando la diferencia de la energía media, <∆Vel X−s > definida como <∆Vel X−s >B - <∆Vel X−s >A , entre la contribución de las interacciones no polares a la energía potencial entre la sustancia química X y la que la rodea (denominada S) en dos estados, un estado (A) en donde la sustancia química está rodea por el disolvente y el otro estado (B) en donde la sustancia química, unida al sitio en la chaperona periplásmica molecular o unida a un sitio análogo de tal sitio, está rodea por el disolvente, evaluando la diferencia de la energía media, <∆Vvdw X−s > definida como <∆Vvdw X−s >B - <∆Vvdw X−s >A , entre la contribución de las interacciones no polares a la energía potencial entre la sustancia química X y la que la rodea (denominada S) en dos estados, un estado (A) en donde la sustancia química está rodea por el disolvente y el otro estado (B) en donde la sustancia química, unida al sitio en la chaperona periplásmica molecular o unida a un sitio análogo de tal sitio, está rodea por el disolvente, y calculando la energía libre de unión absoluta como combinación ajustada de las dos diferencias de la energía media mencionadas anteriormente. 15 11. El procedimiento de acuerdo con la reivindicación 10, en el que el sitio en la chaperona molecular periplásmica es el sitio de unión a la subunidad de pilus de una chaperona molecular periplásmica seleccionada entre el grupo constituido por PapD, FimC, SfaE, FaeE, FanE, Cs3-1, F17D, ClpE, EcpD, Mrkb, FimB, SefB, HifB, MyfB, PsaB, PefD, YehC, MrpD, CssC, NfaE, AggD y Caf1M, o un análogo de tal sitio de unión a la subunidad de pilus. 20 12. El procedimiento de acuerdo con la reivindicación 11, en el que el sitio en la chaperona molecular periplásmica es un sitio de unión a la subunidad de pilus de PapD o análogo del mismo. 25 13. El procedimiento de acuerdo con cualquiera de las reivindicaciones 1-12, en el que la sustancia A se elige realizando las siguientes etapas: 30 - co-cristalizar la chaperona periplásmica molecular o el análogo de la misma con un ligando capaz de interactuar con un sitio en la chaperona periplásmica molecular o el análogo de la misma y establecer la conformación tridimensional de la chaperona periplásmica molecular o del análogo de la misma y el ligando cuando interactúan por medio de cristalografía de rayos X, - usar la conformación establecida anteriormente de la chaperona periplásmica molecular o del análogo de la misma para establecer la representación tridimensional del sitio en la chaperona periplásmica molecular o el análogo de la misma que interactúa con el ligando durante la unión, 35 - seleccionar un número de grupos químicos distintos, X1, y determinar las posibles distribuciones espaciales de los grupos químicos X1 que maximizan la energía libre de unión entre los grupos químicos y el sitio en la chaperona o el análogo que interactúa con el ligando, 40 - extraer, de una base de datos que comprende representaciones tridimensionales de moléculas, una molécula que tenga los grupos químicos X1 en las posibles distribuciones espaciales determinadas anteriormente, - modificar opcionalmente la representación tridimensional de la molécula extraída de la base de datos, 45 e identificar la molécula opcionalmente modificada como la sustancia A. 14. Un procedimiento de acuerdo con la reivindicación 13, en el que el ligando es una subunidad de pilus o una parte de la misma cuya chaperona molecular periplásmica normalmente interactúa durante el transporte de la subunidad de pilus a través del espacio periplásmico y/o durante el ensamblaje de pilus. 50 15. Un compuesto nuevo de fórmula general: 55 60 65 71 ES 2 225 834 T3 en la que V1 es O, S, SO, SO2 , CH2 , C(OH)H, CO o CS; 5 W1 es O, S, SO2 , SO3 , CH2 o NH; R1 es H; alquilo C1−24 , alquenilo C1−24 o alquinilo C1−24 , donde el alquilo, alquenilo y alquinilo pueden estar sustituidos con uno o más sustituyentes seleccionados entre OH, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO2 NH2 y -SO2 NH2 ; acilo; o -(CH2 CH2 O)s -H, en el que s = 1, 2, 3; 10 R2 es un grupo de la fórmula 15 20 25 en la que A es -CH-(CH2 )n - o -CH=CH-(CH2 )n−1 - (n > 0) o -C(=Y2 )H-(CH2 )n -; 30 B es -(CH2 )m - o =CH-(CH2 )m−1 - (m > 0); X2 es N, CH o C (donde B es =CH-(CH2 )m−1 -; e 35 Y2 es O, S, NH, H2 o H (n = 1); y 4 > m + n > 0, n < 3 y m < 3; o R2 es un grupo de fórmula 40 45 50 en la que 55 A es -CH-(CH2 )n - o -CH=CH-(CH2 )n−1 - (n > 0) o -C(=Y2 )H-(CH2 )n -; B es -(CH2 )m - o =CH-(CH2 )m−1 - (m > 0); y X’2 es O, NH, CH2 o S (cuando p = 0); N o CH (p = 1); o C (cuando p = 1 y B es =CH-(CH2 )m−1 -); 60 V2 , Z2 y W2 son independientemente H, OH, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO2 NH, -SO2 NH2 , o V2 y Z2 , o Z2 y W2 juntos forman -NHC(O)NH-, -C(O)NHC(O)-, -NHS(O2 ) NH-, -C(O)NHO-, -C(S)NHO-, -S(O2 )NHO- o -S(O2 )NHC(O)-; 65 4 > m + n > 0, n < 3 y m < 3; o R2 es un grupo -W5 -(alquilo C1−5 o alquenilo C2−5 o alquinilo C2−5 ) donde W5 es un enlace o se selecciona entre 72 ES 2 225 834 T3 -O-, -S-, -SO2 - y -NHC(O)- y el resto alquilo C1−5 , alquenilo C2−5 o alquinilo C2−5 puede estar sustituido con hasta tres grupos seleccionados independientemente entre OH, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, NHCONH2 , -NHCSNH2 , -NHSO2 NH2 y -SO2 NH2 ; 5 -Z1 -R3 es -SO2 (OH), -PO(OH)2 , -OSO2 (OH), -NHSO2 (OH), -SPO(OH)2 , -CH2 COOH, tetrazol-5-ilo o tetrazol-5ilmetilo, o sales del mismo; o Z1 es -O-, -S-, -NH- o -CH2 - y R3 es un grupo de fórmula: 10 15 20 25 30 35 D es -CH2 -, -CO-, -SO2 -, -NH-SO2 -, -NH-CO-, -O-PO(OH)- o una sal del mismo; 40 45 Z3 es H, OH, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO2 NH2 , SO2 NH2 , -SO2 (OH), -PO(OH)2 , -OSO2 (OH), -NHSO2 (OH), -COOH, tetrazolil-5-ilo o tatrazolil-5-ilmetilo o una sal del mismo, con la condición de que cuando D es -CH2 , -CO-, -SO2 -, -NHSO2 - o -NHCO-, entonces Z3 es -SO2 (OH), -PO (OH)2 , -OSO2 (OH), -NHSO2 (OH), -COOH, tetrazolil-5-ilo o tetrazolil-5-ilmetilo o una sal del mismo; X3 e Y3 son independientemente H, NO2 , SO2 NH2 , CONH2 , CF3 o F; y 50 U4 -W4 es -CHCH-, -CH2 CH2 -, -C(OH)CH-, -CHC(OH)-, -CH(OH)CH2 -, -CH2 CH(OH)-, -CH(OH)CH(OH)-, -C (O)NH-, -NHC(O)-; Y1 es -OH- o -S-; 55 60 R4 es H o, cuando Y1 es S, S(CH2 )q N(R9 )3 + y q es un número entero de 2-4, donde R9 es H o CH3 ; R5 es H; alquilo C1−6 , alquenilo C2−6 o alquinilo C2−6 , y el resto alquilo C1−6 , alquenilo C2−6 o alquinilo C2−6 puede estar sustituido con OH, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO2 NH2 o -SO2 NH2 ; o arilo, aril-alquilo (C1−2 ), heterociclilo o heterociclil-alquilo (C1−2 ) que pueden estar opcionalmente sustituidos en los restos arilo o heterociclilo con uno, dos o tres sustituyentes seleccionados independientemente entre OH, F, Cl, NH2 , CONH2 , NHCOH y SO2 NH2 ; X1 es -O-, -S- o -NH-; 65 R6 es H o, cuando X1 es NH, acilo, HOCNH-Val-Met-, HOCNH-Ile-(S,S)-dioxo-metionil- o HOCNH-Val-(piran4-on-2-il)-alanil-; o una sal del mismo. 73 ES 2 225 834 T3 16. Un compuesto nuevo de fórmula general 5 10 15 en la que X es O, P, P(O), S, SO, SO2 , CH2 , C(OH)H o un grupo NQ11 , donde Q11 es H, OH, acilo C1−24 o alquilo C1−24 ; 20 Z11 es un enlace, O, CH2 , S, SO, SO2 o un grupo NQ12 , donde Q12 es H, acilo C1−24 o alquilo C1−24 ; R11 es H; alquilo C1−24 , alquenilo C2−24 o alquinilo C2−24 , que puede estar sustituido con uno o más sustituyentes seleccionados independientemente entre -OH, -COOH, -F, -Cl, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO3 NH2 y -SO2 NH2 ; acilo; o -(CH2 CH2 O)s -H, donde s = 1, 2 ó 3; 25 o R11 es CH=CH-(CH2 )n0 -Q13 o -(CH2 )n0 -Q13 , donde Q13 es un grupo arilo o heteroarilo sustituido con -OH, -COOH, -F, -Cl, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO3 NH2 y -SO2 NH2 , y donde n’ ≥ 0; 30 R12 y R13 son independientemente OH, H, F, Cl, OW11 u O(CO)W11 , donde W11 es alquilo C1−24 , alquenilo C2−24 o alquinilo C2−24 , o un grupo arilo o heteroarilo sustituido con -OH, -COOH, -F, -Cl, -CNH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO3 NH2 y -SO2 NH2 ; Z12 es un enlace, O, S o CH2 ; 35 R14 es -(CH2 )n00 -Q14 , donde Q14 es un grupo arilo o un grupo heteroarilo, dicho grupo arilo o heteroarilo está sustituido con -OH, -COOH, -F, -Cl, -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO3 NH2 y -SO2 NH2 , y donde n” = 0, 1, 2 ó 3; 40 45 Z13 es un enlace, O, CH2 , S, SO, SO2 , NQ14 Q15 , donde Q14 es H, acilo C1−24 o alquilo C1−24 , y Q15 es CO o -C(O) W12 , donde W12 es O o NW13 , donde W13 es H, OH, acilo C1−24 o alquilo C1−24 ; R15 es H; alquilo C1−24 , alquenilo C2−24 o alquinilo C2−24 , donde el alquilo, alquenilo o alquinilo pueden estar sustituidos con uno o más sustituyentes seleccionados independientemente entre -OH, -COOH, -F, -Cl -CONH2 , -CSNH2 , -CONHOH, -CSNHOH, -NHCHO, -NHCONH2 , -NHCSNH2 , -NHSO3 NH2 y -SO2 NH2 ; acilo o -(CH2 CH2 O)s -H, donde s = 1, 2 ó 3; o R15 es CH=CH-(CH2 )n0 -Q13 o -(CH2 )n0 -Q13 , donde Q3 es como se ha definido anteriormente y donde n’ ≥ 0; 50 55 o una sal del mismo. 17. Un compuesto de la reivindicación 15 ó 16, que provoca la prevención, inhibición o mejora de la unión de G1’19’WT a PapD, y/o provoca la prevención, inhibición o mejora de la unión de MBP-G140’ a PapD, y/o provoca la prevención, inhibición o mejora de la unión de G125’-140’ a PapD, y/o provoca la prevención, inhibición o mejora de una subunidad de pilus a PapD. 18. Un nuevo piranósido o una sal del mismo, que se selecciona entre el grupo constituido por 2,3-O-Dibenzoil-4-O-bencil-1-tio-β-D-glucohexopiranósida de etilo; 60 6-O-Acetil-2,3-O-dibenzoil-4-O-bencil-1-tio-β-D-glucohexopiranósido de etilo; 6-O-Acetil-2,3-O-dibenzoil-4-O-bencil-β-D-glucohexopiranósido de metilglicolilo; 65 4-O-Bencil-β-D-glucopiranósido de 2-(hidroxi) etilo; 4-O-Bencil-β-D-glucohexopiranósido de glicolil sódico; 74 ES 2 225 834 T3 2-O-Etil-4,6-O-(4’-metoxi)fenilmetilen-α-D-mannohexopiranósido de metilo; 2-O-Etil-3-O-dimetil-t-butilsilil-4,6-O-(4’-metoxi)fenilmetilen-α-D-mannohexopiranósido de metilo; 5 2-O-etil-3-O-dimetil-t-butilsilil-4,-O-(4’-metoxi)-bencil-α-D-mannohexopiranósido de metilo; 2-O-Etil-3-O-dimetil-t-butilsilil-6-O-(4’-metoxi)-bencil-α-D-mannohexopiranósido de metilo; 2-O-Etil-3-O-dimetil-t-butilsilil-4,-O-(4’-metoxi)-bencil-6(S)-fenil-α-D-mannohexopiranósido de metilo; 10 2,3-Anhidro-4,6-O-p-metoxibenciliden-α-D-mannopiranósido de metilo; 3-Azido-4,6-O-p-metoxibenciliden-α-D-altropiranósido de metilo; 15 3-Azido-2-O-etil-4,6-O-p-metoxibenciliden-α-D-altropiranósido de metilo; 3-Azido-3-desoxi-2-O-etil-4-O-p-metoxibencil-α-D-altropiranósido de metilo; 3-Azido-6-O-benzoil-3-desoxi-2-O-etil-4-O-p-metoxi-bencil-α-D-altropiranósido de metilo; 20 Sal sódica de 6-O-benzoil-3-desoxi-2-O-etil-4-O-p-metoxibencil-3-sulfamino-α-D-altropiranósido de metilo; Sal de amonio de 6-O-benzoil-3-desoxi-2-O-etil-3-sulfamino-α-D-altropiranósido de metilo; 25 3-Azido-6-O-pivaloil-3-desoxi-2-O-etil-4-O-p-metoxi-bencil-α-D-altropiranósido de metilo; Sal sódica de 6-O-pivaloil-3-desoxi-2-O-etil-4-O-p-metoxibencil-3-sulfamino-α-D-altropiranósido de metilo; Sal de amonio de 6-O-pivaloil-3-desoxi-2-O-etil-3-sulfamino-α-D-altropiranósido de metilo; 30 6-O-pivaloil-3-desoxi-2-O-etil-4-O-p-metoxibencil-3-t-butiloxamido-α-D-altropiranósido de metilo; Sal de amonio de 6-O-pivaloil-3-desoxi-2-O-etil-3-oxamido-α-D-altropiranósido de metilo; 35 3-Azido-6-O-pirrol-3’-ilcarboxil-3-desoxi-2-O-etil-4-O-p-metoxibencil-α-D-altropiranósido; y Sal de amonio de 6-O-pirrol-3’-ilcarboxil-3-desoxi-2-O-etil-3-sulfamino-α-D-altropiranósido de metilo. 40 45 50 19. Una a una composición farmacéutica, que comprende, como compuesto activo, una sustancia que puede interactuar con al menos un tipo de chaperona molecular periplásmica que une las subunidades de pilus durante el transporte de esas subunidades de pilus a través del espacio periplásmico y/o durante el procedimiento de ensamblaje de los compuestos de pilus intacto, de tal forma que la unión de las subunidades de pilus a la chaperona molecular periplásmica se previene, inhibe o mejora, donde la prevención, inhibición o mejora de la unión se realiza por la interacción con, en la chaperona molecular periplásmica, un sitio de unión que es un sitio de unión al que la parte carboxilo terminal de una subunidad de pilus es capaz de unirse, y que comprende puntos de sitio sustancialmente idénticos a los residuos invariantes Arg-8 y Lys-112 en PapD y un fragmento polipeptídico que es capaz de interactuar con una cadena β de la parte carboxilo terminal de la subunidad de pilus, estabilizando por lo tanto la unión de dicha subunidad en los puntos de sitio Arg-8 y Lys-112 del sitio de unión, en combinación con al menos un vehículo o excipiente farmacéuticamente aceptable, siendo la sustancia un compuesto de acuerdo con cualquiera de las reivindicaciones 1518. 20. Una composición farmacéutica de acuerdo con la reivindicación 19, que además comprende al menos una sustancia farmacéutica adicional. 55 21. Una composición farmacéutica de acuerdo con la reivindicación 20, donde al menos una sustancia farmacéutica adicional es un agente antibacteriano seleccionado entre el grupo constituido por penicilinas, cefalosporinas, aminoglicósidos, sulfonamidas, tetraciclinas, cloranfenicol, polimixinas, fármacos anti-micobacterianos y antisépticos urinarios. 60 22. Una sustancia para uso farmacéutico, siendo la sustancia capaz de interactuar con al menos un tipo de chaperona molecular periplásmica que une las subunidades de pilus durante el transporte de esas subunidades de a través del espacio periplásmico y/o durante el procedimiento de ensamblaje de los compuestos de pilus intacto, de tal forma que la unión de las subunidades de pilus a la chaperona molecular periplásmica se previene, inhibe o mejora, donde el sitio de unión es un sitio de unión al que la parte carboxilo terminal de una subunidad de pilus es capaz de unirse, y que comprende puntos de sitio sustancialmente idénticos a los residuos invariantes Arg-8 y Lys-112 en PapD y un fragmento polipeptídico que es capaz de interactuar con una cadena β de la parte carboxilo terminal de la subunidad de pilus, estabilizando por lo tanto la unión de dicha subunidad en los puntos de sitio Arg-8 y Lys-112 del sitio de unión, donde la sustancia es un compuesto de acuerdo con cualquiera de las reivindicaciones 15-18. 65 75 ES 2 225 834 T3 23. La sustancia de acuerdo con la reivindicación 22, en el que la sustancia provoca la prevención, inhibición o mejora de la unión de G1’-19’WT a PapD, y/o causa la prevención, inhibición o mejora de la unión de MBP-G140’ a PapD, y/o provoca la prevención, inhibición o mejora de la unión de G125’-140’ a PapD, y/o provoca la prevención, inhibición o mejora de una subunidad de pilus a PapD. 5 10 15 20 25 24. El uso de una sustancia para la preparación de una composición farmacéutica para el tratamiento y/o profilaxis de enfermedades provocadas por bacterias que forman pilus que se adhieren al tejido, siendo dicha sustancia capaz de prevenir, inhibir o mejorar la unión entre al menos un tipo de subunidad de pilus y al menos un tipo de chaperona molecular periplásmica en las bacterias que forman pilus, siendo la chaperona molecular periplásmica una que une subunidades de pilus durante el transporte de dichas subunidades de pilus a través del espacio periplásmico y/o durante el procedimiento de ensamblaje del compuesto de pilus intacto, donde la prevención, inhibición o mejora de la unión se realiza por la interacción con, en la chaperona molecular periplásmica, un sitio de unión al que la parte carboxilo terminal de una subunidad de pilus es capaz de unirse, y que comprende puntos de sitio sustancialmente idénticos a los residuos invariantes Arg-8 y Lys-112 en PapD y un fragmento polipeptídico que es capaz de interactuar con una cadena β de la parte carboxilo terminal de la subunidad de pilus, estabilizando por lo tanto la unión de dicha subunidad en los puntos de sitio Arg-8 y Lys-112 del sitio de unión, siendo la sustancia un compuesto de acuerdo con cualquiera de las reivindicaciones 15-18. 25. El uso de acuerdo con la reivindicación 24, en el que las bacterias se seleccionan entre el grupo constituido por Haemophilus spp, Helicobacter spp, Pseudomonas aeruginosa, Mycoplasma spp y todos los miembros de la familia Enterobacteriacieae, incluyendo Escherichia spp, Salmonella spp, Bordetella spp, Yersinia spp, Klebsiella spp., y Proteus spp. 26. El uso de acuerdo con las reivindicaciones 24-25, en el que la chaperona molecular periplásmica es una proteína seleccionada entre el grupo constituido por PapD, FimC, SfaE, FaeE, FanE, Cs3-1, F17D, ClpE, EcpD, Mrkb, FimB, SefB, HifB, MyfB, PsaB, PefD, YehC, MrpD, CssC, NfaE, AggD y Caf1M. 27. El uso de acuerdo con la reivindicación 24, en el que el sitio de unión es uno al que es capaz de unir el péptido G125’-140’ y/o el péptido de fusión MBP-140’ y/o el péptido G1’-19’. 30 35 40 28. El uso de una sustancia para la preparación de una composición farmacéutica para el tratamiento y/o profilaxis de enfermedades provocadas por infecciones bacterianas, siendo dicha sustancia capaz de interactuar con al menos un tipo de chaperona molecular periplásmica que une subunidades de pilus durante el transporte de dichas subunidades de pilus a través del espacio periplásmico y/o durante el procedimiento de ensamblaje del compuesto de pilus intacto, de tal forma que la unión de las subunidades de pilus a la chaperona molecular periplásmica se previene, inhibe o mejora, siendo el sitio de unión un sitio de unión al que la parte carboxilo terminal de una subunidad de pilus es capaz de unirse, y que comprende puntos de sitio sustancialmente idénticos a los residuos invariantes Arg-8 y Lys-112 en PapD y un fragmento polipeptídico que es capaz de interactuar con una cadena β de la parte carboxilo terminal de la subunidad de pilus, estabilizando de dicha forma la unión de dicha subunidad en los puntos de sitio Arg-8 y Lys-112 del sitio de unión, siendo la sustancia un compuesto de acuerdo con cualquiera de las reivindicaciones 15-18. 45 50 55 60 65 76 ES 2 225 834 T3 77 ES 2 225 834 T3 78 ES 2 225 834 T3 79 ES 2 225 834 T3 80 ES 2 225 834 T3 81 ES 2 225 834 T3 82 ES 2 225 834 T3 83 ES 2 225 834 T3 84 ES 2 225 834 T3 85 ES 2 225 834 T3 86 ES 2 225 834 T3 87 ES 2 225 834 T3 88 ES 2 225 834 T3 89 ES 2 225 834 T3 90 ES 2 225 834 T3 91 ES 2 225 834 T3 92 ES 2 225 834 T3 93 ES 2 225 834 T3 94 ES 2 225 834 T3 95 ES 2 225 834 T3 96 ES 2 225 834 T3 97 ES 2 225 834 T3 98 ES 2 225 834 T3 99 ES 2 225 834 T3 100 ES 2 225 834 T3 LISTA DE SECUENCIAS (1) INFORMACIÓN GENERAL: 5 (i) SOLICITANTE: (A) NOMBRE: Washington University (B) CALLE: 724 South Euclide Avenue 10 (C) CIUDAD: St. Louis (D) ESTADO: Missouri (E) PAÍS: Estados Unidos de América (F) CÓDIGO POSTAL: MO 63110 15 (i) SOLICITANTE: (A) NOMBRE: Symbicom Aktiebolag (B) CALLE: Tvistevaegen 48 20 (C) CIUDAD: Umea (E) PAÍS: Suecia (F) CÓDIGO POSTAL: S-907 36 Umea 25 (ii) TÍTULO DE LA INVENCIÓN: TRATAMIENTO O PROFILAXIS DE ENFERMEDADES CAUSADAS POR BACTERIAS QUE FORMAN PILUS (iii) NÚMERO DE SECUENCIAS: 27 30 (iv) FORMA LEGIBLE POR ORDENADOR: (A) TIPO DE MEDIO: Disco flexible (B) ORDENADOR: PC IBM compatible (C) SISTEMA OPERATIVO: PC-DOS/MS-DOS 35 (D) SOFTWARE: PatentIn Release Nº 1.0, Versión Nº 1.25 (EPO) (2) INFORMACIÓN PARA LA SEC ID Nº: 1: 40 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 874 pares de bases (B) TIPO: ácido nucleico (C) CADENA: sencilla 45 (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: ADN (genómico) (iii) HIPOTÉTICO: NO 50 (iii) ANTI-SENTIDO: NO (vi) FUENTE ORIGINAL: 55 (A) ORGANISMO: Escherichia coli (ix) CARACTERÍSTICAS: (A) NOMBRE/CLAVE: CDS (B) LOCALIZACIÓN: 1..720 60 (ix) CARACTERÍSTICAS: (A) NOMBRE/CLAVE: sig_peptide (B) LOCALIZACIÓN: 1..63 65 (ix) CARACTERÍSTICAS: (A) NOMBRE/CLAVE: matpeptide 1 ES 2 225 834 T3 (B) LOCALIZACIÓN: 64..717 (x) INFORMACIÓN DE LA PUBLICACIÓN: 5 (A) AUTORES: Nogren, M. Baga, M. Tennet, J.M. Normark, S. (C) REVISTA: Mol. Biol. Rep. 10 (D) VOLUMEN: 12 (F) PÁGINAS: 169-178 (G) FECHA: 1987 15 (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 1: 20 25 30 35 40 45 50 55 60 65 2 ES 2 225 834 T3 5 10 15 20 25 (2) INFORMACIÓN PARA LA SEC ID Nº: 2: 30 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 239 aminoácidos (B) TIPO: aminoácido (D) TOPOLOGÍA: lineal 35 (ii) TIPO DE MOLÉCULA: proteína (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 2: 40 45 50 55 60 65 3 ES 2 225 834 T3 5 10 15 20 25 30 35 40 45 50 55 (2) INFORMACIÓN PARA LA SEC ID Nº: 3: (i) CARACTERÍSTICAS DE LA SECUENCIA: 60 (A) LONGITUD: 30 pares de bases (B) TIPO: ácido nucleico (C) CADENA: sencilla 65 (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: ADN (sintético) 4 ES 2 225 834 T3 (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 3: GTCAAACACC GCCGGAACTC GTCCAGGCGA 5 30 (2) INFORMACIÓN PARA LA SEC ID Nº: 4: (i) CARACTERÍSTICAS DE LA SECUENCIA: 10 (A) LONGITUD: 30 pares de bases (B) TIPO: ácido nucleico (C) CADENA: sencilla (D) TOPOLOGÍA: lineal 15 (ii) TIPO DE MOLÉCULA: ADN (sintético) (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 4: 20 CGGGCGATAA AAAAGAGCTA TTTTGGTCTG 30 (2) INFORMACIÓN PARA LA SEC ID Nº: 5: 25 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 27 pares de bases (B) TIPO: ácido nucleico (C) CADENA: sencilla 30 (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: ADN (sintético) (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 5: 35 GCGATAAAAA AGCATTATTT TCCTCTG 27 (2) INFORMACIÓN PARA LA SEC ID Nº: 6: 40 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 19 aminoácidos (B) TIPO: aminoácido 45 (C) CADENA: sencilla (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: péptido 50 55 (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 6: Gly Lys Arg Lys Pro Gly Glu Leu Ser Gly Ser Met Thr Met Val Leu 1 5 10 15 Ser Phe Pro (2) INFORMACIÓN PARA LA SEC ID Nº: 7: 60 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 16 aminoácidos (B) TIPO: aminoácido 65 (C) CADENA: sencilla (D) TOPOLOGÍA: lineal 5 ES 2 225 834 T3 (ii) TIPO DE MOLÉCULA: péptido (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 7: 5 Lys Pro Gly Glu Leu Ser Gly Ser Met Thr Met Val Leu Ser Phe Pro 1 5 10 15 (2) INFORMACIÓN PARA LA SEC ID Nº: 8: 10 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 11 aminoácidos 15 (B) TIPO: aminoácido (C) CADENA: sencilla (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: péptido 20 (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 8: 25 Ser Gly Ser Met Thr Met Val Leu Ser Phe Pro 1 5 10 (2) INFORMACIÓN PARA LA SEC ID Nº: 9: 30 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 7 aminoácidos (B) TIPO: aminoácido (C) CADENA: sencilla 35 (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: péptido (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 9: 40 Thr Met Val Leu Ser Phe Pro 1 5 45 (2) INFORMACIÓN PARA LA SEC ID Nº: 10: (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 19 aminoácidos 50 (B) TIPO: aminoácido (C) CADENA: sencilla (D) TOPOLOGÍA: lineal 55 (ii) TIPO DE MOLÉCULA: péptido (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 10: 60 65 Gly Lys Arg Lys Pro Val Glu Leu Ser Gly Ser Met Thr Met Val Leu 1 5 10 15 Ser Ser Pro (2) INFORMACIÓN PARA LA SEC ID Nº: 11: (i) CARACTERÍSTICAS DE LA SECUENCIA: 6 ES 2 225 834 T3 (A) LONGITUD: 19 aminoácidos (B) TIPO: aminoácido (C) CADENA: sencilla 5 (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: péptido (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 11: 10 15 Gly Lys Arg Lys Pro Gly Glu Leu Ser Gly Ser Met Thr Met Val Leu 1 5 10 15 Ser Phe Pro (2) INFORMACIÓN PARA LA SEC ID Nº: 12: 20 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 20 aminoácidos (B) TIPO: aminoácido 25 (C) CADENA: sencilla (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: péptido 30 35 (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 12: Glu Glu Gly Lys Arg Lys Pro Gly Glu Leu Ser Gly Ser Met Thr Met 1 5 10 15 Val Leu Ser Phe 20 (2) INFORMACIÓN PARA LA SEC ID Nº: 13: 40 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 19 aminoácidos (B) TIPO: aminoácido 45 (C) CADENA: sencilla (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: péptido 50 55 (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 13: Gln Asn Leu Ile Ala Gly Pro Phe Ser Ala Thr Ala Thr Leu Val Ala 1 5 10 15 Ser Tyr Ser (2) INFORMACIÓN PARA LA SEC ID Nº: 14: 60 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 19 aminoácidos (B) TIPO: aminoácido 65 (C) CADENA: sencilla (D) TOPOLOGÍA: lineal 7 ES 2 225 834 T3 (ii) TIPO DE MOLÉCULA: péptido (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 14: 5 Lys Lys Leu Glu Ala Gly Asn Tyr Phe Ala Val Leu Gly Phe Arg Val 1 5 10 15 Asp Tyr Glu 10 (2) INFORMACIÓN PARA LA SEC ID Nº: 15: (i) CARACTERÍSTICAS DE LA SECUENCIA: 15 (A) LONGITUD: 19 aminoácidos (B) TIPO: aminoácido (C) CADENA: sencilla 20 (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: péptido (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 15: 25 Lys Ser Val Val Pro Gly Asp Tyr Glu Ala Thr Ala Thr Phe Glu Leu 1 5 10 15 Thr Tyr Arg 30 (2) INFORMACIÓN PARA LA SEC ID Nº: 16: (i) CARACTERÍSTICAS DE LA SECUENCIA: 35 (A) LONGITUD: 19 aminoácidos (B) TIPO: aminoácido (C) CADENA: sencilla 40 (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: péptido (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 16: 45 Gly Ile Leu Asn Gly Gly Asp Phe Gln Thr Thr Ala Ser Met Ala Met 1 5 10 15 Ile Tyr Asn 50 (2) INFORMACIÓN PARA LA SEC ID Nº: 17: (i) CARACTERÍSTICAS DE LA SECUENCIA: 55 (A) LONGITUD: 16 aminoácidos (B) TIPO: aminoácido (C) CADENA: sencilla 60 (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: péptido (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 17: 65 Tyr Ala Leu Ala Pro Asn Ala Val Ile Pro Thr Ser Leu Ala Leu Leu 1 5 10 15 8 ES 2 225 834 T3 (2) INFORMACIÓN PARA LA SEC ID Nº: 18: (i) CARACTERÍSTICAS DE LA SECUENCIA: 5 (A) LONGITUD: 19 aminoácidos (B) TIPO: aminoácido (C) CADENA: sencilla (D) TOPOLOGÍA: lineal 10 (ii) TIPO DE MOLÉCULA: péptido (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 18: 15 Lys Ser Val Val Pro Gly Asp Tyr Glu Ala Thr Ala Thr Phe Glu Leu 1 5 10 15 Thr Tyr Arg 20 (2) INFORMACIÓN PARA LA SEC ID Nº: 19: (i) CARACTERÍSTICAS DE LA SECUENCIA: 25 (A) LONGITUD: 16 aminoácidos (B) TIPO: aminoácido (C) CADENA: sencilla 30 (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: péptido (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 19: 35 Leu Pro Ala Thr Asn Thr Leu Met Leu Ser Phe Asp Asn Val Gly Gly 1 5 10 15 40 (2) INFORMACIÓN PARA LA SEC ID Nº: 20: (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 16 aminoácidos 45 (B) TIPO: aminoácido (C) CADENA: sencilla (D) TOPOLOGÍA: lineal 50 (ii) TIPO DE MOLÉCULA: péptido (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 20: 55 Asp Gln Ile Lys Gln leu Pro Ala Thr Asn Thr Leu Met Leu Ser Phe 1 5 10 15 (2) INFORMACIÓN PARA LA SEC ID Nº: 21: 60 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 16 aminoácidos (B) TIPO: aminoácido (C) CADENA: sencilla 65 (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: péptido 9 ES 2 225 834 T3 (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 21: 5 Tyr Lys Met Pro Tyr Asp Gln Ile Lys Gln Leu Pro Ala Thr Asn Thr 1 5 10 15 (2) INFORMACIÓN PARA LA SEC ID Nº: 22: 10 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 15 aminoácidos (B) TIPO: aminoácido (C) CADENA: sencilla 15 (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: péptido (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 22: 20 Gln His His Tyr Tyr Asp Leu Trp Gln Asp His Tyr Lys Met Pro Tyr 1 5 10 15 25 (2) INFORMACIÓN PARA LA SEC ID Nº: 23: (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 33 pares de bases 30 (B) TIPO: ácido nucleico (C) CADENA: sencilla (D) TOPOLOGÍA: lineal 35 (ii) TIPO DE MOLÉCULA: ADN (sintético) (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 23: 40 CCCCCCTGCA GATCAGATTA AGCAGCTACC TGC 33 (2) INFORMACIÓN PARA LA SEC ID Nº: 24: (i) CARACTERÍSTICAS DE LA SECUENCIA: 45 (A) LONGITUD: 34 pares de bases (B) TIPO: ácido nucleico (C) CADENA: sencilla 50 (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: ADN (sintético) (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 24: 55 CCCCTGCAGT AAAAATATCT CTGCTCAGAA ATAC (2) INFORMACIÓN PARA LA SEC ID Nº: 25: 60 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 28 pares de bases (B) TIPO: ácido nucleico 65 (C) CADENA: sencilla (D) TOPOLOGÍA: lineal 10 34 ES 2 225 834 T3 (ii) TIPO DE MOLÉCULA: ADN (sintético) (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 25: 5 CCATCGATGA ACAGCCAGTC AGATAATC 28 (2) INFORMACIÓN PARA LA SEC ID Nº: 26: 10 (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 61 pares de bases (B) TIPO: ácido nucleico (C) CADENA: sencilla 15 (D) TOPOLOGÍA: lineal (ii) TIPO DE MOLÉCULA: ADN (sintético) 20 (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 26: GGAAAGAGAA AACCCGGGGA GCTATCTGGT TCTATGACTA TGGTTCTGAG TTTCCCCTGA T 25 60 61 (2) INFORMACIÓN PARA LA SEC ID Nº: 27: (i) CARACTERÍSTICAS DE LA SECUENCIA: (A) LONGITUD: 67 pares de bases 30 (B) TIPO: ácido nucleico (C) CADENA: sencilla (D) TOPOLOGÍA: lineal 35 (ii) TIPO DE MOLÉCULA: ADN (sintético) (xi) DESCRIPCIÓN DE LA SECUENCIA: SEC ID Nº: 27: 40 CGATCAGGGG AAACTCAGAA CCATAGTCAT AGAACCAGAT AGCTCCCCGG GTTTTCTCTT TCCTGCA 45 50 55 60 65 11 60 67