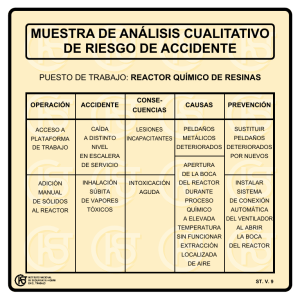
UNIVERSIDAD SIMÓN BOLÍVAR
Anuncio

UNIVERSIDAD SIMÓN BOLÍVAR DECANATO DE ESTUDIOS PROFESIONALES COORDINACIÓN DE INGENIERÍA QUÍMICA SÍNTESIS DE METANOL EN REACTOR DE MICROCANALES Por: Andrea Carolina Machado Miguens Realizado con la asesoría de: Prof. Susana Zeppieri Prof. Hilde Venvik Prof. Anders Holmen PROYECTO DE GRADO Presentado ante la Ilustre Universidad Simón Bolívar como requisito parcial para optar al título de Ingeniero Químico Sartenejas, Noviembre de 2008 UNIVERSIDAD SIMÓN BOLÍVAR DECANATO DE ESTUDIOS PROFESIONALES COORDINACIÓN DE INGENIERÍA QUÍMICA SÍNTESIS DE METANOL EN REACTOR DE MICROCANALES Por: Andrea Carolina Machado Miguens Realizado con la asesoría de: Prof. Susana Zeppieri Prof. Hilde Venvik Prof. Anders Holmen PROYECTO DE GRADO Presentado ante la Ilustre Universidad Simón Bolívar como requisito parcial para optar al título de Ingeniero Químico Sartenejas, Noviembre de 2008 RESUMEN A través de los años, los científicos han tratado de sintetizar catalizadores que permitan que la reacción de síntesis de metanol se lleve a cabo a condiciones menos severas (50-80 bar y 200-280 °C). Con cada logro obtenido en la mejora de la actividad catalítica se ha presentado la necesidad de desarrollar nuevos procesos para aprovechar las ventajas alcanzadas. Los reactores de microcanales tienen el potencial para transformar la economía de estos procesos. Es por esto que el objetivo general del proyecto fue evaluar la actividad catalítica de un catalizador de CuO/ZnO/Al2O3 para la síntesis de metanol. Y los objetivos específicos fueron: sintetizar el catalizador para la síntesis del metanol, siguiendo el método patentado por Imperial Chemical Industries Limited [20]; caracterizar los materiales del catalizador a través de técnicas como difracción de rayos X, adsorción/desorción N2, reducción a temperatura programada e inducción de plasma acoplada-espectroscopía de emisión atómica; evaluar la reproducibilidad de la técnica de síntesis aplicada mediante la caracterización de los materiales de otro lote de catalizador; realizar pruebas de corrida en blanco, sin catalizador, tanto en el reactor de microcanales como en el reactor tubular para verificar la existencia de actividad del acero inoxidable, material de construcción de los reactores; estudiar la desactivación del catalizador en el reactor tubular con el catalizador diluido en SiC y sin diluir, y en el reactor de microcanales; realizar pruebas de actividad catalítica en el reactor tubular con el catalizador diluido y con el catalizador sin diluir; realizar pruebas de actividad catalítica en el reactor de microcanales; estudiar el efecto de variar el flujo de gas de síntesis alimentado a los reactores y comparar las conversiones obtenidas para la producción de metanol con el reactor tubular y el reactor de microcanales. Los resultados obtenidos fueron: no se concluyó acerca de las fases presentes en los catalizadores debido a sus estructuras amorfas; las áreas superficiales específicas resultaron ser 173 m2/g para el primer lote y 170 m2/g para el segundo; los perfiles de reducción a temperatura programada mostraron que el primer lote era más homogéneo que el segundo; no se detectó actividad del acero inoxidable; el catalizador diluido en SiC en el reactor tubular presentó menor desactivación que el catalizador sin diluir, el catalizador en el reactor de microcanales no presentó desactivación; se obtuvo que la conversión de CO disminuyó a medida que el flujo de gas se incrementó y finalmente, se obtuvo que las mayores conversiones se alcanzaron con el reactor de microcanales. Palabras claves: síntesis metanol, reactor microcanales, catalizador. ÍNDICE GENERAL RESUMEN ..................................................................................................................................... iv ÍNDICE DE FIGURAS ................................................................................................................ viii ÍNDICE DE TABLAS.................................................................................................................... xi LISTA DE SÍMBOLOS Y ABREVIATURAS ............................................................................ xii INTRODUCCIÓN........................................................................................................................... 1 CAPÍTULO 1: FUNDAMENTOS TEÓRICOS ............................................................................. 4 1.1 Metanol................................................................................................................................... 4 1.2 Termodinámica del metanol ................................................................................................... 5 1.3 Gas de síntesis para la producción de metanol ....................................................................... 8 1.4 Síntesis de metanol ................................................................................................................. 9 1.5 Rol del dióxido de carbono en la síntesis del metanol ......................................................... 12 1.6 Caracterización de los materiales de catalizadores............................................................... 13 1.6.1 Difracción de rayos X........................................................................................................ 13 1.6.2 Adsorción/desorción de N2 ................................................................................................ 14 1.6.3 Reducción a temperatura programada ............................................................................... 17 1.7 Microreactor ......................................................................................................................... 18 CAPÍTULO 2: METODOLOGÍA EXPERIMENTAL ................................................................. 21 2.1 Catalizador para la síntesis de metanol ................................................................................ 21 2.1.1 Síntesis del catalizador ...................................................................................................... 21 2.1.2 Caracterización de los materiales del catalizador.............................................................. 22 2.1.2.1 Difracción de rayos X..................................................................................................... 22 2.1.2.2 Adsorción/desorción de N2 ............................................................................................. 24 2.1.2.3 Inducción de plasma acoplada-espectroscopía de emisión atómica............................... 24 2.1.2.4 Reducción a temperatura programada ............................................................................ 24 2.2 Montaje experimental para la síntesis de metanol................................................................ 24 2.2.1 Medidores y controladores de flujo másico....................................................................... 27 2.2.2 Medidor y controlador de presión ..................................................................................... 27 2.2.3 Cromatógrafo de gases ...................................................................................................... 27 2.2.3.1 Análisis de la fase gaseosa ............................................................................................. 27 2.2.3.2 Análisis de la fase líquida............................................................................................... 28 2.2.4 Reactor tubular de lecho fijo ............................................................................................. 31 2.2.5 Reactor de microcanales.................................................................................................... 32 2.3 Experimentos ........................................................................................................................ 34 2.3.1 Corridas en blanco ............................................................................................................. 35 2.3.2 Reducción de catalizador................................................................................................... 36 2.3.3 Pruebas de actividad catalítica........................................................................................... 36 CAPÍTULO 3: RESULTADOS Y DISCUSIONES ..................................................................... 41 3.1 3.1.1 Catalizador para la síntesis de metanol ................................................................................ 41 Caracterización de los materiales del catalizador.............................................................. 41 3.1.1.1 Difracción de rayos X..................................................................................................... 41 3.1.1.2 Adsorción/desorción de N2 ............................................................................................. 44 3.1.1.3 Inducción de plasma acoplada-espectroscopía de emisión atómica............................... 44 3.1.1.4 Reducción a temperatura programada ............................................................................ 45 3.2 Experimentos ........................................................................................................................ 47 3.2.1 Corridas en blanco ............................................................................................................. 47 3.2.2 Pruebas de actividad catalítica........................................................................................... 49 3.2.2.1 Realizada en el reactor tubular con el catalizador diluido en Sic................................... 49 3.2.2.2 Realizada en el reactor tubular con el catalizador sin diluir........................................... 54 vi 3.2.2.3 Realizada en el reactor de microcanales con el catalizador sin diluir ............................ 58 CONCLUSIONES......................................................................................................................... 62 REFERENCIAS BIBLIOGRÁFICAS .......................................................................................... 65 APÉNDICES ................................................................................................................................. 67 vii ÍNDICE DE FIGURAS Figura 1.1. ∆G0 como una función de la temperatura de los productos de la reacción de CO con H2 [8] ............................................................................................................................................... 6 Figura 1.2. Conversión de equilibrio de CO a metanol; alimentación de H2/CO=2 [8].................. 7 Figura 1.3. Concentración de equilibrio de metanol a la salida como función de la fracción molar de H2 y CO en la alimentación [9] ........................................................................................ 8 Figura 1.4. Isoterma de adsorción de BET [17] ............................................................................ 16 Figura 2.1. Esquema de la síntesis del catalizador para la producción de metanol....................... 23 Figura 2.2. Diagrama del montaje experimental para la síntesis de metanol ................................ 26 Figura 2.3. Configuración y dimensiones del reactor tubular de lecho fijo .................................. 31 Figura 2.4. Reactor de microcanales con intercambiador de calor, fabricado por FZK................ 32 Figura 2.5. Lámina de metal.......................................................................................................... 33 Figura 2.6. Lámina para la transferencia de calor ......................................................................... 33 Figura 2.7. Ensamblaje de las láminas del reactor de microcanales.............................................. 34 Figura 2.8. Disposición del catalizador sin diluir en el reactor tubular......................................... 38 Figura 3.1. Patrones de difracción de rayos X del primer lote (línea negra) y segundo lote (línea gris) del catalizador CuO/ZnO/Al2O3 mostrando picos de difracción de CuO (♦), ZnO (●) y Al2O3 (■)........................................................................................................................................ 42 Figura 3.2. Patrones de difracción de rayos X del segundo lote del catalizador CuO/ZnO/Al2O3 antes de la calcinación (línea gris) y después de la calcinación (línea negra) mostrando picos de difracción de Cu(NO3)2 (♦), Al(NO3)3 (●), Zn(NO3)2 (■) y ZnCO3 (▲)...................................... 43 Figura 3.3. Perfiles de TPR del primer lote de Cu/ZnO/Al2O3 y del segundo lote de Cu/ZnO/Al2O3. Condiciones: ~0,15 g de muestra, mezcla de gas de 7 % de H2 en Ar, 25-350 °C a 2 °C/min...................................................................................................................................... 46 Figura 3.4. Conversión de CO en función del tiempo durante la prueba en blanco realizada en el reactor de microcanales. Condiciones: 80 bar y 255 °C............................................................ 47 Figura 3.5. Conversión de CO en función del tiempo durante la prueba en blanco realizada en el reactor tubular. Condiciones: 80 bar y 255 °C .......................................................................... 48 Figura 3.6. Conversión de CO y CO+CO2 en función del flujo volumétrico de gas de síntesis alimentado al reactor para el experimento realizado en el reactor tubular de lecho fijo con el catalizador diluido en SiC. Condiciones de reacción: 80 bar y 255 °C......................................... 50 Figura 3.7. Conversión de CO y CO+CO2 en función de la temperatura de la reacción para el experimento realizado en el reactor tubular de lecho fijo con el catalizador diluido en SiC. Condiciones de reacción: 80 bar y 75 nmL/min de gas de síntesis alimentado ............................ 51 Figura 3.8. Conversión de CO y CO+CO2 en función de la presión de la reacción para el experimento realizado en el reactor tubular de lecho fijo con el catalizador diluido en SiC. Condiciones de reacción: 255 °C y 75 nmL/min de gas de síntesis alimentado ........................... 52 Figura 3.9. Desactivación del catalizador durante el experimento realizado en el reactor tubular de lecho fijo con el catalizador diluido en SiC. Condiciones: 80 bar, 255 °C y 75 nmL/min de gas de síntesis alimentado ............................................................................................................. 53 Figura 3.10. Conversión de CO y CO+CO2 en función del flujo volumétrico de gas de síntesis alimentado al reactor para el experimento realizado en el reactor tubular de lecho fijo con el catalizador sin diluir. Condiciones de reacción: 80 bar y 255 °C ................................................. 55 Figura 3.11. Desactivación del catalizador durante el experimento realizado en el reactor tubular de lecho fijo con el catalizador sin diluir. Condiciones de reacción: 80 bar, 255 °C y 150 nmL/min de gas de síntesis alimentado.................................................................................. 56 Figura 3.12. Perfil de temperatura del lecho del catalizador sin diluir durante el experimento realizado en el reactor tubular. Condiciones de reacción: 80 bar.................................................. 57 Figura 3.13. Conversión de CO y CO+CO2 en función del flujo volumétrico de gas de síntesis alimentado al reactor para el experimento realizado en el reactor de microcanales. Condiciones de reacción: 80 bar y 255 °C ......................................................................................................... 59 Figura 3.14. Desactivación del catalizador durante el experimento realizado en el reactor de microcanales. Condiciones de reacción: 80 bar, 255 °C y 150 nmL/min de gas de síntesis alimentado ..................................................................................................................................... 60 ix Figura 3.15. Conversión de CO en función del flujo volumétrico de gas de síntesis alimentado al reactor para todos los experimentos de actividad catalítica. Condiciones de reacción: 80 bar y 255 °C .............................................................................................................. 61 x ÍNDICE DE TABLAS Tabla 1.1. Datos de conversiones de equilibrio de CO y CO2 [7]................................................... 5 Tabla 1.2. Procesos Heterogéneos para la Síntesis de CH3OH [1]................................................ 12 Tabla 2.1. Compuestos químicos utilizados en la preparación del catalizador ............................. 21 Tabla 2.2. Tiempo de retención de la 3-hexanona (al 98% de pureza). Volumen de inyección de 0,06 µl............................................................................................................................................ 29 Tabla 2.3. Tiempo de retención del metilformato, metanol, etanol, propanol y butanol. Volumen de inyección de 0,06 µl.................................................................................................. 29 Tabla 2.4. Tabla de calibración de la fase líquida del GC............................................................. 30 Tabla 2.5. Condiciones experimentales de las pruebas de actividad catalítica ............................. 39 Tabla 3.1. Áreas superficiales específicas de los dos lotes de catalizador .................................... 44 Tabla 3.2. Composición de los dos lotes de catalizador comparado con la composición teórica del catalizador propuesto por ICI para la síntesis de metanol ....................................................... 45 Tabla 3.3. Datos de conversión de CO y de CO+CO2 para cada flujo volumétrico de gas de síntesis alimentado al reactor tubular con catalizador diluido en SiC........................................... 50 Tabla 3.4. Datos de conversión de CO y de CO+CO2 para cada flujo volumétrico de gas de síntesis alimentado al reactor tubular con catalizador sin dilución ............................................... 54 Tabla 3.5. Datos de conversión de CO y de CO+CO2 para cada flujo volumétrico de gas de síntesis alimentado al reactor de microcanales.............................................................................. 58 LISTA DE SÍMBOLOS Y ABREVIATURAS ∆G Cambio de la energía libre de Gibbs ∆G0 Cambio de la energía libre de Gibbs estándar de formación ∆H0298 Cambio de la entalpía estándar de formación a 298 K β Ancho del pico del patrón de difracción de rayos X λ Longitud de onda de los rayos X θ Ángulo entre los rayos incidentes y los planos de dispersión a Peso molecular de la especie adsorbida A Pendiente de la isoterma de adsorción de BET Ai Área del pico del compuesto i, obtenido de los análisis del GC Ar Argón BASF Badische Anilin und Soda Fabrik BET Brunauer Emmett Teller c Constante de BET conv. Conversión CO Monóxido de Carbono CO2 Dióxido de Carbono d Distancia entre los planos de la red cristalina DMT DiMetil Tereftalato DRX Difracción de rayos X etc. et cetera (y así sucesivamente) E1 Calor de adsorción de la primera capa EL Calor de adsorción para la segunda y mayores capas FBR Reactor tubular de lecho fijo FZK Forschung Zentrum Karlsruhe GC Cromatógrafo de gases I Intercepción con el eje y de la isoterma de adsorción de BET ICI Imperial Chemical Industries Limited K Constante (usualmente 1) <L> Dimensión de la partícula en dirección perpendicular al plano reflectante MSR Reactor de microcanales n Constante llamada orden de la reflección nml Mililitros normales, mililitros a 0 °C y 1 bar N Coeficiente estequiométrico de la reacción Na Número de Avogadro pi Presión parcial del componente i P Presión P* Presiones de equilibrio de adsorbatos a la temperatura de adsorción P0 Presiones de saturación de adsorbatos a la temperatura de adsorción xiii R Constante universal de los gases s Sección transversal de adsorción SBET Área superficial específica SBET, Total Área superficial total T Temperatura TPR Reducción a temperatura programada v Volumen adsorbido vm Volumen de la monocapa V Volumen molar de gas adsorbente Xi Conversión del compuesto i xiv INTRODUCCIÓN El metanol es un compuesto químico sumamente importante para la industria, usado principalmente en la manufactura de formaldehído y dimetil tereftalato (DMT). Es empleado como disolvente y como refrigerante gracias a su bajo punto de ebullición. En años más recientes, el metanol ha sido utilizado como combustible sintético considerándose un combustible alternativo [1]. La síntesis de metanol continúa atrayendo a los investigadores científicos a pesar de que la tecnología para la manufactura comercial del mismo ha estado disponible desde principios del siglo XX. A través de los años, los científicos han tratado de sintetizar mejores catalizadores que permitan que la reacción de síntesis se lleve a cabo a condiciones menos severas. Con cada pequeño logro obtenido en la mejora de la actividad catalítica se ha presentado la necesidad de desarrollar nuevos procesos para aprovechar estas ventajas alcanzadas. El deseo de permanecer competitivos en un mundo que está consciente de la necesidad de consumir adecuadamente los recursos energéticos, estimuló a los científicos a estudiar aspectos fundamentales del diseño del proceso con el fin de mejorar la eficiencia de éste. A medida que la dimensión de las unidades comerciales aumentaba, se realizaban mayores esfuerzos para optimizar el proceso. Esto ha impulsado nuevas investigaciones en la transferencia de masa y de calor, termodinámica, cinética, diseño de reactores, modelaje, control de la estabilidad térmica y optimización energética de la síntesis de metanol [1]. Una de las mayores limitaciones en la aplicación comercial de los catalizadores es la disponibilidad de reactores capaces de explotar todo el potencial de los catalizadores más activos. Cuando las reacciones son altamente endotérmicas o exotérmicas, la transferencia de calor es el factor limitante y la actividad del catalizador con frecuencia tiene que ser reducida para adaptarse a la capacidad del reactor. Los reactores son grandes, complejos y costosos, y distan mucho de ser óptimos en la productividad por unidad de volumen. A pesar de los progresos realizados en 2 reactores de lecho fluidizado y otras configuraciones, la mayoría de los procesos comerciales a grandes escalas siguen sacrificando la actividad de los catalizadores debido al diseño del reactor. La síntesis de metanol es una reacción exotérmica. Termodinámicamente, mientras menor es la temperatura y mayor es la presión, más favorables son las condiciones para la reacción. En el proceso de producción convencional de metanol las condiciones de reacción oscilan entre 50 bar a 100 bar en términos de la presión y entre 200 a 300 °C en términos de la temperatura. Los problemas presentes en el proceso de producción convencional son: obtener un perfil de temperatura uniforme, mantener constante la transferencia de calor y mejorar la actividad catalítica (disminuyendo la temperatura de reacción) [2]. Los reactores de microcanales tienen el potencial para transformar la economía de estos procesos. Las pequeñas dimensiones de los canales permiten altas relaciones de superficievolumen, mejorando así la transferencia de masa y de calor. La ventaja incluye un aumento en la conversión del producto, la mejora de la eficiencia energética y la reducción de los costos de capital [3]. Además, los reactores de microcanales ofrecen mayor seguridad operativa, principalmente por dos razones: en primer lugar, las reacciones en cadena pueden ser detenidas cuando las moléculas de la fase gaseosa chocan con las paredes de los canales. En microreactores, el área superficial específica es mayor que en reactores macroscópicos, dándole así importancia a las reacciones en la superficie del catalizador y potencialmente, permitiendo mayor seguridad al detener reacciones en cadena. En segundo lugar, las explosiones térmicas son prevenidas gracias a la eficiente transferencia de calor existente en los microreactores, cierta energía térmica puede ser sustraída tan rápido que se impide un aumento significativo en la temperatura, y las explosiones térmicas son evitadas [4]. El presente trabajo de investigación se realizó en la Universidad de Ciencia y Tecnología en Trondheim, Noruega. El objetivo general del proyecto fue evaluar la actividad catalítica de un catalizador de CuO/ZnO/Al2O3 para la síntesis de metanol. Y los objetivos específicos fueron: • Sintetizar un catalizador de CuO/ZnO/Al2O3 para la síntesis del metanol, siguiendo el método patentado por Imperial Chemical Industries Limited. 3 • Caracterizar los materiales del catalizador de CuO/ZnO/Al2O3 a través de técnicas como difracción de rayos X (DRX), adsorción/desorción N2 (BET), reducción a temperatura programada (TPR) e inducción de plasma acoplada-espectroscopía de emisión atómica (ICP-AES). • Evaluar la reproducibilidad de la técnica de síntesis aplicada mediante la caracterización de los materiales de otro lote de catalizador de CuO/ZnO/Al2O3 sintetizado por una estudiante de doctorado, quien aplicó el mismo procedimiento. • Realizar pruebas de corrida en blanco tanto en el reactor de microcanales como en el reactor tubular para verificar la existencia de actividad del acero inoxidable, material de construcción de los reactores. • Estudiar la desactivación del catalizador de CuO/ZnO/Al2O3 en el reactor tubular con el catalizador diluido en SiC y sin diluir, y en el reactor de microcanales. • Realizar pruebas de actividad catalítica en el reactor tubular con el catalizador diluido en (SiC) (1:20) y con el catalizador sin diluir. • Realizar pruebas de actividad catalítica en el reactor de microcanales. • Estudiar el efecto de variar el flujo de gas de síntesis alimentado a los reactores. • Comparar las conversiones obtenidas para la producción de metanol con el reactor tubular y el reactor de microcanales. CAPÍTULO 1 FUNDAMENTOS TEÓRICOS 1.1 Metanol El metanol, también llamado alcohol metílico, alcohol de madera y carbinol, es el primero de los alcoholes. Su fórmula química es CH3OH. Fue descubierto por Robert Boyle en 1661. En condiciones normales es un líquido incoloro, de escasa viscosidad, miscible en agua y como la mayoría de los solventes orgánicos, muy tóxico e inflamable. De los puntos de ebullición y de fusión se deduce que el metanol es un líquido volátil a temperatura y presión atmosféricas. El metanol es muy buen solvente de sustancias polares, pudiéndose disolver sustancias iónicas como el cloruro de sodio en cantidades apreciables. Es considerado como un producto o material inflamable de primera categoría; ya que puede emitir vapores que mezclados en proporciones adecuadas con el aire, originan mezclas combustibles. El metanol es un combustible con un gran poder calorífico, de hecho durante mucho tiempo fue usado como combustible de autos de carrera. Para finalizar con las propiedades y características podemos decir que el metanol es un compuesto orgánico muy importante ya que el grupo hidroxilo se convierte con facilidad en cualquier otro grupo funcional. Así, el metanol se oxida para obtener formaldehído (formol) y ácido fórmico; mientras que por su reducción obtenemos metano. Igualmente importantes son las reacciones de eterificación y esterificación. Es considerado como un producto petroquímico básico, a partir del cual se obtienen varios productos secundarios como dimetil tereftalato, ácido acético, entre otros [5]. 5 1.2 Termodinámica del metanol Las reacciones principales para formación de metanol a partir de gas de síntesis son: ⎯⎯ → CH 3OH CO + 2 H 2 ←⎯ ⎯ ∆H0298= - 90,8 kJ/mol (1.1) ⎯⎯ → CH 3OH + H 2O CO2 + 3H 2 ←⎯ ⎯ ∆H0298= - 49,6 kJ/mol (1.2) Las dos reacciones de formación de metanol están acopladas a su vez con la reacción de desplazamiento de gas de agua (water-gas-shift reaction) [6]: ⎯⎯ → CO2 + H 2 CO + H 2O ←⎯ ⎯ ∆H0298= - 41 kJ/mol (1.3) La Tabla 1.1 muestra valores típicos de conversiones de equilibrio para las reacciones de formación de metanol. Tabla 1.1. Datos de conversiones de equilibrio de CO y CO2 [7] T [K] Conversión de CO Conversión de CO2 Presión [bar] Presión [bar] 50 100 300 10 100 300 525 0,524 0,769 0,951 0,035 0,052 0,189 575 0,174 0,440 0,825 0,064 0,081 0,187 625 0,027 0,145 0,600 0,100 0,127 0,223 675 0,015 0,017 0,310 0,168 0,186 0,260 6 Claramente, la temperatura debe ser baja para que la conversión de CO sea mayor. También debe notarse que la conversión de CO2 aumenta con la temperatura siendo esto una consecuencia del efecto de la reacción de desplazamiento de gas de agua. En comparación con la síntesis de amoníaco, el desarrollo de catalizadores para la síntesis de metanol es más difícil porque además de la actividad, la selectividad del mismo es crucial. Es de esperarse que en la hidrogenación del CO se puedan formar sub-productos como alcoholes superiores e hidrocarburos. La termodinámica muestra que esto, sin duda, es posible. La Figura 1.1 muestra los datos termodinámicos para la formación de metanol y de algunos posibles subproductos de la reacción de CO con H2. Evidentemente el metanol es menos estable termodinámicamente y en ese sentido es menos probable que se forme a partir de la reacción del CO con H2 en comparación con otros posibles productos, como el metano, que puede formarse a través de la reacción de metanización. Por lo tanto, el catalizador debe ser muy selectivo. La selectividad de los catalizadores modernos (Cu/ZnO/Al2O3) es superior al 99%, lo que significa un logro excepcional considerando el gran Cambio de la energía libre de Gibbs estándar de formación (KJ/mol°C) número posible de sub-productos que pueden formarse [8]. Temperatura (K) Figura 1.1. ∆G0 como una función de la temperatura de los productos de la reacción de CO con H2 [8] 7 Los catalizadores utilizados originalmente en la síntesis de metanol (ZnO-Cr2O3) eran activos solamente a altas temperaturas. Por lo tanto, la presión tenía que ser alta (250-350 bar) para alcanzar valores de conversión aceptables. Hasta finales de 1960 este tipo de catalizador clásico fue utilizado. En la Figura 1.2 se muestran conversiones de equilibrio de CO a metanol como función de la temperatura y la presión. Los rangos típicos de temperatura de los procesos clásicos y modernos para la síntesis del metanol se indican en dicha figura. La misma ilustra que el desarrollo de catalizadores que son activos a bajas temperaturas hacen posible la operación a bajas presiones (50-100 bar), manteniéndose las mismas conversiones que en los procesos clásicos. La temperatura es una variable crítica. Bajas temperaturas son favorables desde el punto de vista termodinámico y para la economía del proceso, pero los catalizadores altamente activos son sensibles a la sinterización (pérdida de área superficial activa). La desactivación del catalizador siempre va a ocurrir. El método más utilizado para la regeneración de la actividad del catalizador es el aumento de la temperatura, éste se realiza normalmente para mantener la velocidad de reacción, pero para ello también es necesario aumentar la presión. La temperatura no debe exceder los 570 K porque a partir de ese valor Conversión de equilibrio de CO (-) ocurrirá sinterización [8]. Procesos Clásicos Procesos Modernos Temperatura (K) Figura 1.2. Conversión de equilibrio de CO a metanol; alimentación de H2/CO=2 [8] 8 1.3 Gas de síntesis para la producción de metanol Hoy en día, la mayor parte del gas de síntesis, también conocido como syngas, para la producción de metanol es obtenido a través de la reformación de vapor del gas natural [6]. El syngas ideal para la producción de metanol posee una mezcla de H2 y CO en una relación de 2:1. Por razones termodinámicas, sería deseable que el CO pudiese ser hidrogenado directamente vía la ecuación (1.1) en vez de involucrar dos reacciones más que están acopladas (ecuaciones (1.2) y (1.3)), ya que esto produciría mayor concentración de equilibrio de metanol a la salida del reactor. En la Figura 1.3 se presenta las concentraciones de equilibrio del metanol para diferentes mezclas del gas alimentado. Es notable que la máxima concentración de metanol ocurre para una mezcla de CO+H2 en una relación de 2:1 (relación estequiométrica). En teoría, una mezcla de H2 y CO puede ser utilizada agregando una pequeña proporción de CO2 para producir la máxima cantidad de metanol. Sin embargo, no sólo es importante la constante de equilibrio, también lo es la velocidad de formación del metanol y por esto es relevante recordar que el metanol se forma F ra c ci ó en la n molar de alim e n ta CO ción Fr a en cció la n m al i o m l ar en d ta e H ció n 2 equilibrio Fracción molar de de metanol del CO2 y no del CO. Figura 1.3. Concentración de equilibrio de metanol a la salida como función de la fracción molar de H2 y CO en la alimentación [9] La curva sólida mostrada en la Figura 1.3 es la curva de equilibrio del metanol sin CO2 en la mezcla de gases. Esta curva se ve proyectada en el plano de la izquierda [9]. 9 La composición del gas de síntesis depende de la materia prima utilizada para su elaboración. Cuando la nafta es la materia prima, la estequiometría es aproximadamente correcta. Sin embargo, cuando el CH4 es utilizado, hay H2 en exceso. En la práctica, el exceso de H2 debe ser quemado como combustible, o el contenido de óxidos de carbono en el gas de síntesis se incrementa. Esto puede ser realizado mediante dos métodos: 1. Cuando hay disponibilidad de CO2, éste se agrega al proceso de una manera simple y efectiva para balancear el hidrógeno y los óxidos de carbono contenidos en el gas de síntesis. La adición de CO2 al proceso puede ser implementada mediante la inyección del mismo, ya sea a la corriente de alimentación o al gas de síntesis previamente. En ambos casos se logra la relación estequiométrica en la síntesis del metanol, a pesar de que las composiciones son un poco diferentes. 2. Utilizando la reformación autotérmica. En este caso, el gas de síntesis es muy rico en óxidos de carbono por lo que se requiere hacer ajustes en el sistema para remover el CO2 [6]. 1.4 Síntesis de metanol La identidad química o molecular del metanol fue establecida por primera vez por Dumas y Peligot en el año 1834. Desde entonces, se han realizado muchos esfuerzos por diversos investigadores para sintetizar el metanol. Según Ullmann [10], el proceso de síntesis de metanol puede ser clasificado en tres grupos basados en la presión de operación; 1. Procesos a presión alta: 250-300 atm. 2. Procesos a presión media: 100-250 atm. 3. Procesos a presión baja: 50-100 atm. 10 Una patente alemana fue emitida por BASF en 1913, la cual consistía en un proceso de síntesis de metanol a una temperatura de 300 a 400 °C y a una presión de 100 a 250 atm sobre un catalizador de óxidos de zinc y cromo. La primera planta comercial para la síntesis de metanol a partir de gas de síntesis fue construida por BASF en 1923; el proceso operando a 300 °C y a 200 atm utilizaba un catalizador de óxido de zinc/óxido de cromo. El proceso fue exitoso durante un largo período de tiempo, hasta que una tecnología más eficiente de síntesis a baja presión lo fue reemplazando. Este proceso desarrollado por BASF es comúnmente llamado “síntesis de metanol a presión media”, al ser comparado con el proceso que se introdujo después, el cual requiere presiones de operación más bajas para la síntesis. La corporación Commercial Solvents y Dupont iniciaron la experimentación con la síntesis de metanol en 1927; la producción comercial de metanol con tecnología de alta presión fue aplicada por Commercial Solvents, el CO2 producido en la planta de fermentación de butanol de esta compañía era hidrogenado a metanol a 300 atm en la presencia de catalizadores a base de óxidos de metal [11]. En 1927, DuPont comenzó la operación de una planta en Belle, Francia, tanto metanol como amoníaco eran producidos en esta planta. La materia prima era un gas compuesto por una mezcla de CO, CO2, H2 y N2 que se producía a partir de carbón o coque por la reacción de desplazamiento de gas de agua y que se purificaba para luego ser pasado sobre un catalizador para la síntesis de metanol. El catalizador utilizado era a base de zinc/cromo o cromo/cobre. La planta continuó la producción de metanol y amoníaco hasta finales de 1940, ya que para ese momento se hizo disponible un suministro abundante de gas natural y el carbón fue abandonado como materia prima del proceso. El uso de óxidos de metales, especialmente óxido de cobre, como promotores de catalizadores de ZnO/Cr2O3 fueron ampliamente investigados por muchos científicos. Además se realizaron muchas otras investigaciones acerca de la utilización del óxido de cobre como un ingrediente básico del catalizador más no como un promotor. Imperial Chemical Industries Limited (ICI) presentó e introdujo al mercado el catalizador más activo a base de Cu/ZnO en 1966, poniendo fin a los procesos de síntesis de metanol a altas presiones. Este catalizador operaba a 11 temperaturas entre 250 a 300 °C y a presiones entre 50 a 100 atm, condiciones que eran considerablemente más bajas que aquellas de los procesos a alta presión. El uso de estos catalizadores más activos fue posible porque empezaron a estar disponibles procesos de purificación del gas de síntesis más eficientes, con los cuales se lograba remover químicos que envenenaban el catalizador incluyendo carbonilos de metales y azufre. Los catalizadores a base de Cu/ZnO son mucho más susceptibles a envenenamiento por compuestos de azufre, y pueden ser permanentemente desactivados fácilmente a altas temperaturas o por la presencia de puntos calientes locales, por lo que es necesario un control adecuado de la temperatura del reactor; sin embargo, a pesar de utilizar los catalizadores preparados de la forma más cuidadosa, pequeñas cantidades de metano, dimetil éter, alcoholes superiores, entre otros compuestos, aparecen entre los productos de la síntesis de metanol [12]. Actualmente, el proceso a baja presión de ICI utiliza un catalizador de Cu/ZnO/Al2O3 muy estable, el cual opera en un rango de presión entre 50 a 80 atm y de temperatura entre 250 y 300 °C. El reactor es un recipiente a presión que contiene un solo lecho de catalizador [1]. El proceso a baja presión de la compañía Lurgi para la síntesis de metanol también utiliza un catalizador basado en cobre (Cu/ZnO). La temperatura de reacción (entre 250 y 260 °C) es controlada con agua caliente que se hace pasar por la coraza del reactor. En general, el proceso de Lurgi presenta una alta economía y alta fiabilidad operacional [13]. Hoy en día, la síntesis comercial del metanol se basa exclusivamente en sistemas heterogéneos. Sin embargo, cabe destacar que el metanol también puede ser sintetizado vía catálisis homogénea. Algunos procesos heterogéneos y sus condiciones de reacción se presentan en la Tabla 1.2. En la Tabla 1.2, las formulas químicas de los catalizadores están presentadas en su forma estable, es decir, sin haber sido reducidas. Para el uso de estos catalizadores en el proceso de síntesis de metanol, es necesario realizar el proceso de reducción apropiado al catalizador. El proceso de reducción cambia dependiendo del proceso y la configuración del sistema de reacción. 12 Tabla 1.2. Procesos Heterogéneos para la Síntesis de CH3OH [1] 1.5 Proceso Catalizador T [°C] P [atm] Nissui-Topsoe CuO/ZnO/Cr2O3 230-260 100-150 BASF CuO/ZnO/Al2O3 200-350 50-250 ICI CuO/ZnO/Al2O3 220-280 50-100 Lurgi CuO/ZnO 230-250 40-50 Chem. Systems CuO/ZnO/Al2O3 250-275 50-120 Rol del dióxido de carbono en la síntesis del metanol Es importante resaltar que existe una controversia concerniente a cuál es el rol del dióxido de carbono en la síntesis de metanol. Muchos artículos han discutido el tema debido a que tiene importancia tanto a nivel científico como industrial. La literatura disponible se puede clasificar como sigue [1]: 1. Artículos que apoyan que el metanol es producido predominantemente por la hidrogenación del CO2. 2. Artículos que apoyan la alternativa de que el metanol es producido predominantemente por la hidrogenación de CO. Kagan et al [14] se encuentran entre los que apoyan la idea de que el metanol es producido por la hidrogenación del CO2. Klier et al [15] han publicado una serie de artículos sobre estructuras de catalizadores y su relación en la síntesis de metanol, ellos consideran que el metanol es producido predominantemente por la hidrogenación de CO y que mientras pueda darse lugar la hidrogenación del CO2, la velocidad de ésta será mucho más lenta que la velocidad de reacción de hidrogenación de CO. 13 1.6 Caracterización de los materiales de catalizadores La caracterización de los materiales de un catalizador es un campo sumamente importante en catálisis. La difracción, los métodos basados en adsorción y desorción o las reacciones de bulbo (reducción, oxidación) ofrecen las herramientas necesarias para investigar la naturaleza de un catalizador activo. Con esos conocimientos es posible entender mejor los catalizadores, de forma tal que nuevos diseños puedan ser creados o los viejos puedan ser mejorados. 1.6.1 Difracción de rayos X La difracción de rayos X (DRX) es una de las técnicas más antiguas que se aplicada con mayor frecuencia en la caracterización de catalizadores. Se utiliza para identificar las fases cristalinas dentro de los catalizadores y para obtener una indicación del tamaño de las partículas [9]. La base de la fuente de rayos X es un proceso llamado fluorescencia de rayos X. En este proceso, la fuente consta de un objetivo: un ánodo que es bombardeado con electrones de alta energía emitidos por un cátodo. El resultado de esto es que el ánodo emite rayos X proveniente de dos procesos. En primer lugar los electrones emitidos por el haz de electrones del cátodo crean un fondo continuo del espectro de Bremsstrahlung. En segundo lugar, los agujeros del núcleo son ocupados por los electrones de transición desde los niveles más altos de energía. Este proceso conduce a la generación de fotones de rayos X [16]. La cristalografía de rayos X es una técnica que consiste en hacer pasar un haz de rayos X a través de un cristal de la sustancia sujeta a estudio. El haz se divide en varias direcciones debido a la simetría de la agrupación de átomos y, por difracción, da lugar a un patrón de intensidades que puede interpretarse según la ubicación de los átomos en el cristal, aplicando la ley de Bragg, mostrada en la ecuación (1.4) [9]. nλ = 2 d s en (θ ); n = 1, 2,... (1.4) Donde λ es la longitud de onda de los rayos X, d es la distancia entre los planos de la red cristalina, θ es el ángulo entre los rayos incidentes y los planos de dispersión y n una constante llamada orden de la reflección y es un número entero. 14 En la práctica, los patrones de difracción de rayos X de una muestra en polvo se pueden medir con rayos X estacionarios o con un detector movible. La intensidad de la radiación difractada es escaneada como una función del ángulo 2θ existente entre el rayo entrante y el difractado. Esta configuración permite la determinación de la distancia entre los planos de la red cristalina y en consecuencia las fases cristalográficas presentes en el catalizador. Los tamaños de las partículas también pueden ser estimados a partir de los patrones de DRX. En la fórmula de Scherrer el tamaño del cristal está relacionado con el ancho del pico como se muestra en la ecuación (1.5) [16]. L = Kλ β cos(θ ) (1.5) Donde <L> es la dimensión de la partícula en dirección perpendicular al plano reflectante, K es una constante (usualmente 1), λ es la longitud de onda de los rayos X, β es el ancho del pico y θ es el ángulo entre los rayos incidentes y los planos de dispersión La DRX es una de las técnicas que goza de mayor prestigio entre la comunidad científica para dilucidar estructuras cristalinas, debido a su precisión y a la experiencia acumulada durante décadas, elementos que la hacen muy fiable. Sus mayores limitaciones se deben a la necesidad de trabajar con sistemas cristalinos, por lo que no es aplicable a disoluciones, a sistemas amorfos o a gases. Por lo tanto, no se puede estar seguro de que no hay otras fases presentes en el material aparte de las detectadas con la DRX [9]. 1.6.2 Adsorción/desorción de N2 La teoría de BET (Brunauer, Emmett, y Teller) es una regla conocida para estudiar la adsorción física de moléculas gaseosas sobre una superficie sólida. Es la base para una importante técnica de análisis utilizada para la medición del área superficial específica de un material. 15 En 1938, Stephen Brunauer, Paul Hugh Emmett, y Edward Teller publicaron un artículo sobre la teoría de BET por primera vez; “BET” consiste en las iniciales de los apellidos de los investigadores. El concepto de la teoría de BET es una extensión de la teoría de Langmuir, la cual propone, partiendo de la adsorción molecular en monocapa, las siguientes hipótesis para la adsorción molecular en multicapas [17]: (a) Las moléculas de gas se adsorben físicamente sobre el sólido en capas infinitas. (b) No hay interacción entre cada capa de adsorción. (c) La teoría de Langmuir puede ser aplicada para cada capa. La ecuación resultante de la teoría de BET se muestra en la ecuación (1.6): 1 υ[( P0 P * ) − 1] = c − 1 ⎛ P* ⎞ 1 ⎜ ⎟+ υm c ⎝ P0 ⎠ υm c (1.6) Donde P* y P0 son las presiones de equilibrio y de saturación de adsorbatos a la temperatura de adsorción respectivamente, v es el volumen adsorbido, vm es el volumen de la monocapa y c es la constante de BET la cual se expresa como se muestra en la ecuación (1.7): ⎛ E − EL ⎞ c = exp ⎜ 1 ⎟ ⎝ RT ⎠ (1.7) Donde E1 es el calor de adsorción de la primera capa y EL es el calor de adsorción para la segunda y mayores capas. E1-EL es igual al calor de licuefacción. 16 La ecuación (1.6) es una isoterma de adsorción y puede ser graficada como se muestra en la Figura 1.4. El resultado de graficar valores experimentales es un línea recta donde en el eje x se tiene la variable φ=P/P0 y en el eje y la variable 1/v[φ/(φ−1)]. Figura 1.4. Isoterma de adsorción de BET [17] El valor de la pendiente (A) y de la intercepción con el eje y (I) de la isoterma de adsorción, son usados para calcular el volumen de la monocapa (vm) y la constante de BET (c). Las siguientes ecuaciones (1.8) y (1.9) son las utilizadas para los respectivos cálculos [17]: υm = 1 A+ I (1.8) A I (1.9) c = 1+ El método de BET se utiliza ampliamente en la ciencia de las superficies para el cálculo de las áreas superficiales de sólidos a través de la adsorción física de las moléculas de gas. El área 17 superficial total SBET, Total y el área superficial específica SBET son calculadas utilizando las ecuaciones (1.10) y (1.11) [17]: S BET ,Total = S BET = υm N a s V S BET ,Total a (1.10) (1.11) Donde Na es el número de Avogadro, s es la sección transversal de adsorción, V es el volumen molar de gas adsorbente, vm es el volumen de la monocapa y a es el peso molecular de la especie adsorbida. 1.6.3 Reducción a temperatura programada Los métodos de reacción a temperatura programada son una técnica en la cual una reacción química es monitoreada mientras se aumenta la temperatura linealmente en el tiempo. Existen diversas reacciones que son utilizadas en la caracterización de catalizadores como lo son: la reducción a temperatura programada (TPR), la oxidación a temperatura programada (TRO) y la sulfidación a temperatura programada (TRS). La instrumentación utilizada para investigaciones a temperatura programada es relativamente simple. El reactor es llenado con el catalizador y luego es controlado por un procesador, el cual calienta el reactor a una tasa lineal. Mientras la temperatura aumenta se hace fluir una mezcla de un gas reductor sobre el catalizador. Comúnmente se utiliza hidrógeno diluido en un gas inerte. Un detector de conductividad térmica mide la composición de H2 en el gas de salida [9]. La ecuación que describe la reducción de un óxido de metal utilizando hidrógeno se muestra en la ecuación (1.12) [16]. 18 MOn + nH 2 → M + nH 2O (1.12) Con el fin de tener una reducción termodinámica favorable, la energía libre de Gibbs de la reacción (∆G) debe tener un valor negativo. Como se indica en la ecuación (1.13) la energía libre de Gibbs depende de la presión y de la temperatura. ∆G = ∆G 0 + NRT ln pH 2O pH 2 (1.13) Donde ∆G es el cambio de la energía libre de Gibbs para la reducción, ∆G0 es el cambio de energía libre de Gibbs a condiciones estándar, N es el coeficiente estequiométrico de la reacción, R es la constante universal de los gases, T es la temperatura y pi es la presión parcial de cada componente [16]. La reducción a temperatura programada proporciona información útil sobre las temperaturas necesarias para la reducción completa del catalizador. El área bajo la curva de TPR representa el consumo total de hidrógeno [9]. 1.7 Microreactor El término “microreactor” tradicionalmente ha sido utilizado para denominar a los pequeños reactores tubulares usados en las pruebas de rendimiento de catalizadores, pero con la actual utilización de tecnologías de microfabricación, el prefijo “micro” se utiliza para designar, en general, sistemas químicos fabricados con técnicas originalmente desarrolladas para circuitos electrónicos. Dichos sistemas tienen dimensiones en orden de magnitud de submilímetros. La reducción en tamaño y la integración de múltiples funciones tienen el potencial para producir estructuras con capacidades que superan a los sistemas macroscópicos convencionales y al mismo tiempo añaden nuevas funcionalidades que disminuyen potencialmente los costos de producción en masa [18]. 19 Se espera que la tecnología de microreacción tenga una serie de ventajas para la producción de sustancias químicas. Las pequeñas dimensiones implican la existencia de flujo laminar, por lo que es posible caracterizar completamente la transferencia de masa y calor. La alta tasa de transferencia de masa y calor permite que las reacciones se lleven a cabo bajo condiciones uniformes de temperatura y bajo condiciones más severas obteniéndose mayor rendimiento del que se podría obtener con reactores convencionales. Nuevas vías de reacción que se han considerado muy difíciles de llevar a cabo en reactores convencionales, podrían realizarse en equipos microscópicos. Los experimentos a escala convencional está limitada por los altos costos de los reactivos y la preocupación por cumplir estándares de seguridad, los pequeños volúmenes y características inherentes de seguridad que poseen los microreactores elimina dichas limitaciones. La introducción de nuevos productos químicos está también limitada por el riesgo y los altos costos de escalar una planta piloto de laboratorio a una planta real de producción. El reescalamiento de una planta piloto a una planta de producción se podrá realizar con la repetición de unidades de microreacción usadas en el laboratorio, lo cual eliminará los costos por rediseño y experimentación en planta piloto, y acortará el tiempo de desarrollo de unidades comerciales a partir de la producción en el laboratorio. Estos sistemas requerirán menos espacio y servicios, y producirán menos desechos. Además permitirán el alto rendimiento de procesos químicos bajo condiciones controladas, que a menudo han sido difíciles de lograr con los sistemas macroscópicos convencionales [18]. La tecnología de microreacción incluye dispositivos con canales y otras estructuras en el rango de dimensión de 1 a 1000 µm [19]. Los reactores de microcanales son arreglos paralelos de canales, con dimensiones críticas de 0,25 a 5 mm. Las altas tasas de transferencia de masa y calor, debidas a las altas relaciones de superficie-volumen del reactor, permiten la utilización de los catalizadores más activos con velocidades de reacción de 100 a 500 veces más altas que los reactores convencionales [3]. Los principales pioneros en el desarrollo de reactores de microcanales son Forschungzentrum Karlsruhe, Institute for Microtechnology Mainz y Pacific North West National Laboratories [19]. Ellos están liderando el camino, pero otras empresas están comenzando a desarrollar aplicaciones. Entre estas empresas se encuentran Degussa, FMC Corporation y Velocys Inc. 20 Degussa lideriza un proyecto piloto para la producción de oxido de propileno a través de la oxidación catalítica en fase gaseosa del propileno utilizando peróxido de hidrógeno en un reactor de microcanales. FMC Corporation está trabajando con Stevens Institute of Technology en el desarrollo de una ruta directa para la producción de peróxido de hidrógeno a partir de hidrógeno y oxígeno llevada a cabo en un reactor de microcanales. Velocys Inc está desarrollando reactores de microcanales para una variedad de procesos incluyendo la producción de hidrógeno, la conversión de gas natural en combustible sintético y la producción de varios compuestos químicos básicos [18]. A pesar de los éxitos y el interés cada vez mayor que se está teniendo en la tecnología de microreacción, aún hace falta desarrollar nuevos tipos de catalizadores para aplicaciones en reactores de microcanales. Los pequeños canales requieren nuevas formas de los catalizadores incluyendo microlechos empacados, revestimiento de las paredes de los microcanales, etc. Los mayores retos para el uso comercial de los reactores de microcanales son: obtener una estabilidad en el revestimiento que proporcione suficientes sitios activos, asegurar la uniformidad dentro de los microcanales y entre ellos y colocar el catalizador dentro de los microcanales del reactor [3]. CAPÍTULO 2 METODOLOGÍA EXPERIMENTAL Capitulo 2 2.1 Catalizador para la síntesis de metanol A continuación se explica en detalle la metodología experimental utilizada para sintetizar y caracterizar el catalizador CuO/ZnO/Al2O3 para la producción de metanol. 2.1.1 Síntesis del catalizador Diferentes métodos han sido utilizados para la preparación de catalizadores de mezclas de óxidos de metales a base de cobre. Este sub-capítulo contiene los detalles de la síntesis del catalizador CuO/ZnO/Al2O3 para la producción de metanol. Los compuestos químicos utilizados en la preparación del catalizador se presentan en la Tabla 2.1. Tabla 2.1. Compuestos químicos utilizados en la preparación del catalizador Compuesto Fórmula Química Productor Pureza [%] Aluminato Sódico NaAlO2 Riedel-de Haën® 99,95 Ácido Nítrico HNO3 Merck 65,00 Nitrato de Zinc Tetrahidratado Zn(NO3)2·4H2O Merck 99,99 Carbonato de Sodio Na2CO3 VWRTM BDH Prolabo 99,90 Nitrato Cúprico Trihidratado Cu(NO3)2·3H2O Prolabo 99,41 22 Un segundo lote de catalizador a base de cobre fue sintetizado siguiendo el método patentado por Imperial Chemical Industries Limited [20]. El primer lote de catalizador fue sintetizado por la estudiante de doctorado Xuyến Kim Phan usando el mismo procedimiento. Para este método de co-precipitación, se siguió el procedimiento mostrado en la Figura 2.1. Para todo el proceso de síntesis del catalizador se utilizó agua destilada. 2.1.2 Caracterización de los materiales del catalizador Los dos lotes de catalizador fueron caracterizados empleando los siguientes métodos: difracción de rayos X, adsorción/desorción de N2, inducción de plasma acoplada – espectroscopia de emisión atómica y reducción de temperatura programada. 2.1.2.1 Difracción de rayos X Las pruebas de DRX fueron realizadas a una muestra del primer lote de catalizador, a una muestra del segundo lote de catalizador y a una muestra del segundo lote de catalizador antes de ser calcinado. Los patrones de rayos X de los catalizadores fueron obtenidos utilizando un difractómetro D8 Focus de Bruker AXS con radiación CuKα. El aparato D8 Focus estaba equipado con un ángulo de análisis de 2θ/θ y un detector LynxEye. La rotación se activo para los análisis. Las muestras del primer lote de catalizador y el segundo lote de catalizador fueron escaneadas en un rango de 2θ desde 25° hasta 90° con un tamaño del paso de 0,06. Las muestras del segundo lote de catalizador antes y después de ser calcinado fueron escaneadas en un rango de 2θ desde 25° hasta 90° con un tamaño del paso de 0,02. Los patrones de difracción fueron analizados y las fases fueron identificadas con el programa de análisis cualitativo DIFFRACplus EVA (2006) el cual posee la base de datos de Powder Diffraction (1999). 23 1 Solución de Aluminato Sódico [NaAlO2] (193,5 g) en Agua [H2O] (1000 mL) 2 Ácido Nítrico [HNO3] (510,7 mL 65 % p/p) Agregar 2 a 1 Agitar hasta disolver el precipitado de Hidróxido de Aluminio [Al(OH)3] 4 Solución de Nitrato de Zinc Tetrahidratado [Zn(NO3)2·4H2O] (262,3 g) en Agua [H2O] (250 mL) 3 Solución Agregar 4 a 3 Agregar Agua [H2O] hasta llegar a un volumen de (3000 mL) Calentar solución hasta 85 ºC 5 Solución molar de Carbonato de Sodio [Na2CO3] 6 Solución Calentar solución hasta 85 ºC Zona de mezclado 5y6 Ajustar los flujos para que la mezcla tenga un pH=6,5 mediado a 65 ºC Filtrar y lavar el precipitado resultante Disolver 697 g del precipitado en Agua [H2O] (1500 mL) 7 Solución de Nitrato Cúprico Trihidratado [Cu(NO3)2·3H2O] (217,5 g) y Nitrato de Zinc Tetrahidratado [Zn(NO3)2·4H2O] (58,9 g) en Agua [H2O] (600 mL) 8 Solución Mezclar 7 y 8 10 Calentar solución hasta 85 ºC Solución molar de Carbonato 9 Solución Calentar solución hasta 85 ºC de Sodio [Na2CO3] Zona de mezclado 9 y 10 Ajustar los flujos para que la mezcla tenga un pH=6,5 mediado a 65 ºC Calentar el slurry hasta 85 ºC en 10 min, luego mantener la temperatura de 85 ºC por 20 min Agitar suavemente durante el calentamiento Filtrar el precipitado, luego lavarlo y secarlo durante toda la noche a 110 ºC Calcinar la torta seca a 300 ºC por 6 h Figura 2.1. Esquema de la síntesis del catalizador para la producción de metanol 24 2.1.2.2 Adsorción/desorción de N2 Las áreas superficiales de BET de los dos lotes de catalizador fueron analizadas en el instrumento Micromeritics TriStar 3000: analizador de área superficial y porosidad. La cantidad de muestra de catalizador colocado en el instrumento fue aproximadamente 0,05 g. Un flujo de gas a 300 °C se dejó pasar sobre el catalizador durante la noche y luego se analizó la adsorción de N2 obteniéndose el área superficial de BET. 2.1.2.3 Inducción de plasma acoplada-espectroscopía de emisión atómica La inducción de plasma acoplada – espectroscopía de emisión atómica (ICP-AES) fue utilizada para determinar la composición actual del primer y segundo lote de catalizador. Los análisis fueron realizados por Molab AS. Las muestras fueron disueltas en ácido clorhídrico antes de ser analizadas y no fue observado ningún residuo. 2.1.2.4 Reducción a temperatura programada Los análisis de la reducción de temperatura programada (TPR) fueron realizados a una muestra del primer lote de catalizador y a una muestra del segundo lote de catalizador. Los experimentos de TPR fueron realizados en el instrumento Quantachrome CHEMBET 3000, en el cual el gas efluente es analizado a través de un detector de conductividad térmica. Para cada muestra, una cantidad aproximada de 0,10-0,15 g de catalizador fue colocada en un reactor de cuarzo y se utilizó lana de cuarzo para mantener el catalizador como un lecho fijo. La reducción fue llevada a cabo con 7% H2 en Ar desde temperatura ambiente hasta 350 °C utilizando una tasa de calentamiento de 2 °C/min. 2.2 Montaje experimental para la síntesis de metanol El montaje experimental consistía en una sección de alimentación, los reactores y una sección de análisis. Cada parte del montaje experimental estaba elaborada en acero inoxidable. Las tuberías eran de ¼”. Todos los equipos estaban diseñados para soportar un máximo de presión de 25 100 bar y un máximo de temperatura de 500 °C. Todos los equipos (exceptuando el cromatógrafo de gases y las computadoras) estaban localizados dentro de una cabina cerrada de policarbonato con ventilación separada. El montaje estaba equipado con dos líneas de reactores, una línea con un reactor tubular de lecho fijo y la otra con un reactor de microcanales. Estos tenían tres líneas comunes de alimentación de gases: H2, He y el gas de síntesis (H2/CO/CO2/N2). Las líneas de alimentación de gases estaban equipadas con manómetros, válvulas de reducción de presión, válvulas manuales (on/off) y válvulas de ventilación. El H2 y el He eran pasados a través de filtros de oxígeno y humedad. Después de la purificación y despresurización del gas de síntesis, parte de éste era enviado a la sección de análisis. Antes de ingresar al reactor, el gas de síntesis era precalentado a condiciones cercanas a las necesarias para la reacción. El producto a la salida del reactor era condensado en un separado caliente o frío. Existía un segundo separador frío para condensar los productos que eventualmente no fueran condensados en el primer separador. Parte del producto gaseoso era enviado a la sección de análisis. Un esquema más detallado del proceso se muestra en la Figura 2.2. Algunos elementos claves del montaje eran: · Control de temperatura para ambos reactores y bandas de calentamiento para las líneas de gas de alimentación. · Control y monitoreo de todos los flujos de gas y líquido, así como temperatura y presión del sistema. 26 Figura 2.2. Diagrama del montaje experimental para la síntesis de metanol 27 2.2.1 Medidores y controladores de flujo másico El sistema de alimentación de gases constaba de tres líneas de alta presión para H2, He y gas de síntesis (H2/CO/CO2/N2). Las líneas estaban equipadas con manómetros. Los flujos de gases hacia el reactor disponían controladores digitales de flujo másico de Bronkhorst. Los flujos de gas eran ajustados por la computadora. Los controladores de flujo másico de las líneas de H2 y He fueron calibrados en febrero de 2007 por los estudiantes de doctorado Hamidreza Bakhtiary y Xuyến Kim Phan. El controlador de flujo másico de la línea de gas de síntesis fue calibrado en septiembre de 2007 por el estudiante de doctorado Hamidreza Bakhtiary. Las curvas de calibración de los controladores de flujo másico se muestran en el apéndice A. 2.2.2 Medidor y controlador de presión La presión del reactor estaba controlada por un controlador digital de presión de Bronkhorts. El controlador de presión soportaba una presión máxima de 100 bar y un flujo máximo de 500 nml/min. La presión a la salida del reactor era 1 bar. La presión del reactor era ajustada por la computadora. 2.2.3 Cromatógrafo de gases El cromatógrafo de gases (GC) era de modelo Agilent 6890N con detector de conductividad térmica (TCD) y detector por ionización de llama (FID) instalados. El equipo era capaz de realizar análisis en línea del gas alimentado y el gas producto de la reacción, así como análisis del producto líquido. Era posible seleccionar entre análisis de gas o líquido mediante una válvula. 2.2.3.1 Análisis de la fase gaseosa Para los análisis de la fase gaseosa se utilizó una columna 10ft Carbosieve. La columna estaba equipada con un TCD que identificaba la presencia de H2, N2, CO, CO2 y CH4 en el gas 28 alimentado y en el gas producto de la reacción. El N2 era un gas inerte en la reacción por lo que fue utilizado como estándar en los análisis. La calibración de la fase gaseosa del GC fue realizada por el investigador científico Rune Myrstad en diciembre de 2005. Las curvas de calibración de cada compuesto identificado por el detector TCD se muestran en el apéndice B. 2.2.3.2 Análisis de la fase líquida Para los análisis de la fase líquida se utilizó una columna HP-FFAP (50 m, 0,32 mm, 0,5 µm). La columna estaba equipada con un FID que identificaba la presencia de metilformato, metanol, etanol, propanol, butanol y 3-hexanona en el producto líquido de la reacción. La 3-hexanona fue utilizada como estándar en los análisis. Con este detector no era posible identificar la presencia de agua en el producto líquido. Para realizar el análisis de la muestra líquida era necesario inyectar el líquido en la columna utilizando una inyectadora. La calibración de la fase líquida del GC fue realizada en febrero de 2008. Para calibrar el GC primero se inyectó 3-hexanona (al 98% de pureza) en la columna. El GC analizaba la muestra líquida y generaba un cromatograma donde era posible observar un pico que correspondía al compuesto 3-hexanona. El tiempo en el cual el pico aparecía en el cromatograma es llamado tiempo de retención. El tiempo de retención de la 3-hexanona fue calculado realizando un promedio de los valores obtenidos en cuatro corridas como se muestra en la Tabla 2.2. 29 Tabla 2.2. Tiempo de retención de la 3-hexanona (al 98% de pureza). Volumen de inyección de 0,06 µl Corrida Tiempo de retención [min] 1 9,101 2 8,972 3 8,986 4 9,133 Promedio 9,048 Luego de inyectar 3-hexanona y analizar los resultados, una mezcla de 20 % de metilformato, 20 % de metanol, 20 % de etanol, 20 % de propanol y 20 % de butanol fue inyectada a la columna. El GC analizaba la mezcla de líquidos y generaba un cromatograma donde era posible observar cinco picos, cada uno correspondiente a cada compuesto de la mezcla. Los tiempos de retención del metilformato, metanol, etanol, propanol y butanol fueron calculados realizando un promedio de los valores obtenidos en tres corridas como se muestra en la Tabla 2.3. Tabla 2.3. Tiempo de retención del metilformato, metanol, etanol, propanol y butanol. Volumen de inyección de 0,06 µl Tiempo de retención [min] Corrida Metilformato 1 2,373 3,855 4,660 8,481 11,948 2 2,383 3,877 4,618 8,312 11,841 3 2,384 3,829 4,586 8,307 11,845 Promedio 2,380 3,854 4,621 8,367 11,878 Metanol Etanol Propanol Butanol 30 Una nueva mezcla fue inyectada a la columna del cromatógrafo de gases. La mezcla contenía 50 % de metanol y 10 % del resto de los compuestos previamente analizados (3-hexanona, metilformato, etanol, propanol y butanol). La composición volumétrica de la mezcla y el tiempo de retención promedio obtenido anteriormente de cada compuesto fueron colocados como datos en el software del GC. Cuando la mezcla líquida fue inyectada a la columna, el GC generó un cromatograma y calculó el área de cada pico correspondiente a los compuestos inyectados. Con el área y la composición volumétrica, el software calculó el factor de respuesta como la relación entre la composición y área del pico. Finalmente, el GC generó los datos de calibración que se muestran en la Tabla 2.4. Tabla 2.4. Tabla de calibración de la fase líquida del GC Factor de Tiempo de Composición retención [min] [% vol.] Metil-formato 2,380 10 2672,00 0,00374 Metanol 3,854 50 18774,30 0,00266 Etanol 4,621 10 5568,60 0,00179 Propanol 8,367 10 7509,25 0,00133 3-hexanona 9,048 10 8438,50 0,00119 Butanol 11,878 10 9229,40 0,00108 Compuesto Área Respuesta Utilizando la tabla de calibración presentada en la Tabla 2.4, el software del GC calculó las curvas de calibración para cada compuesto identificado por el detector FID las cuales se muestran en el apéndice C. 31 Para todas la inyecciones de líquido realizadas en la columna, la temperatura del horno se mantenía a 40 °C por 8 min, luego se incrementaba a razón de 10 °C/min hasta 200 °C y finalmente la temperatura se mantenía a 200 °C durante 11 min. 2.2.4 Reactor tubular de lecho fijo El reactor tubular de lecho fijo (FBR) era un tubo de 1/2” fabricado de acero inoxidable. Tenía un diámetro interno de 9,2 mm y una longitud total de 43,4 cm. En el fondo del reactor se insertaba un cilindro de acero inoxidable con un tejido de acero para mantener el catalizador en su lugar. El diseño del reactor se muestra en la Figura 2.3. 43,4 cm 38 cm ½”x 0,070” Figura 2.3. Configuración y dimensiones del reactor tubular de lecho fijo El FBR se encontraba fijo dentro de un bloque de aluminio para mejorar la transferencia y distribución de calor. El bloque de aluminio estaba compuesto por dos mitades de un cilindro con un espacio en el medio para colocar el reactor. Un tubo, de diámetro externo de 2,0 mm y diámetro interno de 1,2 mm, contenía en su interior una termocupla con un diámetro de 1 mm y con 3 puntos de medición. Dicho tubo era colocado en el centro del reactor. La temperatura era controlada con un controlador Eurotherm. El FBR era calentado con un horno. El horno eléctrico era de 35 cm de longitud y constaba de dos módulos cilíndricos que se cerraban para contener adentro al reactor. La temperatura del lecho del catalizador era medida en 3 lugares dentro del reactor y se regulaba variando la temperatura de calentamiento del horno eléctrico. 32 El gas alimentado a la reacción era precalentado hasta valores cercanos a la temperatura de reacción utilizando bandas eléctricas de calentamiento que recubrían las tuberías adyacentes al reactor. El precalentamiento afectaba el perfil de temperaturas en el reactor. 2.2.5 Reactor de microcanales El reactor de microcanales (MSR) fue diseñado y fabricado por la compañía alemana Forschungzentrum Karlsruhe (FZK). Está fabricado en acero inoxidable y diseñado para un máxima presión de operación de 100 bar. El volumen dentro de los microcanales, donde el catalizador es colocado, es de 2 ml. El cuerpo, la salida y la entrada del reactor son mostrados en la Figura 2.4. a) Cuerpo del reactor b) Entrada del reactor c) Detalle de los microcanales d) Salida del reactor Figura 2.4. Reactor de microcanales con intercambiador de calor, fabricado por FZK 33 Las láminas del reactor se construyen grabando una estructura cilíndrica en un arreglo hexagonal de profundidad de 0,4 mm en una lámina metálica de acero inoxidable de 0,8 mm. Ensamblando dos láminas cara a cara le da una profundidad de 0,8 mm a la estructura cilíndrica. Una de las láminas del reactor le falta una fila donde queda el espacio necesario para introducir un termopar de 0,5 mm de diámetro dentro del reactor para medir la temperatura. La lámina de metal es mostrada en la Figura 2.5. Figura 2.5. Lámina de metal Además, para el intercambio de calor, canales de 0,25 x 0,50 mm con aletas de 0,25 x 0,25 mm son colocados en una lámina metálica de acero inoxidable de 0,5 mm como se muestra en la Figura 2.6. 0,25x0,25 mm 0,25x0,50 mm 0,50 mm 60 mm Figura 2.6. Lámina para la transferencia de calor Un esquema básico del ensamblaje de las láminas de los microcanales del reactor es mostrado en la Figura 2.7. 34 Figura 2.7. Ensamblaje de las láminas del reactor de microcanales El reactor de microcanales es calentado con aceite entre 70 y 400 °C, el cual circula a través de los canales del intercambiador de calor. De esta forma la temperatura del reactor es regulada. 2.3 Experimentos En la presente investigación se realizaron cinco experimentos para la síntesis del metanol, los cuales fueron: • Una corrida en blanco en el reactor tubular. • Una corrida en blanco en el reactor de microcanales. • Una prueba de actividad catalítica en el reactor tubular con el catalizador diluido en carburo de silicio (SiC). • Una prueba de actividad catalítica en el reactor tubular con el catalizador sin diluir. • Una prueba de actividad catalítica en el reactor de microcanales. Para todos los experimentos se calculó la conversión de CO y la conversión de CO+CO2 en la síntesis de metanol como se muestra en el apéndice D. 35 2.3.1 Corridas en blanco Las corridas en blanco fueron realizadas tanto en el reactor tubular como en el reactor de microcanales. Se realizaron en ausencia de catalizador para determinar la actividad del acero inoxidable que era el material de construcción de ambos reactores. En ambos experimentos la composición del gas de síntesis alimentado fue 25 % de CO, 5 % de CO2, 5 % de N2 y 6 5% de H2. En los dos experimentos se realizó la prueba de fugas al sistema antes de introducir el gas de síntesis, para ello se introdujo H2 y N2 al reactor y se aumentó poco a poco la presión hasta llegar a 80 bar. Con detectores de H2 y N2 se verificó, a medida que se aumentaba la presión, que no hubiese ninguna fuga. Una vez culminada la prueba de fugas, la presión se mantuvo en 80 bar y la temperatura de reacción se llevó a 255 °C. Durante ambos experimentos, el flujo del gas de síntesis alimentado al reactor se varió: primero fue 150 nml/min y luego 250 nml/min. El producto gaseoso, en ambos experimentos, fue continuamente analizado utilizando el cromatógrafo de gases en línea y el gas alimentado al reactor fue precalentado con mantas de calentamiento. Para el experimento realizado en el reactor de microcanales, se colocó una cantidad de 2,88 g de SiC dentro del reactor, el SiC tenía un tamaño de partícula entre 75-150 µm. El termopar fue insertado dentro del canal del medio del reactor para medir la temperatura. Para el experimento realizado en el reactor tubular, se colocó una cantidad de 21 g de SiC dentro del reactor, el SiC tenía un tamaño de partícula entre 75-150 µm. El SiC fue colocado en el tope del cilindro de acero inoxidable sobre un poco de lana de vidrio para mantener fijo el lecho de reacción. La temperatura del SiC fue medida en tres puntos con un termopar. 36 2.3.2 Reducción de catalizador El proceso de reducción del catalizador se realizó utilizando un flujo de gas de síntesis de 350 nml/min. El mismo se llevo a cabo a presión ambiental. La temperatura se aumentó de 25 °C a 160 °C a razón de 1 °C/min, luego se aumentó hasta 200 °C con un incremento de 2 °C/h, se mantuvo a 200 °C por 2 h, seguidamente se incrementó hasta 220 °C a razón de 2 °C/h y se mantuvo en 220 °C por 4 h. Finalmente, la temperatura se aumentó hasta 250 °C con un incremento de 2 °C/h y se mantuvo en 250 °C por 4 h. Durante la reducción del catalizador, el gas de síntesis alimentado fue precalentado con mantas de calentamiento. 2.3.3 Pruebas de actividad catalítica Se realizaron tres pruebas de actividad catalítica utilizando el segundo lote del catalizador para la síntesis del metanol. Dos pruebas en el reactor tubular y una prueba en el reactor de microcanales. En todos los experimentos la composición del gas de síntesis alimentado fue 25 % de CO, 5 % de CO2, 5 % de N2 y 65 % de H2. En todos experimentos se realizó la prueba de fugas al sistema antes de introducir el gas de síntesis, para ello se introdujo H2 y N2 al reactor y se aumentó poco a poco la presión hasta llegar a 80 bar. Con detectores de H2 y N2 ser verificó, a medida que se aumentaba la presión, que no hubiese ninguna fuga. Una vez culminada la prueba de fugas, se procedió a realizar el proceso de reducción del catalizador. Cuando culminó la reducción del catalizador, la presión del reactor se llevó a 80 bar y la temperatura de reacción se llevó a 255 °C. El producto gaseoso, en todos los experimentos, fue continuamente analizado utilizando el cromatógrafo de gases en línea y el gas alimentado al reactor fue precalentado con mantas de calentamiento. También, en todos los experimentos, el producto líquido fue recolectado para su posterior análisis, el mismo no podía ser analizado en línea durante el proceso de reacción porque el GC estaba realizando los análisis de la fase gaseosa continuamente. 37 El experimento realizado en el reactor tubular con el catalizador diluido en SiC con una relación (1:20) tuvo una duración de tres semanas y media. El modo de operación de reactor era downflow. Se mezcló 1 g de catalizador que tenía un tamaño de partícula entre 50-200 µm con 20 g de SiC que tenía un tamaño de partícula entre 75-150 µm y se colocó dentro del reactor en el tope del cilindro de acero inoxidable con tejido de acero sobre un poco de lana de vidrio para mantener fijo el lecho de reacción. Durante este experimento el flujo de gas de síntesis alimentado al reactor se varió usando los siguientes valores: 75, 150, 250, 375, 500 y 750 nml/min. Al final del experimento el flujo de gas de síntesis se fijó en 75 nml/min de nuevo para estudiar la desactivación del catalizador. También se varió la presión a 60 y 70 bar, manteniendo la temperatura a 255 °C y el flujo de gas de síntesis a 75 nml/min, para estudiar el efecto de la presión en la síntesis de metanol. Finalmente, se varió la temperatura de la reacción a 245 y 265 °C, manteniendo la presión a 80 bar y el flujo de gas de síntesis a 75 nml/min, para estudiar el efecto de la temperatura en la síntesis de metanol. La temperatura del lecho del catalizador fue medida en tres puntos con un termopar. Se monitoreó la temperatura durante todo el experimento y se verificó la existencia de condiciones isotérmicas. El experimento realizado en el reactor tubular con el catalizador sin diluir tuvo una duración de dos semanas. El modo de operación de reactor era downflow. Se colocó 1 g de catalizador con tamaño de partícula entre 50-200 µm dentro del reactor como se muestra en la Figura 2.8. Entre el cilindro de acero inoxidable y el SiC, y entre el SiC y el catalizador se colocó lana de vidrio. Durante este experimento el flujo de gas de síntesis alimentado al reactor se varió usando los siguientes valores: 75, 150, 250, 375, 500, 750, 1000 y 1500 nml/min. Al final del experimento el flujo de gas de síntesis se fijó en 150 nml/min de nuevo para estudiar la desactivación del catalizador. La temperatura del lecho del catalizador fue medida en tres puntos con un termopar. Se monitoreó la temperatura durante todo el experimento y se realizó un perfil de temperaturas para cada condición para poder mantener la temperatura de la reacción en 255°C y la presión a 80 bar. 38 43,4 cm Catalizador 3 cm 15 cm 12 cm SiC 3 cm Cilindro de 9 cm acero inoxidable 0 cm Figura 2.8. Disposición del catalizador sin diluir en el reactor tubular El experimento realizado en el reactor de microcanales tuvo una duración de dos semanas. Se colocó 1 g de catalizador con tamaño de partícula entre 50-200 µm dentro del reactor. El termopar fue insertado dentro del canal del medio del reactor para medir la temperatura del lecho del catalizador. Durante este experimento el flujo de gas de síntesis alimentado al reactor se varió usando los siguientes valores: 75, 150, 250, 375, 500, 750, 1000 y 1500 nml/min. Al final del experimento el flujo de gas de síntesis se fijó en 150 nml/min de nuevo para estudiar la desactivación del catalizador. La temperatura durante la reacción se mantuvo a 255 °C y la presión a 80 bar. A continuación, en la Tabla 2.5, se presenta un resumen de las condiciones experimentales de las pruebas de actividad catalítica realizadas en ambos reactores. 39 Tabla 2.5. Condiciones experimentales de las pruebas de actividad catalítica No de condición T [°C] P [bar] Q [nml/min] mcat [g] mSiC [g] Reactor tubular de lecho fijo 1 255 80 75 1 20 2 255 80 150 1 20 3 255 80 250 1 20 4 255 80 375 1 20 5 255 80 500 1 20 6 255 80 750 1 20 7 255 60 75 1 20 8 255 70 75 1 20 9 245 80 75 1 20 10 265 80 75 1 20 11 255 80 75 1 - 12 255 80 150 1 - 13 255 80 250 1 - 14 255 80 375 1 - 15 255 80 500 1 - 16 255 80 750 1 - 17 255 80 1000 1 - 18 255 80 1500 1 - Reactor de microcanales 1 255 80 75 1 - 2 255 80 150 1 - 3 255 80 250 1 - 40 Continuación de la tabla 2.5 4 255 80 375 1 - 5 255 80 500 1 - 6 255 80 750 1 - 7 255 80 1000 1 - 8 255 80 1500 1 - CAPÍTULO 3 RESULTADOS Y DISCUSIONES ¡ 3.1 Catalizador para la síntesis de metanol En este sub-capítulo se presentan y se discuten los resultados de la caracterización de los materiales de los dos lotes de catalizador para la síntesis del metanol. 3.1.1 Caracterización de los materiales del catalizador Dos lotes de catalizador fueron caracterizados utilizando difracción de rayos X, adsorción/desorción de N2, reducción a temperatura programada e inducción de plasma acopladaespectroscopía de emisión atómica. Mediante la caracterización de los materiales de los dos lotes del catalizador CuO/ZnO/Al2O3 se estudió la reproducibilidad del procedimiento presentado por Imperial Chemical Industries Limited en la patente Methanol Synthesis Catalyst [20]. Los resultados de dichos experimentos son presentados y discutidos a continuación. 3.1.1.1 Difracción de rayos X Las diferentes fases presentes en los catalizadores fueron estudiadas a través de la difracción de rayos X (DRX). Los patrones de DRX para el primer y segundo lote de catalizador calcinado son mostrados en la Figura 3.1 y los patrones para el segundo lote de catalizador antes y después de ser calcinado son mostrados en la Figura 3.2. 42 En la Figura 3.1, los patrones de DRX para el primer y segundo lote de catalizador, indican la presencia de una estructura amorfa de los materiales en el catalizador, en vez de una estructura cristalina porque los picos de difracción para cada compuesto no están definidos claramente. Es decir, existe un solapamiento de diversos picos lo cual no permite la identificación de cada pico perteneciente a cada compuesto presente en el catalizador. La ventaja de caracterizar el catalizador utilizando la DRX es que provee información precisa acerca de las estructuras cristalinas permitiendo estimar el tamaño de las partículas. La limitación es que no se pueden detectar partículas muy pequeñas o amorfas [9]. Como es el caso, por lo que no se puede concluir Intensidad acerca de las fases presentes en los dos lotes de catalizador ni calcular el tamaño de las partículas. Ángulo 2θ existente entre el rayo entrante y el difractado Figura 3.1. Patrones de difracción de rayos X del primer lote (línea negra) y segundo lote (línea gris) del catalizador CuO/ZnO/Al2O3 mostrando picos de difracción de CuO (♦), ZnO (●) y Al2O3 (■) 43 En la Figura 3.1, los picos del patrón de DRX del segundo lote del catalizador tienen mayor altura que los picos del patrón del primer lote del catalizador. La altura de los picos indica la intensidad con que cada compuesto de la muestra reflecta el haz de luz que incide sobre ellos. Debido a esto, es posible considerar que se obtuvo un pico de difracción claro para el segundo lote del catalizador. Pero cuando, en la Figura 3.2, se compara el patrón de DRX del segundo lote del catalizador con el patrón del mismo lote antes de la calcinación, se puede observar más notoriamente que no se obtienen picos de difracción claros para el segundo lote después de la Intensidad calcinación sino solapamiento de los picos. Ángulo 2θ existente entre el rayo entrante y el difractado Figura 3.2. Patrones de difracción de rayos X del segundo lote del catalizador CuO/ZnO/Al2O3 antes de la calcinación (línea gris) y después de la calcinación (línea negra) mostrando picos de difracción de Cu(NO3)2 (♦), Al(NO3)3 (●), Zn(NO3)2 (■) y ZnCO3 (▲) La calcinación es un proceso térmico aplicado a materiales sólidos para lograr una descomposición térmica, una transición de fase o para remover fracciones volátiles. El objetivo de la calcinación es remover agua, dióxido de carbono y otros componentes volátiles presentes en la muestra, así como oxidar los compuestos presentes en el catalizador. Durante la calcinación los nitratos de metales y los carbonatos de metales son transformados en óxidos de metales [21]. Para 44 la muestra del segundo lote del catalizador antes de la calcinación se obtuvieron picos claros de difracción. Se logró identificar la presencia de fases como Cu(NO3)2, Al(NO3)3, Zn(NO3)2 y ZnCO3 antes de la calcinación. Estos compuestos fueron identificados utilizando el programa DIFFRACplus EVA. En esta base de datos se buscó el CuCO3 pero no se encontró la presencia del mismo en la muestra. 3.1.1.2 Adsorción/desorción de N2 Las áreas superficiales específicas de los dos lotes de catalizador fueron determinadas a través del estudio de la adsorción de N2. Los resultados se presentan en la Tabla 3.1. Tabla 3.1. Áreas superficiales específicas de los dos lotes de catalizador Catalizador SBET [m2/g] Primer lote de Cu/ZnO/Al2O3 173 Segundo lote de Cu/ZnO/Al2O3 170 En las mediciones del área superficial específica de ambos lotes de catalizador se obtuvieron valores altos de áreas específicas con respecto a los reportados por Meland et al. [22] para un catalizador de CuO/ZnO/Al2O3. Esto se debe a que el óxido de zinc está íntimamente asociado al óxido de cobre, es decir, el soporte posee tanto óxido de zinc como óxido de cobre [9]. 3.1.1.3 Inducción de plasma acoplada-espectroscopía de emisión atómica La composición del primer y segundo lote de catalizador fue determinada utilizando la técnica de inducción de plasma acoplada. Los resultados se muestran en la Tabla 3.2. 45 Tabla 3.2. Composición de los dos lotes de catalizador comparado con la composición teórica del catalizador propuesto por ICI para la síntesis de metanol Catalizador Primer lote de Cu/ZnO/Al2O3 Segundo lote de Cu/ZnO/Al2O3 ICI Cu/ZnO/Al2O3 %p/p Na2O nAl nCu nZn 0,045 0,23 0,30 0,47 0,003 0,24 0,32 0,44 0,000 0,24 0,33 0,43 Nota: nAl, nCu y nZn son las fracciones normalizadas de Al, Cu y Zn respectivamente en el catalizador y %m/m Na2O es el porcentaje en peso de Na2O en CuO.ZnO.Al2O3.Na2O. Los resultados mostrados en la Tabla 3.2 muestran un máximo de 6 % de diferencia entre la fracción molar de Al, Cu y Zn entre el primer y el segundo lote de catalizador. Los resultados se ajustan a la composición teórica del catalizador para la síntesis de metanol reportada por ICI [20]. El porcentaje másico de Na2O del primer y segundo lote de catalizador es mayor que el porcentaje reportado en la composición teórica del catalizador por ICI [20]. La posible razón por la que los dos lotes de catalizador poseen un porcentaje de Na2O a diferencia de la composición teórica en la cual es 0,000 %p/p es que los lotes de catalizador no fueron lavados suficientes veces. El Na2O es soluble en agua y reacciona para formar hidróxido de sodio por lo que al lavar bien el catalizador con agua el Na2O es eliminado de la composición. 3.1.1.4 Reducción a temperatura programada Las pruebas de reducción de temperatura programada fueron realizadas a una muestra del primer y segundo lote del catalizador para determinar la temperatura necesaria para la reducción completa del mismo. Los perfiles de TPR se presentan en la Figura 3.3. 46 Los perfiles de la TPR muestran que los intervalos de reducción para el catalizador son entre 185-280 °C para el primer lote el cual presenta un pico en 263 °C. Y en el intervalo (bimodal) de 185-340 °C para el segundo lote, el cual presenta dos picos: uno a 255 y el otro a 293 °C. 200 Primer lote del catalizador Cu/ZnO/Al2O3 Segundo lote del catalizador Cu/ZnO/Al2O3 Consumo de H2 150 100 50 0 0 50 100 150 200 250 300 350 Temperatura [°C] Figura 3.3. Perfiles de TPR del primer lote de Cu/ZnO/Al2O3 y del segundo lote de Cu/ZnO/Al2O3. Condiciones: ~0,15 g de muestra, mezcla de gas de 7 % de H2 en Ar, 25-350 °C a 2 °C/min Los perfiles de TPR muestran que el primer lote del catalizador es más homogéneo en su estructura que el segundo lote. La presencia de dos picos en el perfil de TPR del segundo lote de catalizador muestra un producto no homogéneo. Estos perfiles coinciden con los presentados por Meland et al. [20] para un catalizador de CuO/ZnO/Al2O3. 47 3.2 Experimentos Los resultados de los cinco experimentos realizados con el montaje experimental para la síntesis del metanol se presentan y se discuten a continuación. 3.2.1 Corridas en blanco En ausencia de catalizador, se realizaron corridas tanto en el reactor tubular como en el reactor de microcanales para determinar la actividad del acero inoxidable que era el material de construcción de ambos reactores. Se realizaron las corridas en blanco en ambos reactores metálicos, ya que según la literatura se cumple la siguiente secuencia de actividad no deseada de materiales: acero>aluminio>titanio [4]. Es decir, que el material que tiene mayor tendencia a intervenir en la reacción es el acero. El experimento realizado en el reactor de microcanales duró aproximadamente 47 h y durante este período el gas producto de la reacción fue analizado en el cromatógrafo de gases (GC) para luego poder calcular la conversión de CO. Los resultados se muestran en la Figura 3.4. 6 Flujo de gas de síntesis alimentado = 250 nmL/min Flujo de gas de síntesis alimentado = 150 nmL/min Conversión de CO [%] 5 4 3 2 1 0 0 10 20 30 40 50 Tiempo [h] Figura 3.4. Conversión de CO en función del tiempo durante la prueba en blanco realizada en el reactor de microcanales. Condiciones: 80 bar y 255 °C 48 El experimento realizado en el reactor tubular de lecho fijo duró aproximadamente 87 h y durante este período el gas producto de la reacción fue analizado en el GC para luego poder calcular la conversión de CO. Los resultados se muestran en la Figura 3.5. 30 Flujo de gas de síntesis alimentado = 250 nmL/min Flujo de gas de síntesis alimentado = 150 nmL/min Conversión de CO [%] 25 20 15 10 5 0 0 20 40 60 80 100 Tiempo [h] Figura 3.5. Conversión de CO en función del tiempo durante la prueba en blanco realizada en el reactor tubular. Condiciones: 80 bar y 255 °C En la Figura 3.4 se observa que para el reactor de microcanales la conversión inicial de CO es de 5 % la cual decrece hasta 0 % en 15 h, manteniéndose constante en ese valor durante el resto de la prueba. La actividad obtenida al principio de la prueba puede ser causa de la incertidumbre introducida a través de los análisis del gas producto y el gas de alimentación realizados con el cromatógrafo de gases, los cuales se utilizan en el cálculo de la conversión. Por otra parte, en la Figura 3.5 se observa que para el reactor tubular la conversión inicial de CO es de 28 % la cual decrece hasta 0 % en 23 h, manteniéndose constante en ese valor durante el resto de la prueba. Una actividad aparente es detectada durante las primeras 23 h del experimento. Esta actividad puede ser debida a dos razones: 49 1. Un experimento para la síntesis de metanol fue llevado a cabo días antes de realizar la corrida en blanco en el reactor tubular y trazas del gas producto de la reacción pudieron quedar en la columna del cromatógrafo de gases, lo cual afectó los análisis arrojados por el equipo durante las primeras 23 h del experimento, hasta que las trazas salieron de la columna. 2. El material del reactor tubular (acero inoxidable) es activo al principio de la prueba y después de 23 h se desactiva. El resultado obtenido fue que el acero inoxidable del reactor de microcanales y del reactor tubular no interviene en la actividad de la reacción durante largos períodos de operación del sistema. 3.2.2 Pruebas de actividad catalítica Se realizaron tres pruebas de actividad catalítica en la síntesis del metanol. Debido a la controversia sobre el rol del monóxido y dióxido de carbono en el mecanismo de producción de metanol discutido en el Capítulo 1, en todos los resultados experimentales presentados a continuación, se graficó tanto la conversión de CO como la conversión de CO+CO2 en la síntesis de metanol. 3.2.2.1 Realizada en el reactor tubular con el catalizador diluido en Sic Para el experimento, realizado en el reactor tubular con el catalizador diluido en SiC se analizaron las siguientes variables. Primero, se varió el flujo de gas de síntesis alimentado al reactor y se calculó la conversión de CO y de CO+CO2 utilizando las ecuaciones (D.1) y (D.2) presentadas en el apéndice D. Los resultados del porcentaje de conversión de CO y CO+CO2 en función del flujo de alimentación se muestran en la Tabla 3.3 y en la Figura 3.6. 50 Tabla 3.3. Datos de conversión de CO y de CO+CO2 para cada flujo volumétrico de gas de síntesis alimentado al reactor tubular con catalizador diluido en SiC Flujo volumétrico de gas de síntesis Conversión de CO Conversión de alimentado [nmL/min] [%] CO+CO2 [%] 75 57 47 150 39 33 250 27 23 375 21 18 500 15 13 750 8 7 60 Conversión de CO Conversión de CO+CO2 Conversión [%] 50 40 30 20 10 0 200 400 600 800 Flujo volumétrico del gas de síntesis alimentado [nmL/min] Figura 3.6. Conversión de CO y CO+CO2 en función del flujo volumétrico de gas de síntesis alimentado al reactor para el experimento realizado en el reactor tubular de lecho fijo con el catalizador diluido en SiC. Condiciones de reacción: 80 bar y 255 °C 51 Estos resultados muestran que el porcentaje de conversión de CO y CO+CO2 disminuye a medida que el flujo volumétrico de gas de síntesis aumenta. Este comportamiento o tendencia se obtiene porque cuando se aumenta el flujo de gas alimentado, el tiempo de residencia o tiempo de contacto disminuye, por lo que el gas tiene menos tiempo para adsorberse en el catalizador y reaccionar, por esta razón la conversión disminuye. Luego se varió la temperatura de la reacción manteniendo constante la presión a 80 bar y el flujo volumétrico de gas de síntesis a 75 nmL/min. Los resultados se muestran en la Figura 3.7. 60 Conversión de CO Conversión de CO+CO2 Conversión [%] 55 50 45 40 35 240 245 250 255 260 265 270 Temperatura [°C] Figura 3.7. Conversión de CO y CO+CO2 en función de la temperatura de la reacción para el experimento realizado en el reactor tubular de lecho fijo con el catalizador diluido en SiC. Condiciones de reacción: 80 bar y 75 nmL/min de gas de síntesis alimentado En la Figura 3.7 se puede observar que la conversión de CO a 245 °C es 44 %, luego ésta aumenta hasta 57 % a una temperatura de 255 °C y finalmente decrece hasta 55% cuando la temperatura es 265 °C. Para esta prueba, la conversión de CO más alta se obtuvo cuando la temperatura de reacción es 255 °C. La conversión presenta un máximo con la temperatura. 52 Por último, se varió la presión de la reacción manteniendo constante la temperatura a 255 °C y el flujo volumétrico de gas de síntesis en 75 nmL/min. Los resultados se muestran en la Figura 3.8. 60 Conversión de CO Conversión de CO+CO2 Conversión [%] 55 50 45 40 35 30 55 60 65 70 75 80 85 Presión [bar] Figura 3.8. Conversión de CO y CO+CO2 en función de la presión de la reacción para el experimento realizado en el reactor tubular de lecho fijo con el catalizador diluido en SiC. Condiciones de reacción: 255 °C y 75 nmL/min de gas de síntesis alimentado Es posible notar que en la Figura 3.8 el porcentaje de conversión aumenta con la presión. Para una presión de 60 bar la conversión de CO es 38 %, para 70 bar es de 46 % y para 80 bar la conversión de CO es 57 %. La conversión de CO+CO2 sigue la misma tendencia que la conversión de CO. La Tabla 1.1 del sub-capítulo 1.2, en el cual se discutió la termodinámica de la síntesis de metanol, muestra valores típicos de conversiones de equilibrio para las reacciones de formación de metanol. Claramente, la temperatura debe ser baja para que la conversión de CO sea mayor. También debe notarse que la conversión de CO2 aumenta con la temperatura hasta un punto y 53 luego disminuye siendo esto una consecuencia del efecto de la reacción de desplazamiento de gas del agua (water-gas-shift reaction). Los datos presentados en la literatura corresponden con los obtenidos experimentalmente. Al final del experimento el flujo de gas de síntesis se fijó nuevamente en 75 nmL/min, la presión en 80 bar y la temperatura en 255 °C para estudiar la desactivación del catalizador. Esta condición se repitió después de 450 h de reacción y se mantuvo por 120 h. El resultado se muestra en la Figura 3.9. 58 Conversión de CO Conversión de CO+CO2 56 54 Conversión [%] 52 50 48 46 44 42 40 38 0 100 200 300 400 500 600 Tiempo de reacción [h] Figura 3.9. Desactivación del catalizador durante el experimento realizado en el reactor tubular de lecho fijo con el catalizador diluido en SiC. Condiciones: 80 bar, 255 °C y 75 nmL/min de gas de síntesis alimentado Durante el uso y hasta en períodos de inactividad, los catalizadores algunas veces pierden la integridad de sus propiedades físicas y químicas. Sufren alteraciones que conllevan a su desactivación. La velocidad, la severidad y la seriedad de la desactivación varía según las circunstancias [23]. Es por ello que en la Figura 3.9 se puede observar que después de 570 h de reacción la conversión de CO disminuyó desde 57 % hasta 47 %. La conversión de CO+CO2 en 54 ese tiempo de reacción disminuyó desde 47 % hasta 39 %. Por esto se puede decir que el catalizador diluido en SiC sufrió una leve desactivación. 3.2.2.2 Realizada en el reactor tubular con el catalizador sin diluir Para el experimento, realizado en el reactor tubular con el catalizador sin diluir se analizaron las siguientes variables. Primero, se varió el flujo de gas de síntesis alimentado al reactor y se calculó la conversión de CO y de CO+CO2 utilizando las ecuaciones (D.1) y (D.2) presentadas en el apéndice D. Los resultados del porcentaje de conversión de CO y CO+CO2 en función del flujo de alimentación se muestran en la Tabla 3.4 y en la Figura 3.10. Tabla 3.4. Datos de conversión de CO y de CO+CO2 para cada flujo volumétrico de gas de síntesis alimentado al reactor tubular con catalizador sin dilución Flujo volumétrico de gas de síntesis Conversión de CO Conversión de alimentado [nmL/min] [%] CO+CO2 [%] 75 57 47 150 37 31 250 26 22 375 18 15 500 14 12 750 7 6 1000 5 4 1500 2 2 55 60 Conversión de CO Conversión de CO+CO2 50 Conversión [%] 40 30 20 10 0 200 400 600 800 1000 1200 1400 Flujo volumétrico del gas de síntesis alimentado [nmL/min] Figura 3.10. Conversión de CO y CO+CO2 en función del flujo volumétrico de gas de síntesis alimentado al reactor para el experimento realizado en el reactor tubular de lecho fijo con el catalizador sin diluir. Condiciones de reacción: 80 bar y 255 °C Al final del experimento el flujo de gas de síntesis se fijó nuevamente en 150 nmL/min, la presión en 80 bar y la temperatura en 255 °C para estudiar la desactivación del catalizador. Esta condición se repitió después de 235 h de reacción y se mantuvo por 100 h. En la Figura 3.11 se puede observar que después de 335 h de reacción la conversión de CO disminuyó desde 37 % hasta 26 %. La conversión de CO+CO2 en ese tiempo de reacción disminuyó desde 31 % hasta 22 %. Por esto se puede decir que el catalizador sin diluir sufrió una leve desactivación. 56 40 Conversión de CO Conversión de CO+CO2 Conversión [%] 35 30 25 20 0 100 200 300 Tiempo de reacción [h] Figura 3.11. Desactivación del catalizador durante el experimento realizado en el reactor tubular de lecho fijo con el catalizador sin diluir. Condiciones de reacción: 80 bar, 255 °C y 150 nmL/min de gas de síntesis alimentado De acuerdo a estos resultados, el catalizador diluido en SiC presenta menor porcentaje de desactivación que el catalizador sin diluir. Esto se debe a que se presentan puntos calientes en el catalizador cuando éste no está diluido en el reactor tubular. La dilución del catalizador ayuda a que exista una mejor transferencia de calor y que las condiciones en el lecho fijo de reacción sean isotérmicas. Cuando el catalizador no se encontraba diluido en el reactor fue más difícil controlar la temperatura del lecho y no se alcanzó la condición isotérmica (ver Figura 3.12). Al existir puntos calientes locales en el catalizador puede ocurrir la sinterización del mismo. El proceso de sinterización es un fenómeno causado por altas temperaturas que conllevan a la desactivación térmica y a la pérdida de área superficial activa del catalizador [6]. Para cada flujo volumétrico de gas de síntesis, se realizó un perfil de temperatura del lecho de reacción. Los resultados se muestran en la Figura 3.12. 57 Temperatura del lecho [°C] 255 250 245 240 Dirección del Flujo de Gas 235 12,0 12,5 13,0 13,5 14,0 14,5 15,0 Posición a lo largo del reactor [cm] Fujo de gas de síntesis alimentado = 1500nmL/min Fujo de gas de síntesis alimentado = 1000nmL/min Fujo de gas de síntesis alimentado = 750nmL/min Fujo de gas de síntesis alimentado = 500nmL/min Fujo de gas de síntesis alimentado = 375nmL/min Fujo de gas de síntesis alimentado = 250nmL/min Fujo de gas de síntesis alimentado = 150nmL/min Fujo de gas de síntesis alimentado = 75nmL/min Figura 3.12. Perfil de temperatura del lecho del catalizador sin diluir durante el experimento realizado en el reactor tubular. Condiciones de reacción: 80 bar En la Figura 3.12 la posición a lo largo del reactor va desde 12 cm hasta 15 cm. Esos 3 cm es donde se encontraba el lecho del catalizador. Una termocupla fue utilizada para medir la temperatura dentro del reactor, al mover la termocupla a lo largo del reactor fue posible desarrollar un perfil de temperatura del lecho del catalizador. El perfil muestra que no existía condición isotérmica de reacción. En la Figura 3.12 la temperatura máxima de reacción (255 °C) se mueve de derecha a izquierda a medida que el flujo volumétrico de gas de síntesis alimentado aumenta. Esto sucede porque para un menor flujo de gas, mayor es el tiempo de residencia, por lo que el gas tiene mayor tiempo para adsorberse y reaccionar al principio del lecho. Mientras el flujo de gas alimentado aumenta, el tiempo de residencia decrece y la temperatura más alta del lecho se encuentra más abajo en dicho lecho, porque el tiempo que tiene el gas para reaccionar es 58 menor. El experimento en el reactor tubular con el catalizador sin diluir fue realizado para evaluar el perfil de temperatura en el lecho y poder hacer comparaciones con los experimentos en el reactor de microcanales ya que el mismo utiliza el lecho del catalizador sin dilución. 3.2.2.3 Realizada en el reactor de microcanales con el catalizador sin diluir Para el experimento, realizado en el reactor de microcanales con el catalizador sin diluir se analizaron las siguientes variables. Primero, se varió el flujo de gas de síntesis alimentado al reactor y se calculó la conversión de CO y de CO+CO2 utilizando las ecuaciones (D.1) y (D.2) presentadas en el apéndice D. Los resultados del porcentaje de conversión de CO y CO+CO2 en función del flujo de alimentación se muestran en la Tabla 3.5 y en la Figura 3.13. Tabla 3.5. Datos de conversión de CO y de CO+CO2 para cada flujo volumétrico de gas de síntesis alimentado al reactor de microcanales Flujo volumétrico de gas de síntesis Conversión de CO Conversión de alimentado [nmL/min] [%] CO+CO2 [%] 75 65 53 150 54 44 250 39 33 375 30 23 500 22 18 750 12 11 1000 8 7 1500 5 5 59 70 Conversión de CO Conversión de CO+CO2 60 Conversión [%] 50 40 30 20 10 0 200 400 600 800 1000 1200 1400 Flujo volumétrico del gas de síntesis alimentado [nmL/min] Figura 3.13. Conversión de CO y CO+CO2 en función del flujo volumétrico de gas de síntesis alimentado al reactor para el experimento realizado en el reactor de microcanales. Condiciones de reacción: 80 bar y 255 °C Al final del experimento el flujo de gas de síntesis se fijó nuevamente en 150 nmL/min, la presión en 80 bar y la temperatura en 255 °C para estudiar la desactivación del catalizador. Esta condición se repitió después de 225 h de reacción y se mantuvo por 110 h. Los resultados se muestran en la Figura 3.14. 60 55 Conversión [%] 50 45 40 Conversión de CO Conversión de CO+CO2 35 0 100 200 300 Tiempo de reacción [h] Figura 3.14. Desactivación del catalizador durante el experimento realizado en el reactor de microcanales. Condiciones de reacción: 80 bar, 255 °C y 150 nmL/min de gas de síntesis alimentado En la Figura 3.14 se puede observar que después de 335 h de reacción la conversión de CO disminuyó desde 54 % hasta 52 %, disminuyó 2 %. La conversión de CO+CO2 en ese tiempo de reacción disminuyó 3 %. Por esto se puede decir que el catalizador no sufrió desactivación en el reactor de microcanales. Finalmente, en la Figura 3.15 se presenta un gráfico comparativo de los porcentajes de conversión de CO en función del flujo volumétrico de gas de síntesis alimentado a la reacción, para los tres experimentos realizados; en el reactor de microcanales, en el reactor tubular con el catalizador sin dilución y diluido en SiC. 61 70 Reactor de microcanales Reactor tubular con catalizador sin dilución Reactor tubular con catalizador diluido en SiC 60 Conversión [%] 50 40 30 20 10 0 200 400 600 800 1000 1200 1400 Flujo volumétrico del gas de síntesis alimentado [nmL/min] Figura 3.15. Conversión de CO en función del flujo volumétrico de gas de síntesis alimentado al reactor para todos los experimentos de actividad catalítica. Condiciones de reacción: 80 bar y 255 °C Se puede observar que la mayor conversión de CO para los diversos flujos de alimentación se alcanza en el reactor de microcanales, por lo que la eficiencia de la síntesis de metanol es mayor en éste que en el resto de los sistemas evaluados. Además es importante recordar que la temperatura del reactor de microcanales es controlada fácilmente a través del sistema de recirculación de aceite caliente, por lo que al evaluar el perfil de temperaturas se obtuvo que las condiciones eran isotérmicas. CONCLUSIONES Las conclusiones de esta investigación se presentan a continuación. • Se sintetizó un segundo lote de catalizador de CuO/ZnO/Al2O3, a base de cobre, para la síntesis del metanol, siguiendo el método patentado por Imperial Chemical Industries Limited. • Se realizó la caracterización de los materiales de dos lotes de catalizador de CuO/ZnO/Al2O3. Se estudió la reproducibilidad del procedimiento utilizado para sintetizar el catalizador a través de técnicas como DRX, adsorción/desorción N2, TPR e ICP-AES. • El estudio a través de la difracción de rayos X mostró que el primer y segundo lote de catalizador poseen una estructura amorfa en vez de cristalina. Para ambos lotes de catalizador no se puede concluir acerca de las fases presentes en el mismo ni calcular el tamaño de las partículas debido a que son estructuras amorfas. Se identificaron el Cu(NO3)2, Al(NO3)3, Zn(NO3)2 y ZnCO3 en el segundo lote de catalizador antes de la calcinación. • Las áreas superficiales específicas del primer y segundo lote de catalizador de CuO/ZnO/Al2O3 resultaron ser altas con respecto a los reportados por Meland et al. [22] para un catalizador de CuO/ZnO/Al2O3. El área superficial del primer lote de catalizador fue 173 m2/g y el del segundo lote fue 170 m2/g. • La composición del primer y segundo lote de catalizador fueron determinadas a través de la inducción de plasma acoplada-espectroscopia de emisión atómica. Los resultados mostraron una pequeña diferencia en la fracción molar de Al, Cu y Zn entre el primer y el segundo lote de catalizador. Además los resultados coincidían con la composición reportada por ICI para el catalizador para la producción de metanol. 63 • Los perfiles de reducción a temperatura programada mostraron que el primer lote de catalizador era más homogéneo que el segundo La presencia de dos picos en el perfil de TPR del segundo lote del catalizador señala la no homogeneidad del producto. Estos perfiles coinciden con los presentados por Meland et al. [20] para un catalizador de CuO/ZnO/Al2O3. • Se llevaron a cabo pruebas de corrida en blanco (sin catalizador) tanto en el reactor de microcanales como en el reactor tubular para verificar si existía considerable actividad del acero inoxidable durante la reacción. La actividad detectada al principio de las pruebas fue menor en el reactor de microcanales que en el reactor tubular. Para largos períodos de operación del sistema no se detectó actividad del acero inoxidable ni en el microreactor ni en el reactor tubular. • Se estudio la desactivación del segundo lote de catalizador de CuO/ZnO/Al2O3 en el reactor tubular con el catalizador diluido en SiC y sin diluir, y en el reactor de microcanales. El catalizador diluido en el reactor tubular presentó una desactivación menor que el del catalizador sin diluir. El catalizador en el reactor de microcanales no presentó considerable desactivación. • En el reactor tubular con el catalizador diluido en (SiC) (1:20), el porcentaje de conversión de CO y CO+CO2 disminuyó a medida que el flujo de gas alimentado se incrementó. La mayor conversión de CO y CO+CO2 se obtuvo a una temperatura de 255 °C. El porcentaje de conversión de CO y CO+CO2 aumentó a medida que la presión de la reacción aumentó. • Se estudió el efecto de variar el flujo de gas de síntesis alimentado al reactor tubular con el catalizador sin dilución. Se obtuvo la misma tendencia que con el experimento con el catalizador diluido. Se elaboró un perfil de temperatura del lecho de reacción obteniéndose una condición no isotérmica. 64 • Se estudió el efecto de variar el flujo de gas de síntesis alimentado al reactor de microcanales. El porcentaje de conversión de CO y CO+CO2 disminuyó a medida que el flujo de gas alimentado se incrementó. • Se comparó las conversiones obtenidas para la producción de metanol con el reactor tubular y el reactor de microcanales. Se obtuvo que las mayores conversiones se alcanzan con el reactor de microcanales, siendo éste más eficiente en la síntesis de metanol. Finalmente se recomienda analizar las muestras del producto líquido de la reacción para realizar el balance de masa y así conocer la producción de metanol en cada sistema. Y a pesar de los éxitos y el interés cada vez mayor que se está teniendo en la tecnología de microreacción, aún hace falta desarrollar nuevos tipos de catalizadores para aplicaciones en reactores de microcanales. REFERENCIAS BIBLIOGRÁFICAS [1] Lee, S. Methanol Synthesis Technology. USA: CRC Press, Inc, p.p. 1-154. (1990). [2] Reporte técnico de Nippon Steel. Development of Highly Efficient Methanol Synthesis Process with New Catalysts. No. 92, p.p. 83-84. (2005). [3] Brophy, J. The Microchannel Revolution. Focus on Catalyst, Vol. 1, p.p. 1-2. (2005). [4] Hessel, V., Hardt, S., y Löwe, H. Chemical Micro Process Engineering: Fundamentals, Modelling and Reactions. Weinheim, Alemania: Wiley-VCH, p.p. 43-58. (2004). [5] Olah, G. A., Goeppert, A. y Surya Prakash, G. K. Beyond Oil and Gas: The Methanol Economy. Inglaterra: Wiley-VCH, p.p. 25-46. (2006). [6] Moulijn, J. A., Makkee, M. y Van Diepen, A. Chemical Process Technology. Inglaterra: Wiley-VCH, p.p. 67-78. (2001). [7] Chang, T., Rousseau, R. W. y Kilpatrick, P. K. Methanol Synthesis Reactions: Calculation of Equilibrium Conversions using Equations of State. Industrial and Engineering Chemistry Process Design and Development, 25, p.p. 477-481. (1986). [8] Hansen, J. B. Methanol Synthesis en: Ertl G, Knözinger H. y Weitkamp J. (eds) Handbook of Heterogeneous Catalysis. Weinheim, Alemania: VCH, p.p. 1856-1876. (2004). [9] Chorkendorff, I. y Niemantsverdriet, J.W. Concept of Modern Catalysis and Kinetics. Weinheim, Alemania: Wiley-VCH, p.p. 78-99. (2005). [10] Ullmann, F. Ullmann’s Encyclopedia of Industrial Chemistry. 5ta edición, Weinheim, Alemania, Vol. A16, p.p. 465-486. (1996). [11] Wender, I. y Sayari, A. Chemistry and Uses of Carbon Dioxide. RP-25635, Final Rep., Electrical Power Research Institute, Palo Alto, California, p.p. 1-8. (1985). [12] Natta, G. Synthesis of Methanol, Catalysis, Emmet, P. H., Ed. Reinhold, New York, Vol. 3, p.p. 1-10. (1955). 66 [13] Lurgi Corporation Ltd. Hydrocarbon Process. Vol. 60 (3), p.p. 71. (1981). [14] Kagan, Y. B., Rozovskii, A., Lin, G. I., Slivinski, E. V., Loktev, S. M., Liberov, L. G. y Bashkirov, A. N. Mechanism for the synthesis of methanol from carbon dioxide and hydrogen. Kinet. Katal., 16(3), Plenum Press. USA, New York, p.p. 809. (1975). [15] Klier, K., Chatikvanij, V., Herman, R. G. y Simmons, G. W. Catalytic synthesis of methanol from CO/H2 IV. The effects of carbon dioxide. J. Catal., 74, p.p. 343. (1982). [16] Niemantsverdriet, J.W. Spectroscopy in Catalysis: An Introduction. Weinheim, Alemania: Wiley-VCH, p.p. 34-59. (1993). [17] Brunauer S., Emmett P. H. y Teller E. Adsorption of Gases in Multimolecular Layers. Journal of the American Chemical Society, 60 (2), p.p. 309-319. (1938). [18] Jensen, K. F. Microreaction engineering - is small better?. Chemical Engineering. Science, 56, p.p. 293-303. (2001). [19] Ehrfeld, W., Hessel, V. y Löwe, H. Microreactors: New technology for modern chemistry. Weinheim, Alemania: Wiley-VCH, p.p. 76-79. (2000). [20] Cornthwaite, D. (Imperial Chemical Industries Ltd. ICI). Methanol synthesis catalyst. Número de Patente: 3923694. Fecha de envío: Jul 26, 1974. Fecha de aprobación: Dec 1975. [21] Hagen, J. Industrial Catalysis. 2da edición, Mannheim: Wiley-VCM, p.p. 66-89. (2006). [22] Meland, H., Sletengen, K., Lind, A. M., Rønning M., Venvik H. J. y Holmen A. Preparación de catalizadores por spray drying para la reacción de water-gas shift a baja temperatura. Departmento de Ingeniería Química, Universidad Noruega de Ciencia y Tecnología. Trondheim, Noruega, p.p. 5-13. (2005). [23] Trambouze P. y Euzen J. P. Chemical Reactors: from design to operation. Institut Français du Pétrole Publications. Francia, París: Ediciones TECHNIP, p.p. 7-27. (2004). [24] Fogler, H. S. Elements of Chemical Reaction Engineering. USA: Prentice Hall, p.p. 15-19. (1999). APÉNDICES Apéndice A: Curvas de calibración de los controladores de flujo másico del montaje experimental para la síntesis de metanol. Apéndice B: Curvas de calibración de los compuestos de la fase gaseosa del GC del montaje experimental para la síntesis de metanol. Apéndice C: Curvas de calibración de los compuestos de la fase líquida del GC del montaje experimental para la síntesis de metanol. Apéndice D: Cálculo de las conversiones de CO y CO+CO2 en los experimentos para la síntesis del metanol. 68 Apéndice A: Curvas de calibración de los controladores de flujo másico del montaje experimental para la síntesis de metanol 3000 y = 57,406x - 75,255 R2 = 0,9993 Flujo de gas [nmL/min] 2500 2000 1500 1000 500 0 0 10 20 30 40 50 60 70 80 90 100 Punto de consigna del controlador [%] Figura A.1. Curva de calibración del controlador de flujo másico del gas de síntesis 300 Flujo de gas [nmL/min] 250 y = 2.8059x - 13.886 2 R = 0.9976 200 150 100 50 0 0 10 20 30 40 50 60 70 80 90 100 Punto de consigna del controlador [%] Figura A.2. Curva de calibración del controlador de flujo másico del helio 69 300 y = 2,7214x - 4,5386 R2 = 0,9966 Flujo de gas [nmL/min] 250 200 150 100 50 0 0 10 20 30 40 50 60 70 80 90 100 Punto de consigna del controlador [%] Figura A.3. Curva de calibración del controlador de flujo másico del hidrógeno 70 Apéndice B: Curvas de calibración de los compuestos de la fase gaseosa del GC del montaje experimental para la síntesis de metanol Figura B.1. Curva de calibración del H2 Figura B.2. Curva de calibración del N2 71 Figura B.3. Curva de calibración del CO Figura B.4. Curva de calibración del CO2 72 Figura B.5. Curva de calibración del CH4 73 Apéndice C: Curvas de calibración de los compuestos de la fase líquida del GC del montaje experimental para la síntesis de metanol Figura C.1. Curva de calibración del metilformato Figura C.2. Curva de calibración del metanol 74 Figura C.3. Curva de calibración del etanol Figura C.4. Curva de calibración del propanol 75 Figura C.5. Curva de calibración del butanol Figura C.6. Curva de calibración del 3-hexanona 76 Apéndice D: Cálculo de las conversiones de CO y CO+CO2 en los experimentos para la síntesis del metanol La conversión Xi es el número de moles de i que reaccionan por cada mol de i alimentado al sistema [24]. La conversión de CO y la conversión de CO+CO2 fueron calculadas utilizando los análisis de la fase gaseosa del cromatógrafo de gases. El N2 era un gas inerte en la reacción por lo que fue utilizado como estándar en los análisis. Analizando el gas alimentado al reactor y el gas producto de la reacción, fue posible obtener el área de los picos de N2, CO y CO2 en el cromatógrafo. La conversión fue calculada correlacionando estos resultados como se muestra a continuación: X CO = 1 − X CO +CO2 ( ACO / AN2 ) producto ( ACO / AN2 ) alimentación ×100 ⎡( ACO / AN ) + ( ACO / AN ) ⎤ − ⎡( ACO / AN2 ) + ( ACO / AN2 ) ⎤ 2 2 ⎦ 2 2 ⎣ ⎣ ⎦ producto alimentación = ×100 ⎡( ACO / AN ) + ( ACO / AN ) ⎤ 2 2 2 ⎣ ⎦ alimentación Donde, Xi = Conversión de compuesto i. Ai = Área del pico del compuesto i, obtenido del análisis del GC. (D.1) (D.2)


![A↔ B (-rA) = 0.04CA-0.01CR, [=] mol*L](http://s2.studylib.es/store/data/005357341_1-6e8dd554fb791e1c2c9f555a9c29f5b3-300x300.png)