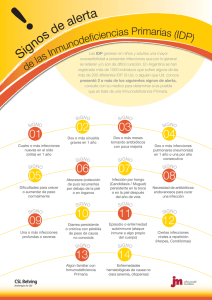
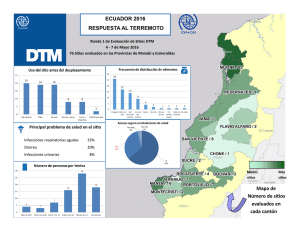
Capítulo de ejemplo
Anuncio

36 El niño con infeccio n e s r e cu r r e n t e s. Inmunodeficiencias p r ima r ia s A. Delgado Rubio 36.1. Introducción Es frecuente que el pediatra reciba mensajes por parte de las madres del tipo: “el niño está siempre malo”, “no salimos de la consulta”, “tiene una infección tras otra”, “está continuamente con infecciones”, etc. Se trata de una realidad que los pediatras viven a diario, especialmente en el ámbito de la Atención Primaria. Las madres pretenden obligar así al pediatra a solicitar estudios (análisis de sangre, radiografías, etc.) o emprender medidas terapéuticas, utilizando muchas veces antibióticos que son innecesarios y con frecuencia muy costosos, o supuestos activadores de las defensas, lo que ocasiona un gasto farmacéutico tan elevado como contraproducente. Se puede afirmar que suele existir una sobreestimación tanto del número como de la gravedad de los procesos infecciosos que sufren los niños. Por otro lado, durante los primeros años de vida, un niño sano, inmunocompetente, presenta entre 8 y 10 infecciones al año, que se reducen a 3-4 infecciones al año en la época de la adolescencia. Es decir, se calcula que entre el nacimiento y los 10 años de edad un niño sano padece en torno a 80-100 procesos infecciosos, la mayoría de ellos autolimitados, moderados o leves, sin afectación general, sin compromiso del crecimiento ni el desarrollo, con frecuencia subclínicos, carentes de importancia y habitualmente de etiología vírica. En principio, no cabe preocuparse cuando se trate de infecciones de las vías respiratorias superiores (otitis media aguda, OMA) o gastroenteritis agudas (GEA), el niño se encuentre bien entre los episodios infecciosos, muestre un crecimiento y un desarrollo adecuados, y las determinaciones analíticas basales se hallen dentro de los valores normales. Por el contrario, sí deben tenerse en cuenta en los siguientes casos: cuando el niño haya sufrido dos más infecciones graves que hayan requerido hospitalización o tratamiento antibiótico intravenoso en un intervalo de tiempo igual o inferior a 5 años; cuando se trate de infecciones prolongadas o que no responden al tratamiento adecuado; cuando las infecciones hayan sido causadas por gérmenes poco habituales y oportunistas; cuando las infecciones interfieren en el crecimiento y el desarrollo del niño, o cuando las infecciones se presentan en niños con antecedentes familiares similares, hermanos fallecidos a corta edad o afectados de inmunodeficiencia. En la Tabla 36.1 se muestran los signos de sospecha de una inmunodeficiencia primaria (IDP) en niños que propone la Cruz Roja Americana, similares a los propuestos por la Jeffrey Modell Foundation. En efecto, estos procesos, denominados catarrales, se producen con mayor frecuencia en niños que acuden a guarderías, jardines de infancia o escuelas primarias, así como en entornos urbanos superpoblados, razón por la que se observan preferentemente en hijos de familias numerosas, en los meses fríos, en hogares con padres fumadores, en ambientes con condiciones socioeconómicas precarias, etcétera. Se trata, por tanto, de una patología con un componente ambiental y social importante. 689 1. 2. 3. 4. 5. 6. Ocho o más episodios de OMA en 1 año Dos o más infecciones graves de senos paranasales en 1 año Dos o más meses de tratamiento con antibióticos con poca mejoría Dos o más neumonías en 1 año Fallo de medro Infecciones recurrentes de piel, tejidos blandos y abscesos de órganos 7. Muguet o candidiasis cutánea persistente desde el año de vida 8. Necesidad de antibióticos intravenosos para curar las infecciones 9. Dos o más infecciones graves (sepsis, meningitis, osteomielitis) 10. Historia familiar de inmunodeficiencia Tabla 36.1. Los diez signos de sospecha de inmunodeficiencia primaria de la Cruz Roja americana Tratado de pediatría Sección III - Patología infecciosa Las funciones del sistema inmunitario son: defensa contra los agentes infecciosos (bacterias, virus, protozoos, etc.), discriminación entre lo propio y lo extraño, y destrucción de las células tumorales que pueden surgir en el organismo. Ésta es la razón por la que las inmunodeficiencias, tanto primarias como secundarias, se manifiestan mediante infecciones graves y de repetición causadas por los gérmenes más diversos, se acompañan a menudo de enfermedades autoinmunitarias y tienden a la aparición de neoplasias (Tabla 36.2). En la Figura 36.1 se muestran los cambios de la función de los linfocitos T (L-T) durante el desarrollo, indicando la escasez de linfocitos Th1 en la primera infancia con niveles elevados de Th2. Al aumentar la edad, se produce una maduración del sistema Th1 hasta la edad adulta, seguida de un descenso en las edades avanzadas. Aunque no se sabe con certeza, los linfocitos T reguladores (Treg) predominan en los jóvenes, perdiendo gradualmente la función con el envejecimiento; se cree que esta inmunosenescencia explicaría el incremento de las enfermedades autoinmunitarias y de las neoplasias en las personas mayores. Función Naturaleza del estímulo inmunitario Ejemplo Trastornos hiper Defensa Exógena Agentes infecciosos Alergias Homeostasis Endógena/exógena Destrucción de células dañadas o lesionadas Enfermedades autoinmunitarias Vigilancia Endógena/exógena Destrucción de células mutantes Trastornos hipo Inmunodeficiencias Tumores malignos Tabla 36.2. Funciones del sistema inmunitario en la salud y en la enfermedad Figura 36.1. Representación esquemática de los cambios en el desarrollo de la función de los L-T (tomado de Bellanti JA, 2012) El sistema inmunitario puede definirse como la suma de todos los procesos fisiológicos que permiten que el huésped, es decir, el organismo, tenga la capacidad de reconocer elementos ajenos o extraños, y por tanto no propios, y neutralizarlos, eliminarlos o metabolizarlos, produciendo o no lesiones en los propios tejidos. Esta capacidad de diferenciar lo propio de lo ajeno es una de las principales funciones del sistema inmunitario. Cuando el organismo detecta una sustancia extraña, porque no ha podido ser bloqueada por las barreras naturales como la piel o las secreciones mucosas, esa sustancia entra en contacto con el sistema inmunitario, lo que puede dar lugar o no a una respuesta inmunitaria; cuando Figura 36.2. Posible resultado del contacto de una sustancia extraña (patógeno) con el sistema inmunitario (tomado de Bellanti JA, 2012) sucede lo segundo, es cuando se habla de la existencia de tolerancia inmunológica o inmunotolerancia. La respuesta inmunitaria puede ser, a su vez, que se pueden producir tras el contacto de una sustancia con el innata o inespecífica o bien adaptativa o específica. Los resultados sistema inmunitario se esquematizan en la Figura 36.2. 690 El niño con infecciones recurrentes. Inmunodeficiencias primarias 36.2. Composición y fisiología del sistema inmunitario El sistema inmunitario consta de dos componentes: la inmunidad innata y la inmunidad específica o adaptativa (Tabla 36.3); ambas reconocen las sustancias extrañas gracias a los receptores de superficie de las células de cada sistema. El sistema inmunitario innato usa un número relativamente pequeño de genes (102-103) en la línea germinal, que ya están programados en el genoma. Por el contrario, el sistema inmunitario adaptativo utiliza menos genes preprogramados heredados, pero tienen una gran expansión por recombinación somática (1014-1018). Inmunidad innata · · · · · Células presentadoras de Ag o CPA Células dendríticas, macrófagos Sistema fagocítico Sistema del complemento Células natural killer (NK) Inmunidad específica o adaptativa · Linfocitos B (inmunidad humoral): 1014 clonos · Linfocitos T (inmunidad celular): 1018 clonos CD4: linfocitos cooperadores: (helper) CD8: linfocitos citotóxicos o supresores Tabla 36.3. Sistema inmunitario La primera respuesta ante la presencia de una sustancia extraña es la respuesta innata (ya presente al nacer), que es primaria, estereotipada y carente de memoria. El sistema inmunitario innato reconoce en estas sustancias determinadas estructuras conocidas como PAMP (Pathogen-Associated Molecular Patterns, patrones moleculares 36 asociados a patógenos), que utilizan receptores denominados PRR (Pattern Recognition Receptors, receptores de reconocimiento de patrones), localizados en la superficie de distintas células del sistema inmunitario innato como los macrófagos. La respuesta inmunitaria innata desencadena tres mecanismos: fagocitosis, o capacidad de determinadas células para ingerir sustancias extrañas, inflamación, o respuesta del organismo a las lesiones, y citotoxicidad, o eliminación de células infectadas o transformadas mediante apoptosis. La inmunidad adaptativa, adquirida o específica es el segundo componente del sistema inmunitario, e incluye un conjunto complejo de respuestas interdependientes e interactivas, genéticamente controladas. En comparación con la respuesta inmunitaria innata, la respuesta adaptativa es más expansiva (1013- 1024) y se caracteriza por su: • Especificidad. Reconocimiento de una sustancia extraña (p. ej., antígeno) por moléculas reconocedoras de antígenos en la superficie de los linfocitos. • Heterogeneidad. Las células y los productos celulares que comprenden el sistema inmunitario adaptativo constituyen una diversidad de tipos diferentes. • Capacidad para generar memoria. Lo que la hace más eficaz al permitir que se produzca una mayor respuesta del huésped contra patógenos específicos cuando éste entra en contacto con ellos en una segunda ocasión, incluso muchos años después del contacto inicial. La capacidad del sistema inmunitario es la suma de sus capacidades de actuación frente a las sustancias extrañas, tal como se esquematiza en la Figura 36.3. La primera fase supone la respuesta inmunitaria innata; cuando es suficiente, se elimina el agente y la inmunidad innata no interviene. Si este encuentro primario se Figura 36.3. Relación entre la respuesta inmunitaria innata y adaptativa. Representación esquemática de la relación interactiva de las respuestas inmunitarias, innata y adaptativa, a la infección microbiana. Los agentes infecciosos pueden considerarse como mosaicos de PAMP y antígenos. La respuesta inmunitaria innata está inducida precozmente por PAMP (patrones moleculares asociados a patógenos) y ayuda a la respuesta inmunitaria adaptativa dirigida al antígeno a través de los procesos de presentación y coestimulación del antígeno. A pesar de que ambas respuestas son habitualmente eficaces en controlar las infecciones, a veces una respuesta no controlada o exagerada puede dar lugar a un daño propio por una respuesta inmunitaria innata no controlada (p. ej., un síndrome de respuesta inflamatoria sistémica) o un cuadro inmunopatológico por una respuesta inmunitaria adaptativa incontrolada (p. ej., enfermedad autoinmunitaria) (tomado de Bellanti JA, 2012) 691 Tratado de pediatría Sección III - Patología infecciosa produce con una sustancia extraña más compleja (bacteria, virus, etc.), ésta viene procesada (antígeno procesado), lo que produce una respuesta inmunitaria adaptativa más sofisticada, estimulándose los dos sistemas básicos: la inmunidad humoral mediada por linfocitos B, con la elaboración de los cinco tipos de inmunoglobulinas (IgM, IgG, IgA, IgD, IgE) con propiedades físicas, químicas y biológicas diferentes, y unión específica a antígenos extracelulares, y la inmunidad celular mediada por los linfocitos T. Si el antígeno se elimina con éxito, la respuesta inmunitaria finaliza en esta fase. Habitualmente, la mayoría de las sustancias extrañas se eliminan mediante estos dos primeros mecanismos, lo que supone el resultado beneficioso del sistema inmunitario en la persona sana. Si, por el contrario, el antígeno no puede eliminarse en estos dos primeros procesos, persiste y pueden producirse dos situaciones: supresión o desarrollo de daño tisular. En algunos casos, la persistencia de un antígeno puede hacer que se silencie una respuesta inmunitaria (p. e.j , supresión), y el papel inmunosupresor de los linfocitos Treg (T reguladores) parece ser preeminente. Estas res- puestas pueden ser beneficiosas, como sucede con la inmunodepresión que se produce durante el embarazo para preservar la supervivencia del embrión y del feto hasta el nacimiento, o nocivas, como la supresión de la inmunidad frente a tumores malignos, lo que les permite evadir la vigilancia del sistema inmunitario, con la consiguiente progresión del proceso neoplásico. Por otro lado, la persistencia del antígeno puede causar daño tisular. En esta tercera fase, la persistencia del antígeno puede deberse a la naturaleza de éste que, debido a su adaptación, puede resistir la degradación (p. ej., el bacilo tuberculoso en la infección tuberculosa latente), o a defectos genéticos en el procesamiento del antígeno, como ocurre en las inmunodeficiencias primarias. Como resultado de todo ello, si el antígeno persiste, se pueden producir cuatro tipos de interacciones inmunopatológicas de la clasificación de Gellis y Coombs: tipos I, II, III y IV. Estas respuestas no son beneficiosas a largo plazo para el huésped y se pueden manifestar como trastornos con una base inmunitaria, lo que representa el sistema inmunitario en la enfermedad (Figura 36. 4). Figura 36.4. El sistema inmunitario en la salud y en la enfermedad (tomado de Bellanti JA, 2012) 692 El niño con infecciones recurrentes. Inmunodeficiencias primarias La duración de esta fase de lesión tisular variará según la efectividad de la eliminación del antígeno, y puede ser pasajera o temporal (si el antígeno puede eliminarse) o más prolongada (si el antígeno persiste). Si el antígeno puede eliminarse, la respuesta en esta tercera fase acabará con unas molestias mínimas para el paciente (p. ej., contraindicar la administración de penicilina a un paciente alérgico a ésta). Sin embargo, si la respuesta en esta tercera fase es ineficaz y el antígeno sigue persistiendo, emergen otras expresiones nocivas del sistema inmunitario, y pueden manifestarse clínicamente en forma de enfermedades alérgicas, procesos autoinmunitarios o tumores malignos La inmunidad celular es el otro componente de la inmunidad adaptativa y depende de los linfocitos T, que son capaces de reconocer infecciones intracelulares (virus y bacterias que pueden sobrevivir dentro de las células que las han digerido). Los linfocitos T (L-T) desempeñan un papel principal en las respuestas de inmunidad celular y humoral del sistema inmunitario adaptativo. Estas respuestas están encomendadas a los L-T, que interactúan con los antígenos a través de receptores TCR (T- cell receptor). El procesamiento del antígeno puede producirse en dos lugares: en las células presentadoras de antígeno (APC) y en las células diana. 36 proteínas que han sido en las células a través de la vía endógena se presentan asociados a MHC-I y linfocitos T CD8 (8 x 1 = 8). La inmunidad celular es un proceso llevado a cabo por los linfocitos T, que actúan a través de citocinas o induciendo la muerte celular (citotoxicidad) sin la participación de anticuerpos (Tabla 36.4). Tradicionalmente, las citocinas han recibido su denominación según las células de origen o sus funciones específicas, lo que conlleva evidentes inconvenientes, ya que con frecuencia se producen solapamientos en ambos casos. En la Tabla 36.5 se clasifican las citocinas según las propiedades funcionales, pudiéndose agrupar en distintas categorías: interleucinas (IL), interferones (IFN), citocinas proinflamatorias y antiinflamatorias, factores de crecimiento (GF, grow factors) y quimiocinas. Las citocinas representan las moléculas por excelencia que mantienen el balance inmunológico, ya que pueden promover o inhibir la inflamación (Figura 36.6). En la Figura 36.5 se representan de forma esquemática los dos modos de procesamiento del antígeno que determina el MHC (maior histocompatibility complex, complejo principal de histocompatibilidad) con el que reaccionan. En el caso de antígenos procesados por APC, por la vía exógena, los linfocitos T CD4 reconocen el antígeno que ha sido procesado en fragmentos de péptidos (epítopos), que se colocan posteriormente en un canal de la molécula MHC II, y se presentan a los TCR y a los linfocitos T helper (células CD4). Otros antígenos se procesan a través de la vía endógena y son captados por los L-T CD8 en el marco del MHC-I. Según Bellanti, una regla nemotécnica útil para recordar qué molécula MHC se asocia a su linfocito T correspondiente es la regla del 8. La molécula procesada por APC a través de la vía exógena presenta péptidos asociados a MHC-II y linfocitos T CD4 (2 x 4 = 8). Los fragmentos peptídicos de Figura 36.5. Modos de procesamiento del antígeno Tipo Células efectoras Mecanismo efector Resultados Humoral Linfocitos B Anticuerpos Neutralización de antígenos extraños y revestimiento de sustancias para opsonización Celular Linfocitos T · Citocinas, interacción · Promoción o inhibición de la inflamación y/o función humoral célula-célula · Lisis de células infectadas · Actividad citotóxica Tabla 36.4. Componentes del sistema inmunitario adaptativo 693 Tratado de pediatría Sección III - Patología infecciosa Toda actividad celular que interviene en el mantenimiento del equilibrio entre el entorno externo e interno depende del inicio de señales extracelulares que las células reciben a través de sus membranas y transmiten después al ADN nuclear y, posteriormente, al ARN citoplasmático. Esta secuencia de acontecimientos en cascada constituye el núcleo de la biología celular y molecular (Figura 36.7), y se concreta en lo que Bellanti denomina las tres T: Transduction (Transducción), Transcription (Transcripción) y Translation (Traducción). En estas vías están implicados distintos sustratos, entre ellos JAK (Janus Kinasa) y transductores de señales y activadores de transcripción (STA). En la actualidad se sabe que las respuestas inmunitarias, innata y adaptativa, son interdependientes e interactivas, y actúan conjuntamente (véase Figura 36.3). En la Tabla 36.6 se presentan las diferencias principales entre la respuesta de la inmunidad innata y la de la inmunidad adaptativa, y en la Tabla 36.7 y Tabla 36.8, las células que las componen y sus principales funciones. En la inmunidad innata intervienen las células presentadoras de antígenos o ACP (células dendríticas, macrófagos), el sistema fagocítico (macrófagos y neutrófilos), los reactantes de fase aguda, el sistema del complemento y células citolíticas naturales (NK, natural killer). Figura 36.6. Representación esquemática de las funciones de las citocinas proinflamatorias y antiinflamatorias que mantienen el equilibrio inmunológico (tomado de Bellanti JA, 2012) Grupo Interleucinas (IL1-IL37) Producidas sobre todo por Funciones/dianas principales Células dendríticas, macrófagos, células NK, L-T, L-B y otros tipos de células somáticas Regulan la comunicación entre células dendríticas, macrófagos, L-T, L-B, células NK, células inflamatorias y otras células somáticas · Células dendríticas plasmocitoides, macrófagos · Fibroblastos Antiviral · Th1 y células Tc1, células T γδ, células NKT y NK · L-T y L-B, células NK, macrófagos, fibroblastos, células endoteliales y epiteliales, osteoblastos y otras Inmunorregulación Antiviral e inmunomodulación Citocinas proinflamatorias: · TNF- · IL-1 · IL-6 · IL-17 · TNF-/linfotoxina · · · · · · · · · Citocinas antiinflamatorias: · IL-10 · TGF- · Tr1 · Th3, IL-35? Inflamación, inmunosupresión Factores crecimiento: · G-CSF · GM-CSF · M-CSF · TGF- · · · · · · · · Interferones (IFN): · Tipo I: IFN- IFN- · Tipo II (IFN-) · Tipo III (INF-) Quimocinas: · CL · CCL · CXCL · CX3CL Macrófagos, mastocitos Macrófagos, queratinocitos y otras células nucleadas Macrófagos, células linfoides Células T memoria, Th 17 y NK Células Th1 Fibroblastos, macrófagos Fibroblastos, macrófagos L-T y otras Plaquetas, células nucleadas Muchos tipos diferentes de células Proapoptótica, promueve inflamación La mayor parte de células nucleadas L-B, hepatocitos, células hematopoyéticas APC, DC y células estromales, promueve la autoinmunidad · Proapoptótica, promueve la inflamación, organogénesis linfoide Células hematopoyéticas, granulocitos Monocitos, células progenitoras, células dendríticas Monocitos Muchas células Neutrófilos, monocitos, NK, dendríticas, L-B, L-T, eosinófilos, basófilos, mastocitos, células endoteliales Tabla 36.5. Nomenclatura de los principales grupos de citocinas (tomado de Bellanti JA, 2012) 694 El niño con infecciones recurrentes. Inmunodeficiencias primarias Características Respuesta inmunidad innata 36 Respuesta inmunidad adaptativa Momento Respuesta rápida (minutos-horas) Respuesta primaria (3-10 días); respuesta de memoria (1-2 días) Inductores moleculares PAMP: estructuras encontradas en muchos agentes infecciosos, ausentes en el huésped Antígenos: macromoleculares (proteínas y polisacáridos), formados por epítopos, presentes en moléculas extrañas pero, en ocasiones, compartidas por el huésped Receptores PRR: codificadas por genes Genes receptores específicos (receptor células T y receptor células B) en pequeño número Distribución filogenética En algunas plantas, invertebrados y vertebrados 2 Sobre todo en vertebrados 3 Número de receptores Entre 10 -10 Entre 1014-1018 Afinidad de receptores Baja afinidad Ka = 105 Alta afinidad Ka = 108 Células Neutrófilos, monocitos/macrófagos, eosinófilos, basófilos, Linfocitos T y B células epiteliales, endoteliales, dendríticas, NK y mastocitos Mecanismos efectores Producción de citocinas y quimocinas, fagocitos asesinos, Producción de citocinas, citotoxicidad celular, anticuerpos inflamación, apoptosis mediada por células neutralizantes, opsonizantes y activadores del complemento PAMP: patrones moleculares asociados a patógenos; PRR: receptores reconocedores de patrones Tabla 36.6. Principales diferencias entre las respuestas de la inmunidad innata y la adaptativa (tomado de Escobar-Gutiérrez A et al, 2012) Célula Funciones Neutrófilos Fagocitosis, inflamación; habitualmente es la primera célula que sale del torrente sanguíneo y llega al foco infeccioso Monocitos Sale del torrente sanguíneo y entra en los tejidos convirtiéndose en macrófago Macrófagos Es el fagocito más eficaz; importante en los estadios finales de la infección y en la reparación tisular; intercepta las sustancias extrañas, procesa antígenos, involucrados en la activación de los linfocitos B y T Basófilos Célula móvil que sale del torrente sanguíneo entre los tejidos y libera sustancias químicas que promueven la inflamación Mastocitos Células inmóviles presentes en el tejido conectivo que promueven la inflamación al liberar sustancias químicas Eosinófilos Salen del torrente sanguíneo y liberan productos químicos que inhiben la inflamación Natural killer (NK) Destruye las células infectadas por virus y las tumorales Figura 36.7. Las 3 T: Transducción, Transcripción y Traducción Tabla 36.7. Células de la inmunidad innata y sus funciones Célula Funciones Linfocito B Tras la activación, se diferencia y convierte en célula plasmática o linfocito B de memoria Linfocito plasmático Produce anticuerpos que son directa o indirectamente responsables de la destrucción de antígenos Linfocito B de memoria Respuesta rápida y efectiva contra un antígeno frente al cual el sistema inmunitario había previamente reaccionado Linfocito T citotóxico Responsable de la destrucción de células por lisis o por producción de citocinas Linfocito T de hipersensibilidad retardada Productoras de citocinas que promueven la inflamación Linfocito T “helper” Activa los linfocitos B y T efectores Linfocito T “supresor” Inhibe los linfocitos B y T efectores Linfocito T de memoria Respuesta rápida y eficaz contra un antígeno frente al cual el sistema inmunitario había previamente reaccionado Célula dendrítica Procesadora de antígenos e involucrada en la activación de los linfocitos B y T Tabla 36.8. Células de la inmunidad adaptativa y sus funciones 695 Tratado de pediatría Sección III - Patología infecciosa En la Tabla 36.9 se presentan los distintos componentes del sistema de reconocimiento de antígenos. El complemento es otro elemento importante de la inmunidad innata. Puede activarse de forma inespecífica cuando la manosa de la pared de algunas bacterias se fija a las proteínas fijadoras de manosa, que son reactantes de fase aguda y se sintetizan y vierten al plasma en caso de infección. La inmunidad innata se produce en pocas horas, e incluye las funciones de reconocimiento, quimiotaxis, fagocitosis, digestión intracelular y presentación antigénica, además de la lisis mediada por el complemento. Como ya se ha mencionado, los linfocitos B (inmunidad humoral) y T (inmunidad celular) son los dos componentes de la inmunidad adaptativa o específica. El reconocimiento del antígeno se realiza a través del receptor específico que se encuentra en la superficie de los linfocitos: la inmunoglobulina de superficie, en el caso de los linfocitos B, y el receptor TCR (receptor de linfocitos T), en el caso de los linfocitos T. En ambos casos, la enorme cantidad de clonos de linfocitos T y B para reconocer cualquier posible antígeno se genera mediante el mecanismo de recombinación del ADN celular denominado reordenamiento. Se ha calculado que existen 1014 clonos de linfocitos B y 1018 clonos de linfocitos T, lo que garantiza el reconocimiento de cualquier antígeno. La activación de los linfocitos B hace que se transformen en células plasmáticas y conlleva la secreción de inmunoglobulinas, mientras que la activación de los linfocitos T conduce a la producción de respuestas que amplifican y hacen más efectiva la inmunidad innata y adaptativa (linfocitos TCD4+ cooperadores), o a respuestas citotóxicas que destruyen células infectadas o con cambios tumorales (linfocitos TCD8+ citotóxicos o supresores). La función completa de los linfocitos B requiere la colaboración de los linfocitos CD4+ (helper). Por esta razón, en la mayoría de las inmunodeficiencias de células T existen alteraciones de la función de los linfocitos B. Componente celular Funciones Ontogénesis de la inmunidad A partir de la célula madre pluripotencial (pluripotential stem cell) derivan, por una parte, la célula madre o célula estaminal linfoide, y por otra, la célula estaminal no linfoide. De la célula estaminal no linfoide derivan las plaquetas, los eritrocitos y las células mielomonocíticas (granulocitos, monocitos, macrófagos, y células citolíticas naturales o NK). En el ambiente microquímico del timo, la célula estaminal linfoide da lugar a los linfocitos T cooperadores (helper) y los linfocitos T supresores, que serán los responsables de la inmunidad celular. Por otro lado, en los órganos bursoequivalentes en el hombre, la célula estaminal linfoide da lugar a las células B precursoras, que posteriormente se convertirán en linfocitos B, los cuales se transforman en células plasmáticas y darán lugar a las distintas inmunoglobulinas (IgG, IgA, IgM, IgD, IgE), que se encargarán de la inmunidad humoral (Figura 36.8). Dependiendo del lugar en el que se produzca el bloqueo, se producirán los distintos tipos de inmunodeficiencias primarias (IDP) (Figura 36.9). Clasificación de las inmunodeficiencias primarias A lo largo de los años, se ha ido sucediendo una serie de clasificaciones de las inmunodeficiencias primarias (IDP). La última clasificación disponible es la propuesta por el Comité de Expertos de la International Union of Immunological Societes (IUIS), que se reunieron en la ciudad de Nueva York los días 31 de mayo y 1 de junio de 2011. La clasificación actual propone ocho grupos distintos; sin embargo, cualquier clasificación de las IDP es arbitraria y alguno de los trastornos podrían incluirse en más de un grupo. 1. Inmunodeficiencias combinadas 2. Síndromes bien definidos con inmunodeficiencias 3. Deficiencias predominantemente de anticuerpos 4. Enfermedades de desregulación inmunitaria 5. Defectos congénitos del número o de la función de los fagocitos, o de ambas cosas Ejemplos de tipos de células efectoras Células presentadoras de antígenos Presentación de antígeno Macrófagos, células dendríticas, L-B Linfocitos T Inmunidad celular Linfocitos CD4 “helper”: · Células Th1 · Células Th2 · Células T reguladoras · Células Th17 Mecanismos Captación, digestión y exposición del antígeno en la superficie de la célula MHC · Promoción de la inflamación por producción de citocinas · Proliferación linfocitos B, producción de anticuerpos y células B de memoria · Respuestas reguladoras inmunosupresoras · Promoción de producción de citocinas inflamatorias Linfocitos CD8 citotóxicos: Destrucción de células diana · Células Tc1 · Células Tc2 Linfocitos B Inmunidad humoral Células plasmáticas Síntesis de anticuerpos Tabla 36.9. Componentes celulares, funciones y mecanismos del sistema de reconocimiento de antígenos 696 El niño con infecciones recurrentes. Inmunodeficiencias primarias 36 Figura 36.8. Ontogénesis del sistema inmunitario. Representación esquemática de la ontogénesis del sistema inmunitario, mostrando la diferenciación de células progenitoras en células hematopoyéticas y células linfopoyéticas inmunocompetentes a partir de una población común de células madre hematopoyéticas pluripotenciales CD34+ de la médula ósea (HSC: hematopoietic stems cells). Los precursores mieloides se diferencian en linajes, eritroides, megacariocíticos y granulocitos/monocitos, mientras que los precursores linfoides dan lugar a las células natural killer (NK) y linfocitos T y B. Las células progenitoras linfoides pueden diferenciarse a lo largo de dos vías adicionales. El desarrollo de los linfocitos T requiere la influencia del timo, mientras los linfocitos B se desarrollan en el microambiente de los órganos “bursodependientes”. Tras la diferenciación, los L-T y L-B se localizan en las regiones T y B de los ganglios linfáticos. respectivamente (tomado de Bellanti JA, 2012) Figura 36.9. Principales inmunodeficiencias primarias, según localización del bloqueo. Representación esquemática de las fases del desarrollo inmunológico en el que la disfunción o la deficiencia dan lugar a distintas inmunodeficiencias primarias. 1. Disgenesia reticular. 2. Anemia aplásica. 3. Enfermedad granulomatosa crónica. 4. Inmunodeficiencia combinada severa (IDCS). 5. Síndrome de Di George. 6. Deficiencia de coronina-1A. 7. Agammaglobulinemia ligada al X. 8. Inmunodeficiencia común variable (IDCV) (tomado de Bellanti JA, 2012) 697 Tratado de pediatría Sección III - Patología infecciosa dicina molecular, a un diagnóstico correcto, un asesoramiento genético y un tratamiento, con lo que podrá en muchas ocasiones curar, mejorar la calidad de vida del paciente o influir sobre su evolución y pronóstico. 6. Defectos de la inmunidad innata 7. Enfermedades autoinflamatorias 8. Deficiencias del complemento Según el Registro Europeo de IDP, estas inmunodeficiencias presentan distintas frecuencias (Figura 36.10). Las IDP constituyen, por tanto, un grupo muy heterogéneo de enfermedades del sistema inmunitario, individualmente inusuales (a excepción del déficit de IgA), pero relativamente frecuentes en su conjunto. Debido a que suelen debutar en la edad infantojuvenil, el pediatra tiene que conocerlas bien, ya que es su deber sospecharlas para poder llegar, con la colaboración de Unidades de Inmunología y laboratorios especializados en genética y meDéficit anticuerpos El síndrome clínico con el que se manifiestan las IDP va depender del aspecto de la inmunidad que esté alterado (Tabla 36.10 y Tabla 36.11). Las inmunodeficiencias humorales se caracterizan por comenzar en el segundo semestre de la vida, una vez que han desaparecido los anticuerpos de origen materno transmitidos a través de la placenta. Cursan con infecciones recurrentes por bacterias encapsuladas que producen manifestaciones respiratorias (neumo- Déficit fagocitosis Inmunodeficiencias combinadas Déficit complemento Tipo de infecciones Infecciones recurrentes por bacterias (sepsis, broncopulmonía) Infección recurrente o grave (sepsis, abscesos) por bacterias y hongos Infecciones graves (neumopatía, enteritis, sepsis) bacterianas, víricas, por hongos Meningitis por Neisseria (déficit factores tardíos) o (déficit de C3) por piógenos Edad de comienzo Después del sexto mes Desde los primeros meses Desde los primeros meses En época neonatal Distrofia, fallecimiento precoz Lupus eritematoso sistémico (déficit factores precoces) Complicaciones Bronconeumopatía crónica Bronconeumopatía hepatoesplenomegalia osteomielitis Tabla 36.10. Evolución natural de las principales formas de inmunodeficiencias primarias 2011 2010 Diagnóstico 2011 Trastorno predominantemente de los anticuerpos 2010 56,23% (n = 8.464) 55,60% (n = 7.238) Deficiencias predominantemente de linfocitos T 7,71% (n = 1.160) 7,62% (n = 992) Trastornos de la fagocitosis 8,10% (n = 1.219) 10,22% (n = 1.330) Deficiencias del complemento 4,27% (n = 643) 4,73% (n = 616) 15,46% (n = 2.327) 16,83% (n = 2.191) S. autoinmunitarios y de desregulación inmunitaria 3,77% (n = 567) 1,39% (n = 181) Enfermedades autoinflamatorias 1,93% (n = 291) 1,88% (n = 245) Defectos de la inmunidad innata 0,94% (n = 141) 0,00% (n = 0) Otras inmunodeficiencias primarias inclasificables 1,59% (n = 240) 1,72% (n = 224) Otras inmunodeficiencias bien definidas Número total de pacientes: 100,00% (n = 15.052) Figura 36.10. Inmunodeficiencias primarias en Europa (fuente: European Society For Immunodeficiencies) 698 100,00% (n = 13.017) El niño con infecciones recurrentes. Inmunodeficiencias primarias Alteración 36 Síndrome clínico Linfocitos B · Infecciones sinupulmonares por bacterias encapsuladas. Otitis media crónica · Giardiasis. Episodios repetidos de enfermedades víricas · Enfermedades víricas de grave evolución. Encefalitis crónica por enterovirus Linfocitos T · · · · Complemento · Infecciones sinupulmonares por bacterias encapsuladas. Otitis crónica · Meningitis y bacteriemias por Neisseria. S. autoinmunitaros. Glomerulonefritis Infecciones diseminadas por patógenos intracelulares. Infecciones por hongos y protozoos oportunistas Infecciones por virus ADN, habitualmente benignas. Diarrea prolongada. Eczema Endocrinopatía Enf. injerto contra huésped después de transfusión. Neoplasías malignas Enfermedad granulomatosa · Infección debida a bacterias catalasa positivas (estafilococo, enterobacterias) crónica · Nocardia, Cándida, Aspergillus · Celulitis, adenitis supurativa, sinusitis, osteomielitis, periodontitis, abscesos viscerales y cerebrales, granulomas recurrentes Deficiencia de mieloperoxidasa Niños asintomáticos. No aumento de susceptibilidad a las infecciones. Infección fúngica asociada a enfermedad sistémica (p. ej.: niño diabético que desarrolla una candidiasis invasiva) Síndrome de Chediak-Higashi · Neutropenia moderada. Albinismo oculocutáneo parcial. Anomalías neurológicas · Identificación de gránulos gigantes en los neutrófilos Síndrome de hiper-IgE Dermatitis atópica “atípica”. Facies peculiar. Cabello rojizo. Eosinofilia. Elevación IgE Neutropenia congénita grave · Neutropenia intensa (< 500 polimorfonucleares/mm3) y persistente · Infecciones graves y recurrentes (gingivitis, celulitis, adenitis) Deficiencia adhesividad leucocitaria · Hiperleucocitosis 20.000-30.000 leucocitos/mm3, con 80-90% neutrófilos) en condiciones basales · Retraso caída cordón umbilical Tabla 36.11. Síndromes clínicos asociados a inmunodeficiencias nía, sinusitis) crónicas o recurrentes, sepsis y, con menos frecuencia, meningitis, osteomielitis, etc., junto con otras características que se recogen en la Tabla 36.12. En la Tabla 36.13 se muestran los agentes infecciosos más habituales en las IDP por déficis de la inmunidad humoral. de injerto contra huésped debidas a transfusiones de sangre o al paso de linfocitos T de la madre al niño durante el parto; en este Las inmunodeficiencias celulares suelen debutar en el primer semestre de la vida con la aparición de infecciones recurrentes causadas por virus, hongos y micobacterias, así como por microorganismos oportunistas (P. jiroveci, Cryptosporidium, citomegalovirus, etc.). Es habitual el fallo de medro en los niños afectados. Estos pacientes pueden presentar infecciones muy graves e incluso mortales tras la administración de la vacuna BCG o de vacunas con virus vivos atenuados. También son frecuentes las reacciones Presentación Infecciones Ejemplos Laboratorio Bacterias · · · · · · · Haemophilus Streptococcus Staphylococcus Salmonella Meningococos Pseudomonas Campylobacter Otros · Mycoplasma · Ureaplasma Virus · · · · · ECHO Polio Rotavirus Hepatitis B Hepatitis C Protozoos · Lamblia · Cryptosporidium · P. jiroveci Tabla 36.13. Agentes infecciosos en los déficits de anticuerpos Infecciones recurrentes (OMA, sinubroncopulmonares), diarrea crónica , malabsorción intestinal, fallo de medro, afectación articular crónica, aumento procesos autoinmunitarios y alérgicos, comienzo de los síntomas a los 7-9 meses Bacterias encapsuladas (p. ej., S. pneumoniae, H. influenzae), virus (enterovirus) Síndrome de Bruton, inmunodeficiencia común variable Screening Determinación cuantitativa de niveles de inmunoglobulinas (IgG, IgA, IgM, IgE) y recuento de L-B por citometría de flujo Avanzado Determinación de subclases de IgG (IgG1, IgG2, IgG3 e IgG4) y respuesta de anticuerpos específicos preinmunización a vacunas de difteria, tétanos, neumococo y H. influenzae; si no hay niveles protectores revacunar y controlar títulos, tras la vacunación 3-4 semanas después y determinar la respuesta linfoproliferativa a mitógenos; fitohemaglutimina (PHA), concanavalina A (Con A) y forbomiristil acetato (PMA/ionomicina) Especializado Biopsia ganglio linfático, análisis de citocinas y medición de deficiencias moleculares específicas Tabla 36.12. Características y estudios a practicar en pacientes con defectos de los linfocitos B (tomado de Bellanti JA, 2012) 699 Tratado de pediatría Sección III - Patología infecciosa · · · · · · caso, el recién nacido presenta exantemas generalizados a los pocos días de nacer. Existe, además, una incidencia elevada de tumores malignos y una disminución de la supervivencia. En la Tabla 36.14 se presentan las principales manifestaciones clínicas de las inmunodeficiencias celulares. Neisseria Streptococcus H. influenzae E. coli Brucella Staphylococcus aureus Tabla 36.16. Agentes infecciosos en los deficit de complemento En muchas inmunodeficiencias celulares se afecta la función de los linfocitos B por la ausencia de la función cooperadora de los linfocitos T helper, lo que da lugar a las inmunodeficiencias combinadas, que son sin duda alguna las formas más graves de IDP. En el caso de las IDP debidas a defectos de la inmunidad innata, cuando la causa es un déficit de los primeros factores del complemento, es decir, C1Q, C1R, C1S, C2, C3 y C4, los pacientes suelen presentar infecciones recurrentes por bacterias piógenas y síndromes reumatoides por inmunocomplejos. Sin embargo, cuando el déficit corresponde a factores tardíos del complemento, es decir, C5, C6, C7, C8, C9, lo habitual es que los pacientes afectados presenten infecciones recurrentes por Neisseria spp y enfermedades por inmunocomplejos. El déficits de inhibidor C1 se asocia a edema angioneurótico. En la Tabla 36.15 y Tabla 36.16 se resumen los datos clínicos sugestivos de los déficits de complemento, así como los agentes infecciosos implicados con más frecuencia. La deficiencia del número o la función de las células NK se manifiesta por infecciones recurrentes y graves causadas por virus del grupo herpes, sobre todo virus del herpes simple y virus de la varicela-zóster. Presentación Infecciones Ejemplos Laboratorio Aunque existen distintas formas de expresión clínica de los déficit de la función fagocitaria, según se trate de una enfermedad granulomatosa crónica, de una neutropenia cíclica, de una agranulocitosis congénita, de deficiencias de moléculas de adhesión leucocitaria, etc., cada una de ellas con sus peculiaridades, en general este tipo de IDP se caracteriza por infecciones recurrentes de piel y mucosas, lesiones bucales, abscesos de repetición, neumonías con formación de neumatoceles, enfermedad periodontal y retraso en la caída del cordón (deficiencia de moléculas de adhesión leucocitaria), leucopenia, leucocitosis, neutrofilia, eosinofilia, etc. (Tabla 36.17), y los pacientes afectados van a presentar infecciones por determinados gérmenes (Tabla 36.18). Además de las infecciones recurrentes causadas por distintos microorganismos, existen otras manifestaciones (dependiendo del tipo de IDP) que traducen el déficit de otras funciones inmunitarias: • Enfermedades malignas. Los pacientes con IDP tienen un riesgo de desarrollar procesos malignos entre 10 y 200 veces superior al de las personas inmunocompetentes, especialmente en las deficiencias de linfocitos T. • Autoinmunidad. En las IDP se han descrito múltiples síndromes autoinmunitarios, entre ellos anemia hemolítica inmunitaria, púrpura trombocitopénica inmunitaria (PTI), lupus eri- Fallo de medro, diarrea crónica, infecciones recurrentes por bacterias, virus, hongos y por oportunistas, poliendocrinopatías, muguet oral y generalizado, infecciones mortales por BCG o por vacuna de virus vivos, supervivencia acortada, comienzo sintomatología a los 4-5 meses, enfermedad de injerto contra huésped por transfusiones, mayor incidencia de tumores malignos Virales (p. ej., herpes simple, CMV); Candida; protozoos (p. ej., Pneumocystis jiroveci) Síndrome de inmunodeficiencia combinada grave. Síndrome de Di George Screening Serie blanca con recuento y morfología de linfocitos (linfopenia marcada), test de hipersensibilidad retardada (cándida, Trichophyton) Avanzado Determinación cuantitativa de niveles de inmunoglobulinas (IgG, IgA, IgM, IgE) y L-T por citometría de flujo (CD3, CD4, CD8, CD56 y CD25) Especializado Respuesta linfoproliferativa a mitógenos (PHA) Tabla 36.14. Características y estudios a practicar en pacientes con defectos de los linfocitos T (tomado de Bellanti JA, 2012) Presentación Infecciones Ejemplos Laboratorio Infecciones bacterianas recurrentes (meningitis y gonococias) , enfermedades autoinmunitarias (p. ej., LES y LES-like) y angioedema de distinta localización, infecciones piógenas cutáneas , historia de herencia autosómica recesiva Causadas por grampositivos (p. ej., S. aureus) o gramnegativos (p. ej., Neisseria sp) Pacientes con defectos de C3 o C5/C6/C7: LES o esclerodermia; angioedema hereditario Screening Niveles de CH50, C3 y C4 , hipergammaglobulinemia Avanzado Cuantificación de componentes individuales del complemento Especializado Pruebas capacidad opsónica y quimiotáctica del suero Tabla 36.15. Características y estudios a practicar en pacientes con defectos del complemento (tomado de Bellanti JA, 2012) 700 El niño con infecciones recurrentes. Inmunodeficiencias primarias • • tematoso sistémico (LES), hepatitis crónica activa, miastenia grave, enfermedad inflamatoria intestinal, etc. Atopia y dermatitis. En forma de rinitis, asma, eczema, urticaria, anafilaxia, exantemas, etc. Infecciones víricas no habituales. Los pacientes con IDP con un defecto predominantemente de la inmunidad humoral, en especial los afectados por un síndrome de Bruton, son muy propensos a las infecciones víricas por virus ECHO, por enterovirus (poliovirus) o por otros virus hepatotrópicos, por lo que estas infecciones pueden tener una evolución grave. La mayoría de los niños con infecciones recurrentes, especialmente respiratorias, no tienen una IDP, ya que estas infecciones de repetición también pueden observarse en la fibrosis quística, el síndrome de cilios inmóviles (discinesia ciliar), alteraciones anatómicas del aparato respiratorio, algunos errores congénitos del metabolismo, fístulas comunicantes, etc. Como siempre, la anamnesis desempeña un papel fundamental y en ella debe reflejarse la naturaleza, la frecuencia, la duración y la gravedad de las infecciones. Asimismo, es primordial obtener una información detallada sobre los antecedentes familiares, averiguando si los padres presentan comportamientos de riesgo para la infección por el VIH (causa principal de inmunodeficiencia secundaria), si el niño ha tenido hermanos con procesos similares o si existen familiares que hayan fallecido afectados por cuadros parecidos. Todos estos datos pueden ser orientativos. El antecedente de la administración de corticoesteroides o de otros inmunosupresores puede ser la causa de una inmunodeficiencia secundaria. Por ello, es importante interrogar sobre los facPresentación Infecciones Ejemplos Laboratorio 36 tores ambientales para conocer el círculo social del niño, si asiste a guarderías, jardín de infancia o colegio, la posibilidad real de contaminación ambiental si los padres son fumadores, así como las condiciones de hacinamiento, bienestar, temperatura, humedad, etc., relacionadas con el ambiente del hogar en el que el niño vive. El inicio de la sintomatología también puede ser orientador: generalmente, las IDP combinadas y por déficit de la inmunidad celular, así como los trastornos de la fagocitosis, se manifiestan en los primeros meses de vida, mientras que las inmunodeficiencias debidas a alteraciones de la inmunidad humoral debutan en el segundo semestre, una vez que han desaparecido los anticuerpos procedentes de la madre recibidos por el niño a través de la placenta. Debe valorarse también la localización de las infecciones y determinadas características semiológicas. Las anomalías anatómicas o malformativas, así como la hipertrofia de adenoides y el reflujo a través de las trompas de Eustaquio, favorecen la otitis media aguda (OMA) de repetición; las neumonías recidivantes junto a bronquiectasias y atelectasia apuntan hacia una posible fibrosis quística; las sinusitis recidivantes junto a la presencia de dextrocardia deben hacer pensar en un síndrome de cilios inmóviles (síndrome de Kartagener); la presencia de abscesos cutáneos fríos en un niño con facies peculiar y dientes supernumerarios es característica del síndrome de hiper-IgE (SHIE) o síndrome de Job. El tipo de agente infeccioso proporcionará pistas valiosas. Las infecciones causadas por hongos, protozoos y virus del grupo herpes se producen preferentemente en las IDP por afectación de la inmunidad celular. Las infecciones causadas por bacterias capsuladas (neumococo, S. aureus, S. pyogenes, Pseudomonas spp) apuntan Abscesos de repetición, infecciones de localización profunda, úlceras orales, eczema, neumonías de repetición (neumatoceles), mala cicatrización de las heridas, gingivitis, enfermedad periodontal , retraso en la caída del cordón umbilical Causadas por bacterias catalasa positiva (S. aureus, Pseudomonas, Serratia, Klebsiella, Candida, Nocardia, Aspergillus) · Defectos cuantitativos: neutropenias congénitas · Defectos cualitativos: enfermedad granulomatosa crónica (EGC) Screening Leucocitos y fórmula (leucopenia, leucocitosis, eosinofilia), niveles de inmunoglobulinas (hipergammaglobulinemia) y de IgE Avanzado Prueba de la dihidrorodamina Especializado Prueba de adherencia, quimiotaxis, fagocitosis y actividad microbicida Tabla 36.17. Características y estudios a practicar en pacientes con defectos de la fagocitosis (tomado de Bellanti JA, 2012) Organismo Cuadro clínico Comentarios S. aureus Adenitis, abscesos de partes blandas, osteomielitis Aparece en todos los trastornos de la función fagocítica Serratia sp Abscesos, osteomielitis, bacteriemia En la enfermedad granulomatosa crónica (EGC) Salmonella sp Abscesos, bacteriemia En la EGC y en déficit de IL-12 Nocardia sp Abscesos de partes blandas y cerebrales, neumonía En la EGC Burkholderia cepacia Bacteriemia En la EGC Aspergillus sp Abscesos cerebrales y de partes blandas, neumonía En la EGC Micobacterias atípicas Osteomielitis, infección diseminada En el déficit de cadena del receptor del interferón Tabla 36.18. Agentes infecciosos en las deficiencias de la función fagocítica (tomado de Bellanti JA, 2012) 701 Tratado de pediatría Sección III - Patología infecciosa Infecciones urinarias hacia IDP preferentemente humorales. Cuando se trata de bacterias de escasa virulencia, hay que pensar en una posible disfunción de los neutrófilos; las infecciones por P. jiroveci sugiere una infección por el VIH o alteraciones graves de la inmunidad celular; la meningitis meningocócica recidivante hace sospechar una alteración del complemento y, por el contrario, la meningitis neumocócica de repetición señala hacia la presencia de una fístula del líquido cefalorraquídeo. · · · · · Manifestaciones cutáneas S. Wiskott-Aldrich Telangiectasias S. ataxia-telangiectasia Albinismo oculocutáneo S. Chediak-Higashi Exantema dermatomiositis “like” Disfunción linfocitos B (S. Bruton) Dermatitis crónica S. hiper IgE Exantema lupus “like” Déficit complemento (c. precoces) Cicatrices cutáneas Defectos de fagocitosis Molusco contagioso generalizado Déficit linfocitos T Verrugas generalizadas Déficit linfocitos T Candidiasis Déficit linfocitos T Socialización (guardería, escuela, etc.) Inhalación pasiva de humo de tabaco Contaminación atmosférica Bajo nivel socioeconómico familiar Elevado número de convivientes (hacinamiento) Con patología de base · Anomalías anatómicas y malformaciones de vías aéreas (neumonías recidivantes) · Fibrosis quística (neumonías recidivantes) · Asma bronquial · Hipertrofia de adenoides (otitis media recidivante) · Aspiración árbol respiratorio (reflujo gastroesofágico, hernia hiatal, fístula traqueoesofágica, etc.) · Síndrome cilios inmóviles (infección sinupulmonar recidivante) · Déficit de anticuerpos (infecciones recidivantes diversas) · Cuerpos extraños Tabla 36.20. Etiologías de infecciones respiratorias recurrentes en el niño (tomado de Geha RS y cols., modificado, 1986) · · · · · · · · · Infecciones de distinta localización Diabetes mellitus Galactosemia Déficit α1-antitripsina Drepanocitosis Anesplenia funcional u orgánica Leucemias, linfomas, neoplasias Dermatitis atópica Quemaduras Malnutrición 36.3. Inmunodeficiencias combinadas Síndromes de inmunodeficiencia combinada severa o grave Sin embargo, como ya se ha adelantado, no siempre las infecciones intercurrentes se deben a IDP. En la Tabla 36.20 y Tabla 36.21 se resumen las principales etiologías de las infecciones respiratorias recurrentes y de otras localizaciones, respectivamente, en los niños. · · · · · Infecciones vías aéreas Fístula de líquido cefalorraquídeo Malformaciones Válvulas Fracturas De entre todas las IDP referidas en la clasificación de la IUIS (2011), se analizan a continuación las que tienen mayor interés clínico. Tabla 36.19. Manifestaciones cutáneas características de las inmunodeficiencias primarias Por factores ambientales en ausencia de patología de base · · · · Tabla 36.21. Etiologías no inmunológicas de infecciones recurrentes de distinta localización en el niño Defectos inmunitarios Eczema y petequias Estenosis uretral Litiasis renal Reflujo vesicoureteral Compresiones extrínsecas Cuerpos extraños · Hipertrofia adenoides · Estrechez vías aéreas · Dificultad evacuación secreciones · S. cilios inmóviles · Cuerpos extraños · Fibrosis quística En la mayoría de los casos, la exploración física detallada proporciona indicaciones valiosas, como se analizará en el apartado correspondiente al diagnóstico. Existe también una serie de manifestaciones cutáneas que apuntan hacia determinadas IDP (Tabla 36.19). Infecciones del SNC Los síndromes de inmunodeficiencia combinada grave (IDCG) constituyen un conjunto de enfermedades de base genética, en las que existe una afectación de la linfogénesis T y B. Presentan una frecuencia de 1/30.000 – 1/50.000, y entre ellas se deben considerar la IDCG T–B+ y la IDCS T–B–, dentro de las cuales existen distintos subtipos, cada uno con su defecto genético. La IDCG es un trastorno hereditario caracterizado por un defecto grave o una ausencia de función de los linfocitos T y B, lo que conlleva una predisposición importante a las infecciones. Suele ser mortal en los primeros meses de vida, salvo que se realice un trasplante de células madre hematopoyéticas o, en el caso del déficit de adenosina-desaminasa (ADA), de la enzima correspondiente. La identificación precoz del cuadro, antes de que aparezcan infecciones oportunistas, es fundamental para la evolución de estos pacientes. Se debe sospechar la presencia de una IDCG en lactantes con linfopenia < 1.500/mm3 (intervalo normal de 4.000 a 13.500/mm3), < 20% de linfocitos T CD3+ e hipogammaglobulinemia intensa (< 150 mg/dl). A pesar de la heterogeneidad genética de la IDCG, estos pacientes presentan infecciones bacterianas (sepsis por gramnegativos, diseminación de la BCG), por hongos, protozoos y virus, que son causa de muerte en numerosas ocasiones. 702 El niño con infecciones recurrentes. Inmunodeficiencias primarias Estos niños están asintomáticos al nacer, y muchas veces fallecen entre los 6 y los 12 meses de vida sin haberse establecido un diagnóstico y, por supuesto, sin tratamiento. El diagnóstico precoz es fundamental para mejorar el pronóstico y para instaurar las posibles medidas terapeúticas antes de que el paciente presente infecciones graves. Por este motivo, se propone incluir en el cribado neonatal habitual de los errores innatos del metabolismo, utilizando para ello la misma sangre seca del talón, la prueba denominada TREC (T-cell receptor excision circles, círculos de escisión de receptores de linfocitos T) para investigar la producción de linfocitos T en el timo. A esta prueba se puede añadir otra para detectar los defectos de desarrollo de los linfocitos B mediante la k-deletion Recombination Excision Circles (KREC). Algunos autores consideran que el cribado neonatal de la IDCG debiera ser obligatorio en la Unión Europea (como lo es en la actualidad en 13 estados de Estados Unidos) por dos razones fundamentales: porque es más frecuente que algunos errores innatos del metabolismo contemplados actualmente, y porque el diagnóstico precoz influye de forma decisiva en el tratamiento y en el pronóstico. El diagnóstico se sospechará en un niño que presente cualquiera de las manifestaciones siguientes, sobre todo si están asociadas: linfopenia inexplicable, fallo de medro, diarrea crónica, episodios graves recurrentes de infecciones por virus respiratorio sincitial (VRS), virus de la varicela-zóster (VVZ) o virus del herpes simple (VHS), sarampión, virus de la gripe o parainfluenza, antecedentes familiares de IDCG y candidiasis oral recurrente. Según el fenotipo de linfocitos T, B y células NK en sangre periférica, la IDCG se puede dividir en cinco variedades fundamentales (Tabla 36.24). T– B+ NK– SIDCS SIDCS ligado al X, cadena gamma común (gamma C) Janus Kinasa 3 Receptor cadena alfa receptor de interleucina 7 (CD127) Corina 1A proteína reguladora-activa (CORO1A) Componente cadena CD3 · CD3 alfa · CD3 epsilon · CD3 theta Componentes cadena CD 45 · · · · · Tabla 36.22. Inmunodeficiencia grave combinada. Características · · · · Anemia grave. Linfopenia marcada Ausencia o disminución de las inmunoglobulinas séricas Ausencia de respuesta de AC tras estímulo vacunal Compromiso de los test de funcionalidad de los T-linfocitos (in vivo: DNCB, BCG; in vitro; PHA, Con A, rosetas E) · Ausencia de linfocitos, células plasmáticas y linfoblastos a nivel medular · Depleción de los ganglios linfáticos periféricos (áreas T-dependientes y no T-dependientes) · Timo de pequeñas dimensiones con ausencia de linfocitos y corpúsculo de Hassal PATOGENIA: Compromiso de la diferenciación de las células progenitoras en fase muy precoz Tabla 36.23. Inmunodeficiencia grave combinada. Datos de laboratorio IL7RA CORO1A CD3D CD3E CD3Z PTPRC T– B– NK+ SIDCS Gen 1 y 2 activador recombinasa ARTEMIS Subunidad catalítica de la ADN proteína-cinasa RAG1, RAG2 DCLRE1C PRKDC T– B– NK– SIDCS Adenosina-deaminasa (ADA) ADA T– B (bajo) NK+ SIDCS Disgenesia reticular Herencia: A-R, L-X, esporádica Inicio muy precoz, en los primeros meses de la vida Distrofia, anemia, síndrome de malabsorción Infecciones recurrentes de etiología bacteriana, vírica, micótica (Candida), protozoaria (P. jiroveci) Graves complicaciones tras vacunaciones con bacterias o virus vivos atenuados (BCG, etc.) Enfermedad injerto contra huésped después de transfusiones con sangre fresca Ausencia de sombra tímica en la RX tórax Amígdalas y ganglios linfáticos pequeños Fallecimiento habitualmente en el primer año de vida IL2RG JAK3 T– B+ NK+ SIDCS Con frecuencia, los niños afectados presentan fallo de medro por diarrea y malabsorción. La aparición de un eritema cutáneo precoz indica una manifestación de enfermedad de injerto contra huésped como consecuencia de la presencia de linfocitos T en el círculo de procedencia materna, que pasaron a través de la placenta. En la radiografía de tórax suele observarse una ausencia de sombra tímica (Tabla 36.22). En sangre periférica, estos pacientes tienen una cifra de linfocitos T CD3+ < 500/mm3 (intervalo normal de 3.000 a 6.500/mm3), y un número variable de linfocitos B y de células NK (Tabla 36.23). · · · · 36 AK2 Tabla 36.24. Síndrome de inmunodeficiencia combinada grave. Clasificación y defectos genéticos Déficit de adenosina-deaminasa El déficit de adenosina-deaminasa (ADA) produce una acumulación de metabolitos tóxicos de la vía de las purinas y de la vía de la metilación de la S-adenosilhomocisteína en las células, lo que dificulta su proliferación y, como consecuencia, se afectan los L-T y los L-B. Esta afección se debe a mutaciones que codifican la ADA en el cromosoma 20q13-ter, se hereda de forma autosómica recesiva y, en raras ocasiones, da lugar a un cuadro clínico moderado, por lo que el debut del déficit de ADA es más tardío. Disgenesia reticular Esta rara enfermedad autosómica recesiva es la forma más inusual de IDCG, ya que afecta a la inmunidad celular y humoral, y suele ser mortal tras el nacimiento. Se debe a un fallo en la maduración de los precursores linfoides y mieloides, y se caracteriza por linfopenia importante, granulocitopenia, trombocitopenia e hipoplasia tímica, lo que conlleva la aparición de infecciones graves que causan la muerte precoz del paciente. 703 Tratado de pediatría Sección III - Patología infecciosa Deficiencia de RAG1 y RAG2 Déficit de purina-nucleósido-fosforilasa En 1996 se describieron por primera vez los pacientes con déficit del gen que codifica la recombinasa específica de los linfocitos (RAG-1 o RAG-2). Este gen es esencial para la generación de receptores antigénicos de los linfocitos T y B, y su déficit causa, por tanto, la ausencia o la disminución de ambos tipos celulares. Se trata de una inmunodeficiencia combinada poco frecuente en la que los defectos inmunológicos se asocian a síntomas neurológicos como retraso del desarrollo, problemas de comportamiento y anomalías motoras. Aunque a veces el cuadro clínico es similar al de una IDCS, en otras ocasiones se incluye entre las inmunodeficiencias combinadas con defecto moderado de los linfocitos B. En general, estos pacientes presentan linfopenia (< 1.000/mm3), 1-5% de linfocitos T en sangre periférica, y niveles bajos de IgG e IgA. A pesar de una hipofunción de los linfocitos B, más del 30% de estos pacientes desarrollan procesos autoinmunitarios (anemia hemolítica, trombocitopenia, vasculitis), y en ocasiones fallecen a causa de linfomas y otros tumores. Síndrome de Omenn Se trata de una forma poco frecuente de IDCG, descrita por este autor en 1965. Desde el punto de vista clínico, se caracteriza por la presencia de eritrodermia, adenopatías, hepatoesplenomegalia, fallo de medro secundario a diarrea, edemas generalizados y fiebre. Desde el punto de vista analítico, se observa: hipoalbuminemia, eosinofilia (> 1.000/mm3), cifras variables de linfocitos con aumento o disminución de L-T CD3+, ausencia de linfocitos B y un número normal de linfocitos NK; cursa también con una hipogammaglobulinemia policlonal con disminución importante de IgG, IgM e IgA y un aumento de IgE (> 1.000 U/mL). IDCG tipo Navajo Es una forma de IDCG que se produce en 1 de cada 2.000 nacidos vivos entre los indios navajos. El cuadro clínico es similar al de otras IDCG, salvo en que los niños afectados presentan úlceras genitales y orales no asociadas a herpesvirus en los primeros meses de vida. Se observa una afectación grave del número y la función de los linfocitos T y B, con normalidad de los linfocitos NK, por lo que esta afección se incluye en las IDCG de tipo T-B-NK+. Síndrome de hipoplasia cartílago-pelo El síndrome de hipoplasia cartílago-pelo es una enfermedad autosómica recesiva inusual, que cursa con hipocrecimiento (condrodisplasia metafisaria) y otros rasgos fenotípicos característicos, como pelo ralo y escaso, enfermedad de Hirschsprung, inmunodeficiencia humoral y celular, bronquiectasias, enfermedades autoinmunitarias, y un mayor riesgo de aparición de neoplasias malignas cutáneas y hematológicas. Se debe a un defecto del gen RMRP (ribonuclease mitochondrial RNA-processing). El tratamiento de la IDCG se basa en el trasplante de progenitores hematopoyéticos (TPH) y la terapia génica (en casos concretos, como el déficit de ADA y el déficit de cadenas γ del receptor de la IL-2). IDCG ligada al cromosoma X Esta forma representa el 30 al 40% de todas las IDCG, y se observa en 1-2 de cada 100.000 nacimientos. Los pacientes varones con IDCG ligada al cromosoma X tienen una cifra de linfocitos < 2.000/mm3 en sangre periférica, con < 2.000 L-T CD3+, < 100/mm3 linfocitos NK y un 75% de L-B, por lo que se incluyen en las ICDG de tipo T-B+NK-. Los niveles de IgG e IgA son extremadamente bajos, y no existe producción de anticuerpos específicos. Se debe a la ausencia de la cadena gamma común que forma parte de los receptores de IL-2, IL-7, IL-4 y 9, e IL-15. Déficit enzimático de JAK3 Entre las formas de IDCG de tipo T-B+NK-, se incluye también este déficit que se hereda de forma autosómica recesiva y que se debe a la mutación de ambos alelos del gen de JAK3. Ésta es una cinasa citosólica que forma parte de la vía de la activación de los receptores de IL-2, IL-7, IL-4, IL-6 e IL-15, por lo que el fenotipo linfocitario es similar a la IDCG ligada al cromosoma X. 36.4. Síndromes bien definidos de inmunodeficiencias Síndrome de Wiskott-Aldrich El síndrome de Wiskott-Aldrich (SWA) es una afección que se hereda de forma recesiva ligada al cromosoma X, y que debuta en los primeros meses de vida con la tríada clásica de otitis recidivantes, eczema y púrpura trombocitopénica, con diarrea sanguinolenta. Aunque los megacariocitos son normales, las plaquetas son pequeñas y defectuosas. Estos pacientes tienen una gran predisposición a sufrir infecciones. El gen anómalo se sitúa cerca del centrómero del brazo corto del cromosoma X (Xp11.22-11.23), y codifica una proteína de 501 aminoácidos rica en prolina, cuya expresión está limitada a la línea de linfocitos y megacariocitos, y que se denomina proteína del SWA (PSWA). 704 El niño con infecciones recurrentes. Inmunodeficiencias primarias Estos pacientes presentan infecciones de repetición en diversas localizaciones, hemorragias y procesos diarreicos. Casi nunca superan la adolescencia, y fallecen por infecciones, hemorragia o neoplasias malignas del tipo de linfomas asociados a infección por el virus de Epstein-Barr (VEB). En la Tabla 36.25 se especifican los criterios para el diagnóstico del síndrome de Wiskott-Aldrich. presenta niveles elevados de alfafetoproteína sérica. Los niveles de inmunoglobulinas son variables, aunque generalmente se observa una disminución de IgG2, IgG4, IgA e IgE. En la Tabla 36.26 se muestran los criterios para el diagnóstico del síndrome ataxia-telangiectasia. Estos pacientes y sus familiares presentan una tendencia elevada al desarrollo de tumores malignos. Diagnóstico definitivo* Hombre o mujer con aumentada fragilidad cromosómica inducida por radiación en cultivos Diagnóstico celulares o ataxia cerebelosa progresiva y que definitivo* presentan mutaciones en ambos alelos para el gen de la ataxia telangiectasia (ATM) 3 Paciente varón con trombopenia congénita (< 70.000 plaquetas/mm ), plaquetas pequeñas y al menos uno de los siguientes: 1. Mutación en WASP 2. Ausencia ARNm WASP en análisis Northern blot de linfocitos 3. Ausencia de WASP proteína en linfocitos 4. Primos maternos, tíos o sobrinos con plaquetas pequeñas y trombopenia Hombre o mujer con ataxia cerebelosa progresiva y tres de los siguientes hallazgos: 1. Telangiectasia facial u ocular 2. Niveles de IgA al menos 2 desviaciones estándar Diagnóstico por debajo de lo normal para la edad probable* 3. Alfafetoproteína al menos 2 desviaciones estándar por encima de lo normal para la edad 4. Aumento de la fragilidad cromosómica inducida por radiación en cultivos celulares Diagnóstico probable* Paciente varón con trombopenia congénita (< 70.000 plaquetas/mm3), plaquetas pequeñas y al menos uno de los siguientes: 1. Eczema 2. Respuesta de anticuerpos anormal a antígenos polisacáridos 3. Infecciones bacterianas o víricas recurrentes 4. Enfermedades autoinmunitarias 5. Linfoma, leucemia o tumor cerebral Hombre o mujer con ataxia cerebelosa progresiva y al menos uno de los siguientes hallazgos: 1. Telangiectasia facial u ocular 2. Niveles de IgA al menos 2 desviaciones estándar Diagnóstico por debajo de lo normal para la edad posible* 3. Alfafetoproteína superior a 2 desviaciones estándar para la edad 4. Aumentada fragilidad cromosómica tras exposición a radiaciones Diagnóstico posible* Paciente varón con trombopenia congénita (< 70.000 plaquetas/mm3), plaquetas pequeñas o paciente varón esplenectomizado por trombopenia, que tiene al menos uno de los siguientes: 1. Eczema 2. Respuesta de anticuerpos anormal a antígenos polisacáridos 3. Infecciones bacterianas o virásicas recurrentes 4. Enfermedades autoinmunitarias 5. Linfoma, leucemia o tumor cerebral * Pacientes con un diagnóstico definitivo o probable tienen más del 98 y 85% de probabilidades, respectivamente de que en 20 años tengan todavía el mismo diagnóstico. Los pacientes con un diagnóstico posible son aquellos que tienen algunas pero no todas las características clínicas o hallazgos de laboratorio de un determinado trastorno * Pacientes con un diagnóstico definitivo o probable tienen más del 98 y 85% de probabilidades, respectivamente, de que en 20 años tengan todavía el mismo diagnóstico. Los pacientes con un diagnóstico posible son aquellos que tienen algunas pero no todas las características clínicas o hallazgos de laboratorio de un determinado trastorno Tabla 36.25. Criterios diagnósticos del síndrome de Wiskott-Aldrich (tomado de Conley ME et al. Clin Immunol 1999; 93: 190) Síndrome de ataxia-telangiectasia (Louis-Barr) El síndrome de ataxia-telangiectasia (SAT) o síndrome de Louis-Barr (SLB) es un cuadro complejo que asocia anomalías inmunológicas, neurológicas, cutáneas, endocrinológicas y hepáticas. El gen responsable se localiza en el brazo largo del cromosoma 11 (11q22-23), y la afección se hereda de forma autosómica recesiva. Cursa de forma característica con telangiectasias y ataxia cerebelosa progresiva (Figura 36.11), junto con infecciones sinubronquiales recurrentes en el 85% de los casos. El 95% de los pacientes 36 Tabla 36.26. Criterios diagnósticos de la ataxia-telangiectasia (tomado de Conley ME et al. Clin Immunol 1999; 93: 190) Existe un síndrome, conocido como síndrome de NijmegenBreakage, que se asemeja al síndrome de ataxia-telangiectasia, ya que los pacientes presentan radiosensibilidad, inmunodeficiencia y tendencia a desarrollar tumores. Se debe a una Figura 36.11. Telangiectasias conjuntivales y ataxia en un paciente con síndrome de Louis-Barr 705 Tratado de pediatría Sección III - Patología infecciosa translocación cromosómica recíproca que afecta a los cromosomas 7 y 14, y es un síndrome que no cursa con ataxia ni con telangiectasia, y los pacientes sufren microcefalia y retraso mental. Síndrome de inestabilidad cromosómica y anomalías faciales Este síndrome, denominado también ICF (Immunodeficiency Centromeric instability and Facial anomalies), cursa con retraso mental. Se hereda de forma autosómica recesiva, y se debe, en el 50% de los casos, a un defecto de Figura 36.12. Presumible localización del defecto que da lugar a la secuencia de Di George (tomado de Smith D, 1982) metilación del gen DNMT3B, que codifica una ADN-metiltransferasa; en otras · Herencia: ? Defecto III y IV arcos branquiales ocasiones, la mutación se localiza en el gen ZBTB24, aunque el · Ausencia completa, o grave displasia de timo y paratiroides fenotipo resultante es el mismo en ambas mutaciones. · Malformaciones asociadas: hipertelorismo, anomalías auriculares retromicrognatia, malformaciones cardiovasculares, retraso psicomotor, convulsiones hipocalcémicas · Comienzo en edad neonatal con manifestaciones de tetania · Infecciones recurrentes de comienzo en el primer año de vida (virus y hongos) · Ausencia radiológica de sombra tímica · Inmunoglobulinas séricas en límites normales · Buena respuesta de anticuerpos para los antígenos no T-dependientes · Linfopenia con aumento de los linfocitos B · Alteración de los test de funcionalidad de los linfocitos T · Deplección de las áreas T-dependientes de los ganglios linfáticos con número normal o aumentado de centros germinativos y células plasmáticas Los pacientes afectados presentan hipertelorismo, aplanamiento de la raíz nasal, epicanto, implantación baja de las orejas y una facies expresiva de retraso mental. Debido a la inmunodeficiencia, tanto celular como humoral, son frecuentes las infecciones sinubronquiales, gastrointestinales y cutáneas. La inestabilidad centromérica suele afectar a los cromosomas 1, 16 y 9, y de forma menos habitual, a los cromosomas 2 y 10. También se han descrito deleciones, roturas, e intercambios entre cromosomas homólogos y no homólogos. El síndrome ICF se distingue de otros tipos de síndrome de inestabilidad cromosómica en que no presenta hipersensibilidad a los agentes clastogénicos, por lo que no debiera considerarse un síndrome de fragilidad cromosómica. Tabla 36.27. Características de la secuencia de Di George Síndrome de hiper-IgE (síndrome de Job) Secuencia de Di George Esta afección se debe a una dismorfogénesis de las bolsas faríngeas 3ª y 4ª, lo que provoca una hipoplasia o aplasia del timo y de las glándulas paratiroideas (Figura 36.12); también se afectan otras estructuras craneofaciales y cardiovasculares. La hipocalcemia secundaria al hipoparatiroidismo puede producir convulsiones en el periodo neonatal. En la Tabla 36.27 se muestran las características de la secuencia de Di George. En el 95% de los casos se encuentran microdeleciones en el cromosoma 22q11.2 o 22q11-ter. Existen otros síndromes con deleciones localizadas en la misma área, como el síndrome velocardiofacial (SVCF), el síndrome de Cayler y el síndrome facial con anomalías conotroncales (SFACT). Dado que en todos ellos la deleción es en 22q11-ter, se han englobado en el acrónimo CATCH22: Cardiac Abnormalities, Thymic hipoplasia, Cleft palate and Hypocalcemia Se conocen dos tipos de este síndrome que describieron Davis y cols. en 1966: el tipo 1, autosómico dominante o esporádico, que se debe a mutaciones en el STAT3 (transductor de señal y activador de transcripción 3), y el tipo 2. Se caracteriza por: presencia de abscesos fríos diseminados causados por S. aureus, infecciones sinubronquiales de repetición con tendencia a la formación de neumatoceles, dermatitis eczematoide; candidiasis mucocutánea, facies peculiar, alteraciones esqueléticas y articulares, dientes supernumerarios, eosinofilia en sangre y esputo, deficiencias en la quimiotaxis y cifras séricas de IgE elevadas, habitualmente superiores a 2.000 UI/ml (Figura 36.13). En el tipo 2 también se observa eczema grave, infecciones recurrentes y valores elevados de IgE, aunque no cursa con neumonía, neumatoceles ni anomalías óseas 706 El niño con infecciones recurrentes. Inmunodeficiencias primarias Figura 36.13. Síndrome de Job En la Tabla 36.28 se resumen las principales características clinicobiológicas del síndrome de hiper-IgE, y en la Tabla 36.29 se plantea el diagnóstico diferencial entre este cuadro y la dermatitis atópica. · Abscesos “fríos” diseminados por S. aureus · Infecciones sinubronquiales de repetición con tendencia a formación de neumatoceles · Dermatitis eczematoide. Candidiasis mucocutánea · Facies peculiar · Alteraciones dentales, esqueléticas, articulares, etc. · Eosinofilia sanguínea y en esputo · Deficiencias en la quimiotaxis · Elevadas cifras en suero de IgE 36 sos procesos infecciosos que desaparecieron al iniciarse un tratamiento con gammaglobulina intramuscular (Figura 36.14). Se trata de un cuadro ligado al cromosoma X, causado por la mutación de la btk (tirosina-cinasa de Bruton) localizada en el brazo largo del cromosoma X (Xq21.3-22). Constituye el prototipo de los defectos de la inmunidad humoral, y el cuadro clínico suele iniciarse a partir del segundo semestre de vida, cuando los niveles de inmunoglobulina de procedencia materna que atravesaron la placenta ya han desaparecido. Estos pacientes sufren infecciones de repetición, generalmente bacterianas (S. pneumoniae, S. aureus, Streptococcus pyogenes, Pseudomonas spp, etc.), localizadas en las vías respiratorias, el oído medio, los senos paranasales, las meninges y la piel, sepsis, etc. A pesar del gran número de afecciones respiratorias de vías superiores que presentan los afectados, el anillo linfático de Waldeyer se muestra atrófico, y no se observa hipertrofia ni hiperplasia de los ganglios linfáticos laterocervicales. Tabla 36.28. Síndrome hiper-IgE. Características clínicas y biológicas Síndrome hiper-IgE Presente en las primeras semanas de vida Dermatitis atópica grave No aparece hasta los 3-4 meses El exantema generalmente aparece El exantema se localiza en cara y en las superficies de extensión preferentemente en zonas flexoras Abscesos “fríos”, infecciones secundarias bacterianas o micóticas No abscesos ni furunculosis, aunque puede haber sobreinfección cutánea Facies peculiar (leonina) Alteraciones dentales, óseas No facies característica Cifras muy elevadas de IgE En ocasiones aumento moderado de IgE Sin manifestaciones alérgicas ni respiratorias (asma) Frecuentes manifestaciones alérgicas y respiratorias (asma) Tabla 36.29. Diagnóstico diferencial entre S. hiper-IgE y dermatitis atópica 36.5. Deficiencias predominantemente de anticuerpos Agammaglobulinemia congénita (síndrome de Bruton) Figura 36.14. Caso “princeps” descrito, en 1952, por el Dr. Bruton, de agammaglobulinemia congénita ligada al cromosoma X La agammaglobulinemia congénita fue descrita en 1952, por el Dr. Bruton, en un paciente con antecedentes de numero- En estos pacientes, las enfermedades víricas cursan de forma prácticamente normal, salvo las infecciones causadas por 707 Tratado de pediatría Sección III - Patología infecciosa enterovirus, que pueden dar lugar a infecciones diseminadas y meningoencefalitis crónica grave. La administración de la vacuna oral contra la poliomielitis (de virus vivos atenuados) puede causar una infección diseminada y mortal. En un discreto porcentaje de casos, y una vez superada la infancia, se observa en la adolescencia una sintomatología articular que recuerda a la artritis idiopática juvenil, relacionada con infecciones por Mycoplasma y Ureaplasma, y que suele responder bien al tratamiento sustitutivo con gammaglobulina intravenosa (IGIV). En la Tabla 36.30 se resumen las principales características clínicas y analíticas que permiten establecer el diagnóstico de agammaglobulinemia de Bruton. Diagnóstico definitivo* Varón con menos del 2% de linfocitos B CD19 y al menos uno de los siguientes: 1. Mutación en Btk 2. Btk ARNm ausente en Northern blot de neutrófilos o monocitos 3. Proteína Btk ausente en monocitos o plaquetas 4. Tíos maternos, tíos o sobrinos con < 2% de linfocitos B CD19 Diagnóstico probable* Varón con menos del 2% de linfocitos B CD19 en los que todos los siguientes son positivos: 1. Inicio de infecciones bacterianas recurrentes en los 5 primeros años de vida 2. Niveles de IgG, IgM e IgA en suero por debajo de 2 desviaciones estándar para la edad 3. Ausencia de isohemaglutininas y/o pobre respuesta a las vacunas 4. Otras causas de hipogammaglobulinemia han sido excluidas Diagnóstico posible* Varón con menos del 2% de linfocitos B CD19 en las que otras causas de hipogammaglobulinemia han sido excluidas y al menos una de las siguientes es positiva: 1. Inicio de infecciones bacterianas recurrentes en los 5 primeros años de vida 2. Niveles de IgG, IgM e IgA en suero por debajo de 2 desviaciones estándar para la edad 3. Ausencia de isohemaglutininas * Pacientes con un diagnóstico definitivo o probable tienen más del 98 y 85% de probabilidades, respectivamente, de que en 20 años tengan todavía el mismo diagnóstico. Los pacientes con un diagnóstico posible son aquellos que tienen algunas pero no todas las características clínicas o hallazgos de laboratorio de un determinado trastorno. Además de la agammaglobulinemia Tabla 36.30. Criterios diagnósticos de la agammaglobulinemia de Bruton (tomado de Conley ME et al. Clin Immunol 1999; 93: 190) ligada al cromosoma X, que representa el 85-90% de los casos de las agammaglobulinemias congénitas, existen otras formas, La IDCV se define por: de herencia autosómica recesiva, cuyas manifestaciones • Reducción de los valores séricos de IgG, con niveles bajos de IgA o IgM, o ambas. y datos analíticos pueden superponerse a los de la forma • Presencia de linfocitos B. ligada al X. • Respuesta escasa o reducida a las vacunas. • Ausencia de cualquier otro tipo de inmunodeficiencia priSíndrome de hiper-IgM maria. También conocido como inmunodeficiencia con IgM normal o elevada, en el síndrome de hiper-IgM existe un déficit de IgG, IgA Suele observarse después de los dos años de edad, si bien puee IgE, que se manifiesta en forma de infecciones piógenas recu- de aparecer en cualquier época de la vida, detectándose dos rrentes y gastrointestinales, neutropenia recurrente y trastornos picos o puntos máximos: entre los 5 y los 10 años, y entre los 20 y los 30 años. Suele ser una afección esporádica, aunque un autoinmunitarios. 10-20% de los casos son familiares, de tipo autosómico domiLa mayoría de los pacientes varones con este síndrome tienen nante o recesivo. Hay ocasiones en las que se observan en la una mutación del gen CD40L en el cromosoma X. La interac- misma familia casos de IDCV y de déficit de IgA, por lo que se ción entre CD40 y el linfocito B y CD40L en el linfocito T acti- cree que ambos procesos pueden tener una base genética covado es esencial para que se produzca el paso de IgM a IgG. mún. Se trata de una de las inmunodeficiencias primarias más Existen otros síndromes de hiper-IgM de herencia autosómica frecuentes. por mutaciones en el gen del CD40 u otros defectos intrínsecos del linfocito B. Muchos de los niños afectados de IDCV presentan fallo de medro o parecen crónicamente enfermos. Presentan tos crónica y afectación respiratoria con rinitis purulenta, y el tamaño de Inmunodeficiencia común variable amígdalas y adenoides suele ser normal o incluso estar auLa inmunodeficiencia común variable (IDCV) es un cuadro hete- mentado. Son frecuentes las sinusitis (75%), la OMA (67%), la rogéneo, no muy bien definido, en el que existe una disfunción neumonía (58%) e incluso las bronquiectasias. También puede los linfocitos B y T, y de las células dendríticas. El defecto inmu- den observarse enfermedades alérgicas, alergias alimentarias, nológico característico es la incapacidad de los linfocitos B para eczema, urticaria, rinitis, asma, etc. Son frecuentes las enferdiferenciarse en células plasmáticas capaces de secretar todo medades autoinmunitarias como la púrpura trombocitopénica inmunitaria (PTI), el síndrome de Evans (anemia hemolítica tipo de inmunoglobulinas. 708 El niño con infecciones recurrentes. Inmunodeficiencias primarias 36 autoinmunitaria y PTI), así como una tendencia a desarrollar procesos linfoproliferativos y neoplásicos (linfomas, cáncer gástrico, etc.) (Figura 36.15). Los niveles de IgG están muy reducidos (por debajo de 2DS para la edad), la cifra de IgA también está disminuida y en el 50% de los casos se detectan niveles bajos de IgM. Se observa una ausencia de isohemaglutininas y la respuesta de producción de anticuerpos tras la vacunación es escasa. En la Tabla 36.31 se muestran los criterios para el diagnóstico de la IDCV. El diagnóstico diferencial deberá plantearse con la agammaglobulinemia de Bruton, la hipogammaglobulinemia transitoria del lactante, el déficit selectivo de anticuerpos con niveles normales de inmunoglobulinas, los déficit de subclases de IgG, el síndrome de hiper-IgE, el síndrome linfoproliferativo ligado al cromosoma X y las inmunodeficiencias combinadas. Hipogammaglobulinemia transitoria del lactante Figura 36.15. Representación esquemática de las enfermedades infecciosas y de la afectación autoinmunitaria de órganos en pacientes con inmunodeficiencia común variable (tomado de Park MA et al. Lancet 2008; 372: 489-502) Diagnóstico probable* Hombre o mujer que presenta un marcado descenso (al menos por debajo de 2 desviaciones estándar de media para la edad) en suero de IgG e IgA y absolutamente todos los siguientes criterios: 1. Comienzo de la inmunodeficiencia después de los 2 años 2. Ausencia de isohemaglutininas y/o pobre respuesta a las vacunas 3. Se han excluido otras causas de hipogammaglobulinemia Es una afección bien definida dentro Hombre o mujer que presenta un marcado descenso (al menos por debajo de las IDP, aunque puede considerarse de 2 desviaciones estándar de media para la edad) en uno de los isotipos mayores como una situación parafisiológica. Se Diagnóstico (IgM, IgG, IgA) y absolutamente todos los siguientes criterios: trata de una disminución muy intensa de 1. Comienzo de la inmunodeficiencia después de los 2 años posible* 2. Ausencia de isohemaglutininas y/o pobre respuesta a las vacunas los niveles de IgG por la pérdida y el ca3. Se han excluido otras causas de hipogammaglobulinemia tabolismo de las inmunoglobulinas (IgG) de origen materno transplacentarias y * Pacientes con un diagnóstico definitivo o probable tienen más del 98 y 85% de probabilidades, el retraso del comienzo de la síntesis de respectivamente, de que en 20 años tengan todavía el mismo diagnóstico. Los pacientes con un diagnóstico posible son aquellos que tienen algunas pero no todas las características clínicas o hallazgos de laboratorio inmunoglobulinas endógenas. Estos pade un determinado trastorno cientes tienen unos niveles muy bajos y prolongados de IgG, con cifras normales Tabla 36.31. Criterios diagnósticos de la inmunodeficiencia comun variable (tomado de Conley ME et al. Clin Immunol 1999; 93: 190) de IgM e IgA, y un número normal de linfocitos B circulantes, lo que permite diferenciar este cuadro de la agammaglobulinemia de Bruton. Cuando estos pacientes sufren procesos infecciosos de repetiEn la Figura 36.16 se muestra la evolución del desarrollo de las ción, la duda estriba en administrar o no IGIV, ya que con ello podría retrasarse incluso más la síntesis endógena de anticuerinmunoglobulinas en las distintas fases de la infancia. pos. Hay quien considera que sólo se debe administrar IGIV si la Desde el punto de vista patogénico, se cree que lo que suce- cifra de IgG es muy baja y las infecciones muy frecuentes, y quien de en la hipogammaglobulinemia transitoria del lactante es un propone administrar vacunas proteicas o conjugadas para poner retraso en la puesta en marcha de los procesos de síntesis de en marcha y estimular la síntesis de inmunoglobulinas. Se debe anticuerpos de tipo IgG por parte del lactante. En la Tabla 36.32 controlar la cifra de IgG cada 6 meses. Algunos pacientes con se resumen los niveles normales de IgG y sus subclases en las esta hipogammaglobulinemia transitoria o sus familiares pueden desarrollar otros tipos de IDP. distintas edades de la infancia. 709 Tratado de pediatría Sección III - Patología infecciosa • Figura 36.16. Representación esquemática del desarrollo de las inmunoglobulinas séricas en las distintas edades. El RN no tiene virtualmente IgA, IgM ni IgE. El paso de IgG a través de la placenta de la madre al feto se produce en las últimas semanas del embarazo. En el RN a término, los niveles de IgG son similares a los maternos. Después del nacimiento disminuye la IgG transferida por vía transplacentaria asistiéndose a los 4-6 meses a una hipogammaglobulinemia fisiológica transitoria. Los niveles del adulto de IgM, IgG e IgA se alcanzan al año, a los 5-6 años y a los 10 años, respectivamente (tomado de Fireman, P. Ed. Chapter 21: Primary immunodeficiency disease, Atlas of Allergies and Clinical Immunology. Philadelphia, PA: Mosby, Inc., 2006, 332) Déficit de subclases de IgG Puede ser aislado o ir asociado a otras IDP, como ataxia-telangiectasia o déficit de IgA. Puede existir un déficit de IgG2, IgG3 e IgG4, sin que haya una disminución de IgG total, y no se observa relación alguna entre la gravedad de las manifestaciones clínicas y la intensidad del déficit de subclases de IgG. La determinación de las distintas subclases de IgG no es fácil, y deben controlarse periódicamente, cada 3-6 meses, ya que es posible que existan formas transitorias. En la Tabla 36.32 se muestran las cifras normales de las distintas subclases de IgG en las diferentes edades. • Déficit de subclase IgG1. Como esta fracción constituye el 60-75% del total de IgG, un déficit produce hipogammaglobulinemia. En estos pacientes, los niveles del resto de subclases son normales. El tratamiento es similar al de la hipogammaglobulinemia. Edad (años) Número de sujetos Independientemente del nivel total de IgG, los pacientes con deficiencia documentada de subclases o déficits específicos de anticuerpos son candidatos a recibir IGIV sustitutiva a dosis de 200-300 mg/kg cada 3 semanas (pero los requerimientos pueden ser superiores, así que la dosificación debe ser individualizada). Déficit selectivo de anticuerpos con inmunoglobulinas normales Se sabe que algunas personas presentan fallos al responder a antígenos polisacáridos, y mientras que muchas se encuentran asintomáticas, otras sufren infecciones sinubronquiales recurrentes. Para establecer este diagnóstico, es necesario demostrar que existe un fallo de la respuesta de producción de anticuerpos frente a antígenos específicos, como la vacuna IgG IgG1 IgG2 ∆ • Déficit de subclase IgG2. La IgG2 supone el 20-25% de la IgG. Es un grupo heterogéneo de anomalías, con distinto grado de déficit de IgG2, que puede ser aislado o asociarse a un déficit de IgG4 e IgA. El déficit de IgG2 es una de las anomalías observada con más frecuencia en individuos que sufren infecciones de repetición. Estos pacientes suelen desarrollar infecciones sinubronquiales recurrentes, OMA, y enfermedad neumocócica diseminada o invasora. Déficit de subclases IgG3 e IgG4. Suponen el 5-10% y < 5%, respectivamente, de los déficits de IgG. El déficit de IgG3 también se ha descrito en pacientes con infecciones sinubronquiales recurrentes; suele ser transitorio. IgG3 IgG4† 0-1 22 420 (250-690) 340 (190-620) 59 (30-140) 39 (9-62) 19 (6-63) 1-2 42 470 (270-810) 410 (230-710) 68 (30-170) 34 (11-98) 13 (4-43) 2-3 36 540 (300-980) 480 (280-830) 98 (40-240) 28 (6-130) 18 (3-120) 3-4 52 600 (400-910) 530 (350-790) 120 (50-260) 30 (9-98) 32 (5-180) 4-6 31 660 (440-1000) 540 (360-810) 140 (60-310) 39 (9-160) 39 (9-160) 6-8 24 890 (560-1400) 560 (280-1120) 150 (30-630) 48 (40-250) 81 (11-620) 8-10 21 1000 (530-1900) 690 (280-1740) 210 (80-550) 85 (22-320) 42 (10-170) 10-13 33 910 (500-1660) 590 (270-1290) 240 (110-550) 58 (13-250) 60 (7-530) 13-16 19 910 (580-1450) 540 (280-1020) 210 (60-790) 58 (14-240) 60 (11-330) Los niveles fueron determinados mediante difusión radial utilizando antisueros monoespecíficos † Niveles de IgG4 están ausentes en el 10% de los individuos Las medias geométricas están presentadas para cada Ig en las distintas edades Δ Tabla 36.32. Niveles de las subclases de IgG en suero en individuos sanos según la edad (tomado de Stiehm ER et al. Immunology disorders in infants and children, 5th edition. Elsevier, Philadelphia, 2004) 710 El niño con infecciones recurrentes. Inmunodeficiencias primarias neumocócica de polisacáridos o la vacuna con el polisacárido capsular de H. influenzae de tipo b, una respuesta normal a otros antígenos y unos niveles normales de IgG e IgM. En algunos de estos pacientes las cifras de IgG2 son bajas. Algunas personas que no responden a las vacunas pueden incluirse en esta categoría de inmunodeficiencias. 36 transfusionales anafilácticas por formación de anticuerpos anti IgA. Los niveles séricos de IgA no están relacionados con la presencia ni la intensidad de las manifestaciones clínicas. Déficit selectivo de IgA El déficit selectivo de IgA (DSIgA) se define como una deficiencia aislada de IgA sérica, con valores normales de IgG e IgM, en un individuo mayor de 4 años en el que se han descartado otras causas de hipogammaglobulinemia. Las manifestaciones clínicas son variables, y oscilan desde la afección totalmente asintomática hasta la aparición de infecciones intercurrentes o enfermedades autoinmunitarias. Se trata de la IDP más frecuente, ya que se presenta en 1 de cada 100/700 caucásicos, si bien es un déficit muy raro en Japón (1/18.500). Los pacientes muestran unos niveles de IgA < 7 mg/dl, pero las cifras de IgG e IgM son normales, así como la respuesta de producción de anticuerpos tras la vacunación. A pesar de que, incluso en condiciones normales, la cifra de IgA es baja, esta Ig supone más del 70% de las inmunoglobulinas del organismo, ya que también se encuentra en secreciones nasales y pulmonares, la saliva, las lágrimas, la leche materna, así como en las secreciones del tracto genitourinario y el tubo digestivo, en donde ejerce una función protectora de las mucosas impidiendo la fijación y la penetración de los microorganismos. Figura 36.17. Estructuras de la IgA sérica monomérica y de la IgA secretora dimérica Hay que tener en cuenta que algunos fármacos, como la ciclosporina, los anticonvulsivos (fenitoína, ácido valproico, carbamazepina), la D-penicilamina, la aurotioglucosa, etc., pueden causar un déficit transitorio de IgA, y que la ciclosporina puede producir un déficit permanente. En la Tabla 36.33 se presentan las principales características del DSIgA, y en la Tabla 36.34, las enfermedades con las que suelen asociarse. Existen dos subclases de IgA: la IgA1 y la IgA2. La IgA2 predomina en la forma secretora. La IgA se encuentra bajo forma monomérica, que corresponde a la IgA sérica, y bajo forma dimérica, que constituye la IgA secretora (Figura 36.17). La IgA sérica, monomérica, interactúa con el brazo fagocítico del sistema inmunitario. Las moléculas de IgA monomérica poseen una fracción Fab que se une a los antígenos extraños. Una vez ligada a un antígeno, la porción Fc de la IgA se une al receptor Fc-α (CD89) localizado en la superficie de neutrófilos macrófagos y eosinófilos. La IgA unida al receptor inicia la ingestión y destrucción de los microorganismos por fagocitosis. La IgA secretora, dimérica, consiste en dos moléculas de IgA unidas por una cadena J. Es secretada por células plasmáticas que se localizan en el tejido conjuntivo por debajo de la membrana basal epitelial, y su función principal es la protección de las distintas mucosas frente a diferentes antígenos. La mayoría de los pacientes con déficit de IgA selectivo se encuentran asintomáticos, y sólo un 30-35% presentará infecciones sinubronquiales recurrentes, infecciones y trastornos gastrointestinales (celiaquía, enfermedad inflamatoria intestinal, hiperplasia nodular linfoide), trastornos alérgicos y reacciones 711 · Herencia variable · Frecuencia elevada. La ID primaria más frecuente (1/500-1/3.000) · Ausencia o marcada disminución de IgA en suero y en las secreciones · Número normal o elevado de linfocitos portadores de IgA de superficie · Ausencia de células plasmáticas productoras de IgA a nivel mucoso · Formas clínicas: Asintomáticas Asociadas a infecciones recurrentes del aparato respiratorio Asociadas a manifestaciones autoinmunitarias, tipo LED Asociadas a celiaquía Asociadas a reacciones alérgicas, fármacos o trofoalérgenos · Patogenia: bloqueo madurativo o hipercatabolismo de IgA (?) Tabla 36.33. Déficit selectivo de IgA. Características Infecciones respiratorias (IRA, OMA, neumonía) (43%) Enfermedades alérgicas (20%) Enfermedades autoinmunitarias (14%) Enfermedades digestivas (12%) Tumores (1%) Los pacientes con DSIgA tienen un mayor riesgo de infecciones GI, sobre todo por Giardia lamblia Tabla 36.34. Déficit selectivo de IgA. Asociación a otras enfermedades Tratado de pediatría Sección III - Patología infecciosa 36.6. Alteraciones del número y la función de las células fagocíticas 50% de los niños afectados presentan síntomas durante los primeros meses de vida, y el 90% durante 6 meses. Es frecuente la aparición de onfalitis, infecciones de vías respiratorias superiores e inferiores, infecciones cutáneas y abscesos hepáticos. Los neutrófilos maduros se desarrollan en la médula ósea, a partir de las células madre mieloides, y permanecen sólo 6-10 h en el torrente circulatorio antes de salir, por diapédesis, hacia zonas inflamadas. En el adulto, una cifra > 7.500/mm3 indica una neutrofilia, y se debe a causas extrínsecas en los neutrófilos, como infección aguda o crónica, esteroides, epinefrina. La neutropenia puede ser leve (< 1.500/mm3), moderada (1.000-1.500/mm3) o grave (< 500/mm3), y puede ser intrínseca o extrínseca a los neutrófilos o sus progenitores. La neutropenia puede deberse a múltiples causas, ya que puede acompañar a inmunodeficiencias primarias y secundarias, infecciones víricas y por gramnegativos, y algunos fármacos (quimioterapia). No deben descartarse defectos inmunitarios mieloides en pacientes con infecciones bacterianas y fúngicas graves y recurrentes causadas por microorganismos poco frecuentes (Burkholderia cepacia complex, Chromobacterium violaceum) o en localizaciones inusuales (abscesos viscerales). Las infecciones víricas y parasitarias graves no son típicas de los defectos de las células fagocíticas. El neutrófilo normal (Figura 36.18) presenta precozmente en la médula ósea gránulos primarios que contienen numerosas enzimas, entre los que se encuentran la elastasa granulocítica (GE), la catepsina G (CG), así como proteinasa 3 (PR3), mieloperoxidasa (MPO) y defensinas. Los gránulos secundarios aparecen posteriormente en la médula ósea, y contienen lactoferrina y vitamina B12 unida a proteínas, así como proteínas del tipo Mac 1 (CD18/CD11) y el complejo citocromo B (p91phox y p22phox). Por otro lado, p47phox y p67phox están unidas en el citoplasma, pero no fosforiladas. Los neutrófilos ingieren bacterias, hongos y partículas complejas dentro de un fagosoma. Dentro del grupo de alteraciones del número y la función de las células fagocíticas, se analizarán aquí la neutropenia congénita grave (NCG), la neutropenia clínica (NC) y el síndrome de Kostman. Neutropenia congénita grave La neutropenia congénita grave (NCG) comprende un grupo de trastornos heterogéneos que presentan un patrón hereditario distinto. Se produce la detención del paso de maduración de promielocito a mielocito, lo que causa una neutropenia grave (< 200 neutrófilos/mm3). En ocasiones, se asocia a una mayor predisposición al desarrollo de una leucemia mieloide. Las manifestaciones clínicas aparecen poco después del nacimiento. El Figura 36.18. Representación esquemática de un neutrófilo normal (tomado de Bellanti JA, 2012) El tratamiento con G-CSF (factor estimulante de colonias de granulocitos) ha mejorado espectacularmente la calidad de vida de estos niños, al disminuir el número de infecciones y las hospitalizaciones, y aumentar la supervivencia. Neutropenia cíclica Denominada también hematopoyesis cíclica, la neutropenia cíclica (NC) es una enfermedad autosómica dominante, que se caracteriza por fluctuaciones cíclicas, regulares, de todas las líneas hematopoyéticas, aunque los síntomas sólo se producen como consecuencia de la disminución del número de neutrófilos. Cada 14-36 días (promedio de 21 días), se produce una neutropenia grave (< 200/mm3) que dura 3-10 días. Los pacientes presentan aftas bucales, gingivitis, abscesos periapicales que causan pérdida de dientes permanentes, adenopatías, faringitis o lesiones cutáneas. De forma natural, las infecciones van disminuyendo con la edad. El tratamiento consiste en la administración de G-CSF. 712 El niño con infecciones recurrentes. Inmunodeficiencias primarias Síndrome de Kostman En 1956, Kostman describió en un niño sueco una neutropenia crónica grave de herencia autosómica dominante. El número de neutrófilos absoluto era < 200/mm3, con mayor predisposición a sufrir infecciones bacterianas. En el caso original y en una amplia cohorte de pacientes kurdos se ha descrito una mutación recesiva en el gen HAX-1(1q 21.3). 36 ba de un trastorno autosómico recesivo, del que se han descrito tres tipos, que puede incluir inmunodeficiencia y trastornos neurológicos. En la Tabla 36.35 se presentan las características de cada uno de estos tipos, así como la diferenciación con el síndrome de Chediak- Higashi. Los tipos I y II tienen mal pronóstico, y los pacientes fallecen en la infancia por infecciones recurrentes y afectación neurológica. El tipo III tiene un pronóstico mejor ya que no presenta complicaciones inmunológicas ni neurológicas. 36.7. Defectos en el contenido y la formación de gránulos Síndrome de Chediak-Higashi Este trastorno autosómico recesivo cursa con albinismo oculocutáneo, hepatoesplenomegalia, adenopatías, infecciones bacterianas frecuentes, enfermedad periodontal, hemorragias, alteraciones neurológicas con fotofobia, nistagmo y neuropatía periférica progresiva, retraso mental en ocasiones y, con frecuencia, una forma de linfohistiocitosis hemofagocítica. Los pacientes tienen un color de pelo que varía entre el rubio y el ligeramente castaño, con un resplandor plateado o en forma de mechón. El síndrome se debe a una mutación del gen LYST(CHS1), que se localiza en el brazo largo del cromosoma 1 (1q42.1.1q 42.2), y se caracteriza por infecciones bacterianas recurrentes por grampositivos, como S. aureus y estreptococo betahemolítico. Consiste en una alteración de los neutrófilos, que contienen granulaciones gigantes intracitoplasmáticas a causa de una fusión inadecuada de múltiples gránulos primarios, que también se observa en eosinófilos, basófilos y otras células que contienen gránulos, como las plaquetas, los hepatocitos, las neuronas, las células de Schwann, etc. (Figura 36.19), razón por la que la sintomatología afecta a distintos órganos y sistemas Figura 36.19. Representación esquemática de un neutrófilo de un paciente con síndrome de Chediak-Higashi mostrando gránulos primarios gigantes por fusión de muchos gránulos primarios y secundarios más pequeños. Este cuadro se debe a una mutación en el gen del LYST (lysosome trafficking) que provoca una incapacidad de los gránulos primarios y secundarios para fusionarse con el fagosoma. A pesar de que el dímero inicial gp91phox y p22phox se forman, hay un fallo de la adición posterior del p47phox y del p67phox. Estos gránulos anormales no sólo impiden la capacidad de los neutrófilos de atravesar el endotelio por su tamaño, sino también la capacidad de destrucción de las bacterias por los neutrófilos, debido al ensamblaje incompleto del complejo NADPH oxidasa (tomado de Bellanti JA, 2012) Los pacientes afectados suelen fallecer antes de los 10 años de edad por infecciones o hemorragias. Los que sobreviven presentan una marcha atáxica, como neuropatía periférica, y un cuadro similar a un linfoma, si se infectan por el VEB. La quimiotaxis está disminuida, pero la fagocitosis es normal e incluso está incrementada. Los gránulos gigantes evidencian el defecto basal en el transportador lisosomal LYST (lysosomal trafficking). La liberación de granulocitos en la médula ósea es escasa y existe una destrucción intramedular de neutrófilos. Síndrome de Griscelli En el año 1978, Griscelli y cols. describieron un síndrome que recordaba al síndrome de Chediak-Higashi, pero en el que no se observaban los gránulos gigantes en los granulocitos. Se trata- Desde el punto de vista clínico, cursa con albinismo parcial, neutropenia, trombocitopenia y linfohistiocitosis hemofagocítica. La ausencia de gránulos gigantes en los granulocitos y las características histológicas de la hipopigmentación permiten distinguir este síndrome del de Chediak-Higashi. Se debe considerar el diagnóstico de síndrome de Griselli en niños con cabellos de color grisplateado, hepatoesplenomegalia e inmunodeficiencia. El examen microscópico del cabello permitirá confirmar el diagnóstico. 713 Tratado de pediatría Sección III - Patología infecciosa Hallazgos GS1 GS2 GS3 MLPH en Ch 2q37.3 F-exon de Myosin Va SCH Gen afectado Myosin Va en Ch 15q21 Rab 27a en Ch 15q21 LYST en Ch 1q 42-43 Herencia AR AR AR AR Color cabello Gris plateado con reflejos metálicos Gris plateado con reflejos metálicos Gris plateado con reflejos metálicos Gris plateado con reflejos metálicos Estructura cabello con el microscopio óptico Grueso, agregado de melanina irregularmente distribuida Grueso, agregado de melanina irregularmente distribuida Grueso, agregado de melanina irregularmente distribuida Fino, agregados de melanina regularmente distribuidos Piel con el microscopio óptico Pigmento aumentado en melanocitos con pigmento disperso en queratinocitos Pigmento aumentado en melanocitos con pigmento disperso en queratinocitos Pigmento aumentado en melanocitos con pigmento disperso en queratinocitos Menor pigmentación en melanocitos y queratinocitos Defectos neurológicos Grave (retraso mental, hipotonía, convulsiones) Puede haber por infiltración linfo-histiocítica, menos grave Ausente Frecuentes (convulsiones y déficit neurológico progresivo) Defectos inmunitarios Ausente Hipogammaglobulinemia, defecto células NK, supresión hipersensibilidad retardada Ausente Defectos quimiotaxis Defectos células NK Tabla 36.35. Tipos de síndrome de Griscelli y diferencias con el síndrome de Chediak-Higashi (tomado de Holland SM et al. Bellanti JA, 2012) Enfermedad linfoproliferativa ligada al cromosoma X La enfermedad linfoproliferativa ligada al cromosoma X (ELPL-X), también llamada enfermedad de Duncan (nombre de la familia en la que se describió), es una enfermedad recesiva ligada al cromosoma X y caracterizada por una respuesta inadecuada a la infección por el virus de Epstein-Barr (VEB). El gen defectuoso se localiza en el brazo largo del cromosoma X (Xq25) y se conoce como SH2D1A. En estos pacientes, la ausencia de SH2D1A puede provocar una respuesta inmunitaria descontrolada de los linfocitos T frente al VEB. Los varones afectados suelen estar sanos hasta que sufren una infección por este virus. La edad media de presentación suele ser antes de los 5 años, y existen tres fenotipos clínicos principales: • Mononucleosis infecciosa fulminante, a veces mortal (50% de los casos). • Linfoma de línea B (25% de los casos). • Hipogammaglobulinemia adquirida (25% de los casos). El pronóstico de esta enfermedad es desfavorable, ya que más del 70% de los casos no llegan a alcanzar la edad de 10 años. Deficiencia de gránulos específicos de neutrófilos Se trata de una enfermedad autosómica recesiva poco frecuente, caracterizada por una reducción importante o una ausencia de gránulos específicos y de su contenido, así como de defensinas. Se debe a una mutación de C/EBPE (Figura 36.20), y cursa con infecciones piógenas recurrentes de la piel, los oídos, los pulmones, los ganglios linfáticos, etc. 714 Figura 36.20. Representación esquemática de un neutrófilo en la deficiencia de gránulo específico-neutrófilo debido a mutación C/EBPE. Obsérvese la ausencia de lactoferrina y de gránulos secundarios, pero también la ausencia de defensinas, dado que estos productos granulares primarios están controlados transcripcionalmente por el mismo gen que regula productos granulares secundarios como la lactiferrina-like (tomado de Bellanti JA, 2012) El niño con infecciones recurrentes. Inmunodeficiencias primarias La morfología de los neutrófilos es anómala, con núcleos bilobulados (anomalía pseudo-Pelger-Huët). El microscopio electrónico evidencia la ausencia de gránulos secundarios en algunos pacientes y la presencia de gránulos vacíos en otros. En ocasiones, se observan anomalías plaquetarias y hemorrágicas. 36.8. Defectos del metabolismo oxidativo 36 La prueba del nitroazul de tetrazolio (NBT) es esencial para el diagnóstico de la EGC, ya que valora la capacidad de los neutrófilos para reducir al amarillo el azul formazán. En una persona sana, más del 90% de los neutrófilos reducen la tinción, pero en un paciente con EGC sólo el 0-2% lo hace (Figura 36.22). Otras pruebas utilizadas en el diagnóstico de esta afección son la bioluminiscencia leucocitaria, la explosión de oxígeno en los fagocitos y la generación de radicales libres. Dentro de este grupo se considerarán exclusivamente los cuadros que se desarrollan seguidamente. Déficit de mieloperoxidasa El déficit de mieloperoxidasa (MPO) es el más frecuente de los trastornos de la fagocitosis, con una incidencia de 1/2.000 personas. Los neutrófilos con déficit de MPO muestran una quimiotaxis y una fagocitosis normales, así como una producción normal o aumentada de superóxido, pero la capacidad de destruir bacterias patógenas y hongos está disminuida. Afortunadamente, existen otros mecanismos auxiliares no dependientes de la MPO para destruir estos patógenos, por lo que la mayoría de estos pacientes están asintomáticos y no se detectan. En los niños diabéticos con déficit de MPO se observa una candidiasis masiva. Enfermedad granulomatosa crónica La enfermedad granulomatosa crónica (EGC) es el trastorno más frecuente de los síndromes asociados a un defecto del metabolismo oxidativo y uno de los trastornos de los neutrófilos mejor estudiados. Suele heredarse ligado al cromosoma X, aunque también existen formas autosómicas recesivas. En la Figura 36.21 se representan las alteraciones de los neutrófilos en la enfermedad granulomatosa crónica. El pronóstico de esta enfermedad ha mejorado de forma significativa en los últimos años. Los niños afectados presentan infecciones recurrentes en piel, ganglios linfáticos, mucosas y pulmones. También suelen presentar abscesos en múltiples órganos, incluso en órganos huecos (píloro, etc.), que pueden producir obstrucciones. Casi nunca se produce sepsis ni meningitis. Este trastorno se caracteriza por la incapacidad de los neutrófilos para generar la cantidad suficiente de superóxido, H2O2, ácido hipoclórico y radicales de oxígeno, todos ellos con una actividad antibacteriana importante. Sin estas armas químicas, los fagocitos no pueden destruir los microorganismos catalasa positivos como S. aureus, Serratia spp, Salmonella spp, Nocardia spp, Aspergillus spp y otros. 715 Figura 36.21. Representación esquemática de un neutrófilo en la enfermedad granulomatosa crónica (EGC). La NADPH oxidasa está ensamblada ya sea en la pared del fagolisosoma o en la membrana dependiendo de si el invasor ha sido fagocitado o si es demasiado grande para ser ingerido. Tras la activación celular el gránulo secundario se fusiona con el fagolisosoma insertando los componentes del citocromo en la pared de la vesícula. p47phox y p67phox se fosforilizan y se asocian estrechamente en el citosol, seguido de un desplazamiento al complejo del citocromo transmembrana. Cuando el p40phox y el RAC se unen al complejo, se forma la oxidasa madura NADPH y es capaz de captar un electrón del NADPH oxidándolo a NADP+ y lleva ese electrón como oxígeno molecular en el fagosoma generando superóxido (O2-). El superóxido se convierte en peróxido de hidrógeno (H2O2) por la superóxido dismutasa (SOD). A su vez éste se convierte en ácido hipoaloso (HOCl o lejía) por la mieloperoxidasa (MPO). Mutaciones en los 4 genes estructurales de la oxidasa NADPH (gp91phox, p22phox, p47phox y p67phox) causan la enfermedad granulomatosa crónica. El gp91phox es recesivo ligado al X mientras el resto son autosómicos recesivos (tomado de Bellanti JA, 2012) Tratado de pediatría Sección III - Patología infecciosa Figura 36.23. Adhesión de los leucocitos a través de las integrinas. Los neutrófilos se adhieren al endotelio a través de receptores PMN de superficie, compuestos por moléculas heterodiméricas CD18 y las relacionadas CD11a, CD11b y CD11c. A su vez, éstas se fusionan con moléculas de la superficie de la célula en el endotelio, compuesto de moléculas de adhesión intercelular (ICAM) 1 y 2. La mutación de CD18 conduce a la incapacidad de todos los heterodímeros, desactivando la adhesión y la salida de los neutrófilos de los vasos. Por tanto, la deficiencia de CD18 conduce a una deficiencia de adhesión de leucocitos tipo I (LAD-I) (tomado de Bellanti JA, 2012) Figura 36.22. Test del nitroazul de tetrazolio. Permite evaluar la capacidad de los neutrófilos para reducir una tinción amarilla a azul formazán. A: en un paciente con enfermedad granulomatosa crónica, sólo el 0,2% de los neutrófilos tienen azul formazán en su superficie. B: en un individuo sano, el 90% de los neutrófilos reduce la tinción a azul tras estimulación con forbomilistato acetato 36.9. Deficiencias de la adhesión leucocitaria En estos últimos años se han descrito una serie de defectos de los movimientos de los leucocitos desde la sangre hasta los tejidos, lo que predispone a la aparición de infecciones recurrentes. Este grupo de enfermedades se incluyen bajo el término general de deficiencias de la adhesión leucocitaria (DAL). Se trata de defectos moleculares responsables de manifestaciones clínicas debidas a una disminución de los procesos inflamatorios y a la emigración de los leucocitos desde los vasos sanguíneos hasta el foco infeccioso, lo que requiere la adhesión de los leucocitos al endotelio (Figura 36.23). En los pacientes con DAL, los neutrófilos presentan una dificultad importante para la migración al foco infeccioso, y los pocos que llegan son incapaces de llevar a cabo la fagocitosis. Como consecuencia de ello se producen, ya durante la lactancia, infecciones graves bacterianas y micóticas de los tejidos blandos (piel, mucosas, OMA, vías respiratorias), así como meningitis y sepsis. Las infecciones suelen ser necróticas y no responden al tratamiento antibiótico por vía oral. Estos pacientes tienen, en condiciones basales, cifras de leucocitos de 20.000-30.000/mm3 con un 80-90% de neutrófilos, que ascienden a 50.000-120.000/mm3 durante las infecciones, por lo que debe realizarse un diagnóstico diferencial con las leucemias y las reacciones leucemoides. En la actualidad se conocen tres tipos de DAL: • Tipo I. Se trata de una enfermedad autosómica recesiva debida a mutaciones en el CD18. Las principales manifestaciones clínicas de estos pacientes son las infecciones bacterianas recurrentes, localizadas fundamentalmente en piel y mucosas. La gravedad de las complicaciones infecciosas en el tipo I parecen estar relacionadas con el grado de déficit de CD18. Se han descrito dos fenotipos, uno de deficiencia grave y otro de deficiencia moderada. Los pacientes con DAL de tipo I grave tienen menos del 1% de la expresión normal del CD18, mientras que los 716 El niño con infecciones recurrentes. Inmunodeficiencias primarias pacientes con fenotipo moderado pueden presentar unos valores entre el 1 y el 30%. En esta forma de deficiencia grave, las infecciones aparecen desde el momento del nacimiento, incluyendo onfalitis, con retraso de la caída del cordón umbilical, leucocitosis persistente (> 15.000/mm3) en ausencia de infección activa, y gingivitis y periodontitis grave con pérdida de la dentición. Son habituales las infecciones recurrentes de la piel, los pulmones, el intestino y el área perirrectal, y suelen deberse a cocos grampositivos o bacilos gramnegativos. En los pacientes con fenotipo moderado, la caída del cordón umbiFigura 36.24. Representación esquemática de la adhesión débil que permite al neutrófilo desplazarse y lical es normal, tienen un número adherirse al endotelio a través del PMNCD15S y selectinas endoteliales. A: muestra una adhesión normal. menor de infecciones y de menor B: mutaciones de la fucosil transferasa necesaria para una glicosilación (o adición de la SLex tetrasácarida) gravedad, la leucocitosis es más apropiada, conducen a defectos en la adhesión correcta (adecuada) al endotelio e infecciones recurrentes observadas en la LADII (tomado de Bellanti JA, 2012) moderada y existe una tendencia a establecer el diagnóstico de forma más tardía. Sin embargo, también se observa enfermedad Otras inmunodeficiencias primarias asociadas a defectos de periodontales y retraso en la cicatrización de las heridas. la quimiotaxis son la periodontitis juvenil localizada asociada La ausencia de formación de pus en los lugares de in- al gen FRP1, el síndrome de Papillon-Lefévre (periodontitis e fección es una de las características de la DAL de tipo 1. hiperqueratosis palmoplantar) y el síndrome de ShwachmanLa gingivitis grave y la periodontitis también son rasgos Diamond (pancitopenia, insuficiencia pancreática exocrina y característicos de estos pacientes que sobreviven a la in- condrodisplasia). fancia. La incapacidad para cicatrizar heridas traumáticas o quirúrgicas es asimismo una característica de este síndrome. La transfusión de neutrófilos puede ser útil en las infecciones agudas graves, si bien el único tratamiento definitivo es el trasplante de médula ósea. Tipo II. Conocida también como trastorno congénito de glicosilación tipo IIc (CDG-IIc), se trata de una enferme- Las entidades incluidas en este grupo de IDP se han comentado dad autosómica recesiva muy poco frecuente, causada ya anteriormente, y su exposición detallada va más allá del objepor mutaciones en el transportador de la GDP-fucosa, tivo de este capítulo. FUCT1 (11p.11.2). Esto produce un defecto de adherencia a las células endoteliales y las infecciones recurrentes que se observan en estos pacientes (Figura 36.24). Entre las manifestaciones clínicas de este trastorno se encuentran infecciones cutáneas, gingivitis, escasa formación de pus y leucocitosis, así como retraso mental, talla baja y facies peculiar. En algunos casos la suplementación con fucosa ha sido útil. Muchas de estas enfermedades se han reconocido recienteTipo III. Es también una enfermedad autosómica recesiva mente, y suelen cursar con fiebres periódicas. Debido a la iminusual, causada por una mutación en el gen KIND LIN 3 portancia de estos procesos, a su aparición habitualmente en (FERMT3). El cuadro clínico es similar a la DAL de tipo I, la edad pediátrica y al hecho de que muchos de ellos siguen pero también incluye defectos en la activación plaquetaria sin conocerse lo suficiente, se expondrán con más detalle en y tendencia a presentar hemorragias graves. el Capítulo 98. 36.10. Defectos de la inmunidad innata • 36.11. Enfermedades autoinflamatorias • 36 717 Tratado de pediatría Sección III - Patología infecciosa 36.12. Déficit del complemento • Dentro de las IDP por déficit de los componentes del complemento se han descrito trastornos de sus diferentes fracciones, que se resumen en la Tabla 36.36. Cursan con distintas manifestaciones clínicas y tienen diversos mecanismos de herencia. 36.13. Diagnóstico de las infecciones recurrentes en el niño • Como ocurre en cualquier tipo de proceso, el diagnóstico de las infecciones recurrentes en los niños debe basarse en: Enfermedad Anamnesis completa, con un árbol genealógico donde se pueden evidenciar procesos similares en otros familiares, el tipo de herencia, etc. En la anamnesis hay que valorar los antecedentes familiares como: afecciones similares en hermanos u otros familiares, consanguinidad de los padres, factores de riesgo paternos para la infección por elVIH (ADVP, promiscuidad sexual, etc.), país de procedencia, transfusiones recibidas (fecha, país, etc.). También se valorará la asistencia a guarderías, las familias numerosas, el tabaquismo, los contaminantes ambientales, el posible hacinamiento, etc. Asimismo, hay que descartar la presencia de una inmunodeficiencia secundaria que, si bien la más frecuente y grave es la infección por VIHSIDA, también puede ser secundaria a múltiples causas (Tabla 36.37). Exploración física detallada donde se valoren las distintas manifestaciones clínicas con que pueden cursar las diversas formas de IDP o las distintas entidades clínicas que pueden asociarse a inmunodeficiencia. Cuadro asociado Herencia C1q S. LES-like; AR; infecciones C1r S. LES-like; AR; EAM; infecciones AR AR C1s S. LES-like; EAM AR C4 S. LES-like; AR; infecciones; DM tipo 1; meningitis bacterianas AR C2 S. LES-like vasculitis; aterosclerosis; polimiositis; glomerulonefritis; infecciones piógenas AR C3 Infecciones piógenas graves; S. LES-like; glomerulonefritis; SHU atípico, degeneración macular AR C5 LES; infecciones por Neisseria AR C6 LES; infecciones por Neisseria AR C7 LES; infecciones por Neisseria; vasculitis AR C8a LES; infecciones por Neisseria AR C8b LES; infecciones por Neisseria AR C9 Infecciones por Neisseria AR C1 inhibidor Angioedema hereditario AD Factor D Infección grave por Neisseria AR Properdina Infección grave por Neisseria LX Factor I Infecciones piógenas recurrentes; LES; glomerulonefritis; SHU, preeclampsia grave AR Factor H SHU; glomerulonefritis membranoproliferativa; infección por Neisseria; preeclampsia grave AR MASP1 Dismorfia facial; labio leporino y/o fisura palatina; craneosinostosis; trastornos aprendizaje; anomalías miembros , genitales, vesicorrenales AR COLEC11 Síndrome 3MC Dismorfia facial; labio leporino y/o fisura palatina; craneosinostosis; trastornos aprendizaje; anomalías miembros , genitales, vesicorrenales AR MASP2 Infecciones piógenas; enfermedad pulmonar inflamatoria AR CD46 Glomerulonefritis; SHU, atípico; preeclampsia grave AD CD59 Anemia hemolítica. Trombosis AR HPN Hemolisis recurrente; hemoglobinuria; dolor abdominal; distonía musculatura lisa; cansancio, trombosis Mutación adquirida L-X Ficolin 3 Infecciones piógenas graves y recurrentes en pulmón, enterocolitis necrosante y falta respuesta de AC selectiva a polisacaridos neumocócicos Herencia AR: autosómica recesiva; AD: autosómica dominante; LX: ligada al sexo; LES: lupus eritematoso sistémico; AR: artritis reumatoide; EAM: enfermedades autoinmunitarias múltiples; DM: diabetes mellitus; SHU: síndrome hemolítico-urémico; HPN: hemoglobinuria paroxística nocturna Tabla 36.36. Deficiencias del complemento 718 El niño con infecciones recurrentes. Inmunodeficiencias primarias • 36 exactamente el tipo de IDP entre los distintos grupos acEn la exploración física hay que valorar una serie de datos tualmente aceptados. que pueden ser de gran interés en el diagnóstico. La presencia de telangiectasias en las conjuntivas, las mejillas o los Distintas enfermedades Enfermedades genéticas pabellones auriculares junto con una marcha atáxica orieno anomalías cromosómicas tan hacia una ataxia-telangiectasia o síndrome de Louis-Barr; · Uremia · Fibrosis quística · Diabetes mellitus una dismorfia craneofacial asociada a convulsiones hipocal· Síndrome Down · Malnutrición y defi ciencias cémicas y cardiopatía congénita sugiere una secuencia de Di en vitaminas y minerales Edad George; la tríada de eczema, otitis y petequias por púrpura · Enteropatía con pérdida trombocitopénica es típica del síndrome de Wiskott-Aldrich; · Prematuros y recién nacidos de proteínas · Personas mayores · Síndrome nefrótico la presencia de abscesos fríos recidivantes,pelo de color ro· Síndrome miotónico jizo, facies peculiar y dientes supernumerarios sugiere un Cirugía traumatismos · Anemia de células falciformes síndrome de Job o síndrome de hiper-IgE; los cabellos platea· Enfermedad alérgica y asma · Esplenectomía dos, junto a hepatoesplenomegalia y trastornos neurológi· Enfermedades · Quemaduras · Anestesia autoinmunitarias cos apuntan hacia un síndrome de Griscelli; la presencia de · Estrés cuadros respiratorios de sinusitis crónica con dextrocardia Enfermedades malignas · Embarazo linfoproliferativas debe hacer pensar en un síndrome de cilios inmóviles o sín· Leucemias Distintas enfermedades drome de Kartagener; las infecciones bacterianas de repeti· Linfomas ción después de los 6 meses junto con un cuadro articular · Infección VIH-SIDA · Mieloma múltiple · Viriasis exantemáticas que recuerda a la artritis idiopática juvenil se observa en Tratamiento (sarampión, varicela) con inmunodepresores la agammaglobulinemia de Bruton; la presencia de fallo de · Infección medro, hepatoesplenomegalia, eritrodermia, edema gene· Radioterapia por citomegalorivus (CMV) · Fármacos inmunodepresores ralizado, diarrea crónica, hipoalbuminemia, eosinofilia, etc., · Infección por virus · Corticoides de Epstein-Barr (VEB) sugieren un síndrome de Omenn; el retraso de la caída del · Globulina antilinfocito · Rubéola congénita cordón umbilical y la enfermedad periodontal son típicos o antitimocito · Infección bacteriana de los déficit de moléculas de adhesión leucocitaria de tipo I; · Modificadores de la respuesta · Enfermedades biológica (AC-monoclonales) la aparición de úlceras y aftas bucales es característica de por micobacterias, hongos y parásitos la neutropenia cíclica y de la agranulocitosis congénita; la presencia de albinismo, un mechón blanco de cabello, nisTabla 36.37. Inmunodeficiencias secundarias. Principales causas tagmo y granulaciones gigantes en los neutrófilos sugiere un síndrome de Chediak-Higashi; la presencia de abcesos de Como ya se ha comentado, cuando exista una sospecha funlocalización múltiple junto con obstrucciones de órganos dada de que pueda tratarse de una IDP, se pondrá en marcha huecos debe orientar hacia la enfermedad granulomatosa una serie de estudios basados en la presunción clínica. En la crónica; las úlceras/aftas bucales recidivantes con descenso Tabla 36.38 se recogen las exploraciones analíticas basales de los neutrofilos cada 3 semanas se observa en la neutrope- que se deben realizar en los niños con infecciones recurrentes nia cíclica; el fallo de medro con diarrea crónica asociado a en los que se sospecha la presencia de una IDP. Este estudio enfisema, acropaquias, tórax enfisematoso, prolapso rectal y debe ser escalonado y estructurado en tres distintas fases de sabor salado del sudor es muy sugestivo de fibrosis quística; complejidad creciente: screening, avanzado y especializado la presencia de anomalías anatómicas malformativas, hiper- (Tablas 36.10, 36.12, 36.13 y 36.15). trofia de adenoides o Hemograma con recuento leucocitario y fórmula Linfocitos totales/mm3 (CD3) reflujo a través de la (neutrófilos y linfocitos/mm3) trompa de Eustaquio Proteinograma IgG, IgA, IgM, IgD, IgE Linfocitos T4, T4, T4/T8 explicaría las OMA de Subfracciones de IgG (1, 2, 3, 4) Respuesta a mitógenos: repetición; la presencia Fitohemaglutina (L-T) Isohemaglutininas (antiA, antiB) de fístulas traqueoesoConcanavalina A (L-T) (T8) IgA secretora Pokeweed (L-T y L-B) fágicas y malformacioAC antitétanos, difteria, pertussis en vacunados con DTP Lipopolisacáridos (L-B) nes bronquiales explicaría las neumonías Ac antisarampión, paperas, rubéola en vacunados con SRP Antígenos cutáneos recidivantes. Complemento hemolítico total (CH50). AC anti VIH Estudio hematológiOtros componentes del complemento co, bioquímico, inmuExploración de los fagocitos (NTB) nológico, genético y Otras: Rx tórax y senos. Ecografía abdominal. Cistografía. Test del sudor. Determinación de alfa-1-antitripsina. Examen ORL. Evaluación psicológica de biología molecular, Tabla 36.38. Infecciones recurrentes en el niño. Exploraciones analíticas de base (tomado de Bellanti JA,1985) que permita definir 719 Tratado de pediatría Sección III - Patología infecciosa Figura 36.25. Infecciones recurrentes en el niño. Enfoque etiológico general (tomado de Young M y Bellanti A, 1988) En cualquier caso, hay que recordar que las infecciones recurrentes en el niño muchas veces no tienen una etiología inmunológica, sino que pueden ser secundarias y no debidas a inmunodeficiencias primarias. En la Figura 36.25 se presenta el enfoque etiológico general de las infecciones recurrentes en el niño. 36.14. Pronóstico y tratamiento Ante una presunta, IDP celular es primordial instaurar una profilaxis con trimetoprim-sulfametoxazol, en dosis de 150 mg/m2 en días alternos. Se debe aislar al paciente y mantenerle en un ambiente lo más aséptico posible. Las vacunas de virus vivos están absolutamente contraindicadas por la posibilidad de diseminación. También está contraindicada la vacunación con la vacuna oral de la poliomielitis en los familiares que conviven, ya que el virus se excreta en las heces y puede contagiar al paciente. Una de las medidas de protección más eficaz es la creación de barreras inmunes vacunando frente a la gripe y la varicela a todos los familiares que conviven, y poniendo al día su calendario vacunal. Si el paciente necesita una transfusión, se efectuará con sangre radiada para evitar la reacción de injerto contra huésped. En los últimos años ha mejorado significativamente el pronóstico de las IDP, al poder contar con distintas medidas terapéu- Una inmunodeficiencia de linfocitos T o combinada requiere el ticas, como la gammaglobulina intravenosa (IGIV), el levamisol, tratamiento con trasplante de progenitores hemopoyéticos, lo la cimetidina, el ácido ascórbico, los factores estimulantes de que obliga a la búsqueda rápida de un posible donante entre los colonias, el trasplante de médula ósea y, por supuesto, medi- hermanos, familiares y, eventualmente, bancos de progenitores das de profilaxis y tratamientos antibióticos cada vez más efi- hemopoyéticos. caces. El tratamiento de las distintas IDP debe conducirse en colaboración con la Unidad de Inmunología que haya realizado En las deficiencias de anticuerpos es necesario el tratamiento el diagnóstico definitivo de la inmunodeficiencia concreta. En sustitutivo con gammaglobulina endovenosa, en dosis que osla Tabla 36.39 se recogen las medidas · No realice transfusiones con hemoderivados, a no ser que estén irradiados y sean negativos terapéuticas generales para las IDP. De todas formas, las actitudes terapéuticas son muy distintas en función del tipo de IDP, y oscilan desde una actitud expectante (como en la mayoría de los déficit de IgA asintomáticos) hasta la IGIV o subcutánea sustitutiva (que dará unos excelentes resultados en la agammaglobulinemia de Bruton), y hasta los trasplantes de médula ósea (en las formas más graves de inmunodeficiencias combinadas). para citomegalovirus · Evite vacunas de virus vivos, especialmente en pacientes con deficiencias graves de células T o agammaglobulinemia y en los miembros de la unidad familiar · Use profilaxis para Pneumocystis jiroveci (carinii) en inmunodeficiencia de linfocitos T y en síndrome de hiper-IgM ligado al cromosoma X · Controle la función pulmonar en pacientes con neumonía recidivante · Emplee fisioterapia respiratoria y drenaje postural en pacientes con neumonía recidivante · Administre antibióticos profilácticos, ya que las infecciones menores pueden diseminarse rápidamente · Examine en la heces diarreicas la presencia de Giardia lamblia y Clostridium difficile · Evite la exposición innecesaria a individuos infectados · Utilice inmunoglobulinas para estados de deficiencias de anticuerpos graves en dosis de 400-600 mg/kg cada 3-4 semanas i.v. Tabla 36.39. Tratamiento general de los pacientes con inmunodeficiencias 720 El niño con infecciones recurrentes. Inmunodeficiencias primarias cila entre 300-400 mg/kg/día cada 4 semanas. El objetivo es mantener al paciente con pocas infecciones o sin ellas, lo que suele lograrse con concentraciones plasmáticas de 300-400 mg/dl en el período valle (inmediatamente antes de la administración de la dosis). En ocasiones, al inicio del tratamiento o cuando el paciente tiene infecciones graves, con el consiguiente aumento del catabolismo, es necesario administrar dosis semanales hasta alcanzar el equilibrio. En la Tabla 36.40 se resume el pronóstico de las IDP. Mal pronóstico1 Pronóstico reservado2 36 Buen pronóstico3 Inmunodeficiencia combinada grave4 Inmunodeficiencia variable común Hipogammaglobulinemia transitoria Otras inmunodeficiencias combinadas Síndrome hiper-IgM4 Agammaglobulinemia L-X (síndrome de Bruton) Síndrome de Wiskott-Aldrich4 Enfermedad granulomatosa crónica4 Déficit selectivo de IgA Síndrome ataxia-telangiectasia Secuencia de Di George4 Candidiasis mucocutánea crónica Déficit de adhesión de leucocitos4 Síndrome hiper-IgE Déficit de subclases de IgG Síndrome linfoproliferativo L-X4 Déficit del complemento 1 Duración de la vida muy acortada Duración de la vida a menudo acortada Duración de la vida normal con tratamiento óptimo 4 Curable con trasplante 2 3 Tabla 36.40. Pronósticos de las inmunodeficiencias primarias (tomado de Manual Merck. 10.ª ed.) Como corolario, se pueden extraer las siguientes conclusiones: • Las infecciones de repetición en el niño son un motivo fre(radiografía de tórax con alteraciones, diarrea crónica, fallo de cuente de consulta pediátrica y una causa importante de medro, dermopatías crónicas, etc.) deben estudiarse a fin de preocupación por parte de los familiares. descartar la presencia de un cuadro de inmunodeficiencia pri• Entre las infecciones de repetición en la edad infantil, las de maria o secundaria o de etiología no inmunológica. las vías respiratorias superiores son, con diferencia, las de • Si existe una sospecha fundada de que el paciente sufra una inmayor incidencia. Un niño sano puede sufrir en los primeros munodeficiencia, se programará una serie de estudios (orienaños de vida hasta 8-10 procesos respiratorios infecciosos o tados y escalonados) hematológicos, bioquímicos, inmunolócatarrales al año, sin que esto deba ser motivo de preocupagicos, genéticos y moleculares, que permitirán precisar el tipo ción. de inmunodeficiencia primaria o secundaria de que se trate. • La mayor parte de la infecciones respiratorias recurrentes no • La colaboración con las unidades de Genética, Inmunología, tienen un sustrato inmunológico, sino ambiental (guardería, Medicina molecular, etc., será fundamental para el estudio de hacinamiento, tabaquismo, contaminantes) o una etiología una presunta IDP, para precisar el diagnóstico definitivo, y para anatómica o disfuncional instaurar el tratamiento oportuno y proporcionar, cuando sea necesario, el correspondiente consejo genético a los familiares • Una anamnesis cuidadosa y una exploración física exhaustiva permitirán determinar, en la mayoría de las ocasiones, si se trata de infecciones recurrentes banales o de una inmunodeficiencia real. • Los niños con infecciones recurrentes pero con un crecimiento y desarrollo normales, que presentan cuadros infecciosos víricos autolimitados, localizados generalmente en las vías respiratorias superiores, con una radiografía de tórax • Al-Herz W, Bousfiha A, Casanova JL, et al. Primary immunodeficiency disnormal y periodos de completo bienestar entre las infeccioease: an update on the classification from the International Union of Imnes no deben ser motivo de preocupación. En estos casos, munological Societies Expert Committee for Primary Immunodeficiency. se debe tranquilizar a la familia y abstenerse de practicar esDisponible en: <www.frontiersin.org>. November 2011/vol 2/article 54. tudios complejos y de administrar tratamientos, que suelen ser ineficaces, caros y potencialmente peligrosos. El término • Alsina l, LLobet-Agulló P, Soler-Palacín P. Ampliación del cribado neo“aumentar las defensas” tiene una aceptación muy buena por natal a la detección de inmunodeficiencias combinadas graves. Un parte de los familiares y, lamentablemente, se oye con exceimperativo moral. An Pediatr (Barc) 2014; 81(5): 273-274. siva frecuencia en las consultas en boca de los profesionales. • Los pacientes con infecciones recurrentes en los que se objeti- • Bellanti JA. Immunology III. Philadelphia. WB Saunders Company, 1985. va crecimiento y desarrollo deficientes, presencia de infecciones graves (bacteriemias, meningitis, sepsis, etc.) o enfermeda- • Bellanti JA. Immunology IV. Clinical Applications in Health and Diseases. des graves por virus habituales o por agentes poco virulentos Bethesda, Maryland. I Care Press, 2012. (infecciones oportunistas), con escasos períodos de bienestar entre las infecciones, necesidad de ingreso hospitalario y trata- • Bonilla FA. Primary humoral immune deficiencies: An overview. Dismiento antibiótico i.v., y asociación de otras manifestaciones ponible en: <www.uptodate>. May 2013. Bibliografía 721 Tratado de pediatría Sección III - Patología infecciosa • Bonilla FA. Severe combined immunodeficiency (SCID): An overview. Disponible en: <www.uptodate>. May 2013. • Hogan MB, Wilson NW. Common variable immunodeficiency in children. Disponible en: <www.uptodate>. May 2013. • Bonilla FA. Combined immunodeficiencies. Disponible en: <www.uptodate>. May 2013. • Hostoffer R. Selective IgA deficiency: clinical manifestations, pathophysiology, and diagnosis. Disponible en: <www.uptodate>. May 2013. • Dalal I, Roifman CM. Transiet hypogammaglobulinemia of infancy. Disponible en: <www.up todate.com>. May 2013. • Ming JE. Syndromic immunodeficiencies. Disponible en: <www.uptodate>. 2013. • Delgado Rubio A, Ruiz Contreras J. Infecciones recurrentes en el niño. En: Enfermedades infecciosas en Pediatría. Delgado A (ed). Madrid. McGraw-Hill, 2009; 27-56. • Seroogy CM. Di George syndrome: Clinical features and diagnosis.Disponible en: <www.uptodate>. May 2013. • • Delgado Rubio A. El niño con infecciones recurrentes. Temas de Pediatría Vol 17. Bilbao. UPV, 2005. Stiehm ER. Approach to the child with recurrent infections. Disponible en: <www.uptodate>. May 2013. • • Delgado Rubio A, Galán Gómez E, Baca Cots M. Infecciones recurrentes (Síndrome hiper IgE). En: Pediatría Clínica Vol 12. Bilbao. UPV, 2003. Verbsky JW, Growsman WJ. Celular and genetic basis of primary immunodeficiencies. Pediatr Clin North Am 2006; 53: 649-684. • • Gaspar HB, Hammarström L, Mahlaoui N, et al. The case for mandatory newborn screening for severe combined immunodeficiency (SCID). J Clin Immunol 2014; 34: 393-397. Woroniecka M, Ballow M. Office evaluation of children with recurrent infection. Ped Clin North Amer 2000; 47(6): 1211-1224. 722