Hemofilia de A a Z
Anuncio

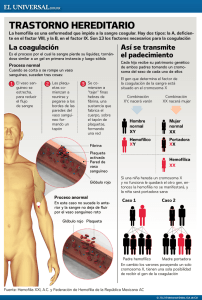
A|B|C|D|E|F|G|H|I|J|K|L|M|N|O|P|Q|R|S|T|U|V|W|X|Y|Z A ADN (ácido desoxirribonucléico) Una compleja molécula que transmite la información genética de la mayoría de los organismos vivientes (instrucciones y programas de vida del cuerpo). Albúmina La Albúmina es la proteína predominante en la sangre. Se usa como estabilizador durante el proceso de fabricación de varios concentrados del factor VIII, también en la preparación o elaboración de factor VIII recombinante actualmente en el mercado o en investigación clínica. Anticuerpo Es una proteína producida por las células plasmáticas que se une con sustancias que el cuerpo reconoce como externas.Una vez unido, el anticuerpo puede destruir o inactivar la sustancia extraña. Los inhibidores que surgen como respuesta a las terapias de reemplazo para la hemofilia son anticuerpos contra el factor VIII. Anticuerpo Monoclonal Es un anticuerpo producido mediante cultivo de un único tipo de célula, producida en el laboratorio, fusionando células que pueden producir anticuerpos con otras células que pueden dividirse y crecer sostenidamente. Este tipo de célula produce un anticuerpo que se une con una región específica (epitope) de una proteína también específica. topArtropatía Enfermedad crónica de las articulaciones (artritis) causada por el repetido y descontrolado sangrado interno en las mismas. Esta es una de las causas principales de minusvalía en los afectados de hemofilia. <Volver a la parte superior Atención Integral Es una forma de brindar cuidado de la salud en la que un equipo de profesionales trabaja con el paciente para mejorar su estado físico, emocional y mental. La atención se lleva a cabo en un mismo lugar, un centro para el tratamiento de la hemofilia (CTH) para que el equipo de profesionales tenga la posibilidad de trabajar en forma conjunta. El equipo cuenta entre sus miembros con hematólogos pediátras, hematólogos, psicólogos, asistentes sociales, odontólogos, terapéutas físicos y asesores.Un laboratorio de análisis de sangre y radiografías se encuentra cerca del centro de tratamiento. Si fuera necesaria la consulta de un especialista no integrante del equipo, como un cirujano o psiquiátra, el equipo podrá enviar al paciente con el especialista. El objetivo de la atención integral es el de ayudar a la persona con hemofilia a llevar una vida saludable. El paciente hemofílico recibe apoyo para superar todo tipo de problemas y aprender a cuidarse a sí mismo dentro de sus posibilidades. Costos de la Atención Integral A pesar de que la atención integral parece tener un costo muy alto, en realidad permite economizar al ayudar a evitar problemas más costosos relacionados con hemofilia. Una primera visita incluirá un control completo del paciente que comprende historia clínica y análisis de sangre. La proporción del costo total a cargo del paciente variará según dónde viva y de acuerdo al tipo de cobertura médica. <Volver a la parte superior Miembros del Equipo de Atención Integral. Todos los integrantes son especialistas en hemofilia. La siguiente es una corta descripción de cada miembro del equipo y el rol que él o ella desempeña en la atención de la hemofilia: C Cascada de Coagulación Es el proceso que se da cuando los factores de coagulación sanguínea trabajan juntos para producir la fibrina, necesaria para la formación de un coágulo. La sangre circula por el cuerpo a través de conductos llamados vasos sanguíneos como venas, arterias y capilares. Cuando uno de estos vasos se daña (o sea cuando alguien se lastima) se pierde sangre. Cuando este daño ocurre en la parte externa del cuerpo, al herirnos o rasguñarnos, la sangre es visible. Pero si el daño ocurre en la parte interna del cuerpo la sangre no se ve. Visible o no, de todos modos el cuerpo debe trabajar para parar el sangrado y reparar el vaso sanguíneo. El primer paso en la reparación es el de contraer los vasos sanguíneos para reducir el flujo sanguíneo. Luego las plaquetas, que son células que existen en la sangre, se apuran a crear un agregado provisorio sobre la herida. Pero este agregado durará sólo unas pocas horas. En todo caso es necesario un agregado más resistente y perdurable. La coagulación sanguínea normal depende de múltiples factores, la mayoría son proteínas que circulan por la sangre en diversas concentraciones. A los factores de coagulación se les da un nombre o se les denomina con un número romano específico. Estos factores hacen que una fuerte malla de proteína - llamada fibrina - se teja sobre el agregado de plaquetas. Esta malla se llama coágulo de fibrina.Una vez que el coágulo detuvo la hemorragia, el cuerpo puede reparar los vasos dañados. Una vez reparados, el coágulo de fibrina es re-absorbido hacia el interior del cuerpo o se desprende externamente en forma de costra. Los primeros dos pasos descritos aquí son iguales se padezca o no de hemofilia: el vaso sanguíneo dañado se contrae y las plaquetas se agrupan para formar un tapón . Pero en las personas con hemofilia el tercer paso no se dá porque la sangre no contiene suficientes factores específicos de coagulación como para crear un coágulo resistente de fibrina. Cuando el factor coagulante no existe en cantidad suficiente, el coágulo de fibrina no se forma o es de tan poca consistencia que el sangrado continua. El hemofílico entonces no sangra más rápido que otras personas sino en forma más prolongada. El factor coagulante que le falta a una persona con hemofilia es el factor VIII o el factor IX. Según el factor carente será el tipo de hemofilia que padece: la deficiencia del factor VIII se llama hemofilia A y la deficiencia del factor IX se llama hemofilia B. <Volver a la parte superior Clonación de gen Es un proceso para hacer copias de un gen. Primero, un gen (molécula de ADN ) es introducido en un vector. Por medio del vector se hace llegar el gen hasta la célula huésped capaz de fabricar un gran número de copias del gen. Coagulación Es el proceso por el cual la sangre líquida se transforma en una sustancia gelatinosa para sellar o taponar un vaso sanguíneo dañado. La sangre de las personas con hemofilia tiene disminuidos los niveles de la proteína funcional que es necesaria para crear un coágulo de sangre efectivo. Coágulo (de sangre) Un tapón plaquetario que se forma sobre la herida para impedir que el cuerpo sangre. <Volver a la parte superior Cromosoma X En los humanos, uno de los dos cromosomas del sexo (X e Y) que contienen los genes que determinan el sexo del bebé. E Enfermedad de Christmas Es otro nombre para la Hemofilia B, un tipo de hemofilia que no es común. A las personas que padecen hemofilia B les falta o no tienen suficiente cantidad de factor IX coagulante. Tomó su nombre de una de las primeras personas diagnosticadas con hemofilia B. Enfermedad Hereditaria Una enfermedad que se pasa genéticamente de padres a hijos. A pesar de que la hemofilia puede resultar de una mutación espontánea del gen, lo más frecuente es que sea un trastorno de tipo hereditario. <Volver a la parte superior F Factor VIII Es una de las proteínas coagulantes encargadas de acelerar el proceso de crear una red eficaz de fibrina. Factor Antihemofílico (FAH) Otro término para el factor VIII, que es uno de los factores coagulantes de la sangre necesarios para fabricar la fibrina que a su vez favorece la interrupción del sangrado. Factor de Coagulación Es uno de los varios compononentes de la sangre que interviene en el proceso de coagulación. <Volver a la parte superior Factor Recombinante El factor recombinante es concentrado del factor VIIa, del factor VIII, o del factor IX creado en un laboratorio. Estos concentrados del factor no se derivan de sangre humana o de plasma. Usando tecnología de ADN recombinante, el gen del factor humano se introduce en una célula no humana, quien le da facultades para producir factores de coagulación humanos. Aunque los concentrados del factor recombinante no se extraen de plasma humano, el factor que producen es factor humano. Los factores recombinantes y derivados del plasma son casi idénticos en sus estructuras físicas y químicas, y ambos concentrados se hacen según las instrucciones genéticas contenidas dentro del gen del factor humano VIII. También tienen el mismo comportamiento una vez infundidos en el cuerpo. Así es cómo se fabrica el factor recombinante: • Los genes humanos que producen el código del factor VIII o del factor IX se insertan en porciones de ADN viral genéticamente modificado llamado vector viral (haga click aquí para aprender más sobre vectores en el módulo de terapia génica). Este vector se introduce luego en las células animales que reconocerán y transmitirán el gen. • Las células animales que contienen el gen del factor VIII o del factor IX se colocan en un recipiente junto con un flúido llamado 'medio', que contiene los nutrientes que las células necesitan para crecer. • Las células animales crecen y se multiplican. • El gen del factor de las células hace que las mismas produzcan factor VIII o factor IX . El factor se secreta en el 'medio' del recipiente. • El 'medio' que contiene el factor se retira del recipiente y se filtran las células. • El factor recombinante es sometido a múltiples pasos de purificación. • Luego se congela en seco, se envasa en botellas y se etiqueta. <Volver a la parte superior Factor VIII recombinante G Gen Una unidad de información que transmite las instrucciones para crear una proteína en el cuerpo. Cada proteína, incluso el factor VIII, se forma a partir de un conjunto de instrucciones transmitidas en el gen. Si falta o se daña un gen que transmite las instrucciones para una proteína específica , el cuerpo no puede fabricar esa proteína apropiadamente. Este es el caso de la hemofilia. <Volver a la parte superior H Hematoma Una marca oscura y azulada bajo la superficie de la piel causada por sangrado subcutáneo. Hemofilia La hemofilia es un trastorno hemorrágico genético que impide que la sangre forme un coágulo eficaz. Sin un proceso de coagulación normal, las lesiones internas y las heridas externas no pueden curarse correctamente. La hemofilia A y B se heredan como enfermedades recesivas ligadas al sexo y ocurren casi exclusivamente en varones. Se estima que la hemofilia tiene una incidencia de 1 cada 5.000 varones nacidos en los Estados Unidos y Canadá. Se encuentra en cualquier grupo étnico del mundo. Aproximadamente 13.500 norteamericanos tienen hemofilia A (también llamada hemofilia clásica), en la cual el factor de coagulación VIII está ausente o no está presente en cantidad suficiente. Aproximadamente 3.500 personas en los Estados Unidos tienen hemofilia B (también llamada Enfermedad del Christmas), en la cual el factor de coagulación IX está ausente o no está presente en cantidad suficiente. En Canadá, alrededor de 2.000 personas tienen hemofilia A, mientras que aproximadamente 450 tienen hemofilia B. La única manera de detectar la hemofilia es mediante un análisis de sangre para medir el nivel de coagulación del factor. Si se sabe que existe hemofilia en una familia, a los bebés recién nacidos se les deben hacer los análisis correspondientes. En el caso de la hemofilia A el control se puede realizar al nacer el bebé, pudiéndose extraer la sangre del cordón umbilical. En el caso de la hemofilia B, el análisis puede indicar que el nivel del factor IX está más alto en el bebé recién nacido que más tarde, porque parte del factor de la madre pudo haber pasado al bebé antes del nacimiento. Si el análisis se hace después, la prueba anticipará el nivel del factor IX que el bebé puede tener el resto de su vida. Una persona puede tener hemofilia leve, moderada, o severa, dependiendo de cuánto factor de coagulación está funcionando en su cuerpo. Si una persona tiene hemofilia leve, moderada, o severa nada cambia. Sin embargo, el número y el tipo de sangrados puede variar de acuerdo a cómo es de sana y activa esa persona. Los miembros de una misma familia con hemofilia tienen generalmente el mismo nivel de coagulación del factor. A los padres de un niño con hemofilia: Es muy importante saber qué tipo de hemofilia tiene su niño. Infórmese si tiene deficiencia del factor VIII, deficiencia del factor IX, u otro tipo menos comúnl. Después conozca el grado de seriedad. No toda las personas con hemofilia sufren el mismo tipo de sangrado, y según el grado de gravedad se puede determinar qué tipos de hemorragias tendrá su niño. Los niveles siguientes se refieren a cuánto factor está funcionando en la circulación sanguínea: • hemofilia severa: Menos de 1% del factor está activo. • Hemofilia moderada: de 1% a 5% del factor está activo. • Hemofilia leve: de 5% a 50% del factor está activo. Técnicamente, las diferencias entre los tres niveles de deficiencia del factor se relacionan con el nivel de actividad del factor, no con I Infusión Proceso de infundir/inyectar el medicamento directamente en una vena. Inhibidores A veces el sistema inmunológico del cuerpo confunde al factor infundido con una sustancia desconocida, entonces crea anticuerpos que destruyen el factor, lo cual impide la coagulación rápida. Cuando el cuerpo crea estos anticuerpos, se dice que la persona tiene un inhibidor. Nadie nace con un inhibidor y no se sabe con seguridad porqué se generan. Los inhibidores ocurren más a menudo en las personas con hemofilia severa. Las personas con hemofilia leve o moderada tienen más factor normal activo en sus cuerpos y generalmente no desarrollan un inhibidor del factor infundido. Se estima que entre 20% y 30% de las personas con deficiencia severa del factor VIII y hasta 4% de aquellas con deficiencia severa del factor IX generarán inhibidores alguna vez en su vida. Algunos pacientes desarrollan inhibidores después de varias infusiones; algunos los desarrollan después de muchos años de administración continua del factor. El número de infusiones que una persona recibe por día, semana, o mes, probablemente no aumente la posibilidad de desarrollar un inhibidor. En algunos casos, el inhibidor simplemente desaparece o llega a ser bastante leve como para no afectar infusiones frecuentes del factor. Sin embargo, muchos individuos con inhibidores requieren terapia alternativa. Es más probable que una persona desarrolle un inhibidor cuando: • Tiene una deficiencia del factor VIII. • Tiene menos de 1% del nivel normal del factor (hemofilia severa). • Tiene un pariente con inhibidores. Los centros de tratamiento de la hemofilia deben controlar la existencia de inhibidores cuando se extrae sangre en un control médico integral. Siempre se deben hacer análisis antes de una cirugía o de o biopsias y extracciones de dientes. Un análisis mostrará lo siguiente: • Si los inhibidores están presentes • Los niveles reales de inhibidor • La fuerza de la respuesta (baja o alta) del inhibidor al factor infundido El título o nivel del inhibidor se mide en unidades Bethesda (UB) . El título/nivel del inhibidor se expresa como un número; cuanto más alto el UB, más fuerte es el inhibidor y menos eficaz será una infusión del factor para detener el sangrado. Este número indica la capacidad relativa del inhibidor para destruir el factor infundido. Un inhibidor de bajo título es menor de 10 UB; un inhibidor de alto nivel indica entre 10 UB y más de 1.000 UB. La respuesta alta o baja del inhibidor está determinada por la respuesta a las infusiones del factor . Se indica una respuesta baja del inhibidor cuando el factor es infundido y los niveles del inhibidor son de P Plaquetas Pequeños fragmentos celulares de la sangre que se agregan a los vasos sanguíneos dañados y facilitan la coagulación. <Volver a la parte superior Plasma Componente líquido de la sangre donde se encuentran los factores de coagulación. Plásmido Una molécula circular de ADN que se usa como vector para hacer llegar un gen a la célula huésped donde no existe naturalmente. Esta es una de las técnicas usadas para producir proteínas recombinantes. Los vectores plásmidos son usados para producir algunas terapias del factor VIII. Portador Es una persona que tiene un gen que altera un rasgo como es el caso de la Hemofilia, pero que no lo expresa. Los genes son programas bioquímicos diseñados para el cuerpo.Están a cargo de proporcionar al cuerpo las instrucciones para producir proteínas que determinan rasgos como el color del pelo, el color de los ojos y el color de la piel.También proveen al cuerpo de información para producir proteínas que realizan funciones específicas. Si uno de estos genes tiene un defecto, se deteriora la función corporal. En los afectados de hemofilia, el gen encargado de informar al cuerpo la creación de un factor de coagulación sanguínea es defectuoso por lo cual produce una alteración en la coagulación de la sangre. Los humanos tenemos entre 50.000 y 100.000 genes microscópicos que se agrupan en fragmentos de ADN llamados cromosomas. Cada célula del cuerpo contiene 46 cromosomas, ordenados en 23 pares. Dos de los genes que intervienen en la coagulación de la sangre se encuentran en el mismo par de cromosomas que determinan si el bebé será mujer u hombre. A estos se los denomina genes “vinculados al sexo”. Por encontrarse los genes que producen hemofilia en el mismo par de cromosomas que los genes que determinan el sexo, la aparición de hemofilia presenta un modelo definido según si el bebé es varón o mujer. A continuación se describe el proceso: • Se concibe un niño en el momento en que el esperma fertiliza el óvulo. • El esperma contiene 23 cromosomas que son copia de la mitad de la compaginación genética paterna. • El óvulo también contiene 23 cromosomas que son copia de la mitad de la compaginación genética materna. • Cuando el esperma fecunda el óvulo, la nueva célula contiene 46 cromosomas -la mitad heredada del padre y la otra mitad heredada de la madre-. • El óvulo siempre contiene un cromosoma X. (Todos los seres humanos tenemos por lo menos un cromosoma X) • El esperma puede contener un cromosoma X o uno Y. • Se concibe una niña cuando existe un cromosoma X en el esperma y otro en el óvulo. Las mujeres son XX. • Se concibe un niño cuando existe un cromosoma Y en el esperma y un cromosoma X en el óvulo. Los hombres son XY. El gen de la hemofilia reside en el cromosoma X. Si un cromosoma X que contiene el gen de la hemofilia (denominado Xh) se combina con un cromosoma X normal, la criatura concebida será una niña que porta el gen de la hemofilia ( el sexo de sus cromosomas será XhX). • Si un cromosoma X que contiene el gen de la hemofilia (Xh) se combina con un cromosoma Y, la criatura será un niño con hemofilia (el sexo de sus cromosomas será XhY). En una madre portadora la mitad de todos sus cromosomas X contienen el gen de la hemofilia en tanto que la otra mitad es normal. La madre probablemente no padecerá hemofilia debido a que su único cromosoma X normal le proporcionará suficiente factor de coagulación en su sangre como para compensar el gen de la hemofilia. Pero puede transmitirle un cromosoma X conteniendo el de la hemofilia a su hijo/hija. Cuando un varón recibe un cromosoma defectuoso de su madre, tendrá hemofilia porque su único cromosoma X contiene el gen de la hemofilia. No tendrá un cromosoma X sano para compensar el gen de la hemofilia. Todos los hijos de portadoras tienen un 50% de posibilidades de recibir un cromosoma modificado. La hija de una mujer portadora tiene un 50% de posibilidades de ser portadora ella misma, dependiendo de si recibe un cromosoma X sano o un gen con hemofilia. El hijo de una mujer portadora tendrá hemofilia si recibe un cromosoma X defectuoso de su madre. Si a su vez tiene descendencia con una mujer no-portadora, sus hijas recibirán un cromosoma X con el gen de la hemofilia y serán portadoras (todas serán XhX), debido a que todos sus cromosomas X son defectuosos. Sus hijos no tendrán hemofilia, sin embargo, porque van a recibir un cromosoma Y y los cromosomas Y no contienen el gen de la hemofilia. Si un hombre con hemofilia tiene descendencia con una mujer noportadora y sólo tiene hijos varones la hemofilia desaparecerá de sus descendientes directos. • • <Volver a la parte superior Profilaxis Profilaxis es una infusión programada de factor de coagulación que mantiene los niveles del factor suficientemente altos en la circulación sanguínea como para evitar la mayoría de los sangrados. En 1994, el Consejo de Consulta Médica y Científica de la Fundación Nacional de Hemofilia, llamado MASAC (Medical and Scientific Advisory Council of the National hemofilia Foundation), recomendó que la profilaxis fuera considerada como terapia óptima para los hemofílicos jóvenes graves. Las infusiones profilácticas se dan para evitar hemorragias Profilaxis primaria se refiere a infusiones frecuentes en un horario determinado. MASAC recomienda que la profilaxis comience entre las edades de 1 y 2 años después de haber evaluado el médico al modelo de sangrado del niño. Profilaxis secundaria es un período más amplio. Puede significar infundir el factor una sola vez o con interrupciones. Por ejemplo, el factor de protección puede ser administrado antes de una actividad física, o de un procedimiento médico como cirugía o extracción de dientes, o por el período de duración de una terapia como la de rehabilitación. La profilaxis secundaria también se puede aplicar para evitar daño adicional en una articulación en el caso de un niño que ya haya experimentado daño de este tipo.