TEMA 8: MEDIDA DE LA CONTAMINACIÓN ATMOSFÉRICA II. 8.1
Anuncio

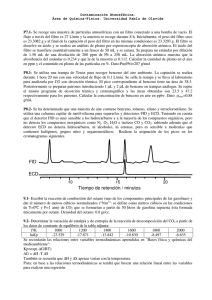
TEMA 8: MEDIDA DE LA CONTAMINACIÓN ATMOSFÉRICA II. 8.1. Métodos de referencia. A la hora de medir la contaminación de una determinada especie, desde un punto de vista legal deben seguirse los denominados métodos de referencia. Estos métodos son los establecidos por la ley, y se consideran métodos patrón respecto de los que se comprueba la eficiencia de otros métodos. Para que un método se catalogue como de referencia debe cumplir tres características además de no presentar interferencias: precisión (desviación estándar pequeña) y exactitud (valor promedio determinado en una gran serie de repeticiones de la medida muy cercano al valor real), y robustez (el resultado se ve poco afectado por otros parámetros no especificados en el protocolo del método: realización por un operario distinto, historial de la preparación de la muestra, concentraciones de otras especies, temperatura, ...). En la UE y en E.E.U.U. (EPA, Environmental Protection Agency) los métodos de referencia para algunos contaminantes son: Contaminante Método UE Método EPA SO2 Fluorescencia UV Método de West-Gaeke Reacción con O3 Reacción con O3 + + quimiluminiscencia quimiluminiscencia NOx Reacción con etileno O3 Espectrofotometría UV + quimiluminiscencia CO PM10 Pb COV NDIR NDIR Captación en filtro Captación en filtro + + gravimetría gravimetría Captación en filtro Captación en filtro + + absorción atómica absorción atómica Ionización en llama (FID) Ionización en llama (FID) 8.2. Medidas de SO2. 8.2.1. Fluorescencia UV. Una de las formas más clásicas de detectar SO2 es por fluorescencia ultravioleta. En esta técnica se ilumina con luz de 214 nm. A esta longitud de onda, el SO2 emite fotones de fluorescencia en el visible (350 nm). La intensidad de fluorescencia es proporcional a la concentración de SO2 de la muestra según la conocida ley de LambertBeer. Nota: se trata de otro ejemplo más de un método basado en la fluorescencia (ver el recuadro en el Tema 7). A diferencia de las fluorescencias vistas anteriormente, en este caso se trata de una fluorescencia molecular, lo que explica que las bandas observadas sean mucho más anchas que en el caso comparable de fluorescencia atómica. Las energías de la radiación estan en el UV/visible, correspondiendo a electrones de valencia. 214 nm absorción 200 250 300 emisión 350 400 FIGURA 8.1: Espectros de absorción y de emisión UV del ozono 8.2.2. Método de West-Gaeke. Otra forma es el método de West-Gaeke o de la pararosanilina. Este método consta de tres pasos: 1. Burbujeo de la muestra en una solución de tetracloromercurato de 2. Complejación con pararosanilina. 3. Medida espectrofométrica del complejo. sodio. Detector fotométrico de llama 8.2.3. Medida de SO2 por quimiluminiscencia y cromatografía de gases. monocromador Tubo fotomultiplicador (detector) aire H2 Componentes separados procedentes de columna de cromatografía de gases FIGURA 8.2: Detección de SO2 mediante detector fotométrico de llama También se puede cuantificar mediante un equipo CG con un detector fotométrico de llama (que usa el principio de la quimiluminiscencia) en tándem. En este detector se lleva a cabo la combustión del H2 con oxígeno del aire en condiciones reductoras (es decir que hay un ligero exceso de H2, necesario para reducir el SO2) para alcanzar las temperaturas altas necesarias para realizar la reacción de formación de la especie quimiluminiscente. El SO2, separado de los demás gases mediante el CG, se reduce con H2 a alta temperatura, generándose azufre molecular excitado S2*, que emite luz de 394 nm detectado en un fotomultiplicador. La intensidad de quimiluminiscencia detectada es proporcional al cuadrado de la concentracion de SO2 de la muestra. (1) H2+1/2 O2 (2) 4 H2+2 SO2 H2O S2*+ 4 H2O Reacción de la combustión Reacción de formación de la especie quimiluminiscente (3) S2* S2 (+h ) Reacción de (quimi)luminiscencia De las tres reacciones detalladas arriba, las reacciones (2) y (3) son las reacciones implicadas en la quimiluminiscencia. 8.2.4. NDIR. Divisor de haz Fuente de IR Celda de referencia SO2 Celda de muestra SO2 Sensor de diferencias de presión Cortador rotatorio de haz FIGURA 8.3: Infrarrojo no dispersivo (NDIR) en el caso de la medida de dióxido de azufre. Otro método muy usado y fácilmente portátil es el del infrarrojo no dispersivo (NDIR). En esta técnica se calienta un filamento metálico para que produzca radiación IR. Este haz pasa por un cortador rotatorio de haz (chóper), un disco mitad opaco mitad transparente que gira, haciendo que tanto las variaciones de la intensidad de la fuente y de la respuesta del detector afecten en la misma medida a la referencia como a la muestra. Los dos haces IR se hacen pasar por sendas celdas, una con la muestra problema y otra con una muestra de concentración conocida. Los haces pasan a continuación por otras dos celdas de SO2 puro. La diferencia de intensidad entre ambos se traduce en una diferencia de presión (como consecuencia del calentamiento de la muestra por la energía absorbida, veáse ec. de estado de los gases) correlacionable con la concentración de SO2 de la muestra. Como se miden presiones, los equipos NDIR son mecánicamente robustos. En el caso del SO2, NO2.es un interferente. 8.3. Medida del ozono y oxidantes totales. El ozono puede medirse directamente por absorción UV porque posee una banda de absorción importante a 254 nm. Para la medida de absorción UV se utiliza un método diferencial de doble haz para comparar la absorbancia en presencia de ozono y sin partículas interferentes (eliminadas con un filtro de teflón, inerte al O3) con la absorbancia en ausencia de ozono tras pasar por un eliminador de este gas, normalmente óxido de molibdeno que convierte ozono a oxígeno molecular. El ozono también se puede medir por absorbancia en el visible de la especie I3que es la forma en la que el yodo es soluble en disolución, tras reacción previa con yoduro (práctica 2). Opcionalmente, se puede añadir almidón y detectar el complejo que éste forma con I3-. A notar que este método detecta la suma de los oxidantes capaces de oxidar el yoduro, por lo que se le llama más correctamente un método de determinación de oxidantes totales, aunque generalmente el oxidante más importante es el ozono. El método establecido por la EPA es como visto el de quimiluminiscencia tras reacción con etileno. Esta reacción produce formaldehído excitado que emite fotones en el visible (430 nm), de forma que la intensidad de quimiluminiscencia será proporcional a la concentración de ozono. 8.4. Medida de CO. Para medidas de CO puede usarse: a).- NDIR, con celdas de CO puro. b).- CG, en la que se acoplan una columna polar que retiene el CO2 y un tamiz molecular de 13-X que separa los otros gases atmosféricos importantes (O2, N2, CH4 y CO) uno de otro y los cuantifica mediante un detector de conductividad térmica. Este detector consta de un filamento al que se aplica un voltaje constante. La temperatura del filamento, y con ello su resistencia eléctrica, depende de la temperatura a la que se encuentra, y ésta depende de la conductividad térmica de los gases que salen de la columna. 8.5. Medida de los óxidos de nitrógeno. 8.5.1. Reacción con ozono+quimiluminiscencia. El NO reacciona con el O3 para dar NO2 excitado que emite fotones IR con un pico en 1200 nm. Si el aire contiene NO2 no lo detectaremos con este sistema. Para ello se descompone previamente todo el NO2 con radiación UV o haciendo pasar la muestra por un convertidor catalítico heterogéneo antes de la reacción con ozono. Por diferencia con la absorbancia de la muestra sin tratar se determina la concentración de NO2. 8.5.2. Reacción con agua oxígenada+potenciometría. La corriente de aire se puede hacer pasar por una disolución oxidante de agua oxigenada. Los NOx se oxidan a nitratos que se miden por potenciometría y amperometría. 8.6. Medida de compuestos orgánicos volátiles. Existen dos alternativas. a).- Adsorción en trampa y desorción con separación e identificación cromatográfica. La adsorción se hace en trampas con adsorbentes adecuados a los compuestos que se desean analizar. Por ejemplo: Tenax = óxido de poli-2,6-difenil-pfenileno para absorción de COV no polares, con posibilidad de desorción térmica. * * O n Estas trampas suelen ser pequeñas y transportables, permiten eliminar interferencias y concentran la muestra en la etapa de muestreo, pero no retienen todos los COV con la misma eficacia y pueden incluso contaminar la muestra. Además, todas las trampas tienen un volumen máximo de adsorción, por encima del cual la determinación deja de ser cuantitativa. Este volumen se denomina volumen de escape. Para comprobar que no se ha superado el límite de escape, las trampas llevan un separador tras el material adsorbente, seguido de más material adsorbente (como un bocadillo: adsorbente-separador-adsorbente). Así, si en el análisis de la segunda zona de adsorbente se detectan COV, indicaría que hemos rebasado el volumen de escape. Una vez que el analito está en la trampa se puede resorber de dos formas: 1).- Desorción térmica, éste se resorbe calentando la trampa hasta una temperatura determinada mientras se hace pasar gas inerte (gas de purga) a su través- el analito es arrastrado por el gas de purga y se preconcentra en un adsorbente secundario. Este adsorbente secundario tiene un volumen más pequeño que la trampa, con lo cual es posible calentarlo en mucho menos tiempo y la desorción es rápida, los contaminantes salen concentrados. Mediante una válvula rotatoria, se consigue resorber otra vez el gas diluyendo con un cierto flujo de gas portador (como aire sintético). Esta mezcla se lleva a una trampa criogénica (opcional) donde se adsorbe el compuesto de interés a baja temperatura. El gas resultante, mucho más concentrado, se analiza por cromatografía de gases. Recordemos que los rellenos de los CG retienen mejor los compuestos que se parecen a él. Por ejemplo, si la columna es polar, retiene los compuestos polares. En cuanto a los detectores, hay infinidad. Los más corrientes son los detectores de ionización en llama y de captura electrónica. Los detectores de ionización en llama (FID) se fundamentan en la pirolisis de los compuestos de carbono en una llama de hidrógeno/aire. Las especies cargadas son atraídas hacia un colector y la corriente que resulta se amplifica y se registra. El número de iones producidos es proporcional al número de átomos de carbono reducidos en la llama. Los grupos carbonilo, amino y halógeno son insensibles (o casi), así como los gases incombustibles como el vapor de agua, dióxido de carbono y óxidos de azufre y nitrógeno. En los detectores de captura electrónica (ECD), el eluato pasa por un emisor como el 63 Ni o el 3H adsorbidos sobre Pt o Ti. Las partículas (electrones) ionizan el gas portador que, en ausencia de muestra, provoca una corriente estacionaria. Cuando una muestra orgánica electronegativa está presente, la intensidad de corriente disminuye porque ésta captura electrones. Por tanto, este detector es selectivo para compuestos electronegativos como halogenados, peróxidos, quinonas y grupos nitrilo, siendo insensible a aminas, alcoholes e hidrocarburos. 2).- Desorción con disolventes; mucho más sencillo que el método anterior, pero menos preciso. Sólo se utiliza cuando hay altas concentraciones de analito o éste o el adsorbente se destruyen a alta temperatura. Consiste simplemente en vaciar el adsorbente en un disolvente apropiado como CS2 y análisis posterior por CG con detector FID que minimiza la interferencia del disolvente. b).- Recogida de muestras y separación e identificación cromatográfica. Las muestras de aire se recogen en cámaras (botellas) de acero inoxidable recubiertas internamente de un material inerte como el SiO2 con un compresor o un sistema controlador de flujo; o bien en bolsas de teflón con cubierta interna de aluminio (para que el teflón no retenga ningún compuesto ni reaccione con él), que se hace expandir con una bomba de succión. La muestra recogida es llevada a una trampa criogénica para su concentración y se detectan por cromatografía de gases. RESOLUCIÓN DE PROBLEMAS. T8.1: El análisis de una muestra de aire de la que se ignora su contenido en NOx se efectúa por el método de la quimiluminiscencia. Haciendo pasar un flujo de aire de 500 ml/min. directamente a la cámara de reacción se obtiene una lectura en el detector de 321 mW/m2. Cuando el flujo de aire se hace previamente pasar por el convertidor catalítico la lectura que se obtiene es 958 mW/m2. Con el objetivo de obtener la concentración de NOx en aire se utiliza un patrón de NO de 100 µg/m3. El paso directo del patrón a la cámara de reacción con un flujo de 500 ml/min satura la señal, por lo que se opta por diluirlo con aire sintético. Se prueban con distintas relaciones de flujo en ml/min. Los resultados son: Patrón (mL/min) Aire sintético (mL/min) Señal (mW/m2) 100 400 8560 50 450 4232 25 475 2105 10 490 838 5 495 428 2.5 497.5 231 1 499 88 ¿Cuál es la concentración de NO y NO2 en aire? Calculamos la concentración de NO para cada dilución del patrón y hacemos una recta de calibrado que represente la intensidad de quimiluminiscencia frente a la concentración de NO. Para calcular la concentración de patrón tomamos como base de cálculo 1 min. Multiplicamos el volumen de patrón en la mezcla con aire sintético en mL por su concentración en g/mL y dividimos por el volumen total (500 mL), obteniendo los siguientes resultados: Patrón (mL/min) Concentración (10-5 g/mL) Señal (mW/m2) 100 2 8560 50 1 4232 25 0.5 2105 10 0.2 838 5 0.1 428 2.5 0.05 231 1 0.04 88 La recta que se obtiene es la siguiente: I/(mW/m2) Recta de calibrado para análisis de NOx 9000 8000 7000 6000 5000 4000 3000 2000 1000 0 0.00E+00 5.00E-06 1.00E-05 1.50E-05 2.00E-05 2.50E-05 [NO]/µ µg/mL La ecuación de la recta es I=4.27⋅108 [NO]−7.6. Cuando el flujo pasa directamente a la cámara, la intensidad es proporcional a la concentración de NO. Sustituyendo en la ecuación correspondiente el valor de 321 mW/m2 tenemos que [NO]=⋅7.7·10-7 g/mL=0.77 g/m3. Cuando lo hacemos pasar por el convertidor catalítico, medimos los NOx totales, con lo que sustituyendo el valor del detector tenemos [NOx]=2.26 g/m3, y entonces [NO2]=[NOx]-[NO]=1.49 g/m3. T8.2: Se está probando un nuevo adsorbente para el muestreo de benceno en aire ambiente. Dos mL de un patrón de 10 mL/m3 se hacen pasar por una trampa que contiene dicho adsorbente. Diez litros de aire limpio se hacen fluir por la trampa después del patrón. Posteriormente el benceno se desorbe y se determina por C.G. Por comparación del área del pico cromatográfico del benceno con el de un patrón, se comprueba que se recupera el 60% del benceno. Al repetir el experimento con 6 L de aire se recupera el 98% ¿Qué límites de volumen de escape podemos deducir de esta experiencia? El benceno total que está entrando en la trampa es de 20 nL. Es fácil ver que la cantidad de benceno que queda retenida en cada experimento es de 12 nL para la elución con 10 L y 19.6 nL para la elución con 6 L. por tanto, los límites del volumen de escape serán 6 y 10 L: en el primer caso, se adsorbe casi todo el benceno (nunca se adsorbe un 100 %); mientras que en el segundo caso aún queda mucho por adsorber. T8.3: Queremos analizar los siguientes COV en aire a 20 oC: pentano, hexano, hexanol y pentanol. a) ¿Puede realizarse la captación con una trampa adsorbente de carbotrap? b) si no fuera así ¿podriamos captar eficientemente estos compuestos a alguna otra temperatura con el mismo adsorbente? c) En caso de que pueda realizarse la captación, ¿a qué temperatura deberíamos realizar la desorción para análisis por CG? Dato:Volumen de escape en L/g de adsorbente. a) Según las tablas, los volúmenes de escape de hexano, pentanol y hexanol son superiores a 10 L/g, lo que indica que la adsorción es factible en la práctica. Sin embargo, para pentano es de 5.89 L/g, con lo cual no se puede utilizar Carbotrap a 20ºC para este conjunto de compuestos. b) Sin embargo, a 0ºC, el volumen de escape es superior a 10 L/g, con lo cual la adsorción si se puede llevar a cabo. c) Las temperaturas de desorción son: heptano: a partir de 200ºC, hexano: 260ºC, pentanol: 260ºC, hexanol: 300ºC. De modo que a partir de 300ºC, se puede desorber cualquier de estos compuestos. P8.1- Se utiliza una trampa de Tenax para recoger benceno del aire ambiente. La captación se realiza durante 1 hora 20 mn con una velocidad de flujo de 0.1 L/min. Se sella la trampa y se lleva al laboratorio para analizarla por CG con desorción térmica. El pico correspondiente al benceno tiene un área de 38.3. Posteriormente se preparan patrones introduciendo 1 µL y 2 µL de benceno en trampas análogas. Se repite el mismo programa de desorción térmica y cromatográfico y las áreas obtenidas son 23.5 y 47.2 respectivamente para los patrones. Calcular la concentración de benceno en aire en ppbv. Dato: benc=0.88 g/mL A partir de los datos de los patrones podemos plantear un sistema de ecuaciones para determinar la ecuación de la recta de calibrado. 23.5 = a + b 47.2 = 2a + b La solución es A=23.7 V-0.2, donde V se expresa en L. Resolviendo para el área problema obtenemos 1.62 L C6H6. Como el benceno de los patrones es líquido, estos L son, sabiendo que la densidad del benceno es 0.879 kg/L y su peso molecular 78 g/mol, 1.83 10-5 moles. Usando la ecuación de los gases ideales, el volumen ocupado en las condiciones del muestreo (P=1atm, 25ºC), son 447 L. Dividiendo por los 8 L de aire que han entrado en la trampa y multiplicando por 109 son 55900 pbbv. P8.2- Se ha determinado que una muestra de aire contiene benceno, tolueno, xileno y tetracloroetileno. Se utiliza una columna capilar de dimetil-silicona para separarlos y detectores FID y ECD. Teniendo en cuenta que el detector FID es muy sensible a los hidrocarburos y a la mayoría de los compuestos orgánicos, pero no detecta los compuestos inorgánicos como N2, O2, H2O e incluso CO y CO2; sabiendo además que el detector ECD no detecta hidrocarburos, ni alcoholes, ni cetonas, pero es sensible a moléculas que contienen halógenos, grupos nitro y organometálicos; Realizar la asignación de los picos en los cromatogramas siguientes. FID ECD 10 15 Tiempo de retención / minutos 20 En un detector de captura electrónica ECD, únicamente podemos observar los compuestos halogenados. Por ello, el pico que únicamente aparece en el cromatograma ECD corresponde al tetracloroetileno. La columna (dimetil-silicona) siendo apolar, el resto de los analitos debe tener un tiempo de retención que dependerá de sus pesos moleculares y sus características dipolares. Benceno (C6H6), tolueno (C6H5CH3), y xilenos (C6H4(CH3)2) son hidrocarburos, son todos muy apolares, la separación entre estos compuestos depende fundamentalmente de los pesos moleculares. Primero saldrá el benceno, después tolueno y como último xileno.