Diagnóstico de th >
Anuncio

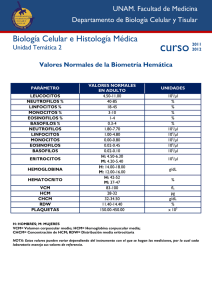
Diagnóstico de th > Hemograma Morfología de los eritrocitos: Marcada anisocitosis con abundantes micro y macrocitos. Marcada poiquilocitosis. Marcada hipocromía. Targets cells, leptocitos. Policromatofilia. Regular cantidad de esquistocitos. Punteado basófilo. Anemia de Cooley Diagnóstico molecular anemia de Cooley En Rosario y zona de influencia los pacientes son homocigotas o doble heterocigotas para alguna de las siguientes mutaciones: IVS1-1 GA, IVS1-6 TC, IVS1-110 GA, IVS2-l GA , CD 39 CT, IVS2-745 CG, que son las más frecuentes en la zona del Mediterráneo. Dosaje de Hb F Aumentada o disminuída MÉTODO DE DESNATURALIZACIÓN ALCALINA Singer y Chernoff Betke V.N: menor de 2% Ligeramente aumentada: menor de 5%(HPFH heterocelular),Th menor, embarazo Muy aumentada: 20-50-90% heterocelular (Anemia de Cooley, βδ Th, RN, neonato. Muy aumentada pancelular: HPFH Test de Kleihauer y Betke adulto normal Test de kleihauer y Betke adulto normal HPFH heterocelular Indices Hematimétricos en Beta-Talasemia Indices en Normal COOLEY PORTADOR sangre periférica VCM fl HCM pg Hb g/dl Hombre Mujer ß-Tal Mayor ß-Tal Minor 89.1±5.01 87.6±5.5 50-70 <79 30.9±1.9 30.2±2.1 12-20 <27 15.9±1.0 14.0±0.9 <7 : 11.5-15.3 : 9.1-14 Talasemia intermedia Se sospecha en individuos que presentan anemia moderada a una edad mas tardía y que raramente requieren transfusiones. Los individuos con Talasemia Intermedia tienen riesgo de sobrecarga de hierro secundaria a absorción intestinal aumentada como consecuencia de la eritropoyesis inefectiva. Talasemia intermedia Hb entre 7 y 10 g/dl Talasemia intermedia Hb: 7-10g /dl VCM disminuído HCM disminuída Hb A2 aumentada (5%), para las doble heterocigosis u homocigosis de formas leves de beta+ Th, Hb F aumentada: 3070% Si se trata de talasemias aumenta la Hb F pero no la Hb A2. Talasemia intermedia El genotipo puede ser variable: Beta+/beta+ (negros) Betaº/( Lepore) Otros -Talasemia menor. Se caracteriza por eritrocitosis microcítica con anemia leve, rara vez se aprecia esplenomegalia, su diagnóstico suele ser casual y facilitado por el empleo de analizadores hematológicos que suministran de forma sistemática los valores de VCM y HCM. -talasemia menor -talasemia menor. diagnóstico º Th: Hb: 8-10 g/dl GR: eritrocitosis VCM y HCM disminuídos Hb A2: 4,5-5,5% Hb F: 0,5-4% Hb A: 90-95% Genetica molecular · Análisis de mutaciones conocidas: las ßtalasemias pueden ser causadas por mas de 200 mutaciones en el gen beta. Comúnmente las mutaciones son detectadas por procedimientos basados en PCR. Los métodos mas usados son reverse dot blot o amplificación primer-específica con un set de primers complementarios a las mutaciones mas frecuentes en la población de la cual el individuo afectado es originario Beta-Talasemia Genética Molecular: Métodos Clínicos PCR-ARMS Mutación 0 IVS-II-nt 1 (634 bp) 861 pb 634 pb Calle 1: blanco de H2O (control de contaminación). Calles 2 y 3: individuo normal (A/ A) con primers normal y mutado respectivamente. Calle 4: marcador de peso molecular (100 bp ladder). La banda más brillante corresponde a 500 pb. Calles 5 y 6: individuo heterocigoto ( A/ T) con primers normal y mutado respectivamente. δβ Talasemias δβ deleción δβ Lepore-fusión δβ HPFH δβ Talasemias Las delta-beta talasemias heterocigotas presentan un cuadro hematológico y clínico similar a las beta Th heterocigotas. Presentan Hb F aumentada 5-15% (heterocelular), la Hb A2 está normalLas delta-beta talasemias homocigotas o doble heterocigotas beta-delta/ beta Th presentan un cuadro clínico y hematológico de Th intermedia o mayor con Hb F muy aumentada y Hb A2 disminuída. Persistencia hereditaria de Hb fetal- HPFH Es más frecuente en la etnia nergra, sólo el homocigota presenta Hb F de 100% pancelular y VCM disminuído. El heterocigota puede no presentar anemia pero presenta HB F de alrededor del 30% siempre de distribución pancelular. δβ talasemias-Lepore La Hb Lepore corre como la Hb S En el heterocigota la morfología yel hemograma son similares a la beta Th menor, pero presentan entre 8-15 % de Hb Lepore, Hb F algo aumentada 5-7%, Hb A2 disminuída. El homocigota clínica y hematológicamente es una Th mayor, con Hb Lepore de 2530%, el resto es Hb F. Alfa talasemias. La patología molecular de las alfa talasemias está determinada por una serie heterogenea de deleciones de diversa extensión: º elimina todo el complejo de genes alfa de uno o de los dos cromososmas + elimina uno de los genes alfa , parte de los mismos o puede ser una mutación puntual. Deleciones causantes de -Talasemia - 70 kpb 5’HVR HS-40 0 kpb 2 inter HVR 10 kpb 1 1 20 kpb 2 1 1 30 kpb 3’HVR 5’ 3’ 3.7 - -3.7 4.2 - -4.2 5.2 - -5.2 MED ---MED SEA ---SEA THI ---THI BO ---BO 60 kpb 230 kpb DUCH ---DUCH 35 kpb 30 kpb Mutaciones Puntuales causantes de -Talasemia - 70 kpb 5’HVR HS-40 0 kpb 10 kpb 2 inter HVR 1 1 2 20 kpb 2 1 30 kpb 3’HVR 1 1) Procesamiento del ARNm Mutaciones en el sitio de Splicing CAP Señal POLI A 1 31 32 99 100 Señal de Poliadenilación 141 5' 3' CAT ATA box box 2) Traducción del ARNm Mutaciones en el codon de iniciación Mutaciones en el codon de terminació Nco Hph T Saudí CS Cambio del marco de lectura Mutación sin sentido 3) Inestabilidad Postranscripcional Secuenciación: nueva mutación IVS2-142 GEN 22 Patient: cctgggccgcactgaccctcttctctgcacaactc ||||||||||||||||||||||||||||||||||| Normal sequence: cctgggccgcactgaccctcttctctgcacagctc Alfa talasemias. Genotipo Hg g/dl Hties X1012/l VCM fl HCM pg Hb H % 15.5 / 14.0 5.20 / 4.60 90 30 0 14.3 / 12.6 5.42 / 4.88 81.2 26.2 0 14.5 / 12.5 5.76 / 5.21 75.5 24.8 0 13.9 / 12.0 5.98 / 5.30 71.6 22.9 0 13.7 / 12.1 6.28 / 5.65 69.1 21.7 0 12.3 / 10.6 5.78 / 5.10 66.1 21.0 0 11.2 / 9.9 5.82 / 5.21 60.5 18.9 10.5 11.1 / 9.4 6.10 / 5.14 64.8 19.1 7.0 10.5 / 8.5 5.10 / 4.68 68.0 18.7 22.3 Alfa talasemias. Deleción alfa 3,7 ó 4,2 Talasemia alfa Sìndrome Genotipo Clínica RN Despues del 1° año Hidrops Fetal - - / - - Muerte fetal Anemia grave HbBart>80% HbH HbPortland Enfermedad por Hb H - - / -a Anemia Hemolitica crònica Hb Bart 20 a HbH 5-20% 40% Trazas de Bart Talasemia Menor -- /aa -a / -a Anemia leve O inexistente HbBart 220% Portador silente -a /aa Sin alteraciones Clínicas ni hematologicas Ninguna Enfermedad de la Hb H Enfermedad de la Hb H Alfa Talasemias - -/- - Alfa talasemia mayor Alfa Talasemias - -/- - Alfa talasemia mayor Alfa Talasemias Diagnóstico Hemograma: cursan sin anemia pero el VCM está siempre disminuído, aún en la deleción 3,7 heterocigota. La morfología es variable y característica para algunas formas de Th. Sólo las formas -/ - y las - -/ presenten morfología más notoria, sin PB, y a veces es posible detectar un cuerpo de inclusión cada varios campos. En los casos leves es imprescindible efectuar la biología molecular, ya que el estudio de Hb generalmente es normal Deleción -3.7 ASO-PCR 1,76 kpb 0,5 kpb 1,76 kpb 0,5 kpb 1 3 4 6 y y y y 2 8 5 7 control Heterocigosis - marcador 100 bp ladder Cordón homocigota - 3.7 Padre - 9 y 10 Madre. 3.7 Alfa Talasemias. -/- homocigota + - -/ heterocigota º Enfermedad Por Hb H Enfermedad Por Hb H: --/- PARÁMETROS HEMATIMÉTRICOS Hb Hb H g/dl % G.R. Hb F mm3 % Hcto % (fl) (pg) 6,6 3.68x106 23 62 17,9 16 VCM HCM MORFOLOGÍA ERITROCITARIA 1,8 % CHCM (g/dl) 28.9 Hb A2 % 0,8 ENFERMEDAD POR Hb H Beta-Talasemia ASOCIACIÓN DELECIÓN -3.7 DE -TALASEMIA HETEROCIGOTA GR Hb Hto VCM HCM CHCM (1012/l) (g/dl) (%) (fl) (pg) (g/dl) 4.9 11,2 37 75 22,8 30,3 039 / -3,7 1 2 3 4 5 6 1,76 KB CON Muñoz Rojas et. al. Protocolo diagnóstico anemias microcíticas Medicine 2008; 10(20):1363-5 ABORDAJE MÉDICO (Unidades de Atención a pacientes con Talasemia) Laboratorio Convencional Investigació Investigación Banco de Sangre HEMATÓLOGO Diagnóstico Tratamiento y Prevención Transfusió Transfusión Tratamiento con Desferal u otros medicamentos Vacunas Apoyo Médico Enfermeras Especializadas Dietista Dentista MEDICOS ESPECIALISTAS (Tratamiento y prevenció prevención) Endocrinó Endocrinólogo Cardió Cardiólogo Oftalmó Oftalmólogo Psicó Psicólogo Genetista (Asesoramiento Gené Genético) CIBO-GENETICA 2004 MUCHAS GRACIAS