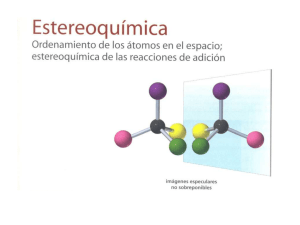
reactivos quirales
Anuncio

ESTRATEGIAS BASADAS EN LA ESTEROQUÍMICA ¿SE DEBERÍA CONSIDERAR LA ESTEREOQUÍMICA? Por razones prácticas y estéticas, ahora es práctica común planificar la síntesis de tal manera con el fin de producir una OM (TGT) enantioméricamente puro (o enriquecido). Esto se ha convertido en una necesidad virtual en los laboratorios de investigación farmaceútica, en donde la estereoquímica es el denominador común entre la química y la biología. Hanessian, S. Pure & Appl. Chem. 1993, 1189. Alrededor del 80% de los compuestos activos que las empresas farmacéuticas tienen en proyecto son quirales, y se estima que esta fracción se incrementará ya que el desarrollo de compuestos activos sigue mejorando. Las autoridades competentes para el registro de nuevos compuestos activos demandan cada vez síntesis selectivas de un estereoisómero. También se están utilizando, cada vez más, compuestos enantioméricamente puros en los productos en la industria agroquímica. La síntesis específica del enantiómero activo puede mejorar la economía del proceso y conducir a la reducción de las cantidades solicitadas y por lo tanto a una reducción del impacto ambiental. Hauer, B. Angew. Chem. Int. Ed. 2004, 788 Hay básicamente tres estrategias principales para adoptar cuando se considera la síntesis de un enantiómero puro molécula : 1) Resolución de un compuesto final racémica o un intermedio 2) Uso de un material de partida enantioméricamente puro, que se puede obtener por resolución, un proceso asimétrico o confiando en la "piscina quiral" 3) A través de una síntesis asimétrica La quiralidad es una propiedad dimensional en el sentido de que se refiere a la orden de dimensión. Un objeto puede ser quiral en uno, dos y tres dimensiones del sistema. Un sistema quiral en una dimensión es aquiral o proquiral en una dimensión de orden superior . espejo espejo Objeto quiral en el sistema unidimensional . aquiral en sistemas bi o tridimensionales objeto quiral en el sistema bidimensional . aquiral o proquiral en los sistemas tridimensionales espejo Objeto quiral en el sistema tridimensional . aquiral o proquiral en sistemas de dimensiones superiores La Naturaleza produce una enorme variedad de compuestos quirales. A menudo cada enantiómero tiene muy diferentes efectos, propiedades y usos Se debe controlar la estereoquímica Nicotina Toxina / estimulante L-alanina Aminoácido esencial en mamíferos D-alanina Aminoácido pared celular de bacterias (-)-Propanolol b-bloquedor para enfermedades del corazón Etambutol Tuberculostático (anti-TB) (+)-Propanolol contraceptivo Enantiómero (R,R) causa ceguera Aproximadamente un tercio de los productos farmacéuticos son quirales; 90 % de los medicamentos que más se venden (top 10), el ingrediente activo es quiral Nombre Ventas globales 2003 (Billones de $ usa) Ingrediente activo Forma de los ingredientes activos Efecto terapeútico Lipitor 10.3 Astrovastatina Enantiómero Agente que disminuye lípidos Zocor 6.1 Simvastatina Enantiómero Agente que disminuye lípidos Ziprexa 4.8 Olanzapina Aquiral Agente sicotrópico Norvaso 4.5 Amlodipina Racemato Bloqueador de los canales de calcio Procrit 4.0 Epoetina Proteína Estimulante de la producción de células rojas Prevacid 4.0 Lansoprazol Racemato Inhibidor de la secreción gástrica Nexium 3.8 Esomeprazol Enantiómero Inhibidor de la secreción gástrica Plavix 3.7 Clopidogrel Enantiómero Inhibidor de la agregación de plaquetas Advair 3.7 Salmeterol Racemato Broncodilatador b-adrenérgico Enantiómero Agente antiinflamatorio Enantiómero Inhibidor de la retoma de serotonina Fluticasona Zoloft 3.4 Total 46.3 Sertalina Atorvastatina Amlodipine Sertralina Simvastatina Lansoprazol Fluticasona Olanzapina Clopidogrel Top Ten Drugs by 2016 proprietary name Generic Name Company Small Molecular Weight (conventional chemical) 1 Crestor rosuvastatin AstraZeneca 2 Advair/Seretide Salmeterol /fluticasone GlaxoSmithKline High Molecular Weight (biopharmaceuticals) 1 Humira adalimumab Abbott 2 Enbrel etanecerpt Amgen 3 Prolia denosumab Amgen 4 Rituxan rituximab Roche/Biogen Idec 5 Avastatin bevacizumab Roche 6 Herceptin trastuzumab Roche 7 Remicade infliximab J&J/Merck & Co. 8 Lantus insulin glargine Sanofi-Aventi Crestor usado para disminuir el colesterol y prevenir enfermedades cardiovasculares Fluticasonas/almeterol Manejo de astma y la enfermedad crónica obstructiva de los pulmones El objetivo directo de las estrategias de estereoquímicas es la reducción de la complejidad estereoquímica por la eliminación retrosintética de elementos estereogénicos en una OM (TGT). La complejidad de la estereoquímica depende en el número de elementos estereogénicos presentes en una molécula y su ubicación espacial y topológica relativas entre sí. Elemento estereogénico es el origen de estereoisomerismo (centro estereogénico , eje o plano) en una molécula de tal manera que el intercambio de dos ligandos (es decir 1 y 2 ) unido a un átomo en una molécula tal conduce a un esteroisomero diferente Estereoisómeros - isómeros que difieren sólo por la disposición de los sustituyentes en el espacio Elemento estereogénico- el origen de estereoisomerismo, ya sea un centro estereogénico, eje o plano, dentro de la molécula. El cambio de dos sustituyentes sobre este elemento conduce a diferentes estereoisómeros Compuesto quiral - simplemente una molécula (o un objeto) que no se pueden superponer sobre su imagen especula. La quiralidad es una propiedad de todo el objeto y no de una parte de ella. El ejemplo más obvio es nuestras manos Imagen especular Manos derecha e izquierda No superponibles Molécula con un único estereocentro o estereounidad, se tolera la antigua definición de centro quiral. En una estructura tetraédrica (Xabcd) o piramidal trigonal (Xabc), el átomo X a la que los cuatro (o tres, respectivamente) abc(d) sustituyentes se unen. Los pares de electrones libres son considerados como un sustituyente con la más baja prioridad d estereocentro OH Unidades estereogénicas diferentes a carbono: átomos de N, S y P FÓSFORO Definición de configuración absoluta: 1) Definir las prioridades de acuerdo a CIP 2) Coloque la prioridad más baja (4) lo más alejado del observador 3) Dibuje una línea de 1 a 3 4) Si el sentido de 1 a 3 es de acuerdo al movimiento de las manecillas de un reloj, el descriptor es (R). En caso contrario es (S) Óxido de (S)-(4-metoxifenil)metilfenilfosfina NITRÓGENO Nitrógeno / aminas tienen el potencial de ser quirales. Sin embargo en los N tetraédricos se presenta una inversión piramidal rápida, lo que normalmente evita el aislamiento de cualquiera de los dos enantiómeros. Si hay dentro de la estructura sustituyentes que limitan la inversión, e.g. un anillo, que implica tener una estructura rígida, se evita la inversión como en el caso de la base de Troger . Base de troger El fósforo (III) presenta una configuración trigonal piramidal, la cual es estable por debajo de 200 °C (S)-ciclohexil-(4metoxifenil)(metil)fosfina El Fósforo (V) tetraédrico tiene una configuración estable ter-butil(fenil)fosfinato de (S)naftalen-1-ilociclohexil-(4metoxifenil)(metil)fosfina Los sulfóxidos de ésteres tiosulfinicos y sulfinamidas tienen un átomo de azufre tetraédrico que posee un par de electrones libre como sustituyente. Tienen una configuración estable a temperatura ambiente pero ciertos aniones (e.g. cloruro), pueden causar la racemización (interconversión entre los enantiómeros ) (R)-1-metil-4(metilsulfinil)benceno (R)-S-ter-butil-2metilpropano-2sulfinotioato (S)-N-bencilidén-2metilpropano-2sulfinamida Las moléculas quirales con elementos de simetría de primer orden (eje de rotación simple) Moléculas con simetría C2, es decir, sólo con un eje de simetría C2 Compuestos espiro espejo Eje C2 oleano (R)-1,7-dioxaespirop[5.6]undecano oleano (S)-1,7-dioxaespirop[5.6]undecano Eje quiral en las definiciones de CIP Moléculas con simetría C2, es decir, sólo con un eje de simetría C2 Eje C2 atropoisomerismo (S)-2-(difenilfosfino)-1-(2-(difenilfosfino)naftalén-1-il)naftaleno Otros sistemas quirales Sistema Helicoidal: moléculas trenzadas (como un sacacorchos) La hélice diestra se denota P (hacia la derecha a medida que se van alejando del espectador) y M para la hélice zurda Quiralidad hélicoidal en las definiciones de CIP espejo P-[8]heliceno M-[8]heliceno Compuestos organometálicos quirales: la quiralidad se da como resultado de la disposición de los grupos fuera de plano con respecto a un plano Quiralidad planar en las definiciones de CIP espejo (S)-2-fenil-1(difenilfosfanil)ferroceno (S)-2-fenil-1(difenilfosfanil)ferroceno Ceir = cheir, mano El científico francés Dominique François Jean Arago observó que la luz que se refleja de un cristal bajo un cierto ángulo tiene propiedades especiales. Experimentos posteriores lo convencieron de que estas propiedades provenían porque la luz es una onda transversa. El nombró a estas propiedades polarización. Campo eléctrico Campo magnético En 1816 Jean-Baptiste Biot comprobó que ciertas sustancias de origen natural como "el aceite esencial del laurel" hacía "girar la luz de derecha a izquierda, al igual que la trementina (aguarras)" mientras que, por el contrario, "el aceite esencial del limón y la disolución de alcanfor en alcohol" lo hacían "de izquierda a derecha" Las hojas de laurel contienen un aceite esencial cuyos principales componentes son el cineol y el eugenol cineol eugenol Propiedades de la luz • Luz Polarizada – La luz ordinaria se encuentra vibrando en muchos planos Campo eléctrico Onda eléctríca Campo magnético Onda magnética Dirección del movimiento del rayo de luz TARTRATO DOBLE DE AMONIO Y SODIO Actividad Óptica La luz que esta restringida para pasar a través de un plano, está polarizada en un plano La luz polarizada en un plano que pasa a través de soluciones de compuestos aquirales, permanece en ese plano Las soluciones de compuestos quirales rotan el plano de la luz polarizada y se dice que las moléculas son ópticamente activas Este fenómeno fue descubierto por Biot a principios del siglo XIX Actividad Óptica La luz pasa a través de un plano del polarizador El plano de la luz polarizada se rota en soluciones de compuestos con actividad óptica Esto se mide con un polarímetro La rotación específica, en grados, es [] Una rotación en el sentido de las manecillas de un reloj análogico se denomina dextrorrotatoria Una rotación en el sentido contrario a las manecillas de un reloj, es levorrotatoria Medición de la rotación óptica • Un polarímetro mide la rotación del plano de la luz polarizada que ha pasado a través de una solución • La fuente pasa a través de un polarizador y luego es detectada en un segundo polizador, el analizador • El ángulo entre los planos de entrada y de salida es la rotación óptica Luz no polarizada Luz polarizada Fuente luminosa Polarizador Tubo con la muestra conteniendo moléculas orgánicas Analizador Observador Un Polarímetro Simple • Mide el grado de rotación del plano de la luz polarizada • El operador alinea el analizador de la polarización y mide el ángulo entre la luz que entra y la luz que sale Polarizador Prisma de Nicol (se encuentra fijo) Tubo conteniendo la solución Analizador Prisma de Nicol (movible) Luz lámpara de sodio Vernier con lectura microscópica Escala graduada Prisma de Nicol: En 1828, el fabricante de instrumentos escocés William Nicol (1768-1851) ideó los prismas que acabaron siendo conocidos con su nombre. Se trataba de dos porciones de espato de Islandia, una variedad incolora de la calcita CaCO3.H2O, unidas por una de sus caras con bálsamo de Canadá Rotación Específica • Para tener una base de comparación, se define la rotación específica, []D para un compuesto ópticamente activo • []D = rotación observada /(longitud del paso x concentración) = /(l x C) = grados/(dm x g/mL) • La rotación específica es la que se observa para una concentración de 1 g/mL en solución en una celda con 10 cm de paso usando la luz de una fuente de vapor de sodio metálico (589 nanómetros) Rotación específica • • • • E Notación (+) y (-) Dextrorrotatoria (+) Levorrotatoria (-) (): mezcla racémica de los enantiómeros El Er El Descubrimiento de Pasteur de los Enantiómeros (1849) • Louis Pasteur descubrió que las sales de amonio del ácido tartárico cristalizan en formas dextrógiras y levógiras Exceso enantiomérico 90 % (+) enantiómero 10 % (-) enantiómero 10 % levógiro 90 % dextrógiro 50 % (+) enantiómero 50 % (-) enantiómero 50 % levógiro 50 % dextrógiro 10 % del enantiómero principal se “cancela” 50 % del enantiómero principal se “cancela” 80 % de rotación máxima es observada 80% e.e. 0 % de rotación máxima es observada 0% e.e. Racemato ( mezcla racémica ) - mezcla 1 a 1 de enantiómeros ( 50 % de cada uno) racemización - la conversión de 1 enantiómero de una mezcla 1: 1 de enantiómeros Polarímetro mide diferencia en la cantidad de cada enantiómero. Nuevos métodos más fiables y pureza medidos en términos de exceso enantiomérico (e.e.) Exceso enantiomérico (% ee) = Compuestos con dos centros estereogénicos Una molécula con 1 centro estereogénico existe como 2 estereoisómeros o enantiómeros Los enantiómeros tienen propiedades físicas idénticas en un entorno aquiral) Una molécula con 2 o más centros estereogénicos puede existir como 4 o 2n estereoisómeros Los enantiómeros (imágenes especulares) todavía tienen propiedades físicas idénticas. Los diastereoisómeros (imágenes no-especulares) tienen propiedades diferentes enantiómeros Epóxido trans p.f. = 98 oC diastereoisómeros p.f. diferente ¿? enantiómeros Epóxido cis p.f. = 98 oC Compuestos con dos centros estereogénicos Los enantiómeros difieren sólo por su estereoquímica absoluta (R o S etc ) y los diastereoisómeros difieren en su estereoquímica relativa. Estereoquímica relativa - define la configuración con respecto a cualquier otro elemento estereogénico dentro de la molécula, pero no diferencia enantiómeros. Quiral Solubilidad 0.1 g/100 mL EtOH meso Solubilidad 3.3 g/100 mL EtOH Una molécula sólo puede tener un enantiómero pero cualquier número de diastereoisómeros. Las propiedades físicas diferentes de los diastereoisómeros nos permiten purificarlos Compuestos meso La regla 2n da el número máximo de estereoisómeros pero en el caso especial de los compuestos meso, el número de posibles estereoisómeros es menor Ácido tartárico Plano de simetría Un compuesto meso es una estructura aquiral de una serie de diastereoisómeros que también incluye al menos un miembro quiral. En forma simplificada se puede decir que es una molécula que contiene al menos un centro estereogénico pero que tiene un elemento de simetría de segundo orden (plano de simetría) por lo que es aquiral Los compuestos meso tienen un plano de simetría que divide la molécula en dos subunidades (cada una con estereocentros) que cada una es la imagen especular de la otra, una con configuración (R) en un lado y la otra con configuración (S) en el otro lado Eje C2 Quiral No hay plano de simetría Forma imágenes especulares no superponibles pero debido a la presencia del eje de simetría C2 es una molécula asimétrica espejo Aquiral hay plano de simetría forma imágenes especulares superponibles: compuesto meso La diferencia en los diastereómeros permite que los agentes de derivatización quirales puedan resolver enantiómeros. Recuerde que un buen agente quiral de derivatización deberá: • Ser enantioméricamente puro (o no tendrá sentido usarlo, será inútil) • La reacción de acoplamiento de ambos enantiómeros debe alcanzar el 100% (si está midiendo ee) • Las condiciones de acoplamiento no deben epimerizar los centros estereogénicos Los enantiómeros deben contener punto de unión La lista anterior es probable que este influenciada dependiendo de si usted está midiendo % ee o separando los enantiómeros a escala preparativa Agente de derivatización enantioméricamente puro Mezcla racémica Enantiómero puro Mezcla de diatereoisómeros Diatereoisómeros separables Ruptura diastereoisómero Diastereoisómero puro Los agentes de derivatización más usados para alcoholes y aminas son el ácido α-metoxi-αtrifluorometilfenilacético (APTM) o ácido de Mosher ácido α-metoxi-α-trifluorometilfenilacético (MPTA) (ácido de Mosher) Una diferencia típica en los desplazamientos químicos de RMN-1H 0,15 ppm y RMN-19F da una señal para cada diastereoisómero No poseen α-hidrógeno de modo que tienen una configuración estable Los diastereoisómeros se pueden separar con frecuencia En muchos casos el uso de ambos enantiómeros de MTPA se puede utilizar para determinar la configuración absoluta de un estereocentro DCC = DICICLOHEXILCARBODIIMIDA Diferencia en las señales de RMN entre los diastereoisómeros: RMN-1H Dd = 0.08 (CH3) RMN-19F Dd =0.17 (CF3) (JACS, 1973 , 512 , JOC 1973 , 2143 y JACS 1991 , 4092) Formación de sales iónicas diastereoisoméricas • No hay necesidad de unir covalentemente un grupo de derivatización quiral • Beneficio - normalmente es más fácil de recuperar y reusar el agente • El uso de interacciones no covalentes permite otros métodos de resolución de enantiómeros El diastereoisómero S es insoluble. Se separa por medio de una filtración (-)-propanolol b-bloqueador Resolución de enantiómeros: cromatografía sobre columna quiral Resolución - la separación de enantiómeros de una mezcla cualquiera racémica o mezcla enantioméricamente enriquecida Cromatografía quiral - Normalmente HPLC o GC Una solución racémica se hace pasar sobre una fase estacionaria quiral Compuesto tiene una interacción diastereotópica, rápida y reversible, con la fase estacionaria Se espera que cada complejo tiene una estabilidad diferente que permite la separación Coincidencia Enantiómero – más estable (3 interacciones) Mezcla racémica en disolución Fase quiral estacionaria No hay coincidencia Enantiómero – menos estable (1 interacciones) Coincidencia Enantiómero – eluye más lentamente (hay retención) No hay coincidencia Enantiómero – eluye con facilidad (no hay retención) Resolución de enantiómeros: cromatografía sobre columna quiral Las mediciones de ee por HPLC o GC son rápidas y precisas ( ± 0,05 %) Fase estacionaria quiral sólo puede trabajar un tipo limitado de compuestos Las columnas son caras ( > € 1500) Se necesitan los dos enantiómeros para configurar un método preciso Se introduce la Mezcla racémica en disolución a la columna Columna quiral preparada a partir de una fase estacionaria quiral apropiada (hay de varios tipos diferentes) sílica Amina quiral fase estacionaria quiral Espectroscopía de RMN: reactivos de desplazamiento quiral Complejos de lantánidos paramagnéticos quirales pueden unirse reversiblemente a ciertas moléculas quirales través del centro metálico. El compuesto debe tener una base de Lewis con un par de electrones libre (OH, NH2, C=O, CO2H. etc). El proceso de coordinación debe ser más rápido que escala de tiempo de la RMN, es normal observar un desplazamiento a campo bajo (d ppm alto) Dos complejos diastereoméricos se forman en la coordinación; éstos pueden tener diferente señal en RMN. PROBLEMAS - como complejos son paramagnéticos, se observa un ensanchamiento de la línea (sobre todo en equipos de campo alto). La precisión es de sólo ± 2 % sustrato sustrato Se están desarrollando nuevos reactivos los cuales pueden superar algunos de estos problemas Los espectros de RMN 1H (400 MHz) de valina (0,06 M , [D ]/[ L ] = 1/2.85) en D2O a pH 9.4 y en el presencia de complejos de samario. La señal no muestra un ensanchamiento paramagnético. Relación experimental estimada D/L: 1/3.02 vs 1/2.85 Sin reactivo L-Valina Desimetrización: proceso que transforma un objeto simétrico o proquiral en uno no simétrica o bien en uno quiral Desde un punto de vista sintético, la introducción de nuevos centros estereogénicos en una OM (TGT) es normal conseguirla por medio de dos procesos fundamentalmente distintos: 1) Es más común a través de la adición a una u otra de las caras estereoheterotópicas de un doble enlace (enantio- o diastereotópicos) 2) También por la modificación selectiva o la sustitución de ligandos estereoheterotópicos Objeto simétrico conteniendo un elemento de simetría de orden II (plano conteniendo 1, 2, 3 y carbono Objeto simétrico conteniendo un elemento de simetría de orden II (plano conteniendo 1, 2 y carbono sustitución adición carbono carbono Quiral en un sistema bidimensional Proquiral en un sistema tridimensional Quiral en un sistema tridimensional Quiral en un sistema bidimensional Proquiral en un sistema tridimensional Sustrato: estereocontrol debido a una característica estereoquímica del sustrato El resultado estereoquímico de una amplia gama de reacciones no esta controlado por cuestiones mecanísticas. Por el contrario, depende de la estructura del sustrato o reactivo. La generación de un nuevo estereocentro puede ser controlado por el factor estérico de estereocentros preexistentes. Este tipo de estereocontrol es frecuente en las estructuras cíclicas, conformacionalmente no flexibles. En sistemas acíclicos, la situación es mucho más complicada, teniendo en cuenta que los nuevos estereocentros suelen ser creados por la adición a un carbono sp2, se puede lograr un estereocontrol alto si la molécula adopta una conformación reactiva definida en la que una de los dos diastereocaras está protegida de una manera eficiente por efectos estéricos de los sustituyentes: 1) En forma pasiva por protección estérico de una o las dos caras diastereotópicas en el centro reactivo. 2) En forma activa mediante la unión del reactivo en forma de interacciones no covalentes y dirigiéndolo hacia una de las caras diastereotópicas Los efectos estérico y estereoelectrónicos juegan un papel crucial para diseñar un potente análisis retrosintético. Aspectos conformacionales que deben ser considerados Sistema acíclicos Sistema cíclicos ¿Cuál es la conformación más estable? ¿Cuál conformación es la más reactiva? Interacciones sin-pentano Se deben evitar Tensión Alílica Se deben evaluar Efectos anoméricos Son recompensados También se deben de considerar: interacciones ácido – base de Lewis, coordinación (quelación), puentes de hidrógeno Efecto estereoelectrónico: es cualquier efecto que determina las propiedades o la reactividad de una especie que depende de la orientación de los orbitales electrónicos (llenos o vacíos) en el espacio. Deslongchamps, P. Stereoelectronic Effects in Organic Chemistry Repulsión débil pares de electrones Más estable Repulsión media pares de electrones Repulsión fuerte pares de electrones Menos estable Mecanismo: transformadas intrínsecamente estereocontroladas Hay reacciones que muestran estereoselectividad principalmente a causa de mecanismo: procesos SN2, hidroboración, epoxidación, OsO4 en la oxidación de alquenos etc … Son especialmente importantes las desconexiones que involucran enlaces C -C La estereoquímica de bis-epóxido controla el resultado final en cuanto a la estereoquímica Estereoselectividad en síntesis orgánica Reacción estereoespecífica - una reacción donde el mecanismo y la estereoquímica del material de partida determinar la estereoquímica del producto; no hay otra opción. Por ejemplo Reacciones SN2 . Reacción diastereospecifica – permite que solo se forme un diastereoisómero, control estereoquímica relativa no absoluta. Por ejemplo, la reacción de yodolactonisación transcurre a través de una especie cíclica yodonio, seguida de una reacción de apertura de anillo intramolecular. La geometría del alqueno controla la estereoquímica relativa. Si hay un centro estereogénico pre-existente, entonces la reacción puede ser estereoselectiva. En tales reacciones se podrían formar dos diastereoisómeros, pero solo uno se ve favorecido anti sin I2 Posición no favorecida carbocatión 2o 5-exo-tet favorecido 6-endo-tet desfavorecido Posición favorecida carbocatión 3o Único producto No se forma Cis, producto cinético trans, producto termodinámico Reacciones estereoselectivas - una reacción en la que se forma el estereoisómero de un producto preferentemente sobre otro. El mecanismo no impide la formación de dos o más estereoisómeros , pero uno predomina. Reacciones diastereoselectiva - un centro estereogénico se introduce en una molécula, de tal manera que se producen diastereoisómeros en cantidades desiguales Reacciones enantioselectivas - una reacción que produce dos enantiómeros de un producto, en cantidades desiguales Reacciones estereoselectivas Adición nucleofílica a C = O y Nomenclatura proquirales Los carbonos trigonales que no son unidades estereogénicas pero pueden transformarse en ellos, se llaman proquiral, a cada cara del carbonilo se le asigna la etiqueta Si o Re basado en las normas de la CIP. Si la función carbonilo está en una molécula quiral se llama unidad de proestereogenica y las caras se dice que son diastereotópicas. Observar desde esta cara Sentido del giro de las manecillas Cara Re Observar desde esta cara Sentido contrario al giro de las manecillas Cara Si En el caso de moléculas aquirales las caras de carbonilo se nombran enantiotópicas y la adición de nucleófilos a la función carbonilo puede ocurrir con enantioselección o la reacción es enantioselectiva si una cara proquiral se une preferentemente sobre la otra. La reacción de un nucleófilo con un carbonilo en un sustrato donde otros están presentes otros estereocentros, da dos posibles diastereoisómeros. La reacción es estereoselectiva si un diastereoisómero predomina sobre el otro Posibles modelos propuestos a lo largo del tiempo para aproximación de un nucleófilo a la función carbonilo con un estereocentro en Traslapo máximo con el ataque p* - nucleófilo a 90º al grupo C=O Modelo acíclico de Cram (1952) Estérico Modelo rígido de Cram (1959) Quelato Modelo acíclico de Cornforth (1959) Electrostático Modelo de Karabatsos (1967) Estado basal, estérico Modelos propuestos con un enfoque perpendicular de la adición de Nu al grupo carbonilo Los nucleófilos atacan al grupo carbonilo a lo largo del ángulo Burghi-Dunitz de ~107 ° Repulsión del orbital p lleno - nucleófilo ataque a través de un ángulo obtuso El ángulo Burghi - Dunitz (107 °) es el compromiso entre la interacción electrostática y la superposición optimizada de los orbitales Compromiso, ataque nucleófilo orbital p* en un ángulo de 107 o Posteriormente otros modelos han sido propuestos con la trayectoria Burghi-Dunitz del Nu al grupo carbonilo Modelo Felkin-Anh (1977) Estérico, torsional, BürghiDunitz Modelo Felkin-Anh polar (1977) Electrónico, torsional, Bürghi-Dunitz Posteriormente otros modelos han sido propuestos con la trayectoria Burghi-Dunitz del Nu al grupo carbonilo Modelo Cieplak (1981) Electrónico, torsional, Bürghi-Dunitz Modelo Evans (2001) Electrostático, torsional, Bürghi-Dunitz Importancia del análisis conformacional Proyección de Newman: dos sustituyentes (C=O y Ph) están eclipsados - desfavorecido Otros posibles confórmeros: dos favorecidos por que el sustituyente más grande (Ph) se encuentra alejado de O y H Cara Re Uno de los confórmeros más estables por que el sustituyente más grande (Ph) se encuentra alejado de O y H Cara Si Uno de los confórmeros más estables por que el sustituyente más grande (Ph) se encuentra alejado de OyH Modelo Felkin-Anh (1977) Estérico, torsional, Bürghi-Dunitz Modelo Felkin – Ahn En la conformación más estable, la aproximación preferida es por donde se encuentra el sustituyente más pequeño (H) Bürghi-Dunitz Ángulo: 107 o Aproximación sin impedimento Cercano al Ph Cercano al Ph Cercano al Me Características Generales para la adición a un carbonilo con un estereocentro: Modelo Felkin-Ahn Para explicar o predecir la estereoselectividad de la adición nucleofílica a un grupo carbonilo con un centro estereogénico adyacente, se puede utilizar el modelo Felkin-Ahn: • Dibujar la proyección de Newman con el sustituyente mas grande (L) perpendicular al C = O; el Nucleófilo (Nu) atacará a lo largo de la trayectoria Bürghi-Dunitz pasando por el sustituyente estéricamente menos demandante (sustituyente S). • Dibuje la proyección de Newman del producto. • Redibujar la molécula en la representación normal. • Mientras que el modelo Felkin-Ahn predice la orientación de ataque, no da ninguna información sobre el grado de selectividad pero si sobre cual será el estereoisómero predominante. • El tamaño del nucleófilo afecta en gran medida la diastereoselectividad de la adición: los nucleófilos más grandes generalmente darán lugar a mayores diastereoselectividades. • La elección del metal también afectará la selectividad, aunque este también puede ser un efecto estérico. • El tamaño de los sustituyentes sobre el sustrato también afectará la diastereoselectividad. Una vez más, los grupos más grandes darán como resultado una mayor selectividad. • Cabe señalar que sustituyentes más grandes normalmente dan como resultado una menor rapidez de reacción. El efecto de átomo electronegativo en a un grupo El impedimento estérico no es el único factor que justifica la elevada estereoselectividad observada y una reacción más rápida en la adición de enolatos de éster a aldehídos -amino sustituidos. Otros factores, como los electrónicos, pueden desempeñar una regla importante como en el caso de aldehídos -dibencilamino sustituidos. El grupo Bn2N debe estar perpendicular a C = O ya que de esta manera hay una mejor interacción entre el CN y el doble enlace del grupo carbonilo. Aplicando el modelo Fenkin-Ahn, la aproximación del enolato se ve favorecida por el sitio opuesto al Bn2N (repulsión electrónica entre un grupo rico en electrones como el ion enolato y el par libre del grupo Bn2N ) . Modelo Fenkin-Ahn Cuando un grupo electronegativo es perpendicular al grupo C = O es posible conseguir un traslapo del orbital p* orbital y el orbital σ*, lo cual se traduce en un nuevo orbital molecular con una energía menor, más susceptible a un ataque nucleofílico, por lo que si el grupo electronegativo esta perpendicular, el grupo C=O, este será más reactivo Z = grupo electronegativo El nucleófilo interacciona con el orbital p* Nuevo orbital LUMO p* + s* Nuevo orbital LUMO p* + s* El nuevo orbital de energía más baja se forma a partir de los obitales de antienlace C=O y C-Z con lo que se favorece el ataque nucleofílico sobre el C=O El efecto de un átomo electronegativo y control por formación de quelato Otro ejemplo de control por efecto electrónico ataque Fenkin-Ahn El efecto de un átomo electronegativo y control por formación de quelato Si el heteroátomo (Z) es capaz de coordinarse y si esta presente un metal es capaz de formar quelato con 2 heteroátomos se observará un control por formación de quelato. En los quelatos metálicos carbonilo y heteroátomo que se fijan juntos, su conformación proporcionan una mayor selectividad y la reacción ocurre con una mayor rapidez. El metal que se quelata actúa como un ácido de Lewis y activa al grupo carbonilo que va a ser atacado. La formación de un quelato puede revertir la selectividad. Adiciones controladas por formación de quelato son fáciles de predecir y normalmente no es necesario dibujar una proyección de Newman El nucleófilo ataca por la cara menos impedida Z = heteroátomo capaz de coordinarse M = metal capaza de coordinarse con más de un heteroátomo El efecto de un átomo electronegativo y control por formación de quelato Rotación para permitir la formación del quelato Control de Cram (quelato) Control por formación de quelato en la adición de -carbonilos: otros ejemplos El siguiente ejemplo muestra una selectividad normal de Felkin–Ahn a través de la cual se forma un diastereoisómero. El grupo fósforo (electronegativo y voluminoso) en posición perpendicular, permite el control por fomación de quelato, y se forma el diastereoisómero opuesto y se forma a través de la formación de un anillo de 6 miembros. Las temperaturas de reacción más bajas son típicas en los sistemas carbonílicos quelatados activados Aplicación del modelo Fenkin-Ahn en síntesis total Un ejemplo de la reacción de Sakurai (adición de alilsilano a un grupo carbonilo) a partir de la síntesis de Preswinholida A, la cual es el monómero de Swinholida A (el regulador de luz, aislado de una esponja del Mar Rojo), un compuesto que muestra una potente actividad citotóxica La síntesis total fue reportada por Ian Paterson Preswinholida Tetrahedron,1995,51,9437 La síntesis de Canadensolido, un agente fungicida es otro ejemplo de la reacción del aldol Mukaiyama (adición de éster sililenoléter a carbonilos) Canadensolido Tetrahedron,2006,62,9713 Reacción estereoselectiva de enolatos La estereoselectividad de las reacciones de enolatos depende de la presencia de centros estereogénicos en R1, R2 y frecuentemente sobre la geometría del enolato (pero no siempre) R2 Geometría del enolato: Los términos cis y trans en relación a la disposición del grupo con la más alta prioridad en el átomo de carbono con el enlace -O-M C-, cara si C-, cara re (Z)-enolato (cis) C-, cara si (E)-enolato (trans) C-, cara re Formación de enolato y geometría Enolato – es normal formarlo por desprotonación, esto se ve favorecido cuando el enlace C- H es perpendicular al enlace C=O ya que esto permite que el orbital σ se pueda traslapar con el orbital p orbital. Al final el orbital σ C-H se convierte en un orbital p en el C-α del orbital p del enlace del enolato Orbtal C-H s enolato Orbtal p Orbtal El proceso de desprotonación y la geometría del enolato C=O p Formación de enolato y geometría Dos posibles conformaciones: 1) En la conformación inicial se presenta poca interacción estérica entre R1 y R2 (proyección de Newman) similar al estado de transición que da como resultado la formación de enolato cis Estado de transición Enolato (Z) (cis) Formación de enolato y geometría 2) Segunda conformación: C-H perpendicular a C=O, que difiere en la posición relativa de R1 y R2 y da el enolato trans Enolato (E) (trans) Formación de enolato y geometría La interacción estérica de R1 y R2 da como resultado la formación del enolato cis, el cual normalmente es el predomina, pero la estereoselectividad está influenciada por el tamaño de R Formación de enolato y geometría La selectividad observada se puede explicar a través del estado de transición silla, como el paso de la desprotonación en cetonas en las que se favorece la formación del enolato-cis si R es grande, pero se favorece el enolato-trans si R es pequeño Si R es grande, este ET‡ es desestabilizado por la interacción R-Me y predomina el enolato-cis Si R es pequeño, la interacción 1,3-diaxial es importante ya que desestabiliza este ET‡ y predomina el enolatotrans R = grande R = pequeño Formación de enolato y geometría Con los ésteres, las interacciones R vs OMe se aligera y la interacción 1,3-diaxial controla la geometría del enolato, por lo tanto predomina el enolato-trans predomina Formación de enolato y geometría Las amidas invariablemente dan el enolato-cis; recordar la rotación restringida del enlace C-N. El anterior argumento es una buena generalización, muchos factores tienen efecto sobre la geometría. El uso de la HMPA como aditivo reduce la coordinación y favorece la formación del enolato termodinámicamente más estable predomina En el éster se observa el efecto inverso: predomina Adición electrofílica a un enolato: alquilación Es importante conocer la trayectoria de aproximación del enolato y el electrófilo La reacción es el traslapo de los orbitales HOMO del enolato y LUMO del electrófilo Por lo tanto , la nueva unión que se forma se encuentra más o menos perpendicular al grupo carbonilo. El ejemplo muestra una reacción SN2 sencillo con X = grupo saliente X= grupo saliente Orbital s* (LUMO del electrófilo) Orbital p (HOMO del enolato) Alquilación estereoselectiva (diastereoselectiva) de enolatos proquirales La alquilación de enolatos proquirales de un ácido, está normalmente preformada con el uso de derivados quirales tales como amidas quirales (la aproximación de Meyer, aminoalcohol enantiopuro) o imidas (la aproximación de Evans de oxazolidinonas enantiopuras) Aproximación de Evans A partir de fenilalanina enolato-cis La formación del quelato es importante para la geometría del enolato y para la aproximación del electrófilo, M = Li (JACS, 1990, 112,8215) d.e. > 95 % a 100 %. Después de eliminar el auxiliar quiral, el ácido final se obtiene con un alto e.e. Aproximación de Myers A partir de seudoefedrina d.e. > 94 % Rendimientos > 80 % enolato-cis JACS, 1994, 116, 9361 1995, 117, 8488 ALQUILACIÓN ESTEREOSELECTIVA (DIASTEREOSELECTIVA) DE ENOLATOS QUIRALES La alquilación simple de un enolato quiral ocurre generalmente con muy alta diastereoselectividad. Dado que el enolato-cis se forma normalmente con confórmero reactivo dando una alta diastereoselectividad, el teniendo en cuenta la tensión en el alqueno A(1,3) (A = Alílica) La geometría del enolato no es importante Alquilación por la cara opuesta a R Conformación: C-H paralelo al enlace C=C Más estable Enolato-cis Con los sustituyentes más grandes, R, mayor es la selectividad y el diastereoisómero minoritario probablemente se forma por la aproximación del electrófilo por la posición en donde esta el grupo R y no reacciona con el enolatotrans. Es posible cambiar la diastereoselectividad simplemente usando el protón como electrófilo en el tratamiento final de la reacción para aislar el enolato alquilado REACCIÓN ALDOLICA ESTEREOSELECTIVA (DIASTEREOSELECTIVA) La reacción aldólica es una reacción de formación de C-C muy valiosa. Además, se pueden formar dos nuevos centros estereogénicos de una manera diastereoselectiva. La mayoría de las reacciones aldólicas tienen lugar a través de un estado de transición muy ordenado conocido como Zimmerman–Traxler, el cual es un estado de transición de 6 miembros (silla). Al contrario de la alquilación, los efectos de la geometría enolato presentan diastereoselectividad Enolato-cis Aldol-sin Enolato-trans Solo es posible un enolato Enolato-trans Aldol-anti Aldol-sin Aldol-sin Enolato-cis Aldol-anti REACCIÓN ALDOLICA ESTEREOSELECTIVA (DIASTEREOSELECTIVA) ESTADO DE TRANSICIÓN ZIMMERMAN-TRAXLER Interacción 1,3diaxial Enolato-cis Enolato-cis R pseudoecuatorial R pseudoecuatorial desfavorecido Sustituyentes en el enolato están fijos debido al doble enlace, de este modo la orientación del aldehído en relación con el enolato es crucial para determinar la estereoselectividad final (diastereo y enantioselectividad) en la reacción aldólica. Un aldehído con un sustituyente voluminoso debe estar en una posición pseudoequatorial en el estado de transición Zimmerman-Traxler a fin de evitar las interacciones 1,3-diaxiales Aldol-sin enolato-cis Aldol-sin TS enantiomérico Me y OH apuntan hacia el observador H Cara si del enolato ataca Cara re del aldehído Cara re del enolato ataca Cara si del aldehído El ataque a través del estado de transición enantiomérico (cara re del aldehído) da el producto aldol enantiomérico. Esto difiere sólo por la estereoquímica absoluta pero la estereoquímica relativa es el mismo para el Me y OH en el mismo sitio Para observar la estereoquímica relativa se debe considerar la secuencia de carbonos de color azul en un plano y ver cuales grupos están arriba y cuales están abajo. En este caso Me y OH están lo más alejados del observador Reacción aldólica estereoselectiva (diastereoselectiva) Estado de transición Zimmerman-Traxler enolato-trans. La estereoquímica opuesta del enolato da la estereoquímica relativa opuesta en el producto enolato-trans Cara re del enolato ataca Cara re del aldehído Aldol -anti Visualizando la estereoquímica relativa En este caso los dos H deben ser axiales Reacción aldólica estereoselectiva (diastereoselectiva): geometría enolato En los enolatos de litio de cetonas, el tamaño del sustituyente no enolizado, R, es importante La geometría del enol en cetonas está determinada por el tamaño de R Con enolatos de boro se puede seleccionar la geometría alterando el reactivo de boro utilizado Sustituyentes voluminosos enolato-trans Aldol anti (>90 % de) Los grupos voluminosos en el B forzan a que el enolato adopte la geometria trans 9-BBN (9-borabiciclononano) parece voluminoso, pero la mayor parte tiene la «espalda atada» detrás del boro, permitiendo así la formación del cis-enolato enolato-cis Aldol sin (>98 % de) Control del sustrato en la síntesis total de la aglicona Oleandomicina Cara re favorecida vs. Enolato trans Da el producto aldólico anti Desfavorecida ¿Interacción par iónico? Aglicona Oleandomicina R = H, pero puede ser un azúcar Control de Cram por formación de quelato Control del sustrato en la síntesis total de aglicona Oleandomicina Ataque por la cara re Favorecido vs. Enolato cis Da el producto aldólico sin Las oportunidades que ofrece la reacción aldólica: por reacción se crea un enlace C-C y 2 centros estereogénicos. Enlaces C-C formados por una reacción aldólica Centros estereogénicos formados por reacción aldólica J. Am. Chem. Soc. 1994.116,11287 Reacción Aldolica: control de sustrato en la síntesis total de Swinholida A ESTERIFICACIÓN 2 FGI Reacción aldólica LACTONIZACIÓN FRAGMENTO A FRAGMENTO B FRAGMENTO B Tetrahedron 1995 ,9393-9467 REACCIÓN ALDÓLICA VINÍLOGA FORMACIÓN C=C FRAGMENTO A ENOLATO QUIRAL REACCIÓN ALDÓLICA CICLIZACIÓN 6-endo-trig REARREGLO DE FERRIER REACCIÓN ALDÓLICA FORMACIÓN C=C RESOLUCIÓN CINÉTICA A TRAVÉS DE EPOXIDACIÓN ESTEREOSELECTIVA FRAGMENTO B C-GLICOSIDACIÓN Transposiciónde Ferrier SÍNTESIS ESTEREOSELECTIVA; AUXILIAR QUIRAL Quiral auxiliar - permite la síntesis enantioselectiva mediante una reacción diastereoselectiva . Se adiciona una unidad quiral al sustrato para controlar la reacción estereoselectiva. Puede actuar como un sistema incorporado como un agente de resolución (si reacción no es diastereoselectiva). Problemas: • Se necesita un punto de unión • Se agregan pasos adicionales (economía atómica) • Las condiciones de ruptura no deben dañar al producto ACOPLAR PARA FORMAR SUSTRATO (AQUIRAL) + AUXILIAR QUIRAL UN NUEVO COMPUESTO QUIRAL SUSTRATO (AQUIRAL) AUXILIAR QUIRAL Resolver otros diastereomeros REACCIÓN GLOBAL SUSTRATO (QUIRAL) REACCIÓN DIASTEREOSELECTIVA PRODUCTO QUIRAL Ruptura + AUXILIAR QUIRAL SUSTRATO (QUIRAL) Auxiliar quiral AUXILIAR QUIRAL AUXILIAR QUIRAL Un auxiliar quiral ideal tiene que cumplir varios criterios: i) debe ser barato, y ambos enantiómeros deben ser fácilmente disponibles ii) La unión con del sustrato con el auxiliar debe proceder con alto rendimiento mediante métodos simples, aplicables a una amplia variedad de sustratos iii) debe haber muchos tipos diferentes de reacciones que se puedan llevar a cabo iv) el auxiliar debe ser estable bajo las condiciones de la reacción diastereoselectiva v) debe haber un alto grado de diastereoselección vi) los derivados del auxiliar quiral deberían ser preferentemente cristalinos, lo que permitirá una purificación más fácil, y la separación de otras impurezas diastereoisoméricas mediante una simple cristalización vii) la ruptura del auxiliar debe ser posible con un alto rendimiento en condiciones suaves, y los procedimientos deben ser de aplicación general viii) el auxiliar no debe ser destruido en las condiciones aplicadas para la ruptura, permitiendo así el reciclaje ix) el aislamiento del producto enantioméricamente puro y la recuperación del auxiliar debería ser posible mediante métodos lo más simple posibles Seebach, D. Helvetica Chimica Acta 1998, 2093 Auxiliar quiral y la adición al grupo carbonilo Hay descritos muchos ejemplos de control del sustrato en adición nucleofílica al grupo (Felkin - Ahn y control por formación de quelato). Si la molécula no contiene un centro estereogénico entonces se puede utilizar un auxiliar quiral. El auxiliar quiral se puede retirar en un paso posterior Diastereoisómero opuesto se puede obtener a partir de la reducción de la cetona con una menor diastereoselectividad ... ' H ' es mas pequeño Auxiliar quiral en síntesis El auxiliar quiral, 8-fenilmentol, se ha utilizado para formar la feromona Frontalina. Es una feromona de agregación del escarabajo del pino del sur - el escarabajo más destructivo para bosques de pino en el sudeste de Estados Unidos Frontalina 100 % ee Síntesis estereoselectiva: reactivos quirales Quiral reactivo – la estereoquímica reside inicialmente en el reactivo • Ventajas I. No hay etapas de acoplamiento / pasos de ruptura son requeridos II. Con frecuencia se anula el control del sustrato. III. Puede ser mucho más suave que los auxiliares quirales • Desventajas – I. Se necesita una cantidad estequiométrica (no hay economía atómica) II. Con frecuencia son caros III. Los procedimientos de aislamiento son problemáticos Reactivo quiral SUSTRATO (AQUIRAL) + AUXILIAR QUIRAL Interacciona con el sustrato aquiral SUSTRATO (AQUIRAL) AUXILIAR QUIRAL Complejo quiral reacción PRODUCTO QUIRAL + REACTIVO “MUERTO ” REACTIVOS QUIRALES En general se prefieren los reactivos quirales a los auxiliares quirales, ya que funcionan en forma independiente de la quiralidad del sustrato o sobre sustratos proquirales. Un gran número han sido desarrollados para la reducción de carbonilos. La mayoría implican la adición de un elemento quiral sobre un reactivo estándar alpinoborano (+)--pineno Se puede reusar alpinoborano Grupo pequeño lineal Procede a través de un estado de transición tipo bote La selectividad esta gobernada por interacciones 1,3-diaxiales Derivado Binol de LiAlH4 La reducción con un reactivo a base de BINOL y el hidruro doble de litio aluminio: • La selectividad se cree que proviene de un estado de transición de 6 miembros • El sustituyente más grande (RL) adopta la posición pseudo-ecuatorial y el sustituyente pequeño (RS) es axial para minimizar las interacciones 1,3diaxiales Estado de transición 1,1'-Bi-2-naftol (BINOL) REACTIVO QUIRAL EN SÍNTESIS TOTALES (+)-Ipc2BCl (Diisopinocamfenilborano) es una versión más reactiva, ácido de Lewis de Alpino-borano • Reacción de Mitsunobu (paso 2 ) > 99% ee 1 recristalización R-(-)-fluoxeteno Prozac J. Org. Chem.,1988,53,2916 Síntesis estereoselectiva: catálisis quiral Catálisis quiral - idealmente es un reactivo que acelera una reacción (sin ser destruido) en un ambiente quiral, con lo cual una molécula quiral genera millones de nuevas moléculas quirales. Es frecuente que la reacción se lleve a cabo en sustratos aquirales o bien proquirales (como por ejemplo el grupo funcional carbonilo). Sustrato (aquiral) Sustrato (aquiral) Catalizador quiral Catalizador quiral Producto (quiral) Producto (quiral) Catalizador quiral Reducción catalítica enantioselectiva Un catalizador eficiente para la reducción de cetonas es el catalizador de CoreyBakshi-Shibata (CBS). El reactivo se prepara a partir de un derivado de prolina: difenilpronilol. La enantioselección es determinada por la naturaleza del reactivo quiral y se produce por la formación de estados de transición diastereoisoméricos con diferentes energías. La reacción utiliza ~ 10 % del heterociclo y una cantidad estequiométrica de borano y trabaja en forma más efectiva si hay una gran diferencia entre los dos sustituyentes en la cetona Catalizador CBS Derivado de la prolina Catalizador activo Grupo carbonilo proquiral Catalizador CBS El mecanismo es bastante elegante: este catalizador trae una cetona y borano juntos en un medio quiral • • • Regeneración del catalizador Interacción de amina y borano: activa al borano El borano se posiciona Se incrementa la acidez de Lewis en el boro endo El boro como centro ácido que se coordina con el C=O La coordinación del C=O activa al aldehído y lo coloca cerca del borano Estado de transición tipo silla. El sustituyente mas grande es pseudo-ecuatorial Adición nucleófila catalitica enantioselectiva Hay descritos muchos métodos diferentes para llevar a cabo reacciones enantioselectivas catalíticas. Es conocido que amino-alcoholes simples catalizan la adición de reactivos dialquilzinc a aldehídos a través de un mecanismo que implica una especie bifuncional de zinc, donde un zinc se convierte en el centro ácido Lewis y activa al aldehído y el segundo equivalente del reactivo de zinc en realidad ataca al aldehído. Una vez más un anillo de 6 miembros está involucrado e interacciones 1,3-diaxiales controlan la selectividad observada Interacción 1,3 - diaxial desfavorecido REACCIÓNES ESTEREOESPECÍFICAS 98.4 % ee 66 % ee HIDROGENACIÓN Es otra reacción importante que se puede llevar a cabo bajo enantioselectividad, emplenado un metal como catalizadorl. Un ejemplo bien conocido por su enorme importancia desde el punto de vista industrial, es la hidrogenación catalítica de derivados deshidroaminoácidos (preparados por Knovenagel como la reacción de la glicina). Se utilizan difosfinas como ligandos de rutenio y es esencial que haya un segundo grupo corrdinante (la amida en el deshidroaminoácido). H2 (g) [((S)-DIMAP)RhL2]+ L = disolvente (S,S)-DIMAP En la coordinación, se forman dos complejos diastereoisoméricos. La estabilidad / proporción de cada uno de estos complejos no es importante en la determinación final de la estereoselección, pero si la proporción de coordinación de hidrógeno Cara Re Cara Si Mecanismo propuesto para la hidrogenación catalítica H2 lento Adición oxidativa de hidrógeno Transferencia de hidrógeno Eliminación reductiva Enantiómero menor Eliminación reductiva Enantiómero mayor H2 lento Adición oxidativa rápida Complejo más reactivo Otros sistemas pueden ser hidrogenados con el mismo catalizador quiral: síntesis industrial de Candoxatril Se utiliza en la síntesis de Candoxatril, un potente factor natriurético auricular ( ANF) potenciador (fármaco cardiovascular desarrollado por Pfizer). Proceso utilizado a escala de toneladas Org. Process Res. Dev.,2001,5,438 Reacciones de los alquenos Reacción de epoxidación diastereoespecifica La reacción permite que solo se forme un diastereoisómero. Hay control relativo de la estereoquímica relativa no de la estereoquímica absoluta. La reacción de epoxidación electrófila a través de un proceso concertado es un buen ejemplo anti sin Transferencia concertada de oxígeno La reacción de epoxidación es irreversible y está bajo control cinético La conformación es importante en determinar la estereoselección observada: la conformación de energía más baja tiene la mayor separación de sustituyentes voluminosos. El control de la conformación en sistemas alílicos se llama tensión alílica o tensión A(1,3) Si el sustituyente no es cis solo hay pequeña diferencia en energía Tensión alílica o tensión A(1,3) Energía más baja: H-H El H eclipsa el plano del alqueno Ligeramente alta energía: Me-H El Me eclipsa el plano del alqueno Sustituyente cis presente solo en una conformación Energía más baja: H-Me El H eclipsa el plano del alqueno Alta energía: Me-Me Esta interacción no favorece la conformación Aproximación preferida S = grupo pequeño L = grupo grande favorecido Adición selectiva a alquenos-cis Desestabilizado por repulsión entre los sustituyentes C1 y C3 ó tensión A(1,3) En el alqueno trans las diferencias de energía entre los dos conformeros es sensiblemente inferior y el d.e. es menor de (61/39) Conformación de más baja energía da el producto principal El MCPA ataca por la cara menos impedida Ambas conformaciones son bajas en energía. Mezcla de productos 40:60 Grupo hidroxilo puede dirigir la epoxidación en compuestos acíclicos , así: • Una vez más, el producto principal es formado a partir de la conformación más estable • Así, el grupo metilo cis es muy importante • El producto menor se forma ya sea por el ataque no dirigido o por medio de la conformación menos favorecida Puente de hidrógeno Conformación favorecida Conformación desfavorecida Epoxidación Dirigida: efecto del sustituyente C- 2 La presencia de un sustituyente en la posición C- 2 (Me) facilita una reacción con una diastereoselectividad muy alta • La conformación preferida minimiza la interacción entre los dos grupos Me-Me • Con sustituyente C- 2 (H) hay una pequeña diferencia de energía entre las conformaciones: por lo tanto se obtiene una baja selectividad Interacción estérica Conformación favorecida Solo están eclipsados CH3 yH Conformación desfavorecida Están eclipsados CH3 y CH3 H muy pequeño para diferenciar las conformaciones Control del sustrato en síntesis totales Epoxidación dirigida para la síntesis de aglicona Oleandomicina • glicosilada versión (R = azúcar) es un potente antibiótico de Streptomyces antibioticus David A. Evans y Annette S. Kim , J.Am.Chem.Soc.,1996 ,118,11323 aglicona Oleandomicina (R = H, pero puede ser un azúcar) Interacción estérica Un grupo hidroxilo puede revertir la selectividad normal y la epoxidación directa • La epoxidación con un perácido, tal como m-CPBA, está dirigida por el puente de hidrógeno y favorece el ataque por la misma cara que el grupo hidroxilo • La reacción con un reactivo de vanadio da como resultado una estereoselectividad más alta conforme se une a través de un quelato con el oxígeno Reactivo Reactivo: sin Puente de hidrógeno Acetilacetonato de vanadilo anti Epoxidación asimétrica de Sharpless (SAE) de alcoholes alílicos • • • Epoxidación asimétrica de Sharpless. El catalizador empleado fue el primer catalizador asimétrico general. Hay un gran número de consideraciones prácticas que no vamos a discutir. Basta con decir que funciona para una amplia gama de compuestos de una manera muy predecible. Los compuestos deben ser alcoholes alílicos , como se muestra por epoxidación de la diolefina (D)-(-)-tartrato de dietilo (D-(-)-DET) (i-Pr-O)4Ti, tBuOOH CH2Cl2, malla molecular de 4 Å (L)-(+)-tartrato de dietilo (L-(+)-DET) JACS,1980, 5974 La SAE es bastante predecible. Mecanismo Debe ser un alcohol alílico Mecanismo de la SAE Se piensa que la especie activa sea 2 x Ti puenteado por 2 x tartratos Es normal que los reactivos se dejen reaccionar antes de la adición del sustrato con lo que se favorece una limpia formación del dímero Debe entregar «O» por la cara inferior SAE funciona bien para una amplia gama de alcoholes alílicos. Sólo los alquenos-cis disustituidos muestran una menor enantioselección Buenos sustratos, altos rendimientos y ee > 90% Es normal que sean buenos sustratos, pocos ejemplos y ee > 90% Reacciones problemáticas, lentas. ee moderados. En especial con R3 voluminoso En una SAE se puede superar (tiene la prioridad) la selectividad inherente de un sustrato. Además, se demuestra el concepto de coinciden y no coinciden. Cuando el catalizador y sustrato refuerzan entre sí (coinciden) se logran buenos resultados condiciones El uso de SAE en síntesis: Fluoxetina La fluoxetina es un antidepresivo comercial (más conocido como Sarafem® o Prozac). Se puede sintetizar a través de varios métodos • Uno de ellos implica el uso de la reacción SAE ... fluoxetina Y. Gao ; K. B. Sharpless, J. Org. Chem .,1988,53,4081. Yun Gao , Robert M. Hanson , Janice M. Klunder , Soo Y. Ko, Hiroko Masamune , y K. Barry Sharpless , J. Am .Chem .Soc.,1987,109 ,5165 Resolución cinética Lento Impedimento estérico Como mezcla racémica Rápido Si se quiere obtener el alcohol alílico: utilizar 0.6 eq. TBHP Si se quiere obtener el epoxialcohol: utilizar 0.45 eq TBHP Utilizando el mismo tartrato de dietilo, ambos enantiómeros se deben epoxidar por la misma cara, pero la rapidez con la que ocurre la epoxidación es diferente y las diferencias son suficientes para que se epoxide sólo un enantiómero, si la reacción se detiene en la conversión de 50 %. Si la reacción se deja llegar 100 % de rendimiento, se obtiene una mezcla 1: 1 de los dos diastereoisómeros Rapidez de epoxidación (S) : (R) 700 : 1 Resolución cinética La resolución cinética es normal que funcione de manera eficiente, pero el problema con la resolución cinética es que se puede dar solamente un rendimiento máximo de 50 % en la formación del epóxido. La desimetrización de un compuesto meso permite alcanzar un rendimiento del 100 %. Este proceso es el mismo que dos resoluciones cinéticas, en la primera el compuesto se desimetrisa y en la segunda se elimina enantiómero no deseado. Se incrementa el e.e del producto deseado con el tiempo de reacción ( 84 % ee 3 h ➔ > 97% 140 h ) lento Rápido OM lento Rápido meso se elimina con facilidad Adición conjugada estereoselectiva (1,4 ) El ataque nucleofílico sobre el enlace C=C normalmente requiere de un alqueno deficiente electrones, como en el caso de derivados de carbonilos ,b- insaturados. La reacción se conoce como adición-1,4 o una adición conjugada de Michael. Después de la adición del nucleófilo, se forma un enolato, con lo que se abre la posibilidad de formar dos centros estereogénicos. Control de sustrato - la adición inicial del nucleófilo a la cara menos impedida de la enona, la adición del electrófilo normalmente ocurre por la cara opuesta Primer estereocentro Segundo estereocentro Las prostaglandinas son técnicamente hormonas con efectos fisiológicos muy fuertes. Por ejemplo, han sido utilizadas para prevenir y tratar las úlceras pépticas, como un vasodilatador, para tratar hipertensión pulmonar e inducir el parto / o el aborto Este estereocentro controla la adición del doble enlace i. ii. Pyr-HF (HF)x hidrólisis Noyori , J. Am.Chem.Soc.,1988 ,110 ,4718 Adiciones conjugadas diastereoselectivas Es posible utilizar un auxiliar quiral para controlar la adición nucleofílica-1,4. La formación de un quelato por los oxígenos de la sultama con el Mg, restringe la rotación y favorece la conformación cis, la adición del nucleófilo (Et) ocurre por el lado estéricamente más accesible. El auxiliar quiral se rompe con facilidad (y reutilizarse) para dar un compuesto enantioméricamente puro a través de la reacción diastereoselectiva Alconforsulfama de Oppolzer Conformación trans no favorecida Quelato restringe la rotación conformación cis favorecida Auxiliar quiral para controlar dos estereocentros Es posible utilizar la adicón-1,4 para introducir dos centros estereogénicos. La primera adición (BuMgBr) se produce como antes, para generar un enolato. El enolato puede entonces ser atrapado por un electrófilo apropiado. Una vez más el auxiliar quiral sultama controla la cara de la adición (de Me) estrategias El electrófilo se aproxima por la cara inferior Adición conjugada enantioselectiva catalítica Mucho esfuerzo se ha realizado para tratar de desarrollar catalizadores enantioselectivos para adiciones conjugadas. Mientras muchos de ellos son muy exitosos para ciertos sustratos, pocos son capaces de actuar sobre un amplia variedad de compuestos. El sistema anterior da excelentes enantioselectividades para ciclohexenona, pero no presenta selectividad para ciclopentenona Adición conjugada enantioselectiva catalítica Eje de simetría C2 Una vez que se habían desarrollado adiciones conjugadas estereoselectivas con radicales en presencia de auxiliares, la variante catalítica enantioselectiva se propuso rápidamente. El siguiente ácido de Lewis quiral catalizó la reacción. La mayoría del trabajo en esta área ha sido promovida por Sibi Eje de simetría C2 Transposiciones sigmatrópicas-[3,3] Un tipo de reacciones pericíclicas cuyo resultado estereoquímico se rige por los requerimientos geométricos de un estado de transición cíclico. Las reacciones generalmente proceden a través de estado de transición con una conformación de silla, en el que se minimizan las interacciones 1,3-diaxiales. El tipo de activación (térmica o fotoquímica) y la estereoquímica, con frecuencia se pueden predecir por las reglas de Woodward-Hoffmann las cuales se basan en el número total de electrones (aquellos en el sistema-p + los de un enlace sencillo) que participan en el proceso de transposición: 4n electrones, es fotoquímicamente permitido a partir del estado excitado; 4n + 2 electrones, la migración es térmicamente permitido. Muchas similitudes con la reacción aldólica. La estereoquímica absoluta - controlada por un estereocentro ya existente Estereoquímica relativa – controlada por la geometría del alqueno/enolato. calor Ambas unidades son estereogénicas o proquirales potenciales, lo cual dependerá de la naturaleza de a, b , c, d Transposición de Cope Un ejemplo muy simple de un sustrato controlado por una transposición sigmatrópica-[3,3] es la transposición de Cope. Para minimizar las interacciones 1,3-diaxiales el grupo fenilo es pseudo-ecuatorial. Nota: el estereocentro original se destruyó medida que se forma el nuevo centro. Este proceso es denomina "transferencia de quiralidad” Interacciones 1,3-diaxiales desfavorecido Classifications 333 Transposición de Claisen Uno de las transposiciones sigmatrópicas más útiles es la transposición de Claisen y todas sus variantes . En color azul el nuevo enlace C -C formado. Transposición de Claisen calor Transposición de Johnson-Claisen calor Transposición de Eschenmoser-Claisen calor Transposición de Ireland-Claisen calor Síntesis Enantioconvergente Ambos enantiómeros del alcohol inicial se pueden convertir en el mismo enantiómero de producto. Este proceso (Eschenmoser - Claisen) muestra la importancia de la geometría del alqueno en una transposición sigmatrópica [3 + 3] La reducción SET da el alqueno más estable Misma configuración H2 catalizador de Lindlar La hidrogenación heterogénea da la adición sin de H2 Transposición de Ireland-Claisen La geometría del enolato controla la estereoquímica relativa, por lo tanto, el paso de enolización controla el estereoquímica del producto final. Como hemos visto es relativamente fácil de controlar la geometría del enolato y en consecuencia la estereoquímica final Control del sustrato en la transposición de Ireland-Claisen En una manera similar a la transposición de Cope, en la transposición de Claisen-Ireland se produce con "Transferencia de quiralidad". El centro estereogénico inicial controla la conformación de la silla en el estado de transición: el sustituyente más grande adoptará la posición pseudo-ecuatorial. Una vez más, la estereoquímica relativa entre los dos nuevos estereocentros es determinada por la geometría de la enolato El grupo Me es pseudo-ecuatorial 98 % sin 91 % ee Transposición controlada con auxiliar quiral en síntesis total Con el uso de auxiliares quirales y enantiopuros es posible llevar a cabo la transposición en forma enantioselectiva. Una aplicación se llevó a cabo en la síntesis de (-) – Malingolido, el cual es un antibiótico aislado de las algas marinas Lyngbya majuscule azul - verde. Esta síntesis utiliza la RAMP hidrazona de Enders como auxiliar quiral para configurar el centro cuaternario . Hidrazina de Ender (-) – Malingolido Tetrahedron,,1996, 52, 5805 Transposición controlada con auxiliar quiral en síntesis total Es posible llevar a cabo la reacción bajo el control de un " reactivo ", como en el caso de boroenolates quirales. Aunque se podría argumentar que esto es sólo una forma de control auxiliar temporal en la formación del enolato (geometría del enolato) determina la estereoquímica relativa calor calor Et3N Tolueno / Hexano - 78 oC Uso de un reactivo quiral en síntesis total El Dolabellatrienona es un diterpenoide marino aislado de octocorales gorgonias como Eunicea calyculata y los otros organismos marinos Su síntesis enantioselectiva basa en la química de un enolato de boro para establecer la estereoquímica de la molécula final A través de un enolato de boro Dolabellatrienona J.Am.Chem.Soc.1996 ,118,1229 Enantioselectividad en la transposición 2,3-Wittig Reactivo de control utilizando un reactivo de boro quiral, de manera similar a la observada en la transposición de Ireland-Claisen. La enamina se puede utilizar como anión y con el uso de una amina quiral se observa una enantioselección significativa Reacción de transposición [2,3 ]-aza-Wittig en síntesis total La reacción de transposición [2,3]-aza-Wittig es menos común. El liberación de la tensión anular del anillo acelera la reacción. La transposición aza- Wittig se ha utilizado en la síntesis total de la indolizidina 209B de Dendrobates pumilio o la rana fresa dardo del veneno . tensión anular del anillo de aziridina indolizidina 209B Tetrahedron,,1995,51,9741 Reacción de Diels -Alder estereoselectiva La reacción de Diels -Alder (DA) es un método muy valioso para la síntesis de anillos de 6 -miembros y presenta una alta regioselectividad. principal Tolueno, 120 oC, sin catalizador Benceno, 25 oC, SnCl4 menor Ácido de Lewis Incrementa la selectividad Está controlada por los "tamaños relativos" de los orbitales p en los orbitales LUMO y HOMO involucrados o en el valor de sus coeficientes orbitales. En presencia de un ácido de Lewis, el dienófilo se polariza para dar una mayor regioselectividad y una reacción más rápida Selectividad endo vs exo El estado de transición endo y su aducto, se encuentra más estéricamente congestionados, por lo que es termodinámicamente menos estable, pero es normal que sea el producto predominante. La razón es que el estado de transición endo se estabiliza por el traslapo del orbital p del grupo en C o D con el HOMO del dieno; un efecto llamado " traslapo orbital secundario". La reacción es suprafacial y la geometría del dieno y dienófilo se conservan. Por último, recuerde que el dienófilo reacciona invariablemente desde el cara menos impedida. traslapo orbital secundario favorecido El método 'cubo' es una buena manera de visualizar la estereoquímica relativa Dibujar un cubo Adicionar el dieno Hacer la reacción (hacer los nuevos enlaces) Adicionar el dienofilo (el sustituyente endo tiene sustituyentes directamente abajo del dieno) Tomar en cuenta otros sustituyentes presentes Deberá ser capaz de ver la estereoquímica relativa Auxiliares quirales en el dienófilo Se forma un diastereoisómero - el producto endo, pero como una mezcla de enantiómeros, Si se adiciona un auxiliar quiral, entonces se forman dos posibles diastereoisómeros endo, pero uno predomina, es así que se puede preparar un solo enantiómero No hay enantioselección dienófilo aquiral + dieno aquiral mezcla de enantiómeros 1:1 Auxiliares quirales en el dienófilo Versión enantioselectiva usando un auxiliar quiral Derivado de la (S)-Valina Dienófilo quiral Dieno aquiral Un solo diastereoisómero R = H 86 % de R = Me 90 % de > 96 % endo Un solo enantiómero Catálisis quiral y la reacción de Diels -Alder El hecho de que la reacción de Diels -Alder es mediada o catalizada por ácidos de Lewis, posibilita que se puedan hacer modificaciones que permitan hacerlas enantioselectivas. Los catalizadores de aluminio, utilizados en la reacción aldólica son muy efectivos Ligandos de bis(oxazolina) se encuentran entre los ligandos más versátiles conocidos hasta el momento. Se preparan simplemente a partir de aminoalcoholes (y por lo tanto, amino- ácidos). Puede ser utilizado tanto en la reacción Diels-Alder (DA) y en las reacciones Hetero-Diels-Alder lo mismo que Reacción Hetero-Diels-Alder (HDA) HAD Catalítica enantioselectiva en síntesis total La (+)-Ambruticina es un agente antifúngico extraído de la mixobacteria Polyangium Cellulsoum , ha presentado actividad contra Coccidioides immitis la causa de coccidioimicosis (+)-Ambruticina J.Am.Chem.Soc.,2001,123,10772 Reacción estereoselectiva mediada con metal La reacción de Heck es un método versátil para los centros de acoplamiento para carbonos con hibridación sp2 Adición oxidativa Eliminación de Hidruro-b Adición sin Isomerización de alqueno El paso de la eliminación del b-hidruro es reversible, por lo que el doble enlace puede "caminar" o migrar para dar el alqueno más estable. La única restricción es que cada paso debe ser sin Adición sin Eliminación de Hidruro-b Hidro paladación Reacción de Heck enantioselectiva Con el uso de ligandos quirales la reacción de Heck puede ser enantioselectiva Una modificación intramolecular permite la construcción de sistemas de anillos. La sal de plata acelera la reacción y evita la isomerización del alqueno lig Derivado de aminoácido N,N-dimetilanilina Review of asymmetric Heck: Chem. Rev. 2003,103,2945 Reacción de Heck enantioselectiva en síntesis total La (+)-Xestoquinona fue aislada de la esponja Xestospongia sapra Pacífico y es un potente inhibidor irreversible tanto de la proteína tirosina quinasa oncogénica pp60v -src codificada por el Rous virus del sarcoma y la quinasa del factor de crecimiento epidérmico humano (EGF). La primera síntesis total involucra dos reacciones de Heck; la primera es enantioselectiva para dar un centro cuaternario y la segunda da un segundo anillo de 6 miembros. (+)-Xestoquinona J.Am.Chem.Soc.,1996,118,10766 ENANTIOSELECTIVIDAD Un problema con la inducción de la selectividad es que el ligando está en el lado opuesto al nucleófilo. Los ligandos voluminosos pueden ayudar a resolver este problema y tener un centro estereogénico en el sustrato o en el nucleófilo. Estereocentro en el sustrato Estereocentro en el nucleófilo Sustitución alılica en síntesis total La (-)-Swainsonina se puede aislar de locoweeds; en el ganado que causa síntomas similares al de las vacas locas, al consumir plantas enfermas (EEB) y de ahí el nombre de la española por loco. En los seres humanos presenta actividad anticáncer, antiviral e inmuno-reguladoras. Meso Proceso de desimetrización (-)-Swainsonina J.Org.Chem.,2002,67,4325. Para la desimetrización véase también J.Org.Chem., 1998,63,1339 Otras reacciones enantioselectivas catalíticas En la actualidad hay un gran número de reacciones enantioselectivas más las que se inventen/desarrollen todo el tiempo. Es muy poco probable que la investigación en este campo vasto y fascinante disminuirá en un futuro previsible. Debería ser posible el poder desarrollar variaciones enantioselectiva de la mayoría de las reacciones. Un ejemplo de una variación quiral del catalizador de Schrock en la reacción de metátesis. La reacción implica una desimetrización por una reacción selectiva en un alqueno disustituido Resumen de métodos para la síntesis estereoselectiva Método Resolución Síntesis asimétrica (chiral pool) Auxiliar quiral Reactivo quiral Catalizador quiral Ventajas Disponibles ambos enantiómeros Se garantiza un 100% ee Con frecuencia excelentes ee Se basa en el agente de resolución Con frecuencia excelentes ee La estereoselectividad puede ser independiente del control en el sustrato Económica, solo pequeñas cantidades de material usado son reciclables Desventajas Rendimiento máximo 50 % Ejemplos Síntesis de (-)-propranodiol Con frecuencia solo está disponible un enantiómero Se necesitan pasos extra para introducir y remover al auxiliar quiral Síntesis de (R)-sulcatol Oxazolidinonas Solo unos pocos reactivos Alpino-borano, reactivos de son exitosos y con alilación de Brown frecuencia lo mismo pasa con solo unos pocos sustratos En realidad solo unas pocas Hidrogenación asimétrica; reacciones son exitosas; con epoxidación de Sharpless frecuencia a una falta de generalidad del sustrato Reservorio quiral (chiral pool) AMINOÁCIDOS HIDROXIÁCIDOS CARBOHIDRATOS TERPENOS ALCALOIDES L-alanina L-arginina Ácido L-láctico Ácido D-láctico D-arabinosa Ácido L-ascórbico Cincodina Cinconina D-asparagina L-asparagina Ácido (S)-málico Ácido 3(R)-hidroxibutírico Ácido D-ascórbico a-cloralosa l-borneol Endo-3-Bromo-dalcanfor d-canfeno d-alcanfor D-(+)-efedrina l-nicotina Ácido L-aspártico Ácido L-tartárico Diacetona-a-glucosa Ácido d-canfórico Quinidina L-cisteína Ácido D-tartárico D-fructosa Quinina Ácido L-glutámico D-treonina Ácido Ácido d-10canforsulfónico d-3-careno D-(+)-seudoefedrina L-isoleucina L-treonina D-galactónico l-carvona L-(-)- seudoefedrina L-glutamina L-leucina L-lisina g-lactona D-galactosa Ácido D-glucoheptónico d-citronelal d-fencona d-isomentol L-metionina Ácido a-Dglucoheptónico Ácido D-glucónico Ácido L-glucónico D-glucosamina D-glucosa D-glucorona d-limoneno Ácido D-glucónico L-glutamina ÁcidoD-quínico D-manitol D-manosa D-ribolactona (-)-a-felandreno (-)-a-pineno (+)-a-pineno (-)-b-pineno (R)-(+)-pulegona L-omitina L-fenilalanina D-fenilglicina L-prolina Ácido L-piroglutárico L-serina L-triptofano L-tirosina L-valina l-limoneno l-mentol d-mentol l-mentona Nopol ORGANOCATALISIS En química orgánica, el término Organocatálisis (una concatenación de los términos "orgánico" y “catalizador") se refiere a una forma de catálisis, por lo que la rapidez de una reacción química se incrementa por un catalizador orgánico, lo que se conoce como un "organocatalizador" que consiste en carbono, hidrógeno, azufre y otros elementos no metálicos que se encuentran en los compuestos orgánicos. Debido a su similitud en composición y descripción, que se confunden a menudo como un nombre poco apropiado para las enzimas debido a su efectos comparables sobre la rapidez de reacción y las formas de la catálisis involucradas. Los organocatalizadores que muestran la funcionalidad de amina secundaria pueden ser descritos al realizar su catálisis como una enamina (al formar cantidades catalíticas de una enamina activa como nucleófilo) o la catálisis de una sal de iminio (por la formación de cantidades catalíticas de una sal de iminio que funciona como un electrófilo activado). Este mecanismo es típico para una organocatálisis covalente. La unión covalente del sustrato normalmente requiere emplear altas cantidades del catalizador (para la catálisis de la prolina es usual usar 20-30 en % mol). Interacciones no covalentes tales como puentes de hidrógeno facilita el emplear bajas cantidades del catalizador (hasta 0.001 % en moles). La organocatálisis ofrece varias ventajas: 1) No hay necesidad para la catálisis empleando metales, por lo que esto representa una contribución a la química verde. En este contexto, se han utilizado ácidos orgánicos simples como catalizadores para la modificación de celulosa en agua, es escala de varias toneladas. 2) Cuando el organocatalizador es quiral se abre la posibilidad a la catálisis asimétrica, por ejemplo el uso de prolina en las reacciones aldólicas 1. Berkessel, A., Groeger, H. (2005). Asymmetric Organocatalysis. Weinheim: Wiley-VCH. List, B. (2007). 2. "Organocatalysis". Chem. Rev. 107 (12): 5413–5883. 3. P. I. Dalko, L. Moisan, Angew.Chem. Int. Ed . 2001, 40, 3726 -3748 and Angew. Chem. Int. 4. Ed. 2004, 43, 5138–5175. 5. M.J. Gaunt, C. C.C. Johansson, A. McNally, N.T. Vo, "Enantioselective organocatalysis" Drug Discovery Today, 2007, 12(1/2), 8-27. 6. D. Enders, C. Grondal, M. R. M. Hüttl, review: "Asymmetric Organocatalytic Domino 7. Reactions", Angew. Chem. Int. Ed. 2007, 46, 1570–1581. Síntesis de Justus von Liebig de oxamida de diciano y agua representa la primera reacción organocatalítica, empleando acetaldehído como el primer "organocatalizador" puro descubierto, el cual actúa de manera similar a los entonces llamados "fermentos" , las cuales ahora se conocen como enzimas. Justus von Liebig, Annalen der Chemie und Pharmacie,1860,113,246-247 Organocatalizadores para la síntesis asimétrica se pueden agrupar en varios tipos: Biomoléculas: en particular, prolina, fenilalanina. Las aminas secundarias en general.Los alcaloides cincona, y cietos oligopéptidos. Catalizadores sintéticos derivados de biomoléculas. Catalizadores de enlace de hidrógeno: incluyendo TADDOLS , derivados de BINOL como Nobin y organocatalizadores basados en tioureas (S. Bertelsen , K. A. Jørgensen, Chem.Soc. Rev.,2009,38,2178-2189). Derivados de imidazolidinona (también llamados organocatalizadores MacMillan): son catalizadores adecuados para muchas reacciones asimétricas, tales como reacciones asimétricas DA. Catalizadores de imidazolidinona 1ª. generación 2ª. generación El primero de estos compuestos fue un derivado de la biomolécula fenilalanina la cual se obtuvo en dos pasos químicos (amidación con metilamina seguido de una reacción de condensación con acetona), lo cual deja intacta la quiralidad J.Am.Chem.Soc., 2000,122,4243-4244 Este catalizador funciona por formación de una sal de iminio con los grupos carbonilos de aldehídos (enales) (enonas) α,β-insaturados, en un equilibrio rápido. La activación de este ion iminio es similar a la activación de grupos carbonilo por un ácidos de Lewis y mabos catalizadores disminuyen la energía del LUMO del sustrato Aldrichimica Acta 39 (3),79 Angew. Chem. Int. Ed. 42 (40): 4955–4957 J. Am. Chem. Soc. 129 (50): 15438–15439. Organocatalizadores aquirales regulares se basan en compuestos conteniendo nitrógeno: 1) Como la piperidina, la cual es utilizada en la Condensación de Knoevenagel. 2) DMAP empleada en esterificaciones. 3) DABCO utilizada en la reacción de Baylis-Hillman. 4) Sales de tiazolio, se emplean en la reacción de Stetter. 1) Condensación de Knoevenagel 2) DMAP empleada en esterificaciones actúa como un agente de transferencia de acilo Esterificación de Steglich 3) DABCO utilizada en la reacción de Baylis-Hillman. DABCO = 1,4-Diazabiciclo[2.2.2]octano Para arilaldehídos bajo condiciones polares, no polares, y próticas, se ha determinado que el paso determinante de la rapidez es de segundo orden en el aldeh´dio y de primer orden en DABCO y acrilato. Con base en estos datos de la rapidez, se ha propuesto el siguiente mecanismo, que involucra la formación de un hemoiacetal como intermediario: J. Org. Chem. 2005, 70, 3980. The First One-Pot Synthesis of Morita-Baylis-Hillman Adducts Starting Directly from Alcohols L. D. S. Yadav, V. P. Srivasta, R. Patel Synlett, 2010, 1047-1050 Synthesis of 1,3-Dialkyl-1,2,3-triazolium Ionic Liquids and Their Applications to the Baylis-Hillman Reaction Y. Jeong, J.-S. Ryu J. Org. Chem., 2010, 75, 4183-4191. Octanol-Accelerated Baylis-Hillman Reaction K.-S. Park, J. Kim, H. Choo, Y. Chong, Synlett, 2007, 395-398 4) Las sales de tiazolio son reactivos umpolong muy versátiles (equivalentes del anion acilio), por ejemplo se han aplicado en la reacción de Stetter Reaction: The First Asymmetric Intramolecular Stetter Reaction D. Enders, K. Breuer, J. Runsink, Helv. Chim. Acta, 1996, 79, 1899-1902. Una reacción pionera desarrollada en la década de 1970 se llama Reacción de Hajos-Parrish: Prolina catalizador natural quiral 99 %, 93 % ee Reacción Hajos–Parrish–Eder–Sauer–Wiechert J. Org. Chem.; 1974, 39,1615-1621 Organocatalysis with proline derivatives: improved catalysts for the asymmetric Mannich, nitroMichael and aldol reactions A. J. A. Cobb, D. M. Shaw, D. A. Longbottom, J. B. Gold, S. V. Ley, Org. Biomol. Chem., 2005, 3, 84-96 Mecanismos de reacción propuestos Mecanismo de reacción propuesto por el grupo de Barbas en 2000 Ataque por la cara Si no favorecido enamina Ataque Re facial Hidrogenación organocatalítica Un desarrollo reciente es hacer uso de pequeñas moléculas orgánicas para lograr hidrogenación • Inspirar por la naturaleza • Basado en la formación de un ion de iminio altamente reactivo (esta es la base de muchas reacciones organocatalíticas) Catalizador 10 % Fuente de hidrógeno 1 eq. Epoxidaciones organocatalíticas Al igual que con la mayoría de las reacciones químicas, la epoxidación se ha movido hacia la química "verde" y el uso de sistemas catalíticos que no implican metales de transición. Hay diversos sistemas, y en particular los catalizadores de Shi y Armstrong se basan en la conversión in situ de cetonas a especies activas: derivados de dioxirano, quienes son las que en realidad llevan a cabo la reacción de epoxidación Cat. Oxono, K2CO3 DME / H2O, -15 oC Dioxirano Reactivo de epoxidación Epoxidaciones organocatalíticas La Compañía Japonesa Tanabe Seiyaku Co., utiliza organocatalisis en la síntesis de Diltiazem-LMR, un agente usado para la reducción de la presión Cat. (5 % mol) Oxono (1 eq), NaHCO3 dioxano/ H2O, Diltiazem-L J. Org. Chem., 2002, 67, 4599 Organocatálisis con ácidos de Lewis El puente de hidrógeno intermolecular actúa como un ácido de Lewis y activa al grupo carbonilo El puente de hidrógeno intramolecular actúa como catalizador, derivado de un producto natural simple: ácido tartárico. Limpio, verde y eficaz Puente de H Intramolecular Grupo voluminoso El ataque es por una cara Organocatálisis en la adición de Michael Ahora nuevos catalizadores orgánicos formados por moléculas pequeñas están logrando resultados notables. La enone es activada por la formación de las especies de sales de iminio cargadas El catalizador también bloquea una cara de la enona permitiendo ataque selectivo Organocatalisis en la adición de Michael Los anillos aromáticos, ricos en electrones, se pueden emplear en la adición Michael El impedimento estérico da como resultado que predomine una conformación Organocatalisis en la adición de Michael Organocatalisis en la adición de Michael Una reacción interesante es la reacción de Stetter - esta consiste en la adición conjugada de un grupo acilo sobre un alqueno activado, y procede a través de la química umpolong (la polaridad inversa del grupo carbonilo) Organocatalisis en la adición de Michael Organocatalisis en la adición de Michael La funcionalidad tiourea actúa como un ácido de Lewis a través de dos puentes de hidrógeno La amina activa al nucleófilo y lo posiciona para permitir una buena selectividad Tolueno, 25 oC, 24 h Organocatalisis en la adición de Michael Ejemplo de una adición conjugada enantioselectiva en una síntesis total Síntesis de un alcaloide marino del foliacea Bryozoa, Flustramina (-)-Flustramina B Catalisis en síntesis totales La (R)-Muscona es el principal contribuyente al olor del almizcle, una secreción glandular del almizcle de ciervos. • Una versión racémica, sintética, se utiliza en perfumes. Adición a la cara Si Reacción de Simmons Smith dirigida por el OH (control del susutrato) Adición catalítica asimétrica sobre el carbonilo i. (COCl)2, DMSO; luego Et3N (R)-Muscona J. Am. Chem. Soc.,1993,115,1593 Organocatálisis y la reacción de Diels-Alder Aminas secundarias pueden catalizar ciertas reacciones de Diels-Alder. La reacción transcurre a través de la formación de una sal de iminio. Esta especie cargada disminuye la energía del LUMO, con lo que se cataliza la reacción. La adición se lleva sobre la cara del dienófilo que esta bloqueada, lo cual permite una alta selectividad Un ejemplo de una reacción de hetero-Diels-Alder El aldehído es el dienófilo y la contraparte es un dieno rico en electrones. El catalizador es una amina, la cual actúa como un ácido de Lewis a través de dos enlaces de hidrógeno Otra reacción hetero-Diels-Alder Este ejemplo es muy similar a la reacción anterior, pero ... Se cree que sólo un enlace de hidrógeno se coordina con el aldehído y el otro se utiliza para formar un ambiente rígido quiral para la reacción