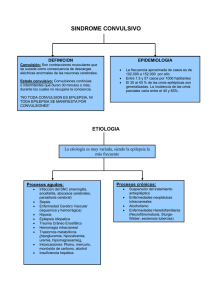
Errores congénitos del metabolismo con crisis epilépticas en
Anuncio

ERRORES CONGÉNITOS DEL METABOLISMO Errores congénitos del metabolismo con crisis epilépticas en los primeros años de vida J. Campistol CONGENITAL ERRORS OF METABOLISM WITH EPILEPTIC SEIZURES DURING THE FIRST YEARS OF LIFE Summary. Objective. In this paper we review the main aetiologies of metabolic origin which cause epilepsy in children aged between 1 and 10 years. Development. There are many aetiological causes of convulsive seizures. Seizures and epilepsies due to congenital errors of metabolism are a minority but should be known. Their identification is not easy although at present there is a wide range of diagnostic methods to confirm the diagnosis when this is suspected. In this review we have only considered the congenital errors of metabolism which start between the ages of 12 months and 10 years, with convulsive seizures. We have divided the conditions into two subgroups depending on whether epilepsy was one of the main symptoms or only part of a set of neurological symptoms and signs. Finally we establish the possible diagnoses of congenital errors of metabolism with seizures occurring in this age group. Conclusions. In neuropaediatrics the paediatrician and epileptolgist must be aware of congenital errors of metabolism as being responsible for epilepsy , especially in cases of drug-resistant epilepsy or when accompanied by other systemic features, neurological deterioration or unexplained biochemical alterations. [REV NEUROL 2002; 35 (Supl 1): S3-20] Key words. Children. Congenital errors of metabolism. Diagnostic protocol. Epilepsy. INTRODUCCIÓN La identificación de una crisis convulsiva carece generalmente de dificultad, mayores dudas surgen en el momento de clasificarla y muy especialmente en la investigación etiológica. Es frecuente que las convulsiones sean una manifestación de una enfermedad sistémica. Procesos como infecciones del sistema nervioso central, patología estructural intracraneal, enfermedades degenerativas, enfermedades sistémicas o incluso trastornos hidroelectrolíticos pueden ser responsables de la aparición de crisis convulsivas en mayor o menor intensidad o preponderancia [1]. Algunas son fácilmente identificables y tratables como la hiponatremia, hipoglucemia, meningitis o intoxicación por drogas. A menudo el tratamiento adecuado permite controlar definitivamente las crisis sin que reaparezcan. Las convulsiones pueden también tener una base genética, como la epilepsia mioclónica juvenil, las convulsiones neonatales familiares benignas o las convulsiones febriles. Encefalopatías estáticas secundarias a infecciones congénitas, daño cerebral perinatal, accidentes vasculares cerebrales o traumatismos también pueden presentar como manifestación principal la epilepsia, entre otros signos y síntomas neurológicos [2]. Sin embargo, existen otras muchas causas de crisis convulsivas cuya identificación no es tan fácil y, en general, mediante un simple interrogatorio no pueden sospecharse. Queda como siempre el cajón de sastre de las epilepsias idiopáticas, cuya incidencia se está reduciendo con la aplicación de nuevas técnicas de neuroimagen, estudios moleculares o con los nuevos exámenes neurometabólicos [3-5]. Dentro del grupo de epilepsias secundarias está el gran grupo de los errores congénitos del metabolismo (ECM). Sus primeras descripciones datan de principios del siglo pasado, sin embargo Recibido: 25.03.02. Aceptado: 27.03.02. Servicio de Neurología. Unitat Integrada de Pediatria Hospital Sant Joan de Déu-Clínic. Universitat de Barcelona. Barcelona, España. Correspondencia: Dr. Jaume Campistol. Servei de Neurologia. Hospital Sant Joan de Déu. Passeig Sant Joan de Déu, 2. E-08950 Esplugues de Ll., Barcelona. E-mail: [email protected] 2002, REVISTA DE NEUROLOGÍA REV NEUROL 2002; 35 (Supl 1): S3-S20 los progresos en los últimos años han sido notables y se han incrementado enormemente el número de nuevos ECM [5,6]. Sigue considerándose excepcional que el origen de una epilepsia sea un ECM. Sin embargo, del total de ECM conocidos en la actualidad, aproximadamente unos 450, entre el 40 y el 60%, pueden manifestar en algún momento de su evolución crisis convulsivas aisladas o una verdadera epilepsia. La incidencia de estos trastornos es baja (1:5.000 recién nacidos vivos), sin embargo hoy en día se diagnostican y tratan más precozmente con lo que aumenta su supervivencia y, por tanto, sus posibilidades de desarrollar a lo largo de los años complicaciones neurológicas. El 25% de los ECM se manifiestan ya en el período neonatal [7] y más del 90% se pueden expresar antes del final de la pubertad. Corresponde pues al pediatra, y muy especialmente al neuropediatra, el conocer estos trastornos, intentar establecer la relación entre la epilepsia que manifiesta el paciente y un posible ECM. Ante la mínima sospecha de ECM deberá buscarlos, identificarlos y proceder al tratamiento oportuno. En algunos casos el tratamiento no pasa por el empleo de fármacos antiepilépticos (FAE). Serán precisos otros medios como el empleo de cofactores, piridoxina, biotina, ácido folínico o una dieta especial, para tratar y controlar las manifestaciones del ECM [5,8]. Fisiopatología Los mecanismos por lo cuales se producen las crisis convulsivas en los ECM, entre otras muchas manifestaciones clínicas, son variados. Lógicamente las neuronas y las células gliales dependen de un adecuado suministro de energía para su normal funcionamiento. Es de sobras conocido que mínimos cambios en su normal funcionamiento pueden dar lugar a manifestaciones diversas y, entre ellas, las crisis convulsivas. Se han implicado como factores favorecedores de las crisis convulsivas en los ECM las alteraciones en la membrana neuronal, las alteraciones en el funcionamiento de la bomba de sodio potasio, defectos energéticos, alteraciones en los neurotransmisores o en las concentraciones de determinados aminoácidos en el líquido cefalorraquídeo entre otros mecanismos, algunos aún poco conocidos (Tabla I) [8-10]. Lógicamente en esta revisión no podemos hablar de todos y cada uno de los 450 ECM conocidos, y que en un momento u otro S3 J. CAMPISTOL Tabla I. Mecanismos de producción de convulsiones en los errores congénitos del metabolismo. Tabla II. Errores congénitos del metabolismo (ECM) con crisis convulsivas en niños de 1-10 años. Defecto suministro energía ECM que presentan crisis convulsivas como una de las manifestaciones predominantes Alteraciones de la integridad de la membrana neuronal Trastornos iónicos intraextracelular Alteraciones de la bomba Na+/K+ Alteración de la permeabilidad de la membrana Desequilibrio de los neurotransmisores Desequilibrio de los aminoácidos Alteraciones en el transporte de moléculas Cúmulo de polímeros Alteración de las características eléctricas de los circuitos neuronales Desequilibrios hidroelectrolíticos 1. Enfermedades neurodegenerativas Enfermedad de Alpers Ceroidolipofuscinosis 2. Trastornos de los metales Enfermedad de Menkes Déficit de sulfito oxidasa 3. Enfermedades lisosomales GM1 GM2 Leucodistrofia de Krabbe Enfermedad de Schindler de su evolución pueden manifestar crisis convulsivas. Nos hemos limitado a las crisis convulsivas que pueden manifestarse en los primeros años de vida secundarias a los ECM. En una anterior revisión analizamos los ECM implicados en los síndromes epilépticos del período neonatal y de los primeros meses de vida (síndrome de Ohtahara, EM precoz de Aicardi, síndrome de West) [7]. Tampoco analizaremos los convulsiones que se manifiestan en niños mayores y adolescentes debidas a ECM, incluidas principalmente dentro de las epilepsias mioclónicas progresivas, por corresponder mayoritariamente a un grupo de edad diferente y a su vez muy numeroso, que requeriría prácticamente otro capítulo. Así pues, nos limitaremos a los ECM que pueden manifestar crisis convulsivas en el período comprendido entre los 12 meses y los 10 años de vida aproximadamente. La distinción entre ECM y enfermedades degenerativas es algo arbitraria, se solapan a menudo. El clásico término ‘degenerativas’ esconde en realidad la ignorancia sobre la verdadera causa de la mayoría de ellas, si bien su carácter habitualmente genético, la evolución progresiva y su semejanza con enfermedades de origen metabólico bien establecido hacen sospechar un ECM [2,5]. El listado de ECM que pueden manifestar convulsiones-epilepsia es muy numeroso y hemos establecido una clasificación absolutamente arbitraria en ECM que presentan crisis convulsivas como una de las manifestaciones principales y ECM que se pueden presentar con convulsiones asociadas a otras manifestaciones neurológicas o extraneurológicas más preponderantes (Tabla II). A su vez, al tratar un ECM nos centraremos solamente en aquellas formas o subtipos que puedan manifestar epilepsia. GRUPO A: ECM QUE PRESENTAN CRISIS CONVULSIVAS COMO UNA DE LAS MANIFESTACIONES PREDOMINANTES Enfermedades neurodegenerativas Constituyen un amplio grupo de encefalopatías progresivas, de tipo familiar, con afectación grave y progresiva del sistema nervioso, que cursan con signos y síntomas neurológicos y que suelen abocar a un grado variable de incapacidad o incluso la muerte. El carácter hereditario es bien conocido en alguna de ellas y de más S4 4. Defectos congénitos de la glicosilación 5. Metabolismo intermediario Mitocondriales: síndrome de Kearn-Sayre, MELAS, MERRF, déficit de fumarasa Transporte de glucosa Hiperglicinemia no cetósica Trastorno de purinas y pirimidinas Defectos de los neurotransmisores ECM que presentan crisis convulsivas como una manifestación más 1. Enfermedades lisosomales Niemann-Pick Gaucher LDM MPS Glicoproteinosis Fucosidosis Sialidosis 2. Peroxisomales ADL-X 3. Metabolismo intermediario Aminoacidopatías: PKU, ciclo urea, leucinosis, homocistinuria, histidinemia, hiperprolinemia, HHH Acidurias orgánicas Déficit vitamínicos difícil interpretación en otras. Los progresos en el conocimiento de la enzimología y especialmente de la biología molecular van aclarando el terreno en este grupo de enfermedades, sacándolas del ‘cajón de sastre’ donde estaban ubicadas, para encuadrarlas mejor REV NEUROL 2002; 35 (Supl 1): S3-S20 ERRORES CONGÉNITOS DEL METABOLISMO Figura 1. Enfermedad de Alpers. RM craneal en T 1. Atrofia corticosubcortical difusa con pérdida de volumen de la sustancia gris y blanca. como encefalopatías genéticas progresivas metabólicas, algunas de ellas aún sin una base bioquímica sólida [1-3]. La clínica de estas afecciones, presidida por el deterioro mental y la afectación neurológica progresiva, no siempre presenta características específicas para sentar un diagnóstico exacto; otras veces es más difícil definir el cuadro clínico. Las convulsiones y la demencia se consideran más características de la afectación de la sustancia gris, mientras que la espasticidad y la ataxia lo serían de la sustancia blanca, pero a menudo coexisten demencia progresiva, convulsiones, ataxia, trastornos del tono y motricidad, defectos sensoriales, alteraciones retinianas e incluso afectación extraneurológica [3]. Poliodistrofia cerebral progresiva o síndrome de Alpers El síndrome de Alpers es un trastorno de carácter multisistémico y de presentación infrecuente, caracterizado por la afectación de la sustancia gris cerebral y del hígado, conocida también como ‘poliodistrofia infantil progresiva o distrofia glioneuronal espongiforme’. El comienzo de esta enfermedad ocurre generalmente antes de los 2 años de edad. Las convulsiones y el retraso del desarrollo aparecen de forma insidiosa en un niño que muestra un desarrollo y adquisición de funciones algo lento. La presencia de mioclonías y la epilepsia parcial continua pueden sugerir el diagnóstico. La microcefalia progresiva y la ceguera son hallazgos frecuentes; esta última puede deberse a atrofia óptica, o bien a la afectación cortical occipital. La espasticidad y el fallecimiento antes de los 5 años constituyen la evolución natural de la enfermedad. Entre los exámenes complementarios el EEG está siem- REV NEUROL 2002; 35 (Supl 1): S3-S20 pre alterado con actividad paroxística multifocal. El electrorretinograma (ERG) no suele mostrar alteraciones mientras que los PEV están gravemente afectados. Asimismo puede existir hiperproteinorraquia y signos de disfunción hepática, que si bien pueden ser mínimos o estar ausentes al inicio de la enfermedad se hacen más patentes en el curso de la misma. Desde el punto de vista neurorradiológico son constantes los signos de atrofia cerebral progresiva de predominio en sustancia gris, sin grandes cambios de señal en sustancia blanca (Fig. 1) [4,10]. Los hallazgos histopatológicos constituyen una ayuda diagnóstica. El SNC se constata una grave pérdida neuronal, cambios espongiformes y proliferación astrocitaria. Se ha podido comprobar cierta predilección por la corteza occipital. Los cambios histológicos hepáticos incluyen esteatosis microvesicular, proliferación de los conductos biliares, inflamación focal, fibrosis periportal y necrosis celular. El diagnóstico, siempre difícil, se basa en la ausencia de una patología previa que justifique el cuadro, los antecedentes familiares y los datos clínicos de regresión psicomotriz, una epilepsia rebelde asociada o no a hepatopatía y una neuroimagen compatible. Raras veces se recurre a la biopsia cerebral para el diagnóstico. La etiología del síndrome de Alpers continúa siendo una incógnita. Se ha podido comprobar una clara incidencia familiar, sugiriéndose un patrón de herencia autosómico recesivo. Han sido referidos en la bibliografía diversos trastornos metabólicos como déficit de la cadena respiratoria mitocondrial (complejos I y IV), déficit de PDH y trastornos del ciclo del ácido cítrico. Estas anomalías se detectan mayormente en tejido hepático que en músculo [11]. Debe tenerse presente también la posible toxicidad de la medicación antiepiléptica, en especial el valproato, en la etiología de la disfunción hepática que aparece en el curso de la enfermedad, si bien ha quedado establecido en diferentes publicaciones la afectación primaria del hígado. El tratamiento es sintomático de la epilepsia (procurando evitar el valproato sódico), del deterioro neurológico y especialmente el consejo genético. Ceroidolipofuscinosis Son encefalopatías progresivas, conocidas también como enfermedad de Batten, de herencia autosómica recesiva, caracterizadas por el cúmulo intra y extracerebral de un material ceroidelipofuscinófilo. Se trata de un grupo heterogéneo de enfermedades, con distintos genes implicados y expresividad clínica variada. Se distinguen cuatro formas distintas según la edad de aparición, clínica, exámenes complementarios y biología molecular. En la forma infantil el defecto se localiza en el cromosoma 1p33-p35, en la forma infantil tardía en el cromosoma 11p15, en la variante infantil tardía en el cromosoma 13q12, en la forma juvenil se localiza en cromosoma 16p12 y, finalmente, en la forma del adulto no se ha identificado el gen (Tabla II). Se han comunicado formas atípicas de la enfermedad con evolución más tórpida. La distribución de estas enfermedades es universal, si bien el tipo infantil precoz predomina en Finlandia. Son afecciones raras con una incidencia comprendida entre 1/50.000 y 1/100.000 [1,3,6,12]. Cursan con epilepsia rebelde y progresiva acompañada de un deterioro intelectual más o menos rápido y afectación visual con retinitis pigmentaria precoz. El diagnóstico se basa en la clínica, EEG (desorganización del trazado de base, complejos de puntaonda lenta), los potenciales evocados visuales (Fig. 2) y muy especialmente el ERG, con abolición progresiva de las respuestas (Fig. 3) [1,4,12], la presencia de linfocitos vacuolados en sangre periférica y sobre todo por la existencia de inclusiones en la piel, S5 J. CAMPISTOL conjuntiva o apéndice cecal, autofluorescentes y apreciables con la microscopía electrónica (Tabla III). No existe un cúmulo específico para cada subtipo; se han descrito algunos cuadros atípicos de evolución lenta, de aparición posiblemente tardía (enfermedad de Kufs) con crisis epilépticas, mioclonías y deterioro intelectual. En ellos solamente la biopsia apendicular y rectal visualiza inclusiones anómalas. La TC/RM suelen demostrar atrofia progresiva del vermis cerebeloso y cortical especialmente [4,12]. La patogenia es desconocida y se sospecha una alteración lisosomal del metabolismo de los ácidos grasos no saturados o menos probablemente de las glucoproteínas; se ha demostrado en algún caso el defecto enzimático (Tabla III). No existe tratamiento definitivo, se ha propuesto el tratamiento con antioxidantes con pobres resultados; para la epilepsia suelen ser más eficaces el valproato sódico, clonacepam, lamotrigina, valproato más clonacepam y lamotrigina más clonacepam. En general, la carbamacepina y la fenitoína pueden exacerbar las convulsiones y se deben evitar. Se postula una selección del FAE según el tipo de crisis y la fisiopatología de las neuronas afectadas por las diferentes mutaciones. Para el cuadro distónico se ha empleado con relativo éxito el trihidroxifenidrilo. Se está trabajando en terapia enzimática sustitutiva y reemplazamiento enzimático, con algunos resultados preliminares bastante esperanzadores [12]. Es posible el diagnóstico prenatal en la forma infantil, infantil tardía y juvenil. En algunos casos (forma infantil) puede realizarse estudio de portadores. Trastornos de los metales Enfermedad de Menkes Figura 2. A la izquierda, potenciales evocados visuales (PEV) por flash normal, y a la derecha, PEV de una paciente con lipofuscinosis ceroidea, donde se evidencia una respuesta anómala de alto voltaje al estímulo visual en córtex occipital. Figura 3. Electrorretinograma de un paciente con lipofuscinosis ceroidea –abolición de la respuesta– Se trata de una enfermedad ligada al cro- (derecha), comparado con un electrorretinograma normal de la misma edad (izquierda). mosoma X resultado de un defecto en la absorción del cobre. Con ello las enzimas dependientes del cobre (citocromo c oxidasa, entre otras) muestran una actividad enzimática deficiente, y repercuten negativamente en la producción de energía. Se inicia en los primeros meses de vida, pero puede empezar más tardíamente, a partir del primer año. No siempre se manifiesta la enfermedad con convulsiones neonatales o con un síndrome de West [1,3,7]. Suelen iniciar crisis mioclónicas, que pueden evolucionar a espasmos infantiles, crisis parciales o generalizadas. Otra característica de la enfermedad son las anomalías del cabello, que se vuelve ensortijado, quebradizo y adquiere una coloración grisácea [13]. Con un simple examen del cabello mediante microscopio se demuestra pili torti, tricorrexis y moniletrix, hallazgos muy sugestivos de esta enfermedad y fácilmente evidenciables. El trazado electroencefalográfico traduce paroxismos multifocales (Figs. 3 y 4) y el diagnóstico se confirma con la Figura 4. EEG de un paciente de 5 años con enfermedad de Menkes, tratado disminución plasmática de cobre y ceruloplasmina. El gen alte- con sulfato de cobre. Actividad de base irregular y desorganizada. Anomalías rado es una ATPasa transportadora de cobre y se localiza en paroxísticas casi continuas bilaterales y asíncronas de muy alto voltaje. S6 REV NEUROL 2002; 35 (Supl 1): S3-S20 ERRORES CONGÉNITOS DEL METABOLISMO Tabla III. Ceroidolipofuscinosis que comienzan antes de los 10 años. Ceroidolipofuscinosis infantiles Infantil (Hagberg-Santavuori) NCL1 Infantil-tardía (Jansky-Bielchowsky) NCL2 Variante infantil-tardía NCL5 Juvenil (Spielmeyer-Vogt) NCL3 Edad de inicio 8-18 meses 2-4 años 5-7 años 4-7 años Primeras manifestaciones Hipotonía Regresión psicomotriz Excitabilidad autismo Epilepsia Deterioro Regresión psicomotriz Déficit visual Ataxia Déficit visual Trast. psiquiátricos Microcefalia + +/– – – Déficit visual ++ (precoz) ++ (precoz) ++ +++ Epilepsia +/- +++ + ++ (tardía) Mioclonías ++ (precoz) ++ + (tardía) +/– Ataxia + ++ ++ ++ Semiología extrapiramidal + + + (disartria) + Exitus 3-10 años 5-15 años 15-20 años 20 años Deterioro intelectual ++ +++ ++ + (tardío) Retinopatía ++, no pigmentaria ++, no pigmentaria ++, no pigmentaria ++, pigmentaria EEG Tendencia hacia el aplanamiento Punta-onda lenta, ondas lentas Aumenta con +/– ELI Punta-onda lenta, ondas lentas Aumenta con +/– ELI Punta-onda lenta, ondas lentas Linfocitos vacuolados – – – ++ Biopsia piel, conjuntiva, apéndice cecal Inclusiones curvilíneas y formas granulosas Inclusiones curvilíneas Inclusiones curvilíneas Formas granulosas Déficit enzimático PPT 1 TPP 1 Desconocido Desconocido Genética. Localización cromosómica 1p 32 11 p 15 13 q 12 16 p 12 PPT1: palmitoil-proteína tioesterasa 1; TPP1: tripeptidil-peptidasa 1. Xq12-q13. El tratamiento con histidinato de cobre puede mejorar algo el pronóstico, especialmente en las primeras fases de la enfermedad [14]. una acidosis láctica, con aciduria fumárica muy característica. No existe tratamiento específico de la enfermedad. Enfermedades mitocondriales Las enfermedades mitocondriales debidas a defectos del ADN mitocondrial (ADNmt) y del ADN nuclear (ADNn) se pueden presentar con manifestaciones multisistémicas que incluyen síntomas neurológicos. Dentro de éstos, las convulsiones no constituyen el síntoma principal [1,15-17]. La mayoría de las citopatías mitocondriales no suelen presentar convulsiones como manifestación común y solamente cuatro tienen como una de sus características clínicas la epilepsia. Predominan en la segunda década de la vida (MERRF, MELAS y Kearns-Sayre), por lo que no las trataremos en este capítulo. Todas ellas son debidas a defectos del ADNmt [8,19,20]. La otra enfermedad es el déficit de fumarasa (fumarasa hidratasa; enzima del ciclo de Krebs, codificada por ADNn), que puede presentarse a cualquier edad, especialmente en edades tempranas, con una epilepsia rebelde en forma de crisis parciales, espasmos infantiles y mioclonías. El trazado electroencefalográfico muestra anomalías multifocales y complejos punta-onda. La neuroimagen puede en algunos casos demostrar alteraciones estructurales del sistema nervioso de origen prenatal y desde el punto de vista bioquímico es frecuente hallar Existe un grupo de enfermedades debidas a una incapacidad genética para sintetizar las enzimas indispensables para la degradación de las macromoléculas. Estas enzimas se hallan localizadas en el lisosoma y su déficit trae como consecuencia el cúmulo intralisosomial de un conjunto de sustancias, muchas de las cuales serán perjudiciales para la célula y darán lugar a diferentes enfermedades denominadas lisosomiales. Actualmente se conocen unas 50 enfermedades dentro de este grupo, aun cuando su incidencia es escasa. Algunas son muy raras como la aspartil glucosaminuria (prevalencia de 0,047 por 100.000 recién nacidos); otras son más frecuentes, como la gangliosidosis GM2 (prevalencia de 0,41 por 100.000 recién nacidos) [21]. Suelen presentarse como encefalopatías progresivas, con inicio en diferentes edades de la vida. En muchas de ellas se asocia un deterioro mental y motor junto a crisis convulsivas y visceromegalia. Algunas comportan una mayor afectación motora y en otras la afectación es mayoritariamente cognitiva. La progresión de la enfermedad es muy variable y el empeoramiento suele ponerse en evidencia tras cuadros infecciosos respiratorios o digestivos [1,3,8]. REV NEUROL 2002; 35 (Supl 1): S3-S20 Enfermedades lisosomales S7 J. CAMPISTOL Se deben sospechar estas enfermedades en función de los hallazgos clínicos, radiológicos y neurofisiológicos, pero son los datos bioquímicos los que nos permitirán identificar el déficit enzimático correspondiente. Es importante el diagnóstico precoz de estos trastornos para establecer un pronóstico, un consejo genético, ofrecer la posibilidad de un diagnóstico prenatal (a excepción de las enfermedades de Salla) y especialmente para intentar una terapia que hoy en día, en fase aún experimental, pasa por el tratamiento enzimático sustitutivo, el trasplante de médula ósea alogénica, el trasplante hepático e incluso la implantación de células epiteliales amnióticas. Sin embargo, hasta el momento ninguna de estas técnicas ha dado lugar a mejorías espectaculares y muchas de ellas se encuentran en estadio muy inicial [22]. Tabla IV. Enfermedades que pueden presentar mancha rojo cereza en mácula. Gangliosidosis GM2 (Tay-Sachs, Sandhoff) Gangliosidosis GM1 infantil Esfingomielinosis (Niemann-Pick tipo A) Sialidosis I (cherry-red spot myoclonus) Sialidosis II Leucodistrofia metacromática Galactosialidosis Gangliosidosis GM1 Es debida a un déficit de la enzima lisosomial beta-galactosidasa. Se trata de una enfermedad hereditaria, autosómica recesiva (prevalencia de 0,23 por 100.000 recién nacidos) [21]. Existen diferentes formas de presentación de la enfermedad, si bien la forma infantil o tipo I es con mucho la más común. En ella la sintomatología aparece al nacer, con disnea, infecciones de las vías aéreas superiores, succión débil, junto a una hipotonía y escasa actividad espontánea. El fenotipo va adquiriendo rasgos más toscos, con orejas de implantación baja, hipertrofia de encías, macroglosia e infiltración cutánea con edema subcutáneo, que se traduce con un tacto especial de la piel. Lentamente se va instaurando una hepatomegalia y en la mitad de los casos se demuestra la presencia de una mancha rojo cereza en la retina. Suelen aparecer anomalías radiológicas con disóstosis múltiple de predominio en cuerpos vertebrales, huesos largos, pelvis y silla turca [1,3,5]. El deterioro neurológico es evidente con una tetraparesia espástica grave, crisis epilépticas (mioclónicas especialmente) e hiperacusia, para posteriormente evolucionar hacia un estado de rigidez de descerebración con ceguera y sordera añadidas. Suelen fallecer estos pacientes antes de los 2 años por problemas respiratorios [1,3,5]. En la forma juvenil o tipo 2 el inicio es más tardío (6-20 meses), con aparición de hipotonía axial, regresión motora e intelectual, y desconexión ambiental progresiva que evoluciona hacia una tetraparesia espástica y un estado vegetativo. A diferencia de la anterior forma no existen rasgos toscos, visceromegalia ni mancha rojo cereza, y las anomalías radiológicas son menos marcadas localizándose especialmente en la columna lumbar (L1 -L2 ) [23]. El diagnóstico se basa en la clínica, los hallazgos radiológicos, oftalmológicos y neurofisiológicos; en las dos formas se pueden hallar células espumosas en la médula ósea muy características. La sobrecarga en gangliósidos en los lisosomas en una muestra de piel o conjuntiva evidencia la presencia de cuerpos membranosos citoplasmáticos que adoptan el aspecto de cebra al microscopio electrónico. Los gangliósidos se acumulan especialmente en el cerebro e hígado. El patrón anormal de excreción de oligosacáridos puede ser un dato bioquímico orientativo. La confirmación se efectúa mediante la determinación de la actividad de la beta-galactosidasa en leucocitos o fibroblastos, que está muy disminuida o ausente. El diagnóstico prenatal es posible en estas enfermedades y no existen alternativas terapéuticas eficaces en la actualidad [22,24]. Gangliosidosis GM2 Se trata de un grupo de enfermedades lisosomiales debidas al cúmulo del gangliósido GM2, especialmente en el cerebro y el hígado. S8 La enfermedad de Tay-Sachs es la de mayor incidencia dentro de las gangliosidosis (1:2.000 en judíos ashkenazis) (prevalencia en la población general 0,41 por 100.000 recién nacidos) [21]. Es debida al déficit de hexosaminidasa A, mientras que la otra enfermedad de este grupo (Sandhoff) es debida al déficit de hexosaminidasa A y B. La enfermedad de Tay-Sachs empieza entre los 3 y 6 meses de vida con hipotonía, pérdida de las adquisiciones y desconexión progresiva. La alteración del tono evoluciona hacia una hipertonía con tetraparesia espástica grave, amaurosis y megacefalia. En el 95% de los pacientes se puede observar una mancha rojo cereza en la región macular; otro hallazgo bastante característico es la presencia de clonías audiógenas (muy espectaculares) ante el mínimo ruido. Las crisis convulsivas suelen manifestarse a partir de los 12 meses de vida (Tabla IV), inicialmente en forma de crisis sutiles, parciales complejas o en forma de ausencias o crisis predominantes mioclónicas, cada vez más frecuentes y rebeldes a la medicación. El trazado electroencefalográfico puede mostrar paroxismos multifocales. Alrededor de los 3 años sobreviene la muerte debido a infecciones intercurrentes. Los estudios anatomopatológicos ponen de manifiesto una sobrecarga lipídica en las neuronas del córtex (responsables de la megacefalia progresiva), en el sistema nervioso autónomo y en los plexos nerviosos del tubo digestivo. Pueden evidenciarse por microscopio electrónico, en fibroblastos y células endoteliales, la presencia de cuerpos membranosos lamelares compuestos por gangliósidos. La forma juvenil se inicia a los 2-6 años con ataxia, trastornos del lenguaje, deterioro intelectual, distonías y espasticidad progresiva junto a crisis convulsivas de tipo mioclónico, focales o clonías audiógenas; en esta forma es mucho más frecuente la atrofia óptica que la mancha rojo cereza. La enfermedad de Sandhoff por déficit de hexosaminidasa A y B es menos frecuente (prevalencia de 0,23 por 100.000 recién nacidos). Se presenta como un cuadro clínico superponible a la enfermedad de Tay-Sachs, si bien la afectación visceral es más evidente [24]. La afectación neurológica y la evolución final son similares [23]. Se han descrito formas de Sandhoff juvenil con inicio a los 3-5 años con un síndrome cerebeloso progresivo, deterioro intelectual, convulsiones y signos piramidales. Algunos presentan mancha rojo cereza. Existe una gangliosidosis GM2 por deficiencia de la proteína activadora de la hexosamisidasa, que se manifiesta por un cuadro semejante al de la enfermedad de Tay-Sachs. En todas estas enfermedades la herencia es autosómica recesiva, se ha localizado el genHEXA en el cromosoma 15q23-q24. El diagnóstico se confirma también mediante la determinación de la actividad de la hexosaminidasa A y B en leucocitos o fibroblas- REV NEUROL 2002; 35 (Supl 1): S3-S20 ERRORES CONGÉNITOS DEL METABOLISMO tos. No existe terapia eficaz a pesar del trasplante de médula ósea y es posible el diagnóstico prenatal [5,23,24]. Leucodistrofias Leucodistrofia de Krabbe La leucodistrofia de células globoides o enfermedad de Krabbe es debida al déficit de la enzima beta-galactocerebrosidasa y se hereda con carácter autosómico recesivo [25]. Se presenta bajo dos formas que corresponden a mutantes alélicas diferentes. Se ha localizado el gen(GALC)en el cromosoma 14q31; existen múltiples mutaciones del gen, de las cuales cinco son comunes en casi el 65% de los pacientes [22,25,26]. La forma infantil precoz suele iniciarse alrededor de los 4 meses (1-12 meses), con aumento progresivo del tono muscular, deterioro global, signos piramidales y arreflexia. Aparecen espasmos tónicos desencadenados por estímulos variados. La epilepsia suele empezar tempranamente, incluso con espasmos infantiles, o en fases más avanzadas en forma de mioclonías o sobresaltos. Es muy característico de la enfermedad la irritabilidad extrema que presentan estos pacientes junto al estancamiento pondoestatural, del perímetro craneal y la atrofia óptica. Contribuyen a la sospecha diagnóstica una disminución de la velocidad de conducción motora y la existencia de una hiperproteinorraquia. La baja actividad de la enzima beta-galactocerebrosidasa en leucocitos o fibroblastos es confirmativa de la enfermedad [1,26-28]. Los estudios anatomopatológicos demuestran la desmielinización en el plano del centro semioval y de los oligodendrocitos, con presencia de macrófagos de origen mesodérmico multinucleado que acumulan en su interior galactocerebrósido y que se denominan células globoides, que han dado nombre a la enfermedad [28,29]. En el nervio periférico no suelen encontrarse células globoides pero en cambio aparecen cambios degenerativos axonales, fibrosis endoneural y cúmulo de histiocitos. No existe tratamiento para la enfermedad y es posible el diagnóstico prenatal [30]. Enfermedad de Schindler Se conoce que la causa etiológica de esta enfermedad es el déficit lisosomial de alfa-N-acetilgalactosaminidasa. El cuadro clínico se inicia alrededor de los 8 m eses con regresión, epilepsia mioclónica o incluso con crisis tonicoclónicas generalizadas, ceguera cortical y visceromegalia [31,32]. El trazado electroencefalográfico demuestra la presencia de complejos punta-onda multifocales. Los pacientes contraen una grave hipotonía con hiperreflexia, espasticidad y atrofia óptica. El cuadro recuerda al de la distrofia neuroaxonal de Seitelberger. No existe tratamiento y el diagnóstico se confirma en leucocitos o fibroblastos. La enfermedad se hereda con carácter autosómico recesivo y es p osible el diagnóstico prenatal [31,33]. Déficit de biotinidasa El déficit de biotinidasa, también denominado déficit múltiple de carboxilasas, se puede presentar de dos formas [34]. La forma precoz empieza en el período neonatal, y la forma de inicio algo más tardío se caracteriza por un inicio a partir de los primeros meses de vida, con un cuadro de crisis generalizadas tonicoclónicas, focales o mioclonías multifocales refractarias a los tratamientos antiepilépticos habituales [35]. El trazado electroencefalográfico muestra un patrón de puntas multifocales o en ocasiones de salvas-supresión. El diagnóstico se basará en la sospecha clínica, las determinaciones analíticas con acidosis láctica y un patrón anormal de ácidos orgánicos en orina [36]. Cuando no se sospecha y trata con rapidez la enfermedad se produce un rápido dete- REV NEUROL 2002; 35 (Supl 1): S3-S20 rioro neurológico con déficit auditivo asociado [24,37]. Por ello, ante la mínima duda, deberemos recoger muestras de sangre y orina e iniciar aun sin diagnóstico el tratamiento con 20 mg de biotina por vía oral. La confirmación de la enfermedad se efectúa mediante la determinación en suero de la actividad de la biotinidasa. El genBTD se ha localizado en el cromosoma 3p25 [38]. La respuesta al tratamiento con biotina oral es espectacular y los pacientes, con el suplemento vitamínico, pueden mantenerse asintomáticos toda la vida [39]. Déficit de transporte de glucosa Se trata de un síndrome descrito recientemente, que se inicia en el período de lactante con crisis mioclónicas o generalizadas, ausencias atípicas o estados confusionales. Se afecta el desarrollo psicomotor, el lenguaje y aparecen problemas de comportamiento, junto a microcefalia. El hallazgo más común es una disminución de la concentración de glucosa en líquido cefalorraquídeo (LCR), con valores normales en plasma. El trazado electroencefalográfico puede ser normal en las primeras etapas y rápidamente se altera con presencia de anomalías punta-onda generalizadas o puntas multifocales. Puede manifestarse también con crisis focales. El diagnóstico se efectúa con los datos clínicos y bioquímicos y mediante el estudio del transporte de glucosa en hematíes, recientemente se ha localizado el gen responsable de la enfermedad. Las crisis son refractarias a los FAE pero pueden mejorar con la dieta cetógena [40]. Deficiencia de sulfito oxidasa Entre las alteraciones del metabolismo de los aminoácidos sulfurados hay que citar el defecto de sulfito oxidasa, que puede presentarse aislado o combinado (debidos a una deficiencia de una pterina específica dependiente de molibdeno) [1]. Se diagnostica por la elevación de sulfito en orina y el cúmulo de aminoácidos sulfurados, particularmente S-sulfocisteína en plasma y orina. Es una enfermedad hereditaria autosómica recesiva, con una frecuencia de presentación muy baja; sin embargo, muestra síntomas neurológicos graves en su formaneonatalconconvulsionesrebeldesenausenciadeantecedentes y trazado electroencefalográfico de salvas-supresión. Existe una forma de inicio más tardío con cuadros repetidos de pseudoencefalitis y hemiparesia, junto a la afectación extrapiramidal, crisis convulsivas de tipo atónico y luxación del cristalino [1,3,5]. El mecanismo patogénico no se conoce bien, pero se ha demostrado que el sulfito es muy tóxico para el sistema nervioso y que posee propiedades neuroexcitadoras notables. No existe un tratamiento específico para disminuir la concentración de sulfito; se ha ensayado dextrometorfano con resultados dispares [5]. Hiperglicinemia no cetósica La hiperglicinemia no cetósica es causada por un defecto de actividad del sistema de degradación de la glicina, complejo multienzimático presente en hígado y cerebro pero no en fibroblastos [2]. Existen dos formas de presentación: la forma neonatal y otra de inicio tardío. La forma tardía se presenta después de un período neonatal normal y a partir de los 6-12 meses aparece retraso del desarrollo en la primera infancia, epilepsia o problemas intelectuales [1,5,41]. Existe una forma de inicio más tardío con afectación cognitiva, delirio, corea, parálisis de la mirada vertical y crisis convulsivas. Por último, se ha descrito una forma de inicio tardío con diplejía espástica, atrofia óptica, sin afectación cognitiva ni epilepsia. Se diagnostica la enfermedad por el aumento de glicina en plasma y particularmente en líquido cefalorraquídeo (LCR), de forma que la relación de las concentraciones de glicina en LCR S9 J. CAMPISTOL y en plasma (valor de referencia: 0,02) puede estar incrementada más de 10 veces en los pacientes afectos. Es la única forma de poder llegar al diagnóstico, y ante la mínima sospecha de la forma tardía de esta entidad deberemos proceder a la cuantificación de aminoácidos en plasma y en LCR [42]. El análisis de ácidos orgánicos en orina es necesario para el diagnóstico diferencial con las hiperglicinemias cetósicas (acidurias orgánicas), en las que el aumento de glicina plasmática (normal en LCR) se produce por inhibición del sistema de degradación hepático de la glicina a causa del cúmulo de ácidos orgánicos tóxicos (acidurias propiónica, metilmalónica, isovalérica, tratamiento con VPA) [1,5]. El tratamiento pasa por la disminución de la concentración plasmática de glicina mediante restricción proteica, reducción de la ingesta de glicina y serina, benzoato para favorecer la eliminación renal y exsanguinotransfusión. También se han empleado estricnina, dextrometorfano, diacepam, suplementos de metionina o colina con pobres resultados. El diagnóstico enzimático se realiza en biopsia hepática y el diagnóstico prenatal es posible en biopsia de corion (no en amniocitos), y también mediante la valoración de la relación glicina/serina en líquido amniótico [41,42]. Síndrome de glicoproteínas deficientes en carbohidratos Los defectos congénitos de la glicosilación (CDG) son un grupo de enfermedades genéticas de reciente conocimiento, causadas por diversos trastornos en la biosíntesis o procesamiento de glicoproteínas. En ellos un amplio número de glicoproteínas que incluye proteínas séricas, enzimas lisosomales y glicoproteínas de membrana muestran estructuras y funciones anómalas. Todo ello explicaría las manifestaciones multisistémicas de este grupo de enfermedades. La transferrina es la glicoproteína más ampliamente utilizada para la detección de los síndromes CDG, pudiéndose estudiar las isoformas por isoelectroenfoque o por inmunoturbidimetría [1-3,42]. Los síndromes CDG tipo Ia son los más frecuentes y mejor conocidos. Periódicamente se están descubriendo nuevos síndromes relacionados con la glicosilación de las proteínas del organismo que tiene lugar en el retículo endoplásmico y en el aparato de Golgi. Es interesante señalar que en la forma de CDG tipo Ia los pacientes pueden presentar ya en el período neonatal accidentes vasculares cerebrales isquémicos o hemorrágicos y desarrollar posteriormente epilepsia. Sin embargo, existen otras formas del síndrome, que aparte de un notable retraso psicomotor, hipotonía, microcefalia y trastornos de coagulación, pueden presentar síndrome de West con mala respuesta terapéutica. Lógicamente, frente a un paciente con síndrome de West y retraso psicomotor, hipotonía y microcefalia deberemos pensar en otras causas mucho más frecuentes, pero en ausencia de antecedentes claros de agresión sobre el sistema nervioso o ante la mínima duda es relativamente sencillo determinar la disialotransferrina en suero. Pueden aparecer también alteraciones de la coagulación y disminución de la ceruloplasmina [5,7,8]. En muchas de estas formas no se conoce bien el defecto enzimático ni genético y en estos casos se prefiere denominarlas CDG-x. Mejor conocida es la forma CDG Ib, debida al déficit de fosfomanomutasa (PMM), cuyo gen se ha cartografiado en 22q13 y que no suele cursar con epilepsia. Alteraciones del metabolismo de las purinas y pirimidinas El conocimiento de este grupo de enfermedades se inicia a partir de la descripción del síndrome de Lesch-Nyhan. Su diagnóstico se basa en el estudio de metabolitos en orina por HPLC-espectrometría de masas. Muchas de estas enfermedades pueden dar lugar a convulsiones en los primeros años de vida, y destaca el mismo S 10 síndrome de Lesch-Nyhan, la forma grave del déficit de adenilosuccinasa, el déficit de dihidropirimidina deshidrogenasa o de dihidropirimidasa y finalmente el déficit de beta-alanina alfacetoglutarato aminotransferasa [5,42]. Pueden asociar microcefalia, retraso mental, autismo, ataxia, afectación hepática y renal (litiasis) y alteraciones inmunológicas, entre otras variadas manifestaciones. Trastornos de los neurotransmisores Las alteraciones del metabolismo monoaminérgico no suelen cursar con epilepsia como síntoma principal excepto en las anomalías del metabolismo de las biopterinas y en la aciduria N-acetilaspártica. Los trastornos del metabolismo de los neurotransmisores aminoacidérgicos incluyen la hiperglicinemia no cetósica tratada en otro apartado. Por último cabe citar los errores innatos del metabolismo del GABA, que incluyen el déficit de glutamato descarboxilasa (convulsiones sensibles a piridoxina), de GABA transaminasa (convulsiones rebeldes, hipotonía, retraso mental, coreoatetosis y elevación del GABA en LCR) y succínico semialdehído deshidrogenasa o aciduria 4-hidroxibutírica [1,3,59]. GRUPO B: ECM QUE PUEDEN MANIFESTAR EPILEPSIA Hemos establecido este segundo subgrupo de ECM en los cuales las convulsiones no son el síntoma más frecuente. Se trata de ECM que en algún momento de su evolución pueden manifestar entre otros síntomas neurológicos convulsiones o una verdadera epilepsia (Tabla II). Enfermedades lisosomales Enfermedad de Niemann-Pick Esta enfermedad es debida al cúmulo de esfingomielina en diferentes tejidos. Su heterogeneidad clínica es notable ya que se han descrito hasta cinco subtipos. El fenotipo C de la enfermedad suele empezar más allá de los 5 años con ataxia, distonía, deterioro neurológico con signos piramidales, oftalmoplejía supranuclear vertical y epilepsia, que se hace más rebelde a partir de la segunda década de la vida [43]. Muchos de los pacientes afectos tienen antecedentes de ictericia colostática prolongada en el período neonatal y primeros meses de vida asociada a visceromegalia. La sobrecarga de esfingomielina es moderada y el déficit de esfingomielinasa sólo se evidencia en el 80% de los casos en fibroblastos [5,43]. En las tres formas descritas de la enfermedad existen células de depósito en la médula ósea, que nos pueden ayudar al diagnóstico [44,45]. El déficit enzimático para el tipo C no es conocido y en las células cultivadas se detectan alteraciones específicas del metabolismo del colesterol exógeno [46]. Se han descrito dos genes implicados en la enfermedad. El más común es elNPC1 localizado en 18q11-q12, que corresponde al 95% de los pacientes afectos, y el gen NPC2, localizado en 14q24.3 en el 5% restante. Es posible el diagnóstico prenatal por técnicas de biología molecular [46-48]. Enfermedad de Gaucher Junto a la GM2 es una de las esfingolipidosis de presentación más frecuente (l:2.000 en los judíos ashkenazis), si bien es menos frecuente en otras etnias (prevalencia de 1,16 por 100.000 nacimientos) [21]. REV NEUROL 2002; 35 (Supl 1): S3-S20 ERRORES CONGÉNITOS DEL METABOLISMO Es debida al déficit de la actividad de la beta-glucocerebrosidasa y se presenta bajo tres tipos distintos [49]. El tipo II o forma neuronopática aguda afecta principalmente a lactantes de 4-8 meses y se inicia con hipotonía axial que se convierte en poco tiempo en una hipertonía de miembros, con visceromegalia enorme (a expensas del bazo especialmente) y epilepsia de difícil control. El cuadro evoluciona con rapidez y los pacientes fallecen antes de los 2 años de vida [49]. El tipo III tiene una clínica muy heterogénea, afecta a niños mayores y adolescentes. Se presenta con visceromegalia, lento deterioro intelectual, y signos cerebelosos y extrapiramidales, además de una epilepsia rebelde. Es característico de esta forma la apraxia oculomotriz y la parálisis de la mirada vertical. Son frecuentes las deformidades esqueléticas y las fracturas espontáneas. El cuadro se complica con la afectación respiratoria y hemorragias debidas a trombocitopenia por hiperesplenismo [50]. El diagnóstico se sospecha por la forma clínica de presentación, la elevación de las fosfatasas ácidas en suero y por la presencia en médula ósea de las células espumosas de Gaucher; se confirma el déficit enzimático en leucocitos o fibroblastos. Son posibles la detección de heterocigotos y el diagnóstico prenatal. No existe tratamiento específico, pero es importante vigilar el hiperesplenismo, la epilepsia y prevenir las fracturas óseas. Se debería posponer el mayor tiempo posible la esplenectomía pues al extirpar el bazo aumenta el depósito de galactocerebrósido en esqueleto, hígado y pulmón, con lo que la enfermedad progresa aún más rápidamente [1,5,8]. Leucodistrofia metacromática El déficit de arilsulfatasa A en los lisosomas da lugar a un cúmulo de sulfátidos con alteración del metabolismo de la mielina y a una enfermedad denominada leucodistrofia metacromática. Existen dos formas de presentación. La forma infantil tardía, que es la más frecuente (1:40.000) [21], suele comenzar una vez adquirida la marcha, entre los 12 y 24 meses, con un trastorno progresivo de la deambulación, hipotonía y arreflexia, para desarrollar posteriormente una clara espasticidad de predominio en miembros inferiores. Posteriormente aparece un deterioro intelectual, atrofia óptica, irritabilidad con dolor a la manipulación de las piernas y dificultades de alimentación [51]. El cuadro progresa con rapidez hacia una tetraparesia, epilepsia con crisis focales, anomalías electroencefalográficas inespecíficas y estado vegetativo; el fallecimiento se produce entre los 3 y 7 años [52]. La forma juvenil suele iniciarse entre los 4 y 15 años con disminución del rendimiento escolar junto a trastorno progresivo de la deambulación, con caídas frecuentes, crisis focales, regresión del lenguaje y aparición de signos piramidales y arreflexia. El cuadro tiene una progresión más lenta que en la forma infantil [53]. En ambas formas se encuentra una disminución de la velocidad de conducción motora e hiperproteinorraquia. Los exámenes neurorradiológicos suelen ser poco específicos hasta muy avanzada la enfermedad, en que se pueden apreciar imágenes TC/RM de atrofia y desmielinización especialmente en centro semioval [51]. El diagnóstico se confirma en cultivo de fibroblastos o leucocitos al demostrar una baja actividad de la enzima arilsulfatasa A. Sin embargo, existen formas de la enfermedad con actividad normal in vitro de la enzima y que son debidas a un déficit de la proteína activadora. En ocasiones es difícil determinar la diferencia entre homo y heterocigotos para la enfermedad por el solapamiento de valores de la arilsulfatasa A. REV NEUROL 2002; 35 (Supl 1): S3-S20 Se trata de enfermedades con herencia autosómica recesiva. Es posible el diagnóstico prenatal por técnicas enzimáticas o moleculares [1,5,8]. Mucopolisacaridosis El defecto de actividad de las enzimas que intervienen en el catabolismo de los mucopolisacáridos (dermatán sulfato, heparán sulfato o keratán sulfato) da lugar a un grupo de enfermedades lisosomiales denominadas mucopolisacaridosis (MPS). Se han descrito siete formas principales, cuatro de las cuales se acompañan indefectiblemente de retraso mental. Pocas formas de MPS suelen manifestar epilepsia: la enfermedad de Sanfilippo en sus diferentes subtipos y la enfermedad de Hurler (prevalencia de 0,71 por 100.000 recién nacidos) [21], de la que hemos descrito recientemente una observación con inicio de la enfermedad en forma de epilepsia tipo síndrome de West [7,54]. La prevalencia de la enfermedad es de 0,36 por 100.000 recién nacidos [21]. La enfermedad de Sanfilippo es la forma de mucopolisacaridosis con más variantes (cuatro subtipos: A, B, C y D), y que a su vez condiciona un mayor déficit intelectual. Por el contrario, los rasgos dismórficos tan característicos de este grupo de enfermedades pueden ser mínimos o no existir. En más de la mitad de los pacientes afectos se manifiestan crisis convulsivas generalizadas, sin un patrón electroencefalográfico característico. Desde el punto de vista clínico es difícil distinguir las cuatro formas. Todas ellas cursan con grave deterioro intelectual, rasgos dismórficos, visceromegalia y sin opacidades corneales. Los pacientes afectos suelen fallecer alrededor de la segunda década de la vida y no existe tratamiento específico, si bien se está trabajando en trasplante de médula ósea y en terapia génica. En todas ellas se encuentra en orina heparán sulfato y solamente el déficit enzimático nos permitirá diferenciar los cuatro subtipos [56]. Todas las MPS se heredan con carácter autosómico recesivo, excepto la enfermedad de Hunter (recesiva ligada al sexo). No existe tratamiento específico para ninguna de ellas y sí paliativos en función de algunas de las complicaciones asociadas. El trasplante de médula ósea en edades tempranas puede ser una opción terapéutica en algunas formas. Otra futura vía de solución podría residir en la terapia génica a nivel de médula ósea [1,5,56]. Glicoproteinosis Las glicoproteínas están constituidas por péptidos enlazados con los oligosacáridos. Están distribuidos por todo el organismo y predominan especialmente en el sistema nervioso. Las alteraciones en el catabolismo de estas sustancias debidas a un déficit enzimático dan lugar a su acumulación en los tejidos y a la excreción de oligosacáridos en orina. Son enfermedades poco frecuentes y se heredan con carácter autosómico recesivo. Fucosidosis Existen dos formas de presentación, I y II. La forma tipo I empieza antes del año con deterioro neurológico progresivo, fenotipo hurleriano, hipotonía en las fases iniciales que evoluciona hacia una hipertonía con posterior rigidez de descorticación, epilepsia, cardiomegalia, disóstosis múltiple y estancamiento pondoestatural. Suelen fallecer por cuadros respiratorios antes de los 6 años. En ambas formas existe un cúmulo de glucolípidos en hígado, piel y linfocitos. Las concentraciones de cloro y sodio están elevadas en el sudor en la forma tipo I. El funcionalismo de la vesícula biliar se afecta progresivamente y el diagnóstico se confirma en leucocitos/fibroblastos al hallar disminuida la actividad de la S 11 J. CAMPISTOL alfa-fucosidasa. El gen responsable de la enfermedad FUCA se ha localizado en 2q 24-q32 [50,55,56]. Sialidosis Debido al déficit de la enzima alfa-neuraminidasa se produce esta enfermedad con muy diferente expresividad clínica. Se han descrito dos formas, tipo I y II. La prevalencia estimada es de 0,02 por 100.000 recién nacidos [21]. La sialidosis tipo I o síndrome mioclono-mancha rojo cereza inicia los síntomas a partir de los 8 años, con disminución progresiva de la agudeza visual, mioclonías generalizadas desencadenadas por estímulos acústicos y afectación de la marcha [58-60]. Suele aparecer una neuropatía periférica desmielinizante, opacidades en cristalino y la característica mancha de color rojo cereza en la retina (Tabla IV). No hay afectación intelectual. Pueden aparecer también crisis epilépticas generalizadas y afectación renal [56]. El trazado electroencefalográfico demuestra una notable lentificación del ritmo de base. La sialidosis tipo II, también conocida por mucolipidosis tipo I, se presenta con un fenotipo hurleriano, mioclonías, disminución progresiva de la agudeza visual, mancha rojo cereza en retina, retraso mental, visceromegalia y anomalías esqueléticas. La forma infantil se inicia antes de los 10 meses mientras que la forma juvenil tiene un curso más lento [56]. Enfermedades peroxisomales Constituyen un grupo de enfermedades genéticas, recientemente identificadas, en las cuales es deficiente la formación o la función del peroxisoma. Éste es un órgano intracelular limitado por una sola membrana, desprovista de ADN y presente en todas las células de los mamíferos y de las plantas [58]. Predominan en el hígado y riñón. En el sistema nervioso central son particularmente abundantes en la glia, células de Schwann, neuronas corticales y células de Purkinje. Sus funciones bioquímicas son numerosas y aún mal conocidas; entre éstas destacan especialmente: biosíntesis de plasmalógenos, beta-oxidación de los ácidos grasos de cadena muy larga, oxidación del ácido fitánico, degradación del ácido pipecólico, síntesis de ácidos biliares, respiración peroxisomal y síntesis de colesterol y dolicoles [61,62]. Las enfermedades del peroxisoma han sido objeto de diversas clasificaciones en función de los datos disponibles en cada momento. Nos limitaremos a las enfermedades del peroxisoma que pueden manifestar convulsiones en pacientes de 1-10 años. Adrenoleucodistrofia ligada al cromosoma X Es una de las más frecuentes enfermedades degenerativas del SNC. Su forma de presentación es muy variada, incluso dentro de una misma familia [61]. En la forma infantil, la más frecuente, el deterioro se inicia entre los 4 y 8 años de vida, con disminución del rendimiento escolar y de la memoria seguidos de trastornos de la marcha, del lenguaje y dificultades en la deglución. Es frecuente la afectación sensorial. El cuadro progresa con aparición de crisis convulsivas (que en ocasiones pueden ser el primer síntoma de la enfermedad); predominan las crisis generalizadas, focales o incluso el estado de mal epiléptico. El trazado electroencefalográfico evidencia la presencia de ondas lentas hipervoltadas. Se instaura un síndrome cerebeloso y piramidal; los pacientes suelen fallecer en 2-4 años después del inicio de los síntomas [62,63]. El diagnóstico bioquímico se basa en el estudio de los ácidos grasos de cadena muy larga en plasma o fibroblastos. En la ma- S 12 Figura 5. RM en T 2 de un paciente de 11 años con ADL-X. Afectación de la sustancia blanca de predominio occipital. yoría de estos pacientes se demuestra una disminución de la respuesta del cortisol plasmático al ACTH. En la mitad de los casos puede aparecer una hiperproteinorraquia oligoclonal. Los exámenes neurorradiológicos confirman la desmielinización de predominio parietoccipital y en centro semioval. Mediante TC puede visualizarse una mayor captación del contraste que traduciría la presencia de la reacción inflamatoria [61,62]. La RM craneal demuestra la afectación de la sustancia blanca (Fig. 5) [4]. Los estudios familiares sistemáticos han demostrado una variable expresividad intrafamiliar de la enfermedad, con casos presintomáticos, y por el contrario, familiares con el déficit bioquímico evidente y totalmente asintomáticos [63,64]. No existe tratamiento eficaz y solamente el trasplante de médula ósea en fases muy precoces puede ser una alternativa [61,63]. Kemp revisó las mutaciones en la adrenoleucodistrofia ligada al cromosoma X y no halló correlación entre genotipo y fenotipo [64]. En heterocigotos obligados la determinación de ácidos grasos de cadena muy larga dio falsos negativos, los análisis mutacionales son los únicos disponibles para la identificación de heterocigotos [64]. Aminoacidopatías Lógicamente en este capítulo no podemos referirnos a todos los ECM de los aminoácidos que en un momento u otro de su evolución puedan presentar crisis epilépticas. Por ello nos limitaremos a los que tienen una mayor incidencia y repercusión sobre el sistema nervioso. Fenilcetonuria La hiperfenilalaninemia es causada por la deficiencia de la enzima fenilalanina hidroxilasa hepática, que transforma la fenilala- REV NEUROL 2002; 35 (Supl 1): S3-S20 ERRORES CONGÉNITOS DEL METABOLISMO Figura 6. EEG de un paciente de 3 años con fenilcetonuria. En sueño se evidencia la presencia de descargas generalizadas de elevado voltaje, sin clínica acompañante. nina en tirosina. Cuando el bloqueo enzimático es suficientemente importante se producen metabolitos alternativos, como el ácido fenilacético, que confiere a la orina un olor peculiar a moho/ ratón, y el ácido fenilpirúvico, que reacciona con el cloruro férrico dando un color verde característico (prueba de Fölling). Fue este autor quien describió por primera vez la enfermedad en 1934, denominándola ‘idiocia fenilpirúvica’, aunque posteriormente fue denominada fenilcetonuria (PKU), y como tal se la conoce en la actualidad [65]. Al principio solamente se consideraba la PKU, pero existen otras causas menos comunes de hiperfenilalaninemia, cuyo diagnóstico diferencial hay que considerar, ya que de él depende el tratamiento y el pronóstico [66-69]. La hiperfenilalaninemia causada por mutaciones en el gen que codifica la fenilalanina 4-monoxigenasa representa un amplio espectro de fenotipos metabólicos debido a la posible combinación de un elevado número de mutaciones alélicas. La forma grave o fenilcetonuria clásica no tratada causa retraso mental y motor graves junto a epilepsia [65,67,68]. Estos trastornos se ponen de manifiesto a partir de los 6-8 meses de vida, por lo que actualmente en un gran número de paises existen programas de detección precoz en el período neonatal aplicables a toda la población, que permiten el diagnóstico y el tratamiento precoz, indispensable y efectivo para evitar la aparición de los síntomas. La PKU clásica no diagnosticada y no tratada cursa con un retraso mental progresivo, microcefalia, epilepsia, eccema y rasgos psicóticos en forma de hiperactividad, tendencias destructivas, automutilaciones, impulsividad, ataques incontrolados de agresividad y aislamiento social [8]. Los pacientes presentan epilepsia generalizada (25% de casos) o síndrome de West, junto con anomalías en el EEG (70-95%), rasgos físicos característicos y olor corporal especial. Los estudios patológicos en estos pacientes demuestran una alteración en la mielinización, con microcefalia y atrofia cerebral [65]. Por su parte, los niños tratados tempranamente siguen una evolución normal, con un cociente intelectual (CI) en los límites de la normalidad, pero algo inferior a los hermanos sanos o grupos control, y con pequeñas dificultades en los aprendizajes, hiperactividad, trastornos del sueño, torpeza motriz e incluso anomalías electroencefalográficas (Fig. 6) así como epilepsia en algún caso excepcional [66,67,69]. Cuando no siguen una dieta estricta y por tanto las cifras plasmáticas de Phe se elevan, manifiestan síntomas REV NEUROL 2002; 35 (Supl 1): S3-S20 neurológicos en forma de hiperactividad, problemas de comportamiento y finalmente deterioro neurológico; también pueden aparecer en la RM craneal imágenes de aumento de señal en T 2 en la sustancia blanca periventricular, que desaparecen en unos meses después de reanudar la dieta. Esta elevada concentración de Phe en el sistema nervioso central también puede evidenciarse mediante RME, siempre que las cifras de Phe sean superiores a 900µmol/L. El tratamiento de la PKU es para toda la vida y se basa en la reducción del aporte de alimentos que contienen fenilalanina, junto a suplementos de tirosina y de carnitina si fuera preciso. La interrupción del tratamiento hacia los 8-10 años conduce a una disminución del CI en 6 puntos, junto a problemas de carácter y comportamiento en forma de agitación, trastornos del sueño y temblores [65,69,70]. Finalmente, existe una variante de hiperfenilalaninemia, que no responde al tratamiento restrictivo en Phe. Se trata de un trastorno del metabolismo de la tetrahidrobiopterina, cofactor que interviene en la hidroxilación de la Phe, tirosina y triptófano [71,72]. Su defecto puede estar en la síntesis o en el proceso de reciclaje de la BH4 . Los pacientes afectos (1% de los casos con PKU) manifiestan, a pesar de un adecuado tratamiento, síntomas de deterioro neurológico debido al déficit de neurotransmisores (catecolaminas y serotonina) [72]. La clínica puede ya iniciarse en los primeros meses de vida con microcefalia, retraso del desarrollo y deterioro neurológico progresivo, con dificultad respiratoria, parkinsonismo, mioclono, corea, distonía, signos piramidales, epilepsia rebelde (espasmos infantiles o epilepsia generalizada) e hipertermia. En estos casos el tratamiento se basa en la restricción dietética junto a los suplementos en levodopa, 5 hidroxitriptófano y en ocasiones ácido folínico [73,74]. La confirmación del defecto de la actividad enzimática de fenilalanina hidroxilasa sólo es posible en biopsia hepática. Se ha localizado el gen que codifica la enzima en el cromosoma 12 (12q22-q24), sin embargo las mutaciones encontradas son muy numerosas (más de 300) [73,74]. El análisis molecular permite conocer mejor el genotipo y establecer correlaciones con el fenotipo, el diagnóstico prenatal y la detección de portadores [67,68,73,74]. La deficiencia de dihidropteridina reductasa puede determinarse mediante la valoración de la actividad enzimática en eritrocitos, fibroblastos y linfocitos. Defectos del ciclo de la urea y alteraciones relacionadas causantes de hiperamonemia La alteración de la ureagénesis, tanto por enzimopatías del ciclo de la urea como por otras causas secundarias que interfieren en este proceso, provoca una hiperamonemia. Es importante el diagnóstico bioquímico diferencial de las alteraciones causantes de hiperamonemia ya que éste no puede realizarse desde el punto de vista clínico ni por la gravedad de la hiperamonemia, sino únicamente en función de los metabolitos acumulados o excretados en fluidos biológicos antes del tratamiento. El análisis de metabolitos se puede llevar a cabo rápidamente; evita la realización de biopsias hepáticas, que pueden ser fatales en pacientes con hiperamonemias graves, y permite la instauración del tratamiento específico que dependerá del proceso desencadenante de la hiperamonemia [75]. El análisis enzimático es importante en algunos casos para la confirmación del diagnóstico y la caracterización de la enzima deficiente, pero no siempre es necesario para establecer el diagnóstico [76]. Considerando los procesos que causan un notable incremento del amonio plasmático destacaremos en primer lugar los defectos de actividad enzimática del ciclo de la urea. Tres de ellos pueden S 13 J. CAMPISTOL distinguirse por el perfil característico de aminoácidos en plasma u orina (citrulinemia, aciduria argininsuccínica e hiperargininemia), y la deficiencia de ornitina carbamil transferasa. Esta última por la presencia en orina de un metabolito específico, el ácido orótico, que constituye la clave del diagnóstico diferencial con la deficiencia de carbamil fosfato sintetasa (ambos defectos tienen un perfil de aminoácidos similar: aumento de glutamina y lisina, y disminución de los niveles de citrulina, arginina y ornitina). Los estudios enzimáticos sólo son indispensables para diagnosticar las deficiencias de carbamil fosfato sintetasa y N-acetil glutamato sintetasa, cuyo perfil de aminoácidos es menos específico; sin embargo, en todos los casos estará indicado llegar hasta el final con todas las posibilidades diagnósticas disponibles [76,77]. Las manifestaciones clínicas son muy variadas, aunque suelen presentarse en el período neonatal (una tercera parte de los casos), con síntomas de intoxicación e hiperamonemia, o bien más tardíamente coincidiendo con infecciones, ingesta proteica exagerada o situaciones de estrés. Las primeras manifestaciones a cualquier edad consisten en el rechazo del alimento, vómitos, alteraciones del tono muscular, convulsiones, letargia y coma. Por su parte, en pacientes con un curso más crónico los síntomas neurológicos dominantes son retraso mental, ataxia y estados confusionales. En la acidemia arginosuccínica se detectan alteraciones del cabello (50% casos) en forma de tricorrexis nodosa, y los pacientes manifiestan irritabilidad, hipertonía muscular, convulsiones rebeldes, ataxia y atetosis [1,8]. Se han comunicado casos de accidentes cerebrovasculares isquémicos en la deficiencia de carbamil fosfato sintetasa [76,77]. La deficiencia de ornitina carbamil transferasa es el defecto del ciclo de la urea más común; su herencia está ligada al cromosoma X. Existe un defecto total de la actividad enzimática en los varones hemicigotos, mientras que las hembras heterocigotas muestran diferentes grados de deficiencia, dependiendo de la inactivación al azar del cromosoma X. La detección de portadoras, más o menos asintomáticas, es muy importante, por el riesgo de crisis de hiperamonemia, para el consejo genético y diagnóstico prenatal. Como el amonio y la excreción de ácido orótico en condiciones basales pueden ser normales, se recurre a la prueba de sobrecarga proteica para poner de manifiesto la alteración de ambos parámetros. La prueba de sobrecarga de alopurinol se utiliza para elevar la excreción urinaria de ácido orótico inhibiendo su catabolismo, lo que evita el peligro de la hiperamonemia causada por la sobrecarga proteica en las heterocigotos de esta deficiencia [77,78]. Alteraciones del metabolismo de los aminoácidos ramificados Leucinosis. Enfermedad de la orina con olor a jarabe de arce La sintomatología clínica de la enfermedad del jarabe de arce o leucinosis es debida al cúmulo de leucina, isoleucina, valina y de 2-oxoisocaproato, 2-oxoisovalerato y 2-oxometilvalerato en plasma y tejidos. Existe una forma grave de presentación durante la primera semana de vida y formas intermitentes que aparecen en los estados catabólicos, manifestadas por vómitos cíclicos, convulsiones generalizadas y afectación del sensorio junto a un olor especial de la orina, y por último una forma más moderada. En estas formas intermitentes y moderadas, sin un tratamiento adecuado los pacientes pueden fallecer en el curso de la descompensación, especialmente en los primeros años de vida [76,79]. El diagnóstico de la leucinosis se basa en el estudio de los aminoácidos plasmáticos y de los ácidos orgánicos en orina, ya que muestra un perfil característico, junto a las anomalías bioquímicas: acidosis metabólica con aumento del anión gap, cetonuria, cetoacidosis, hipocalcemia moderada, hiperlactatemia, hipo o hi- S 14 perglucemia, neutropenia, trombocitopenia, pancitopenia, etc. El tratamiento en la fase aguda debe ser radical, con diálisis peritoneal o exsanguinotransfusión para eliminar rápidamente los aminoácidos ramificados acumulados, al tiempo que se debe aportar energía en forma de glucosa y lípidos; posteriormente se reintroducen las proteínas (con bajo aporte de aminoácidos ramificados) muy lentamente y con estricta monitorización de las concentraciones plasmáticas de los aminoácidos implicados [79]. La confirmación del defecto enzimático se realiza en general con la incorporación de leucina marcada con C14 en leucocitos, fibroblastos y mediante la determinación de la actividad enzimática en fibroblastos. El diagnóstico prenatal es posible en muchos casos mediante análisis de los metabolitos en líquido amniótico y estudio enzimático en amniocitos o biopsia de corion. Defectos del metabolismo de los aminoácidos sulfurados Homocistinuria La alteración más frecuente del metabolismo de los aminoácidos sulfurados es la homocistinuria, que puede ser causada por bloqueo enzimático a diferentes niveles. La incidencia aproximada de la homocistinuria clásica, causada por defecto de la actividad de la enzima cistationina beta sintetasa, es de 1:335.000 recién nacidos vivos. La fisiopatología es aún poco conocida y la afectación intelectual, ocular o esquelética tienen un origen oscuro. En todo caso se trata de un trastorno multisistémico con manifestaciones progresivas, siendo la más constante la luxación del cristalino a partir de los 2 años, también presentan atrofia óptica, desprendimiento de retina, cataratas y opacidades corneales. Son hallazgos frecuentes la osteoporosis, escoliosis u otras deformidades esternales, pies cavos o las anomalías vertebrales. La enfermedad afecta al sistema nervioso con retraso mental como manifestación frecuente (un tercio de los pacientes tienen un CI normal y la media del CI de la población de pacientes con la enfermedad es de 80, siendo más bajo en las formas que no responden a vitamina B 6 ), epilepsia (21%) y trastornos psiquiátricos (cuadros obsesivocompulsivos) (76,81). Los accidentes cerebrovasculares son frecuentes en estos pacientes y pueden manifestarse en varias esperas, en especial en ojos y cerebro con síntomas como hemiparesia, distonía, epilepsia o temblor. El riesgo de los fenómenos tromboembólicos parece superior después de infecciones, estrés o intervenciones. El cuadro clínico y el tratamiento variarán en cada caso, por lo que será necesario realizar el diagnóstico diferencial. La valoración de la homocisteína total es indispensable, ya que incluye las diferentes formas de este aminoácido que se hallan en equilibrio en el plasma: homocisteína ligada a proteínas (hasta un 70-80% de la total), cisteín:homocisteín disulfuro y homocisteína libre (en muy baja concentración en el individuo sano), que es rápidamente oxidada a homocistina [1,5,76,81]. La confirmación del defecto enzimático de la cistationina beta-sintasa se realiza en fibroblastos y linfocitos estimulados con fitohemaglutinina. La detección de heterocigotos es posible mediante sobrecarga oral de metionina y por análisis molecular; es posible establecer el diagnóstico prenatal [82]. El tratamiento pasa por disminuir la concentración plasmática de homocisteína al tiempo que se aumenta la cistina. Los pacientes que responden a vitamina B 6 (100-500 mg/día) tienen un mejor pronóstico. Por el contrario, cuando no responden a la piridoxina requieren entonces dieta baja en metionina con suplementos de cisteína y betaína. Es muy importante la prevención de los fenómenos tromboembólicos mediante un control dietético estricto, una buena hidratación y en ocasiones el empleo de antiagregantes plaquetarios [83]. REV NEUROL 2002; 35 (Supl 1): S3-S20 ERRORES CONGÉNITOS DEL METABOLISMO La deficiencia de metileno tetrahidrofolato reductasa es el más común de lo trastornos del metabolismo del folato y se caracteriza por un deterioro neurológico progresivo de inicio en los primeros años de vida, microcefalia y convulsiones que van desde espasmos infantiles a crisis generalizadas atónicas, mioclónicas o motoras focales. El registro electroencefalográfico demuestra un enlentecimiento difuso, complejos puntaonda y puntas multifocales. Los hallazgos bioquímicos más notables son la elevación de la homocisteína y metionina plasmática, y la presencia de homocistinuria elevada en orina. Los defectos de la biosíntesis de la metionina también pueden asociarse a convulsiones generalizadas, mioclónicas e incluso espasmos infantiles. Su diagnóstico se realiza por la presencia de una anemia megaloblástica, homocistinuria y disminución de lametionina en ausencia de aciduria metilmalónica [84]. La malabsorción del folato debido a un problema digestivo se acompaña de un defecto del transporte de folato y en este caso las dosis elevadas de folato o de ácido folínico pueden mejorar el cuadro. Es importante el diagnóstico diferencial entre éstas y otras entidades causantes de homocistinuria, ya que el tratamiento será muy distinto (vitamina B 6 en CBS y folato en MTHFR) [82]. Histidinemia e hiperprolinemia Se trata de dos trastornos del metabolismo de los aminoácidos que pueden manifestar crisis convulsivas refractarias. La histidinemia debida a una deficiencia de histidinasa (enzima citosólica que cataliza la primera reacción del catabolismo de la histidina, cuyo gen se ha localizado en 12q22-q23) y la hiperprolinemia tipo I, que pueden cursar con deterioro neurológico, mioclonías, espasmos infantiles, crisis focales e incluso reacción de sobresalto; el trazado electroencefalográfico puede mostrar desde un trazado hipsarrítmico a la presencia de ondas delta irregulares. La elevación en plasma de la histidina y de la prolina orientarán el diagnóstico. El tratamiento es sintomático, con restricción de histidina en la primera situación [5,76,77]. Otra causa rara convulsiones y epilepsia es la tirosinemia de tipo III debida al déficit de 4-hidroxifenilpiruvato dioxigenasa [5]. Defectos del transporte Dentro de los defectos de transporte, hay que diferenciar el síndrome de HHH (hiperornitinemia, homocitrulinuria e hiperamonemia), causado por un defecto de transporte intramitocondrial de la ornitina, de la hiperornitinemia con defectos de visión por la atrofia girada de coroides y retina, debido a una deficiencia de actividad enzimática de ornitina amino transferasa, que cursa sin hiperamonemia ni homocitrulinuria [76,77]. Finalmente, el déficit de serina que cursa con microcefalia, deterioro neurológico progresivo y epilepsia rebelde, incluso síndrome de West si no se instaura un tratamiento sustitutivo con este aminoácido. Acidurias orgánicas Las acidurias orgánicas (AO) son trastornos bioquímicos del metabolismo intermediario que afectan a diferentes vías metabólicas de los aminoácidos, ácidos grasos, cetogénesis, cetólisis, vía del piruvato, carbohidratos y del ciclo de Krebbs [75,85]. El número de enfermedades identificadas ha aumentado mucho en los últimos años gracias al empleo de nuevas técnicas bioquímicas. Se trata de un grupo de enfermedades genéticas de carácter autosómico recesivo en las que la disminución o ausencia de actividad de la enzima dará lugar a un cúmulo de ácidos orgánicos con las consiguientes manifestaciones clínicas [75]. Su incidencia es variable, se calcula entre 1:5.000-10.000 recién na- REV NEUROL 2002; 35 (Supl 1): S3-S20 cidos vivos según las series. El diagnóstico se efectúa por la alteración en el perfil de los ácidos orgánicos en los líquidos corporales (orina, plasma, LCR, líquido peritoneal, etc.), mediante cromatografía de gases y posterior identificación de los metabolitos anómalos mediante espectrometría de masas. Es muy importante disponer de un protocolo de estudio para recoger muestras y proceder al análisis de las mismas ante la sospecha de una AO. Muchos pacientes con ECMI fallecen y en la mayoría de las ocasiones los estudios post mortem no ofrecen datos positivos de interés para llegar al diagnóstico definitivo. Por ello es imprescindible disponer de un protocolo de recogida de muestras en caso de fallecimiento de causa desconocida [75]. Una característica común de estas enfermedades es la pobre especificidad clínica; la mayoría se presentan en los primeros días o semanas de vida, pero pueden manifestarse en edades más avanzadas e incluso de forma intermitente, con lo que el diagnóstico es aún más complicado. Si la clínica es poco definida y difícil de identificar, los hallazgos post mortem son inespecíficos, de ahí que el diagnóstico deba efectuarse siempre a partir de las muestras biológicas. Existen un conjunto de signos clínicos y bioquímicos que nos pueden ser útiles para sospechar que estamos delante de una aciduria orgánica, y a partir de estos datos se deberá proceder a una serie de exámenes complementarios, bien pautados, que se completarán finalmente con el análisis cualitativo y posteriormente cuantitativo de los ácidos orgánicos en orina/plasma/LCR. La expresividad clínica de una misma enfermedad es variada y depende en parte del déficit enzimático, de la edad de presentación, de los metabolitos acumulados y del factor desencadenante. Atendiendo a la edad de presentación, las acidurias orgánicas se pueden dividir en las de inicio neonatal, del lactante, formas intermitentes y finalmente formas de lenta evolución [1,6,8,85,86]. En las formas intermitentes es común la fluctuación con períodos de descompensación coincidiendo con infecciones, traumatismos e intervenciones, alternando con fases de aparente normalidad clínica [1,86]. En la fase aguda aparecen vómitos, convulsiones generalizadas o parciales y mioclonías, afectación del estado general con alteración progresiva del sensorio, estado confusional, letargia y coma, o episodios de ataxia intermitente con acidosis metabólica. En las fases de descompensación puede aparecer pancitopenia, acidosis metabólica y cetonuria. Por ello, algunos de estos episodios son etiquetados de vómitos cíclicos acetonémicos cuando en realidad corresponden a formas intermitentes de una aciduria orgánica. Es importante conocerlas y ante la mínima duda recoger una muestra de orina y proceder a su análisis por cromatografía de gases [85]. Las formas de lenta evolución se caracterizan por la práctica ausencia de períodos de descompensación y por la poca especificidad en los síntomas neurológicos: retraso mental, microcefalia, crisis convulsivas rebeldes a la medicación y deterioro lento [86]. Otro dato característico de estas enfermedades es el olor peculiar de los líquidos biológicos, en particular el sudor y la orina, que aparece especialmente en las fases de descompensación de alguna de ellas. La terapia debe iniciarse lo antes posible, aunque no se disponga de la confirmación diagnóstica definitiva. Es importante previo al inicio del tratamiento recoger muestras para los estudios bioquímicos y proceder inmediatamente a las medidas de desintoxicación, al tiempo que se aconseja reducir el aporte proteico al mínimo, suministrando energía no proteica, corrigiendo la acidosis con bicarbonato y empleando cofactores que puedan aumentar la actividad enzimática [37,85,87]. En todas las formas, emplea- S 15 J. CAMPISTOL mos inicialmente una mezcla de cofactores que incluyen biotina, tiamina, riboflavina y piridoxina; una vez confirmado el diagnóstico se selecciona el cofactor adecuado. Si a pesar de estas medidas no se logra una mejoría clinicobiológica debe procederse a la exsanguinotransfusión o diálisis peritoneal como último recurso. Se pueden ensayar suplementos minerales, la extracción del sustrato con algún producto como la L-carnitina (100-500 mg/kg/ día), para favorecer la excreción de los ácidos conjugados con la misma en forma de acilcarnitinas [87]. Una vez estabilizado el cuadro y confirmado el diagnóstico debe procederse al tratamiento dietético específico y al empleo selectivo de cofactores procurando al mismo tiempo evitar las descompensaciones o tratándolas enérgicamente de entrada. A pesar de estas medidas algunas de estas enfermedades siguen un curso tórpido, como por ejemplo la acidemia glutárica tipo I en la que los déficit neurológicos (extrapiramidales) suelen ser permanentes, o algunas formas de acidemia propiónica, en la que las descompensaciones pueden conducir alexitus. En cambio otras, y en función especialmente de una mayor actividad enzimática residual, pueden evolucionar muy favorablemente. La mayoría de las acidurias orgánicas diagnosticadas hasta el momento se heredan con carácter autosómico recesivo. Es posible efectuar el diagnóstico prenatal en muchas de ellas mediante estudios en vellosidades coriónicas o por amniocentesis valorando el cúmulo de ácidos orgánicos y la actividad enzimática, e incluso mediante análisis moleculares. No podemos entrar en detalles con todas y cada una de las más de 80 AO identificadas, algunas de ellas sin manifestaciones en el sistema nervioso. Nos limitaremos a citar las AO que con mayor frecuencia pueden dar lugar a convulsiones en el período de 1-10 años de vida. Estas AO, que por su especial repercusión y manifestaciones clínicas en forma de crisis epilépticas tienen un interés especial para el neuropediatra, vienen referidas en la tabla V [75,88]. Convulsiones piridoxindependientes Se trata de un trastorno metabólico autosómico recesivo resultante de un defecto de la descarboxilasa del ácido glutámico, que condiciona una disminución del GABA en sistema nervioso y que se puede demostrar mediante estudio del LCR [1,5,89]. Habitualmente empiezan en el período neonatal con crisis generalizadas, espasmos tónicos o mioclonías rebeldes a toda medicación antiepiléptica. Existen casos atípicos de convulsiones piridoxindependientes que pueden comenzar más tardíamente, incluso con un síndrome de West como primera manifestación, de aquí el interés en conocer este síndrome y de perseguirlo ante todo paciente con epilepsia de inicio precoz o con síndrome de West de etiología poco clara. El trazado electroencefalográfico puede ayudar al diagnóstico con paroxismos generalizados de elevado voltaje asíncronos o bien anomalías paroxísticas multifocales e incluso complejos punta-onda lenta o trazado hipsarrítmico. En la mayoría de los casos encontraremos una disminución del GABA y elevación del glutamato en LCR como características bioquímicas esenciales [1,2,4]. El déficit enzimático puede confirmarse en fibroblastos y se ha localizado el gen 2q31, pero en otras familias el gen responsable de la descarboxilasa del ácido glutámico se ha localizado en 10p23 y en otras en 5q31. Algunos pacientes responden inicialmente a los FAE y después manifiestan una epilepsia rebelde que solamente se controlará con piridoxina. Cuando responden a la piridoxina debe proseguirse con la medicación por vía oral toda la vida, con excelente respuesta y tolerancia. Existen otras formas atípicas, incluidas dentro de los errores del metabo- S 16 Tabla V. Acidurias orgánicas que pueden acompañarse de epilepsia. Aciduria isovalérica Aciduria propiónica Aciduria metilmalónica, variante CBLC Aciduria 3 metilcrotónica Aciduria 3 metilglutacónica Aciduria 3 hidroxi 3 metil glutárica Aciduria 3-hidroxibutírica Aciduria glutárica I Aciduria N-acetil aspártica Aciduria L-2 hidroxiglutárica Aciduria D-glicérica Déficit de biotinidasa lismo del GABA, que no responden a las dosis habituales y precisan de dosis más altas de piridoxina [42,89]. Ya se ha comentado el déficit de biotinidasa como responsable de convulsiones y epilepsia rebelde si no se instaura un tratamiento sustitutivo con las vitaminas correspondientes. Dieta cetogénica y errores congénitos del metabolismo Desde hace más de 30 años se viene preconizando, con resultados dispares, la dieta cetogénica como alternativa terapéutica en las epilepsias farmacorresistentes. Nosotros mismos la hemos empleado con éxito en algunos casos y con resultados desalentadores en otros, pero no deja de ser una opción terapéutica. Sin embargo, debemos tener muy presente que en algunas formas de ECM el empleo de la dieta cetogénica puede ser extremadamente peligroso para el paciente. No hablamos aquí de la posible deshidratación, hipoglucemia, vómitos o del retraso ponderal, cálculos renales, dislipemia, hiperlipidemia, cardiomiopatía, prolongación QT, neuropatía óptica, elevación de los ácidos grasos de cadena muy larga, déficit de vitamina D, osteomalacia, estreñimiento o exacerbación de reflujo gastroesofágico, complicaciones descritas en pacientes sometidos a dieta cetogénica. Existen una serie de ECM que pueden descompensarse con la aplicación de la dieta cetogénica: porfiria, déficit de piruvato carboxilasa, déficit del transporte de carnitina, defectos de la oxidación de los ácidos grasos y citopatías mitocondriales [90]. En todas estas situaciones, la aplicación de la dieta cetogénica debería estar contraindicada e intentar otras opciones terapéuticas. ORIENTACIÓN DIAGNÓSTICA FRENTE A UN NIÑO CON EPILEPSIA Y SOSPECHA DE UN ERROR CONGÉNITO DEL METABOLISMO Las convulsiones pueden obedecer a múltiples causas etiológicas y pocas veces se relacionan con un ECM. No obstante, es una hipótesis que se debe barajar, especialmente cuando no se pueda demostrar una causa clara de la epilepsia (antecedentes familiares, tumor, malformación vascular, displasia cortical, antecedentes de encefalopatía hipóxica o de infección del SNC). Lógicamente, ante la mínima duda del origen metabólico de una epilepsia se debe profundizar en su conocimiento, sin dejar de lado las demás hipótesis. Se debe tener en cuenta que la normalidad en los REV NEUROL 2002; 35 (Supl 1): S3-S20 ERRORES CONGÉNITOS DEL METABOLISMO Tabla VI. Datos sugestivos de ECM frente a un paciente con epilepsia. Anamnesis Familiares próximos con síntomas similares Familiares próximos fallecidos en circunstancias poco claras Consaguinidad Epilepsia rebelde en familiares próximos Inicio precoz de crisis en ausencia de antecedentes Inicio de epilepsia idiopática en edades no habituales Ausencia de antecedentes personales Epilepsia más deterioro neurológico Epilepsia más afectación sistémica Crisis convulsivas rebeldes a la medicación habitual Crisis convulsivas cíclicas Retraso del crecimiento intrauterino Olor corporal especial Regresión motora y de los aprendizajes Examen físico Macrocefalia, microcefalia Ataxia Demencia Afectación de la oculomotricidad Miopatía Neuropatía periférica Cataratas Alteración del fondo de ojo Sordera Dismorfia Alteraciones del cabello Retraso del crecimiento Organomegalia Datos bioquímicos Hiperamonemia Hiperlactatemia Acidosis metabólica Hipoglucemia Cetonuria Elevación transaminasas Aciduria orgánica Pancitopenia Datos neurofisiológicos Patrón EEG salvas-supresión Anomalías multifocales Hipsarritmia Enlentecimiento difuso Ritmos rápidos Anomalías PEV Anomalías PEATC Alteración ERG Alteración EMG Enlentecimiento VCM, VCS Exámenes complementarios Alteración neuroimagen (sustancia gris, blanca, ganglios basales, cerebelo) RME Punción médula ósea Serie esquelética Ecografía abdominal REV NEUROL 2002; 35 (Supl 1): S3-S20 exámenes complementarios o en los datos analíticos no excluye en absoluto un ECM. Ciertamente, la normalidad de los datos bioquímicos en plena crisis cuestiona el diagnóstico de un ECM, pero nunca lo excluye [5,8,75,85]; se podría establecer un paralelismo con los hallazgos del EEG intercrítico y crítico. La sospecha diagnóstica se basará siempre en el interrogatorio, la edad de aparición de los síntomas, las manifestaciones clínicas y el curso evolutivo. Será imprescindible descartar enfermedades metabólicas mediante los estudios bioquímicos correspondientes. Son de gran utilidad una serie de exploraciones que no siempre serán sistemáticas o imprescindibles, pero cuyos resultados son esenciales: los estudios radiológicos clásicos y estudios por la imagen (TC, RM y RME) se realizan casi siempre, pero sólo en algunas ocasiones serán demostrativos: hipodensidad de la sustancia blanca en las leucodistrofias o de los núcleos grises en el síndrome de Leigh o de Kearn-Sayre; calcificaciones, alteraciones en la mielinización, de la sustancia gris o del cerebelo [4]. Los estudios de LCR serán casi siempre negativos, si bien en algunas entidades (MELAS, Leigh, Alpers) puede existir una elevación del lactato o hiperproteinorraquia, en otros casos la elevación de algún aminoácido, ácido orgánico, del GABA, o simplemente el descenso de la glucorraquia pueden ser muy demostrativos. Los estudios hematológicos comprueban vacuolización o inclusiones anómalas en algunas afecciones metabólicas. Los estudios electrofisiológicos ayudan a descartar enfermedades puramente psiquiátricas y evidencian en ciertas formas de epilepsia una desorganización progresiva de los trazados basales. Los potenciales evocados visuales, auditivos o somatosensitivos son anormales en la mayoría de los procesos y las velocidades de conducción nerviosa motora o sensitiva están retardadas en algunas leucodistrofias; su normalidad, junto a signos de denervación crónica, son típicas de la distrofia neuroaxonal. Finalmente, el examen de muestras biópsicas constituye en algunas de estas enfermedades el único medio diagnóstico. La biopsia cerebral es de aplicación cada vez más restringida; la biopsia de plexo rectal no suele ser superior a la biopsia de piel o conjuntiva; la de tejido muscular o hepático, si bien más cruentas, cada vez se practican con menos riesgos y complicaciones. Actualmente, la biopsia apendicular mediante laparoscopia nos ha permitido obtener muestras de este tejido, para muchas enfermedades por cúmulo, con cierta facilidad. Finalmente, la biopsia de nervio periférico será útil sobre todo para descartar otras afecciones. Los avances en el campo de la biología molecular son espectaculares y permiten hoy en día diagnosticar bastantes enfermedades metabólicas, ofrecer consejo genético y diagnóstico prenatal [75]. Hay que hacer un énfasis especial en la necesidad de una estrecha colaboración entre el neuropediatra, que maneja básicamente un paciente con epilepsia en el que se sospecha un ECM, y el bioquímico. De la evaluación conjunta de los datos clínicos, neurofisiológicos y bioquímicos dependerá en gran parte el diagnóstico. El clínico deberá aportar un resumen de la historia clínica, de los antecedentes, forma de presentación, momento de la recogida de las muestras, dieta y tratamiento efectuado. Con todo ello deberá disponer de una hipótesis diagnóstica que deberá valorarse conjuntamente con el bioquímico experto en metabolopatías. Con los datos clínicos y bioquímicos se deberá establecer un plan diagnóstico (pruebas basales, de sobrecarga, de ayuno, estudio en tejidos, molecular, etc.). Sin todos estos datos y trabajando en solitario es muy difícil, por no decir imposible, llegar al diagnóstico de la mayoría de los ECM que pueden ser la causa etiológica de la epilepsia [1,8,9,75]. Existen una serie de datos de la anamnesis, clíni- S 17 J. CAMPISTOL cos, neurofisiológicos, neurorradiológicos y sobre la respuesta terapéutica que nos podrán poner en guardia sobre una posible hipótesis metabólica como responsable de las convulsiones (Tabla VI). El estudio desde el punto de vista bioquímico a partir de la sospecha clínica obtenida con los datos de la anamnesis, exploración física y con los resultados bioquímicos disponibles, se basa en tres puntos principales: 1. Análisis e identificación de los metabolitos acumulados en los fluidos biológicos (sangre, orina y LCR). 2. Estudio de la proteína alterada para confirmar el defecto de actividad enzimática o del transporte detectado por el análisis de los metabolitos (caracterización de la enzima, electroforesis de la proteína, etc.). 3. Estudio de la mutación del ADN responsable de la proteína alterada. Tabla VII. Pauta de actuación ante un paciente que fallece con epilepsia y sospecha de ECM. En los dos grandes grupos de ECM que tratamos se debe tener en cuenta que en los trastornos del metabolismo de los aminoácidos una sola magnitud puede ser suficiente para orientar y llegar al diagnóstico (p. ej., la determinación plasmática de la fenilalanina en la PKU o la elevación del metilmalonato en orina en la aciduria metilmalónica). Por el contrario, en otras acidurias orgánicas, una sola magnitud bioquímica no será suficiente para llegar al diagnóstico y será preciso evaluar numerosos parámetros biológicos para contribuir a la correcta interpretación de los hallazgos y en especial para llegar al diagnóstico final. También ocurre algo similar en las enfermedades lisosomales o en el síndrome de glicoproteínas deficientes en carbohidratos. En otras ocasiones, una simple radiografía de rodilla puede demostrar las epífisis punteadas características del síndrome de Zellweger o una RME con importante elevación del N-acetil-aspartato, patognomónico de la enfermedad de Canavan [1,4,9]. Hay que tener muy presente que antes de obtener las muestras debemos ya tener una orientación diagnóstica sobre la base de la experiencia clínica, del tipo de crisis epiléptica, de los antecedentes familiares, de la edad de inicio de las convulsiones, de los hallazgos neurofisiológicos y de la neuroimagen, y finalmente de la exploración neurológica. Con los datos de sospecha de un ECM trazaremos un plan estratégico con determinaciones analíticas en sangre/orina/LCR. Es importante tener unas hipótesis diagnósticas, efectuar un diagnóstico diferencial y evaluar qué magnitud debemos determinar y dónde se podrá expresar mejor. Así, por ejemplo, frente a un niño con crisis tónicas rebeldes, deterioro neurológico, alteraciones de cabello y un trazado electroencefalográfico muy desorganizado, una de las hipótesis debe ser la enfermedad de Menkes, y si no determinamos el cobre y la ceruloplasmina plasmáticos es probable que por más análisis y exploraciones complementarias que realicemos, no podamos llegar al diagnóstico, fallezca el paciente y en los estudios patológicos tampoco encontraremos nada específico. Por ello es muy importante disponer de un protocolo de estudio de un paciente que fallece por causas desconocidas o ante un exitus con sospecha de ECM no filiado, procedimiento que venimos aplicando sistemáticamente en nuestro centro desde hace más de 12 años (Tabla VII) [75]. Junto a las determinaciones bioquímicas dirigidas en función de la sospecha diagnóstica se deben solicitar otra serie de exploraciones para completar el abanico de opciones diagnósticas y que podrían pasar, por ejemplo, por un examen del fondo de ojo para descartar la presencia de la mancha rojo cereza o una retinitis pigmentaria; analizar mediante microscopio el cabello, confirmar una 3. Biopsia de piel estéril (0,5 cm diámetro), previa limpieza con alcohol, para cultivo de fibroblastos; colocada en medio cultivo enriquecido/ suero salino estéril a temperatura ambiente (máximo de 24 horas). S 18 1. Inmediatamente después del exitus (no más de una hora) recoger (si no se dispone previamente): 5 mL sangre heparinizada (punción cardíaca), congelar plasma a –20 ºC. Sangre seca en papel de filtro para estudios del ADN 10 mL orina (sondaje/punción suprapúbica) y congelar a –20 ºC Si no se puede obtener orina se debe puncionar el globo ocular para obtener humor acuoso y congelar a –20 ºC Si fuera posible, 10 mL sangre total con EDTA (5%) y congelar a –20 ºC (estudios ADN) 2. Biopsia de hígado (3 cm3) y músculo (2 cm3) envueltos en papel aluminio y congelados a –70 ºC 4. Estudio anatomopatológico neuropatía periférica o detectar depósitos de grasa en la parte baja de la espalda. Otro punto importante es analizar la sintomatología clínica, los antecedentes familiares, la edad de inicio de las convulsiones, el tipo, la respuesta a la medicación y todos los datos que nos pueda ofrecer el examen neurológico, ya de por sí limitado en este grupo de edad. Para el clínico es dificil decidir cuándo hay que proceder a la batería completa de exámenes complementarios ante la posibilidad remota de que pueda tratarse de un ECM. A lo largo de la exposición se han ido detallando algunos de los ECM responsables de manifestaciones epilépticas y por ello debe tener siempre presente que, si bien raras, no son tan excepcionales estas enfermedades. Si pensamos en ellas y las buscamos ante la mínima sospecha de un ECM podremos intentar llegar a una orientación y con la ayuda del bioquímico culminar el diagnóstico. Hemos definido una serie de signos y síntomas generales de sospecha (Tabla VI), y que deben tenerse en cuenta frente a todo niño con epilepsia de etiología poco clara. Con toda la experiencia clínica y el arsenal diagnóstico disponible, podremos llegar en muchas ocasiones al diagnóstico del ECM responsable de una forma u otra de epilepsia de los primeros años de vida. Este diagnóstico puede ser en algunos casos salvador para el niño, como pueden ser las convulsiones piridoxindependientes, el déficit de biotinidasa, la fenilcetonuria o algunas acidurias orgánicas. En otras ocasiones no existe un tratamiento definitivo, pero la confirmación diagnóstica nos permite dar un pronóstico, confirmación molecular, estudiar los portadores, brindar un consejo genético y ofrecer en muchos casos un diagnóstico prenatal en la siguiente gestación. Así pues, sigue siendo prioritario un buen enfoque clínico y una adecuada presunción diagnóstica apoyada en una experiencia y en los exámenes complementarios disponibles y orientados. A pesar de todo ello, bastantes casos de encefalopatías progresivas quedan sin diagnóstico, aun con los métodos de estudio más sofisticados, y constituye un verdadero reto para el neuropediatra el conocer mejor estas enfermedades que se pueden manifestar con convulsiones como uno de lo síntomas capitales, así como investigar conjuntamente con el bioquímico para el mejor enfoque clinicobioquímico y terapéutico. REV NEUROL 2002; 35 (Supl 1): S3-S20 ERRORES CONGÉNITOS DEL METABOLISMO BIBLIOGRAFÍA 1. Aicardi J. Diseases of the nervous system in childhood. 2 ed. London: MacKeith Press; 1998. 2. Allen RJ. Degenerative disorders of the central nervous system. The Practice of Pediatric Neurology. Sant Louis: Mosby CV Eds.; 1989. 3. Arthuis M, Pinsard N, Ponsot G. Neurologie Pediatrique. 2 ed. Paris: Flammarion Medecine Sciences; 1998. 4. Campistol J. Aproximación al diagnóstico de la enfermedades neurometabólicas por la neuroimagen. Rev Neurol 1999; 28: 16-23. 5. Fernández J, Saudubray JM, van den Berghe G, eds. Clinical approach to the inherited metabolic diseases. Berlin: Springer Verlag; 1995. 6. Fejerman N, Fernández Álvarez E. Neurología Pediátrica. 2 ed. Buenos Aires: El Ateneo; 1997. 7. Campistol J. Síndromes epilépticos del período neonatal y errores congénitos del metabolismo. Rev Neurol 2000; 30 (Supl 1): S60-74. 8. Lyon G, Evrard PH. Neuropediatría. Barcelona: Masson; 1990. 9. Cohen BH. Metabolic and degenerative diseases associated with epilepsy. Epilepsia 1993; 34 (Suppl 3): S62-70. 10. Herranz JL, de las Cuevas I. Manifestaciones epilépticas de causa metabólica. Rev Neurol 1999; 28 (Supl 1): S23-8. 11. Valnot I, Ormond S, Gigarel N. Mutations of the SCO 1 gene in mitochondrial cytochrome c oxidase (COX) deficiency with neonatal-onset hepatic failure and encephalopathy. Am J Hum Genet 2000; 67: 1104-9. 12. Wisniewski KE, Zhong N, Philippart M. Pheno/genotypic correlations of neuronal ceroid lipofuscinosis. Neurology 2001; 57: 576-81. 13. Tümer Z, Horn N. Menkes disease: recent advances and new aspects. J Med Genet 1997; 34: 265-74. 14. Guitet M, Campistol J, Medina M. Enfermedad de Menkes: experiencia en el tratamiento con sales de cobre. Rev Neurol 1999; 29: 127-30. 15. Taylor RW, Hayes CM, Chinnery PF, Johnson MA, Kler RS, et al. Epidemiology of pathogenic mitochondrial DNA mutations. Ann Neurol 2000; 48: 188-93. 16. DiMauro S, Schon EA. Mitochondrial DNA mutations in human disease. Am J Med Genet 2001; 10: 18-26. 17. Hirano M, Ricci E, Koenigsberger MR, Defendini R, Pavlakis SG, DeVivo DC, et al. Melas: an original case and clinical criteria for diagnosis. Neuromusc Disord 1992; 2: 125-35. 18. Poulton J, Turnbull DM. 74th ENMC International workshop: mitochondrial diseases. Neuromusc Disord 2000; 10: 460-2. 19. Schon EA, Bonilla E, DiMauro S. Mitochondrial DNA mutations and pathogenesis. J Bioenerget Biomemb 1997; 29: 131-49. 20. Thorburn DR, Dahl HHM. Mitochondrial disorders: genetics, counseling, prenatal diagnosis and reproductive options. Am J Med Genet 2001; 106: 102-14. 21. Meikle PJ, Hopwood JJ, Claque ME, Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999; 281: 249-54. 22. Hobbs JR. Displacement bone marrow transplantation for some inborn errors. J Inher Metab Dis l990; 13: 572-96. 23. Vanier MT, Rousson R. Les sphingolipidoses. Ann Biol Clin l988; 46: 461-8. 24. Soper BN, Lessard MD, Vogler CA, Levy B, Beamer WG, Sly WS. Nonablative neonatal marrow transplantation attenuates functional and physiological defects of beta-glucuronidase deficiency. Blod 2001: 197: 1498-504. 25. Hagberg B, Kollberg H, Sourander P, Akesson HO Infantile globoid cell leukodystrophy (Krabbe’s disease): a clinical and genetic study of 32 Swedish cases 1953-1967. Neuropaediatrie 1970; 1: 74-88. 26. Fu L, Inui K, Nishigaki T, Tatsumi N, Tsukamoto H, Kokubu C, Muramatsu T, Okada S. Molecular heterogeneity of Krabbe disease. J Inherit Metab Dis 1999; 22: 155-62. 27. Loonen MC, van Diggelen OP, Janse HC, Kleijer WJ, Arts WF. Late-onset globoid cell leucodystrophy (Krabbe’s disease). Clinical and genetic delineation of two forms and their relation to the early-infantile form. Neuropediatrics 1985; 16: 137-42. 28. Lyon G, Hagberg B, Evrard P, Allaire C, Pavone L, Vanier M. Symptomatology of late onset Krabbe’s leukodystrophy: the European experience. Dev Neurosci 1991; 13: 240-4. 29. Verdru P, Lammens M, Dom R, van Elsen A, Carton H. Globoid cell leukodystrophy: a family with both late-infantile and adult type. Neurology 1991; 41: 1382-4. 30. Wenger DA. Krabbe disease (globoid cell leukodystrophy). In Rosenberg RN, Prusiner SB, DiMauro S, Barchi RL, eds. The molecular and genetic basis of neurological disease. Boston: Butterworth-Heinemann; 1997. p. 421-31. 31. Van Diggelen OP, Schindler D, Kleijer WS. Lysosomal alpha-Nacetylgalactosaminidase deficiency: a new inherited metabolic disease. Lancet 1987; 2: 804. 32. Desnick RJ, Wang AM. Schindler disease: an inherited neuroaxonal dys- REV NEUROL 2002; 35 (Supl 1): S3-S20 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. 58. 59. trophy due to alpha-N-acetylgalactosaminidase Deficiency. J Inher Metab Dis l990; 13: 549-59. Desnick R, Bishop D. Fabry disease and Schindler disease. In Scriver CH, Beaudet A, Sly W, Valle D, eds. The metabolic basis of inherited disease. New York: McGraw Hill; 1995. Campistol J, Vilaseca MA, Ribes A, Riudor E. Déficit de biotinidasa. Forma de presentación y respuesta al tratamiento. An Esp Ped 1996; 44: 389-92. Salbert BA, Pellock JM, Wolf B. Characterization of seizures associated with biotinidase deficiency. Neurology 1993; 43: 1351-5. McVoy JR, Levy HL, Lawler M, Schmidt MA, Ebers DD, Hart PS, et al. Partial biotinidase deficiency: clinical and biochemical features. J Pediatr 1990; 116: 78-83. Taitz LS, Leonard JV, Bartlett K. Long-term auditory and visual complications of biotinidase deficiency. Early Hum Dev 1985; 11: 325-31. Cole H, Weremowicz S, Morton CC, Wolf B. Localization of serum biotinidase (BTD) to human chromosome 3 in band p25. Genomics 1994; 22: 662-3. Wolf B. Disorders of biotin metabolism. In Scriver CR, Beaudet AL, Sly WS, Valle D, eds. 7 ed. The molecular and metabolic bases of inherited disease. New York: McGraw-Hill; 1995. p. 3151-77. De Vivo, Trifiletti RR, Jacobson RI. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorachia, seizures and developmental delay. N Engl J Med 1991; 325: 703-9. Toone JR, Applegarth AD, Levy HL. Prenatal Dx of non-ketotic hyperglycinemia. J Inher Met Dis 1994; 17: 342-4. García Silva MT. Nuevas enfermedades neurometabólicas: signos clínicos de sospecha. Rev Neurol 1999; 28: 161-8. Philippart M, Engel J Jr, Zimmerman EG. Gelastic cataplexy in Niemann-Pick disease group C and related variants without generalized sphingomyelinase deficiency. Ann Neurol 1993; 14: 492-3. Tedeschi G, Bonavita S, Barton NW, Betolino A, Frank JA, Patronas NJ, et al. Proton magnetic resonance spectroscopic imaging in the clinical evaluation of patients with Niemann-Pick type C disease. J Neurol Neurosurg Psychiatry 1998; 65: 72-9. Patterson MC, Vanier MT, Suzuki K, Morris JA, Carstea E, Neufeld EB, et al. Niemann-Pick disease, type C: a lipid trafficking disorder. In Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Vogelstein B, eds. The metabolic and molecular bases of inherited disease, 8 ed. New York: McGraw-Hill; 2001. Vanier MT, Wenger DA, Comly ME, Rousson R, Brady RO, Pentchev PG. Niemann-Pick disease group C: clinical variability and diagnosis based on defective cholesterol esterification. A collaborative study on 70 patients. Clin Genet 1988; 33: 331-48. Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, et al. Identification of HE1 as the second gene of Niemann-Pick C disease. Science 2000; 290: 2298-301. Greer WL, Dobson MJ, Girouard GS, Byers DM, Riddell DC, Neumann PE. Mutations in NPC1 highlight a conserved NPC1specific cysteinerich domain. Am J Hum Genet 1999; 65: 1252-60. Giraldo P, Giralt M, Pérez-Calvo JJ, Pocoví M, Roca M, Rubio-Feliu D. Enfermedad de Gaucher. In Sanjurjo P, Baldellou A, eds. Errores congénitos del metabolismo. Madrid: Ergón; 2001. Sandhoff K, Conzelmann E. The biochemical basis of gangliosidoses. Neuropediatrics 1984; 15: 85-90. Campistol J, Colomer J, Póo P, Vernet A, Fernández-Álvarez E . Leucodistrofia metacromática. Forma infantil tardía. Revisión de 6 observaciones An Esp Ped 1983; 19: 41-8. Mc Faul R, Cavanagh H, Lake BD, Stephens R, Whitfield AE. Metachromatic leucodystrophy: review of 38 cases. Arch Dis Child 1982; 57: 168-75. Austin JH, McAffec D, Sheares L. Metachromatic form of diffuse cerebral sclerosis. Low sulfatase activity in the urine of nine living patients with metachromatic leukodystrophy. Arch Neurol 1965; 12: 447-50. Nidiffer FD, Kelly TE. Developmental and degenerative patterns associated with cognitive, behavioral and motor difficulties in Sanfilippo syndrome. An epidemiology study. J Ment Defic Res 1983; 27: 185-94. Kaplan P, Wolfe LS. Sanfilippo syndrome type D. J Pediatr 1987; 110: 268-71. Beaudet AL, Thomas GH. Disorders of glycoprotein degradation. In Scriver CH, Beaudet AL, Sly W, Valle D, eds. The metabolic basis of inherited disease. New York: McGraw Hill Inc.; 1995. Vamos E, Libert J, Elkhazen N, Jauniaux E, Hartun J, Wilkin P, et al. Prenatal diagnosis confirmation of infantile sialic storage disease. Prenat Diagn 1986; 6: 437-9. Till JC, Roach ES, Burton BK. Sialidosis types I and II: Neuro-ophthalmic manifestation. J Clin Neuro Ophthalmol 1987; 7: 40-6. Cooper A, Hatton CE, Thornley M, Sardharwalla IB. Alfa and beta mannosidosis. J Inher Metab Dis 1990; 13: 538-48. S 19 J. CAMPISTOL 60. Durand P, Rosanna G, Borrone G. Fucosidosis. In Durand P, O’Brien JS, eds. Genetic errors of glycoprotein metabolism. Berlin: SpringerVerlag; 1982. 61. Goldfisher S, Reddy JK. Peroxisomes in cell pathology. Int Rev Exp Pathol 1984; 26: 45-84. 62. Moser HW, Moser AB. Adrenoleukodystrophy (X-linked) in the metabolic basis of inherited diseases. In Scriver CH, Beaudet AL, Sly WS, Valle D, eds. New York: McGraw-Hill Inc.; 1995. 63. Aubourg P, Blanche S, Jambaque I, Rocchiccioli F, Kalifa G, NaudSaudreau C, et al. Reversal of early neurologic and neuroradiologic manifestations of X-linked adrenoleukodystrophy by bone marrow transplantation. N Engl J Med 1990; 322: 1860-6. 64. Kemp S, Pujol A, Waterham HR, van Geel BM, Boehm CD, Raymond GV, et al. ABCD1 mutations and the X-linked adrenoleukodystrophy mutation database: role in diagnosis and clinical correlations. Hum Mutat 2001; 18: 499-515. 65. Campistol J, Vilaseca MA, Lambruschini N. Hiperfenilalaninemias. In Sanjurjo P, Baldellou A, eds. Errores congénitos del metabolismo. Madrid: Ergón; 2001. 66. Brenton DP, Tarn AC, Cabrera-Abreu JC, Lilburn M. Phenylketonuria: treatment in adolescence and adult life. Eur J Pediatr 1996; 155 (Suppl 1): S93-6. 67. Levy HL. Phenylketonuria: old disease, new approach to treatment. Proc Natl Acad Sci U S A 1999; 96: 1811-3. 68. Scriver CR, Kaufman S. The hyperphenylalaninemias. In Scriver CR, Kaufman S, Eisensmith E, Woo SLC, Vogelstein B, Childs B, eds. The metabolic and molecular bases of inherited disease. 8 ed. New York: McGraw Hill; 2001. 69. Villasana D, Butler IJ, Williams JC, Roongta SM. Neurological deterioration in adult phenylketonuria. J Inherit Metab Dis 1989; 12: 451-7. 70. Pietz J, Dunckelmann R, Rupp A, Rating D, Meinck HM, Schmidt H, et al. Neurological outcome in adult patients with early-treated phenylketonuria. Eur J Pediatr 1998; 157: 824-30. 71. Blau N, Thony B, Cotton RGH, Hyland K. Disorders of tetrahydrobiopterin and related biogenic amines. In Scriver CR, Kaufman S, Eisensmith E, Woo SLC, Vogelstein B, Childs B, eds. The metabolic and molecular bases of inherited disease. 8 ed. New York: McGraw Hill; 2001. 72 Weglage J, Ullrich K, Pietsch M, Funders B, Zass R, Koch HG. Untreated non-phenylketonuric-hyperphenylalaninaemia: intellectual and neurological outcome. Eur J Pediatr 1996; 155 (Suppl 1): S26-8. 73. Guldberg P, Rey F, Zschocke J, Romano V, Francois B, Michiels L, et al. A European multicenter study of phenylalanine hydroxylase deficiency: classification of 105 mutations and a general system for genotype-based prediction of metabolic phenotype. Am J Hum Genet 1998; 63: 71-9. 74. Kwok SC, Ledley FD, DiLella AG, Robson KJ, Woo SL. Nucleotide ERRORES CONGÉNITOS DEL METABOLISMO CON CRISIS EPILÉPTICAS EN LOS PRIMEROS AÑOS DE VIDA Resumen. Objetivo. En el presente trabajo se revisan las principales etiologías de origen metabólico responsables de epilepsia en pacientes pediátricos con edades comprendidas entre 1 y 10 años. Desarrollo. Son muchas las causas etiológicas de las crisis convulsivas. Las convulsiones y epilepsias debidas a errores congénitos del metabolismo constituyen una minoría, sin embargo deben conocerse. La identificación de estos trastornos no es fácil, si bien hoy en día disponemos de un amplio arsenal de métodos diagnósticos para confirmar la sospecha. En esta revisión hemos analizado solamente los errores congénitos del metabolismo que debutan entre los 12 meses y los 10 años con crisis convulsivas. Además los hemos clasificado en dos subgrupos según que la epilepsia constituya uno de los síntomas principales o por el contrario forme parte del cortejo de síntomas y signos neurológicos. Finalmente se establece una aproximación diagnóstica de los errores congénitos del metabolismo con crisis en esta franja de edad. Conclusiones. El neuropediatra, el pediatra y el epileptólogo deben tener muy presentes los errores congénitos del metabolismo como responsables de epilepsia especialmente frente a epilepsias farmacorresistentes o que se presentan acompañadas de otras manifestaciones sistémicas, deterioro neurológico o alteraciones bioquímicas inexplicadas. [REV NEUROL 2002; 35 (Supl 1): S3-20] Palabras clave. Epilepsia. Errores congénitos del metabolismo. Niños. Protocolo diagnóstico. S 20 75. 76. 77. 78. 79. 80. 81. 82. 83. 84. 85. 86. 87. 88. 89. 90. sequence of a full-length complementary DNA clone and amino acid sequence of human phenylalanine hydroxylase. Biochemistry 1985; 24: 556-61. Campistol J. Errores congénitos del metabolismo intermediario en la infancia. Formas de presentación y orientación diagnóstica. Canarias Pediátrica 1997; 7: 99-106. Vilaseca MA. Aminoácidos y derivados. In Fuentes Arderiu X, Castrineiros Lacambra MJ, Queraltó Compañó JM, eds. Bioquímica clínica y patología molecular. 2 ed. Barcelona: Reverté SA; 1998. p. 703-19. Brusilow S, Horwich A. Urea cycle enzymes. In Scriver C, Beaudet A, Sly W, et al, eds. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 1995. Sanjurjo P, Rubio V. Diagnóstico y tratamiento de las enfermedades del ciclo de la urea. In Sanjurjo P, Baldellou A, eds. Errores congénitos del metabolismo. Madrid: Ergón; 2001. Frezal J, Amedee-Manesme O, Mitchell G, Hevertz S, Rey J, Saudubray JM. Maple syrup urine disease: two different forms within a single family. Hum Genet 1985; 71: 89-92. Gallagher PM, Naughten E, Hanson NQ, Schwichtenberg K, Bignell M, Juan M, et al. Characterization of mutations in the cystathionine beta-synthase gene in Irish patients with homocystinuria. Molec Genet Metab 1998; 65: 298-302. Yap S, Rushe H, Howard PM, Naughten ER. The intellectual abilities of early-treated individuals with pyridoxine-nonresponsive homocystinuria due to cystathionine beta-synthase deficiency. J Inherit Metab Dis 2001; 24: 437-47. Kraus JP. Molecular basis of phenotype expression in homocystinuria. J Inherit Metab Dis 1994; 17: 383-90. Mandel H, Brenner B, Berant M, Rosenberg N. Coexistence of hereditary homocystinuria and factor V Leiden: effect on thrombosis. N Engl J Med 1996; 334: 763-8. Couce ML, Fraga JM. Homocistinuria. In Sanjurjo P, Baldellou A, eds. Errores congénitos del metabolismo. Madrid: Ergón; 2001. Hoffman GF, Gibson KM, Trefz KF, Nyhan WL, Bremer HJ, Bignell M, et al. Neurological manifestations of organic acid disorders. Eur J Pediatr 1994; 153 (Suppl 1): S94-100. Brett J. The organic acidurias en pediatric neurology. Edimbourgh: Churchill Livingston; 1991. Roth KS. Inborn errors of metabolism: the essentials of clinical diagnosis. Clin Pediatr 1991; 30: 183-90. Barth PG, Hoffmann GF, Jaeken J, Lehnert W, Hanefeld F, Van Gennip AH, et al. L-2 hydroxyglutaric acidemia: a novel inherited neurometabolic disease. Ann Neurol 1992; 32: 66-71. Gordon N. Pyridoxine dependency: update. Dev Med Child Neurol 1997; 39: 63-5. Wheless JW. The ketogenic diet: an effective medical therapy with side effects. J Child Neurol 2001; 16: 633-5. ERROS CONGÉNITOS DO METABOLISMO COM CRISES EPILÉPTICAS NOS PRIMEIROS ANOS DE VIDA Resumo. Objectivo. No presente trabalho são revistas as principais etiologias de origem metabólica responsáveis pela epilepsia em doentes pediátricos com idades compreendidas entre 1 e 10 anos. Desenvolvimento. São muitas as causas etiológicas das crises convulsivas. As convulsões e epilepsias devidas a erros congénitos do metabolismo constituem uma minoria, e contudo devem ser conhecidas. A identificação destas perturbações não é fácil, embora hoje disponhemos, de um amplo arsenal de métodos de diagnóstico para confirmar a suspeita. Nesta revisão, analisámos apenas os erros congénitos do metabolismo que ocorrem pela primeira vez entre os 12 meses e os 10 anos com crises convulsivas. Além disso, classificámo-los em dois subgrupos, considerando a epilepsia como um dos principais sintomas ou, pelo contrário, como parte do conjunto de sintomas e sinais neurológicos. Por fim, estabelece-se uma aproximação diagnostica dos erros congénitos do metabolismo com crises nesta faixa etária. Conclusões. O neuropediatra, o pediatra e o epileptólogo devem ter sempre presentes os erros congénitos do metabolismo como responsáveis pela epilepsia, especialmente perante epilepsias farmaco-resistentes ou que se apresentem acompanhadas de outras manifestações sistémicas, deterioração cognitiva ou alterações bioquímicas inexplicáveis. [REV NEUROL 2002; 35 (Supl 1): S3-20] Palavras chave. Crianças. Epilepsia. Erros congénitos do metabolismo. Protocolo diagnóstico. REV NEUROL 2002; 35 (Supl 1): S3-S20