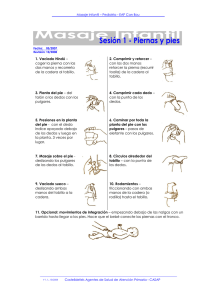
Dismorfias faciales, pulgares y primeros dedos de los pies anchos e
Anuncio

Dermatología Pediátrica Latinoamericana Volumen 10 • Número 2. Mayo/Agosto 2012 haga su diagnóstico Dismorfias faciales, pulgares y primeros dedos de los pies anchos e hipertricosis 75 Leticia Lara-Mendoza Residente de Dermatología Pediátrica Carola Durán-McKinster Jefe del Servicio Luz Orozco-Covarrubias Marimar Sáez-de-Ocariz Carolina Palacios-López Dermatólogas Pediatras Adscritas Ramón Ruiz-Maldonado Investigador Emérito Servicio de Dermatología, Instituto Nacional de Pediatría, México D.F., México CASO CLÍNICO Paciente de sexo femenino, de 12 años de edad, que fue referida al Servicio de Dermatología por presentar exceso de vello. Fue producto de un primer embarazo normoevolutivo, de 38 semanas de gestación. No existían antecedentes heredofamiliares relevantes. A los 3 meses de edad le diagnosticaron ceguera congénita y retraso mental profundo. En la exploración física encontramos los siguientes hallazgos: fisuras palpebrales inclinadas hacia abajo, puente nasal ancho, narinas antevertidas, paladar alto, maxilar inferior hipoplásico, cabeza pequeña, hipertricosis generalizada con predominio en cara, dorso y hombros (Figura 1) conformada por pelo negro terminal. Llamaba la atención que los pulgares y los primeros dedos de los pies así como las uñas eran muy anchos con respecto al resto de los dedos (Figuras 2 A y B). 2A 2B Figura 2A. Dedos pulgares y aparato ungueal anchos. Figura 2B. Primeros dedos de los pies anchos en forma de raqueta, muy característicos de este síndrome. Diagnóstico: Síndrome de Rubinstein-Taybi COMENTARIO Figura 1 Hipertricosis generalizada de pelo terminal. Correspondencia: El síndrome de Rubinstein-Taybi (SRT)1 se caracteriza por retraso en el crecimiento y el desarrollo, cabeza pequeña, dismorfias faciales y retraso mental profundo. La mitad de los pacientes tiene hipertricosis generalizada. Los pulgares y primeros dedos de los pies anchos con un aparato ungueal grande son característicos. La frecuencia del SRT se estima en un caso por cada 100.000 nacidos vivos.2 Su herencia es autosómica dominante, sin Dermatol Pediatr Latinoam (En línea). 2012; 10 (2): 75-6. Carola Durán-McKinster Insurgentes Sur 3700-C, Col. Insurgentes Cuicuilco, México D.F., México CP: 04530 E-mail: [email protected] Dermatología Pediátrica Latinoamericana Volumen 10 • Número 2. Mayo/Agosto 2012 haga su diagnóstico Pulgares y primeros dedos de los pies anchos Leticia Lara-Mendoza et al. embargo, la mayoría de los casos son esporádicos.2 Ocurre por una mutación en el gen CREBBP (50-70% de los casos) o en el gen EP300 (3%).3 Existen también comunicaciones de mosaicismo somático.2 En nuestra paciente se consideró que se trataba de una mutación de novo ya que fue el primer caso en la familia. Cuando existe hipertricosis esta se observa desde el nacimiento, predomina en la cara, el dorso y los hombros y persistirá toda la vida como pelo terminal.2 La apariencia facial es característica: cejas arqueadas, pestañas largas, fisuras palpebrales inclinadas hacia abajo, puente nasal ancho, punta nasal alargada con extensión del tabique nasal por debajo de las alas nasales, paladar alto y micrognatia leve.4 Otros hallazgos son una prominencia ósea triangular en la cara lingual de los incisivos superiores de la dentición permanente, así como dientes supernumerarios que apoyan el diagnóstico, particularmente cuando las manifestaciones 5 faciales son parciales. La hipertricosis es un signo muy importante, sin embargo, el dato clínico que más ayuda a sospechar el diagnóstico de SRT es la presencia de los pulgares y los primeros dedos de los pies anchos.4 De la misma manera que la anoniquia congénita se acompaña de ausencia de falange terminal, los pulgares anchos tienen un aparato ungueal grande y una falange ancha; en caso de duda, una radiografía corroborará el diagnóstico. No es raro que estos pacientes consulten por paroniquia. Otras alteraciones posibles en el SRT son la obstrucción del conducto nasolagrimal, ptosis palpebral, glaucoma, errores en la refracción y ceguera.2 Un tercio de los casos presenta malformaciones cardíacas como persistencia del conducto arterioso, defectos del tabique interventricular e in- terauricular y coartación o estenosis pulmonar.6 Los pacientes con SRT pueden tener anormalidades renales como hidronefrosis, duplicaciones, reflujo vesico-ureteral, cálculos renales y síndrome nefrótico, así como un riesgo mayor de desarrollo de meningioma y leucemia antes de los 15 años7 y de cicatrices queloides8 y pilomatrixomas.2,6 En el caso de los varones, pueden presentar criptorquidia. A pesar de que el SRT se conoce como el síndrome de los pulgares anchos, en la gran mayoría de casos este dato clínico valioso es pasado por alto provocando retraso en el diagnóstico, como sucedió en nuestra paciente. Las anormalidades de los dedos y pulgares pueden observarse también en síndromes con craneosinostosis relacionados con la mutación del gen FGFR2, como el síndrome de Pfeiffer y el síndrome de Apert, que se diferencian del SRT por la craneosinostosis.9 Los pulgares anchos también se observan como característica aislada en la braquidactilia tipo D.9 Aunque las uñas anchas y cortas “en raqueta” pueden presentarse en forma aislada o asociada a síndromes, en ninguno de ellos la uña se acompaña de una falange ancha y grande como en el SRT. Como característica asociada, pueden presentar desviación y ocasionalmente duplicación de las falanges afectadas. Por lo anterior, debemos reconocer las características clínicas relevantes de cada entidad para realizar un abordaje adecuado de nuestros pacientes y evitar retraso en el diagnóstico así como exámenes innecesarios. En la paciente con SRT que presentamos la presencia de pulgares y primeros dedos de los pies anchos fue el marcador clínico más valioso para la sospecha diagnóstica de esta genodermatosis. En todos los casos deberá realizarse el estudio y asesoramiento genético. REFERENCIAS BIBLIOGRÁFICAS 1. Rubinstein JH, Taybi H. Broad thumbs and toes and facial abnormalities. A possible mental retardation syndrome. Am J Dis Child. 1963;105:588-608. 2. Roelfsema JH, Peters DJ. Rubinstein-Taybi syndrome: clinical and molecular overview. Expert Rev Mol Med. 2007; 9:1-16. 3. Bartsch O, Kress W, Kempf O, Lechno S, Haaf T, Zechner U. Inheritance and variable expression in Rubinstein-Taybi syndrome. Am J Med Genet A. 2010; 152A:2254-61. 4. Hennekam RC. Rubinstein-Taybi syndrome. Eur J Hum Genet. 2006; 14:981-5. 5. Gunashekhar M, Hameed MS, Bokhari SK. Oral and dental manifestations in Rubinstein-Taybi syndrome: report of a rare case. Prim Dent Care. 2012; 19:35-8 6. Wiley S, Swayne S, Rubinstein JH, Lanphear NE, Stevens CA. Rubinstein-Taybi syndrome medical guidelines. Am J Med Genet A. 2003; 119A:101-10. 7. Miller RW, Rubinstein JH. Tumors in Rubinstein-Taybi syndrome. Am J Med Genet. 1995; 56:112-5. 8. Goodfellow A, Emmerson RW, Calvert HT. Rubinstein-Taybi syndrome and spontaneous keloids. Clin Exp Dermatol. 1980; 5:369-70. 9. Stevens CA. Rubisntein-Taybi syndrome. [En línea], GeneReviews™, agosto 2009. http://www.ncbi.nlm.nih.gov/books/NBK1526/ [consulta: septiembre 2012]. 76