- Ninguna Categoria
LAS AMILOIDOSIS HUMANAS: CUANDO LAS PROTEÍNAS
Anuncio
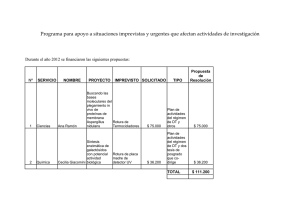
Bustos Jaimes I, Castañeda Patlán C, Oria Hernández J, Rendón Huerta E, Reyes Vivas H, Romero Álvarez I, (eds). Mensaje Bioquímico, Vol XXXII. Depto de Bioquímica, Fac de Medicina, Universidad Nacional Autónoma de México. Cd Universitaria, México, DF, MÉXICO (2008). (http://bq.unam.mx/mensajebioquimico) (ISSN-0188-137X) LAS AMILOIDOSIS HUMANAS: CUANDO LAS PROTEÍNAS MUESTRAN SU LADO OSCURO Luis Del Pozo Yauner1,2*, Sandra Leticia Rodríguez Ambriz3 y Baltazar Becerril1** 1. Instituto de Biotecnología, UNAM. Av. Universidad #2001, Col. Chamilpa C.P. 62210 Cuernavaca, Morelos 2. Instituto Nacional de Medicina Genómica, Periférico Sur No. 4124, Torre Zafiro II, Piso 6 Col. Ex Rancho de Anzaldo, Álvaro Obregón, C.P. 01900, México, D.F. 3. Centro de Desarrollo de Productos Bióticos /IPN. San Isidro Km. 8.5 Carretera YautepecJojutla, Yautepec. C.P. 62730, Morelos *[email protected], **[email protected] Resumen Las proteínas son esenciales para la vida debido a que ejecutan la mayoría de las funciones que le dan soporte. Para cumplir estas funciones, las proteínas deben plegarse en su correspondiente estructura nativa y conservar este estado, aun en las condiciones de estrés físico, químico y funcional que enfrentan tanto en el interior como en el exterior de la célula. Se ha demostrado que numerosas enfermedades humanas y de los animales, reunidas bajo el término de amiloidosis, están asociadas a alteraciones del plegamiento de un grupo particular de proteínas que se depositan en el espacio extracelular en forma de agregados fibrilares insolubles. Los factores y condiciones que determinan este comportamiento patológico son muy diversos, como diversas en secuencia y estructura son las proteínas implicadas. Sin embargo, los agregados fibrilares que forman, denominados amiloides, comparten numerosas propiedades biofísicas y estructurales. Este trabajo trata de los fundamentos de la agregación amiloide de las proteínas y de las características generales de esta clase de agregados. Se analizan las condiciones y factores relacionados con su formación y se describen los elementos básicos de los diferentes modelos estructurales propuestos para explicar las propiedades de las fibras amilolides. Palabras clave: amiloidosis, plegamiento de proteinas, agregación de proteínas. 79 MENSAJE BIOQUÍMICO, Vol. XXXII (2008) Abstract Proteins are essential for life because they carry out most of the cellular functions. In order to display its structural or catalytic roles, proteins must fold to its native structural state and keep this conformation, even in the adverse physicochemical conditions that may face either inside or outside the cell. It is well known that many human and animal diseases, classified together as amyloidosis, are produced by defects in the folding of proteins. These misfolded proteins accumulate as aggregates as insoluble fibrils in the extracellular space. The conditions that determine this pathological behaviour are diverse as is the nature of proteins. However, these fibrils aggregates, also called amyloids, share many biophysical and structural properties. In this work we review the fundamentals of amyloid aggregation of proteins as well as the general properties of the amyloid state. The conditions and factors promoting the amyloid formation are also reviewed in addition to the current structural models and theories to explain the physicochemical properties of amyloid fibrils. Keywords: amyloidosis, protein folding, protein aggregation. Introducción Las proteínas, protagonistas fundamentales de la vida: propiedades estructurales generales Las proteínas son polímeros lineales constituidos por subunidades o bloques menores denominados aminoácidos. Aunque en la naturaleza se han identificado cientos de aminoácidos diferentes, sólo veinte de estos, todos -aminoácidos de la serie L, forman parte de las proteínas [1]. La característica distintiva de una proteína es su estructura primaria, que es el orden lineal o secuencia de sus aminoácidos; ésta representa un código esencialmente unidimensional en el que, sin embargo, está contenida la información necesaria para que la molécula adopte la estructura tridimensional estable y funcional [2,3]. Tanto en las proteínas como en los ácidos nucleicos se convierte la información unidimensional de la estructura primaria en un tipo de información cualitativamente diferente, que es la contenida en la estructura tridimensional de la molécula y que apreciamos en forma de sus propiedades funcionales [2-5]. Sin embargo, es en las proteínas donde este fenómeno, denominado plegamiento, alcanza el más alto grado de complejidad, característica que se sustenta en la mayor diversidad estructural de sus elementos constituyentes y que se corresponde con la gran diversidad de sus funciones [1-3,6]. Las proteínas son las moléculas efectoras por excelencia y ejecutan todas las funciones que requieren carácter informacional, con excepción de la de almacenar y transferir información sobre la secuencia de otras proteínas, función privativa de los ácidos nucleicos [1]. Algunas proteínas pueden plegarse de forma correcta in vitro si se establecen las condiciones adecuadas de pH, temperatura, fuerza iónica, balance redox, concentración de ligandos específicos, entre otras [2,3,6,7]. El plegamiento in vitro de las proteínas ha sido extensamente estudiado mediante el empleo de una amplia variedad de métodos espectroscópicos y bioquímicos, así como mediante simulaciones computacionales [8-10]. Estos estudios han permitido identificar las fuerzas que estabilizan la estructura nativa de las proteínas y describir, en lo general, sus mecanismos de plegamiento [7-14]. Notablemente, se ha observado que muchas proteínas tienden a formar agregados cuando se intenta plegarlas in vitro. Dado que los agregados que forman carecen generalmente de actividad, se concluye que están formados por moléculas que no están plegadas, o que 80 Del Pozo Yauner y cols. poseen un plegamiento diferente del nativo, lo cual sugiere una relación entre los estados de plegamiento no nativo y la proclividad a la agregación [15,16]. La agregación de las proteínas no ocurre únicamente en las condiciones in vitro, ni es una propiedad de un grupo particular de estas moléculas. Abundante evidencia experimental indica que es un fenómeno frecuente, que puede ocurrir tanto dentro como fuera de la célula y que puede afectar, en mayor o menor medida, a casi cualquier proteína [17]. La tendencia de las proteínas a agregarse ha jugado un papel relevante en la evolución y su huella puede observarse a nivel de la estructura de las proteínas mismas y en los numerosos sistemas y grupos de factores, presentes en todos los tipos celulares, cuya función primaria es la de evitar la formación y/o acumulación de agregados proteicos en el interior de la célula, así como fuera de esta [17-20]. El análisis comparativo de la secuencia de cientos de proteínas y los estudios estructurales y biofísicos han aportado evidencias que sugieren que, a través de la evolución, estas moléculas han incorporado elementos de secuencia y/o motivos estructurales cuya función es la de protegerlas de la agregación [18,21]. El efecto protector de estos elementos puede ejercerse tanto durante el plegamiento como luego de que la molécula ha adquirido su estado nativo. En algunas proteínas se ha demostrado que la sustitución de ciertos aminoácidos se asocia a la acumulación de intermediarios estables con plegamiento no nativo que muestran tendencia a la agregación. Estas especies pueden ser componentes de la vía cinética normal o encontrarse fuera de ella, y su acumulación puede responder tanto a variaciones de las constantes cinéticas del plegamiento como de sus equilibrios termodinámicos [14,22,23]. Estos hallazgos pueden interpretarse en términos de cómo las proteínas evolucionaron no sólo para plegarse en forma funcional y estable, sino también para asegurar que este estado se alcance a través de una vía que no represente riesgo de acumulación de formas no nativas con propiedades potencialmente perjudiciales para la célula [24]. Por otra parte, se han identificado motivos estructurales comunes en las proteínas con estructura de tipo a los que se les atribuye función inhibidora de la agregación, debido a su potencial capacidad para impedir o desfavorecer las interacciones intermoleculares que pueden causarla [18,21,25]. Los sistemas de control del plegamiento Todos los organismos vivos poseen complejos sistemas multiproteicos, presentes en los diferentes compartimientos celulares donde ocurre síntesis de proteína, los cuales controlan y asisten el plegamiento de estas moléculas y eliminan a las que no consiguen plegarse correctamente, así como a las que habiendo alcanzado previamente su estado nativo, lo han perdido por el efecto de condiciones ambientales adversas [19,20,26]. Estos sistemas de control del plegamiento de las proteínas se caracterizan por ser redundantes y muy dinámicos, pudiendo responder a los cambios en el microambiente intracelular, ajustando la concentración de varios de sus componentes de acuerdo a la demanda de la célula. Aunque varias de las proteínas que componen estos sistemas son expresadas constitutivamente a alta concentración aún en condiciones fisiológicas, la expresión de otras es inducida intensamente cuando la cantidad de moléculas de proteínas que no pueden alcanzar su plegamiento nativo tiende a incrementarse. Esto último ocurre bajo ciertas condiciones de estrés celular. Este complejo proceso de ajuste de ciertos componentes en respuesta a condiciones de estrés celular se denomina “respuesta a las proteínas no plegadas” [27,28]. Uno de los componentes fundamentales de los sistemas de control del plegamiento son las chaperonas moleculares [20]. Esta clase de proteínas, muy conservadas entre las diferentes especies, unen reversiblemente a las moléculas de proteína de reciente síntesis y, mediante mecanismos diversos que pueden requerir la hidrólisis de ATP, las ayudan a plegarse en su estado nativo [29-31]. Algunas chaperonas pueden revertir el plegamiento de las moléculas que están plegadas incorrectamente, dándoles una nueva oportunidad de iniciar el proceso y potencialmente alcanzar su estado nativo funcional [32]. 81 MENSAJE BIOQUÍMICO, Vol. XXXII (2008) Además de las chaperonas moleculares, los sistemas de control del plegamiento se componen de varias proteasas muy especializadas cuya distribución intracelular semeja a la de las chaperonas. Estas proteasas, cuya función está coordinada con la de las chaperonas, degradan a las moléculas de proteínas plegadas incorrectamente, generalmente una vez que chaperonas específicas las han desplegado convenientemente [33]. La vía fundamental de eliminación de las proteínas incorrectamente plegadas en el compartimiento citoplasmático es el complejo del proteosoma. Este sistema degrada a las proteínas que han sido marcadas para tal fin mediante la unión covalente a su estructura de un número variable de moléculas de ubiquitina [33,34]. La vía del proteosoma es también el destino de algunas de las proteínas sintetizadas en los ribosomas unidos al retículo endoplásmico y que por alguna razón no alcanzaron su estado nativo [19,26,35]. Las enfermedades por plegamiento anormal de las proteínas La existencia en todos los organismos vivos de complejos sistemas de control del plegamiento es evidencia de que las proteínas enfrentan condiciones hostiles en el medio intracelular, que con frecuencia impiden que se plieguen exitosamente. También sugieren la magnitud de los riesgos que entraña para las células la acumulación de moléculas plegadas incorrectamente [6,14,18,36]. El plegamiento incorrecto puede ser causado por mutaciones en la proteína que, como se mencionó antes, pueden modificar la cinética del plegamiento o disminuir la estabilidad del estado nativo [37,38]. Adicionalmente, pueden acumularse moléculas con plegamiento incorrecto en ciertas condiciones de estrés celular que cursan con el incremento sostenido de la síntesis de proteínas y/o provocan la disminución de la capacidad funcional de los sistemas de control del plegamiento [36,39]. Los estados de plegamiento incorrecto generalmente se asocian a la pérdida parcial o total de la función de la proteína involucrada, lo cual puede ser causa de enfermedad; como en los errores congénitos del metabolismo [36,40,41]. Además, como se comentó antes, estos estados se relacionan con frecuencia a la acumulación de agregados en los diferentes compartimientos celulares, así como en el espacio extracelular. En ciertas circunstancias, las proteínas pueden formar agregados que son muy diversos, tanto por su morfología al microscopio electrónico como por su orden interno. Estas diferencias se reflejan en sus propiedades bioquímicas y en su capacidad para interactuar, con muy diversas consecuencias, con diferentes estructuras y componentes celulares [6,14,17,18,24,36]. En los últimos 20 años se ha acumulado abundante evidencia que indica que numerosas enfermedades humanas tienen su causa, o están relacionadas de alguna forma, con alteraciones del plegamiento de proteínas específicas y su acumulación en forma de agregados insolubles. Tanto la pérdida de la función debido al plegamiento incorrecto como la ganancia de propiedades citotóxicas, una vez agregadas, son dos mecanismos patogénicos fundamentales de estas enfermedades, que se reúnen bajo el término de “enfermedades por plegamiento incorrecto de las proteínas” [17,24,36]. Al lector interesado en ampliar sus conocimientos sobre este grupo de enfermedades se le recomienda leer tres magnificas revisiones recientemente publicadas [4244]. Dentro de este grupo de enfermedades, las amiloidosis han sido extensamente estudiadas debido a que muchas de ellas representan problemas de salud de primer orden para los humanos [45]. A continuación describiremos en sus características generales, a este grupo de enfermedades. Amiloidosis: características generales Las amiloidosis son un grupo de enfermedades degenerativas del hombre y los animales, clínica y bioquímicamente muy heterogéneas, que se caracterizan por la deposición 82 Del Pozo Yauner y cols. extracelular de agregados fibrilares insolubles de naturaleza proteica. A este grupo pertenecen enfermedades muy relevantes de la patología humana como la diabetes mellitus tipo II, las enfermedades de Parkinson y Alzheimer, la enfermedad de Creutzfeldt-Jacob -variante en humanos de la encefalopatía espongiforme bovina-, la polineuropatía familiar amiloidótica, la amiloidosis asociada a la diálisis renal crónica y la amiloidosis derivada de las cadenas ligeras (AL), entre otras [45,46]. La heterogeneidad clínica de las amiloidosis deriva de la amplia y diversa distribución tisular de los depósitos amiloides. En las variantes localizadas, como el término lo indica, estos se limitan a un único órgano o tipo de tejido; por ejemplo, en la diabetes mellitus tipo II y la enfermedad de Alzheimer los órganos afectados son el páncreas y el cerebro, respectivamente. En contraste, varios órganos están involucrados en las variantes sistémicas, como la amiloidosis AL [45,46]. La deposición amiloide se asocia a la aparición de un conjunto de signos y síntomas que reflejan el grado de disfunción del órgano u órganos involucrados, condición que generalmente se profundiza con el tiempo si no se modifica mediante acciones terapéuticas. La distribución y cuantía de los depósitos varían notablemente de un tipo de amiloidosis a otro, e incluso, pueden diferir entre pacientes afectados por la misma variante, lo cual determina considerable heterogeneidad en la evolución clínica de los pacientes afectados por estas entidades. Muchas amiloidosis afectan órganos vitales, como el corazón, los riñones, el hígado y el cerebro, por lo que su evolución suele ser generalmente fatal [45,46]. Los precursores de amiloide El componente fundamental de los depósitos en cada tipo de amiloidosis son agregados fibrilares insolubles de una proteína particular o sus fragmentos. Al presente, se han identificado veinticuatro proteínas diferentes cuya agregación fibrilar se asocia a alguna forma clínica de amiloidosis. A estas proteínas se les denomina precursores de amiloide y es notable que no compartan similitud de función, secuencia ni estructura (Tabla I) (45-48). Algunos, como la transtiretina, o prealbúmina, la cual está asociada a diversas formas de amiloidosis, tanto de aparición esporádica como hereditaria [49], son proteínas con plegamiento estable en condiciones fisiológicas (Figura 1). En contraste, otros, como el péptido insular amiloide, o amilina, asociado a la diabetes mellitus tipo II, carecen de plegamiento estable, aun en condiciones fisiológicas. A esta clase singular de proteínas se les denomina “péptidos desordenados en condiciones nativas” [50]. Los precursores de amiloide también difieren en la proporción de elementos de estructura secundaria que caracteriza su plegamiento. Por ejemplo, la insulina, sólo posee estructura secundaria de tipo -hélice, mientras otros, como la 2-microglobulina, son moléculas todo-; un tercer grupo, como la lisozima y la anteriormente mencionada transtiretina, se caracterizan por una proporción diferente de ambas formas de plegamiento [45-49] (Figura 1). Características generales de los amiloides Al margen de la diversidad estructural de los precursores, los amiloides poseen propiedades de apariencia microscópica y sub-microscópica, así como tintoreales y espectroscópicas comunes, lo cual sugiere que comparten elementos estructurales fundamentales [47]. Al microscopio óptico los depósitos tisulares de amiloide tienen aspecto hialino y homogéneo y producen una característica birrefringencia verde manzana cuando son observados al microscopio de luz polarizada, una vez teñidos con Rojo Congo [51]. La unión del colorante rojo Congo a los amiloides determina un cambio en sus propiedades ópticas, lo cual puede usarse para cuantificar los agregados mediante espectroscopia convencional [52] (Figura 83 MENSAJE BIOQUÍMICO, Vol. XXXII (2008) 2A). Otra característica común de los amiloides es la de unir al colorante tioflavina T, lo cual modifica las propiedades de fluorescencia de esta molécula [53] (Figura 2B). Al microscopio electrónico, los depósitos de amiloide están compuestos por manojos de fibras no ramificadas, de aspecto recto y rígido, de longitud variable, pero con diámetro en el orden de los 75 a los 120 Å [54] (Figura 2C). Tabla I. Amiloidosis humanas. Precursor Tipo de plegamiento (Estructura secundaria) Amiloide Sistémica (S) o localizada (L) Cadena ligera de inmunoglobulinas Inmunoglobulinas (Todo-) AL S, L Cadena pesada de inmunoglobulinas Inmunoglobulinas (Todo-) AH S, L 2 microglobulina Inmunoglobulinas (Todo-) A2M S ¿L? (Articulaciones) Transtiretina (Mutantes) (Silvestre) Prealbúmina (+) ATTR Apoproteína sérica AA Desconocido (Todo-) AA S Secundario a inflamación crónica No estructurado en condiciones nativas AApoAI S Familiar Desconocido AApoAII S Familiar Desconocido AApoAIV S Esporádico, asociado al envejecimiento Gelsolina (Mutantes) No estructurado en condiciones nativas AGel S Familiar (Variante finlandesa) Lisozima (Mutantes) Lisozima (+) ALys S Familiar Cadena del Fibrinógeno Desconocido AFib S Familiar Cistatina C (Mutantes) Cistatina (+) ACys S Familiar Polipéptido ABri No estructurado en condiciones nativas ABri S Demencia familiar (Variante Británica) Polipéptido ADan No estructurado en condiciones nativas ADan L Demencia familiar (Variante Danesa) Proteína precursora A No estructurado en condiciones nativas A L Enfermedad de Alzheimer, demencia senil Proteína prion No estructurado en condiciones nativas (segmento 1-120) y hélice (121-230) APrP L Encefalopatías espongiformes transmisibles Apolipoproteína AI (Fragmento Nterminal) Apolipoproteína II (Fragmento Nterminal) Apolipoproteína IV (Fragmento Nterminal) 84 S Síndrome Primario y secundario a mieloma múltiple Primario y secundario a mieloma múltiple Asociado a hemodiálisis Familiar Senil sistémico Del Pozo Yauner y cols. Pro-Calcitonina No estructurado en condiciones nativas ACal L Carcinoma medular del tiroide Polipéptido amiloide insular (Amilina) No estructurado en condiciones nativas AIAPP L Diabetes mellitus tipo II Polipéptido atrial natriurético No estructurado en condiciones nativas AANF L Deposición en Aurículas (Atrios) Prolactina Cuatro hélices de citoquinas (Todo-) APro L Senil (Glándula pituitaria) Prolactinomas Insulina Insulina (Todo-) AIns L Iatrogénico (Sitio de inyección) Lactaderina (Medina) Desconocido AMed L Senil aórtico Keratoepitelina Desconocido AKer L Familiar (Córnea) Lactoferina Proteína periplásmica de unión (+) ALac L Córnea Figura 1. Estructura tridimensional de precursores de amiloide. A) Transtiretina o prealbúmina (PDB 1G1O). B) Lisozima (PDB 1LYY). C) 2 microglobulina (PDB 2F8O) y D) Insulina (PDB 2C8Q). Las regiones plegadas en hebras y hélice se representan en forma de cintas, en color amarillo y rojo, respectivamente. El resto de la molécula, incluyendo las regiones plegadas en asa, se representa como cordones. Nótese que las moléculas se representan con diferente escala. 85 MENSAJE BIOQUÍMICO, Vol. XXXII (2008) El carácter insoluble y la talla de las fibras amiloides ha impedido hasta el presente dilucidar su estructura interna a nivel atómico, pues no son aplicables en su análisis los métodos estructurales convencionales como la difracción de rayos X de monocristales y la resonancia magnética nuclear (RMN) en fase líquida. Los estudios de difracción de rayos X de fibras alineadas, obtenidas tanto de muestras ex vivo como producidas in vitro, dan un patrón común aunque poco informativo que se caracteriza por reflexiones perpendiculares, situadas alrededor de los 4.7 Å en la dirección meridional y alrededor de los 10-12 Å en la dirección ecuatorial. Este patrón denominado “ cruzado”, indica que las fibras amiloides están formadas por hojas extensas orientadas paralelamente al eje longitudinal de la fibra, mientras las hebras que las forman se disponen perpendicularmente a éste eje. Las reflexiones meridionales en 4.7 Å corresponden al espaciado de las hebras adyacentes, mientras que las reflexiones ecuatoriales a los 10-12 Å corresponden a la separación cara-cara de las hojas y sólo se presentan si la unidad estructural menor de la fibra posee dos o más de estas hojas [55] (Figura 3). El estudio, mediante diversos métodos bioquímicos y espectroscópicos, incluyendo la RMN en fase sólida, de los agregados fibrilares producidos in vitro por varios precursores ha aportado información que soporta este modelo estructural de la fibra amiloide [56-59]. Figura 2. Propiedades espectroscópicas y morfológicas de los agregados fibrilares obtenidos in vitro. Espectro de absorción de luz de rojo Congo (A) y de emisión de fluorescencia de tioflavina T (B) en presencia de agregados fibrilares () y de la forma soluble () de un dominio variable recombinante de cadena ligera. Nótese en (A) el incremento de la absorción del colorante en presencia de las fibras, con el característico corrimiento del máximo de absorción de 500 nm a 550 nm. En (B) es evidente el aumento de la emisión de fluorescencia de tioflavina T unido a las fibras, con un máximo en 482 nm. En (C) se muestra micrografía electrónica de los agregados fibrilares analizados en (A) y (B). Además del péptido o la proteína precursora respectivos, otras proteínas y compuestos de naturaleza no proteica están generalmente asociados a las fibras amiloides, por lo que son parte integral de los depósitos. A estas sustancias, entre las que destacan la polipoproteína E, la proteína sérica del amiloide (SAP, por sus siglas en inglés) y los glicosaminoglicanos, entre otros, se les denomina “moléculas accesorias” y se les han atribuido diversas funciones. Se cree que algunos pueden actuar como moduladores de la cinética de agregación, aportando sitios de 86 Del Pozo Yauner y cols. anclaje en la matriz extracelular para los agregados fibrilares. Otros, pueden estabilizar de los agregados, incrementando su resistencia a la acción de las proteasas extracelulares. También se ha sugerido que pueden contribuir a su citotoxicidad [60]. Figura 3. Patrón de difracción de rayos X y estructura cruzada de las fibras amiloides. (A) Bosquejo simple del patrón de difracción de rayos X cruzado. Los componentes típicos del patrón son reflecciones perpendiculares, situadas a aproximadamente 4.7 Å y 10 Å en las direcciones meridional y ecuatorial, respectivamente. La flecha vertical indica el eje longitudinal de la fibra. (B) Representación esquemática de la estructura -cruzada de las fibras amiloides. Ver texto para descripción más detallada. En el caso particular de las moléculas accesorias de naturaleza glucídica glucosaminoglicanos y proteoglicanos- su presencia en los depósitos indujo de inicio a una apreciación errónea sobre la naturaleza química de los amiloides. Por su naturaleza, estos componentes confieren propiedades tintoreales similares a los depósitos de almidón, como la de colorearse de azul pálido cuando se les trata con soluciones de iodo y cambiar subsecuentemente esta coloración a violeta cuando se les adiciona ácido sulfúrico. Esta propiedad llevó al médico alemán Rudolph Virchow a introducir el término “amiloide” –derivado del Latín “amylum” y del Griego “amylon”- para identificar los depósitos anormales, con las propiedades antes descritas, que él observó en lesiones del cerebro de pacientes afectados de enfermedades neurodegenerativas, no bien identificadas entonces. Él supuso que la sustancia responsable de tales propiedades era del tipo de la celulosa o el almidón [61,62]. La amiloidogénesis como consecuencia del plegamiento anormal de las proteínas Las evidencias aportadas por numerosos estudios acerca de los factores y condiciones que modifican la fibrilogénesis in vitro de las proteínas globulares, unido al conocimiento sobre las características clínico-biológicas de los pacientes con amiloidosis, han dado lugar a la “hipótesis conformacional”. El postulado fundamental de esta hipótesis refiere que la agregación amiloide implica la ganancia de un estado de plegamiento total o parcialmente diferente del nativo, el cual puede ser alcanzado a través de vías diferentes dependiendo de la proteína implicada [6,18,24,45-48]. En soporte de esta hipótesis, se ha demostrado que la fibrilogénesis de ciertas proteínas puede promoverse in vitro si se les somete a condiciones que desestabilizan su estructura nativa, como la adición de sustancias de efecto desnaturalizante (cloruro de guanidinio y urea), altas temperaturas, pH extremos, proteólisis parcial, presencia de iones metálicos, etc. [64-68]. Además, se han identificado numerosas mutaciones que están asociadas 87 MENSAJE BIOQUÍMICO, Vol. XXXII (2008) a formas prematuras y/o de evolución más grave de ciertas amiloidosis, las cuales, como regla general, desestabilizan de la estructura nativa de la proteína precursora. Se propone que estas mutaciones favorecen formas de plegamiento no nativo que pueden ser proclives a la agregación fibrilar [14,17,24,69-71]. Como cabría esperase, las mutaciones que estabilizan el plegamiento nativo de la proteína precursora, así como la unión de ligandos específicos o la presencia de ciertos solutos que ejercen el mismo efecto, por lo general disminuyen su tendencia a agregarse en forma fibrilar in vitro [72-75]. El estudio sistemático del efecto de las mutaciones sobre las propiedades biofísicas de los precursores de amiloide permitió reconocer la relación entre la estabilidad termodinámica y la amiloidogénesis. Algunas observaciones sugieren que las mutaciones también podrían promover la agregación a través de su influencia en otras propiedades de la molécula como la solubilidad, las propiedades ácido-básicas, la distribución de cargas de su superficie y la propensión de ciertas regiones a adoptar estructura secundaria de tipo [76]. En algunas formas de amiloidosis los agregados fibrilares están compuestos exclusiva o mayoritariamente por fragmentos de la proteína precursora. Por ejemplo, en la amiloidosis AL, si bien la cadena ligera monoclonal puede estar formando parte de los depósitos fibrilares, es más frecuente encontrar que el componente fundamental de estos son fragmentos de la proteína que incluyen el dominio variable (VL), o este más una porción del dominio constante (CL) [77]. Se ha demostrado, mediante experimentos en un modelo in vivo, que el potencial fibrilogénico de las cadenas ligeras reside en el VL y que los fragmentos que incluyen este dominio poseen generalmente mayor propensión a la agregación fibrilar in vitro que la molécula íntegra [78]. Estos hallazgos sugieren que, en el caso de la amiloidosis AL, la escisión del precursor podría ser un evento importante en el mecanismo de agregación. Sin embargo, al presente no se demostrado inequívocamente el origen de estos fragmentos, o si resultan de la acción de alguna proteasa sobre el precursor. Otro caso en el que la proteólisis del precursor parece jugar un papel trascendental es en la enfermedad de Alzheimer. Acorde a la hipótesis de la cascada amiloide [79], el complejo proceso patológico que da lugar a esta enfermedad se inicia con la deposición extracelular amiloide de los péptidos A, fundamentalmente el A42. Estos depósitos forman el núcleo de la placa senil, lesión que caracteriza la neocorteza y el hipocampo de los pacientes afectados por esta enfermedad neurodegenerativa. Los péptidos A se producen durante el procesamiento proteolítico secuencial de la proteína precursora APP, por las y secretasas, complejos proteicos asociados a la membrana de las células de los mamíferos [80]. APP es una proteína de membrana de 695 residuos de aminoácidos que es escasamente amiloidogénica in vitro, en contraste con la alta tendencia de los péptidos A a formar agregados fibrilares [81]. Se propone que el incremento en la concentración local de los péptidos A, particularmente el A42, el de mayor potencial fibrilogénico, inicia la cadena de eventos que causan la enfermedad [79,80]. En apoyo de esta hipótesis, se ha observado que mutaciones cerca de los sitios de procesamiento de la APP y/o en los genes de las presenilinas 1 y 2, componentes catalíticos del complejo de la secretasa , se asocian al incremento en la producción del péptido A42 y causan una forma hereditaria, autosómica dominante, de la enfermedad de Alzheimer [82-84]. Similarmente, los individuos con síndrome de Down, causado por trisomía del cromosoma 21, donde se localiza en gen de APP, sufren de enfermedad de Alzheimer de inicio temprano, debido a la sobreexpresión del precursor [85]. En algunas formas de amiloidosis de aparición esporádica, la proteína precursora posee mutación alguna ni ha sufrido escisión proteolítica, lo cual indica que este tipo modificaciones no son la única causa de agregación amiloide. En algunas de estos estados, la alteración de la vía normal de catabolismo del precursor y el subsecuente incremento de concentración sistémica, lo que parece exacerbar su propensión a la agregación fibrilar [46]. 88 no de es su Del Pozo Yauner y cols. En los pacientes que sufren de insuficiencia renal crónica que han sido sometidos a hemodiálisis por muchos años, es frecuente encontrar depósitos amiloides que se localizan preferentemente en la membrana sinovial y los tejidos periarticulares, aunque eventualmente puede detectarse deposición en algunos órganos [86,87]. El componente fibrilar de estos depósitos es la 2 microglobulina, proteína que forma parte del antígeno mayor de histocompatibilidad de clase I (MHC-I) y que es catabolizada en los riñones, por lo que su concentración se incrementa varias veces la normal en estos pacientes [86,87]. Algunos hallazgos sugieren que, además del incremento en su concentración, la interacción de la 2 2+ microglobulina con iones de Cu presentes en las membranas y las soluciones de diálisis y probablemente con componentes específicos de la sustancia extracelular, como glucosaminoglicanos y el colágeno, podrían promover la agregación [88-91]. En el caso de los 2+ iones de Cu , se ha demostrado que disminuyen la estabilidad del estado nativo del precursor, lo cual se propone como base molecular de su efecto [88,89]. La amiloidogénesis es un proceso complejo en el que, además de las causales antes mencionadas, influyen probablemente muchos otros factores como los estados que cursan con insuficiencia de los mecanismos celulares de control del plegamiento de las proteínas y de remoción de los agregados que estas tienden a formar cuando no se pliegan correctamente. Las mutaciones que alteran el funcionamiento de proteínas relacionadas al metabolismo y/o la fisiología de los precursores de amiloide también han sido señaladas como parte de las causas moleculares de este grupo de enfermedades [17,24,36,39,46,48]. La demostración de que algunos precursores pueden adoptar estados de plegamiento no nativo bajo condiciones que disminuyen su estabilidad termodinámica y promueven su agregación fibrilar in vitro ha dado lugar a la teoría sobre los intermediarios semi-plegados como componentes claves de la cinética de agregación amiloide, la cual ha constituido el paradigma central en este campo en los últimos 15 años [65-68,92-94]. Modelos moleculares de los amiloides No existe un modelo estructural único que explique las propiedades individuales de todas las fibras amiloides. En base a la información estructural obtenida mediante diversos métodos biofísicos, se propusieron tres modelos básicos, con sus variantes, que explican las propiedades identificadas en algunos tipos particulares, desde la perspectiva de los posibles mecanismos de conversión del estado de plegamiento inicial, representado por el estado nativo del precursor, en el estado fibrilar [95]. Uno de estos modelos, denominado “de replegamiento”, postula que el estado nativo y el fibrilar del precursor son esencialmente diferentes, siendo el primero una entidad definida por las interacciones mediadas por las cadenas laterales de sus residuos constituyentes, mientras que en el segundo son las interacciones intra e intermoleculares, dependientes del esqueleto peptídico, las determinantes. Para que esta transición ocurra, la proteína debe desplegarse totalmente para luego adoptar el estado fibrilar, rico en estructura . En algunos casos, como en el de la proteína prion, se cree que el proceso de replegamiento sólo afecta una región de la molécula [96]. Este modelo también se ha propuesto también para explicar la estructura de la fibra amiloide de la insulina, cuya deposición amiloide se ha observado en el sitio de inyección en pacientes con diabetes mellitus [97,98]. La insulina posee plegamiento mayoritariamente de tipo hélice- (Figura 1C), sin embargo, forma agregados fibrilares de naturaleza amiloide cuando es incubada in vitro a altas temperatura y pH ácido [99]. El análisis de estos agregados mediante espectroscopia de infrarrojo (FTIR) indica que su contenido de estructura alcanza el 90% [100]. Como se aprecia en la Figura 4A, a pH 2.0 y o 60 C, condiciones que promueven su agregación fibrilar, la insulina adopta una conformación parcialmente desplegada caracterizada por pérdida del plegamiento en hélice- del extremo Nterminal de ambas cadenas, fundamentalmente de la A [99]. Fink y cols. han obtenido evidencias sobre la presencia de al menos dos poblaciones de intermediarios no nativos, enriquecidos en 89 MENSAJE BIOQUÍMICO, Vol. XXXII (2008) estructura que participan en la vía cinética de formación de las fibras de insulina [101]. Estos hallazgos en su conjunto dan soporte al modelo de fibrilogénesis por replegamiento de esta proteína. Algunos de los péptidos y proteínas que clasifican como desordenados, o de estructura no definida en condiciones nativas, como el péptido A, la amilina, la huntintina y los priones de levadura Ure2p y Sup35p, forman amiloides in vivo. Se ha propuesto que la incorporación en la estructura -cruzada fibrilar implica que las regiones desordenadas de esta clase de moléculas, o parte de ellas, adopten previamente un plegamiento compacto [95]. En los modelos propuestos para la estructura de las fibras A40 y A42 , basados en datos aportados por diferentes métodos, incluyendo la RMN en fase sólida, ambos péptidos adoptan una conformación denominada “hebra - giro -hebra ”, diferente a la colapsada, pero poco definida que los caracteriza en solución acuosa [56,57]. Estudios experimentales indican que una especie de estructura relativamente compacta, con plegamiento de hélice , es un intermediario clave en la transición entre la forma desordenada en solución y la fibrilar [102]. En el caso de la amilina, los estudios in vitro sugieren que su agregación fibrilar cursa a través de intermediarios oligoméricos ricos en estructura , sin embargo, se ha propuesto que la interacción del precursor con las membranas lipídicas puede modificar la cinética, con la participación de intermediarios ricos en estructura [103]. Un tercer grupo de modelos, reunidos bajo la denominación común de “modelos de ganancia de interacciones”, propone que la formación de la fibra amiloide requiere un reajuste estructural en el precursor que se limita a un segmento menor de su estructura, mientras que el resto de la molécula conserva su estado nativo [95]. Figura 4. Estructura de intermediario proamiloidogénicos de la insulina (A) y la 2microglobulina (B). Ver explicación en el texto para mayor detalle. La estructura en A fue determinada mediante RMN (PDB 1SF1) y en B mediante cristalografía de rayos X (PDB 2F8O). Las regiones con diferente tipo de plegamiento de ambas moléculas se representan como se describe en la Figura 1. La flecha horizontal en B señala la región de la molécula que adopta un plegamiento diferente. Se ha propuesto que la consecuencia es la exposición de regiones de la cadena polipeptídica que normalmente no son accesibles en el estado nativo y que, por la naturaleza de su secuencia, pueden establecer interacciones autocomplementarias, caracterizadas por la interdigitación estrecha de las cadenas laterales de los residuos implicados, formándose un 90 Del Pozo Yauner y cols. “zipper estérico”. Se afirma, basado en datos cristalográficos, que interacciones de esta naturaleza, denominadas “espina ”, son la base de la estructura -cruzada [104]. La ganancia de interacciones parece ser importante en la agregación amiloide de la 2-microglobulina. Se demostró, mediante RMN y cristalografía de rayos X, que dos mutantes altamente fibrilogénicas de este precursor poseen plegamiento muy similar al nativo pero con reajustes locales que implican la eliminación de un motivo protector de la agregación, denominado bulbo-, y la formación de una nueva región de interacción potencial en forma de una hebra externa [93,105,106] (comparar Figuras 1C y 4B). Un mecanismo similar ha sido propuesto para la transtiretina [107]. Se ha demostrado que otras proteínas no asociadas a las enfermedades por deposición amiloide también forman agregados in vitro que por sus propiedades son indistinguibles de los agregados amiloides recuperados ex vivo y post-mortem. Este descubrimiento ha llevado a sugerir que la formación de este tipo de agregados es una propiedad intrínseca de la cadena polipeptídica [108,109]. Consideraciones finales Aunque se ha avanzado en la comprensión de los factores que se asocian a la agregación amiloide de las proteínas, aún no se han esclarecido en detalle el, o los mecanismos moleculares de formación de las fibras y tampoco se ha determinado la estructura a nivel atómico de estos agregados. Esta carencia de información ha dificultado el desarrollo de estrategias terapéuticas eficaces que modifiquen el carácter crónico y el pronóstico sombrío que típicamente acompaña a este grupo de enfermedades. Los aspectos clínicos y moleculares de las amiloidosis y los fenómenos asociados a la amiloidegénesis son, en el presente, campos muy activos de investigación científica. Esto se debe, como se menciona, a que la evolución de la mayoría de estas enfermedades es irremediablemente mortal, o cuando no, provocan un alto grado de invalidez. Ambas características las convierte en una prioridad para los sistemas de salud y estimulan la inversión de cuantiosos recursos en su investigación. Algunas de estas enfermedades, como la diabetes mellitus tipo II, la enfermedad de Alzheimer y la encefalopatía espongiforme bovina son además causa de pérdidas económicas de consideración. Por otra parte, el reconocimiento de las alteraciones del plegamiento como parte fundamental del mecanismo molecular de amiloidogénesis y el reto que el mismo estado amiloide -como forma alternativa de plegamiento- implica para la conceptos tradicionales en el campo, han atraído la atención de muchos investigadores interesados en desentrañar las causas que llevan a una proteína a adoptar un plegamiento diferente del nativo, condición última que se supone es la favorecida por la evolución. Referencias 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. nd Creighton, T.E. (1993) Proteins: structure and molecular properties, 2 Ed., W. H. Freeman & Co; NY Anfinsen, C.B., Haber, E., Sela, M., White, F.H. (1961) Proc. Natl. Aca. Sci. USA 47, 1309-1314 Creighton, T.E. (1990) Biochem. J. 270, 1-16 Qin, Y., Hurley, L.H. (2008) Biochimie [Epub ahead of print]. Geis, M., Flamm, C., Wolfinger, M.T., Tanzer, A., Hofacker, I.L., Middendorf, M., Mandl, C., Stadler, P.F., Thurner, C. (2008) J. Mol. Biol. 379,160-173 Jahn, T.R. & Radford, S.E. (2005) FEBS 272, 5962-5970 Pandit, A.D., Jha, A., Freed, K.F., Sosnick, T.R. (2006) J. Mol. Biol. 361, 755-770 Onuchic J.N. (1997) Proc. Natl. Acad. Sci. USA 94, 7129-7131 Dopson, C.M. (2004) Methods 34, 4-14 Oliveberg, M., y Wolynes, P.G. (2005) Q. Rev. Biophys. 38, 245-288 Murphy, K.P., y Freire, E. (1993) Pure & Appl. Chem. 65, 1939-1946 Fersht, A.R. (1997) Curr. Opin. Struct. Biol. 7, 3-9 Capaldi, A.P., y Radford, S.E. (1998) Curr. Opin. Struct. Biol. 8, 86-92 91 MENSAJE BIOQUÍMICO, Vol. XXXII (2008) 14. 15. 16. 17. 18. 19. 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. 58. 59. 60. 61. 62. 63. 64. 65. 66. Dobson C.M. (2004) Sem. Cell & Develop. Biol. 15, 3-16 Vermeer, A.W.P., y Norde, W. (2000) Biophys. J. 78, 394-404 Chi, E.Y., Krishnan, S., Randolph, T.W., y Carpenter, J.F. (2003) Pharm. Res. 20, 1325-1336 Stefani, M., y Dobson, C.M. (2003) J. Mol. Med. 81, 678–699 Thirumalai, D., Klimov, D.K., Dima, R.I. (2003) Curr. Opin. Struct. Biol. 13, 146-159 Sitia, R., y Braakman, I. (2003) Nature 426, 891-894 Lee, S., y Tsai F.T.F. (2005) J. Biochem. Mol. Biol. 38, 259-265 Richardson, J.S., y Richardson D.C. (2002) Proc. Natl. Acad. Sci. USA 99, 2754-2759 Brockwell, D.J., y Radford, S.E. (2007) Curr. Opin. Struct. Biol. 17, 30-37 Singh, D., Raman, B., Ramakrishna, T., Rao, C.M. (2006) Mol. Vision 12, 1372-1379 Dobson, C.M. (1999) Trends Biochem. Sci. 24, 329-332 Chow, M.K.M., Lomas, D.A., Bottomley, S.P. (2004) Curr. Med. Chem. 11, 491-499 Anelli, A., y Sitia, R. (2008) EMBO J. 27, 315-327 Zhang, K., y Kaufman, R.J. (2006) Neurology 66, S102-109 Bernales, S., Papa, F.R., Walter, P. (2006) Annu. Rev. Cell. Dev. Biol. 22, 487-508 Hartl, F.U., y Hayert-Hartl, M. (2002) Science 295, 1852-1858 Young, J.C., Agashe, V.R., Siegers, K., Hartl, F.U. (2004) Nat. Rev. Mol. Cell. Biol. 5, 781-791 Clarke, A.R. (2006) Mol. Cell. 24, 165-167 Bosl, B., Grimminger, V., Walter, S. (2006) J. Struct. Biol. 156, 139-148 Esser, C., Alberti, S., Höhfeld, J. (2004) Biochim. Biophys. Acta. 1695, 171-188 Smith, D.M., Benaroudj, N., Goldberg, A. (2006) J. Struct. Biol. 156, 72-86 McCracken, A.A., y Brodsky, J.L. (2005) Curr. Top. Microbiol. Immunol. 300, 17-40 Gregersen, N., Bross, P., Vang, S., Christensen, J.H. (2006) Annu. Rev. Genom. Human. Genet. 2006 7, 103-124 Bross, P., Andresen, B.S., Gregersen, N. (2008) Prog. Nucleic. Acid. Res. Mol. Biol. 58, 301-337 Sanchez, I.E., Tejero, J., Gomez-Moreno, C., Medina, M., Serrano, L. (2006) J. Mol. Biol. 363, 422-432 Edor, K., y Durham, H.D. (2006) Biochim. Biophys. Acta 1762, 1038-1050 Gregersen, N., Bross, P., Andresen, B.S., Pedersen, C.B., Corydon, T.J., Bolund, L. (2001) J. Inherit. Metab. Dis. 24, 189-212 Waters, P.J. (2001) Curr. Issues Mol. Biol. 3, 57-65 Herczenik, E., y Gebbink, M.F. (2008) FASEB J. doi: 10.1096/fj.07-099671. Luheshi, L.M., Crowther, D.C., Dobson, C.M. (2008) Curr. Opin. Chem. Biol. 12, 25-31 Soto, C., y Estrada, L.D. (2008) Arch. Neurol. 65, 184-189 Hirschfield, G.M. (2004) Sem. Cell. & Develop. Biol. 15, 39-44 Buxbaum J. (2006) Genes & Immunity 7, 439-449 Sunde, M., y Blake, C.C.F. (1998) Q. Rev. Biophys. 31, 1-39 Chiti, F., y Dobson, C.M. (2006) Annu. Rev. Biochem. 75, 333-366 Hund, E., Linke, R.P., Willig, F., Grau, A. (2001) Neurology 56, 431-435 Uversky, V.N. (2002) Prot. Sci. 11, 739–756 Missmahl, H. P., y Hartwig, M. (1953) Virchows Arch. Path. Anat. 324, 489-508 Klunk, W.E., Jacob, R.F., Mason, R.P. (1999) Methods Enzymol. 309, 285-305 Naiki, H., Higuchi, K., Hosokawa, M., Takeda, T. (1989) Anal. Biochem. 177, 244-249 Cohen, A.S., y Calkins, E. (1959) Nature 183, 1202-1203 Eanes, E.D., y Glenner, G.G. (1968) J. Histochem. Cytochem. 16, 673-677 Petkova, A., Ishii, Y., Balbach, J.J., Antzutkin, O.N., Leapman, R.D., Delaglio, F., Tycko, R. (2002) Proc. Natl. Acad. Sci. USA 99, 16742-16747 Lührs, T., Ritter, C., Adrian, M., Riek-Loher, D., Bohrmann, D., Döbeli, H., Schubert, D., Riek, R. (2005) Proc. Natl. Acad. Sci. USA 102, 17342-17347 Fergunson, N., Becker, J., Tidow, H., Tremmel, S., Sharpe, T.D., Krause, G., Flinders, J., Petrovich, M., Berriman, J., Oschkinat, H., Fersht, (2006) Proc. Natl. Acad. Sci. USA 103, 16248-16253 Iwata, K., Fujiwara, T., Matsuki, I., Akutsu, H., Takahashi, S., Naiki, H., Goto, Y. (2006) Proc. Natl. Acad. Sci. USA 103, 18119-18124 Alexandrescu, A.T. (2005) Prot. Sci. 14,1-12 Virchow, R. (1854) Virchows Arch. 6, 415-426 Sipe, J.D., y Cohen, A.S. J. Struct. Biol. 130, 88-98 Ferreira, S.T., y De Felice, F.G. (2001) FEBS Lett. 498, 129-134 Chitti, F., Webster, P., Taddei, N., Clark, A., Stefani, M., Ramponi, G. Dobson; M. (1999) Proc. Nat. Acad. Sci. USA 96, 3590-3594 Khurana, R., Gillespie, J.R., Talapatra, A., Minert, L.J., Ionescu-Zanetti, C., Millet, I., Fink, A. (2001) Biochemistry 40, 3525-3535 Uversky, V.N., y Fink, A.L. (2004) Biochem. Biophys. Acta, 1698, 131-153 92 Del Pozo Yauner y cols. 67. Vernaglia, B.A., Huang, J., Clark, E.D. (2004) Biomacromol. 5, 1362-1370 68. Ricchelli, F., Buggio, R., Drago, D., Salmona, M., Forloni, G, Negro, A., Tognon, G., Zatta, P. (2006) Biochemistry 45, 6724-6732 69. Stevens, F.J. Pokkuluri, P.R., Schiffer, M. (2000) Biochemistry 39, 15291-15292 70. Terry C.J., Damas, A.M., Oliveira, P., Saraiva, M.J., Alves, I.L., Costa, P.P., Matias, P.M., Sakaki, Y., Blake, C.C. (1993) EMBO J. 12, 735-741 71. Hurle, M.R., Helms, L.R., Li, L., Chan, W., Wetzel, R. (1994) Proc. Natl. Acad. Sci. USA 91, 5446-5450 72. Soldi, G., Bemporad, F., Chiti, F. (2008) J. Am. Chem. Soc. 130, 4295-4302 73. Hammarström, P., Wiseman, R.L., Powers, E.T., Kelly, J.W. (2003) Science 299, 713-716 74. Kim, Y-S., Wall, J.S., Meyer, J., Murphy, C., Randolph, T.W., Manning, M. C., Solomon, A., Carpenter, J.F. (2000) J. Biol. Chem. 275, 1570-1574 75. Kim, Y-S., Cape, S.P., Chi, E., Raffen, R., Wilkins-Stevens, P., Stevens, F.J., Manning, M.C., Randolph, T.W., Solomon, A., Carpenter, J.F. (2000) J. Biol. Chem. 276, 1626-1633 76. Chiti, F., Stefani, M., Taddei, N., Ramponi, G., Dobson, C.M. (2003) Nature 424, 805-808 77. Glenner, G.G., Harbaugh, J., Ohms, J.I., Harada, M., Cuatrecasas, P. (1970) Biochem. Biophys. Res. Comm. 41, 1287-1289 78. Solomon, A., Weiss, D., T., Williams, T., K. (1992) Curr. Top. Microb. & Immunol. 182, 261-267 79. Hardy, J.A., y Higgins, G.A. (1992) Science 286, 184–185 80. Eckman, C.B., y Eckman, E.A. (2007) Neurol. Clin. 25, 669–682 81. Jarrett, J.T., Berger, E.P., Lansbury, P.T., Jr. (1993) Ann. N. Y. Acad. Sci. 695, 144–148 82. Scheuner, D., Eckman, C., Jensen, M., Song, X., Citron, M., Suzuki, N., Bird, T.D., Ardí, J., Hutton, M., Kukull, W., Larson, E., Levy-Lahad, E., Viitanen, M., Peskind, E., Poorkaj, P., Schellenberg, G., Tanzi, R., Wasco, W., Lannfelt, L., Selkoe, D., Younkin, S. (1996) Nat. Med. 2, 864–870 83. Murrell, J., Farlow, M., Ghetti, B., Benson, M.D. (1991) Science 254, 97–99 84. Wolfe, M.S., Xia, W., Ostaszewski, B.L., Diehl, T.S., Kimberly, W.T., Selkoe, D.J. (1999) Nature 398, 513–517 85. Glenner, G.G., y Wong, C.W. (1984) Biochem. Biophys. Res. Comm. 122, 1131–1135 86. Saito, A., y Gejyo, F. (2006) Ther. Apher. Dial. 10, 316–320 87. Takayama, F., Miyazaki, S., Morita, T., Hirasawa, Y., Niwa, T. (2001) Kidney Int. 78, S172-176 88. Eakin, C.M., y Miranker, A.D. (2005) Biochim. Biophys. Acta 1753, 92-99 89. Morgan, C.J., Gelfand, M., Atreya, C., Miranker, A.D. (2001) J. Mol. Biol. 309, 339-345 90. Borysik, A.J., Morten, I.J., Radford, S.E., Hewitt, E.W. (2007) Kidney Int. 72, 174-181 91. Relini, A., De Stefano, S., Torrassa, S., Cavalleri, O., Rolandi, R., Gliozzi, A., Giorgetti, S., Raimondi, S., Marchese, L., Verga, L., Rossi, A,. Stoppini, M., Bellotti, V. (2008) J. Biol. Chem. 283, 4912-4920 92. Wiseman, R.L., Powers, E.T., Kelly, J.W. (2005) Biochemistry 44, 16612-16623 93. Jahn, T.R., Parker, M.J., Homan, S.W., Radford, S.E. (2006) Nat. Struct. Mol. Biol. 13, 195-201 94. McPharland, V.J., Kalverda, A.P., Homans S.W., Radford, S.E. (2002) Nat. Struct. Biol. 9, 326-331 95. Nelson, R., y Eisenberg, E. (2006) Adv. Prot. Chem. 73, 235-282 96. Govaerts, C., Wille, H., Prusiner, S.B., Cohen, F.E. (2004) Proc. Natl. Acad. Sci. USA 101, 8342-8347 97. Jiménez, J.L., Nettleton, E.J., Bouchard, M., Robinson, C.V., Dodson, C.M., Saibil, H.R. (2002) Proc. Natl. Acad. Sci. USA 99, 9196-9201 98. Dische, F.E., Wernstedt, C., Westermark, G.T., Westermark, P., Pepys, M.B., Rennie, J.A., Gilbey, S.G., Watkins, P.J. (1988) Diabetologia 31, 158-161 99. Hua, Q-X., y Weiss, M.A. (2004) J. Biol. Chem. 279, 21449-21460 100. Nielsen, L., Frokjaer, S., Carpenter, J.F., Brange, J. (2001) J. Pharm. Sci. 90, 29-37 101. Ahmad, A., Uversky, V.N., Hong, D., Fink, A.L. (2005) J. Biol. Chem. 280, 42669-42675 102. Kirkitadse, M.D., Condron, M.M., Teplow, D.B. (2001) J. Mol. Biol. 312, 1103-1119 103. Jayasinghe, S.A., y Langen, R. (2007) Biochim. Biophys. Acta 1768, 2002-2009 104. Sawaya, M.R., Sambashivan, S., Nelson, R., Ivanova, M.I., Sievers, S.A., Apostol, M.I., Thompson, M.J., Balbirnie, M., Wiltzius, J.J.W., McFarlane, H.T., Madsen, A., Riekel, C., Eisenberg, D. (2007) Nature 447, 453-457 105. Dobson, C.M. (2006) Nat. Struct. Mol. Biol. 13, 295-297 106. Eakin, C.M., Berman, A.J., Miranker, A.D. (2006) Nat. Struct. Mol. Biol. 13, 202-208 107. Laidman, J., Forse, G.J., Yeates, T.O. (2006) Acc. Chem. Res. 39, 576-583 108. Fändrich, M., Fletcher, M.A., y Dobson, C.M. (2001) Nature 410, 165-166 109. Jiménez, J.L., Guijarro, J.L., Orlova, E., Zurdo, J., Dodson, C.M., Sunde, M., Saibil, H.R. (1999) EMBO J. 18, 815-821 93 MENSAJE BIOQUÍMICO, Vol. XXXII (2008) Semblanza del Dr. Baltazar Becerril El Dr. Baltazar Becerril se tituló de la licenciatura en Biología en la Universidad Autónoma de México en 1979. Posteriormente hizo estudios de Maestría y Doctorado en la misma institución. Es autor de más de 30 artículos científicos y patentes. Durante varios años ha trabajado con anticuerpos para la generación de sistemas de diagnóstico y terapéuticos basados en estas proteínas. El Dr. Becerril también ha estudiado la estabilidad termodinámica de la región variable de anticuerpos con el fin de establecer correlaciones entre la estabilidad y la capacidad de ciertas proteínas para producir fibras amiloides, así como para estudiar las propiedades biofísicas de estas últimas. El Dr. Becerril es investigador del Instituto de Biotecnología de la UNAM. 94
 0
0
Anuncio




Descargar
Anuncio
Añadir este documento a la recogida (s)
Puede agregar este documento a su colección de estudio (s)
Iniciar sesión Disponible sólo para usuarios autorizadosAñadir a este documento guardado
Puede agregar este documento a su lista guardada
Iniciar sesión Disponible sólo para usuarios autorizados