Instrucciones para los titulares de autorización de comercialización
Anuncio

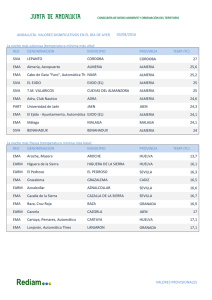
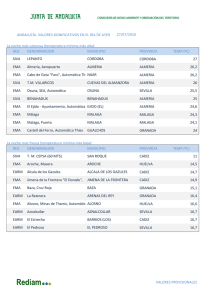
DEPARTAMENTO DE MEDICAMENTOS DE USO HUMANO División de Farmacoepidemiología y Farmacovigilancia NOTIFICACIÓN DE CASOS ESPAÑOLES DE SOSPECHAS DE REACCIONES ADVERSAS PUBLICADOS EN LA LITERATURA MÉDICA INSTRUCCIONES PARA LOS TITULARES DE AUTORIZACIÓN DE COMERCIALIZACIÓN TRAS LA PUESTA EN MARCHA DE LA REVISIÓN DE LA LITERATURA MÉDICA POR LA AGENCIA EUROPEA DE MEDICAMENTOS Fecha de publicación: 18 de junio de 2015 De acuerdo con el artículo 27 del Reglamento nº 726/2004 por el que se establecen los procedimientos comunitarios para la autorización y el control de los medicamentos de uso humano y veterinario y por el que se crea la Agencia Europea de Medicamentos (EMA), “La Agencia (EMA) hará un seguimiento de la bibliografía médica seleccionada en busca de notificaciones de sospechas de reacciones adversas a medicamentos que contengan determinados principios activos. Publicará la lista de los principios activos sometidos al seguimiento y de la bibliografía médica objeto de ese seguimiento”. Por este motivo la EMA pondrá el próximo 1 de julio de 2015 en funcionamiento el servicio de revisión de la literatura médica, Monitoring of Medical Literature (MLM), con el fin de detectar casos de sospechas de reacciones adversas a determinados principios activos incluidos en el listado publicado en su página web. De esta manera, los casos de sospechas de reacciones adversas detectados en la literatura objeto de seguimiento y que cumplan los criterios establecidos en el documento “Monitoring of medical literature and the entry of relevant information into the EudraVigilance database by the European Medicines Agency. Inclusion and exclusion criteria for processing of Individual Case Safety Reports” serán incluidos en EudraVigilance con el formato electrónico adecuado y, posteriormente, serán enviados a las Autoridades Nacionales Competentes del país de origen en la Unión Europea. Asimismo, los Titulares de Autorización de Comercialización (TAC) podrán descargarse los casos individuales en sus bases de datos a través de las herramientas que la EMA ha diseñado a tal efecto. El 12 de mayo de 2015 la EMA ha incluido en su página web un espacio dedicado a MLM donde se encuentra disponible toda la información relacionada con este servicio. Para acceder a esta información puede pinchar aquí. Este servicio entrará en funcionamiento en una primera fase el 1 de julio de 2015 con la revisión inicial de 50 principios activos, siendo a partir del 1 de septiembre de 2015 cuando está prevista la revisión del listado completo de 400 principios activos. CORREO ELECTRÓNICO [email protected] Página 1 de 2 C/ CAMPEZO, 1 – EDIFICIO 8 28022 MADRID TEL: 91 822 53 30/31 FAX: 91 822 53 36 Por lo tanto, a partir del 1 de julio, cuando un TAC identifique un caso en la literatura cuyo país de origen, entendiendo éste como el país del primer autor firmante, sea España deberá comprobar si el caso ya ha sido registrado en las bases de datos de las Autoridades, para lo cual deberá: 1. Revisar el listado de seguimiento de la literatura de la EMA. 2. Si el caso está incluido en el listado, el TAC NO está obligado a notificarlo al Sistema Español de Farmacovigilancia de Medicamentos de uso humano (SEFV-H). 3. Si el caso no está incluido, el TAC revisará en la web de la AEMPS el listado de casos de la literatura notificados al SEFV-H. En este listado de la AEMPS no se registrarán los casos que han sido revisados por la EMA. 4. Si no está incluido en ninguna de las listas anteriores, el TAC deberá notificar el caso al SEFV-H de la manera habitual. A continuación se muestra el proceso resumido en forma de esquema. Página 2 de 2 MINISTERIO DE SANIDAD, SERVICIOS SOCIALES E IGUALDAD Agencia Española de Medicamentos y Productos Sanitarios