Retículo endoplasmático, Aparato de Golgi y Lisosomas Archivo
Anuncio

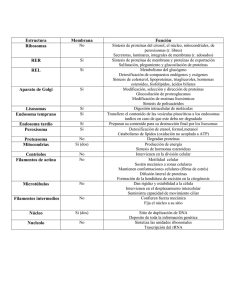
UNIDAD 5. RETÍCULO ENDOPLASMÁTICO Documento elaborado con fines docentes por: GUSTAVO LOZANO CASABIANCA Biólogo M. Sc. Profesor asociado Escuela de Nutrición y Dietética Universidad de Antioquia VIVIANA MARTÍNEZ BETANCUR Bióloga. M. Sc. Universidad de Antioquia SANDRA MILENA JARAMILLO JARAMILLO Nutricionista Dietista Universidad de Antioquia El Retículo Endoplasmático (RE) es una organela dinámica esencial para la vida celular (1). Es un compartimento que comprende una red de túbulos y sacos (cisternas) rodeados de membrana que se extiende desde la membrana nuclear a través del citoplasma (Figura 5.1) (2). Estos túbulos y sacos están interconectados, así que la membrana del RE forma una lámina continua que encierra un único espacio intermembrana llamado lumen (3). La membrana del RE separa el lumen del citosol y media la transferencia selectiva de moléculas entre estos dos compartimentos. La membrana del retículo constituye típicamente la mitad de la membrana total de una célula animal promedio (3). El RE es una organela clave en la denominada ruta de secreción de proteínas (Figura 5.2). Algunas de las proteínas recién sintetizadas entran a esta ruta ingresando al RE desde el citosol. Posteriormente se transportan por medio de vesículas desde el RE hacia el aparato de Golgi y desde allí hacia el exterior celular (3). Figura 5.1 Esquema de una célula donde se muestra el Retículo endoplásmico Figura 5.2 Ruta secretora La membrana del RE es el sitio de producción de las proteínas transmembrana y lípidos para la mayoría de las organelas celulares. Además, casi todas las proteínas que serán secretadas al exterior celular o destinadas al lumen del RE, aparato de Golgi o lisosomas son inicialmente situadas en el lumen del RE (3). RETÍCULO ENDOPLASMÁTICO LISO Estructura Se caracteriza porque no posee ribosomas adheridos a su superficie. En la mayoría de las células estas regiones son escasas y frecuentemente son parcialmente lisas y parcialmente rugosas (3). Funciones El Retículo Endoplasmático liso es el principal sitio en el cual se sintetizan lípidos en las células eucariotas. Las membranas de los eucariotas se componen de tres tipos principales de lípidos: los fosfolípidos, los glicolípidos y el colesterol (2). El RE liso también contiene enzimas que catalizan una serie de reacciones para detoxificar drogas liposolubles y varios compuestos dañinos producidos por el metabolismo (3). En ciertas células especializadas el RE liso es abundante y tiene funciones adicionales. En particular, es usualmente prominente en células que se especializan en el metabolismo lipídico (3). Síntesis de derivados lipídicos Síntesis de hormonas esteroideas: el primer paso se realiza en el interior de las mitocondrias y consiste en la rotura de las cadenas laterales de colesterol para producir pregnenolona. Los siguientes pasos se realizan gracias a enzimas del RE liso y conducen a la formación de testosterona, estrona, estradiol, corticosterona y desoxicortisol. Estas dos últimas pueden penetrar en la mitocondria para transformarse en aldosterona y cortisol respectivamente (4). Síntesis de quilomicrones intestinales: la lipasa pancreática degrada la grasa de la luz intestinal hasta glicerol, ácidos grasos y monoglicéridos. Así la absorben los enterocitos, mediante proteínas de transporte de ácidos grasos (FATP) presentes en la membrana plasmática. Dentro de las células los ácidos grasos quedan unidos a las proteínas de unión de ácidos grasos (FABP), que forman una bolsa en la que se introducen los ácidos grasos, que interaccionan de forma no covalente con la proteína. Los enterocitos transforman los ácidos grasos y el glicerol en triglicéridos, que son exportados unidos a proteínas en forma de quilomicrones. En su síntesis participan tanto el RE rugoso como le RE liso, formando membranas que van a parar al Aparato de Golgi (AG), desde donde las lipoproteínas son segregadas como vesículas con un contenido denso, al espacio extracelular lateral de los enterocitos. Desde allí penetran en los extremos ciegos de los vasos linfáticos, que los conducen a la sangre. Síntesis de lipoproteínas en el hígado: en la sangre, los quilomicrones segregados por los enterocitos son convertidos por las lipasas de lipoproteínas de los vasos sanguíneos en ácidos grasos y glicerol (unidos a transportadores extracelulares de lípidos como la albúmina) y remanentes de quilomicrones (enriquecidos en colesterol), que alcanzan los sinusoides hepáticos. Los ácidos grasos y el glicerol son transportados al citosol del hepatocito mediante las proteínas FATP presentes en la membrana plasmática. Dentro de las células los ácidos grasos quedan unidos a las proteínas FABP. Los remanentes de quilomicrones son endocitados y degradados por los hepatocitos. En los hepatocitos se sintetizan lipoproteínas hepáticas, principalmente las de muy baja densidad (VLDL), que contienen triglicéridos y colesterol. En este proceso intervienen tanto el RE liso como el rugoso y el AG, donde se reúnen ambos componentes para emigrar en vesículas que se vierten por exocitosis del lado de un sinusoide, terminando en sangre (4). Síntesis de ácidos biliares: son derivados del colesterol y se sintetizan a partir de éste en el RE liso de los hepatocitos. Los ácidos biliares se difunden por el citoplasma, ligados a proteínas de unión de lípidos tipo FABP hasta la membrana plasmática (no se desplazan dentro de vesículas), donde hay transportadores de membrana del tipo ABC que los vierten a los canalículos biliares (no al espacio sinusoidal) (4). Destoxificación Un ejemplo de células con abundante RE liso son los hepatocitos, donde el RE liso es extenso y consiste de una red de túbulos interconectados (1). Las enzimas en el RE liso del hígado modifican o detoxifican químicos hidrofóbicos tales como pesticidas y carcinógenos por medio de una conversión química que los hace más solubles al agua y pueden ser secretados del cuerpo. Altas dosis de esos compuestos resultan en una gran proliferación del RE liso de las células de hígado (5). Regulación del nivel de calcio Otra función del RE liso en la mayoría de células eucariotas es el secuestro de iones calcio (Ca2+) del citosol. La liberación de calcio desde el retículo al citosol y su posterior retención, está involucrada en muchas respuestas rápidas a señales extracelulares (3, 6). RETÍCULO ENDOPLASMÁTICO RUGOSO Estructura El RE rugoso se denomina de esta manera debido a que poseen ribosomas unidos a su membrana. Estos ribosomas están encargados de sintetizar ciertas proteínas de membrana y de las organelas, y virtualmente todas las proteínas que van a ser secretadas de la célula (5). Muchas células sintetizan también proteínas para la exportación, como son los componentes estructurales extracelulares de los tejidos. Funciones Síntesis de proteínas La síntesis proteica es una actividad esencial y continua de todas las células (7). Las principales organelas que intervienen en la síntesis proteica son el núcleo y los ribosomas. La síntesis de proteínas consta de dos pasos (8): Transcripción: se sintetiza una molécula de ARN complementaria a la cadena de ADN transcrita o patrón. Las moléculas de ARN mensajero (ARNm) contienen información que especifican las secuencias de aminoácidos de las cadenas polipeptídicas. Traducción: una vez que el ARNm atraviesa el núcleo hacia citoplasma, se une a los ribosomas los cuales traducen el ARNm a una secuencia específica de aminoácidos. Los ribosomas que fabrican proteínas destinadas a algunas organelas como los lisosomas, la membrana plasmática o las que van a ser secretadas, se unen al retículo endoplasmático rugoso. La cadena polipeptídica naciente atraviesa la membrana del retículo con ayuda de proteínas de membrana específicas y entran a la luz del retículo endoplasmático a medida que se produce la traducción (9). En células de mamífero, la importación de proteínas al RE comienza antes de que la cadena polipeptídica esté completamente sintetizada; un proceso denominado co-traduccional. Así, en contraste a la importación de proteínas post-traduccional dentro de las mitocondrias y los cloroplastos, las proteínas no son liberadas al citosol y por lo tanto no sufren plegamiento antes de alcanzar el translocador del RE (3). Existen, por lo tanto 2 poblaciones separadas de ribosomas en el citosol: Ribosomas unidos a membrana: adheridos al lado citosólico de la membrana del RE. Están involucrados en la síntesis de proteínas que están siendo translocadas al RE, el destino de estas proteínas puede ser la membrana plasmática, vesículas secretoras, el RE, el aparato de Golgi o lisosomas (3). Ribosomas libres: no están unidos a ninguna membrana. Sintetizan otras proteínas codificadas por el genoma nuclear que tendrán destinos como el núcleo, las mitocondrias, los cloroplastos o los peroxisomas (Figura 5.3) (3). No obstante es importante señalar que un ribosoma puede sintetizar una proteína adherido al RE y en otro momento sintetizar una proteína libre en el citosol. La síntesis de todas las proteínas comienza en el citosol, sin embargo, las proteínas que deben seguir su síntesis en el RE rugoso tienen una señal molecular en forma de una secuencia peptídica sintetizada al inicio del extremo amino terminal. Cuando existe esta secuencia peptídica llamada péptido señal se interrumpe la síntesis proteica en el citosol gracias a su reconocimiento por una SRP (Partícula de Reconocimiento de la Señal RE) y no prosigue sino hasta que el ribosoma encuentra el RE rugoso y se organiza el translocón en la membrana; el cual permite que la cadena peptídica naciente pase del citosol al lumen del RE. Figura 5.3 Posible destino de las proteínas sintetizadas en ribosomas unidos a membrana y ribosomas libres. Este translocón es un complejo proteico dinámico que forma un poro acuoso que atraviesa la membrana del RE rugoso facilitando el paso de proteínas a través de dicha membrana (10). Por el contrario, si la proteína carece del péptido señal, el ribosoma permanece libre, la síntesis proteica continúa en el citosol y el producto es descargado allí (3). Todos los eucariotas contienen RE rugoso debido a que este es necesitado para la síntesis de proteínas de la membrana plasmática y proteínas de la matriz extracelular. El RE rugoso es particularmente abundante en las células que se especializan en producir proteínas secretadas. Por ejemplo, las células acinares pancreáticas sintetizan enzimas digestivas, las cuales posteriormente son transportadas al intestino. En este tipo de células, una gran parte del citosol está ocupado por el RE rugoso (5). Otras células que tienen mucha demanda en cuanto a síntesis de proteínas y por lo tanto tienen un RE de gran extensión, son las células plasmáticas y las células del hígado (11). El plegamiento de las cadenas polipeptídicas en sus conformaciones tridimensionales correctas, el ensamblaje de los polipéptidos en proteínas constituidas por varias subunidades y las modificaciones covalentes implicadas en el procesamiento de proteínas, puede ocurrir durante la translocación a través de la membrana del RE o en el lumen. Uno de estos procesos es el rompimiento proteolítico de la secuencia señal a medida que la cadena polipeptídica se transloca (2). Las proteínas se translocan a través de la membrana a modo de cadenas polipeptídicas sin plegarse mientras prosigue su traducción. Por lo tanto, estos péptidos se pliegan en su conformación tridimensional en el RE, asistidos por unas proteínas especiales denominadas chaperonas que facilitan el plegamiento de los polipéptidos (Figura 5.4) (2). Únicamente las proteínas que están correctamente plegadas son capaces de alcanzar su destino (12). Figura 5.4 Plegamiento de las proteínas en el RE El RE es también el sitio donde tiene lugar el ensamblaje de proteínas de varias subunidades, la formación de los puentes disulfuro, las primeras etapas de la glicosilación y la adición de anclajes de glicolípidos a algunas proteínas de la membrana plasmática (2). Uno de los fenómenos más conocidos de modificación post-traduccional es la formación de puentes disulfuro, los cuales permiten a la cadena polipeptídica replegarse y adquirir su estructura terciaria (10). Estos puentes se forman entre las cadenas laterales de los residuos cisteína. Estos enlaces no se forman en el citosol, que se caracteriza por ser un ambiente reductor que mantiene los residuos de cisteína en su estado reducido (-SH) (2). El lumen del RE, sin embargo, tiene un ambiente altamente oxidante que facilita la formación de puentes disulfuro (S-S) (13). Adicionalmente, muchas de las proteínas sintetizadas en el RE rugoso sufren un proceso de glicosilación que es de vital importancia para su plegamiento (10, 14). En este proceso, se añaden unidades de oligosacáridos a los aminoácidos asparagina de las cadenas polipeptídicas en crecimiento, mientras están siendo translocadas al RE (2). La glicosilación tiene varias implicaciones importantes: la naturaleza hidrofílica de los carbohidratos aumenta la solubilidad de las glicoproteínas y estabiliza su conformación (15, 16). Eficacia de la síntesis de proteínas La síntesis de proteínas requiere un control que asegure una función adecuada de estas en su destino final (10). Mientras las proteínas que son adecuadamente plegadas y ensambladas salen del RE y progresan a través de la ruta secretora; las proteínas plegadas incorrectamente son retenidas en el RE para que se plieguen correctamente o son marcadas para sufrir un proceso de degradación (15, 16). En ocasiones, los procesos de modificación y plegamiento de las proteínas en el RE presentan errores que llevan a una incorrecta estructura final produciendo proteínas inactivas aberrantes (10). En el caso específico de proteínas no plegadas, se ha encontrado que estas exponen sus aminoácidos hidrofóbicos y tienden a formar agregados proteicos tóxicos que perturban la homeostasis en el RE y crean una condición de estrés (6). Se ha estimado que alrededor del 30% de las proteínas nacientes no se pliegan o son mal plegadas, por lo que deben ser degradadas (12). Un sistema de control de la eficacia de la síntesis en el RE detecta las proteínas mal plegadas o con errores en la glicosilación y las dirige hacia sistemas de eliminación presentes en el citosol (10). Estas proteínas aberrantes del RE sufren un proceso de eliminación dependiente del sistema citosólico ubiquitina-proteasoma por lo que deben translocarse a través de la membrana de esta organela hacia el citosol. Una vez en este sitio, las proteínas son marcadas por medio de la ubiquitinación para su posterior degradación (10). Las ubiquitinas son proteínas pequeñas altamente conservadas que son añadidas a las proteínas que deben ser eliminadas por proteólisis en el proteasoma. En general es requerida una cadena que contenga por lo menos cuatro ubiquitinas para la activación de la degradación. El proteasoma es un complejo proteolítico en forma de cilindro, donde son dirigidas las proteínas aberrantes poliubiquitinadas; una vez la proteína entra, la cadena de poliubiquitinas es hidrolizada, la proteína es degradada y los pequeños péptidos resultantes salen por el otro extremo del proteasoma (10). BIBLIOGRAFIA 1. Lavoie C, Paiement J. Topology of molecular machines of the endoplasmic reticulum: a compilation of proteomics and cytological data. Histochem Cell Biol. 2008; 129:117–128. 2. Cooper GM, Hausman RE. La célula. 4a ed. Madrid: Marbán; 2008. 3. Alberts B, Bray D, Hopkin K, Jonson A, Lewis J, Raff M, Roberts K, Walter P. Introducción a la biología celular. 2a ed. España: Panamericana; 2006. 4. Paniagua R, Nistral M, Sesma P, Álvarez-Uría M, Fraile B, Anadón R, Sáez FJ. Biología Celular. 3a ed. España: McGraw-Hill; 2007. 5. Lodish H, Berk A, Zipursky LS, Matsudaira P, Baltimore D, Darnell J. Biología celular y molecular. 5a ed. Madrid: Panamericana; 2005. 6. Ji C. Dissection of endoplasmic reticulum stress signaling in alcoholic and nonalcoholic liver injury. J Gastroenterol Hepatol. 2008; 23(Suppl 1): S16–S24. 7. Young B, Heath JW. Wheater´s Histología funcional texto y atlas en color. 4 ed. España: Elsevier. 2000. 8. Solomon EP, Berg LR, Martin DW. Biología. 8a ed. México: McGraw-Hill; 2008. 9. Mataix Verdú J. Nutrición y alimentación humana Situaciones fisiológicas y patológicas. Vol 2. España: Oceáno; 2005. 10. Patiño Grajales PJ, Ossa Londoño JE, McEwen JG. Biología de la célula. Medellín: Biogénesis. 2006. 11. Federovitch CM, Ron D, Hampton RY. The dynamic ER: experimental approaches and current questions. Curr Opin Cell Biol. 2005; 17: 409–414. 12. Schubert U, Anton LC, Gibbs J, Norbury CC, Yewdell JW, Bennink JR. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000; 404:770–774. 13. Rajan SS, Srinivasan V, Balasubramanyam M, Tatu U. Endoplasmic reticulum (ER) stress & diabetes. Indian J Med Res. 2007; 125: 411-424. 14. Wormald MR, Dwek RA. Glycoproteins: glycan presentation and protein-fold stability. Struct Fold Des. 1999; 7: 155-60. 15. Ellgaard L, Molinari M, Helenius A. Setting the standards: quality control in the secretory pathway. Science. 1999; 286: 1882–1888. 16. Ellgaard L, Helenius A. ER quality control: towards an understanding at the molecular level. Curr Opin Cell Biol. 2001; 13: 431-7. UNIDAD 6. APARATO DE GOLGI Documento elaborado con fines docentes por: GUSTAVO LOZANO CASABIANCA Biólogo M. Sc. Profesor asociado Escuela de Nutrición y Dietética Universidad de Antioquia VIVIANA MARTÍNEZ BETANCUR Bióloga. M. Sc. Universidad de Antioquia SANDRA MILENA JARAMILLO JARAMILLO Nutricionista Dietista Universidad de Antioquia El aparato de Golgi (AG) es el principal sitio de síntesis de carbohidratos; es considerada una estación de clasificación y envío de productos proteicos a diferentes lugares celulares. La mayoría de proteínas sintetizadas en el retículo endoplasmático (RE) pueden ser incorporadas en vesículas de transporte para ser enviadas al AG (1). Muchos de los polisacáridos de las células son hechos en el aparato de Golgi, incluyendo la pectina y la hemicelulosa de la pared celular de las plantas y la mayoría de los glucosaminoglicanos de la matriz extracelular en animales. Muchos de los oligosacáridos sintetizados en el AG se unen a proteínas y lípidos que son enviados desde el RE (2). Algunos de estos oligosacáridos sirven como etiquetas para dirigir proteínas específicas a vesículas que luego las transportarán a diferentes destinos como lisosomas, membrana plasmática o secreción (3, 4). ESTRUCTURA Morfológicamente el AG está compuesto de sacos aplanados rodeados de membranas (cisternas) y asociados a vesículas (4). Cada cisterna se caracteriza por poseer una región central (lumen) y por tener además espacios entre las cisternas (5). Adicionalmente, cada cisterna cuenta con un set de enzimas importantes para la función del AG (6). El AG tiene una característica sorprendente: su polaridad en cuanto a estructura y función. Las proteínas provenientes del RE entran por la cara cis, la cual es convexa y usualmente orientada hacia el núcleo; son transportadas a través de Golgi por la región medial y salen desde la cara trans que es cóncava (Figura 6.1) (4, 6). Tanto la cara cis como la trans están asociadas con compartimentos especiales, cada uno compuesto de una red de estructuras tubulares y cisternales: la red cis Golgi y la red trans Golgi, respectivamente. Ambas redes son importantes para la clasificación de las proteínas; las proteínas entran a la red cis y se pueden mover a través del AG o regresar al RE (Figura 6.2). Similarmente, las proteínas que salen de la red trans pueden ser clasificadas de acuerdo a si son destinadas a lisosomas, vesículas secretoras o a la superficie celular; o pueden ser devueltas al compartimento anterior (2). Figura 6.1 Aparato de Golgi Figura 6.2 Movimiento de las proteínas a través del RE y del AG La asimetría de las caras del AG se refleja en la morfología de las membranas de las cuales se forma. La cara cis está hecha de membranas de 5.5 nm de grueso como las del RE; mientras que las de la cara trans tienen 10 nm (5). Una de las características del AG es su posición adyacente al núcleo, lo cual es debido a la acción de los microtúbulos (7). Esta organela es especialmente prominente en las células que son especializadas en la secreción, tal como las células del epitelio intestinal, las cuales secretan grandes cantidades de sustancias mucosas ricas en polisacáridos. En tales células, se encuentran grandes vesículas en la cara trans del AG, la cual está orientada hacia la membrana plasmática donde ocurre la secreción (2). FUNCIONES Glicosilación de proteínas A medida que las proteínas se mueven a través del AG, los oligosacáridos que ya están unidos pueden ser modificados y oligosacáridos adicionales pueden ser adheridos. Esta glicosilación desempeña un rol importante ya que está involucrada en la decisión de clasificación en la red trans-Golgi, para el envío de las proteínas a su destino final (5). La modificación de las proteínas ocurre en una secuencia organizada, con cada cisterna conteniendo varias enzimas involucradas. Las diferencias funcionales entre las regiones cis, medial y trans del Golgi yacen en las diferentes enzimas relacionadas con el proceso de glicosilación (2). Las proteínas son modificadas en el RE por la adición de un oligosacárido que tiene 14 residuos de azúcar; de estos, 3 residuos de glucosa y uno de manosa son removidos mientras los polipéptidos todavía se encuentran en el RE. Dentro del AG los oligosacáridos N-ligados de estas glicoproteínas están sujetos a modificaciones adicionales (4). Diferentes glicoproteínas son modificadas en distinto grado durante su paso a través de Golgi dependiendo tanto de la estructura de la proteína como de la cantidad de enzimas involucradas en el procesamiento que están presentes en el complejo de Golgi de diferentes tipos de células. Consecuentemente, las proteínas pueden emerger del AG con una variedad de oligosacáridos N-ligados (4). ¿Cuál es el propósito de la glicosilación? Mientras los ácidos nucléicos y las proteínas son copiadas a partir de un molde en una serie repetida de pasos idénticos usando la misma enzima o set de enzimas, los carbohidratos complejos requieren diferentes enzimas en cada paso. La glicosilación N-ligada es prevalente en todos los eucariotas pero ausente en los procariotas. Debido a que uno o más oligasacáridos N-ligados están presentes en la mayoría de las proteínas transportadas a través del RE y el AG, se sospecha que ellos funcionan ayudando en el plegamiento y el proceso de transporte (2). Debido a que las cadenas de azúcares tienen flexibilidad limitada, incluso un pequeño oligosacárido que sobresale de la superficie de la glicoproteína puede limitar la aproximación de otras macromoléculas a la superficie de la proteína. De esta manera, la presencia de oligosacáridos tiende a hacer las proteínas más resistentes a la digestión por proteasas. Adicionalmente, algunos oligosacáridos adheridos a proteínas de superficie celular son reconocidos por las selectinas, unas proteínas que están involucradas en el proceso de adhesión célula a célula (2). Clasificación de las proteínas La red trans-Golgi es la principal estación de clasificación del tráfico intracelular de proteínas y de lípidos (8). Aunque hay algún procesamiento final de las proteínas en la red trans-Golgi, la mayoría que llegan a este punto han recibido todas las modificaciones necesarias para hacerlas completamente funcionales y especificar su destino final. Además, la red trans-Golgi es el lugar donde las proteínas son clasificadas en varias vesículas apropiadas y enviadas a una de las siguientes rutas principales: secreción, membrana plasmática, lisosomas y recuperación de proteínas residentes en el RE o en el AG (5, 9). En los dos primeros casos; las vesículas son dirigidas hacia la membrana plasmática y cuando entra en contacto con ella, se fusiona. En el punto de fusión la membrana se rompe y el contenido de la vesícula sale al espacio extracelular. Este proceso se denomina exocitosis e implica que la membrana de la vesícula se incorpore también en la membrana plasmática; consecuentemente las proteínas integrales y los lípidos de la vesícula llegan a pertenecer a la membrana plasmática. Esta es la principal manera en que las proteínas integrales fabricadas en el retículo endoplasmático son añadidas a la membrana plasmática (5). Existen dos tipos de secreción: Secreción constitutiva: esta ruta siempre está “encendida”, es decir, las vesículas que contienen proteínas de secreción continuamente realizan exocitosis. Secreción regulada: la vesícula que contiene proteínas de secreción se acumula en el citoplasma hasta que reciben una señal específica, usualmente un incremento en la concentración de iones calcio en torno al citosol, y es entonces cuando se lleva a cabo la exocitosis de forma rápida (Figura 6.3). Figura 6.3 Trasporte de proteínas desde el AG La ruta mejor caracterizada de clasificación de proteínas en el AG es el transporte selectivo de proteínas a los lisosomas. En levaduras y células vegetales, con carencia de lisosomas, las proteínas son transportadas del AG a un destino adicional: la vacuola, la cual asume las funciones del lisosoma y además ejecutan otro tipo de tareas, tal como almacenamiento de nutrientes. La adición de grupos fosfato a la posición 6 de residuos de manosa es la señal que dirige las proteínas lisosómicas, en contraste las proteínas que van dirigidas a vacuolas son marcadas por una secuencia peptídica corta en lugar de carbohidratos (4). Las proteínas residentes en el RE y en el AG tienen secuencias cortas de aminoácidos que sirven de señal de retención o recuperación de estas proteínas desde compartimentos posteriores. Metabolismo de los lípidos y los polisacáridos En adición a las actividades relacionadas con el procesamiento y clasificación de las glicoproteínas, el AG participa en el metabolismo lipídico, en particular en la síntesis de glicolípidos y esfingomielina. Los glicerol-fosfolípidos, el colesterol y la ceramida son sintetizados en el RE; por su parte, la esfingomielina y los glicolípidos son sintetizados a partir de la ceramida en el AG (4). Los lípidos unidos a proteínas (lipoproteínas), como los quilomicrones producidos en los enterocitos y las lipoproteínas hepáticas, pasan por el AG, desde allí emigran en vesículas que se vierten por exocitosis al exterior. Lo que se ignora es si, en estos casos, el AG sirve únicamente de embalaje para el transporte o si desempeña algún papel bioquímico importante en la liberación de estas sustancias (10). En las plantas, el AG tiene una tarea adicional de servir como el sitio de síntesis de polisacáridos complejos de la pared celular. La hemicelulosa y la pectina, componentes importantes de la pared celular, son moléculas sintetizadas en el AG que luego son transportadas en vesículas a la superficie de la célula. La síntesis de estos polisacáridos de las plantas es una tarea importante de esta organela: Aproximadamente el 80% de la actividad metabólica del AG en las células vegetales puede ser dedicada a la síntesis de polisacáridos (4). BIBLIOGRAFIA 1. Bannykh SI, Nishimura N, Balch WE. Getting into the Golgi. Trends Cell Biol. 1998; 8: 21–25. 2. Alberts B, Bray D, Hopkin K, Jonson A, Lewis J, Raff M, Roberts K, Walter P. Introducción a la biología celular. 2a ed. España: Panamericana; 2006. 3. Farquhar MG, Palade GE. The Golgi-apparatus-100 years of progress and controversy. Trends Cell Biol. 1998; 8: 2–10. 4. Cooper GM, Hausman RE. La célula. 4a ed. Madrid: Marbán; 2008. 5. Bolsover SR, Hyams JS, Shephard EA, White HA, Wiedemann CG. Cell Biology. 2a ed. John Wiley & sons; 2004. 6. Pfeffer SR. Constructing a Golgi complex. J Cell Biol. 2001; 155(6): 873–875. 7. Barr FA, Egerer J. Golgi positioning: are we looking at the right MAP? J Cell Biol. 2005; 168(7): 993–998. 8. Keller P, Simons K. Post-Golgi biosynthetic trafficking. J Cell Sci. 1997; 110: 3001-3009. 9. Luini A, Mironov AA, Polishchuk EV, Polishchuk RS. Morphogenesis of postGolgi transport carriers. Histochem Cell Biol. 2008; 129: 153-161. 10. Paniagua R, Nistral M, Sesma P, Álvarez-Uría M, Fraile B, Anadón R, Sáez FJ. Biología Celular. 3a ed. España: McGraw-Hill; 2007. UNIDAD 7. LISOSOMAS Documento elaborado con fines docentes por: GUSTAVO LOZANO CASABIANCA Biólogo M. Sc. Profesor asociado Escuela de Nutrición y Dietética Universidad de Antioquia VIVIANA MARTÍNEZ BETANCUR Bióloga. M. Sc. Universidad de Antioquia SANDRA MILENA JARAMILLO JARAMILLO Nutricionista Dietista Universidad de Antioquia Son organelas que funcionan como el sistema digestivo de las células, sirviendo tanto para degradar material proveniente del exterior de la célula, como componentes obsoletos de la misma célula (1). ESTRUCTURA Los lisosomas son compartimentos encerrados por una única membrana, estos contienen enzimas hidrolíticas que se utilizan para la digestión intracelular controlada de macromoléculas: proteínas, ácidos nucleicos, carbohidratos y lípidos (2, 1). El transporte de proteínas a través de esta membrana permite que los productos finales de la digestión de las macromoléculas, tales como aminoácidos, azúcares y nucleótidos, sean transportados hacia el citosol desde donde pueden ser excretados o reutilizados por la célula (2, 3). Este transporte hacia fuera del lisosoma se realiza mediante difusión pasiva y transportadores específicos (3, 4, 5). Esta organela fue descubierta a mediados de los años 50 por Christian de Duve quién la reconoció bioquímicamente en hígados de rata como una estructura vacuolar que contiene varias enzimas hidrolíticas que funcionan óptimamente a un pH ácido (6, 7). Actualmente se reconoce un sistema lisosomal/vacuolar en el que se incluyen estructuras carentes de hidrolasas como los endosomas tempranos (8). En su forma más simple los lisosomas son visualizados como vacuolas esféricas densas, pero pueden mostrar una variación considerable en tamaño y forma como un resultado de las diferencias en los materiales tomados para la digestión (1). Esto contrasta con las estructuras relativamente uniformes de la mayoría de las organelas (2). De esta manera, los lisosomas representan morfológicamente diversas organelas definidas por la función común de degradar material celular (1). Los lisosomas están presentes en muchas células eucariotas y contienen alrededor de 50 tipos de enzimas hidrolíticas incluyendo proteasas, nucleasas, glicosidasas, lipasas, fosfolipasas, fosfatasas y sulfatasas (1, 2). Para su actividad óptima las enzimas requieren un ambiente ácido y el lisosoma lo provee por medio de una bomba de protones que mantiene un pH interior de alrededor de 5.0 (Figura 7.1). La membrana del lisosoma mantiene las enzimas digestivas fuera del citosol; adicionalmente el citosol de la célula posee un pH neutro aproximadamente de 7.2, por lo cual es un ambiente protector contra las enzimas del lisosoma; estos dos fenómenos evitan que el resto de la célula sea atacado por el sistema digestivo propio (2). Figura 7.1 Organización del lisosoma La bomba de protones ubicada en la membrana lisosomal utiliza la energía proveniente de la hidrólisis del ATP para bombear iones H+ al interior del lisosoma, manteniendo de esta manera el lumen con un pH ácido. La mayoría de proteínas de la membrana lisosomal están inusualmente altamente glicosiladas, lo cual les ayuda a protegerlas de las proteasas lisosomales del lumen (2). Mutaciones en los genes que codifican las enzimas del lisosoma son responsables de alrededor de 50 enfermedades genéticas diferentes causadas por la acumulación de material no degradado dentro de los lisosomas, debido a que se afectan enzimas involucradas directa o indirectamente en la degradación de varios compuestos (1, 9). La disfunción lisosomal resultante conduce a una patología celular y luego cambios en la estructura y función de tejidos y órganos. En muchos casos, estas enfermedades son fatales con un promedio de vida corto (a veces tan poco como algunos meses) (9). ENDOCITOSIS Y FORMACIÓN DE LOS LISOSOMAS Una de las principales funciones de los lisosomas es la digestión de material tomado desde el exterior celular por medio de endocitosis. Particularmente los lisosomas son formados por la fusión de vesículas de transporte originadas desde la red trans-Golgi con endosomas formados por moléculas tomadas por endocitosis en la membrana plasmática. Es así como la formación de los lisosomas representa una intersección entre la vía secretora en la cual las proteínas lisosomales son procesadas, y la vía endocítica en la que moléculas extracelulares son tomadas de la superficie celular (Figura 7.2). El material proveniente desde el exterior es tomado en vesículas endocíticas que surgen de la membrana plasmática y luego se fusionan con endosomas tempranos. Los componentes de la membrana plasmática son luego reciclados y los endosomas tempranos gradualmente maduran a endosomas tardíos (1). En momento su interior es medianamente ácido (pH ~ 6) y es aquí donde el material endocitado comienza su digestión. Los lisosomas maduros se forman a partir de los endosomas tardíos, acompañados por una disminución adicional en el pH interno. Se cree que los lisosomas se producen por un proceso gradual de maduración, durante el cual las proteínas de la membrana endosomal son selectivamente recuperadas del lisosoma en desarrollo por vesículas de transporte que las regresan a los endosomas o a la red trans-Golgi (2). En la endocitosis mediada por receptores, moléculas específicas se combinan con proteínas receptoras incluidas en la membrana plasmática. Los receptores se concentran en depresiones recubiertas por una capa de la proteína denominada clatrina, una vez que una molécula se une de manera específica a un receptor (ligando), la depresión forma una vesícula por endocitosis (10). Las células animales captan el colesterol sanguíneo mediante endocitosis mediada por receptor, el colesterol se transporta por la sangre unido a proteínas formando las lipoproteínas de baja densidad (LDL). Las moléculas de colesterol quedan rodeadas por una monocapa lipídica que contiene una única molécula proteica, uno de cuyos extremos sobresale de la esfera para unirse a los receptores de la membrana plasmática, los individuos con carencia congénita de estos receptores presentan hipercolesterolemia, que produce aterosclerosis prematura, lo que aumenta el riesgo de ataque cardiaco (11). Figura 7.2 Endocitosis y formación de los lisosomas Luego de que la vesícula entra al citoplasma, el recubrimiento de clatrina se disocia de ella y deja libre en el citoplasma una vesícula descubierta, llamada endosoma, que forma dos vesículas, una con los receptores y otra con la partícula de LDL. Los receptores regresan a la membrana plasmática, donde se les recicla, y la vesícula restante se fusiona con un lisosoma, de modo que su contenido se digiere y se libera en el citosol (10). TRANSPORTE DE HIDROLASAS A LOS LISOSOMAS Tanto las hidrolasas como las proteínas de membrana de los lisosomas son sintetizadas en el RE rugoso y transportadas a través del AG a la red trans-Golgi (2). Proteínas receptoras ubicadas en este sitio reconocen grupos manosa 6fosfato que son marcadores que dirige las hidrolasas ácidas a los lisosomas, por lo que las proteínas son empaquetadas en vesículas que se transportan y se fusionan con los endosomas tardíos. Las hidrolasas son así liberadas en el lumen del endosoma, mientras que las proteínas receptoras son eventualmente recicladas al Golgi. Los endosomas tardíos luego maduran a lisosomas a medida que adquieren un complemento completo de hidrolasas ácidas, las cuales digieren las moléculas originalmente tomadas por endocitosis (1). FAGOCITOSIS Y AUTOFAGIA Los lisosomas digieren material derivado de otras diferentes rutas, dos de ellas son fagocitosis y autofagia (Figura 7.3). En la fagocitosis, células especializadas, tales como macrófagos y neutrófilos, toman y degradan partículas grandes incluyendo bacterias, desechos celulares y células viejas que deben ser eliminadas del cuerpo. Estas partículas son tomadas en vacuolas fagocíticas (fagosomas) que luego se fusionan con lisosomas llevándose a cabo la digestión de sus contenidos. Los lisosomas también son responsables de la autofagia, un proceso en el que se eliminan las partes obsoletas de la célula (1, 2). Las proteínas de vida corta son conjugadas con ubiquintina y degradadas por el proteosoma; sin embargo, más del 90% de las proteínas celulares son de larga vida (12, 13). Estas proteínas, otras macromoléculas, membranas biológicas, retículo endoplasmático, ribosomas y otras organelas como mitocondrias, y peroxisomas son degradadas por autofagia (14). De esta manera, tanto el lisosoma como el proteosoma trabajan en conjunto en el desdoblamiento de las proteínas incorrectamente plegadas ilustrando la interdependencia de las funciones de diferentes compartimentos subcelulares (15). Figura 7.3 Rutas de degradación en lisosomas El primer paso de la autofagia parece ser el secuestro de una organela o parte celular en una membrana derivada del RE. La vesícula resultante (autofagosoma) se fusiona luego con un lisosoma, y sus contenidos son digeridos (1). Este proceso es altamente regulado y los componentes celulares seleccionados pueden de alguna manera ser marcados para su destrucción lisosomal (2). ALGUNOS LISOSOMAS PUEDEN PRODUCIR EXOCITOSIS El direccionamiento de material a los lisosomas no es necesariamente el final de la ruta. La secreción lisosomal del contenido no digerido posibilita a las células eliminar los desechos no digeribles. Para la mayoría de las células, esto parece ser una ruta de poca importancia, utilizada sólo para las células que están bajo estrés. Algunos tipos de células, sin embargo, contienen lisosomas especializados que han adquirido la maquinaria necesaria para la fusión con la membrana plasmática. Los melanocitos en la piel, por ejemplo, producen y almacenan pigmentos en sus lisosomas que son liberados al espacio extracelular por exocitosis. El pigmento es tomado luego por los queratinocitos, conduciendo la pigmentación normal de la piel (2). BIBLIOGRAFIA 1. Cooper GM, Hausman RE. La célula. 4a ed. Madrid: Marbán; 2008. 2. Alberts B, Bray D, Hopkin K, Jonson A, Lewis J, Raff M, Roberts K, Walter P. Introducción a la biología celular. 2a ed. España: Panamericana; 2006. 3. Winchester B. Lysosomal membrane proteins. Eur J Paed Neurol. 2001; 5(suppl A): 11–19. 4. Winchester B. Lysosomal metabolism of glycoproteins. Glycobiology. 2005; 15(6): 1R–15R. 5. Lloyd JB. Metabolite efflux and influx across the lysosmal membrane. Subcell Biochem. 1996; 27: 361–386. 6. de Duve C, Gianetto R, Appelmans F, Wattiaux R. Enzymic content of the mitochondria fraction. Nature (London). 1953; 172: 1143-1144. 7. Gianetto R, de Duve C. Tissue fractionation studies 4. Comparative study of the binding of acid phosphatase, b-glucoronidase and cathepsin by rat liver particles. Biochem J. 1955; 59: 433-438. 8. Ciechanover A. Intracellular Protein Degradation: From a Vague Idea thru the Lysosome and the Ubiquitin-Proteasome System and onto Human Diseases and Drug Targeting. Cell Death Differ. 2005; 12: 1178–1190. 9. Jakóbkiewicz-Banecka J, Wêgrzyn A, Wêgrzyn G. Substrate deprivation therapy: a new hope for patients suffering from neuronopathic forms of inherited lysosomal storage diseases. J Appl Genet. 2007; 48(4): 383–388. 10. Solomon EP, Berg LR, Martin DW. Biología. 8a ed. México: McGraw-Hill; 2008. 11. Paniagua R, Nistral M, Sesma P, Álvarez-Uría M, Fraile B, Anadón R, Sáez FJ. Biología Celular. 3a ed. España: McGraw-Hill; 2007. 12. Myung J, Kim KB, Crews CM. The ubiquitin-proteasome pathway and proteasome inhibitors. Med Res Rev. 2001; 21: 245-273. 13. Hilt W, Wolf DH. A field guide to ubiquitylation. Cell Mol Life Sci. 2004; 61: 1546–1561. 14. Cuervo AM. Autophagy: many paths to the same end. Mol Cell Bioche. 2004; 263: 55-72. 15. Suzuki T, Lennarz WJ. Hypothesis: a glycoprotein-degradation complex formed by protein–protein interaction involves cytoplasmic peptide: N-glycanase. Biochem Biophys Res Commun. 2003; 302: 1–5.