Contrato de estudio post-autorización de tipo observacional prospectivo con medicamentos en las organizaciones del servicio vasco de salud
Anuncio

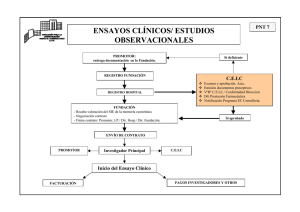
CONTRATO DE ESTUDIO POSTAUTORIZACIÓN DE TIPO OBSERVACIONAL PROSPECTIVO En a ................de..................... de 200.. REUNIDOS (Promotor) D/Dña. ......................................................................en ..............................................................................., en su nombre y calidad de representación de..............................................................con C.I.F. nº..................................., y con domicilio social en............................................................................. (Fundación Vasca de Innovación e Investigación Sanitarias/BIO Eusko Fundazioa), Dña Carmen Garaizar en su calidad de Directora Organización con domicilio y en representación de esa y C. l. F. nº (Centro) D. en su calidad de Director Gerente del centro sanitario representación de esa Organización con domicilio en la calle C. l. F. nº (Investigador con C. y en de y . Principal) D.................................... ..........................del centro sanitario con D.N.I...........................adscrito al Servicio en calidad de Investigador Principal y actuando en su propio nombre. CONSIDERANDO Lo dispuesto en la legislación española vigente en materia de estudios post-autorización de tipo observacional con medicamentos y productos sanitarios y acatando las normas éticas aplicables a la realización de estos estudios. MANIFlESTAN Las partes se reconocen respectivamente la capacidad necesaria y suficiente para obligarse por el presente contrato. El presente contrato tiene por OBJETO la realización en el Centro sanitario ………………………………del ESTUDIO POST-AUTORIZACIÓN DE TIPO OBSERVACIONAL titulado ....................................................................................................................................................... 1 ..............................................................................................................código...............promovido por……................................................................ y que será dirigido por el Dr. ....................................................... (denominado lnvestigador Principal) del Servicio............................ del centro sanitario …………………….. (denominado Centro o Centro de investigación), de acuerdo con el Protocolo de Estudio Post-autorización de tipo observacional código ....................(denominado Protocolo). I. Que para ello, el PROMOTOR ha seleccionado a los investigadores más adecuados según su calificación y medios disponibles para realizar, dirigir y supervisar el estudio en las instalaciones de los CENTROS, de acuerdo con el contrato y el Protocolo que se acompañan como Anexo al presente contrato. II. Que dicho estudio tiene por objeto determinar la efectividad, seguridad, obtener información sobre los patrones de utilización del medicamento……............. o conocer su efecto desde la perspectiva del paciente; todo ello de acuerdo con el Protocolo código................... que se acompaña como Anexo, y que describe detalladamente los procedimientos y alcance del estudio observacional a realizar. III. Que el estudio se realizará tras la obtención del informe favorable del Comité Ético de Investigación Clínica de Euskadi, la autorización de de la Dirección de Farmacia de Gobierno Vasco y la conformidad de la Dirección del centro………………………. Que en base a los anteriores principios y objetivos, las partes acuerdan celebrar el presente contrato bajo las siguientes: ESTIPULACIONES PRIMERA.- Objeto Por el presente contrato el CENTRO autoriza la realización en sus instalaciones del Estudio PostAutorización de tipo observacional prospectivo al que se refieren los anexos I (Protocolo) y II (memoria económica), que será realizado, dirigido y, supervisado personalmente con el INVESTIGADOR a quien se confiere expresamente la labor de investigación. Por otra parte, el Estudio se realiza en un mínimo de ....... sujetos, de conformidad a lo dispuesto en el protocolo, a los que previamente el investigador habrá informado de los riesgos y alcance del estudio y habrá obtenido su consentimiento informado por escrito. Cuando se estime necesario modificar el número de sujetos, se precisará la aprobación previa y por escrito de las partes. SEGUNDA.- Condiciones de realización 2.1. - Protocolo.Las condiciones de realización del estudio serán las establecidas en la legislación vigente y en el presente contrato con su protocolo anexo. Las partes cumplirán con lo estipulado en el Protocolo, incluidas las enmiendas o modificaciones que puedan introducir en él en cada momento siempre que hayan sido firmadas y aceptadas por el INVESTIGADOR y el PROMOTOR, los cuales conservarán en sus archivos copias de las enmiendas y modificaciones que vayan introduciéndose en el Protocolo. 2.2.- Periodo de vigencia y duración.El estudio se iniciará en el centro con la inclusión del primer paciente y tendrá una duración estimada de …………….meses a partir de la fecha de inclusión del primer paciente, según lo estipulado en el protocolo del estudio. En el supuesto de que o bien el inicio o la duración del estudio sean modificados, deberá ser comunicado por el PROMOTOR al CENTRO. 2.3. - Modificación.- 2 El Protocolo no podrá ser modificado unilateralmente por el INVESTIGADOR sino que requerirá el consentimiento y la aprobación previa del PROMOTOR. Cualquier modificación en las condiciones autorizadas para un estudio que se considere relevante no podrá llevarse a efecto sin el previo dictamen favorable del Comité Ético de Investigación Clínica de Euskadi y la autorización de la Dirección de Farmacia. La modificación deberá contar con el visto bueno del Investigador Principal del estudio. Las modificaciones o enmiendas del protocolo deberán ser comunicadas al CENTRO, pudiendo éste, si las considera como una modificación o enmienda relevante, rescindir el contrato o, de mutuo acuerdo con el promotor, proceder a las modificaciones oportunas del contrato y/o anexos. 2.4. – Normas ético-legales.- Todas las partes se comprometen a cumplir la legislación española vigente en materia de estudios observacionales con medicamentos: Ley 25/1990, de 20 de diciembre, del Medicamento, Real Decreto 711/2002 de 19 de Julio, Decreto 102/2005, por el que se regula la realización de estudios post-autorización de tipo observacional con medicamentos, Convenio de 4 de Abril de 1.997, para la Protección de los Derechos Humanos y la Dignidad del ser humano con respecto a las obligaciones de la Biología y la medicina, ratificado por instrumento de 23 de Julio de 1999- fecha de entrada en vigor en España el día 1 de Enero de 2.000, y demás normas concordantes por las que se establecen los requisitos para la realización de estudios observacionales con medicamentos. - Se acuerda su realización conforme a las Disposiciones de la Declaración de Helsinki, en su última versión. - En particular, el CENTRO cuidará de que en la realización del estudio se respeten íntegramente los derechos fundamentales de la persona, de acuerdo con las normas esenciales de la Bioética, normas sanitarias, éticas y de buena práctica aplicables al estudio, sin sustituir las funciones encomendadas a PROMOTOR, INVESTIGADOR y COMITÉ ÉTICO DE INVESTIGACIÓN CLÍNICA. 2.5. - Consentimiento informado del paciente.De conformidad con lo previsto en la Ley 41/2002, de 14 de noviembre, reguladora de la autonomía de los pacientes, en los estudios post-autorización de tipo observacional prospectivos es imprescindible que el sujeto otorgue libre y voluntariamente su consentimiento informado antes de ser incluido en el estudio. Este consentimiento debe obtenerse preferentemente por escrito o, en situaciones muy excepcionales, de forma oral ante testigos independientes del equipo investigador que lo declararán por escrito bajo su responsabilidad. Si en el estudio se va a recoger información de sujetos menores de edad o incapaces, el consentimiento lo otorgará siempre por escrito su representante legal, tras haber recibido y comprendido la información mencionada. Cuando las condiciones del sujeto lo permitan y, en todo caso, cuando el menor tenga doce o más años, deberá prestar además su consentimiento para participar en el estudio, después de haberle dado toda la información pertinente adaptada a su nivel de entendimiento. En la historia clínica del paciente se archivará una copia del consentimiento informado. 2.6. - Acceso.El CENTRO tendrá acceso en cualquier momento a la documentación relativa al Estudio, especialmente al consentimiento informado de los pacientes que participen en el mismo. El Monitor del estudio también tendrá acceso a documentación clínica pertinente de los pacientes incluidos en el estudio, en cada visita que realice. En todo caso deberá respetar la confidencialidad de los datos de conformidad con la legislación vigente. 3 El promotor del estudio post-autorización de tipo observacional con medicamentos, directamente o a través del monitor, deberá realizar el seguimiento directo de la realización del estudio. Igualmente las Autoridades Sanitarias competentes podrán tener acceso a la documentación clínica del paciente, al realizar las inspecciones correspondientes. 2.7. - Publicación de resultados.El promotor se compromete a la publicación de los resultados del presente estudio. El Investigador Principal podrá presentar los resultados en una reunión científica apropiada y/o publicarlos en una revista de reconocido prestigio, comprometiéndose a suministrar al Promotor una copia del manuscrito u original, con la suficiente antelación, a efectos de que éste tenga oportunidad de conocer dicha información o material informativo para la realización de sus comentarios sobre el contenido de tales comunicaciones/publicaciones en un plazo de 30 días a contar desde la recepción de los mismos. Si el Promotor así lo solicita, con el fin de asegurar apropiadamente la protección de invenciones o desarrollos derivados del estudio, el Investigador Principal acepta retrasar la presentación de la publicación propuesta, durante un plazo no superior a 6 meses. 2.8.- Confidencialidad y Protección de datosTodas las informaciones relativas a la realización del estudio observacional prospectivo, sean anteriores o posteriores al mismo, suministradas u obtenidas, son confidenciales. Las partes de este contrato y el personal colaborador deberán tomar las medidas oportunas para guardar la confidencialidad de los datos de carácter personal de los que tuvieran conocimiento como consecuencia de la realización del estudio observacional prospectivo, impidiendo el acceso a los mismos a terceros no autorizados, en el marco de lo dispuesto en la Ley Orgánica 15/99 de 13 de diciembre, de protección de Datos de Carácter Personal, y la Ley 41/2002, de 14 de noviembre, básica reguladora de la autonomía del paciente y de derechos y obligaciones en materia de información y documentación clínica. El CENTRO, junto al INVESTIGADOR PRINCIPAL, restringirá el acceso a la información a aquellos supuestos necesarios para la correcta ejecución del protocolo. Los informes y memorias que se elaboren por el centro y el investigador principal con destino al promotor no contendrán datos personales de pacientes, procediéndose con carácter previo a someter estos datos a un proceso de disociación de forma que los mismos no puedan asociarse a persona identificada o identificable. Siempre y cuando se respeten los postulados del artículo 2.7, el CENTRO no estará facultado para desvelar o difundir por cualquier medio los resultados, datos e informaciones que resulten directa o indirectamente de la realización del estudio ni siquiera con fines científicos, salvo autorización escrita del PROMOTOR. 2.9.- Archivo de la documentación.- Las Historias Clínicas de los pacientes y demás datos originales se conservaran durante el periodo mínimo de QUINCE AÑOS, conforme lo establecido en el Decreto 45/1998 (B.O.P.V. nº 67 de fecha 8 de abril de 1998), modificado por Decreto 102/2005 de 26 de abril, por el que se regula la realización de estudios post-autorización de tipo observacional con medicamentos. Las historias clínicas de los pacientes dispondrán de un sistema ágil y rápido para identificar que un paciente participa o ha participado en un estudio post-autorización de tipo observacional. TERCERA.- Participantes y lugar de realización 3.1.- Participantes 3.1.1.- Promotor.3.1.2.- Investigador Principal.- El INVESTIGADOR PRINCIPAL cuidará y garantizará que todos los participantes en el estudio post-autorización de tipo observacional y, especialmente, los 4 colaboradores cumplen fielmente con este contrato y sus anexos, habiendo sido informados suficientemente sobre el mismo. Asimismo se compromete a incluir un mínimo de ……pacientes en el estudio. 3.1.3. - Equipo colaborador.- El equipo de colaboradores del INVESTIGADOR deberá ser suficiente para cumplir con éxito el estudio post-autorización previsto. Dicho equipo estará formado por: • D............................................................................................................................... • D............................................................................................................................... • D............................................................................................................................... La retribución de colaboradores y demás obligaciones legales accesorias serán realizadas por (PROMOTOR/INVESTIGADOR) 3.1.4.Monitor.El Promotor, designa como monitor del Estudio a D................................................... de la empresa …………………………………………… En caso de sustitución del mismo, el Promotor informará de la identidad del nuevo monitor designado. 3.1.5.- Colaborador del monitor.- 3.2.- Lugar de realización El estudio objeto de este contrato se realizará en el Servicio/s de ………………. del centro sanitario ……………………………………………………………… ……………... CUARTA.- Relaciones económicas PRESUPUESTO.Según memoria económica que se adjunta como anexo II al presente contrato: El presupuesto inicial del estudio, deberá comprender todas las remuneraciones del mismo, es decir, los pagos al CENTRO y a BIO (gestión del estudio y costes indirectos) y al equipo investigador, e irá desglosado en los siguientes apartados: • Pago a la Fundación Vasca de Innovación e Investigación Sanitarias/BIO Eusko Fundazioa en concepto de evaluación y gestión del estudio • Costes indirectos • Costes de ejecución (por paciente): Compensación a investigadores 1. Pago a la Fundación Vasca de Innovación e Investigación Sanitarias/BIO Eusko Fundazioa en concepto de EVALUACIÓN Y GESTIÓN DELESTUDIO Por evaluación y gestión, se establece un módulo de 1.350€ + 216€ (IVA) = 1566€, a abonar por la realización del estudio en centros hospitalarios y otro módulo de idéntica cuantía cuando se lleve a cabo, en su caso, en centros extrahospitalarios Este pago se realizara contra la presentación de la factura correspondiente antes de la evaluación del estudio por el Comité Ético de Investigación Clínica de Euskadi en la siguiente dirección y cuenta corriente: ………………………………… ………………………………… ……………………………….. 5 2. En concepto de colaboración general y cesión de locales para la realización del estudio se abonará la cantidad de .......... €, correspondiente al 18 % del presupuesto inicial del estudio. Dicho importe se entenderá que cubre los costes indirectos, e irá desglosado en dos partes: • 1/3 se ingresará en el centro: ……………….€ por paciente • 2/3 partes se facturarán a la Fundación Vasca de Innovación e Investigación Sanitarias/BIO Eusko Fundazioa: ……………….€ por paciente Se emitirá tanto por parte del centro investigador como de la fundación, la factura correspondiente, a la que se le repercutirá el I.V.A Las cantidades ingresadas a la Fundación Vasca de Innovación e Investigación Sanitarias/BIO Eusko Fundazioa se destinarán a promover la investigación biomédica, a propuesta y de acuerdo con la comisión de investigación del centro Los plazos de pagos se establecerán de forma consensuada entre el Promotor del estudio, la Fundación BIO y el Centro. (SE ESTABLECERA AL DETALLE LOS PLAZOS DE LOS PAGOS SI ES POSIBLE) 3. Retribuciones DEL INVESTIGADOR: El abono de las compensaciones al INVESTIGADOR, así como las obligaciones legales adicionales (retenciones por I.R.P.F. ) corresponderán directamente al PROMOTOR. El promotor……………..se compromete a facilitar a la dirección económica del centro el estudio código ………………………. y ……………..una vez finalizado titulado”…………………………………………………………………………………..……” una copia de la liquidación de gastos correspondientes al citado estudio. La Entidad Promotora hace constar que no se han establecido ni se establecerán acuerdos ajenos al presente contrato por la realización de este estudio con el Investigador Principal, sus colaboradores ni con ninguna institución implicada directa o indirectamente con la realización de este estudio, de los que deriven retribuciones económicas adicionales o contraprestaciones en especie. En caso de terminación prematura del estudio, los costes indirectos y de ejecución deberían ser proporcionales al número de pacientes incluidos en el mismo y al número de visitas realizadas QUINTA.- Obligaciones del promotor: Comunicación del Inicio del estudio:. (se considerará fecha de inicio la fecha de reclutamiento del primer paciente). El promotor del estudio deberá comunicar al Comité Ético de Investigación Clínica de Euskadi la fecha de inicio del estudio. En el caso de estudios que vayan a ser realizados en centros sanitarios que tengan un CEIC acreditado, el promotor deberá comunicar también la fecha de inicio del estudio a este comité. (párrafo a aplicar en centros con CEIC local). Información sobre el seguimiento y finalización de los estudios post-autorización con medicamentos. 6 Anualmente el promotor del estudio deberá informar al Comité Ético de Investigación Clínica de la Comunidad Autónoma de Euskadi acerca del desarrollo y posibles incidencias que tengan lugar durante la realización del mismo. El promotor deberá informar al Comité Ético de Investigación Clínica de Euskadi de la finalización del estudio en el plazo de 90 días. El plazo se reducirá a 15 días en el caso de terminación anticipada. En el caso de estudios que vayan a ser realizados en centros sanitarios que tengan un CEIC acreditado, el promotor deberá remitir también el informe de seguimiento y la comunicación de la finalización a este comité. Comunicación de Reacciones adversas. El promotor del estudio post-autorización de tipo observacional con medicamentos deberá notificar a la Unidad de Farmacovigilancia de la Comunidad Autónoma del País Vasco las sospechas de reacciones adversas de acuerdo con lo establecido en el Real Decreto 711/2002 y en el Decreto 239/2002. SEXTA.- Obligaciones del Investigador Principal: - - Supervisar todos los aspectos médicos del Estudio. Velar porque las actividades relacionadas con el estudio se ejecuten de acuerdo con las directrices establecidas en el protocolo, con las establecidas por el Comité Ético de Investigación Clínica de Euskadi, con las estipulaciones del presente contrato y demás legislación vigente aplicable a la realización de estudios clínicos sobre personas. Proponer a un sustituto en la dirección de este Estudio, si por excedencia, traslado u otra causa similar no pudiese seguir como Investigador Principal. Notificar al Promotor, las sospechas de reacciones adversas de acuerdo con lo establecido en el Real Decreto 711/2002 y en el Decreto 239/2002. Asegurarse de que todos los sujetos incluidos en el estudio han otorgado el consentimiento informado Realizar el presente estudio de acuerdo a la práctica habitual. SÉPTIMA.- Archivo de Documentación del Estudio Observacional. 7.1. - El promotor del estudio es responsable del archivo de la documentación del estudio. 7.2. - El investigador principal se ocupará de que los códigos de identificación de los sujetos se conserven durante al menos quince años después de concluido o interrumpido el estudio. 7.3. - Las historias clínicas de los pacientes y demás datos originales se conservarán de acuerdo a la legislación vigente en la Comunidad Autónoma Vasca (decreto 272/1986 de 25 de Noviembre por el que se regula el uso de la Historia Clínica de los centros hospitalarios de la Comunidad Autónoma del País Vasco; y decreto 45/1998, de 17 de Marzo, por el que se establece el contenido y se regula la valoración, conservación y expurgo de los documentos del Registro de Actividades Clínicas de los Servicios de Urgencias de los hospitales y de las historias clínicas hospitalarias publicados en el BOPV) 7.4. – El promotor o propietario de los datos conservará toda la restante documentación relativa al estudio durante el período de validez del medicamento. Estos documentos incluirán: a/ El protocolo, incluyendo su justificación, objetivos, diseño estadístico y metodología del estudio, con las condiciones en las que se efectúe y gestione, así como los pormenores de los medicamentos objeto de estudio.. b/ Los procedimientos normalizados de trabajo. c/ Todos los informes escritos sobre el protocolo y los procedimientos. 7 d/ LA FICHA TECNICA del medicamento a estudio e/ El cuaderno de recogida de datos de cada paciente. f/ Los documentos administrativos correspondientes a las autorizaciones del protocolo y posteriores modificaciones. g/ El informe final: El promotor o propietario subsiguiente conservará el informe final hasta cinco años después de haberse agotado el plazo de validez del medicamento. h/ El certificado de auditoria, cuando proceda. 7.5. - La recogida de datos se realizará de acuerdo a las normas de Buena Práctica Clínica. 7.6. - Se documentará todo cambio que se produzca en la posesión de los datos. 7.7. - Todos los datos y documentos se pondrán a disposición de las autoridades competentes si éstas así lo solicitan. 7.8. - Se asegurará, en todo caso, la confidencialidad de los datos y documentos contenidos en el archivo. OCTAVA.- Informes y propiedad de los resultados - El promotor del estudio post-autorización de tipo observacional con medicamentos deberá elaborar el informe final, que contará con la aprobación del investigador responsable de la coordinación del estudio en la Comunidad Autónoma del País Vasco, y deberá remitir una copia del mismo en el plazo de seis meses desde la finalización del mismo al Comité Ético de Investigación Clínica de Euskadi. El informe será enviado independientemente de la finalización anticipada del estudio. - En el caso de estudios post-autorización de tipo observacional prospectivos con medicamentos que vayan a ser realizados en centros sanitarios que tengan un CEIC acreditado, el promotor deberá remitir también el informe final o el anual cuando proceda a este comité. - Propiedad de los resultados.- Las partes acuerdan que todos los derechos, datos, resultados y descubrimientos o inventos, patentables o no, realizados, obtenidos o generados en relación con el Estudio serán propiedad exclusiva del PROMOTOR. NOVENA - Seguros y responsabilidades Al ser un estudio observacional el investigador deberá limitarse a observar la realidad sin modificarla, sin introducir activamente la intervención farmacológica, y sin realizar visitas o pruebas extraordinarias. En ningún caso podrán utilizarse los medicamentos cuya observación se realiza en este estudio para indicaciones no autorizadas o en condiciones de uso diferentes a las establecidas en la ficha técnica de los mismos, garantizando el promotor y el investigador que así se cumplirá. DÉCIMA.- Representación de las partes El CENTRO no ostenta representación alguna del PROMOTOR frente a terceros. 8 UNDÉCIMA.- Facultad de inspección y supervisión El CENTRO y el INVESTIGADOR PRINCIPAL posibilitarán a las autoridades sanitarias y al auditor externo designado por el PROMOTOR, auditar o inspeccionar sus Registros del estudio cuando se solicite. DUODÉCIMA.- Regulación 12.1. - Contractual.- Ambas partes convienen que sus relaciones se regulan por el contenido del presente documento, sin perjuicio de la regulación contenida en el Protocolo y demás documentos concordantes que se firmen en relación con este documento, siendo nulo y quedando sin efecto, cualquier acuerdo anterior, expreso o tácito, documentado o no. El presente contrato solo se entenderá modificado o enmendado por acuerdo escrito de las partes. 12.2. - Legislativa.- El presente contrato se somete a las leyes y normas españolas. Para solventar cualquier discrepancia que pudiera surgir en la aplicación o interpretación de lo establecido en el presente contrato, ambas partes se someten, con renuncia expresa al fuero que pudiera corresponderles, a la Jurisdicción de los Juzgados y Tribunales de Vitoria-Gasteiz. DECIMOTERCERA.- Causas de terminación 13.1.- Ordinaria.- El contrato finalizará con el cumplimiento del protocolo de estudio. 13.2.- Resolución.- Este contrato podrá ser resuelto por cualquiera de las Partes con efecto inmediato mediante notificación por escrito, a no ser que la parte incumplidora subsane sus actos en el plazo de los 30 días siguientes recibir la notificación, si se da una de las circunstancias siguientes: a) El INVESTIGADOR finalizara o suspendiera su relación profesional con el CENTRO. b) El incumplimiento de las cláusulas del contrato o de la normativa legal aplicable c) Imposibilidad de reclutar el número de pacientes previstos. d) Suspensión o finalización anticipada del estudio por cualquier circunstancia debidamente justificada. La resolución o modificación del contrato entre el INVESTIGADOR y el PROMOTOR deberá ser notificada al CENTRO a los efectos de la liquidación del presente contrato. 13.3.- En el supuesto de suspensión o finalización anticipada del estudio, la resolución del contrato tendrá efectos inmediatos; en los restantes casos la resolución deberá comunicarse con un mes de antelación. En todos los casos de resolución del contrato, el centro deberá entregar al promotor toda la información y documentación que haya acordado en este contrato, y la información correspondiente a todos los trabajos realizados hasta la fecha efectiva de la resolución. El promotor sólo vendrá obligado a satisfacer las cantidades debidas por los trabajos realizados hasta la fecha de resolución. En el caso que el saldo resulte negativo, el Centro deberá devolver el importe adelantado por el promotor”. En señal de conformidad y después de leído el presente contrato, todas las partes lo firman por cuadriplicado en el lugar y fecha indicados en el encabezamiento. 9 POR EL CENTRO POR EL PROMOTOR Fdo.: Dr. DIRECTOR GERENTE Fdo.:............................... POR EL INVESTIGADOR POR FUNDACIÓN VASCA DE INNOVACIÓN E INVESTIGACIÓN SANITARIAS/BIO EUSKO FUNDAZIOA Fdo.: ..................................... Fdo.: ..................................... 10 ANEXOS Se acompaña: Anexo I.- Protocolo. Anexo II.- Memoria económica: Se incluye el precio por paciente incluido y los plazos de pago. 11